· web viewcalcineurin-nfat signaling pathway helps to promote osteoblast differentiation)...
TRANSCRIPT

Giles Kisby GE Y1 Musculoskeletal
Musculoskeletal:
Spring Term:
LECTURES:
07/03/14: Bone Development and Metabolism: Duncan Bassett
Los (from booklet):Define bone structure.Explain the difference between intramembranous and endochondral ossification.Define the role of the osteoblast, osteoclasts and chondrocyte.Recall the bone remodeling cycle in adult bone.Define osteoporosis, list its common causes and outline treatment strategies that may be used.Define Paget’s disease and the bone pathology seen in this disorder.
Los (from slides):1. Contrasting endochondral and intramembranous ossification2. Understand the role of chondrocyte, osteoblasts, osteoclasts and osteocytes3. Describe the bone remodelling cycle4. Contrast the affect of intermittent and continuous PTH on the skeleton5. Define age related osteoporosis and list the common risk factors6. Describe the causes of secondary osteoporosis7. Describe DXA BMD analysis with particular reference to the T and Z scores8. List the main medications used to treat osteoporosis and describe their action9. Compare and contrast the uses of PTH and the bisphosphonates10. Describe the skeletal consequences of long term glucocorticoid treatment11. Describe the indications for treatment of Paget’s disease and the mechanism ofaction of the therapy
Notes1

Giles Kisby GE Y1 Musculoskeletal
- Skeletal Physiologyo Bone Structure
Osteoid: is a protein mixture secreted by osteoblasts that forms the organic matrix of bone. Bone is formed when osteoid mineralizes
There are two types of osseous tissue that form bones: Cortical bone = compact bone:
Forms the cortex, or outer shell, of most bones Osteon: refers to the functional unit of most compact bone
comprised of a central blood vessel [in a “Haversian canal”] and concentric rings of new bone encircling the vessel
o Hydroxyapatite crystals [Ca10(PO4)6(OH)2] pack in between Type 1 collagen fibrils within and between each ring
o Each ring is from successive remodelling cycleso Osteon structure limits fracture propagationo Collagen fibres orientated in all various directions within
each ringo Type I collagen molecule:
Triple helical collagen molecule 2x alpha 1 (COL1A1) and 1x alpha2 (COL1A2)
o 200 non-collagenous proteins are also present in boneo Human bone is 60% mineralised: increased mineralisation
increases stiffness but reduces flexibilityo Cortical bone accounts for 80% of the total bone mass of an
adult skeleton The microscopic difference between compact and cancellous bone is
that compact bone consists of haversian sites and osteons, while cancellous bones do not.
Also, bone surrounds blood in the compact bone, while blood surrounds bone in the cancellous bone
Cancellous bone = trabecular bone = spongy bone: Filling the interior of the bone is the trabecular bone tissue (make
the overall organ lighter and allow room for blood vessels and marro)
Trabecular bone accounts for the remaining 20% of total bone mass but has nearly ten times the surface area of compact bone. Its porosity is 30–90%
Compared to compact bone, cancellous bone has a higher surface area to mass ratio because it is less dense
The primary anatomical and functional unit of cancellous bone is the trabecula.
Cancellous bone is highly vascular and frequently contains red bone marrow where haematopoiesis, occurs.
2

Giles Kisby GE Y1 Musculoskeletal
Cancellous bone is typically found at the ends of long bones, proximal to joints and within the interior of vertebrae.
o Bone development: Long bones form by endochondral ossification
Type II collagen is found in cartilage (tensile strength) Aggrecan (Aggrecan is the major proteoglycan in articular cartilage)
is negative so draws in water for compressive strength CHONDROCYTES ARE THE ONLY CELLS FOUND IN HEALTHY
CARTILAGE; undergo proliferation differentiation hypertrophy (chondrocytes can expand 10x just before they die; helps extend bone) death as they migrate out from center of the bone; this leaves a tunnel through which blood vessels can form; bone can then be laid down from these centers to form cortical bone from these centers (primary ossification center)
o Sox9: Master transcriptional regulator in chondrocyte
3

Giles Kisby GE Y1 Musculoskeletal
o FGF/FGFRs: Inhibit chondrocyte proliferation and differentiation
o Indian hedgehog (Ihh): Promotes chondrocyte proliferation and induces PTHrP
o PTHrP/PTHR1: Inhibit chondrocyte differentiation Secondary ossification center is at the epiphysis growth plate; the
growth plate is supported by trabecular bone and will fuse with the ‘head’ bone only when growth is complete
Achondroplasia:o Cause: Constitutively active FGF/FGFR3 signalling inhibits
chondrocyte proliferation and differentiation Gain of function mutation FGFR3
o Most common form of dwarfism o Macrocephaly [head is abnormally large], frontal bossing,
midface hypoplasia, small chest, rhizomelia [shortened limbs]
Craniofacial bones by intramembranous ossification Fontanelles form Mesenchymal cells differentiate into osteoblasts Bone (osteoblasts) is formed directly without a cartilage scaffold
o Osteoblasts, chondrocytes and adipocytes all derive from mesenchymal cells
o Osteoblasts are formed from the mesenchymal cells in the context of Runx2, Osterix and high B-Catenin[other factors also have pos / neg roles: Wnt, BMPs and FGFsSystemic hormones: GH/IGF1, glucocorticoids, E2: Estradiol, PTH and 1,25(OH)2D]
o Wnt signalling role: Promotes osteoblast differentiation, proliferation
and mineralisation On absence of Wnt
GSK3/APC/Axin degradation complex targets B-catenin for degradation by phosphorylation
On presence of Wnt: Wnt binds Frizzled with co-receptors LRP5/6 and inhibits GSK3
Preventing B-catenin degradation B-catenin enters nucleus regulating target
genes (but is not a TF itself) Negative regulation of Wnt signalling [ie these neg
regulators are targets for new anabolic agents for treatment of osteoporosis]:
4

Giles Kisby GE Y1 Musculoskeletal
Target Wnt: Wnt binding (WIF-1 , cereberus and sFRP)
Target coreceptor: LRP5/6 degradation (Sclerostin (SOST gene in nature) and Dickkopf (Dkk))
Relavant info from endo:
• PTH regulates calcium via PTHR1 in bone:• PTHR1 expressed in osteoblasts and osteocytes but NOT osteoclasts• PTH has anabolic and catabolic actions:
• PTH can stimulate bone resorption or formation• Intermittent PTH (net trabecular formation)• Continuous PTH (net cortical resorption)
• PTH increases osteoclast differentiation indirectly by action in osteoblasts
• Increased expression of M-CSF/RANKL• Reduced expression of OPG
5

Giles Kisby GE Y1 Musculoskeletal
• PTH regulates maturation of preosteoblasts• Intermittent PTH increases Runx2• Continuous PTH represses Runx2
• PTH also increases bone formation by paracrine mechanisms:
• Increased FGF release at osteocytes• Increased signalling for osteoblast lineage:
Increased IGF-1 Increasing Wnt signallingReduced inhibition of Wnt signalling: dec dickkopf and Sclerostin, the product of the SOST gene
o Maintenance of adult bone Terminally differentiated osteoblasts act as Mechanosensors & regulators of
bone remodeling The bone remodelling cycle:
Functions:o Maintain homeostasis of Ca 2+ and PO4o Repair damaged matrix and micro-fractureso Adapt to mechanical stress and strain
Cells involved:o Osteocytes make up 90-95% of all adult bone cellso Osteoblasts 5% of all adult bone cellso Osteoclasts 1-2% of all adult bone cells
Osteocytes regulate bone turnover
o In response to bone loading state During bone loading osteocytes inhibit
osteoclast resorption (via inc OPG and TGFB)
Unloading, hypoxia or apoptosis initiates resorption (via inc RANKL)
o In response to endocrine phosphate needs Low phosphate:
Osteocyte Sclerostin binds LRP5 ( dec Wnt signaling and dec bone formation)
Vs High phosphate:
Osteocyte Matrix extracellular phosphoglycoprotein (MEPE) inhibits phosphate resorption
Osteocyte Dmp1 and Phex decrease leading to inc FGF23 for increase to phosphate levels
6

Giles Kisby GE Y1 Musculoskeletal
Osteoclasts o Development:
Osteoclasts derive from the myeloid lineage; therefore:
Osteoclasts are multinucleated cells M-CSF regulates proliferation, survival and
differentiation of precursors RANKL is key osteoclastogenic cytokine sufficient for
differentiation OPG is a ‘decoy receptor’ (binds and blocks RANKL:
ie physiological inhibitor of RANKL/RANK signaling) [RANK is on the osteoclast precursor]
Denosumab acts like OPG therefore can be used as a drug to reduce bone resorption via dec osteoclast levels
PTH, 1,25(OH)2D and pro-inflammatory cytokines increase RANKL expression and suppress OPG
o Function: Active osteoclasts are polarised cells: Attach to the
bone surface via integrin α5β3 (this forms the sealing zone that degradatory components are secreted into; nb lacunae = an unfilled space; a gap, as created under site of osteoclast binding):
Requires action of small GTPases (inhibited by bisphosphonates)
Secrete hydrogen and chloride ions that dissolve bone mineral
Matrix metalloproteinases (MMPs) and Cathepsin K degrade the collagen matrix: Cathepsin K inhibited by Cathepsin K inhibitors
7

