· the insulin and insulin-like growth factor 1 (igf-1) pathways. last data suggest that klotho...
TRANSCRIPT




Spis treści
Agnieszka Szymczyk, Ewa Forma ................................................................. 151
Struktura i funkcje białka Klotho
Structure and functions of Klotho protein
Agnieszka Szymczyk, Ewa Forma, Anna Krześlak, Paweł Jóźwiak,
Adam Madej, Waldemar Różański, Magdalena Bryś .................................... 189
Genetic polymorphism of Klotho gene and bladder cancer risk
Polimorfizm genu Klotho a ryzyko raka pęcherza moczowego
Ewa Forma, Magdalena Bryś ....................................................................... 207
Rola metalotionein i kadmu w rozwoju raka piersi Role of metalothioneins and cadmium in breast carcinogenesis
Paweł Jóźwiak ................................................................................................ 245
Rola transporterów GLUT1 i GLUT3 w pobieraniu glukozy
i kwasu dehydroaskorbinowego przez komórki nowotworowe
The role of transporters GLUT1 and GLUT3 in glucose and dehydroascorbic acid
uptake in cancer cells
Piotr K. Zakrzewski ....................................................................................... 265
Zaburzenia kaskady transformujących czynników wzrostu typu β
w wybranych patologiach człowieka
Aberrations in the signalling cascade of transforming growth factor β type
in selected human pathologies
Magdalena Bryś, Magdalena Laskowska, Ewa Forma, Anna Krześlak ....... 293
Telomeraza – struktura i funkcja oraz regulacja ekspresji genu
Telomerase – structure, function and the regulation of gene expression
Od Redakcji .................................................................................................... 330
Lista Recenzentów w latach 2011-2012
List of Reviewers in 2011-2012
Informacja dla autorów ................................................................................. 331


Folia Medica Lodziensia, 2012, 39/2:151-187
Adres do korespondencji: dr Ewa Forma, Katedra Cytobiochemii, Uniwersytet Łódzki;
90-236 Łódź, ul. Pomorska 141/143; e-mail: [email protected];
Tel.:+ 48 42 635 4371, Fax: +48 42 6354484
Struktura i funkcje białka Klotho
Structure and functions of Klotho protein
AGNIESZKA SZYMCZYK, EWA FORMA
Katedra Cytobiochemii, Wydział Biologii i Ochrony Środowiska
Uniwersytet Łódzki
Streszczenie Gen Klotho, odkryty został w roku 1997, a jego nazwa wywodzi się od imienia
greckiej bogini Klotho, która przędła nić ludzkiego żywota. Myszy z inakty-
wowanym genem Klotho wykazują cechy przedwczesnego starzenia się,
natomiast nadekspresja Klotho skutkuje wydłużeniem czasu ich życia. Białko
Klotho występuje w dwóch formach - transbłonowej oraz sekrecyjnej,
którym przypisuje się odmienne funkcje. Najwyższą ekspresję transbłonowej
formy Klotho obserwuje się w nerkach i splotach naczyniówkowych
komór mózgowych. Forma ta, funkcjonuje jako koreceptor dla czynnika
wzrostu fibroblastów 23 (FGF23), który uczestniczy w utrzymywaniu
homeostazy fosforanowej oraz regulacji metabolizmu witaminy D. Sekrecyjna
postać białka, której obecność wykazano w osoczu, płynie mózgowo-
rdzeniowym oraz w moczu, funkcjonuje jako czynnik humoralny. Reguluje ona
aktywność kanałów jonowych, transporterów błonowych, a także receptorów
dla czynników wzrostu. Poprzez modyfikację N-glikanów kanałów TRPV5,
Klotho sekrecyjne bierze udział w utrzymywaniu homeostazy jonów wapnia.
Ponadto, sekrecyjna postać białka uczestniczy w hamowaniu szlaku
insuliny/insulinopodobnego czynnika wzrostu. Ostatnie doniesienia sugerują,
iż Klotho spełnia także rolę supresora procesu nowotworzenia.
Obniżenie ekspresji genu Klotho wykazano m.in. w raku piersi, trzustki, żołądka,
jelita grubego, płuc oraz w raku szyjki macicy. Spadek ekspresji genu Klotho
skorelowany jest z bardziej agresywnym fenotypem badanych nowotworów.
Wśród mechanizmów leżących u podstaw obniżenia ekspresji Klotho
wyróżnia się m.in. hipermetylację wysp CpG w obrębie regionu promotorowego
oraz deacetylację histonów.

Struktura i funkcje białka Klotho 152
Słowa kluczowe: białko Klotho, TRPV, wapń, fosfor, witamina D, czynniki wzrostu fibroblastów, nowotwory.
Abstract
Klotho gene was identified in 1997, and named after a Greek goddess Klotho, who spun the thread of life. The inactivation of Klotho gene in mice leads to a syndrome resembling aging, whereas the overexpression of Klotho extends their life span. Protein Klotho exists in two forms: membrane and secreted Klotho which play different functions. The highest expression of transmembrane form of Klotho is observed in the kidney and choroid plexus in the brain. The transmembran form of Klotho acts as a coreceptor for fibroblast growth factor 23 (FGF23) and regulates phosphate homeostasis band vitamin D metabolism. The secreted form of Klotho, which was detected in plasma, cerebrospinal fluid and urine functions as a humoral factor that regulates the activity of several ion channels, transporters, and growth factor receptors. Moreover, this form of Klotho protein can modify N-glycans of TRPV5 channel and regulate calcium homeostasis. The secreted Klotho can also inhibit the insulin and insulin-like growth factor 1 (IGF-1) pathways. Last data suggest that Klotho can act as a tumor supressor gene. The decrease of Klotho expression was observed in the breast, pancreas, stomach, colon, lung and cervical cancer. Moreover, the decrease of Klotho expression was correlated with the more aggressive phenotype of examined cancers. Downregulation of Klotho gene was associated with CpG hypermethylation of promoter region and histones deacetylation.
Key words: Klotho protein, TRPV, calcium, phosphorus, vitamin D, fibroblast growth factors, neoplasms. Wstęp
Gen Klotho (KL, określany również jako αKL) został po raz pierwszy
zidentyfikowany w 1997 roku u myszy transgenicznych, które otrzymano
w wyniku przypadkowej insercji transgenu do fragmentu 5’ regionu

A. Szymczyk, E. Forma 153
promotorowego genu KL, co doprowadziło do jego inaktywacji [1]. Początkowo
linia myszy KL–/– uważana była za bezużyteczną, gdyż wprowadzony do
genomu myszy transgen nie powodował zmian fenotypowych. Pogląd ten uległ
zmianie, gdy u myszy, które do 3-4 tygodnia życia rozwijały się prawidłowo,
niemal tak samo jak heterozygoty KL–/+ lub osobniki typu dzikiego, w kolejnych
tygodniach obserwowano zahamowanie wzrostu oraz występowanie szeregu
zmian charakterystycznych dla procesu starzenia, takich jak: miażdżyca tętnic,
zwapnienie naczyń i tkanek miękkich, rozedma płuc, dysplazja gonad,
bezpłodność, atrofia skóry, zaburzenia słuchu, przedwczesny zanik grasicy,
osteopenia, sarkopenia, hipoglikemia oraz hiperfosfatemia. Myszy Klotho
(KL–/–) umierały przedwcześnie około 8-9 tygodnia życia [1-3]. Z kolei
nadekspresja Klotho skorelowana jest ze zwiększeniem długości życia myszy.
W odniesieniu do myszy typu dzikiego długość życia samców z nadekspresją
Klotho ulega wydłużeniu o około 31%, zaś samic o około 19% [3, 4].
Struktura genu i białka Klotho
Nazwa genu i białka wywodzi się od greckiej bogini Klotho, która wraz
z siostrami Lachesis i Atropos przędła nić ludzkiego życia i decydowała o jego
długości [4, 6]. Gen KL człowieka obejmujący około 50 kpz zlokalizowany jest
na chromosomie 13q12 (u myszy na chromosomie 5) i składa się z 5 eksonów
[2, 7]. W regionie promotorowym genu KL nie stwierdzono obecności typowej
kasety TATA oraz kasety CAAT, natomiast w regionie tym znajduje się pięć
potencjalnych miejsc wiązania czynnika transkrypcyjnego Sp1, które często
występują w promotorach nie zawierających kasety TATA [2; 8]. Ponadto
w regionie obejmującym fragment sekwencji promotora oraz pierwszy ekson
genu KL (łącznie 1304 pz) stwierdzono obecność wyspy CpG [8]. Na podstawie

Struktura i funkcje białka Klotho 154
podobieństwa sekwencji zidentyfikowano kolejne 2 geny należące do rodziny
Klotho, tj. βKlotho oraz γKlotho, który określany jest jako KLPH
(ang. KL lactase phlorizin hydrolase) lub LCTL (ang. lactase-like protein) [9].
Analiza sekwencji cDNA KL wykazała istnienie dwóch transkryptów
powstających w wyniku alternatywnego składania mRNA. Pierwszy transkrypt
obejmujący otwartą ramkę odczytu (ORF, ang. open reading frame) o długości
3036 pz koduje białko transbłonowe składające się z 1012 reszt amino-
kwasowych. Drugi transkrypt KL różni się od pierwszego obecnością insercji
o długości 50 pz zawierającej kodon STOP, który sprawia, że ORF jest krótsza
(1647 pz), a powstające białko składa się z 549 reszt aminokwasowych. Jest
to sekrecyjna forma białka Klotho (sKL). Miejsce alternatywnego składania
znajduje się w obrębie 3 eksonu [2, 8, 10].
Transbłonowa forma białka Klotho człowieka składa się z 1012
aminokwasów i wykazuje 86% homologię w stosunku do białka myszy
(1014 aa) [2]. Masa cząsteczkowa białka Klotho wynosi 130 kDa i na jej
wartość ma wpływ N-glikozylacja. Traktowanie komórek CHOTM2
N-glikozydazą oraz tunikamycyną, będącą inhibitorem procesu glikozylacji,
pozwoliło na detekcję białka Klotho o masie cząsteczkowej 110 kDa, która
wydaje się być zgodna z masą cząsteczkową przewidywaną na podstawie
sekwencji aminokwasowej [11]. W strukturze białka KL wyróżnia się trzy
domeny: krótką, wewnątrzkomórkowa domenę C-końcową (11 aa), domenę
transbłonową (21 aa), która tylko raz przebija błonę komórkową oraz długą,
zewnątrzkomórkową domenę N-końcową (980 aa) [12]. W domenie
zewnątrzkomórkowej wyróżnia się dwa powtórzenia sekwencji składające się
z 440 aminokwasów każde, które określane są jako KL1 i KL2 (Ryc. 1).

A. Szymczyk, E. Forma 155
Ryc. 1. Struktura białka Klotho, w którym wyróżnia się N-końcową domenę
zewnątrzkomórkową z powtórzeniami KL1 i KL2,
domenę transmembranową (TM) oraz cytoplazmatyczną (CYT).
Pomiędzy powtórzeniami KL1 i KL2 znajduje się odcinek łącznikowy
(linkerowy), w obrębie którego znajduje się sekwencja Lys-Lys-Arg-Lys,
stanowiąca potencjalne miejsce cięcia proteolitycznego [2, 8, 12].
Powtórzenia KL1 i KL2 wykazują aktywność enzymatyczną i na
podstawie podobieństwa strukturalnego białko Klotho zostało zaliczone
do rodziny 1 glikozydaz. Zarówno KL1 jak i KL2 charakteryzują się 20-40%
podobieństwem do β-glukozydaz występujących u bakterii i roślin oraz do
hydrolazy florydzynowej ssaków [4, 12]. Kluczową rolę dla aktywności
β-glukozydaz pełnią dwie reszty kwasu glutaminowego. Pierwsza z nich
funkcjonuje jako nukleofil, druga zaś spełnia rolę kwasu/zasady. W przypadku
białka Klotho reszty te zostają jednak zastąpione resztami innych aminokwasów,
przy czym w obu powtórzeniach zmiana dotyczy tylko jednej reszty kwasu
glutaminowego. W przypadku KL1 reszta kwasu glutaminowego, pełniąca
funkcję nukleofila, zastąpiona jest asparaginą, natomiast w KL2 kwas
glutaminowy funkcjonujący jako kwas/zasada zastąpiony jest przez serynę.
Wykazano, że hydrolizie przy udziale białka Klotho podlegają m.in.
glukoronidy estradiolu, estronu i estriolu [12, 13]. Najwyższy poziom ekspresji
transbłonowego białka Klotho obserwuje się w kanalikach nerkowych i splocie
naczyniówkowym w mózgu. Ponadto ekspresję Klotho wykazano w przysadce,
uchu wewnętrznym, mózgu, przytarczycach, trzustce, jelicie grubym, mięśniach

Struktura i funkcje białka Klotho 156
szkieletowych, pęcherzu moczowym, jajnikach, jądrach oraz w komórkach
nabłonkowych piersi [4, 14, 15].
Forma sekrecyjna białka Klotho, to druga obok transbłonowej postać tego
białka [2, 8, 12, 16]. Powstaje ona w wyniku alternatywnego składania
i obejmuje 549 reszt aminokwasowych i dopowiada fragmentowi KL1 białka
błonowego [2, 8]. Wykazano, że sKL może powstawać z transbłonowego białka
Klotho w wyniku cięcia proteolitycznego, dzięki aktywności enzymów
należących do rodziny ADAM (ang. a disintegrin and metalloproteinase
domain-containing protein), tj. ADAM10 i ADAM17. Białka te dokonują cięcia
białka Klotho na dwa sposoby, które nazwane są cięciem α oraz β i generują
odpowiednio fragmenty o masie cząsteczkowej 130 i 68 kDa. Cięcie α zachodzi
tuż za domeną transbłonową białka Klotho a powstający w jego wyniku
fragment zawiera zarówno KL1 jak i KL2. Cięcie β zachodzi natomiast
w obrębie odcinka o łączącego fragment KL1 i KL2. Na skutek różnej
wydajności obu procesów fragment powstający w wyniku cięcia α stanowi
dominującą formę sKL. Obecność sekrecyjnego białka Klotho wykazano
w osoczu, płynie mózgowo-rdzeniowym oraz w moczu [17].
Funkcje sekrecyjnej formy białka Klotho
Udział białka Klotho w homeostazie jonów wapnia
Wapń pełni istotną rolę w skali całego organizmu. Funkcjonuje on
zarówno jako czynnik zewnątrzkomórkowy, jak i wewnątrzkomórkowy [18].
Dorosły człowiek spożywa w ciągu dnia około 1000 mg wapnia. Resorpcja
jonów wapnia przebiega w komórkach nabłonka jelita cienkiego, gdzie
wchłaniane jest około 400 mg wapnia pobranego z pożywieniem oraz
w dystalnych kanalikach nerkowych [19]. Wchłanianie jonów wapnia stanowi

A. Szymczyk, E. Forma 157
proces trzystopniowy. Pierwszy etap obejmuje wniknięcie jonów Ca2+ przez
błonę luminalną bądź apikalną zgodnie z gradientem stężeń. Pasywny transport
jonów wapniowych odbywa się przy udziale selektywnych kanałów
zmiennopotencjałowych receptorów waniloidowych (TRPV, ang. transient
receptor potential vanilloid), tj. receptorów typu piątego (TRPV5) w nerkach
oraz zlokalizowanych w jelicie cienkim receptorów typu szóstego (TRPV6).
Jony wapniowe dyfundują następnie przez cytozol, gdzie wiążą się z białkami
pomocniczymi: kalbindyną 28 (D28K, ang. calbindin D-28, vitamin D-dependent
calcium-binding protein) i kalbindyną 9 (D9K, ang. calbindin D-9). Obecność
kalbindyny D28K stwierdza się głównie w nabłonku nerek, natomiast kalbindyna
D9K zlokalizowana jest przede wszystkim w jelicie. W ostatnim etapie jony
wapnia przenoszone są do krwioobiegu przez błonę podstawną komórek nerek
bądź jelita. Zachodzący wbrew gradientowi stężeń transport jonów Ca2+,
odbywa się w nerkach przy udziale białka NCX1 (ang. Na+/Ca2+exchanger-1),
ponadto w nerkach i w jelicie w transport jonów wapnia zaangażowana jest
ATPaza Ca2+ (PMCA1b, ang. plasma membrane calcium-ATPase 1b) [20-22].
Zmiennopotencjałowe receptory waniloidowe TRPV5 i 6 są białkami
składającymi się z około 730 aminokwasów. Geny kodujące wyżej wymienione
receptory zlokalizowane są u człowieka na chromosomie 7q35 i obejmują
15 eksonów [20, 22]. Receptory TRPV5 i 6 ulegają N-glikozylacji w obrębie
odcinka łączącego domenę 1 i 2, znajdującego się w przestrzeni
zewnątrzkomórkowej. Miejsce glikozylacji stanowi asparagina w pozycji
358 oraz 357 [23]. W regionie N-końcowym białek TRPV5 i 6 odnotowano
obecność powtórzeń ankirynowych, zaangażowanych w proces tworzenia
tetrametrów, w postaci których kanały te spełniają swoje fizjologiczne
funkcje [20]. Receptory TRPV5 i 6 są najbardziej selektywnymi kanałami dla

Struktura i funkcje białka Klotho 158
jonów wapnia, spośród wszystkich poznanych dotąd kanałów zmienno-
potencjałowych. Właściwość ta uwarunkowana jest obecnością reszty kwasu
asparaginowego w regionie formującym kanał jonowy. W przypadku TRPV5
reszta kwasu asparaginowego znajduje się w pozycji 542, natomiast w TRPV6
w pozycji 541 [20, 22, 24].
W nerkach poziom ekspresji TRPV5 podlega regulacji m.in. przez
hormony kalcytropowe, tj. parathormon (PTH), kalcytriol oraz dostarczane
z pożywieniem jony Ca2+. Ekspresja TRPV5 kontrolowana jest również przez
hormony płciowe (estrogeny, testosteron), a także równowagę kwasowo-
zasadową [19, 22]. Oprócz wzrostu ekspresji TRPV5, parathormon może
również stymulować transport jonów Ca2+ poprzez aktywację szlaku kinazy
białkowej A (PKA, ang. protein kinase A). Uruchomienie tego szlaku
sygnalizacyjnego prowadzi do zależnej od PTH fosforylacji TRPV5, w wyniku
której dochodzi do zwiększenia aktywności kanałów. Parathormon poprzez szlak
kinazy białkowej C (PKC, ang. protein kinase C) może również przyczyniać się
do hamowania endocytozy TRPV5. Inhibicja endocytozy skutkuje z kolei
zwiększeniem liczności kanałów na powierzchni komórek [19].
U myszy pozbawionych kanałów TRPV5, pomimo wysokiego stężenia
kalcytriolu odnotowuje się obniżenie nerkowej resorpcji jonów wapnia, która
z kolei jest po części rekompensowana przez jelita, na terenie których obserwuje
się zwiększenie ekspresji TRPV6 oraz kalbindyny D9K. Mimo to, myszy
wykazują nieprawidłowości w strukturze kości, dochodzi u nich m.in.
do zmniejszenia grubości kości korowej i beleczkowej. U myszy TRPV5−/−
obserwowano również obniżenie nerkowej ekspresji białka Klotho. Wspólna
lokalizacja białka Klotho oraz TRPV5 na terenie kanalików dystalnych była
podstawą sugestii, iż białko to zaangażowane jest w regulację gospodarki

A. Szymczyk, E. Forma 159
wapniowej poprzez wpływ na aktywność kanałów TRPV [22, 24]. W wyniku
traktowania komórek HEK293 (ang. human embryonic kidney 293) sekrecyjną
formą białka Klotho dochodziło do nagromadzenia kanałów TRPV5
na powierzchni badanych komórek. W przypadku podstawienia, kluczowej
dla N-glikozylacji TRPV5, reszty asparaginy innym aminokwasem
nie obserwowano takiego efektu. [12, 24, 25]. Wykazująca aktywność
β-glukuronidazy sekrecyjna forma białka Klotho, przyczynia się zatem do
zwiększenia resorpcji jonów wapnia na terenie nerek. Białko Klotho uczestniczy
w modyfikacji N-glikanów kanałów TRPV5 poprzez usuwanie reszt kwasu
sjalowego. N-glikany glikoprotein takich jak TRPV5 składają się z nie więcej
niż czterech ramion. Utworzenie owych ramion inicjowane jest poprzez dołącze-
nie czterech cząsteczek N-acetyloglukozaminy do rdzenia pentasacharydowego
Man3GlcNAc2 poprzez wiązania β2-, β4- i β6-glikozydowe. W następnej
kolejności dochodzi do przyłączenia reszt galaktozy i utworzenia disacharydów:
galaktoza-N-acetyloglukozamina, określanych jako N-acetylolaktozamina
(LacNAc, ang. N-acetyl lactosamine). W przypadku N-glikanów kanałów
TRPV5 cząsteczkę końcową stanowi kwas sjalowy przyłączony do galaktozy
wiązaniami α2,3- bądź α2,6-glikozydowym. Enzymami odpowiedzialnymi za
syntezę wiązań są sialotransferazy: α2,3- oraz α2,6-sjalotransferaza [12, 25].
Modyfikację N-glikanów kanałów TRPV5 przez białko Klotho
zaobserwowano m.in. w komórkach HEK293. W przypadku komórek CHO
(ang. chinese hamster ovary), w których α2,6-sialotransferaza nie ulega ekspresji
endogennie, Klotho sekrecyjne nie wpływało na poziom ekspresji kałów TRPV5
w błonie komórkowej. Wyniki te sugerują zatem, iż reszta kwasu sjalowego
przyłączona do galaktozy wiązaniem α2,6- a nie α2,3-glikozydowym stanowi
substrat dla białka Klotho [12, 25, 26].

Struktura i funkcje białka Klotho 160
Mechanizm, za sprawą którego modyfikacja N-glikanów prowadzi do
zwiększenia liczby kanałów TRPV5 na powierzchni komórki wyjaśniony został
przez Cha i wsp. [26]. Usunięcie reszty kwasu sjalowego z N-glikanów kanału
TRPV5 przez białko Klotho umożliwia wiązanie się TRPV5 z galektyną 1.
Substratem dla galektyny 1 jest N-acetylolaktozamina połączona z kwasem
sjalowym wiązaniem α2,3-glikozydowym. Usunięcie przez białko Klotho
reszty kwasu sjalowego z N-glikanów TRPV5 prowadzi do odsłonięcia
N-acetylolaktozaminy, stanowiącej substrat dla galektyny 1. Za sprawą
galektyny 1 dochodzi następnie do sieciowania N-glikanów sąsiadujących
kanałów TRPV5. Połączenie to ogranicza z kolei ich endocytozę,
w konsekwencji czego dochodzi do zwiększenia retencji TRPV5 na
powierzchni komórek [12, 25, 26]. Sugeruje się, że regulacja gospodarki
wapniowej podlega również kontroli przez transbłonową formę białka. Klotho
zaangażowane jest bowiem w regulację aktywności ATPazy sodowo-potasowej.
ATPaza Na+K+ jest szeroko rozpowszechnionym enzymem stanowiącym
integralny składnik błon wielu komórek. Białko to składa się z podjednostki α
(ok. 113 kDa) oraz β (ok. 35 kDa). Podjednostka α odpowiada za transport
jonów oraz aktywność katalityczną, natomiast podjednostka β zaangażowana
jest w regulację aktywności podjednostki α [21]. ATPaza Na+K+ jest ważnym
regulatorem wewnątrzkomórkowego stężenia jonów sodu i potasu. Gradient
sodowo-potasowy stanowi z kolei siłę napędową dla transportu wielu cząsteczek
przez błonę komórkową. Sugeruje się, że wytwarzany przez ATPazę Na+K+
gradient jonów sodu zaangażowany jest również w regulację transepitelialnego
transportu jonów wapniowych na terenie splotów naczyniówkowych,
przytarczyc oraz nerek [21, 27]. Wykorzystując metodę frakcjonowania
subkomórkowego udowodniono, iż ATPaza sodowo-potasowa ma zdolność

A. Szymczyk, E. Forma 161
wiązania się z niedojrzałą formą białka Klotho (120 kDa) w obrębie siateczki
śródplazmatycznej, natomiast w obrębie aparatu Golgiego i wczesnych
endosomów ATPaza Na+K+ oddziałuje z dojrzałą formą białka Klotho o masie
cząsteczkowej 135 kDa. Sugeruje się, że oddziaływanie ATPazy Na+K+
z białkiem Klotho umożliwia jej rekrutację do błony komórkowej [28].
Badania in vitro prowadzone na komórkach nabłonkowych splotów
naczyniówkowych dowiodły, iż aktywność ATPazy Na+K+ wzrasta w przypadku
niedoborów jonów wapnia. Wykazano, że w wyniku obniżenia stężenia wapnia
dochodziło do wzrostu ilości ATPazy Na+K+ w błonie komórkowej o około
12%. Wysoka podaż jonów Ca2+ skutkowała z kolei zmniejszeniem poziomu
tego enzymu w błonie komórkowej o około 8% [21, 27]. Nie wykazano
natomiast wpływu zewnątrzkomórkowego stężenia jonów wapnia na poziom
ekspresji ATPazy Na+K+ w błonie komórkowej komórek nabłonkowych splotów
naczyniówkowych myszy Klotho. Wydaje się to potwierdzać sugestię, iż białko
Klotho jest kluczowym czynnikiem wpływającym na rekrutację ATPazy Na+K+
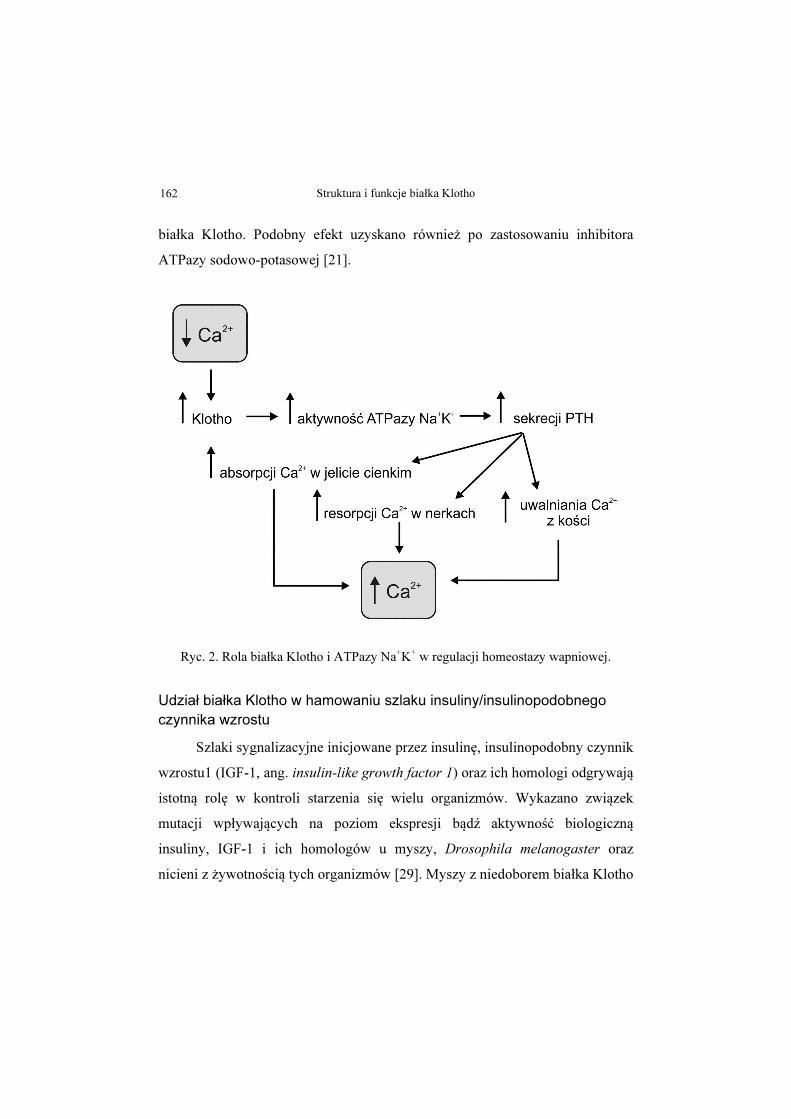
do błony komórkowej [21]. Sugeruje się, iż w odpowiedzi na spadek
zewnątrzkomórkowego poziomu wapnia, Klotho wzmaga rekrutację
ATPazy Na+K+ do błony komórkowej, a wytwarzany przez enzym gradient
jonów sodu i potasu stymuluje uwalnianie PTH w przytarczycach. W przypadku
niedoboru jonów wapnia, hormon ten poprzez zwiększanie syntezy kalcytriolu
w nerkach stymuluje wchłanianie wapnia w jelicie cienkim. Działając na
komórki kości zwiększa również uwalniania wapnia z zasobów szkieletowych
oraz wzmaga ich resorpcję w nerkach (Ryc. 2) [21, 28]. Potwierdzeniem tezy,
iż kompleks Klotho–ATPaza Na+K+ odpowiedzialny jest za stymulowanie
uwalniania PTH może być fakt, iż obniżenie wydzielania PTH odnotowano
m.in. u myszy, u których dochodziło do zaburzenia ekspresji bądź aktywności
Struktura i funkcje białka Klotho 162
białka Klotho. Podobny efekt uzyskano również po zastosowaniu inhibitora
ATPazy sodowo-potasowej [21].
Ryc. 2. Rola białka Klotho i ATPazy Na+K+ w regulacji homeostazy wapniowej.
Udział białka Klotho w hamowaniu szlaku insuliny/insulinopodobnego
czynnika wzrostu
Szlaki sygnalizacyjne inicjowane przez insulinę, insulinopodobny czynnik
wzrostu1 (IGF-1, ang. insulin-like growth factor 1) oraz ich homologi odgrywają
istotną rolę w kontroli starzenia się wielu organizmów. Wykazano związek
mutacji wpływających na poziom ekspresji bądź aktywność biologiczną
insuliny, IGF-1 i ich homologów u myszy, Drosophila melanogaster oraz
nicieni z żywotnością tych organizmów [29]. Myszy z niedoborem białka Klotho

A. Szymczyk, E. Forma 163
wykazują hipoglikemię i wysoką wrażliwość na insulinę, natomiast przy
nadekspresji Klotho obserwowano umiarkowaną oporność na działanie insuliny
i IGF-1. Mimo insulinooporności myszy te wykazują jednak prawidłowy poziom
glukozy we krwi i nie chorują na cukrzycę. Dane te, były podstawą twierdzenia,
iż białko Klotho zaangażowane jest w tłumienie szlaku insuliny/IGF-1 [4-5].
Należące do podrodziny kinaz tyrozynowych receptory insuliny oraz IGF-1 są
tetramerycznymi białkami składającymi się z dwóch podjednostek α oraz
dwóch β. Funkcjonują one jako enzymy allosteryczne, w których w przypadku
braku liganda, podjednostki α hamują aktywność kinazową podjednostek β.
W wyniku połączenia podjednostek α z ligandem, receptory te ulegają
autofosforylacji w obrębie pętli aktywacyjnych. Aktywacja receptorów skutkuje
również fosforylacją reszt tyrozynowych, stanowiących miejsce przyłączenia
substratów. Jak dotąd zidentyfikowano co najmniej dziewięć substratów
przyłączanych do aktywnego receptora dla insuliny/IGF-1. Cztery z nich należą
do rodziny IRS (ang. insulin-receptor substrate), wśród pozostałych wyróżnić
można substraty takie jak: Gab-1 (ang. Grb2-associated binder 1), p60dok
(ang. p60 docking protein), Cbl (ang. casitas B-lineage lymphoma), APS
(ang. adapter protein with pleckstrin homology and Src homology 2 domains)
oraz izoformy Shc (ang. src homology/collagen homology). Kolejnym
komponentem na szlaku insuliny/IGF-1 jest 3-kinaza fosfatydyloinozytolu
(PI3K). Kinaza ta składa się z podjednostki katalitycznej (110 kDa)
oraz regulatorowej (85 kDa), w obrębie której znajdują się dwie domeny SH2
(ang. SRC homology 2) umożliwiające wiązanie z białkami IRS. PI3K katalizuje
reakcję fosforylacji fosfatydyloinozytolo-4,5-bisfosforanu (PIP2) do
fosfatydyloinozytolo-3,4,5-trisfosforanu (PIP3). PIP3 zaangażowany jest z kolei
w aktywację trzech klas cząsteczek sygnalizacyjnych: serynowo-treoninowych

Struktura i funkcje białka Klotho 164
kinaz z podrodziny AGC (ang. cAMP-dependent, cGMP-dependent and protein
kinase C), białek Rho (ang. Ras homologous), a także kinaz tyrozynowych TEC
(ang. tyrosine kinase expressed in hepatocellular carcinoma) [30]. Kinaza PI3K
zaangażowana jest również w aktywację szlaku sygnalizacyjnego mTOR/FRAP
(ang. mammalian target of rapamycin kinase/FKBP12-rapamycin associated
protein). Jedną z najlepiej scharakteryzowanych kinaz AGC jest PDK1
(ang. phosphoinositide-dependent kinase 1). Aktywna kinaza PDK1 fosforyluje
i aktywuje serynowo-treoninową kinazę Akt/PKB (ang. protein kinase B).
Białko to nie jest zakotwiczone w błonie komórkowej, w związku z czym może
ono swobodnie przemieszczać się fosforylując liczne białka docelowe. Białka
aktywowane na szlaku PI3K/Akt zaangażowane są w regulację procesów
związanych ze wzrostem, metabolizmem, przeżyciem oraz proliferacją komórek
[30, 31]. Mutacje genów kodujących ortologi receptora insulinowego, jego
substratów (IRS) oraz PI3K skutkowały wydłużeniem czasu przeżycia
u C. elegans oraz D. melanogaster [32-35]. Występujący u nicieni
i D. melanogaster pojedynczy szlak insuliny/IGF-1 w toku ewolucji u ssaków
został rozdzielony na dwie ścieżki sygnalizacyjne. Pomimo, iż ich funkcje
nakładają się, szlak inicjowany przez insulinę zaangażowany jest głównie
w regulację metabolizmu, z kolei oś IGF-1/hormonu wzrostu (GH, ang. growth
hormone) warunkuje wzrost, rozwój i prawdopodobnie długowieczność. Wpływ
insuliny na metabolizm podlega kontroli przez hormony o działaniu
przeciwstawnym. IGF-1 jest składową większego systemu, który obejmuje
hormon wzrostu oraz liczne białka wiążące, biorące udział w regulacji funkcji
oraz wzrostu komórek. Pomimo widocznych różnic w funkcji owych szlaków,
mechanizm, za sprawą którego IGF-1 sprzyja długowieczności zależny jest
w dużej mierze od prawidłowego metabolizmu insuliny [35]. Interesujących

A. Szymczyk, E. Forma 165
danych dostarczyły m.in. badania przeprowadzone na karłowatych myszach
szczepów Ames i Snell. W odniesieniu do myszy typu dzikiego, myszy
z homozygotyczną mutacją w genach Prop1 (ang. prophet of Pit-1) oraz Pit-1
(ang. POU domain, class 1, transcription factor 1) były o 66% mniejsze,
jednakże czas ich życia ulegał znacznemu wydłużeniu (42-70%). Myszy te
cierpiały jednak na bezpłodność [36]. Na skutek zaburzeń wydzielania hormonu
wzrostu przez przysadkę, karłowate myszy wykazywały niskie stężenie IGF-1
w surowicy krwi. Obserwowano u nich również obniżone stężenie insuliny,
a także zwiększoną wrażliwość na insulinę podawaną egzogennie [34, 36].
Dłuższą przeżywalność odnotowano również u heterozygotycznych myszy
pozbawionych genu receptora dla IGF-1. Myszy homozygotyczne pod
względem tej mutacji umierały tuż po urodzeniu. W przypadku heterozygot
wydłużenie czasu przeżycia uzależnione było od płci, w odniesieniu do myszy
typu dzikiego samice żyły o 33% dłużej, natomiast samce o 16%. Myszy te nie
wykazywały zmian metabolicznych, ani też zaburzeń płodności. U osobników
dorosłych obserwowano jednak zwiększone stężenie IGF-1, które
prawdopodobnie niwelowało efekty mutacji receptora dla tego czynnika
wzrostu. Myszy wykazywały prawidłowy poziom glukozy na czczo, jednakże
po posiłku w porównaniu do kontroli u samców poziom ten był o 12% wyższy,
u samic z kolei obserwowano spadek o 4,4% [34, 37, 38]. Ciekawych
spostrzeżeń dostarczyły również badania prowadzone na linii myszy FIRKO
(ang. fat-specific insulin receptor knockout mice), u których dokonano
wyłączenia ekspresji receptora insulinowego na terenie tkanki tłuszczowej.
Myszy te charakteryzowały się niską masą ciała, mimo że spożywały więcej
pokarmu. Ponadto wykazywały one mniejszą insulinemię i nie rozwijała się
u nich związana z wiekiem insulinooporność. Długość życia myszy FIRKO

Struktura i funkcje białka Klotho 166
ulegała zwiększeniu o ok. 18% [36, 38, 39]. Fakt, iż szlaki aktywowane przez
insulinę i IGF-1 są silnie zakonserwowane ewolucyjnie może sugerować, że
odgrywają one istotną rolę w regulacji długowieczności u ludzi. Badania
wykazały, iż wraz z wiekiem dochodzi do zmniejszenia wydzielania GH,
a w konsekwencji obniżenie stężenia IGF-1. Fenotyp starzenia obejmuje m.in.
zwiększenie tkanki tłuszczowej, insulinooporność, choroby układu krążenia
(miażdżyca, nadciśnienie). Wszystkie spośród wyżej wymienionych cech
obserwuje się również u osób z niedoborem GH [34]. U osób cierpiących na
zespół Larona (niewrażliwość na GH) odnotowuje się wysokie stężenie hormonu
wzrostu oraz niskie stężenia IGF-1. Chorych charakteryzuje niski wzrost,
otyłość, opóźnienie umysłowe, a także nietolerancja glukozy. Fenotyp ten, nie
skutkuje jednak krótkim czasem przeżycia, gdyż pacjenci z zespołem Larona
dożywają sędziwego wieku (niekiedy ponad 90 lat) [34, 40]. Osoby cierpiące na
akromegalię (nadprodukcja GH) mają zwiększone ryzyko zgonu z powodu
rozwijających się u nich chorób układu krążenia, cukrzycy oraz nowotworów
[34]. Ponadto terapia substytucyjna GH u osób z niedoborem tego hormonu
prowadziła do rozwoju insulinooporności, nietolerancji glukozy, bólów stawów,
obrzęków, retencji sodu oraz nadmiernej aktywacji mitogenów, skutkującej
najprawdopodobniej rozwojem nowotworów [34]. Mechanizm, za sprawą
którego sekrecyjna forma białka Klotho przyczynia się do inhibicji szlaku
insuliny i IGF-1 nie został do końca poznany. Wiadomo jednak, że funkcja
Klotho opiera się na hamowaniu autofosforylacji receptorów, nie zaś na
blokowaniu ich interakcji z ligandem. Inhibicja autofosforylacji receptorów
insuliny i IGF-1 skutkuje obniżeniem aktywności IRS, blokowaniem ich
oddziaływań z PI3K, a w konsekwencji hamowaniem aktywacji dalszych białek
szlaku. W badaniach in vitro udowodniono, iż białko to może również hamować

A. Szymczyk, E. Forma 167
fosforylację receptorów uprzednio aktywowanych przez insulinę bądź IGF-1.
Aktywność sekrecyjnego białka Klotho jest specyficzna dla receptorów dla
insuliny i IGF-1, ponieważ nie wykazywało ono podobnego efektu w stosunku
do receptorów płytkowego czynnika wzrostu (PDGF, ang. platelet-derived
growth factor) i czynnika wzrostu naskórka (EGF, ang. epidermal growth
factor), które również posiadają aktywność kinaz tyrozynowych [5].
Aktywność enzymatyczna Klotho, skłania również do sugestii, iż białko
to może modyfikować N-glikany receptorów insuliny i IGF-1, wynikiem czego
jest blokowanie ich aktywności lub/i zmniejszenie liczby tych receptorów na
powierzchni komórek Potwierdzeniem tej tezy może być fakt, iż traktowanie
hodowli komórek sialidazą indukuje insulinooporność. Ponadto nadekspresja
sialidazy na powierzchni komórek, skutkuje u myszy rozwojem
insulinooporności oraz nietolerancji glukozy [4]. Sugeruje się, iż białko Klotho
przyczynia się do przedłużenia żywotności dzięki dwóm współdziałającym ze
sobą mechanizmom, tj. hamowaniu sygnalizacji IGF-1 oraz zwiększaniu
oporności na stres oksydacyjny. Zablokowanie transmisji sygnału na szlaku
zależnym od insuliny/IGF-1 prowadzi do zahamowania aktywacji kinazy Akt,
która fosforyluje czynnik transkrypcyjny FOXO (ang. forkhead transcription
factor). Nieufosforylowany czynnik FOXO zlokalizowany jest w jądrze
komórkowym gdzie spełnia rolę czynnika transkrypcyjnego [4, 41].
Występujące u ssaków czynniki transkrypcyjne FOXO stanowią ortologi
obecnego u C. elegans czynnika DAF-16 (ang. abnormal dauer formation
protein 16). U gryzoni zidentyfikowano do tej pory cztery czynniki z rodziny
FOXO: FOXO1, FOXO3a FOXO4 oraz FOXO6 [42]. W swej strukturze
czynniki transkrypcyjne z rodziny forkhead posiadają silnie zakonserwowaną
domenę wiążącą DNA (ang. winged helix/forkhead domain). Białka należące do

Struktura i funkcje białka Klotho 168
rodziny FOXO odgrywają istotną rolę w procesach różnicowania komórek,
embriogenezie, nowotworzeniu oraz w regulacji metabolizmu [43]. Czynniki
transkrypcyjne FOXO zaangażowane są w regulację ekspresji wielu genów,
w tym enzymów antyoksydacyjnych takich jak katalaza czy mitochondrialna
manganowa dysmutaza ponadtlenkowa (SOD2, ang. superoxide dismutase 2).
Enzymy te usuwając reaktywne formy tlenu przyczyniają się do redukcji stresu
oksydacyjnego. Redukcja stresu oksydacyjnego może być zatem czynnikiem
kluczowym w procesie hamowania starzenia się organizmów, u których doszło
do inhibicji szlaku insuliny/IGF-1 [4].
W badaniach prowadzonych na komórkach HeLa wykazano, iż forma
sekrecyjna białka Klotho zmniejsza peroksydację lipidów, powstającą na skutek
traktowania komórek herbicydem generującym nadtlenki (paraquat). Na przy-
kładzie linii komórek HeLa oraz CHO udowodniono również, iż białko to może
chronić komórki przed apoptozą indukowaną stresem oksydacyjnym [41].
Myszy z nadekspresją Klotho wykazują zwiększoną ekspresję SOD2 na terenie
mięśni, a także niższy poziom fosforylacji czynników FOXO w porównaniu do
myszy typu dzikiego. U myszy tych odnotowuje się również spadek stężenia w
moczu 8-hydroksydeoksyguanozyny (8-OHdG, ang. 8-hydroxydeoxyguanosine),
będącej markerem oksydacyjnych uszkodzeń DNA. Ponadto śmiertelna dla
myszy typu dzikiego dawka paraquatu, nie okazała się być letalną dla myszy
z nadekspresją Klotho, co wskazuje na udział Klotho w redukcji stresu
oksydacyjnego [4, 41]. Antyapoptotyczna aktywność Klotho potwierdzona
została również w badaniach na komórkach śródbłonka żyły pępowinowej
człowieka (HUVEC, ang. human umbilical vascular endothelial cells). Apotozę
w komórkach HUVEC indukowano nadtlenkiem wodoru. W odniesieniu do
kontroli w komórkach HUVEC traktowanych Klotho obserwowano znacznie

A. Szymczyk, E. Forma 169
niższą aktywność kaspazy 3 i kaspazy 9. Białko Klotho wykazywało również
aktywność antyapoptotyczną w stosunku do komórek, którym podawano
epoksyd (inhibitor topoizomerazy II), stymulujący cięcie DNA i kierujący
komórki na drogę apoptozy. Przypuszcza się zatem, że obok inhibicji szlaku
insuliny/IGF-1/SOD2 istnieje jeszcze inny mechanizm, za sprawą którego białko
Klotho chroni komórki przed apoptozą [44].
Rola transbłonowej formy białka Klotho w homeostazie
fosforanów
Fosfor stanowi ważne ogniwo w regulacji przemian śródkomórkowych
organizmu. Zaangażowany jest on m.in. w biosyntezę nukleotydów,
fosfolipidów oraz 2,3-difosfoglicerolu, który ułatwia uwalnianie tlenu
z hemoglobiny. Fosfor odpowiedzialny jest również za utrzymywanie
integralności morfologicznej i czynnościowej układu ruchu. Jest czynnikiem
niezbędnym dla prawidłowego przebiegu wielu szlaków metabolicznych [45].
Dorosły człowiek spożywa w ciągu dnia około 1,5 g fosforu, z czego
2/3 wchłaniane jest w jelitach [45]. Fosfor w organizmie człowieka
zgromadzony jest głównie w kościach (ponad 80%), mięśniach (około 9%).
Około 10,9% fosforu obecne jest w innych tkankach, natomiast fosforany
nieorganiczne (Pi) stanowią jedynie ok. 0,1% całkowitej puli fosforu [21].
Kluczową rolę w regulacji stężenia fosforanów w surowicy krwi odgrywają
nerki. Około 90% fosforanów zawartych w osoczu podlega filtracji
w kłębuszkach nerkowych, przy czym 15% z nich przy prawidłowej diecie
wydalane zostaje z moczem. Resorpcja fosforanów ma miejsce głównie
w kanalikach proksymalnych i zachodzi ona przy udziale dwóch kotranspor-
terów sodowo-fosforanowych, tj. NaPi-2a (ang. sodium-phosphate cotransporter

Struktura i funkcje białka Klotho 170
type-2a) oraz NaPi-2c (ang. sodium-phosphate cotransporter type-2c) [45].
Prawidłowe stężenie fosforanów we krwi utrzymywane jest dzięki równowadze
zachodzącej pomiędzy procesami wchłaniania fosforanów zawartych w diecie
przez jelita, mobilizacją z kości oraz resorpcją i wydalaniem przez nerki.
Procesy te z kolei regulowane są przez szereg czynników endokrynnych takich
jak kalcytriol, parathormon, czy FGF23 (ang. fibroblast growth factor 23) [4,
16, 47, 49]. Dowodów na to, w jaki sposób białko Klotho wpływa na regulację
gospodarki fosforanowej dostarczyły m.in. badania przeprowadzone na myszach
pozbawionych genu FGF23. U myszy tych, podobnie jak u myszy Klotho,
zaobserwowano wystąpienie przedwczesnych objawów przypominających
starzenie się człowieka. U myszy FGF23−/− obserwowano m.in. zahamowanie
wzrostu, kifozę, osteopenię, atrofię gonad, inwolucję grasicy, rozedmę płuc,
zaniki mięśniowe i skórne. Myszy FGF23−/− wykazywały również nieprawidło-
wą resorpcję fosforanów w nerkach, a także zwiększoną syntezę kalcytriolu.
Przyczyną takiego stanu rzeczy okazał się fakt, iż transbłonowa forma białka
Klotho stanowi koreceptor dla FGF23 [5, 9, 50]. Gen FGF23 został pierwotnie
zidentyfikowany u pacjentów z autosomalną dominującą krzywicą hipofosfate-
miczną (ADHR, ang. autosomal dominant hypophosphatemic rickets). U osób
z ADHR stwierdzono występowanie mutacji zmiany sensu w genie FGF23,
w wyniku której reszta argininy w pozycji 176 lub 179 zostaje zamieniona na
glutaminę. Mutacja ta skutkuje opornością FGF23 na trawienie endopeptydazą,
a w konsekwencji zwiększeniem jego stężenia w surowicy krwi. Zwiększony
poziomy FGF23 prowadzi natomiast do nadmiernej utraty fosforanów, rozwoju
hipofosfatemii oraz krzywicy [3, 9, 16, 48, 51]. Kolejnym schorzeniem
objawiającym się zwiększonym poziomem FGF23 w surowicy krwi
jest hipofosfatemia sprzężona z chromosomem X (XLH ang. X-linked

A. Szymczyk, E. Forma 171
hypophosphataemia). XLH występuje z częstością 1 na 20 000 żywych urodzeń
i dziedziczona jest jako cecha dominująca sprzężona z chromosomem X.
Jej przyczyną jest mutacja genu PHEX (ang. phosphateregulating gene
with homologies to endopeptidases on the X chromosome) kodującego
endopeptydazę, która rozpoznaje motyw 176RXXR179 w białku FGF23
i dokonuje jego proteolitycznej inaktywacji [51-53]. Czynnik wzrostu
fibroblastów 23 jest białkiem o masie cząsteczkowej 32 kDa zbudowanym z 251
aminokwasów, który ulega ekspresji głównie w komórkach tkanki kostnej,
tj. osteocytach i osteoblastach. Nieznaczną ekspresję odnotowano również
w nerkach, wątrobie, tarczycy i przytarczycach, jelicie cienkim oraz mięśniach
szkieletowych. W wyniku cięcia proteolitycznego pomiędzy resztą Arg179
a Ser180 białko to ulega rozszczepieniu na dwa fragmenty: osiągający masę
cząsteczkową 18 kDa fragment N-końcowy oraz fragment C-końcowy o masie
cząsteczkowej 12 kDa [45, 54].
FGF23 należy do rodziny 22 czynników wzrostu fibroblastów
zgrupowanych w siedmiu podrodzinach określanych jako: FGF1, FGF4, FGF7,
FGF8, FGF9, FGF11 oraz FGF19. FGF23 wraz z FGF19 i FGF21, należy do
podrodziny FGF19 [21, 55]. Owe trzy czynniki określane są również mianem
czynników endokrynnych, gdyż w przeciwieństwie do pozostałych, klasycznych
czynników wzrostu fibroblastów, działających w sposób auto- lub parakrynny,
funkcjonują one, jako czynniki dokrewne [16, 48]. Czynniki wzrostu
fibroblastów pełnią swoje biologiczne funkcje po związaniu się z odpowiednim
receptorem o aktywności kinazy tyrozynowej zlokalizowanym w błonie
komórkowej. Aktywne receptory funkcjonują w formie dimerów. W strukturze
receptorów dla czynników wzrostu fibroblastów (FGFR, ang. fibroblast growth
factor receptor) wyróżnia się domenę zewnątrzkomórkową zawierającą trzy

Struktura i funkcje białka Klotho 172
pętle immunoglobulino-podobne (D1-D3), odcinek transbłonowy oraz
wykazującą aktywność katalityczną domenę wewnątrzkomórkową [51, 55, 56].
Immunoglobulino-podobne pętle D2 i D3 stanowią miejsce wiązania ligandów.
W obrębie odcinka linkerowego pomiędzy pętlą D1 a D2 znajduje się odcinek
bogaty w aminokwasy kwaśnie oraz reszty seryny określany jako AB (ang. acid
box) [57, 58]. Wśród receptorów dla FGF wyróżnić można 7 izoform,
tj. FGFR1b, FGFR1c, FGFR2b, FGFR2c, FGFR3b, FGFR3c oraz FGFR4.
Izoformy b i c powstają na skutek alternatywnego składania w obrębie eksonów
kodujących trzecią z immunoglobulino-podobnych pętli [21, 45, 49, 55, 56].
Proces dimeryzacji receptorów dla FGF oraz stabilność kompleksu FGFR-FGF
regulowane są przez siarczan heparanu (HS, ang. heparan sulfate) bądź
heparynę. Czynniki wzrostu fibroblastów z podrodziny 19 (FGF19, 21, 23)
w odróżnieniu od pozostałych FGF, wykazują małe powinowactwo do HS,
w wyniku czego do aktywacji kinazy tyrozynowej i transdukcji sygnału
wymagają dodatkowego koreceptora. Zmniejszenie powinowactwa do HS
wynika ze zmian w obrębie domeny wiążącej proteoglikan (nie dochodzi w tym
przypadku do utworzenia wiązań wodorowych pomiędzy siarczanem heparanu
a aminokwasami domeny wiążącej). Dzięki tej właściwości czynniki te mogą
przedostawać się do układu krążenia i funkcjonować jako hormony [47, 48, 55,
56, 59]. Z przeprowadzonych badań wynika, iż C-końcowy region FGF
z podrodziny 19 odpowiada za oddziaływanie z koreceptorem (β-Klotho
w przypadku FGF19 i FGF21, oraz Klotho w przypadku FGF23), natomiast
poprzez N-koniec oddziałują one z odpowiednim receptorem FGFR [59].
Czynniki wzrostu fibroblastów mogą stanowić ligandy dla kilku izoform FGFR.
FGF23 wykazuje powinowactwo do izoform FGFR1c, 3c oraz 4. Z owymi
izoformami FGFR oddziałuje również białko Klotho, które poprzez tworzenie

A. Szymczyk, E. Forma 173
binarnych kompleksów Klotho-FGFR zwiększa ich powinowactwo do FGF23
[5, 16, 21, 45, 47, 48]. FGF23 spełnia swoje biologiczne funkcje poprzez
aktywację szlaku MAP kinaz (MAPK, ang. mitogen-activated protein kinase),
który uczestniczy w regulacji wzrostu i różnicowania komórek. Wykazujące
aktywność kinazy tyrozynowej receptory FGFR po przyłączeniu ligandu
(FGF23) w obecności Klotho, ulegają aktywacji i fosforylują kolejne białka.
Na szlaku tym dochodzi do aktywacji małych białek Ras (ang. rat sarcoma, Ras
protein). Małe białka Ras dokonują fosforylacji kinazy serynowo-treoninowej
Raf (ang. rapidly accelerated fibrosarcoma, Raf kinase), która uruchamia
kaskadę kinaz MAP, w wyniku czego dochodzi do aktywacji kinazy MEK
(ang. mitogen-activated protein kinase kinase, MAPKK), a następnie fosforylacji
ERK1/2 (ang. extracellular signal regulated kinases 1/2). Za sprawą białek Ras
dochodzi również do aktywacji MEKK (ang. MAP kinase kinase kinase,
MAPKKK), a w konsekwencji fosforolylacji kinazy JNK (ang. jun N-terminal
kinase), p38 (ang. p38 mitogen-activated protein kinase) oraz CREB
(ang. cAMP response element-binding) [50]. Wykazano ponadto, że kompleks
FGF23-FGFR1-Klotho aktywuje również szlak 3-kinazy fosfatydyloinozytolu,
który prowadzi do aktywacji kinazy Akt, fosforylującej między innymi kinazę
IKK (ang. I kappa B kinase) oraz GSK3β (ang. glycogen synthase kinase 3β).
Szlak ten sprzyja przeżyciu komórek oraz zaangażowany jest w kontrolę ich
proliferacji. Szlak PI3K przeciwdziała również apoptozie poprzez hamowanie
indukowanej przez witaminę D aktywacji kaspaz [50].
Przy udziale białka Klotho, FGF23 może pełnić funkcję czynnika
fosfaturycznego, którego aktywność polega na hamowaniu resorpcji fosforanów
w jelitach i nerkach. Badania in vivo wykazały, że FGF23 jest jednym
z głównych regulatorów metabolizmu witaminy D. Aktywna postać witaminy D

Struktura i funkcje białka Klotho 174
(1,25(OH)2D3, kalcytriol) syntetyzowana jest głównie przez nerki i odgrywa
istotną rolę w regulacji gospodarki mineralnej organizmu. Miejscem działania
kalcytriolu są jelita, gdzie promując ekspresję transportera sodowo-
fosforanowego NaPi-2b stymuluje on wchłanianie wapnia i fosforu, wpływając
tym samym na dodatni bilans fosforanów. W odpowiedzi na zwiększony poziom
fosforanów, FGF23 reguluje ekspresję dwóch genów, tj. Cyp27b1 oraz Cyp24,
których produkty białkowe uczestniczą w syntezie witaminy D. Gen Cyp27b1
koduje 1α-hydroksylazę, która katalizując reakcję hydroksylacji 25(OH)D3
prowadzi do powstania kalcytriolu. Poprzez zahamowanie ekspresji Cyp27b1,
FGF23 hamuje syntezę 1,25(OH)2D3, ograniczając tym samym jelitową
resorpcję fosforanów. Promuje on natomiast ekspresję genu Cyp24 kodującego
24-hydroksylazę. Enzym ten katalizuje reakcję hydroksylacji 25(OH)D3
w pozycji 24, której produktem jest nieaktywna forma witaminy D [16, 47,
48, 60].
Utrzymywanie homeostazy fosforanowej odbywa się na zasadzie szeregu
sprzężeń zwrotnych. FGF23 hamuje syntezę kalcytriolu, a aktywna postać
witaminy D stymuluje ekspresję FGF23 (oś kości–nerki). Kalcytriol wiąże się
z receptorem witaminy D (VDR, ang. vitamin D receptor), który tworzy
heterodimer wraz z receptorem retinoidowym (RXR, ang. retinoid X receptor).
Heterodimer VDR-RXR funkcjonuje jako czynnik transkrypcyjny regulujący
ekspresję wielu genów w tym m.in. zwiększający ekspresję FGF23
w osteocytach [9]. Ponadto kalcytriol może być również zaangażowany
w regulację ekspresji Klotho, gdyż w regionie promotorowym genu Klotho
człowieka zidentyfikowano sekwencję VDRE (ang. vitamin D responsive
element), z którą wiąże się VDR-RXR [60].

