1-month practical course genome analysis lecture 4: pair-wise alignment centre for integrative...
Post on 21-Dec-2015
219 views
TRANSCRIPT

1-month Practical CourseGenome Analysis
Lecture 4: Pair-wise alignment
Centre for Integrative Bioinformatics VU (IBIVU)Vrije Universiteit AmsterdamThe Netherlands
CENTR
FORINTEGRATIVE
BIOINFORMATICSVU
E

Pair-wise alignment
Combinatorial explosion- 1 gap in 1 sequence: n+1 possibilities- 2 gaps in 1 sequence: (n+1)n - 3 gaps in 1 sequence: (n+1)n(n-1), etc.
2n (2n)! 22n
= ~ n (n!)2 n 2 sequences of 300 a.a.: ~1088 alignments 2 sequences of 1000 a.a.: ~10600 alignments!
T D W V T A L KT D W L - - I K

Technique to overcome the combinatorial explosion:Dynamic Programming
• Alignment is simulated as Markov process, all sequence positions are seen as independent
• Chances of sequence events are independent– Therefore, probabilities per aligned position need
to be multiplied– Amino acid matrices contain so-called log-odds
values (log10 of the probabilities), so probabilities can be summed

•To perform statistical analyses on messages or sequences, we need a reference model.
•The model: each letter in a sequence is selected from a defined alphabet in an independent and identically distributed (i.i.d.) manner.
•This choice of model system will allow us to compute the statistical significance of certain characteristics of a sequence, its subsequences, or an alignment.
•Given a probability distribution, Pi, for the letters in a i.i.d. message, the probability of seeing a particular sequence of letters i, j, k, ... n is simply Pi Pj Pk···Pn.
•As an alternative to multiplication of the probabilities, we could sum their logarithms and exponentiate the result. The probability of the same sequence of letters can be computed by exponentiating log Pi +
log Pj + log Pk+ ··· + log Pn.
•In practice, when aligning sequences we only add log-odds values (residue exchange matrix) but we do not exponentiate the final score.
To say the same more statistically…

