69451 weinheim, germany - wiley-vch from heat and poured into a beaker of hot water (300 ml, 100 c)...
TRANSCRIPT

Supporting Information
© Wiley-VCH 2008
69451 Weinheim, Germany

S-1
Design and Formation of a Large, Tetrahedral Supramolecular
Cluster Using 1,1′-Binaphthyl Ligands
Shannon M. Biros, Robert M. Yeh and Kenneth N. Raymond*
Contribution from the Department of Chemistry, University of California,
Berkeley, California 94720-1460.
SUPPORTING INFORMATION
Experimental Section. General Experimental. Dichloromethane, tetrahydrofuran, and diethyl ether were
passed through columns of activated alumina as described by Grubbs and coworkers.1
Deuterated solvents were used as purchased from Cambridge Isotope Laboratories. All
other chemicals were used as obtained from Sigma-Aldrich or Acros Organics and used
without further purification unless otherwise noted. 1H and 13C{1H} NMR spectral data
were recorded on either a Brüker AV-300, AVQ-400, AVB-400, AV-500 or DRX-500,
as noted. Chemical shifts are expressed as parts per million (δ) relative to SiMe4 (TMS, δ
= 0), and referenced internally with respect to the protio solvent impurity. Fast atom
bombardment low resolution (FABLR), high resolution electrospray ionization
quadrupole time-of-flight (HR ESI-QTOF) mass spectra were obtained at the University
of California, Berkeley, Microanalytical Facility. Elemental analyses were performed at
the University of California, Berkeley, Microanalytical Facility on a Perkin-Elmer Series
II CHNO/S analyzer. Molecular modeling (molecular mechanics calculations) was
carried out using the MM3 forcefield as employed by the CAChe 6.1 program from
Fujitsu, Inc.

S-2
NH2
Br
1-amino-5-bromonaphthalene (4). This is a modification of a literature preparation.2
(1) Bromination. A round bottom flask was filled with 1-nitronaphthalene (25.0 g, 0.145
mol) and fresh acid-washed fine iron powder (0.30 g). The organic solid was melted and
maintained in the liquid phase by raising the temperature to 85 °C. Bromine (23 g, 0.14
mol) was added dropwise over a period of 2 h to the hot liquid 1-nitronaphthalene. After
an additional h of heating, the dark mixture was cooled to room temperature. The solid
was dissolved in ethanol at reflux, filtered hot through a plug of celite to remove residual
iron powder, and crystallized by cooling in an ice bath for several h. The isolated solid
(5-bromo-1-nitronaphthalene) was recrystallized once from hot acetone as yellow
crystalline needles and dried under a stream of air overnight (14 g, 40 % yield). m.p.
found120-122 °C. lit. 122.5 °C. 1H NMR (400 MHz, CDCl3) δ 8.61 (1H); 8.47 (1H);
8.23 (1H); 7.94 (1H); 7.67 (1H); 7.55 (1H).
(2) Reduction. A round bottom flask was filled with water (200 mL) and fresh iron
powder (8.0 g, 0.15 mol). The suspension was heated to reflux and glacial acetic acid
(0.5 mL) was carefully added. 5-bromo-1-nitronaphthalene (10 g, 0.04 mol) was added
in small portions to the vigorously stirred hot suspension. More iron powder (4.0 g) was
added and the mixture was stirred for 1h at reflux. The biphasic reaction mixture
(organic / aqueous) was cooled to room temperature, diluted with water (300 mL) and
CH2Cl2 (200 mL) and filtered through a plug of celite to remove residual iron powder.
The aqueous layer was placed in a separatory funnel and sufficient 1N NaOH was added
to raise the pH to about 9 at which point a large amount of precipitate formed. The
organic phase was added to the separatory funnel and the precipitate was extracted into
the organic layer. The organic layer was dried with sodium sulfate and evaporated to
dryness to yield a brown microcrystalline powder (3.7 g, 40 %). 1H NMR (400 MHz,
CDCl3) δ 7.81 (2H), 7.74 (1H); 7.42 (1H), 7.31 (1H), 6.85 (1H), 4.20 (2H). This
literature compound was used without further characterization.