Giles Kisby GE Y1 Musculoskeletal
- Skeletal Pathologyo Osteoporosis
Traits: Increased bone resorption relative to formation Low bone mass Micro-architectural deterioration Fragility fractures Diagnosis of osteoporosis: Fragility fracture and decreased bone
mineral density (BMD) Peak bone mass
Achieved at 20 - 30 years of age major genetic component to peak bone mass Estrogens is critical in both male and females for peak bone mass Peak influenced by: Physical exercise, alcohol excess, smoking,
eating disorders, systemic illness Increased progressive loss of bone mass occurs from 45 years of age;
Mechanism: Estrogens deficiency at the menopause
o More rapid loss in women due to estrogens deficiency at menopause
o Increased expression of skeletal cytokines especially IL-6 inc [“pro-inflammatory cytokines”]
o Reduced expression of OPG and thus increased osteoclastogenesis
Investigationo Ca2+, Pi, ALP [high bone alkaline phosphatase in
osteoporosis], Cre [looking for potential liver problem giving dec hydroxylase activity], PTH, 25-OH-vitD, DEXA, Urinary
8

Giles Kisby GE Y1 Musculoskeletal
NTX [N-terminal telopeptide (NTx) molecules are mobilized from bone by osteoclasts and subsequently excreted in the urine]
o DEXA = Dual-energy X-ray absorptiometry: assesses BMD (Bone mineral density)
Results are interpreted according to the standard deviation from the mean to give T and Z score:
a) Sex matched peak bone mass (T-score) [ie no. SD from those in 20s with a max BMD]
b) Sex and age matched BMD (Z-score) [ie no. SD from those of same age as the person]
WHO diagnostic criteria: Osteoporosis
o T score ≤ - 2.5 lumbar spine, femoral neck or total hip
Osteopeniao T score ≤ - 1.0 lumbar spine, femoral
neck or total hip Treatment of age related osteoporosis
o Simple advice: inc weight bearing exercise, dec smoking and alcohol
o Optimise vitamin D status: Calcium and vitamin D supplementation
o Antiresorptive agents: Bisphosphonates (impaired sealing zone
attachment) Denosumab (monoclonal ab to RANKL) (OPG like
activity) Selective estrogens receptor modulators (inc OPG,
dec IL6)o Anabolic agents
Strontium ranelate (now limited use due to cardiac risk)
Teriparatide (PTH 1-34) (Intermittent PTH: intermittent use activates osteoblasts more than osteoclasts, which leads to an overall increase in bone)
continuous PTH - decrease Runx2 Intermittent - increase runx2
9

Giles Kisby GE Y1 Musculoskeletal
Secondary Osteoporosis Endocrine
o Thyrotoxicosis (increased bone turnover)o Hyperprolactinemia (reduced gonadotrophins and sex
hormones Eg E2!!)o Primary hyperparathyroidism (Increased resorption)o Hypogonadism (increased resorption due to low E2 etc)o Cushing’s Syndrome (impaired bone formation due to high
cortisol) Nutritional [but not severe enough to give osteomalacia]
o Vitamin D deficiency (impaired mineralisation)o Coeliac disease (impaired mineralisation)o Chronic liver disease [ie loss of liver vit d role]
Iatrogenico High dose glucocorticoids (Glucocorticoid induced
osteoporosis)o GnRH agonists (eg for prostate cancer: after about ten days,
a profound hypogonadal effect (i.e. decrease in FSH and LH) is achieved through receptor downregulation by internalization of receptors. Generally this induced and reversible hypogonadism is the therapeutic goal.)
o Aromatase inhibitors (eg for Breast cancer)o Thyroid hormone excess (Excessive replacement or Thyroid
cancer)o Anticoagulants and Anticonvulsants [mech is unknown]o Immunosuppression (inhibits calcineurin and NFAT to give
dec IL2 so dec im response but calcineurin-NFAT signaling pathway helps to promote osteoblast differentiation)
10

Giles Kisby GE Y1 Musculoskeletal
o Thiazolidinediones (PPARϒ agonists; as on prev diagram this is a inducer of the adipocyte lineage: dec osteoblastogenesis, inc adipogenesis)
Glucocorticoid induced osteoporosis in detail [Commonest iatrogenic cause of osteoporosis]:
As prev, inhibits osteoblast differentiation and encourages osteoblast apoptosis:
Also: Decreased osteoclastogenesis but prolonged survival
Treatmento Bisphosphonateso Teriparatide
o Paget’s Disease Traits:
Localised disorder of bone remodelling) Osteoclast abnormality: Increased osteoclast numbers Osteoblast abnormality: Disorganised rapid bone formation
o Replacement by sclerotic boneo Bone marrow cavity replaced by vascular fibrous
connective tissueo Increase in bone size and bone deformity
Increased markers of formation and resorptiono bALP: Bone alkaline phosphataseo P1NP: Procollagen type I N-terminal propeptideo uNTX: Urinary N-terminal telopeptide
Aetiology Predominantly unknown Family history in 15%
11

Giles Kisby GE Y1 Musculoskeletal
Declining in incidence : Reason for decline in frequency is unknown Clinical features
Bone pain, joint pain, deformity, fracture increased temperature [high metabolic activity] Deafness (due to cranial nerve compression) Abnormal x-ray
Complications Osteoarthritis due to deformity Cranial nerve palsy and spinal stenosis [abnormal narrowing
(stenosis) of the spinal canal] Hypercalcaemia if are also immobilised (immobility is associated
with hypercalcaemia in a variety of diseases, as here, but unknown mech
Fracture Osteosarcoma (very rare)
Diagnosis Raised alkaline phosphatase: ALP Abnormal x-ray (osteolysis, osteosclerosis and bone expansion) 99Tc bone scan: is far more sensitive than plain X-ray; is taken up in
the areas of high turnover [Technetium-99m] Treatment: Bone pain is the indication for treatment
Ensure patients are vitamin D and calcium replete Simple analgesia (NSAIDs) Physio/hydrotherapy Bisphosphonates: reduce pain, do not prevent deformity or
deafness Zolendronic acid (Alk Phos normalises in 90%) Surgery for severe deformity or osteoarthritis
12

Giles Kisby GE Y1 Musculoskeletal
07/03/14: Connective tissue and articulations: Matthew Pickering
Los (from slides/booklet):List the main components of the extracellular matrixRecall the principal type of collagen in bone and articular cartilageList the main collagen-cleaving enzymes (collagenases)Explain what is meant by synarthrosis, diarthrosis, amphiarthrosisDescribe the structure of articular cartilage and synoviumList the key features of the heritable collagen disorders:
Osteogenesis imperfectaMarfan’s syndromeEhlers-Danlos syndrome
Notes
- Extracellular matrix [ie are initially just looking generally at ECM during this lec]o ECM functions:
Mechanical: tensile and compressive strength and elasticity Protection: buffering against extracellular change and retention of water Organisation: control of cell behaviour by binding of growth factors and
interaction with cell-surface receptorso ECM contains:
Proteins: Collagen Elastin Fibrillin Adhesin
Proteoglycans: Are proteins bound to glycosaminoglycans (GAGs)
o ECM contains: Proteins:
Collageno 20-30% of body mass: is the most abundant protein in the
human bodyo Types I, II, III, V and XI are the most abundant o Bone = type I collageno Cartilage = type II collagen
13

Giles Kisby GE Y1 Musculoskeletal
o Triple helix: = tropocollagen 3 polypeptide chains twisted into right handed
major helix Type 1 = 2x alpha 1 and 1x alpha2 Type 2 = 3x alpha 1 repeating triplet amino acid Glycine-X-Y 300nm long and 1.5nm wide
o During synthesis polypeptide chains are hydroxylated Enzymes: prolyl hydroxylase and lysine hydroxylase The enzymatic reactions need cofactors including
VITAMIN C In vitamin C deficiency (scurvy) collagen becomes
unstableo Degradation: Collagenases binds to triple helical collagen,
unwinds polypeptide chains and then cleaves -chains by peptide hydrolysis
Certain Matrix metalloproteinases [eg 1/8/13/ gelatinases/stromelysins] are collagenases
o type 1 collagen 2x alpha 1, 1x alpha 2 bone, tendon, ligaments, skin, joint
capsule/synovium, cornea/sclera [= type 5]o type 2 collagen
3x alpha1 cartilage, intervertebral discs, vitreous humour [is
the clear gel that fills the space between the lens and the retina of the eyeball] [= type 11]
o type 3 collagen blood vessels
o type 4 collagen basement membrane, lens capsule [ie is the
basement membrane of the lens]
14

Giles Kisby GE Y1 Musculoskeletal
Elastino Present at ligaments, vessels and skino Elastin is polymer of tropoelastin monomers [lysine
residues in adjacent monomers cross link by forming desmosine]
o Elastin = Fibres that can stretch when hydrated and return to original length after being stretched
o Mutations in elastin gene cause autosomal dominant cutis laxa
Cutis laxa is a group of disorders that share common finding of lax, redundant skin.
skin does not recoil when stretched: ‘appears to have lost elasticity’
Fibrillin
o Present at ligaments, vessels and skino Is a glycoprotein that is essential for formation of elastic
fibres: elastin must be deposited around a fibrillin core therefore loss gives similar phenotype to elastin deficiency
o Fibrillin-1 mutations → Marfan’s syndrome o Fibrillin-2 mutations → Contractural arachnodactyly
Adhesino Is a group term: refers to cell-binding glycoproteins present
in matrix and basement membranes o attach to cells via cell integrinso Include:
Fibronectin in connective tissue Laminin in basement membrane Chondroadherin in cartilage Osteoadherin in bone
15

