studentsrepo.um.edu.mystudentsrepo.um.edu.my/4831/1/development_of_a_rapid... · web viewnosocomial...
TRANSCRIPT

DEVELOPMENT OF RAPID LAMP ASSAYS FOR THE
DETECTION OF POTENTIAL NOSOCOMIAL PATHOGENS
DONG HONG

FACULTY OF SCIENCE
UNIVERSITY OF MALAYA
KUALA LUMPUR
2013

DEVELOPMENT OF RAPID LAMP ASSAYS FOR THE
DETECTION OF POTENTIAL NOSOCOMIAL PATHOGENS
DONG HONG
DISSERTATION SUBMITTED IN FULFILLMENT OF THE
REQUIREMENTS FOR THE DEGREE OF MASTER OF
BIOTECHNOLOGY
INSTITUTE OF BIOLOGICAL SCIENCES
FACULTY OF SCIENCE

UNIVERSITY OF MALAYA
KUALA LUMPUR
2013

UNIVERSITI MALAYA
ORIGINAL LITERARY WORK DECLARATION
Name of Candidate: Dong Hong (I.C/Passport No: G45811942 )
Registration/Matric No: SGF110009
Name of Degree: Master of Biotechnology
Title of Project Paper/Research Report/Dissertation/Thesis (“this Work”):
Development of rapid LAMP assays for the detection of potential nosocomial pathogens
Field of Study: Microbial Biotechnology
I do solemnly and sincerely declare that:
(1) I am the sole author/writer of this Work;(2) This Work is original;(3) Any use of any work in which copyright exists was done by way of fair
dealing and for permitted purposes and any excerpt or extract from, or reference to or reproduction of any copyright work has been disclosed expressly and sufficiently and the title of the Work and its authorship have been acknowledged in this Work;
(4) I do not have any actual knowledge nor do I ought reasonably to know that the making of this work constitutes an infringement of any copyright work;
(5) I hereby assign all and every rights in the copyright to this Work to the University of Malaya (“UM”), who henceforth shall be owner of the copyright in this Work and that any reproduction or use in any form or by any means whatsoever is prohibited without the written consent of UM having been first had and obtained;
(6) I am fully aware that if in the course of making this Work I have infringed any copyright whether intentionally or otherwise, I may be subjected to legal action or any other action as may be determined by UM.

Candidate’s Signature Date
Subscribed and solemnly declared before,
Witness’s Signature Date
Name:Designation

ABSTRACT
Nosocomial pathogens are organisms that cause diseases in a patient during his/her
stay in a hospital or health care center. These pathogens can spread easily in the hospital
and cause an outbreak because of the low immune system of the hospitalized patients.
Even so, they are resistant to most of the antibiotics. Rapid detection of these pathogens
would be useful to trace the source of an outbreak. In this study, a new detection
method, the loop-mediated isothermal amplification (LAMP) was developed for the
rapid detection of three nosocomial pathogens which are highly multidrug resistant.
They are Acinetobacter baumannii, Pseudomonas aeruginosa and Klebsiella
pneumoniae.
Acinetobacter baumannii is a Gram-negative bacterium which can cause serious
infection. It can cause wound infection, pneumonia, urinary tract infection, and etc. The
LAMP primers for Acinetobacter baumannii were based on gltA gene and amplified in
62℃ for 90 min. The assay was evaluated on 50 bacterial strains, including 30
Acinetobacter baumannii and 20 non-Acinetobacter baumannii. All the positive strains
were correctly identified, while the negatives were true negative. The sensitivity of
LAMP was 5.5×104 CFU/ml, and it was 10-fold more sensitive than the normal PCR
method (5.5×105 CFU/ml). The sensitivity of both LAMP and PCR were the same at
5.5×105 CFU/ml in spiked blood samples.
Pseudomonas aeruginosa causes pneumonia, otitis, endocarditis, septicemia and
keratitis. The LAMP primers for P. aeruginosa were based on 16S rRNA-processing
protein rimM gene and amplified at 65℃ for 60 min. The specificity of the primers was
i

also evaluated on 50 strains (30 P. aeruginosa, 20 non-P. aeruginosa). The detection
limit of this assay on bacterial culture was 3.6×104 CFU/ml, and it was 1000-fold more
sensitive than PCR (3.6×107 CFU/ml). For spiked blood samples, the detection limit for
LAMP was 7.7×104 CFU/ml which was 1000 fold higher than PCR (7.7×107 CFU/ml).
Klebsiella pneumoniae is a bacterial pathogen which causes pneumonia, bacteremia
and meningitis. The LAMP primers were based on ABC transport permease gene and
amplified at 65℃ for 90 min. The assay was evaluated on 50 bacterial strains, including
30 Klebsiella pneumoniae and 20 non-Klebsiella pneumoniae. There was no false
positive or false negative result. The detection limit of LAMP was 7×103 CFU/ml,
which was the same with normal PCR method. For spiked blood samples, the detection
limit of both LAMP and PCR was the same at 1.4×104 CFU/ml.
Overall, LAMP is a rapid, effective and efficient assay which would contribute to
the efficient detection of nosocomial pathogens.
ii

ABSTRAK
Patogen nosocomial adalah organisma yang menyebabkan penyakit dalam pesakit
semasa beliau di hospital atau pusat penjagaan kesihatan. Patogen ini boleh merebak
dengan mudah di hospital dan menyebabkan wabak kerana sistem imun yang rendah
pada pesakit ke hospital. Tambahan, pathogen bakteria adalah menentang terhadap
banyak antibiotik. Dalam kajian ini, kaedah pengesanan baru telah dioptimumkan untuk
tiga patogen bakteria, iaitu, Acinetobacter baumannii, Pseudomonas aeruginosa dan
Klebsiella pneumoniae. Kaedah ini dipanggil ‘loop-mediated isothermal amplification’
(LAMP) dan ia boleh mengesan jangkitan dalam fasa awal.
Acinetobacter baumannii adalah bakteria Gram-negatif yang boleh menyebabkan
jangkitan yang serius. Ia boleh menyebabkan jangkitan luka, pneumonia, jangkitan
saluran kencing, dan lain-lain. LAMP untuk Acinetobacter baumannii adalah
berdasarkan gen gltA dan bertindak balas dalam 62℃ selama 90 min. Ujian ini dinilai
dengan 50 strain bakteria, termasuk 30 Acinetobacter dan 20 bukan Acinetobacter.
Semua strain Acinetobacter dibukti positif manakala strain bukan Acinetobacter adalah
negative. Had pengesanan adalah 5.5×104 CFU/ml, dan ia adalah 10 kali ganda lebih
sensitif daripada kaedah PCR normal (5.5×105 CFU/ml). Sensitiviti kedua-dua LAMP
dan PCR adalah sama sebanyak 5.5×105 CFU/ml bila diuji dengan sampel darah yang
dicemari dengan strain AC090215.
Pseudomonas aeruginosa adalah bakteria yang boleh menyebabkan pneumonia,
otitis, endokarditis, septisemia dan keratitis. The primer LAMP untuk P. aeruginosa
adalah berdasarkan 16S rRNA pemprosesan protein rimM dan bertindak balas dalam
iii

65℃ selama 60 minit. Specificity primers untuk P. aeruginosa juga dinilai dengan 50
strain bakteria (30 P. aeruginosa, 20 bukan P. aeruginosa). Had pengesanan adalah
3.6×104 CFU/ml untuk LAMP, dan ia adalah 1000 kali ganda sensitif daripada PCR
(3.6×107 CFU/ml). Had pengesanan adalah 7.7×104 CFU/ml dan kepekaan assay LAMP
adalah lebih tinggi daripada PCR (1000-kali ganda) bila diuji dengan sampel darah yang
dicemari dengan strain PS19.
Klebsiella pneumoniae adalah bakteria patogen yang menyebabkan pneumonia,
bacteremia dan meningitis. Primer untuk ujian LAMP adalah berdasarkan gen tonB
gene dan tindakbalas boleh diselesai pada suhu 65℃ dalam 90 min. Ujian ini dinilai
dengan 50 strain termasuk 30 Klebsiella pneumoniae dan 20 strain bukan Klebsiella
spp. Tiada positif palsu atau negative palsu dikesan. Had pengesanan adalah 7×103
CFU/ml, dan sama dengan kaedah PCR biasa. Kepekaan LAMP dan PCR adalah sama
(1.4×104 CFU/ml) bila ujian dijalankan dengan sampel yang dicemari dengan strain
K10-04.
Secara keseluruhan, LAMP adalah ujian yang cepat, cekap dan berkesan yang akan
menyumbang kepada pengesanan jangkitan nosocomial
iv

ACKNOWLEDGEMENT
I would like to express the deepest appreciation to my supervisor Professor Dr.
Thong Kwai Lin for her patience guidance. And also thanks to all the lab mates in
Laboratory of Biomedical Science and Molecular Microbiology.
I would like to express my special thanks to my parents for the long time's care and
love. Without their support and help, this project would never be finished.
Thanks to University of Malaya for providing the PPP grants (P0024/2012A) for
the research materials and reagents.
At last, thanks to all the people who have ever helped me during my research.
v

TABLE OF CONTENT
Page
ABSTRACT....................................................................................................................i
ABSTRAK....................................................................................................................iii
ACKNOWLEDGEMENT.............................................................................................v
TABLE OF CONTENT................................................................................................vi
LIST OF FIGURES......................................................................................................ix
LIST OF TABLES..........................................................................................................x
LIST OF APPENDIX....................................................................................................xi
ABBREVIATIONS AND SYMBOLS.......................................................................xiii
CHAPTER 1: INTROUCTION.....................................................................................1
1.1 General introduction.........................................................................................1
1.2 Objectives.........................................................................................................3
CHAPETER 2: LITERATURE REVIEW.....................................................................4
2.1 Definition of nosocomial infection..................................................................4
2.2 Klebsiella pneumoniae.....................................................................................5
2.3 Pseudomonas aeruginosa.................................................................................6
2.4 Acinetobacter baumannii.................................................................................6
2.5 Conventional detection methods for nosocomial pathogens............................7
2.6 Loop-mediated isothermal amplification (LAMP) method.............................8
CHAPETER 3: METHODOLOGY.............................................................................12
3.1 Materials.........................................................................................................12
3.1.1 Bacterial strains...................................................................................12
vi

3.1.2 Chemicals and reagents...............................................................................12
3.2 Methods..........................................................................................................12
3.2.1 DNA template......................................................................................12
3.2.2 Primer design......................................................................................13
3.2.3 LAMP reaction....................................................................................13
3.2.4 Data analysis.......................................................................................14
3.2.5 PCR.....................................................................................................15
3.2.6 Evaluation of sensitivity of LAMP assay in culture and blood samples17
CHAPTER 4: RESULTS.............................................................................................18
4.1 Primer designed for LAMP............................................................................18
4.2 Acinetobacter baumannii...............................................................................18
4.2.1 Optimized LAMP assay......................................................................18
4.1.2 Sensitivity and specificity of the method............................................19
4.1.3Evaluation of LAMP on spiked blood sample.....................................24
4.2 P. aeruginosa..................................................................................................25
4.2.1 Optimized LAMP assay......................................................................25
4.2.2 Sensitivity and specificity of the method............................................26
4.2.3Evaluation of LAMP on Spiked blood.................................................29
4.3 K. pneumoniae................................................................................................31
4.3.1 Optimized LAMP assay......................................................................31
4.3.2 Sensitivity and specificity of the method............................................31
4.3.3Evaluation of LAMP on spiked blood..................................................34
CHAPTER 5: DISCUSSION.......................................................................................36
CHAPTER 6: CONCLUSION.....................................................................................41
vii

viii

LIST OF FIGURES
List of figures Page
Figure 2.1: Principle of LAMP 10
Figure 4.1: Comparison of LAMP results at different temperature of A. baumannii 19
Figure 4.2: Detection of the LAMP products using SYBR Green Ι 22
Figure 4.3: Visualization of LAMP products by agarose gel electrophoresis 22
Figure 4.4: Sensitivity of the LAMP assay (a) and PCR (b) for A. baumannii AC090213
23
Figure 4.5: The detection limit of LAMP (a) and PCR (b) using A. baumannii spiked
blood sample 24
Figure 4.6: Comparison of LAMP results of P. aeruginosa PS19 at different
temperature 25
Figure 4.7: Comparison of LAMP results with or without loop primer on PS19 26
Figure 4.8: Sensitivity of the LAMP assay (a) and PCR (b) for P. aeruginosa PS19
29
Figure 4.9: The detection limit of LAMP (a) and PCR (b) using P. aeruginosa spiked
blood sample 30
Figure 4.10: Optimized temperature (65℃) of K. pneumoniae LAMP detection 31
Figure 4.11: Sensitivity of the LAMP assay (a) and PCR (b) for K. pneumoniae K10-04
34
Figure 4.12: The detection limit of LAMP (a) and PCR (b) using K. pneumoniae spiked
blood sample 35
ix