A. Szymczyk, E. Forma 175
FGF23 hamuje również ekspresję i wydzielanie syntetyzowanego przez
przytarczyce parathormonu. W celu utrzymania homeostazy mineralnej PTH
wiąże się z receptorami PTH1R (ang. parathyroid hormone 1 receptor)
zlokalizowanymi w komórkach kości (osteocyty, osteoblasty, chondrocyty)
i kanalikach nerkowych [61]. Mechanizm działania PTH jest złożony i opiera się
na promowaniu zarówno syntezy kalcytriolu, jak i wydalania fosforanów
z moczem. W przeciwieństwie do kalcytriolu, PTH może zatem selektywnie
zwiększać stężenie wapnia we krwi, nie przyczyniając się jednocześnie do
wzrostu stężenia fosforanów. FGF23 poprzez oddziaływanie z kompleksem
FGFR1c-Klotho w komórkach przytarczyc obniża ekspresję PTH, hamując
tym samym nerkową biosyntezę 1,25(OH)2D3. Ekspresja PTH regulowana
jest również przez kalcytriol, który na zasadzie sprzężenia zwrotnego
(oś nerki-przytarczyce) przyczynia się do obniżenie stężenia parathormonu [4, 9,
45, 47, 61]. W badaniach przeprowadzonych na szczurach, Lavi-Moshayoff
i wsp. [61] wykazali wpływ PTH na poziom ekspresji FGF23. Stwierdzono, iż
PTH zwiększa ekspresję FGF23 w indukowanej doświadczalnie niewydolności
nerek. U zwierząt, którym podawano pokarmy bogate w fosforany i adeninę
obserwowano podwyższony poziom FGF23, który ulegał obniżeniu wraz
z usunięciem przytarczyc [61]. FGF23 poprzez hamowanie ekspresji
kotransporterów sodowo-fosforanowych (NaPi-2a i NaPi-2c) ogranicza również
nerkową resorpcję fosforanów. Owe dwa transportery zlokalizowane są
na biegunie apikalnym komórek cewkowych. Transportują one do wnętrza
komórek kanalika proksymalnego jon wodorofosforanowy (HPO4
2-) oraz jony
sodowe. Transporter NaPi-2a przenosi trzy jony Na+, z kolei NaPi-2c dwa [45].

Struktura i funkcje białka Klotho 176
Z badań przeprowadzonych na myszach wynika, iż mechanizm
hamowania ekspresji transporterów odbywa się głównie poprzez interakcje
FGF23-Klotho-FGFR1c. Mniejsze znaczenie przypisuje się izoformie FGFR3,
natomiast udział FGFR4 w tym procesie określa się jako niewielki [51].
Do obniżenia ekspresji NaPi-2a i 2c przyczynia się również pochodzący
z przytarczyc PTH (Ryc. 3) [45]. Hamowanie nerkowej resorpcji fosforanów
odbywa się również przy udziale sekrecyjnej formy białka Klotho. Białko to,
dzięki aktywności β-glukuronidazy modyfikuje N-glikany transporterów
NaPi-2a. W wyniku owej modyfikacji transportery te ulegają inaktywacji
i kierowane są na drogę proteolitycznej degradacji [9, 62].
Ryc. 3. Regulacja gospodarki fosforanowej: oś sprzężeń zwrotnych kości – przytarczyce
– nerki – jelito. Przerywanymi liniami oznaczono wzrost, ciągłymi zaś spadek ekspresji
określonego czynnika.

A. Szymczyk, E. Forma 177
Klotho a nowotwory
Liczne badania kliniczne i laboratoryjne wskazują, iż u podstaw rozwoju
wielu typów nowotworów leżą zaburzenia sygnalizacji inicjowanej przez
insulinę, insulinopodobny czynnik wzrostu, a także czynniki wzrostu
fibroblastów [63, 64]. Zdolność Klotho do regulacji owych szlaków, była
podstawą sugestii, iż białko to może odgrywać znaczącą rolę w inhibicji procesu
nowotworzenia [63]. Dane te potwierdzone zostały m.in. w badaniach
prowadzonych na komórkach gruczolakoraka trzustki. U ludzi rak ten jest
czwartą pod względem częstości przyczyną zgonów z powodu chorób
nowotworowych w krajach zachodnich. Gruczolakorak trzustki należy do
nowotworów o najgorszym rokowaniu, średni okres przeżycia od postawienia
rozpoznania nie przekracza 6 miesięcy. Istotną rolę w rozwoju owego
nowotworu przypisuje się szlakom sygnalizacyjnym inicjowanym przez
insulinopodobny czynnik wzrostu oraz czynniki wzrostu fibroblastów.
Nadmierna aktywacja receptora IGF-1 skutkuje transformacją nowotworową,
wzrostem agresywności guza, a także hamowaniem procesu apoptozy.
W nowotworach trzustki często odnotowuje się wzrost ekspresji
insulinopodobnego czynnika wzrostu i jego receptora. Inhibicja ścieżki
sygnalizacyjnej IGF-1 prowadzi natomiast do zahamowania wzrostu guza,
a także zwiększa jego wrażliwość na promieniowanie jonizujące i chemio-
terapię. Szlak sygnalizacyjny FGF zaangażowany jest w regulację różnych
etapów rozwoju nowotworów, tj. mitogenezy, różnicowania komórek czy
angiogenezy. Zwiększoną ekspresję FGFR oraz ligandów takich jak FGF1, 2, 5
i 7 odnotowano zarówno w przypadku linii komórkowych, jak i komórek
nowotworowych pobranych od pacjentów. Wysoka ekspresja FGF2,
określanego także jako zasadowy czynnik wzrostu fibroblastów

Struktura i funkcje białka Klotho 178
(bFGF, ang. basic fibroblast growth factor), powiązana była również
ze stopniem zaawansowania nowotworu oraz krótszym czasem przeżycia
chorych [63].
Wyniki badań przeprowadzonych przez zespół Wolfa, wykazały, że
białko Klotho pełni funkcję supresora w procesie nowotworzenia trzustki.
W odniesieniu do zdrowej tkanki, gruczolakoraki trzustki charakteryzowały się
obniżonym poziomem ekspresji tego białka. Nadekspresja Klotho prowadziła
natomiast do hamowania wzrostu komórek nowotworowych, zarówno
w warunkach in vitro, jak i in vivo. Sugeruje się, iż za ograniczenie aktywności
białka Klotho w nowotworach trzustki odpowiadają dwa mechanizmy,
tj. epigenetyczne wyciszanie genu KL oraz ekspresja białka o zmniejszonej
aktywności [63]. Aktywność supresorowa białka Klotho uwarunkowana jest
głównie jego zdolnością do inhibicji szlaku IGF-1. Przypuszcza się jednak, iż
nie mniej istotną rolę może odgrywać także blokowanie przez niego ścieżki
sygnalizacyjnej bFGF, mechanizm ten odnotowano m.in. w komórkach
HEK293, komórkach raka trzustki, a także w nowotworach wywodzących się
z innych tkanek [63]. Szlak sygnalizacyjny insuliny/IGF-1 odgrywa także
znaczącą rolę w rozwoju raka piersi, który jest najczęstszym nowotworem
złośliwym kobiet. Badania immunohistochemiczne wykazały, iż w odniesieniu
do tkanki prawidłowej, komórki przedinwazyjnego oraz inwazyjnego
przewodowego raka piersi wykazują znacznie niższy poziom ekspresji Klotho
[4, 65]. Rubinek i wsp. [66] wykazali, iż obniżenie ekspresji Klotho
w nowotworach piersi następuje w wyniku hipermetylacji wysp CpG
zlokalizowanych w regionie promotora genu Klotho, a także deacetylacji białek
histonowych. Liczne badania przeprowadzone m.in. na linii komórek raka piersi
MCF-7 dowiodły, iż wyłączenie ekspresji Klotho skutkuje wzrostem proliferacji

A. Szymczyk, E. Forma 179
komórek. Natomiast nadekspresja białka Klotho była związana z zahamowaniem
fosforylacji receptora IGF-1 oraz kolejnych białek szlaku, czego skutkiem było
ograniczenie proliferacji komórek nowotworowych. Następstwem wyciszenia
szlaku IGF-1 przez Klotho w przypadku nowotworów piersi jest podwyższenie
ekspresji czynników transkrypcyjnych C/EBP określanych również jako białka
wiążące się z sekwencją CCAAT (ang. CCAAT enhancer binding proteins,
CCAAT binding factor). Wykazano, że w przypadku nowotworów piersi
czynniki te spełniają funkcję supresorów wzrostu [65]. Poziom ekspresji Klotho
okazał się być także niezwykle istotny w przebiegu nowotworów płuc,
tj. drobnokomórkowego raka płuc (SCLC, ang. small cell lung cancer) oraz
wielkokomórkowego raka o cechach neuroendokrynnych (LCNEC, ang. large
cell neuroendocrine carcinoma). Nowotwory te, charakteryzuje krótki czas
przeżycia, pięcioletnie prognozowane przeżycie chorych sięga średnio 40,3%
w przypadku LCNEC oraz 35,7% w SCLC. Wyniki badań histochemicznych
dowiodły, iż poziom ekspresji Klotho może spełniać rolę markera
prognostycznego w przypadku pacjentów poddanych resekcji zmienionych
nowotworowo tkanek. Ekspresja Klotho w guzach pierwotnych raków płuc,
związana była bowiem z lepszym rokowaniem i dłuższym czasem przeżycia
chorych. Ocena poziomu ekspresji Klotho, może zatem okazać się w przyszłości
niezwykle pomocną w doborze terapii dla pacjentów z LCNEC i SCLC [67, 68].
Supresorową funkcję genu Klotho odnotowano również w komórkach raka
żołądka, drugiego pod względem śmiertelności nowotworu na świecie.
Udowodniono, iż w przypadku raka żołądka, obniżenie ekspresji Klotho
następuje w wyniku hipermetylacji wysp CpG w regionie promotorowym tego
genu (46% pacjentów). Ocena hipermetylacji promotora Klotho okazała się być
również istotnym czynnikiem prognostycznym czasu przeżycia chorych na

Struktura i funkcje białka Klotho 180
nowotwory żołądka [64]. Obniżenie ekspresji Klotho, bądź też całkowite jej
wyciszenie skorelowane jest również z rozwojem raka jelita grubego,
stanowiącego trzecią pod względem częstości przyczynę zgonów z powodu
chorób nowotworowych na świecie. Podobnie jak w innych typach
nowotworów, zmiana ekspresji Klotho w komórkach tego nowotworu jest
konsekwencją hipermetylacji wysp CpG. Metylację CpG odnotowano
w przypadku 83,3% badanych linii komórkowych oraz 85% preparatów
tkankowych jelita grubego. Nadekspresja białka Klotho poprzez indukcję
apoptozy oraz zatrzymanie cyklu komórkowego powodowała zahamowanie
proliferacji komórek nowotworowych [69]. Nowe spojrzenie na mechanizmy
leżące u podstaw supresorowej funkcji Klotho przyniosły badania
przeprowadzone przez Lee i wsp. [70]. Wykazali oni, iż w przypadku raka szyjki
macicy, który jest drugą najczęstszą przyczyną zgonów wśród kobiet na całym
świecie, Klotho działa jako inhibitor kanonicznej ścieżki szlaku Wnt
(ang. wingless and Int-1). Liczne badania epidemiologiczne wskazują, iż
czynnikiem inicjującym rozwój tego nowotworu jest infekcja wirusem
brodawczaka ludzkiego HPV (ang. human papilloma virus). Przyjmuje się,
iż zależna od β-kateniny ścieżka szlaku Wnt może stanowić kolejny krok
w wieloetapowym procesie karcynogenezy szyjki macicy [70]. Szlak Wnt
zaangażowany jest w regulację wielu procesów biologicznych, takich jak:
samoodnowa komórek macierzystych, proliferacja i różnicowanie komórek, ich
migracja czy apoptoza. Aktywacja owego szlaku rozpoczyna się od związania
liganda Wnt z obecnym w błonie komórkowej receptorem Fzd (ang. Frizzeled)
oraz koreceptorami w postaci białek LRP5 i LRP6 (ang. low-density lipoprotein
receptor-related proteins). Połączenie liganda i receptora skutkuje fosforylacją
cytoplazmatycznego białka Dishevelled oraz jego translokacją do błony

A. Szymczyk, E. Forma 181
komórkowej. Dochodzi wówczas do zablokowania fosforylacji β-kateniny,
dzięki czemu może ona ulec translokacji do jądra komórkowego, gdzie pełni
rolę czynnika transkrypcyjnego. Przy nieobecności liganda Wnt, β-katenina
fosforylowana jest przez kinazę kazeiny γ (CK1γ, ang. casein kinase 1γ) oraz
kompleks białek takich jak GSK3β, aksyna i białko APC (ang. adenomatosis
polyposis coli). Ufosforylowana β-katenina rozpoznawana jest przez ligazę
ubikwityny E3 β-TRCP (ang. β-transducin repeat-containing protein) oraz
kierowana na drogę degradacji przez proteasomy [71]. Badania prowadzone na
linii komórek nowotworowych szyjki macicy CaSki dowiodły, iż na skutek
ektopowej ekspresji sKL dochodzi do zmniejszenia całkowitego poziomu
β-kateniny, a także znacznej redukcji aktywnej formy białka. Nadekspresja
Klotho prowadzi zatem do spadku ekspresji genów regulowanych przez
β-kateninę, takich jak onkogen c-MYC, czy gen CCND1 (ang. cyclin D1)
kodujący cyklinę D1 [70]. W przypadku wielu linii komórkowych raka szyjki
macicy, a także w preparatach raka inwazyjnego odnotowano spadek poziomu
mRNA Klotho. Zmian tych nie wykazano natomiast w raku przedinwazyjnym.
Obserwowany spadek ekspresji Klotho był wynikiem metylacji wysp CpG
(41%), a także deacetylacji histonów w obrębie regionu promotorowego genu
KL. Badania prowadzone na liniach komórkowych raka szyjki macicy SiHa
wykazały, że kluczową rolę w procesie epigenetycznego wyciszania genu
Klotho odgrywa deacetylacja histonu H3. Dane te sugerują, iż białko Klotho
może stanowić w przyszłości cel w terapii raka szyjki macicy, oraz posłużyć
jako marker prognostyczny u chorych na ten nowotwór [70].

Struktura i funkcje białka Klotho 182
Podsumowanie
Gen Klotho zaangażowany jest w regulację procesu starzenia u ssaków.
Za sprawą istnienia dwóch form transbłonowej oraz sekrecyjnej tego białka
uczestniczy ono w regulacji licznych procesów komórkowych. Spełnia ono m.in.
rolę koreceptora dla FGF23, odgrywając istotną rolę w regulacji homeostazy
fosforanów oraz metabolizmu witaminy D. Jest inhibitorem szlaku
insuliny/insulinopodobnego czynnika wzrostu przez co przyczynia się m.in. do
redukcji stresu oksydacyjnego. Dzięki aktywności enzymatycznej modyfikuje
N-glikany kanałów TRPV5, zwiększając nerkową resorpcję jonów wapnia.
Molekularny mechanizm działania Klotho nie został jednak do końca poznany.
Jego przyszłe poznanie może przyczynić się do lepszego zrozumienia procesu
starzenia się organizmów, a także umożliwić zapobieganie chorób związanych
z wiekiem. Ostatnie doniesienia o supresorowej funkcji Klotho w wielu typach
nowotworów, sugerują, że białko to może posłużyć w przyszłości jako marker
molekularny bądź też stać się nowym celem w terapii nowotworów.
Piśmiennictwo
1. Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T i wsp.
Mutation of the mouse klotho gene leads to a syndrome resembling ageing.
Nature 1997; 390: 45-51.
2. Wang Y, Sun Z. Current understanding of Klotho. Ageing Res Rev. 2009;
8: 43-51.
3. Kurosu H, Kuro-o M. The Klotho gene family as a regulator of endocrine
fibroblast growth factors. Mol Cell Endocrinol. 2009; 299: 72-78.
4. Kuro-o M. Klotho and aging. Biochim Biophys Acta 2009; 1790: 1049-1058.
5. Kurosu H, Yamamoto M, Clark JD, Pastor JV, Nandi A, Gurnani P i wsp.
Suppression of aging in mice by the hormone Klotho. Science 2005;
309: 1829-1833.
6. Kuro-o M. Klotho and βKlotho. Adv Exp Med Biol. 2012; 728: 25-40.

A. Szymczyk, E. Forma 183
7. Manya H, Akasaka-Manya K, Endo T. Klotho protein deficiency and aging.
Geriatr Gerontol Int. 2010; 10 Suppl 1: 80-87.
8. Matsumura Y, Aizawa H, Shiraki-Iida T, Nagai R, Kuro-o M, Nabeshima Y.
Identification of the human klotho gene and its two transcripts encoding
membrane and secreted klotho protein. Biochem Biophys Res Commun. 1998;
242: 626-630.
9. Kuro-o M. Klotho and aging process. Korean J Intern Med 2011; 26:113-122.
10. Forster RE, Jurutka PW, Hsieh JC, Haussler CA, Lowmiller CL, Kaneko I i wsp.
Vitamin D receptor controls expression of the anti-aging klotho gene in mouse
and human renal cells. Biochem Biophys Res Commun. 2011; 414: 557-562.
11. Imura A, Iwano A, Tohyama O, Tsuji Y, Nozaki K, Hashimoto N i wsp.
Secreted Klotho protein in sera and CSF: implication for post-translational
cleavage in release of Klotho protein from cell membrane. FEBS Lett. 2004;
565: 143-147.
12. Huang CL. Regulation of ion channels by secreted Klotho: mechanisms and
implications. Kidney Int. 2010; 77: 855-860.
13. Tohyama O, Imura A, Iwano A, Freund JN, Henrissat B, Fujimori T, Nabeshima
Y. Klotho is a novel beta-glucuronidase capable of hydrolyzing steroid
beta-glucuronides. J Biol Chem 2004; 279: 9777-9784.
14. Ben-Dov IZ, Galitzer H, Lavi-Moshayoff V, Goetz R, Kuro-o M, Mohammadi M
i wsp. The parathyroid is a target organ for FGF23 in rats. J Clin Invest. 2007;
117: 4003-4008.
15. Kamemori M, Ohyama Y, Kurabayashi M, Takahashi K, Nagai R, Furuya N.
Expression of Klotho protein in the inner ear. Hear Res 2002; 171: 103-110.
16. Kuro-o M. Klotho. Pflugers Arch. 2010; 459:333-343.
17. Chen CD, Podvin S, Gillespie E, Leeman SE, Abraham CR. Insulin stimulates
the cleavage and release of the extracellular domain of Klotho by ADAM10
and ADAM17. Proc Natl Acad Sci U S A. 2007; 104: 19796-19801.
18. Lewin E, Olgaard K. Klotho, an important new factor for the activity
of Ca2+ channels, connecting calcium homeostasis, ageing and uraemia. Nephrol
Dial Transplant 2006; 21: 1770-1772.
19. Dimke H, Hoenderop JG, Bindels RJ. Molecular basis of epithelial Ca2+ and
Mg2+ transport: insights from the TRP channel family. J Physiol. 2011; 589:
1535-1542.
20. Hoenderop JG, Bindels RJ. Calciotropic and magnesiotropic TRP channels.
Physiology (Bethesda) 2008; 23: 32-40.

Struktura i funkcje białka Klotho 184
21. Razzaque MS. Klotho and Na+,K+-ATPase activity: solving the calcium
metabolizm dilemma? Nephrol Dial Transplant 2008; 23: 459-461.
22. van Abel M, Hoenderop JG, Bindels RJ. The epithelial calcium channels TRPV5
and TRPV6: regulation and implications for disease. Naunyn Schmiedebergs
Arch Pharmacol. 2005; 371: 295-306.
23. Lu P, Boros S, Chang Q, Bindels RJ, Hoenderop JG. The β-glucuronidase klotho
exclusively activates the epithelial Ca2+ channels TRPV5 and TRPV6. Nephrol
Dial Transplant 2008; 23: 3397-3402.
24. Chang Q, Hoefs S, van der Kemp AW, Topala CN, Bindels RJ, Hoenderop JG.
The β-Glucuronidase Klotho hydrolyzes and activates the TRPV5 channel.
Science 2005; 310: 490-493.
25. Huang CL, Moe OW. Klotho: a novel regulator of calcium and phosphorus
homeostasis. Pflugers Arch. 2011; 462: 185-193.
26. Cha SK, Ortega B, Kurosu H, Rosenblatt KP, Kuro-O M, Huang CL. Removal of
sialic acid involving Klotho causes cell-surface retention of TRPV5 channel via
binding to galectin-1. Proc Natl Acad Sci USA 2008; 105: 9805-9810.
27. Imura A, Tsuji Y, Murata M, Maeda R, Kubota K, Iwano A i wsp. α-Klotho
as a regulator of calcium homeostasis. Science. 2007; 316: 1615-1618.
28. Nabeshima Y, Imura H. α-Klotho: a regulator that integrates calcium
homeostasis. Am J Nephrol 2008; 28: 455-464.
29. Bartke A. Long-lived Klotho mice: new insights into the roles of IGF-1 and
insulin in aging. Trends Endocrinol Metab. 2006; 17: 33-35.
30. Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid
metabolism. Nature 2001; 414: 799-806.
31. Krześlak A. Kinaza Akt: kluczowy regulator metabolizmu i progresji
nowotworów. Postępy Hig Med Dośw. 2010; 64: 490-503.
32. Kaletsky R, Murphy CT. The role of insulin/IGF-like signaling in C. elegans
longevity and aging. Dis Model Mech. 2010; 3: 415-419.
33. Clancy DJ, Gems D, Harshman LG, Oldham S, Stocker H, Hafen E i wsp.
Extension of life-span by loss of CHICO, a Drosophila insulin receptor substrate
protein. Science. 2001; 292: 104-106.
34. Rincon M, Rudin E, Barzilai N. The insulin/IGF-1 signaling in mammals and its
relevance to human longevity. Exp Gerontol. 2005; 40: 873-877.
35. Tatar M, Bartke A, Antebi A. The endocrine regulation of aging by insulin-like
signals. Science. 2003; 299: 1346-1351.
36. Brown-Borg HM. Hormonal regulation of longevity in mammals. Ageing Res
Rev. 2007; 6: 28-45.

A. Szymczyk, E. Forma 185
37. Holzenberger M, Dupont J, Ducos B, Leneuve P, Géloën A, Even PC i wsp.
IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice.
Nature. 2003; 421: 182-187.
38. Klöting N, Blühera M. Extended longevity and insulin signaling in adipose
tissue. Exp Gerontol. 2005; 40: 878-883.
39. Bluher M, Michael MD, Peroni OD, Ueki K, Carter N, Kahn BB i wsp.
Adipose tissue selective insulin receptor knockout protects against obesity
and obesity-related glucose intolerance. Dev. Cell 2002; 3: 25–38.
40. Laron Z. The essential role of IGF-1: Lessons from the long-term study and
treatment of children and adults with Laron syndrome. J Clin Endocrinol Metab.
1999; 84: 4397–4404.
41. Yamamoto M, Clark JD, Pastor JV, Gurnani P, Nandu A, Kurosu H i wsp.
Regulation of oxidative stress by the anti-aging hormone Klotho. J Biol Chem.
2005; 280: 38029–38034.
42. Carter ME, Brunet A. FOXO transcription factors. Curr Biol. 2007;
17: R113-114.
43. Imae M, Fu Z, Yoshida A, Noguchi T, Kato H. Nutritional and hormonal factors
control the gene expression of FoxOs, the mammalian homologues of DAF-16.
J Mol Endocrinol. 2003; 30: 253-262.
44. Ikushima M, Rakugi H, Ishikawa K i wsp. Anti-apoptotic and anti-senescence
effects of Klotho on vascular endothelial cells. Biochem Biophys Res Commun.
2006; 339: 827-832.
45. Gattineni J, Baum M. Regulation of phosphate transport by fibroblast growth
factor 23 (FGF23): conduplicatio for disorders of phosphate metabolism.
Pediatr Nephrol. 2010; 25: 591-601.
46. Razzaque MS. FGF23-mediated regulation of systemic phosphate homeostasis:
is Klotho an essential player? Am J Physiol Renal Physiol. 2009; 296:
F470-F476.
47. Kuro-o M. A potential link between phosphate and aging – lessons from. Mech
Ageing Dev. 2010; 131: 270–275.
48. Kuro-o M. Overview of the FGF23-Klotho axis. Pediatr Nephrol. 2010;
25: 583-590.
49. Kurosu H, Ogawa Y, Miyoshi M, Yamamoto M, Nandi A, Rosenblatt KP i wsp.
Regulation of fibroblast growth factor-23 signaling by Klotho. J Biol Chem.
2006; 281: 6120-6123.

Struktura i funkcje białka Klotho 186
50. Medici D, Razzaque MS, Deluca S, Rector TL, Hou B, Kang K i wsp.
FGF-23-Klotho signaling stimulates proliferation and prevents vitamin
D-induced apoptosis. J Cell Biol. 2008; 182: 459-465.
51. Gattineni J, Bates C, Twombley K, Dwarakanath V, L. Robinson M, Goetz R
i wsp. FGF23 decreases renal NaPi-2a and NaPi-2c expression and induces
hypophosphatemia in vivo predominantly via FGF receptor 1. Am J Physiol
Renal Physiol. 2009; 297: F282-F291.
52. Amatschek S, Haller M, Oberbauer R. Renal phosphate handling in human –
what can we learn from hereditary hypophosphataemias? Eur J Clin Invest. 2010;
40: 552-560.
53. Perwad F, Portale A. Vitamin D metabolism in the kidney: Regulation
by phosphorus and fibroblast growth factor 23. Mol Cell Endocrinol. 2011; 347:
17-24.
54. Mazzaferro S, Pasquali M, Pirrò G, Rotondi S, Tartaglione L. The bone and
the kidney. Arch Biochem Biophys. 2010; 503: 95-102.
55. Fon Tacer K, Bookout AL, Ding X, Kurosu H, John GB, Wang L i wsp.
Research resource: Comprehensive expression atlas of the fibroblast growth
factor system in adult mouse. Mol Endocrinol. 2010; 24: 2050-2064.
56. Goetz R, Beenken A, Ibrahimi OA, Kalinina J, Olsen SK, Eliseenkova AV i wsp.
Molecular insights into the Klotho-dependent, endocrine mode of action
of FGF19 subfamily members. Mol Cell Biol 2007; 27: 3417-3428.
57. Kalinina J, Dutta K, Ilghari D, Beenken A, Goetz R, Eliseenkova AV i wsp.
The alternatively spliced acid box region plays a key role in FGF receptor
autoinhibition. Structure. 2012; 20: 77-88.
58. Yie J, Wang W, Deng L, Tam LT, Stevens J, Chen MM, i wsp. Understanding
the physical interactions in the FGF21/FGFR/β-Klotho complex: structural
requirements and implications in FGF21 signaling. Chem Biol Drug Des. 2012;
79: 398-410.
59. Wu X, Lemon B, Li X, Gupte J, Weiszmann J, Stevens J, i wsp. C-terminal tail
of FGF19 determines its specificity toward Klotho co-receptors. J Biol Chem.
2008; 283: 33304-33309.
60. Prie D, Friedlander G. Reciprocal Control of 1,25-dihydroxyvitamin D and
FGF23 formation involving the FGF23/Klotho system. Clin J Am Soc Nephrol
2010; 5: 1717-1722.

A. Szymczyk, E. Forma 187
61. Lavi-Moshayoff V, Wasserman G, Meir T, Silver J, Naveh-Many T. PTH
increases FGF23 gene expression and mediates the high-FGF23 levels
of experimental kidney failure: a bone parathyroid feedback loop. Am J Physiol
Renal Physiol 2010; 299: F882-F889.
62. Hu MC, Shi M, Zhang J, Pastor J, Nakatani T, Lanske B, i wsp. Klotho: a novel
phosphaturic substance acting as an autocrine enzyme in the renal proximal
tubule. FASEB J. 2010; 24: 3438-3450.
63. Abramovitz L, Rubinek T, Ligumsky H, Bose S, Barshack I, Avivi C i wsp.
KL1 internal repeat mediates klotho tumor suppressor activities and inhibits
bFGF and IGF-I signaling in pancreatic cancer. Clin Cancer Res. 2011;
17: 4254-4266.
64. Wang L, Wang X, Wang X, Jie P, Lu H, Zhang S, i wsp. Klotho is silenced
through promoter hypermethylation in gastric cancer. Am J Cancer Res. 2011;
1: 111-119.
65. Wolf I, Levanon-Cohen S, Bose S, Ligumsky H, Sredni B, Kanety H, i wsp.
Klotho: a tumor suppressor and a modulator of the IGF-1 and FGF pathways
in human breast cancer. Oncogene. 2008; 27: 7094-7105.
66. Rubinek T, Shulman M, Israeli S, Bose S, Avraham A, Zundelevich A i wsp.
Epigenetic silencing of the tumor suppressor klotho in human breast cancer.
Breast Cancer Res Treat. 2012; 133: 649-657.
67. Usuda J, Ichinose S, Ishizumi T, Ohtani K, Inoue T, Saji H i wsp. Klotho predicts
good clinical outcome in patients with limited-disease small cell lung cancer who
received surgery. Lung Cancer. 2011; 74: 332-337.
68. Usuda J, Ichinose S, Ishizumi T, Ohtani K, Inoue T, Saji H i wsp. Klotho is
a novel biomarker for good survival in resected large cell neuroendocrine
carcinoma of the lung. Lung Cancer. 2011; 72: 355-359.
69. Pan J, Zhong J, Gan LH, Chen SJ, Jin HC, Wang X, Wang LJ. Klotho, an anti-
senescence related gene, is frequently inactivated through promoter
hypermethylation in colorectal cancer. Tumour Biol. 2011; 32: 729-735.
70. Lee J, Jeong DJ, Kim J, Lee S, Park JH, Chang B, i wsp. The anti-aging gene
KLOTHO is a novel target for epigenetic silencing in human cervical carcinoma.
Mol Cancer. 2010; 9: 109.
71. Kharaishvili G, Simkova D, Makharoblidze E, Trtkova K, Kolar Z, Bouchal J.
Wnt signaling in prostate development and carcinogenesis. Biomed Pap Med Fac
Univ Palacky Olomouc Czech Repub. 2011; 155: 11-18.


Folia Medica Lodziensia, 2012, 39/2:189-205
Corresponding author: Dr hab. Magdalena Bryś, prof. nadzw. UŁ; Katedra Cytobiochemii,
Uniwersytet Łódzki, 90-236 Łódź, ul. Pomorska 141/143; e-mail: [email protected];
Tel.:+ 48 42 6354371; Fax: +48 42 6354484
Genetic polymorphism of Klotho gene
and bladder cancer risk
Polimorfizm genu Klotho a ryzyko raka pęcherza moczowego
AGNIESZKA SZYMCZYK1, EWA FORMA1, ANNA KRZEŚLAK1, PAWEŁ JÓŹWIAK1, ADAM MADEJ2, WALDEMAR RÓŻAŃSKI2,
MAGDALENA BRYŚ1
1Katedra Cytobiochemii, Wydział Biologii i Ochrony Środowiska Uniwersytet Łódzki
2II Klinika Urologii Uniwersytetu Medycznego w Łodzi
Abstract
Introduction: Bladder cancer is the most frequent tumor of the urinary tract
in Poland. Klotho gene can act as an suppressor gene. Therefore,
variability of this gene might be implicated in the carcinogenesis of urinary
bladder. The aim of the study was analysis of the association between
the g.33590184 G>A (rs1207568), g.33634983 C>T (rs564481), g.33628193
G>C (rs9527025) polymorphisms of the Klotho gene and bladder cancer risk.
Materials and methods: The study included 96 patients diagnosed with
transitional cell carcinoma of the bladder (TCC) and 114 healthy, cancer-free
individuals. Three selected polymorphisms were typed by PCR with confronting
two-pair primers (PCR-CTPP) and Real Time PCR with TaqMan probes.
Results: The GA and AA genotypes of the rs1207568 polymorphism increased
the risk of bladder cancer (OR = 1.86, 95% CI [1.04-3.33], p = 0.03 and
OR = 6.58, 95% CI [1.27-34.02], p = 0.01, respectively). Individuals who were
heterozygous and homozygous for the A variant had 2.10-fold higher risk
of bladder cancer (OR = 2.10, 95% CI [1.20-3.65], p = 0.009). On the other
hand, heterozygous subjects and homozygous carriers of the wild-type allele (G)
had a decreased bladder cancer risk (OR = 0.19, 95% CI [0.04-0.95], p = 0.043).
Also, the occurrence of bladder cancer was positively correlated with
the presence of the GC genotype of the rs9527025 polymorphism (OR = 2.84,
95% CI [1.57-5.15], p = 0.0001).

Klotho gene and bladder cancer 190
Conclusions: Two polymorphisms of Klotho gene (rs1207568 and rs9527025)
may play a role in susceptibility to bladder cancer.
Key words: genetic polymorphism, Klotho, bladder cancer, SNP
Streszczenie
Wstęp: Rak pęcherza moczowego jest najczęściej występującym nowotworem układu moczowego w Polsce. Sugeruje się, że gen Klotho może pełnić funkcje genu supresorowego. W związku z tym, polimorfizm genu Klotho może mieć wpływ na proces transformacji nowotworowej pęcherza moczowego. Celem przedstawionych badań był analiza związku pomiędzy występowaniem wybranych polimorfizmów pojedynczych nukleotydów g.33590184 G>A (rs1207568), g.33634983 C>T (rs564481), g.33628193 G>C (rs9527025) a ryzykiem zachorowania na raka pęcherza moczowego. Materiały i metody: Do badań włączono 96 pacjentów ze zdiagnozowanym przejściowokomórkowym rakiem pęcherza moczowego (TCC; transitional cell carcinoma) oraz 114 osób zdrowych, u których nie stwierdzono choroby nowotworowej. Występowanie trzech wybranych polimorfizmów analizowano przy użyciu techniki PCR z dwiema parami przeciwstawnych starterów (PCR-CTPP; PCR with confronting two-pair primers) oraz metody Real Time PCR z sondami fluorescencyjnymi TaqMan. Wyniki: Genotypy GA i AA polimorfizmu rs1207568 wpływają na wzrost ryzyka zachorowania na przejściowokomórkowego raka pęcherza moczowego (OR = 1,86, 95% PU [1,04-3,33], p = 0,03 oraz OR = 6,58, 95% CI [1,27-34,02], p = 0,01, odpowiednio). U osób będących heterozygotami lub homozygotami pod względem allela A wykazano ponad 2-krotnie wyższe ryzyko zachorowania na raka pęcherza moczowego (OR = 2,10, 95% PU [1,20-3,65], p = 0,009). Natomiast w przypadku nosicieli allela G, w układzie homozygotycznym lub heterozygotycznym, obserwowano spadek ryzyka zachorowania na badany nowotwór (OR = 0,19, 95% PU [0,04-0,95], p = 0,043). W przypadku polimorfizmu rs952705 wykazano, że genotyp GC zwiększa ryzyko zachorowania na przejściowokomórkowego raka pęcherza moczowego (OR = 2,84, 95% PU [1,57-5,15], p = 0,0001).

A. Szymczyk et al. 191
Wnioski: Dwa spośród badanych polimorfizmów genu Klotho (rs1207568 i rs9527025) mogą mieć wpływ na predyspozycję do zachorowania na przejściowokomórkowego raka pęcherza moczowego.
Słowa kluczowe: polimorfizm genów, Klotho, rak pęcherza, SNP Introduction
In Europe, bladder cancer is the 5th most commonly diagnosed cancer type
and the 9th leading cause of cancer mortality [1]. In addition, bladder cancer
is the most frequent tumor of the urinary tract in Poland [2]. It affects men more
frequently than women. Typical of solid tumors, bladder cancer incidence
increases with age. Tumor of the bladder rarely occur before the age of 40 – 50,
arising most commonly in the seventh decade of life [1]. The most important risk
factors for the development of bladder cancer are smoking and occupational
exposure to toxic chemicals, such as aromatic amines and polycyclic aromatic
hydrocarbons [3]. Recent studies have facilitated even more rapid progress in the
identification of the molecular mechanisms involved in bladder cancer initiation
and progression. Many transforming and tumor suppressor genes, such as
FGFRs (fibroblast growth factor receptors), Ras, p53 and RB (retinoblastoma)
may be involved in bladder carcinogenesis [4, 5]. The fibroblast growth factor
receptors (FGFRs) play a role in tumor associated angiogenesis, as some
receptors (FGFR3) are mutated and others (FGFR 1, 2, and 4) are abnormally
expressed in bladder cancers. Overexpression of wild-type FGFR4 in bladder
tumors can be associated with progression and aggressive clinical behavior of
this cancer [5]. Recent studies indicate that Klotho (KL) protein can modulate
FGF/FGFR signaling and act as tumor suppressor [6]. The Klotho gene named
after a Greek goddess who spins the thread of life, was originally identified as

Klotho gene and bladder cancer 192
a gene mutated in a mouse strain that inherits a premature aging syndrome
in an autosomal recessive manner [7, 8]. The Klotho gene is composed of five
exons and encodes a type I single-pass transmembrane protein of 1,012 amino
acid long (~130 kDa). The intracellular part of Klotho protein is very short
(10 amino acid) and has no known functional domains. The extracellular domain
is composed of two internal repeats with homology to family 1 glycosidases that
hydrolyze β-glucosidic linkage in saccharides, glycoproteins, and glycolipids
[9, 10]. The extracellular domain is also subject to ectodomain shedding.
As a result, the entire extracellular domain is released into the extracellular space
and is detectable in blood, urine, and cerebrospinal fluid [11, 12]. Thus,
the Klotho protein exist in two forms: membrane Klotho and secreted Klotho.
Membrane Klotho functions as a coreceptor for endocrine FGF23 that regulates
excretion of phosphate and synthesis of active vitamin D [13-16]. Secreted
Klotho functions as a humoral factor with pleiotropic activities, including
suppression of growth factor signaling, suppression of oxidative stress, and
regulation of ion channels and transporters [17, 18]. Furthermore, secreted
Klotho protein may modify glycans of receptor tyrosine kinases, such as insulin-
and IGF-1 receptors and FGFRs, which inhibits their activity and/or alters cell
surface abundance [19]. Goetz et al. [20] suggested that the association of
Klotho protein with FGFR not only enhances the binding affinity of the receptor
for endocrine FGFs such as FGF23 but at the same time also suppresses the
binding and activation of FGFR by FGF8 subfamily ligands and possibly other
paracrine FGFs [20].
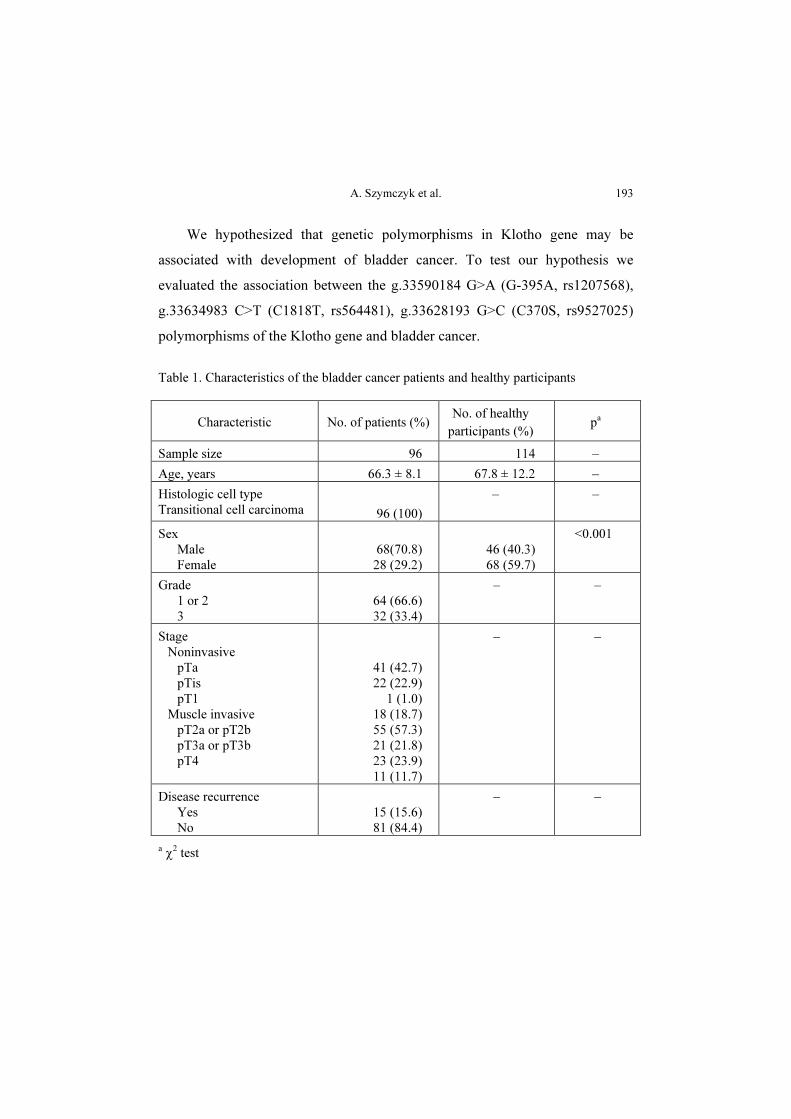
A. Szymczyk et al. 193
We hypothesized that genetic polymorphisms in Klotho gene may be
associated with development of bladder cancer. To test our hypothesis we
evaluated the association between the g.33590184 G>A (G-395A, rs1207568),
g.33634983 C>T (C1818T, rs564481), g.33628193 G>C (C370S, rs9527025)
polymorphisms of the Klotho gene and bladder cancer.
Table 1. Characteristics of the bladder cancer patients and healthy participants
Characteristic No. of patients (%) No. of healthy
participants (%) pa
Sample size 96 114 –
Age, years 66.3 ± 8.1 67.8 ± 12.2 –
Histologic cell type
Transitional cell carcinoma
96 (100)
–
–
Sex
Male
Female
68(70.8)
28 (29.2)
46 (40.3)
68 (59.7)
<0.001
Grade
1 or 2
3
64 (66.6)
32 (33.4)
– –
Stage
Noninvasive
pTa
pTis
pT1
Muscle invasive
pT2a or pT2b
pT3a or pT3b
pT4
41 (42.7)
22 (22.9)
1 (1.0)
18 (18.7)
55 (57.3)
21 (21.8)
23 (23.9)
11 (11.7)
– –
Disease recurrence
Yes
No
15 (15.6)
81 (84.4)
– –
a χ2 test

Klotho gene and bladder cancer 194
Material and methods
Patients
The study included 96 patients diagnosed with transitional cell carcinoma
of the bladder at the 2nd Clinic of Urology, Medical University of Łódź, Poland
between 2009 and 2012. The gender was 68 male and 28 female. The mean age
of patients was 66.3 years. Clinicopathological characteristics of participants are
shown in Table 1. The control group consisted of 114 healthy individuals
(46 male and 68 female) recruited from the 2nd Clinic of Urology, Medical
University of Łódź, Poland, from periodic health check-ups. The controls were
genetically unrelated cancer-free individuals and frequency matched to the cases
based on age (mean age 67.8 years). This study was approved by the Ethical
Committee of the Medical University of Łódź. Each subject donated a venous
blood samples of ~ 2 mL, 250 µL of which was used for genomic DNA
extraction. Blood samples of all patients and controls were collected
into ethylenediaminetetraacetic acid (EDTA) tubes and store at -20°C until
further use.
DNA preparation and storage
Genomic DNA was obtained from a 250 µl aliquot of blood using
a commercially available AxyPrepTM Blood genomic DNA Miniprep Kit
(Axygen Biosciences, Union City, CA, USA), according to the manufacturer’s
instructions. DNA purity and concentration were determined spectro-
photometrically at 260 and 280 nm. The purified genomic DNA was stored
in TE buffer at -20°C.

