chaptershodhganga.inflibnet.ac.in/bitstream/10603/4295/16/16_chapter 7.pdf · chapter – vii ....
TRANSCRIPT


CHAPTER – VII
FTIR, FT−RAMAN, NBO AND QUANTUM CHEMICAL
INVESTIGATIONS OF 4,5−DIMETHYL−1,3−DIOXOL−2−ONE
7.1. Introduction
Harmonic force fields of polyatomic molecules play a vital role in the
interpretation of vibrational spectra and in the prediction of other vibrational
properties. The cyclic carbonates and their derivatives have been widely used as
starting materials in a vast amount of chemicals, pharmaceuticals, dyes,
electro−optical and many other industrial processes [1−4]. The understanding of their
structure, molecular properties as well as nature of reaction mechanism they undergo
has great importance and has been the subject of many experimental and theoretical
studies. The 1,3−dioxolan−2−one is a conformationally interesting molecule because
there is a balance between the π−bonding favoring a planar ring conformation and the
H∙∙∙∙H non−bonded repulsions which favor a twist conformation. The crystal structure
of 1,3−dioxolan−2−one showed that the ring was non−planar and that the molecules
had crystallographic C2 symmetry. X−ray studies of 1,3−dioxolan−2−one indicate that
the ring is bent, the two ethylene carbon atoms forming an angle of 20° with the plane
in which the carbonate group is located [5−7]. This non−planarity has been confirmed
by microwave studies. 4,5−dimethyl−1,3−dioxol−2−one is used for the synthesis of
4−chloro−4−methyl−5−methylene−1,3−dioxolane−2−one which is useful as an
intermediate for the synthesis of 4−chloromethyl−5−methyl−1,3−dioxolene−2−one
which, in turn, finds use as a modifying agent for making various prodrugs [8].
Due to their high energy density, high voltage, and long cycle life, lithium−ion
batteries have been a popular power source for advanced portable electronics [9]. A
typical lithium−ion battery consists of a graphite anode, a transition metal oxide (such
as LiMn2O4, LiCoO2, LiNiO2) cathode and a non−aqueous organic electrolyte, which
acts as an ion conductor between electrodes and separates the two electrode materials.
The electrolytes used in commercial lithium−ion batteries are prepared by dissolving
LiPF6 into binary or ternary organic carbonates, including dimethyl carbonate (DMC),
ethylmethyl carbonate (EMC) and diethyl carbonate (DEC), as well as ethylene
carbonate (EC) [9,10]. During the initial cycles, a solid electrolyte interface (SEI) is

239
formed on the surface of the graphite anode which consists of reductive
decomposition products of the electrolyte [9,11–13]. Many studies have focused on
improving the properties of the SEI since the structure of the SEI plays an important
role in cycle life, power capability, and safety of the battery [14−17]. Commercial
cells with LiPF6−EC based electrolyte have several problems, including loss of power
and capacity upon storage or prolonged use, especially at elevated temperature and
poor lower−temperature performance due to the high melt point (36oC) of EC. These
problems limit the use of lithium−ion batteries for many large power applications,
such as hybrid electric vehicles or plug−in hybrid electric vehicles (PHEVs). Due to
the importance of SEI formation, many electrolyte additives have been investigated
with the goal of controlling the structure of the electrode/electrolyte interface via
surface modification. The additives usually have a higher reduction potential than the
solvents and are preferably reduced on the graphite electrode forming insoluble solid
products, which subsequently cover the surface of graphite and minimize further
reduction of solvent.
Various electrolyte additives have been developed to assist the formation of a
stable SEI on the graphite anode and improve the performance of lithium−ion
batteries, including vinylene carbonate (VC) [17–18], ethylene sulfite (ES) [19],
1,4−butane sultone (BS) [20], 1,3−propane sultone (PS) [21,22], and boron−based
compounds [22–25]. Among these additives, VC is one of the most widely
investigated additives since it can form a more stable SEI film on the surface of the
graphite anode than the electrolyte solvents. However, VC is not a stable compound
because it tends to polymerize, which may restrict its application [26,27].
A novel VC−derivative, 4,5−dimethyl−[1,3]dioxol−2−one (DMDO), as
electrolyte additive for lithium−ion batteries. DMDO is supposed to be more stable
than VC due to the steric hindrance of the methyl substituents resulting in decreased
the reactivity of the double bond. It is shown that the addition of 2% DMDO to LiPF6
in EC/DMC/DEC can significantly improve the cyclic performance of
LiNi0.8Co0.2O2/graphite cell [28]. DMDO is supposed to be more stable than VC due
to the steric hindrance of the methyl substituents resulting in decreased the reactivity
of the double bond. N−(5−methyl−2−oxo−1, 3−dioxol−4−yl)methyl derivatives of
imidacloprid and 1−chlorothiazolylmethyl−2−nitroimino−imidazolidine were used as
proinsecticide [29].

240
The quantum chemical ab initio, DFT and normal coordinate analysis give
information regarding the nature of structure, the functional groups, and orbital
interactions and mixing of skeletal frequencies. The introduction of one or more
substituents in the five membered 1,3−dioxol−2−one ring leads to the variation of
charge distribution in the molecule, and consequently, this greatly affects the
structural, electronic and vibrational parameters [30,31]. The structural and
vibrational characteristics of the compounds under investigation,
4,5−dimethyl−1,3−dioxol−2−one have not been analysed in detail. Thus, considering
the industrial and biological importance of these compounds, an extensive
experimental and theoretical ab initio and DFT studies were carried out to obtain a
complete reliable and precise vibrational assignments and structural characteristics of
the compounds. The DFT calculations with the hybrid exchange−correlation
functional B3LYP (Becke’s three parameter (B3) exchange in conjunction with the
Lee−Yang−Parr’s (LYP) correlation functional) which are especially important in
systems containing extensive electron conjugation and/or electron lone pairs [32].
7.2. Experimental
The compound 4,5−dimethyl−1,3−dioxol−2−one were obtained from
Shanghai Boyle Chemicals Co., Ltd, China and used as such to record FTIR and
FT−Raman spectra. The FTIR spectrum has been recorded in the region between
4000 and 400 cm−1 using Bruker IFS 66V spectrometer equipped with a Globar
source, Ge/KBr beam splitter, and TGS detector. The frequencies for all sharp bands
are accurate to 2 cm−1. The FT−Raman spectrum was also recorded in the range
between 4000 cm−1 and 100 cm−1 by the same instrument with FRA 106 Raman
module equipped with Nd:YAG laser source with 200 mW powers operating at 1.064
m and the spectral resolution is 2 cm−1. A liquid nitrogen cooled−Ge detector was
used.
7.3. Computational details
The combination of vibrational spectroscopy with quantum chemical
calculations is effective for understanding the fundamental mode of vibrations of the
compounds. The structural characteristics, stability, thermodynamic properties and
energy of the compounds under investigation are determined by LCAO−MO−SCF
restricted Hartree−Fock (HF) and the gradient corrected density functional theory

241
(DFT) [33] with the three−parameter hybrid functional (B3) [34] for the exchange
part and the Lee−Yang−Parr (LYP) [35] correlation functional, using 6−31G(d,p),
6−311++G(d,p) and cc−pVTZ basis sets with Gaussian−03 program package [36],
invoking gradient geometry optimisation on Intel core i5/3.03 GHz processor. The
energy minima with respect to the nuclear coordinates were obtained and the initial
geometry generated from standard geometrical parameters was minimised without
any constraint, adopting the 6−31G(d,p), 6−311++G(d,p) and cc−pVTZ basis sets.
The optimised structural parameters were used in the vibrational frequency
calculations resulting in IR and Raman frequencies together with intensities and
Raman depolarization ratios, thermodynamic properties and energies of the optimised
structures. The force constants obtained from the ab initio basis sets have been
utilised in the normal coordinate analysis by Wilson’s FG matrix method [37−39] and
the potential energy distribution corresponding to each of the observed frequencies
were calculated with the program of Fuhrer et al. [40].
7.4. Results and discussion
7.4.1. Molecular Geometry
The structure and atom numbering of 4,5−dimethyl−1,3−dioxol−2−one is
shown in Figure 7.1. The 4,5−dichloro−l,3−dioxolan−2−one molecule possess C2V
point group symmetry. The planar 4,5−dimethyl−1,3−dioxol−2−one molecule with
C2V symmetry has 36 normal modes of vibration, composed of 12 of A1 symmetry
(transforming as x2, y
2, z
2), 9 of A2 symmetry (transforming as xy), 7 of B1 symmetry
(transforming as xz) and 8 of B2 symmetry (transforming as yz), choosing the x axis to
be perpendicular to the molecular plane. Therefore, in the C2V point group, all of the
fundamental vibrations are Raman active and all but the A2 vibrations are infrared
active. The vibrations of the A1 and B2 species are the in−plane modes and those of
A2 and B1 species are out of plane vibrations. Thus, all the 36 fundamental modes of
vibrations of the compound are distributed into the irreducible representations under
C2v point group as vib = 12A1 + 9A2 + 7B1 + 8B2. All the frequencies are assigned in
terms of fundamental, overtone and combination bands.

242
Figure 7.1. Optimised structure of 4,5−dimethyl−1,3−dioxol−2−one
7.4.2. Structural properties
The optimised structural parameters bond lengths, bond angles and dihedral
angles for the thermodynamically preferred geometry of
4,5−dimethyl−1,3−dioxol−2−one determined at B3LYP level with 6−31G(d,p),
6−311++G(d,p) and cc−pVTZ basis sets are presented in Table 7.1. From the
structural data, it is observed that the influence of the substituent on the molecular
parameters, particularly in the C–C bond distance of ring carbon atoms seems to be
negligibly small. The O1–C2 and O3–C2 bond distances of the ring of
4,5−dimethyl−1,3−dioxol−2−one are same. The other C4–O3 and C5–O1 bond
distances of are also same. The shorter C2–O6 bond distance 1.19 Å is due to the
double bond. All the structural parameters determined from this study are well agreed
with the experimental values of twist conformer of 1,3−dioxolan−2−one [5,41]. But