Sequence alignmentHistory of the Dynamic
Programming (DP) algorithm
1970 Needleman-Wunsch global pair-wise alignment
Needleman SB, Wunsch CD (1970) A general method applicable to the search for similarities in the amino acid sequence of two proteins, J Mol Biol. 48(3):443-53.
1981 Smith-Waterman local pair-wise alignment
Smith, TF, Waterman, MS (1981) Identification of common molecular subsequences. J. Mol. Biol. 147, 195-197.
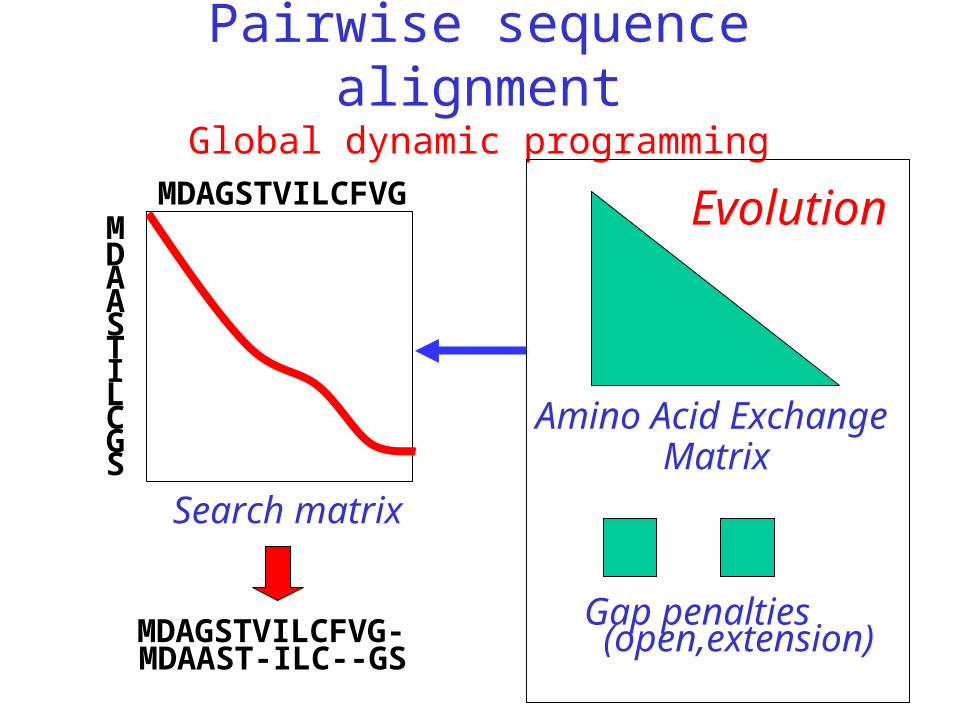
Pairwise sequence alignmentGlobal dynamic programming
MDAGSTVILCFVGMDAASTILCGS
Amino Acid Exchange
Matrix
Gap penalties (open,extension)
Search matrix
MDAGSTVILCFVG-MDAAST-ILC--GS
Evolution
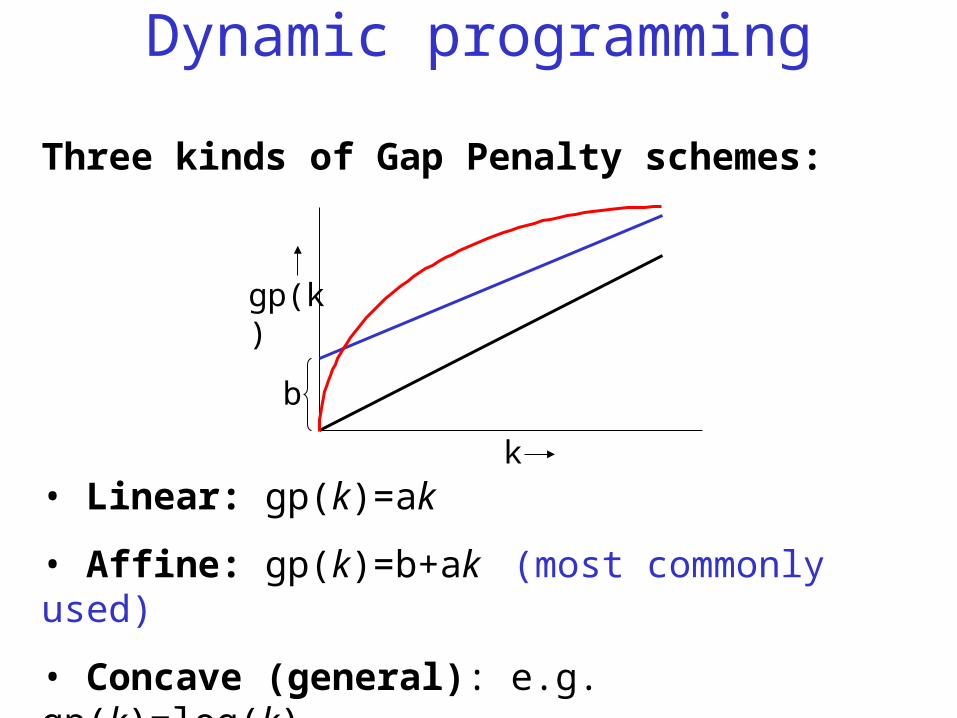
Three kinds of Gap Penalty schemes:
• Linear: gp(k)=ak
• Affine: gp(k)=b+ak (most commonly used)
• Concave (general): e.g. gp(k)=log(k)k is the gap length
Dynamic programming
b
k
gp(k)
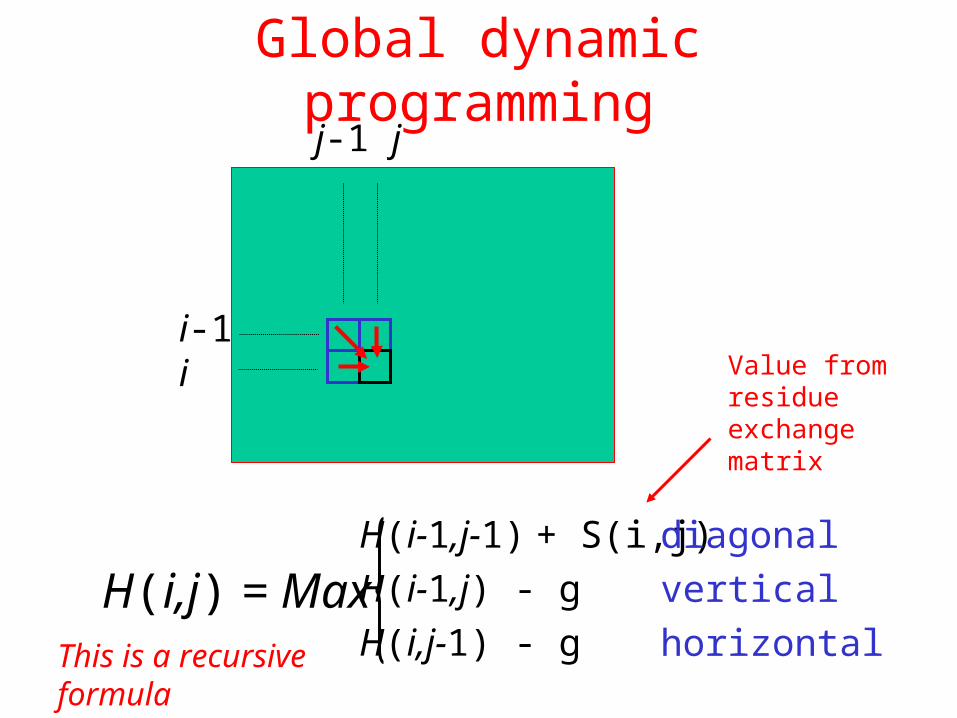
Global dynamic programming
i-1i
j-1 j
H(i,j) = Max H(i-1,j-1) + S(i,j)H(i-1,j) - gH(i,j-1) - g
diagonalverticalhorizontal
Value from residue exchange matrix
This is a recursive formula