A. Szymczyk et al. 195
Table 2. Sequence of primers and probes used for the genotyping of Klotho gene
SN
P
Method of
genotyping Sequence (5’-3’) Reference
rs1
20
75
68
PCR-CTPP
F1:GTTTCGTGGACGCTCAGGTTCATTCTC
(21) F2:GAGAAAAGGCGCCGACCAACTTTC
R1:GATCCCGCCCC CAAGTCGGGA
R2:GTCCCTCTAGGATTTCGGCCAG
rs5
64
48
1
PCR-CTPP
F1:CTCAGTTTACCGACCTGAATGTTTACCTG
(21) F2:CAGATCGCTTTACTCCAGGAAATGCAC
R1:GTCCAGGGAGAAGCGAAAATGTGTAACA
R2:5’- GAGCTCTTGAAAGCACAGTCGGGC-3’
rs9
52
70
25
TaqMan®
assay
F:AGTTCATCAAAGGAACTGCT GACTT
(22)
R:CTTCATGTGAGGGTCCAAAA GTT
S1 (allele G) FAM-
TTGCTCTTTGCTTTGGACCCACCT-BHQ1
S2 (allele C) VIC-
TTGCTCTTTCCTTTGGACCCACCTT-BHQ1

Klotho gene and bladder cancer 196
PCR-CTPP
The genotyping of g.33590184 G>A (G-395A, rs1207568) in the promoter
region and g.33634983 C>T (C1818T, rs564481) in exon 4 was performed using
PCR with confronting two-pair primers (PCR-CTPP) assay (21). In this assay,
confronting pairs of primers (four primers in all) are used as shown in Table 2.
The regions containing studied polymorphism were amplified by PCR with these
primers with the initial denature at 95°C for 1 min, at 65°C (rs1207568) or 69°C
(rs564481) for 1 min, at 72°C for 1 min and additionally at 72°C for 10 min.
PCR products were visualized on a 2% agarose gel with ethidium bromide
staining. Genotyping was performed as follows: rs1207568 – 252, 175 bp for
GG genotype, 252, 175, 121 bp for GA genotype, 252, 121 bp for AA genotype
(Fig. 1) and rs564481 – 416, 291 bp for CC genotype, 416, 291, 179 bp for CT
genotype, and 416, 179 bp for TT genotype (Fig. 2).
Fig. 1. Genotypes of the g.33590184 G>A (G-395A) polymorphism (rs1207568)
determined by PCR-CTPP method and analyzed by 2% agarose gel electrophoresis
stained with ethidium bromide and viewed under UV.

A. Szymczyk et al. 197
Fig. 2. Genotypes of the g.33634983 C>T (C1818T) polymorphism (rs564481)
determined by PCR-CTPP method and analyzed by 2% agarose gel electrophoresis
stained with ethidium bromide and viewed under UV.
TaqMan® Assay
Genotyping for g.33628193 G>C (C370S, rs9527025) allelic variant
in exone two was performed with using TaqMan® genotyping assay in which
a fluorogenic probe, consisting of an oligonucleotide labeled with both
a fluorescent reporter dye (FAM or VIC) and a quencher dye, is included
in a typical PCR. Amplification of the probe-specific product causes cleavage
of the probe, generating an increase in reporter fluorescence. Sequences of pair
of primers and two probes are shown in Table 2 (22). The PCR thermal cycling
was as follows: initial denaturing at 95°C for 10 min; 45 cycles of 95°C for
30 sec and 60°C for 60 sec were carried out and fluorescence was measured
at the end of each cycle and at endpoint. Detection was carried out by qPCR
in an Eppendprf realplex thermocycler (Eppendorf, Germany). All samples were
determined in duplicate and genotypes were assigned by gene identification
software (RealPlex 2.0, Eppendorf, Germany).

Klotho gene and bladder cancer 198
Statistical data analysis
Genotype distributions were evaluated for agreement with Hardy–Weinberg
equilibrium by the Chi-square test. All genotype distributions of Klotho gene fit
Hardy–Weinberg equilibrium. Unconditional multiple logistic regression models
were used to calculate odds ratios (ORs) and 95 % confidence intervals (CIs) for
the association of genotype with bladder cancer risk. Genotype data were
analyzed with the homozygote of the common allele as the reference group.
Variants of homozygotes and heterozygotes were combined to evaluate
the dominant effect. Reported p values were two sided. Probabilities were
considered significant whenever p value was lower than 0.05. All analyses were
completed using STATA software (version 11.0 Stata-Corp., Texas, USA).
Results
The PCR-CTPP and Real Time TaqMan® PCR analyses were successful
for all cases and controls. The characteristics of the patients and controls are
presented in Table 1. There were significantly more male among the patients
than controls (70.8% vs 40.3%, p<0.001). The genotype and allele distributions
of the g.33590184 G>A (rs1207568), g.33634983 C>T (rs564481) and
g.33628193 G>C (rs9527025) Klotho polymorphisms in bladder cancer patients
and controls are summarized in Table 3. In our study, all the observed genotype
frequencies were in agreement with Hardy-Weinberg equilibrium calculated
for the cases and controls (p>0.05). As shown in Table 3 the differences
in the frequency distributions of genotypes of the rs1207568 and
rs9527025 polymorphisms between the cases and controls were statistically
significant (p<0.05).
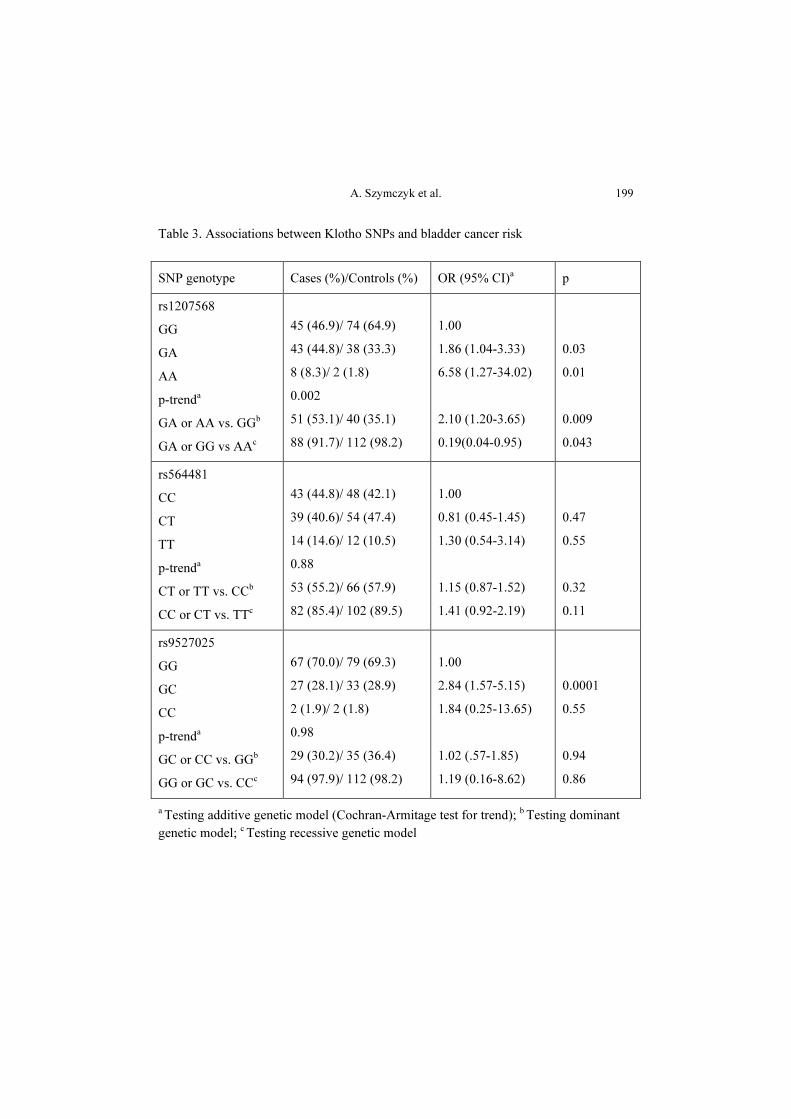
A. Szymczyk et al. 199
Table 3. Associations between Klotho SNPs and bladder cancer risk
SNP genotype Cases (%)/Controls (%) OR (95% CI)a p
rs1207568
GG
GA
AA
p-trenda
GA or AA vs. GGb
GA or GG vs AAc
45 (46.9)/ 74 (64.9)
43 (44.8)/ 38 (33.3)
8 (8.3)/ 2 (1.8)
0.002
51 (53.1)/ 40 (35.1)
88 (91.7)/ 112 (98.2)
1.00
1.86 (1.04-3.33)
6.58 (1.27-34.02)
2.10 (1.20-3.65)
0.19(0.04-0.95)
0.03
0.01
0.009
0.043
rs564481
CC
CT
TT
p-trenda
CT or TT vs. CCb
CC or CT vs. TTc
43 (44.8)/ 48 (42.1)
39 (40.6)/ 54 (47.4)
14 (14.6)/ 12 (10.5)
0.88
53 (55.2)/ 66 (57.9)
82 (85.4)/ 102 (89.5)
1.00
0.81 (0.45-1.45)
1.30 (0.54-3.14)
1.15 (0.87-1.52)
1.41 (0.92-2.19)
0.47
0.55
0.32
0.11
rs9527025
GG
GC
CC
p-trenda
GC or CC vs. GGb
GG or GC vs. CCc
67 (70.0)/ 79 (69.3)
27 (28.1)/ 33 (28.9)
2 (1.9)/ 2 (1.8)
0.98
29 (30.2)/ 35 (36.4)
94 (97.9)/ 112 (98.2)
1.00
2.84 (1.57-5.15)
1.84 (0.25-13.65)
1.02 (.57-1.85)
1.19 (0.16-8.62)
0.0001
0.55
0.94
0.86
a Testing additive genetic model (Cochran-Armitage test for trend); b Testing dominant
genetic model; c Testing recessive genetic model

Klotho gene and bladder cancer 200
An association between bladder cancer and the GA and AA genotypes of
the rs1207568 polymorphism was found. Both genotypes increased the risk
of bladder cancer occurence (OR = 1.86, 95% CI [1.04-3.33], p = 0.03 and
OR = 6.58, 95% CI [1.27-34.02], p = 0.01, respectively). Moreover, we found
that individuals who were heterozygous and homozygous for the A variant had
2.10-fold higher risk of bladder cancer (OR = 2.10, 95% CI [1.20-3.65],
p=0.009). On the other hand, heterozygous subjects and homozygous carriers of
the wild-type allele (G) had a decreased bladder cancer risk (OR = 0.19, 95% CI
[0.04-0.95], p=0.043). In addition, the occurrence of bladder cancer was
positively correlated with the presence of the GC genotype of the rs9527025
polymorphism (OR=2.84, 95% CI [1.57-5.15], p = 0.0001).
Discussion
In recent years, the role of genetic factors in bladder cancer development
has been extensively investigated. Numerous case-control studies have
confirmed the association between single nucleotide polymorphisms (SNPs)
and bladder cancer [23-25]. We analyzed three single nucleotide polymorphisms
in Klotho gene. One SNP (rs1207568) is located in the promoter region of
Klotho gene. We also analyzed a G to C transversion at the position g.33628192
(rs9527025) located in the second exon of this gene. This polymorphism causes
change of cysteine to serine at the amino acid position 370 (C370S) of Klotho
protein. The third SNP (rs564481) is located within exon 4 and is synonymous
mutation (His589His) [26]. We found significant association between rs1207568
and rs9527025 polymorphisms and bladder cancer risk in Polish population.
To our knowledge, this is the first study to report an association of SNPs
in Klotho with the risk of bladder cancer. Polymorphisms of Klotho gene have

A. Szymczyk et al. 201
been studied for their associations with risk for a variety of disease, such as
osteoarthritis, ischemic stroke, coronary artery disease, metabolic syndrome and
type 2 diabetes [22, 26-30]. There are a few studies analyzing the correlation
between Klotho SNPs and breast and ovarian cancer risk [31, 32]. A functional
variant of Klotho, termed KL-VS, contains six sequence variants in complete
linkage disequilibrium, two of which result in amino acid substitutions F352V
and C370S. The presence of phenylalanine at a position equivalent to position
352 in the human Klotho gene is highly conserved among species and its
substitution to valine may alter the excretion and enzymatic activity
of the protein [31]. Wolf et al. [31] analyzed the association between KL-VS and
cancer risk among BRCA1/BRCA2 mutation carriers of Ashkenazi origin.
Among BRCA1 carriers, heterozygosity for the KL-VS allele was associated
with increased breast and ovarian cancer risk and younger age at breast cancer
diagnosis [31]. The effect of KL-VS variant on breast and ovarian cancer risk
in non-Jewish BRCA1/BRCA2 mutation carriers has not been reported [32].
Moreover, recent studies indicate that Klotho protein can act as tumor
suppressor [6]. Analysis of KL protein expression in breast tissue revealed high
Klotho expression in normal breast samples but very low expression in breast
cancer [33, 34]. Klotho gene expression was also downregulated in cell lines and
tissue samples of human gastric, colorectal, cervical and lung cancer [35-39].
In the case of cervical cancer samples, loss of Klotho mRNA was observed
in invasive carcinoma but not during the early, preinvasive phase of primary
cervical tumorigenesis. Abramovitz et al. [6] showed that Klotho expression
is downregulated in pancreatic adenocarcinoma. Overexpression of KL, or
treatment with soluble Klotho, reduced growth of pancreatic cancer cells in vitro
and in vivo [6].

Klotho gene and bladder cancer 202
In conclusion, our work shows that genetic polymorphisms of the Klotho
gene may be associated with development of bladder cancer. These findings may
be helpful in increasing our understanding of carcinogenesis of urinary bladder.
References
1. Letasiova S, Medve'ova A, Sovcikova A, Dusinska M, Volkovova K, Mosoiu C
et al. Bladder cancer, a review of the environmental risk factors. Environ Health.
2012; 11 Suppl 1: 11.
2. Nawrocki S, Milecki P, Skacel T, Skoneczna I, Kwias Z. Treatment of bladder
cancer: present and perspectives. Współczesna Onkologia 2002; 7: 465-472.
3. Griffiths TR. Current perspectives in bladder cancer management. Int J Clin
Pract. 2012; doi: 10.1111/ijcp.12075.
4. Al Hussain TO, Akhtar M. Molecular basis of urinary bladder cancer. Adv Anat
Pathol. 2013; 20: 53-60.
5. Czerniak B. Molecular pathology and biomarkers of bladder cancer. Cancer
Biomark. 2010; 9: 159-176.
6. Abramovitz L, Rubinek T, Ligumsky H, Bose S, Barshack I, Avivi C et al. KL1
internal repeat mediates klotho tumor suppressor activities and inhibits bFGF and
IGF-I signaling in pancreatic cancer. Clin Cancer Res. 2011; 17: 4254-4266.
7. Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T et al.
Mutation of the mouse klotho gene leads to a syndrome resembling ageing.
Nature 1997; 390: 45-51.
8. Kuro-o M. Overview of the FGF23-Klotho axis. Pediatr Nephrol. 2010;
25: 583-590.
9. Kuro-o M. Klotho. Pflugers Arch. 2010; 459: 333-343.
10. Mian IS. Sequence, structural, functional, and phylogenetic analyses of three
glycosidase families. Blood Cells Mol Dis. 1998; 24: 83-100.
11. Imura A, Iwano A, Tohyama O, Tsuji Y, Nozaki K, Hashimoto N et al. Secreted
Klotho protein in sera and CSF: implication for post-translational cleavage
in release of Klotho protein from cell membrane. FEBS Lett. 2004; 565: 143-147.
12. Bloch L, Sineshchekova O, Reichenbach D, Reiss K, Saftig P, Kuro-o M et al.
Klotho is a substrate for alpha-, beta- and gamma-secretase. FEBS Lett. 2009;
583: 3221-3224.

A. Szymczyk et al. 203
13. Kurosu H, Ogawa Y, Miyoshi M, Yamamoto M, Nandi A, Rosenblatt KP et al.
Regulation of fibroblast growth factor-23 signaling by klotho. J Biol Chem.
2006; 281: 6120-6123.
14. Urakawa I, Yamazaki Y, Shimada T, Iijima K, Hasegawa H, Okawa K et al.
Klotho converts canonical FGF receptor into a specific receptor for FGF23.
Nature 2006; 444: 770-774.
15. Kuro-o M. Klotho as a regulator of fibroblast growth factor signaling
and phosphate/calcium metabolism. Curr Opin Nephrol Hypertens. 2006;
15: 437-441.
16. Martin A, David V, Quarles LD. Regulation and function of the FGF23/klotho
endocrine pathways. Physiol Rev. 2012; 92: 131-155.
17. Kuro-o M. Klotho and the aging process. Korean J Intern Med. 2011;
26: 113-122.
18. Yamamoto M, Clark JD, Pastor JV, Gurnani P, Nandi A, Kurosu H et al.
Regulation of oxidative stress by the anti-aging hormone klotho. J Biol Chem.
2005; 280: 38029-38034.
19. Kuro-o M. Klotho and aging. Biochim Biophys Acta 2009; 1790: 1049-1058.
20. Goetz R, Ohnishi M, Ding X, Kurosu H, Wang L, Akiyoshi J et al. Klotho
coreceptors inhibit signaling by paracrine fibroblast growth factor 8 subfamily
ligands. Mol Cell Biol. 2012; 32: 1944-1954.
21. Shimoyama Y, Nishio K, Hamajima N, Niwa T. KLOTHO gene polymorphisms
G-395A and C1818T are associated with lipid and glucose metabolism, bone
mineral density and systolic blood pressure in Japanese healthy subjects. Clin
Chim Acta 2009; 406: 134-138.
22. Rhee EJ, Oh KW, Lee WY, Kim SY, Jung CH, Kim BJ et al. The differential
effects of age on the association of KLOTHO gene polymorphisms with coronary
artery disease. Metabolism 2006; 55: 1344-1351.
23. Dodurga Y, Tataroglu C, Kesen Z, Satiroglu-Tufan NL. Incidence of fibroblast
growth factor receptor 3 gene (FGFR3) A248C, S249C, G372C, and T375C
mutations in bladder cancer. Genet Mol Res. 2011; 10: 86-95.
24. Liu ZH, Bao ED. Quantitative assessment of the association between TP53
Arg72Pro polymorphism and bladder cancer risk. Mol Biol Rep. 2012; doi:
10.1007/s11033-012-2319-z
25. Zhi Y, Yu J, Liu Y, Wei Q, Yuan F, Zhou X et al. Interaction between
polymorphisms of DNA repair genes significantly modulated bladder cancer
risk. Int J Med Sci. 2012; 9: 498-505.

Klotho gene and bladder cancer 204
26. Zhang F, Zhai G, Kato BS, Hart DJ, Hunter D, Spector TD et al. Association
between KLOTHO gene and hand osteoarthritis in a female Caucasian
population. Osteoarthritis Cartilage. 2007; 15: 624-629.
27. Tsezou A, Furuichi T, Satra M, Makrythanasis P, Ikegawa S, Malizos KN.
Association of KLOTHO gene polymorphisms with knee osteoarthritis in Greek
population. J Orthop Res. 2008; 26: 1466-1470.
28. Majumdar V, Nagaraja D, Christopher R. Association of the functional KL-VS
variant of Klotho gene with early-onset ischemic stroke. Biochem Biophys Res
Commun. 2010; 403: 412-416.
29. Majumdar V, Christopher R. Association of exonic variants of Klotho with
metabolic syndrome in Asian Indians. Clin Chim Acta 2011; 412: 1116-1121.
30. Freathy RM, Weedon MN, Melzer D, Shields B, Hitman GA, Walker M et al.
The functional "KL-VS" variant of KLOTHO is not associated with type 2
diabetes in 5028 UK Caucasians. BMC Med Genet. 2006; 7: 51.
31. Wolf I, Laitman Y, Rubinek T, Abramovitz L, Novikov I, Beeri R et al.
Functional variant of KLOTHO: a breast cancer risk modifier among BRCA1
mutation carriers of Ashkenazi origin. Oncogene 2010; 29: 26-33.
32. Laitman Y, Kuchenbaecker KB, Rantala J, Hogervorst F, Peock S, Godwin AK,
Arason A et al. The KL-VS sequence variant of Klotho and cancer risk in
BRCA1 and BRCA2 mutation carriers. Breast Cancer Res Treat. 2012; 132:
1119-1126.
33. Wolf I, Levanon-Cohen S, Bose S, Ligumsky H, Sredni B, Kanety H et al.
Klotho: a tumor suppressor and a modulator of the IGF-1 and FGF pathways
in human breast cancer. Oncogene 2008; 27: 7094-7105.
34. Rubinek T, Shulman M, Israeli S, Bose S, Avraham A, Zundelevich A et al.
Epigenetic silencing of the tumor suppressor klotho in human breast cancer.
Breast Cancer Res Treat. 2012; 133: 649-657.
35. Wang L, Wang X, Wang X, Jie P, Lu H, Zhang S et al. Klotho is silenced
through promoter hypermethylation in gastric cancer. Am J Cancer Res. 2011; 1:
111-119.
36. Pan J, Zhong J, Gan LH, Chen SJ, Jin HC, Wang X et al. Klotho, an anti-
senescence related gene, is frequently inactivated through promoter
hypermethylation in colorectal cancer. Tumour Biol. 2011; 32: 729-735.
37. Lee J, Jeong DJ, Kim J, Lee S, Park JH, Chang B et al. The anti-aging gene
KLOTHO is a novel target for epigenetic silencing in human cervical carcinoma.
Mol Cancer. 2010; 9: 109.

A. Szymczyk et al. 205
38. Usuda J, Ichinose S, Ishizumi T, Ohtani K, Inoue T, Saji H et al. Klotho is a
novel biomarker for good survival in resected large cell neuroendocrine
carcinoma of the lung. Lung Cancer 2011; 72: 355-359.
39. Usuda J, Ichinose S, Ishizumi T, Ohtani K, Inoue T, Saji H et al. Klotho predicts
good clinical outcome in patients with limited-disease small cell lung cancer who
received surgery. Lung Cancer 2011; 74: 332-337.


Folia Medica Lodziensia, 2012, 39/2: 207-244
Adres do korespondencji: dr Ewa Forma; Katedra Cytobiochemii, Uniwersytet Łódzki; 90-236 Łódź; ul. Pomorska 141/143; e-mail: [email protected], Tel.: + 48 42 6354371; Fax: +48 42 6354484
Rola metalotionein i kadmu w rozwoju raka piersi
Role of metalothioneins and cadmium in breast carcinogenesis
EWA FORMA, MAGDALENA BRYŚ
Katedra Cytobiochemii, Wydział Biologii i Ochrony Środowiska Uniwersytet Łódzki
Streszczenie Metalotioneina (MT) została po raz pierwszy odkryta ponad 50 lat temu, jako
białko wiążące kadm w nerce konia. Metalotioneiny są grupą powszechnie
występujących białek o niskiej masie cząsteczkowej, wysokiej zawartości reszt
cysteiny, których grupy tiolowe wiążą jony takich metali jak kadm (Cd), cynk (Zn)
i miedź (Cu). U ssaków wyróżnia się cztery izoformy metalotioneiny oznaczone
jak MT-1 – MT-4. Do podstawowych funkcji metalotionein zalicza się utrzymanie
homeostazy metali ciężkich, ochronę przed ich toksycznym działaniem, a także
ochronę przed stresem oksydacyjnym. Kadm, który został zakwalifikowany do
substancji karcynogennych I klasy, może między innymi stymulować proliferację
komórek, hamować naprawę uszkodzeń DNA, zaburzać proces apoptozy oraz
promować rozwój nowotworów w licznych tkankach. Kadm znajdujący się
w środowisku naturalnym pochodzi ze źródeł naturalnych, jak i antropogenicz-
nych, przy czym ilość kadmu dostająca się do środowiska w wyniku działalności
człowieka od 3 do 10 razy przekracza ilości kadmu pochodzące ze źródeł
naturalnych. Kadm akumuluje się w organizmie człowieka, a jego okres
biologicznego półtrwania wynosi od 10 do 30 lat. Zarówno akumulacja Cd, jak
i zaburzenia ekspresji metalotionein mogą mieć związek z rozwojem wielu
typów nowotworów, w tym raka piersi.
Słowa kluczowe: metalotioneiny, kadm, rak piersi.
Abstract
Metallothionein (MT) was first discovered over 50 years ago, as a cadmium-binding protein in the kidney of a horse. Metallothioneins are a class of

Metalotioneiny i kadm a rak piersi 208
ubiquitously occurring cysteine and metal-rich proteins of low molecular weight, containing sulfur-based metal clusters formed with zinc (Zn), cadmium (Cd), and copper (Cu). In mammals, four distinct MT isoforms designated MT-1 – MT-4 are identified. The major physiological functions of metallothioneins include the homeostasis of essential metals Zn and Cu and the protection against oxidative stress and toxic effects of heavy metals such as Cd. Cadmium ,which has been classified as class I carcinogen, can stimulate cell proliferation, inhibit DNA repair, impair apoptosis and promote the development of cancer in a number of tissues. Significant quantities of Cd are introduced into the environment both by natural and anthropogenic activities, with anthropogenic activities contributing 3–10 times more Cd to the environment than natural activities. Cadmium accumulates in the human body with a long biological half-life ranging from 10 to 30 years. Cd accumulation and the aberrant expression of MTs have been found to be associated with the development of many types of cancers, including breast cancer.
Key words: metallothioneins, cadmium, breast cancer. Wstęp
Metalotioneiny (MTs, ang. metallothioneins) to grupa wewnątrz-
komórkowych białek charakteryzujących się niską masą cząsteczkową
(6 – 7 kDa) oraz wysoką zawartością reszt cysteiny, które stanowią około 30%
wszystkich aminokwasów budujących łańcuch polipeptydowy. W białkach tych
brak jest aminokwasów aromatycznych oraz histydyny [1, 2]. MT mają zdolność
do wiązania różnych jonów metali, w tym cynku, kadmu, rtęci, miedzi, ołowiu
i srebra [2]. Białka te odpowiadają za homeostazę jonów cynku i miedzi,
detoksykację metali ciężkich (zwłaszcza Cd i Hg) oraz ochronę przed
uszkodzeniami oksydacyjnymi i apoptozą [3, 4]. Kadm jest związkiem
karcynogennym wykazującym negatywny wpływ na wiele procesów
komórkowych. Jony kadmu biorą udział we wszystkich etapach transformacji

E. Forma, M. Bryś 209
nowotworowej [5-7]. Związek metalotionein i kadmu z procesem
karcynogenezy analizowano w odniesieniu do wielu typów nowotworów, w tym
raka płuc, prostaty, jelita grubego, wątroby oraz nerek [8-13].
Klasyfikacja metalotionein
Podział metalotionein na rodziny, podrodziny, podgrupy i izoformy opiera
się na podobieństwie sekwencji i pokrewieństwie filogenetycznym.
Metalotioneiny podzielono na 4 grupy: MT-1, MT-2, MT-3 oraz MT-4. Geny
MT-1 kodują szereg izoform (MT-1A, -B, -E, -F, -G, -H, -I, -J, -K, -L, -X),
natomiast pozostałe białka z rodziny metalotionein kodowane są przez
pojedyncze geny (MT-2A, MT-3 i MT-4) [3].
Struktura metalotionein
Metalotioneina została po raz pierwszy wyizolowana z nerki konia przez
Margoshesa i Vallee’a w 1957 r., jako niskocząsteczkowe białko wiążące kadm.
W 1990 roku dzięki zastosowaniu spektroskopii NMR i krystalografii
rentgenowskiej określono trójwymiarową strukturę metalotioneiny [14, 15].
Geny metalotionein znajdują się na długim ramieniu chromosomu 16 (16q13)
[15]. MT-1 i MT-2A zbudowane są z 61, MT-3 z 68, a MT-4 z 62
reszt aminokwasowych. MT-3 jest najdłuższą izoformą metalotionein
w ludzkich komórkach. Różnica w długości łańcucha polipeptydowego między
MT-1/MT-2A a MT-3 związana jest z obecnością w tej ostatniej izoformie
reszty treoniny w pozycji 5 i heksapeptydu o charakterze kwaśnym poniżej
pozycji 53, których brak jest w MT-1 i MT-2A. Podobnie jak w MT-3,
tak i w MT-4 w pozycji 5 znajduje się reszta kwasu glutaminowego, której brak
jest w pozostałych izoformach. MT-3 wykazuje 70% zgodność sekwencji

Metalotioneiny i kadm a rak piersi 210
z MT-1/MT-2A, natomiast MT-4 różni się od MT-1 i MT-2A większością
aminokwasów niebędących resztami cysteiny [14, 16-19]. W cząsteczkach MT
nie występują aminokwasy aromatyczne [1, 2]. Wszystkie izoformy MT
zawierają w swojej strukturze 20 konserwatywnie zachowanych reszt cysteiny,
znajdujących się w obrębie charakterystycznych sekwencji: Cys-x-Cys, Cys-x-y-
Cys i Cys-Cys, gdzie x i y to aminokwasy inne niż Cys [14, 20, 21]. Obecność
dwóch konserwatywnych reszt proliny, znajdujących się w charakterystycznej
dla MT-3 sekwencji Cys-Pro-Cys-Pro, zawartej między pozycją 6 i 9 łańcucha
białkowego decyduje o aktywności biologicznej MT-3 [14, 17, 18]. Atomy
siarki grup tiolowych reszt cysteiny biorą udział w koordynacyjnym wiązaniu
jonów metali dwuwartościowych (Zn2+, Cd2+, Hg2+, Pb2+) i jednowartościowych
(Cu+, Ag+) [3]. Pierwszą izoformą MT, której struktura trójwymiarowa została
poznana była pochodząca z wątroby szczura MT-2A [14, 18]. Późniejsze
badania wykazały, że w analogiczny do MT-2A sposób zbudowane są pozostałe
izoformy MT. W cząsteczce metalotioneiny wyróżnia się dwie globularne
domeny odpowiadające za wiązanie metali (Ryc. 1). W N-końcowej części
MT-2A znajduje się domena β, odpowiadająca resztom aminokwasowym
od 1 do 30. W domenie tej znajduje się 9 reszt cysteiny, które uczestniczą
w koordynacyjnym wiązaniu trzech jonów metali, co można opisać
stechiometrycznie M3IIS9, gdzie MII to jon metalu na drugim stopniu utlenienia,
a S to atom siarki grupy tiolowej Cys biorący udział w wiązaniu metali.
C-końcową część białka stanowi domena α składająca się z aminokwasów
od 31 do 61. W domenie tej znajduje się 11 reszt cysteiny wiążących cztery jony
metali grupy II – stechiometrycznie M4IIS11. Wymiana metali w domenie β
zachodzi szybciej niż w domenie α, ponieważ jest ona bardziej reaktywna.

E. Forma, M. Bryś 211
Ryc. 1. Schemat genu, mRNA oraz białka metalotioneiny 2A.
Domeny MT połączone są konserwatywnym segmentem, składającym się
z dwóch reszt lizyny znajdujących się w pozycjach 30 i 31 i pełniącym funkcję
ruchomego zawiasu. W MT-4 w pozycji 30 (region zawiasu), zamiast
występującej w innych izoformach reszty lizyny, znajduje się reszta argininy.
W MT 20 reszt cysteiny pozwala na związanie 7 jonów Zn2+, Cd2+, Hg2+ lub
12 jonów Cu+ (jony Cu2+ nie są wiązane przez MT). MT niezawierająca jonów

Metalotioneiny i kadm a rak piersi 212
nosi nazwę apo-MT lub tioneiny. Cechuje ją nieuporządkowana struktura,
wysoka niestabilność i podatność na utlenianie oraz proteolizę. Związanie jonów
metali przez apo-MT powoduje wzrost stabilności termodynamicznej
i oporności na trawienie proteolityczne, co w konsekwencji prowadzi do
akumulacji metali związanych przez MT [18, 20, 22-24].
Charakterystyka poszczególnych izoform metalotionein
Metalotioneiny MT-1 i MT-2
MT-1 i MT-2 ulegają ekspresji we wszystkich tkankach ssaków, przy czym
najwyższe ich stężenie obserwuje się w hepatocytach [3, 25]. Wysoki poziom
ekspresji MT-1 i MT-2 charakterystyczny jest również dla komórek nerki,
trzustki i jelita [26]. Wysokie stężenie tych białek w wątrobie sugerować może,
iż główną funkcją MT-1 i MT-A2 jest detoksykacja metali ciężkich (zwłaszcza
Cd), utrzymanie homeostazy cynku i miedzi oraz ograniczenie uszkodzeń
oksydacyjnych [27]. MT-2A wykazuje wyższy poziom ekspresji niż MT-1
w tkankach ludzkich [20]. Stężenie MT-1 i MT-2A wzrasta w czasie proliferacji
i różnicowania komórek, co może być związane ze wzrostem zapotrzebowania
na cynk, który jest niezbędnym składnikiem enzymów i czynników
transkrypcyjnych, kluczowych dla tych procesów. Metalotioneiny są białkami
cytoplazmatycznymi, jednakże w czasie proliferacji i różnicowania ulegają
przejściowej translokacji do jądra komórkowego dostarczając cynk do miejsc
zwiększonego zapotrzebowania [15, 20, 25, 27, 28]. Najwyższe stężenie MT-1
i MT-2A w jądrze komórkowym występuje w późnej fazie G1 oraz w fazie S
[29]. Ekspresja MT-1 i MT-2A, w przeciwieństwie do ekspresji dwóch
pozostałych lizoform, tj. MT-3 i MT-4, indukowana jest przez wiele czynników

E. Forma, M. Bryś 213
chemicznych i fizycznych, w tym przez metale, stres oksydacyjny, hormony
(głównie glukortykoidy), cytokiny oraz promieniowanie [19, 25, 30, 31].
Metalotioneina MT-3
Izoforma MT-3 po raz pierwszy została zidentyfikowana jako czynnik
hamujący wzrost komórek nerwowych (GIF, ang. growth inhibitory factor).
W wyniku dalszych badań czynnik GIF zaliczono do rodziny metalotionein jako
izoformę MT-3 [3, 32]. Ekspresję tego białka obserwuje się przede wszystkim
w neuronach i astrocytach mózgu, szczególnie w korze mózgu, hipokampie oraz
ciele migdałowatym [2]. Obniżony poziom ekspresji MT-3 neuronach mózgu
występuje u osób cierpiących na chorobę Alzheimera 3, 14]. Bardzo niski
poziom ekspresji MT-3 charakterystyczny jest dla komórek trzustki, jelita,
żołądka, serca i nerki. [20, 25, 32, 33]. Jak wykazały badania prowadzone
na myszach transgenicznych przez Quaife i wsp. [34] nadekspresja MT-3
w tkankach zlokalizowanych poza ośrodkowym układem nerwowym wywołuje
efekt letalny w wyniku atrofii trzustki. MT-3 uważana jest za niespecyficzny
inhibitor procesu proliferacji [14, 35].
Metalotioneina MT-4
Izoforma MT-4 jest najpóźniej odkrytym białkiem zaliczanym do
metalotionein i najmniej poznanym. MT-4 ulega ekspresji w nabłonku
wielowarstwowym płaskim, gdzie prawdopodobnie pełni funkcje w regulacji
stężenia jonów cynku w czasie procesu różnicowania się komórek [14, 15, 27,
32, 36]. Ekspresję MT-4 zaobserwowano w komórkach błony śluzowej jamy
ustnej, języka i nosogardzieli, górnej części żołądka oraz w skórze [2, 27].

Metalotioneiny i kadm a rak piersi 214
Funkcje metalotionein
Zasadniczą funkcją metalotionein, wynikającą z ich budowy, jest
utrzymywanie homeostazy jonów metali niezbędnych dla prawidłowego
metabolizmu, detoksykacja ustroju z metali toksycznych oraz ochrona przed
uszkodzeniami oksydacyjnymi [1, 19, 25].
Udział metalotionein w homeostazie cynku
Cynk odgrywa ważną rolę w metabolizmie i jest niezbędny dla
prawidłowego funkcjonowania organizmu człowieka [33]. Cynk jest kofaktorem
ponad 300 enzymów, w tym dehydrogenazy mleczanowej, fosfatazy zasadowej,
anhydrazy węglanowej, dysmutazy ponadtlenkowej i dehydrogenazy alkoholo-
wej. Bierze on udział w tworzeniu motywu palca cynkowego (ZF, ang. zinc
finger motif), który występuje w białkach wiążących się z DNA, w tym
w czynnikach transkrypcyjnych oraz receptorach dla hormonów sterydowych,
hormonów tarczycy, kalcytriolu i retinoidów, które po związaniu ligandu pełnią
funkcje czynników transkrypcyjnych [4, 23, 27, 37-39]. Najwyższe stężenie
cynku występuje w wątrobie, nerce, siatkówce, prostacie i mięśniach [4, 40].
Głównymi wewnątrzkomórkowymi białkami wiążącymi cynk są metalotioneiny,
które pełnią ważną funkcję w regulacji pobierania, dystrybucji, magazynowania
i uwalniania Zn [3, 4, 33]. W utrzymaniu homeostazy Zn2+ bierze udział układ
metalotioneina/tioneina. Wzrost dostępności cynku indukuje syntezę tioneiny
i prowadzi do formowania metalotioneiny. Cynk uwalniany jest z MT wówczas,
gdy obniża się stężenie wolnego Zn [4, 23, 27]. Uwalnianie cynku z MT może
zachodzić na drodze utleniania grup tiolowych reszt cysteiny. W procesie tym
ważną rolę pełni glutation. Układ zredukowany/utleniony glutation
(GSH/GSSG) moduluje uwalnianie cynku z Zn7-MT w zależności od stężenia

E. Forma, M. Bryś 215
Zn i statusu redoks [19, 23, 27, 38, 41]. Liczne badania in vitro wykazały, że
układ metalotioneina/tioneina poprzez uwalnianie lub wiązanie Zn2+ wpływa na
aktywność białek zależnych od cynku i może modulować zdolność wiązania się
czynników transkrypcyjnych, zawierających motyw palca cynkowego, z DNA
[33, 41, 42]. Wiązanie jonów cynku przez tioneinę prowadzi również do
aktywacji enzymów, których aktywność hamowana jest przez ten metal [41, 42].
Wiązanie kadmu przez metalotioneiny
Metalotioneiny uczestniczą w detoksykacji metali ciężkich dzięki zdolności
wiązania różnych metali, w tym toksycznych metali ciężkich, do których
zaliczamy kadm i rtęć. MT wykazują wyższe powinowactwo do metali takich
jak Cd i Hg niż do Zn. Dzięki temu metale ciężkie zatrzymywane są we wnętrzu
cząsteczek tych białek, co chroni komórkę przed ich toksycznym działaniem
[2-4, 25, 35, 43]. Kadm jest metalem toksycznym występującym w środowisku
naturalnym w glebie, w wodzie i powietrzu. Do organizmu kadm dostaje się
wraz z pożywieniem, dymem tytoniowym i zanieczyszczonym powietrzem.
Również wykonywanie pewnych zawodów wiąże się z narażeniem na wyższe
stężenia Cd w miejscu pracy [5, 6, 44-49]. W 1993 roku Cd został uznany za
związek karcynogenny dla ludzi [45, 46, 50-53]. Kadm charakteryzuje się
bardzo długim okresem biologicznego półtrwania ocenianym u ludzi na
10 – 30 lat [50, 54, 55]. W następstwie długotrwałej ekspozycji na kadm, metal
ten gromadzi się w organizmie - głównie w wątrobie i nerkach. Nerki są
najważniejszym narządem docelowego działania kadmu. W komórkach cewek
nerkowych kadm gromadzi się w postaci związanej z metalotioneiną,
a po przekroczeniu tzw. stężenia krytycznego powoduje dysfunkcje cewek
nerkowych, a następnie zwłóknienie śródmiąższowe nerek [48, 55, 56]. Kadm

Metalotioneiny i kadm a rak piersi 216
wpływa na proliferację, różnicowanie się komórek, apoptozę oraz inne procesy
komórkowe. Zaburzenia tych mechanizmów są bezpośrednio zaangażowane
w proces karcynogenezy [7, 50]. Liczne badania wykazały wpływ kadmu na
rozwój raka płuc, prostaty, nerki, wątroby i żołądka. [50, 53, 57]. Ponieważ
kadm cechuje wyższe powinowactwo do metalotionein niż cynk dlatego też
wypiera cynk z tych białek [2, 4, 18, 20]. W konsekwencji prowadzi to do
zaburzenia homeostazy cynku, co wpływa niekorzystnie na proces transkrypcji,
proliferacji i różnicowania komórek [5, 6, 30, 39, 50, 58]
Regulacja ekspresji genów metalotionein
Ekspresja najlepiej poznanych MT-1 i MT-2 jest indukowana przez liczne
metale, cytokiny i hormony [20]. Wzrost stężenia MT-1 i MT-2 wiąże się
z odpowiedzią na różnego typu czynniki stresowe pochodzenia wewnętrznego
np. stany zapalne, jak i zewnętrznego np. ekspozycja na działanie metali
ciężkich takich jak Cd [2, 25].
Struktura regionów regulatorowych genów MT-1 i MT-2
Rdzeń promotora genów MT-1 i MT-2 zawiera sekwencję TATA oraz
region inicjatorowy, do których przyłącza się czynnik transkrypcyjny IID
(TFIID, ang. transcription factor IID) będący częścią kompleksu
preinicjacyjnego uczestniczącego w transkrypcji [25]. W regionie
promotorowym znajdują się sekwencje odpowiedzi na: (1) glukokortykoidy
(GRE; ang. glucocorticoid responsive element), (2) jony metali (MRE; ang.
metal responsive element), (3) stres oksydacyjny (ARE; ang. antioxidant
responsive element) oraz sekwencja aktywowana przez czynnik transkrypcyjny
STAT (ang. signal transducers and activator of transcription). W komórkach

E. Forma, M. Bryś 217
człowieka region promotorowy MT zawiera sześć sekwencji MRE [2, 25, 35].
W regionie promotorowym wszystkich genów MT znajduje się motyw bogaty
w pary GC (GGGGCGGGG), z którym łączą się czynniki transkrypcyjne
z rodziny Sp, w tym białko Sp1 (ang. specificity protein 1) [25].
Regulacja ekspresji genów MT-1 i MT-2 przez jony metali
Geny MT-1 i MT-2 w przeciwieństwie do genów MT-3 i MT-4 są silnie
indukowane w komórkach ssaków przez jony metali włączając w to Zn2+, Cd2+,
Hg2+ i Cu+. Metale te wiążą się z MT. Nikiel i kobalt również zdolne są do
indukcji transkrypcji MT jednakże nie wiążą się z tymi białkami [25, 59].
W regulacji ekspresji genów MT-1 i MT-2 przez metale ciężkie biorą udział
sekwencje MRE, które są również konieczne do transkrypcji tych genów na
poziomie podstawowym. Sekwencje MRE (CTNTGC(G/A)CNCGGCCC)
obecne są w wielu kopiach w regionach promotorowych genów MT u ssaków.
Z sekwencjami MRE oddziałuje zależny od cynku czynnik transkrypcyjny
MTF-1 (ang. metal responsive transcription factor 1) [2, 25, 54, 60-64]. MTF-1
po raz pierwszy został opisany przez Westina i Schaffnera w 1988 roku jako
białko wiążące się z sekwencją MRE, wymagające podwyższonego stężenia Zn
do optymalnego wiązania się z DNA. Brak czynnika MTF-1 daje efekt letalny
na poziomie rozwoju zarodkowego. Czynnik ten niezbędny jest do rozwoju
wątroby w życiu płodowym oraz odpowiedzi na stres [25, 64-69]. Gen kodujący
ludzki czynnik MTF-1 znajduje się na krótkim ramieniu chromosomu 1 (1p33)
i koduje białko zbudowane z 753 aminokwasów. W N-końcowej części MTF-1
znajduje się sześć motywów palca cynkowego (ZF, ang zinc finger) typu
Cys2-His2, które odpowiadają za wiązanie się z sekwencjami MRE.
W C-końcowej części białka znajduje się domena transaktywacyjna obejmująca

Metalotioneiny i kadm a rak piersi 218
region bogaty w aminokwasy kwaśne, region bogaty w reszty proliny oraz
region bogaty w reszty seryny i treoniny. W N-końcowej części MTF-1,
bezpośrednio przed motywami ZF znajduje się sekwencja lokalizacji jądrowej
(NLS, ang. nuclear localization signal), uczestnicząca w translokacji czynnika
MTF-1 z cytoplazmy do jądra komórkowego. W regionie bogatym
w aminokwasy kwaśne zlokalizowany jest sygnał eksportu jądrowego (NES,
ang. nuclear export signal) [25, 60, 65-68, 70]. W wyniku działania czynników
stresowych takich jak hipoksja, stres oksydacyjny czy ekspozycja na działanie
metali, MTF-1 ulega translokacji z cytoplazmy do jądra komórkowego, gdzie
bierze udział w regulacji transkrypcji MT-1 i MT-2 poprzez bezpośrednie lub
pośrednie oddziaływanie z czynnikami transkrypcyjnymi związanymi z RNA
polimerazy II [25, 62, 68, 70, 71]. Wiązanie MTF-1 do MRE wymaga
wysycenia cynkiem motywów palca cynkowego. Wysokie stężenie metali
ciężkich (Cd, Hg) może powodować wypieranie cynku z wewnątrz-
komórkowych białek odpowiadających za jego magazynowanie, w wyniku
czego wzrasta pula wolnego cynku zdolnego do aktywacji MTF-1. Wolne jony
Zn wiążą się z MTF-1 dzięki czemu wzrasta zdolność wiązania się tego
czynnika z DNA, co z kolei prowadzi do aktywacji transkrypcji genów MT [25,
62, 65, 67, 72, 73]. Ekspresja metalotionein na poziomie podstawowym
regulowana jest także przez czynnik transkrypcyjny Sp1, który zawiera trzy
motywy palca cynkowego, a jego aktywność zależy od stężenia wolnych jonów
Zn2+ [25]. Kadm silniej indukuje transkrypcję genów MT niż wynikałoby to ze
wzrostu siły wiązania MTF-1 z DNA. Sugeruje to, że Cd aktywuje ekspresję MT
przez mechanizmy niezależne od wzmocnienia oddziaływania MTF-1/DNA.
Kadm ma wyższy potencjał oksydacyjny niż cynk i indukuje zjawisko stresu
oksydacyjnego oraz spadek stężenia glutationu. Stres oksydacyjny wywołany

E. Forma, M. Bryś 219
ekspozycją na Cd może powodować aktywację transkrypcji genów MT-1
i MT-2A bezpośrednio przez sekwencję ARE, na którą nachodzi sekwencja
wiązania czynnika transkrypcyjnego USF (ang. upstream stimulatory factor).
W warunkach stresu oksydacyjnego dochodzi do zwiększenia dostępności Zn
w wyniku utlenienia grup tiolowych MT, co powoduje uwolnienie jonów tego
metalu. Kadm wypiera Zn z MT zwiększając tym samym pulę dostępnego cynku
zdolnego do wiązania się z motywami palca cynkowego MTF-1 [25, 41, 74, 75].
Oprócz Cd również jony Cu, Pb, Ag, Hg i Bi mogą wypierać Zn z MT [20].
Ważną rolę w regulacji ekspresji genów MT-1 i MT-2 odgrywa fosforylacja
czynnika MTF-1 [61, 62, 70, 72]. W warunkach fizjologicznych MTF-1 ulega
konstytutywnej fosforylacji na resztach seryny i treoniny [60, 65, 71, 73, 76].
Jony metali ciężkich, takie jak Zn i Cd, powodują wzrost poziomu fosforylacji
MTF-1. W proces ten zaangażowane są kinaza białkowa C (PKC, ang. protein
kinase C), kinaza białkowa fosforylyjąca N-koniec białka Jun (JNK, ang. Jun
N-terminal kinase), 3-kinaza fosfatydyloinozytolu (PI3K, ang. phosphoinositol-3
kinase), kinaza tyrozynowa, II kinaza kazeiny (CK II, ang. casein kinase II) oraz
szlak sygnałowy z udziałem jonów Ca2+ [25, 61, 62, 68, 77]. Analiza sekwencji
MTF-1 wykazała obecność konserwatywnie zachowanych potencjalnych miejsc
fosforylacji przez PKC, CK II, kinazę tyrozynową i JNK [62, 76, 78]. Indukcja
ekspresji genów MT-1 i MT-2 przez metale obejmuje trzy zasadnicze etapy, tj.
translokację MTF-1 z cytoplazmy do jądra komórkowego, związanie się MTF-1
z sekwencjami MRE oraz aktywację transkrypcji [62, 65]. Wyniki badań
sugerują, że poziom fosforylacji MTF-1 może mieć wpływ na translokację tego
czynnika transkrypcyjnego do jądra komórkowego oraz na aktywację
transkrypcji genów, których ekspresja regulowana jest przez MTF-1. W wyniku
ekspozycji komórek na działanie Cd2+ i Zn2+ dochodzi do nagromadzenia

Metalotioneiny i kadm a rak piersi 220
ufosforylowanej formy MTF-1 w jądrze komórkowym. Zjawisko to może mieć
związek z fosforylacją tyrozyny w pozycji 140 łańcucha polipeptydowego, która
znajduje się za sekwencją NLS, obejmującą aminokwasy od 133 do 139
(KRKEVKR) [62, 70, 71]. Wzrost poziomu fosforylacji MTF-1 może odgrywać
również ważną rolę w indukcji ekspresji genów MT-1 i MT-2 przez metale
ciężkie. Potwierdzeniem roli fosforylacji w aktywacji transkrypcji genów przez
MTF-1 są wyniki badań, których celem było określenie wpływu inhibitorów
kinaz białkowych na poziom ekspresji MT-1 i MT-2. Zastosowanie inhibitorów
PKC, II kinazy kazeiny i kinazy tyrozynowej prowadziło do zablokowania
indukowanej cynkiem i kadmem transkrypcji genów MT-1 i MT-2. Inhibitory
kinaz białkowych nie wpływały natomiast na wiązanie się MTF-1 z DNA.
Sugeruje to, że wiązanie się białka MTF-1 z MRE jest konieczne, ale
niewystarczające do aktywacji transkrypcji genów MT-1 i MT-2A [62, 71, 76].
Kadm poprzez stymulację aktywności licznych kinaz białkowych prowadzi do
zaburzenia wielu szlaków transdukcji sygnałów. Wynikiem tych zmian może
być wzrost aktywności czynników transkrypcyjnych i nieprawidłowa ekspresja
wielu genów. Do czynników transkrypcyjnych, których aktywność wzrasta
w wyniku fosforylacji indukowanej przez kadm należy między innymi MTF-1
[68, 73, 77]. Czynnik MTF-1 uczestniczy również w regulacji ekspresji
ZnT1 (ang. zinc transporter 1), α-fetoproteiny, C/EBPα (ang. CCAAT-enhancer
binding protein α), syntazy γ-glutamylocysteiny (γ-GCS, ang.
γ-glutamylocystein synthase) oraz γ-glutamylotranspeptydazy [37, 65, 67, 69].