S-3
NH
Br
O
OMe
OMe
1-bromo-5-(2',3′-dimethoxybenzamido)naphthalene (6). A Schlenk flask was charged
with 1-amino-5-bromonaphthalene (6.0 g, 27 mmol), 2,3-dimethoxybenzoic acid chloride
(6, 5.5 g, 28 mmol), NEt3 (4.3 mL, 30 mmol) and anhydrous CH2Cl2 (200 mL). The
solution was filtered after stirring for 12 h at room temperature. The filtrate was washed
with 1 M NaOH (3 x 100 mL) and 1 M HCl (3 x 100 mL). Upon drying with sodium
sulfate, the CH2Cl2 solution was evaporated to yield a crude product. The solid was
dissolved in a small volume of ethanol at reflux and allowed to cool slowly to room
temperature. The product was isolated as large, off-white crystals. A second crop of
product was isolated from a reduced volume of the ethanol. (Total yield: 9.0 g, 86 %). 1H
NMR (500 MHz, CDCl3) δ 10.65 (bs, 1H), 8.51 (d, J = 7.5Hz, 1H), 8.13 (d, J = 8.7 Hz,
1H), 8.03 (d, J = 8.4 Hz, 1H), 7.88 (dd, J = 8.1, 1.4 Hz, 1H), 7.84 (d, J = 7.3 Hz, 1H),
7.66 (dd, J = 8.7, 8.1 Hz, 1H), 7.38 (dd, J = 8.6, 7.5 Hz, 1H), 7.28 (dd, J = 8.5, 8.1 Hz,
1H), 7.16 (dd, J = 8.2, 1.4 Hz, 1H), 4.09 (s, 3H), 3,99 (s, 3H); FAB MS m/z (%) 386 (50)
[M]+; Calcd. (Anal.) for C19H16BrNO3: C, 59.08 (59.09); H, 4.18 (4.28), N; 3.63 (3.49).
NHO
OMe
OMe
HN O
MeO
MeO 5,5′-(2′′,3′′-dimethoxybenzamido)-1,1′-binaphthalene (7). A dry Schlenk flask was
charged with 1-bromo-5-(2′,3′-dimethoxybenzamido)naphthalene (4.0 g, 10 mmol), bis-

S-4
pinacolatodiborane (1.3 g, 5.0 mmol, 0.5 equivalent with respect to the aryl bromide),
PdCl2(dppf)·CH2Cl2 (0.25 g, 3 mol %), Pd(PPh3)4 (0.25 g, 2 mol %), and an excess of
anhydrous K3PO4 (3.0 g) and anhydrous K2CO3 (2.0 g) under a dinitrogen atmosphere.
Degassed DMF (100 mL) was added to the solid. Under vigorous stirring, degassed
water (2 mL) was added to DMF solution to aid the dissolution of the base. The mixture
was stirred for 2 h at 90 °C and 20 h at 80 °C. Water (500 – 800 mL) was added to the
solution after it had cooled to room temperature to precipitate the product. The large
amount of precipitate was isolated by filtration through a medium glass frit. The solid on
the frit was washed repeatedly with water to remove residual DMF and base. After air-
drying, the solid was dissolved in CH2Cl2 : ethyl acetate (50:1) and passed through a
silica gel plug (4 cm) to remove dark palladium impurities. Repeated crystallization (3x)
from hot ethanol yielded off-white microcrystalline product (1.6 g, 95 %). 1H NMR (500
MHz, CDCl3) δ 10.74 (s, 2H), 8.43 (d, J = 7.1 Hz, 2H), 8.18 (d J = 8.7 Hz, 2H), 7.92 (dd,
J = 7.9, 1.5 Hz, 2H), 7.67 (dd, J = 8.7, 6.9 Hz, 2H), 7.54 (dd, J = 6.8, 0.8 Hz, 2H), 7.35
(dd, J = 8.7, 7.8 Hz, 2H), 7.30 (dd, J = 8.5, 8.1 Hz, 2H), 7.22 (apparent d, J = 8.7 Hz,
2H), 7.19 (dd, J = 8.2, 1.4 Hz, 2H), 4.15 (s, 6H), 4.02 (s, 6H); FAB MS m/z (%) 613 (50)
[M]+; Calcd. (Anal.) for C38H32N2O6: C, 74.49 (74.20); H, 5.26 (5.37); N, 4.57 (5.68).
NHO
OH
OH
HN O
HO
HO
5,5’-(2′′,3′′-dihydroxybenzamido)-1,1′-binaphthalene (8). A round bottom flask was
charged with 5,5′-(2′′,3′′-dimethoxybenzamido)-1,1′-binaphthalene (1.6 g, 2.6 mmol),
anhydrous CH2Cl2 (150 mL), and BBr3 (7.4 mL, 74 mmol, 30 equivalents). The yellow
suspension was stirred for 5 days. Water was added very slowly to the ice cooled yellow