Dynamic programming steps
MDAASTILCGS
Search matrix
MDAGSTVILCFVG-MDAAST-ILC--GS
MDAGSTVILCFVG 1. Forward step: Fill in search matrix using appropriate scoring system (exchange matrix and gap penalties) and place back-pointers
2. Get alignment score from ‘last’ cell:
1. Lower right cell (global and semi-global DP)
2. Highest scoring cell anywhere in matrix (local alignment – see later slides)
3. Perform trace-back (using back-pointers placed during forward step) and get alignment

Global dynamic programmingPAM250, Gap =6 (linear)
S P E A R E
0 -6 -12 -18 -24 -30 -36
S -6 2 -4 -10 -16 -22 -28
H -12 -4 2 -3 -9 -14 -20 -24
A -18 -10 -3 0 -1 -7 -13 -18
K -24 -16 -9 -3 -1 2 -4 -12
E -30 -22 -15 -5 -3 -2 6 -6
-30 -24 -18 -12 -6 0
S P E A R E
S 2 1 0 1 0 0
H -1 0 1 -1 2 1
A 1 1 0 2 -2 0
K 0 -1 0 -1 3 0
E 0 -1 4 0 -1 4
These values are copied from the PAM250 matrix (see earlier slide) The extra bottom row and rightmost column give the
penalties that would need to be applied due to end gaps
Higgs & Attwood, p. 124
S-HAKESPEARE

Global dynamic programming
i-1i
j-1 j
H(i,j) = Max H(i-1,j-1) + S(i,j)H(i-1,j) - gH(i,j-1) - g
diagonalverticalhorizontal
Problem with simple DP approach:
•Can only do linear gap penalties
•Not suitable for affine and concave penalties

