162005/fulltext01.pdffig. 1. schematic illustrations of a) a red blood cell, b) a typical animal...
TRANSCRIPT


Chromatographic Studies of SoluteInteractions with Immobilized Red Blood
Cells and Biomembranes
BY
INGO GOTTSCHALK
UPPSALA UNIVERSITY 2002

Comprehensive Summaries of Uppsala Dissertationsfrom the Faculty of Science and Technology 755
Chromatographic Studies of SoluteInteractions with Immobilized Red Blood
Cells and Biomembranes
BY
INGO GOTTSCHALK
ACTA UNIVERSITATIS UPSALIENSISUPPSALA 2002

Dissertation for the Degree of Doctor of Philosophy in Biochemistry presented at UppsalaUniversity in 2002
ABSTRACT
Gottschalk, I., 2002. Chromatographic Studies of Solute Interactions with Immobilized RedBlood Cells and Biomembranes. Acta Universitatis Upsaliensis. Comprehensive Summaries ofUppsala Dissertations from the Faculty of Science and Technology 755. 43 pp. Uppsala. ISBN91-554-5413-5.
Specific and non-specific interactions of solutes with immobilized biomembranes were studiedusing chromatographic methods. Liposomes, proteoliposomes and red blood cell (RBC)membrane vesicles were immobilized by a freeze-thawing procedure, whereas whole RBCs wereadsorbed in the gel beds using electrostatic interaction, binding to wheat germ agglutinin (WGA)or the streptavidin-biotin interaction.
Superporous agarose gel with coupled WGA was the most promising matrix for RBCadsorption and allowed frontal chromatographic analyses of the cells for about one week.Dissociation constants for the binding of cytochalasin B and glucose to the glucose transporterGLUT1 were determined under equilibrium conditions. The number of cytochalasin B-bindingsites per GLUT1 monomer was calculated and compared to corresponding results measured onfree and immobilized membrane vesicles and GLUT1 proteoliposomes. This allowed conclusionsabout the protein´s binding state in vitro and in vivo.
Partitioning of drugs into biomembranes was quantified and the system was suggested as ascreening method to test for possible intestinal absorption of drug candidates. We also studiedhow membrane partitioning of drugs is affected by the presence of integral membrane proteins orof charged phospholipids.An attempt to combine the theory for specific binding and membrane partitioning of solutes in asingle equation is briefly presented.
Keywords: Affinity, Binding, Biomembrane, Biotin, Chromatography, Cytochalasin B,Dissociation constant, Drug absorption, Equilibrium, Glucose, GLUT1, Immobilization,Immobilized biomembrane affinity chromatography, Immobilized liposome chromatography,Interaction, Liposome, Membrane protein, Membrane vesicle, Partitioning, Phospholipidbilayer, Proteoliposome, Quantitative, Red blood cell, Solute, Specific, Streptavidin, Wheatgerm agglutinin.
Ingo Gottschalk, Department of Biochemistry, Uppsala University, Box 576, SE-751 23 Uppsala,
Sweden
© Ingo Gottschalk 2002
ISSN 1104-232XISBN 91-554-5413-5
Printed in Sweden by University Printers, Ekonomikum, Uppsala 2002

Das Herz pumpt schwerträge blubberndes Blut -und das ist gut.Extrabreit, 1980
To the memory of my fatherTo my mother

PAPERS INCLUDED IN THE THESIS
This thesis is based on the following papers, which will be referred to in the text by theirroman numerals.
I. Beigi, F., Gottschalk, I., Lagerquist Hägglund, C., Haneskog, L., Brekkan, E., Zhang,Y., Österberg, T. and Lundahl, P. (1998) Immobilized liposome and biomembranepartitioning chromatography of drugs for prediction of drug uptake.Int. J. Pharm. 164, 129–137.
II. Lundahl, P., Zeng, C.-M., Lagerquist Hägglund, C., Gottschalk, I. and Greijer, E.(1999) Chromatographic approaches to liposomes, proteoliposomes and biomembranevesicles.J. Chromatogr. B 722, 103–120.
III. Lagerquist Hägglund, C., Gottschalk, I. and Lundahl, P. (2000) Cytochalasin B-binding and transport properties of the Glut1 human red cell glucose transporter.Recent Res. Devel. Bioener. 1, 117–129.
IV. Gottschalk, I., Li, Y.-M. and Lundahl, P. (2000) Chromatography on cells: analyses ofsolute interactions with the glucose transporter Glut1 in human red cells adsorbed onlectin-gel beads.J. Chromatogr. B 739, 55–62.
V. Gottschalk, I., Lundqvist, A., Zeng, C.-M., Lagerquist Hägglund, C., Zuo, S.-S.,Brekkan, E., Eaker, D. and Lundahl, P. (2000) Conversion between two cytochalasinB-binding states of the human GLUT1 glucose transporter.Eur. J. Biochem. 267, 6875–6882.
VI. Gottschalk, I., Lagerquist, C., Zuo, S.-S., Lundqvist, A. and Lundahl, P. (2002)Immobilised biomembrane affinity chromatography for binding studies of membraneproteins.J. Chromatogr. B 768, 31–40.
VII. Gottschalk, I., Gustavsson, P.-E., Ersson, B. and Lundahl, P., Improved lectin-mediated immobilization of human red blood cells in superporous agarose beads.Submitted for publication in J. Chromatogr. B.
Reprints were made with permission from the publishers
Other publications
Zhao, Q., Gottschalk, I., Carlsson, J., Arvidsson, L.-E., Oscarsson, S., Medin, A.,Ersson, B. and Janson, J.-C. (1997) Preparation and purification of an end to endcoupled mEGF-dextran conjugate.Bioconjugate Chem. 8, 927–934.

TABLE OF CONTENTS
1. Biological membranes - life in itself ............................................................................. 5
2. Solute interactions with biomembranes ...................................................................... 5
3. Red blood cells ............................................................................................................... 6
4. The human glucose transporter GLUT1; a molecular fuel tap ................................ 7
5. Immobilized-cell technologies ...................................................................................... 10
6. In vitro prediction of drug uptake ............................................................................... 11
7. Aims of the study .......................................................................................................... 13
8. Membrane preparations .............................................................................................. 13
8.1. RBCs ................................................................................................................ 14
8.2. RBC membrane vesicles .................................................................................. 14
8.3. GLUT1 proteoliposomes ................................................................................. 15
8.4. Liposomes ........................................................................................................ 15
9. Immobilization of membrane materials ..................................................................... 15
9.1. RBC immobilization ......................................................................................... 15
9.1.1. Electrostatic RBC immobilization .................................................... 16
9.1.2. WGA-mediated RBC immobilization................................................. 16
9.1.3. Streptavidin/biotin RBC immobilization........................................... 18
9.2. Entrapment of membrane vesicles and (proteo)liposomes ............................. 19
9.3. Attempts on immobilization of cancer cells and yeast .................................... 19
10. Analytical methods .................................................................................................... 20
10.1. Immobilized biomembrane affinity chromatography (IBAC) ....................... 20
10.2. Hummel and Dreyer SEC ........................................................................... 23
10.3. Ultracentrifugal assay .................................................................................. 24
10.4. Transport retention chromatography (TRC) ................................................ 24
10.5. Immobilized biomembrane partitioning chromatography (IBiPaC)............. 24
11. Discussion of results .................................................................................................. 26
11.1. Can RBCs be adsorbed for chromatographic analyses? ............................. 26
11.2. Implications about GLUT1 .......................................................................... 29
11.3. IBiPaC for prediction of drug uptake .......................................................... 32
12. Conclusions ................................................................................................................ 34
Acknowledgements .......................................................................................................... 36
References ........................................................................................................................ 38

Abbreviations
CB Cytochalasin BCB/mon Number of CB binding sites per GLUT1 monomerGLUT1 Human erythrocyte-type glucose transporterIAM Immobilized artificial membraneIBAC Immobilized biomembrane affinity chromatographyIBiPaC Immobilized biomembrane partitioning chromatographyOG n-Octyl-β-D-glucopyranosidePC Phosphatidyl cholinePS Phosphatidyl serineSEC Size exclusion chromatographySPA Superporous agaroseRBC Red blood cellWGA Wheat germ agglutinin

5
1. Biological membranes - life in itself
Living cells and their organelles are usually enclosed by biological membranes that play key
roles in their function. Integral membrane proteins in the membranes show an enormous functional
diversity and contribute to the fact that biomembranes are more than just envelopes that surround and
protect the cells’ life functions. Instead, "life" is focussed very much within the membranes
themselves. For example, the respiratory chain in the inner mitochondrial membrane or the
photosynthetic membrane assemblies in the chloroplast membrane are the main sources of chemical
energy for animals and plants, respectively. Membranes are permeability barriers for larger polar
molecules (Mr > approx. 100) and for ions, making control of the critical cytoplasmatic solute balance
possible. This means also that mechanisms for transport of essential metabolites or signals across the
membrane are required. Transport channels and pores as well as signal receptors at the cell surface are
therefore further functional features of the biological membranes that make life possible. In addition,
membrane transport of protons is an important feature of the respiratory and photosynthetic
machineries. As a consequence, for the chemistry of life it is just as important to study biomembranes
and membrane proteins as it is to focus on processes that occur in the inner cell compartments.
2. Solute interactions with biomembranes
The term biomembrane as it appears in the title of this thesis is not well-defined in the
literature. Sometimes it may designate only natural membranes of certain cells and organelles,
whereas other authors include lipid bilayers in general. In the latter meaning bio- may be understood
as composed of biological molecules as opposed to artificial membranes of, e.g., nylon or
nitrocellulose. In this thesis, the more general meaning of biomembranes is usually used, although the
exact nature of the materials is often specified. For example, the term immobilized biomembrane
affinity chromatography (IBAC, cf. Section 10.1 and Paper VI) covers experiments on both cell
membranes, membrane vesicles and proteoliposomes, and biomembranes in the title of the thesis is
meant to include membrane vesicles and proteoliposomes.
Solute interactions with biomembranes are of major importance in various respects and
usually non-specific interactions can be distinguished from specific binding. In the non-specific case,
mainly hydrophobic interactions of a solute within the membrane core and ionic and polar interactions
with charged and hydrophilic membrane components, respectively, determine the strength of the
solute-membrane contact. An equilibrium constant is usually not measurable and the binding is not
saturable at high solute concentrations. These non-specific interactions determine the solute
partitioning into and diffusion across biomembranes, which were analyzed in Paper I and summarized
in Section 6. Specific solute (or ligand) binding to biomembranes, on the other hand, involves a
defined contact between the ligand and a membrane protein or lipid and is, in the scope of this thesis,

6
an important part of protein-mediated solute transport across membranes. The ligand (may it be a
transported substrate or a transport inhibitor) binds in a certain steric orientation and with a defined
affinity to a definite number of binding sites located on a transport protein in the membrane and the
binding is usually saturable at high ligand concentrations.
3. Red blood cells
The human body is composed of an astonishing number (approx. 100 trillion (1014)) of cells
(Guyton 1992) of presumably thousands of different types. As many as one fourth of these smallest
living units of our bodies are red blood cells (RBCs), the cell type, which by that measure, is
apparently our most abundant one (Guyton 1992; Dixon 1997). Although very flexible, RBCs are
biconcave discs with a diameter of 8 µm and a height of 2 µm (Fig. 1A). Provided with this
Fig. 1. Schematic illustrations of A) a red blood cell, B) a typical animal cell (reprinted from Guytonet al. (1992) with permission from W. B. Saunders).
information, how high would a stack of all the RBCs from only one human being be? Yes, that is
right, 50 000 km! The reason that there are so many RBCs is their essential task to provide virtually all
other of our cells with O2, a gas that within the RBCs becomes reversibly bound to the protein
hemoglobin. Therefore, during RBC-maturation from reticulocytes, all organelles except the cell
membrane are reduced to give room for as much hemoglobin as possible. This leaves RBCs with a
distinctly simpler intracellular structure in comparison to the highly compartmentalized construction
of other typical tissue cells (Fig. 1B). At least 110 other water-soluble proteins and enzymes (Pennell

7
1964) are present in RBCs, but their amount is small compared to that of hemoglobin, which crowds
the cell volume at about 0.34 gram per milliliter (Dixon 1997). The relatively small size and the
flexibility of RBCs allow them to enter even the thinnest capillary vessels. Their life span is limited to
120 days, which means that every day 0.8% of our RBCs are replaced by maturing stem cells, which
in adults originate mainly from the ribs and the sternum. Considering that RBCs cannot reproduce or
synthesize proteins and lack most of the metabolism common to other cells, their energy (ATP)
requirements are relatively low and can be covered by anaerobic glycolysis. However, intracellular
ATP depletion leads, among other things, to decreased cell membrane deformability, loss of
membrane lipids and disturbance of the ion balance (Weed and Lacelle 1969). Therefore, to maintain
their function and the essential flexibility of their membrane, RBCs are dependent on a constant
supply of energy which originates from glucose.
The only membrane structure of RBCs is their cell membrane, which consists, by weight
fraction, of 49% proteins, 43% small amphiphiles (mainly phospholipids), and 8% oligosaccharides
(Guidotti 1972). The phospholipids form a bilayer with their hydrophilic parts pointing toward the
outside and their hydrophobic parts pointing toward the inside of the membrane, as illustrated below
(e.g., Fig. 4). The distribution of phospholipids between the two leaflets of the bilayer is
assymmetrical, e.g., phosphatidyl choline (PC) and sphingomyelin are located predominantly within
the outer monolayer, whereas phosphatidyl serine (PS) and phosphatidyl ethanolamine are situated
mainly within the inner layer (Devaux 1993). Cholesterol is evenly distributed at about 20 weight % of
the lipids. At least 30 different integral membrane proteins (Anstee 1990) are embedded in the
membrane bilayer, either floating freely or physically coupled to a meshwork of fibrous peripheral
membrane proteins called the cytoskeleton. The latter structure is associated with the cytoplasmic face
of the membrane and provides the cell with the necessary stability without loss of flexibility.
Peripheral proteins can usually be dissociated from the membrane by relatively mild treatments, such
as increased ionic strength or pH, whereas extraction of integral proteins usually requires dissolution
of the membrane with detergents. The outer membrane face is essentially characterized by the
presence of a coat of oligosaccharides linked to the integral membrane proteins or lipids. This
increases the hydrophilicity of the cell surface. A high content of sialic acid in the oligosaccharide
chains of glycophorin A adds a negative charge, causing the cells to repel each other gently in the
blood stream. The sugars also offer lectin-binding sites and sialic acid is known (Adair and Kornfeld
1974) to be the main target for binding of wheat germ agglutinin (WGA).
4. The human glucose transporter GLUT1; a molecular fuel tap
D-glucose is the common major fuel resource for most cells in the human body. The brain
alone steadily consumes about 120 g of glucose per day (Stryer 1989). The diffusion of this
tremendous number (4 x 1023) of hydrophilic sugar molecules across the blood-brain barrier is