S-5
suspension which contained a large excess of unreacted BBr3. After vigorous bubbling
had subsided, the slurry was filtered. The solid was washed with chloroform and hot
ethyl acetate until the filtrate was colorless. The air dried solid was dissolved in hot
methanol (200-300 mL) and kept at reflux for 3 h. Hot water (5 mL) was added to the
solution and the solution was heated to reflux for an additional 8 h. The solution was
removed from heat and poured into a beaker of hot water (300 mL, 100 °C) and allowed
to cool to room temperature. The off-white precipitate was isolated by filtration and
dried under vacuum. (1.1 g, 76 %). 1H NMR (500 MHz, DMSO-d6) δ 11.94 (s, 2H),
10.94 (s, 2H), 9.51 (bs, 2H), 8.16 (d, J = 8.6 Hz, 2H), 7.83 (d, J = 7.3 Hz, 2H), 7.74 (dd, J
= 8.9, 6.9 Hz, 2H), 7.65 (dd, J = 7.9, 1.2 Hz, 2H), 7.58 (dd, J = 6.9, 0.8 Hz, 2H), 7.41 (dd,
J = 8.7, 7.5 Hz, 2H), 7.05 (dd, J = 7.6, 1.4 Hz, 2H), 6.86 (dd, 8.2, 7.9 Hz, 2H); FAB-MS:
m/z 556 [M]+; Calcd. (Anal.) for C34H24N2O6·1.5 H2O : C, 69.98 (69.55); H, 4.66 (4.33);
N, 4.80 (4.69).
Pr4N+
HN
O
O
O
NH
O
O
O
[(6Pr4N)(Pr4N+ ⊂ BINAP Ga4L6.)(5K)]. A 100 mL round bottom schlenk flask was
charged with 75 mg BINAP H4L (8, 0.135 mmol) and 300 mg Bu4NBr (1.12 mmol). The
solids were suspended in ca. 20 mL methanol, and the mixture was degassed by bubbling
through N2 gas for ~15 minutes. A solution of 1N KOH/MeOH was added slowly (540
μL, 0.540 mmol) and was again degassed for ~10 minutes at which time all of the solids
had dissolved. In one portion, 33 mg Ga(acac)3 (0.090 mmol) was added, and the
reaction was heated to 65 °C and stirred for 22 hours under an atmosphere of nitrogen.
The reaction was allowed to cool to room temperature, and the volatiles were removed
under reduced pressure. The crude product was triturated with ca. 10 mL acetone and
filtered under a stream of nitrogen using a Büchner funnel. The resulting solid was
S-6
further washed with acetone (2 x 10 mL) to give 140 mg of product as an olive green
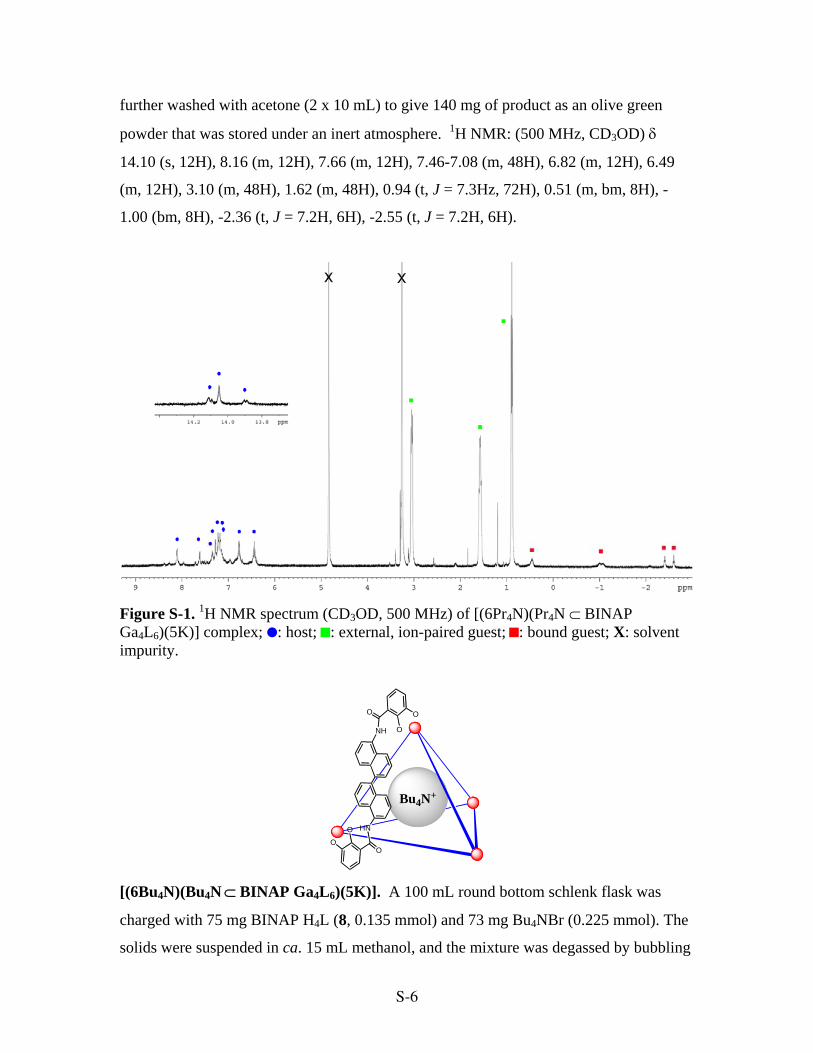
powder that was stored under an inert atmosphere. 1H NMR: (500 MHz, CD3OD) δ