E. Forma, M. Bryś 221
Indukcja ekspresji genów MT-1 i MT-2 w wyniku stresu oksydacyjnego
Indukcja ekspresji MT-1 i MT-2 w wyniku stresu oksydacyjnego zachodzi
z udziałem sekwencji ARE (TGACNNNGC). Na sekwencję ARE nakłada się
sekwencja odpowiedzialna za wiązanie się czynnika transkrypcyjnego USF,
zwanego także MLTF (ang. adeno major late transcription factor). USF/MLTF
jest białkiem wiążącym się z DNA dzięki obecności motywu heliks-pętla-heliks
(HLH) [25, 59, 67, 79]. Występują trzy izoformy USF, tj. USF-1, USF-2a
i USF-2b. USF-1 (42 kDa) i USF-2 (44 kDa) kodowane są przez odrębne geny.
W wyniku alternatywnego składania pierwotnego transkryptu powstają dwie
izoformy USF-2 (USF-2a i USF-2b) [75, 80]. Sekwencje ARE biorą udział
w regulacji ekspresji genów związanych z odpowiedzią na stres oksydacyjny
poprzez oddziaływanie z czynnikiem Nrf-2 (ang. NF-E2- related factor 2).
Związanie Nrf-2 z sekwencją ARE powoduje ekspresję genów regulowanych
przez tę sekwencję, w tym MT. Ekspresja tych genów ulega represji w wyniku
przyłączenia białek Fos (ang. Finkel-Biskis-Jinkins murine osteosarcoma)
i Fra-1 (ang. Fos-related antygen-1). Innymi genami, których transkrypcja
regulowana jest przez sekwencję ARE jest gen podjednostki Ya S-transferazy
glutationowej, gen UDP-glukuronylotransferazy oraz inne geny kodujące
enzymy biorące udział w drugiej fazie detoksykacji ksenobiotyków [25, 59, 81].
Indukcja ekspresji genów MT-1 i MT-2 przez glukokortykoidy
Receptory glukortykoidów (GR, ang. glucocorticoid hormone receptor)
znajdują się na terenie cytoplazmy i związane są z białkami szoku cieplnego Hsp
(ang. heat shock protein), które utrzymują GR w nieaktywnej formie.
Glukokortykoidy przechodzą przez błonę komórkową i łączą się z
nieaktywnymi monomerami GR, co powoduje odłączenie białek Hsp i

Metalotioneiny i kadm a rak piersi 222
aktywację GR. Aktywowane monomery GR tworzą aktywne homodimery
ulegające translokacji do jądra komórkowego, gdzie łączą się z sekwencjami
GRE i aktywują transkrypcję. W ludzkich komórkach ekspresja MT-2A jest
silniej indukowana przez glukokortykoidy niż MT-1 [2, 25].
Rola matolotionein i kadmu w rozwoju nowotworów
Wpływ kadmu na regulację ekspresji genów i szlaki przekaźnictwa sygnałów w komórce
Zaburzenia regulacji ekspresji genów stanowią podstawę procesu
nowotworzenia. Jony kadmu indukują geny natychmiastowej odpowiedzi na
mitogeny (IERG, ang. immediate early response gene), które kodują czynniki
transkrypcyjne zaangażowane w proces proliferacji i różnicowania się komórek.
Produkty białkowe genów IERG stymulują podziały komórek, a ich
nadekspresja jest często obserwowana w wielu typach nowotworów [7, 49, 77].
Do genów IERG, których ekspresja indukowana jest przez kadm zalicza się
komórkowe onkogeny Fos, Jun, Myc [7, 52, 61, 74]. Protoonkogeny Fos i Jun
kodują białka tworzące czynnik transkrypcyjny AP-1 (ang. activating protein 1),
który reguluje ekspresję wielu genów kontrolujących wzrost i różnicowanie się
komórek [7, 53, 74, 82, 83]. Aktywacja protoonkogenów Jun i Fos przez
kadm uważana jest za główny mechanizm transformacji nowotworowej
indukowanej przez ten metal [7]. W wyniku ekspozycji na kadm dochodzi
również do aktywacji innych czynników transkrypcyjnych takich jak
MTF-1, USF, NF-κB (ang. nuclear factor κB), Nrf-2 i TFIIH (ang. transcription
factor IIH) [7, 52, 61, 74].

E. Forma, M. Bryś 223
Jony kadmu indukują również ekspresję genów odpowiedzi na stres, do
których zalicza się geny metalotionein i białek szoku cieplnego, białek
biorących udział w odpowiedzi na stres oksydacyjny i kontrolujących syntezę
glutationu [7, 44, 52]. Hsp to grupa zróżnicowanych białek o masie
cząsteczkowej w granicach 25 – 110 kDa, odpowiadających za przeżycie
komórki w warunkach stresu [7, 83]. Kadm powoduje powstawanie
zdenaturowanych lub nieprawidłowo zwiniętych białek w wyniku zmiany ich
struktury na drodze wiązania się z grupami tiolowymi lub poprzez podstawienie
cynku. Pojawienie się w komórce białek o nieprawidłowej strukturze jest
sygnałem prowadzącym do indukcji ekspresji białek Hsp [7].
Glutation oraz białka zawierające liczne grupy tiolowe odgrywają kluczową
rolę w obronie komórki przed toksycznym działaniem kadmu [7, 49]. Kadm
nie uczestniczy w reakcji Fentona, ponieważ w warunkach fizjologicznych
nie jest ani donorem ani akceptorem elektronów, jednakże powoduje wzrost
stężenia reaktywnych form tlenu (ROS, ang. reactive oxygen species) w sposób
pośredni poprzez wpływ na system antyoksydacyjny komórki [7, 84]. Ważną
rolę w usuwaniu ROS, powstających w wyniku działania kadmu, odgrywa
peroksydaza glutationowa i reduktaza glutationowa, odpowiadające za cykl
oksydoredukcyjny GSH. Jednakże kadm silnie hamuje aktywność peroksydazy
glutationowej i innych enzymów, takich jak dysmutaza ponadtlenkowa
i katalaza, które uczestniczą w usuwaniu wolnych rodników powstających w
komórce w wyniku procesów fizjologicznych. Pod wpływem działania kadmu w
komórce dochodzi do indukcji ekspresji genu γ-GCS oraz S-transferazy
glutationowej. Enzymy te odpowiadają za syntezę glutationu, którego
podwyższone stężenie umożliwia szybką i efektywną eliminację kadmu [6, 7,
44, 49, 74, 77]. Jony kadmu powodują również wzrost wewnątrzkomórkowego

Metalotioneiny i kadm a rak piersi 224
stężenia Ca2+. Jony wapnia wpływają bezpośrednio na ekspresję wielu genów
poprzez czynnik transkrypcyjny CREB (ang. cAMP-response element binding
protein). Kadm może również pośrednio uczestniczyć w aktywacji kinaz
białkowych, które poprzez fosforylację określonych czynników
transkrypcyjnych powodują wzrost ich aktywności. Do kinaz tych zalicza się
kinazę białkową C, kinazę tyrozynową, CK II oraz kinazy rodziny MAPK (ang.
mitogen activated protein kinase), tj. kinazy ERK (ang. extracellular signal
regulated kinase), JNK i p38 [7, 49, 74, 77, 83].
Kadm a apoptoza
Apoptoza odgrywa istotną rolę w homeostazie komórkowej i embrio-
genezie. Prawidłowy przebieg tego procesu zapewnia właściwy wzrost i rozwój
narządów oraz różnicowanie komórek. W wyniku apoptozy dochodzi również
do eliminacji uszkodzonych i nieprawidłowych komórek [85, 86]. Apoptoza
obejmuje szereg zmian morfologicznych, do których zaliczamy obkurczanie się
komórek, kondensację chromatyny, fragmentację jądra komórkowego,
powstawanie ciałek apoptotycznych oraz biochemicznych takich jak
fragmentacja DNA, proteoliza białek strukturalnych i enzymatycznych [86-88].
Do inicjacji programowanej śmierci komórki może dojść w wyniku działania
czynników uszkadzających DNA i zaburzających cykl komórkowy, aktywacji
określonych receptorów błonowych, szoku cieplnego, stresu oksydacyjnego oraz
promieniowania jonizującego [86]. Również jony kadmu mogą prowadzić do
apoptozy [5, 7, 84, 89, 90]. Podatność na apoptozę indukowaną kadmem zależy
od stężenia metalotionein, które wiążąc ten metal chronią komórkę przed jego
toksycznym działaniem [90, 91]. W czasie apoptozy indukowanej jonami kadmu
dochodzi do zwiększenia przepuszczalności błony mitochondrialnej, uwolnienia

E. Forma, M. Bryś 225
cytochromu c oraz aktywacji prokaspazy 9. Kadm powoduje wzrost ekspresji
proapoptotycznego białka Bad (ang. Bcl-2 antagonist of cell death), które
powoduje niestabilność błony mitochondrialnej. Kadm zwiększa
przepuszczalność błony mitochondrialnej w wyniku koordynacyjnego wiązania
się z grupami tiolowymi białek budujących tę błonę oraz zaburza działanie
łańcucha oddechowego, przez co wzrasta w komórce stężenie reaktywnych form
tlenu. Antyoksydanty komórkowe, do których zaliczamy również MT chronią
komórkę przed apoptozą indukowaną kadmem [7, 90, 92]. Zmiany zachodzące
w czasie indukcji apoptozy przez kadm charakterystyczne są dla szlaku
wewnętrznego, zwanego inaczej mitochondrialnym [85-87]. Ponadto, Aimola
i wsp. [93] wykazali, że w komórkach nowotworowych prostaty kadm indukuje
apoptozę zależną od białka p53.
Kadm a naprawa uszkodzeń DNA
Uszkodzenia DNA mogą powstawać w wyniku działania czynników
egzogennych takich jak mutagenne związki chemiczne oraz promieniowanie
jonizujące i UV. Endogennymi przyczynami uszkodzeń DNA są ROS
powstające w łańcuchu oddechowym oraz błędy w procesie replikacji.
Uszkodzenia te jeżeli nie zostaną usunięte powodują powstawanie mutacji, które
mogą być przyczyną rozwoju nowotworu [7]. W każdej komórce istnieje szereg
mechanizmów naprawy DNA, których zadaniem jest zachowanie integralności
genomu. Kadm może przyczyniać się do wzrostu częstości powstawania mutacji
poprzez hamowanie systemów naprawy DNA [7, 49, 53, 94]. Metal ten
uniemożliwia rozpoznanie uszkodzeń DNA usuwanych za pomocą systemu
NER (ang. nucleotide excision repair) poprzez wiązanie się z białkiem XPA
(ang. xeroderma pigmentosum, complementation group A) odpowiedzialnym za

Metalotioneiny i kadm a rak piersi 226
ten etap [7, 95, 96]. W wyniku utlenienia dGTP powstaje 8-okso-dGTP, którego
obecność w DNA jest przyczyną transwersji AT→ CG w czasie replikacji.
Kadm może prowadzić do wzrostu liczby mutacji i rozwoju nowotworu poprzez
hamowanie aktywność 8-okso-dGTPazy, odpowiadającej za usuwanie 8-okso-
dGTP z DNA. [7, 49].
Hamowanie metylacji DNA przez kadm
Metylacja wysp CpG jest procesem epigenetycznym prowadzącym do
wyciszenia ekspresji określonych genów. Enzymem katalizującym metylację
cytozyny w pozycji 5’ jest metylotransferaza DNA [25, 97, 98]. Kadm powoduje
spadek aktywności tego enzymu, co prowadzi do obniżenia poziomu metylacji
DNA i nadekspresji protoonkogenów. Aktywacja onkogenów komórkowych jest
jednym z podstawowych mechanizmów transformacji nowotworowej [7, 49].
Wpływ kadmu na adhezję międzykomórkową
E-kadheryna jest charakterystyczną dla komórek epitelialnych
transbłonową glikoproteiną wiążącą jony wapnia i odpowiadającą za adhezję
międzykomórkową. Domena wewnątrzkomórkowa E-kadheryny wiąże się
z kateniną β, która oddziałuje z cytoszkieletem aktynowym. Część zewnątrz-
komórkowa E-kadheryny zawiera miejsca wiązania jonów wapnia, które są
niezbędne dla prawidłowego funkcjonowania tego białka oraz domeny
odpowiedzialne za oddziaływanie kadheryna-kadheryna w przylegających
komórkach [7, 99]. Jony Ca2+ w domenie zewnątrzkomórkowej mogą być
wypierane przez Cd2+. W wyniku tego dochodzi do zmiany konformacji tej
domeny i zaburzenia adhezji międzykomórkowej. Osłabienie przylegania
komórek ułatwia proces przerzutowania nowotworu. Zmiana konformacji

E. Forma, M. Bryś 227
E-kadheryny w wyniku wyparcia jonów wapnia prowadzi również do odłączenia
kateniny β od cytoszkieletu i jej translokację do jądra komórkowego, gdzie
łącząc się z czynnikami transkrypcyjnymi wpływa na ekspresję protoonkogenów
takich jak Jun i Myc. Zaburzenie ekspresji onkogenów komórkowych w wyniku
uwolnienia kateniny β z kompleksów z E-kadheryny może stanowić ważny krok
w procesie nowotworzenia [7, 100-104].
Udział kadmu i metalotionein w regulacji aktywności
czynników transkrypcyjnych
Rola motywów palca cynkowego i jonów cynku w procesie karcynogenezy
Motyw palca cynkowego występuje w wielu grupach białek, do których
zalicza się czynniki transkrypcyjne oraz białka systemów naprawy uszkodzeń
DNA. Palce cynkowe stanowią domeny białkowe składające się najczęściej
z 30 – 40 aminokwasów, a przestrzenna struktura tych domen utrzymywana jest
przez koordynacyjne wiązanie cynku przez grupy tiolowe cystein i/lub
pierścienie imidazolowe histydyn. Cynk nie oddziałują bezpośrednio z DNA, ale
jest niezbędny dla utrzymania prawidłowej konformacji motywów ZF [96].
Struktura palców cynkowych może zostać zaburzona w wyniku podstawienia
cynku przez kadm, który tworzy wysoce stabilne wiązania z grupami tiolowymi
wchodzącymi w skład tych domen. Konsekwencją tego jest utrata zdolności
wiązania się białek zawierających palce cynkowe z DNA i zaburzenia ekspresji
genów i naprawy uszkodzeń DNA [89, 96]. Ważnym czynnikiem wpływającym
na aktywność białek zawierających motyw ZF jest deficyt jonów cynku.
W przypadku niedoboru tego pierwiastka obserwuje się wzrost częstości
uszkodzeń DNA oraz ryzyka rozwoju nowotworu w wyniku działania związków
karcynogennych [39]. Objawy niedoboru Zn w komórce niekoniecznie wynikają

Metalotioneiny i kadm a rak piersi 228
z braku tego metalu w diecie, ale mogą być związane z nadekspresją MT,
która powoduje obniżenie stężenia wolnych jonów cynku poprzez silne
ich wiązanie [105].
Regulacja aktywności białka p53 przez kadm i metalotioneiny
Białko p53, będące produktem genu supresorowego, uczestniczy
w odpowiedzi komórki na uszkodzenia DNA, hipoksję, stres oksydacyjny
i aktywację onkogenów [106-108]. Białko p53 nazywane jest „strażnikiem
genomu” ponieważ główną jego funkcją jest zachowanie integralności genomu.
Jeżeli uszkodzenia DNA powstające w wyniku działania czynników stresowych
są możliwe do usunięcia przez komórkę, to białko p53 prowadzi do zatrzymania
cyklu komórkowego w fazie G1 lub G2 dając tym samym komórce czas na
naprawę DNA. W przeciwnym wypadku białko p53 kieruje komórkę na drogę
apoptozy [39, 106, 108-111]. Białko p53 jest czynnikiem transkrypcyjnym
odgrywającym istotną rolę w regulacji cyklu komórkowego, naprawie
uszkodzeń DNA oraz apoptozie [39, 112-115]. Białko p53 pełni funkcję
inhibitora proliferacji komórek poprzez nasilenie ekspresji genów regulujących
cykl komórkowy, np. genu p21, którego produkt jest inhibitorem kinaz
zależnych od cyklin – CDK (ang. cyclin-dependent kinases) i decyduje
o zatrzymaniu cyklu komórkowego w punkcie kontrolnym G1/S [107, 109,
115-118]. Białko p21 prowadzi do zahamowania replikacji oraz wpływa na
naprawę uszkodzeń DNA w wyniku oddziaływania z białkiem PCNA
(ang. proliferating cell nuclear antigen), które jest ważnym czynnikiem
biorącym udział w systemach naprawy DNA poprzez wycinanie nukleotydów
i zasad azotowych. [115, 117]. Ekspozycja komórek na działanie kadmu
prowadzi do nasilenia ekspresji białka p53, a co za tym idzie również białka p21.

E. Forma, M. Bryś 229
W wyniku wzrostu stężenia białka p21 dochodzi do obniżenia ekspresji CDK1,
CDK2 oraz cykliny A i B, a tym samym do zatrzymania cyklu komórkowego
w punkcie kontrolnym G2/M [116; 118, 119]. Białko p53 indukuje również
ekspresję białka GADD45 (ang. growth arrest and DNA damage 45)
odpowiedzialnego za zahamowanie wzrostu komórki i odpowiedź na
uszkodzenia DNA w punkcie kontrolnym G2/M. Kolejnymi białkami, których
ekspresja zależy od białka p53 jest białko 14-3-3σ, uczestniczące w transdukcji
sygnału prowadzącego do zatrzymania cyklu komórkowego w czasie przejścia
komórki z fazy G2 do M oraz szereg białek proapoptotycznych, do których
zalicza się białko Bax (ang. Bcl-2- associated x protein) regulujące
przepuszczalność błony mitochondrialnej oraz receptor śmierci – Fas [29, 119].
Cynk ma zasadnicze znaczenie dla utrzymania prawidłowej konformacji
i stabilności białka p53. Jony tego metalu wpływają również na powinowactwo
p53 do specyficznych sekwencji DNA oraz na aktywność transkrypcyjną tego
białka [29, 107, 108, 113, 120, 121]. Niedobór Zn może prowadzić do powstania
uszkodzeń DNA indukujących ekspresję p53. Jednakże zbyt niski poziom cynku
może prowadzić do nieprawidłowego formowania motywów ZF białka p53, co
znacznie obniża zdolność wiązania się tego białka z DNA i prowadzi do
zaburzeń ekspresji genów niezbędnych do naprawy powstałych uszkodzeń
DNA. Badania prowadzone przez Méplan i wsp. [122] wykazały, że ekspozycja
komórek na działanie kadmu prowadzi do zaburzenia prawidłowej struktury
białka p53 i zahamowania jego wiązania z DNA. Inaktywacja p53 jest wynikiem
podstawienia cynku przez kadm [29, 39, 58, 112]. Zahamowanie aktywności
białka p53 przez kadm może odgrywać kluczową rolę w procesie
nowotworzenia z udziałem tego metalu [122]. Do deficytu cynku w komórce
może dochodzić na skutek nadekspresji apo-MT wywołanej stresem

Metalotioneiny i kadm a rak piersi 230
oksydacyjnym i działaniem metali ciężkich [97, 113, 123]. Apo-MT-1 wiąże się
z białkiem p53, co uniemożliwia wiązanie się tego ostatniego z DNA oraz chroni
tioneinę przed degradacją. Apo-MT wiąże się za pomocą grup tiolowych
z cynkiem obecnym w cząsteczce p53. Tioneina, poprzez chelatowanie Zn2+,
hamuje również aktywność innych czynników transkrypcyjnych takich jak
TFIIA (ang. transcription factor IIA) i Sp1. Istnieją również doniesienia
o bezpośrednim oddziaływaniu apo-MT z motywami palca cynkowego TFIIA
[113]. Utrata funkcji białka p53 (w wyniku mutacji lub niedoboru cynku)
prowadzi do niestabilności genetycznej, wzrostu aktywności proliferacyjnej
komórki, braku kontroli cyklu komórkowego w punkcie kontrolnym G1/S oraz
oporności na apoptozę [115].
Regulacja aktywności czynnika NF-κB przez metalotioneiny
NF-κB/Rel (ang. nuclear factor κB/ viral oncogene v-rel homolog -
reticuloendotheliosis) jest to rodzina dimerycznych czynników
transkrypcyjnych, charakteryzujących się obecnością homologicznej domeny
Rel (RHD, ang. Rel homology domain). Domena RHD składa się z około
300 aminokwasów. W jej obrębie znajduje się miejsce wiązania się z DNA,
sekwencja odpowiedzialna za dimeryzację, sygnał lokalizacji jądrowej oraz
miejsce wiązania inhibitora IκB (ang. inhibitor κB) [124-126]. Klasyczny
czynnik NF-κB jest heterodimerem składającym się z podjednostki p65 i p50
[125-127]. Czynnik jądrowy κB występuje w cytoplazmie w formie
nieaktywnego kompleksu ze specyficznym białkiem inhibitorowym określanym
jako IκB. Aktywacja NF-κB zachodzi w wyniku fosforylacji i ubikwityno-
zależnej degradacji IκB. Aktywny dimer p65/p50 ulega translokacji do jądra
komórkowego, gdzie wiąże się z sekwencjami κB w DNA [30, 74, 126].

E. Forma, M. Bryś 231
Czynnikami aktywującymi NF-κB są między innymi czynnik martwicy
nowotworu (TNFα, ang. tumor necrosis factor α), promieniowanie,
chemioterapeutyki, erytropoetyna, interleukina 1 [124-126].Czynnik NF-κB
chroni komórkę przed apoptozą oraz pełni ważną rolę w kontroli proliferacji
komórek i onkogenezie [30, 74, 126, 127]. Obok aktywności antyapoptotycznej
czynnik NF-κB wykazuje również działanie proapoptyczne poprzez nasilanie
ekspresji p53 [128].
Zaburzenia procesu aktywacji NF-κB w wyniku destabilizacji IκB
zaobserwowano we wczesnych etapach transformacji nowotworowej. Białko
NF-κB uczestniczy w przekazywaniu sygnałów indukujących proliferację
komórek. Czynnik ten bezpośrednio aktywuje protoonkogen myc. Interleukiny
i czynniki wzrostu wpływają na podziały komórek poprzez aktywację NF-κB
[126]. Opisywany czynnik transkrypcyjny stymuluje proliferację komórek
poprzez indukcję ekspresji genu cykliny D1 [124]. Ważnymi regulatorami
aktywności NF-κB są jony cynku i metalotioneiny. Badania prowadzone przez
Kim i wsp. [30] wykazały, że wysokie stężenie Zn2+ hamuje wiązanie się NF-κB
z DNA. Wzrost ekspresji MT wywołuje spadek stężenia Zn, co prowadzi
do przywrócenia zdolności wiązania się NF-κB z DNA. Czynniki indukujące
ekspresję MT przyczyniają się do aktywacji NF-κB, a co za tym idzie
do stymulacji proliferacji komórek i hamowania apoptozy. MT wiąże się
z podjednostką p50 i ulega translokacji do jądra komórkowego w połączeniu
z aktywnym czynnikiem NF-κB. MT stabilizuje wiązanie czynnika jądrowego
κB z odpowiednimi sekwencjami DNA [30, 33, 78, 113]. Czynniki wpływające
na poziom wolnych jonów cynku i ekspresję metalotionein pośrednio
uczestniczą w regulacji aktywności transkrypcyjnej NF-κB [105]. Do czynników

Metalotioneiny i kadm a rak piersi 232
tych zaliczamy jony kadmu, reaktywne formy tlenu i azotu powstające
w warunkach stresu oksydacyjnego [25, 35].
Metalotioneiny a nowotwory piersi
Spośród siedmiu funkcjonalnych izoform MT-1 w komórkach nowotworów
piersi wykryto mRNA dla MT-1A, MT-1E, MT-1F, MT-1G, MT-1H i MT-1X.
W guzach piersi ekspresji nie ulega MT-1B. Jednakże spośród wyżej wymienionych
izoform metalotionein najwyższe stężenie osiągały MT-1E i MT-1F i te białka
uznaje się za charakterystyczne dla przewodowego raka piersi [15, 21, 58, 129,
130]. Ekspresja MT-1E jest negatywnie skorelowana z obecnością receptora
estrogenowego (ER, ang. estrogen receptor). Stężenie mRNA MT-1E w komórkach
nowotworowych charakteryzujących się brakiem funkcjonalnego receptora
estrogenowego jest wyższe niż w komórkach z prawidłowym ER [15, 58, 131,
132]. Ekspresja receptora estrogenowego jest ważnym czynnikiem prognostycznym
w nowotworach piersi. Brak prawidłowo funkcjonującego receptora dla estrogenów
świadczy o złym rokowaniu i oporności na terapię hormonalną. Estrogeny
odgrywają istotną rolę w prawidłowym rozwoju piersi oraz wpływają na rozwój
raka piersi [131, 133]. Aktywne receptory estrogenowe pełnią funkcję czynników
transkrypcyjnych regulujących ekspresję genów mających wpływ na wzrost
i proliferację komórek [131, 133, 134]. Przyjmuje się, że w przypadku ekspresji
nieprawidłowego receptora estrogenowego, jego funkcje w regulacji proliferacji
komórki przejmuje MT-1E. Stąd może wynikać podwyższona ekspresja tej
izoformy w nowotworach piersi posiadających zmutowany ER [131]. MT-1F obok
MT-2 jest główną izoformą wpływająca na proliferację komórek przewodowego
raka piersi [15, 58]. Znacząco wyższy poziom ekspresji MT-1F charakterystyczny
jest dla nowotworów o większej złośliwości histologicznej. Sugeruje to, że MT-1F

E. Forma, M. Bryś 233
wpływa na procesy różnicowania się komórek w przewodowym raku piersi.
Wysoki poziom ekspresji MT-1F w komórkach nowotworowych wskazuje na
gorsze rokowanie [130]. MT-2A osiąga najwyższe stężenie spośród wszystkich
izoform MT ulegających ekspresji w nowotworach piersi [15, 21, 58]. Poziom
ekspresji MT-2 jest pozytywnie skorelowany z aktywnością proliferacyjną
komórek. Wykazano, że nadekspresja MT-2A powoduje dwukrotny wzrost liczby
podziałów komórkowych, podczas gdy w przypadku nadekspresji MT-1E i MT-3
nie obserwowano takiego efektu [21]. Potwierdzeniem wpływu MT-2A
na proliferację komórek jest korelacja między poziomem ekspresji tej izoformy
metalotionein a ekspresją Ki-67, markera proliferacji [123]. Zarówno stężenie
MT-2A jak i mRNA dla tej izoformy jest wyższe w nowotworach bardziej
zaawansowanych niż w nowotworach w niższych stadiach rozwoju. Silna korelacja
między ekspresją MT-2A a aktywnością proliferacyjną nowotworów piersi
wskazuje na potencjalne znaczenie tej izoformy w procesie karcynogenezy [123].
Wojnar i wsp. [135] wykazali wyższy poziom ekspresji MT-1 i MT-2A w rakach
piersi w najwyższym stopniu zaawansowania w porównaniu do pozostałych
nowotworów. Ponadto ekspresja MT-1 i MT-2 była pozytywnie skorelowana
z ekspresją markerów proliferacji takich jak Ki-67 i MCM-2 [135, 136]. Na ryzyko
zachorowania na raka piersi wydają się mieć również wpływ polimorfizmy
pojedynczych nukleotydów stwierdzone w genach metalotionein [137]. MT-4 nie
ulega ekspresji ani w prawidłowych ani w nowotworowych komórkach raka piersi.
Natomiast około 90% przypadków raka piersi wykazuje ekspresję MT-3, której nie
obserwuje się w tkance prawidłowej [123, 138]. Obecność MT-3 w komórkach guza
świadczy o złym rokowaniu [15, 58, 138]. Nadekspresja tego białka jest przyczyną
wysokiego stężenia apo-MT, co prowadzi do niedoboru cynku i zaburzenia
fundamentalnych procesów komórkowych [15].

Metalotioneiny i kadm a rak piersi 234
Podsumowanie
Metalotioneiny to grupa powszechnie występujących białek, które
uczestniczą w utrzymaniu homeostazy jonów metali ciężkich głównie cynku
i kadmu, ochronie przed ich toksycznym działaniem oraz ochronie przed stresem
oksydacyjnym. Zmiany ekspresji metalotionein oraz akumulacja jonów kadmu
mogą prowadzić do zaburzenia wielu procesów komórkowych, takich jak
proliferacja komórek, apoptoza, transmisja sygnału, odpowiedź na stres
oksydacyjny, a tym samym przyczyniać się do rozwoju nowotworów, w tym
raka piersi.
Praca finansowana w ramach Grantu Prezydenta Miasta Łodzi na realizację
pracy magisterskiej o Łodzi dla Ewy Formy na projekt pt.: „Udział kadmu
i polimorfizmu genu metalotioneiny w nowotworzeniu piersi kobiet z Łodzi
i regionu łódzkiego”
Piśmiennictwo
1. Arriaga JM, Levy EM, Bravo AI, Bayo SM, Amat M, Aris M i wsp. Metallothionein expression in colorectal cancer: relevance of different isoforms for tumor progression and patient survival. Hum Pathol. 2012; 43: 197-208.
2. Sabolic I, Breljak D, Skarica M, Herak-Kramberger CM. Role of metallothionein in cadmium traffic and toxicity in kidneys and other mammalian organs. Biometals. 2010; 23: 897-926.
3. Babula P, Masarik M, Adam V, Eckschlager T, Stiborova M, Trnkova L i wsp. Mammalian metallothioneins: properties and functions. Metallomics. 2012; 4: 739-750.
4. Chasapis CT, Loutsidou AC, Spiliopoulou CA, Stefanidou ME. Zinc and human health: an update. Arch Toxicol. 2012; 86: 521-534.
5. Hartwig A. Mechanisms in cadmium-induced carcinogenicity: recent insights. Biometals. 2010; 23: 951-960.

E. Forma, M. Bryś 235
6. Joseph P. Mechanisms of cadmium carcinogenesis. Toxicol Appl Pharmacol. 2009; 238: 272-279.
7. Waisberg M, Joseph P, Hale B, Beyersmann D. Molecular and cellular mechanisms of cadmium carcinogenesis. Toxicology. 2003; 192: 95-117.
8. Werynska B, Pula B, Muszczynska-Bernhard B, Gomulkiewicz A, Piotrowska A, Prus R i wsp. Metallothionein 1F and 2A overexpression predicts poor outcome of non-small cell lung cancer patients. Exp Mol Pathol. 2012; 94: 301-308.
9. Forma E, Krzeslak A, Wilkosz J, Jozwiak P, Szymczyk A, Rozanski W i wsp. Metallothionein 2A genetic polymorphisms and risk of prostate cancer in a Polish population. Cancer Genet. 2012; 205: 432-435.
10. Gumulec J, Masarik M, Krizkova S, Hlavna M, Babula P, Hrabec R i wsp. Evaluation of alpha-methylacyl-CoA racemase, metallothionein and prostate specific antigen as prostate cancer prognostic markers. Neoplasma. 2012; 59: 191-201.
11. Yan DW, Fan JW, Yu ZH, Li MX, Wen YG, Li DW, Zhou CZ, Wang XL, Wang Q, Tang HM, Peng ZH. Downregulation of metallothionein 1F, a putative oncosuppressor, by loss of heterozygosity in colon cancer tissue. Biochim Biophys Acta. 2012; 1822: 918-926.
12. Schöpfer J, Drasch G, Schrauzer GN. Selenium and cadmium levels and ratios in prostates, livers, and kidneys of nonsmokers and smokers. Biol Trace Elem Res. 2010; 134: 180-187.
13. Sakamoto LH, DE Camargo B, Cajaiba M, Soares FA, Vettore AL. MT1G hypermethylation: a potential prognostic marker for hepatoblastoma. Pediatr Res. 2010; 67: 387-393.
14. Vašák M, Meloni G. Chemistry and biology of mammalian metallothioneins. J Biol Inorg Chem. 2011; 16: 1067-1078.
15. Cherian MG, Jayasurya A, Bay BH. Metallothioneins in human tumors and potential roles in carcinogenesis. Mutat Res. 2003; 533: 201-209.
16. Palacios O, Atrian S, Capdevila M. Zn- and Cu-thioneins: a functional classification for metallothioneins? J Biol Inorg Chem. 2011; 16: 991-1009.
17. Cai B, Zheng Q, Teng XC, Chen D, Wang Y, Wang KQ i wsp. The role of Thr5 in human neuron growth inhibitory factor. J Biol Inorg Chem. 2006; 11: 476-482.
18. Romero-Isart N, Vašák M. Advances in the structure and chemistry of metallothioneins. J Inorg Biochem. 2002; 88: 388-396.
19. Vašák M. Advances in metallothionein structure and functions. J Trace Elem Med. Biol. 2005; 19: 13-17.

Metalotioneiny i kadm a rak piersi 236
20. Coyle P, Philcox JC, Carey LC, Rofe AM. Metallothionein: the multipurpose protein. Cell Mol Life Sci. 2002; 59: 627-647.
21. Jin R, Huang J, Tan PH, Bay BH. Clinicopathological significance of metallothioneins in breast cancer. Pathol Oncol Res. 2004; 10: 74-79.
22. Sutherland DE, Summers KL, Stillman MJ. Noncooperative metalation of metallothionein 1a and its isolated domains with zinc. Biochemistry. 2012; 51: 6690-700.
23. Maret W. The function of zinc metallothionein: a link between cellular zinc and redox state. J Nutr. 2000; 130(5S Suppl): 1455-1458
24. Rigby KE, Stillman MJ. Structural studies of metal-free metallothionein. Biochem Biophys Res Commun. 2004; 325: 1271-1278.
25. Haq F, Mahoney M, Koropatnick J. Signaling events for metallothionein induction. Mutat Res. 2003; 533: 211-226.
26. Davis SR, Cousins RJ. Metallothionein expression in animals: a physiological perspective on function. J Nutr. 2000; 130: 1085-1088.
27. Tapiero H, Tew KD. Trace elements in human physiology and pathology: zinc and metallothioneins. Biomed Pharmacother. 2003; 57: 399-411.
28. Jayasurya A, Bay BH, Yap WM, Tan NG, Tan BK. Proliferative potential in nasopharyngeal carcinoma: correlations with metallothionein expression and tissue zinc levels. Carcinogenesis. 2000; 21: 1809-1812.
29. Méplan C, Richard MJ, Hainaut P. Metalloregulation of the tumor suppressor protein p53: zinc mediates the renaturation of p53 after exposure to metal chelators in vitro and in intact cells. Oncogene. 2000; 19: 5227-5236.
30. Kim CH, Kim JH, Lee J, Ahn YS. Zinc-induced NF-kappaB inhibition can be modulated by changes in the intracellular metallothionein level. Toxicol Appl Pharmacol. 2003; 190: 189-196.
31. Tanji K, Irie Y, Uchida Y, Mori F, Satoh K, Mizushima Y i wsp. Expression of metallothionein-III induced by hypoxia attenuates hypoxia-induced cell death in vitro. Brain Res. 2003; 976: 125-129.
32. Thirumoorthy N, Shyam Sunder A, Manisenthil Kumar K, Senthil Kumar M, Ganesh G, Chatterjee M. A review of metallothionein isoforms and their role in pathophysiology. World J Surg Oncol. 2011; 9: 54.
33. Vasák M, Hasler DW. Metallothioneins: new functional and structural insights. Curr Opin Chem Biol. 2000; 4: 177-183.
34. Quaife CJ, Kelly EJ, Masters BA, Brinster RL, Palmiter RD. Ectopic expression of metallothionein-III causes pancreatic acinar cell necrosis in transgenic mice. Toxicol Appl Pharmacol. 1998; 148: 148-157.

E. Forma, M. Bryś 237
35. Sato M, Kondoh M. Recent studies on metallothionein: protection against toxicity of heavy metals and oxygen free radicals. Tohoku J Exp Med. 2002; 196: 9-22.
36. Quaife CJ, Findley SD, Erickson JC, Froelick GJ, Kelly EJ, Zambrowicz BP i wsp. Induction of a new metallothionein isoform (MT-IV) occurs during differentiation of stratified squamous epithelia. Biochemistry. 1994; 33: 7250-7259.
37. Eide DJ. Zinc transporters and the cellular trafficking of zinc. Biochim Biophys Acta. 2006; 1763: 711-722.
38. Giles NM, Watts AB, Giles GI, Fry FH, Littlechild JA, Jacob C. Metal and redox modulation of cysteine protein function. Chem Biol. 2003; 10: 677-693.
39. Ho E. Zinc deficiency, DNA damage and cancer risk. J Nutr Biochem. 2004; 15: 572-578.
40. Bryś M, Nawrocka AD, Miekoś E, Zydek C, Foksiński M, Barecki A, Krajewska WM. Zinc and cadmium analysis in human prostate neoplasms. Biol Trace Elem Res. 1997; 59: 145-152.
41. Maret W. Redox biochemistry of mammalian metallothioneins. J Biol Inorg Chem. 2011; 16: 1079-1086.
42. Maret W, Jacob C, Vallee BL, Fischer EH. Inhibitory sites in enzymes: zinc removal and reactivation by thionein. Proc Natl Acad Sci U S A. 1999; 96: 1936-1940.
43. Namdarghanbari M, Wobig W, Krezoski S, Tabatabai NM, Petering DH. Mammalian metallothionein in toxicology, cancer, and cancer chemotherapy. J Biol Inorg Chem. 2011; 16: 1087-1101.
44. Cuypers A, Plusquin M, Remans T, Jozefczak M, Keunen E, Gielen H i wsp. Cadmium stress: an oxidative challenge. Biometals. 2010; 23: 927-940.
45. Pappas RS. Toxic elements in tobacco and in cigarette smoke: inflammation and sensitization. Metallomics. 2011; 3: 1181-1198.
46. Fatur T, Tusek M, Falnoga I, Scancar J, Lah TT, Filipic M. DNA damage and metallothionein synthesis in human hepatoma cells (HepG2) exposed to cadmium. Food Chem Toxicol. 2002; 40: 1069-1076.
47. Himeno S. Application of metallothionein null cells to investigation of cadmium transport. J Inorg Biochem. 2002; 88: 207-212.
48. Mlynek V, Skoczyńska A. Prozapalne działanie kadmu, Postępy Hig. Med. Dośw. 2005; 59: 1–8.
49. Stavrides JC. Lung carcinogenesis: pivotal role of metals in tobacco smoke. Free Radic Biol Med. 2006; 41: 1017-1030.

Metalotioneiny i kadm a rak piersi 238
50. Templeton DM, Liu Y. Multiple roles of cadmium in cell death and survival. Chem Biol Interact. 2010; 188: 267-275.
51. Kakkar P, Jaffery FN. Biological markers for metal toxicity. Environ Toxicol Pharmacol. 2005; 19: 335-349.
52. Liu J, Kadiiska MB, Corton JC, Qu W, Waalkes MP, Mason RP i wsp. Acute cadmium exposure induces stress-related gene expression in wild-type and metallothionein-I/II-null mice. Free Radic Biol Med. 2002; 32: 525-535.
53. Waalkes MP. Cadmium carcinogenesis in review. J Inorg Biochem. 2000; 79: 241-244.
54. Kita K, Miura N, Yoshida M, Yamazaki K, Ohkubo T, Imai Y i wsp. Potential effect on cellular response to cadmium of a single-nucleotide A --> Gpolymorphism in the promoter of the human gene for metallothionein IIA. Hum Genet. 2006; 120: 553-560.
55. Lu J, Jin T, Nordberg G, Nordberg M. Metallothionein gene expression in peripheral lymphocytes and renal dysfunction in a population environmentally exposed to cadmium. Toxicol Appl Pharmacol. 2005; 206: 150-156.
56. Garrett SH, Phillips V, Somji S, Sens MA, Dutta R, Park S i wsp. Transient induction of metallothionein isoform 3 (MT-3), c-fos, c-jun and c-myc in human proximal tubule cells exposed to cadmium. Toxicol Lett. 2002; 126: 69-80.
57. Klaassen CD, Liu J, Diwan BA. Metallothionein protection of cadmium toxicity. Toxicol Appl Pharmacol. 2009; 238: 215-220.
58. Theocharis SE, Margeli AP, Klijanienko JT, Kouraklis GP. Metallothionein expression in human neoplasia. Histopathology. 2004; 45: 103-118.
59. Bi Y, Palmiter RD, Wood KM, Ma Q. Induction of metallothionein I by phenolic antioxidants requires metal-activated transcription factor 1 (MTF-1) and zinc. Biochem J. 2004; 380: 695-703.
60. Laity JH, Andrews GK. Understanding the mechanisms of zinc-sensing by metal-response element binding transcription factor-1 (MTF-1). Arch Biochem Biophys. 2007; 463: 201-210.
61. LaRochelle O, Gagné V, Charron J, Soh JW, Séguin C. Phosphorylation is involved in the activation of metal-regulatory transcription factor 1 in response to metal ions. J Biol Chem. 2001; 276: 41879-41888.
62. Saydam N, Adams TK, Steiner F, Schaffner W, Freedman JH. Regulation of metallothionein transcription by the metal-responsive transcription factor MTF-1: identification of signal transduction cascades that control metal-inducible transcription. J Biol Chem. 2002; 277: 20438-20445.

E. Forma, M. Bryś 239
63. Vasconcelos MH, Tam SC, Hesketh JE, Reid M, Beattie JH. Metal- and tissue-dependent relationship between metallothionein mRNA and protein. Toxicol Appl Pharmacol. 2002; 182: 91-97.
64. Wang Y, Wimmer U, Lichtlen P, Inderbitzin D, Stieger B, Meier PJ, Hunziker L, Stallmach T, Forrer R, Rülicke T, Georgiev O, Schaffner W. Metal-responsive transcription factor-1 (MTF-1) is essential for embryonic liver development and heavy metal detoxification in the adult liver. FASEB J. 2004; 18: 1071-1079.
65. Günther V, Lindert U, Schaffner W. The taste of heavy metals: gene regulation by MTF-1. Biochim Biophys Acta. 2012; 1823: 1416-1425.
66. Chen X, Zhang B, Harmon PM, Schaffner W, Peterson DO, Giedroc DP. A novel cysteine cluster in human metal-responsive transcription factor 1 is required for heavy metal-induced transcriptional activation in vivo. J Biol Chem. 2004; 279: 4515-4522.
67. Günes C, Heuchel R, Georgiev O, Müller KH, Lichtlen P, Blüthmann H, Marino S, Aguzzi A, Schaffner W. Embryonic lethality and liver degeneration in mice lacking the metal-responsive transcriptional activator MTF-1. EMBO J. 1998; 17: 2846-2854.
68. Lichtlen P, Schaffner W. The "metal transcription factor" MTF-1: biological facts and medical implications. Swiss Med Wkly. 2001; 131: 647-652.
69. Wimmer U, Wang Y, Georgiev O, Schaffner W. Two major branches of anti-cadmium defense in the mouse: MTF-1/metallothioneins and glutathione. Nucleic Acids Res. 2005; 33: 5715-5727.
70. Rutherford JC, Bird AJ. Metal-responsive transcription factors that regulate iron, zinc, and copper homeostasis in eukaryotic cells. Eukaryot Cell. 2004; 3: 1-13.
71. Adams TK, Saydam N, Steiner F, Schaffner W, Freedman JH. Activation of gene expression by metal-responsive signal transduction pathways. Environ Health Perspect. 2002; 110 Suppl 5: 813-817.
72. Bourdineaud JP, Baudrimont M, Gonzalez P, Moreau JL. Challenging the model for induction of metallothionein gene expression. Biochimie. 2006; 88: 1787-1792.
73. Vergani L, Lanza C, Borghi C, Scarabelli L, Panfoli I, Burlando B i wsp. Effects of growth hormone and cadmium on the transcription regulation of two metallothionein isoforms. Mol Cell Endocrinol. 2007; 263: 29-37.
74. Thévenod F. Cadmium and cellular signaling cascades: to be or not to be? Toxicol Appl Pharmacol. 2009; 238: 221-239.
75. Andrews GK. Regulation of metallothionein gene expression by oxidative stress and metal ions. Biochem Pharmacol. 2000; 59: 95-104.

Metalotioneiny i kadm a rak piersi 240
76. Jiang H, Fu K, Andrews GK. Gene- and cell-type-specific effects of signal transduction cascades on metal-regulated gene transcription appear to be independent of changes in the phosphorylation of metal-response-element-binding transcription factor-1. Biochem J. 2004; 382: 33-41.
77. Lau AT, Zhang J, Chiu JF. Acquired tolerance in cadmium-adapted lung epithelial cells: roles of the c-Jun N-terminal kinase signaling pathway and basal level of metallothionein. Toxicol Appl Pharmacol. 2006; 215: 1-8.
78. Rao PS, Jaggi M, Smith DJ, Hemstreet GP, Balaji KC. Metallothionein 2A interacts with the kinase domain of PKCmu in prostate cancer. Biochem Biophys Res Commun. 2003; 310: 1032-1038.
79. Ray PD, Huang BW, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012; 24: 981-990.
80. Corre S, Galibert MD. USF as a key regulatory element of gene expression. Med Sci (Paris). 2006; 22: 62-67.
81. Venugopal R, Jaiswal AK. Nrf1 and Nrf2 positively and c-Fos and Fra1 negatively regulate the human antioxidant response element-mediated expression of NAD(P)H: quinone oxidoreductase1 gene. Proc Natl Acad Sci USA. 1996; 93: 14960-14965
82. Ozanne BW, McGarry L, Spence HJ, Johnston I, Winnie J, Meagher L i wsp. Transcriptional regulation of cell invasion: AP-1 regulation of a multigenic invasion programme. Eur J Cancer. 2000, 36: 1640-1648.
83. Beyersmann D, Hechtenberg S. Cadmium, gene regulation, and cellular signaling in mammalian cells. Toxicol Appl Pharmacol. 1997; 144: 247-261.
84. Pathak N, Khandelwal S. Role of oxidative stress and apoptosis in cadmium induced thymic atrophy and splenomegaly in mice. Toxicol Lett. 2007; 169: 95-108.
85. Bednarek J, Kiliańska ZM.Białka przestrzeni międzybłonowej mitochondriów uczestniczące w procesie apoptozy. Postępy Biochem. 2005; 51: 447–458.
86. Kiliańska ZM, Miśkiewicz A. Kaspazy kręgowców: ich rola w przebiegu apoptozy. Postępy Biol Kom. 2003; 30: 129–152.
87. Łabędzka K, Grzanka A, Izdebska M. Mitochondrium a śmierć komórki. Postępy Hig Med Dośw. 2006; 60: 439–446.
88. Rogalińska M. Alterations in cell nuclei during apoptosis. Cell Mol Biol Lett. 2002; 7: 995-1018.
89. Hamada T, Tanimoto A, Sasaguri Y. Apoptosis induced by cadmium. Apoptosis.1997; 2: 359-367

E. Forma, M. Bryś 241
90. Pulido MD, Parrish AR. Metal-induced apoptosis: mechanisms. Mutat Res. 2003; 533: 227-241.
91. Kim MS, Kim BJ, Woo HN, Kim KW, Kim KB, Kim IK i wsp. Cadmium induces caspase-mediated cell death: suppression by Bcl-2. Toxicology. 2000; 145: 27-37.
92. Kondo Y, Rusnak JM, Hoyt DG, Settineri CE, Pitt BR, Lazo JS. Enhanced apoptosis in metallothionein null cells. Mol Pharmacol. 1997; 52: 195-201.
93. Aimola P, Carmignani M, Volpe AR, Di Benedetto A, Claudio L, Waalkes MP, van Bokhoven A, Tokar EJ, Claudio PP. Cadmium induces p53-dependent apoptosis in human prostate epithelial cells. PLoS One. 2012; 7: e33647.
94. Hengstler JG, Bolm-Audorff U, Faldum A, Janssen K, Reifenrath M, Götte W i wsp. Occupational exposure to heavy metals: DNA damage induction and DNA repair inhibition prove co-exposures to cadmium, cobalt and lead as more dangerous than hitherto expected. Carcinogenesis. 2003; 24: 63-73.
95. Asmuss M, Mullenders LH, Eker A, Hartwig A. Differential effects of toxic metal compounds on the activities of Fpg and XPA, two zinc finger proteins involved in DNA repair. Carcinogenesis. 2000; 21: 2097-2104.
96. Witkiewicz-Kucharczyk A, Bal W. Damage of zinc fingers in DNA repair proteins, a novel molecular mechanism in carcinogenesis. Toxicol Lett. 2006; 162: 29-42.
97. Jacob ST, Majumder S, Ghoshal K. Suppression of metallothionein-I/II expression and its probable molecular mechanisms. Environ Health Perspect. 2002; 110 Suppl 5: 827-30.
98. Morahan JM, Yu B, Trent RJ, Pamphlett R. Are metallothionein genes silenced in ALS? Toxicol Lett. 2007; 168: 83-87.
99. Beavon IR. The E-cadherin-catenin complex in tumour metastasis: structure, function and regulation. Eur J Cancer. 2000; 36: 1607-1620.
100. Edwards JR, Kolman K, Lamar PC, Chandar N, Fay MJ, Prozialeck WC. Effects of cadmium on the sub-cellular localization of β-catenin and β-catenin-regulated gene expression in NRK-52E cells. Biometals. 2012 DOI 10.1007/s10534-012-9592-0
101. Chakrabarty S, Radjendirane V, Appelman H, Varani J. Extracellular calcium and calcium sensing receptor function in human colon carcinomas: promotion of E-cadherin expression and suppression of beta-catenin/TCF activation. Cancer Res. 2003; 63: 67-71.
102. Conacci-Sorrell M, Zhurinsky J, Ben-Ze'ev A. The cadherin-catenin adhesion system in signaling and cancer. J Clin Invest. 2002; 109: 987-991.