LIST OF TABLE
List of tables Page
Table 3.1: The information of PCR primers 16
Table 4.1: PCR and LAMP result of A. baumannii strains 20
Table 4.2: PCR and LAMP result of P. aeruginosa strains 27
Table 4.3: PCR and LAMP result of K. pneumoniae strains 32
x

LIST OF APPENDIX
List of appendix page
Appendix 1: Bacterial strains used for optimization 50
Appendix 2: Chemicals and reagents 53
Appendix 3: In silico PCR amplification of gltA F3, B3 from A. baumannii for LAMP
primer 56
Appendix 4: In silico PCR amplification of gltA FIP from A. baumannii for LAMP
primer 57
Appendix 5: In silico PCR amplification of gltA BIP from A. baumannii for LAMP
primer 58
Appendix 6: In silico PCR amplification of ABC transporter permease F3, B3 from K.
pneumoniae for LAMP primer 59
Appendix 7: In silico PCR amplification of ABC transporter permease FIP from K.
pneumoniae for LAMP primer 60
Appendix 8: In silico PCR amplification of ABC transporter permease BIP from K.
pneumonia for LAMP primer 61
Appendix 9: In silico PCR amplification of rimM F3, B3 from P. aeruginosa for
LAMP primer 62
Appendix 10: In silico PCR amplification of rimM FIP from P. aeruginosa for LAMP
primer 63
Appendix 11: In silico PCR amplification of rimM BIP from P. aeruginosa for LAMP
primer 64
xi

Appendix 12: Presentation and publication 65
xii

ABBREVIATION AND SYMBOLS
List of abbreviations and symbols
xiii
PCR
DNA
RNA
NCBI
BLAST
LB
TBE
MgCl2
dNTPs
A. baumannii
E. coli
P. aeruginosa
K. pneumoniae
S. Typhi
ddH2O
UV
et al
s
min
-Polymerase Chain Reaction
-Deoxyribonucleic acid
-Ribonucleic acid
-National center for Biotechnology Information
-Basic Local Alignment Search Tools
-Luria Bertani
-Tris Borate EDTA
-Magnesium chloride
-deoxyribonucleotide triphosphates (dATP, dTTP, dCTP and dGTP)
-Acinetobacter baumannii
-Escherichia coli
-Pseudomonas aeruginosa
-Klebsiella pneumoniae
-Salmonella Typhi
-Double distill/Deionized water
-Ultraviolet
-Et alia (and other)
-seconds
-minutes

xv
℃
µM
µL
mL
mM
-degree celsius
-micromolar
-microliter
-milliliter
-millimolar

CHAPTER 1: INTRODUCTION
1.1 General introduction
Nosocomial infection (NI) is also known as hospital-acquired infection. It poses a
significant problem worldwide. In the USA, there are roughly 1.7 million hospital-
associated infections from all types of microorganisms, and that cause or contribute to
99,000 deaths each year. In Europe, Gram-negative infections are estimated to two-third
of the 25,000 deaths each year (Pollack, 2010). The rapid spread of nosocomial
pathogens has eventually increased the difficulty in treatment due to delayed detection
and diagnosis. The difficult in treatment of nosocomial infection is also because of the
multidrug resistance of the pathogen.
Usually, the detection and identification of nosocomial bacterial pathogen is
performed by culture methods. A selective medium will be used for the identification,
like the CHROMagar. However, the conventional culture methods are time consuming,
usually need more than one day to detect up to genus level, and longer time is needed
for species level identification. Because of the time consuming limitation of culture
detection method, molecular method like Polymerase Chain Reaction (PCR) which is
faster than conventional culture methods were introduced in medical diagnostics.
However, PCR also has some disadvantages. It needs additional steps like to run
agarose gel electrophoresis continues with visualization of the products. So, it will take
around 4 hours to finish the whole detection. The delay in detection may provide
opportunity for pathogens to spread in the hospital. Therefore, it is essential to develop a
more rapid and easier approach for the detection of nosocomial pathogens. 1

In this study, a relatively new approach based on the loop-mediated isothermal
amplification (LAMP) method is adopted, optimized and evaluated to determine its
usefulness in detecting clinically important nosocomial bacterial pathogens. LAMP is
one of the isothermal nucleic acid amplification methods and has received a lot of
attention during the last decade because of its simplicity. It is an auto-cycling DNA
synthesis which performed by four LAMP primers and DNA polymerase. Since the
invention of LAMP method, it has already been developed for a lot of pathogens, like
Mycobacterium (Iwamoto et al., 2003), Plasmodium falciparum (Poon et al., 2006),
Shigella, Escherichia coli (Song et al., 2006), Streptococcus pneumoniae (Seki et al.,
2005), Staphylococcus aureus (Lim et al., 2013) and many other bacterial pathogens.
With the help of Loopamp EXIA machine, the detection time can be shortened to 1.5
hours, or even 1 hour. This method can detect the bacteria in a really short time with
less equipment and steps.
This thesis will focus on the development of LAMP assay for detection of three
main nosocomial pathogens-A. baumannii, P. aeruginosa and K. pneumoniae, and the
specificity and sensitivity were tested on bacterial cultures and spiked blood samples.
This LAMP assays were compared with the conventional PCR.
2

1.2 Objectives
Overall, the goal of the study was to develop loop-mediated isothermal
amplification (LAMP) assays for detection of Acinetobacter baumannii, Klebsiella
pneumoniae and Pseudomonas aeruginosa.
Specifically, the objectives were:
i) To optimize the conditions for the developed LAMP assays.
ii) To determine the sensitivity and specificity of the assays.
iii) To compare the LAMP assays with conventional PCR.
iv) To evaluate the LAMP assays on spiked blood samples.
3

CHAPETER 2: LITERATURE REVIEW
2.1 Definition of nosocomial infection
Nosocomial infection (NI) is also known as hospital-acquired infection. This
infection usually cause high morbidity and mortality rates in hospitals or healthcare
facilities worldwide (Hughes et al., 2005). The etiologic agents of NI can spread easily
in hospitals through the air, medical equipment or hands of healthcare workers and can
cause infection because of the low immune system of hospitalized patients. Among all
major complications that happened in the hospital, nosocomial infections take amount
of 50%; the infection reasons are medication errors, patient falls, and other events
(Becker et al., 1987). In the USA, there are roughly 1.7 million NI and 99,000 deaths
each year (Pollack, 2010). In Europe, Gram-negative infections are estimated to two-
thirds of the 25,000 deaths each year (Pollack, 2010). According to Hughes et al.
(2005), the rate of NI is 13.9% among 535 patients surveyed in the University of
Malaya Medical Center. Bacterial agents, viruses, fungi, and parasites are recognized as
sources of nosocomial infections, among them, bacterial agents are the most commonly
recognized cause of hospital-acquired infections. Some of the nosocomial bacteria are
antibiotic resistant and a majority of the antimicrobial resistance problems are typically
associated with gram-positive nosocomial pathogens (Singh et al., 2006). There are lots
of bacteria species in the group of nosocomial pathogens, and these include
Enterococcus spp., Escherichia coli, Pseudomonas spp., Staphylococcus aureus.
Klebsiella pneumoniae, Pseudomonas aeruginosa, and Acinetobacter baumannii are
4

three common pathogens that are often associated with NI.
2.2 Klebsiella pneumoniae
Klebsiella pneumoniae (K. pneumoniae) is an encapsulated, rod-shape, non-motile,
Gram-negative bacterium of the family of Enterobacteriaceae, causes pneumonia,
bacteremia and meningitis. It was first found in the sputum of lobar pneumonia in 1882
by Friedlande, so, it is also called Friedlande bacillus. K. pneumoniae is the most
important member in the Klebsiella spp. About 95% of the Klebsiella infections are
caused by K. pneumoniae. A survey on the data of an adult medical-surgical ICU ward
of a University Hospital and two governmental hospitals in Malaysia from October
2003 to December 2006 showed that the most common causative pathogen was K.
pneumoniae (Katherason et al., 2009). K. pneumoniae is responsible for 4% - 8%
proportion for the respiratory NI (Diancourt et al., 2005). It can not only cause
pneumonia, but also can cause urinary tract infection, biliary tract infection, septicemia
and meningitis. This infection is more common among elderly, malnutrition, chronic
alcoholism, and chronic bronchial-lung disease patients. A study in Malaysia and Japan
estimated that the rate of this infection in elderly persons is 15%-40% (Umeh et al.,
2002). K. pneumoniae often exists in human upper respiratory tract and intestinal tract,
when the immunity of human body is reduced, the pathogen can go into the lungs
through respiratory tract and cause big leaf lesions or lobular fusion. K. pneumoniae is
the fourth or fifth most common cause of pneumonia and bacteremia, respectively
(National Nosocomial Infections Surveillance (NNIS) System Report, 2003). This
5

bacterium even have the cephalosporin resistance strains which caused an outbreak in
New York City previously, this kind of strain is resistant to almost every common
antibiotics and the control of this strain is very crucial (Bratu et al., 2005). The
mortality due to pneumonia caused by K. pneumoniae is 50% (Umeh et al., 2002). For
the detection of K. pneumoniae, there are a lot of published papers based on Polymer
Chain Reaction (PCR). The genes that used for PCR primer target were rpoB (Chander
et al., 2011), pehX (Kovtunovych et al., 2003), and gyrA (Brisse and Verhoef, 2001).
2.3 Pseudomonas aeruginosa
Pseudomonas aeruginosa (P. aeruginosa) is a rod-shaped Gram-negative obligatory
aerobic bacterium, belongs to the family of Pseudomonadaceae (Schwartz et al., 2006).
It is one of the top three opportunistic pathogens which are capable of causing NI when
the host's resistance is low especially when there is a burnt-wound in the patient’s body
(Stover et al., 2000) and is associated with cystic fibrosis (Filho et al., 1999). A lot of
serious infections like pneumonia, otitis, endocarditis, septicemia and keratitis are
caused by P. aeruginosa (Lavenir et al., 2007). Most P. aeruginosa are multiple drug-
resistant and it exists widely in nature. P. aeruginosa strains also have developed
resistance against antibiotics such as fluoroquinolones and even disinfectant (Schwartz
et al., 2006).
6

2.4 Acinetobacter baumannii
Acinetobacter baumannii (A. baumannii) is a Gram-negative bacterium, which lives
freely in water, soil and human skin, especially hands (Rungruanghiranya et al., 2005).
The simplicity of its growth requirements and its high tolerance of environmental
conditions results in more outbreaks caused by this pathogen (Chang et al., 2009). In
Malaysia, the prevalence rate of nosocomial infection was 13.9% (Hughes et al., 2005).
It can cause pneumonia, urinary tract infection, otitis media, catheter-related infection,
central nervous system infection, peritonitis, and primary bloodstream infection (Levin
et al., 1999). The main pathway of the infection is through the hospital equipment,
especially the equipment which has direct contact with blood. The resistance of A.
baumannii in fluoroquinolones, aminoglycosides and broad-spectrum b-lactams has
been reported (Koeleman et al., 2001) and from the surveillance carried out from 2004
to 2009 in 36 countries, the resistance rates of A. baumannii are all above 50%
(Rosenthal et al,. 2012).
2.5 Conventional detection methods for Nosocomial pathogens
Nowadays, the treatment of NI is more difficult due the multidrug resistance property
of these bacterial pathogens and the rapid spread of nosocomial pathogens will
eventually increase the difficulty in treatment due to delayed detection and diagnosis.
Therefore, there is a need to develop a more rapid detection system to avoid further
complications. Hence, different detection methods such as conventional culture method,
7