243
shorter C4−C5 bond distance (1.33 Å) of 4,5−dimethyl−1,3−dioxol−2−one than that
of l,3−dioxolan−2−one (1.54) indicates the presence of localised double bond in
4,5−dimethyl−1,3−dioxol−2−one. The study also confirms that the compound under
investigation is present in the form of planar structure and is the most stable. The ideal
planarity around the carbonate grouping may not lost in the sterically crowded
substituted ring systems as in the case of 4,5−dimethyl−1,3−dioxol−2−one. The
planarity of the compounds under investigation have been confirmed by present DFT
studies from the ring dihedral angle O1−C5−C4−O3 of 0o and the other dihedral
angles are also 0o and 180o. The twisting is not possible due to the presence of
C4=C5 double bond. But in the case of 4−methyl−1,3−dioxolan−2−one and
4,5−dichloro−1,3−dioxolan−2−one the non−planar twist conformer is more stable
[42]. In these cases the twisting around C4−C5 single bond is possible. The
comparison of the dihedral angles of these compounds determined by
B3LYP/6−311++G(d,p) method with that of the dihedral angles of
4,5−dimethyl−1,3−dioxol−2−one clearly confirm the planarity of the molecule under
investigation.
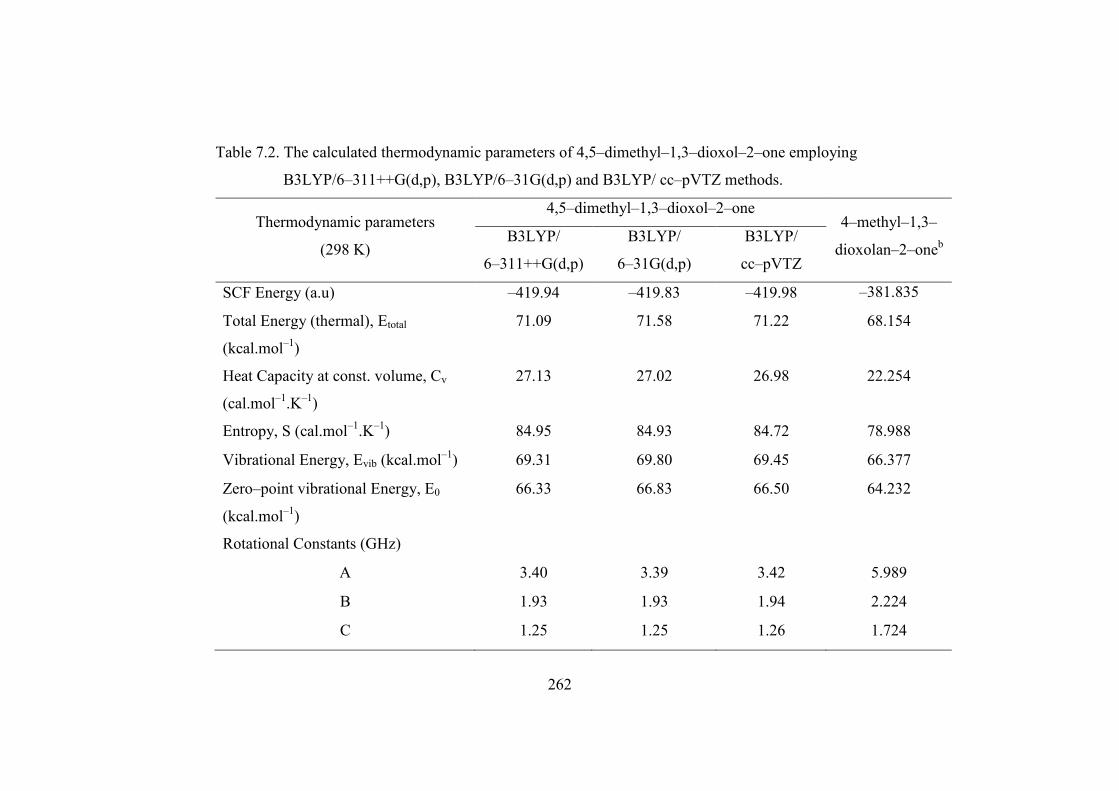
The thermodynamic parameters namely heat capacity, entropy, rotational
constants, dipole moments, vibrational and vibrational zero point energies of the
compounds have also been computed at DFT−B3LYP levels using6−31G(d,p),
6−311++G(d,p) and cc−pVTZ basis sets and are presented in Table 7.2. The energy
of 4,5−dimethyl−1,3−dioxol−2−one is more (−419.94 a.u) than that of
4−methyl−1,3−dioxolan−2−one it is equal to –381.835 a.u. This reveals that
4,5−dimethyl−1,3−dioxol−2−one is more stable than 4−methyl−1,3−dioxolan−2−one
and is due to the planarity of 4,5−dimethyl−1,3−dioxol−2−one molecule. The
thermodynamic data provides helpful information for the further study on the title
compounds, when these may be used as a reactant to take part in a new reaction.
These standard thermodynamic functions can be used as reference thermodynamic
values to calculate changes of entropies (ΔST), changes of enthalpies (ΔHT) and
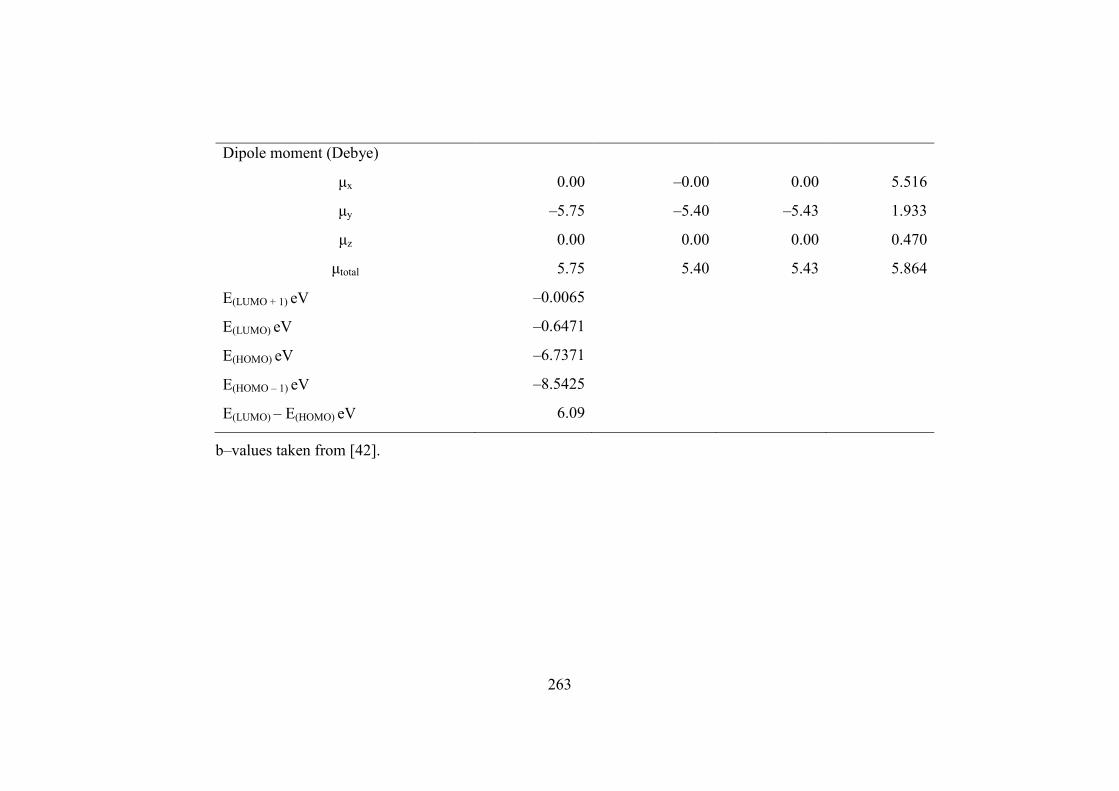
changes of Gibbs free energies (ΔGT) of the reaction. The dipole moment and its
principal inertial axes are strongly depend upon the conformation of the molecule.
The 4−methyl−1,3−dioxolan−2−one has higher dipole moment (5.864 debye) than
that of 4,5−dimethyl−1,3−dioxol−2−one (5.75 debye). The zero dipole moment in the
z and x axis is due to the C2V point group symmetry of the molecule.
244
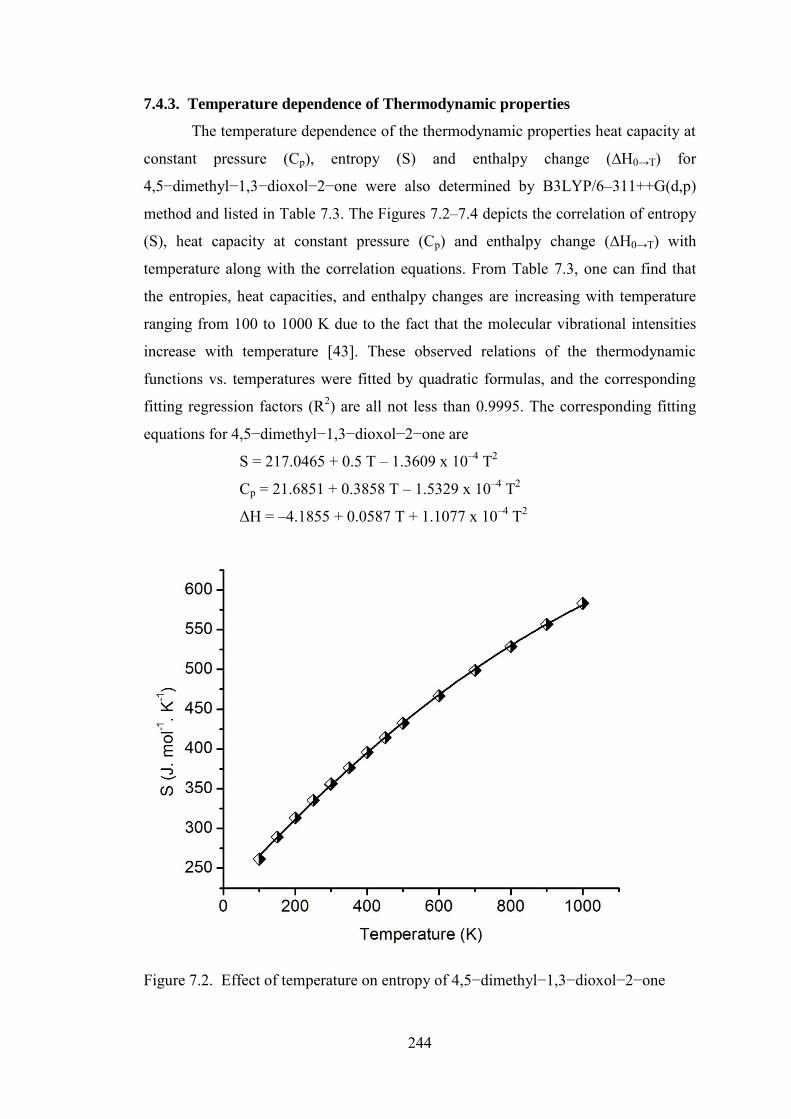
7.4.3. Temperature dependence of Thermodynamic properties
The temperature dependence of the thermodynamic properties heat capacity at
constant pressure (Cp), entropy (S) and enthalpy change (∆H0→T) for
4,5−dimethyl−1,3−dioxol−2−one were also determined by B3LYP/6–311++G(d,p)
method and listed in Table 7.3. The Figures 7.2–7.4 depicts the correlation of entropy
(S), heat capacity at constant pressure (Cp) and enthalpy change (∆H0→T) with
temperature along with the correlation equations. From Table 7.3, one can find that
the entropies, heat capacities, and enthalpy changes are increasing with temperature
ranging from 100 to 1000 K due to the fact that the molecular vibrational intensities
increase with temperature [43]. These observed relations of the thermodynamic
functions vs. temperatures were fitted by quadratic formulas, and the corresponding
fitting regression factors (R2) are all not less than 0.9995. The corresponding fitting
equations for 4,5−dimethyl−1,3−dioxol−2−one are
S = 217.0465 + 0.5 T – 1.3609 x 10–4 T2
Cp = 21.6851 + 0.3858 T – 1.5329 x 10–4 T2
ΔH = –4.1855 + 0.0587 T + 1.1077 x 10–4 T2
Figure 7.2. Effect of temperature on entropy of 4,5−dimethyl−1,3−dioxol−2−one