Metalotioneiny i kadm a rak piersi 242
103. Curran S, Murray GI. Matrix metalloproteinases: molecular aspects of their roles in tumour invasion and metastasis. Eur J Cancer. 2000; 36: 1621-1630.
104. Heimann R, Hellman S. Individual characterisation of the metastatic capacity of human breast carcinoma. Eur J Cancer. 2000; 36: 1631-1639.
105. Beattie JH, Owen HL, Wallace SM, Arthur JR, Kwun IS, Hawksworth GM i wsp. Metallothionein overexpression and resistance to toxic stress. Toxicol Lett. 2005; 157: 69-78.
106. Fan LZ, Cherian MG. Potential role of p53 on metallothionein induction in human epithelial breast cancer cells. Br J Cancer. 2002; 87: 1019-1026.
107. Méplan C, Richard MJ, Hainaut P. Redox signalling and transition metals in the control of the p53 pathway. Biochem Pharmacol. 2000; 59: 25-33.
108. Reaves SK, Fanzo JC, Arima K, Wu JY, Wang YR, Lei KY. Expression of the p53 tumor suppressor gene is up-regulated by depletion of intracellular zinc in HepG2 cells. J Nutr. 2000; 130: 1688-1694.
109. Agarwal ML, Agarwal A, Taylor WR, Stark GR. p53 controls both the G2/M and the G1 cell cycle checkpoints and mediates reversible growth arrest in human fibroblasts. Proc Natl Acad Sci USA. 1995; 92: 8493-8497.
110. Murphy KL, Rosen JM. Mutant p53 and genomic instability in a transgenic mouse model of breast cancer. Oncogene. 2000; 19: 1045-1051.
111. Wani MA, Zhu QZ, El-Mahdy M, Wani AA. Influence of p53 tumor suppressor protein on bias of DNA repair and apoptotic response in human cells. Carcinogenesis. 1999; 20: 765-772.
112. Koedrith P, Seo YR. Advances in carcinogenic metal toxicity and potential molecular markers. Int J Mol Sci. 2011; 12: 9576-9595.
113. Ostrakhovitch EA, Olsson PE, Jiang S, Cherian MG. Interaction of metallothionein with tumor suppressor p53 protein. FEBS Lett. 2006; 580: 1235-1238.
114. Lozano G. The oncogenic roles of p53 mutants in mouse models. Curr Opin Genet Dev. 2007; 17: 66-70.
115. Elledge RM, Allred DC. Prognostic and predictive value of p53 and p21 in breast cancer. Breast Cancer Res Treat. 1998; 52: 79-98.
116. Svechnikova I, Ammerpohl O, Ekström TJ. p21waf1/Cip1 partially mediates apoptosis in hepatocellular carcinoma cells. Biochem Biophys Res Commun. 2007; 354: 466-471.
117. Thor AD, Liu S, Moore DH 2nd, Shi Q, Edgerton SM. p(21WAF1/CIP1) expression in breast cancers: associations with p53 and outcome. Breast Cancer Res Treat. 2000; 61: 33-43.

E. Forma, M. Bryś 243
118. Xie J, Shaikh ZA. Cadmium induces cell cycle arrest in rat kidney epithelial cells in G2/M phase. Toxicology. 2006; 224: 56-65.
119. Taylor WR, DePrimo SE, Agarwal A, Agarwal ML, Schönthal AH, Katula KS, Stark GR. Mechanisms of G2 arrest in response to overexpression of p53. Mol Biol Cell. 1999; 10: 3607-3622.
120. Jiang H, Fu K, Andrews GK. Gene- and cell-type-specific effects of signal transduction cascades on metal-regulated gene transcription appear to be independent of changes in the phosphorylation of metal-response-element-binding transcription factor-1. Biochem J. 2004; 382: 33-41.
121. Fan LZ, Cherian MG. Potential role of p53 on metallothionein induction in human epithelial breast cancer cells. Br J Cancer. 2002; 87: 1019-1026.
122. Méplan C, Mann K, Hainaut P. Cadmium induces conformational modifications of wild-type p53 and suppresses p53 response to DNA damage in cultured cells. J Biol Chem. 1999; 274: 31663-31670.
123. Jin R, Chow VT, Tan PH, Dheen ST, Duan W, Bay BH. Metallothionein 2A expression is associated with cell proliferation in breast cancer. Carcinogenesis. 2002; 23: 81-86.
124. Cogswell PC, Guttridge DC, Funkhouser WK, Baldwin AS Jr. Selective activation of NF-kappa B subunits in human breast cancer: potential roles for NF-kappa B2/p52 and for Bcl-3. Oncogene. 2000; 19: 1123-1131.
125. Doszczak MM, Kaszubowska L, Pierzchalski A, Bigda J. Mechanizm aktywacji czynnika transkrypcyjnego NF-κB przez czynnik martwicy nowotworu (TNF). Postępy Biochem. 2002; 48: 54–65.
126. Kim DW, Sovak MA, Zanieski G, Nonet G, Romieu-Mourez R, Lau AW i wsp. Activation of NF-kappaB/Rel occurs early during neoplastic transformation of mammary cells. Carcinogenesis. 2000; 21: 871-879.
127. Liu P, Kimmoun E, Legrand A, Sauvanet A, Degott C, Lardeux B i wsp. Activation of NF-kappa B, AP-1 and STAT transcription factors is a frequent and early event in human hepatocellular carcinomas. J Hepatol. 2002; 37: 63-71.
128. Gruber BM. Czynnik transkrypcyjny NF-κB – nowa perspektywa w leczeniu nowotworów. Postępy Biochem. 2004; 50: 118–130.
129. Jin R, Bay BH, Chow VT, Tan PH, Dheen T. Significance of metallothionein expression in breast myoepithelial cells. Cell Tissue Res. 2001; 303: 221-226.
130. Jin R, Bay BH, Chow VT, Tan PH. Metallothionein 1F mRNA expression correlates with histological grade in breast carcinoma. Breast Cancer Res Treat. 2001; 66: 265-272.

Metalotioneiny i kadm a rak piersi 244
131. Jin R, Bay BH, Chow VT, Tan PH, Lin VC. Metallothionein 1E mRNA is highly expressed in oestrogen receptor-negative human invasive ductal breast cancer. Br J Cancer. 2000; 83: 319-323.
132. Barnes NL, Ackland ML, Cornish EJ. Metallothionein isoform expression by breast cancer cells. Int J Biochem Cell Biol. 2000; 32: 895-903.
133. Platet N, Cathiard AM, Gleizes M, Garcia M. Estrogens and their receptors in breast cancer progression: a dual role in cancer proliferation and invasion. Crit Rev Oncol Hematol. 2004; 51: 55-67.
134. Osborne CK. Steroid hormone receptors in breast cancer management. Breast Cancer Res Treat. 1998; 51: 227-238.
135. Wojnar A, Pula B, Piotrowska A, Jethon A, Kujawa K, Kobierzycki C i wsp. Correlation of intensity of MT-I/II expression with Ki-67 and MCM-2 proteins in invasive ductal breast carcinoma. Anticancer Res. 2011; 31: 3027-3033.
136. Gomulkiewicz A, Podhorska-Okolow M, Szulc R, Smorag Z, Wojnar A, Zabel M i wsp. Correlation between metallothionein (MT) expression and selected prognostic factors in ductal breast cancers. Folia Histochem Cytobiol. 2010; 48: 242-248.
137. Krześlak A, Forma E, Jóźwiak P, Szymczyk A, Smolarz B, Romanowicz-Makowska H i wsp. Metallothionein 2A genetic polymorphisms and risk of ductal breast cancer. Clin Exp Med. 2012; DOI 10.1007/s10238-012-0215-4
138. Somji S, Garrett SH, Zhou XD, Zheng Y, Sens DA, Sens MA. Absence of Metallothionein 3 Expression in Breast Cancer is a Rare, But Favorable Marker of Outcome that is Under Epigenetic Control. Toxicol Environ Chem. 2010; 92: 1673-1695.

Folia Medica Lodziensia, 2012, 39/2:245-264
Adres do korespondencji: mgr Paweł Jóźwiak, Katedra Cytobiochemii, Wydział Biologii i Ochrony Środowiska, Uniwersytet Łódzki; 90-236 Łódź, ul. Pomorska 141/143; Tel.: 0 42 635 44 03, Fax.: 0 42 635 44 84, e-mail: [email protected]
Rola transporterów GLUT1 i GLUT3 w pobieraniu
glukozy i kwasu dehydroaskorbinowego przez komórki
nowotworowe
The role of transporters GLUT1 and GLUT3 in glucose and dehydroascorbic acid uptake in cancer cells
PAWEŁ JÓŹWIAK
Katedra Cytobiochemii Wydział Biologii i Ochrony Środowiska Uniwersytetu Łódzkiego
Streszczenie Charakterystyczną cechą komórek nowotworowych jest zwiększony metabolizm
połączony z zahamowaniem fosforylacji oksydacyjnej. Oddychanie prze-
biegające w warunkach beztlenowych prowadzi do przyspieszonej glikolizy
i wzrostu zapotrzebowania na glukozę. W nasilonym pobieraniu glukozy przez
komórki pośredniczą transportery glukozy – GLUT (ang. glucose transporters).
Nadekspresję białek GLUT, w szczególności regulowanych warunkami hipoksji -
GLUT1 i GLUT3 zaobserwowano w wielu typach nowotworów. Ponieważ
transportery te wykazują wysokie powinowactwo do glukozy i przenoszą ją
z dużą wydajnością, stanowią one kluczowy czynnik limitujący metabolizm
glukozy w komórkach guza. Dotychczasowe badania koncentrowały się na roli
jaką odgrywają transportery GLUT1 i GLUT3 w transporcie glukozy pomijając
aspekt związany z metabolizmem witaminy C. Transportery GLUT pośredniczą
w pobieraniu przez komórkę kwasu dehydroaskorbinowego (DHA), który
wewnątrz komórki ulega redukcji do biologicznie aktywnej formy witaminy C.
W warunkach fizjologicznych witamina C ma właściwości antyoksydacyjne,
chroniąc komórki przed stresem oksydacyjnym. Jednakże w zależności od
warunków, witamina C może również wywoływać efekty pro-oksydacyjne
związane z generowaniem reaktywnych form tlenu. Stwierdzono, że niski
poziom wolnych rodników i nadtlenku wodoru stymuluje żywotność i proliferację
komórek, podczas gdy wysokie stężenie reaktywnych form tlenu prowadzi do
apoptozy lub nekrozy. W pracy przedstawiono aktualny stan wiedzy na temat

Rola transporterów GLUT1 i GLUT3 w nowotworach 246
transportu glukozy i kwasu dehydroaskorbinowego z udziałem transporterów
GLUT1 i GLUT3 w komórkach nowotworowych
Słowa kluczowe: GLUT1, GLUT3, glukoza, kwas dehydroaskorbinowy, kwas
askorbinowy, nowotwory
Abstract
Cancer cells are characterized by acceleration in energy consumption, together with the inhibition of oxidative phosphorylation. The anaerobic respiration leads to the enhancement of glycolysis and the higher rate of glucose uptake into cancer cells. The upregulation of glucose transport across the plasma membrane is mediated by facilitative glucose transporters named GLUTs. The overexpression of GLUTs, especially the hypoxia responsive GLUT1 and GLUT3, has been frequently observed in the variety of human malignancies. Since GLUT1 and GLUT3 have a high transporting efficiency and affinity to glucose, these proteins are key rate-limiting factors in glucose uptake by cancer cells. The reported data focus on the glucose transport mediated by GLUT1 and GLUT3, ignoring the participation of both carriers in vitamin C metabolism. The GLUT proteins transport dehydroascorbic acid (DHA) which intracellularly is reduced to an antioxidant, protecting cells from oxidative stress. However, vitamin C may also have pro-oxidant effects, via its ability to generate reactive oxygen species (ROS). It has been shown, that the low level of free radicals and hydrogen peroxide stimulates cell proliferation and viability, whereas the high concentration of reactive oxygen species may induce apoptosis or necrosis. This paper presents the current state of knowledge concerning the transport of glucose and dehydroascorbic acid mediated by hypoxia-induced facilitative glucose transporters in cancer cells.
Key words: GLUT1, GLUT3, glucose, dehydroascorbic acid, ascorbic acid, cancers

P. Jóźwiak 247
Wprowadzenie
Powszechnie wiadomo, że komórki nowotworowe charakteryzują się
odmiennym metabolizmem w porównaniu z komórkami prawidłowymi.
W komórkach nowotworowych obserwuje się zahamowanie procesu fosforylacji
oksydacyjnej i transportu elektronów na tlen, co prowadzi do nasilonej produkcji
reaktywnych form tlenu, które zwiększają poziom mutacji w obrębie proto-
onkogenów [1, 2]. Warunki hipoksji nowotworu przesuwają metabolizm
w stronę glikolizy beztlenowej, której kontynuacja wymaga regeneracji
koenzymu NAD+. W tym celu komórki nowotworowe preferują przemianę
pirogronianu do mleczanu poprzez indukcję izoformy A dehydrogenazy
mleczanowej. W reakcji produkcji mleczanu odtworzony zostaje koenzym
NAD+ umożliwiający cykliczność procesu glikolizy, a całkowity zysk
energetyczny oddychania beztlenowego stanowią dwie cząsteczki ATP [3, 4].
Wysokie wymagania energetyczne komórek nowotworowych prowadzą
do kompensacji niskiego zysku energetycznego oddychania beztlenowego
poprzez 20-30-krotne zintensyfikowanie procesu glikolizy. Nasileniu procesu
glikolizy towarzyszy zwiększone pobieranie glukozy przez komórki
nowotworowe, które odbywa się za pośrednictwem transporterów glukozy
określanych jako GLUT [2, 5]. Oprócz transportu glukozy, białka GLUT
uczestniczą w metabolizmie witaminy C, która w zależności od stężenia
w komórce wywołuje efekty anty- lub pro-oksydacyjne [6]. Zależne od
warunków hipoksji transportery GLUT1 i GLUT3 mogą odgrywać istotną rolę
w regulacji stanu czynnościowego komórek nowotworowych.

Rola transporterów GLUT1 i GLUT3 w nowotworach 248
Transportery GLUT
Rodzina transporterów GLUT składa się z 14 białek, które zostały
zgrupowane w trzech klasach. Klasa I zawiera najlepiej scharakteryzowane
transportery GLUT1-GLUT4 i niedawno poznany transporter GLUT14.
Do klasy II należy transporter fruktozy GLUT5 oraz transportery GLUT7,
GLUT9 i GLUT11. Pozostałe transportery: GLUT6, GLUT8, GLUT10,
GLUT12 i transporter HMIT (HMIT, ang. H+-coupled myo-inositol transporter)
wchodzą w skład klasy III [7, 8]. Wszystkie transportery GLUT zawierają
12 hydrofobowych α-helikalnych transmembranowych domen z dużą wewnątrz-
komórkową pętlą (pomiędzy domeną 6 i 7) (Ryc. 1).
Regiony N- i C- końcowe tych białek zlokalizowane są po stronie
cytoplazmatycznej. Białka GLUT klasy I i II mają pomiędzy domeną
transmembranową 1 i 2 zewnątrzkomórkową pętlę z miejscem N-glikozylacji
[9, 10]. N-glikozylacja GLUT1 na Asn45 zwiększa jego stabilność i aktywność
transportową [11]. Analogiczne miejsce N-glikozylacji w przypadku
transporterów klasy III występuje pomiędzy domeną transmembranową 9 i 10.
Wszystkie białka GLUT transportują heksozy niezależnie od energii, zgodnie
z gradientem stężeń, z różną kinetyką i powinowactwem względem substratu.
Największe powinowactwo do glukozy mają indukowane warunkami hipoksji
transportery GLUT1 i GLUT3 oraz insulino-zależny transporter GLUT4.
Poszczególne tkanki organizmu cechują się odmiennym profilem ekspresji
transporterów GLUT.

P. Jóźwiak 249
Ryc. 1 Schemat budowy transporterów glukozy GLUT (opis w tekście – wg. [12]; w modyfikacji własnej)
W tej samej tkance można obserwować ekspresję kilku izoform GLUT, jak
również wiele tkanek może transportować heksozy z udziałem tego samego
transportera [7,12]. Nadekspresję transporterów GLUT, w szczególności
zależnych od warunków hipoksji nowotworu GLUT1 i GLUT3 zaobserwowano
w wielu typach nowotworów. Obecnie sugeruje się, że specyficzny profil
ekspresji białek GLUT mógłby stanowić prognostyczny wskaźnik złośliwości

Rola transporterów GLUT1 i GLUT3 w nowotworach 250
oraz stopnia zaawansowania klinicznego nowotworu, który umożliwiłby
wyodrębnienie pacjentów wymagających agresywniejszego leczenia [13, 14].
Transport glukozy z udziałem transporterów GLUT1 i GLUT3
w nowotworach
Transportery GLUT1 i GLUT3 charakteryzują się wyższym
powinowactwem do glukozy w porównaniu z pozostałymi białkami rodziny
GLUT. Stwierdzono, że nadekspresja obu izoform, wynikająca ze wzmożonego
zapotrzebowania komórek nowotworowych na energię, koreluje ze wzrostem
rozmiarów i inwazyjnością guza [15]. Proces zwiększonego transportu glukozy
do komórek nowotworowych znalazł kliniczne zastosowanie w diagnostyce
wielu typów nowotworów i ich przerzutów przez znakowanie guzów analogiem
glukozy [18F]-fluoro-2-deoksy-D-glukozą (18FDG) z wykorzystaniem techniki
pozytonowej tomografii emisyjnej (PET) [16]. Wysoka maksymalna
standardaryzowana wartość wychwytu 18FDG (SUV(max)) związana
z podwyższonym poziomem ekspresji GLUT1 w płaskonabłonkowych rakach
płuc w porównaniu z gruczolakami, korelowała ze stopniem zróżnicowania
komórek nowotworu oraz przerzutami do węzłów chłonnych [17]. Podobnie
w rakach pleomorficznych płuc zaobserwowano ścisły związek pomiędzy
poziomem ekspresji transporterów GLUT1 i GLUT3, a wychwytem 18FDG [18].
Warunkiem aktywnego transportu cukru z udziałem białek GLUT jest ich
translokacja z przestrzeni wewnątrzkomórkowej do błony komórkowej. Ponad
dwukrotnie niższy wychwyt 18FDG skorelowany z translokacją GLUT3 z błony
plazmatycznej do cytozolu stwierdzono w liniach komórkowych wrażliwych na
gefitynib – inhibitor kinazy EGFR (EGFR, ang. epidermal growth factor
receptor) [19]. Z kolei, stymulacja insuliną linii komórkowych mięsaków

P. Jóźwiak 251
kościopochodnych podnosiła konsumpcję glukozy w wyniku przemieszczenia
GLUT1 do błony komórkowej [20]. Regulacja ekspresji transporterów GLUT1
i GLUT3 jest złożonym procesem, w którym bierze udział wiele czynników
takich jak: hipoksja, czynniki wzrostu, hormony, onkogeny i geny supresorowe
[21-25]. Z drugiej strony nadekspresja transporterów GLUT, w szczególności
indukowanych warunkami hipoksji nowotworu GLUT1 i GLUT3, umożliwia
lepszą akumulację w komórkach nowotworowych pochodnych glukozy oraz
skoniugowanych z nimi związków o potencjale terapeutycznym. Jednym
z takich analogów jest 2-deoksyglukoza (2-DG), której działanie prowadzi do
kompetycyjnej inhibicji glikolizy [26]. Wewnątrz komórki 2-DG podlega
fosforylacji przez heksokinazę, ale nie stanowi ona substratu dla izomerazy
glukozo-fosforanowej. W rezultacie, nagromadzenie fosforylowanej
2-deoksyglukozy w komórce prowadzi do zatrzymania glikolizy, cyklu
komórkowego i uruchomienia apoptozy [27]. Ponadto, 2-DG hamuje
N-glikozylację transporterów GLUT, która jest niezbędna dla ich translokacji do
błony komórkowej [28]. Szereg badań przeprowadzonych na wielu typach linii
nowotworowych wskazuje na fakt, że 2-DG wspomaga stosowaną powszechnie
radioterapię. Sugeruje się, że efekt ten ma związek ze wzrostem stresu
oksydacyjnego wynikającego z zahamowania metabolizmu tioli (związków
zawierających grupę –SH, np.: cysteina) [29]. Co więcej, liczne badania
wykazały, że 2-DG uwrażliwia komórki nowotworowe na chemioterapię.
Traktowanie komórek glejaka oraz nowotworu złośliwego skóry 2-DG
w połączeniu z kamptotecyną (inhibitor topoizomerazy I) i etopozydem
(inhibitor topoizomerazy II) znacząco podnosiło cytotoksyczność terapii, co
skutkowało nasileniem apoptozy lub śmierci mitotycznej komórek [30].
Podobne wspomagające działanie 2-DG związane ze wzrostem cytotoksyczności

Rola transporterów GLUT1 i GLUT3 w nowotworach 252
zaobserwowano w przypadku chemioterapii innych linii nowotworowych
z zastosowaniem adriamycyny i paklitakselu [31]. Ponadto, 2-DG hamuje
transkrypcję wirusa brodawczaka ludzkiego, co sugeruje, że związek ten może
uwrażliwiać na chemioterapię lekooporne nowotwory szyjki macicy [29]. Obok
rozwoju terapii łączonej z wykorzystaniem niemetabolizowanych analogów
glukozy, rośnie zainteresowanie syntezą koniugatów konwencjonalnych
cytotoksycznych leków z cząsteczkami glukozy. Głównym celem takich
połączeń jest zwiększenie biodostępności i efektywności stosowanej obecnie
chemioterapii. Za przykład może posłużyć 2-GluSNAP – koniugat powstały
z połączenia glukozy z donorem tlenku azotu S-nitrozo-N-acetyl-penicyllaminą
(SNAP), którego cytotoksyczność koreluje z poziomem ekspresji transportera
GLUT1 [32]. W odróżnieniu od komórek prawidłowych, głównym substratem
energetycznym komórek nowotworowych jest glukoza. Fakt ten sprzyja
postępowi badań nad zastosowaniem niemetabolizowanych analogów glukozy
w terapii nowotworów.
Transport witaminy C do komórek nowotworowych
W dostępnych danych literaturowych dotyczących roli transporterów
GLUT w patogenezie i terapii nowotworów autorzy skupiają uwagę na
aspektach związanych z transportem glukozy i jej analogów pomijając znaczenie
jakie odgrywają transportery GLUT w metabolizmie witaminy C. Transport
kwasu askorbinowego (AA) do komórek może zachodzić na drodze dwóch
różnych mechanizmów: transportu czynnego z udziałem transporterów
zależnych od jonów sodu SVCT (ang. sodium-dependent vitamin C transporter)
lub dyfuzji ułatwionej za pośrednictwem transporterów glukozy GLUT.
Wysokie stężenie kwasu askorbinowego hamuje ekspresję transporterów SVCT

P. Jóźwiak 253
na zasadzie sprzężenia zwrotnego, natomiast transport z udziałem białek GLUT
wymaga utlenienia kwasu askorbinowego do kwasu dehydroaskorbinowego
(DHA), który następnie wewnątrz komórki jest redukowany do aktywnej
biologicznie witaminy C (Ryc. 2).
Ryc. 2. Transport i rola witaminy C w degradacji podjednostek α czynnika transkrypcyjnego HIF.
SVCT – transporter witaminy C zależny od jonów sodu (ang. sodium-dependent vitamin C transporter);
GLUT – transporter glukozy (ang. glucose transporter); AA – kwas askorbinowy (ang. ascorbic acid); DHA – kwas dehydroaskorbinowy (ang. dehydroascorbic acid); HIF-α – podjednostka α czynnika transkrypcyjnego indukowanego warunkami hipoksji
(ang. hypoxia-inducible factor α); vHL – białko von Hippel-Lindau (von Hippel-Lindau); 2-OG – 2-oksoglutaran (2-oxoglutarate); PHD – hydroksylaza prolinowa (ang. prolyl hydoxylase); FIH – hydroksylaza asparaginianowa hamująca ekspresję czynnika HIF
(ang. asparaginyl hydroxylase, factor inhibiting HIF) (opis w tekście)

Rola transporterów GLUT1 i GLUT3 w nowotworach 254
Ze względu na podobieństwo strukturalne DHA do glukozy, oba substraty,
w zależności od typu komórek, mogą rywalizować o wiązanie do transportera
[6, 33]. Przeprowadzone badania sugerują, że w dyfuzji ułatwionej DHA do
komórki pośredniczą izoformy pierwszej klasy białek GLUT, tj. GLUT1,
GLUT3 oraz GLUT4, a wydajność transportu DHA z udziałem wymienionych
białek, określona na podstawie stałych kinetycznych, jest zbliżona do
wydajności transportu glukozy [34, 35]. Do tej pory nie wiadomo, który
z mechanizmów odgrywa istotniejszą rolę w akumulacji askorbinianu
w komórkach nowotworowych. Komórki chłoniaka Nb2 transportują zarówno
AA jak i DHA, podczas gdy komórki białaczki szpikowej HL-60 transportują
tylko utlenioną formę witaminy C [36, 37]. Badania przeprowadzone na liniach
komórkowych czerniaka i prawidłowych melanocytach człowieka wykazały, że
obie linie transportują witaminę C na drodze zależnej i niezależnej od
jonów sodu. Jednakże, komórki czerniaka gromadzą ponad sto razy
więcej askorbinianu w wyniku transportu DHA drogą dyfuzji ułatwionej
w porównaniu z komórkami prawidłowymi [38]. Przypuszcza się, że istnieją
mechanizmy, które prowadzą do utlenienia AA do DHA w mikrośrodowisku
komórek nowotworowych. Prawdopodobnie w procesie zewnątrzkomórkowego
utleniania askorbinianiu uczestniczy γ-glutamylotransferaza – enzym
pro-oksydacyjny zakotwiczony w błonie komórkowej [39]. Opublikowane przez
KC i wsp. [40] wyniki badań, wykazały że oprócz transportu przez błonę
plazmatyczną GLUT1 pośredniczy w transporcie DHA do mitochondriów.
Stwierdzono, że niskie stężenia AA chronią mitochondria przed stresem
oksydacyjnym, natomiast wysokie stężenia indukują spadek potencjału błony
mitochondrialnej i uwolnienie cytochromu c z mitochondrium do cytozolu, co
promuje apoptozę [40].

P. Jóźwiak 255
Biologiczne funkcje kwasu askorbinowego w nowotworach
Kwas askorbinowy uczestniczy w wielu procesach biochemicznych
katalizowanych przez dioksygenazy zależne od Fe2+ i 2-oksoglutaranu.
Askorbinian stabilizuje żelazo na II stopniu utlenienia dzięki czemu
dioksygenazy uzyskują pełną aktywność enzymatyczną [41]. Stwierdzono, że
kwas askorbinowy działa stymulująco na produkcję kolagenu typu I i III
w fibroblastach człowieka. Białka kolagenowe wydzielane do macierzy
zewnątrzkomórkowej hamują inwazyjność i metastazę komórek
nowotworowych [42, 43].
Witamina C jest ważnym kofaktorem hydroksylaz, które uczestniczą
w regulacji ekspresji aktywowanego niedotlenieniem czynnika transkrypcyjnego
HIF-1, [44]. HIF-1 reguluje transkrypcję wielu genów, których produkty
białkowe są zaangażowane w metabolizm glukozy, angiogenezę i metastazę
komórek nowotworowych. Białko HIF jest heterodimerem składającym się
z podjednostki HIF-α, oraz konstytutywnie stabilnej podjednostki HIF-β [5].
W warunkach fizjologicznych podjednostki HIF-α podlegają hydroksylacji przez
hydroksylazy prolinowe (PHD1, PHD2 i PHD3) oraz hydroksylazę
asparaginianową, których aktywność jest uwarunkowana obecnością tlenu.
Oprócz tlenu, hydroksylazy prolinowe i asparaginianowe, podobnie jak inne
dioksygenazy zależne od Fe2+ i 2-oksoglutaranu, wymagają askorbinianu
niezbędnego do utrzymania stopnia utlenienia żelaza i osiągnięcia pełnej
aktywności enzymatycznej (Ryc. 2). Hydroksylacja umożliwia wiązanie
podjednostek HIF-α z białkiem vHL (von Hippel-Lindau) rozpoznawanym przez
ligazę ubikwitynową, a następnie proteolityczną degradację powstałego
kompleksu [45]. Badania wskazują na związek pomiędzy poziomem
askorbinianu a ekspresją czynnika HIF-1 w nowotworach. Askorbinian obniża

Rola transporterów GLUT1 i GLUT3 w nowotworach 256
poziom ekspresji HIF-1 zmniejszając złośliwość guza nowotworowego [46].
Kolejną grupą enzymów, których kofaktorem jest kwas askorbinowy stanowią
demetylazy histonowe [47]. Zaobserwowano zwiększoną acetylację histonów
oraz ekspresję demetylaz histonowych po suplementacji embrionalnych
komórek macierzystych człowieka askorbinianem [48]. Sugeruje to, że
wewnątrzkomórkowy poziom kwasu askorbinowego może wpływać na status
epigenetyczny komórek. Ponieważ rozwój nowotworu jest związany
z hipermetylacją DNA, przypuszcza się, że witamina C i inne suplementy diety
pośrednio uczestnicząc w mechanizmach epigenetycznych mogą chronić przed
nowotworzeniem [49].
Anty- i pro-oksydacyjne właściwości kwasu askorbinowego
Antyoksydacyjne działanie kwasu askorbinowego obniża peroksydację
lipidów, chroni przed mutacjami w obrębie genów oraz zmniejsza oksydację
reszt aminokwasowych zwiększając integralność białek komórkowych [50].
Badania dowodzą, że suplementacja witaminą C ogranicza ryzyko wystąpienia
przewlekłych chorób w tym także nowotworów. Bogata w kwas askorbinowy
dieta poprzez niszczenie wolnych rodników tlenowych produkowanych
intensywnie w trakcie przebiegu zakażenia Helicobacter pylori oraz hamowanie
procesu nitrozowania amin biogennych chroni przed rozwojem raka żołądka
[51]. W warunkach fizjologicznych stężenie kwasu askorbinowego w surowicy
krwi człowieka wynosi od 40 do 80 µM/L. Kwas askorbinowy redukując rodniki
takie jak: hydroksylowy (OH•), nadtlenolipidowy (LOO•), alkoksylowy (RO•)
czy tiolowy (GS•) ulega utlenieniu do znacznie mniej reaktywnego rodnika
askorbylowego (Asc•–). Ponadto, w wyniku redukcji, powstałego w reakcji
z produktami peroksydacji lipidów, rodnika α-tokoferoksylowego (TO•) kwas

P. Jóźwiak 257
askorbinowy istotnie zwiększa poziom witaminy E w komórce. Regeneracja
rodnika askorbylowego do reaktywnej cząsteczki askorbinianu następuje pod
wpływem działania NADH- lub NADPH-zależnej reduktazy występującej
w siateczce śródplazmatycznej i błonie komórkowej [6]. Paradoksalnie, kwas
askorbinowy może wywoływać efekty pro-oksydacyjne. Właściwości te
wynikają z redukcji jonów metali przejściowych takich jak żelaza lub miedzi,
które następnie reagując z nadtlenkiem wodoru generują wysoce reaktywne
rodniki hydroksylowe. W warunkach fizjologicznych metale przejściowe
występują głównie w formie związanej z białkami. Uważa się, że w wysokich
terapeutycznych stężeniach pro-oksydacyjne działanie kwasu askorbinowego
wynika z produkcji nadtlenku wodoru i może przebiegać niezależnie od jonów
metali przejściowych. W komórkach nowotworowych obserwuje się
zahamowanie działania glutationu oraz aktywności enzymów neutralizujących
stres oksydacyjny, na przykład katalazy i dysmutazy ponadtlenkowej [52]. Stres
oksydacyjny odgrywa istotną rolę na każdym etapie rozwoju nowotworu.
W wielu typach komórek niskie stężenie wolnych rodników i nadtlenku wodoru
stymulowało proliferację, podczas gdy wysoki poziom stresu oksydacyjnego
stawał się cytotoksyczny i prowadził do indukcji apoptozy lub nekrozy. Wolne
rodniki hamują aktywność fosfataz komórkowych prawdopodobnie poprzez
interakcję z grupami sulfhydrylowymi cystein, których utlenienie prowadzi do
powstania wewnątrz- lub międzycząsteczkowych wiązań dwusiarczkowych.
Zmiany konformacyjne białek wynikające z zaburzeń statusu redox powodują
stymulację kaskad sygnalizacyjnych z udziałem kinaz MAPK (ang. mitogen-
activated protein kinases), PI3K (ang. phosphatidylinositol-3-kinase) oraz
kinazy src/Abl (ang. sarcoma/Abelson murine leukemia tyrosine kinase), które
pośredniczą w kontroli wzrostu komórek guza. Sygnalizacja z udziałem

Rola transporterów GLUT1 i GLUT3 w nowotworach 258
wymienionych kinaz prowadzi do aktywacji czynników transkrypcyjnych
odpowiedzialnych za zmianę statusu redox komórki takich jak: AP-1
(ang. activator-protein 1), NF-ӄB (ang. nuclear factor kappa-light-chain-
enhancer of activated B cells), p53 (ang. protein 53), HIF-1 (ang. hypoxia-
inducible factor-1), NFAT (ang. nuclear factor of activated T cells) [53, 54].
Witamina C w terapii nowotworów
Początkowe badania kliniczne polegające na doustnej suplementacji
pacjentów wysokimi dawkami kwasu askorbinowego nie przyniosły
oczekiwanych rezultatów z powodu niskiej biodostępności witaminy C
w krwiobiegu [55]. Stężenie kwasu askorbinowego w surowicy zależy od dwóch
procesów, w których pośredniczą transportery SVCT: absorpcji z przewodu
pokarmowego oraz wchłanianiu zwrotnemu w nerkach. Ponieważ ekspresja
transporterów SVCT jest hamowana wysokim stężeniem substratu, nie można
poprzez doustną suplementację osiągnąć wysokiego stężenia witaminy C
w surowicy [56]. Doustne przyjmowanie kwasu askorbinowego w dawce 3 g
odpowiadało 206 µM/L stężeniu askorbinianu w surowicy, a w dawce 1,25 g
odpowiednio 187 µM/L stężeniu w surowicy [57]. Ponadto, poziom
askorbinianu w organizmie jest regulowany przez składniki krwi takie jak:
limfocyty, monocyty i płytki krwi, które gromadzą około 4 mM witaminy C.
Wysoki poziom askorbinianu występuje również w niektórych narządach.
Na przykład, milimolowe stężenie witaminy C chroni tkanki oka przed
radiacyjnym działaniem promieni słonecznych [6]. Osiągnięcie wysokich
terapeutycznych stężeń witaminy C w surowicy jest możliwe jedynie drogą
dożylnej iniekcji. Zainteresowanie witaminą C w terapii nowotworów, wynika
z postępu badań klinicznych, które dowodzą, że wysokie dawki kwasu

P. Jóźwiak 259
askorbinowego hamują rozwój guza i wspomagają konwencjonalną chemio-
i radioterapię. Efekt ten wynika z pro-oksydacyjnych właściwości witaminy C.
Terapeutyczne stężenia kwasu askorbinowego obniża poziom białka
C-reaktywnego i prozapalnych cytokin w komórkach nowotworowych.
Stwierdzono, że w nowotworach złośliwych prostaty zmniejszenie stanu
zapalnego koreluje z obniżoną ekspresją markera PSA [58]. W badaniach
Espeya i wsp. [59] stosowanie wysokich dawek askorbinianu w połączeniu
z gemcytabiną zwiększało cytotoksyczność chemioterapii w komórkach
nowotworowych trzustki. W innym przypadku terapeutyczne dawki witaminy C
poprawiały jakość życia pacjentek z rakiem piersi podczas oraz po stosowanej
radio- i chemioterapii [60].
Podsumowanie
Zależne od warunków hipoksji transportery GLUT1 i GLUT3 transportują
zarówno glukozę jak i kwas dehydroaskorbinowy odgrywając tym samym
istotną rolę w regulacji statusu czynnościowego komórki. Ponieważ
nadekspresja obu białek jest obserwowana w wielu typach nowotworów,
w przyszłości dokładniejsze poznanie struktury i regulacji transporterów GLUT
może wpłynąć na rozwój nowych metod diagnostyki i terapii nowotworów.
Podziękowania
Autor pragnie podziękować Pani prof. dr hab. Annie Lipińskiej za cenne
uwagi w trakcie pisania niniejszego artykułu.
Praca została wykonana w ramach dotacji na finansowanie działalności
polegającej na prowadzeniu badań naukowych lub prac rozwojowych oraz zadań

Rola transporterów GLUT1 i GLUT3 w nowotworach 260
z nimi związanych służących rozwojowi młodych naukowców oraz uczestników
studiów doktoranckich (Rozporządzenie MNiSW z 5 listopada 2010 r.).
Piśmiennictwo
1. Ferreira LMR. Cancer metabolism: the Warburg effect today. Exp Mol Pathol., 2010; 89: 372-380.
2. Ganapathy V, Thangaraju M, Prasad PD. Nutrient transporters in cancer: relevance to Warburg hypothesis and beyond. Pharmacol Ther. 2009; 121: 29-40.
3. Thangaraju M, Carswell KN, Prasad PD, Ganapathy V. Colon cancer cells maintain low levels of pyruvate to avoid cell death caused by inhibition of HDAC1/HDAC3. Biochem J., 2009; 417: 379-389.
4. Xie H, Valera VA, Merino MJ, Amato AM, Signoretti S, Linehan WM i wsp. LDH-A inhibition, a therapeutic strategy for treatment of hereditary leiomyomatosis and renal cell cancer. Mol Cancer Ther. 2009; 8: 626-635.
5. Airley RE, Mobasheri A. Hypoxic regulation of glucose transport, anaerobic metabolism and angiogenesis in cancer: novel pathways and targets for anticancer therapeutics. Chemotherap. 2007; 53: 233-256.
6. Du J, Cullen JJ, Buettner GR. Ascorbic acid: Chemistry, biology and the treatment of cancer. Biochi Biophys Acta. 2012; 1826: 443-457.
7. Wood IS, Trayhurn P. Glucose transporters (GLUT and SGLT): expanded families of sugar transport proteins. Br J Nutr. 2003; 89: 3-9.
8. Wu X, Freeze HH. GLUT14, a duplicon of GLUT3, is specifically expressed in testis as alternative splice forms. Genomics. 2002; 80: 553-557.
9. Joost HG, Thorens B. The extended GLUT-family of sugar/polyol transport facilitators: nomenclature, sequence characteristics, and potential function of its novel members (Review). Mo Membr Biol. 2001; 18: 247-256.
10. Zhao FQ, Keating AF. Functional properties and genomics of glucose transporters. Curr Genomics. 2007; 8: 113-128.
11. Samih N, Hovsepian S, Notel F, Prorok M, Zattara-Cannoni H, Mathieu S i wsp. The impact of N- and O-glycosylation on the functions of Glut-1 transporter in human thyroid anaplastic cells. Biochim Biophys Acta. 2003; 1621: 92-101.
12. Macheda ML, Rogers S, Best JD. Molecular and cellular regulation of glucose transporter (GLUT) proteins in cancer. J Cell Physiol. 2005; 202: 654-662.

P. Jóźwiak 261
13. Younes M, Brown RW, Stephenson M, Gondo M, Cagle PT. Overexpression of GLUT1 and GLUT3 in stage I nonsmall cell lung carcinoma is associated with poor survival. Cancer. 1997; 80: 1046-1051.
14. Ayala FR., Rocha RM, Carvalho KC, Carvalho AL, da Cunha IW, Lourenco SV, i wsp. GLUT1 and GLUT3 as potential prognostic markers for oral squamous cell carcinoma. Molecules. 2010; 15: 1046-1051.
15. Furuta E, Okuda H, Kobayashi A, Watabe K. Metabolic genes in cancer: their roles in tumor progression and clinical implications. Biochim Biophys Acta, 2010; 1805: 141-152.
16. Maschauer S, Prante O, Hoffmann M, Deichen JT, Kuwert T. Characterization of 18F-FDG uptake in human endothelial cells in vitro. J Nucl Med. 2004; 45: 455-460.
17. Usuda K, Sagawa M, Aikawa H, Ueno M, Tanaka M, Machida Y i wsp. Correlation between glucose transporter-1 expression and 18F-fluoro-2-deoxyglucose uptake on positron emission tomography in lung cancer. Gen Thorac Cardiovasc Surg. 2010; 58: 405-410.
18. Kaira K, Endo M, Abe M, Nakagawa K, Ohde Y, Okumura T i wsp. Biologic correlates of ¹⁸F-FDG uptake on PET in pulmonary pleomorphic carcinoma. Lung Cancer. 2011; 71: 144-150
19. Su H, Bodenstein C, Dumont RA, Seimbille Y, Dubinett S, Phelps ME. Monitoring tumor glucose utilization by positron emission tomography for the prediction of treatment response to epidermal growth factor receptor kinase inhibitors. Clin Cancer Res. 2006; 12: 5659-5667.
20. Cifuentes M, García MA, Arrabal PM, Martínez F, Yañez MJ, Jara N i wsp. Insulin regulates GLUT1-mediated glucose transport In MG-63 human osteosarcoma cells. J Cell Physiol. 2011; 226: 1425-1432.
21. Hayashi M, Sakata M, Takeda T, Yamamoto T, Okamoto Y, Sawada K i wsp. Induction of glucose transporter 1 expression through hypoxia-inducible factor 1α under hypoxic conditions in trophoblast-derived cells. J Endocrinol. 2004; 183: 145-154.
22. Frolova A, Flessner L, Chi M, Kim ST, Foyouzi-Yousefi N, Moley KH. Facilitative glucose transporter type 1 is differentially regulated by progesterone and estrogen in murine and human endometrial stromal cells. Endocrinology. 2009; 150: 1512-1520.
23. Díaz M, Vraskou Y, Gutiérrez J, Planas JV. Expression of rainbow trout glucose transporters GLUT1 and GLUT4 during in vitro muscle cell differentiation and

Rola transporterów GLUT1 i GLUT3 w nowotworach 262
regulation by insulin and IGF-I. Am J Physiol Regu Integr Comp Physiol. 2009; 296: R794-R800.
24. Schwartzenberg-Bar-Yoseph F, Armoni M, Karnieli E. The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res. 2004; 64: 2627-2633.
25. Yun J, Rago C, Cheong I, Pagliarini R, Angenendt P, Rajagopalan H i wsp. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science. 2009; 325: 1555-1559
26. Maher JC, Savaraj N, Priebe W, Liu H, Lampidis TJ. Differential sensitivity to 2-deoxy- D-glucose between two pancreatic cell lines correlates with GLUT1 expression. Pancreas. 2005; 30: e34-e39.
27. Aft RL, Zhang FW, Gius D. Evaluation of 2-deoxy-D-glucose as a chemotherapeutic agent: mechanizm of cell death. Br J Cancer. 2002; 87: 805-812.
28. Kurtoglu M, Gao N, Shang J, Maher JC, Lehrman MA, Wangpaichitr M, i wsp. Under normoxia, 2-deoxy-D-glucose elicits cell death in select tumor types not by inhibition of glycolysis but by interfering with N-linked glycosylation. Mol Cancer Ther. 2007; 6: 3049-3058.
29 Dwarakanath BS. Cytotoxicity, radiosensitization, and chemosensitization of tumor cells by 2-deoxy-D-glucose in vitro. J Cancer Res Ther. 2009; 5 Suppl. 1: 27-31.
30. Dwarakanath BS, Khaitan D, Ravindranath T. 2-deoxy-D-glucose enhances the cytotoxicity of topoisomerase inhibitors in human tumor cell lines. Cancer Biol Ther. 2004; 3: 864-870.
31. Maschek G, Savaraj N, Priebe W, Braunschweiger P, Hamilton K, Tidmarsh GF. 2-deoxy-D-glucose increases the efficacy of adriamycin and paclitaxel in human osteosarcoma and non-small cell lung cancers in vivo. Cancer Res. 2004; 64: 31-34.
32. Subbarayan PR, Wang PG, Lampidis TJ, Ardalan B, Braunschweiger P. Differential expression of GLUT1 mRNA and protein levels correlates with increased sensitivity to the glyco-conjugated nitric oxide donor (2-glu-SNAP) in different tumor cell types. J Chemother. 2008; 20: 106-111.
33. Corti A, Casini AF, Pompella A. Cellular pathways for transport and efflux of ascorbate and dehydroascorbate. Arch Biochem Biophys. 2010; 500: 107-115
34. Rumsey SC, Kwon O, Xu GW, Burant CF, Simpson I, Levine M. Glucose transporter isoforms GLUT1 and GLUT3 transport dehydroascorbic acid. J Biol Chem. 1997; 272: 18982-18989.

P. Jóźwiak 263
35. Rumsey SC, Daruwala R, Al-Hasani H, Zarnowski MJ, Simpson IA, Levine M. Dehydroascorbic acid transport by GLUT4 in Xenopus oocytes and isolated rat adipocytes. J Biol Chem. 2000; 275: 28246-28253.
36. Bode AM, Liang HQ, Green EH, Meyer TE, Buckley DJ, Norris A. Ascorbic acid recycling in Nb2 lymphoma cells: implications for tumor progression. Free Radic Biol Med. 1999; 26: 136-147.
37. Vera JC, Rivas CI, Zhang RH, Farber CM, Golde DW. Human HL-60 myeloid leukemia cells transport dehydroascorbic acid via the glucose transporters and accumulate reduced ascorbic acid. Blood. 1994; 84: 1628-1634.
38. Spielholz C, Golde DW, Houghton AN, Nualart F, Vera JC. Increased facilitated transport of dehydroascorbic acid without changes in sodium-dependent ascorbate transport in human melanoma cells. Cancer Res. 57: 2529-2537.
39. Corti A, Raggi C, Franzini M, Paolicchi A, Pompella A, Casini AF. Plasma membrane gamma-glutamyltransferase activity facilitates the uptake of vitamin C in melanoma cells. Free Radic Biol Med. 2004; 37: 1906-1915.
40. KC S, Cárcamo JM, Golde DW. Viatmin C enters mitochondria via facilitative glucose transporter 1 (Glut1) and confers mitochondrial protection against oxidative injury. FASEB J. 2005; 19: 1657-1667
41. Ozer A, Bruick RK. Non-heme dioxygenases: cellular sensors and regulators jelly rolled into one? Nat Chem Biol. 2007; 3: 144-153.
42. Tajima S, Pinnell SR. Ascorbic acid preferentially enhances type I and III collagen gene transcription in human skin fibroblasts. J Dermato Sci. 1996; 11: 250-253.
43. Groblewska M, Mroczko B, Szmitkowski M. The role of selected matrix metalloproteinases and their inhibitors in colorectal cancer development. Postepy Hig Med Dosw. 2010; 64: 22-30.
44. Bruick RK, McKnight SL. A conserved family of prolyl-4-hydroxylases that modify HIF. Science. 2001; 294: 1337-1340.
45. Pagé EL, Chan DA, Giaccia AJ, Levine M, Richard DE. Hypoxia-inducible factor-1α stabilization in nonhypoxic conditions: role of oxidation and intracellular ascorbate depletion. Mol Biol Cell 2008; 19: 86-94.
46. Li SH, Ryu JH, Park SE, Cho YS, Park JW, Lee WJ i wsp. Vitamin C supplementation prevents testosterone-induced hyperplasia of rat prostate by down-regulating HIF-1alpha. J Nutr Biochem. 2010; 21: 801-808.
47. Cascella B, Mirica LM. Kinetic analysis of iron-dependent histone demethylases: α-ketoglutarate substrate inhibition and potential relevance to the regulation of histone demethylation in cancer cells. Biochemistry. 2012; 51: 8699-8701.

Rola transporterów GLUT1 i GLUT3 w nowotworach 264
48. Esteban MA, Pei D. Vitamin C improves the quality of somatic cell reprogramming. Nat Genet. 2012; 44: 366-367.
49. Ong TP, Moreno FS, Ross SA. Targeting the epigenome with bioactive food components for cancer prevention. J Nutrigenet Nutrigenomics. 2011; 4: 275-292.
50. Li Y, Schellhorn HE. New development and novel therapeutic perspectives for vitamin C. J Nutr. 2007; 137: 2171-2184.
51. Aditi A, Graham DY. Vitamin C, gastritis, and gastric disease: a historical review and update. Gig Dis Sci. 2012; 57: 2504-2515.
52. Verrax J, Calderon PB. The controversial place of vitamin C in cancer treatment. Biochem Pharmacol. 2008; 76: 1644-1652
53. Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact. 2006; 160: 1-40
54. Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007; 39: 44-84.
55. Cameron E, Campbell A. The orthomolecular treatment of cancer. II. Clinical trial of high-dose ascorbic acid supplements in advanced human cancer. Chem Biol Interact. 1974; 9: 285-315.
56. Mandl J, Szarka A, Bánhegyi G. Vitamin C: update on physiology and pharmacology. Br J Pharmacol. 2009; 157: 1097-1110.
57. Padayatty SJ, Sun H, Wang Y, Riordan HD, Hewitt SM, Katz A, i wsp. Vitamin C pharmacokinetics: implications for oral and intravenous use. Ann Intern Med. 2004; 140: 533-537.
58. Mikirova N, Casciari J, Rogers A, Taylor P. Effect of high-dose intravenous vitamin C on inflammation in cancer patients. J Transl Med. 2012; 10:187.
59. Espey MG, Chen P, Chalmers B, Drisko J, Sun AY, Levine M, i wsp. Pharmacologic ascorbate synergizes with gemcitabine in preclinical models of pancreatic cancer. Free Radic Biol Med. 2011; 50: 1610-1619.
60. Vollbracht C, Schneider B, Leendert V, Weiss G, Auerbach L, Beuth J. Intravenous vitamin C administration improves quality of life in breast cancer patients during chemo-/radiotherapy and aftercare: results of a retrospective, multicentre, epidemiological cohort study in Germany. In Vivo. 2011; 25: 983-990.