14.10 (s, 12H), 8.16 (m, 12H), 7.66 (m, 12H), 7.46-7.08 (m, 48H), 6.82 (m, 12H), 6.49
(m, 12H), 3.10 (m, 48H), 1.62 (m, 48H), 0.94 (t, J = 7.3Hz, 72H), 0.51 (m, bm, 8H), -
1.00 (bm, 8H), -2.36 (t, J = 7.2H, 6H), -2.55 (t, J = 7.2H, 6H).
Figure S-1. 1H NMR spectrum (CD3OD, 500 MHz) of [(6Pr4N)(Pr4N ⊂ BINAP Ga4L6)(5K)] complex; : host; : external, ion-paired guest; : bound guest; X: solvent impurity.
Bu4N+
HN
O
O
O
NH
O
O
O
[(6Bu4N)(Bu4N ⊂ BINAP Ga4L6)(5K)]. A 100 mL round bottom schlenk flask was
charged with 75 mg BINAP H4L (8, 0.135 mmol) and 73 mg Bu4NBr (0.225 mmol). The
solids were suspended in ca. 15 mL methanol, and the mixture was degassed by bubbling
S-7
through N2 gas for ~15 minutes. A solution of 1N KOH/MeOH was added slowly (540
μL, 0.540 mmol) and was again degassed for ~10 minutes at which time all of the solids
had dissolved. In one portion, 33 mg Ga(acac)3 (0.090 mmol) was added, and the
reaction was heated to 65 °C and stirred for 22 hours under an atmosphere of nitrogen.
The reaction was allowed to cool to room temperature, and the volatiles were removed
under reduced pressure. The crude product was triturated with ca. 10 mL acetone and
filtered under a stream of nitrogen using a Büchner funnel. The resulting solid was
further washed with acetone (2 x 10 mL) to give 100 mg of product as an olive green
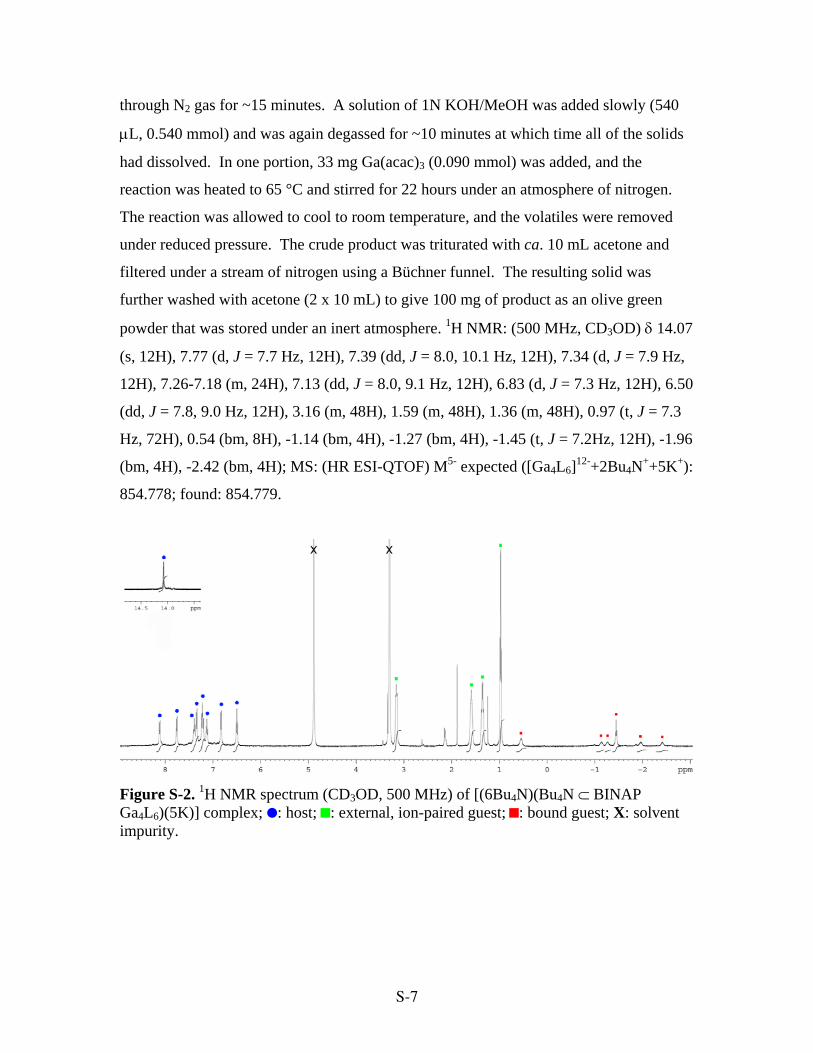
powder that was stored under an inert atmosphere. 1H NMR: (500 MHz, CD3OD) δ 14.07
(s, 12H), 7.77 (d, J = 7.7 Hz, 12H), 7.39 (dd, J = 8.0, 10.1 Hz, 12H), 7.34 (d, J = 7.9 Hz,
12H), 7.26-7.18 (m, 24H), 7.13 (dd, J = 8.0, 9.1 Hz, 12H), 6.83 (d, J = 7.3 Hz, 12H), 6.50
(dd, J = 7.8, 9.0 Hz, 12H), 3.16 (m, 48H), 1.59 (m, 48H), 1.36 (m, 48H), 0.97 (t, J = 7.3
Hz, 72H), 0.54 (bm, 8H), -1.14 (bm, 4H), -1.27 (bm, 4H), -1.45 (t, J = 7.2Hz, 12H), -1.96
(bm, 4H), -2.42 (bm, 4H); MS: (HR ESI-QTOF) M5- expected ([Ga4L6]12-+2Bu4N++5K+):
854.778; found: 854.779.
Figure S-2. 1H NMR spectrum (CD3OD, 500 MHz) of [(6Bu4N)(Bu4N ⊂ BINAP Ga4L6)(5K)] complex; : host; : external, ion-paired guest; : bound guest; X: solvent impurity.

