1.3 schedule of activities - transceleratebiopharmainc.com · web viewconfirmation of...
TRANSCRIPT
Common Protocol Template Alzheimer’s Disease Library V003
Common Protocol Template Alzheimer’s Disease Library V003
Section in Common Protocol Template (CPT)
Library Content
1.3. Schedule of Activities (SoA) Instructional text regarding the following:
Medical History/ Disease History
Baseline magnetic resonance imaging (MRI) and/or positron emission tomography (PET) studies
Cerebrospinal fluid (CSF) and plasma neurochemical biomarkers
Genetics (blood sample)
Laboratory assessments to rule out other disorders
3. Objectives and [Estimands/Endpoints] Objectives and corresponding estimands/endpoints utilizing Alzheimer’s Disease (AD) specific instruments for cognitive, functional, behavioral, global function, quality of life (QOL), and resource utilization assessments
5. Study Population
5.1. Inclusion Criteria
5.2. Exclusion Criteria
Instructional text and suggested inclusion and exclusion criteria
8. Study Assessments and Procedures
8.1. Efficacy Assessments
Instructional text on screening assessments
Descriptions of the AD specific instruments for cognitive, functional, behavioral, global function, QOL, and resource utilization assessments
9. Statistical Considerations
9.3. Populations for Analyses
Capping strategy for target subgroups, if any.
Study populations for efficacy and safety analyses
Brief description of analyses for the primary endpoint(s)
10.5 Appendix 5: Genetics Instructions for obtaining and handling the blood sample for Apolipoprotein E (ApoE) Pharmacogenomic Sample(s)
© 2018 TransCelerate BioPharma 1
Common Protocol Template Alzheimer’s Disease Library V003
10.[X] Appendix [X]: Diagnostic and Statistical Manual of Mental Disorders- (DSM) Criteria
Additional appendix relevant to Alzheimer’s Disease which can be added to the Core
10.[X] Appendix [X]: National Institute of Neurological and Communicative Disorders and Stroke – Alzheimer’s Disease and Related Disorder Association (NINCDS-ADRDA) Criteria
Additional appendix relevant to Alzheimer’s Disease which can be added to the Core
10.[X] Appendix [X]: Modified Hachinski Ischemic Score
Additional appendix relevant to Alzheimer’s Disease which can be added to the Core
11. References References applicable to the Alzheimer’s library
© 2018 TransCelerate BioPharma 2

Common Protocol Template Alzheimer’s Disease Library V003
1.3 Schedule of Activities (SoA)
[X]
3. Objectives and [Estimands/Endpoints]
Primary Objectives Primary [Estimands/Endpoints]
To assess the effect of [study intervention X] [vs. comparator, if applicable] on the [cognitive scale] at Week [X] in participants with [severity] Alzheimer’s Disease
The change from baseline to Week [X] in the Alzheimer’s Disease Assessment Scale – Cognitive Assessment (ADAS-Cog) [subscore type number] total score
To assess the effect of [study intervention X] [vs. comparator, if applicable] on the [functional scale] at Week [X] in participants with [severity] Alzheimer’s Disease
The change from baseline to Week [X] in the Alzheimer’s Disease Cooperative Study –Activity in Daily Living [Mild Cognitive Impairment or Severe Impairment Version, as applicable] (ADCS-ADL-[MCI or SIV]) total score
Secondary Objectives Secondary [Estimands/Endpoints]
To assess the effect of [study intervention X] [vs. comparator X, if applicable] on the [measure of behavioral/neuropsychiatric symptoms] at Week [X] in participants with [severity] Alzheimer’s Disease
The change from baseline to Week [X] in the Neuropsychiatric Inventory (NPI) total score
To assess the effect of [study intervention X] [vs. comparator X, if applicable] on the [ Mini-Mental State Examination (MMSE) / Disability Assessment for Dementia (DAD)] at Week [X] in participants with [severity] Alzheimer’s Disease
The change from baseline to Week [X] in the MMSE total score
The change from baseline to Week [X] in the DAD total score
To assess the effect of [study intervention X] [vs. comparator X, if applicable] on the [measure of global function] at Week [X] in participants with [severity] Alzheimer’s Disease
The change from baseline to Week [X] in the Clinical Dementia Rating Scale Sum of Boxes (CDR-SB)
© 2018 TransCelerate BioPharma 3