245
Figure 7.3. Effect of temperature on heat capacity at constant pressure of
4,5−dimethyl−1,3−dioxol−2−one
Figure 7.4. Effect of temperature on enthalpy change (∆H0→T) of
4,5−dimethyl−1,3−dioxol−2−one

246
7.4.4. Analysis of Molecular electrostatic potential (MESP)
The molecular electrostatic potential surface (MESP) which is a method of
mapping electrostatic potential onto the iso–electron density surface simultaneously
displays electrostatic potential (electron + nuclei) distribution, molecular shape, size
and dipole moments of the molecule and it provides a visual method to understand
the relative polarity. Electrostatic potential maps illustrate the charge distributions of
molecules three dimensionally. These maps allow us to visualize variably charged
regions of a molecule. Knowledge of the charge distributions can be used to
determine how molecules interact with one another. One of the purposes of finding
the electrostatic potential is to find the reactive site of a molecule. In the electrostatic
potential map, the semi–spherical blue shapes that emerge from the edges of the
above electrostatic potential map are hydrogen atoms. The molecular electrostatic
potential (MEP) at a point r in the space around a molecule (in atomic units) can be
expressed as:
rr
drr
rR
ZrV
AA
A
'
')'()(
where, ZA is the charge on nucleus A, located at RA and ρ(r′) is the electronic density
function for the molecule. The first and second terms represent the contributions to
the potential due to nuclei and electrons, respectively. V(r) is the resultant at each
point r, which is the net electrostatic effect produced at the point r by both the
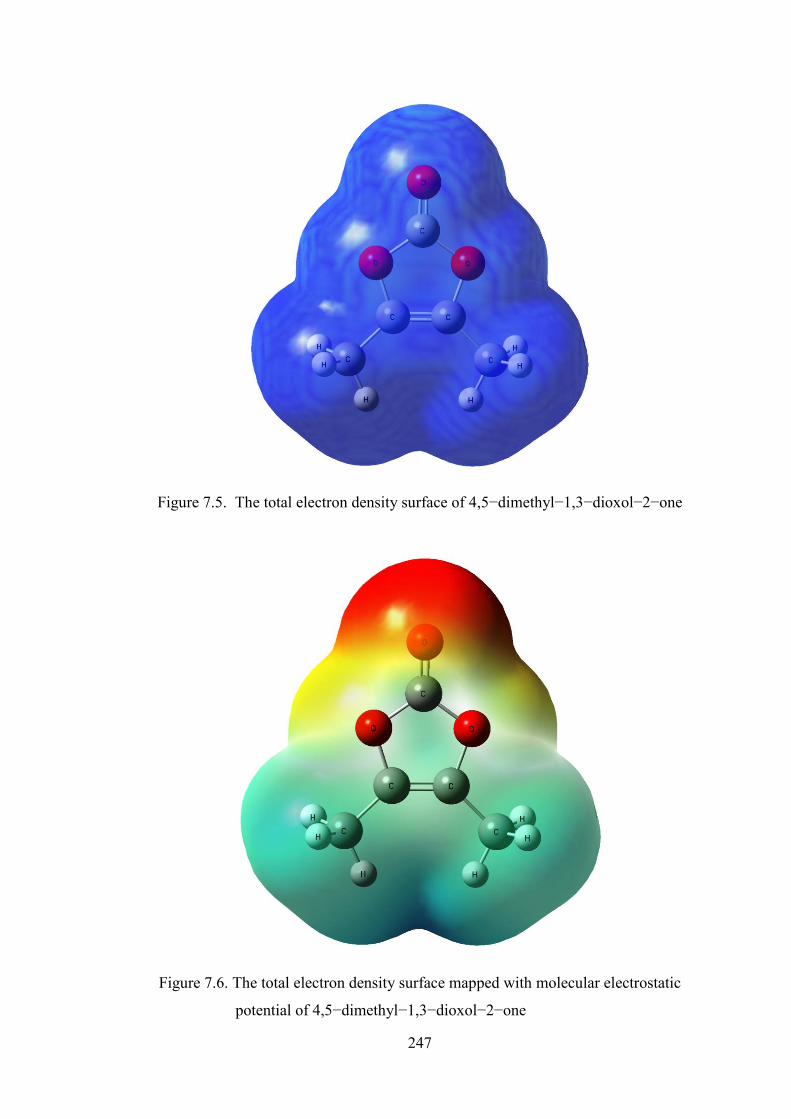
electrons and nuclei of the molecule. The total electron density and MESP surfaces of
the molecules under investigation are constructed by using B3LYP/6–311++G(d,p)
method. These pictures illustrate an electrostatic potential model of the compounds,
computed at the 0.002 a.u isodensity surface. The total electron density surface of
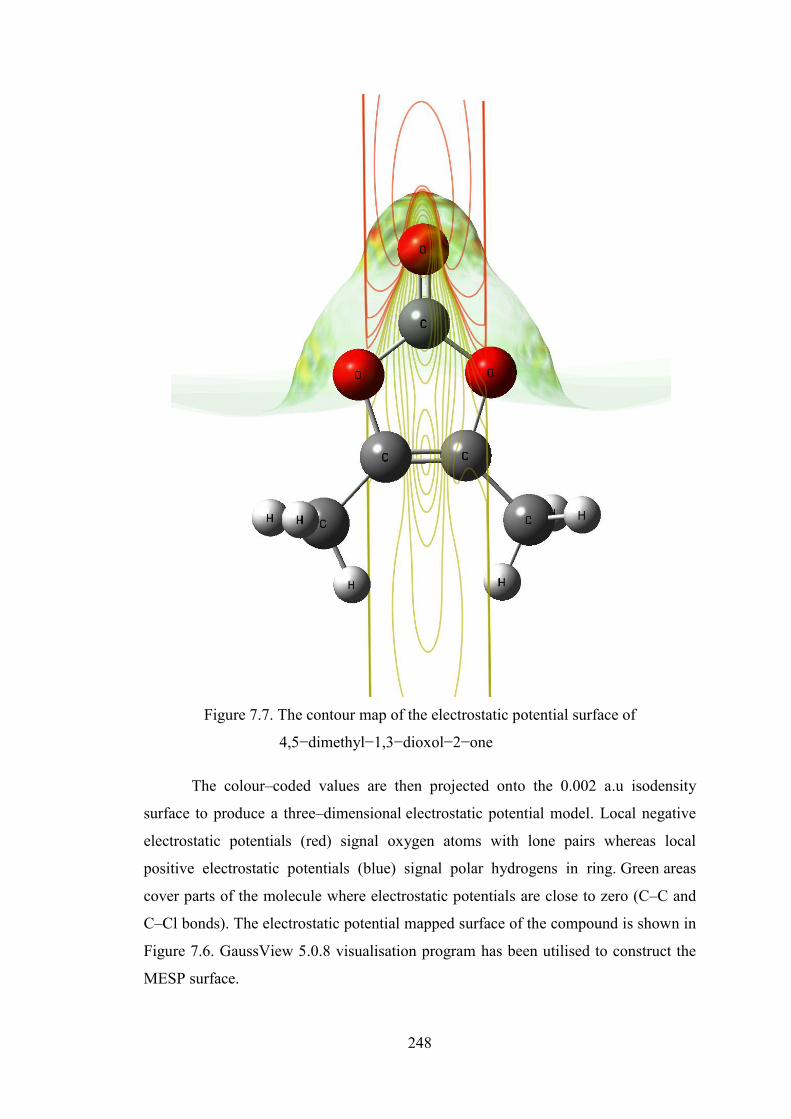
4,5−dimethyl−1,3−dioxol−2−one is depicted in Figure 7.5 while the MESP mapped
surface of the compound and electrostatic potential contour map for positive and
negative potentials are shown in Figures 7.6 and 7.7. The colour scheme of MESP is
the negative electrostatic potentials are shown in red, the intensity of which is
proportional to the absolute value of the potential energy, and positive electrostatic
potentials are shown in blue while Green indicates surface areas where the potentials
are close to zero. The Figure 7.8 shows the molecular electrostatic potential surface of
4,5−dimethyl−1,3−dioxol−2−one.
247
Figure 7.5. The total electron density surface of 4,5−dimethyl−1,3−dioxol−2−one
Figure 7.6. The total electron density surface mapped with molecular electrostatic
potential of 4,5−dimethyl−1,3−dioxol−2−one
248
Figure 7.7. The contour map of the electrostatic potential surface of
4,5−dimethyl−1,3−dioxol−2−one
The colour–coded values are then projected onto the 0.002 a.u isodensity
surface to produce a three–dimensional electrostatic potential model. Local negative
electrostatic potentials (red) signal oxygen atoms with lone pairs whereas local
positive electrostatic potentials (blue) signal polar hydrogens in ring. Green areas
cover parts of the molecule where electrostatic potentials are close to zero (C–C and
C–Cl bonds). The electrostatic potential mapped surface of the compound is shown in
Figure 7.6. GaussView 5.0.8 visualisation program has been utilised to construct the
MESP surface.

249
Figure 7.8. The molecular electrostatic potential surface of
4,5−dimethyl−1,3−dioxol−2−one
7.4.5. Natural bond orbital analysis
Natural bond orbital (NBO) methods encompass a suite of algorithms that
enable fundamental bonding concepts to be extracted from Hartree‐Fock (HF),
Density Functional Theory (DFT), and post‐HF computations. NBO analysis
originated as a technique for studying hybridisation and covalency effects in
polyatomic wave functions, based on local block eigen vectors of the one–particle
density matrix. NBOs would correspond closely to the picture of localised bonds and
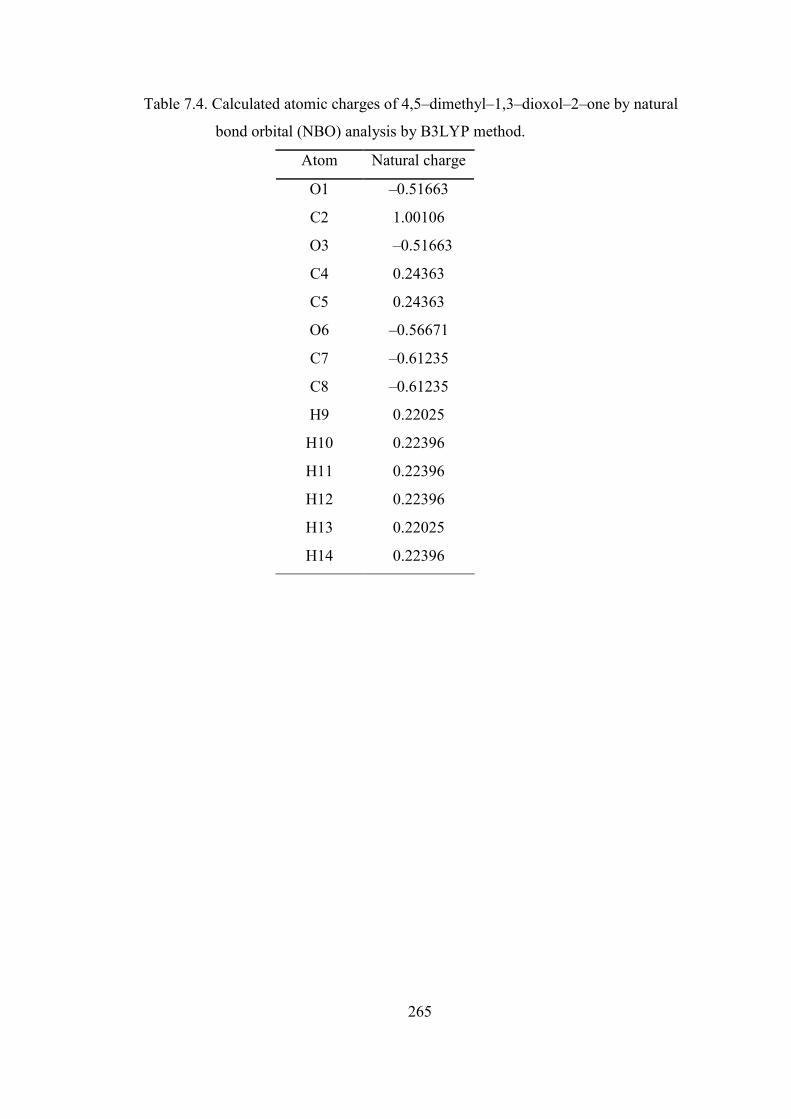
lone pairs as basic units of molecular structure. The atomic charges of
4,5−dimethyl−1,3−dioxol−2−one calculated by NBO analysis using the
B3LYP/6–311++G(d,p) method are presented in Table 7.4. Among the ring oxygen
atoms O1 and O3 have same negative charges. But O6 has more negative charge due
to the resonance of carbonyl group. The charges of C4 and C5 are also same. The
methyl carbon atoms C7 and C8 have negative charge due to hyperconjugative effect
of methyl groups.
In Table 7.5, the natural atomic orbitals, their occupancies and the
corresponding energy of 4,5−dimethyl−1,3−dioxol−2−one were described. In a given
molecular environment the natural atomic orbitals reflect the chemical give and take
of electronic interactions, with variations of shape (e.g., angular deformations due to
steric pressures of adjacent atoms) and size (e.g., altered diffuseness due to increased