8
facilitated by the glucose transporter GLUT1, a specific gate for passive diffusion of glucose down a
concentration gradient. The richest occurrence of this protein in our bodies is found in the membranes
of RBCs, but there is a moderate expression of the transporter in many other tissues. With about 5 x
105 copies per cell (Mascher and Lundahl 1988; Anstee 1990) GLUT1 is the third most abundant
protein of the RBC membrane, just after the anion transporter and glycophorin A (each about 106
copies per cell). Most of the research on GLUT1 has been performed on protein from this source and
GLUT1 is frequently called the erythrocyte glucose transporter.
The first functional reconstitution of GLUT1 purified on DEAE-cellulose was done by
Kasahara and Hinkle in 1977, who used Triton X-100 for solubilization of the protein. The preparation
protocol has been modified many times and the standard procedure employed octylglucoside (OG)
(Baldwin et al. 1982; Cairns et al. 1984; Mascher and Lundahl 1988; Baldwin and Lienhard 1989), but
octaethylene glycol dodecyl ether (C12E8, e.g., Haneskog et al. 1996) can be used instead. A recent
protocol involved decylmaltoside detergents (Boulter and Wang, 2001) and on the basis of a direct
comparison the authors claim that their preparation procedure yields more stable and monodispersely
solubilized GLUT1 than did earlier ones. Maltoside detergents may therefore be promising in future
attempts to crystallize GLUT1.
In 1985, Mueckler and coworkers cloned and sequenced a glucose transporter from human
HepG2 hepatoma cells which is similar or identical to GLUT1. The transporter turned out to consist of
492 amino acid residues corresponding to a Mr of 54 117 and an N-linked heterogeneous
oligosaccharide was located at Asn 45. The same paper also presented the 12 α-helix topology model
for GLUT1 (Fig. 2), which is now generally accepted and supported by additional experimental
evidence, including proteolytic digestion (Cairns et al. 1984 and 1987), circular dichroism (Chin et al.
1987), antibody binding (Davies et al. 1990), Fourier transform infrared spectroscopy (Alvarez et al.
1987) as well as glycosylation- and cysteine-scanning mutagenesis (Hresko et al. 1994, Olsowski et
Fig. 2. The 12 α-helix topology model for GLUT1. Reprinted with permission from (Mueckler et al.1985). Copyright [1985] American Association for the Advancement of Science.

9
al. 1998). Computational 3D-structural models for a glucose channel within GLUT1 (Zeng et al.
1996) or the whole GLUT1 monomer (Zuniga 2001) were published, based mainly on the alignment
of polar amino acid residues within the 12 proposed transmembrane α-helices and the supposed α-
helix arrangement in the closely related and thoroughly studied E. coli lactose permease (Frillingos et
al. 1998; Weinglass et al. 2002). Another model for the arrangement of the α-helices to form a
glucose transport channel based on cysteine-scanning mutagenesis had been presented by Hruz and
Mueckler in 2000. Although these three models have in common some α-helices which are supposed
to be part of the glucose channel (e.g., H7, H8 and H11), a detailed convergence towards the true
GLUT1 3D-structure remains to be seen, possibly pending future crystallization of the protein.
Alternative GLUT1 topology models containing 16 transmembrane β-sheets (Fischbarg et al. 1993) or
a combination of 10 α-helices and 4 β-sheets (Ducarme et al. 1996) have not gained any further
support.
GLUT1 is a member of a family of homologous but distinctive facilitative sugar transporters
which operate more or less specifically in different organs, and the corresponding gene family is
termed solute carriers 2A (SLC2A). GLUT2–GLUT5 were known and cloned by the end of the 80s
(Bell et al. 1990, Mueckler et al. 1994) and the recent availability of the human genome data led to a
rapid identification of GLUT6–GLUT12 as well as the proton-myoinositol symporter HMIT1, which
was included in the GLUT family, all as reviewed by Joost and Thorens (2001).
GLUT1 transport is stereospecific for D-glucose, although some substituted forms (e.g.,
deoxy-D-glucose) and a collection of other hexoses are transported (cf. Paper III) at different
efficiencies. Dehydroascorbic acid, the oxidized form of vitamin C, has also been shown to be a
GLUT1 substrate (Vera et al. 1993). One of the most frequently used and potent inhibitors of GLUT1
transport is cytochalasin B (CB), an antibiotic from the fungus Drechslera dematiodeum. CB binds to
the cytoplasmic face of GLUT1 close to its C-terminus (Cairns et al. 1984) by hydrogen bonds and
hydrophobic interactions. The CB-binding affinity of GLUT1 is almost 106-fold higher than that for D-
glucose and the binding is competitive. CB has been widely used to study the mechanism and kinetics
of glucose transport and, in particular, the association state of GLUT1 monomers. Originally, an
alternating conformation model was described (Barnett et al. 1975; Baldwin et al. 1982; Lienhard et
al. 1992; Barrett et al. 1999) whereby glucose bound to GLUT1 monomers at only one binding site.
After a conformational change the glucose was released on the opposite side of the membrane. The
number of CB-binding sites per GLUT1 monomer (CB/mon, r in Papers V and VII) is 1.0 in this
model. The alternating conformation model is compatible with saturable glucose transport with
Michaelis-Menten kinetics as first reported by Widdas in 1952. Already in 1970, GLUT1 tetramers
were discussed (Lieb and Stein 1970). During the 80s and 90s CB/mon values of ≈ 0.5 were measured
for GLUT1 in cholate solution (Hebert and Carruthers 1992) and maltose- and glucose-binding
measurements led to the evolution of the fixed-site carrier model for GLUT1 (Sulzman and Carruthers

10
1999). In this model, both in vitro and in vivo (Cloherty et al. 2001), GLUT1 acts as a dimer of
dimers, e.g., a tetramer, that exposes two binding sites for CB (CB/mon=0.5) and in the absence of CB
simultaneously shows two sugar export and import sites (Fig. 3). Also in the fixed-site carrier model,
Fig. 3. The GLUT1 fixed-site carrier model representing the transporter as a dimer of independentdimers. Panel A shows a view through the plane of the membrane and Panel B shows the four possibletransporter configurations. Reprinted from Hamill et al. (1999), with permission from BlackwellScience.
sugar transport is accomplished by a conformational change that enables substrate to diffuse through a
channel within GLUT1 towards another binding site at the opposite face of the protein. The two
monomers within a GLUT1 dimer can only change conformation simultaneously (Fig. 3; Paper III,
Fig. 3). Alkaline reduction in 10 mM DTT during purification was reported to dissociate the GLUT1
tetramers into two dimers (Hebert and Carruthers, 1992), indicating the presence of disulfide bridges
between the dimers. However, such an effect of DTT has not been confirmed by any other research
group.
5. Immobilized-cell technologies
The small size of biological cells often makes it desirable to facilitate their handling, e.g., by
attaching them to a support of choice. In addition to this technical advantage, immobilization can also
protect the cells from unfavourable environmental conditions and high cell densities can be maintained
for repeated experiments or for biocatalysis. Therefore, cell adsorption onto surfaces and
immobilization of cells in gel matrices are rapidly growing scientific fields. Adsorption or growing of
cells on microchips, membranes or other surfaces (Falsey et al. 2001; Bisson et al. 2002; Prechtel et
al. 2002) has become a widespread method and enables time- and space-saving analyses. For large-
scale use both prokaryotic and eukaryotic cells have been grown in bioreactors (Park and Chang
2000). Some interesting applications are: the degradation of phenol by Pseudomonas (Amanda Wu et
al. 1991), the production of penicillin by Penicillium chrysogenum (Mussenden et al. 1993), ethanol

11
fermentation on adsorbed Saccharomyces cerevisiae (Holeberg and Margalith 1981; Nagashima et al.
1984) and highly efficient production of monoclonal antibodies on hybridoma cells photo-crosslinked
to BIX12 gel beads (Kamihira et al. 1993). The methods for immobilization of these and other cell
types are many, but entrapment of, e.g., yeast cells in calcium-alginate beads (Galazzo and Bailey
1990) is very mild and has received particular attention. In a simple variant of the method a sodium
alginate solution is dropped into a yeast culture containing CaCl2. Cell-containing beads are formed
which are then incubated in growth medium and finally in the fermentation medium. Within the beads,
cells are vital and continue proliferating to a high density and even exhibit ethanol production rates 1.5
times higher than do cells grown in suspension (Galazzo and Bailey 1990). Cells can also be
encapsulated in micro suspension droplets enclosed by alginate membranes (Park and Chang 2000) to
prevent leakage of cells into the growth medium, while still allowing release of products.
Immobilization of proliferating cells is usually done under optimized conditions regarding
temperature, growth medium, etc. Even though the complex structure of these cells may make them
vulnerable in many respects, their capability to grow may also be of advantage to keep the number of
immobilized cells at a certain saturation level, as compared to immobilized cell organelles or RBCs.
Cells that are lost or die can be replaced continuously. Moreover, the viability of many cell types is
even dependent on adhesion to a matrix or other cells, for example, mediated by the integrin protein
family (Miranti 2002).
In the work of this thesis, RBC adsorption onto agarose gel beads was mediated by the
interaction with WGA or streptavidin-biotin, in 10 mM phosphate buffered saline containing 3 mM
NaN3, 50 mM mannitol. The effect of adding low concentrations of different preservatives was studied
(Paper IV). The RBCs are relatively robust, but do have a limited life-span in buffer suspension at
room temperature. Anyhow, the conditions of first choice were those that retained the highest
comparability to previous ligand-binding data for GLUT1 in membrane-vesicle- or proteoliposome
systems.
6. In vitro prediction of drug uptake
Orally administered drugs have to enter the blood circulation and this absorption process
usually takes place in the small intestine. The main obstacles to this are biomembranes, e.g., of the
mucous epithelial cells or cells of the underlying blood vessels. The different possible routes for the
drugs to cross these barriers are simple diffusion (the passive route), transport by a membrane protein
(the active route), leakage through the tight junctions in between the cells (the paracellular route) or
passage by transcytosis (Artursson et al. 1996). In the general case, without involvement of a specific
membrane transporter, the passive pathway is the most common. Compounds that are too hydrophilic
may therefore never be able to partition into the membranes and pass unabsorbed through the body,
whereas very hydrophobic substances instead may accumulate in membranes or fatty tissues.

12
During the development of new drugs, a large number of different candidates are synthesized
and considerable effort and resources can be spared if the compounds with low chances of intestinal
absorption may be excluded at an early stage of the process.
One of the first and most frequently used methods to determine the molecules’ ability to
partition into lipid phases is to dissolve the analyte in a system of two immiscible phases, e.g., n-
octanol and buffer (Hansch and Fujita 1964). The ratio of the analyte concentrations in the two phases
gives the well known partition coefficient P. This method is routinely used in the pharmaceutical
industry. A theoretical disadvantage is that the n-octanol phase lacks structural similarity to
biomembranes, which motivated numerous studies of drug partitioning into suspended liposomes
(Betageri and Rogers 1987; Ma et al. 1991). Experiments on monolayers of cultured human epithelial
cells, such as the Caco-2 cell line, are another commonly used method, which allows valuable
determinations of true permeability values rather than just partition data. However, these analyses are
quite time-consuming and results from different laboratories are not comparable due to differences in
experimental conditions or the cell line itself (Artursson et al. 1996). Within a given laboratory, fairly
accurate comparisons between different drug candidates are possible.
At least two chromatographic approaches to prediction of drug absorption have been made.
One of them was the system of immobilized artificial membranes (IAMs), which involves monolayers
of phospholipid analogues covalently coupled to a silica support (Pidgeon and Vantarekum 1989;
Yang et al. 1996). HPLC columns of more than 20 different IAM supports have been prepared and
several are commercially available. The bonded phospholipids of an IAM share many of the physical
and chemical properties of a biomembrane, despite differences regarding the size and flexibility of
phospholipid bilayers. In addition to drug screening, they have been applied for preparative purposes
(Pidgeon et al. 1991; Liu et al. 1997) and the immobilization and quantitative chromatographic
analyses of integral membrane proteins, such as the nicotinic acetylcholine receptor (Wainer et al.
1999), P-glycoprotein (Zhang et al. 2000) and the estrogen receptor (Moaddel et al. 2002).
Apparently, a broad range of detergent solubilized membrane proteins are able to arrange themselves
in the phospholipid monolayers in a way that retains specific ligand-binding activity. The other
method was originally called immobilized liposome chromatography (ILC, Beigi et al. 1995; Lundahl
and Beigi 1997) and was applied to screening of drugs on liposomes immobilized in gel beads. The
method is here, as in Paper I, referred to as immobilized biomembrane partitioning chromatography
(IBiPaC) to include biomembranes other than liposomes. The preferred way of liposome
immobilization was steric entrapment by freeze-thawing (cf. Section 9.2.), but also liposomes
absorbed by streptavidin-biotin interaction (Yang et al. 1998) or covalently bound to CNBr activated
gel (Yang et al. 1999) have been used for IBiPaC. The latter two immobilization methods allow the
immobilization of unilamellar (proteo)liposomes, which may be of advantage for certain experiments.
A strength of IBiPaC is that a multitude of different biomembranes can be immobilized (cf. Section 9;
Paper I; Lagerquist et al. 2001) and studied repeatedly with the ease and accuracy of a