Common Protocol Template Alzheimer’s Disease Library V003
Secondary Objectives Secondary [Estimands/Endpoints]
To assess the effect of [study intervention X] [vs. comparator X, if applicable] on the [measure of AD-related psychosis] at Week [X] in participants with [severity] Alzheimer’s Disease
The change from baseline to Week [X] in the sum of the delusions and hallucinations subdomain scores of the NPI
To assess the effect of [study intervention X] [vs. comparator X, if applicable] on the [measure of resource utilization] at Week [X] in participants with [severity] Alzheimer’s Disease
The change from baseline to Week [X] in the primary and secondary caregiver time components of the Resource Utilization in Dementia (RUD[-Lite])
The change from baseline to Week [X] in the (RUD[-Lite]) total score
To assess the effect of [study intervention X] [vs. comparator X, if applicable] on the [quality of life scale] at Week [X] in participants with [severity] Alzheimer’s Disease
The change from baseline to Week [X] in the EuroQol 5-Dimensional Health Related Quality of Life Scale (EQ-5D) total score and domain scores
To assess the effect of [study intervention X] [vs. comparator X, if applicable] on the [measure of clinician assessment/rating of global function] at Week [X] in participants with [severity] Alzheimer’s Disease
The change from baseline to Week [X] in the Clinician’s Interview-Based Impression of Change plus caregiver input (CIBIC+)
To evaluate the safety and tolerability of [study intervention X] [vs. comparator X, if applicable] over [X] weeks in participants with [severity] AD who are on a stable background regimen of [background treatment]
The proportion of participants with adverse events, serious adverse events (SAEs), and adverse events leading to study intervention discontinuation over the [X]-week study intervention period
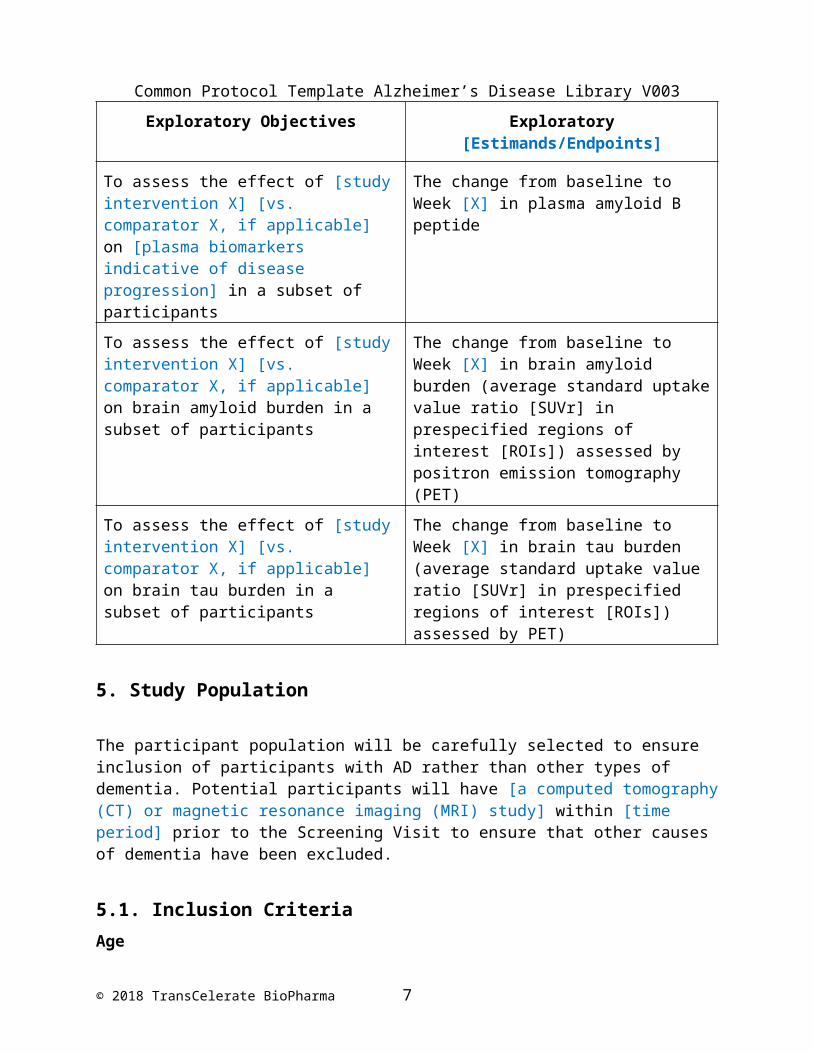
Exploratory Objectives Exploratory [Estimands/Endpoints]
To assess the effect of [study intervention X] [vs. comparator X, if applicable] on [cerebrospinal fluid (CSF) biomarkers indicative of disease progression] in a subset of participants
The change from baseline to Week [X] in [CSF A1-42, total tau protein (T-tau), and/or phosphorylated tau protein (P-tau)]
To assess the effect of [study intervention X] [vs. comparator X, if applicable] on [plasma biomarkers indicative of disease progression]
The change from baseline to Week [X] in plasma amyloid B peptide
© 2018 TransCelerate BioPharma 4
Common Protocol Template Alzheimer’s Disease Library V003
Exploratory Objectives Exploratory [Estimands/Endpoints]
in a subset of participants
To assess the effect of [study intervention X] [vs. comparator X, if applicable] on brain amyloid burden in a subset of participants
The change from baseline to Week [X] in brain amyloid burden (average standard uptake value ratio [SUVr] in prespecified regions of interest [ROIs]) assessed by positron emission tomography (PET)
To assess the effect of [study intervention X] [vs. comparator X, if applicable] on brain tau burden in a subset of participants
The change from baseline to Week [X] in brain tau burden (average standard uptake value ratio [SUVr] in prespecified regions of interest [ROIs]) assessed by PET)
5. Study Population
The participant population will be carefully selected to ensure inclusion of participants with AD rather than other types of dementia. Potential participants will have [a computed tomography (CT) or magnetic resonance imaging (MRI) study] within [time period] prior to the Screening Visit to ensure that other causes of dementia have been excluded.
5.1. Inclusion CriteriaAge
1. Participants must be men and women [> 50 years of age] at the time of signing the informed consent with a diagnosis of probable AD.
Type of Participant and Disease Characteristics
2. [Severity] AD as characterized by the following clinical, cognitive, and functional criteria.2
a. Diagnosis of a clinical syndrome of cognitive impairment consistent with [severity] AD per International Working Group (IWG) diagnostic criteria or [severity] AD per National Institute on Aging - Alzheimer’s Association (NIAA-AA) diagnostic criteria
b. Self or informant report of memory decline c. MMSE scores between [X to X] within the last [X] months d. Objective memory loss by education-adjusted Wechsler Memory Scale Logical
Memory IIe. Absence of significant levels of impairment in other cognitive assessments
© 2018 TransCelerate BioPharma 5
Common Protocol Template Alzheimer’s Disease Library V0033. Meets the Diagnostic and Statistical Manual of Mental Disorders Version [X] (DSM-X)
criteria (see Appendix 10.[X]) and/or National Institute of Neurological and Communicative Disorders and Stroke-Alzheimer’s Disease and Related Disorder Association’s (NINCDS-ADRDA) criteria for probable AD (see Appendix 10. [X]))
4. Modified Hachinski Ischemic Score <= 4 (see Appendix 10.[X])5. Brain imaging study, such as magnetic resonance imaging (MRI) and/or computed
tomography (CT), consistent with a diagnosis of probably AD without any other clinically significant co-morbid pathologies within 12 months prior to the Screening Visit. If there has been a significant change in clinical status suggestive of stroke or other possible central neurological disease with onset between the time of the last MRI or CT and the Screening evaluation, the scan should be repeated during Screening procedures if considered appropriate by the Investigator.
6. Screening CSF results consistent with the presence of amyloid pathology.7. Absence of other (non-AD) types of dementia.8. Willing and able to give informed consent. If the study participant is not competent, a
legally acceptable representative must provide informed consent on their behalf, and the participant must provide assent.
9. Either currently or previously (in pre-AD condition) literate and capable of reading, writing, and communicating effectively with others.
10. [May/Must] be using the following concomitant medications for management of AD at Screening and during the study: [list medications]. These medications must be started at least [X months] before the first dose of [study intervention] and the regimens must remain constant throughout the study.
11. Have a caregiver who assists the participant at least 5 days per week for at least [X] hours per day and has intimate knowledge of the participant’s cognitive, functional, and emotional states and of the participant’s personal care. The caregiver must be willing to accompany the participant to all study visits and to supervise study intervention administration as well as report adverse events. The caregiver must be willing and able to give informed consent for their own participation, be able to read and write, and be capable of providing responses to the [list assessments that require caregiver written or oral input/response/other participation/activity].
12. Participants previously enrolled in an AD clinical trial involving a disease modifying or symptomatic therapeutic agent may enroll in this study if treatment with the symptomatic therapeutic agent ended more than [X months] before the first dose of [study intervention] in this study.
5.2. Exclusion Criteria
Medical Conditions
1. Major structural brain disease (e.g., ischemic infarcts, subdural hematoma, hemorrhage, hydrocephalus, brain tumors, multiple subcortical ischemic lesions, or a single lesion in a critical region [e.g., thalamus, hippocampus])
© 2018 TransCelerate BioPharma 6
Common Protocol Template Alzheimer’s Disease Library V0032. Any other CNS trauma (e.g., contusion), or infections that affect brain function (e.g.,
HIV, syphilis)3. Diagnosis of schizophrenia4. History or current suicide risk 5. Major medical illness or unstable medical condition within 6 months before Screening
that in the opinion of the investigator may interfere with the participant’s ability to comply with study procedures and abide by study restrictions, or with the ability to interpret safety data, including any physical disability (e.g., blindness, deafness, non-AD-related speed impairment, sensory or motor dysfunction) that would prevent completion of study procedures or assessments
6. Diagnosis of a dementia-related central nervous system disease other than AD (e.g., Parkinson’s Disease, Huntington’s Disease, frontotemporal dementia, multi-infarct dementia, dementia with Lewy bodies, normal pressure hydrocephalus)
Prior/Concomitant Therapy
7. Concomitant [list of medications] within [X] days before the first dose of [study intervention] and throughout the study
8. Use of narcotic analygesics more frequently than [X] days per week as needed for pain within [X] days before the first dose of [study intervention] and throughout the study
9. Prescription medical food (e.g., AxonaTM) or prescription neutriceuticals marketed for AD or cognitive impairment within [X] days before the [Screening MMSE or other cognitive assessment] and throughout the study
Other Exclusions
10. Reside in a nursing home or assisted care facility with need for direct continuous medical care and nursing supervision. Participant may reside in such facilities provided continuous direct medical care is not required and a qualified caregiver is available for co-participation and the participant is physically able to attend all required study visits.
8. Study Assessments and Procedures
8.1. Efficacy Assessments
The primary efficacy assessment scale(s) selected for this study are the [list scales for the primary efficacy endpoints].
Every effort should be made to ensure that the protocol required tests and procedures are completed as described. However, it is anticipated that, from time to time, there may be
© 2018 TransCelerate BioPharma 7