250
anionic or cationic character) that distinguish them appreciably from free atom forms.
The NAOs orbital energies εi(A) are calculated by using Kohn–Sham operator (F) as
)*()()( A
i
A
i
A
i F
The NAOs deals the molecular properties in terms of inter atomic and intra
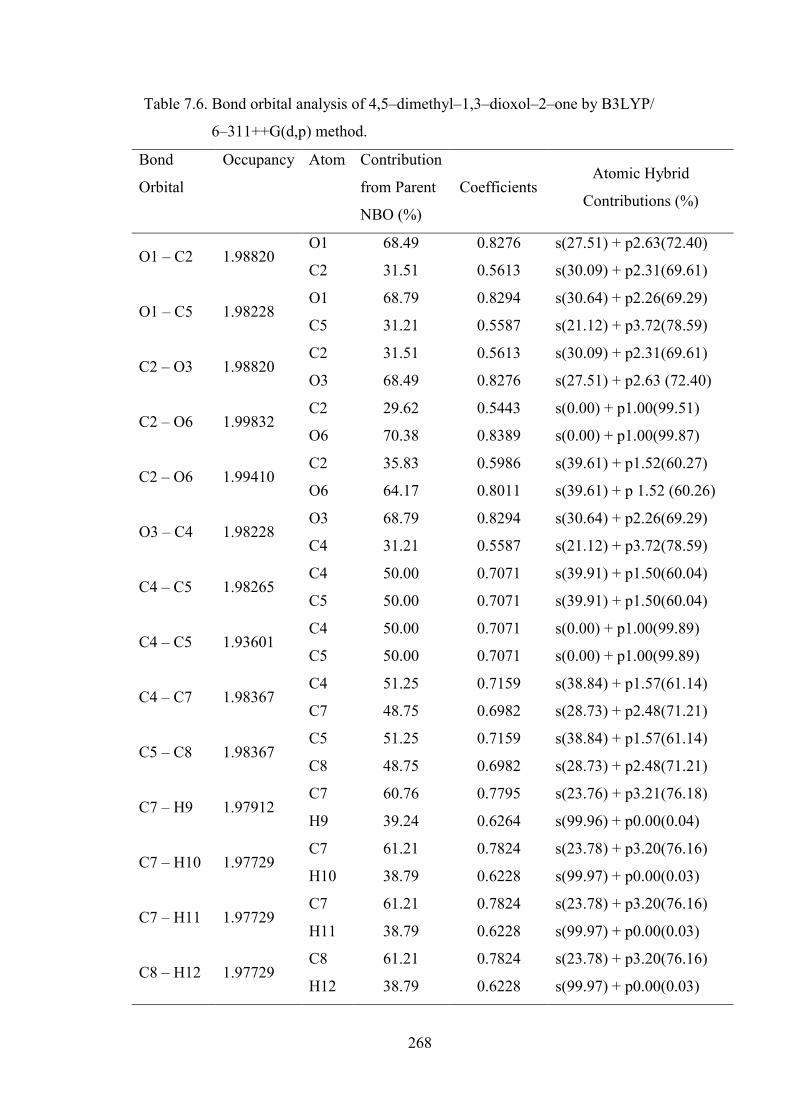
atomic contributions. The Table 7.6 depicts the bonding concepts such as type of bond
orbital, their occupancies, the natural atomic hybrids of which the NBO is composed,
giving the percentage of the NBO on each hybrid, the atom label, and a hybrid label
showing the hybrid orbital (spx) composition (the amount of s–character,
p–character, etc.,) of 4,5−dimethyl−1,3−dioxol−2−one molecule determined by
B3LYP/6–311++G(d,p) method with respectable accuracy. The occupancies of NBOs
in 4,5−dimethyl−1,3−dioxol−2−one reflecting their exquisite dependence on the
chemical environment. The NBO energy values show the corresponding spatial
symmetry breaking in the direction of unpaired spin. The Lewis structure that is
closest to the optimised structure is determined. The hybridisation of the atoms and
the weight of each atom in each localised electron pair bond is calculated in this
idealised Lewis structure and presented in Table 7.6. For
4,5−dimethyl−1,3−dioxol−2−one, no antibonding orbitals are listed so that the
structure is adequately explained by normal Lewis electron pair orbitals.
For example, the bonding orbital for both O1–C2 and O3−C2 with 1.9882
electrons has 68.49% O1 character in a sp2.63 hybrid and has 31.51% C2 character in a
sp2.31 hybrid orbital of 4,5−dimethyl−1,3−dioxol−2−one. This clearly reveals the same
nature of bonding. The bonding orbital C2–O6 with 1.9941 electrons has 29.62% C2
character in a sp1.0 hybrid and has 70.38% O6 character in a sp1.0 hybrid orbital. The
same bonding orbital of C2–O6 with 1.9941 electrons has 35.83% C2 character and
has 64.17% O6 character with sp1.52. The C4–C5 with 1.9827 electrons has 50%
character of C4 and C5 each in a sp1.15 hybrid.
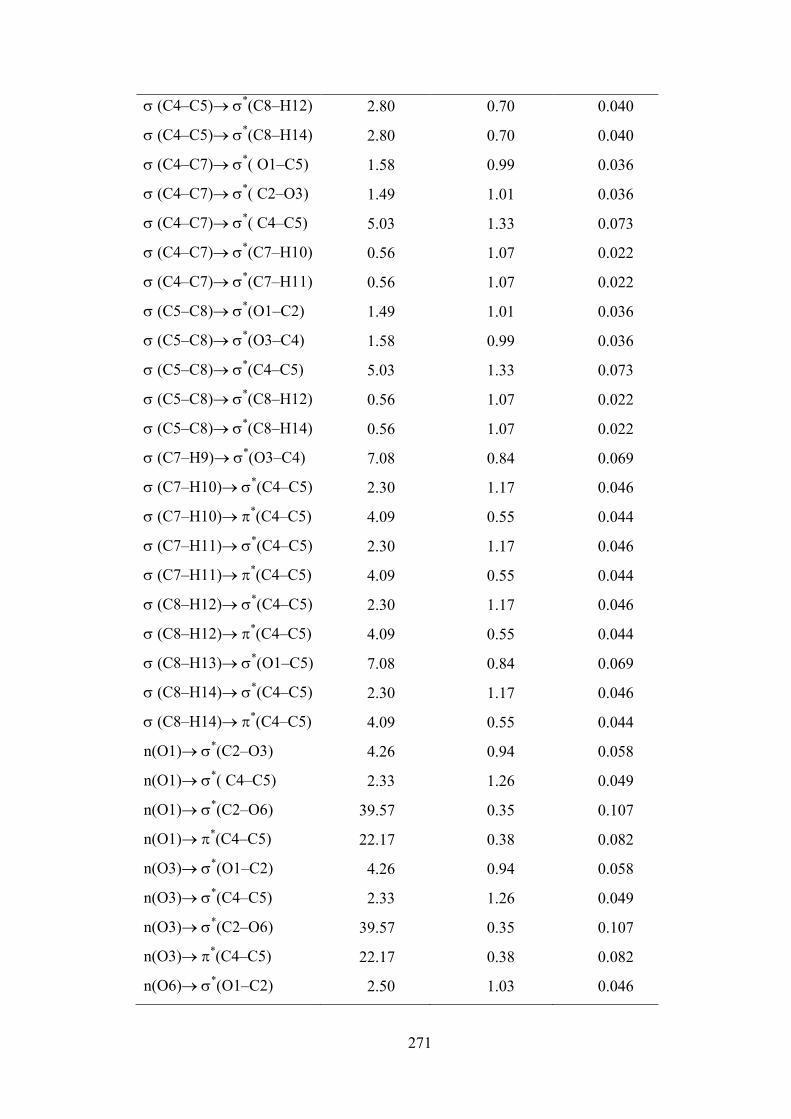
7.4.6. Donor Acceptor Interactions: Perturbation Theory Energy Analysis
The localised orbitals in the Lewis structure of
4,5−dimethyl−1,3−dioxol−2−one can interact strongly. A filled bonding or lone pair
orbital can act as a donor and an empty or filled bonding, antibonding, or lone pair
orbital can act as an acceptor. These interactions can strengthen and weaken bonds.
For example, a lone pair donor → antibonding acceptor orbital interaction may