13
chromatographic system. In Paper I, various fundamental properties of the system itself were
elucidated and a comparison with IAM chromatography was made. The effect on drug partitioning of
certain biomembrane components, such as membrane proteins or cholesterol, could also be studied, as
reported initially in Paper I and, more extensively, by Lagerquist et al. (2001).
7. Aims of the study
The fundamental chromatographic properties of the newly introduced IBiPaC system for
prediction of drug uptake (Beigi et al. 1995, Lundahl and Beigi 1997) were to be elucidated as well as
the influence of some physical and chemical properties of the mobile phase (cf. Section 10.5). Also,
the consistency of IBiPaC results with those from other systems, such as the related IAM-
chromatography, was to be tested and measured absorption values for a number of drugs were to be
correlated with IBiPaC capacity factors (Paper I).
Ligand binding to the glucose transporter GLUT1 had been studied in our laboratory in
immobilized RBC membrane-vesicles and proteoliposomes of different compositions. For
comparison, GLUT1 in a more natural membrane, e.g., in immobilized RBCs, should be studied using
the IBAC method (Paper IV).
Using the first immobilized RBC systems developed (Zeng et al. 1997; Paper IV), attempts to
improve cell adsorption stability and capacity were made (Papers V and VII), while at the same time
monitoring possible effects of the new cell environments on GLUT1. Regarding the importance of
RBC research and cytology in general, future analytical applications of immobilized cell systems seem
very probable.
8. Membrane preparations
A membrane protein can be studied in membrane systems of different compositions and complexities.
Usually, one attempts to avoid possible disturbances from other membrane proteins in the natural
biomembrane by purifying the desired protein in detergent solution and reinserting (reconstituting) it
into proteoliposomes composed of lipids of choice. Furthermore, attempts to crystallize a protein are
only promising with a highly pure preparation. The first IBAC studies of GLUT1 (Yang and Lundahl
1995; Brekkan et al. 1996) were accordingly done on purified protein. However, one generally
suspects that detergents involved during purification, reconstitution or crystallization may harm or
alter the investigated membrane protein, which potentially complicates conclusions about its in vivo
properties. Beyond that, it is understood that various membrane proteins are influenced by changes in
their lipid environment (Romsicki and Sharom 1999; Haruna et al. 2000; Hu et al. 2000; Bogdanov et
al. 2002). Therefore, examination of the protein at different stages of preparation may reveal crucial
information. Figs. 4A–C show sections of the different membrane materials used for IBAC studies of

14
GLUT1 in Papers IV, V and VII. Materials A and B and, above all, plain liposomes (Fig. 4D) were
used for partition chromatography of drugs in Paper I. The preparation of these membrane materials is
briefly outlined below.
Fig. 4. Sections of the membrane materials used for this study: A) RBC membrane, B) RBCmembrane vesicles, C) GLUT1 proteoliposomes and D) liposomes. Reprinted with modifications fromPaper VI, with permission from Elsevier Science.
8.1. RBCs
Fresh human blood or RBC concentrate were washed 3 times in phosphate-buffered saline.
This removed the buffy coat of the whole blood. The final cell pellet was diluted two-fold used for
electrostatic immobilization (Paper I), or five-fold for coupling to WGA-gel beads (Papers IV and
VII), or biotinylation (Paper V). Fig. 4A depicts different glycosylated membrane proteins, some of
which make contact with the cytoskeleton consisting mainly of spectrin linked to the membrane by
peripheral membrane proteins. The phospholipid asymmetry of the membrane double leaflet is also
outlined, but it is likely that this feature is lost upon immobilization and long-time analysis of the cells
in buffer solution.
8.2. RBC membrane vesicles
Membrane vesicles from RBCs were prepared as in Lundahl et al. (1986). Hypotonic lysis of
a batch of RBCs (Dodge et al. 1962) yielded membrane ghosts which were separated from soluble
proteins by size exclusion chromatography (SEC) at pH 8.2 and pH 10.5. Washing the membranes at
pH 12 in 0.2 mM DTE split off the cytoskeleton due to electrostatic repulsion of its protein
components and the membranes formed smaller membrane vesicles. The DTE concentration during
the pH 12-washing was optionally raised to 10 mM in Paper V to reveal the relevance of disulfide

15
bridges for GLUT1 oligomerization that had been proposed by others (Hebert and Carruthers, 1992;
Hamill et al. 1999). Fig. 4B shows a section of a cytoskeleton-stripped membrane vesicle containing
only integral membrane proteins and a certain degree of peripheral membrane proteins. Membrane
vesicles were used for IBiPaC studies (Paper I), Hummel and Dreyer chromatography (Papers II and
V) and for IBAC analyses (Paper V).
8.3. GLUT1 proteoliposomes
GLUT1 in membrane vesicles was solubilized with OG and purified on DEAE-cellulose at pH
8, making use of the fact that GLUT1 has one of the highest pI values (i.e. 8.0 (Englund et al. 1995))
among the integral RBC-membrane proteins. A substantial fraction of GLUT1 is eluted within the
total volume of the column, whereas most other RBC-integral membrane proteins became adsorbed to
the ion exchanger. The purity attained by this procedure (Mascher and Lundahl 1988) was estimated to
be 90 ± 5% by amino acid analyses of GLUT1, consistent with data in Rampal et al. (1986). A major
contaminant is the nucleoside transporter that also has been studied by IBAC (Haneskog et al. 1998).
The purified GLUT1 was reconstituted by SEC on Sephadex G-50 medium (Amersham Biosciences)
together with co-purified endogenous RBC-membrane lipids or with a 40-fold excess of egg yolk
phospholipids. IBAC on GLUT1 proteoliposomes was described in Paper V and compared to identical
experiments on proteoliposomes with GLUT1 that had been solubilized in C12E8 or that had been
reconstituted by dialysis as in Baldwin and Lienhard (1989).
8.4. Liposomes
Protein-free liposomes composed of synthetic PC, PC/PS, egg phospholipids (prepared from
hens’ eggs as in Yang and Lundahl (1994)) or endogenous RBC membrane lipids (extracted in 2:1
chloroform:methanol as in Folch et al. (1957)) were used for IBiPaC in Paper I. The relevant lipids
were dissolved in organic solvents, which were rotary evaporated and the final lipid film was
suspended in buffer to attain large multilamellar liposomes, a leaflet of which is shown in Fig. 4D.
9. Immobilization of membrane materials
9.1. RBC immobilization
Three different methods for RBC immobilization in chromatographic gel beds were used for
IBAC or IBiPaC analyses, e.g., adsorption mediated by electrostatic interaction, binding to WGA, or
streptavidin-biotin interaction, as illustrated in Fig. 5 and described below.

16
Fig. 5. Schematic illustration of RBC immobilization techniques. A) electrostatic immobilization, andadsorption mediated by B) WGA or C) Streptavidin-biotin interaction.
9.1.1. Electrostatic RBC immobilization.
Positively charged gel particles were prepared as described by Zeng et al. (1997) by
polymerization of methacrylamide/piperazine diacrylamide in the presence of N-allyldimethylamine.
The gel was crushed with a glass rod and the resulting particles of sizes of 40 - 100 µm were incubated
with RBCs and packed into plastic columns (1 cm i.d.). Electrostatically immobilized cells were used
for IBiPaC in Paper I.
9.1.2. WGA-mediated RBC immobilization.
WGA (Goldstein and Hayes 1976) is a lectin dimer of Mr 36 000 that binds specifically to N-
acetylglucosamine (Kd = 760 µM (Nagata and Burger 1974)) and a number of other sugars, but its
affinity for D-glucose is negligible. WGA-agarose gel beads are commercially available (WGA-
Sepharose 4B, bead diameter 45–165 µm, [WGA] = 1.96 mg/ml, Amersham Biosciences) with the
original purpose to separate cells or cell organelles, rather than to bind them more permanently.
Accordingly, when RBCs were incubated together with WGA-agarose beads, almost all of the cells
detached from the gel beads upon packing a column. However, pumping the RBCs at 0.15 ml/min into
packed gel beds (0.5 – 1.5 ml) of WGA-agarose and incubation for half an hour effected more
permanent retention, at least for a few days, during which affinity chromatographic analyses were
possible (Papers IV and VII). We assumed that multiple binding of many lectins to one cell was
established during the incubation and caused the adsorption of the cells. Five-fold cell application and
incubation were usually employed for RBC-coupling to WGA-agarose columns and were sufficient to
approach saturation of the gel bed with cells (Fig. 6). Before IBAC analyses (cf. Section 10.1) the

17
Number of RBC applications0 1 2 3 4 5
Fron
tal C
B e
lutio
n vo
lum
e [m
l]0
2
4
6
8
Fig. 6. The following procedure was performed repeatedly: RBCs (5-fold diluted cell pellet) wereapplied to a column of WGA-SPA (Vt=0.73 ml) at 0.15 ml/min followed by 30 min incubation and 30min washing at 0.5 ml/min and a frontal run of 2 nM [3H]CB. The elution volumes were supposed tobe proportional to the number of adsorbed cells.
columns were washed overnight to remove insufficiently adsorbed cells.
Paper IV describes the use of WGA-Sepharose 4B for IBAC analyses and the effects of
different additives in the running buffer, e.g., fructose, pyruvate, adenine, glutamine and inosine, on
the stability of the columns were studied. Coating of the immobilized cells with a positively charged
polymer (poly-Lys) was also intended initially to improve column stability, but soon we instead
focussed on the apparent effects of this treatment on GLUT1. Paper VII describes the use of
superporous agarose (SPA) beads (Gustavsson and Larsson 1996) with coupled WGA. These beads
had a diameter of approx. 200 µm, and in addition to the diffusion pores of common agarose gel, they
possessed 30-µm superpores. The pores were thus large enough to host RBCs in a hopefully more
protected environment as compared to cells attached to the surface of WGA-Sepharose 4B beads. A
chromatographic buffer flow through the superpores of these gel beads is documented (Gustavsson
and Larsson 1996). A SPA bead with a few bound RBCs is shown in Fig. 7., showing a clearly
Fig. 7. A WGA-SPA gel bead. The length of the scale bar is 50 µm.

18
rougher surface than do Sepharose 4B beads (cf. Fig 1A of Paper IV). WGA was coupled to the SPA
beads after in situ CNBr-activation (Nandakumar et al. 2000; Paper VII). In brief, packed beads were
activated by circulating through the gel bed a solution of 0.5 g CNBr dissolved in acetonitrile and
diluted in 15 ml of 1 M K3PO4-HCl, pH 11, for 5 min at 10 ml/min to form cyanate esters and cyclic
imidocarbonate (Carlsson et al. 1989). WGA was then covalently attached to the activated matrix by
overnight circulation through the gel in 0.1 M NaHCO3, pH 8.2. Fig. 8A shows a WGA-Sepharose 4B
bead covered with some RBCs, panel B a SPA bead filled with immobilized RBCs and Fig. 8C
illustrates a column of SPA beads with WGA-immobilized RBCs.
Fig. 8.A) A WGA-Sepharose 4B gel bead. Reprinted with modifications from Paper IV, withpermission from Elsevier Science; B) A WGA-SPA gel bead; C) A typical WGA-SPA column (1 cmi.d.), all with immobilized RBCs. The length of the scale bars is 50 µm.
9.1.3. Streptavidin-biotin immobilization.
The extraordinarily high affinity (Kd ≈ 10-15 M) of the Mr 244 vitamin biotin for binding to the
tetrameric proteins avidin or streptavidin (Mr 67 000) is commonly utilized for crosslinking, labelling,
purification or immobilization of biomolecules. Paper V describes coupling of the reagent Sulfo-NHS-
LC-biotin (Pierce, USA) to primary amines on the RBC surface (Fig. 9) by 45-min incubation in a
five-fold diluted RBC-pellet. The N-hydroxysulfosuccinimide was released and the biotin remained
bound to the cell via a spacer arm. Possible biotinylation targets on the RBC surface are, e.g., PS,
phosphatidyl ethanolamine or amino groups on membrane proteins. The biotinylated cells were
washed and pumped into a column of streptavidin-derivatized Sepharose 4B (Amersham Biosciences)
at 0.15 ml/min. The process had to be stopped after a few minutes to prevent the column from
clogging due to the high binding affinity of the cells. Sulfo-NHS-LC-biotin concentrations of 1-5 mM
in the incubation mixture were found to be sufficient to retain the RBCs in the gel bed.

19
RBC NH2
NHNH
SN
O
OO
NH
O
O
SO
OO O
HH
NHNH
SN
O
OO
NH
O
SO
OO O
HHO
NH
RBCH
NHNH
SNH
O
O
O
HH
NH2
N
O
OOH
SOO O
RBC
- -
-
+
-
Fig. 9. Covalent coupling of Sulfo-NHS-LC-biotin to primary amines on the RBC surface.
9.2. Entrapment of membrane vesicles and (proteo)liposomes.
The observation that very large liposomes can be prepared by freezing and thawing of a
sufficiently concentrated liposome suspension (Pick 1981; Mayer et al. 1985) was the basis for a
method for sterical entrapment of liposomes and proteoliposomes in gel beads (Yang and Lundahl,
1994). During subsequent years the method was improved several times and extended (Brekkan et al.
1996; Lundqvist et al. 1997). For this thesis, liposomes (Paper I), GLUT1 proteoliposomes (Paper V)
or RBC membrane vesicles (Papers I and V) were immobilized. The membranes were mixed with
dried Superdex 200 prepgrade gel beads, degassed and incubated for at least 30 min. The beads
swelled and 70–80 % of their inner volume became accessible to the membranes (Lundqvist et al.
1998b). The mixtures were frozen in -70°C (EtOH containing CO2(s)) and thawed in a 25°C water
bath, which presumably caused the membranes to fuse and become entrapped in the cavities within the
gel beads. After removing non-entrapped membranes by repeated centrifugal washings, the gel beads
(0.5–1.0 ml) were packed into glass columns (5 mm i.d.).
9.3. Attempts on immobilization of cancer cells and yeast.
Immobilization of cells other than RBCs on agarose gel beads was attempted. Epidermoid
carcinoma cells of the human line A-431 that express 106 epidermal growth-factor receptors (EGFR,
Carpenter 2000) per cell were grown under sterile conditions and harvested mechanically by scraping
off from the culture flasks. Adsorption of these cells onto agarose gel was attempted using the
streptavidin-biotin interaction and binding to WGA or concanavalin A. However, IBAC analysis of125I-EGF binding to the receptors never revealed any specific binding. Vinblastine binding to P-
glycoprotein in K-562 cells (human chronic myelogenous leukemia) immobilized in similar systems
also failed.