Common Protocol Template Alzheimer’s Disease Library V003circumstances, outside of the control of the investigator, that make it not feasible to perform the test. In these cases, the investigator will take all steps necessary to ensure the safety and well being of the participant. When a protocol required assessment cannot be performed, the investigator will document the reason for this and any corrective and preventive actions which he/she has taken/will take to ensure that normal processes are adhered to as soon as possible. The study team will be informed of these incidents in a timely fashion.
8.1.1. Cognitive Scales
8.1.1.a. Alzheimer’s Disease Assessment Scale – Cognitive Assessment (ADAS-cog)
The ADAS-cog is a structured scale that evaluates memory, orientation, attention, reasoning, language, and constructional praxis. Higher scores indicate greater impairment. The ADAS-cog will be administered at the Screening and Baseline visits before study intervention administration, and at the [Week(s) X] study visits for all participants. These assessments will be used as a [primary/secondary] cognitive measure of clinical effect. The ADAS-cog will be administered by a trained member of the investigational team. At each site, the same individual, whenever possible, will perform the ADAS-cog evaluation on a specific participant throughout the study.
8.1.1.b. Mini-Mental State Examination (MMSE)
The MMSE is a brief 30-point questionnaire used to assess cognitive impairment with lower scores indicating greater impairment. The MMSE assesses 11 categories of cognition including orientation to time, memory, attention, concentration, naming, repetition, comprehension, and the ability to create a sentence and to copy two intersecting polygons. MMSE scores outside of the target severity of disease and score range, inclusive, will be excluded from participation in this study. The MMSE will be administered at the Screening and Baseline visits before study intervention administration, and at the [Week(s) X] study visits for all participants. These assessments will be used as a [primary/secondary] cognitive measure of clinical effect. The MMSE will be administered by a trained member of the investigational team. At each site, the same individual, whenever possible, will perform the MMSE evaluation on a specific participant throughout the study. The Screening MMSE will be used for purposes of determining participant enrollment and does not need to be reconciled with the Baseline MMSE score
8.1.1.c. Severe Impairment Battery (SIB)
The SIB is a 51-item scale developed to assess people who have dementia so severe that they cannot complete conventional neuropsychological testing. The SIB has been shown to be a
© 2018 TransCelerate BioPharma 8