251
weaken the bond associated with the antibonding orbital. Conversely, an interaction
with a bonding pair as the acceptor may strengthen the bond. Strong electron
delocalisation in the Lewis structure also shows up as donor–acceptor interactions.
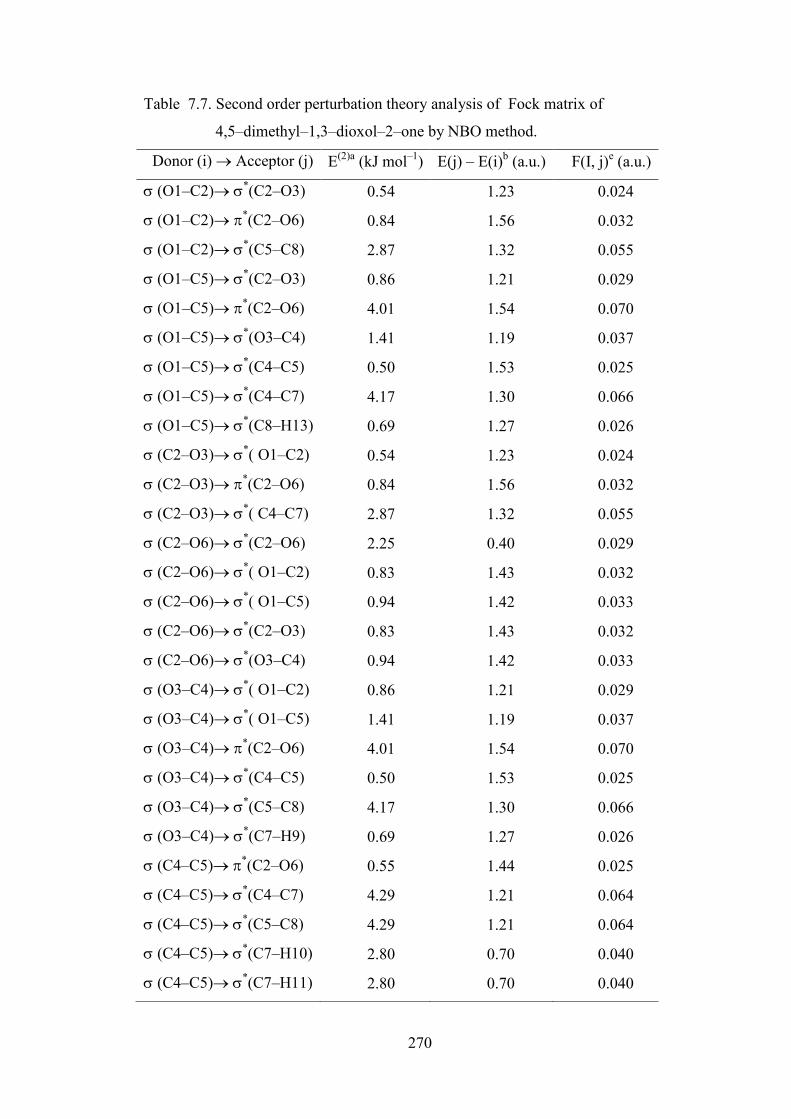
The stabilisation energy of different kinds of interactions are listed Table 7.7. This
calculation is done by examining all possible interactions between ‘filled’ (donor)
Lewis–type NBOs and ‘empty’ (acceptor) non–Lewis NBOs, and estimating their
energetic importance by 2nd–order perturbation theory. Since these interactions lead to
loss of occupancy from the localised NBOs of the idealised Lewis structure into the
empty non–Lewis orbitals (and thus, to departures from the idealised Lewis structure
description), they are referred to as ‘delocalisation’ corrections to the natural Lewis
structure.
The NBO method demonstrates the bonding concepts like atomic charge,
Lewis structure, bond type, hybridization, bond order, charge transfer and resonance
weights. Natural bond orbital (NBO) analysis is a useful tool for understanding
delocalisation of electron density from occupied Lewis–type (donor) NBOs to
properly unoccupied non–Lewis type (acceptor) NBOs within the molecule. The
stabilisation of orbital interaction is proportional to the energy difference between
interacting orbitals. Therefore, the interaction having strongest stabilisation takes
place between effective donors and effective acceptors. This bonding–anti bonding
interaction can be quantitatively described in terms of the NBO approach that is
expressed by means of second–order perturbation interaction energy E(2) [65–68]. This
energy represents the estimate of the off–diagonal NBO Fock matrix element. The
stabilisation energy E(2) associated with i (donor) → j (acceptor) delocalisation is
estimated from the second–order perturbation approach as [68] given below
ij
i
jiFqE
),(2)2(
where, qi is the donor orbital occupancy, εi and εj are diagonal elements (orbital
energies) and F(i,j) is the off–diagonal Fock matrix element.
In 4,5−dimethyl−1,3−dioxol−2−one molecule, the bond pair donor
orbital, σOC → π*CO interaction between the O1–C5 bond pair and the antiperiplanar
C2–O6 antibonding orbital give stabilisation of 4.01 kJ. mol–1. The bond pair donor
orbital, σCC → σ*CC interaction between the C4–C5 bond pair and the antiperiplanar
C4–C7 and C5−C8 antibonding orbital give equal stabilisation of 2.8 kJ. mol–1. The

252
lone pair donor orbital, nO → σ*CO interaction between the O1 and O3 oxygen lone
pair and the antiperiplanar C2–O6 antibonding orbital is seen to give a strong
stabilisation, 39.57 kJ. mol−1. The lone pair donor orbital, nO → σ*OC interaction
between the oxygen O6 lone pair and the antiperiplanar O1–C2 and C2−O3
antibonding orbital is seen to give a strong and equal stabilisation, 32.48 kJ. mol−1.
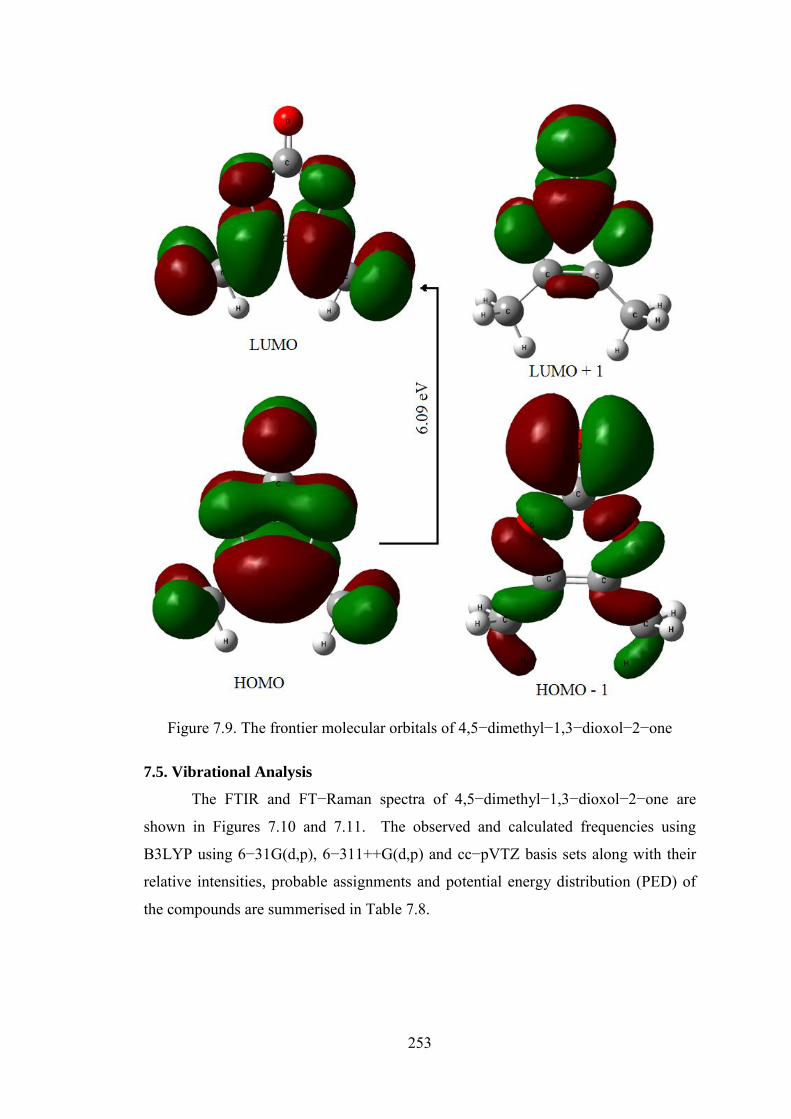
7.4.7. Frontier molecular orbitals
Highest occupied molecular orbital (HOMO) and lowest unoccupied
molecular orbital (LUMO) are very important parameters for quantum chemistry. The
HOMO is the orbital that primarily acts as an electron donor and the LUMO is the
orbital that largely acts as the electron acceptor [44]. The MOs are defined as eigen
functions of the Fock operator, which exhibits the full symmetry of the nuclear point
group, they necessarily form a basis for irreducible representations of full point–
group symmetry. The energies of HOMO, LUMO, LUMO+1 and HOMO–1 and their
orbital energy gaps of 4,5−dimethyl−1,3−dioxol−2−one are calculated using
B3LYP/6–311++G(d,p) method and the pictorial illustration of the frontier molecular
orbitals and their respective positive and negative regions are shown in Figure 7.9.
Molecular orbitals, when viewed in a qualitative graphical representation, can provide
insight into the nature of reactivity, and some of the structural and physical properties
of molecules. Well known concepts such as conjugation, aromaticity and lone pairs
are well illustrated by molecular orbitals.
The positive and negative phase is represented in red and green colour,
respectively. From the plots we can see that the region of HOMO and LUMO levels
spread over the entire molecule and the calculated energy gap of HOMO–LUMO’s
explains the ultimate charge transfer interface within the molecule. The frontier
orbital energy gaps (ELUMO – EHOMO) in case of 4,5−dimethyl−1,3−dioxol−2−one is
found to be 6.09 eV. Gauss View 5.0.8 visualisation program [45] has been utilised to
construct the shapes of frontier molecular orbitals.
253
Figure 7.9. The frontier molecular orbitals of 4,5−dimethyl−1,3−dioxol−2−one
7.5. Vibrational Analysis
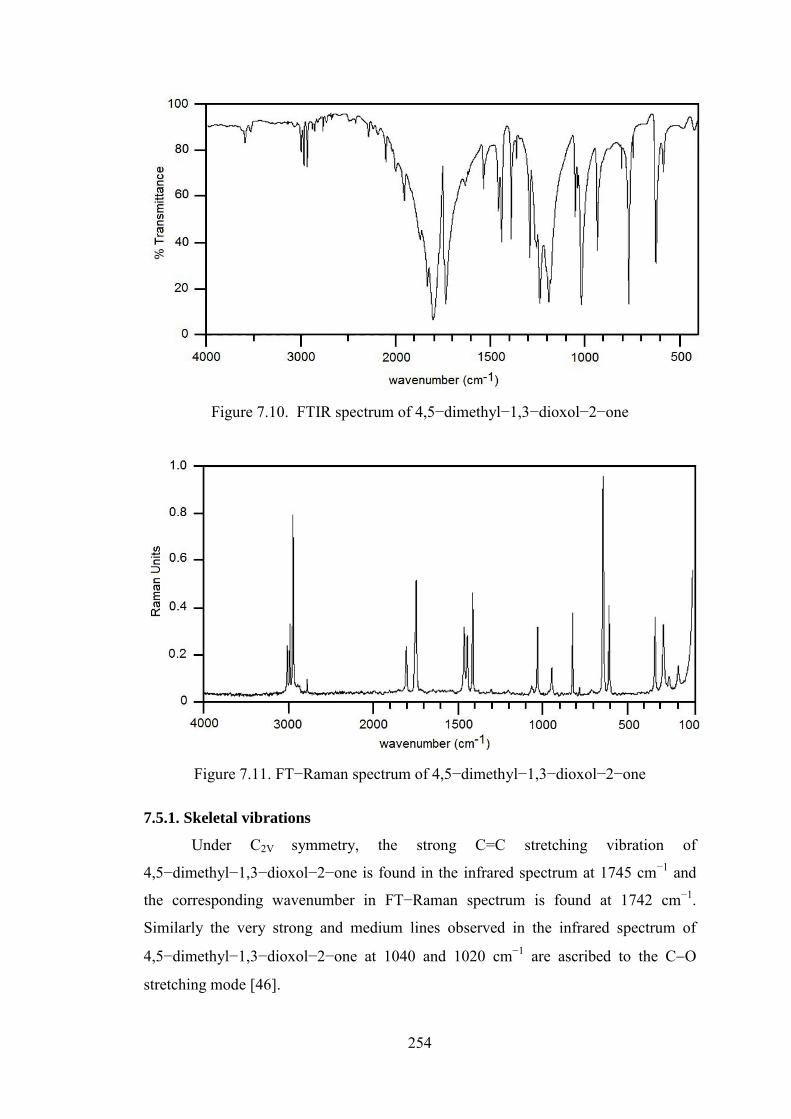
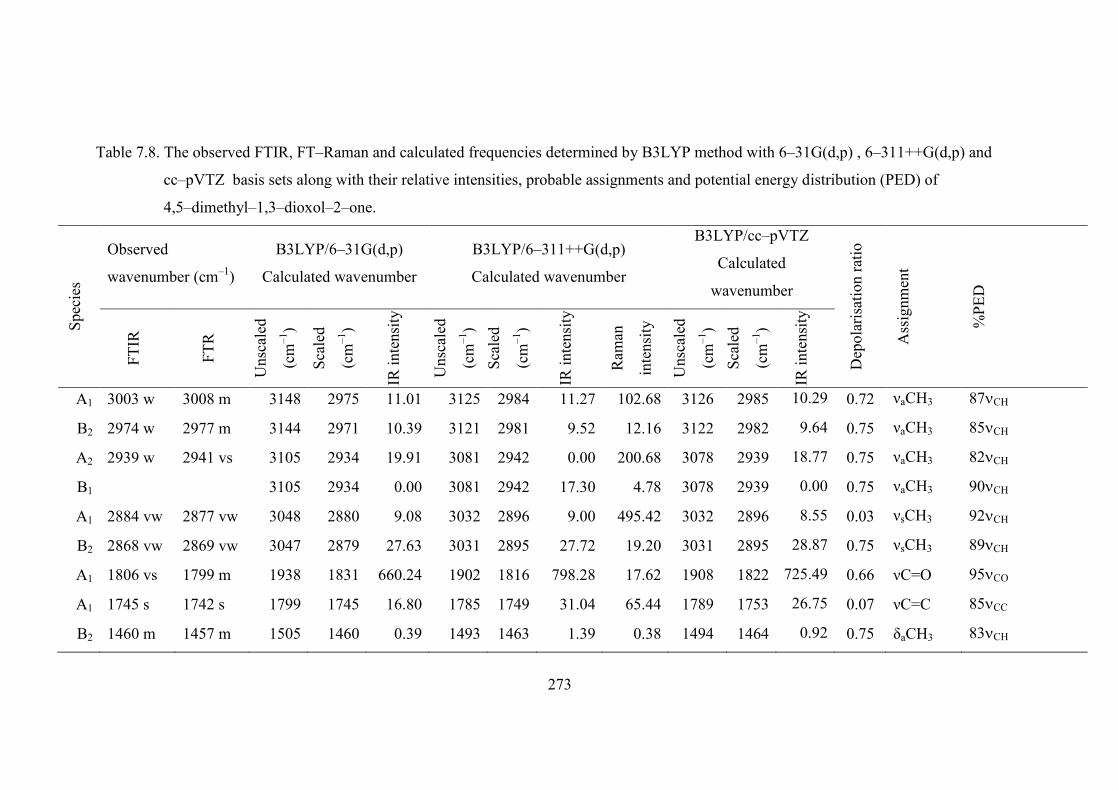
The FTIR and FT−Raman spectra of 4,5−dimethyl−1,3−dioxol−2−one are
shown in Figures 7.10 and 7.11. The observed and calculated frequencies using
B3LYP using 6−31G(d,p), 6−311++G(d,p) and cc−pVTZ basis sets along with their
relative intensities, probable assignments and potential energy distribution (PED) of
the compounds are summerised in Table 7.8.
254
Figure 7.10. FTIR spectrum of 4,5−dimethyl−1,3−dioxol−2−one
Figure 7.11. FT−Raman spectrum of 4,5−dimethyl−1,3−dioxol−2−one
7.5.1. Skeletal vibrations
Under C2V symmetry, the strong C=C stretching vibration of
4,5−dimethyl−1,3−dioxol−2−one is found in the infrared spectrum at 1745 cm−1 and
the corresponding wavenumber in FT−Raman spectrum is found at 1742 cm−1.
Similarly the very strong and medium lines observed in the infrared spectrum of
4,5−dimethyl−1,3−dioxol−2−one at 1040 and 1020 cm−1 are ascribed to the CO
stretching mode [46].