Giles Kisby GE Y1 Musculoskeletal
Proteoglycans: Are glycoproteins containing one or more sulphated
glycosaminoglycan (GAG) chains GAGs: are repeating polymers of disaccharides [N-acetyl
glucosamine is usually one of the two repeating sugars; the chain is also usually sulfated]:[eg Heparan sulphate, Chondroitin sulphate, Keratan sulphate, Dermatan sulphate, Heparin]
Proteoglycans may be:o Intra-cellularo Cell surface-associatedo Secreted into ECM
Intracellular proteogylcan - Serglycin Cell surface associated - betaglycan,
syndecan Secreted into ECM - aggrecan, decorin,
fibromodulin, lumican, biglycan Examples:
o Aggrecan (in association with hyaluronan) is the major proteoglycan in articular cartilage
Also: aggrecan molecules associate with central hyaluronic acid filament in cartilage
o Hyalurinic acid (by itself) is the only non-sulphated GAG and is major component of synovial fluid where it has an important role in maintaining synovial fluid viscosity
Synchondrosis, syndesmosis, secondary cart/symphysis, synovial
- CLASSIFICATION OF JOINTSo Synarthrosis:
suture lines of skulls where adjoining plates are separated by thin fibrous tissue
o Amphiarthrosis: adjacent bones bound by flexible cartilage (‘fibro-cartilaginous joints’) e.g. pubic symphysis, sacro-iliac joints, intervertebral discs
o Diarthrosis: synovial joints and include:
Ball and socket e.g. hip joint Hinge e.g. inter-phalangeal joint Saddle e.g. first carpo-metacarpal joint Plane e.g. patello-femoral joint
Synovium [=Synovial membrane] contains: macrophage-like phagocytic cells and fibroblast-like cells
16

Giles Kisby GE Y1 Musculoskeletal
- ARTICULAR CARTILAGEo Components:
Collagen II: 90% is type II: tensile strength Chondrocytes Proteoglycan monomers (aggrecan): compressive strength
Is combo of chondroitin sulphate chains and keratan sulphate chains (ie on the protein)
non-covalently linked aggrecan molecules associate with central hyaluronic acid filament
Negatively charged chemical groups of GAGs attract watero Is an avascular and aneural structureo Weight-bearing properties of articular cartilage depend on intact collagen scaffold
and high aggrecan content
- OSTEOGENESIS IMPERFECTA o = ‘brittle bone disease’o mostly due to autosomal dominant collagen type I mutationso variable phenotypic severity [Types I (good) – IV (bad) describe the severity]o Pathogenesis:
Abnormal type I collagen results in bone fragility: osteopenia and fractures Diminished collagen in the sclera (of the eye) leads to increased
translucency and apparent blueness of the sclera
17

Giles Kisby GE Y1 Musculoskeletal
- EHLERS-DANLOS SYNDROMES Ehlers-danlos syndromes:o Majority are autosomal dominant disorders o Involve abnormalities in synthesis or enzymatic modification of collagen o [characterised by joint, tissue/skin laxity and arterial wall abnormality/estensibility]o Clinical features:
Joint hypermobility Skin fragility (easy bruising) Hyper-extensibility in other organs e.g. Gorlin’s sign: hyperextensible tongue Arterial wall laxity → aneurysms
o EDS type IV “= arterial form”: autosomal dominant defect in type III collagen found in blood vessels
little skin and joint disease but arterial or bowel ruptureo Not all are autosomal dominant disorders: EDS type VI (oculo-scoliotic) which is
autosomal recessive in addition to skin and joint hyperextensibility, scoliosis and ocular globe
rupture occur.
18

Giles Kisby GE Y1 Musculoskeletal
- MARFAN’S SYNDROMEo Autosomal dominant condition due to mutation in fibrillin-1 geneo characterised by ocular, skeletal, vascular, lung and skin abnormalitieso Pathogenesis:
Defect is due to fibrillin which forms constituent of extracellular microfibrils that form sub-structure for elastin
Fibrillin important in elastic walls of arteries especially aorta, zonular fibres of the eye, ligaments, skin and lung parenchyma
o Clinical features: Musculoskeletal
tall stature, long thin extremities, arachnodactyly, dolichostenomelia (low ratio of upper to lower body segments) pectus excavatum [“hollowed chest”], scoliosis high arched palate
Other ectopia lentis i.e. upward dislocation of the lens ascending aorta dissection (most common cause of death) mitral valve prolapse, aortic regurgitation cystic lung disease and spontaneous pneumothorax
o “Patients are often unusually tall, their aorta is prone to rupture/displacement and they can have abnormalities of the skeleton and joints and lens. The product of the affected gene is unable to play its normal role in stabilising the elastic fibres that give connective tissue its strength and flexibility.”
19

Giles Kisby GE Y1 Musculoskeletal
- OTHER SYNDROMESo Stickler syndrome
type II collagen Autosomal dominant, ocular and joint disease
o Alport’s syndrome
type IV collagen
autosomal recessive Hereditary glomerulonephritis and deafness “This inherited kidney disease is characterised by dysfunctional glomerular
filtering. The defective protein product of the affected gene cannot form its normal sheet-like structure in the basal lamina where it normally provides tensile strength. “
o Dystrophic Epidermolysis bullosa
type VII collagen
Hereditary skin blisters Dominant or recessive forms
20

Giles Kisby GE Y1 Musculoskeletal
- Junctional Epidermolysis Bullosao “Characterised by extensive mucocutaeous blistering and recurrent infections. The
protein product of the affected gene normally forms heterotrimeric molecules that are crucial in forming the 2-dimensional structure of the basal lamina.”
o Laminin 5 or Type XVII collagen
- Congenital Muscular Dystrophyo Heterogenous group of neuromuscular disorders characterised by muscle weakness
and hypotonia. About half of these patients have mutations in a gene encoding of a key component of the basal lamina of skeletal muscle.
o Laminin 2
21

Giles Kisby GE Y1 Musculoskeletal
07/03/14: ARTICULAR PATHOLOGY: Matthew Pickering
Los (from slides/booklet):- Define what is meant by matrix metalloproteinase and give some examples of their
substrates- Define what is meant by ADAMTS protease and understand that aggrecanases are important
in the turnover of proteoglycan in articular cartilage- Recall that cathepsin K is important protease in bone matrix turnover- Define two abnormalities seen in the synovium of patients with rheumatoid arthritis- Explain the importance of the inflammatory cytokine, tumour necrosis factor-alpha (TNF-α)
in rheumatoid arthritis pathology- Define two abnormalities seen in the cartilage and two abnormalities seen in the bone in the
osteoarthritic jointFrom slides supplementary:
- Summarise the pathogenesis, clinical features and management of rheumatoid arthritis- Explain the significance of a ‘rheumatoid factor’- Explain the importance of anti-CCP antibodies in rheumatoid arthritis
Notes
- Overviewo In brief:
Connective tissue turnover is mediated by matrix proteinases through complex regulatory networks
Rheumatoid arthritis and osteoarthritis represent the two major articular pathologies
Rheumatoid is a disease of synovium whilst osteoarthritis is disease of articular cartilage
o Connective tissue turnover and matrix proteinases Most of joint tissue is extra-cellular matrix and proteinases are key in
extracellular matrix (ECM) degradation Proteases are numerous and combine to form complex regulatory networks
o Articular pathology Rheumatoid arthritis Osteoarthritis
- Connective tissue turnovero Cartilage and bone destruction is mediated by ECM degradationo Modulated by level of:
Proteases MMP collagenases vs cartilage collagens ADAMTS aggrecanases vs cartilage proteoglycans
22

Giles Kisby GE Y1 Musculoskeletal
Cathepsin K vs bone matrix [is in the acidic secretions of osteoclasts] Protease inhibitors Matrix synthesis
o Source of proteinases depends on pathological process: Osteoarthritis = chondrocyte (intrinsic) Rheumatoid arthritis = synovial cells (intrinsic), inflammatory cells (extrinsic),
chondrocytes Infection = inflammatory cells (extrinsic), bacterial proteases (exogenous)
o MATRIX METALLOPROTEINASES Family of calcium-dependent zinc-containing endopeptidases Important functions include tissue remodelling and degradation of
extracellular matrix (ECM) ECM substrates include: [all the main ECM components:]
Collagen and gelatin (=hydrolysed collagen) Elastin Proteoglycans and Matrix glycoproteins
Regulated by hormones, growth factors and cytokines Synthesised in zymogen form by many cell types Inhibited by ‘tissue inhibitors of matrix metalloproteinases’ (TIMPs) Certain MMPs are COLLAGENASES:
MMP-1, -8, -13 (= collagenase -1, -2, -3): cleave fibrillar collagen at a single site to form fragments
stromelysins and gelatinases degrade the collagen fragments [these are also MMPs]
o ADAMTS METALLOPROTEINASES ADAMTS = a disintegrin and metalloproteinase with thrombospondin motifs Are a family with diverse functions but note ADAMTS-4 and 5 are
aggrecanases i.e. degrade aggrecan, the major human cartilage proteoglycan
o CATHEPSIN K Important in turnover of matrix of long bone Active against helical type I collagen Cysteine protease most active at acidic pH; are highly expressed by
osteoclasts; secretions of the osteoclast is acidic; Cathepsin K-deficient osteoclasts cannot degrade bone protein matrix
Human deficiency results in skeletal dysplasia (Pycnodysostosis)
23

