1 integrated genomics steven jones genome sciences centre vancouver
TRANSCRIPT

1
Integrated Genomics
Steven JonesGenome Sciences Centre
Vancouver

2
Integrated Genomics
• How are biological relationships represented?• What is the underlying topology of such
networks?• How do we visualize such networks?• How can we exploit such networks?• How do we go about building such networks?

3
BiologicalNetworks
Peter Yodzis, University of Guelph

4
How to make a biological network
• Protein Interaction – Y2H, TAP tagging• ChIP/chip – Protein/DNA regulatory networks• RNAi screens• Synthetic lethal – epistatic relationships• Gene Expression – SAGE,microarray• Metabolic pathways • Signalling – protein/protein protein/sm. mol.
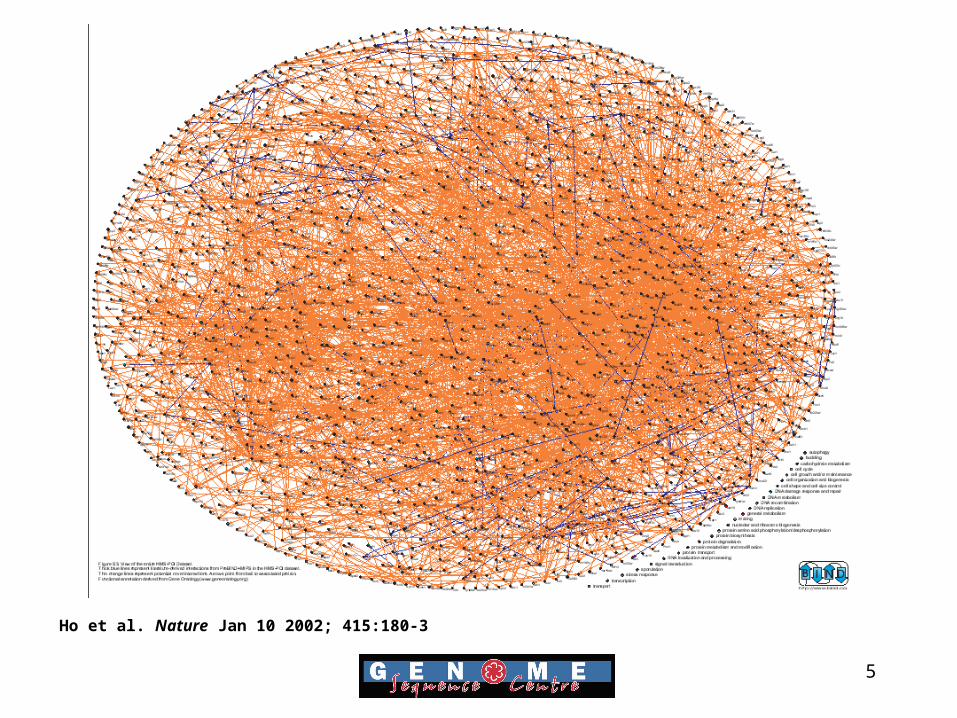
5
Ho et al. Nature Jan 10 2002; 415:180-3

6

7
Studying networks
• How do we start to model or understand biology in the concept of networks.
• What do they look like? How do they behave? How do they evolve?

8
Erdös-Rényi Model
Pál Erdös Pál Erdös (1913-1996)
RRényinyi (1921-1970)
Erdös-Rényi Model:• Classical random network theory

9
Who was Paul Erdös?
• He managed to think about more problems than any other mathematician in history and could recite the details of all 1,475 of the papers he had written or co-authored. Fortified by expresso and amphetamines, Erdös did mathematics 19 hours a day, seven days a week.
• For the last 25 years of his life, Erdös raced against the specter of old age to prove as many mathematical theorems as possible. "The first sign of senility," Erdös often said, "is when a man forgets his theorems. The second sign is when he forgets to zip up. The third sign is when he forgets to zip down."