Folia Medica Lodziensia, 2012, 39/2: 265-292
Adres do korespondencji: Piotr Zakrzewski; Katedra Cytobiochemii, Wydział Biologii i Ochrony Środowiska, Uniwersytet Łódzki 90-236 Łódź, ul. Pomorska 141/143, tel.: 42 635-52-99, fax.: 42 635-44-84, e-mail: [email protected]
Zaburzenia kaskady transformujących czynników wzrostu
typu β w wybranych patologiach człowieka
Aberrations in the signalling cascade of transforming growth factor β type in selected human pathologies
PIOTR K. ZAKRZEWSKI
Katedra Cytobiochemii, Wydział Biologii i Ochrony Środowiska Uniwersytet Łódzki
Streszczenie Kaskada sygnalizacyjna transformujących czynników wzrostu typu β (TGFβ)
stanowi indukowany przez wiele cytokin szlak przekazywania sygnału
w komórce, pod kontrolą którego znajduje się szereg procesów komórkowych
odpowiedzialnych za prawidłowe funkcjonowanie ludzkiego organizmu.
Znaczenie zaburzeń sygnalizacji indukowanej czynnikami TGFβ pozostaje nadal
nie do końca poznane. Niemniej jednak już na obecnym etapie badań stwierdzić
można ich bezsprzeczny udział w patologiach układu kostno-mięśniowego,
układu krwionośnego czy układu rozrodczego.
Słowa kluczowe: transformujące czynniki wzrostu typu β - TGFβ, sygnalizacja
komórkowa, patofizjologia, polimorfizm, mutacje.
Abstract
A transforming growth factor β type (TGFβ) cascade is a multifactorial signalling pathway, which controls the plethora of cellular processes responsible for human organism homeostasis. The importance of alterations of TGFβ-induced signalling remains unknown. Up till now, impaired TGFβ signalling has been observed in pathologies of the musculoskeletal, cardiovascular and reproductive systems. Abnormalities in the TGFβ pathway can be either genetically determined or appear as spontaneous disorders which emerged during embryonic development. Understanding the role of the TGFβ

TGFβ w chorobach człowieka 266
pathway in the aetiology of various diseases appears to be necessary as it may serve in developing new strategies for therapeutic or diagnostic methods.
Key words: transforming growth factor β type - TGFβ, cellular signalling, pathophysiology, polymorphism, mutations. Wstęp
Różnorodność funkcji i procesów komórkowych jest następstwem udziału
wysoce specyficznych szlaków sygnalizacyjnych, dzięki aktywności których
sygnał pochodzący z zewnątrz komórki, zostaje przekazany do jej wnętrza,
czego konsekwencją jest regulacja transkrypcji określonych genów. Inicjacja
sygnalizacji komórkowej zwykle następuje pod wpływem interakcji ligandów
obecnych w macierzy zewnątrzkomórkowej ze specyficznymi receptorami,
będącymi nieodzownymi elementami błony komórkowej. Przekazywanie tak
zainicjowanego sygnału następuje często dzięki aktywności kinazowej
(tyrozynowej lub serynowo/treoninowej) domeny cytoplazmatycznej
receptorów. Funkcja receptorów może być modulowana przez dodatkowe białka
błonowe pełniące rolę receptorów pomocniczych (ang. accessory receptors,
auxilliary receptors, co-receptors), których głównym zadaniem jest
prezentowanie ligandów ich specyficznym receptorom. Przykładem szlaku
sygnalizacyjnego inicjowanego ligandami obecnymi w macierzy
zewnątrzkomórkowej jest szlak transformujących czynników wzrostu typu β
(TGFβ, ang. transforming growth factor β). W szlaku tym przekazywanie
sygnału następuje dzięki aktywności kinazowej serynowo/treoninowej
specyficznych transbłonowych receptorów. Aktywność tych receptorów
dodatkowo modulowana jest przez transbłonowe lub zakotwiczone na
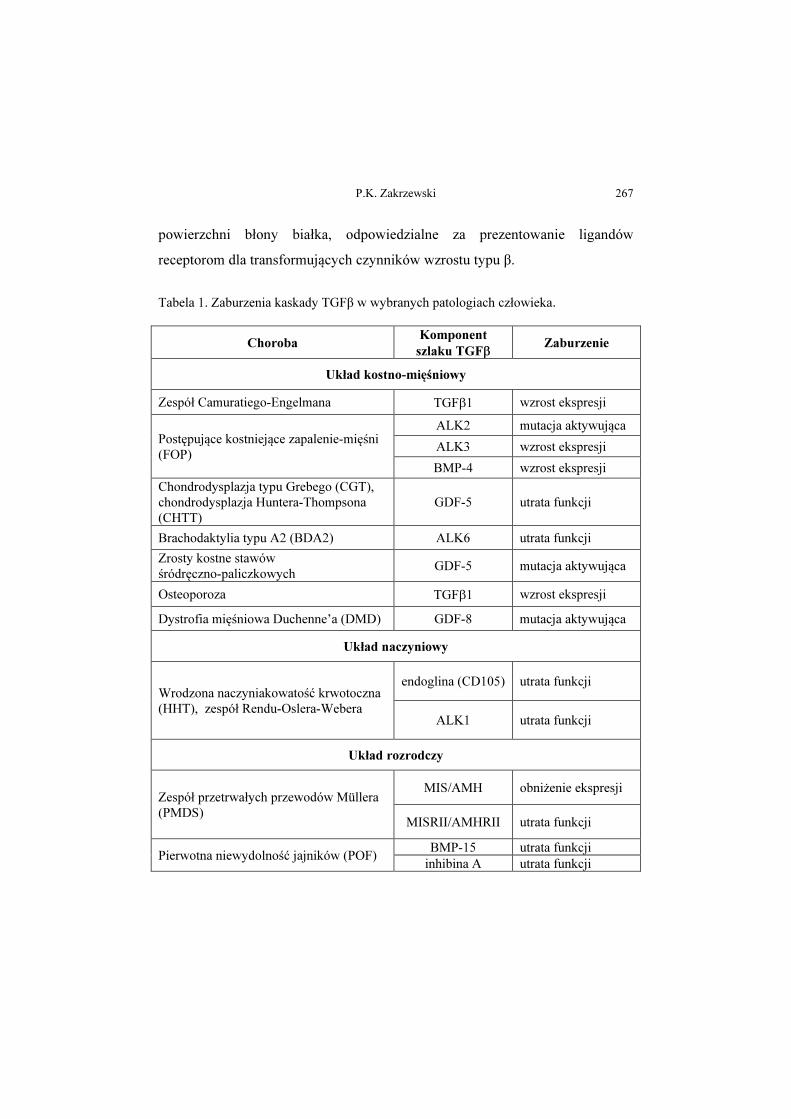
P.K. Zakrzewski 267
powierzchni błony białka, odpowiedzialne za prezentowanie ligandów
receptorom dla transformujących czynników wzrostu typu β.
Tabela 1. Zaburzenia kaskady TGFβ w wybranych patologiach człowieka.
Choroba Komponent
szlaku TGFβ Zaburzenie
Układ kostno-mięśniowy
Zespół Camuratiego-Engelmana TGFβ1 wzrost ekspresji
Postępujące kostniejące zapalenie-mięśni (FOP)
ALK2 mutacja aktywująca
ALK3 wzrost ekspresji
BMP-4 wzrost ekspresji
Chondrodysplazja typu Grebego (CGT), chondrodysplazja Huntera-Thompsona (CHTT)
GDF-5 utrata funkcji
Brachodaktylia typu A2 (BDA2) ALK6 utrata funkcji
Zrosty kostne stawów śródręczno-paliczkowych
GDF-5 mutacja aktywująca
Osteoporoza TGFβ1 wzrost ekspresji
Dystrofia mięśniowa Duchenne’a (DMD) GDF-8 mutacja aktywująca
Układ naczyniowy
Wrodzona naczyniakowatość krwotoczna (HHT), zespół Rendu-Oslera-Webera
endoglina (CD105) utrata funkcji
ALK1 utrata funkcji
Układ rozrodczy
Zespół przetrwałych przewodów Müllera (PMDS)
MIS/AMH obniżenie ekspresji
MISRII/AMHRII utrata funkcji
Pierwotna niewydolność jajników (POF) BMP-15 utrata funkcji
inhibina A utrata funkcji

TGFβ w chorobach człowieka 268
Różnorodność transformujących czynników wzrostu typu β pociąga za sobą
mnogość procesów fizjologicznych regulowanych przez te czynniki. Zaburzenia
przekazywania sygnału w kaskadzie TGFβ są przyczyną wielu chorób człowieka
zarówno dziedzicznych, jak i pozbawionych komponenty genetycznej.
Do chorób tych należą nieprawidłowości w układzie kostno-mięśniowym oraz
tkanki łącznej, choroby układu krwionośnego, nieprawidłowości budowy
i funkcji układu rozrodczego będące przyczyną bezpłodności, jak również
zaburzeń rozwojowych (Tabela. 1).
Kaskada TGFβ
Rodzina transformujących czynników wzrostu typu β to liczna grupa
ewolucyjnie zakonserwowanych (utrwalonych) białek, których cytokinowa
aktywność wykazuje plejotropowy efekt w wielu fizjologicznych i pato-
logicznych procesach. Do rodziny TGFβ zalicza się oprócz trzech izoform
TGFβ, tj. TGFβ1, TGFβ2, TGFβ3, które to uznawane są za białka prototypowe,
ponad 30 polipeptydów takich jak: aktywiny A i B (ang. activins), inhibiny A
i B (ang. inhibins), białka morfogenetyczne kości 1-20 (BMP, ang. bone
morphogenetic proteins), czynniki wzrostu i różnicowania (GDF, ang. growth
differentiation factors), np. miostatyna, czynniki determinujące asymetrię lewo-
prawo, tj. nodal i lefty 1 i 2, oraz MIS/AMH (ang. Müllerian-inhibiting
substance/anti-Müllerian hormone) [1-3]. Na podstawie podobieństwa
w obrębie ich sekwencji aminokwasowej, jak i rodzaju szlaku sygnalizacyjnego,
który może zostać indukowany ich aktywnością, wspomniane czynniki
podzielone zostały na dwie podrodziny – TGFβ/aktywiny/nodal/lefty/miostatyna
oraz BMP/GDF/MIS [4-7]. Cechą wspólną wszystkich czynników należących
do rodziny TGFβ jest siedem ewolucyjnie zakonserwowanych reszt

P.K. Zakrzewski 269
cysteinowych tworzących strukturę rdzeniową (ang. cysteine knot) [8]. Ligandy
te funkcjonujące w formie dimerów syntetyzowane są w formie prekursorowego
pre-probiałka. Aktywną formę TGFβ stanowią dimery o masie cząsteczkowej
25 kDa. W tak złożonym homodimerze, obie podjednostki polipeptydów
połączone są ze sobą wiązaniami disiarczkowymi, a całość struktury
stabilizowana jest oddziaływaniami hydrofobowymi. Prekursor TGFβ w wyniku
działania proteaz pozbawiony zostaje peptydu sygnałowego o długości 112 reszt
aminokwasowych od strony C-końca, czemu towarzyszy uwolnienie dojrzałej
formy białka. Postać dojrzała białka pozostaje związana w sposób
niekowalencyjny z N-końcowym fragmentem probiałka (LAP, ang. latency-
associated peptide). Kompleks formy dojrzałej oraz białka LAP tworzy dimer
nazywany małym kompleksem latentnym (SLC, ang. small latency complex).
SLC łącząc się następnie z białkiem LTBP (ang. latent TGFβ binding protein)
formuje tzw. duży kompleks latentny (LLC, ang. large latent complex). LTBP ze
względu na swoją strukturę, oddziałuje z białkami macierzy zewnątrz-
komórkowej (ECM, ang. extracellular matrix), a tym samym odpowiedzialny
jest za magazynowanie TGFβ w ECM do momentu jego aktywacji [8-10].
Wydzielone do macierzy zewnątrzkomórkowej latentne formy czynnika TGFβ
podlegają różnorodnym procesom, prowadzącym do ich aktywacji [11], tak aby
mogły oddziaływać ze specyficznymi receptorami błonowymi o aktywności
kinaz serynowo/treoninowych. W obrębie receptorów dla czynników TGFβ
wyróżnić można dwie podstawowe grupy, tj. receptory typu I oraz typu II.
Podział ten został dokonany na podstawie właściwości strukturalnych obu typów
receptorów. Receptory typu I cechuje obecność w ich strukturze aminokwasowej
regionu regulatorowego bogatego w reszty glicyny i seryny (ang. GS region),
ulokowanego powyżej domeny kinazowej [12]. W komórkach człowieka

TGFβ w chorobach człowieka 270
zidentyfikowano siedem receptorów typu I, które określa się wspólnym mianem
ALK (ang. activin receptor-like kinases), z jednoczesnym wskazaniem numeru
od 1 do 7 dla odpowiedniego receptora. Natomiast w przypadku receptorów
typu II zidentyfikowano pięć białek, tj. ActRII, ActRIIB (ang. activin receptor),
BMPRII (ang. BMP receptor), MISRII/AMHRII (ang. Müllerian-inhibiting
substance receptor/anti-Müllerian hormone receptor) oraz TGFβRII (ang. TGFβ
receptor II) [9]. Ligandy mogą wiązać się zarówno z receptorem typu I jak
i typu II w sposób niezależny lub jak ma to miejsce w przypadku izoform TGFβ,
w pierwszej kolejności powstaje kompleks ligand-receptor typu II, do którego
następnie przyłączany jest receptor typu I. W przypadku braku ligandów
w macierzy zewnątrzkomórkowej receptor typu II może ulegać homo-
dimeryzacji, jak również heterooligomeryzacji z podjednostkami receptora
typu I [13]. Niektóre czynniki z rodziny TGFβ oddziałują z więcej niż jednym
receptorem typu II, co skutkuje interakcją powstałego kompleksu z różnymi
receptorami typu I (Tabela 2). Aktywowane receptory typu I i II przekazują
sygnał wewnątrzkomórkowym mediatorom, tj. białkom z rodziny Smad (ang.
similar to mother against + mother against decapentaplegic) [14]. Pierwszymi
zidentyfikowanymi białkami będącymi homologami białek z rodziny Smad,
występującymi u kręgowców, były odkryte u Drosphila melanogaster białko
Mad (ang. mother against decapentaplegic) oraz trzy białka występujące
u Caenorhabditis elegans: Sma-2, Sma-3 oraz Sma-4 (ang. similar to mother
against). Białka Smad kręgowców podzielone zostały na trzy podklasy ze
względu na pełnione przez nie w komórce funkcje: receptorowe Smad – R-Smad
(ang. receptor-activated Smad), inhibitorowe Smad – I-Smad (ang. inhibitory
Smad) oraz pośredniczące Smad – Co-Smad (ang. common-mediators Smad).
Aktywacja białek R-Smad, do których należą Smad1, Smad2, Smad3, Smad5
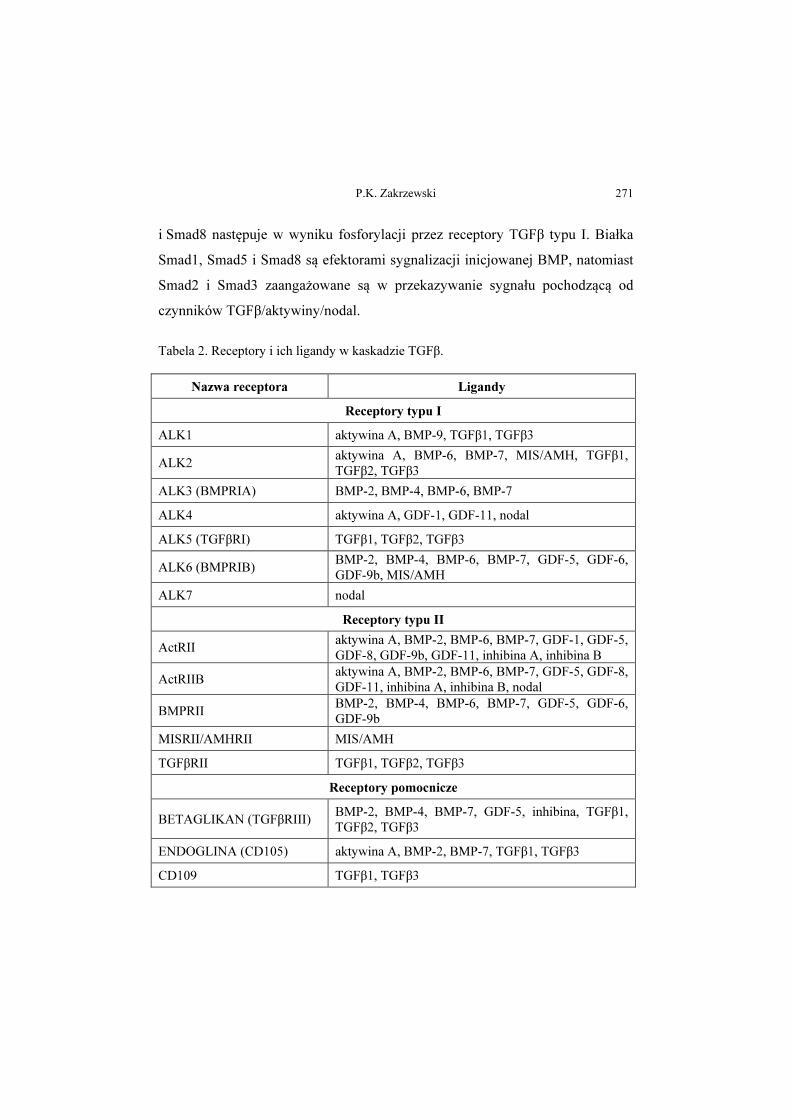
P.K. Zakrzewski 271
i Smad8 następuje w wyniku fosforylacji przez receptory TGFβ typu I. Białka
Smad1, Smad5 i Smad8 są efektorami sygnalizacji inicjowanej BMP, natomiast
Smad2 i Smad3 zaangażowane są w przekazywanie sygnału pochodzącą od
czynników TGFβ/aktywiny/nodal.
Tabela 2. Receptory i ich ligandy w kaskadzie TGFβ.
Nazwa receptora Ligandy
Receptory typu I
ALK1 aktywina A, BMP-9, TGFβ1, TGFβ3
ALK2 aktywina A, BMP-6, BMP-7, MIS/AMH, TGFβ1, TGFβ2, TGFβ3
ALK3 (BMPRIA) BMP-2, BMP-4, BMP-6, BMP-7
ALK4 aktywina A, GDF-1, GDF-11, nodal
ALK5 (TGFβRI) TGFβ1, TGFβ2, TGFβ3
ALK6 (BMPRIB) BMP-2, BMP-4, BMP-6, BMP-7, GDF-5, GDF-6, GDF-9b, MIS/AMH
ALK7 nodal
Receptory typu II
ActRII aktywina A, BMP-2, BMP-6, BMP-7, GDF-1, GDF-5, GDF-8, GDF-9b, GDF-11, inhibina A, inhibina B
ActRIIB aktywina A, BMP-2, BMP-6, BMP-7, GDF-5, GDF-8, GDF-11, inhibina A, inhibina B, nodal
BMPRII BMP-2, BMP-4, BMP-6, BMP-7, GDF-5, GDF-6, GDF-9b
MISRII/AMHRII MIS/AMH
TGFβRII TGFβ1, TGFβ2, TGFβ3
Receptory pomocnicze
BETAGLIKAN (TGFβRIII) BMP-2, BMP-4, BMP-7, GDF-5, inhibina, TGFβ1, TGFβ2, TGFβ3
ENDOGLINA (CD105) aktywina A, BMP-2, BMP-7, TGFβ1, TGFβ3
CD109 TGFβ1, TGFβ3

TGFβ w chorobach człowieka 272
Aktywowane białka R-Smad tworzą wraz białkiem Smad4 (Co-Smad)
heteromeryczne kompleksy, które po translokacji do jądra komórkowego
modulują w sposób bezpośredni lub w kompleksach z innymi czynnikami
transkrypcyjnymi ekspresję genów docelowych (Ryc. 1) [15, 16].
Ryc. 1. Przekazywanie sygnału w kaskadzie TGFβ.
Badania nad mechanizmem przekazywania sygnału indukowanego
czynnikami TGFβ pozwoliły stwierdzić, że oprócz udziału receptorów typu I
i typu II, ważną rolę odgrywają inne białka zlokalizowane na powierzchni błony
komórkowej. W literaturze określane są one jako receptory pomocnicze (ang.
auxiliary, accessory receptors) lub receptory typu III. Receptory pomocnicze są
białkami transbłonowymi lub zakotwiczonymi w błonie komórkowej za

P.K. Zakrzewski 273
pośrednictwem glikolipidów. W swej strukturze posiadają silnie rozbudowaną
domenę zewnątrzkomórkową, zdolną do wiązania ligandów, lecz pozbawione są
domeny funkcjonalnej o aktywności enzymatycznej. Główną ich funkcją jest
modulowanie siły sygnału poprzez tworzenie z ligandami kompleksów oraz
prezentowanie ich stosownym receptorom. Ze względu na brak aktywności
enzymatycznej, termin receptory pomocnicze, nie może zostać uznany za
pojęcie systematyczne, gdyż receptory te nie stanowią aktywnych przekaźników
sygnału. Do tej pory zidentyfikowano trzy główne receptory pomocnicze
kaskady TGFβ, tj. betaglikan (TGFβRIII), endoglinę (antygen CD105) oraz
antygen CD109 (Tabela 2).
Betaglikan wykazuje w komórkach ludzkich powinowactwo do wszystkich
trzech izoform TGFβ, niemniej jednak oddziaływanie to nie zachodzi z taką
samą siłą w stosunku do każdej izoformy. Betaglikan najefektywniej wiąże się
z TGFβ2, tym samym wzmacnia sygnał pochodzący od tej właśnie izoformy.
Niektórzy badacze postulują, że to właśnie tylko za pośrednictwem betaglikanu
następuje przekazywanie sygnału indukowanego czynnikiem TGFβ2, ponieważ
powinowactwo tej izoformy do receptorów TGFβRI i TGFβRII jest stosunkowo
niskie. Potwierdzeniem tego jest fakt, iż komórki niewykazujące ekspresji
betaglikanu, są niewrażliwe na stymulację czynnikiem TGFβ2 [17-20].
Analizując rolę endogliny w przekazywaniu sygnału stwierdzono, że oddziałuje
ona zarówno z receptorem TGFβRI jak i TGFβRII. W przeciwieństwie do
betaglikanu endoglina nie tworzy bezpośrednio kompleksów z izoformami
czynników TGFβ. Tworzenie kompleksów z receptorami następuje niezależnie
od obecność ligandów. Endoglina wykazuje powinowactwo tylko do izoform
TGFβ1 i TGFβ3, przy czym najsilniej oddziałuje z izoformą TGFβ1. Taki
sposób oddziaływania z czynnikami TGFβ wynika ze specyficzności wiązania

TGFβ w chorobach człowieka 274
się receptora TGFβRII ze wspomnianymi izoformami. W literaturze znaleźć
można również informacje wskazujące, na zdolność endogliny do oddziaływania
z kompleksami sygnalizacyjnymi innych czynników z rodziny TGFβ.
Stwierdzono, że endoglina wykazuje powinowactwo do aktywiny i czynnika
BMP-7 związanych z receptorami ActRII lub ActRIIB, jak również
z czynnikiem BMP-2 związanym z receptorem ALK3 lub ALK6 [21].
Zaburzenia w układzie kostno-mięśniowym
Układ kostny pełni w organizmie człowieka różnorodne funkcje wśród
których wyróżnić można: regulowanie stężenia jonów wapnia we krwi,
stabilizację oraz ochronę narządów wewnętrznych, jak i miejsce procesów
krwiotwórczych. Zbudowany jest on w głównej mierze z organicznej macierzy
utworzonej przez kolagen typu I, wysyconej nieorganicznymi kryształami
hydroksyapatytu. Mineralno-organiczna budowa kości determinuje zachodzące
w nich ciągłe procesy mineralizacji i demineralizacji, określane łącznie jako
proces remodelowania, zachodzący przy udziale osteoklastów oraz
osteoblastów. Znaczącą rolę w procesach resorpcji oraz formowania kości,
odgrywa jedna z trzech izoform czynnika TGFβ, tj. izoforma pierwsza. TGFβ1
zaangażowany jest w proces tworzenia kości, tj. stymulację proliferacji
i różnicowanie komórek progenitorowych osteoblastów. Ekspresja TGFβ
w tkance kostnej nie ograniczona jest tylko do okresu rozwoju embrionalnego,
lecz ma miejsce również w organizmach dorosłych, w których to
zaobserwowano podwyższony poziom ekspresji czynników TGFβ zwłaszcza
podczas zrastania się złamań. Ponadto na skutek aktywności TGFβ1
zahamowaniu ulega proces apoptozy osteoblastów [22]. Rolę TGFβ1
w procesach fizjologicznych tkanki kostnej potwierdziły badania na modelu

P.K. Zakrzewski 275
mysim. Zwierzęta, u których zablokowano jego ekspresję wykazywały
zahamowanie wzrostu i mineralizacji kości, a ponadto kości ich posiadały
obniżoną elastyczność oraz aktywność fosfatazy alkalicznej. Innym ważnym
czynnikiem z rodziny TGFβ zaangażowanym w metabolizm tkanki kostnej jest
czynnik BMP, którego aktywność indukuje proces formowania kości.
Selektywne zablokowanie ekspresji dwóch czynników z rodziny BMP,
tj. BMP-2 oraz BMP-4 w listku mezenchymalnym, odpowiedzialnym
za wykształcenie kończyn u myszy powodowało poważne zaburzenia w ich
formowaniu [23]. Jednakże wyciszenie ekspresji powyższych czynników
w sposób niezależny, tj. każdego z czynników z osobna, nie wpływało ujemnie
na proces formowania kości kończyn. Niemniej jednak w przypadku czynnika
BMP-2 przyczyniało się do zwiększenia podatności osobników na złamania,
a także powodowało zahamowanie inicjacji procesu zrastania się złamanych
kończyn. Podobnego efektu nie stwierdzono w przypadku braku ekspresji
czynnika BMP-4 [24, 25]. Równie ważną rolę w procesie kościotworzenia
pełnią odpowiednie receptory dla białek morfogenetycznych kości, tj. BMPR1A
oraz BMPR1B, których wyciszenie ekspresji w chondrocytach jest przyczyną
chondrodysplazji oraz zaburzeń formowania kości, na skutek zaburzonej
proliferacji i różnicowania chondrocytów [26]. Jednostką chorobową związaną
z mutacjami w genie kodującym izoformę TGFβ1 jest zespół wad wrodzonych
Camuratiego-Engelmanna (CED, ang. Camurati-Engelmann disease), określany
inaczej jako postępująca dysplazja kości długich (ang. progressive diaphyseal
dysplasia). Dziedziczony jest on w sposób autosomalny dominujący, natomiast
najczęstszymi objawami tej niezwykle rzadkiej choroby genetycznej jest
zwiększona gęstość oraz stwardnienie kości, zwłaszcza trzonów kości długich
oraz czaszki, upośledzone funkcjonowanie szpiku kostnego, jak również

TGFβ w chorobach człowieka 276
zmniejszenie siły motorycznej mięśni, kaczkowaty chód oraz silne bóle kostne
[22, 27]. Do tej pory zidentyfikowano około 40 mutacji w genie TGFβ1.
Większość z nich dotyczy fragmentu genu kodującego region LAP, podczas gdy
inne mają miejsce w regionie kodującym peptyd sygnałowy. Najczęściej
występującymi mutacjami są mutacje typu „zmiany sensu” (ang. missense
mutation), które prowadzą do zwiększonej ekspresji izoformy TGFβ1, a tym
samym wzmożonej odpowiedzi komórki na sygnał przez nią indukowany [28].
Ponadto badania na liniach komórkowych nabłonka nerki, które charakte-
ryzowały mutacje identyfikowane w zespole Camuratiego-Engelmanna,
wykazały zwiększony efekt autokrynny oraz nasilenie transkrypcji genów
aktywowanych czynnikami TGFβ [27]. Kolejną chorobą związaną z układem
kostno-mięśniowym, u podstaw której leżą zaburzenia przekazywania sygnału
szlaku TGFβ jest postępujące kostniejące zapalenie mięśni (FOP, ang.
fibrodysplasia ossificans progressiva). Jest to choroba autosomalna dominująca,
charakteryzująca się postępującym kostnieniem tkanki mięśniowej, co ma
miejsce w sposób spontaniczny lub na skutek doznanego urazu, zabiegu
chirurgicznego, bądź infekcji wirusowej. Molekularną przyczyną FOP jest
mutacja w genie ACVR1, kodującym receptor typu I ALK2, prowadząca do
podstawienia reszty argininy, zlokalizowanej w domenie GS receptora, resztą
histydyny [29]. Mutacja ta wydaje się być charakterystyczna dla sporadycznych,
jak i rodzinnych postaci choroby. Podobnie jak w przypadku mutacji
obserwowanych w zespole CED, mutacja ta ma charakter aktywujący,
tj. receptor nabiera konstytutywnej aktywności kinazowej w sposób niezależny
od obecności ligandu [29, 30]. Analiza ekspresji genów w limfocytach oraz
w komórkach progenitorowych zębów mlecznych pacjentów cierpiących na
FOP wykazała zwiększony poziom ekspresji czynnika BMP-4, jak również

P.K. Zakrzewski 277
zwiększoną ekspresję genów indukowanych tymi czynnikami. Podwyższony
poziom ekspresji obserwowano też w przypadku receptora typu I ALK3, co
sugeruje jako przyczynę FOP zaburzenia przekazywania sygnału indukowanego
czynnikami BMP. Równolegle stwierdzono również wzmożoną aktywność
szlaku kinaz MAP (ang. mitogen-activated protein), które aktywowane są
czynnikami BMP [31-34]. Zaburzenia w sygnalizacji BMP potwierdzone zostały
na modelu mysim. Transgeniczne osobniki, u których indukowano nadekspresję
czynnika BMP-4 wykazywały cechy fenotypowe zbliżone do ludzkiej postaci
FOP [34]. Chorobami manifestującymi się skróceniem elementów szkieleto-
wych i/lub nieprawidłowo wykształconymi stawami są chondrodysplazja typu
Grebego (CGT, ang. chodrodysplasia Grebe type), bądź chondrodysplazja
Huntera-Thompsona (CHTT, ang. chodrodysplasia Hunter-Thompson type). Pod
względem molekularnym CHTT spowodowana jest mutacją zmiany sensu w obu
allelach genu kodującego czynnik GDF-5, tym samym dziedziczona jest
w sposób autosomalny recesywny. Mutacja ta powoduje całkowitą utratą funkcji
wspomnianego białka [35]. Natomiast w przypadku CGT obserwowana jest
w domenie funkcjonalnej substytucja zachowanej ewolucyjnie reszty cysteiny
resztą tyrozyny. Rezultatem takiej zamiany jest nabycie przez białko GDF-5
zdolności do tworzenia heterokompleksów z białkami należącymi do podrodziny
BMP [35-37]. Eksperymenty na zwierzętach, u których blokowano ekspresję
czynnika GDF-5, potwierdziły jego funkcję w procesie kostnienia. Osobniki
takie wykazywały zaburzenia kondensacji chrząstki oraz proliferacji
chondrocytów [37]. Mutacje w genie GDF-5, których rezultatem jest
zmniejszenie powinowactwa czynnika GDF-5 do receptora typu I ALK6 zostały
zidentyfikowane także w brachodaktylii typu A2 (BDA2, ang. brachodactyly
type A2). Choroba ta charakteryzuje się autosomalnym dominującym modelem

TGFβ w chorobach człowieka 278
dziedziczenia, a objawia się skróceniem i zakrzywieniem paliczków palca
wskazującego oraz pierwszego i drugiego palca u stóp [38]. Ponadto
obserwowane są również mutacje zmiany sensu w regionie GS oraz domenie
kinazowej receptora typu I ALK6. Delecja genu GDF-5 u myszy manifestowała
się silną redukcją kości kończyn oraz nieprawidłowo wykształconymi
połączeniami stawowymi [39]. Przeciwny skutek mutacji w genie GDF-5, tzn.
wzrost powinowactwa receptora typu I ALK6 stwierdzono u osób dotkniętych
zrostami kostnymi stawów śródręczno-paliczkowych (ang. symphalangism)
czyli dziedziczoną autosomalnie chorobą manifestującą się występowaniem
sztywnych połączeń kostnych w stawach stóp i dłoni. Kolejną chorobą związana
z nieprawidłowościami w szlaku sygnalizacyjnym TGFβ jest osteoporoza
(zrzeszotnienie kości). W przeciwieństwie do omawianych wcześniej rzadkich
jednostek chorobowych, ta występuje stosunkowo często, zwłaszcza wśród
kobiet w wieku pomenopauzalnym. Rozwój osteoporozy jest wynikiem
zaburzenia równowagi między procesami resorpcji i kostnienia, które
w warunkach fizjologicznych podlegają wieloczynnikowej regulacji.
Do głównych przyczyn związanych z czynnikami TGFβ należy zaliczyć
występowanie polimorfizmów pojedynczych nukleotydów (SNP, ang. single
nucleotide polymorphism) w genach kodujących izoformę TGFβ1 [22],
receptory TGFβRI oraz TGFβRII, jak również białka efektorowe szlaku TGFβ,
tj. Smad2, Smad3, Smad4 i Smad7 [40]. Polimorfizmy genu kodującego TGFβ1
podzielono ze względu na miejsce ich występowania, tj. na promotorowe,
kodujące, bądź intronowe. Pierwsze z nich przyczyniają się do zmienionego
poziomu ekspresji czynnika TGFβ1. Efektem polimorfizmów w obrębie
sekwencji kodujących najczęściej są zaburzenia struktury czwartorzędowej lub
zmieniony metabolizm kodowanego białka. Natomiast w przypadku

P.K. Zakrzewski 279
polimorfizmów zlokalizowanych w intronach, sugeruje się ich bezpośredni
wpływ na proces składania pierwotnego transkryptu i możliwość powstawania
różnych wariantów funkcjonalnych [22]. Potwierdzeniem powyższych
przypuszczeń jest fakt występowania podwyższonego stężenia TGFβ1 w
surowicy krwi osób, u których zidentyfikowano wspomniane polimorfizmy.
Ponadto, u osób z osteoporozą zaobserwowano obniżony efekt terapeutyczny
witaminy D [41, 42], która wzmaga procesy formowania kości poprzez indukcję
proliferacji osteoblastów oraz mineralizację macierzy zewnątrzkomórkowej [22].
Omówione powyżej zaburzenia funkcji niektórych elementów kaskady TGFβ
skutkowały nieprawidłowościami w procesie resorpcji oraz formowania kości,
niemniej jednak istnieją dowody potwierdzające także ich znaczący udział w
patogenezie chorób związanych z tkanką mięśniową. Za przykład może
posłużyć dystrofia mięśniowa Duchenne’a (DMD, ang. Duchenne muscular
dystrophy), będąca jedną z najczęściej diagnozowanych chorób zanikowych
mięśni. Początkowo dystrofii ulegają mięśnie szkieletowe, lecz wraz z postępem
choroby obserwuje się zmiany także w mięśniu sercowym, prowadzące do
kardiomiopatii. Pod względem etiologicznym pierwotną przyczyną DMD jest
mutacja w genie niezwiązanym ze szlakiem TGFβ, kodującym białko dystrofinę
[43]. Recesywny allel tego genu zlokalizowany jest na chromosomie X, przez co
choroba ta podlega dziedziczeniu sprzężonemu z płcią. Obecność wspomnianej
mutacji przyczynia się do obniżonego poziomu produktu białkowego.
U pacjentów z DMD stwierdzono dodatkowo współistnienie mutacji zmiany
sensu w genie czynnika GDF-8, znanego również z racji pełnionej funkcji jako
miostatyna. GDF-8 to tkankowo specyficzny ligand z rodziny TGFβ, którego
ekspresja charakterystyczna jest dla komórek tkanki mięśniowej. Pod jego
kontrolą znajdują się procesy wzrostu komórek mięśniowych, a dokładniej

TGFβ w chorobach człowieka 280
hamowanie ich wzrostu. Potwierdzeniem potencjalnej roli czynnika GDF-8
są wyniki badań, które wykazały, że u myszy knock-out genu GDF-8
z jednoczesnym współwystępowaniem mutacji genu dystrofiny powoduje
pojawienie się fenotypu przeciwstawnego do obserwowanego w DMD,
tj. zwiększoną masę oraz siłę mięśniową zwierząt [44, 45].
Nieprawidłowości w układzie naczyniowym
Układ naczyniowy człowieka obejmują naczynia krwionośne oraz
limfatyczne, zbudowane z pojedynczej warstwy komórek endotelium,
otoczonych wieloma warstwami komórek tkanki mięśniowej gładkiej, a także
perycytami. Procesy formowania układu naczyniowego noszą nazwę procesów
waskulogenezy i angiogenezy [46]. Pierwszy z wymienionych procesów
dotyczy formowania de novo pierwotnych naczyń w rozwijającym się zarodku,
podczas gdy angiogeneza dotyczy wytwarzania nowych rozgałęzień już
istniejącej funkcjonalnej sieci naczyń. Angiogenezę obserwuje się podczas
całego życia organizmu, jego wzrostu oraz w trakcie gojenia się ran, jak również
w następstwie niektórych procesów patofizjologicznych, tj. progresji
i przerzutowania zmian nowotworowych oraz w przypadku chorób
naczyniowych [47]. Regulacja procesów angiogenezy pozostaje pod ścisłą
kontrolą różnorodnych współoddziałujących ze sobą szlaków sygnalizacyjnych,
wśród których istotną rolę odgrywa kaskada sygnałowa inicjowana czynnikami
TGFβ. Jednakże w przypadku aktywności czynników z rodziny TGFβ zjawisko
regulacji procesu angiogenezy cechuje znacząca złożoność, wynikająca
z plejotropowego wpływu tych czynników na komórki nabłonka. Czynniki te
mogą pełnić rolę inhibitorów i/lub aktywatorów procesu formowania nowych
naczyń. Ich wpływ nie zależy tylko od rodzaju działającego czynnika, lecz

P.K. Zakrzewski 281
również od stopnia zaawansowania całego procesu. Niektóre z nich początkowo
pełnią funkcję ograniczającą rozrost naczyń włosowatych, tak aby
w późniejszych etapach przybrać charakter aktywujący. Rola poszczególnych
komponentów szlaku TGFβ została bliżej poznana dzięki badaniom na modelach
mysich, w których hamowanie ekspresji wybranych elementów kaskady
powodowało ujawnienie się patologicznego fenotypu. Nieprawidłowości
dotyczące układu krwionośnego stwierdzono w przypadku wyciszania genów
kodujących receptory typu I i II ALK1, ALK5 i TGFβRII, receptory pomocnicze
betaglikan i endoglinę oraz białka efektorowe Smad1, Smad2, Smad4, Smad5,
Smad6 i Smad7. Zahamowanie ekspresji receptorów ALK1, ALK5 oraz
TGFβRII przyczyniało się do zaburzeń rozwoju sieci naczyń krwionośnych,
natomiast brak ekspresji białek z rodziny Smad skutkował nieprawidłowościami
w rozwoju i funkcjonowaniu serca. Ponadto stwierdzono komórkowo-
specyficzną ekspresję endogliny w intensywnie proliferujących komórkach
śródbłonka naczyń krwionośnych. Komórki endotelium pochodzące od
embrionów o fenotypie endoglina -/- wykazywały obniżony poziom migracji
oraz niezdolność do prawidłowej proliferacji w warunkach in vitro [47-50].
Dotychczas zidentyfikowane zostało kilkadziesiąt mutacji w genach kodujących
komponenty szlaku TGFβ mających jak się okazało decydujące znaczenie
w rozwoju wybranych chorób układu naczyniowego. Przykładem choroby
w etiologię, której zaangażowana jest kaskada TGFβ jest wrodzona
naczyniakowatość krwotoczna (HHT, ang. hereditary hemorrhagic
telangiectasia), określana również jako zespół Rendu-Oslera-Webera. Jest to
choroba dziedziczona w sposób autosomalny dominujący, charakteryzująca się
częstością występowania około 1/7500 przypadków [51]. Do głównych jej
objawów należą dysplazje naczyń włosowatych, obejmujące zależnie od typu

TGFβ w chorobach człowieka 282
(HHT1, HHT2) różne narządy wewnętrzne. W przypadku HHT1 obserwowana
jest przewaga zaburzeń w wykształceniu połączeń tętniczo-żylnych w obrębie
płuc, wątroby oraz mózgu, podczas gdy w HHT2 występujące malformacje
tętniczo-żylne dotyczą znacznie częściej kanalików nerkowych [52].
Podobieństwo cech klinicznych obu typów HHT sprawia, że jednoznaczna
diagnoza może zostać postawiona jedynie dzięki badaniom genetycznym na
obecność określonych mutacji. Liczne badania potwierdziły bezsporny udział
komponentów szlaku TGFβ, tj. endogliny oraz receptora typu I ALK1
w rozwoju HHT [50]. Mutacje w genie ENG kodującym endoglinę stwierdzono
u chorych na HHT typu pierwszego. Lokalizacja mutacji ograniczona jest do
regionu kodującego domenę zewnątrzkomórkową endogliny. Przyczyniają się
one zatem do nieprawidłowego wiązania ligandów, co skutkuje wadliwym
przekazywaniem sygnału indukowanego czynnikami TGFβ. Mutacje genu
endogliny obejmują szeroki wachlarz zmian, tj. delecje, insercje, mutacje
zmiany sensu, nonsensowne, jak również zmiany składania pierwotnego
transkryptu [53, 54]. Większość z tych mutacji skutkuje biosyntezą białka
posiadającego skrócony łańcuch aminokwasowy, co uniemożliwia jego eksport
do błony komórkowej, miejsca występowania i aktywności endogliny [55].
Natomiast typ drugi HHT związany jest z obecności mutacji genu ACVRL1,
który odpowiedzialny jest za syntezę receptora typu I ALK1. W przeciwieństwie
do mutacji w genie endogliny, zidentyfikowane mutacje dotyczą sekwencji
kodujących domenę zewnątrz- oraz wewnątrzkomórkową, jak również fragment
transbłonowy białka. Największą liczbę mutacji wykryto w domenie cyto-
plazmatycznej, która ma decydujące znaczenie w przekazywaniu sygnału [51,
55, 56]. Głównym mechanizmem, którego zaburzenia prowadzą do rozwoju
wrodzonej naczyniakowatości krwotocznej jest współistnienie w komórkach

P.K. Zakrzewski 283
endotelium dwóch wykluczających się szklaków indukowanych czynnikami
z rodziny TGFβ [57]. W pierwszym z nich przekazanie sygnału ma miejsce za
pośrednictwem receptora typu I ALK1, co skutkuje aktywacją procesów
proliferacji i migracji komórek endotelium. Przeciwstawny efekt obserwowany
jest natomiast w sytuacji, gdy sygnał zewnątrzkomórkowy przekazywany jest do
wnętrza komórki przez receptor typu I ALK5 [51, 55]. Klamrą spinającą oba
mechanizmy jest w tym przypadku endoglina, która może oddziaływać zarówno
z receptorem ALK1 oraz ALK5 [58-60].
Nieprawidłowości w układzie rozrodczym
Rodzina czynników TGFβ odgrywa również znaczącą rolę w rozwoju
i funkcjonowaniu układu rozrodczego. Dzięki aktywności ligandów TGFβ
regulowane są takie procesy jak rozwój pęcherzyków jajnikowych, rekrutowanie
pęcherzyków pierwotnych, proliferacja komórek warstwy ziarnistej i osłonki
oocytu, ekspresja receptorów gonadotropin, dojrzewanie oocytu, owulacja,
luteinizacja oraz formowanie ciałka żółtego, czy rozwoju jąder [61, 62].
Kluczową rolę w wymienionych procesach odgrywają aktywiny oraz inhibiny,
które są wzajemnymi antagonistami. Poprzez wiązanie się z receptorem
pomocniczym kaskady TGFβ betaglikanem, inhibiny działają hamująco na
sygnalizację pochodzącą od aktywin, blokując im tym samym dostępność
receptora typu II. Innym zidentyfikowanym antagonistą aktywin warunkującym
zahamowanie sygnalizacji pochodzącej od czynników BMP jest folistatyna [63,
64]. Wpływ aktywin i inhibin w fizjologicznych warunkach na rozwój
i funkcjonowanie układu rozrodczego potwierdzają badania na modelach mysich
w których wykazano, że nadekspresja inhibiny A prowadzi u osobników
męskich do obniżenia wielkości jąder, podczas gdy u samic skutkuje

TGFβ w chorobach człowieka 284
upośledzeniem dojrzewania pęcherzyków jajnikowych. Zwiększona ekspresja
inhibiny A przyczynia się do zaburzonej sekrecji hormonu folikulotropowego
(FSH, ang. follicle-stimulating hormone) przez przysadkę [65]. Podobny efekt
do nadekspresji inhibiny A uzyskano wyciszając ekspresję genu receptora dla
aktywiny ACVR2, co prowadziło u samic do bezpłodności, podczas gdy
u samców dochodziło do zmniejszenia wielkości jąder [62]. W warunkach
fizjologicznych, podczas rozwoju embrionalnego ssaków, komórki Sertolego
obecne w jądrach wytwarzają hormony odpowiedzialne za prawidłowe
wykształcenie męskiego układu rozrodczego. Pod wpływem testosteronu
przewody Wolffa ulegają przekształceniu w najądrze, nasieniowód oraz kanaliki
nasienne, podczas gdy czynnik z rodziny TGFβ, tj. MIS/AMH warunkuje zanik
przewodów Müllera w rozwijającym się jądrze. W przypadku braku sekrecji
czynnika MIS/AMH przez komórki Sertolego, przewody Müllera rozwijają się
w jajowody, macicę oraz pochwę. Brak regresji przewodów Müllera w zarodku
męskim charakteryzuje bardzo rzadką jednostkę chorobową, uwarunkowaną
genetycznie o recesywnym modelu dziedziczenia, określaną jako zespół
przetrwałych przewodów Müllera (PMDS, ang. persistent Müllerian duct
syndrome). Choroba ta manifestuje się obecnością jajowodów oraz macicy
u osobników fenotypowo męskich na skutek zbyt niskiego poziomu czynnika
MIS/AMH w trakcie rozwoju płodowego. Aktualnie zidentyfikowano 38
mutacji w genie kodującym czynnik MIS/AMH, odpowiedzialnych za obniżenie
jego stężenia. Najczęściej są to mutacje zmiany sensu w obrębie domeny
C-końcowej. Obecność mutacji w genie kodującym MIS/AMH skutkuje
nieprawidłowym transportem syntetyzowanych cząsteczek czynnika z siateczki
śródplazmatycznej, bądź wpływa na tworzenie struktur wyższego rzędu
i obniżenie stabilność produktu białkowego w komórce. Oprócz obniżonego

P.K. Zakrzewski 285
poziomu ekspresji czynnika MIS/AMH u osób z PMDS obserwuje się również
mutacje w genie kodującym receptor typu II dla tego czynnika, tj.
MISRII/AMHRII. Rezultatem mutacji w receptorze MISRII/AMHRII jest
formowanie postaci rozpuszczalnej receptora lub syntetyzowanie skróconego
białka pozbawionego domeny kinazowej. Forma rozpuszczalna receptora nie
wbudowuje się do błony cytoplazmatycznej komórki, podczas gdy brak domeny
kinazowej uniemożliwia prawidłowe wiązanie ligandu przez receptor. Oba
zaburzenia molekularne dają podobny obraz kliniczny i są praktycznie nie do
odróżnienia bez badań molekularnych. Ponadto zidentyfikowano również
przypadki kliniczne, w których pomimo prawidłowej ekspresji czynnika
MIS/AMH i receptora MISRII/AMHRII występują cechy zespołu przetrwałych
przewodów Müllera. Jednakże dotychczasowe badania dotyczące innych
komponentów kaskady TGFβ nie wykazały obecności zaburzeń molekularnych
na tym poziomie [66-69]. Inną jednostką chorobową związaną z zaburzeniami
w sygnalizacji TGFβ, a dotyczącą układu rozrodczego człowieka jest zespół
przedwczesnego wygasania funkcji jajników (POF, ang. premature ovarian
failure). Choroba ta wiąże się z z zanikiem funkcji jajników u kobiet poniżej
czterdziestego roku życia i jest jedną z przyczyn wtórnego braku miesiączki oraz
niepłodności. Czynnikami związanymi z chorobą jest wysokie stężenie
gonadotropin oraz hipoestrogenizm, jak również zanik czynności jajników,
w którym dochodzi do rozwoju tkanki łącznej w miejscu pęcherzyków
jajnikowych. Obecnie zidentyfikowane zostały mutacje w genach kodującym
czynnik BMP-15 oraz podjednostkę α inhibiny A. Wydaje się również, że za
występowanie POF odpowiedzialny jest polimorfizm w eksonie 12 genu
kodującego receptor pomocniczy kaskady TGFβ, tj. betaglikan [70-73].