Global dynamic programming with concave gap penalties
Ngila is a global, pairwise alignment program that uses logarithmic and affine gap costs, i.e. C(g) = a+b*g+c*ln(g).
These gap costs are more biologically realistic than the more popular (and efficient) affine gap cost model.
Ngila version 1.2.1 includes two new, evolutionary alignment models. These models allow you to find the maximum alignment of two sequences based on biological, evolutionary parameters--no more guessing at biological costs. Additional changes are noted on the website.
Website & Manual: http://scit.us/projects/ngila/
Reference: Cartwright RA (2007) Ngila: global pairwise alignments with logarithmic and affine gap costs. Bioinformatics. 23(11):1427-1428
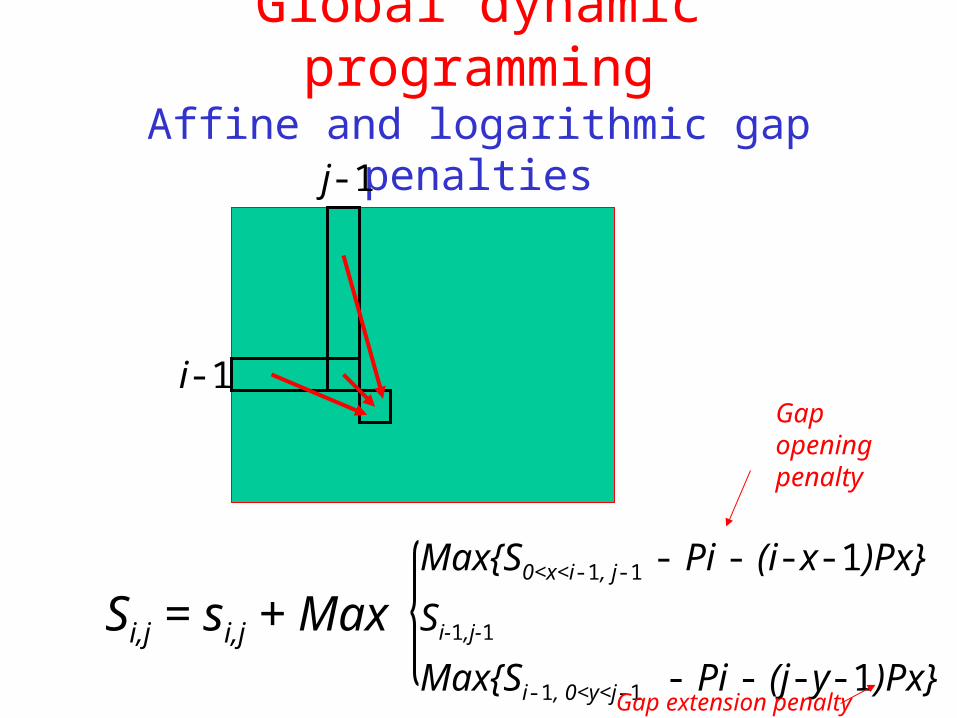
Global dynamic programmingAffine and logarithmic gap penalties
i-1
j-1
Si,j = si,j + Max Max{S0<x<i-1, j-1 - Pi - (i-x-1)Px}
Si-1,j-1
Max{Si-1, 0<y<j-1 - Pi - (j-y-1)Px}
Gap opening penalty
Gap extension penalty
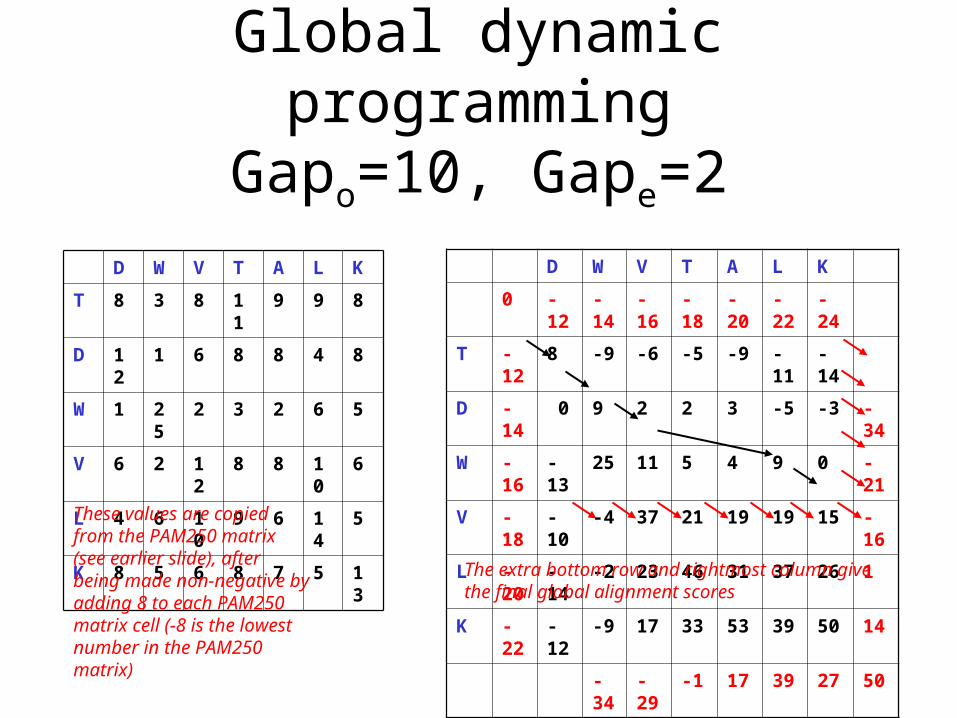
Global dynamic programmingGapo=10, Gape=2
D W V T A L K
0 -12 -14 -16 -18 -20 -22 -24
T -12 8 -9 -6 -5 -9 -11 -14
D -14 0 9 2 2 3 -5 -3 -34
W -16 -13 25 11 5 4 9 0 -21
V -18 -10 -4 37 21 19 19 15 -16
L -20 -14 -2 23 46 31 37 26 1
K -22 -12 -9 17 33 53 39 50 14
-34 -29 -1 17 39 27 50
D W V T A L K
T 8 3 8 11 9 9 8
D 12 1 6 8 8 4 8
W 1 25 2 3 2 6 5
V 6 2 12 8 8 10 6
L 4 6 10 9 6 14 5
K 8 5 6 8 7 5 13
These values are copied from the PAM250 matrix (see earlier slide), after being made non-negative by adding 8 to each PAM250 matrix cell (-8 is the lowest number in the PAM250 matrix)
The extra bottom row and rightmost column give the final global alignment scores