S-8
Figure S-3. HR-ESI-QTOF mass spectrum of Bu4N ⊂ BINAP Ga4L6 complex: the observed spectrum is shown on the bottom along with the predicted isotope patterns for the host-guest complex in the 5- charge state.
Pentyl4N+
HN
O
O
O
NH
O
O
O
[(6n-Pentyl4N)(n-Pentyl4N ⊂ BINAP Ga4L6)(5K)]. A 100 mL round bottom schlenk
flask was charged with 75 mg BINAP H4L (8, 0.135 mmol) and 85 mg n-pentyl4NBr
(0.225 mmol). The solids were suspended in ca. 25 mL methanol, and the mixture was
degassed by bubbling through N2 gas for ~15 minutes. A solution of 1N KOH/MeOH
was added slowly (540 μL, 0.540 mmol) and was again degassed for ~10 minutes at
which time all of the solids had dissolved. In one portion, 33 mg Ga(acac)3 (0.090 mmol)
was added, and the reaction was heated to 65 °C and stirred for 22 hours under an

S-9
atmosphere of nitrogen. The reaction was allowed to cool to room temperature, and the
volatiles were removed under reduced pressure. The crude product was triturated with
ca. 10 mL acetone and filtered under a stream of nitrogen using a Büchner funnel. The
resulting solid was further washed with acetone (2 x 10 mL) to give 126 mg of product as
an olive green powder that was stored under an inert atmosphere. 1H NMR: (500 MHz,
CD3OD) δ 13.93 (s, 12H), 7.98 (d, J = 8.7 Hz, 12H), 7.78 (d, 7.1Hz, 12H), 7.30 (m,
12H), 7.18 (m, 12H), 7.02 (dd, J = 7.7, 8.4 Hz, 12 H), 6.78 (m, 12H), 6.45 (dd, 7.8,
7.9Hz, 12H), 3.10 (bm, 48H), 1.55 (bm, 48H), 1.33 (bm, 48H), 1.24 (bm, 48H), 0.86 (t, J
= 6.9H, 72H), 0.48 (bm, 4H), 0.30 (bm, 4H), 0.01 (t, J = 6.9 Hz, 12H), -0.74 (bm, 4H), -
0.85 (bm, 4H), -1.61 (bm, 4H), -2.30 (bm, 8H), -2.42 (bm, 4H); MS: (ESI-QTOF) M5-
expected ([Ga4L6]12-+2n-pentyl4N++5K+): 877.203; found: 877.232.
Figure S-4. 1H NMR spectrum (CD3OD, 500 MHz) of [(6 n-pentyl4N)(n-pentyl4N ⊂ BINAP Ga4L6)(6K+)] complex; : host; : external, ion-paired guest; : bound guest; X: solvent impurity.