Common Protocol Template Alzheimer’s Disease Library V003reliable and valid measure of cognitive function, particularly in people with moderate to severe AD. The SIB is therefore a useful outcome measure in clinical trials that include participants with more advanced stages of AD.
The items of the SIB are composed of single one-step commands. They are presented in conjunction with gestural cues and instructions to the participant can be repeated a number of times to facilitate comprehension. The SIB is divided into nine subscales as follows: social interaction, memory, orientation, language, attention, praxis, visuospatial ability, construction, and orienting to name. The SIB total score ranges from 0 to 200 with lower score indicating greater cognitive impairment. In addition, a subtotal is computed for each domain in order to assess different cognitive domains separately.
The SIB will be administered at the Screening and Baseline visits before study intervention administration, and at the [Week(s) X] study visits for all participants. These assessments will be used as a [primary/secondary] cognitive measure of clinical effect. The SIB will be administered by a trained member of the investigational team. At each site, the same individual, whenever possible, will perform the SIB evaluation on a specific participant throughout the study.
8.1.2. Functional Scales
8.1.2.a. Alzheimer’s Disease Cooperative Study – Activity in Daily Living (ADCS-ADL)
The ADCS-ADL assesses the competence of participants with AD in basic and instrumental activities of daily living (ADLs). It can be completed by [a caregiver in questionnaire format or administered by a clinician/researcher as a structured interview with a caregiver]. All responses should relate to the 4 weeks prior to the time of rating. Each ADL item takes an ADL (e.g., eating) and provide descriptions of level of competence with the rater selecting the most appropriate option (e.g., ate without physical help and used a knife; used a fork or spoon but not a knife; used fingers to eat; was usually fed by someone else). The ADCS-ADL is available in two versions: one for participants with severe impairment and one for patients with less than severe impairment. This study uses the [severe impairment version (SIV), mild cognitive impairment (MCI) version, or both the severe impairment version (SIV) and the mild cognitive impairment (MCI) version].
The ADCS-ADL-MCI is a 23-item scale that measures basic and instrumental abilities in participants with mild to moderate AD. It has a score ranging from 0 to 53 with lower scores indicating greater functional impairment. It will be administered to the participant’s caregiver by a trained and certified member of the investigational team at the following visits [list visits]. At each site, the same individual, whenever possible, will perform each of the evaluations of a specific participant’s caregiver throughout the study.
© 2018 TransCelerate BioPharma 9