20
Baker’s yeast cells showed no binding affinity for WGA-agarose gel beads. Apparently the
chitin (poly-N-acetylglucosamine) moieties in the cell wall of the yeast cells were not accessible for
WGA-binding. However, biotinylated yeast cells adsorbed on streptavidin-agarose and freeze thaw
immobilization of yeast plasma membrane vesicles enabled preliminary IBiPaC analyses (Suer,
Gottschalk and Lundahl, unpublished data).
10. Analytical methods
10.1. Immobilized biomembrane affinity chromatography (IBAC)
Affinity chromatography is well known as a preparative method in bioseparations, but it can
also be used for quantitative analysis of ligand binding to biomolecules (first applied by Andrews et
al. 1973; Dunn and Chaiken 1974), particularly when frontal sample application is used (Nichol et al.
1974, Kasai and Ishii 1975). In frontal quantitative affinity chromatography a large volume of a
relatively diluted ligand sample is applied to the analytical column containing the immobilized
acceptor molecules. The experimental set-up is sketched in Fig. 10A. During such an experiment the
Fig 10. A) Experimental set-up for IBAC. B) Frontal elution profiles drawn for a series of sampleswith [ligand] = 0.5–2Kd (cf. Paper VI). Both reprinted from Paper VI, with permission from ElsevierScience.
ligand concentration at the top of the column rises until a binding equilibrium is established and the
thereby formed ligand boundary migrates through the column. The characteristic shape of
chromatograms resulting from frontal runs is drawn in Fig. 10B. The plateau height of the elution
pattern corresponds to the ligand concentration of the original sample, which is not diluted during the
analysis. The retention volume in a particular run is read at half the plateau height and depends on the
ligand concentration [B] in the sample, the constant number of immobilized binding sites N and the
dissociation constant Kd. The non-specific retention volume under conditions where specific ligand

21
binding is completely suppressed (Vmin, cf. Fig. 10B; the determination of Vmin is explained below) is
also constant at all ligand concentrations. For known N, Kd and Vmin in a given experiment the
retention volume V can be calculated according to Eq. 1 [Kasai et al. 1986]:
[ ] dmin KB
NVV+
+= (1),
where the term N/([B]+Kd) is equal to the retention volume, Vspec, caused by specific interaction
between B and the immobilized analyte. Some inherent advantages of the method are easy handling of
the immobilized biomolecules in a chromatographic system and that results can be obtained relatively
quickly (compared to dialysis) at equilibrium without separation of free from bound ligand. Both
affinity constants and the number of immobilized binding sites can be determined (see below) by use
of the relevant theory published, e.g., in Winzor et al. (1985), Kasai et al. (1986), Winzor and Jackson
(1993) and Winzor (1998).
The first experiments with frontal quantitative affinity chromatography on membrane proteins
were performed in 1996, by Brekkan et al. using GLUT1 in immobilized proteoliposomes and
membrane vesicles. The method was later described as immobilized biomembrane affinity
chromatography (IBAC, cf. Papers V and VI; Lundqvist and Lundahl 2002). For this thesis, GLUT1
was analyzed in immobilized RBCs, membrane vesicles and proteoliposomes. CB-binding parameters
were basically determined from a series of frontal runs of 1 or 2 nM [3H]CB supplemented to total CB
concentrations of 2–100 nM (Fig. 11B). Signals were detected on-line in a flow scintillation detector
with a six second update-time and the elution fronts were evaluated by curve-fitting of the data to the
Elution volume [ml]0 5 10 15 20 25
Elution volume [ml]0 5 10 15 20 25
Cou
nts [
% o
f pla
teau
leve
l]
0
20
40
60
80
100
Vmin Vmin
A B
Fig. 11. Frontal elution profiles of 2 nM [3H]CB on RBCs immobilized in a WGA-SPA column. Thesamples further contained from the right to the left A) 0, 0, 7.5, 12.5, 25 and 50 mM D-glucose and B)0, 0, 22, 47, 75 and 100 nM cold CB.
22
four parameter logistic function
dxcdaxf b ++
−=)(1
)( (2),
where a and d are the plateau- and background level, respectively; b, the slope coefficient and c, the
elution volume of the front at f(c)=(a+d)/2.
The frontal elution volumes, V, at the different free CB-concentrations, [CB], enabled calculation of
the amount B of bound CB at each [CB],
( )[ ]CBVVB min−= (3).
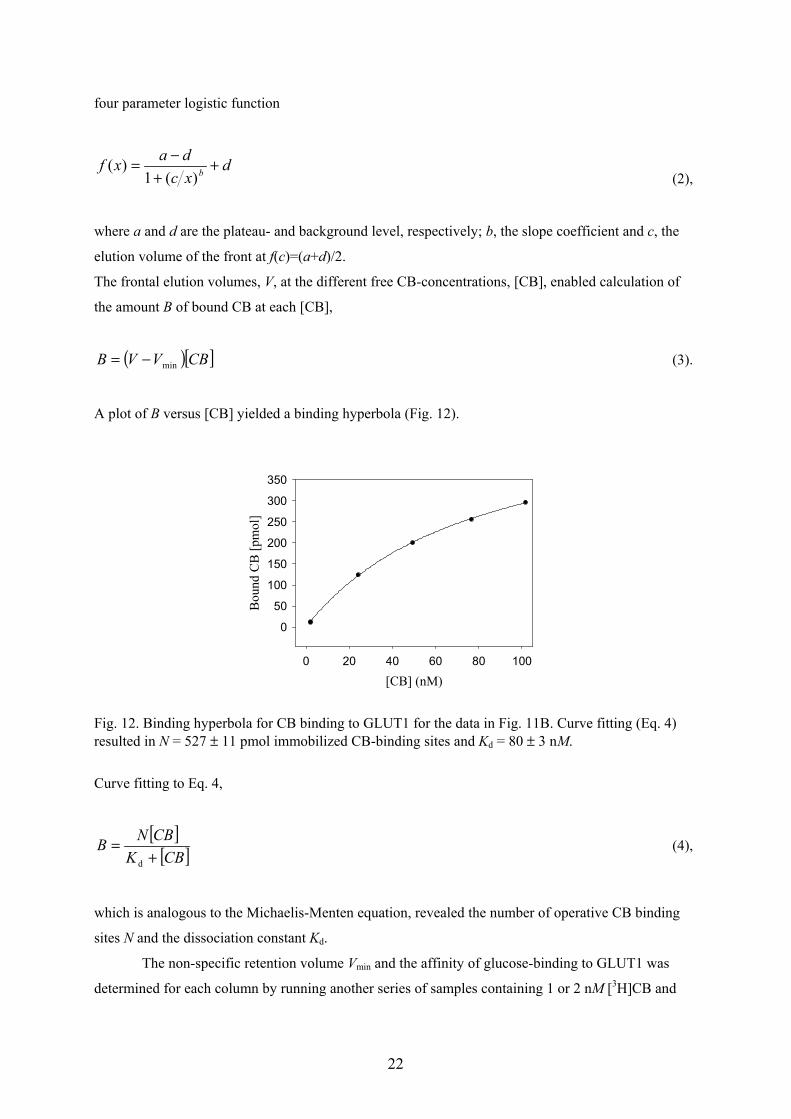
A plot of B versus [CB] yielded a binding hyperbola (Fig. 12).
[CB] (nM)0 20 40 60 80 100
Bou
nd C
B [p
mol
]
0
50
100
150
200
250
300
350
Fig. 12. Binding hyperbola for CB binding to GLUT1 for the data in Fig. 11B. Curve fitting (Eq. 4)resulted in N = 527 ± 11 pmol immobilized CB-binding sites and Kd = 80 ± 3 nM.
Curve fitting to Eq. 4,
[ ][ ]CBK
CBNB+
=d
(4),
which is analogous to the Michaelis-Menten equation, revealed the number of operative CB binding
sites N and the dissociation constant Kd.
The non-specific retention volume Vmin and the affinity of glucose-binding to GLUT1 was
determined for each column by running another series of samples containing 1 or 2 nM [3H]CB and

23
varying concentrations of D-glucose (0-50 mM, Fig. 11A) using linear regression (Eqs. 5 and 6; Fig.
13) as in Brekkan et al. (1996).
[ ] [ ][ ]glc
1)1(1V
1 2
imax CBglc
CB
CB
CB
KNKKCB
NKKCB
V+
++
=−
(5)
[ ]CBNNKVV CB
111
min
+=−
(6),
with [glc] being the free concentration of D-glucose; Kglc and KCB, the association constants for
glucose- and CB-binding to GLUT1, respectively; Vmax, the maximal CB elution volume at
[CB]<<(KCB)-1 and [glc]=0; Vi, the CB-retention volume at i mM [glc]; V, the CB-retention volume at
different [CB] and [glc]=0. Vmin was obtained by extrapolation of the linear plot (Fig. 13A, according
[glc]-1 (mM)-1
0,00 0,02 0,04 0,06 0,08 0,10 0,12 0,14
( Vm
ax- V
i)-1 [m
l-1]
0,15
0,20
0,25
0,30
0,35
0,40
0,45
0,50
0,55
[CB] (nM)0 20 40 60 80 100
( V- V
min)-1
[ml-1
]0,140,160,180,200,220,240,260,280,300,320,340,36
A B
Fig. 13. Linear regression analysis of the data in Fig. 11 using A) Eq. 5 and B) Eq. 6.
to Eq. 5) to infinite glucose concentration. Eq. 6 then gave N and KCB and insertion of these parameters
into Eq. 5 also allowed calculation of Kglc. Hence, although D-glucose itself was not measurably
retarded on GLUT1-containing columns due to its low affinity for the protein, inhibition of CB-
binding at accordingly higher glucose concentrations revealed the glucose binding constants. It is
clear, however, that non-linear regression of binding data, as in Eq. 4, is to be preferred whenever
possible, since extrapolation and compression of the data inherent to reciprocal linear plots is thereby
avoided (Klotz 1983; Leatherbarrow 1990).
10.2. Hummel and Dreyer SEC
The use of Hummel and Dreyer SEC (Hummel and Dreyer 1962) was described in Papers II
and V for the analysis of CB- and glucose binding to GLUT1 in freely suspended membrane vesicles

24
and proteoliposomes. A gel bed of Superdex 75 (Amersham Biosciences) was equilibrated with 2 nM
[3H]CB and 0 – 200 nM cold CB. The biomembranes were incubated with the relevant running buffer
and 20- or 50-µl aliquots were applied to the column. In the typical resulting chromatogram (Fig. 9 in
Paper II) the baseline signal of [3H]CB first rose to a positive peak corresponding to membranes with
bound CB eluting in the void volume and later a negative peak appeared due to depletion of [3H]CB in
the mobile phase upon CB-binding to the membranes. The area of the negative peak was used for
calculating the amount of bound CB, which increased in runs with increasing concentrations of
unlabelled CB included in the mobile phase. The Kd value and the number of binding sites in the
aliquots were determined by curve fitting to an analog of Eq. 4.
10.3. Ultracentrifugal assay
Ultracentrifugation was used as an alternative method for the measurement of CB binding to
free membrane vesicles, as described in Paper V, similarly as by Pinkofsky et al. (1985) or Helgersson
and Carruthers (1987). GLUT1 proteoliposomes were incubated with CB at different concentrations
(0-500 nM) and centrifuged for 60 min at 226 000 g followed by separation of free from bound CB
and scintillation counting. The method may be suitable for preliminary screening of the strength of a
biointeraction because many samples can be handled at the same time, but this method has the obvious
disadvantage that bound ligand is separated from free ligand in a rather time-consuming procedure.
The ligand concentration in the emerging pellet almost certainly differs from that in the incubation
mixture, which causes ligand to detach or bind during the separation.
10.4. Transport retention chromatography
A partial chromatographic separation of D-glucose from L-glucose has been demonstrated on
immobilized GLUT1-proteoliposomes (Lu et al. 1993) or RBCs (Zeng et al. 1997; Paper IV).
Mixtures (20 µl) of D-[14C]glucose and L-[3H]glucose were run through columns containing the
immobilized GLUT1 material and the eluent was monitored on-line with a flow scintillation detector
using two different energy windows. An elution volume difference, ∆Ve, between the enantiomers was
caused by the stereospecificity of GLUT1, which only allowed D-glucose access to the membrane-
enclosed volume. The contribution of D-glucose-binding to GLUT1 is negligible according to Eq. 1.
Zeng et al. (1997) used transport retention chromatography to measure the stability of their
immobilized RBC columns, but the very small ∆Ve involved makes this method less sensitive than
measuring the frontal retention volume of an inhibitor such as CB.
10.5. Immobilized biomembrane partitioning chromatography (IBiPaC)