PCR, real-time PCR and ELISA have been developed for the detection of nosocomial
pathogens. A faster and accurate identification of the etiologic agents would be needed
for prompt treatment of hospitalized patients.
Conventional culture method is time consuming and the results may be subjective,
more than 1 day will be needed with this method. Now, PCR is the most common
method applied in detection of bacterial pathogens. PCR is more sensitive, specific and
faster compared to conventional blood culture methods. However, PCR involves
multiple steps and requires special equipment such as the thermocycler and an
electrophoretic system. Furthermore, this technique is also dependent on the skill of the
person who performs PCR (Yamazaki, 2009). Another advanced diagnostic method,
real-time PCR is more rapid and sensitive compared to PCR but requires an expensive
thermal cycler with a fluorescence detector and the reagents are costly, and therefore,
this method is limited diagnostic laboratories with sufficient resources (Mullah et al.,
1998). For ELISA, the detection requires a high population of the target pathogen
(Chapman et al., 2001).
2.6 Loop-mediated isothermal amplification (LAMP) method
An alternative method called the loop-mediated isothermal amplification (LAMP)
was recently developed to circumvent the problems in identification and detection of
specific bacteria. This method can detect the pathogens rapidly and effectively under
isothermal condition (Notomi et al., 2000). The nucleotide will amplified in a fixed
temperature between 60℃ to 65℃ in even less than 60 min, and the products have 8

many types of structures in large amount (Hara-Kudo et al., 2006). At the beginning,
this method can be done only with a heating block and does not need extra detection
step as the product can be viewed visually (Allison, 2008). As the development of this
method, LAMP machine has been produced, inside the machine there is a turbimeter,
and can show the turbidity graph during the reaction. Also, the LAMP machine does not
need a lot of space, and even small lab can have it easily.
LAMP assay include a DNA polymerase and a set of four specially designed primers
that recognize a total of six distinct sequences on the target DNA. The mechanism of
LAMP method is shown in Figure 2.1. An inner primer (FIP) containing sequence of the
sense and antisense strands of the target DNA initiates LAMP. The following strand
displacement DNA synthesis primed by an outer primer (F3) releases a single-stranded
DNA. This serves as template for DNA synthesis primed by the second inner (BIP) and
outer primers (B3) that hybridize to the other end of the target, which produces a stem-
loop DNA with a stem twice as long. The cycling reaction continues with accumulation
of 109 copies of target in less than an hour (Notomi et al., 2000).
9
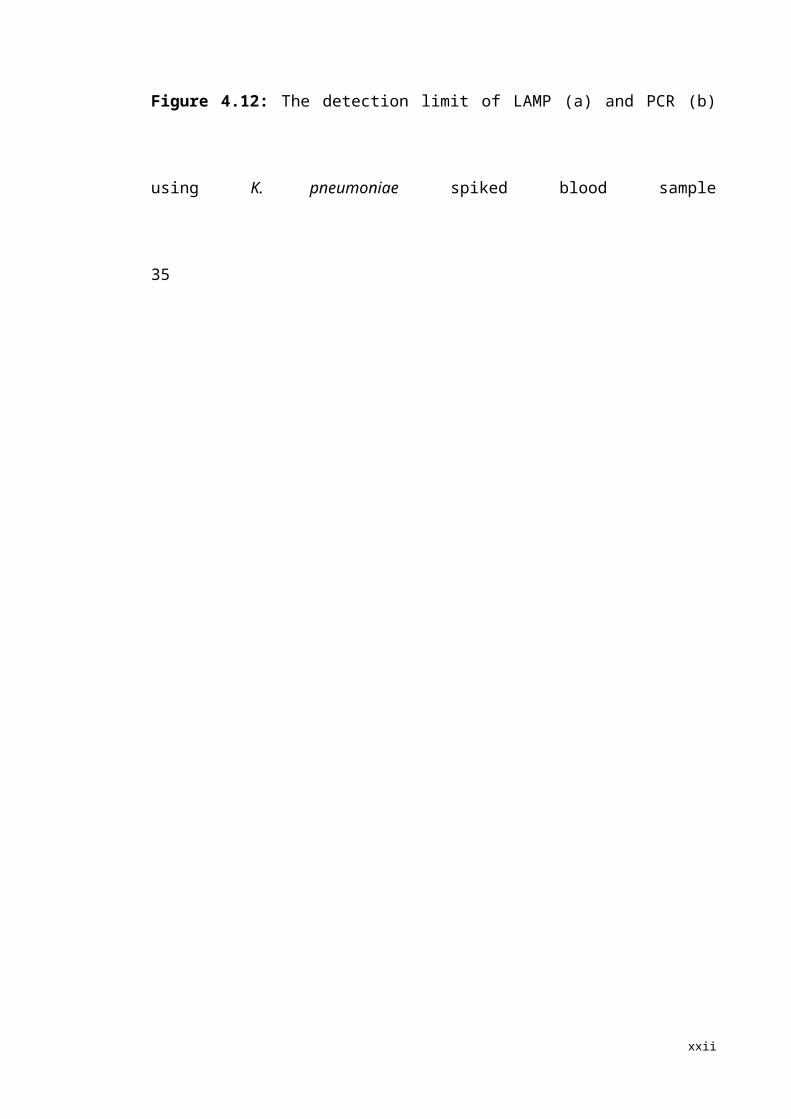
Figure 2.1: Principle of LAMP (adapted from Notomi et al., 2000)
10

LAMP is extensively used in the detection of food-born pathogens (Song et al.,
2006, Zhao et al., 2010), nosocomial pathogens (Hill et al., 2008, Lim et al., 2013),
virus (Paride et al., 2005, Poon et al., 2006), and parasite (Kuboki et al., 2003).
11

CHAPETER 3: METHODOLOGY
3.1 Materials
3.1.1 Bacterial strains
A total of 107 bacterial strains including 30 A. baumannii, 30 P. aeruginosa, 30 K.
pneumoniae, 3 Escherichia coli, 4 Shigella spp., 4 Vibrio spp., and 6 Salmonella spp.
were revived from the glycerol stocks. All the strains were obtained from the culture
collection of the Laboratory of Biomedical Science and Molecular Microbiology,
Institute of Graduate Studies, University of Malaya (Appendix 1). The purity of the
strains was checked by streaking the culture on appropriate selective media. The P.
aeruginosa strains were checked by CHROMagar, A. baumannii and K. pneumoniae
were checked by MacConkey agar.
3.1.2 Chemicals and reagents
All the information about Chemicals and the preparation of the growth media,
buffers and reagent used in this study are listed in Appendix 2.
3.2 Methods
3.2.1 DNA template
DNA extraction was performed on the overnight culture by direct cell lysate method.
Briefly, a loopful of colonies was suspended in 100 µl double distill water. The
12

suspension was boiled at 99℃ for 5 min, snapped cool on ice for 10 min and centrifuge
at 13,400 rpm for 3 min. The supernatant was used as DNA template for LAMP
analysis. And then, the DNA template was keep in fridge at -20℃.
3.2.2 Primer design
Prior to primer design, selected genes would be checked by BLAST
(http:/www.ncbi.nih.gov) to minimize the similarity to the other species. The sequence
of gltA (Assession no. NC_011595.1), tonB (NC_012731.1) and 16S rRNA-processing
protein rimM (NC_009656.1) were retrieved from NCBI Genbank for the
oligonucleotide primers design for A. baumannii, K. pneumoniae and P. aeruginosa,
respectively. Four primers including one forward inner primer (FIP), one backward
inner primer (BIP), one forward outer primer (F3) and one backward outer primer (B3)
were designed by using Primer Explorer V4 (EIKEN CHEMICAL CO., LTD. Japan).
For P. aeruginosa, another two loop primers-LF, LP were generated. The specificity of
the designed primers was determined by using insilico PCR
(http://insilico.ehu.es/PCR/).
3.2.3 LAMP reaction
For A. baumannii: The reaction mixture in a total volume of 25 µl contained 12.5 µl
RM (EIKEN CHEMICAL CO.),40 pmol (each) of FIP and BIP, 5 pmol for each of F3
and B3, 1µl DNA polymerase and 2.5 µl DNA template. The reaction was incubated in 13

Loopamp EXIA machine the real-time turbidimeter (EIKEN CHEMICAL CO., LTD),
under 62 ℃ for 90 min and followed by inactivation at 80 ℃ for 2 min. Each run
contained positive and negative control.
For K. pneumoniae: The reaction mixture in a total volume of 25 µl contained 12.5
µl RM (EIKEN CHEMICAL CO.),40 pmol (each) of FIP and BIP, 5 pmol for each of
F3 and B3, 1µl DNA polymerase and 2.5 µl DNA template. The reaction tube was then
incubated in the Loopamp EXIA machine the real-time turbidimeter (EIKEN
CHEMICAL CO., LTD), under 65 ℃ for 90 min and followed by inactivation at 80 ℃ for 2 min. Each run contained positive and negative controls.
For P. aeruginosa: The reaction mixture in a total volume of 25 µl contained 12.5 µl
RM (EIKEN CHEMICAL CO.),40 pmol (each) of FIP and BIP, 5 pmol for each of F3
and B3, 20 pmol (each) of LF and LB, 1µl DNA polymerase, 1 µl double distilled water
and 2.5 µl DNA template. The reaction was incubate in Loopamp EXIA machine the
real-time turbidimeter (EIKEN CHEMICAL CO., LTD), under 65 ℃ for 60 min and
followed by inactivation at 80℃ for 2 min. Each run contained positive and negative
controls.
3.2.4 Data analysis
The analysis of the LAMP product can be visualized by three methods:
14

3.2.4.1 Real time monitoring by turbidity change
The visualization of the LAMP product was checked by turbidity. Positive results
were indicated by the increase in turbidity above the threshold (0.1) within 70 min. No
changes in turbidity indicated negative result with the Loopamp EXIA machine.
3.2.4.2 Formation of pellet method
The reaction tube was briefly spun for half a min. The presence of a pellet indicated a
positive result, while the absence of a pellet indicated negative result.
3.2.4.3 Dye method
An aliquot of 1 µl of SYBR Green (10×dilution) was added into the LAMP product
and the change of color from orange to green indicated a positive reaction, A negative
result was indicated by no change in the orange color.
3.2.5 PCR
PCR was carried out in parallel with the LAMP assay to compare their specificity and
sensitivity. PCR primers used for each of these three bacterial species are shown in
Table 3.1. The target gene for PCR test of A. baumannii was blaOXA-51 gene since it was
reported this gene is universally present in this bacterium (Turton et al., 2006). PCR
reaction was carried out in a total volume of 25 µl, containing 1×PCR buffer, 1.2 mM
MgCl2, 120 mM each dNTPs, 0.5 µM of each primer, 1 U of Taq DNA polymerase and
5 µl of DNA sample. The cycling conditions consisted of an initial denaturation at of
15

94℃ for 5 min followed by 30 cycles of 25 s at 94℃, 40 s at 52℃, 50 s at 72℃ and
the final extension at 6 min at 72℃.
For K. pneumoniae, the target gene was mdh (Thong et al., 2011). The PCR mix
(25µl) consisted of 1×PCR buffer, 1.4 mM MgCl2, 140 mM each of the four
nucleotides, 5 µl (20ng) DNA sample, 0.3 µM of each primer and 1U Taq DNA
polymerase. The cycling conditions consisted of an initial denaturation at 95℃ for 5
min, 30 cycles of 95℃ for 1 min, 53℃ for 1min and 72℃ for 1 min, followed by an
extension of 72℃ for 5 min.
For P. aeruginosa, the target gene was algD (Da et al., 1999) gene. The PCR mix
(25µl) consisted of 1×PCR buffer, 2 mM MgCl2, 200 mM each of the four nucleotides,
5 µl (20ng) DNA sample, and 0.4 µM of each primer and 1U Taq DNA polymerase. The
cycling conditions consisted of an initial denaturation at 94℃ for 5 min, 30 cycles of
94℃ for 5 min, 60℃ for 1min and 72℃ for 1 min, followed by an extension of 72℃ for 7 min.
The PCR products were monitored by 1.5% agarose gel electrophoresis and then
visualized under a UV transiluminator after staining with ethidium btomide (EtBr) for
30 min.
Table 3.1: The information of PCR primers
Gene Sequence reference
blaOXA-51 F: 5'TAATGCTTTGATCGGCCTTG3'
R: 5'TGGATTGCACTTCATCTTGG3'
Turton et al., 2006
Mdh F: 5'GCGTGGCGGTAGATCTAAGTCATA3' Thong et al., 201116

R: 5'TTCAGCTCCGCCACAAAGGTA3'
algD F: 5'TTCCCTCGCAGAGAAAACATC3'
R: 5'CCTGGTTGATCAGGTCGATCT3'
Da et al., 1999
3.2.6 Evaluation of sensitivity of LAMP assay in culture and blood samples
Culture sample: A colony of fresh bacterial culture was inoculated into 1 ml of LB
broth and incubated at 37℃ for 3 hours with agitation until the OD600 of the cell
cultures reached approximately 1. Then a 10-fold dilution was done. An aliquot of 100
µl of each dilution was spread on both LB agar and selective media for the CFU count
while another 100 µl was subjected to DNA extraction for LAMP and PCR assays.
Blood sample: A colony of fresh bacterial culture was inoculated into 1 ml of LB broth
and incubated at 37℃ for 3 hours with agitation until the OD600 of the broth reached
approximately 1. An aliquot of 100 µl of the culture was spiked in 900 µl blood (healthy
volunteer) and incubated at 37℃ for 2 h, followed by a 10-fold dilution. A 100 µl of
each dilution was spread on both LB agar and selective media for the CFU count while
another 100 µl was subjected to DNA extraction for LAMP and PCR assays.
17

CHAPTER 4: RESULTS
4.1 Primer designed for LAMP
Suitable genes for primer design were found from the literature. More than five
published papers about the detection of these three pathogens were found. After BLAST
analysis, only a few genes were suitable. The LAMP primer design software showed 5
primers for each suitable gene fragment. These primers were then subjected to in silico
test to determine specificity. The result of in silico is shown in Appendix 3-11. Due to
each LAMP primer has two sets, the in silico was run twice to check the specificity. The
nucleotide sequences of the primers were being filed for patent. The primers used for
optimization were synthesized by a commercial company, BIONEER Company
(Korea). For P. aeruginosa, an additional set of loop primer was synthesized to reduce
the amplification time.
4.2 Acinetobacter baumannii
4.2.1 Optimized LAMP assay
The temperature and time of reaction were optimized for A. baumannii. Four different
temperatures were tested for A. baumannii which included 60℃, 62℃, 63℃ and
65℃. At 62℃, the assay took the shortest time to obtain amplification. Generally, the
optimized temperature for A. baumannii was 62℃ (Fig 4.1) and the whole reaction time
was fixed at 90 min. Fig 4.1 shows the temperature optimization for the positive control
strain, AC081229. The curves were obtained directly from the screen of the Loopamp
18