10
Erdös-Rényi Model
Pál Erdös Pál Erdös (1913-1996)
RRényinyi (1921-1970)
Erdös-Rényi Model:• Connectivity follows a Poisson distribution

11
Pál Erdös Pál Erdös (1913-1996)
RRényinyi (1921-1970)
Scale-Free Network:• Deviation from complete randomness.
Scale-Free Model

12
Power Law Distribution
• Research has shown that the distribution of links to all sites on the web approximates a “power law”.
• A small number of sites receive the majority of links and most sites receive very few links.
DM Pennock, GW Flake, S. Lawrence, EJ Glover and CL Giles, “Winners don’t take all: Characterizing the competition for links on the web”, (2002) PNAS 99(8):5207-5211.
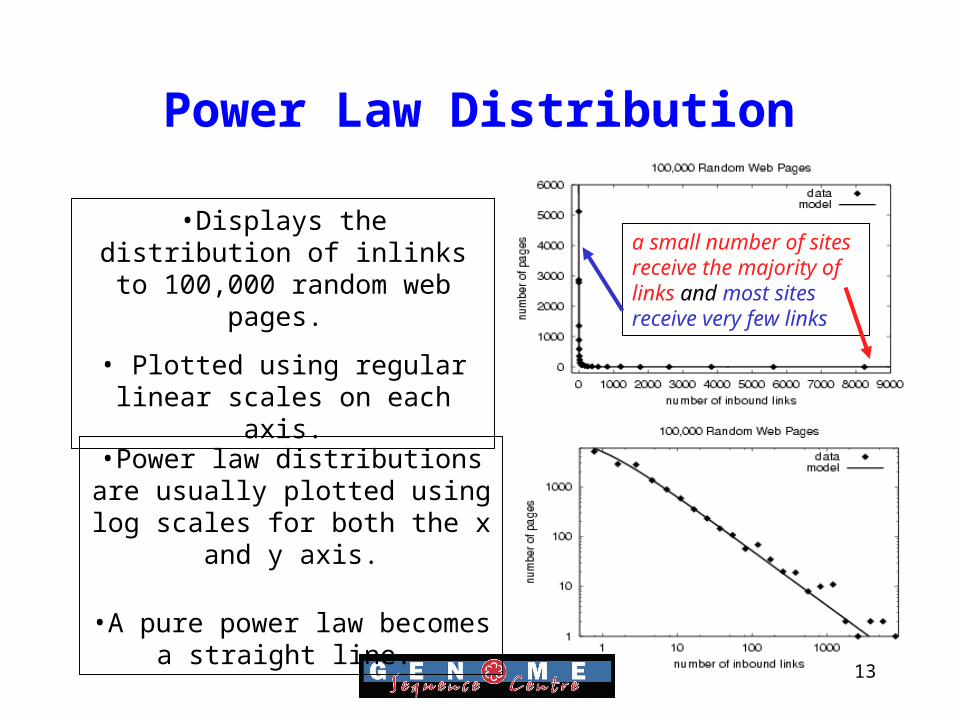
13
Power Law Distribution
•Power law distributions are usually plotted using log scales
for both the x and y axis.
•A pure power law becomes a straight line.
•Displays the distribution of inlinks to 100,000 random web pages.
• Plotted using regular linear scales on each axis.
a small number of sites receive the majority of links and most sites receive very few links
14
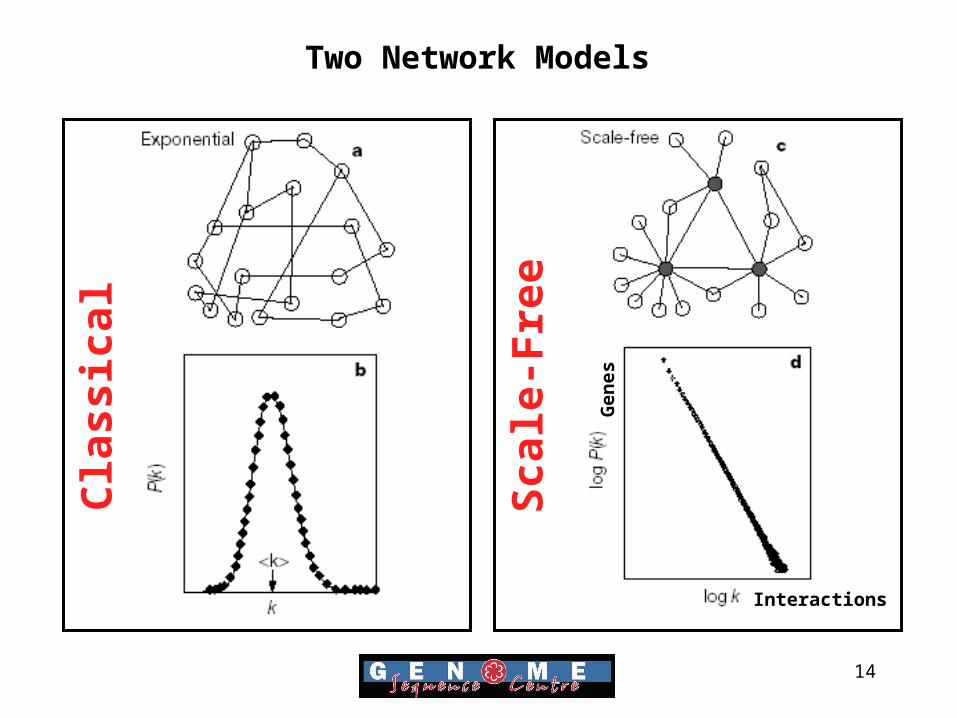
Classical
Scale-Free
Two Network Models
Gen
es
Interactions