Common Protocol Template Alzheimer’s Disease Library V003The ADCS-ADL-SIV is a 19-item scale modified from the ADCS-ADL-MCI that measures the most appropriate basic and instrumental abilities in participants with severe impairment (moderate to severe AD). It has a score ranging from 0 to 54 with lower scores indicating greater functional impairment. It will be administered to the participant’s caregiver by a trained and certified member of the investigational team at the following visits [list visits]. At each site, the same individual, whenever possible, will perform each of the evaluations of a specific participant’s caregiver throughout the study.
8.1.2.b. Disability Assessment for Dementia (DAD)
The DAD is a 40-item scale that addresses a variety of functional abilities in ADLs. Each item is rated as 0 (No) if the participant is unable to perform the described task, 1 (Yes) indicating normal function, or 999 (Not Applicable [N/A]) if the participant never performed the activity/task before AD diagnosis or if the participant did not have the opportunity to perform the activity/task during the assessment period. The total score is the sum of the scores from the individual items not marked as ‘N/A’ and ranges from 0 to 40 with a lower score indicating greater functional impairment.
The DAD will be administered at the Screening and Baseline visits before study intervention administration, and at the [Week(s) X] study visits for all participants. These assessments will be used as a [primary/secondary] cognitive measure of clinical effect. The DAD will be administered by a trained member of the investigational team. At each site, the same individual, whenever possible, will perform the DAD evaluation on a specific participant throughout the study.
8.1.3. Global Function Scales
8.1.3.a. Clinical Dementia Rating (CDR)
The CDR scale is a clinician-rated dementia staging system that tracks the progression of cognitive impairment in 6 categories (memory, orientation, judgment and problem solving, community affairs, home and hobbies, and personal care). Each category is scored on a 5-point scale in which None=0 , Questionable=0.5, Mild=1, Moderate=2, and Severe=3. The global CDR score is established by clinical scoring rules and has values of 0 (no dementia), 0.5, (questionable dementia), 1 (mild dementia), 2 (moderate dementia), and 3 (severe dementia). The CDR-SB is obtained by adding the ratings in each of the 6 categories and ranges from 0 to 18 with higher scores indicative of greater impairment.
The CDR/CDR-SB will be administered at the Screening and Baseline visits before study intervention administration, and at the [Week(s) X] study visits for all participants. These assessments will be used as a [primary/secondary] of clinical effect. The CDR/CDR-SB will be
© 2018 TransCelerate BioPharma 10