S-10
Figure S-5. HR-ESI-QTOF mass spectrum of n-Pentyl4N⊂BINAP Ga4L6 complex: the observed spectrum is shown on the bottom along with the predicted isotope patterns for the host-guest complex in the 5- charge state.
HN
O
O
O
NH
O
O
O
Ph4P+
[(12Ph4P)(Ph4P ⊂ BINAP Ga4L6)]. A 100 mL round bottom schlenk flask was charged
with 100 mg BINAP H4L (8, 0.177 mmol), 44 mg Ga(acac)3 (0.12 mmol) and 332 mg
Ph4PCl (0.885 mmol, 5 eq.). The solids were suspended in ca. 20 mL methanol, and the
mixture was degassed by bubbling through N2 gas for ~15 minutes. A solution of 0.5N
KOH/MeOH was added slowly (1.4 mL, 0.70 mmol, 4eq./H4L) and was again degassed
for ~10 minutes. The reaction flask was equipped with a reflux condenser, heated to 65
°C and stirred for 22 hours under an atmosphere of nitrogen. The reaction was allowed to
cool to room temperature, and the volatiles were removed under reduced pressure. The
crude product was triturated with ca. 20 mL acetone and filtered under a stream of
nitrogen using a Büchner funnel. The resulting yellow solid was further washed with

S-11
acetone (2 x 10 mL) to give 240 mg of product as an orange-yellow powder that was
stored under an inert atmosphere. 1H NMR: (CD3OD, 500 MHz) δ 14.07 (bs, 12H), 8.09
(d, J = 8.5 Hz, 12H), 7.86-7.75 (m, 48H), 7.69-7.58 (m, 108H), 7.58-7.48 (m, 96H), 7.30
(dd, J = 8.1, 1.4 Hz, 12H), 7.01 (m, 24H), 6.88 (d, J = 7.2 Hz, 12H), 6.85 (d, J = 8.5 Hz,
12H), 6.79 (dd, J = 7.3, 1.4 Hz, 12H), 6.43 (dd, J = 9.2, 7.9 Hz, 12H), 5.77 (t, J = 8.1 Hz,
4H), 4.79 (m, 8H, solvent obscured), 4.05 (m, 8H); MS: (HR ESI-QTOF) M5- expected
([Ga4L6]12-+2Ph4P++4K++1H+): 885.926; found: 885.922.
Figure S-6. 1H NMR spectrum (CD3OD, 500 MHz) of [(12Ph4P+)(Ph4P ⊂ BINAP Ga4L6)] complex; : host; : external, ion-paired guest; : bound guest; X: solvent impurity.