255
The fundamental modes observed at 1256 cm−1 in the infrared is attributed to
the OCO asymmetric stretching and at 1241 cm−1 is assigned to the OCO symmetric
stretching modes of 4,5−dimethyl−1,3−dioxol−2−one [47]. The bands occurring at
770 cm−1 in the infrared and the theoretical value of 630 cm−1 are assigned to the OCO
and COC in−plane bending modes of 4,5−dimethyl−1,3−dioxol−2−one. The COC and
OCC out of plane bending modes of 4,5−dimethyl−1,3−dioxol−2−one under C2V
symmetry are attributed to the Raman wavenumbers 320 and 266 cm−1. The in−plane
bending and out of plane vibrations are described as mixed modes as there are about
15 to 25% PED contributions mainly from C=O and CCO in−plane bending and out
of plane bending vibrations, respectively.
7.5.2. C=O vibrations
The C=O stretch lies in the spectral range 1750−1860 cm−1, and is very intense
in the infrared and only moderately active in Raman. In
4,5−dimethyl−1,3−dioxol−2−one the carbonyl stretching frequency is observed in the
high frequency region as a very strong band at 1806 cm−1 in IR and medium band at
1799 m−1 in Raman spectra. In 4−methyl−l,3−dioxolan−2−one, the carbonyl
stretching frequency is observed as a very strong band at 1790 cm−1 in IR and in
Raman spectra [42].The fundamental mode at 587 cm−1 in the infrared spectrum is
attributed to the C=O in−plane bending of 4,5−dimethyl−1,3−dioxol−2−one. The
wavenumber observed at 626 cm−1 in the infrared and at 632 cm−1 in Raman spectra
of are assigned to the C=O out of plane bending vibration. The C=O out of plane
bending of 4,5−dimethyl−1,3−dioxolan−2−one is observed at 820 cm−1 in IR
spectrum as a strong bands. The in−plane and out of plane bending of the carbonyl
group is mixed significantly with the in−plane and out of plane bending vibrations of
OCO vibration.
7.5.3. Methyl group vibrations
The asymmetric stretching and asymmetric deformation modes of the −CH3
group would be expected to be depolarised. The νs(CH3) frequencies are established
at 2884 and 2868 cm−1 in the infrared spectrum and νa(CH3) is assigned at 3033, 2974
and 2939 cm−1 of 4,5−dimethyl−1,3−dioxol−2−one. The –CH3 twist, rock and
deformation motions and the vibrations of the ring moieties are found in the spectral
region 900−1500 cm−1. The symmetrical methyl deformational mode is obtained at

256
1392 and 1293 cm−1 in IR spectrum as strong modes. The asymmetrical methyl
deformational modes are assigned at 1460 and 1442 cm−1 in IR [48,49].
7.5.4. Scale factors
The vibrational frequencies calculated using DFT methods are known to be
overestimated probably because of the neglect of anharmonicity of vibrations in the
real systems. Accepted values of scaling factors for DFT 0.96 and it has been used to
correct the frequency values [50]. A better agreement between the theoretical and
experimental frequencies can be obtained by using different scale factors for different
regions of vibrations.
To determine the scale factors, the procedure used previously [51–60] have
been followed that minimises the residual separating experimental and theoretically
predicted vibrational frequencies. The optimum scale factors for vibrational
frequencies were determined by minimising the residual
N
i
2Expti
Theori νλωΔ
where, Theoiω and
Exptiν are the i
th theoretical harmonic frequency and ith
experimental fundamental frequency (in cm–1), respectively and N is the number of
frequencies included in the optimisation which leads to
NΔRMS
The scale factors used in this study minimised the deviations between the
computed and experimental frequencies. A uniform scaling factor is recommended for
all frequencies < 1800 cm–1 at the B3LYP method with 6–31G(d,p), 6–311++G(d,p)
and cc−pVTZ basis sets and is adopted in this study. Initially, all scaling factors have
been kept fixed at a value of 1.0 to produce the pure DFT calculated vibrational
frequencies (unscaled) which are given in Table 7.8.
The correction factors are used to correlate the experimentally observed and
theoretically computed frequencies for each vibrational modes. Initially, all scaling
factors have been kept fixed at a value of 1.0 to produce the pure ab initio calculated
vibrational frequencies and the potential energy distributions (PED) which are given
in Tables. For 4,5−dimethyl−1,3−dioxol−2−one 0.955 for C−H and C=O vibrations,
0.98 upto 1200 cm−1 and 1.0 for all other lower frequencies were used in B3LYP

257
method with 6−311++G(d,p) and cc−pVTZ basis sets. The scale factors 0.945 for
C−H and C=O, and 0.97 for other vibrations upto 1200 cm−1 and 1.0 for all other
mode observed in the lower frequencies are used in B3LYP/6−31G(d,p) level. The
scale factors minimised the deviations between the computed and experimental
frequencies both at HF and DFT−B3LYP level calculations. DFT−B3LYP correction
factors are all much closer to unity than the HF correction factor.
7.6. Conclusions
The molecular structural parameters, thermodynamic properties and
vibrational frequencies of the fundamental modes of the optimised geometry of
4,5−dimethyl−1,3−dioxol−2−one have been determined from DFT calculations. The
geometry was optimised with the DFT−B3LYP method using 6−31G(d,p),
6−311++G(d,p) and cc−pVTZ basis sets. The complete vibrational assignment and
analysis of the fundamental modes of the compounds were carried out using the
observed FTIR and FT−Raman spectral data. The vibrational frequencies determined
experimentally were compared with those obtained theoretically from DFT−B3LYP
gradient calculations employing 6−31G(d,p), 6−311++G(d,p) and cc−pVTZ basis
sets. The deviation between the experimental and calculated (both unscaled and
scaled) frequencies were reduced with the use of DFT−B3LYP method with
6−311++G(d,p) and cc−pVTZ basis sets.
The total electron density surface, MESP mapped surface of the compound
and electrostatic potential contour map and electrostatic potential surface of
4,5−dimethyl−1,3−dioxol−2−one were presented. The atomic charges of
4,5−dimethyl−1,3−dioxol−2−one calculated by NBO analysis using the
B3LYP/6–311++G(d,p) method. The stabilisation energy of different kinds of
interactions were calculated by examining all possible interactions between ‘filled’
(donor) Lewis–type NBOs and ‘empty’ (acceptor) non–Lewis NBOs, and estimated
their energetic importance by 2nd–order perturbation theory. The energies of HOMO,
LUMO, LUMO+1 and HOMO–1 and their orbital energy gaps of
4,5−dimethyl−1,3−dioxol−2−one are calculated using B3LYP/6–311++G(d,p)
method and the pictorial illustration of the frontier molecular orbitals and their
respective positive and negative regions are shown.

