1 pharmacovigilance lindsey connery pharmacovigilance manager, cancer research uk clinical trials...
TRANSCRIPT

1
Pharmacovigilance
Lindsey ConneryPharmacovigilance Manager, Cancer Research UK Clinical Trials Unit, Glasgow

2
Pharmacovigilance
What is Pharmacovigilance? Why is safety reporting important? Definitions in Pharmacovigilance Responsibilities of the Investigator Responsibilities of the trial Sponsor Why good Pharmacovigilance remains key What you can do to help Examples

3
Definition of Pharmacovigilance
Pharmakon (Greek) = Medical SubstancesVigilia (Latin) = To keep watch
The science of collecting, monitoring, researching, assessing, and evaluating information on theadverse effects of medicinal products.1
Helps ensure the safety, quality and efficacy ofmedicinal products.
1 http://www.mhra.gov.uk/Howweregulate/Medicines/Inspectionandstandards/GoodPharmacovigilancePractice/index.htm

4
Why is Safety Reporting Important?
It is mandated by law Protects trial participants Increases our understanding of
drugs Identifies new unexpected
reactions Identifies unexpected patterns
of events

5
Why is Safety Reporting Important?
Sulfanilamide (1937)
Elixir sulfanilamide was an improperly prepared sulfamide medicine that caused mass poisoning in the US
It caused the death of more than 100 people The public outcry from this and other such incidents
led to the 1938 Federal Food Drug and Cosmetics Act

6
Why is Safety Reporting Important?
Thalidomide (1961- 1962)
Introduced as a safe hypnotic and antiemetic rapidly became a popular to treat morning sickness
Drug proved to be a potent human teratogen that caused major birth defects in an estimated 10,000 children
The thalidomide drug disaster led Europe and elsewhere to the establishment of the drug regulatory mechanisms of today
These mechanisms require that new drugs are licensed by well established regulatory authorities before being introduced for clinical use

7
Pharmacovigilance Rationale
Pre-Approval DataControlledLimitedImmature safety data
Post-Approval DataReal life, uncontrolledOff label useGeneric
Solicited Safety data
Unsolicited safety data
PreclinicalAnimal Phase I II III Trials Phase IV, Post Approval Studies Spontaneous Reporting Studies

8
Pharmacovigilance
Legislation
EU Clinical Trials Directive (2001/20/EC) Medicines for Human Use (Clinical trial Regulations SI
200/1031 (transposed EU Directive into UK law in 2004) Amendment SI2006/1928 the Good Clinical Practice (GCP)
Directive in 2006 Further amendments and EU requirements such at CT3 in
June 2011 and ICH E2F guidelines for Development Safety Update Reports (DSURs) September 2011
New EU Clinical Trials directive became effective on 16 Jun 2014

9
Pharmacovigilance
The Clinical Trial Regulations Cover:
Definitions (IMPs, AEs, SAEs, SUSARs, DSURs etc.) The responsibilities of Investigators for recording Adverse Events
(AEs) and the notification of AEs to trial Sponsors including reporting SAEs immediately (within a maximum of 24 hrs)
The responsibilities of Sponsors for keeping detailed records of all AEs relating to a clinical trial which are reported by Investigators for that trial. Also Sponsors must record and report all relevant information about SUSARs which occur during the course of a clinical to the Licensing Authorities/Competent Authorities of any EEA state where the trial is being conducted and relevant Ethics Committee, Sponsors are also required to provide annual detailed safety reports

10
Pharmacovigilance Definitions
Investigational Medicinal Products (IMPs)- Pharmaceutical form of an active ingredient or placebo tested as a reference in a clinical trial, including a product with a marketing authorisation when used or assembled (formulated or packaged) in a way different from the approved form, or used for an unapproved indication, or used to gain further information about an approved use
Adverse Events (AEs)- any untoward medical occurrence in a subject to whom a medicinal product has been administered, including occurrences which are not necessarily caused by or related to that product
Adverse Reaction (AR)- any untoward and unintended response in a subject to an investigational medicinal product which is related to any dose administered to that subject