15
Inherent Robustness (and Fallibility)
• Can functionality be maintained with errors or failures?• Yes - Lethality correlates with the number of interactions the node
(protein) has.• Random attacks on the network will likely hit poorly connected nodes• Targetted attacks on highly connected nodes will have a much greater
effect
Non-Lethal
Lethal

16
Exploiting Biological Networks
• Topology of biological networks displays similarity to other self-organising networks such as the world-wide-web and the internet
• Much of this work pioneered by Albert Barabási, University of Notre Dame

17
• H.Jeong, B.Tombor, R.Albert, Z.N.Oltvai, A.L.Barabasi, “Lethality and centrality in protein networks”, Nature 407 651 (2000).
Predicting perturbation of a network

18

19
Modeling Network
Behaviours
Ideker T, Ozier O, Schwikowski B, Siegel A. Discovering regulatory and signaling circuits in molecular interaction networks. Bioinformatics 18, S233 (2002).
Cytoscape allows the correlation of protein interaction data With expression data
Hypothesis is that changes in gene expression should Correlate with specific regions of the network (“Hot-Spots”)
http://www.cytoscape.org/

20
Cytoscape can identify active subnetworks Under different conditions in yeast
Protein linkages can be through Physical protein interaction (Blue) or through Protein->DNA interactions (yellow)

21
The Cytoscape Application

22
Also Osprey, a Canadian equivalent. Breitkreutz, BJ., Stark, C., Tyers M. "Osprey: A Network Visualization System." Genome Biology 2003 4(3):R22

23
Are Humans Capable of Understanding or Interpreting Networks?

24
How can we use LSF networks to study Cancer?
• LSF networks are robust. Randomly knocking out a node is unlikely to perturb the network.
• Targeting highly-connected nodes is likely to perturb the network. But these are also more likely to have a severe effect on normal cells.

25
• This can explain how chemotherapeutics have been historically chosen, why they are toxic and why they don’t work.
• Therefore, we need to determine a combination of drugs that will preferentially damage a cancer cells and not the network of normal cells.