EXIA machine. The reaction amplification time for A. baumannii ranged from 60 to 80
min for different strains.
0 10 20 30 40 50 60 70 80 90 1000
0.05
0.1
0.15
0.2
0.25
0.3
0.35
0.4
60℃62℃63℃65℃
Time (min)
T u r b i d it y ( 4 0 0 n m )
Fig 4.1: Comparison of LAMP results at different temperatures of A. baumannii
4.1.2 Sensitivity and specificity of the method
For analysis of the LAMP results, three methods were used for end point detection.
Firstly, when the reaction tube was briefly spun, a white pellet was formed at the bottom
of the tube for the sample which showed positive amplification. For tube with negative
amplification, no precipitate formed was observed. Secondly, a color change was
observed when 1 µl of SYBR Green Ι was added to the reaction tube (Fig 4.2). Thirdly,
when the amplification products were analyzed on agarose gel electrophoresis, a
positive amplification was indicated by smears (multiple bands) (Fig 4.3). If there was
no amplification, no band or smear was shown on the gel.
19

The LAMP assay correctly identified 30 positive strains and no positive reaction was
observed for all 20 non-A. baumannii strains with Loopamp EXIA machine. The non-
A. baumannii strains were Klebsiella pneumoniae, E. coli, Vibrio, Shigella and
Salmonella. In order to know the real specificity of the method, a comparison was made
between LAMP and PCR. The results for these two methods are shown in Table 4.2.
Both the LAMP and PCR had the same specificity that all A. baumannii gave positive
amplification while non-A. baumannii strains were not amplified.
Table 4.1: PCR and LAMP results of A. baumannii strains
Species/SubspeciesIdentification
codeSpecific
PCR resultsLAMP-result
Real Time Gel SYBR Green I
A. baumannii AC/060110 + + + +A. baumannii AC/0612-17 + + + +A. baumannii AC/0701-11 + + + +A. baumannii AC/0702-5 + + + +A. baumannii AC/0702-17 + + + +A. baumannii AC/0703-14 + + + +A. baumannii AC/0703-21 + + + +A. baumannii AC/0711-7 + + + +A. baumannii AC/0801-6 + + + +A. baumannii AC/0801-11 + + + +A. baumannii AC/0801-13 + + + +A. baumannii AC/0802-1 + + + +A. baumannii AC/0802-4 + + + +A. baumannii AC/0802-14 + + + +A. baumannii AC/0802-20 + + + +A. baumannii AC/0803-15 + + + +A. baumannii AC/0804-19 + + + +A. baumannii AC/0812-29 + + + +A. baumannii AC/0901-5 + + + +A. baumannii AC/0901-14 + + + +A. baumannii AC/0901-36 + + + +A. baumannii AC/0901-37 + + + +A. baumannii AC/0902-5 + + + +
20

A. baumannii AC/0902-6 + + + +A. baumannii AC/0902-13 + + + +A. baumannii AC/0902-14 + + + +A. baumannii AC/0902-15 + + + +A. baumannii AC/0903-15 + + + +A. baumannii AC/0904-3 + + + +A. baumannii AC/0905-31 + + + +
Klebsiella pneumoniae
PS81 - - - -
Klebsiella pneumoniae
PS92 - - - -
Klebsiella pneumoniae
PS88 - - - -
E. coli P49 - - - -E. coli P41 - - - -E. coli BS4 - - - -Vibrio VPD21 - - - -Vibrio VPD22 - - - -Vibrio VPD26 - - - -Vibrio VPD27 - - - -
Shigella flexneri TH32/98 - - - -Shigella sonnei TC3/99 - - - -
Shigella flexmeri TH23/97 - - - -Shigella flexneri Y
variantTH5/02 - - - -
Salmonella S.Meto303/94 - - - -Salmonella S.Oke-nara - - - -Salmonella S.LOM - - - -Salmonella S.Bevis - - - -Salmonella S.Hvrt - - - -Salmonella SAB79 - - - -
21

Fig 4.2: Detection of the LAMP products using SYBR Green Ι.N indicates negative sample, P indicates positive sample. Negative result is indicated by no color change while positive result is indicated by a color change to green.
Fig 4.3: Visualization of LAMP products by agarose gel electrophoresis. Lane L, 100-bp DNA ladder; Lane N, negative control; Lane 1, AC071107; Lane 2, AC081229; Lane 3, AC090203; Lane 4, AC090215; Lane 5, BS4 (E. coli); Lane 6, p41( E. coli) ; Lane 7, p49( E. coli) ; Lane 8 PS81 (K. pneumoniae); Lane 9, PS88. (K. pneumoniae). A positive LAMP result is indicated by smearing (lanes 1-4) while negative results have no amplification (lanes 5-9).
22
N N P P
L N 1 32 4 5 6 7 8 99
L

In order to know the sensitivity or the detection limit of the LAMP assay, a parallel
test with PCR was carried out. DNA from an aliquot of each dilution was tested with the
LAMP and PCR. The detection limit of LAMP assay and PCR was 5.5×104 CFU/ml
(equal to 1375 CFU per reaction) and 5×105 CFU/ml, respectively. This shows that the
LAMP method for detection of A.baumannii was 10-fold more sensitive than PCR (Fig
4.4).
(a) 0 20 40 60 80 100 120
0
0.05
0.1
0.15
0.2
0.25
0.3
0.3512345
Time(mins)
Tu
rbi
dit
y( 40 0n m )
(b)
Fig 4.4: Sensitivity of the LAMP assay (a) and PCR (b) for A. baumannii AC090215.1=5.5×107cfu/ml; 2=5.5×106; 3=5.5×105cfu/ml; 4=5.5×104cfu/ml; 5=5.5×104cfu/ml. Fig (a): Amplified product with LAMP is seen for lanes 1 - 4.Fig (b): Amplified product with PCR is seen for lanes 1 - 3.
23
1 2 3 Ladder
353bp
4 5

4.1.3Evaluation of LAMP on Spiked blood sample
The performance of PCR and LAMP assay on spiked blood samples was comparable.
Blood specimen (from the author) was taken by a doctor at the student and staff clinic of
University of Malaya. The detection limit of A. baumannii for both PCR and LAMP
assays was 5.5×105 CFU/ml which equals to 1100 CFU per reaction.
(a)
0 20 40 60 80 100 1200
0.05
0.1
0.15
0.2
0.25
0.3
0.35
123
Time (mins)
T u r b i d it y ( 4 0 0 n m )
(b)
Fig 4.5: The detection limit of LAMP (a) and PCR (b) using A. baumannii spiked blood sample.1 = 5.5×106 CFU/ml; 2 = 5.5×105 CFU/ml; 3 = 5.5×104 CFU/ml.
24
Ladder 1 2
353bp
3

Amplified product is seen for lanes 1-2.
4.2 P. aeruginosa
4.2.1 Optimized LAMP assay
Five temperatures were tested to determine the optimum temperature. These
included 60℃, 62℃, 63℃, 64℃and 65℃ (Fig 4.6). Figure 4.6 shows that at 65℃ the reaction had the shortest amplification time. The primer for P. aeruginosa was
different from the primers for A. baumannii and K. pneumoniae. This set of primer was
combined with loop primers, so the amplification time would be much shorter. Fig 4.7
shows the difference between the amplification with and without loop primer. The
whole reaction was run for 60 min at 65℃. With the inclusion of loop primers, the
amplification of P. aeruginosa began at 25 min, loop primer made a 20 min saving of
the reaction. Compared to the reaction of A. baumannii and K. pneumoniae, this
reaction was faster. This shows that the ability of loop primer was to shorten the
detection time.
25

0 10 20 30 40 50 60 70 80 900
0.1
0.2
0.3
0.4
0.5
0.660℃62℃63℃64℃65℃
Time (min)
Tur
bid-
ity
(40
0nm
)
Fig 4.6: Comparison of LAMP results for P. aeruginosa PS19 at different temperatures
0 10 20 30 40 50 60 70 80 90 1000
0.1
0.2
0.3
0.4
0.5
0.6with loop primerwithout loop primer
Time (mins)
T u r b i d it y ( 4 0 0 n m )
Fig 4.7: Comparison of LAMP results with or without loop primer on PS19
26

4.2.2 Sensitivity and specificity of the method
In order to test the specificity of the assay, 50 bacterial cultures were used. Positive
reaction was observed with 30 P. aeruginosa strains while no amplification was
observed for the 20 non-P. aeruginosa strains which included Klebsiella pneumoniae,
E. coli, Vibrio, Shigella and Salmonella.
PCR was also used on all of these strains. Table 4.2 shows the results of these two
methods. Both the LAMP and PCR had the same specificity, in which all P. aeruginosa
gave positive amplification while non- P. aeruginosa strains were not amplified.
Table 4.2: PCR and LAMP result of P. aeruginosa strains
Species/SubspeciesIdentification
codeSpecific
PCR resultsLAMP-result
Real Time Gel SYBR Green I
P. aeruginosa PS2 + + + +P. aeruginosa PS16 + + + +P. aeruginosa PS19 + + + +P. aeruginosa PS20 + + + +P. aeruginosa PS23 + + + +P. aeruginosa PS67 + + + +P. aeruginosa PS98 + + + +P. aeruginosa PS100 + + + +P. aeruginosa PS102 + + + +P. aeruginosa PS103 + + + +P. aeruginosa PS105 + + + +P. aeruginosa PS108 + + + +P. aeruginosa PS110 + + + +P. aeruginosa PS239 + + + +P. aeruginosa PS339 + + + +
27

P. aeruginosa PS341 + + + +P. aeruginosa PS362 + + + +P. aeruginosa R01 + + + +P. aeruginosa R02 + + + +P. aeruginosa R04 + + + +P. aeruginosa 1182 + + + +P. aeruginosa 1186 + + + +P. aeruginosa 1260 + + + +P. aeruginosa 1288 + + + +P. aeruginosa B14141 + + + +P. aeruginosa B14143 + + + +P. aeruginosa B14128 + + + +P. aeruginosa B14262 + + + +P. aeruginosa B14349 + + + +P. aeruginosa BF2087 + + + +
Klebsiella pneumoniae
PS81 - - - -
Klebsiella pneumoniae
PS92 - - - -
Klebsiella pneumoniae
PS88 - - - -
E. coli P49 - - - -E. coli P41 - - - -E. coli BS4 - - - -Vibrio VPD21 - - - -Vibrio VPD22 - - - -Vibrio VPD26 - - - -Vibrio VPD27 - - - -
Shigella flexneri TH32/98 - - - -Shigella sonnei TC3/99 - - - -Shigella flexneri TH23/97 - - - -
shigella flexneri Y variant
TH5/02 - - - -
Salmonella S.Meto303/94 - - - -Salmonella S.Oke-nara - - - -Salmonella S.LOM - - - -Salmonella S.Bevis - - - -Salmonella S.Hvrt - - - -Salmonella SAB79 - - - -
The detection limit of this assay on bacterial culture was 3.6×104 CFU/ml which
equals to 72 CFU per reaction. For PCR, the limitation of detection was 3.6×107
28

CFU/ml. So, this LAMP assay was 1000-fold more sensitive than conventional PCR
(Fig 4.8).The sensitivity test was based on the 10-fold serial dilution, and the initial
inoculum of P. aeruginosa was 3.6×106 CFU/ml.
29

(a)
0 10 20 30 40 50 600
0.05
0.1
0.15
0.2
0.25
0.3
0.35
0.4
0.4512345
Time(mins)
T u r b i d i t y ( 4 0 0 n m )
(b)
Fig 4.8: Sensitivity of the LAMP assay (a) and PCR (b) for P. aeruginosa.PS19.1 = 3.6×107 CFU/ml; 2 = 3.6×106 CFU/ml; 3 = 3.6×105 CFU/ml; 4 = 3.6×104 CFU/ml; 5 = 3.6×103 CFU/ml; N = negative control (distilled water). Fig (a): Amplified product is seen for lanes 1-4. Fig (b): Amplified product is seen for lanes 1.
4.2.3Evaluation of LAMP on spiked blood
In order to check the sensitivity of the LAMP, the test was carried out with DNA
prepared serially diluted spiked blood samples. The detection limit of LAMP assay for
P. aeruginosa was 7.7×104 CFU/ml which means 154 CFU per reaction (Fig 4.9). The
30
Ladder
N 1 2 3 4 5
520bp

detection limit for PCR was 7.7×107 CFU/ml. Therefore, the sensitivity of LAMP assay
for P. aeruginosa was much higher than conventional PCR.
(a)
0 10 20 30 40 50 60 70-0.010.040.090.140.190.240.290.340.390.440.49
123456
Time (min)
T u r b i d it y ( 4 0 0 n m )
(b)
Fig 4.9: The detection limit of LAMP (a) and PCR (b) using P. aeruginosa spiked blood sample.Fig (a): 1 = negative control (distilled water); 2 = 7.7×107 CFU/ml; 3 = 7.7×106 CFU/ml; 4 = 7.7×105 CFU/ml; 5 = 7.7×104 CFU/ml; 6 = 7.7×103 CFU/ml. Amplified product is seen for lanes 2-5.Fig (b): N = negative control (distilled water); 1 = 7.7×107 CFU/ml; 2 = 7.7×106
CFU/ml; 3 = 7.7×105 CFU/ml; 4 = 7.7×104 CFU/ml; 5 = 7.7×103 CFU/ml.Amplified product is seen for lanes 1.
31
Ladder N 1 2 3 4 5
520bp