Easy DP recipe for using affine gap penalties
• M[i,j] is optimal alignment (highest scoring alignment until [i,j])• Check
– preceding row until j-2: apply appropriate gap penalties– preceding row until i-2: apply appropriate gap penalties– and cell[i-1, j-1]: apply score for cell[i-1, j-1]
i-1
j-1

DP is a two-step process
• Forward step: calculate scores
• Trace back: start at highest score and reconstruct the path leading to the highest score– These two steps lead to the highest scoring
alignment (the optimal alignment)– This is guaranteed when you use DP!

Global dynamic programming

Semi-global pairwise alignment
• Global alignment: all gaps are penalised• Semi-global alignment: N- and C-terminal gaps
(end-gaps) are not penalised
MSTGAVLIY--TS-----
---GGILLFHRTSGTSNS
End-gaps
End-gaps

Semi-global dynamic programming- two examples with different gap penalties -
Global score is 65 –10 – 1*2 –10 – 2*2
These values are copied from the PAM250 matrix (see earlier slide), after being made non-negative by adding 8 to each PAM250 matrix cell (-8 is the lowest number in the PAM250 matrix)

Semi-global pairwise alignmentApplications of semi-global:
– Finding a gene in genome
– Placing marker onto a chromosome
– One sequence much longer than the other
Danger: if gap penalties high -- really bad alignments for divergent sequences
N- or C-terminal amino acids are often small and polar, giving rise to semi-global alignments with tips of sequences matched in cases of low sequence similarity

Local dynamic programming (Smith & Waterman, 1981)
LCFVMLAGSTVIVGTREDASTILCGS
Amino AcidExchange Matrix
Gap penalties (open, extension)
Search matrix
Negativenumbers
AGSTVIVG
A-STILCG

Local dynamic programming (Smith & Waterman, 1981)
i-1
j-1
Si,j = Max
Si,j + Max{S0<x<i-1,j-1 - Pi - (i-x-1)Px}
Si,j + Si-1,j-1
Si,j + Max {Si-1,0<y<j-1 - Pi - (j-y-1)Px}
0
Gap opening penalty
Gap extension penalty
Local dynamic programming

Dot plots
• Way of representing (visualising) sequence similarity without doing dynamic programming (DP)
• Make same matrix, but locally represent sequence similarity by averaging using a window

Comparing two sequences
We want to be able to choose the best alignment between two sequences.
A simple method of visualising similarities between two sequences is to use dot plots. The first sequence to be compared is assigned to the horizontal axis and the second is assigned to the vertical axis.•Comparison is done through scoring diagonals of preset length (gapless fragments)

Comparing two sequences using a dot plot
Central cell in window takes the (average) score over all window cells
Here you see a diagonal (window) of 7 residue pairs, i.e. the window has length 7.

Dot plots can be filtered by window approaches (to calculate running averages) and applying a threshold
They can identify insertions, deletions, inversions