25
A variety of biomembranes were immobilized as described in Section 9 for IBiPaC analysis in
Paper I. The retention volumes VR of drugs (10–50 µl, 0.02–0.5 mg/ml) on the biomembrane-
containing columns were measured using a set-up as in Fig. 10A, except that a correspondingly
smaller sample loop, UV-detection at 220 nm and paper recording were used. In contrast to the frontal
elution mode (cf. Section 10.1.), zonal elution was used in IBiPaC, i.e., the drug samples were eluted
as peaks. The capacity factor Ks is a simple normalization of VR to enable comparison of results
obtained on different columns.
AVV
K s0R −
= (7),
with V0, the elution volume of the reference substance Cr2O72- that does not interact with the
membranes, and A, the molar amount of immobilized phospholipids determined by a phosphorus assay
according to Bartlett (1959). Units were chosen to obtain Ks in M-1.
Drugs of considerably different nature (β-blockers, phenothiazines and benzodiazepines) were
analyzed and compared. VR, the variable in Eq. 7, and accordingly also Ks, are dependent on the degree
of the drug’s partitioning into the immobilized biomembranes. Eq. 7 is a simplified version of a
definition by Lundahl and Beigi (1997) which included the internal volume of the immobilized
biomembranes.
In a first set of experiments on entrapped PC liposomes the effect on Ks of varying some
chromatographic parameters (cf. Section 11.3.), as well as ionic strength, pH and temperature was
investigated. Also the effect of incorporating negatively charged PS into PC liposomes on the
retention of positively charged drugs was studied. A set of drugs was run on IBiPaC and IAM columns
and the log Ks values were correlated to the corresponding log k’IAM values (Ong et al. 1996). A
diagram for prediction of drug uptake was prepared using literature data for absorption of the drugs in
humans.
For molecules with specific binding sites within the examined biomembrane, for example CB
binding to RBC membranes, IBAC and IBiPaC may be combined by substituting Vmin in Eq. 3 with
VR. Eq. 4 then becomes
( )[ ] [ ] d0s CB
CBK
NVAKV+
=−− (8).
For Eq. 8 it is assumed that the zonal and frontal non-specific CB retention volumes (inhibiting
specific binding to GLUT1 at high glucose concentrations) are approximately equal, which seems
likely at least at low drug concentrations. Although Eq. 8 is more complex than Eq. 4 it would

26
facilitate the experimental work, as the determination of Vmin for each column by an experimental
series with glucose inhibition (cf. Section 10.1) becomes unnecessary. Instead, a frontal Ks value for
CB could be carefully determined once, and measurement of the amount of immobilized lipids, A, and
the frontal elution volume, V0, of a reference substance would then enable the use of Eq. 8. The zonal
log Ks value for CB on egg yolk phospholipid liposomes is 1.99 (unpublished results, Lagerquist
Hägglund et al.) indicating a strong partitioning and a good membrane permeability for the molecule
(cf. Section 11.3.). Drawbacks of the approximation are that a Kd value for glucose inhibition of CB-
binding is not obtained and that changes in Vmin caused, for example, by treatment of immobilized
cells with poly amino acids might remain undetected.
11. DISCUSSION OF RESULTS
11.1. Can RBCs be adsorbed for chromatographic analyses?
Mainly two methods for RBC immobilization were developed for this thesis; namely, adsorption to
agarose beads via immobilized WGA or by means of the streptavidin-biotin interaction (cf. Section
9.1; Papers IV, V and VII). Electrostatic adsorption of RBCs to charged bisacrylamide particles (Zeng
et al. 1997) played a minor role in drug partitioning in Paper I. The quality of the immobilization was
judged in different ways, e.g., concerning RBC adsorption capacity, stability, relative errors of results
or the ratio of specific (Vspec) to non-specific (Vmin) CB binding in the columns (cf. Table 1). The
author has chosen to split the data for WGA-Sepharose 4B and WGA-SPA to distinguish plain cells
from poly-Lys treated cells and high ( 3.1 mg/ml) WGA-concentrations from lower ones ( 1.2
mg/ml), respectively.
Table 1. Summary of data on immobilized-RBC column capacity, stability and quality of results.Numbers in parentheses are the numbers of used columns; the percentage data given in columns 2, 4and 5 are relative errors of the respective values.Method 109 RBCs/ml gela T90[days]b 103 CB sites/RBC Vspec/Vmin
WGA-Sepharose 4B (3)c 0.82 ±23% 3–7 (6) 260 ±23% 2.0 ±20%
WGA-Sepharose 4B + poly-K (6)d 0.60 ±17% 4 (1) 500 ±40% 2.9 ±28%
WGA-SPA, 3.1mg WGA/ml (4)e 0.98 ±11% 9 (1) 330 ±12% 4.9 ±14%
WGA-SPA, 1.2mg WGA/ml (8)f 0.58 ±16% 6 (1) 250 ±16% 1.8 ±7%
Streptavidin-biotin (7) 0.53 ±17% (11) 5 (1)g; 8 (1)h 350 ±14% 1.6 ±13%
Electrostatic immobilizationi 1.14 ±11% 10 (1) n.d. n.d.a obtained by automated amino acid analysis after 2 days of IBAC experiments (rows 1–5) or anoptical assay directly after immobilization (row 6); b Number of days after RBC immobilization withmeasured stability ≥ 90%; c Paper IV; d Papers IV and VII; e SPA with 3.1 (1) or 4.3 (3) mg WGA/ml,Paper VII; f SPA with 0.08 (2) or 1.2 (6) mg WGA/ml; g [Biotin]=2 mM, cf. Paper V; h [Biotin]=1 mM;i Zeng et al. (1997).

27
The average number of immobilized RBCs per milliliter gel was about equal for poly-Lys
treated cells on WGA-Sepharose 4B (0.60 ± 0.10 × 109/ml), WGA-SPA at low WGA concentrations
(0.58 ± 0.09 × 109/ml) and biotinylated cells on streptavidin-agarose (0.53 ± 0.09 × 109/ml). It should
be noted that the poly-Lys treatment of the cells on WGA-Sepharose 4B, which was originally meant
to stabilize the immobilization, lowered the cell density by more than 25%, which probably
contributed to the high CB/mon value measured for this material in Paper IV. In general, the
immobilized RBC density on WGA-agarose was dependent on [WGA]. The corresponding plot in Fig.
14A described a hyperbola with saturation at 1.3 × 109 bound cells per milliliter gel (Paper VII).
[WGA] [mg/ml gel bed]0 1 2 3 4
No.
of b
ound
RB
Cs p
er m
l gel
bed
0,0
2,0e+8
4,0e+8
6,0e+8
8,0e+8
1,0e+9
1,2e+9
T90 [days]4 5 6 7 8 9
Rel
ativ
e er
ror f
or C
B/m
on [%
]
10
15
20
25
30
35
40
45A B
2
1
3
45
Fig. 14. A) The number of immobilized RBCs per ml gel bed of WGA-SPA ( , Paper VII) or WGA-Sepharose 4B ( , Paper IV) versus the WGA concentration in the gel. B) Dependence of the relativeerror for the number of CB binding sites per RBC on the T90 values. Numeral marks refer to rows 1–5in Table 1. The data gave the best fit to the equation )%120012()T( 90T
90−×+= ef .
The high RBC density upon electrostatic adsorption (Table 1, row 6) was obtained directly
after immobilization and a different assay was used, which may complicate a direct comparison. In
addition, the interpretation of the results in Zeng et al. (1997) suffers from the parallel statements that
such a high density of intact cells was immobilized and that a large fraction of the cells became lysed
and formed ghosts.
The stability of a series of columns with cells on WGA-Sepharose 4B was assayed by daily
frontal runs of CB (Paper IV). The T90 values, e.g., the number of days during which at least 90%
stability was observed, were not completely reproducible (i.e., 3–7 days) and were not affected by
chromatographic adjustments or cell preservative additives in the mobile phase. For later work on
immobilized RBCs, stability measurements were usually done only once per investigated system and
the best result (T90 = 9 days) was obtained with WGA-SPA at high lectin concentration. For
streptavidin-biotin-immobilized cells (Paper V) the stability decreased for RBCs biotinylated with
increasing concentration of Sulfo-NHS-LC-biotin (1–5 mM). Muzykantov et al. (1996) reported that

28
RBCs biotinylated in 20–700 µM NHS-LC-biotin tended to crosslink and lyse upon addition of
streptavidin. Although massive cell lysis was not observed in our chromatographic system it seemed
that the cells were unfavourably influenced by biotinylation and/or streptavidin binding (Fig. 1 in
Paper V). The stability measurements on the electrostatic immobilization system may not be
completely comparable to the present ones, but the reported T90 of 10 days is similar to that for the
best WGA-system. It is not surprising that none of the so far investigated RBC immobilization
systems had a durability comparable to sterically entrapped GLUT1 proteoliposomes or membrane
vesicles (Section 9.2; Paper V). These latter columns could be used for IBAC analyses for several
months, apparently without variation of results, retaining 80–90 % of the immobilized material
(Lundqvist et al. 1998a). The dextran-coated interior of the Superdex 200 beads (cf. Fig. 5 in Paper
III) was obviously a very benign environment for the immobilized biomembranes.
The measured number of CB-binding sites per cell (cf. Table 1) was 250–350 × 103 for most
materials. Poly-Lys-treated WGA-Sepharose 4B (Table 1 and Paper IV) gave a higher value, which
was later discarded because of the large error limit (cf. Paper VII). The highest accuracy was obtained
for WGA-SPA. The relative errors of 12% or 16% covered values for both plain and poly amino acid
treated cells, whereas the individual values for the different materials had even lower error limits
(Paper VII).
It is evident from Fig. 14B that the quality of the IBAC results was dependent on the
chromatographic stability of the immobilized material. The plot of the relative error for the CB-
binding sites per cell (Table 1) decayed exponentially with increasing T90 for the different materials
(Fig. 14B). Another requirement for good IBAC results seemed to be a high density of immobilized
binding sites giving a high ratio of Vspec to Vmin (cf. Paper VI). Although no correlation between
Vspec/Vmin and the relative errors for CB sites/cell, similar to that above, could be deduced from Table
1, at least the distinctly highest Vspec/Vmin value was obtained for WGA-SPA at high [WGA] which
also had the lowest error limit for the number of CB sites/cell. Immediate suggestions for further
improvement of the IBAC results are to elaborate lectin coupling conditions that always yield high
[WGA] and the use of smaller SPA beads with a larger surface area accessible to RBCs.
Immobilization and/or IBAC analyses of membrane proteins of cancer cells (cf. Section 9.3.)
proved to be difficult in the frame of this thesis, although such mild conditions as running in culture
medium at 37°C were tried. Although methods for immobilization of animal and plant cells exist (cf.
Section 5), these tiny entities may be too sensitive to withstand the non-sterile and non-optimized
growth conditions in a chromatographic system. Better results may be obtained upon improvement of
the harvesting technique or growing the cells directly in the columns. Also, the observation that EGF
binding was retained after fixation of A-434 cells in, e.g., approx. 90% ethanol (unpublished results)
could be the key to chromatographic analysis of that interaction.
To answer the title question of this section: Yes! The system can be optimized further but
chromatographic analyses on immobilized RBCs with reasonable accuracy are already possible.

29
Considering the importance of RBCs for our organism and the existence of parasites and major
diseases that can affect them, the new technique presented in this thesis might well prove useful for
certain specific analytical purposes.
11.2. Implications about GLUT1
The analysis of ligand binding to GLUT1 is an intricate matter. The supply of literature data is
abundant and reported results vary considerably, perhaps depending on the preparation status of the
protein. A generally accepted picture of the salient attributes of the protein, such as its oligomerization
state or transport mechanism, has still not been established (cf. Section 4).
We determined CB/mon values of 0.5 for GLUT1 (designated State 1 in Paper V) in most of
our immobilized biomembranes (RBCs, membrane vesicles and GLUT1 proteoliposomes (Papers IV,
V and VII)). Suspended membrane vesicles, however, showed a CB/mon close to 1 (State 2, Paper V).
The same high value had been obtained with poly-Lys treated RBCs (Paper IV), but an attempt to
reproduce these experiments (Paper VII) led to the inconclusive CB/mon values of 0.77 ± 42% or 0.98
± 40% (corresponding to 500 CB sites per cell, cf. Table 1) without correction of the number of
immobilized cells using the standard curve from Paper IV. The CB/mon for streptavidin-biotin
immobilized cells (0.68 ± 16%, Paper V) was interpreted to be due to the simultaneous presence of
GLUT1 functional dimers and monomers. Considering the results in Paper VII, the author now rather
reasons that the loss of hemoglobin from the columns (cf. Muzykantov et al. 1996) caused the elevated
CB/mon ratio. The use of WGA-SPA in Paper VII finally solved the somewhat uncertain situation,
giving CB/mon values close to 0.5 for both plain cells and those treated with poly-Lys, -Arg and -Glu.
The relative errors were 3–18% with a grand average of 0.54 ± 17%. Together with the results for both
immobilized and suspended membrane vesicles and GLUT1 proteoliposomes (cf. Paper V), an
updated scheme for GLUT1 conversions in the different biomembranes is presented in Fig. 15.
The low CB/mon for free membrane vesicles (0.43 ± 0.03, Paper II) was by mistake obtained
on vesicles that retained most of the cytoskeleton due to washing at a maximum pH of 10.5. CB/mon
measured on regular membrane vesicles was 0.97 ± 0.04. Zhang and Ismail-Beigi (1998) reported a
similar 1.8-fold increase of glucose-sensitive CB binding sites in RBC ghosts upon washing in 0.2 mM
EDTA, pH 12.
The number of CB-binding sites per cell (Table 1) was solely based on IBAC results, whereas
CB/mon values for RBCs were dependent on the assumption of the presence 5.1 × 105 GLUT1 copies
per cell. The calculation of this value presented in Paper IV resulted in a large relative error of 41%.
However, it is likely that the true value is closer to 5 × 105. The determination of Mascher and Lundahl
(1988) of 5.3 × 105 GLUT1 per RBC by analysis of DEAE purification of the protein and the results of
monoclonal-antibody binding by Allard and Lienhard (1985) yielding 5 × 105 copies/cell (cf. Anstee

30
Fig. 15. Overview of the CB-binding state of GLUT1 in different materials as concluded from PapersIV, V and VII. The materials were A) Free cells (according to e.g. Hebert and Carruthers 1992); B)RBCs adsorbed on WGA-SPA; C) free membrane vesicles, determined by Hummel and Dreyer SEC;D) GLUT1 in the presence of OG or C12E8, with cholate oligomers are formed (Hebert and Carruthers1992); E) free proteoliposomes; F) sterically entrapped membrane vesicles or proteoliposomes.Reprinted with modifications from Paper V, with permission from Blackwell Sciences.
1990) gave us the confidence to assume 5.1 × 105 GLUT1 monomers per cell. The results in Paper V
for entrapped GLUT1 proteoliposomes (CB/mon=0.40±0.01) and membrane vesicles (0.50±0.02)
were instead dependent on the purity and the fraction of active transporters, but all supported the
presence of GLUT1 in State 1.
Hence, our CB/mon values support the conclusion that GLUT1 in the RBC membranes works
as a functional dimer that can reversibly dissociate into functional monomers during membrane vesicle
preparation. It is not clear to which degree a physical dissociation of the dimers takes place. The
formation of GLUT1 tetramers, as in the GLUT1 fixed site carrier model (Sulzman and Carruthers
1999; cf. Section 4 and Paper III), is supported by a CB/mon of 0.5, although the formation of larger
aggregates (hexamers, octamers, etc) cannot be excluded. Exposure of membrane vesicles to 10 mM
DTT during the washing at pH 12 did not effect the CB/mon of the immobilized vesicles themselves
nor that of immobilized GLUT1 proteoliposomes prepared from that material (cf. Paper V), in
disagreement with the results of Hebert and Carruthers (1992).
The dissociation constants for CB-binding to GLUT1 seem to be independent of the
preparation state of the protein. For almost all analyzed GLUT1 materials (Papers IV, V, VII) the Kd