Giles Kisby GE Y1 Musculoskeletal
- Articular pathologyo Rheumatoid arthritis
Chronic autoimmune disease characterised by pain, stiffness and symmetrical synovitis (inflammation of the synovial membrane) of synovial (diarthrodial) joints
PATHOGENESIS There is an excess of pro-inflammatory vs. anti-inflammatory
cytokines (‘cytokine imbalance’); immune cells recruited Neovascularization occurs The synovium becomes a proliferated mass of tissue (pannus)
Treatment: Anti TNFα antibodies : TNFα is the dominant pro-inflammatory
cytokine so mAbs against this are usedo But use is linked to inc TB infection risk
Interleukin-6 and interleukin-1 blockade : now available in clinic Anti-B cell antibodies : We can deplete B cells in rheumatoid arthritis
by parenteral (intravenous) administration of an antibody against a B cell surface antigen (are vs CD20)
o But use linked to hepatitis B reactivation DENOSUMAB : monoclonal antibody against RANKL. RANKL
(“receptor activator of nfKB ligand”) is important in bone destruction in rheumatoid arthritis:
o Produced by T cells and synovial fibroblasts in rheumatoid arthritis
o Acts to stimulate osteoclast formation (osteoclastogenesis)o Upregulated by: IL-1, TNF-a IL-17, PTH-rpo Binds to RANK on osteoclast precursors; action antagonized
by OPG too ‘DMARDS’: disease- modifying anti-rheumatic drugs
o may induce remission (not cure) and prevent joint damageo eg methotrexate, sulphasalazine [and its metabolite 5-ASA
nb given that ulcerative colitis produces arthritic symptoms, the benefits may be a product of unrecognized ulcerative colitis]
o all have significant adverse effects Glucocorticoid therapy (‘steroids’, ‘prednisolone’) but:
o preference is to avoid long-term use because of side-effects o useful as short-term treatment options e.g. to control flare
of disease or inflammation of single joint Key features:
Chronic arthritiso Poly arthritis - swelling of the small joints of the hand and
wrists is common
24

Giles Kisby GE Y1 Musculoskeletal
o Symmetricalo Early morning stiffness in and around jointso May lead to joint damage and destruction - ‘joint erosions’
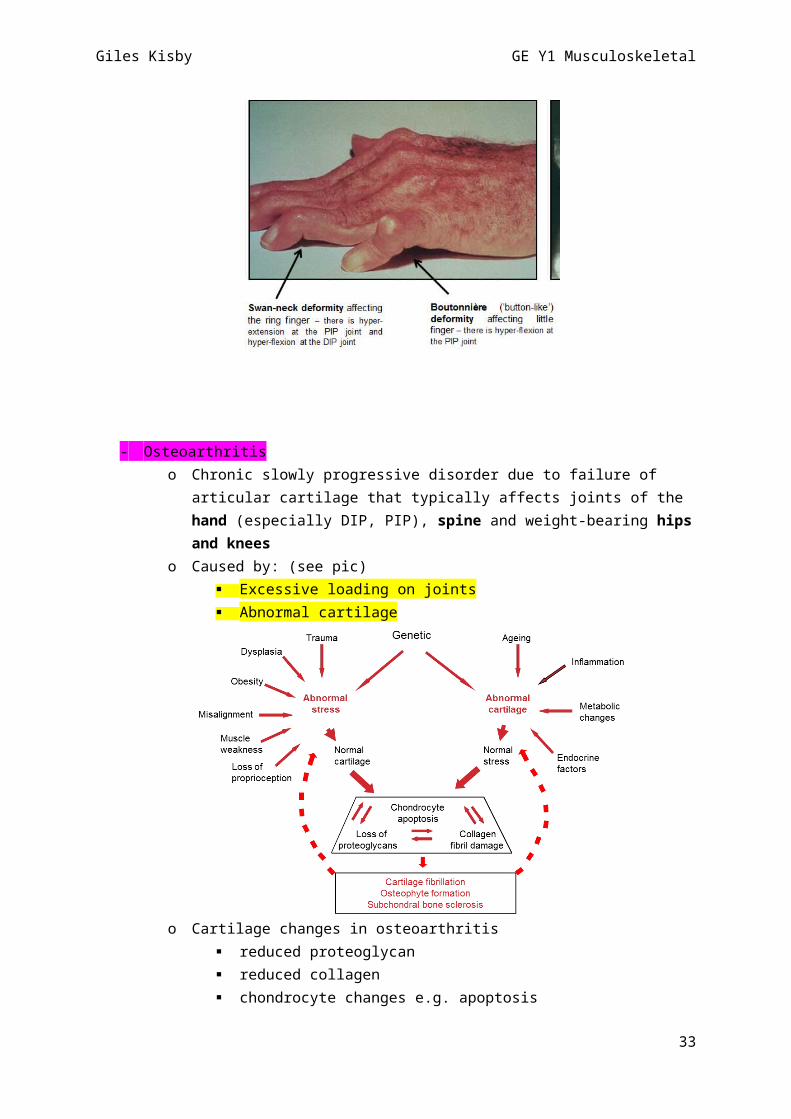
on radiographso Swan-neck deformity [PIP hypext; DIP hypflex]o Boutonnière (‘button-like’) deformity [PIP hypflex]
Extra-articular disease can occuro Fever, weight loss [due to the immune complexes]o Subcutaneous rheumatoid nodules [are associated with
Rheumatoid factor] central area of fibrinoid necrosis [fibrin-rich necrotic
material middle layer of macrophages and fibroblasts Outer layer of connective tissue
o Others rare e.g. vasculitis, episcleritis, Amyloidosis Rheumatoid ‘factor’ may be detected in blood
o = IgM autoantibody against Fc portion of IgG [misnomer: should really be called rheumatoid ‘antibody’ not ‘factor’]
Will deposit elsewhere
Antibodies to citrullinated protein antigens (ACPA)o Antibodies to citrullinated peptides are highly specific for
rheumatoid arthritis and also associated with smoking = Anti-cyclic citrullinated peptide antibody ‘anti-CCP
antibody’ [=ACPA]o Citrullination of peptides is mediated by enzymes termed
Peptidyl arginine deiminases (PADs): perform Arg Citrulline citrullinated peptides present in high concentrations
in neutrophils and monocytes Primary site of pathology is in the synovium [the synovium becomes
a proliferated mass of tissue (pannus)]; synovium includes:o Tenosynovium surrounding tendonso Bursaso Synovial joints
Wrists Knees
25

Giles Kisby GE Y1 Musculoskeletal
Ankles Metacarpophalangeal joints (MCP) Proximal interphalangeal joints (PIP) Metatarsophalangeal joints (MTP)
Important genetic component Specific HLA-DRβ gene variants mapping to amino acids 70-74 of the
T cell DRβ-chains are strongly associated with rheumatoid arthritis = the ‘shared epitope’
nb in the ‘shared epitope’ hypothesis: a common region of the beta chain on the T cells is thought to be integral to presenting auto-immunological peptides.
Important environmental component Smoking: contributes 25% of population-attributable risk and
interacts with ‘shared epitope’ to increase risk Radiographic abnormalities
Earlyo Juxta-articular osteopenia
Latero Joint erosions at margins of the joint
Later stillo Joint deformity and destruction
26

Giles Kisby GE Y1 Musculoskeletal
- Osteoarthritiso Chronic slowly progressive disorder due to failure of articular cartilage that typically
affects joints of the hand (especially DIP, PIP), spine and weight-bearing hips and knees
o Caused by: (see pic) Excessive loading on joints Abnormal cartilage
o Cartilage changes in osteoarthritis reduced proteoglycan reduced collagen chondrocyte changes e.g. apoptosis
o Bone changes in osteoarthritis Proliferation of superficial osteoblasts results in production of sclerotic bone
[pathologic thickening of the bone] e.g. subchondral sclerosis
27

Giles Kisby GE Y1 Musculoskeletal
Focal superficial necrosis resulting from focal stress on sclerotic bone New bone formation at the joint margins (termed osteophytes)
Osteophytes at the PIPJ are called Bouchard's nodes Osteophytes a the DIPJ are called Heberden's nodes
o Schlerosis, cysts, osteophytes, narrowing
28

Giles Kisby GE Y1 Musculoskeletal
14/03/14: Energy pathways in muscle and the Metabolic Myopthies: (Federico Roncaroli)
Los (from slides/booklet):- Recognise the bioenergetics of muscle contraction- Summarise the role of creatine phosphate, creatine kinase and myokinase as a short term
energy source- Summarise the role of anaerobic glycolysis i.e. the break-down of glucose to lactate and
pyruvate and conversion of ADP to ATP (mainly type II fibres that have few mitochondria and many glycogen granules) as an intermediate-term energy source
- Summarise the role of oxidative phosphorylation i.e. aerobic process that generates ATP from fat, carbohydrate and protein (type I fibres are suited to this as thay have many mitochondria and lipid droplets) as a long term energy source
- Distinguish the different types of metabolic myopathy- Summarise the key types of primary metabolic myopathies i.e. (1) glycogen storage
disorders, (2) lipid disorders and (3) mitochondrial disorders (NB detailed knowledge of individual syndromes is not required)
- Summarise the common glycogen storage disorders: Myophosphorylase deficiency (also termed: McArdle’s syndrome, glycogen storage disorder type V) (NB detailed knowledge of individual syndromes is not required)
Notes [lec was v. poor]
- Primary muscle diseaseo Genetico Non genetic
- Secondary muscle disease: muscle involvement in a systemic conditiono Autoimmuneo Cancero Infection
- ESSENTIAL CONCEPTSo Energy pathways are aimed to reconstitute ATP from ADP
ATP is the immediate source of energy: binds myosin and allows sarcomeric contraction and release of actin
o Acetyl-CoA is the essential molecule of all pathways o Muscle fibres need energy for contractiono Slow and fast twitch fibres have different metabolic requirements [ie low but long
term requirements vs high but short term requirements]
29