Common Protocol Template Alzheimer’s Disease Library V003administered by a trained member of the investigational team. At each site, the same individual, whenever possible, will perform the CDR/CDR-SB evaluation on a specific participant throughout the study.
8.1.3.b. Alzheimer's Disease Cooperative Study (ADCS) Clinician’s Interview Based Assessment of Change Plus Caregiver Input (CIBIC+)
The version of the CIBIC+ selected for use in this study is the [specify battery such as Alzheimer's Disease Cooperative Study - Clinical Global Impression of Change (ADCS-CGIC)4]. The trained and certified rater for this assessment will be a clinician whose only role in the study is to conduct these global assessments. This independent rater will evaluate the participant’s overall disease severity prior to the initiation of the study intervention. This assessment, known as the Clinician’s Interview Based Assessment of Severity Plus Caregiver Input (CIBIS+), rates the patient on a 7-point scale from extremely severe AD to no symptoms of AD. The overall impression of change from baseline (CIBIC+) is rated on a 7-point scale relative to the initial assessment of severity at baseline: 1=marked improvement; 2=moderate improvement; 3=minimal improvement; 4=no change; 5=minimal worsening; 6=moderate worsening; 7=marked worsening. The [CIBIC+ (specific battery)] has been previously validated and similar versions of the CIBIC+ have been used in other AD trials to support product registration.
At the baseline (CIBIS+) visit, the independent clinician rater will interview the participant and the caregiver together and review the participant’s medical history and all other relevant data to develop a comprehensive impression of the participant’s overall general condition, and more specifically, the participant’s cognitive abilities and deficits, behavior, and ADLs. At subsequent visits [list visits], the same rater will interview the participant and the caregiver separately to perform the CIBIC+ assessment. This rater, whenever possible, will conduct all CIBIC+ assessments for a given participant throughout the study without knowledge of (i.e., independent of) other endpoint assessments or the adverse events experienced by the participant during the study. The rater will exclusively consider observations of the participant’s cognitive, functional, and behavioral performance obtained through the interview process. The rater will compare the findings at each subsequent visit to the baseline assessment.
8.1.3. Behavioral Scales: Neuropsychiatric Inventory (NPI) and Caregiver Distress (NPI-D)
The NPI is a rater-administered, fully structured interview in which all questions are provided and read verbatim. The sole source of information is the interview with a caregiver who knows the participant well. This study uses the NPI version with [10 or 12] behavioral domains: delusions, hallucinations, agitation/aggression, depression/dysphoria, anxiety, elation/euphoria, apathy/indifference, disinhibition, irritability/lability, and aberrant motor behavior [sleep and nightmare behavior disorders and appetite and eating disorders].
© 2018 TransCelerate BioPharma 11