31
values fell within the interval 70 ± 14 nM determined in Paper VII. Our measured CB-binding affinity
is higher than most others reported in the literature, which partly may be due to the use of non-
equilibrium methods for the ligand binding assays (e.g., centrifugation or ultrafiltration).
The affinity for D-glucose binding to GLUT1, on the other hand, appeared to be higher in the
more natural biomembranes than in proteoliposomes, as determined by displacement of CB. For all
immobilized RBCs studied in this thesis work, the Kd was 12±3 mM, Zeng et al. (1997) reported even
7 mM, whereas for immobilized membrane vesicles and proteoliposomes 18±4 and 41±5 mM,
respectively, were measured.
The partial separation of D-glucose and L-glucose in transport retention chromatography
experiments succeeded on RBCs immobilized on WGA-Sepharose 4B (Fig. 16).
Elution volume [ml]1,0 1,2 1,4 1,6 1,8 2,0
Cou
nts (
%)
0
20
40
60
80
100
120L D
∆Ve
Fig. 16. Simultaneous elution of L-[3H]glucose and D-[14C]glucose on RBCs adsorbed on WGA-Sepharose 4B (Vt = 1.3 ml) at 0.1 ml/min. The difference in elution volume (∆Ve= 64 µl) was read at50% peak height.
An increase in ∆Ve with the number of immobilized RBCs was described (Paper IV). For unknown
reason, the elution volumes of the enantiomers did not differ for columns prepared by any of the other
RBC immobilization methods developed in this thesis work. In principle, this could have been caused
by glucose leakage from the cells (although they retained hemoglobin), inactive GLUT1 or too slow
equilibration of the column with glucose. For the WGA-SPA columns it did not matter whether D-
glucose or deoxy-D-glucose, which is transported into but not metabolized in RBCs, were used.
In conclusion, the direct comparison of IBAC results obtained on a multitude of different
GLUT1 materials as presented here may yield more insight into the protein’s attributes than do
isolated analyses of single preparations. All such knowledge about the properties of GLUT1 may
prove helpful in attempts to crystallize the protein.

32
11.3. IBiPaC for prediction of drug uptake
The typical dimensions of an IBiPaC column containing freeze-thaw immobilized egg yolk
phospholipid or PC liposomes were approx. 5 cm × 0.5 cm (i.d.), corresponding to a total volume of
≈1 ml, and a typical gel bed contained about 50 µmol lipids. The retention volumes of different drugs
on such columns covered a range of ≈1–500 ml, resulting in log Ks values between ≈0.5 (e.g., for
atenolol) and ≈4 (e.g., for chlorpromazine). A remarkable feature of the columns was their
extraordinary stability. Once prepared, they could be run (or stored) for months or even more than a
year (Österberg et al. 2001), without significant decrease of the drugs’ retention volumes.
The log Ks values were found to be independent of the total column volume (0.1–1.0 ml), the
chromatographic flow rate (0.1–1.2 ml/min), the drug concentration and the concentration of
immobilized phospholipids (10–55 mM) thereby justifying the use of Eq. 7. However, departure from
standard experimental conditions, 150 mM NaCl, pH 7.4 and 23°C, did effect drug partitioning into
PC liposomes containing 18.5 mol % PS (cf. Fig. 1 in Paper I). Lowering the salt concentration to 0
increased the log Ks values of the positively charged β-blockers, dramatically for the relatively
hydrophilic drugs atenolol and metoprolol, and more moderately for the more lipophilic propranolol.
The reason was an increased electrostatic interaction of the drugs with the negatively charged PS. On
pure PC liposomes with neutral net charge the effect was only observed with atenolol. Raising the
NaCl concentration to 0.6 M resulted at most in a slightly stronger partitioning on the PS-free
liposomes due to enhanced hydrophobic interactions. The log Ks values for β-blockers on PS/PC
liposomes were essentially constant in the pH interval 6–9, but increased for pH 9–11, probably due to
deprotonation of the model drugs. The drugs showed a heterogeneous partitioning behavior upon a
temperature increase from 10°C to 60°C, e.g., low log Ks values increased linearly, whereas high
values decreased. The analyzed phenothiazines, promethazine and fluphenazine, both showed linearly
decreasing membrane partitioning in response to the temperature rise.
Log Ks values of 15 drugs on egg phospholipid liposomes were plotted versus corresponding data
obtained on PC liposomes, RBC membrane lipid (ML) liposomes, RBC membrane vesicles and
RBCs/ghosts (Fig. 17). All plots were linear with the best correlations (r2 = 0.96) for the membrane
lipids (B) and membrane vesicles (C). The slopes of the lines increased through panels A to D in line
with the complexity of the immobilized membranes. Low log Ks values increased, whereas high values
decreased in the panels to the right. Later work (Lagerquist et al. 2001) showed that the presence of
cholesterol in liposomes generally decreased log Ks values, whereas model transmembrane proteins
(GLUT1 and bacteriorhodopsin) caused electrostatic attraction or repulsion of the drugs. A
combination of these effects may have caused the slope shift in Fig. 17. A complication is that
positively charged drugs may get deprotonized, at the surface of the membrane while partitioning into
the membrane.

33
Fig. 17. Comparison of drug interaction with egg phospholipid (EPL) liposomes with results obtainedon other biomembranes. ML denotes endogenous RBC membrane lipids. The drugs were a, atenolol;b, acebutolol; c, nadolol; d, metoprolol; e, pindolol, f, oxprenolol; g, fluphenazine; h, pyrilamine; i,flunitrazepam; j, alprenolol; k, propranolol; l, diazepam; m, nitrazepam; n, oxazepam; o,desmethyldiazepam; p, promethazine; q, chlorpromazine. Reprinted from Paper I, with permissionfrom Elsevier Science.
The correlation of log Ks values on PC liposomes and log k’IAM values (Fig. 4 in Paper I)
obtained on an IAM.PC.DD column was slightly poorer (r2=0.83) than the above ones, despite the fact
that the same type of phospholipid was present in both columns. Sterical hindrance for diffusion of
some of the drugs within the IAM lipid monolayers may be among the reasons for the difference.
Fig. 18. Oral absorption of drugs versus log Ks values obtained on RBC membrane vesicles. Thedifferent compounds were as listed in Fig. 17 and r, polyethylene glycol (Mw 200); s, mannitol; t,salicylic acid; u, hydrocortisone; v, corticosterone; w, warfarin; x, terbutaline. The absorption datawere adopted from Palm et al. (1997) and Fagerholm et al. (1997).
According to plots of drug absorption data in humans versus log Ks values obtained on
immobilized membrane vesicles (e.g. Fig. 18), it is likely that drugs with intermediate log Ks (≈ 1.25–
2.25) are absorbed well. Molecules with lower log Ks values seem to be too hydrophilic for adequate

34
membrane partitioning. Several drugs in the original plot of Fig. 5 in Paper I have been removed:
acetylsalicylic acid is known to be hydrolyzed to salicylic acid upon membrane transport
(www.fass.se, search for Aspirin) and sulfasalazine is a substrate for effective efflux systems (e.g., P-
glycoprotein) (Österberg et al. 2000). For log Ks values higher than approx. 2.25 the situation appeared
to be more intricate. The removal of sulfasalazine from the plot lead to a shallower decrease of
absorption for high log Ks values. In Österberg et al. (2000) many drugs with log Ks values > 2.5 on
PC liposomes are shown to be almost completely absorbed in humans, making a dramatic absorption
decrease for drugs with such high log Ks values more improbable. Obviously, the course of the line in
this region is dependent on the choice of model drugs. Features other than the model drugs’ passive
diffusion, such as their metabolism, have to be known. A choice of different model compounds
representative of different groups of related drug candidates may be considered.
Another important factor for prediction of drug uptake by IBiPaC is the choice of immobilized
biomembranes. In Paper I, RBC membrane vesicles of high stability containing membrane proteins
and a diversity of lipids were the most reliable and relevant of the available systems. A system of
immobilized brush-border membranes from pig mucosa is currently under development (Engvall et
al., unpublished).
12. Conclusions
Paper I The simple definition of log Ks presented in the paper worked for
normalization of drug partitioning into biomembranes and for prediction of drug uptake
in humans. The log Ks values were independent of the chromatographic flow rate, the
gel bed volume as well as drug and lipid concentrations, but were affected by changes in
ionic strength, pH and temperature. Effects of charge, composition and the complexity
of the immobilized membranes on IBiPaC were, for the first time, systematically
recorded. Among the drugs tested, those with intermediate log Ks values were absorbed
effectively in humans. To make calibration curves that are practically useful for
prediction of drug uptake the model compounds have to be chosen carefully. The study
of more model drugs is necessary and more work is required for immobilization of the
most relevant biomembranes.
Paper II Extension of the size range for chromatographically separable biomembranes
in connection with the development of gels with larger pore sizes was made evident.
Characteristics of biomembranes, such as flexibility, non-specific binding to gel
matrices and light scattering and their impact on chromatography were explained.
Preparative and analytical applications, including a previously unpublished method,
Hummel and Dreyer analysis of ligand binding to GLUT1, were described. Finally,
alternative methods and analyses on immobilized biomembranes were presented briefly.

35
Paper III An overview of some of the known features of GLUT1 was given concerning
its purification, topology and structure. The alternating conformation model and the
fixed-site-carrier model for glucose transport kinetics were described and a possible
connection between the two was explained and reconciled with our own results.
Papers IV, V, Equilibrium constants for ligand binding to membrane proteins in
and VII immobilized RBCs can be determined by IBAC. The most promising results were so far
obtained upon adsorption of the cells on SPA beads containing more than 3.1 mg/ml
coupled WGA (Paper VII). The density of immobilized cells in the gel bed was high
(≈109 cells/ml gel) and the cell immobilization stability seemed to be the most critical
factor for the quality of the IBAC analyses. Results obtained with RBCs on commercial
Sepharose 4B with 1.96 mg/ml coupled WGA showed larger error limits and the column
stability was lower, especially for columns treated with polylysine. Biotinylation of
RBCs with increasing concentrations of biotin reagent followed by adsorption on
streptavidin-agarose seemed to reduce the time available for analysis of the system
(Paper V). The method was thus apparently harmful to the cells and was not developed
further.
The large amount of data for CB binding to GLUT1 in proteoliposomes,
membrane vesicles and cell membranes collected during several years of IBAC analyses
was complemented and summarized. A more general picture regarding the association
state of the transporter monomers arose, suggesting that the dimeric state of CB binding
observed in immobilized proteoliposomes probably also prevailed in the native RBC
membrane. The dissociation constant for CB-binding to GLUT1 was approx. 70 nM for
most of the analyzed materials. D-Glucose showed a higher affinity for GLUT1 in the
more natural the membranes as determined by inhibition of CB binding. The Kd
increased from 12 mM in RBC membranes to 41 mM in proteoliposomes.
In relation to Papers IV–VII, IBAC analyses of human cancer cells attached to
agarose gel beads have hitherto not been successful. More favorable conditions for
chromatographic analyses of these cells remain to be found.
Paper VI Methodology and principles of IBAC were presented. The potential of this
method to analyze ligand binding to a membrane protein in many different
biomembranes was emphasized and exemplified by our studies of GLUT1. Applications
of quantitative affinity chromatography on a variety of other immobilized membrane
proteins were briefly described, and reference to alternative techniques and the related
immobilized-liposome chromatography system was made.

36
Acknowledgements
Financial support for this work from the University of Uppsala, grants to P. Lundahl et al.from the O.E. and Edla Johanssons Science Foundation, the Swedish Natural ScienceResearch Council and the Swedish Research Council for Engineering Sciences is gratefullyacknowledged.
Writing these lines I am about to finish five years of work. It is due to a whole lot of people,colleagues, friends and family that this was a good time of my life, so I think it is about timeto thank those who matter most to me.
I owe deep gratitude to my supervisor Per Lundahl for letting me join his research group,trusting me and sharing his vast knowledge and experience in the field of biomembranes.Maybe I will remember our nice and interesting conference trip to Amsterdam the most.
Thank you Janos Hajdu, my examiner, for support and instructions on membrane proteincrystallization.
Former members of the biomembrane group who introduced me to the laboratory and withwhom I enjoyed to work, discuss and chat are Andreas, Cheng-Ming, Eggert, Eva, Farideh,Lars, Lili, Yanxiao, and Yi-Ming. Thanks Andreas, for support when watching "Saving PrivateRyan" and Farideh for being a good party fellow. I appreciated playing darts with Eggert andtalking (almost watching) birds with Lars. Today, running into the home stretch, I sense apleasant climate in the group being created by Caroline (=> glassfika), Christine (=>kaffefika), Juanjo (Mr. Footbag => fotbollsfika a la catalana), Lisa (=> partyfika) and Shu-Sheng (=> sun flower fika). Neither have I forgotten the undergraduate students in our group:Jan (cool language, idea Fig. 9), Emma (=> torsdagsfika), Chris, Anders and Natalie.
I am especially happy about the doctoral student meetings at Norreda Torp, in 1999 and 2001,which, I think, were first initiated by Ann, Tove and Cynthia. Apart from the good spirits anddiscussions and nice scenery out there, I got to know and make friends with folks in the othergroups of the department. I’d like to name Pelle+Per (håll proggen vid liv!), Nisse (den somklättrar högt behöver inte falla djupt), Peo (YOU introduced me to whitewater-paddling!!),and all regulars at our student pub evenings, such as Sussi, Anna-Karin and Ylva.
Thanks to Christer, who introduced me to teaching at the course lab, and the other nice peoplewith whom I had the pleasure to co-teach and I owe special thanks to Lilian for her kindnessand great back-up at the course lab.
I appreciated quick and skilful technical help from Per-Axel and Hasse P. and the organizingskills of Lasse L.. Thanks Ulla, Birgitta and Inger for kind help in economical andadministrative matters. I am grateful to David, Marie and Karin for amino acid analyses;David and the members of the biomembrane group also for proof reading of manuscripts, inparticular, the thesis summary.