Giles Kisby GE Y1 Musculoskeletal
Slow fibres use/have: (run marathons) low glycogen high lipids [They are the major source of energy for muscle after
long exercise when glycogen is exhausted; Their hydrolysis produces Acetyl-CoA]
more mitochondria oxidative phosphorylation:
o aerobic process that generates ATP from fat, carbohydrate and protein
o long term energy source Fast fibres use/have: (run 100m)
High glycogen level [Synthesised from glucose by Glycogen synthase]
Few mitochondria anaerobic glycolysis,
o the break-down of glucose to lactate and pyruvateo intermediate-term energy source
Creatine kinase reaction [ ], Adenylate kinase reaction [ADP ⇔ ATP + AMP]
o Nb Adenylate kinase = myokinase; don’t confuse with Myophosphorylase
- From wiki; not from the course:
[note that the twitch category reflects the properties of the diff myosin isoforms: one key difference between slow and fast twitch fibres is the ability of myosin ATPase, to speed up
30

Giles Kisby GE Y1 Musculoskeletal
the cross bridge action. There is a small structural difference between FT and ST myosin ATPase, which is enough to cause a difference in cross bridging recycling rates]
- METABOLIC MYOPATHIES o Usually infantile and adult forms – Infants are usually more severely affected o They usually present with
muscle dysfunction induced by exercise rhabdomyolysis may become mildly symptomatic during childhood and emerge late in life
o heterogenous group of disorders characterised by anomalies of energy production: a huge array of the enzymes of the diverse pathways may be the root cause of the myopathy
- Types of myopathy:o Congenitalo Dystrophies [degeneration due to disease or malnutrition]o Metabolic / mitochondrialo Inflammatoryo Neurogenic
- Key types of primary metabolic myopathies:[(NB detailed knowledge of individual syndromes is not required)]
o (1) glycogen storage disorders, Myophosphorylase deficiency = McArdle’s syndrome
Is a glycogen storage disorder type V Myophosphorylase is the muscle isoform of the enzyme glycogen
phosphorylase: o Cytosolic enzymeo Removes glucose residues from α-(1,4)-linkages within
glycogen molecules o Product of reaction: Glucose-1-phosphate. (NOT Glucose)
CLINICAL FEATURES o Exclusively myopathico Presents with cramps and myoglobinuria following exerciseo No respiratory impairment (unlike other myopathies)
Autosomal recessive PYGM gene mutation on ch11[“phosphorylase, glycogen, muscle”]
80 known types of mutation: No protein expression or unstable protein (ie wholly dysfunctional)
31

Giles Kisby GE Y1 Musculoskeletal
Acid maltase deficiency = Pompe’s disease = alpha-glucosidase deficiency Is a glycogen storage disorder type II Defect of acid 1-4 alpha glucosidase [GAA gene; “glucosidase, alpha;
acid”]o Lysosomal enzyme o Hydrolyses linear α1-4 glucosidic linkages on
carbohydrateso Catalytic site: Asp-518
Clinical features o Children:
Muscle weakness and respiratory impairment; especially severe in infants
Common cardiac involvemento Widespread accumulation of glycogen (eg positive in white
blood cells) [excessive accumulation of glycogen within lysosome-derived vacuoles in nearly all types of cells. Excessive quantities of free extralysosomal glycogen also have been described]
o Increase in acid phosphatase activity at lysosomeso Focal expression of MHC class I antigen (misdiagnosed as
possible myositis) o The enzyme may retain some functionality; exact symptoms
depend of type of mutation Subtle changes in adults Is classed as an autosomal recessive mutation to
acid alpha-glucosidase GAA geneo (2) lipid disorders
Result from defects of: transport or endogenous triglycerides catabolism or beta-oxidation
Various clinical presentation: progressive myopathy with muscle weakness or recurrent episodes of rhabdomyolysis causing intolerance to exercise
PATHOLOGICAL FEATURES Accumulation of lipid droplets Fibre necrosis in some patients presenting with rhabdomyolysis
e.g. carnitine deficiency syndromeo (3) mitochondrial disorders
Presumably oxidative phos is defective etc Eg Impairment of respiratory chain eznymes
32
Giles Kisby GE Y1 Musculoskeletal
- Diagnosis techniques:o Examination [weakness, wasting (gait, winging, ptosis), pain]o Biopsy [BUT: if good may still be a myopathy, if bad may still be in normal range]o MRIo Genetic analyseso Blood testso EMG [electromyography], NCV [nerve conduction veloicity] (these are for analysing
nerve problems eg MG diagnosis)o Blood: CK (not raised in all myopathies; only if muscle cell membranes are
damaged), lactate, inflam markers
- Neuropathy results in distal weaknes of muscle e.g. gripping
- Muscle problem results in proximal weakness- e.g. shoulders, problems standing
33

Giles Kisby GE Y1 Musculoskeletal
14/03/14: Molecular basis of muscle contraction (Valentina Caorsi)
Los (from slides/booklet):Recall the actin-myosin cross-bridging cycle.Recall the organisation of individual myocyte and motor unit.Explain the processes involved in depolarisation and intracellular calcium flux.Distinguish the structure of the thin and thick filaments in muscle.
Notes
- The contractile Unit: the Sarcomereo Many in parallel per cello When end to end will form a myofibril; myofibrils continuous between myocytes
positioned end to endo A band = all myosin o I band = actin ONLYo actin is the major component of thin filaments
troponin and tropomyosin etc will also contribute to the light filaments actin filaments made by polymerisation of G-actin Thin filament length: 1.1 µm:
Ie shorter than thick filaments Length regulated by nebulin and tropomodulin
o myosin forms the thick filaments are single molecules but the tails of many diff myosins can interlink as in
muscle myosin molecules are composed of a head, neck (= essential light chain +
regulatory light chain), and tail domain Titin is a large elastic and extensible protein that links the thick filaments to
the Z-line, thus ensuring that the thick filaments remain centred in the sarcomere.
Thick filaments length: 1.6µm Ie longer than thin filaments
34

Giles Kisby GE Y1 Musculoskeletal
35

Giles Kisby GE Y1 Musculoskeletal
- Cross-bridges Cycle detail:o See below:o thin filament moves toward the centre of the sarcomereo Step size is small: sliding produced by one cycle is only about 1% of the sarcomere
length
- Motor Unito Is the Motor Neuron + all the muscle fibres it innervateso 1 motor unit has muscle fibres of all the same type [ie slow vs fast; is because type is
determined by signalling from the nerve]] Ie fast vs slow etc is not just determined by glycogen reserves etc but also
determined by the isoforms of myosin present; these will all be of the same type within a motor unit
o Muscles of a motor unit will contract simultaneously (all or none fashion)o number of muscle fibres 1 motor unit innervates can vary
smaller motor units present in the eye for sensitive movementso Number of motor units that are in a muscle can vary
More present at sensitive muscles eg eye muscleso When a muscle grows only the number of actins and mosins etc within individual
sarcomeres/myofibrils increases; the number of sarcomeres/myofibrils stays the same as does the number of motor units
36

Giles Kisby GE Y1 Musculoskeletal
- Latent period:- Intention to move Movement:
o Intention to move o Activity in cerebral cortex basal ganglia, cerebellumo Motor cortex firingo Impulses in descending motor pathwayso Motoneuron Excitationo Impulse propagation to axon terminalo Neuromuscular transmissiono Impulse propagation along muscle fiber plasmalemmao Impulse into T-tubuleso voltage-gated DHPRs mechanically linked to RyR of SR inducing Ca2+ releaseo Troponin binds Ca2+; tropomyosin moves out myosin binding sites on actino Cross-bridges engagemento Movement
- Contraction:o Cross bridge cycle occurringo Isotonic
No change in tension: tension in the muscle remains constant; Muscle is changing in length
Concentric tension rises to meet the resistance, then remains the same as the
muscle shortens Eccentric
muscle lengthens due to the resistance being greater than the force the muscle is producing
o Isometric Tension rises then remains constant but no shortening occurs
o How force is varied in vivo Frequency of action potentials Recruitments of motor units
37

Giles Kisby GE Y1 Musculoskeletal
Filament overlap Velocity of contraction
Frequency of action potentials:
o Temporal Summation occurs if a second stimulus is applied before completion of relaxation
Additional influx of Ca2+ promotes a second contraction which is added to the first
Stronger overall force results
o Tetanus is rapid multiple stimulations which will fuse into a smooth continuous total contraction
Abundant intracellular calcium provides continually available binding sites on actin for cross-bridge cycling
The maximum force achievable will result from the tetanus due to maximal Ca2+ release
Fatigue will eventually occur as ATP is exhausted /Ca escapes through PM
Recruitments of motor units:
o Greater percent of motor neurone pool recruited with an increasing force requirement
o Henneman's size principle states that in order to move a load motor units are recruited from smallest to largest
o This also equates to slow FFR FF
38