Global or Local Pairwise alignment
Local
Global
A B
A
B
A B
B A
AB
Local
Global
A BA
CC
A B C
A B C
A B
A
C
C
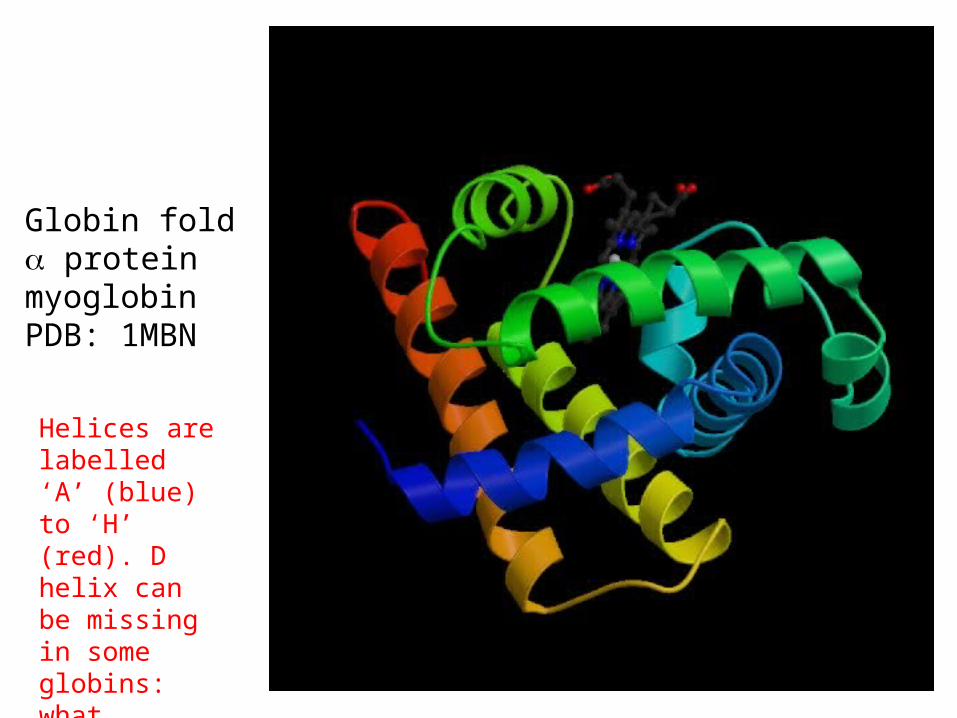
Globin fold proteinmyoglobinPDB: 1MBN
Helices are labelled ‘A’ (blue) to ‘H’ (red). D helix can be missing in some globins: what happens with the alignment?

Pyruvate kinasePhosphotransferase
barrel regulatory domain
barrel catalytic substrate binding domain
nucleotide binding domain

What does this mean for alignments?
• Alignments need to be able to skip secondary structural elements to complete domains (i.e. putting gaps opposite these motifs in the shorter sequence).
• Depending on gap penalties chosen, the algorithm might have difficulty with making such long gaps (for example when using high affine gap penalties), resulting in incorrect alignment.

There are three kinds of pairwise alignments
Global alignment – align all residues in both sequences; all gaps are penalised
Semi-global alignment – align all residues in both sequences; end gaps are not penalised (zero end gap penalties)
Local alignment – align part of each sequence; end gaps are not applicable

Easy global DP recipe for using affine gap penalties (after Gotoh)
M[i,j] is optimal alignment (highest scoring alignment until [i, j]) At each cell [i, j] in search matrix, check Max coming from:
any cell in preceding row until j-2: add score for cell[i, j] minus appropriate gap penalties;
any cell in preceding column until i-2: add score for cell[i, j] minus appropriate gap penalties;
or cell[i-1, j-1]: add score for cell[i, j] Select highest scoring cell in bottom row and rightmost column and do
trace-back
i-1
j-1Penalty = Pi + gap_length*Pe
Si,j = si,j + Max
Max{S0<x<i-1, j-1 - Pi - (i-x-1)Px}
Si-1,j-1
Max{Si-1, 0<y<j-1 - Pi - (j-y-1)Px}

Let’s do an example: global alignment
Gotoh’s DP algorithm with affine gap penalties (PAM250, Pi=10, Pe=2)
Row and column ‘0’ are filled with 0, -12, -14, -16, … if global alignment is used (for N-terminal end-gaps); also extra row and column at the end to calculate the score including C-terminal end-gap penalties.
D W V T A L K
T 0 -5 0 3 1 1 0
D 4 -7 -2 0 0 -4 0
W -7 17 -6 -5 -6 -2 -3
V -2 -6 4 0 0 2 -2
L -4 -2 2 1 -2 6 -3
K 0 -3 -2 0 -1 -3 5
D W V T A L K
0 -12 -14 -16 -18 -20 -22 -24
T -12 0 -17 -14 -13 -17 -19 -22 -22
D -14 -8 -7 -14 -14 -13 -42
W -16 -21 9 -13 -19 -18
V -18 -18 -20 13 -3 -16
L -20 -22 -18 -1 14 -1 -14
K -22 -20 -21 -12
-24 -42 -41 -18 -16 -14 -12 0PAM250
Cell (D2, T4) can alternatively come from two cells (same score): ‘high-road’ or ‘low-road’