TGFβ w chorobach człowieka 286
Podsumowanie
Nieprawidłowości w kaskadzie TGFβ mają charakter zarówno
uwarunkowany genetycznie, jak i są zaburzeniami spontanicznymi, zaistniałymi
podczas rozwoju embrionalnego. Identyfikacja molekularnych przyczyn
dysfunkcji na poziomie przekazywania sygnału w szlaku TGFβ umożliwi
w przyszłości wytypowanie nowych potencjalnych celów dla interwencji
farmakologicznych. Poznanie roli szlaku TGFβ w etiologii różnych chorób
wydaje się być cenne w opracowywaniu nowych strategii terapeutycznych lub
metod diagnostycznych.
Podziękowania
Podziękowania dla prof. dr hab. Wandy M. Krajewskiej za pomoc oraz
cenne uwagi podczas przygotowywania pracy.
Praca finansowana z dotacji na prowadzeniu badań naukowych lub prac
rozwojowych oraz zadań z nimi związanych służących rozwojowi młodych
naukowców oraz uczestników studiów doktoranckich (Rozporządzenie MNiSW
z 5 listopada 2010 r.).
Piśmiennictwo
1. Heldin CH, Miyazono K, ten Dijke P. TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature. 1997; 390: 465-471.
2. Miyazono K, Maeda S, Imamura T. BMP receptor signaling: transcriptional targets, regulation of signals, and signaling cross-talk. Cytokine Growth Factor Rev. 2005; 16: 251-263.
3. De Caestecker M. The transforming growth factor-beta superfamily of receptors. Cytokine Growth Factor Rev. 2004; 15: 1-11.

P.K. Zakrzewski 287
4. Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003; 113: 685-700.
5. Massague J. TGFbeta in Cancer. Cell. 2008; 134: 215-230. 6. Derynck R, Akhurst RJ. Differentiation plasticity regulated by TGF-beta family
proteins in development and disease. Nat Cell Biol. 2007; 9: 1000-1004. 7. Roberts AB, Wakefield LM. The two faces of transforming growth factor beta
in carcinogenesis. Proc Natl Acad Sci USA. 2003; 100: 8621-8623. 8. Rahimi RA, Leof EB. TGF-beta signaling: a tale of two responses. J Cell
Biochem. 2007; 102: 593-608. 9. Padua D, Massague J. Roles of TGFbeta in metastasis. Cell Res. 2009;
19: 89-102. 10. Mokrosinski J, Krajewska WM. TGF beta signalling accessory receptors.
Postepy Biochem. 2008; 54: 264-273. 11. Yang Z, Mu Z, Dabovic B, Jurukovski V, Yu D, Sung J i wsp. Absence
of integrin-mediated TGFbeta1 activation in vivo recapitulates the phenotype of TGFbeta1-null mice. J Cell Biol. 2007; 176: 787-793.
12. Massague J, Gomis RR. The logic of TGFbeta signaling. FEBS Lett. 2006; 580: 2811-2820.
13. Stover DG, Bierie B, Moses HL. A delicate balance: TGF-beta and the tumor microenvironment. J Cell Biochem. 2007; 101: 851-861.
14. Glasgow E, Mishra L. Transforming growth factor-beta signaling and ubiquitinators in cancer. Endocr Relat Cancer. 2008; 15: 59-72.
15. ten Dijke P, Hill CS. New insights into TGF-beta-Smad signalling. Trends Biochem Sci. 2004; 29: 265-273.
16. Miyazawa K, Shinozaki M, Hara T, Furuya T, Miyazono K. Two major Smad pathways in TGF-beta superfamily signalling. Genes Cells. 2002; 7: 1191-1204.
17. Rotzer D, Roth M, Lutz M, Lindemann D, Sebald W i wsp. Type III TGF-beta receptor-independent signalling of TGF-beta2 via TbetaRII-B, an alternatively spliced TGF-beta type II receptor. EMBO J. 2001; 20: 480-490.
18. Bernabeu C, Lopez-Novoa JM, Quintanilla M. The emerging role of TGF-beta superfamily coreceptors in cancer. Biochim Biophys Acta. 2009; 1792: 954-973.
19. Santander C, Brandan E. Betaglycan induces TGF-beta signaling in a ligand-independent manner, through activation of the p38 pathway. Cell Signal. 2006; 18: 1482-1491.
20. Wiater E, Harrison CA, Lewis KA, Gray PC, Vale WW. Identification of distinct inhibin and transforming growth factor beta-binding sites on betaglycan:

TGFβ w chorobach człowieka 288
functional separation of betaglycan co-receptor actions. J Biol Chem. 2006; 281: 17011-17022.
21. Barbara NP, Wrana JL, Letarte M. Endoglin is an accessory protein that interacts with the signaling receptor complex of multiple members of the transforming growth factor-beta superfamily. J Biol Chem. 1999; 274: 584-594.
22. Janssens K, ten Dijke P, Janssens S, Van Hul W. Transforming growth factor-beta1 to the bone. Endocr Rev. 2005; 26: 743-774.
23. Bandyopadhyay A, Tsuji K, Cox K, Harfe BD, Rosen V, Tabin CJ. Genetic analysis of the roles of BMP2, BMP4, and BMP7 in limb patterning and skeletogenesis. PLoS Genet. 2006; 2: e216.
24. Tsuji K, Cox K, Bandyopadhyay A, Harfe BD, Tabin CJ, Rosen V. BMP4 is dispensable for skeletogenesis and fracture-healing in the limb. J Bone Joint Surg Am. 2008; 90 Suppl 1: 14-18.
25. Tsuji K, Bandyopadhyay A, Harfe BD, Cox K, Kakar S, Gerstenfeld L i wsp. BMP2 activity, although dispensable for bone formation, is required for the initiation of fracture healing. Nat Genet. 2006; 38: 1424-1429.
26. Yoon BS, Ovchinnikov DA, Yoshii I, Mishina Y, Behringer RR, Lyons KM. Bmpr1a and Bmpr1b have overlapping functions and are essential for chondrogenesis in vivo. Proc Natl Acad Sci U S A. 2005; 102: 5062-5067.
27. Janssens K, ten Dijke P, Ralston SH, Bergmann C, Van Hul W. Transforming growth factor-beta 1 mutations in Camurati-Engelmann disease lead to increased signaling by altering either activation or secretion of the mutant protein. J Biol Chem. 2003; 278: 7718-7724.
28. Harradine KA, Akhurst RJ. Mutations of TGFbeta signaling molecules in human disease. Ann Med. 2006; 38: 403-414.
29. Groppe JC, Shore EM, Kaplan FS. Functional modeling of the ACVR1 (R206H) mutation in FOP. Clin Orthop Relat Res. 2007; 462: 87-92.
30. Yu PB, Deng DY, Lai CS, Hong CC, Cuny GD, Bouxsein ML i wsp. BMP type I receptor inhibition reduces heterotopic [corrected] ossification. Nat Med. 2008; 14: 1363-1369.
31. Fiori JL, Billings PC, De La Pena LS, Kaplan FS, Shore EM. Dysregulation of the BMP-p38 MAPK signaling pathway in cells from patients with fibrodysplasia ossificans progressiva (FOP). J Bone Miner Res. 2006; 21: 902-909.
32. Billings PC, Fiori JL, Bentwood JL, O'Connell MP, Jiao X, Nussbaum B i wsp. Dysregulated BMP signaling and enhanced osteogenic differentiation of

P.K. Zakrzewski 289
connective tissue progenitor cells from patients with fibrodysplasia ossificans progressiva (FOP). J Bone Miner Res. 2008; 23: 305-313.
33. Kaplan FS, Fiori J, De La Pena LS, Ahn J, Billings PC, Shore EM. Dysregulation of the BMP-4 signaling pathway in fibrodysplasia ossificans progressiva. Ann N Y Acad Sci. 2006; 1068: 54-65.
34. Kan L, Hu M, Gomes WA, Kessler JA. Transgenic mice overexpressing BMP4 develop a fibrodysplasia ossificans progressiva (FOP)-like phenotype. Am J Pathol. 2004; 165: 1107-1115.
35. Massague J, Blain SW, Lo RS. TGFbeta signaling in growth control, cancer, and heritable disorders. Cell. 2000; 103: 295-309.
36. Kawabata M, Imamura T, Miyazono K. Signal transduction by bone morphogenetic proteins. Cytokine Growth Factor Rev. 1998; 9: 49-61.
37. Thomas JT, Lin K, Nandedkar M, Camargo M, Cervenka J, Luyten FP. A human chondrodysplasia due to a mutation in a TGF-beta superfamily member. Nat Genet. 1996; 12: 315-317.
38. Lehmann K, Seemann P, Stricker S, Sammar M, Meyer B, Suring K i wsp. Mutations in bone morphogenetic protein receptor 1B cause brachydactyly type A2. Proc Natl Acad Sci U S A. 2003; 100: 12277-12282.
39. Seemann P, Schwappacher R, Kjaer KW, Krakow D, Lehmann K, Dawson K i wsp. Activating and deactivating mutations in the receptor interaction site of GDF5 cause symphalangism or brachydactyly type A2. J Clin Invest. 2005; 115: 2373-2381.
40. Watanabe Y, Kinoshita A, Yamada T, Ohta T, Kishino T, Matsumoto N i wsp. A catalog of 106 single-nucleotide polymorphisms (SNPs) and 11 other types of variations in genes for transforming growth factor-beta1 (TGF-beta1) and its signaling pathway. J Hum Genet. 2002; 47: 478-483.
41. Yamada Y, Harada A, Hosoi T, Miyauchi A, Ikeda K, Ohta H i wsp. Association of transforming growth factor beta1 genotype with therapeutic response to active vitamin D for postmenopausal osteoporosis. J Bone Miner Res. 2000; 15: 415-420.
42. Yamada Y, Miyauchi A, Goto J, Takagi Y, Okuizumi H, Kanematsu M i wsp. Association of a polymorphism of the transforming growth factor-beta1 gene with genetic susceptibility to osteoporosis in postmenopausal Japanese women. J Bone Miner Res. 1998; 13: 1569-1576.
43. Nishiyama A, Takeshima Y, Saiki K, Narukage A, Oyazato Y, Yagi M i wsp. Two novel missense mutations in the myostatin gene identified in Japanese patients with Duchenne muscular dystrophy. BMC Med Genet. 2007; 8: 19

TGFβ w chorobach człowieka 290
44. Wagner KR, McPherron AC, Winik N, Lee SJ. Loss of myostatin attenuates severity of muscular dystrophy in mdx mice. Ann Neurol. 2002; 52: 832-836.
45. Bogdanovich S, Krag TO, Barton ER, Morris LD, Whittemore LA, Ahima RS i wsp. Functional improvement of dystrophic muscle by myostatin blockade. Nature. 2002; 420: 418-421.
46. Brouillard P, Vikkula M. Genetic causes of vascular malformations. Hum Mol Genet. 2007; 16 Spec No. 2: R140-R149.
47. Goumans MJ, Liu Z, ten Dijke P. TGF-beta signaling in vascular biology and dysfunction. Cell Res. 2009; 19: 116-127.
48. Gordon KJ, Blobe GC. Role of transforming growth factor-beta superfamily signaling pathways in human disease. Biochim Biophys Acta. 2008; 1782: 197-228.
49. Otten J, Bokemeyer C, Fiedler W. Tgf-Beta superfamily receptors-targets for antiangiogenic therapy? J Oncol. 2010; 2010: 317068-
50. ten Dijke P, Arthur HM. Extracellular control of TGFbeta signalling in vascular development and disease. Nat Rev Mol Cell Biol. 2007; 8: 857-869.
51. Fernandez L, Sanz-Rodriguez F, Blanco FJ, Bernabeu C, Botella LM. Hereditary hemorrhagic telangiectasia, a vascular dysplasia affecting the TGF-beta signaling pathway. Clin Med Res. 2006; 4: 66-78.
52. Sabba C, Pasculli G, Lenato GM, Suppressa P, Lastella P, Memeo M i wsp. Hereditary hemorrhagic telangiectasia: clinical features in ENG and ALK1 mutation carriers. J Thromb Haemost. 2007; 5: 1149-1157.
53. van den Driesche S, Mummery CL, Westermann CJ. Hereditary hemorrhagic telangiectasia: an update on transforming growth factor beta signaling in vasculogenesis and angiogenesis. Cardiovasc Res. 2003; 58: 20-31.
54. Prigoda NL, Savas S, Abdalla SA, Piovesan B, Rushlow D, Vandezande K i wsp. Hereditary haemorrhagic telangiectasia: mutation detection, test sensitivity and novel mutations. J Med Genet. 2006; 43: 722-728.
55. Abdalla SA, Letarte M. Hereditary haemorrhagic telangiectasia: current views on genetics and mechanisms of disease. J Med Genet. 2006; 43: 97-110.
56. Park SO, Lee YJ, Seki T, Hong KH, Fliess N, Jiang Z, i wsp. ALK5- and TGFBR2-independent role ofALK1 in the pathogenesis of hereditary hemorrhagic telangiectasia type 2. Blood. 2008; 111: 633-642.
57. Goumans MJ, Valdimarsdottir G, Itoh S, Rosendahl A, Sideras P, ten Dijke P. Balancing the activation state of the endothelium via two distinct TGF-beta type I receptors. EMBO J. 2002; 21: 1743-1753.

P.K. Zakrzewski 291
58. Lebrin F, Deckers M, Bertolino P, ten Dijke P. TGF-beta receptor function in the endothelium. Cardiovasc Res. 2005; 65: 599-608.
59. Lebrin F, Goumans MJ, Jonker L, Carvalho RL, Valdimarsdottir G, Thorikay M i wsp. Endoglin promotes endothelial cell proliferation and TGF-beta/ALK1 signal transduction. EMBO J. 2004; 23: 4018-4028.
60. Blanco FJ, Santibanez JF, Guerrero-Esteo M, Langa C, Vary CP, Bernabeu C. Interaction and functional interplay between endoglin and ALK-1, two components of the endothelial transforming growth factor-beta receptor complex. J Cell Physiol. 2005; 204: 574-584.
61. Knight PG, Glister C. TGF-beta superfamily members and ovarian follicle development. Reproduction. 2006; 132: 191-206.
62. Matzuk MM, Kumar TR, Bradley A. Different phenotypes for mice deficient in either activins or activin receptor type II. Nature. 1995; 374: 356-360.
63. Bilezikjian LM, Blount AL, Donaldson CJ, Vale WW. Pituitary actions of ligands of the TGF-beta family: activins and inhibins. Reproduction. 2006; 132: 207-215.
64. Lewis KA, Gray PC, Blount AL, MacConell LA, Wiater E, Bilezikjian LM i wsp. Betaglycan binds inhibin and can mediate functional antagonism of activin signalling. Nature. 2000; 404: 411-414.
65. Pierson TM, Wang Y, DeMayo FJ, Matzuk MM, Tsai SY, Omalley BW. Regulable expression of inhibin A in wild-type and inhibin alpha null mice. Mol Endocrinol. 2000; 14: 1075-1085.
66. Josso N, Belville C, di Clemente N, Picard JY. AMH and AMH receptor defects in persistent Müllerian duct syndrome. Hum Reprod Update. 2005; 11: 351-356.
67. Belville C, Van Vlijmen H, Ehrenfels C, Pepinsky B, Rezaie AR, Picard JY i wsp. Mutations of the anti-müllerian hormone gene in patients with persistent müllerian duct syndrome: biosynthesis, secretion, and processing of the abnormal proteins and analysis using a three-dimensional model. Mol Endocrinol. 2004; 18: 708-721.
68. Belville C, Josso N, Picard JY. Persistence of Müllerian derivatives in males. Am J Med Genet. 1999; 89: 218-223.
69. Belville C, Marechal JD, Pennetier S, Carmillo P, Masgrau L, Messika-Zeitoun L i wsp. Natural mutations of the anti-Müllerian hormone type II receptor found in persistent Müllerian duct syndrome affect ligand binding, signal transduction and cellular transport. Hum Mol Genet. 2009; 18: 3002-3013.
70. Beck-Peccoz P, Persani L. Premature ovarian failure. Orphanet J Rare Dis. 2006; 1: 9

TGFβ w chorobach człowieka 292
71. Di Pasquale E, Rossetti R, Marozzi A, Bodega B, Borgato S, Cavallo L i wsp. Identification of new variants of human BMP15 gene in a large cohort of women with premature ovarian failure. J Clin Endocrinol Metab. 2006; 91: 1976-1979.
72. Chand AL, Ooi GT, Harrison CA, Shelling AN, Robertson DM. Functional analysis of the human inhibin alpha subunit variant A257T and its potential role in premature ovarian failure. Hum Reprod. 2007; 22: 3241-3248.
73. Shelling AN, Burton KA, Chand AL, van Ee CC, France JT, Farquhar CM i wsp. Inhibin: a candidate gene for premature ovarian failure. Hum Reprod. 2000; 15: 2644-2649.

Folia Medica Lodziensia, 2012, 39/2: 293-326
Adres do korespondencji: dr hab. Magdalena Bryś, prof. nadzw. UŁ; Katedra Cytobiochemii, Uniwersytet Łódzki; 90-236 Łódź; ul. Pomorska 141/143; e-mail: [email protected] Tel.: + 48 42 6354371; Fax: +48 42 6354484
Telomeraza – struktura i funkcja
oraz regulacja ekspresji genu
Telomerase – structure, function and the regulation of gene expression
MAGDALENA BRYŚ, MAGDALENA LASKOWSKA, EWA FORMA, ANNA KRZEŚLAK
Katedra Cytobiochemii, Wydział Biologii i Ochrony Środowiska,
Uniwersytet Łódzki
Streszczenie Telomer jest to fragment chromosomu zlokalizowany na jego końcu, który
zabezpiecza go przed uszkodzeniem podczas kopiowania. Telomery są także
czynnikami kontrolującymi liczbę podziałów komórkowych i dlatego uważane są
za supresory transformacji nowotworowej, ponieważ ograniczona, ściśle
kontrolowana liczba podziałów zapobiega ewentualnemu kumulowaniu się
mutacji w komórce. Przyjęto, że obecność 4-6 mutacji w materiale genetycznym
jest czynnikiem karcynogennym, a po granicznej liczbie podziałów (około 60-70)
komórka wchodzi w fazę spoczynku M1. Enzymem, którego zadaniem jest
dobudowanie 3'-końcowego odcinka nici DNA i tym samym wydłużanie
sekwencji telomerowych jest enzym telomeraza. Białko to jest polimerazą DNA
zależną od RNA, która syntetyzuje telomery na zasadzie odwrotnej transkrypcji.
Unikalną cechą telomerazy jest to, że jej integralnym składnikiem jest matryca
RNA służąca do syntezy DNA. Telomeraza występuje w intensywnie dzielących
się komórkach, a jej aktywność zmniejsza się wraz z wiekiem. W komórkach
prawidłowych zwykle nie stwierdza się aktywności telomerazy, natomiast
w nowotworowych aktywność tego enzymu zwykle jest podwyższona. W pracy
omówiono strukturę sekwencji telomerowych oraz udział białek zaangażowa-
nych w jej utrzymanie. Szczegółowo przedstawiono także strukturę i funkcję
genu/białka telomerazy, z uwzględnieniem regulacji ekspresji genu na poziomie
transkrypcji. Scharakteryzowano ponadto udział telomerazy w procesach
transformacji nowotworowej.

Telomeraza – struktura i funkcja oraz regulacja ekspresji genu 294
Słowa kluczowe: telomeraza, gen, białko, ekspresja genu, czynniki
transkrypcyjne.
Abstract
A telomere is a fragment localized at the end of chromosome which protects the chromosome from damage during replication. Telomeres are also factors that control number of cell divisions and are thought to be a suppressors of carcinogenesis since limited, strictly determined number of cell divisions protects from accumulation of mutations in cell. It is assumed that presence of 4-6 mutation in genetic material is a carcinogenic factor and after about 60-70 divisions, the cell enter the resting phase. Telomerase is an enzyme which adds DNA sequence to the 3’ end of DNA and extends the telomere region. This protein is a DNA polymerase dependent on RNA, which syntheses telomere by reverse transcription. The unique characteristics of telomerase is that RNA matrix for DNA synthesis is an integral component of this enzyme. Telomerase is present in intensively dividing cells and its activity is decreasing with age. In normal cells usually activity of telomerase is undetectable but in cancer cells activity of this enzyme is high. The aim of this work is to present the structure of telomeres and the role of proteins involved in maintaining the structure. In details, the structure and function of the telomerase gene/protein is described, including the regulation of gene expression at the transcriptional level. The involvement of telomerase in the neoplastic transformation has been also characterized.
Key words: telomerase, gene, protein, gene expression, transcription factors. Wstęp
Opisy struktury i funkcji telomeru pojawiły się w literaturze naukowej
w latach 30. XX wieku. Po raz pierwszy w 1978 roku sekwencja telomeru
została określona u jednokomórkowego orzęska Tetrahymena pyriformis [1].

M. Bryś i wsp. 295
W latach 90. XX wieku Jerry Shay i współpracownicy wykryli telomerazę
w 90 na 101 preparatach komórek pobranych z różnych nowotworów człowieka
i nie wykazali obecności telomerazy w żadnej z 50 różnych, prawidłowych
komórek somatycznych. Wyniki tych badań sugerowały, że telomeraza pełni
istotną funkcję w procesie transformacji nowotworowej [2]. W 1997 r. Robert A.
Weinberg wraz z zespołem badawczym sklonowali gen kodujący podjednostkę
telomerazy o aktywności odwrotnej transkryptazy [3]. W 2009 roku Nagrodę
Nobla w dziedzinie fizjologii lub medycyny za odkrycie, w jaki sposób telomery
i enzym telomeraza chronią chromosomy, otrzymała trójka amerykańskich
uczonych - Elizabeth H Blackburn, Jack W Szostak i Carol W Greiner [4].
Zgodnie z prawami natury, celem nadrzędnym wszystkich organizmów jest
utrzymanie ciągłości gatunku i dokonuje się to poprzez przekazywanie materiału
genetycznego organizmom potomnym. Aby organizm mógł prawidłowo
funkcjonować, w czasie jego rozwoju osobniczego muszą zachodzić w ściśle
określonym czasie procesy syntezy i naprawy uszkodzeń materiału
genetycznego oraz programowanej śmierci komórki, czyli apoptozy. Zgodnie
z teorią Kirkwooda oraz w związku z faktem, iż średnia długość życia człowieka
wydłuża się, korzystniejsze jest wydatkowanie większej części energii na
procesy naprawcze organizmu niż reprodukcyjne. Wraz z długością życia
zwiększeniu ulega ryzyko związane z powstawaniem nowotworów, co
bezpośrednio wynika z wydłużenia okresu działania czynników karcynogennych
na komórki oraz z gromadzenia się nieprawidłowości na poziomie
molekularnym [5].
Odmienna specyfika aktywności telomerazy w materiale prawidłowym
i nowotworowym stała się podstawą do rozważania tego enzymu w kontekście
terapii przeciwnowotworowej. Komórki nowotworowe charakteryzują się

Telomeraza – struktura i funkcja oraz regulacja ekspresji genu 296
znacznie krótszymi telomerami niż komórki prawidłowe i zależne są od
aktywności telomerazy. Zablokowanie aktywności tego enzymu może wpływać
na zahamowanie transformacji nowotworowej bez równoczesnego ingerowania
w funkcjonowanie komórek prawidłowych [6, 7]
Odmienna specyfika aktywności telomerazy w materiale prawidłowym
i nowotworowym stała się podstawą do rozważania tego enzymu w kontekście
terapii przeciwnowotworowej. Komórki nowotworowe charakteryzują się
znacznie krótszymi telomerami niż komórki prawidłowe i zależne są od
aktywności telomerazy. Zablokowanie aktywności tego enzymu może wpływać
na zahamowanie transformacji nowotworowej bez równoczesnego ingerowania
w funkcjonowanie komórek prawidłowych [6, 7].
Struktura i funkcje telomerów
Telomery (gr. telos – koniec i meros – część) są to wyspecjalizowane
kompleksy nukleoproteinowe, które chronią zakończenia prawidłowych
chromosomów przed degradacją i fuzją, co w konsekwencji przekłada się na
utrzymanie stabilności genomu [1, 8, 9]. W komórkach człowieka występują
92 telomery, po jednym na każdym końcu chromosomu. Wysoce
konserwatywne sekwencje telomerowe są kilkutysięcznymi, nie kodującymi,
powtarzającymi się motywami nukleotydowymi, bogatymi w reszty guaniny
i tyminy [10]. U człowieka i większości kręgowców powtarzającym się
motywem jest sekwencja TTAGGG [11, 12]. Sekwencje telomerowe wybranych
organizmów zawarte zostały w Tabeli 1.

M. Bryś i wsp. 297
Tabela1. Sekwencje telomerowego DNA u wybranych zwierząt i roślin [wg 13, 14]
ORGANIZM Sekwencja (5’→3’)
człowiek (łac.Homo sapiens) TTAGGG
drożdże (łac. Saccharomyces cerevisiae) TGTGGGTGTGGTG
glista ludzka (łac. Ascaris lumbricoides) TTAGGC
jedwabnik (łac. Bombyx mori) TTAGG
mysz polna (łac. Apodemus agrarius) TTAGGG
pantofelek (łac. Paramecium caudatum) TTGGG(T/G)
rzodkiewnik pospolity (łac. Arabidopsis thaliana) TTTAGGG
tasiemiec uzbrojony (łac. Taenia solium) TTAGGC
zawłotnia (łac. Chlamydomonas) TTTTAGGG
U człowieka powtórzenia (TTAGGG)n mają długość około 15-20 kpz
w chwili narodzin, ale już u osób dorosłych ulegają skróceniu do 8-10 kpz.
Długość sekwencji telomerowych wykazuje zmienność osobniczą, jak również
może wahać się w zależności od komórki czy tkanki [15, 16].
Obowiązujący aktualnie model struktury telomeru uzyskano dzięki
badaniom z użyciem mikroskopu elektronowego. Jest on zdecydowanie bardziej
złożony w swojej przestrzennej strukturze, niż wynikało to z modelu
klasycznego, który zakładał strukturę liniową. W obecnym modelu jednoniciowe
końce 3’ chromosomów, o długości około 75–200 pz, tworzą strukturę
przestrzenną zwaną kwartetem G (G4-DNA), który uniemożliwia dostęp
egzonukleaz do końcowych odcinków telomerów i tym samym zabezpiecza nić
DNA przed degradacją. Różne struktury przestrzenne kwartetu G zostały
przedstawione na ryc. 1 i 2.

Telomeraza – struktura i funkcja oraz regulacja ekspresji genu 298
Ryc. 1. Struktura kwartetu G [wg 20, w modyfikacji własnej].
Oddziaływanie z białkiem G4BP (białko oddziałujące z sekwencją
kwartetu G, ang. G-quadruplex binding protein) dodatkowo wzmacnia
konstrukcję kwartetu G (Ryc. 3) [17-19]. Dwuniciowe fragmenty DNA
na końcach telomerów formują dwie pętle – pętlę T (ang. T-loop) oraz pętlę D
(ang. D-loop). Pętla T zakończona jest jednoniciowym „zwisającym”
fragmentem na końcu 3’. Pętla D natomiast, powstaje kilkaset nuklotydów
wcześniej niż pętla T i jest formowana dzięki oddziaływaniu między
jednoniciowym i dwuniciowym DNA, na zasadzie wyparcia jednej nici dupleksu
(Ryc. 4) [18]. Konfiguracja pętli T jest strukturą zabezpieczającą, dzięki której
koniec 3’ chromosomu jest ukryty. Uniemożliwia to błędne uznanie go przez
systemy naprawcze DNA jako uszkodzenie ciągłości nici [23].

M. Bryś i wsp. 299
Ryc. 2. Przestrzenne struktury kwartetu G. DNA bogate w reszty guaniny może tworzyć
różne struktury kwartetu G. Pojedyncza nić DNA może składać się ku sobie, tworząc wewnętrzny kwartet G, podczas gdy dwie i cztery nici DNA mogą się łączyć
odpowiednio, w struktury dimeryczne lub tetrameryczne. Strzałki wskazują orientację DNA – równoległą i przeciwrównoległą. Reszty guanozyny przedstawione zostały w
różnych kolorach, według konfiguracji wiązania glikozydowego. Wiązanie glikozydowe typu „anty” – kolor szary, typu „trans” – kolor czarny [wg 21; w modyfikacji własnej].

Telomeraza – struktura i funkcja oraz regulacja ekspresji genu 300
Ryc. 3. (A) Schemat sekwencji nukleotydowej oraz przestrzennej organizacji kwartetu G. (B) Model końca telomeru niezabezpieczonego przed trawieniem
egzonukleolitycznym oraz telomer z uformowanym kwartetem G i przyłączonym do niego białkiem G4BP [wg 22; w modyfikacji własnej].

M. Bryś i wsp. 301
Ryc. 4. Schemat obrazujący powstawanie pętli na zakończeniach chromosomów [wg 22; w modyfikacji własnej].
Telomery chronione są przez wyspecjalizowane kompleksy białkowe, które
rozpoznają podwójną nić DNA oraz kwartet G. Taki białkowy kompleks
w komórkach ssaków nazywany jest „shelterin” i składa się z sześciu białek:
TRF1 i TRF2 (czynnik wiążący się do podwójnych powtórzeń TTAGGG 1, 2,
ang. telomeric repeat binding factor 1, 2), RAP1 (białko represorowo-
aktywatorowe, ang. repressor - activator protein 1), TIN2 (czynnik jądrowy
oddziałujący z TRF1 2, ang. TRF1 interacting nuclear factor 2), TPP1
(trójpeptydylopeptydaza 1, ang. tripeptidyl peptidase 1) i POT1 (białko
chroniące telomery 1, ang. protection of telomeres 1) [24]. Białka TRF1 TRF2
łączą się bezpośrednio z telomerowym dwuniciowym DNA, natomiast białka
RAP1, TIN2, TPP1 i POT1 stabilizują strukturę kompleksu „shelterin” [25-27].

Telomeraza – struktura i funkcja oraz regulacja ekspresji genu 302
Układ przestrzenny i charakterystyka wyżej wymienionych białek została
przedstawiona na rycinie 5 oraz w Tabeli 2.
Ryc. 5. Układ przestrzenny kompleksu „shelterin” oraz konfiguracja białek podtrzymująca strukturę pętli T i pętli D telomeru [wg 28, 29; w modyfikacji własnej].
Białko POT1 łącząc się z pojedynczą nicią telomerowego DNA oddziałuje
z białkiem TPP1. Guanina na końcu 3' telomerowego DNA jest „zanurzona”
w białku POT1, co powoduje niedostępność substratu dla telomerazy
i najprawdopodobniej hamuje wydłużanie nici DNA. Z kolei białko Rap1 łączy
się z białkiem TIN2 oraz białkami TRF1 i TRF2. Białko TIN2 oddziałuje
również zarówno z białkiem TRF1, jak i TRF2, a jego zadaniem jest rekrutacja
białek POT1 oraz TPP1 do dwuniciowego fragmentu telomeru [30, 31]
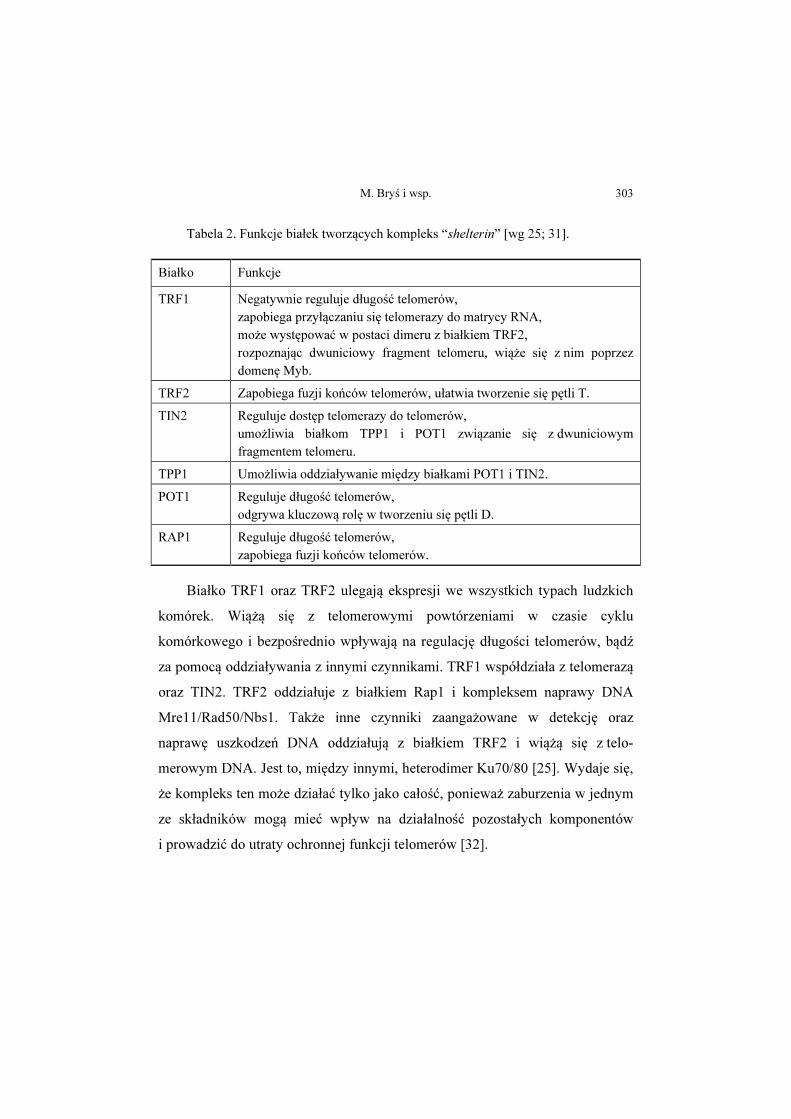
M. Bryś i wsp. 303
Tabela 2. Funkcje białek tworzących kompleks “shelterin” [wg 25; 31].
Białko Funkcje
TRF1 Negatywnie reguluje długość telomerów, zapobiega przyłączaniu się telomerazy do matrycy RNA, może występować w postaci dimeru z białkiem TRF2, rozpoznając dwuniciowy fragment telomeru, wiąże się z nim poprzez domenę Myb.
TRF2 Zapobiega fuzji końców telomerów, ułatwia tworzenie się pętli T.
TIN2 Reguluje dostęp telomerazy do telomerów, umożliwia białkom TPP1 i POT1 związanie się z dwuniciowym fragmentem telomeru.
TPP1 Umożliwia oddziaływanie między białkami POT1 i TIN2.
POT1 Reguluje długość telomerów, odgrywa kluczową rolę w tworzeniu się pętli D.
RAP1 Reguluje długość telomerów, zapobiega fuzji końców telomerów.
Białko TRF1 oraz TRF2 ulegają ekspresji we wszystkich typach ludzkich
komórek. Wiążą się z telomerowymi powtórzeniami w czasie cyklu
komórkowego i bezpośrednio wpływają na regulację długości telomerów, bądź
za pomocą oddziaływania z innymi czynnikami. TRF1 współdziała z telomerazą
oraz TIN2. TRF2 oddziałuje z białkiem Rap1 i kompleksem naprawy DNA
Mre11/Rad50/Nbs1. Także inne czynniki zaangażowane w detekcję oraz
naprawę uszkodzeń DNA oddziałują z białkiem TRF2 i wiążą się z telo-
merowym DNA. Jest to, między innymi, heterodimer Ku70/80 [25]. Wydaje się,
że kompleks ten może działać tylko jako całość, ponieważ zaburzenia w jednym
ze składników mogą mieć wpływ na działalność pozostałych komponentów
i prowadzić do utraty ochronnej funkcji telomerów [32].

Telomeraza – struktura i funkcja oraz regulacja ekspresji genu 304
Telomery jako struktury dynamiczne, ochraniające końce chromosomów
przed degradacją, odpowiadają między innymi za umożliwienie systemom
naprawczym rozpoznania prawidłowych i uszkodzonych zakończeń
chromosomów, przestrzenną organizację jądra komórkowego oraz regulację
transkrypcji genów zlokalizowanych w pobliżu telomerów [33, 34]. Ponadto,
telomery zapobiegają translokacjom, amplifikacjom i delecjom materiału
genetycznego. Zabezpieczając przed fuzją chromosomów, zapewniają
prawidłowy przebieg procesu rekombinacji i zapobiegają powstawaniu
chromosomów dicentrycznych. W przypadku uszkodzenia telomeru często
dochodzi do procesu rekombinacji. W zależności od typu tego zjawiska,
dochodzi do fuzji ramion chromosomów typu p-p, p-q i q-q [35, 36].
Po osiągnięciu krytycznego stanu skrócenia telomerów (3 kpz), przy którym
zakończenia chromosomów nie są w stanie utrzymać charakterystycznej dla
telomerów struktury pętli, telomery nie mogą chronić chromosomów przed
utratą materiału genetycznego i niestabilnością genomu, co prowadzi do śmierci
komórki. Jest to jeden z mechanizmów inicjujących apoptozę i naturalna
konsekwencja starzenia się komórek [37].
Aż do wczesnych lat 60 uważano, że hodowane prawidłowe komórki
człowieka, mogą dzielić się nieskończenie wiele razy, tak długo jak dobre
warunki hodowli zostaną utrzymane. Przełom nastąpił w 1965 roku za sprawą
Leonarda Hayflick’a, który obalił dotychczasową koncepcję i wykazał, iż
prawidłowe komórki człowieka mają ograniczoną liczbę podziałów. To zjawisko
starzenia replikacyjnego prawidłowych komórek ludzkich bardzo często
nazywane jest „limitem Hayflicka” (około 60 podziałów). Każdy podział
komórki związany jest z nieodwracalnym skracaniem sekwencji telomerowej
o ok. 50-150 pz., co jest związane z problem replikacji końców nici opóźnionej.

M. Bryś i wsp. 305
Polimeraza DNA replikuje tylko w kierunku 5’→3’, a do uruchomienia
replikacji wymaga starterowych fragmentów RNA, co powoduje niepełną
replikację nici DNA przy końcu 5’ [38, 39]. Telomer jest tzw. molekularnym
zegarem biologicznym wyznaczającym ściśle określoną liczbę podziałów
komórkowych. Komórki mają więc ograniczony potencjał proliferacyjny,
a co za tym idzie po pewnej liczbie podziałów wkraczają w etap starzenia się.
Tracą wtedy zdolność podziałów, ale nadal są aktywne metabolicznie [40].
Telomery dzięki określonej liczbie podziałów komórkowych stają się
supresorami procesu nowotworzenia, gdyż w ten sposób zapobiegają nagro-
madzeniu się mutacji prowadzących do rozwoju nowotworu [35, 36, 41, 42].
Telomeraza – struktura i funkcje
U większości ssaków za utrzymanie odpowiedniej długości telomerów
odpowiada specyficzna odwrotna tranksyptaza – telomeraza. Jest to enzym
odgrywający kluczową rolę także w takich procesach jak proliferacja komórki
i transformacja nowotworowa [43]. Unikalną cechą telomerazy jest to, że jej
integralnym składnikiem jest matryca RNA służąca do syntezy DNA.
Telomeraza jest polimerazą DNA zależną od RNA, która syntetyzuje telomery
na zasadzie odwrotnej transkrypcji. Jest to duży kompleks rybonukleo-
proteinowy - masa cząsteczkowa holoenzymu telomerazy przekracza 500 kDa
[44]. Telomeraza człowieka składa się z fragmentu nici RNA (hTERC/hTR) oraz
podjednostki białkowej o aktywności odwrotnej transkryptazy (hTERT) [45].
Struktura genu TERC u różnych organizmów różni się pod względem rozmiaru,
jak i sekwencji nukleotydowej. Dojrzała struktura genu TERC u Saccharomyces
cerevisiae składa się z 1167 nukleotydów, podczas, gdy u pierwotniaków i

Telomeraza – struktura i funkcja oraz regulacja ekspresji genu 306
ssaków jest ona znacząco krótsza i wynosi odpowiednio około 150 i 450
nukleotydów [46-49].
Struktura TERC zawiera:
• domenę centralną
• miejsce wiązania białek odpowiedzialnych za dojrzewanie i stabilizację
domeny
• miejsce wiązania z TERT
Ryc. 6. Schemat ludzkiego chromosomu 3 z zaznaczoną lokalizacją genu hTERC (q26).
Gen hTERC zlokalizowany jest na chromosomie 3q26 (Rys 6). Jest on
transkrybowany przez polimerazę RNA II. Składa się z 445 nukleotydów,
z powtarzającą się sekwencją 11 nukleotydową: 5’-(CUAACCCUAAC)-3’,
która jest komplementarna do sekwencji telomeru (TTAGGG) [50]. Sekwencja
nukleotydowa oraz konformacja przestrzenna podjednostki TR przedstawiona
została na rycinie 7.

M. Bryś i wsp. 307
Ryc. 7. Sekwencja nukleotydowa (z zaznaczoną sekwencją komplementarną do sekwencji telomeru) oraz konformacja przestrzenna podjednostki TR telomerazy
[wg Telomerase Database, www.telomerase.asu.edu; w modyfikacji własnej].
Struktura przestrzenna podjednostki o aktywności odwrotnej transkryptazy
to tak zwana „struktura rękawiczki”. Umożliwia ona owinięcie się enzymu
dookoła końca chromosomu w celu dodania jednoniciowych powtórzeń. TERT
jako odwrotna transkryptaza syntetyzuje na matrycy RNA pojedynczy łańcuch
DNA złożony z telomerowych powtórzeń. hTERT jest stosunkowo dużym

Telomeraza – struktura i funkcja oraz regulacja ekspresji genu 308
białkiem, jego masa cząsteczkowa wynosi 127 kDa. Gen dla tej podjednostki
zlokalizowany jest na krótszym ramieniu chromosomu 5 (p15.33) (Ryc.8).
Obejmuje on 37 pz genomowego DNA i składa się z 16 eksonów [43].
Ryc. 8. Schemat ludzkiego chromosomu 5 z zaznaczoną lokalizacją genu hTERT (p15.33).
Chociaż struktura krystaliczna dla białka TERT nie została jeszcze w pełni
poznana, sugeruje się istnienie trzech domen:
• N-końowej (NTE, ang. N-terminal extension) – zawierającej motyw
wiążący RNA oraz DNA
• centralnej (RT, ang. reverse transcriptase) o aktywności katalitycznej
• C-końcowej (CTE, ang. C-terminal extension) [51-53].
Na rycinie 9 przedstawiony został schemat organizacji strukturalnej
podjednostki TERT telomerazy.

M. Bryś i wsp. 309
Ryc. 9. Schemat organizacji strukturalnej podjednostki TERT telomerazy. U większości organizmów domena TERT składa się z długiego N-końca (NTE), domeny centralnej
o aktywności katalitycznej (RT) oraz krótkiego C-końca (CTE). Na schemacie zaznaczono motywy CP, QFP i TS, wchodzące w skład odcinka TRBD
oraz telomerazowo specyficzną domenę IFD (pole zakreślone ukośnie). W obrębie N-końca znajduje się region „linkerowy” łączący domenę TEN i TRBD
[wg 55; w modyfikacji własnej].
Domena N-końcowa zawiera dwa motywy - TEN (N-końcowy motyw
telomerazy, ang. telomerase essential N-terminal) i TRBD (motyw wiążący się
z RNA, ang. telomerase RNA-binding domain). Oba odpowiedzialne są za
fałdowanie białka zaangażowanego w wiązanie się z jednoniciowym kwasem
nukleinowym. Pomiędzy tymi regionami znajduje się stosunkowo długi region
łącznikowy, który może mieć znaczenie w utrzymaniu elastyczności
holoenzymu. N-końcowa część białka TERT zawiera również kilka
specyficznych dla telomerazy motywów, w tym CP, QFP i TS. Regiony te
odgrywają rolę w oddziaływaniach TERT-TERC, a także mają wpływ na
szybkość kopiowania matrycy podczas syntezy telomerów [44]. Katalityczna
domena białka TERT zawiera siedem ewolucyjnie konserwatywnych motywów
RT, istotnych dla aktywności enzymatycznej telomerazy. Domena RT
zorganizowana jest w dwie poddomeny, które przypominają „palce” i „dłoń”.
Regiony te połączone są w pętlę, która zawiera konserwatywny region

Telomeraza – struktura i funkcja oraz regulacja ekspresji genu 310
„primer grip”, determinujący aktywność enzymatyczną. Pętla ta umożliwia
bezpośredni kontakt hybrydy RNA-DNA z końcem 3’ jednoniciowego DNA, co
sugeruje, że region „primer grip” może brać udział w lokowaniu końca 3’ DNA
w centrum aktywnym enzymu. Unikatową cechą domeny RT jest obecność
poddomeny IFD (domena odcinka RT białka TERT, ang. insertion in fingers).
Składa się ona z dwóch antyrównoległych helis α, które umieszczone są
pomiędzy „palcami” a „dłonią”. Poddomena IFD zaangażowana jest
w wewnątrzcząsteczkowe oddziaływania białko-białko, które warunkują
i stabilizują konformację przestrzenną tego regionu domeny RT [44, 54].
Domena C-końcowa w przeciwieństwie do domeny N-końcowej i domeny RT,
jest słabo zachowana ewolucyjnie. Pozwala to wnioskować, iż może ona
spełniać specyficzne gatunkowo funkcje. Jak dotąd nie wyjaśniono kwestii
dlaczego domena C-końcowa jest niezbędna dla aktywności telomerazy u, na
przykład orzęska i człowieka, ale nie jest wymagana dla aktywności tego
enzymu u drożdży i w ogóle nie występuje u innych organizmów [44].
Z wymienionymi podjednostkami telomerazy zasocjowane są liczne białka, jak
na przykład TP1 (białko związane z telomerazą 1, ang. telomerase associated
protein 1), Hsp23, Hsp90 (białka szoku cieplnego 23 i 90, ang. heat shock
protein 23, 90), hStau (białko łączące się z dwuniciowym RNA, ang. human
Staufen), L22 i dyskeryna. Ich obecność nie jest jednak konieczna dla
zachowania aktywności telomerazy [56-59]. Wydaje się, że domena hTERT jest
głównym wyznacznikiem aktywności telomerazy. Zgodnie z danymi
literaturowymi, gen hTERC ulega ekspresji we wszystkich typach tkanek,
natomiast ekspresja genu hTERT ograniczona jest do tak zwanych tkanek
telomerazowo-dodatnich, co sugeruje, że hTERT jest czynnikiem
odpowiedzialnym za enzymatyczną aktywność telomerazy.

M. Bryś i wsp. 311
Ryc. 10. Schemat przedstawia podstawowe etapy cyklu reakcji telomerazy: rozpoznanie i związanie startera, wydłużanie nici DNA oraz translokację enzymu. Telomerazowy
RNA (TR) przedstawiony został jako czarna linia (brak zachowania skali). TR dostarcza matrycy do syntezy telomeru. Synteza DNA katalizowana jest przez TERT
[wg 43; w modyfikacji własnej].