Giles Kisby GE Y1 Musculoskeletal
Filament overlap:
o Maximum force is achieved at intermediate overlapo When overlap is too great the filaments become constrained / not enough space in
the sarcomere for the filaments to move into upon further movemento When overlap is too little there are too few heads engaged to achieve maximum
force Aside: at heart the pericardial sac will prevent overextension therefore the
force is just seen to increase with length / preload
Velocity of contraction:
o The greater the velocity in the flexion direction the weaker the force will beo The greater the velocity in the opposite direction to the flexion direction the
stronger the force will be until a maximum is reached (muscle acting as brake);
39

Giles Kisby GE Y1 Musculoskeletal
[is because more crossbridges can be attached as are not needing to reel in the weight]
“hence bodybuilders do exercises beyond their strength where are acting as brake”
- Relaxation:o Once the membrane has repolarised, the calcium channels in the sarcoplasmic
reticulum close. o The sarcoplasmic Ca-ATPase [SERCA] pumps calcium back into the reticulum, causing
a decrease in calcium concentration in the cytoplasm. o This causes calcium dissociation from troponin, resulting in inactivation of myosin
binding sites on actin, and hence muscle relaxation.
40

Giles Kisby GE Y1 Musculoskeletal
14/03/14: Molecular basis and features of the muscular dystrophies: (Matthew Pickering)
Los (from slides/booklet):Define muscular dystrophy.Explain how muscular dystrophies may be inherited. Give one example of X-linked, autosomal recessive and autosomal dominant muscular dystrophy.Explain how these conditions manifest clinically (NB detailed knowledge of individual syndromes is not required).Explain the histological differences between myopathy and dystrophy.Explain the significance of an elevated blood level of creatine kinase (also termed creatine phosphokinase and referred to commonly by physicians as ‘CK’ or ‘CPK’).
Notes
- Disorder of neurons = neuropathy : typically distal dysfunction (hands / feet)- Disorders of skeletal muscle = myopathy : typically proximal dysfunction (shoulders / hips):
o GENETIC
Muscular dystrophies [the focus of this lec] Dystrophies (or muscular dystrophies) are a subgroup of myopathies
characterized by muscle degeneration and regeneration. Clinically, muscular dystrophies are typically progressive, because the muscles' ability to regenerate is eventually lost, leading to progressive weakness
e.g. Duchenne muscular dystrophy eg Beckers muscular dystrophy eg Limb-girdle dystrophy
Metabolic myopathies defects of glycogen metabolism: e.g. myophosphorylase deficiency defects in lipid metabolism: e.g. carnitine deficiency syndrome Mitochondrial myopathies
o complex multisystem disorderso myopathy is common
Congenital myopathies a cause of ‘floppy infant’
o ACQUIRED Autoimmune inflammatory myopathies
polymyositis dermatomyositis
Toxin / drug induced alcohol steroid myopathy (glucocorticoids)
Endocrine: Cushing’s syndrome
41

Giles Kisby GE Y1 Musculoskeletal
Thyroid disease Acquired metabolic myopathy
K, Mg, Ca, P abnormalities due to diet etc vitamin D deficiency uraemia, hepatic failure
- Creatine phosphokinaseo Abundant in heart, skeletal muscle and brain and raised in blood when:
Skeletal muscle pathology All types of muscular dystrophies (nb usually normal in neurogenic
muscle disease) Rhabdomyolysis [lysis of skeletal muscle cells]
o Alcoholo Crush injuryo Drugs
Muscle injury o trauma, o surgery,o intra-muscular injections
Autoimmune inflammatory muscle diseaseso Dermatomyositiso Polymyositis
Myocardial infarction Some cases of brain injury e.g. stroke
o CRE: = creatinine blood test measures the level of creatinine in the blood. Creatinine is a waste product that forms when creatine breaks down;
creatine, creatinine and CPK will all be raise in muscle injury as above
- Muscular dystrophies:o = Inherited group of disorders characterized by muscle wasting and weaknesso Genetic heterogeneity o Clinical heterogeneity
e.g. childhood vs. adult onset e.g. cardiac vs respiratory involvement
o X-linked dystrophies Duchenne muscular dystrophy
X linked recessive = 2/3; sporadic = 1/3 most common muscular dystrophy; considered a mainly proximal
muscle weakness problem mutations in the dystrophin gene on Xp21(locus 21 on X)
42

Giles Kisby GE Y1 Musculoskeletal
o mutations result in severe reduction or absence of dystrophin in skeletal and cardiac muscle
o many diff mutations possible; typically are out of frameo one third have no family history i.e. de novo mutations
relatively higho Dystrophin is an intracellular protein linking intracellular
actin of sarcomeres with the ECM; dystrophin loss gives loss of protection against contraction induced damage to the cell (so muscular dystrophy results)
o Absence of dystrophin results in muscle fibre damage, abnormal intra-cellular calcium entry, cell death
dystrophin isoforms are expressed in brain (therefore deficiency results in mental retardation in ~30%)
Clinical featureso Young onset: symptoms typically occur before age of 6 years o progressive muscle weakness:
initially legs and pelvis (Gower’s sign: difficulty rising from the floor)
abnormal gait wheelchair bound by teenage years
o spinal scoliosiso muscle contractures [muscle and its tendons shorten,
resulting in reduced flexibility] in the legs due to fibrosis of damaged muscle fibres
43

Giles Kisby GE Y1 Musculoskeletal
o respiratory insufficiency due to respiratory muscle weaknesso feeding difficulties in late stages
o mortality in late teenage years / early twenties [eg due to
cardiomyopathy]
o mental retardation in ~30% Diagnosis
o Examination Clinical signs as above
o Creatine phosphokinase (CPK) massive elevation (since birth) 10-100x normal
rangeo Molecular genetic testing
screen for commonest mutations in 90% genotype-phenotype correlation can be
deducedo Muscle biopsy
In a dystrophy Muscle fibre degeneration and regeneration Fibres of all sizes randomly arranged in
muscle fascicle Accumulation of fibrous tissue and fat
within the muscle In a atrophy
changes are due to motor neurone damage; this only affects fibres belonging to the motor units whose axon has been damaged (ie observe healthy muscle next to wasted motor units)
44

Giles Kisby GE Y1 Musculoskeletal
Management
o Corticosteroidso Management of complications; examples:
Spinal fusion for scoliosis Non-invasive ventilation Feeding aids, gastrotomy ACE inhibitors for cardiomyopathy
o Experimental gene therapy approaches Beckers muscular dystrophy
X linked recessive Less common and less severe than Duchenne muscular dystrophy
o ‘patient walking in their teens’ Many due to ‘in-frame’ dystrophin gene mutations so less
dysfunctiono partial expression of truncated but functional protein o Heterogeneity of presentation due to the exact nature of
the mutation As with other dystrophies: Hypertrophic proximal weakness (ie the
hypertrophy is the fibrosis / lipid deposition)
Cardiomyopathy common (two thirds of cases) Malignant hyperthermia on exposure to muscle relaxants (e.g.
suxamethonium) and halogenated inhaled anaesthetics (e.g. isoflurane)
o Ie due to the overstimulation causing excessive ATP use and thus heat generated
o Autosomal dystrophies Limb-girdle muscular dystrophy
Ie name indicates is a proximal dystrophy has dominant and recessive forms
o Autosomal dominant (LGMD1) formo Autosomal recessive (LGMD2) form
Complex group of disorders as diverse clinical featureso Nomenclature based on the deficient protein is commonly
used: E.g. laminin → ‘laminopathy’, Eg. dysferlin → ‘dysferlinopathy’
Muscle biopsy key investigationo Dystrophic changeso Absence of specific muscle protein
Raised CPK
45

Giles Kisby GE Y1 Musculoskeletal
21/03/14: Musculoskeletal examination and synovial fluid analysis: Matthew Pickering
Los (from booklet):Define and perform the GALS (Gait, Arms, Legs, Spine) examinationDefine the following commonly used rheumatological terms: arthritis, arthralgia, subluxation, synovitisDistinguish the pattern of joint disease in Rheumatoid arthritis, Osteoarthritis and Reactive ArthritisSummarise the importance of arthrocentesis and its general contraindications and potential complicationsSummarise the clinical features of goutDescribe the crystals seen within the synovial fluid in goutRecognise the role of synovial fluid examination in septic arthritis
Notes
Nb Crepitus = grating, crackling or popping sounds and sensations experienced under the skin and joints
- Definitions:o Arthritis - refers to definite inflammation of a joint(s) i.e. swelling, tenderness and
warmth of affected joints o Arthralgia - refers to pain within a joint(s) without demonstrable inflammation by
physical examinationo Dislocation - articulating surfaces are displaced and no longer in contacto Subluxation - Partial dislocationo Valgus deformity - lower limb deformity whereby whereby distal part is directed
away from the midline e.g. hallux valgus o Varus deformity - lower limb deformity whereby whereby distal part is directed
towards the midline e.g. varus knee with medial compartment osteoarthritis
46