Let’s do another example: semi-global alignment
Gotoh’s DP algorithm with affine gap penalties (PAM250, Pi=10, Pe=2)
Starting row and column ‘0’, and extra column at right or extra row at bottom is not necessary when using semi global alignment (zero end-gaps). Rest works as under global alignment.
D W V T A L K
T 0 -5 0 3 1 1 0
D 4 -7 -2 0 0 -4 0
W -7 17 -6 -5 -6 -2 -3
V -2 -6 4 0 0 2 -2
L -4 -2 2 1 -2 6 -3
K 0 -3 -2 0 -1 -3 5
D W V T A L K
T 0 -5 0 3
D 4 -7 -7
W -7 21 -13
V -2 -13 25 9
L
K
PAM250
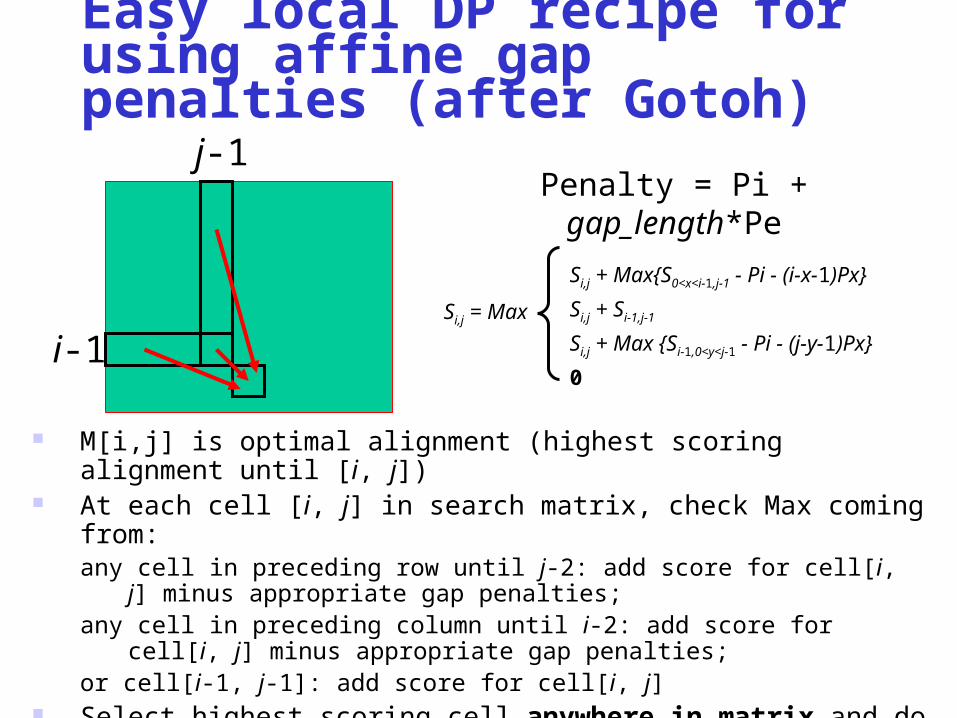
Easy local DP recipe for using affine gap penalties (after Gotoh)
M[i,j] is optimal alignment (highest scoring alignment until [i, j]) At each cell [i, j] in search matrix, check Max coming from:
any cell in preceding row until j-2: add score for cell[i, j] minus appropriate gap penalties;
any cell in preceding column until i-2: add score for cell[i, j] minus appropriate gap penalties;
or cell[i-1, j-1]: add score for cell[i, j] Select highest scoring cell anywhere in matrix and do trace-back until
zero-valued cell or start of sequence(s)
i-1
j-1Penalty = Pi + gap_length*Pe
Si,j = Max
Si,j + Max{S0<x<i-1,j-1 - Pi - (i-x-1)Px}
Si,j + Si-1,j-1
Si,j + Max {Si-1,0<y<j-1 - Pi - (j-y-1)Px}
0

Let’s do yet another example: local alignment
Gotoh’s DP algorithm with affine gap penalties (PAM250, Pi=10, Pe=2)
Extra start/end columns/rows not necessary (no end-gaps). Each negative scoring cell is set to zero. Highest scoring cell may be found anywhere in search matrix after calculating it. Trace highest scoring cell back to first cell with zero value (or the beginning of one or both sequences)
D W V T A L K
T 0 -5 0 3 1 1 0
D 4 -7 -2 0 0 -4 0
W -7 17 -6 -5 -6 -2 -3
V -2 -6 4 0 0 2 -2
L -4 -2 2 1 -2 6 -3
K 0 -3 -2 0 -1 -3 5
D W V T A L K
T 0 0 0 3
D 4 0 0 0
W 0 21 0 0
V 0 0 25 9
L 0 0 11
K 0 0
PAM250

What to align, nucleotide or amino acid sequences?
If ORF then align at protein level– (i) Many mutations within DNA are synonymous, leading to
overestimation of sequence divergence if compared at the DNA level. – (ii) Evolutionary relationships can be more finely expressed using a
20×20 amino acid exchange table than using nucleotide exchanges. – (iii) DNA sequences contain non-coding regions which should be
avoided in homology searches. Still an issue when translating into (six) protein sequences through a codon table.
– (iv) Searching at protein level: frameshifts can occur, leading to stretches of incorrect amino acids and possibly elongation of sequences due to missed stop codons. But frameshifts normally result in stretches of highly unlikely amino acids: can be used as a signal to trace.