26
Can we compare network topologies from tumorous and normal tissues?
• Are there real differences in the networks that we can exploit?
• If differences exist, then is there a combination of drugs which can selectively perturb the cancerous network and not the normal network?

27
Comparative Topologies

28
10000000
1E+09
1E+11
1E+13
1E+15
1E+17
1E+19
1E+21
1E+23
1E+25
1E+27
1E+29
1E+31
1E+33
1E+35
1E+37
1E+39
1E+41
2 3 4 5 6 7 8 9 10
Perturbing a 10,000 node network. Targeting between 2 and 10 random nodes (Atoms on earth 1E+49).
The number of possible permutations

29
Network Perturbation Results

30
Integrative Genomics
• We are along way from being able to simulate the entire biological network of a cell. Although, groups are working on this, e.g. the CyberCell project, Alberta.
• However, we can already combine genetic, expression, interaction, pathway, medical, physiological data into networks to allow us to answer biologically relevant questions.

31
• Show all the genes that are significantly up-regulated in a tumor and which are known cancer genes or are more than 50% identical to a known human cancer gene.
• Show all the proteins that are known to play a role in the process of apoptosis in human or are 70% identical to proteins known to be involved in apoptosis in any other organism.
• Show all the genes which are significantly down-regulated in the tumor and for which mutants are known in either the mouse, Drosophila, or C.elegans
Example Integrative Questions

32
•Show me all the genes that have been implicated in a human disease, or genes that are known to be part of biological pathway for which a disease gene has been determined
•Show me all the Zn-finger proteins that are up-regulated, or any proteins known to bind to these Zn-finger proteins
•Using literature data, show all the genes that are up regulated and which are thought to bind or interact with telomeres in any organism.
•etc…

33
An Example
(c) CGDN

34
• COX deficeincy, also Known as Leigh Syndrome
•COX, functioning in the mitochondria, consists of 13 subunits and many other proteins are required for proper assembly and coordination with co-factors.
•An integrative genomics approach, using bioinformatics, was able to consolidate mitochondrial proteomics, gene expression data and the genetic map to pin-point the exact gene from a 2 megabase pair interval