258
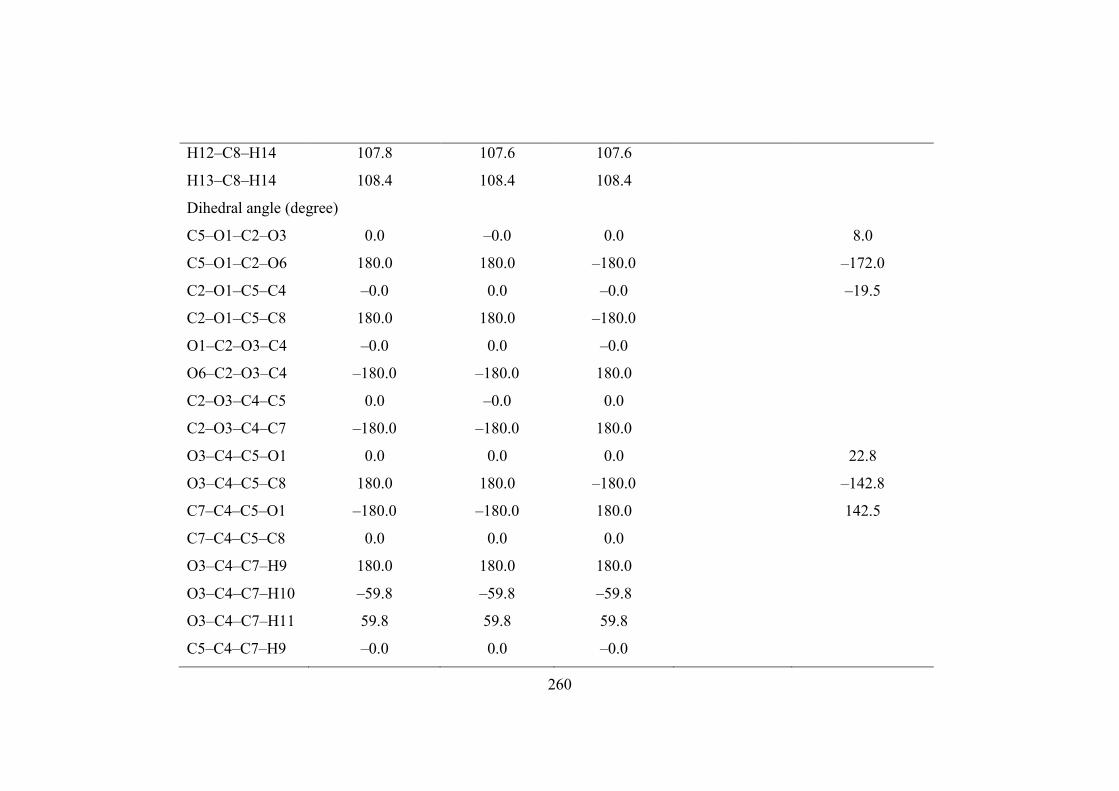
Table 7.1. Structural parameters calculated for 4,5–dimethyl–1,3–dioxol–2–one employing B3LYP/6–311++G(d,p),
B3LYP/6–31G(d,p) and B3LYP/ cc–pVTZ methods.
Structural
Parameters
4,5–dimethyl–1,3–dioxol–2–one
Expermentala
B3LYP/
6–311++G(d,p)
B3LYP/
6–31G(d,p)
B3LYP/
cc–pVTZ
4–methyl–1,3–
dioxolan–2–oneb
Internuclear Distance (Ǻ)
O1–C2 1.37 1.37 1.36 1.358 1.362
O1–C5 1.40 1.40 1.40 1.428 1.436
C2–O3 1.37 1.37 1.36 1.358 1.358
C2–O6 1.19 1.20 1.19 1.200 1.189
O3–C4 1.40 1.40 1.40 1.428 1.448
C4–C5 1.33 1.34 1.33 1.540 1.533
C4–C7 1.48 1.48 1.48 1.514
C5–C8 1.48 1.48 1.48 1.514
C7–H (methyl)c 1.09 1.09 1.09
C8–H (methyl)c 1.09 1.10 1.09
Bond Angle (degree)
C2–O1–C5 107.9 107.8 107.8 109.3
O1–C2–O3 107.8 107.9 107.8 111.67 110.2

259
O1–C2–O6 126.1 126.1 126.1 124.7
O3–C2–O6 126.1 126.1 126.1 124.17 125.1
C2–O3–C4 107.9 107.8 107.8 108.71 109.9
O3–C4–C5 108.2 108.3 108.3 102.16 101.8
O3–C4–C7 117.0 117.0 117.0 110.4
C5–C4–C7 134.8 134.7 134.8 115.7
O1–C5–C4 108.2 108.3 108.3 103.3
O1–C5–C8 117.0 117.0 117.0
C4–C5–C8 134.8 134.7 134.8 115.7
C4–C7–H9 110.3 110.3 110.4
C4–C7–H10 110.9 111.0 110.9
C4–C7–H11 110.9 111.0 110.9
H9–C7–H10 108.4 108.4 108.4
H9–C7–H11 108.4 108.4 108.4
H10–C7–H11 107.8 107.6 107.6
C5–C8–H12 110.9 111.0 110.9
C5–C8–H13 110.3 110.3 110.4
C5–C8–H14 110.9 111.0 110.9
H12–C8–H13 108.4 108.4 108.4
260
H12–C8–H14 107.8 107.6 107.6
H13–C8–H14 108.4 108.4 108.4
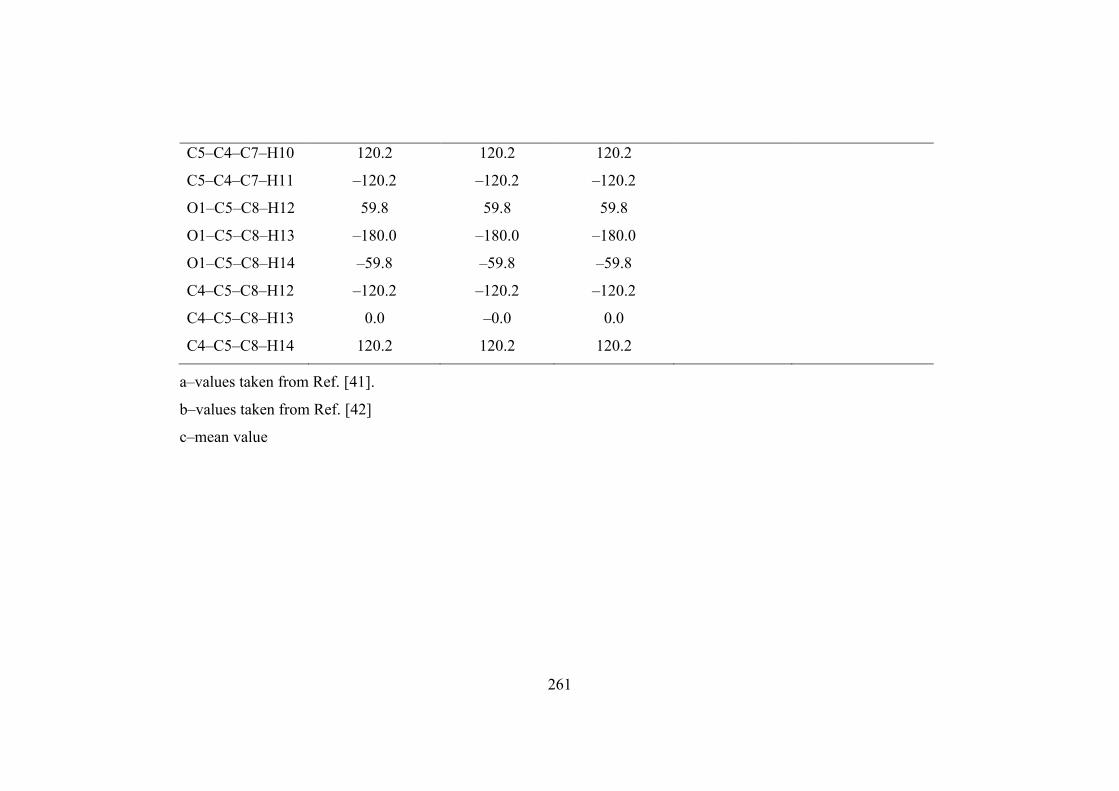
Dihedral angle (degree)
C5–O1–C2–O3 0.0 –0.0 0.0 8.0
C5–O1–C2–O6 180.0 180.0 –180.0 –172.0
C2–O1–C5–C4 –0.0 0.0 –0.0 –19.5
C2–O1–C5–C8 180.0 180.0 –180.0
O1–C2–O3–C4 –0.0 0.0 –0.0
O6–C2–O3–C4 –180.0 –180.0 180.0
C2–O3–C4–C5 0.0 –0.0 0.0
C2–O3–C4–C7 –180.0 –180.0 180.0
O3–C4–C5–O1 0.0 0.0 0.0 22.8
O3–C4–C5–C8 180.0 180.0 –180.0 –142.8
C7–C4–C5–O1 –180.0 –180.0 180.0 142.5
C7–C4–C5–C8 0.0 0.0 0.0
O3–C4–C7–H9 180.0 180.0 180.0
O3–C4–C7–H10 –59.8 –59.8 –59.8
O3–C4–C7–H11 59.8 59.8 59.8
C5–C4–C7–H9 –0.0 0.0 –0.0
261
C5–C4–C7–H10 120.2 120.2 120.2
C5–C4–C7–H11 –120.2 –120.2 –120.2
O1–C5–C8–H12 59.8 59.8 59.8
O1–C5–C8–H13 –180.0 –180.0 –180.0
O1–C5–C8–H14 –59.8 –59.8 –59.8
C4–C5–C8–H12 –120.2 –120.2 –120.2
C4–C5–C8–H13 0.0 –0.0 0.0
C4–C5–C8–H14 120.2 120.2 120.2
a–values taken from Ref. [41].
b–values taken from Ref. [42]
c–mean value
262
Table 7.2. The calculated thermodynamic parameters of 4,5–dimethyl–1,3–dioxol–2–one employing
B3LYP/6–311++G(d,p), B3LYP/6–31G(d,p) and B3LYP/ cc–pVTZ methods.
Thermodynamic parameters
(298 K)
4,5–dimethyl–1,3–dioxol–2–one 4–methyl–1,3–
dioxolan–2–oneb B3LYP/
6–311++G(d,p)
B3LYP/
6–31G(d,p)
B3LYP/
cc–pVTZ
SCF Energy (a.u) –419.94 –419.83 –419.98 –381.835
Total Energy (thermal), Etotal
(kcal.mol–1)
71.09 71.58 71.22 68.154
Heat Capacity at const. volume, Cv
(cal.mol–1.K–1)
27.13 27.02 26.98 22.254
Entropy, S (cal.mol–1.K–1) 84.95 84.93 84.72 78.988
Vibrational Energy, Evib (kcal.mol–1) 69.31 69.80 69.45 66.377
Zero–point vibrational Energy, E0
(kcal.mol–1)
66.33 66.83 66.50 64.232
Rotational Constants (GHz)
A 3.40 3.39 3.42 5.989
B 1.93 1.93 1.94 2.224
C 1.25 1.25 1.26 1.724
263
Dipole moment (Debye)
μx 0.00 –0.00 0.00 5.516
μy –5.75 –5.40 –5.43 1.933
μz 0.00 0.00 0.00 0.470
μtotal 5.75 5.40 5.43 5.864
E(LUMO + 1) eV –0.0065
E(LUMO) eV –0.6471
E(HOMO) eV –6.7371
E(HOMO – 1) eV –8.5425
E(LUMO) – E(HOMO) eV 6.09
b–values taken from [42].