A note on reliabilityA note on reliability
All these matrices are designed using standard evolutionary models.
It is important to understand that evolution is not the same for all proteins, not even for the same regions of proteins.
No single matrix performs best on all sequences. Some are better for sequences with few gaps, and others are better for sequences with fewer identical amino acids.
Therefore, when aligning sequences, applying a general model to all cases is not ideal. Rather, re-adjustment can be used to make the general model better fit the given data.

Alignment summaryAlignment summary If ORF exists, then align at protein level. Amino acid substitution matrices reflect the log-odds ratio
between the evolutionary and random model and therefore help in determining homology via the alignment score. The evolutionary and random models depend on generalized data used to derive them. This is not an ideal
solution. Apart from the PAM and BLOSUM series, a large number of further matrices have been developed. Matrices have been made based on DNA, protein structure, information content, etc. For local alignment, BLOSUM62 is often superior; for distant (global) alignments, BLOSUM50, GONNET, or (still) PAM250 work well. Remember that gap penalties are always a problem; unlike the matrices themselves, there is no formal way to calculate their values -- you can follow recommended settings, but these are based on trial and error and not on a formal framework.
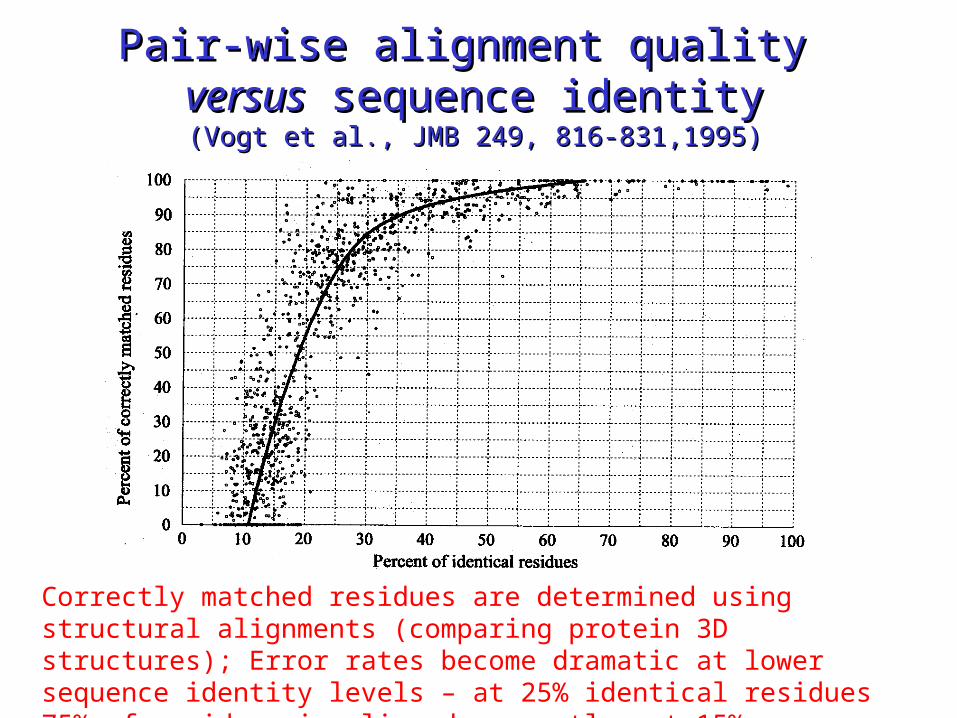
Pair-wise alignment quality Pair-wise alignment quality versusversus sequence identity sequence identity
(Vogt et al., JMB 249, 816-831,1995)(Vogt et al., JMB 249, 816-831,1995)
Correctly matched residues are determined using structural alignments (comparing protein 3D structures); Error rates become dramatic at lower sequence identity levels – at 25% identical residues 75% of residues is aligned correctly, at 15% sequence identity only 30% is aligned correctly!