35
Building Biological relationships and networks
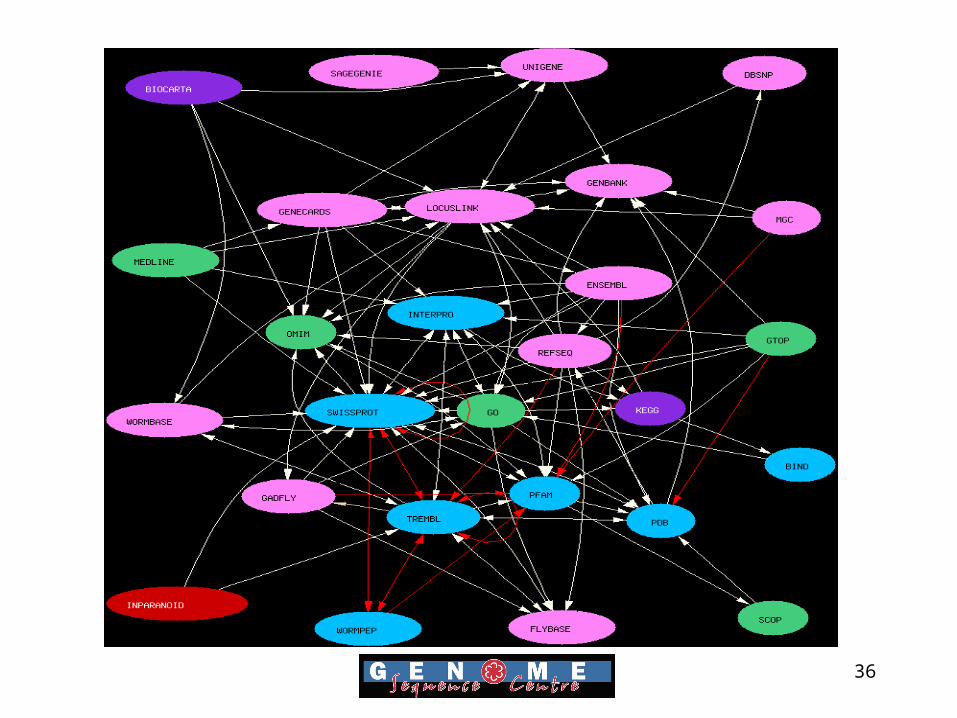
36

37
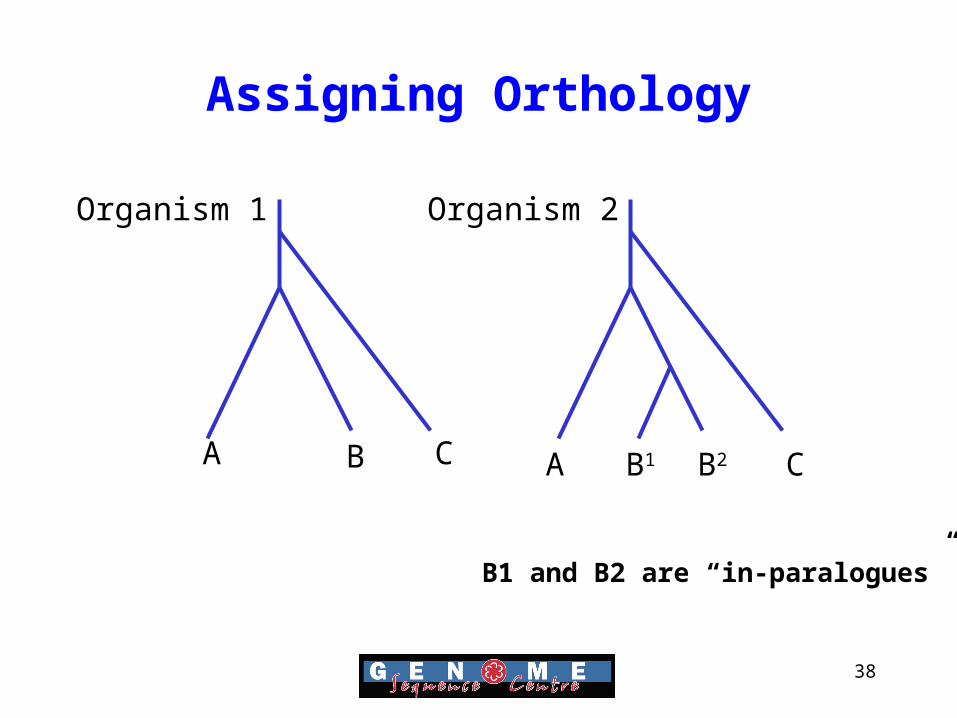
Assigning Orthology
• A key step in integrative genomics is going to be able to infer information between species
• Important if we using the relationship to infer a function.
• Need to consider “In-paralogs” which are also bona-fide orthologs
• Software such as Inparanoid can detect both orthologues and in-paralogs
Automatic clustering of orthologs and in-paralogs from pairwise species comparisons. Remm M, Storm CE, Sonnhammer EL. J Mol Biol. 2001
38
Assigning Orthology
A B C A B2B1 C
Organism 1 Organism 2
B1 and B2 are “in-paralogues”

39
Assigning Function
• PFAM provides a large collection of Hidden-Markov-Models determined from protein multiple sequence alignments.
• Version 6.6 contains over 3071 protein domain families covering 69% of proteins in the SwissProt database
The Pfam Protein Families Database, Bateman et al. Nucleic Acids Research, 2002, Vol. 30, No. 1 276-280.
Through Computational Means

40

41
Using Sequence similarity to assign function
• Exploits the standard hypothesis that the more similar two proteins are the more likely they are to have the same function.
• But how do we know the annotations of the proteins are correct. Many annotations are inferred from other incorrect annotations.
• Is the annotation relevant to the species.• Annotations are inconsistent in their wording and
specificity. • Therefore, need a way to link the computational
output of a blast search with curated annotation.