11
Pharmacovigilance
Unexpected Adverse Reaction (UAR)-
An adverse reaction, the nature, or severity of which is not consistent with the applicable product information about the medicinal product in question set out:
In the case of a product with market authorisation the Summary of Product Characteristics (SmPC) for that product
In the case of any other investigational medicinal product, in the Investigators Brochure (IB) relating to the trial in question

12
Pharmacovigilance Definitions
A Serious Adverse Event (SAE) Serious Adverse Reaction (SAR) or Suspected Unexpected Serious Adverse Reaction(SUSAR) is defined as any untoward medical occurrence, notnecessarily related to protocol treatment, that:
Results in death Is life-threatening Requires hospitalisation or prolongation of existing hospitalisation Results in persistent or significant disability or incapacity Consists of a congenital anomaly or birth defect Is considered medically significant by the Investigator

13
Pharmacovigilance Definitions
Life threatening: The patient is at immediate risk of
death from the event as it occurred. It does not include an event that, had it occurred in a more serious form, might have caused death
Requires in-patient
hospitalisation: Is as a hospital admission required
for treatment of an adverse event even when the adverse event is not related to the protocol treatment

14
Pharmacovigilance Definitions
Development Safety Update Report (DSUR)
Is a comprehensive safety report submitted annually to the Regulatory Authority and EthicsCommittee throughout the duration of a trial. DSURs provide a common standard for periodicreporting on drugs under development (including drugs that are under further study) among ICH
regions and meets national and regional requirements previously met by the US IND Annual Report and the EU Annual Safety Report)

15
Pharmacovigilance Definitions
Reference Safety Information (RSI)
Reference Safety Information (RSI) are thedocuments that are used to assess whether serious adverse reactions are unexpected and meet the criteria of a SUSAR The events that are known reactions for theIMP (based on previous trials and safety reports submitted) are recorded in a specific section of the IB and section 4.8 undesirable effects of SmPC. Any
reported reaction that is not listed in the RSI as expected, requires reporting as a SUSAR

16
Investigator Responsibilities
Investigators are responsible for the following:
The safety of the trial participants recruited from their hospital trial site
Recording adverse events in the patient notes (source data) and Case Report Forms (CRFs), the data collection forms, as detailed in the trial protocol
Reporting SAEs immediately to the Sponsor (within a maximum of 24 hours after their knowledge of the event)
Providing information to report SUSARs
Maintaining the RSI and all other trial documentation, including safety documentation in their trial site and pharmacy file