Giles Kisby GE Y1 Musculoskeletal
NOTE:
- ‘sero-negative spondyloarthropathies’: [each are expanded upon later]: DHTK: (NB detailed knowledge of these conditions is not required): Seronegative spondyloarthropathy (or seronegative spondyloarthritis) is a group of diseases involving the axial skeleton and having a negative serostatus. "Seronegative" refers to the fact that these diseases are negative for rheumatoid factor
o Ankylosing spondylitis a chronic inflammatory disease of the axial skeleton with variable
involvement of peripheral joints and nonarticular structures. AS is a form of spondyloarthritis, a chronic, inflammatory arthritis
spinal fusion; enthesopathy; HLA-B27o Reiter’s syndrome = reactive arthritis
develops in response to an infection in another part of the body lower limb asymmetric oligoarthritis with axial involvement
o Arthritis associated with psoriasis (psoriatic arthritis) psoriasis = chronic relapsing/remitting immune-mediated skin disease
characterized by red, scaly patches, papules, and plaques, which usually itcho Arthritis associated with gastrointestinal inflammation (enteropathic synovitis)
- GALS EXAMINATION:o Gait o Arms o Legs o Spine
- GALS EXAMINATION:
47

Giles Kisby GE Y1 Musculoskeletal
o Gait smoothness and symmetry of leg, pelvis and arm movements normal stride length ability to turn quickly
o Arms Look for normal girdle muscle bulk and symmetry Look to see if there is full extension at the elbows Are shoulder joints normal? Examine hands palms down with fingers straight Observe supination, pronation, grip and finger movements Test for synovitis at the metacarpo-phalangeal joints (MCP joints)
o Legs Look for knee or foot deformity Assess flexion of hip and knee Look for knee swellings Test for synovitis at the metatarso-phalangeal joints (MTP joints) Inspect soles of the feet
o Spine Is paraspinal and shoulder girdle muscle bulk symmetrical? Is the spine straight? Are the iliac crests level? Is the gluteal muscle bulk normal? Are there popliteal swellings? Are the Achilles tendons normal? Are there signs of fibromyalgia? Are spinal curvatures normal? Is lumbar spine and hip flexion normal? Is cervical spine normal?
Then, after the GALS screen should perform a detailed examination of any abnormal joint(s) identified in the GALS screen:
- The fundamentals aspects of locomotion to examine are:o the nature of the joint abnormalityo the extent (distribution) of the joint involvemento Any other features of diagnostic importance
- The fundamentals aspects of locomotion to examine are:o the nature of the joint abnormality
Is there inflammation? Swelling Warmth
48

Giles Kisby GE Y1 Musculoskeletal
Erythema Loss of function Tenderness
Is there irreversible joint damage? Joint deformity Crepitus Loss of joint range or abnormal movement
Is there a mechanical defect? May due to inflammation, degenerative arthritis or trauma identified by:
o painful restriction of motion in absence of features of inflammation
o instability
Examples: Swelling is present in: GOUT
o Acute gout is a good example of arthritiso Tissue deposition of monosodium urate (MSU) crystals
occurs to give: Gouty arthritis Tophi (aggregated deposits of MSU in tissue) eg ear
o Commonly affects the metatarsophalangeal joint of the big toe (‘1st MTP joint’)
Gout is known as podagra when it involves the big toe
o Abrupt onseto Extremely painfulo Joint red, warm, swollen and tender
Swelling is present in: ENTHESOPATHY:o = pathology at the enthesis i.e. the site where ligament or
tendon inserts into boneo examples include:
49

Giles Kisby GE Y1 Musculoskeletal
plantar fasciitis Achilles tendinitis ANKYLOSING SPONDYLITIS REACTIVE ARTHRITIS
Joint deformity may occur in: ANKYLOSING SPONDYLITISo It is a chronic inflammatory disease affecting:
Sacroiliac joints and spine: May lead to spinal fusion and deformity
Entheses resulting in chronic enthesopathy Non-axial joints – usually hips and shoulders
o Strong association with HLA-B27o Rheumatoid factor is negative: It is part of a group of
conditions termed ‘sero-negative spondyloarthropathies
50

Giles Kisby GE Y1 Musculoskeletal
o the extent (distribution) of the joint involvement determine number of joints involved:
polyarthritis > 4 joints involved oligoarthritis 2-4 joints involved monoarthritis single affected joint
note if involvement is symmetrical note the size of the involved joints is there axial involvement?
Examples: rheumatoid arthritis
o bilateral o symmetrical o involvement of large and small joints
reactive arthritis o lower limb (predominantly) asymmetrical oligoarthritis o axial involvement
gout o inflammation of the first metatarsophalangeal joints
[*****************]
51

Giles Kisby GE Y1 Musculoskeletal
o Any other features of diagnostic importance
Examples: RHEUMATOID ARTHRITIS
o SUBCUTANEOUS NODULES
GOUTo TOPHI – subcutaneous deposits of uric acid
Systemic lupus erythematosus o MALAR RASH
52

Giles Kisby GE Y1 Musculoskeletal
- SYNOVIAL FLUID ANALYSISo Used to test for
Infection crystal arthritis [different crystals]
Gouto Needle shaped birefringence negative
o Monosodium Urate Pseudogout
o Brick shaped birefringence positive
o Calcium pyrophosphate dihydrate (CPPD) crystals
o normally: Colourless or pale yellow transparent viscous film covering synovium and cartilage with few cells
o Synthesized by synovial lining cells of synovium Two types of synovial lining cells:
“type A” = macrophage-like “type B” = fibroblast-like: secrete the hyaluronic acid which results
in the increased viscosity of synovial fluid [Synovial fluid is viscous fluid rich in hyaluronic acid]
synovium collagen is Type I collagen
53

Giles Kisby GE Y1 Musculoskeletal
o Abnormal increase in synovial fluid volume is termed ‘synovial effusion’ osteoarthritis
due to abnormal mechanical stimulation with damage to cartilage and bone
synovitis, gout, rheumatoid arthritis due to inflammation
septic arthritis due to infection ie bacteria in joint due to injury, impaired defence
etc
54

Giles Kisby GE Y1 Musculoskeletal
21/03/14: Rheumatoid arthritis, Osteoarthritis and Reactive arthritis: (Matthew Pickering)
Los (from booklet):Define the term ‘reactive arthritis’ and summarise how it may present.Recognise that ‘reactive arthritis’ is part of a family of inflammatory arthritic syndromes termed ‘seronegative spondyloarthroapthies’ (NB detailed knowledge of these conditions is not required).Summarise the pathogenesis, clinical features and management of rheumatoid arthritis.Explain the importance of anti-CCP antibodies in rheumatoid arthritis.
Notes
- REACTIVE ARTHRITISo 20 – 40 YRSo URETHRITIS o sterile inflammation in joints
Reactive arthritis is distinct from infection in joints (septic arthritis)o occurs following/after infection especially urogenital (e.g. Chlamydia trachomatis)
and gastrointestinal (e.g. Salmonella, Shigella, Campylobacter infections) infections Symptoms follow 1-4 weeks after infection and this infection may be mild
o Reactive arthritis may be first manifestation of HIV or hepatitis C infectiono Important extra-articular manifestations include:
Enthesopathy Skin inflammation Eye inflammation
o Associated with HLA-B27o M>F [“males more likely to have HIV”]o musculoskeletal symptoms of reactive arthritis
ARTHRITIS – ie joints Asymmetrical Oligoarthritis (<5 joints) Lower limbs typically affected: large joints
ENTHESITIS – ie tendons Heel pain (Achilles tendonitis) Swollen fingers (dactylitis) Painful feet (metatarsalgia due to plantar fasciitis)
SPONDYLITIS – ie spine Sacroiliitis (inflammation of the sacro-iliac joints) Spondylitis (inflammation of the spine)
o Treatment: Non steroidal: NSAIDs Steroidal: intra-articular corticosteroid therapy
55

Giles Kisby GE Y1 Musculoskeletal
o Rheumatoid arthritis vs. Reactive arthritis:
o Septic arthritis vs. Reactive arthritis
- OSTEOARTHRITISo Chronic slowly progressive disorder o Cartilage changes
reduced proteoglycan reduced collagen chondrocyte apoptosis
o failure of articular cartilage that typically affecting joints of the hand (especially those involved in pinch grip), spine and weight-bearing joints (hips and knees)
Joints of the hand D istal interphalangeal joints (DIP)
o Osteophytes at the DIP joints are termed Heberden’s nodes P roximal interphalangeal joints (PIP)
o Osteophytes at the PIP joints are termed Bouchard’s nodes First carpometacarpal joint (CMC)
56

Giles Kisby GE Y1 Musculoskeletal
Spine Weight-bearing joints of lower limbs
knees and hips First metatarsophalangeal joint (MTP)
o Caused by: Excessive loading on joints Abnormal joint components
o Radiographic features Joint space narrowing Subchondral bony sclerosis
Proliferation of superficial osteoblasts Osteophytes
new bone formation at the joint margins Subchondral cysts
focal superficial necrosis
Radiographic changes in Rheumatoid Arthritis vs. Osteoarthritis:
[Osteopenia refers to bone density that is lower than normal]
57

Giles Kisby GE Y1 Musculoskeletal
o Management physiotherapy Weight loss Exercise Analgesia Joint replacement
- The following Los are covered in the ‘ARTICULAR PATHOLOGY LECTURE’:o Summarise the pathogenesis, clinical features and management of rheumatoid
arthritis.o Explain the significance of a ‘rheumatoid factor’.o Explain the importance of anti-CCP antibodies in rheumatoid arthritis.
21/03/14: The Connective Tissue Disorders: (Matthew Pickering)
Los (from booklet):Summarise the pathogenesis and clinical features of systemic lupus erythematosus (SLE).Recognise the importance of autoantibody measurement in the assessment of connective tissue disease and list the important antibodies associated with
(1) systemic lupus erythematosus (SLE), (2) scleroderma, (3) Sjogren’s syndrome and (4) polymyositis.
List the key features of: (NB detailed knowledge of these conditions is not required)Sjogren’s syndrome, scleroderma polymyositis
Explain what is meant by the term ‘overlap syndrome’ in the setting of connective tissue disease.
58