42
The Gene Ontology
• Provides a controlled vocabulary to describe the roles of genes and proteins.
• GO assigns three ontologies to each gene, molecular function, cellular location and cellular location.
• GO has now been adopted by almost all model organism databases
Gene Ontology: tool for the unification of biology. The Gene OntologyConsortium (2000) Nature Genet. 25: 25-29.

43
But what is an ontology?
The hierarchical structuring of knowledge about things by subcategorising them according to their essential (or at least relevant and/or cognitive) qualities.

44
Representing an Ontology

45
The Gene OntologyConsortium
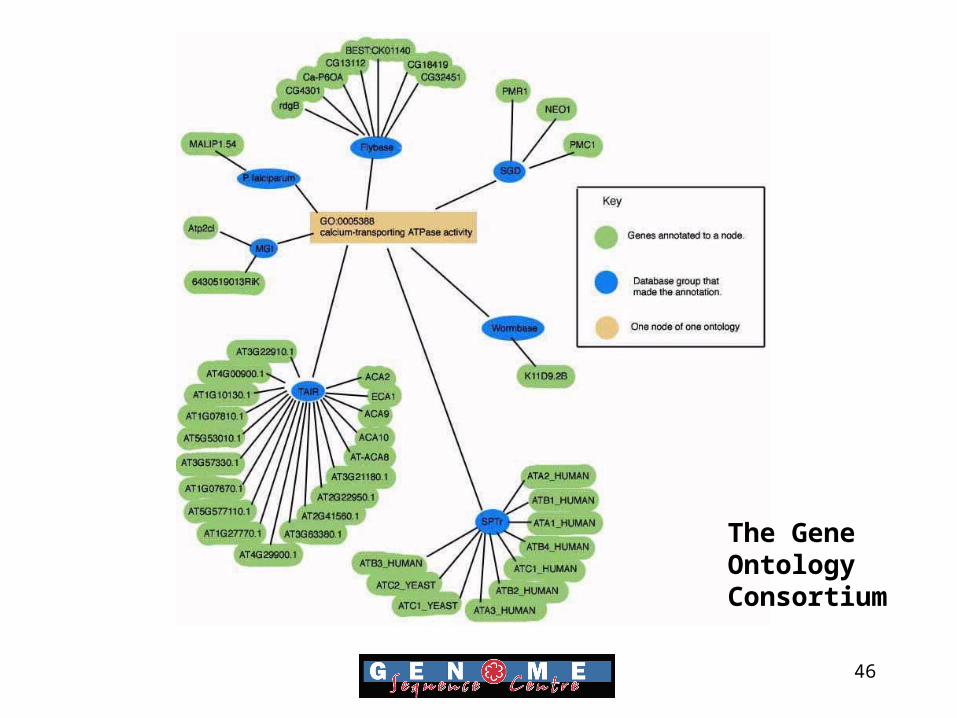
46
The Gene OntologyConsortium

47
Mapping Protein Interactions
• Bind database aims to provide full descriptions of interactions, molecular complexes and pathways
• Data is freely available and in XML, ASN.1 and text format for easy manipulation
• Over 16,000 protein interactions are currently in the database.
Bader GD, Betel D, Hogue CW. (2003) BIND: the Biomolecular
Interaction Network Database. Nucleic Acids Res. 31(1):248-50
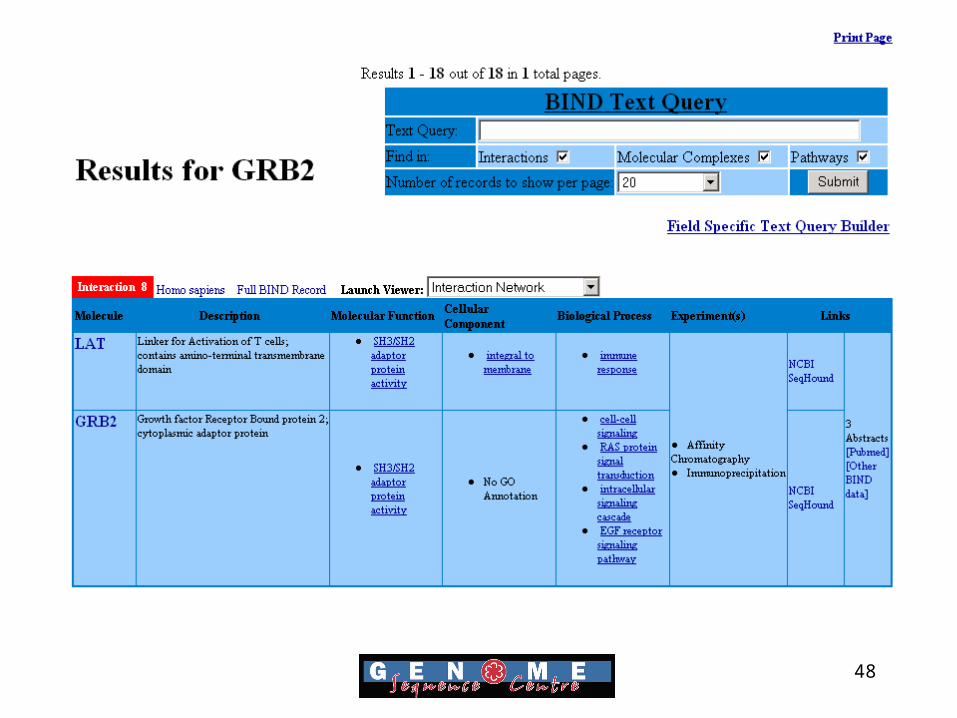
48
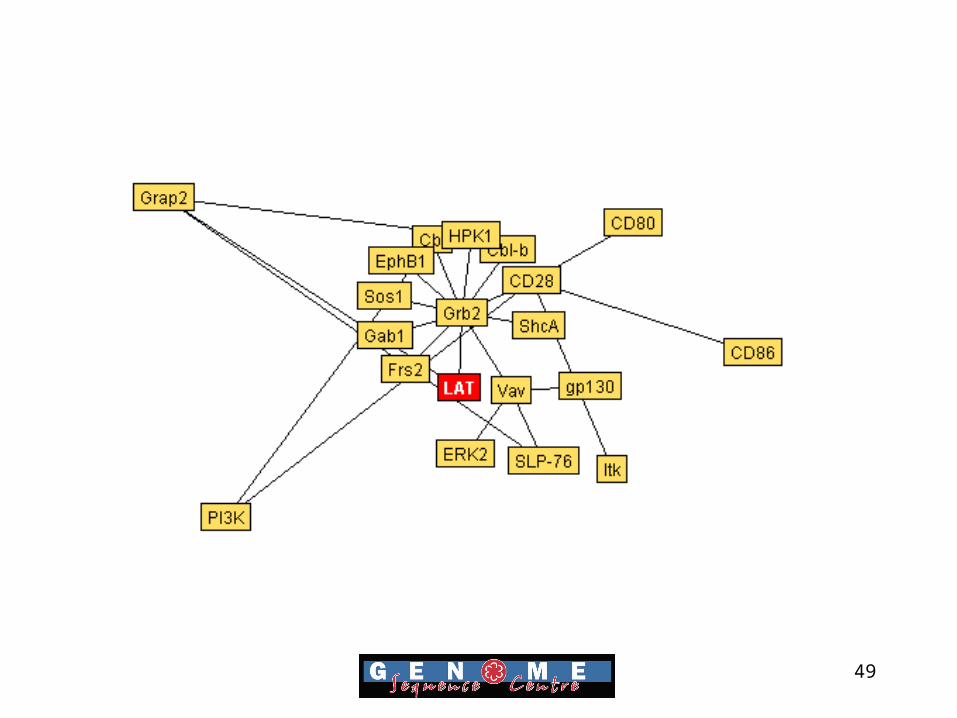
49