37
Among my co-authors I want to specially thank Per-Erik Gustavsson, who kindly sent mesuperporous agarose gel without ever having seen me and Bo Ersson for supplying me withWGA.
I would also like to thank my first mentors in biochemistry, Qinghai Zhao and JörgenCarlsson, as well as Veronika Asplund-Eriksson and Lars Gedda for help with cell culturingat the Division of Biomedical Radiation Sciences during my thesis work.
My dear friends outside the academic world are important to me. I love(d) to spend time withyou around: Sara, Olof, Rebbi, Karl, Benjamin, María-José, Fia, Anette, Alex, all former bandand percussion group mates, Johan, Ransta Trädgård et al. I was and am always happy to seeBritta, Hans, Susan and Morgan in Värmland. My oldest friends live back in Germany, wheremy roots are. I know times are changing and we do not get to see each other too often thesedays, but it is important to me to keep contact and I am first thinking of Helge & family, Ralf& family, Arndt, Katrin, Antje, Kai, Stephan, Silke, Henning and many others.
I am glad to have found my way to Uppsala Paddlarklubb where I found the exercise,recreation, and nature experience that helped me even (especially) through periods ofconcentrated work. Thanks, Hasse, Malin, Peo, Gunnar, Jakob, David, Dan and others forinstructions and good fellowship.
This is also where I met Ebba, you are a great support, a wise and humorous person in allmatters of life. We have such a wonderful time together.
I wish that my dad Heinrich could be around now. After all, it was you and my mother Helgawho enabled my studies and my stay in Sweden. In 1997, after a short but intense struggleagainst a vicious cancer you passed away in Helgas, my brother Carstens and my arms. I willalways keep feeling sorry for your suffering and our inability to help you. Natürlich will ichauch euch danken, Helga und Carsten, für eure Unterstützung und daß ihr in Deutschlandnach dem Rechten seht, sowie für Hilfe, zB. sobald es mir einfällt, mal wieder irgendwas ausder Heimat besorgen zu müssen.
Peace,
Ingo

38
References
Adair, W.L. and Kornfeld, S. (1974) Isolation of the receptors for wheat germ lectin and the Ricinuscommunis lectins from human erythrocytes using affinity chromatography. J. Biol. Chem. 15,4696–4704.
Allard, W.J. and Lienhard, G.E. (1985) Monoclonal antibodies to the glucose transporter from humanerythrocytes, J. Biol. Chem. 260, 8668–8675.
Alvarez, J., Lee, D.C., Baldwin, S.A. and Chapman, D. (1987) Fourier transform infraredspectroscopic study of the structure and conformational changes of the human erythrocyte glucosetransporter. J. Biol. Chem. 262, 3502–3509.
Amanda Wu, K.–Y. and Wisecarver, K.D. (1991) Cell immobilization using PVA crosslinked withboric acid. Biotechnol. Bioeng. 39, 447–449.
Andrews, P., Kitchen, B.J. and Winzor, D.J. (1973) Use of affinity chromatography for thequantitative study of acceptor-ligand interactions: the lactose synthetase system. Biochem. J. 135,897–900.
Anstee, D.J. (1990a) The nature and abundance of human red cell surface glycoproteins. J.Immunogenet. 17, 219–225.
Anstee, D.J. (1990b) Blood group-active surface molecules of the human red blood cell. Vox Sang. 58,1–20.
Artursson, P., Palm, K. and Luthman, K. (1996) Caco-2 monolayers in experimental and theoreticalpredictions of drug transport. Adv. Drug Del. Rev. 22, 67–84.
Baldwin, S.A., Baldwin, J.M. and Lienhard, G.E. (1982) Monosaccharide transporter of the humanerythrocyte. Characterization of an improved preparation. Biochemistry 21, 3836–3842.
Baldwin, S.A. and Lienhard, G.E. (1989) Purification and reconstitution of glucose transporter fromhuman erythrocytes. Methods Enzymol. 174, 39–50.
Barnett, J.E.G., Holman, G.D., Chalkley, R.A. and Munday, K.A. (1975) Evidence for twoasymmetric conformational states in the human erythrocyte sugar-transport system. Biochem. J.145, 417–429.
Barrett, M.P., Walmsley, A.R. and Gould, G.W. (1999) Structure and function of facilitative sugartransporters. Curr. Opin. Cell. Biol. 11, 496–502.
Bartlett, G.T. (1959) Phosphorus assay in column chromatography. J. Biol. Chem. 234, 466–468.Beigi, F., Yang, Q. and Lundahl, P. (1995) Immobilized-liposome chromatographic analysis of drug
partitioning into lipid bilayers. J. Chromatogr. A 704, 315–321.Bell, G.I., Kanyano, T., Buse, J.B., Burant, C.F., Takeda, J., Lin, D., Fukumoto, H. and Seino, S.
(1990) Molecular biology of mammalian glucose transporters. Diabetes Care 13, 198–208.Betageri, G.V. and Rogers, J.A. (1987) Thermodynamics of partitioning of β-blockers in the n-
octanol-buffer and liposome systems. Int. J. Pharm. 36, 165–173.Bisson, I., Hilborn, J., Wurm, F., Meyrat, B. and Frey, P. (2002) Human urothelial cells grown on
collagen adsorbed to surface-modified polymers. Urology 60, 176–180.Bogdanov, M., Heacock, N. and Dowhan, W. (2002) A polytopic membrane protein displays a
reversible topology dependent on membrane lipid composition. EMBO J. 21, 2107–2116.Boulter, J.M. and Wang, D.N. (2001) Purification and characterization of human erythrocyte glucose
transporter in decylmaltoside detergent. Protein Expr. Purif. 22, 337–348.Brekkan, E., Lundqvist, A. and Lundahl, P. (1996) Immobilized membrane vesicle or proteoliposome
affinity chromatography. Frontal analysis of interactions of cytochalasin B and D-glucose with thehuman red cell glucose transporter. Biochemistry 35, 12141–12145.
Cairns, M.T., Elliot, D.A., Scudder, P.R. and Baldwin, S.A. (1984) Proteolytic and chemicaldissection of the human erythrocyte glucose transporter. Biochem. J. 221, 179–188.
Cairns, M.T., Alvarez, J., Panico, M., Gibbs, A.F., Morris, H.R., Chapman, D. and Baldwin, S.A.(1987) Investigation of the structure and function of the human erythrocyte glucose transporter byproteolytic dissection. Biochim. Biophys. Acta 905, 295–310.
Carlsson, J., Janson, J.-C. and Sparrman, M. (1989) Affinity Chromatography. In Protein Purification.Principles, High Resolution Methods, and Applications (Janson, J.-C. and Rydén, L., Eds.), p. 292,VHC Publishers, New York, USA.
Carpenter, G. (2000) The EGF receptor: a nexus for trafficking and signaling. Bioassays 22, 697–707.

39
Chin, J.J., Jung, E.K.Y., Chen, V. and Jung, C.Y. (1987) Structural basis of human erythrocyteglucose transporter function in proteoliposome vesicles: circular dichroism measurements. Proc.Natl. Acad. Sci. USA 84, 4113–4116.
Cloherty, E.K., Hamill, S., Levine, K. and Carruthers, A. (2001) Sugar transporter regulation by ATPand quaternary structure. Blood Cell Mol. Dis. 27, 102–107.
Davies, A., Ciardelli, T.L., Lienhard, G.E., Boyle, J.M., Whetton, A.D. and Baldwin, S.A. (1990) Site-specific antibodies as probes of the topology and function of the human erythrocyte glucosetransporter. Biochem. J. 266, 799–808.
Devaux, P.F. (1993) Lipid transmembrane asymmetry and flip-flop in biological membranes and inlipid bilayers. Curr. Opin. Struc. Biol. 3, 489–494.
Dixon, L.R. (1997) The complete blood count: Physiologic basis and clinical usage. J. Perinat.Neonat. Nurs. 11, 1–18.
Ducarme, P., Rahman, M., Lins, L. and Brasseur, R. (1996) The erythrocyte/brain glucose transportermay adopt a two-channel transmembrane α/β structure. J. Mol. Model. 2, 27–45.
Dunn, B.M. and Chaiken, I. (1974) Quantitative affinity chromatography. Determination of bindingconstants by elution with competitive inhibitors. Proc. Nat. Acad. Sci. USA 71, 2382–2385.
Englund, A.-K., Lundahl, P., Elenbring, K., Ericson, C. and Hjertén, S. (1995) Capillary and rotating-tube isoelectric focusing of a transmembrane protein, the human red cell glucose transporter. J.Chromatogr. A 711, 217–222.
Fagerholm, U., Johansson, M. and Lennernäs, H. (1996) Comparison between permeabilitycoefficients in rat and human jejunum. Pharm. Res. 13, 1336–1342.
Falsey, J.R., Renil, M., Park, S., Li, S. and Lam, K.S. (2001) Peptide and small molecule microarrayfor high throughput cell adhesion and functional assays. Bioconjugate Chem. 12, 346–353.
Fischbarg, J., Cheung, M., Czegledy, F., Li, J., Iserovich, P., Kuang, K., Hubbard, J., Garner, M.,Rosen, O.M., Golde, D.W. and Vera, J.C. (1993) Evidence that facilitative glucose transportersmay fold as β-barrels. Proc. Natl. Acad. Sci. USA 90, 11658–11662.
Folch, J., Lees, M. and Stanley, G.H.S. (1957) A simple method for the isolation and purification oftotal lipids from animal tissues. J. Biol. Chem. 226, 497–509.
Frillingos, S., Sahin-Tóth, M., Wu, J. and Kaback, H.R. (1998) Cys-scanning mutagenesis: a novelapproach to structure-function relationships in polytopic membrane proteins. FASEB J. 121281–1299.
Galazzo, J.L. and Bailey, J.E. (1990) Growing Saccharomyces cerevisiae in calcium-alginate beadsinduces cell alterations which accelerate glucose conversion to ethanol. Biotechnol. Bioeng. 36,417–426.
Goldstein, I.J. and Hayes, C.E. (1978) The lectins: carbohydrate-binding proteins of plants andanimals. Adv. Carbohydr. Chem. Biochem. 35, 127–340.
Griffin, J.F., Rampal, A.L. and Jung, C.Y. (1982) Inhibition of glucose transport in humanerythrocytes by cytochalasins: a model based on diffraction studies. Proc. Natl. Acad. Sci. USA79, 3759–3763.
Gustavsson, P.-E. and Larsson, P.-O. (1996) Superporous agarose, a new material for chromatography.J. Chromatogr. A 734, 231–240.
Guyton, A.C. (1992) Human Physiology and Mechanisms of Disease (Wonciewicz, J.M., Ed.), 5th ed.,W.B. Saunders, Philadelphia, Pennsylvania, USA.
Hamill, S., Cloherty, E.K. and Carruthers, A. (1999) The human erythrocyte sugar transporter presentstwo sugar import sites. Biochemistry 38, 16974–16983.
Haneskog, L., Andersson, L., Brekkan, E., Englund, A.-K., Kameyama, K., Liljas, L., Greijer, E.,Fischbarg, J. and Lundahl, P. (1996) Monomeric human red cell glucose transporter (Glut1) innon-ionic detergent solution and a semi-elliptical torus model for detergent binding to membraneproteins. Biochim. Biophys. Acta 1282, 39–47.
Haneskog, L., Zeng, C.-M., Lundqvist, A. and Lundahl, P. (1998) Biomembrane affinitychromatographic analysis of inhibitor binding to the human red cell nucleoside transporter inimmobilized cells, vesicles and proteoliposomes. Biochim. Biophys Acta 1371, 1–4.
Hansch, C. and Fujita, T. (1964) ρ−σ−π Analysis. A method for the correlation of biological activityand chemical structure. J. Am. Chem. Soc. 86, 1616–1626.