17
AE Assessment
Serious Does the event meet the
regulatory definition of serious?
Causality: Is the event a reaction to the study
medication or not?
Expectedness: Is it a recognized adverse effect of
the study medication?
18
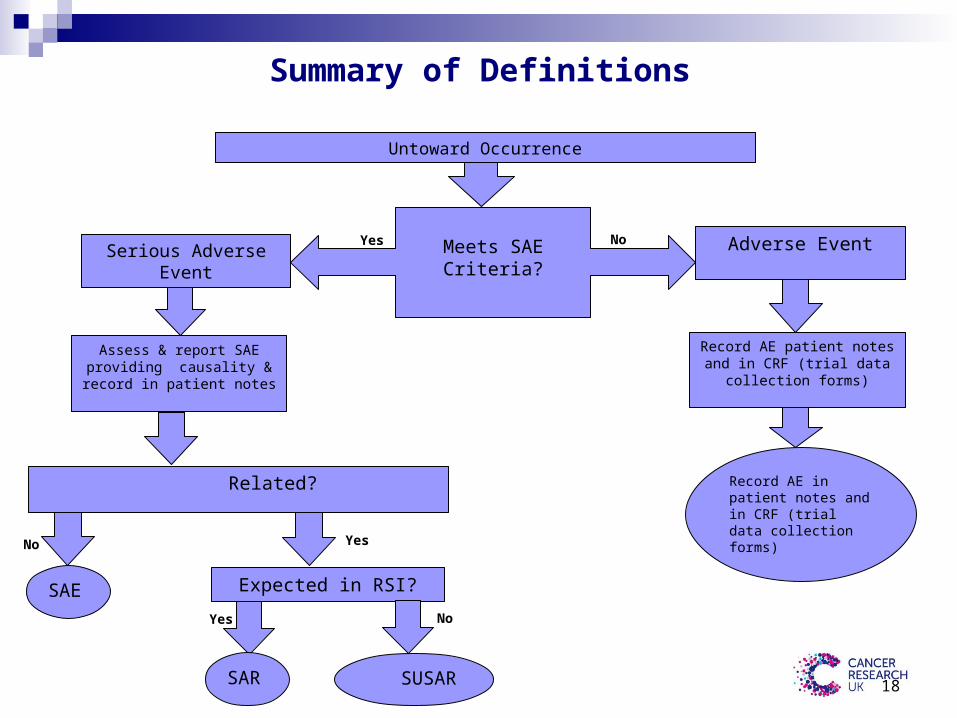
Summary of Definitions
Untoward Occurrence
Meets SAE Criteria?Yes No Adverse Event
Record AE patient notes and in CRF (trial data collection forms)
Assess & report SAE providing causality &record in patient notes
Record AE in patient notes and in CRF (trial data collection forms)
Related?
Serious Adverse Event
YesNo
SAE Expected in RSI?
Yes No
SAR SUSAR
19
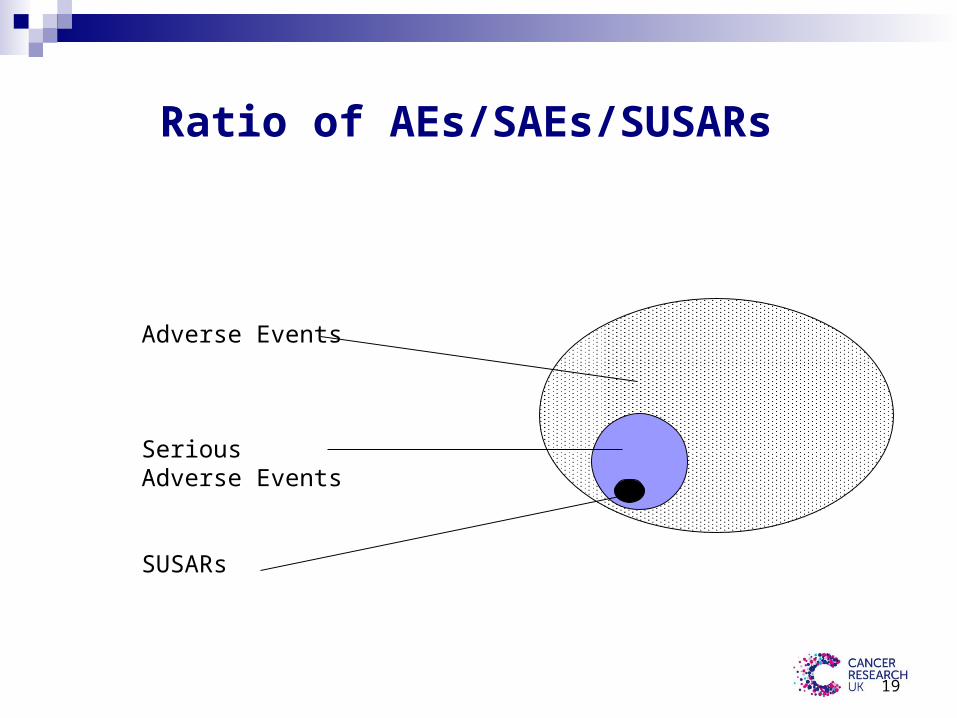
Ratio of AEs/SAEs/SUSARs
Adverse Events
Serious Adverse Events
SUSARs

20
Sponsor Responsibilities
The Sponsor is responsible for the following:
The ongoing safety evaluation of trials of investigational medicinal products (IMPs)
For identifying and the expedited reporting (within 7 & 15 days), of adverse drug reactions that are both serious and unexpected (SUSARs)
Promptly informing all concerned Investigators/trial sites, Regulatory Authorities and Ethics Committees of findings that could adversely affect the safety of trial participants, impact on the trial or alter the continued approval of the trial
For preparing and submitting to the Regulatory Authorities and Research Ethics Committees a detailed annual update on the safety of trial participants for CTIMPs – the Development Safety Update Reports (DSURs)
Managing updates to RSI

21
Delegation
The Sponsor may delegatepharmacovigilance or safetyreporting activities to a third partysuch as a Contract Research trialsOrganisation (CRO) or academic unit however the Sponsor remainsresponsible for the trials ongoingsafety evaluation.