4.3 K. pneumoniae
4.3.1 Optimized LAMP assay
Five temperatures were tested to determine the optimum temperature. These
included 60℃, 62℃, 63℃, 64℃ and 65℃. At 65℃, the reaction had the shortest
amplification time. Fig 4.10 shows the turbidity curve of the reaction which amplified
at 65℃. No loop primer was designed for K. pneumoniae LAMP assay, because the
amplification time was less than 60 min.
0 10 20 30 40 50 60 70 80 90 1000
0.1
0.2
0.3
0.4
0.5
0.6
0.7
Time (min)
T u r bi di ty
( 4 0 0 n m )
Fig 4.10: Optimized temperature (65℃) for K. pneumoniae LAMP detection
4.3.2 Sensitivity and specificity of the method
In order to test the specificity of the assay, 50 bacterial cultures were used. Positive
reaction was observed with 30 K. pneumoniae strains while no amplification was
observed for the 20 non- K. pneumoniae strains which included P. aeruginosa, E. coli,
Vibrio, Shigella and Salmonella.
32

PCR was also used on all of these strains. Table 4.3 shows the result of these two
methods. All the LAMP results were confirmed by three different methods, there were
turbidity method, fluorescence (SYBR Green 1) and precipitation. The results showed
that both LAMP and PCR had highly specificity in the detection of P. aeruginosa.
Table 4.3: Comparison of PCR and LAMP result of K. pneumoniae strains
Species/SubspeciesIdentification
codeSpecific
PCR resultsLAMP-result
Real Time Gel SYBR Green I
K. pneumoniae PS19 + + + +
K. pneumoniae PS23 + + + +
K. pneumoniae PS31 + + + +
K. pneumoniae PS35 + + + +
K. pneumoniae PS36 + + + +
K. pneumoniae PS49 + + + +
K. pneumoniae PS50 + + + +
K. pneumoniae PS51 + + + +
K. pneumoniae PS80 + + + +
K. pneumoniae PS82 + + + +
K. pneumoniae PS83 + + + +
K. pneumoniae PS86 + + + +
K. pneumoniae PS88 + + + +
K. pneumoniae PS90 + + + +
K. pneumoniae PS92 + + + +
K. pneumoniae PS96 + + + +
K. pneumoniae PS138 + + + +
K. pneumoniae PS156 + + + +
K. pneumoniae K09-24 + + + +
K. pneumoniae K09-25 + + + +
K. pneumoniae K10-03 + + + +
K. pneumoniae K10-04 + + + +
K. pneumoniae K10-05 + + + +
K. pneumoniae K11-01 + + + +
K. pneumoniae K11-02 + + + +
K. pneumoniae K11-03 + + + +
K. pneumoniae K11-04 + + + +
K. pneumoniae K11-05 - - - -
33

K. pneumoniae K11-06 - - - -K. pneumoniae K11-09 - - - -P. aeruginosa B14262 + + + +P. aeruginosa B14349 + + + +P. aeruginosa BF2087 + + + +
E. coli P49 - - - -E. coli P41 - - - -E. coli BS4 - - - -Vibrio VPD21 - - - -Vibrio VPD22 - - - -Vibrio VPD26 - - - -Vibrio VPD27 - - - -
Shigella flexneri TH32/98 - - - -Shigella sonnie TC3/99 - - - -Shigella flexneri TH23/97 - - - -
shigella flexneri Y variant
TH5/02 - - - -
Salmonella S.Meto303/94 - - - -Salmonella S.Oke-nara - - - -Salmonella S.LOM - - - -Salmonella S.Bevis - - - -Salmonella S.Hvrt - - - -Salmonella SAB79 - - - -
The detection limit of culture sample was 7×103 CFU/ml, it means 17.5 CFU per
reaction, and it was the same with PCR method (7×103 CFU/ml) (Fig 4.11). The initial
inoculum of K. pneumoniae was 7×107 CFU/ml.
34

(a)
0 10 20 30 40 50 60 70 80 90 100-0.1
0
0.1
0.2
0.3
0.4
0.5
0.6
1
2
3
4
5
6
Time (min)
T ur bi di ty (4 0 0 n m )
(b)
Fig 4.11: Sensitivity of the LAMP assay (a) and PCR (b) for K. pneumoniae K10-04.Lane 1 = negative control (distilled water); lane 2 = 7×107 CFU/ml; lane 3 = 7×106
CFU/ml; lane 4 = 7×105CFU/ml; lane 5 = 7×104 CFU/ml; lane 6 = 7×103 CFU/ml. Amplified products are seen for lanes 2-6.
4.3.3 Evaluation of LAMP on spiked blood
The detection limit of both LAMP and PCR on spiked blood sample was the same at
1.4×104 CFU/ml which equals to 35 CFU per reaction (Fig 4.12). The sensitivity of
LAMP assay was reduced for spiked blood sample.
35
520bp
1 2 3 4 5 6Lader

(a)
0 10 20 30 40 50 60 70 80 90 100-0.1
0
0.1
0.2
0.3
0.4
0.5
0.6123456
Time (min)
T u r b i d it y ( 4 0 0 m n )
(b)
Fig 4.12: The detection limit of LAMP (a) and PCR (b) using K. pneumoniae spiked blood sample.1 = negative control (distilled water); 2 = 1.4×108 CFU/ml; 3 = 1.4×107 CFU/ml; 4 = 1.4×106 CFU/ml; 5 = 1.4×105 CFU/ml; 6 = 1.4×104 CFU/ml. Fig (a): Amplified product is seen for lanes 2-6. Fig (b): Amplified product is seen for lanes 2-5
36
520bp
1 2 3
N
4 5 6 Lader

CHAPTER 5: DISCUSSION
LAMP assay was optimized for the detection for A. baumannii, K. pneumoniae and
P. aeruginosa. These are three of the many clinically important nosocomial pathogens.
Currently, there are many interests to develop rapid and cheap detection for these
pathogens. Since the introduction of the LAMP method, numerous researches on its
application have been reported (Iwamoto et al., 2003; Poon et al., 2006; Song et al.,
2006).
In this study, a lot of genes were checked to determine their suitability for the
design of appropriate primers for LAMP. A. For A. baumannii, genes efp, fumC, mutY,
ppa (Ecker et al., 2006) and gltA (Thong et al., 2011) were checked. For K.
pneumoniae, the genes tested were mdh (Thong et al., 2011), ABC transport permease,
gyrA, parC (Deguchi et al., 1997) and ntrA (Anbazhagan et al., 2010). For P.
aeruginosa, genes tested were oprL (Vos et al., 1997), rimM, proC (Savli et al., 2003)
gyrB, and toxA (Lavenir et al., 2007). However, after analysis with the BLAST
program, most of these genes were not suitable as they lack specificity. Only genes that
showed 100% specificity were used in the primer design for LAMP analysis.
Eventually, the gltA, ABC transport permease and rimM was selected separately for A.
baumannii, K. pneumoniae and P. aeruginosa primer design respectively.
For the DNA extraction, two methods have been used were the extraction kit and
direct boiling method. The procedure DNA extraction kit was more complicated and
took a longer time to obtain DNA. On the other hand, the simply boiling method was
much easier and had higher DNA concentration. For Gram negative bacteria, due to the
37

structure of its cell membrane, it can be simply lysed by boiling method. Therefore,
boiling method was used for the DNA extraction for further analysis.
The gltA gene which codes for the citrate (Si)-synthase of the cell was the target
gene for LAMP primer design for A. baumannii detection. The gltA gene is a
housekeeping gene for A. baumannii. It has been widely used as the target gene in the
PCR (Jiang et al., 2012), Mass Spectrometry (Mencacci et al., 2013) and multilocus
sequence typing (MLST) (Giannouli et al., 2010) for A. baumannii.
ABC transport permease gene is a gene (accession number was NC_011283.1)
codes for the ATP-binding cassette (ABC) transporter permease. The ABC transport
permease helps in the absorption of carbohydrate, peptide, amino acids and some other
nutrition. Other ABC transport related gene like the ycjV has been used as the target
gene in the DNA microarray technology for the detection of K. pneumoniae (Dome
´nech-Sa´nchez et al., 2006).
The rimM gene is the 16S rRNA-processing protein RimM coding gene. The
accession number of this gene is NC_002516.2. The RimM protein is associated with
the maturation of the 30S ribosomal subunit. It binds to ribosomal protein S19 which is
located at the head domain of the 30S subunit (Suzuki et al., 2007). 16s rRNA has
highly stability and specificity, and the size of the gene sequence is long enough for the
primer design. With the development of PCR and nuclear technology, 16S rRNA gene
detection technology has become a powerful tool for bacterial detection and
characterization.
LAMP is a complicated technique and many parameters, such as primers, DNA
38

polymerase, reagents, temperature and reaction time have to be considered for
evaluation. In this assay, only the temperature and reaction time were the parameters
needed to be optimized. The whole assay used the DNA amplification kit which was
purchased from EIKEN CHEMICAL and the concentrations of primers and DNA
polymerase used were pre-determined by the manufacturer.
The suggested condition for LAMP was 12.5µl of 2×Reaction Mix, 40 pmol FIP, 40
pmol BIP, 20 pmol Loop-F, 20 pmol Loop-B, 5 pmol F3, 5 pmol B3, 1.0µl Bst DNA
polymerase, distilled water and 2µl DNA template will be added. For this assay, two of
the pathogens (A. baumannii and K. pneumoniae) did not use the loop primer, and
another one (P. aeruginosa) used the loop primer, so the LAMP conditions for these
three pathogens were slightly different.
Usually, the reaction temperature for LAMP was from 60℃ to 65℃. So, a few
temperatures from 60℃ to 65℃ were tested to find the shortest amplification time. The
reaction time was set based on the amplification time and the slope of turbidity curve.
Overall, the optimization of these three assays was done by evaluating different
temperatures and time. The optimized temperatures for A. baumannii, K. pneumoniae,
and P. aeruginosa were 62℃, 65℃, and 65℃, respectively at the shortest reaction
times.
In this assay, the sensitivity of LAMP using spiked blood samples was lower than
using pure culture sample. This shows that some inhibitors in the blood may have
affected the efficiency in the binding of the primers to the target sites. There are three
major inhibitors identified in blood such as immunoglobulin G in plasma, haemoglobin
39

in erythrocytes and lactoferrin in leukocytes (Waleed 2000) for PCR.
The culture method is still the gold standard in clinical diagnosis. However, the
culture method is time-consuming and usually takes 9 days (Gleaves et al., 1985). The
specificity of culture detection is also not 100%. Ajao et al. (2011) compared various
media in the detection of A. baumannii. They found that MacConkey agar can only
detect 16 out of 18 Acinetobacter strains and the specificity is 89%.
In this study, the LAMP method took one and a half hours for confirmation and the
specificity of LAMP method was 100%. It can be applied either directly from an
unknown bacterial culture for confirmation or directly from a blood specimen for
identification.
Another commonly detection method used is PCR. Many findings have been
published to demonstrate the application of PCR in rapid detection of A. baumannii.
Turton et al. (2006) described a multiplex PCR for the detection of A. baumannii and K.
pneumoniae. Mosca et al. (2013) reported the use of RT-PCR to detect K. pneumoniae
carbapenemase (KPC)-producing strains. Similarly, detection of P. aeruginosa by
qPCR targeting ecfX gene (Colinon et al., 2013) was reported. The conventional PCR
and Multiplex PCR detection require further post PCR manipulation in that there is a
need to run gel electrophoreses to detect the end products. This will make the detection
more complicated and time-consuming. Moreover, the PCR reaction time is usually
more than 2 hours and also depends on the type of the machine. On the other hand,
LAMP reaction will only take less than one and a half hours, and one can see the
amplification curve during the reaction, which means it is a real time detection method.
40

Multiplex PCR is designed for the time saving detection which can detect more than one
pathogen in one single reaction, but this can result in low specificity and false result.
LAMP assay solved this problem with short time and high specificity.
There are also some other detection methods, like nucleic acid hybridization and
DNA microarray. But these methods are not widely used in the clinic or laboratory, due
to its complexity and the need for expertise.
In this study, LAMP assay is a really simple and effective method which has the
advantages of time saving, high sensitivity, high specificity and real time. It is a
potential method in the detection of A. baumannii, K. pneumoniae and P. aeruginosa.
However, the high cost of the machine and reagents is the limitation of this assay.
More research needs to be done to reduce of the cost of machine and reagents.
41