Common Protocol Template Alzheimer’s Disease Library V003The NPI total score is calculated by adding the scores of the domains (each domain scores ranges from 0 to 12). The NPI total score ranges from [0 to 120 or 0 to 144] with higher scores indicating greater behavioral impairment.
The caregiver distress scores in each of the domains are not included in the NPI total score. The caregiver distress (NPI-D) total score is calculated by adding the scores of caregiver distress in each of the domains (score ranges from 0 to 5 in each domain). The NPID total score ranges from [0 to 50 or 0 to 60] with higher scores indicating greater distress.
The NPI will be administered at the Screening and Baseline visits before study intervention administration, and at the [Week(s) X] study visits for all participants. These assessments will be used as a secondary cognitive behavioral measure of clinical effect. The NPI will be administered by an independent, trained, and certified member of the investigational team. At each site, the same individual, whenever possible, will perform the NPI evaluation on a specific participant throughout the study.
8.1.4. Health Outcomes Scales
8.1.4.a. EuroQol 5-Dimensional Health Related Quality of Life Scale (EQ-5D)
The EQ-5D is a standardized instrument for use as a measure of health outcome. Mobility, self-care, usual activities, pain/discomfort, and anxiety/depression are each assessed on 3-point categorical scales ranging from “no problem” to “severe problem”. Two different administrations of the EQ-5D will be performed at baseline and [list post-baseline visits]. First, the participant’s caregiver will provide a proxy-rating of the participant’s health status. Second, the caregiver will provide a self-report of his or her own health status. In both situations, the EQ-5D questionnaire will be completed by the participant’s caregiver.
8.1.4.b. Resource Utilization in Dementia (RUD)
The RUD is an instrument to assess the amount of both formal and informal resources used by the participants and their primary caregiver. The [RUD version] has been selected for this study.
The [RUD version] will be administered at the Screening and Baseline visits before study intervention administration, and at the [Week(s) X] study visits for all participants. The [RUD version] will be administered by a trained member of the investigational team. At each site, the same individual, whenever possible, will perform the RUD evaluation on a specific participant/caregiver throughout the study.
© 2018 TransCelerate BioPharma 12

Common Protocol Template Alzheimer’s Disease Library V0039. Statistical Considerations
9.3. Populations for Analyses
Management of MMSE Inclusion criteria: To ensure an appropriate distribution of participants across the study-allowed spectrum of severity of AD, enrollment targets based on Screening MMSE scores will be enforced through the [IVRS or other system] used to randomize participants. The capping strategy for the [X] randomized participants is as follows:
MMSE Score Category
Allowed Number of Participants in MMSE Score Category
Corresponding Proportion of Target Enrollment
5 to 10 inclusive Identify Cap or Number x %
11 to 14 inclusive Identify Cap or Number y%
Analyses of baseline characteristics and efficacy endpoints are based on data from [all randomized participants or other specifications]. The primary analysis set for efficacy analyses will be the [name the data set], which includes [specify which participants].
The primary endpoint will be analyzed using a [log rank test or other test] stratified by geographic region with a two-sided [α=0.05]. The hazard ratio and a 95% confidence interval (95% CI) will be reported from a Cox proportional hazards model stratified by geographic region with study intervention group as a covariate. Kaplan-Meier estimates will be plotted.
Analyses of safety and the effects of exposure to study intervention include all participants who received at least one dose of study intervention.
© 2018 TransCelerate BioPharma 13

Common Protocol Template Alzheimer’s Disease Library V003
10. Supporting Documentation and Operational Considerations
10.5 Appendix 5: Genetics
Apolipoprotein E (ApoE) Pharmacogenomic Sample(s)
A 3-mL blood sample will be collected at Visit [x] into a plastic di-potassium ethylenediaminetetraacetic acid (K2-EDTA) tube labeled with the participant’s identification number and date and time of sample collection. The whole blood will be frozen and stored upright in the K2-EDTA tube at -20C in a secure freezer at the investigative site until shipped in batches on dry ice for genotyping. The shipment address and assay lab contact information are provided in the [Laboratory Manual].
10.x Appendix X: Diagnostic and Statistical Manual of Mental Disorders-[Version X] (DSM-[X]) CriteriaA. The development of multiple cognitive deficits manifested by both:A1. Memory impairment (impaired ability to learn new information or to recall previously learned information)A2. One (or more) of the following cognitive disturbances:
- Aphasia (language disturbance);- Apraxia (impaired ability to carry out motor activities despite intact
motor function);- Agnosia (failure to recognize or identify objects despite intact sensory
function); - Disturbance in executive functioning (i.e., planning, organizing,
sequencing, abstracting).
B. The cognitive deficits in Criteria A1 and A2 each cause significant impairment in social or occupational functioning and represent a significant decline from a previous level of functioning.C. The course is characterized by gradual onset and continuing cognitive decline.D. The cognitive deficits in Criteria A1 and A2 are not due to any of the following:
- Other central nervous system conditions that cause progressive deficits in memory and cognition (e.g., cerebrovascular disease,
© 2018 TransCelerate BioPharma 14