Telomeraza – struktura i funkcja oraz regulacja ekspresji genu 312
Wykazano, że sztucznie wymuszona ekspresja genu TERT wydłuża
żywotność komórki w ponad dwudziestu różnych typach komórek [50, 60, 61].
Rzeczywisty mechanizm reakcji telomerazy można opisać w trzech etapach.
W pierwszym etapie, krótka telomerowa sekwencja na końcu 3’ chromosomu
wiąże się z matrycowym RNA telomerazy. Kolejny etap stanowi wydłużanie
nici, gdzie podjednostka TERT wykorzystuje fragment RNA jako matrycę do
produkcji nowego zakończenia telomeru. Ostatnim etapem jest translokacja bądź
dysocjacja, gdzie telomeraza przenosi się na nowy koniec 3’ chromosomu w
celu dodawania kolejnych powtórzeń lub opuszcza obszar telomeru [50]. Rycina
10 przedstawia mechanizm telomerazy.
Regulacja ekspresji genu TERT na poziomie mRNA i białka odbywać się
może na poziomie transkrypcji genu poprzez udział czynników transkryp-
cyjnych oraz modyfikacje potranskrypcyjne i potranslacyjne [62, 63]. Ścisła
korelacja między ekspresją genu hTERT na poziomie mRNA a aktywnością
telomerazy sugeruje, że regulacja transkrypcji tego genu jest jednym
z podstawowych mechanizmów regulujących aktywność enzymu [64; 65].
Analiza promotora genu hTERT wykazała obecność szeregu potencjalnych
miejsc dla oddziaływania z czynnikami transkrypcyjnymi, zarówno
wzmacniającymi, jak i wyciszającymi transkrypcję genu. Schemat obszaru
promotorowego genu TERT z zaznaczonymi miejscami oddziaływania
z wybranymi czynnikami transkrypcyjnymi, przedstawia rycina 11. Dwie
sekwencje oddziałujące z białkami c-Myc i Mad/Max oznaczane są jako dalszy
i bliższy motyw E-box (ang. distal/proximal E-box) [58, 65-67].
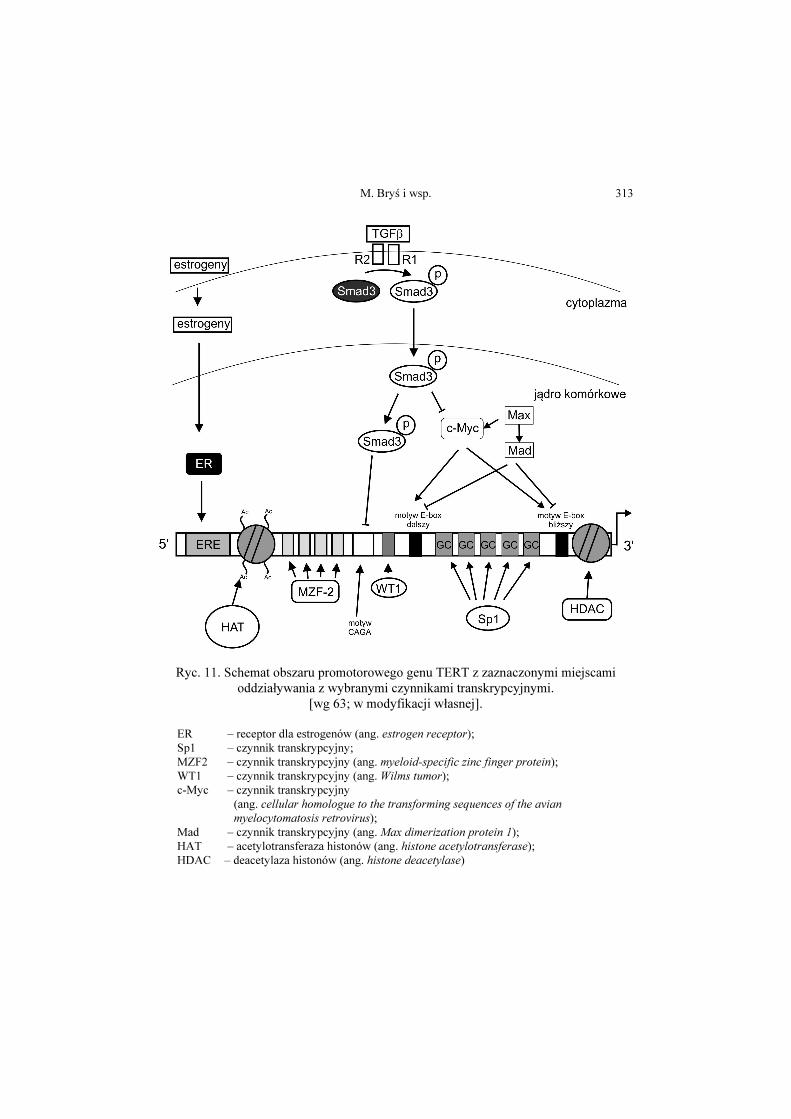
M. Bryś i wsp. 313
Ryc. 11. Schemat obszaru promotorowego genu TERT z zaznaczonymi miejscami oddziaływania z wybranymi czynnikami transkrypcyjnymi.
[wg 63; w modyfikacji własnej].
ER – receptor dla estrogenów (ang. estrogen receptor); Sp1 – czynnik transkrypcyjny; MZF2 – czynnik transkrypcyjny (ang. myeloid-specific zinc finger protein); WT1 – czynnik transkrypcyjny (ang. Wilms tumor); c-Myc – czynnik transkrypcyjny
(ang. cellular homologue to the transforming sequences of the avian myelocytomatosis retrovirus);
Mad – czynnik transkrypcyjny (ang. Max dimerization protein 1); HAT – acetylotransferaza histonów (ang. histone acetylotransferase); HDAC – deacetylaza histonów (ang. histone deacetylase)
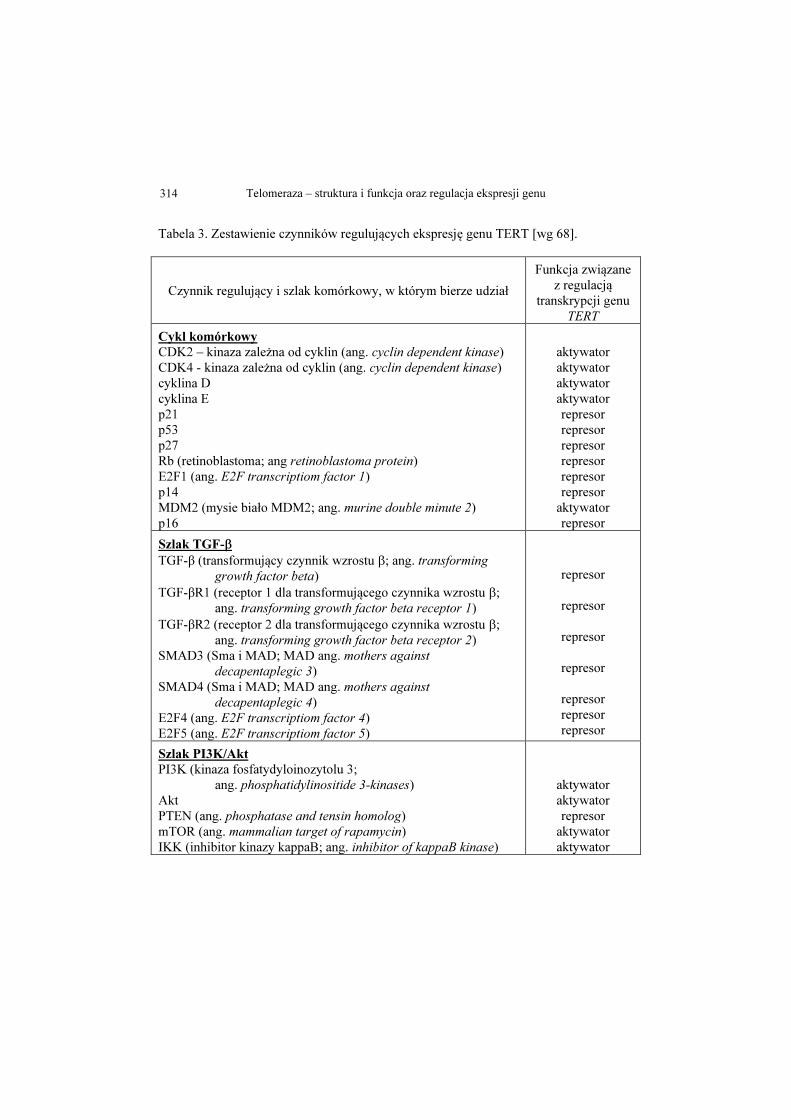
Telomeraza – struktura i funkcja oraz regulacja ekspresji genu 314
Tabela 3. Zestawienie czynników regulujących ekspresję genu TERT [wg 68].
Czynnik regulujący i szlak komórkowy, w którym bierze udział
Funkcja związane z regulacją
transkrypcji genu TERT
Cykl komórkowy
CDK2 – kinaza zależna od cyklin (ang. cyclin dependent kinase) CDK4 - kinaza zależna od cyklin (ang. cyclin dependent kinase) cyklina D cyklina E p21 p53 p27 Rb (retinoblastoma; ang retinoblastoma protein) E2F1 (ang. E2F transcriptiom factor 1) p14 MDM2 (mysie biało MDM2; ang. murine double minute 2) p16
aktywator aktywator aktywator aktywator represor represor represor represor represor represor
aktywator represor
Szlak TGF-β
TGF-β (transformujący czynnik wzrostu β; ang. transforming
growth factor beta) TGF-βR1 (receptor 1 dla transformującego czynnika wzrostu β;
ang. transforming growth factor beta receptor 1) TGF-βR2 (receptor 2 dla transformującego czynnika wzrostu β;
ang. transforming growth factor beta receptor 2) SMAD3 (Sma i MAD; MAD ang. mothers against
decapentaplegic 3) SMAD4 (Sma i MAD; MAD ang. mothers against
decapentaplegic 4) E2F4 (ang. E2F transcriptiom factor 4) E2F5 (ang. E2F transcriptiom factor 5)
represor
represor
represor
represor
represor represor represor
Szlak PI3K/Akt
PI3K (kinaza fosfatydyloinozytolu 3; ang. phosphatidylinositide 3-kinases)
Akt PTEN (ang. phosphatase and tensin homolog) mTOR (ang. mammalian target of rapamycin) IKK (inhibitor kinazy kappaB; ang. inhibitor of kappaB kinase)
aktywator aktywator represor
aktywator aktywator

M. Bryś i wsp. 315
Tabela 3. Zestawienie czynników regulujących ekspresję genu TERT [wg 68]. (cd)
Czynnik regulujący i szlak komórkowy, w którym bierze udział
Funkcja związane z regulacją
transkrypcji genu TERT
Szlak NF-κB (kanoniczny) NF-κB (p50) (ang. nuclear factor kappa-light-chain-enhancer of
activated B cells) NF-κB (p65) IKK (ang. inhibitor of kappaB kinase) IκB (ang. inhibitor of kappa B)
aktywator aktywator aktywator represor
Szlak NF-κB (niekanoniczny) NIK (ang. NF-kappaB-inducing kinase) IKK NF-κB (p100) NF-κB (RelB)
aktywator aktywator aktywator aktywator
Szlak kinaz MAP
RAS (ang. rat sarcoma) MEK (ang. mitogen extracellular kinase) ERK (ang. extracellular signal-regulated kinase)
aktywator aktywator aktywator
Szlak erbB
EGF (ang. epidermal growth factor) EGFR (ang. epidermal growth factor receptor) erB-2 (ang. human epidermal growth factor 2) PI3K
aktywator aktywator aktywator aktywator
Inne
AP2 (ang. activator protein 2) AP4 (ang. activating enhancer-binding protein 4) BRCA1 (ang. breast cancer type 1 susceptibility protein 1) c-Ets-2 (ang. v-ets erythroblastosis virus E26 oncogene
homolog 2) c-Myb (ang. myeloblastosis) CREB/ATF (ang. cAMP responsive element binding protein/
activating transcription factors) CTCF (ang. CCCTC-binding factor) AP1 (ang. activator protein 1) ERβ (ang. estrogen receptor beta) IK2 (ang. I kappaB kinase-like 2)
aktywator aktywator represor
aktywator aktywator
aktywator represor
aktywator/represor aktywator aktywator

Telomeraza – struktura i funkcja oraz regulacja ekspresji genu 316
Tabela 3. Zestawienie czynników regulujących ekspresję genu TERT [wg 68]. (cd)
Czynnik regulujący i szlak komórkowy, w którym bierze udział
Funkcja związane z regulacją
transkrypcji genu TERT
Inne (cd)
INHBC (ang. inhibin beta C chain) MAD3 (ang. mitotic spindle assembly checkpoint protein MAD3) MAZ (ang. Myc-associated zinc finger protein) Menin MNT (ang. MAX binding protein) MyoD (ang. myoblast determination protein) MZF-2 (ang. myeloid zinc finger protein 2) NF1 (ang. nuclear factor 1) NFAT (ang. nuclear factor of activated T-cells) NF-E2 [ang. nuclear factor (erythroid-derived 2)] PITX1 (ang. paired-like homeodomain 1) Sp1 (ang. specificity protein 1) USF1/USF2 (ang. upstream stimulatory factor 1/2) WT1 (ang. Wilms tumor)
aktywator represor
aktywator represor represor
aktywator/represor represor
aktywator aktywator aktywator represor
aktywator aktywator/represor
represor
W Tabeli 3 zestawiono czynniki oddziałujące z sekwencjami regulatoro-
wymi genu TERT, wzmacniające oraz wyciszające jego ekspresję. Czynniki
podzielono na grupy odnośnie do ich udziału w szlakach komórkowych [68].
Szczegółowo opisany został mechanizm regulacji ekspresji genu TERT z udzia-
łem czynnika transkrypcyjnego c-Myc. Białko to jest produktem protoonkogenu
c-MYC i należy do klasy białek posiadających domenę typu helisa-pętla-helisa
(HLH; ang. helix-loop-helix). Czynnik ten oddziałuje z DNA wyłącznie w po-
staci kompleksu z białkiem Max. W przypadku negatywnej regulacji trans-
krypcji z udziałek białka c-Myc, konieczne jest dodatkowe oddziaływanie czyn-
nika transkrypcyjnego Max (ang. myc-associated factor X) z białkiem jądrowym
Mad (ang. MAX dimerization protein). W komórkach nowotworowych

M. Bryś i wsp. 317
konsekwencją tego procesu jest przesunięcie równowagi w kierunku kompleksu
Myc-Max, co prowadzi do ciągłych podziałów komórkowych [69].
Aktywacja ekspresji genu TERT wymaga oddziaływania z DNA, w obrębie
regionu promotorowego, zarówno czynników modyfikujących strukturę
chromatyny, jak i podstawowych czynników transkrypcyjnych. W przypadku
regulacji ekspresji genu TERT za pośrednictwem białka c-Myc,
zidentyfikowano poza kompleksem Mad-Max, białka koregulatorowe takie jak,
koaktywatory transkrypcji o aktywności acetylotransferazy histonów
(p300/CBP; (ang. CREB binding protein and binding protein p300), kompleks
STAGA (SPT3-TAF9-GCN5-acetylaza; SPT3; ang. suppressor of Ty 3, TAF9;
ang. TBP-associated factor 9, GCN5, acetylase) pełniący funkcję koaktywatora
i kompleks Mediator (kompleks białkowy oddziałujący z polimerazą RNA II)
[70-74]. Szczegółowy model składania kompleksu preinicjacyjnego na
promotorze genu TERT, przy udziale czynnika transkrypcyjnego c-Myc,
przedstawia rycina 12.
Czynniki transkrypcyjne, których udział w regulacji ekspresji genu TERT
został stwierdzony, podlegają wzajemnym oddziaływaniom (ang. cross talk).
W wyniku tych procesów mogą one osłabiać lub wzmacniać swoją aktywność
[77]. Rycina 13 przedstawia hipotetyczny schemat wzajemnego oddziaływania
i regulacji białek uczestniczących w szlakach transmisji sygnału z udziałem
estrogenów, czynnika NFkB oraz transformującego czynnika wzrostu β
(TGF-β).
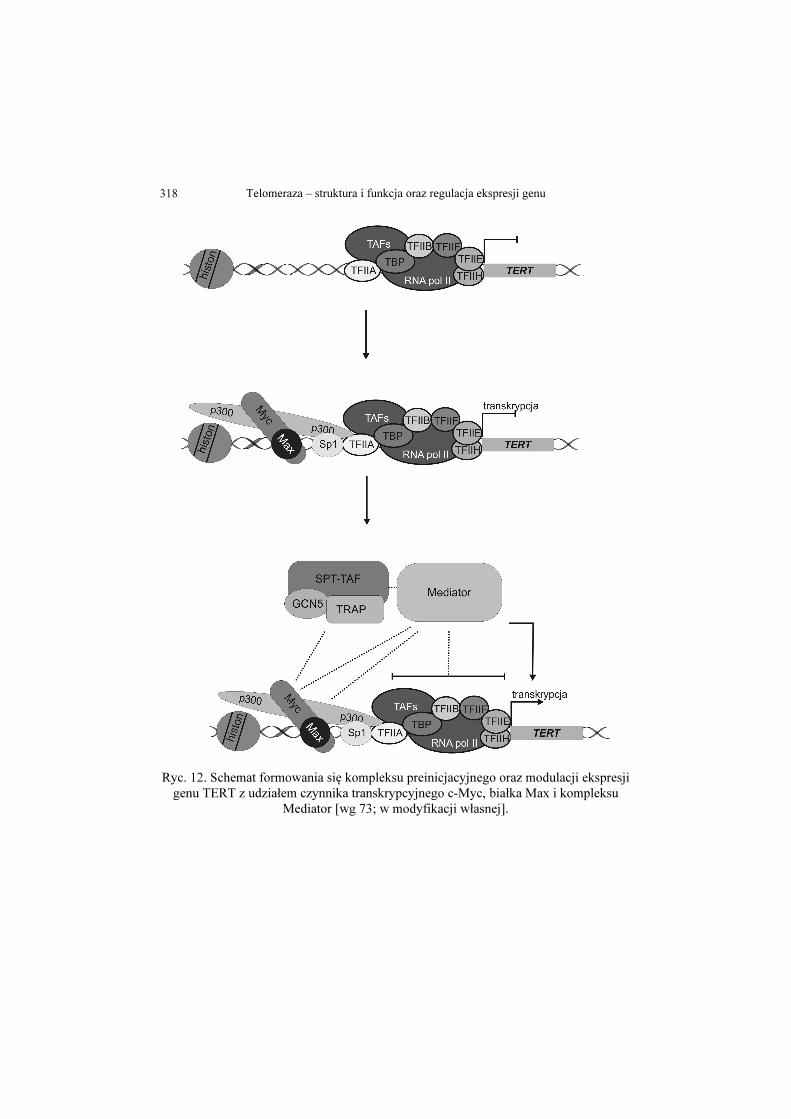
Telomeraza – struktura i funkcja oraz regulacja ekspresji genu 318
Ryc. 12. Schemat formowania się kompleksu preinicjacyjnego oraz modulacji ekspresji genu TERT z udziałem czynnika transkrypcyjnego c-Myc, białka Max i kompleksu
Mediator [wg 73; w modyfikacji własnej].

M. Bryś i wsp. 319
CBP/p300 - koaktywatory transkrypcji o aktywności acetylotransferazy histonów
(ang. CREB binding protein and binding protein p300); c-Myc - czynnik transkrypcyjny
(ang. cellular homologue to the transforming sequences of the avian myelocytomatosis retrovirus);
CREB - czynnik transkrypcyjny oddziałujący z sekwencją CRE aktywowany w wyniku fosforylacji przez PKA (ang. cAMP response element binding);
GCN5 - acetylotransferaza histonów; Max - czynnik transkrypcyjny (ang. myc-associated factor X); p300/CBP - koaktywatory transkrypcji o aktywności acetylotransferazy histonów (ang. CREB
binding protein and binding protein p300); SPT3 - białko wchodzące w skład kompleksu STAGA (ang. suppressor of Ty 3); STAGA - kompleks białkowy o funkcji acetylazy histonów i aktywatora transkrypcji (ang.
SPT-ADA-GCN5 acetylase); TAF - czynnik oddziałujący z białkiem TBP (ang. TBP-associated factor); TATA - sekwencja DNA znajdująca się obszarze promotorowym genów eukariotycznych
(ang. TATA-box); TBP - białko łączące się z sekwencją TATA (ang. TATA-box binding protein); TF - podstawowy czynnik transkrypcyjny (ang. transcription factor); TRRAP - białko o aktywności fosfatazy (ang. tartrate-resistant acid phosphatase).

Telomeraza – struktura i funkcja oraz regulacja ekspresji genu 320
Ryc. 13. Hipotetyczny schemat wzajemnego oddziaływania i regulacji białek uczestniczących w szlakach transmisji sygnału z udziałem estrogenów, czynnika NFκB oraz transformującego czynnika wzrostu β (TGF-β) [wg 77; w modyfikacji własnej].
IL-6 – interleukina 6 (ang. interleukin 6); TAK – kinaza aktywowana przez TGF (ang. TGF-activated kinase); FOXL2 – czynnik transkrypcyjny (ang. forkhead box protein L2).
Udział telomerów i telomerazy w transformacji nowotworowej
Związek pomiędzy telomerami, telomerazą a starzeniem komórkowym
i transformacją nowotworową wydaje się być oczywisty, mimo to, nadal jest
trudny do wyjaśnienia. Ekspresja telomerazy sprzyja nieograniczonej proliferacji
komórek i jest jedną z cech nowotworów [4, 35]. W większości komórek

M. Bryś i wsp. 321
somatycznych człowieka stwierdza się aktywność telomerazy poniżej granicy
wykrywalności, podczas gdy nowotwory złośliwe cechują się wysoką
aktywnością tego enzymu. Ulega ona ekspresji w prawie 90% nowotworów
człowieka (Tabela 4.).
Tabela 4. Aktywność telomerazy w wybranych nowotworach człowieka [wg 75].
UKŁAD NERWOWY
mózg (prawidłowy) 0% siatkówczak (łac. retinoblastoma) 50% glejak wielopostaciowy (łac. glioblastoma multiforme) 75% skąpodrzewiak (łac. oligodendroglioma) 100% gwiaździak anaplastyczny (łac. astrocytoma anaplasticum) 100% oponiaki (łac. meningiomas) 17-100% nerwiak płodowy (łac. neuroblastoma) 94%
GŁOWA I SZYJA
błona śluzowa jamy ustnej (prawidłowa) 3% rak kolczystokomórkowy skóry (łac. carcinoma spinocellulare) 86%
PŁUCA
rak płaskonabłonkowy drobnokomórkowy (łac. carcinoma planoepitheliale microcellulare) 100% rak płaskonabłonkowy niedrobnokomórkowy (łac. carcinoma planoepitheliale non microcellulare) 86%
GRUCZOŁ PIERSIOWY
tkanka prawidłowa 0% gruczolakowłókniak (łac. fibroadenoma) 28% rak nieinwazyjny przewodowy (łac. carcinoma ductale in situ) 75% rak przewodowy lub zrazikowy (łac. carcinoma ductale or lobulare) 88%
WĄTROBA
tkanka prawidłowa 0% nowotwory łagodne 29% rak pierwotny (łac. carcinoma hepatocellulare) 86%

Telomeraza – struktura i funkcja oraz regulacja ekspresji genu 322
Tabela 4. Aktywność telomerazy w wybranych nowotworach człowieka [wg 75]. (cd)
UKŁAD POKARMOWY
gruczolak żołądka (łac. adenoma ventriculi,) 27% rak żołądka (łac. carcinoma ventriculi,) 86% polip gruczolakowaty (łac polypus adenomatosus) 48%
rak jelita grubego (łac. carcinoma coli et recti) 85% rak trzustki (łac. carcinoma pancreatis) 95%
UKŁAD ROZRODCZY - KOBIETY
jajniki 33% błona śluzowa trzonu macicy (prawidłowa) 0% mięśniak gładkokomórkowy (łac. leiomyoma) 0% mięsak gładkokomórkowy (łac. leiomyosarcoma) 100% rak endometrium (łac. carcinoma endometriale) 100% rak jajnika (łac. carcinoma ovarii) 91%
UKŁAD ROZRODCZY - MĘŻCZYŹNI
jądra (łac. testis) 100% gruczoł krokowy (łac. prostata) 0% łagodny rozrost gruczołu krokowego (łac. hyperplasia prostatae) 5% rak gruczołu krokowego (łac. carcinoma prostatae) 90%
SKÓRA
naskórek (łac. epidermis) 44% czerniak (łac. melanoma malignum) 86%
Wysoka aktywność telomerazy może prowadzić do tzw. „unieśmiertel-
nienia” komórki i/lub umożliwić jej nieograniczoną proliferację. W przypadku
braku aktywności tego enzymu, telomery ulegają skracaniu w każdej rundzie
podziału komórkowego, aż do osiągnięcia długości progowej [76].
Nowotwory złośliwe są chorobami charakteryzującymi się niekontrolowaną
proliferacją oraz naciekaniem otaczających tkanek lub odległych narządów.
Większość komórek somatycznych człowieka nie wykazuje aktywności
telomerazy, ma ograniczoną żywotność i w celu wydłużenia długości ich życia
konieczne jest uaktywnienie telomerazy. Doniesienia literaturowe sugerują, że
telomeraza może sprzyjać procesowi transformacji nowotworowej niezależnie

M. Bryś i wsp. 323
od długości telomerów [33, 78]. Poprzez stabilizację długości telomerów
i stymulowanie nieograniczonych podziałów komórek nowotworowych,
telomeraza odgrywa kluczową rolę w etiologii i progresji nowotworów. Uważa
się, iż telomeraza może być aktywowana na różnych etapach procesu
nowotworowego. W niektórych przypadkach ekspresja tego enzymu może być
obserwowana w stanach przednowotworowych, podczas gdy w innych
przypadkach telomeraza może być aktywowany dopiero na etapie progresji
procesu nowotworowego. Różnice te mogą w bardzo istotny sposób rzutować na
kliniczną użyteczność telomerazy jako markera nowotworowego oraz określenie
czy enzym ten może być wykorzystany w przesiewowych badaniach pacjentów
z grupy wysokiego ryzyka, w diagnostyce wczesnego wykrywania, czy też do
celów prognostycznych [79]. Pomimo, iż w większości komórek somatycznych
obserwuje się brak aktywności telomerazy, to jednak niektóre tkanki posiadają
wyspecjalizowane komórki, takie jak limfocyty, komórki rozrodcze, komórki
nabłonkowe, które wykazują niewielki poziom aktywności tego enzymu.
W tkankach zawierających wymienione wyżej komórki, wykrywanie
aktywności telomerazy jest utrudnione i wymaga precyzyjnego określenia czy
aktywność enzymu pochodzi z prawidłowych, telomerazowo-dodatnich
komórek, czy też z komórek zmienionych nowotworowo. Jak już zostało
wspomniane, aktywność ludzkiej telomerazy uwarunkowana jest obecnością
dwóch głównych składowych - fragmentu nici RNA (hTERC) oraz podjednostki
o aktywności odwrotnej tranksryptazy (hTERT). Prowadzone obecnie badania
naukowe i kliniczne koncentrują się na osobnej analizie ekspresji tych dwóch
elementów, jako doskonalszych zamienników w stosunku do oznaczania
aktywności telomerazy [52, 80].

Telomeraza – struktura i funkcja oraz regulacja ekspresji genu 324
Podsumowanie
Poznanie struktury i funkcji telomerów oraz telomerazy przyczyniło się do
opracowania nowych form leczenia przeciwnowotworowego. Przewiduje się, że
blokowanie aktywności telomerazy zaowocuje w niedalekiej przyszłości
opracowaniem skuteczniejszych leków dla pacjentów cierpiących z powodu
chorób nowotworowych, jak również rozszerzy możliwość ich chemoprewencji.
Najbardziej efektywne wydaje się zastosowanie kombinacji klasycznych
chemioterapeutyków z bezpośrednimi inhibitorami aktywności telomerazy, czy
związkami blokującymi oddziaływania telomeraza-telomer. Dzięki takiemu
postępowaniu terapeutycznemu być może uda się zmniejszyć dawki
stosowanych leków, a tym samym zmniejszyć skutki uboczne terapii.
Piśmiennictwo
1. Zvereva MI, Shcherbakova DM, Dontsova OA. Telomerase: structure, functions, and activity regulation. Biochemistry. 2010; 75: 1563-1583.
2. Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL i wsp. Specific association of human telomerase activity with immortal cells and cancer. Science. 1994; 266: 2011-2015.
3. Meyerson M, Counter CM, Eaton EN, Ellisen LW, Steiner P, Caddle SD i wsp. Hest2, the putative human telomerase catalytic subunit gene, is up-regulated in tumor cells and during immortalization. Cell. 1997; 90:785–795.
4. Sikora E. Telomery i telomeraza — starzenie komórkowe nagroda nobla z fizjologii lub medycyny. Kosmos. 2009; 59: 286–287.
5. Wright WE, Shay JW. Telomere biology in aging and cancer. J Am Geriatr Soc. 2005; 53: 292-294.
6. Kowalska A, Kowalik A. Telomer i telomeraza w ontogenezie. Współczesna Onkologia. 2006; 10: 485–496.
7. Shay JW, Wright WE. Telomerase therapeutics for cancer: challenges and new directions. Nat Rev Drug Discov. 2006; 5: 577-584.
8. Cheung AL, Deng W. Telomere dysfunction, genome instability and cancer. Front Biosci. 2008; 13: 2075–2090.

M. Bryś i wsp. 325
9. McGrath M, Wong JY, Michaud D, Hunter DJ, De Vivo I. Telomere length, cigarette smoking, and bladder cancer risk in men and women. Cancer Epidemiol Biomarkers Prev. 2007; 16:815–819.
10. Allshire RC, Dempster M, Hastie ND. Human telomeres contain at least three types of G-rich repeat distributed non-randomly. Nucleic Acids Res. 1989; 17: 4611–4627.
11. Blackburn EH, Gall JG. A tandemly repeated sequence at the termini of the extrachromosomal ribosomal RNA genes in Tetrahymena. J Mol Biol. 1978; 120:33–53.
12. Griffith JD, Comeau L, Rosenfield S, Stansel RM, Bianchi A, Moss H i wsp. Mammalian telomeres end in a large duplex loop. Cell. 1999; 97: 503–514.
13. Moyzis RK, Buckingham JM, Cram LS, Dani M, Deaven LL, Jones MD i wsp. A highly conserved repetitive DNA sequence, (TTAGGG)n, present at the telomeres of human chromosomes. Proc Natl Acad Sci U S A. 1988; 85: 6622-6626.
14. Witzany G. Viral origins of telomeres and telomerases. And their important role in eukaryogenesis and genome maintenance. Biosemiotics. 2008; 1: 191.
15. Aubert G, Lansdorp PM. Telomeres and aging. Physiol Rev. 2008; 88: 557-579. 16. Marión RM, Blasco MA. Telomeres and telomerase in adult stem cells and
pluripotent embryonic stem cells. Advexpmed Biol. 2010; 695:118-131. 17. Bryan TM, Baumann P. G-quadruplexes: from guanine gels to chemo-
therapeutics. Mol Biotechnol. 2011; 49: 198-208. 18. Oeseburg H, de BoerR A, van Gilst WH, van der Harst P. Telomere biology in
healthy aging and disease. Eur J Physiol. 2010; 459: 259–268. 19. Songyang Z. Introduction to telomeres and telomerase. Methods Mol Biol. 2011;
735: 1-11. 20. Oganesian L, Moon IK, Bryan TM, Jarstfer MB. Extension of G-quadruplex
DNA by ciliate telomerase. EMBO J. 2006; 25: 1148-1159. 21. Oganesian L, Karlseder J. Telomeric armor: the layers of end protection. J Cell
Sci. 2009; 122: 4013-4025. 22. Olaussen KA, Dubrana K, Domont J, Spano JP, Sabatier L, Soria JCh. Telomeres
and telomerase as a targets for anticancer drug development. Crit Rev in Oncol/Hematol. 2006; 57:191-214.
23. De Lange T. T-loops and the origin of telomeres. Nat Rev Mol Cell Biol. 2004; 5: 323–329.

Telomeraza – struktura i funkcja oraz regulacja ekspresji genu 326
24. Akbay EA, Peña CG, Ruder D, Michel JA, Nakada Y, Pathak S i wsp. Cooperation between p53 and the telomere-protecting shelterin component Pot1a in endometrial carcinogenesis. Oncogene. 2012; 11. doi: 10.1038/onc.2012.232.
25. O’Sullivan RJ, Karlseder J. Telomeres: protecting chromosomes against genome instability. Nat Rev Mol Cell Biol. 2010; 11: 171–181.
26. Stewart JA, Chaiken MF, Wang F, Price CM. Maintaining the end: roles of telomere proteins in end-protection, telomere replication and length regulation. Mutat Res. 2012; 730: 12-19.
27. Xin H, Liu D, Songyang Z. The telosome/shelterin complex and itsfunctions. Genome Biol. 2008; 9:232.1-232.7.
28. Bayne S, Liu JP. Hormones and growth factors regulate telomerase activity in ageing and cancer. Mol Cell Endocrinol. 2005; 240:11–22.
29. Martinez P, Blasco MA. Telomeric and extra-telomeric roles for telomerase and the telomere-binding proteins. Nat Rev Cancer. 2011; 11: 161-176.
30. Bianchi A, de Lange T. Ku binds telomeric DNA in vitro. J Biol Chem. 1999; 274: 21223-21227.
31. Van Steensel B, de Lange T. Control of telomere length by the human telomeric protein TRF1. Nature. 1997; 385:740-743.
32. De Lange T. How telomeres solve the end-protection problem. Science. 2009; 326: 948-952.
33. Deng Y, Chang S. Role of telomeres and telomerase in genomic instability, senescence, and cancer. Lab Invest. 2007; 87: 1071–1076.
34. Urquidi V, Tarin D, Goodison S. Role of telomerase.in cell senescense and oncogenesis. Annu Rev Med. 2000; 51: 65-79.
35. Artandi SE, DePinho RA. Telomeres and telomerase in cancer. Carcinogenesis. 2010; 31: 9–18.
36. McNees CJ, Tejera AM, Martinez P, Murga M, Mulero F, Fernandez-Capetillo O i wsp. ATR suppresses telomere fragility and recombination but is dispensable for elongation of short telomeres by telomerase. J Cell Biol. 2010; 188: 639–652.
37. Zhu H, Belcher M, van der Harst P. Healthy aging and disease: role for telomere biology? Clinical Science. 2011; 120: 427–440.
38. Lingner J, Cooper JP, Cech TR. Telomerase and DNA end replication: no longer a lagging strand problem? Science. 1995; 269: 1533–1534.
39. Zhong Z-H, Jiang W-Q, Cesare AJ, Neumann AA, Wadhwa R, Reddel R R. Disruption of telomere maintenance by depletion of the MRE11/RAD50/NBS1 complex in cells that use alternative lengthening of telomeres. J Biol Chem. 2007; 282: 29314-29322.

M. Bryś i wsp. 327
40. Georgin-Lavialle S, Aouba A, Mouthon L, Londono-Vallejo JA, Lepelletier Y, Gabet AS i wsp. The telomere/telomerase system in autoimmune and systemic immune-mediated diseases. 2010; 9: 646-651.
41. Azzalin CM, Reichenbach P, Khoriauli L, Giulotto E, Lingner J. Telomeric repeat containing RNA and RNA surveillance factors at mammalian chromosome ends. Science. 2007; 318:798–801.
42. Baur JA, Zou Y, Shay JW, Wright WE. Telomere position effect in human cells. Science. 2001; 292: 2075–2077.
43. Podlevsky JD, Chen JJL. It all comes together at the ends: Telomerase structure, function, and biogenesis. Mutat Res. 2012; 730:3-11.
44. Wyatt HD, West SC, Beattie TL. In TERT preting telomerase structure and function. Nucleic Acids Res. 2010; 38: 5609-5622.
45. Philippi C, Loretz B, Schaefer UF, Lehr CM. Telomerase as an emerging target to fight cancer — Opportunities and challenges for nanomedicine. J Control Release. 2010; 146: 228–240.
46. Chen JL, Blasco MA, Greider CW. Secondary structure of vertebrate telomerase RNA. Cell. 2000; 100: 503-514.
47. Dandjinou AT, Levesque N, Larose S, Lucier JF, Abou Elela S, Wellinger RJ. A phylogenetically based secondary structure for the yeast telomerase RNA. Curr Biol. 2004; 14: 1148-1158.
48. Romero DP, Blackburn EH. A conserved secondary structure for telomerase RNA. Cell. 1991; 67:343-353.
49. Zappulla DC, Cech TR. Yeast telomerase RNA: a flexible scaffold for protein subunits. Proc Natl Acad Sci USA. 2004; 101: 10024-10029.
50. Feng J, Funk WD, Wang SS, Weinrich SL, Avilion AA, Chiu CP i wsp. The RNA component of human telomerase. Science. 1995; 269: 1236–1241.
51. Autexier C, Lue NF. The structure and function of telomerase reverse transcriptase. Annu Rev Biochem. 2006; 75: 493–517.
52. Avendaño C, Menéndez JC. Medicinal Chemistry of Anticancer Drugs. Elsevier, Amsterdam, 2008.
53. Collins K. The biogenesis and regulation of telomerase holoenzymes. Nat Rev Mol Cell Biol. 2006; 7: 484–494.
54. Wyatt HD, Lobb DA, Beattie TL. Characterization of physical and functional anchor site interactions in human telomerase. Mol Cell Biol. 2007; 27:3226-3240.
55. Nicholls C, Li H, Wang JQ, Liu JP. Molecular regulation of telomerase activity in aging. Protein Cell. 2011; 2:726–738.

Telomeraza – struktura i funkcja oraz regulacja ekspresji genu 328
56. Blackburn EH. Switching and signaling at the telomere. Cell. 2001; 106: 661-673.
57. Horikawa L, Oshimura M, Barrett JC. Repression of the telomerase catalytic subunit by a gene on human chromosome 3 that induces cellular senescence. Mol Carcinog. 1998; 22: 65-72.
58. Kyo S, Takakura M, Kanaya T, Zhuo W, Fujimoto K, Nishio Y i wsp. Estrogen activates telomerase. Cancer Res. 1999; 59: 5917–5922.
59. Nakamura TM, Morin GB, Chapman KB, Weinrich SL, Andrews WH, Lingner J i wsp. Telomerase catalytic subunit homologs from fission yeast and human. Science. 1997; 277: 955–959.
60. Nakayama J, Tahara H, Tahara E, Saito M, Ito K, Nakamura H i wsp. Telomerase activation by hTRT in human normal fibroblasts and hepatocellular carcinomas. Nat Genet. 1998; 18: 65–68.
61. Weinrich Sl, Pruzan R, Ma L, Ouellette M, Tesmer Vm, Holt Se i wsp. Reconstitution of human telomerase with template RNA component hterc and the catalytic proteinsubunit htert. Nat Genet. 1997; 17: 498–502.
62. Liu JP. Studies of the molecular mechanisms in the regulation of telomerase activity. FASEB J. 1999; 13: 2091–2104.
63. Liu JP. Telomerase: Not just black and white, but shades of gray. Mol Cell Biol Res Commun. 2000; 3: 129–135.
64. Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB i wsp. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998; 279: 349–352.
65. Xu D, Gruber A, Bjorkhold M, Peterson C, Pisa P. Suppression of telomerase reverse transcriptase (hTERT) expression in differentiated HL-60 cells: regulatory mechanisms. Br J Cancer. 1999; 80: 1156–1161.
66. Horikawa I, Cable PL, Afshari C, Barrett JC. Cloning and characterization of the promoter region of human telomerase reverse transcriptase gene. Cancer Res. 1999; 59: 826–830.
67. Misiti S, Nanni S, Fontemaggi G, Cong YS, Wen J, Hirte HW i wsp. Induction of hTERT expression and telomerase activity by estrogens in human ovary epithelium cells. Mol Cell Biol. 2000; 20: 3764–3771.
68. Daniel M, Peek GW, Tollefsbol TO. Regulation of the human catalytic subunit of telomerase (hTERT). Gene. 2012; 498: 135-46.
69. Ścibior-Bentkowska D, Czeczot H. Komórki nowotworowe a stres oksydacyjny. Postępy Hig Med Dośw. 2009; 63: 58-72.

M. Bryś i wsp. 329
70. Conaway RC, Conaway JW. Origins and activity of the Mediator complex. Semin Cell Dev Biol. 2011; 22: 729-734.
71. Gamper AM, Kim J, Roeder RG. The STAGA subunit ADA2b is an important regulator of human GCN5 catalysis. Mol Cell Biol. 2009; 29: 266-280.
72. Holmlund T, Lindberg MJ, Grander D, Wallberg AE. GCN5 acetylates and regulates the stability of the oncoprotein E2A-PBX1 in acute lymphoblastic leukemia. Leukemia. 2012; doi: 10.1038/leu.2012.265.
73. Liu X, Vorontchikhina M, Wang YL, Faiola F, Martinez E. STAGA recruits Mediator to the MYC oncoprotein to stimulate transcription and cell proliferation. Mol Cell Biol. 2008; 28: 108-121.
74. Taatjes DJ. The human Mediator complex: a versatile, genome-wide regulator of transcription. Trends Biochem Sci. 2010; 35: 315-322.
75. Granger MP, Wright WE, Shay JW. Telomerase in cancer and aging. Crit Rev Oncol Hematol. 2002; 41: 29-40.
76. .Biroccio A, Leonetti C. Telomerase as a new target for the treatment of hormone-refractory prostate cancer. Endocr Relat Cancer. 2004; 11:407-421.
77. Drummond AE, Fuller PJ. Activin and inhibin, estrogens and NFκB, play roles in ovarian tumourigenesis is there crosstalk? Mol Cell Endocrinol. 2012; 359: 85-91.
78. Heaphy CM, Meeker AK. The potential utility of telomere-related markers for cancer diagnosis. J Cell Mol Med. 2011; 15: 1227-1238.
79. Wanat JJ, Johnson FB. Telomere stability and carcinogenesis: an off-again, on-again relationship. J Clin Invest. 2012; 122: 1962-1965.
80. Elmore LW, Forsythe R, Forsythe H, Bright AT, Nasim S, Endo K i wsp. Overexpression of telomerase-associated chaperone proteins in prostatic intraepithelial neoplasia and carcinomas. Oncol Rep. 2008; 20:613-617.

330
Od Redakcji
Redakcja „Folia Medica Lodziensia” składa serdeczne podziękowania
recenzentom prac nadesłanych w latach 2011-2012 i opublikowanych
w tomach 38 i 39
Magdalena Bryś, Ewa Brzezińska-Błaszczyk,
Jan Brzeziński, Jan Chojnacki,
Adam Cygankiewicz, Marian Danilewicz,
Wojciech Dębek, Andrzej Głąbiński,
Anna Głowacka, Małgorzata Karbownik-Lewińska,
Zofia M. Kiliańska, Józef Kobos,
Jolanta Kunert-Radek, Anna Kreślak,
Marek Lipiński, Zbigniew Maziarz,
Beata Olas, Marek Pawlikowski,
Bogusława Pietrzak, Stanisław Sporny,
Katarzyna Starska, Ewa Toporowska-Kowalska,
Elżbieta Waszczykowska, Anna Zalewska-Janowska,
Wojciech Zieleniewski

331
Folia Medica Lodziensia
Informacje dla autorów
Informacje ogólne
Czasopismo Folia Medica Lodziensia (FML) jest wydawnictwem środowiska
medycznego skupionego wokół Łódzkiego Towarzystwa Naukowego, w którym
publikowane są oryginalne prace badawcze (kliniczne i doświadczalne) i artykuły
poglądowe z zakresu nauk medycznych, farmaceutycznych i biologicznych.
Pierwotna wersja czasopisma jest drukowana. Bieżące i archiwalne
numery FML dostępne są w internecie: www.ltn.lodz.pl/index.php?option=
com_content&view=article&id=97&Itemid=64
Zasady recenzowania prac
Każda praca nadesłana do Redakcji jest oceniana pod względem wartości
merytorycznych takich jak: nowatorskie przedstawienie tematu, znaczenia dla
rozwoju badań naukowych i postępowania klinicznego. Manuskrypty oceniane
są wstępnie przez Redakcję FML. Prace niekompletne lub nie spełniające
warunków publikacji ustalonych przez Redakcję są zwracane Autorowi bez
oceny merytorycznej. Po nadesłaniu pracy Autor otrzymuje potwierdzenie jej
wpłynięcia i numer referencyjny manuskryptu, którym powinien posługiwać się
przy kontaktach z Redakcją. Autor pracy może zaproponować nazwiska
recenzentów, jednakże prawo ostatecznego ich wyboru należy do Redakcji.
Przyjęcie pracy do druku odbywa się na podstawie pozytywnych opinii
recenzentów. Redakcja nie informuje autora o nazwiskach recenzentów.
Zasady przygotowania manuskryptu
Zasady publikacji zgłaszanych do druku artykułów opracowano na podstawie
International Committee of Medical Journal Editors - Uniform Requirements for
Manuscripts Submitted to Biomedical Journals (http://www.icmje.org/). Prace

332
zawierające wyniki badań przeprowadzonych na ludziach i/lub zwierzętach
powinny posiadać pozytywną opinię odpowiedniej lokalnej Komisji Etycznej.
Folia Medica Lodziensia ogłasza prace w języku polskim i angielskim.
W pracach oryginalnych preferowany jest język angielski, a w pracach
poglądowych język polski.
Po uprzednim uzgodnieniu z Redakcją istnieje także możliwość publikowania
monografii (np. prac habilitacyjnych) na koszt Autora. Monografie również
podlegają recenzji.
Objętość prac oryginalnych nie powinna przekraczać 3000 słów, a prac
poglądowych (pisanych w zasadzie na zamówienie Redakcji) 6000 słów.
Z limitu wyłączone jest piśmiennictwo, tabele, ryciny i streszczenie. Manuskrypt
powinien być napisany standardową jednolitą czcionką (Times New Roman lub
Arial) o rozmiarze 12, z podwójnym odstępem na arkuszu formatu A4, zawierać
27-30 wierszy na stronie z 2,5 cm marginesami z każdej strony. Tekst winien
być pisany bez żadnych wyróżnień (podkreśleń, spacjowania, pisania dużymi
literami). Nie należy stosować przenoszeń wyrazów.
Struktura nadsyłanych prac
Każda praca powinna zawierać stronę tytułową, streszczenie, właściwy tekst
pracy, piśmiennictwo oraz w zależności od potrzeby ryciny i tabele.
Podziękowania i dodatkowe informacje (np. źródło finansowania) można
umieszczać po właściwym tekście pracy przed spisem piśmiennictwa. Strona
tytułowa powinna zawierać tytuł pracy, imiona i nazwiska wszystkich Autorów,
pełną nazwę instytucji, w której praca została wykonana, skrócony tytuł
(maksimum 45 znaków), listę 5-8 słów lub wyrażeń kluczowych zgodnych
z Medical Subject Headings Index Medicus oraz imię i nazwisko Autora
odpowiedzialnego za korespondencję, a także jego adres, numer telefonu i faksu
oraz adres e-mailowy. Ponadto, należy umieścić informację o grantach i innych
źródłach finansowania oraz aktualne miejsce pracy Autorów.

333
W każdej pracy (zarówno napisanej w języku polskim jak i w języku
angielskim) tytuł pracy, treść streszczenia, słowa i/lub wyrażenia kluczowe oraz
objaśnienia do rycin i tabel powinny być napisane w wersji polskiej
i angielskiej.
Streszczenie winno zawierać od 200 do 250 słów. W streszczeniu pracy
oryginalnej należy wyodrębnić następujące części składowe: wstęp
(zawierający cel pracy), materiały i metody, wyniki, wnioski. Treść
streszczenia powinna umożliwić zorientowanie się w tematyce pracy bez
szczegółowego czytania całego tekstu. Streszczenie wymagane jest także do
pracy poglądowej.
W pracach oryginalnych właściwy tekst powinien zawierać następujące
części: wstęp, materiały i metody, wyniki, dyskusję i piśmiennictwo.
W tekście prac poglądowych należy wyróżnić wstęp, właściwe omówienie
tematu z podziałem na szczegółowe zagadnienia oraz wnioski końcowe lub
podsumowanie.
Piśmiennictwo w pracy powinno być cytowane przez umieszczenie numeru
pozycji w nawiasach zgodnie z kolejnością cytowania w tekście. Spis
piśmiennictwa powinien zawierać pozycje w kolejności cytowania. Pozycja
będąca pracą oryginalną winna zawierać nazwisko autora (autorów), inicjały
imion, tytuł pracy, skrót nazwy czasopisma wg Index Medicus, rok wydania;
tom: strony (od-do). W przypadku więcej niż sześciu autorów należy podać
nazwiska pierwszych sześciu i skrót et al. (lub w wersji polskojęzycznej
- i wsp.) W przypadku monografii należy podać nazwisko autora (autorów),
inicjały imion, tytuł monografii, wydawcę, miejsce i rok wydania. W przypadku
dzieł zbiorowych należy podać nazwiska autorów, inicjały imion, tytuł pracy
(rozdziału). W: tytuł monografii, nazwisko i inicjały imion redaktora
(redaktorów), wydawcę, miejsce i rok wydania: strony (od-do).

334
Przykłady:
Artykuł w czasopiśmie:
1. Axelrod J. The pineal gland: a neurochemical transducer. Science. 1974; 184:
1341-1348.
2. Frisch RE, Wyshak G, Yincent L. Delayed menarche and amenorrhea in
ballet dancers. New Engl J Med. 1980; 303: 17-19.
3. Lardone PJ, Carrillo-Vico A, Naranjo MC, De Felipe B, Vallejo A, Karasek
M et al. Melatonin synthesized by Jurkat human leukemic T cell line is
implicated in IL-2 production. J Cell Physiol. 2006; 206: 273-279.
Monografia:
4. Kokot P, Stupnicki R. Metody radioimmunologiczne i radiokompetycyjne
stosowane w klinice. PZWL, Warszawa, 1985.
Rozdział w książce:
5. Landolt AM, Strebel P. Technique of transsphenoidal operation of pituitary
adenomas. W: Advances and technical standards in neurosurgery, Vol. 7,
Krayenbuhl K. (red.), Springer, Vienna-New York, 1980: 119-177.
Tabele i ryciny
Tabele winny być pisane bez wyróżnień, wytłuszczeń i zmiany rodzaju czcionki,
każda na oddzielnej stronie i numerowane od góry cyframi arabskimi. Opis do
tabeli (w języku polskim i angielskim) winien być umieszczony w oddzielnym
wierszu nad tekstem tabeli.
Ryciny winny być wykonane techniką komputerową w postaci wektorowej lub
rastrowej, względnie stanowić wysokiej jakości fotografie (czarno-białe – min.
300 dpi; kolorowe – min. 600 dpi).
Przekazanie praw autorskich i pozwolenie na druk
Przesłanie manuskryptu do Redakcji jest równoznaczne ze stwierdzeniem, że
praca nie była uprzednio publikowana, ani nie została złożona do oceny

335
w innym czasopiśmie (z wyłączeniem streszczeń nie przekraczających 250
słów). Jeżeli praca zawiera elementy (np. ryciny, tabele itp.) uprzednio
publikowane należy dołączyć pisemną zgodę na ponowne ich wydanie, zarówno
od poprzedniego wydawcy, jak i autorów oryginalnej pracy. Ponadto wszyscy
Autorzy pracy oświadczają, że po zaakceptowaniu materiałów do publikacji
w FML automatycznie i nieodpłatnie przekazują Wydawcy wszelkie prawa
autorskie do wydawania i rozpowszechniania nadesłanych materiałów we
wszystkich znanych formach eksploatacji. Autor korespondujący zobowiązany
jest do podpisania w imieniu własnym i współautorów pracy Umowy
o przeniesieniu praw autorskich na Wydawcę FML - Łódzkie Towarzystwo
Naukowe (wzór umowy na stronie www.ltn.lodz.pl/index.php?option=
com_content&view =article&id=97&Itemid=64).
Konflikt interesów
Złożeniem manuskryptu jest równoznaczne z ujawnieniem wszystkich zobo-
wiązań finansowych oraz wszelkich niezgodności interesów związanych z pracą
badawczą.
Dostarczenie manuskryptu do Redakcji
Prosimy o przesyłanie prac za pomocą poczty elektronicznej na adres
[email protected]. Po uzyskaniu wstępnej akceptacji należy
dostarczyć 3 wydrukowane egzemplarze pracy (1 oryginał i 2 kopie) oraz tekst,
tabele i ryciny zapisane na nośniku CD-ROM. Każda rycina powinna być
wydrukowana na oddzielnej stronie i winna być podpisana nazwiskiem Autora
korespondującego oraz kolejnym numerem z zaznaczeniem góry ryciny.
Na oddzielnej stronie należy dołączyć podpisy do rycin.
Do materiałów ilustracyjnych poprzednio publikowanych powinna być
dołączona pisemna zgoda Wydawcy na ponowną publikację.
Do manuskryptu należy dołączyć List przewodni do Redakcji stwierdzający, że:
− praca nie została opublikowana ani obecnie nie została złożona do innej
redakcji;

336
− wszyscy Autorzy pracy wyrażają zgodę na jej publikację;
− w chwili zaakceptowania manuskryptu do druku Autorzy zobowiązują się
do podpisania Umowy o przeniesieniu praw autorskich na Wydawcę;
− Autorzy zapoznali się z zasadami publikacji prac ogłaszanych w Folia
Medica Lodziensia i zobowiązują się do ich przestrzegania;
− Autorzy są gotowi pokryć koszty druku kolorowych ilustracji.
Schemat Listu przewodniego jest dostępny na stronie:
www.ltn.lodz.pl/index.php?option=com_content&view=article&id=97&Itemid=
64
Uwagi końcowe
Redakcja i Wydawca dokładają wszelkich starań, by informacje opublikowane
w FML były wiarygodne i dokładne. Jednakże treści i opinie zawarte
w artykułach i reklamach są publikowane na wyłączną odpowiedzialność
Autorów i/lub Reklamodawców, a Redakcja i Wydawca FML nie ponoszą
odpowiedzialności za publikowane informacje.