CHAPTER 6 CONCLUSION
Nosocomial infection is a real important problem in hospitals or health care
facilities throughout the world. For the conventional detection method for nosocomial
pathogens, there are so many disadvantages, so a relative new detection method-LAMP
was developed in this study for three main nosocomial bacterial pathogens namely, A.
baumannii, K. pneumoniae and P. aeruginosa.
1. A LAMP assay was developed for the detection of A. baumannii. The optimized
condition was 62℃ and amplified for 90 min. The sensitivity of LAMP was 5.5×104
CFU/ml, and it was 10-fold more sensitive than the normal PCR method (5.5×105
CFU/ml). The sensitivity of both LAMP and PCR were the same at 5.5×105 CFU/ml in
spiked blood samples. The assay shows highly specificity compared with PCR in the
evaluation of 50 strains.
2. A LAMP assay was developed for the detection of K. pneumoniae. The optimized
condition was 65℃ and amplified for 60 min. The sensitivity of LAMP was 7×103
CFU/ml, which was the same with normal PCR method. The sensitivity of both LAMP
and PCR were the same at 1.4×104 CFU/ml in spiked blood samples. The assay shows
highly specificity compared with PCR in the evaluation of 50 strains.
3. A LAMP assay was developed for the detection of P. aeruginosa. The optimized
condition was 65℃ and amplified for 90 min. The sensitivity of LAMP was 3.6×104
CFU/ml, and it was 1000-fold more sensitive than PCR (3.6×107 CFU/ml). For spiked
blood samples, the detection limit for LAMP was 7.7×104 CFU/ml which was 1000 fold
42

higher than PCR (7.7×107 CFU/ml). The assay shows highly specificity compared with
PCR in the evaluation of 50 strains.
4. For the general application, the cost will become the limitation of the method.
More research should be made to develop this method as it is a very rapid test and has
the same specificity and sensitivity as PCR, if not better.
As a conclusion, the three LAMP assays developed in this study were able to detect
A. baumannii, K. pneumoniae and P. aeruginosa.
43

Bibliography
Ajao, A. O., Robinson, G., Lee, M. S., Ranke, T. D., Venezia, R. A., Furuno, J. P. and Johnson, J. K. (2011). Comparison of culture media for detection of Acinetobacter baumannii in surveillance cultures of critically-ill patients. European Journal of Clinical Microbiology & Infectious Diseases 30: 1425-1430
Allison, L. J. and Hanson, M. F. (2008).Laboratory Service: Scottish E. coli O157/VTEC Reference Laboratory. Annual Report April (2007) – March (2008) Scottish E. coli O157/VTEC Reference Laboratory
Anbazhagan, D., Kathirvalu, G. G., Mansor, M., Siok Yan, G. O., Yusof, M. Y. and Sekaran, S. D. (2010). Multiplex polymerase chain reaction (PCR) assays for the detection of Enterobacteriaceae in clinical samples. African Journal of Microbiology Research 4:1186-1191
Becker, P. M., McVey, L. J., Saltz, C. C., Feussner, J. R., and Cohen, H. J. (1987). Hospital-acquired complications in a randomized controlled clinical trial of a geriatric consultation team. JAMA: The Journal of The American Medical Association 257: 2313-2317
Bratu, S., Landman, D., Haag, R., Recco, R., Eramo, A., Alam, M. and Quale, J. (2005). Rapid spread of carbapenem-resistant Klebsiella pneumoniae in New York City: a new threat to our antibiotic armamentarium. Archives of Internal Medicine 165: 1430
Brisse, S., and Verhoef, J. (2001). Phylogenetic diversity of Klebsiella pneumoniae and Klebsiella oxytoca clinical isolates revealed by randomly amplified polymorphic DNA, gyrA and parC genes sequencing and automated ribotyping. International Journal of Systematic and Evolutionary Microbiology 51: 915-924
Chander, Y., Ramakrishnan, M., Jindal, N., Hanson, K., & Goyal, S. M. (2011). Differentiation of Klebsiella pneumoniae and K. oxytoca by Multiplex Polymerase Chain Reaction. International Journal of Applied Research inVeterinary Medicine 9: 138
Chang, H. L., Tang, C. H., Hsu, Y. M., Wan, L., Chang, Y. F., Lin, C. T. and Chang, Y. C. (2009). Nosocomial Outbreak of Infection With Multidrug‐Resistant Acinetobacter baumannii in a Medical Center in Taiwan. Infection Control and Hospital Epidemiology 30: 34-38
Chapman, P. A., Ellin, M. and Ashton, R. (2001). A comparison of immunomagnetic separation and culture, RevealTM and VIPTM for the detection of E. coli O157 in enrichment cultures of naturally‐contaminated raw beef, lamb and mixed meat products.
44

Letters in Applied Microbiology 32: 171-175
Colinon, C., Deredjian, A., Hien, E., Brothier, E., Bouziri, L., Cournoyer, B. and Ranjard, L. (2013). Detection and enumeration of Pseudomonas aeruginosa in soil and manure assessed by an ecfX qPCR assay. Journal of Applied Microbiology 114: 1734-1749
Da Silva Filho, L. V. F., Levi, J. E., Bento, C. N. O., Ramos, S. R. T. D. S. and Rozov, T. (1999). PCR identification of Pseudomonas aeruginosa and direct detection in clinical samples from cystic fibrosis patients. Journal of Medical Microbiology 48: 357-361
Deguchi, T., Fukuoka, A., Yasuda, M., Nakano, M., Ozeki, S., Kanematsu, E. and Kawada, Y. (1997). Alterations in the GyrA subunit of DNA gyrase and the ParC subunit of topoisomerase IV in quinolone-resistant clinical isolates of Klebsiella pneumoniae. Antimicrobial Agents and Chemotherap 41: 699-701
De Vos, D., Lim, A., Pirnay, J.-P., Struelens, M., Vandenvelde, C., Duinslaeger, L. and Cornelis, P. (1997). Direct detection and identification of Pseudomonas aeruginosa in clinical samples such as skin biopsy specimens and expectorations by multiplex PCR based on two outer membrane lipoprotein genes, oprI and oprL. Journal of Clinical Microbiology 35: 1295-1299
Diancourt, L., Passet, V., Verhoef, J., Grimont, P. A.D. and Brisse, Sylvain. (2005). Multilocus sequence typing of Klebsiella pneumoniae nosocomial isolates. Journal of Clinical Microbiology 43: 4178-4182
Dorak, M. T. (2006). Real-time PCR: Taylor & Francis Group
Doménech‐Sánchez, A., Javier Benedí, V., Martínez‐Martínez, L., & Alberti, S. (2006). Evaluation of differential gene expression in susceptible and resistant clinical isolates of Klebsiella pneumoniae by DNA microarray analysis. Clinical Microbiology and Infection 12: 936-940
Ecker, J. A., Massire, C., Hall, T. A., Ranken, R., Pennella, T. T. D., Ivy, C. A. and Scott, P. T. (2006). Identification of Acinetobacter species and genotyping of Acinetobacter baumannii by multilocus PCR and mass spectrometry. Journal of Clinical Microbiology 44: 2921-2932
Eiken genome site. http://loopamp.eiken.co.jp/e/lamp/primer.htm (accessed on 5 April 2013)
Giannouli, M., Cuccurullo, S., Crivaro, V., Di Popolo, A., Bernardo, M., Tomasone, F. and Utili, R. (2010). Molecular epidemiology of multidrug-resistant Acinetobacter
45

baumannii in a tertiary care hospital in Naples, Italy, shows the emergence of a novel epidemic clone. Journal of Clinical Microbiology 48: 1223-1230
Gleaves, C. A., Smith, T. F., Shuster, E. A. and Pearson, G. R. (1985). Comparison of standard tube and shell vial cell culture techniques for the detection of cytomegalovirus in clinical specimens. Journal of Clinical Microbiology 21: 217-221
Go, E. S., Urban, C., Burns, J., Mariano, N., Mosinka-Snipas, K., Rahal, J. J and Eisner, W. (1994). Clinical and molecular epidemiology of Acinetobacter infections sensitive only to polymyxin B and sulbactam. The Lancet 344: 1329-1332
Gordon, F. B., Harper, I. A., Quan, A. L., Treharne, J. D., Dwyer, R. S. C. and Grandland, J. A. (1969). Detection of Chlamydia (Bedsonia) in certain infections of man. I. Laboratory procedures: comparison of yolk sac and cell culture for detection and isolation. The Journal of Infectious Diseases 451-462
Hara-Kudo, Y., Nemoto, J., Ohtsuka, K., Segawa, Y., Takatori, K., Kojima, T., & Ikedo, M. (2007). Sensitive and rapid detection of Vero toxin-producing Escherichia coli using loop-mediated isothermal amplification. Journal of Medical Microbiology 56: 398-406
Handschur, M., Karlic, H., Hertel, C., Pfeilstöcker, M. and Haslberger, A. G. (2009). Preanalytic removal of human DNA eliminates false signals in general 16S rDNA PCR monitoring of bacterial pathogens in blood. Comparative Immunology, Microbiology and Infectious Diseases 32: 207-219
Han, F., Wang, F. and Ge, B. (2011). Detecting potentially virulent Vibrio vulnificus strains in raw oysters by quantitative loop-mediated isothermal amplification. Applied and Environmental Microbiology 77: 2589-2595
Hill, J., Beriwal, S., Chandra, I., Paul, V. K., Kapil, A., Singh, T. and Jahnukainen, T. (2008). Loop-mediated isothermal amplification assay for rapid detection of common strains of Escherichia coli. Journal of Clinical Microbiology 46: 2800-2804
Hughes, A. J., Ariffin, N., Huat, T. L., Molok, H. A., Hashim, S., Sarijo, J. and Kamarulzaman, A. (2005). Prevalence of nosocomial infection and antibiotic use at a university medical center in Malaysia. Infection Control and Hospital Epidemiology 26: 100-104
Iwamoto, T., Sonobe, T. and Hayashi, K. (2003). Loop-mediated isothermal amplification for direct detection of Mycobacterium tuberculosis complex, M. avium, and M. intracellulare in sputum samples. Journal of Clinical Microbiology 41: 2616-2622
46

Jiang, W., Liu, H., Zhong, M., Yang, Y. C., Xiao, D. W. and Huang, W. F. (2012). Study on the Resistant Genes to Carbapenems and Epidemiological Characterization of Multidrug-Resistant Acinetobacter baumannii Isolates. Microbial Drug Resistance 19: 117-123
Katherason, S. G., Naing, L., Jaalam, K., Musa, K. I., Mohamad, N. A. N., Aiyar, S and Ismail, A. (2009). Ventilator-associated nosocomial pneumonia in intensive care units in Malaysia. The Journal of Infection in Developing Countries 3: 704-710.
Koeleman, J. G., Stoof, J., Van Der Bijl, M. W., Vandenbroucke-Grauls, C. M., & Savelkoul, P. H. (2001). Identification of Epidemic Strains of Acinetobacter baumannii by Integrase Gene PCR. Journal of Clinical Microbiology 39: 8-13
Kovtunovych, G., Lytvynenko, T., Negrutska, V., Lar, O., Brisse, S. and Kozyrovska, N. (2003). Identification of Klebsiella oxytoca using a specific PCR assay targeting the polygalacturonase pehX gene. Research in Microbiology 154: 587-592
Kong, B. H., Hanifah, Y. A., Yusof, M. Y. and Thong, K. L. (2011). Research Note Application of amplified ribosomal DNA restriction analysis in identification of Acinetobacter baumannii from a Tertiary Teaching Hospital, Malaysia. Tropical Biomedicine 28: 563-568
Kuboki, N., Inoue, N., Sakurai, T., Di Cello, F., Grab, D. J., Suzuki, H. and Igarashi, I. (2003). Loop-mediated isothermal amplification for detection of African trypanosomes. Journal of Clinical Microbiology 41: 5517-5524
Lavenir, R., Jocktane, D., Laurent, F., Nazaret, S. and Cournoyer, B. (2007). Improved reliability of Pseudomonas aeruginosa PCR detection by the use of the species-specific ecfX gene target. Journal of Microbiological Methods 70: 20-29
Levin, A. S., Barone, A. A., Penço, J., Santos, M. V., Marinho, I. S., Arruda, Erico A. G. and Costa, S. F. (1999). Intravenous colistin as therapy for nosocomial infections caused by multidrug-resistant Pseudomonas aeruginosa and Acinetobacter baumannii. Clinical Infectious Diseases 28: 1008-1011
Lim, K. T., Teh, C. S. J. and Thong, K. Lin. (2013). Loop-Mediated Isothermal Amplification Assay for the Rapid Detection of Staphylococcus aureus. BioMed Research International http://dx.doi.org/10.1155/2013/895816
Matrix-Assisted Laser Desorption Ionization–Time of Flight Mass Spectrometry. Journal of Clinical Microbiology 51: 603-606Mullah, B., Livak, K., Andrus, A. and Kenney, P. (1998). Efficient synthesis of double dye-labeled oligodeoxyribonucleotide probes and their application in a real time PCR assay. Nucleic Acids Research 26: 1026-1031
47