Common Protocol Template Alzheimer’s Disease Library V003Parkinson’s disease, Huntington’s disease, subdural hematoma, normal-pressure hydrocephalus, brain tumor);
- Systemic conditions that are known to cause dementia (e.g., hypothyroidism, vitamin B12 or folic acid deficiency, niacin deficiency, hypercalcemia, neurosyphilis, human immunodeficiency [HIV] infection);
- Substance-induced conditions.
E. The deficits do not occur exclusively during the course of a delirium.F. The disturbance is not better accounted for by another Axis I disorder (e.g., Major Depressive Disorder, Schizophrenia).
© 2018 TransCelerate BioPharma 15

Common Protocol Template Alzheimer’s Disease Library V003
10.X Appendix X: National Institute of Neurological and Communicative Disorders and Stroke – Alzheimer’s Disease and Related Disorder Association (NINCDS-ADRDA) CriteriaI. Criteria for Diagnosis of Probable Alzheimer’s Disease
- Dementia established by clinical examination and documented by a standard test of cognitive function (e.g., Mini-Mental State Examination [MMSE], Blessed Dementia Scale, etc.) and confirmed by neuropsychological tests;
- Significant deficiencies in two or more areas of cognition, for example, word comprehension and task-completion ability;
- Progressive deterioration of memory and other cognitive functions;- No loss of consciousness;- Onset from age 40 to 90, typically after 65;- No other diseases or disorders that could account for the loss of
memory and cognition.
II. Diagnosis of Probably Alzheimer’s Disease is Supported by:
- Progressive deterioration of specific cognitive functions: language (aphasia), motor skills (apraxia), and perception (agnosia);
- Impaired activities of daily living and altered patterns of behavior;- A family history of similar problems, particularly if confirmed by
neurological testing;- The following laboratory results:
-- Normal cerebrospinal fluid (CSF) (lumbar puncture test);-- Normal electroencephalogram test of brain activity;-- Evidence of cerebral atrophy in a series of computed tomography (CT) scans.
III. Other Features Consistent with Alzheimer’s Disease:
- Plateaus in the course of illness progression;- Computed tomography (CT) findings normal for the person’s age;- Associated symptoms including depression, insomnia, incontinence,
delusions, hallucinations, weight loss, sex problems, and significant verbal, emotional, and physical outbursts;
- Other neurological abnormalities, especially in advanced disease, including increased muscle tone and a shuffling gait.
© 2018 TransCelerate BioPharma 16

Common Protocol Template Alzheimer’s Disease Library V003IV. Features that Decrease the Likelihood of Alzheimer’s Disease:
- Sudden onset;- Such early symptoms as seizures, gait problems, and loss of vision and
coordination.
© 2018 TransCelerate BioPharma 17

Common Protocol Template Alzheimer’s Disease Library V003
10.X. Appendix X: Modified Hachinski Ischemic Score
The Hachinski Ischemic Score (HIS) is a simple clinical tool for differentiating major types of dementia such as primary degenerative, vascular or multi-infarct, and mixed type. A high HIS is closely related with cerebrovascular disease and its vascular factors.
The table below is used to derive the Modified Hachinski Ischemic Score. Answers to the following questions are determined from the participant, caregiver, and/or medical record. For each “Yes” response, add the corresponding value to the score column. Then add the scores for all the questions. If the total score is 4 or less, the dementia is not likely to be due to vascular causes and the participant meets the AD inclusion criteria for the Modified Hachinski Ischemic Score.
Question Yes Response Score
Abrupt onset of dementia
2
Stepwise deterioration of dementia
1
Somatic complaints 1
Emotional incontinence
1
History of hypertension
1
History of stroke 2
Focal neurologic symptoms
2
Focal neurologic signs 2
Total Score
© 2018 TransCelerate BioPharma 18

Common Protocol Template Alzheimer’s Disease Library V003
© 2018 TransCelerate BioPharma 19

Common Protocol Template Alzheimer’s Disease Library V003
11. References
© 2018 TransCelerate BioPharma 20

2 Albert MS, DeKoksy ST, Dickson D, Dubois B, Feldman HH, Fox NC, et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging - Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:270-9.
4 Schneider LS, Olin JT, Doody RS, Clark CM, Morris JC, Reisberg B, et al. Validity and reliability of the Alzheimer’s Disease Cooperative Study Clinician Global Impression of Change. The Alzheimer’s Disease Cooperaative Study. Alzheimer Dis Assoc Disord. 1997;11Suppl 2:S22-32.