Giles Kisby GE Y1 Musculoskeletal
Notes
Nb Arthralgia and arthritis is typically non-erosive (though not in RA)
- Key conditionso Systemic Lupus Erythematosus (SLE)o Sjögren’s syndromeo Autoimmune inflammatory muscle disease
Polymyositis Dermatomyositis
o Systemic sclerosis (= scleroderma)o Overlap syndromes
- Raynaud’s phenomenon o Raynaud’s phenomenon is most commonly isolated and benign condition but can be
associated with some of the above conditions (cf for further info)o Painfulo Is Intermittent vasospasm of digits on exposure to cold
o Typical colour changes – white blue red:
Vasospasm leads to blanching of digit Cyanosis as static venous blood deoxygenates Reactive hyperaemia then occurs (transient increase in organ blood flow
that occurs following a brief period of ischaemia)
59

Giles Kisby GE Y1 Musculoskeletal
- SYSTEMIC LUPUS ERYTHEMATOSUSo Prototypic autoimmune disease: B cell hyper-reactivity making autoantibodies
o Associated with HLA-DR3 [same as the secondary HLA cause of coeliac (behind HLADQ2)]
o Clinical manifestations: Malar rash – erythema that spares the nasolabial fold Photosensitive rash Mouth ulcers Hair loss Raynaud’s phenomenon (see above) Arthralgia and sometimes arthritis Serositis (vs serous tissues: pericarditis, pleuritis, less commonly peritonitis) Renal disease – glomerulonephritis (‘lupus nephritis’) Cerebral disease – ‘cerebral lupus’ e.g. psychosis
o Laboratory manifestations: Haematological:
Haemolytic anaemia Lymphopenia Thrombocytopenia [relative decrease of platelets in blood]
Immunological Antinuclear antibodies
o Likely accessed by antibodies by translocation of nuclear antigens to membrane surface in apoptosis and then impaired clearance of apoptotic cells
o Eg Anti-Ro Anti-La Anti-Sm Anti-RNP Anti-Scl-70 Anti-ds-DNA antibodies
o Also some non nuclear!: Anti-tRNA synthetase antibodies Anti-double-stranded DNA antibodies
o Correlate with disease activity Complement consumption
o Low C4 and C3 due to the Abs activating complement
Sick lupus patient commonly has Low complement levels & High serum levels of anti-ds-DNA antibodies
60

Giles Kisby GE Y1 Musculoskeletal
- SJÖGREN’S SYNDROME: sjögren’s syndromeo F>Mo Autoimmune exocrinopathy
lymphocytic infiltration of especially exocrine glands and sometimes of other organs (extra-glandular involvement)
Exocrine gland pathology results in: Dry eyes (xerophthalmia) Dry mouth (xerostomia) Parotid gland enlargement
o Commonest extra-glandular manifestations are: non-erosive arthritis Raynaud’s phenomenon
o Termed ‘secondary’ Sjögren’s syndrome if occurs in context of another connective tissue disorder e.g. SLE
o Associated with autoantibodies: Antinuclear antibody
Anti-Ro (ie as does SLE) Anti-La (ie as does SLE)
Rheumatoid factor
61

Giles Kisby GE Y1 Musculoskeletal
- INFLAMMATORY MUSCLE DISEASEo Proximal muscle weakness due to autoimmune-mediated inflammation
o Either with (=“dermatomyositis”) or without (=“polymyositis”) a rash
o Skin changes in dermatomyositis: Lilac-coloured (heliotrope) rash on eyelids, malar region and naso-labial
folds [nb these folds are spared in SLE] Red or purple flat or raised lesions on knuckles (Gottron’s papules) Subcutaneous calcinosis (ie calcium deposits) Mechanic’s hands (fissuring and cracking of skin over finger pads)
o Associated with autoantibodies: Antinuclear antibody (ie as does SLE) Anti-tRNA synthetase antibodies (ie as does SLE)
o Elevated CPK, abnormal electromyograpghy, abnormal muscle biopsy (polymyositis = CD8 T cells; dermatomyositis = CD4 T cells in addition to B cells)
o Associated with malignancy (10-15%) and pulmonary fibrosis
62

Giles Kisby GE Y1 Musculoskeletal
- SYSTEMIC SCLEROSIS = sclerodermao Raynaud’s phenomenono Thickened skin: fibrotic skin
Signs: Dermal fibrosis cutaneous calcinosis telangiectasia [small dilated blood vessels near the surface of the
skin or mucous membranes]o Skin changes may be limited or diffuse
Diffuse systemic sclerosis Fibrotic skin everywhere including proximal to elbows or knees
(excluding face and neck) Anti-topoisomerase-1 (=anti DNA gyrase = anti-Scl-70) antibodies Pulmonary fibrosis, renal (thrombotic microangiopathy)
involvement Short history of Raynaud’s phenomenon
Limited systemic sclerosis Fibrotic skin hands, forearms, feet, neck and face Anti-centromere antibodies Pulmonary hypertension Long history of Raynaud’s phenomenon
63
Giles Kisby GE Y1 Musculoskeletal
- OVERLAP SYNDROMEo When features of more than 1 connective tissue disorder are present e.g. SLE and
inflammatory muscle disease we can use the term “overlap syndrome”o When incomplete features of a connective tissue disease are present we can use the
term “undifferentiated connective tissue disease”o overlap syndromes include “mixed connective tissue disease” (‘MCTD’):
- Mixed Connective Tissue Disease means Anti-U1-RNP antibody present (ie also in SLE [TRUE])
o Associated with features of seen in SLE, scleroderma, rheumatoid arthritis, and polymyositis
Nb no autoantibodies for:
- Osteoarthritis- Gout - Reactive arthritis- Ankylosing Spondylitis- psoriatic arthritis- enteropathic synovitis
64

Giles Kisby GE Y1 Musculoskeletal
Revision lecture put up by lecturer (can also use slides direct to test self):
- List the key features of rheumatoid arthritiso Morning stiffness in and around jointso Symmetrical polyarthritis typically involving the small joints of the hand and/or
wristso Subcutaneous noduleso Rheumatoid factoro Joint erosions on radiographs
- Define rheumatoid factor
o Antibodies that recognize the Fc portion of IgG as their target antigeno typically IgM antibodies i.e. IgM anti-IgG antibody !
- What substance makes synovial fluid viscous?o Hyaluronic acid: a non-sulphated glycosaminoglycan
- Define Reactive Arthritiso Sterile inflammatory synovitis following an infection whose extra-articular
manifestations may include: Enthesopathy Skin inflammation Eye inflammation
65

Giles Kisby GE Y1 Musculoskeletal
- List two infections associated with Reactive Arthritiso Urogenital infections
E.g. Chlamydia trachomatiso Enterogenic infections
E.g. Salmonella, Shigella, Campylobacter infectionso Reactive arthritis may be first manifestation of HIV or hepatitis C
infection
- Define and give two examples of an enthesopathyo Inflammation where a ligament, tendon, fascia or capsule insert into bone. Examples
include: Achilles tendonitis (painful heel)
inflammation at insertion of Achilles tendon into calcaneum Plantar fasciitis (painful feet)
inflammation at insertion of plantar fascia Dactylitis (swollen digits)
inflammation at insertion of capsule and ligaments in digits Spondylitis (spinal inflammation) in Ankylosing Spondylitis
inflammation where the outer part (annulus fibrosis) of the inter-vertebral disc inserts into the vertebral body
66

Giles Kisby GE Y1 Musculoskeletal
[see above for ans]
- In a single statement summarise the key pathological finding in osteoarthritiso Irreversible loss of articular cartilage
- Define (i) proteoglycan and (ii) glycosaminoglycan and give one example of eacho glycoproteins containing sulphated glycosaminoglycan chains e.g.
Aggrecan o [GAGs: are repeating polymers of disaccharides] repeating polymers of disaccharides
e.g. Hyaluronic acid (= hyaluronate)
disaccharides are: glucuronic acid and N-acetyl glucosamine UNSULFATED!!
Keratan sulphate disaccharides are: galactose and N-acetyl glucosamine
Chondroitin sulphate disaccharides are: glucuronic acid and N-acetyl galactosamine
- What is the major (i) collagen and (ii) proteoglycan found in articular cartilage?o Type II collageno Aggrecan
67

Giles Kisby GE Y1 Musculoskeletal
- List the major HLA association for each of the following diseaseso Ankylosing Spondylitis & Reactive Arthritis HLA-B27
“there be average of 2 words in ‘Ankylosing Spondylitis’ and ‘Reactive Arthritis’ and 7 syllables (Ankylosing spondylitis = 8, Reactive Arthritis = 6, average = 7) so HLA-B27”
o SLE HLA-DR3 “SLE has 3 letters so HLA-DR3”
o Rheumatoid arthritis HLA-DR4 “U will remember that U is fourth letter in rheumatoid so HLA-DR4”
68

Giles Kisby GE Y1 Musculoskeletal
69