Mencacci, A., Monari, C., Leli, C., Merlini, L., De Carolis, E., Vella, A. and Bistoni, F. (2013). Typing of Nosocomial Outbreaks of Acinetobacter baumannii by Use of
Mosca, A., Miragliotta, L., Del Prete, R., Tzakis, G., Dalfino, L., Bruno, F. and Miragliotta, G. (2013). Rapid and sensitive detection of bla KPC gene in clinical isolates of Klebsiella pneumoniae by a molecular real-time assay. SpringerPlus 2: 1-5
Newton, C. R, Graham, A. and Ellison, J. S. (1997). PCR: BIOS Scientific Publishers.
Nordmann, P., Cuzon, G. and Naas, Thierry. (2009). The real threat of Klebsiella pneumoniae carbapenemase-producing bacteria. The Lancet infectious diseases 9: 228-236
Notomi, T., Okayama, H., Masubuchi, H., Yonekawa, T., Watanabe, K., Amino, N., & Hase, T. (2000). Loop-mediated isothermal amplification of DNA. Nucleic Acids Research 28: e63-e63.
Parida, M., Horioke, K., Ishida, H., Dash, P. K., Saxena, P., Jana, A. M. and Morita, K. (2005). Rapid detection and differentiation of dengue virus serotypes by a real-time reverse transcription-loop-mediated isothermal amplification assay. Journal of Clinical Microbiology 43: 2895-2903
Poon, L. L. M., Wong, B. W. Y., Ma, E. H. T., Chan, K. H., Chow, L. M. C., Abeyewickreme, W. and Looareesuwan, S. (2006). Sensitive and inexpensive molecular test for falciparum malaria: detecting Plasmodium falciparum DNA directly from heat-treated blood by loop-mediated isothermal amplification. Clinical Chemistry 52: 303-306
Pollack, A. (2010). Rising threat of infections unfazed by antibiotics. New York Times
Rosenthal, V. D., Bijie, H., Maki, D. G., Mehta, Y., Apisarnthanarak, A., Medeiros, E. A. and Khader, I. A. (2012). International Nosocomial Infection Control Consortium (INICC) report, data summary of 36 countries, for 2004-2009. American Journal of Infection Control 40: 396-407
Rungruanghiranya, S., Somboonwit, C. and Kanchanapoom, T. (2005). Acinetobacter infection in the intensive care unit. Journal of Infectious Diseases and Antimicrobial Agents 22:77-92
Savli, H., Karadenizli, A., Kolayli, F., Gundes, S., Ozbek, U. and Vahaboglu, H. (2003). Expression stability of six housekeeping genes: a proposal for resistance gene quantification studies of Pseudomonas aeruginosa by real-time quantitative RT-PCR. Journal of Medical Microbiology 52: 403-408
48

Seki, M., Yamashita, Y., Torigoe, H., Tsuda, H., Sato, S. and Maeno, M. (2005). Loop-mediated isothermal amplification method targeting the iytA gene for detection of Streptococcus pneumoniae. Journal of Clinical Microbiology 43: 1581-1586
Schwartz, T., Volkmann, H., Kirchen, S., Kohnen, W., Schön‐Hölz, K., Jansen, B. and Obst, U. (2006). Real‐time PCR detection of Pseudomonas aeruginosa in clinical and municipal wastewater and genotyping of the ciprofloxacin‐resistant isolates. FEMS Microbiology Ecology 57: 158-167
Singh, A., Goering, R. V., Simjee, S., Foley, S. L. and Zervos, M. J. (2006). Application of molecular techniques to the study of hospital infection. Clinical Microbiology Reviews 19: 512-530
Stover, C. K., Pham, X. Q., Erwin, A. L., Mizoguchi, S. D., Warrener, P., Hickey, M. J. and Lagrou, M. (2000). Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 406: 959-964
Song, T., Toma, C., Nakasone, N., & Iwanaga, M. (2005). Sensitive and rapid detection of Shigella and enteroinvasive Escherichia coli by a loop‐mediated isothermal amplification method. FEMS Microbiology Letters 243: 259-263
Solomon, S., Horan, T., Andrus, M., Edwards, J., Fridkin, S., Koganti, J. and Tolson, J. (2003). National Nosocomial Infections Surveillance (NNIS) system report, data summary from January 1992 through June 2003, issued August 2003. American Journal of Infection Control 31: 481-498
Suzuki, S., Tatsuguchi, A., Matsumoto, E., Kawazoe, M., Kaminishi, T., Shirouzu, M. and Yokoyama, S. (2007). Structural characterization of the ribosome maturation protein, rimM. Journal of Bacteriology 189: 6397-6406
Thong, K. L., Lai, M. Y., Teh, C. S. J. and Chua, K. H. (2011). Simultaneous detection of methicillin-resistant Staphylococcus aureus, Acinetobacter baumannii, Escherichia coli, Klebsiella pneumoniae and Pseudomonas aeruginosa by multiplex PCR. Tropical Biomedicine 28:21-31
Turton, J. F., Woodford, N., Glover, J., Yarde, S., Kaufmann, M. E. and Pitt, T. L. (2006). Identification of Acinetobacter baumannii by detection of the blaOXA-51-like carbapenemase gene intrinsic to this species. Journal of Clinical Microbiology 44: 2974-2976
Umeh, O. and Berkowitz, L. B. (2002). Klebsiella infections. eMedicine Journal
Vos, D. D., Jr, A. L., Pirnay, J. P., Struelens, M., Vandenvelde, C., Duinslaeger, L., Vanderkelen, A. and Cornelis, P. (1997). Direct detection and identification of
49

Pseudomonas aeruginosa in clinical samples such as skin biopsy specimens and expextorations by multiplex PCR based on two outer membrane lipoprotein genes, oprl and oprL. Journal of Clinical Microbiology 35: 1295-1299
Waleed, A. A. (2000). Optimisation of diagnostic PCR a study of PCR inhibitors in blood and sample pretreatment. Lund University Dissertation
Yamazaki, W., Taguchi, M., Kawai, T., Kawatsu, K., Sakata, J., Inoue, K. and Misawa, N. (2009). Comparison of loop-mediated isothermal amplification assay and conventional culture methods for detection of Campylobacter jejuni and Campylobacter coli in naturally contaminated chicken meat samples. Applied and Environmental Microbiology 75: 1597-1603
Zhao, X., Wang, L., Chu, J., Li, Y., Li, Y., Xu, Z. and Liu, Y. (2010). Rapid detection of Vibrio parahaemolyticus strains and virulent factors by loop-mediated isothermal amplification assays. Food Science and Biotechnology 19: 1191-1197
50

Appendix 1: Bacterial strains used for optimization
No. Species/Subspecies Identification code1 A. baumannii AC/0601102 A. baumannii AC/0612-173 A. baumannii AC/0701-114 A. baumannii AC/0702-55 A. baumannii AC/0702-176 A. baumannii AC/0703-147 A. baumannii AC/0703-218 A. baumannii AC/0711-79 A. baumannii AC/0801-610 A. baumannii AC/0801-1111 A. baumannii AC/0801-1312 A. baumannii AC/0802-113 A. baumannii AC/0802-414 A. baumannii AC/0802-1415 A. baumannii AC/0802-2016 A. baumannii AC/0803-1517 A. baumannii AC/0804-1918 A. baumannii AC/0812-2919 A. baumannii AC/0901-520 A. baumannii AC/0901-1421 A. baumannii AC/0901-3622 A. baumannii AC/0901-3723 A. baumannii AC/0902-524 A. baumannii AC/0902-625 A. baumannii AC/0902-1326 A. baumannii AC/0902-1427 A. baumannii AC/0902-1528 A. baumannii AC/0903-1529 A. baumannii AC/0904-330 A. baumannii AC/0905-3131 K. pneumoniae PS1932 K. pneumoniae PS2333 K. pneumoniae PS3134 K. pneumoniae PS3535 K. pneumoniae PS3636 K. pneumoniae PS4937 K. pneumoniae PS5038 K. pneumoniae PS5139 K. pneumoniae PS80
51

40 K. pneumoniae PS8241 K. pneumoniae PS8342 K. pneumoniae PS8643 K. pneumoniae PS8844 K. pneumoniae PS9045 K. pneumoniae PS9246 K. pneumoniae PS9647 K. pneumoniae PS13848 K. pneumoniae PS15649 K. pneumoniae K09-2450 K. pneumoniae K09-2551 K. pneumoniae K10-0352 K. pneumoniae K10-0453 K. pneumoniae K10-0554 K. pneumoniae K11-0155 K. pneumoniae K11-0256 K. pneumoniae K11-0357 K. pneumoniae K11-0458 K. pneumoniae K11-0559 K. pneumoniae K11-0660 K. pneumoniae K11-0961 P. aeruginosa PS262 P. aeruginosa PS1663 P. aeruginosa PS1964 P. aeruginosa PS2065 P. aeruginosa PS2366 P. aeruginosa PS6767 P. aeruginosa PS9868 P. aeruginosa PS10069 P. aeruginosa PS10270 P. aeruginosa PS10371 P. aeruginosa PS10572 P. aeruginosa PS10873 P. aeruginosa PS11074 P. aeruginosa PS23975 P. aeruginosa PS33976 P. aeruginosa PS34177 P. aeruginosa PS36278 P. aeruginosa R0179 P. aeruginosa R0280 P. aeruginosa R0481 P. aeruginosa 1182
52

82 P. aeruginosa 118683 P. aeruginosa 126084 P. aeruginosa 128885 P. aeruginosa B1414186 P. aeruginosa B1414387 P. aeruginosa B1412888 P. aeruginosa B1426289 P. aeruginosa B1434990 P. aeruginosa BF208791 E. coli P4992 E. coli P4193 E. coli BS494 Vibrio VPD2195 Vibrio VPD2296 Vibrio VPD2697 Vibrio VPD2798 shigella TH32/9899 shigella TC3/99100 shigella TH23/97101 shigella flexneri Y
variantTH5/02
102 Salmonella S.Meto303/94103 Salmonella S.Oke-nara104 Salmonella S.LOM105 Salmonella S.Bevis106 Salmonella S.Hvrt107 Salmonella SAB79
53

Appendix 2: Chemicals and reagents
Sodium chloride AnalaR
Tryptone OXOID
Yeast extract OXOID
Bacteriological agar Becton, Dickinson and Company
Agarose Promega, USA
Tris (base) Ultra Pure, BIO BASIC
Orthoboric acid MERCK
EDTA Ultra Pure, Invitrogen
Potassium chloride AnalaR
Potassium hydrogen phosphate SIGMA-ALDRICH
Sodium hydrogen phosphate SIGMA-ALDRICH
Hydrochloric acid
MacConkey agar
SYSTERM
OXOID
Luria Bertani broth (LBB)
NaCl 0.5g
Tryptone 1g
Yeast extract 0.5g
ddH2O 100ml
To prepare the LBB, all the ingredients were suspended in 100ml of ddH2O, and then
autoclaved at 121℃ for 15 min.
54
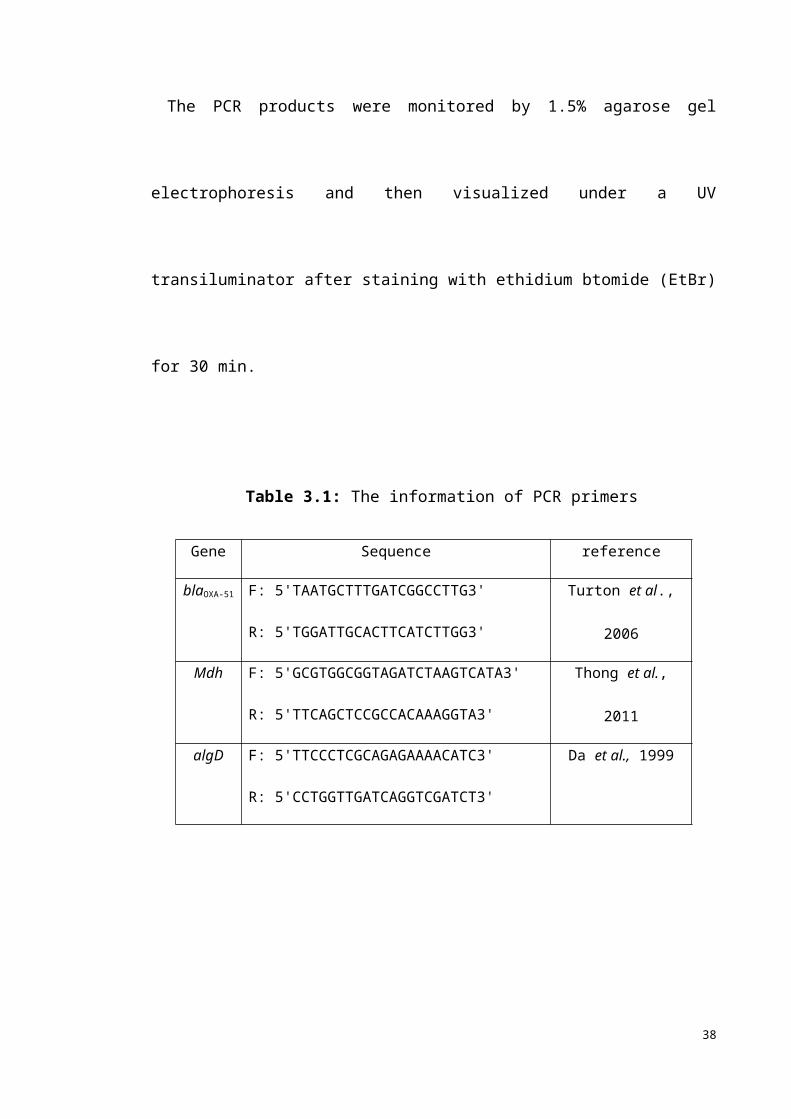
Luria Bertani Agar (LBA)
NaCl 0.5g
Tryptone 1g
Yeast extract 0.5g
Bacto agar 1.5g
ddH2O 100ml
To prepare LBA, all the ingredients were suspended in 100ml of H2O, and then
autoclaved at 121℃ for 15 min. After autoclaving, the agar was cooled and then poured
into petri plates, and kept in the refrigerator until use.
0.5×Tris Borate EDTA buffer (TBE)
The 0.5×TBE was diluted from 10×TBE, the preparation of 10×TBE is showing
below.
Tris(base) 121.2g
Orthoboric acid 61.8g
EDTA 0.745g
ddH2O 1000.0ml
To prepare 10×TBE, all the ingredients were suspended into 500 ml of ddH2O. The
pH of the solution was adjusted to pH 8, before being sterilized by autoclaving at 121℃
for 15 min.
Saline
NaCl 8.5g
ddH2O 1000.0ml55