S-12
Figure S-7. 1H NMR spectrum (DMSO-d6, 500 MHz) of [(12Ph4P)(Ph4P ⊂ BINAP Ga4L6)] complex; : host; : external, ion-paired guest; : bound guest; X: solvent impurity.
Figure S-8. HR-ESI-QTOF mass spectrum of Ph4P ⊂ BINAP Ga4L6 complex: the observed spectrum is shown on the bottom along with the predicted isotope patterns for the host-guest complex in the 5- charge state.

S-13
HN
O
O
O
NH
O
O
O
Ph3PrP+
Ph3PrP+⊂BINAP Ga4L6. This complex was generated and characterized in-situ. An
NMR tube was charged with 5 mg ligand 8 (0.009 mmol, 6 eq.) and 2.4 mg Ph3BuPBr
(0.006 mmol, 4 eq.). The solids were suspended in 700 μL CD3OD, and 72 μL 0.5N
KOH/MeOH (0.036 mmol, 24 eq.) was added by syringe. The tube was shaken, and
Ga(acac)3 (300 μL of 22 mg/3.0 mL CD3OD; 0.006 mmol, 2.2 mg, 4 eq.) was added.
The NMR tube was immediately flame sealed under static vacuum and the tube heated to
65 °C overnight in an oil bath. After cooling to room temperature, the following spectra
were obtained: MS: (HR ESI-QTOF) M5- expected ([Ga4L6]12-+Ph3PrP++6K+): 828.10;
found: 827.90.

S-14
Figure S-9. 1H NMR spectrum (DMSO-d6, 500 MHz) of [(Ph3PrP ⊂ BINAP Ga4L6)(11K)] complex; : host; : free guest; : bound guest; X: solvent impurity.
Figure S-10. HR-ESI-QTOF mass spectrum of [Ph3PrP ⊂ BINAP Ga4L6] complex: the observed spectrum is shown on the bottom along with the predicted isotope patterns for the host-guest complex in the 5- charge state. The peak to the immediate left of the identified peak results from H+/K+ exchange in the gas phase. (This sample was exposed to CD3OD, so there is some H/D exchange at the amide functions. The exact molecular formula is listed in the figure.)

S-15
HN
O
O
O
NH
O
O
O
Ph3BuP+
Ph3BuP+⊂BINAP Ga4L6. This complex was generated and characterized in-situ. An
NMR tube was charged with 5 mg ligand 8 (0.009 mmol, 6 eq.) and 2.3 mg Ph3BuPBr
(0.006 mmol, 4 eq.). The solids were suspended in 700 μL CD3OD, and 72 μL 0.5N
KOH/MeOH (0.036 mmol, 24 eq.) was added by syringe. The tube was shaken, and
Ga(acac)3 (300 μL of 22 mg/3.0 mL CD3OD; 0.006 mmol, 2.2 mg, 4 eq.) was added.
The NMR tube was immediately flame sealed under static vacuum and the tube was
heated to 65 °C overnight in an oil bath. After cooling to room temperature, the
following spectra were obtained: MS: (HR ESI-QTOF) M5- expected ([Ga4L6]12-
+2Ph3BuP++5K+): 867.14; found: 867.15.
Figure S-11. 1H NMR spectrum (DMSO-d6, 500 MHz) of [(Ph3BuP ⊂ BINAP Ga4L6)(11K)] complex; : host; : free guest; : bound guest; X: solvent impurity.

S-16
Figure S-12. HR-ESI-QTOF mass spectrum of [Ph3BuP⊂BINAP Ga4L6]complex: the observed spectrum is shown on the bottom along with the predicted isotope patterns for the host-guest complex in the 5- charge state. The peak to the immediate left of the identified peak results from H+/K+ exchange in the gas phase. (This sample was exposed to CD3OD, so there is some H/D exchange at the amide functions. The exact molecular formula is listed in the figure.)
Literature cited.
(1) Pangborn, A. B.; Giardello, M. A.; Grubbs, R. H.; Rosen, R. K.; Timmers, F. J. Organometallics 1996, 15, 1518-1520.
(2) Klemm, L. H.; Sprague, J. W.; Mak, E. Y. K. Journal of Organic Chemistry 1957, 22, 161-166.