22
Sponsor Responsibilities
Systems
The sponsor must have systems in place to ensurethey meet their responsibilities. The System must allow for:
Recording Notifying Assessing Reporting Analysing Managing
Adverse Events

23
Risk-Adapted Approaches
Based on what is known about the IMPs The phase of the trial (from phase I first in man to phase IV trials in
licensed medication) The level of clinical experience with the trial medication in the population
under study
Depending on the risk associated with a trial it may be reasonable to collect one or all of the following:
All AEs (serious and/or non-serious) Only SAEs (or, in certain circumstances only specific types of SAE) All ARs (serious and/or non-serious) Only SARs All AEs/ARs of a certain grade of severity

24
Safety Reporting for Non-CTIMPs
Non-CTIMPs are required to adhere to the National ResearchEthics Service (NRES) safety reporting requirements and theResearch Governance Framework for Health and Social Care
Systems for managing safety are required Investigators are required to document AEs and report SAEs in accordance
with the protocol SAEs that are considered related by the Investigator to the trial intervention
and listed as unexpected in the protocol are required to be reported to
The Ethics Committee within 15 days and to trial sites
Safety information needs to be included in the trial annual progress report which is submitted to the Ethics Committee

25
Why Good Pharmacovigilance Remains key
List of licensed drugs withdrawnafter marketing for drug safety reasons:
Secholex (polidexide) 1975 Zomax 1983 Halcion 1991 Kava kava 2001-due to liver toxicity Vioxx 2004-due to increased cardiovascular risks Bextra 2005-due to Stevens-Johnson Syndrome

26
How You Can Help
Meet your legal reporting obligations
Report SAEs immediately and within a maximum of 24 hours of your knowledge of the event Provide your contact details when submitting SAE reports Ensure you have received an acknowledgement receipt for the SAE
report Submit timely follow-up information until the SAE resolves When a SAE is identified as a potential SUSAR, provide full and accurate
information as requested to allow proper assessment of the event

27
How You Can Help
Enable the Sponsor to meeting their legal obligations
Provide information for SUSAR reports as quickly as possible to allow the Sponsor to meet their legal obligations of identifying andreporting SUSARs within 7 days for fatal and life threatening events and 15 days for all other events Provide a causality assessments based on the information the Investigator has available regards the patient, using their knowledge of the known toxicities of the IMPs and of the disease
being treated. It is acceptable for causality to be changed based on the information available.

28
How Can You Help
Meet GCP requirements for quality
Check the protocol for details of SAE reporting for each trial, the timeframe for reporting, additional reporting requirements (AEs of special interest), exceptions to reporting
Ensuring that there are the staff and systems in place to ensure the 24 hour reporting requirements for reporting SAEs can always be met including over holiday periods
Ensure the information provided in SAE reports is accurate, consistent and contains detailed relevant information including a diagnosis of the event and causality to the IMPs
Respond to SAE data queries quickly and accurately

29
Safety Reporting Scenarios A 54 year old patient participation in a cardiac disease trial slips and falls
on ice whilst out shopping. He hurts his wrist. An X-ray performed at A&E reveals he has fractured his wrist. He has the support of his daughter and is allowed to return home with an appointment at the Fracture Clinic
Is this an AE? A SAE? Related to trial drugs? What action is required?
A 47 year old women with ovarian cancer has consented to participate in a trial of a novel chemotherapy drug. After 4 weeks on study she is admitted to hospital with acute liver failure. The Investigator Brochure describes mild liver enzyme elevation as a possible side effect of treatment
Is this an AE? A SAE? Related to the trial medication? What action isrequired?