The saline used was 0.85%. the solution was autoclaved at 121℃ for 15 min
Phosphate-buffered Saline(PBS)
NaCl 8g
KCl 0.2g
Na2HPO4 1.44g
KH2PO4 0.24g
ddH2O 1000ml
To prepare PBS, all the ingredients are suspended into 800ml of ddH2O. Adjust the
PH of the solution to 7.4, and then add distill water and constant the volume to 1L.
Autoclaved at 121℃ for 15 min.
DNA amplification Kit
The DNA amplification kit was used for the LAMP reaction, the whole name of this
kit is Loopamp DNA amplification kit. It is produced by EIKEN CHEMICAL CO.,
LTD from Japan.
56

Appendix 3: In silico PCR amplification of gltA F3, B3 from A. baumannii for LAMP primer
No mismatches allowed. info
Selected strains1 - Acinetobacter baumannii 1656-2 chromosome
2 - Acinetobacter baumannii AB0057
3 - Acinetobacter baumannii AB307-0294
4 - Acinetobacter baumannii ACICU
5 - Acinetobacter baumannii ATCC 17978
6 - Acinetobacter baumannii AYE
7 - Acinetobacter baumannii D1279779
8 - Acinetobacter baumannii MDR-TJ
9 - Acinetobacter baumannii MDR-ZJ06
10 - Acinetobacter baumannii SDF
11 - Acinetobacter baumannii TCDC-AB0715
12 - Acinetobacter baumannii TYTH-1
13 - Acinetobacter calcoaceticus PHEA-2
14 - Acinetobacter sp. ADP1
15 - Acinetobacter sp. DR1
57

Appendix 4: In silico PCR amplification of gltA FIP from A. baumannii for LAMP primer
No mismatches allowed. info
Selected strains1 - Acinetobacter baumannii 1656-2 chromosome
2 - Acinetobacter baumannii AB0057
3 - Acinetobacter baumannii AB307-0294
4 - Acinetobacter baumannii ACICU
5 - Acinetobacter baumannii ATCC 17978
6 - Acinetobacter baumannii AYE
7 - Acinetobacter baumannii D1279779
8 - Acinetobacter baumannii MDR-TJ
9 - Acinetobacter baumannii MDR-ZJ06
10 - Acinetobacter baumannii SDF
11 - Acinetobacter baumannii TCDC-AB0715
12 - Acinetobacter baumannii TYTH-1
13 - Acinetobacter calcoaceticus PHEA-2
14 - Acinetobacter sp. ADP1
15 - Acinetobacter sp. DR1
58

Appendix 5: In silico PCR amplification of gltA BIP from A. baumannii for LAMP primer
No mismatches allowed. info
Selected strains1 - Acinetobacter baumannii 1656-2 chromosome
2 - Acinetobacter baumannii AB0057
3 - Acinetobacter baumannii AB307-0294
4 - Acinetobacter baumannii ACICU
5 - Acinetobacter baumannii ATCC 17978
6 - Acinetobacter baumannii AYE
7 - Acinetobacter baumannii D1279779
8 - Acinetobacter baumannii MDR-TJ
9 - Acinetobacter baumannii MDR-ZJ06
10 - Acinetobacter baumannii SDF
11 - Acinetobacter baumannii TCDC-AB0715
12 - Acinetobacter baumannii TYTH-1
13 - Acinetobacter calcoaceticus PHEA-2
14 - Acinetobacter sp. ADP1
15 - Acinetobacter sp. DR1
59

Appendix 6: In silico PCR amplification of ABC transporter permease F3, B3 from K. pneumoniae for LAMP primer
No mismatches allowed. info
Selected strains1 - Klebsiella oxytoca E718
2 - Klebsiella oxytoca KCTC 1686
3 - Klebsiella pneumoniae 342
4 - Klebsiella pneumoniae KCTC 2242
5 - Klebsiella pneumoniae NTUH-K2044
6 - Klebsiella pneumoniae subsp. pneumoniae 1084
7 - Klebsiella pneumoniae subsp. pneumoniae HS11286
8 - Klebsiella pneumoniae subsp. pneumoniae MGH 78578
9 - Klebsiella variicola At-22
60

Appendix 7: In silico PCR amplification of ABC transporter permease FIP from K. pneumoniae for LAMP primer
No mismatches allowed. info
Selected strains1 - Klebsiella oxytoca E718
2 - Klebsiella oxytoca KCTC 1686
3 - Klebsiella pneumoniae 342
4 - Klebsiella pneumoniae KCTC 2242
5 - Klebsiella pneumoniae NTUH-K2044
6 - Klebsiella pneumoniae subsp. pneumoniae 1084
7 - Klebsiella pneumoniae subsp. pneumoniae HS11286
8 - Klebsiella pneumoniae subsp. pneumoniae MGH 78578
9 - Klebsiella variicola At-22
61

Appendix 8: In silico PCR amplification of ABC transporter permease BIP from K. pneumoniae for LAMP primer
No mismatches allowed. info
Selected strains1 - Klebsiella oxytoca E718
2 - Klebsiella oxytoca KCTC 1686
3 - Klebsiella pneumoniae 342
4 - Klebsiella pneumoniae KCTC 2242
5 - Klebsiella pneumoniae NTUH-K2044
6 - Klebsiella pneumoniae subsp. pneumoniae 1084
7 - Klebsiella pneumoniae subsp. pneumoniae HS11286
8 - Klebsiella pneumoniae subsp. pneumoniae MGH 78578
9 - Klebsiella variicola At-22
62

Appendix 9: In silico PCR amplification of rimM F3, B3 from P. aeruginosa for LAMP primer
No mismatches allowed. info
Selected strains1 - Pseudomonas aeruginosa
2 - Pseudomonas aeruginosa B136-33
3 - Pseudomonas aeruginosa DK2
4 - Pseudomonas aeruginosa LESB58
5 - Pseudomonas aeruginosa M18
6 - Pseudomonas aeruginosa NCGM2.S1
7 - Pseudomonas aeruginosa PA7
8 - Pseudomonas aeruginosa UCBPP-PA14
9 - Pseudomonas brassicacearum subsp. brassicacearum NFM421
10 - Pseudomonas denitrificans ATCC 13867
63

Appendix 10: In silico PCR amplification of rimM FIP from P. aeruginosa for LAMP primer
No mismatches allowed. info
Selected strains1 - Pseudomonas aeruginosa
2 - Pseudomonas aeruginosa B136-33
3 - Pseudomonas aeruginosa DK2
4 - Pseudomonas aeruginosa LESB58
5 - Pseudomonas aeruginosa M18
6 - Pseudomonas aeruginosa NCGM2.S1
7 - Pseudomonas aeruginosa PA7
8 - Pseudomonas aeruginosa UCBPP-PA14
9 - Pseudomonas brassicacearum subsp. brassicacearum NFM421
10 - Pseudomonas denitrificans ATCC 13867
64
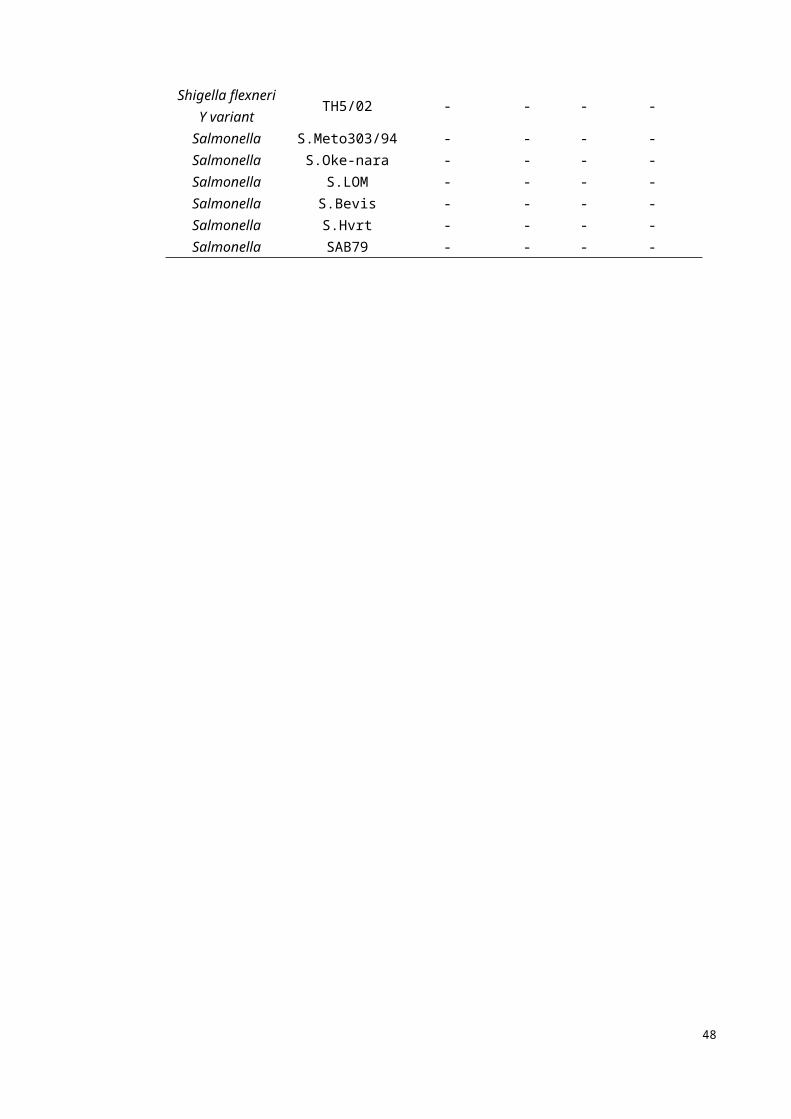
Appendix 11: In silico PCR amplification of rimM BIP from P. aeruginosa for LAMP primer
No mismatches allowed. info
Selected strains1 - Pseudomonas aeruginosa
2 - Pseudomonas aeruginosa B136-33
3 - Pseudomonas aeruginosa DK2
4 - Pseudomonas aeruginosa LESB58
5 - Pseudomonas aeruginosa M18
6 - Pseudomonas aeruginosa NCGM2.S1
7 - Pseudomonas aeruginosa PA7
8 - Pseudomonas aeruginosa UCBPP-PA14
9 - Pseudomonas brassicacearum subsp. brassicacearum NFM421
10 - Pseudomonas denitrificans ATCC 13867
65

Appendix 11: Presentation and publication
Presentation
Hong Dong, Cindy Shuan Ju Teh, Kwai Lin Thong. (2012). Development of a loop-mediated isothermal amplification for detection of Acinetobacter baumannii. National Postgraduate Seminar 2012. Kuala Lumpur, Malaysia. Meeting Abstract: pg 94.
Publication
Dong, H., Teh, C., S., J. and Thong, K., L. Rapid detection for Acinetobacter baumannii using loop-mediated isothermal amplification method. (Manuscript in submission)
66