40
Haruna, T., Horie, M., Takano, M., Kono, Y., Yoshida, H., Otani, H., Kubota, T., Ninomiya, T., Akao,M. and Sasayama, S. (2000) Alteration of the lipid environment by L-palmitoylcarnitinemodulates KATP channels in guinea-pig ventricular myocytes. Pflüglers Arch.-Eur. J. Physiol. 441,200–207.
Hebert, D.N. and Carruthers, A. (1992) Glucose transporter oligomeric structure determinestransporter function. Reversible redox-dependent interconversions of tetrameric and dimericGLUT1. J. Biol. Chem. 267, 23829–23838.
Helgerson, A.L. and Carruthers, A. (1987) Equilibrium ligand binding to the human erythrocyte sugartransporter. Evidence for two sugar binding sites per carrier. J. Biol. Chem. 262, 5464–5475.
Hresko, R.C., Kruse, M., Strube, M. and Mueckler, M. (1994) Topology of the GLUT1 glucosetransporter deduced from glycosylation scanning mutagenesis. J. Biol. Chem. 269, 20482–20488.
Holeberg, I.B. and Margalith, P. (1981) Alcoholic fermentation by immobilized yeast at high sugarconcentrations. European J. Appl. Microbiol. Biotechnol. 13, 133–140.
Hu, K., Sun, Y., Chen, D. and Zhang, Y. (2000) The effect of lipid environment in purple membraneon bacteriorhodopsin. J. Photoch. Photobio. B 58, 163–169.
Hummel, J.P. and Dreyer, W.J. (1962) Measurement of protein-binding phenomena by gel filtration.Biochim. Biophys. Acta 63, 530–532.
Joost, H.G. and Thorens, B. (2001) The extended GLUT-family of sugar/polyol transport facilitators:nomenclature, sequence characteristics, and potential function of its novel members. Mol. Membr.Biol. 18, 247–256.
Kamihira, M., Wang, J., Hata, M., Ijima, S. and Kobayashi, T. (1993) Immobilization of animal cellsusing photo-crosslinkable resin. J. Ferment. Bioeng. 75, 138–144.
Kasahara, M. and Hinkle, P.C. (1977) Reconstitution and purification of the D-glucose transporterfrom human erythrocytes. J. Biol. Chem. 252, 7384–7390.
Kasai, K. and Ishii, S. (1975) Quantitative analysis of affinity chromatography of trypsin. A newtechnique for investigation of protein-ligand interaction. J. Biochem. 77, 261–264.
Kasai, K., Oda, Y., Nishikata, M. and Ishii, S. (1986) Frontal affinity chromatography: theory for itsapplication to studies on specific interactions of biomolecules. J. Chromatogr. 376, 33–47.
Klotz, I.M. (1983) Ligand-receptor interactions: what we can and cannot learn from bindingmeasurements. Trends Pharmacol. Sci. 4, 253–255.
Lagerquist, C., Beigi, F., Karlén, A., Lennernäs, H. and Lundahl, P. (2001) Effects of cholesterol andmodel transmembrane proteins on drug partitioning into lipid bilayers as analysed byimmobilized-liposome chromatography. J. Pharm. Pharmacol. 53, 1477–1487.
Leatherbarrow, R.L. (1990) Using linear and non-linear regression to fit biochemical data. TrendsBiochem. Sci. 15, 455–458.
Lienhard, G.E., Slot, J.W., James, D.E. and Mueckler, M.M. (1992) How cells absorb glucose. Sci.Am. 266, 34–39.
Liu, H., Cohen, D.E. and Pidgeon, C. (1997) Single step purification of rat liver aldolase usingimmobilized artificial membrane chromatography. J. Chromatogr. B 703, 53–62.
Lu, L., Brekkan, E., Haneskog, L., Yang, Q. and Lundahl, P. (1993) Effects of pH on the activity ofthe human red cell glucose transporter Glut 1: transport retention chromatography of D-glucoseand L-glucose on immobilized Glut 1 liposomes. Biochim. Biophys. Acta 1150, 135–146.
Lundahl, P., Greijer, E., Cardell, S., Mascher, E. and Andersson, L. (1986) Improved preparation ofthe integral membrane proteins of human red cells, with special reference to the glucosetransporter. Biochim. Biophys. Acta 855, 345–356.
Lundahl, P. and Beigi, F. (1997) Immobilized liposome chromatography of drugs for analysis of drug–membrane interactions. Adv. Drug Del. Rev. 23, 221–227.
Lundqvist, A., Brekkan, E., Lagerquist, C., Haneskog, L. and Lundahl, P. (1997) Frontal affinitychromatographic analysis of membrane protein reconstitution. Mat. Sci. Eng. C 4, 221–226.
Lundqvist, A., Brekkan, E., Haneskog, L., Yang, Q., Miyake, J. and Lundahl, P. (1998a)Determination of transmembrane protein affinities for solutes by frontal chromatography. InQuantitative analysis of biospecific interactions (Lundahl, P., Lundqvist, A. and Greijer, E., Eds.)pp. 79–93, Chapter 5, Harwood Academic, Amsterdam.

41
Lundqvist, A., Ocklind, G., Haneskog, L. and Lundahl, P. (1998b) Freeze-thaw immobilization ofliposomes in chromatographic gel beads: evaluation by confocal microscopy and effects offreezing rate. J. Mol. Recogn. 11, 52–57.
Lundqvist, A. and Lundahl, P. (2001) Advantages of quantitative affinity chromatography for theanalysis of solute interaction with membrane proteins. J. Biochem. Biophys. Methods 49, 507–521.
Ma, L., Ramachandran, C. and Weiner, N.D. (1991) Partitioning of an homologous series of alkyl p-aminobenzoates into multilamellar liposomes: effect of liposome composition. Int. J. Pharm. 70,209–218.
Mascher, E. and Lundahl, P. (1988) The human red cell glucose transporter in octyl glucoside. Highspecific activity of monomers in the presence of membrane lipids. Biochim. Biophys. Acta 945,350–359.
Mayer, L.D., Hope, M.J., Cullis, P.R. and Janoff, A.S. (1985) Solute distributions and trappingefficiencies observed in freeze-thawed multilamellar vesicles. Biochim. Biophys. Acta 817, 193–196.
Miranti, C.K. (2002) Application of cell adhesion to study signaling networks. Methods Cell Biol. 69,359–383.
Moaddel, R., Lu, L., Baynham, M. and Wainer, I.W. (2002) Immobilized receptor- and transporter-based liquid chromatographic phases for on-line pharmacological and biochemical studies: a mini-review. J. Chromatogr. B 768, 41–53.
Mueckler, M., Caruso, C., Baldwin, S.A., Panico, M., Blench, I., Morris, H.R., Allard, W.J., Lienhard,G.E. and Lodish, H.F. (1985) Sequence and structure of a human glucose transporter. Science 229,941–945.
Mueckler, M. (1994) Facilitative glucose transporters. Eur. J. Biochem. 219, 713–725.Mussenden, P., Keshavarz, T., Saunders, G. and Bucke, C. (1993) Physiological studies related to the
immobilization of Penicillium chrysogenum and penicillin production. Enzyme Microb. Technol.15, 2–7.
Muzykantov, V.R., Muricano, J.C., Taylor, R.P., Atochina, E.N. and Herraez, A. (1996) Regulation ofthe complement-mediated elimination of red blood cells modified with biotin and streptavidin.Anal. Biochem. 241, 109–119.
Nagashima M., Azuma, M., Noguchi, S., Inuzuka, K. and Samejima, H. (1984) Continuous ethanolfermentation using immobilized yeast cells. Biotechnol. Bioeng. 26, 992–997.
Nagata, Y. and Burger, M.M. (1974) Wheat germ agglutinin molecular characteristics and specificityfor sugar binding. J. Biol. Chem. 249, 3116–3122.
Nandakumar, M.P., Pålsson, E., Gustavsson, P.-E., Larsson, P.-O. and Mattiasson, B. (2000)Superporous agarose monoliths as mini-reactors in flow injection systems. On-line monitoring ofmetabolites and intracellular enzymes in microbal cultivation processes. Bioseparation, 9 193–202.
Nichol, L.W., Ogston, A.G., Winzor, D.J. and Sawyer W.H. (1974) Evaluation of equilibriumconstants by affinity chromatography. Biochem. J. 143, 435–443.
Olsowski, A., Monden, I. and Keller, K. (1998) Cystein-scanning mutagenesis of flanking regions atthe boundary between external loop I or IV and transmembrane segment II or VII in the GLUT1glucose transporter. Biochemistry 37, 10738–10745.
Ong, S., Liu, H. and Pidgeon, C. (1996) Immobilized-artificial-membrane chromatography:measurements of membrane partition coefficient and predicting drug membrane permeability. J.Chromatogr. A 728, 113–128.
Österberg, T., Svensson, M. and Lundahl, P. (2001) Chromatographic retention of drug molecules onimmobilised liposomes prepared from egg phospholipids and from chemically pure phospholipids.Eur. J. Pharm. Sci. 12, 427–439.
Palm, K., Stenberg, P., Luthman, K. and Artursson, P. (1997) Polar molecular surface propertiespredict the intestinal absorption of drugs in humans. Pharm. Res. 14, 568–571.
Park, J.K. and Chang, H.N. (2000) Microencapsulation of microbial cells. Biotechnol. Adv. 18, 303–319.
Pennell, R.B. (1964) Composition of normal human red cells. In The Red Blood Cell (Bishop, C. andSurgenor, D.M., Eds.), Academic Press, London, pp. 29–69.

42
Pick, U. (1981) Liposomes with a large trapping capacity prepared by freezing and thawing ofsonicated phospholipid mixtures. Arch. Biochem. Biophys. 212, 186–194.
Pidgeon, C. and Venkataram, U.V. (1989) Immobilized artificial membrane chromatography: supportscomposed of membrane lipids. Anal. Biochem. 176, 36–47.
Pidgeon, C., Stevens, J., Otto, S., Jefcoate, C. and Marcus, C. (1991) Immobilized artificial membranechromatography: rapid purification of functional membrane proteins. Anal. Biochem. 194, 163–173.
Pinkofsky, H.B. and Jung, C.H. (1985) Accessibility of sulfhydryl residues induced by cytochalasin Bbinding and conformational dynamics in the human erythrocyte glucose transporter. Arch.Biochem. Biophys. 240, 94–101.
Prechtel, K., Bausch, A.R., Marchi-Artzner, V., Kantlehner, M., Kessler, H. and Merkel R. (2002)Dynamic force spectroscopy to probe adhesion strength of living cells. Phys. Rev. Lett. 89,028101.
Rampal, A.L., Jung, E.K.Y., Chin, J.J., Deziel, M.R., Pinkofsky, H.B. and Jung, C.Y. (1986) Furthercharacterization and chemical purity assessment of the human erythrocyte glucose transporterpreparation. Biochim. Biophys. Acta 859, 135–142.
Romsicki, Y. and Sharom, J. (1999) The membrane lipid environment modulates drug interactionswith the P-glycoprotein multidrug transporter. Biochemistry 38, 6887–6896.
Seatter, M.J. and Gould, G.W. (1999) The mammalian facilitative glucose transporter (GLUT) family.Pharm. Biotechnol. 12, 201–228.
Stryer, L. (1989) Biochemistry, 3rd ed., p. 634, W.H. Freeman, New York, USA.Sultzman, L.A. and Carruthers, A. (1999) Stop-flow analysis of cooperative interactions between
GLUT1 sugar import and export sites. Biochemistry 38, 6640–6650.Vera, J.C., Rivas, C.I., Fischbarg, J. and Golde, D.W. (1993) Mammalian facilitative hexose
transporters mediate the transport of dehydroascorbic acid. Nature 364, 79–82.Wainer, I.W., Zhang, Y., Xiao, Y. and Kellar, K.J. (1999) Liquid chromatographic studies with
immobilized neuronal nicotinic acetylcholine receptor stationary phases: effects of receptorsubtypes, pH and ionic strength on drug-receptor interactions. J. Chromatogr. B 724, 65–72.
Weed, R.I. and Lacelle, P.L. (1969) ATP dependence of erythrocyte membrane deformability. In RedCell Membrane Structure & Function (Jamieson, G.A. and Greenwalt T.J., Eds.), pp. 318–338,J.B Lippincott, Philadelphia, USA.
Weinglass, A.B., Sondej, M. and Kaback, H.R. (2002) Manipulating conformational equilibria in thelactose permease of Escherichia coli. J. Mol. Biol. 315, 561–571.
Widdas, W.F. (1952) Inability of diffusion to account for placental glucose transfer in the sheep andconsideration of a possible carrier transfer. J. Physiol. 118, 23–29.
Winzor, D.J. (1985) Quantitative characterization of interactions by affinity chromatography. InAffinity Chromatography, A Practical Approach (Dean, P.D.G., Johnson, W.S. and Middle, F.A.,Eds.) pp. 149–168, IRL Press, Oxford, UK.
Winzor, D.J. and Jackson, C.M. (1993) Determination of binding constants by quantitative affinitychromatography. Current and future applications. In Handbook of Affinity Chromatography(Kline, T., Ed.), pp. 253–298, Marcel Dekker, New York, NY, USA.
Winzor, D.J. (1998) Biphasic thermodynamic characterization of interactions by quantitative affinitychromatography and biosensor technology. J. Chromatogr. A 803, 291–297.
Yang, C.Y., Cai, S.J., Liu, H. and Pidgeon, C. (1996) Immobilized artificial membranes – screens fordrug membrane interactions. Adv. Drug Del. Rev. 23, 229–256.
Yang, Q. and Lundahl, P. (1994) Steric immobilization of liposomes in chromatographic gel beads andincorporation of integral membrane proteins into their lipid bilayers. Anal. Biochem. 218, 210–221.
Yang, Q. and Lundahl, P. (1995) Immobilized proteoliposome affinity chromatography forquantitative analysis of specific interactions between solutes and membrane proteins. Interactionof cytochalasin B and D-glucose with the glucose transporter Glut1. Biochemistry 34, 7289–7294.
Yang, Q., Liu, X.-Y., Ajiki, S.-i., Hara, M., Lundahl, P. and Miyake, J. (1998) Avidin–biotinimmobilization of unilamellar liposomes in gel beads for chromatographic analysis of drug–membrane partitioning. J. Chromatogr. B 707, 131–141.

43
Yang, Q., Liu, X.-Y., Yoshimoto, M., Kuboi, R. and Miyake, J. (1999) Covalent immobilization ofunilamellar liposomes in gel beads for chromatography. Anal Biochem. 268, 354–362.
Zeng, C.-M., Zhang, Y., Lu, L., Brekkan, E., Lundqvist, A. and Lundahl, P. (1997) Immobilization ofhuman red cells in gel particles for chromatographic activity studies of the glucose transporterGLUT1. Biochim. Biophys. Acta 1325, 91–98.
Zeng, H., Parthasarathy, R., Rampal, A.L. and Jung, C.Y. (1996) Proposed structure of putativeglucose channel in GLUT1 facilitative glucose transporter. Biophys. J. 70, 14–21.
Zhang, J.-Z. and Ismail-Beigi, F. (1998) Activation of Glut1 glucose transporter in humanerythrocytes. Arch. Biochem. Biophys. 356, 86 –92.
Zhang, Y., Leonessa, F., Clarke, R. and Wainer, I.W. (2000) Development of an immobilized P-glycoprotein stationary phase for on-line chromatographic determination of drug-bindingaffinities. J. Chromatogr. B 739, 33–37.
Zuniga, F.A., Shi, G., Haller, J.F., Rubashkin, A., Flynn, D.R., Iserovich P. and Fischbarg, J. (2001) Athree-dimensional model of the human facilitative glucose transporter GLUT1, J. Biol. Chem. 276,44970–44975.
The cover picture was reprinted with modifications from Stryer (1989), p. 142, with permission fromR. G. Kessel.