50
How do we computerize biological knowledge?
• Most biological facts and inferences are present in the literature and not in accessible databases.
• Literature is represented by free-form text.• Gene names are inconsistent and ambiguous• PreBind database has been generated
through a literature mining approach
Donaldson et al (2003) PreBIND and Textomy - mining thebiomedical literature for protein-protein interactions using a support vector machine. BMC Bioinformatics. 4(1):11.
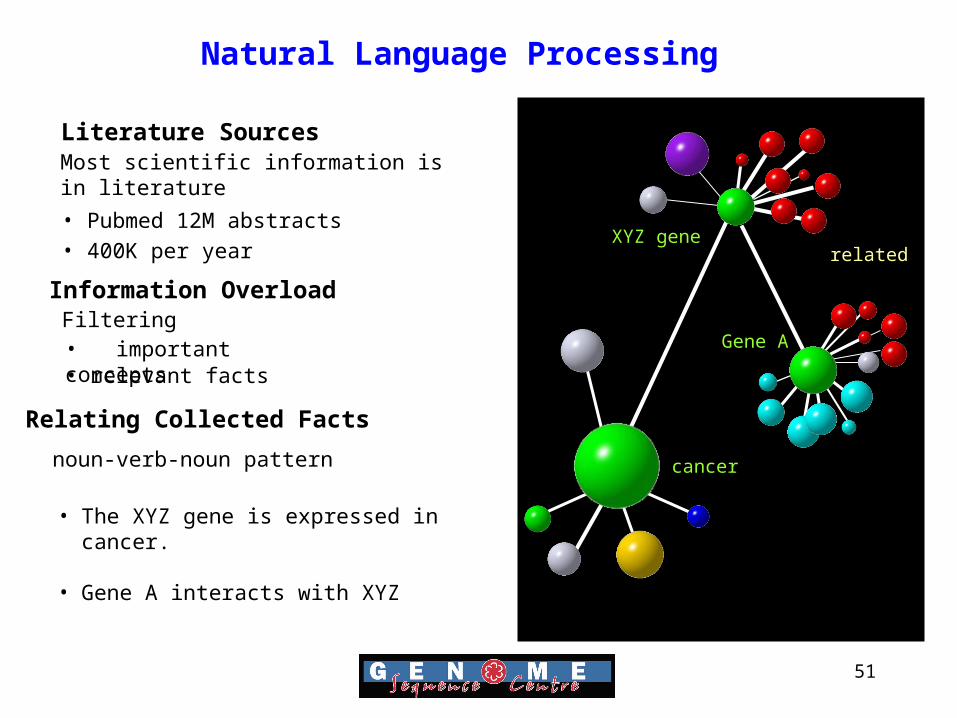
51
Natural Language Processing
Information OverloadFiltering• important concepts• relevant facts
Relating Collected Facts
noun-verb-noun pattern
Literature Sources
• Pubmed 12M abstracts• 400K per year
Most scientific information is in literature
cancer
related
• The XYZ gene is expressed in cancer.
• Gene A interacts with XYZ
XYZ gene
Gene A

52
Data Federation Approaches

53
Further Reading H. Jeong, B. Tombor, R. Albert, Z.N. Oltvai, and A.-L. Barabási The large-scale organization of metabolic networks Nature 407, 651-654 (2000).
Schwikowski, B., et al. 2000. A network ofprotein-protein interactions in yeast. Nature Biotechnology. 18:1257-1261.
Lenhard B, Hayes WS, Wasserman WW. GeneLynx: a gene-centric portal to the humangenome. Genome Res. 2001 Dec;11(12):2151-7.