264
Table 7.3. Thermodynamic properties of 4,5–dimethyl–1,3–dioxol–2–one determined
at different temperatures with B3LYP/6–311++G(d,p) level.
T (K) S (J.mol–1.K–1) Cp (J.mol–1.K–1) ΔH0→T (kJ.mol–1)
100 261.4 60.32 4.23
150 289.1 76.88 7.68
200 313.28 91.9 11.9
250 335.41 107.05 16.87
298.15 355.53 121.83 22.38
300 356.28 122.39 22.61
350 376.29 137.48 29.11
400 395.59 151.86 36.34
450 414.27 165.27 44.28
500 432.33 177.59 52.85
600 466.66 199.03 71.72
700 498.72 216.78 92.53
800 528.66 231.56 114.97
900 556.67 243.99 138.77
1000 582.94 254.51 163.71
265
Table 7.4. Calculated atomic charges of 4,5–dimethyl–1,3–dioxol–2–one by natural
bond orbital (NBO) analysis by B3LYP method.
Atom Natural charge
O1 –0.51663
C2 1.00106
O3 –0.51663
C4 0.24363
C5 0.24363
O6 –0.56671
C7 –0.61235
C8 –0.61235
H9 0.22025
H10 0.22396
H11 0.22396
H12 0.22396
H13 0.22025
H14 0.22396

266
Table 7.5. The atomic orbital occupancies of 4,5–dimethyl–2,3–dioxol–2–one
Atom No. Atomic orbital Type Occupancy Energy
O1
1s Core 1.99971 –18.99204
2s Valence 1.63436 –0.93158
2px Valence 1.76730 –0.35680
2py Valence 1.40965 –0.34915
2pz Valence 1.69000 –0.37852
C2
1s Core 1.99953 –10.25242
2s Valence 0.67550 –0.13403
2px Valence 0.81706 –0.15954
2py Valence 0.75127 –0.02854
2pz Valence 0.70209 –0.05004
O3
1s Core 1.99971 –18.99204
2s Valence 1.63436 –0.93158
2px Valence 1.76730 –0.35680
2py Valence 1.40965 –0.34915
2pz Valence 1.69000 –0.37852
C4
1s Core 1.99873 –10.12217
2s Valence 0.85058 –0.17321
2px Valence 1.06917 –0.14974
2py Valence 0.76377 –0.07091
2pz Valence 1.05195 –0.09131
C5
1s Core 1.99873 –10.12217
2s Valence 0.85058 –0.17321
2px Valence 1.06917 –0.14974
2py Valence 0.76377 –0.07091
2pz Valence 1.05195 –0.09131
O6
1s Core 1.99971 –18.84489
2s Valence 1.70454 –0.90472
2px Valence 1.49898 –0.26153
2py Valence 1.55432 –0.30957

267
2pz Valence 1.79645 –0.26747
C7
1s Core 1.99927 –10.05757
2s Valence 1.08226 –0.26862
2px Valence 1.22580 –0.12832
2py Valence 1.14334 –0.12239
2pz Valence 1.15292 –0.12310
C8
1s Core 1.99927 –10.05757
2s Valence 1.08226 –0.26862
2px Valence 1.22580 –0.12832
2py Valence 1.14334 –0.12239
2pz Valence 1.15292 –0.12310
H9 1s Valence 0.77830 0.00816
H10 1s Valence 0.77396 0.01584
H11 1s Valence 0.77396 0.01584
H12 1s Valence 0.77396 0.01584
H13 1s Valence 0.77830 0.00816
H14 1s Valence 0.77396 0.01584
268
Table 7.6. Bond orbital analysis of 4,5–dimethyl–1,3–dioxol–2–one by B3LYP/
6–311++G(d,p) method.
Bond
Orbital
Occupancy Atom Contribution
from Parent
NBO (%)
Coefficients Atomic Hybrid
Contributions (%)
O1 – C2 1.98820 O1 68.49 0.8276 s(27.51) + p2.63(72.40)
C2 31.51 0.5613 s(30.09) + p2.31(69.61)
O1 – C5 1.98228 O1 68.79 0.8294 s(30.64) + p2.26(69.29)
C5 31.21 0.5587 s(21.12) + p3.72(78.59)
C2 – O3 1.98820 C2 31.51 0.5613 s(30.09) + p2.31(69.61)
O3 68.49 0.8276 s(27.51) + p2.63 (72.40)
C2 – O6 1.99832 C2 29.62 0.5443 s(0.00) + p1.00(99.51)
O6 70.38 0.8389 s(0.00) + p1.00(99.87)
C2 – O6 1.99410 C2 35.83 0.5986 s(39.61) + p1.52(60.27)
O6 64.17 0.8011 s(39.61) + p 1.52 (60.26)
O3 – C4 1.98228 O3 68.79 0.8294 s(30.64) + p2.26(69.29)
C4 31.21 0.5587 s(21.12) + p3.72(78.59)
C4 – C5 1.98265 C4 50.00 0.7071 s(39.91) + p1.50(60.04)
C5 50.00 0.7071 s(39.91) + p1.50(60.04)
C4 – C5 1.93601 C4 50.00 0.7071 s(0.00) + p1.00(99.89)
C5 50.00 0.7071 s(0.00) + p1.00(99.89)
C4 – C7 1.98367 C4 51.25 0.7159 s(38.84) + p1.57(61.14)
C7 48.75 0.6982 s(28.73) + p2.48(71.21)
C5 – C8 1.98367 C5 51.25 0.7159 s(38.84) + p1.57(61.14)
C8 48.75 0.6982 s(28.73) + p2.48(71.21)
C7 – H9 1.97912 C7 60.76 0.7795 s(23.76) + p3.21(76.18)
H9 39.24 0.6264 s(99.96) + p0.00(0.04)
C7 – H10 1.97729 C7 61.21 0.7824 s(23.78) + p3.20(76.16)
H10 38.79 0.6228 s(99.97) + p0.00(0.03)
C7 – H11 1.97729 C7 61.21 0.7824 s(23.78) + p3.20(76.16)
H11 38.79 0.6228 s(99.97) + p0.00(0.03)
C8 – H12 1.97729 C8 61.21 0.7824 s(23.78) + p3.20(76.16)
H12 38.79 0.6228 s(99.97) + p0.00(0.03)

269
C8 – H13 1.97912 C8 60.76 0.7795 s(23.76) + p3.21(76.18)
H13 39.24 0.6264 s(99.96) + p0.00(0.04)
C8 – H14 1.97729 C8 61.21 0.7824 s(23.78) + p3.20(76.16)
H14 38.79 0.6228 s(99.97) + p0.00(0.03)
270
Table 7.7. Second order perturbation theory analysis of Fock matrix of
4,5–dimethyl–1,3–dioxol–2–one by NBO method.
Donor (i) Acceptor (j) E(2)a (kJ mol–1) E(j) – E(i)b (a.u.) F(I, j)e (a.u.)
(O1–C2) *(C2–O3) 0.54 1.23 0.024
(O1–C2) *(C2–O6) 0.84 1.56 0.032
(O1–C2) *(C5–C8) 2.87 1.32 0.055
(O1–C5) *(C2–O3) 0.86 1.21 0.029
(O1–C5) *(C2–O6) 4.01 1.54 0.070
(O1–C5) *(O3–C4) 1.41 1.19 0.037
(O1–C5) *(C4–C5) 0.50 1.53 0.025
(O1–C5) *(C4–C7) 4.17 1.30 0.066
(O1–C5) *(C8–H13) 0.69 1.27 0.026
(C2–O3) *( O1–C2) 0.54 1.23 0.024
(C2–O3) *(C2–O6) 0.84 1.56 0.032
(C2–O3) *( C4–C7) 2.87 1.32 0.055
(C2–O6) *(C2–O6) 2.25 0.40 0.029
(C2–O6) *( O1–C2) 0.83 1.43 0.032
(C2–O6) *( O1–C5) 0.94 1.42 0.033
(C2–O6) *(C2–O3) 0.83 1.43 0.032
(C2–O6) *(O3–C4) 0.94 1.42 0.033
(O3–C4) *( O1–C2) 0.86 1.21 0.029
(O3–C4) *( O1–C5) 1.41 1.19 0.037
(O3–C4) *(C2–O6) 4.01 1.54 0.070
(O3–C4) *(C4–C5) 0.50 1.53 0.025
(O3–C4) *(C5–C8) 4.17 1.30 0.066
(O3–C4) *(C7–H9) 0.69 1.27 0.026
(C4–C5) *(C2–O6) 0.55 1.44 0.025
(C4–C5) *(C4–C7) 4.29 1.21 0.064
(C4–C5) *(C5–C8) 4.29 1.21 0.064
(C4–C5) *(C7–H10) 2.80 0.70 0.040
(C4–C5) *(C7–H11) 2.80 0.70 0.040
271
(C4–C5) *(C8–H12) 2.80 0.70 0.040
(C4–C5) *(C8–H14) 2.80 0.70 0.040
(C4–C7) *( O1–C5) 1.58 0.99 0.036
(C4–C7) *( C2–O3) 1.49 1.01 0.036
(C4–C7) *( C4–C5) 5.03 1.33 0.073
(C4–C7) *(C7–H10) 0.56 1.07 0.022
(C4–C7) *(C7–H11) 0.56 1.07 0.022
(C5–C8) *(O1–C2) 1.49 1.01 0.036
(C5–C8) *(O3–C4) 1.58 0.99 0.036
(C5–C8) *(C4–C5) 5.03 1.33 0.073
(C5–C8) *(C8–H12) 0.56 1.07 0.022
(C5–C8) *(C8–H14) 0.56 1.07 0.022
(C7–H9) *(O3–C4) 7.08 0.84 0.069
(C7–H10) *(C4–C5) 2.30 1.17 0.046
(C7–H10) *(C4–C5) 4.09 0.55 0.044
(C7–H11) *(C4–C5) 2.30 1.17 0.046
(C7–H11) *(C4–C5) 4.09 0.55 0.044
(C8–H12) *(C4–C5) 2.30 1.17 0.046
(C8–H12) *(C4–C5) 4.09 0.55 0.044
(C8–H13) *(O1–C5) 7.08 0.84 0.069
(C8–H14) *(C4–C5) 2.30 1.17 0.046
(C8–H14) *(C4–C5) 4.09 0.55 0.044
n(O1) *(C2–O3) 4.26 0.94 0.058
n(O1) *( C4–C5) 2.33 1.26 0.049
n(O1) *(C2–O6) 39.57 0.35 0.107
n(O1) *(C4–C5) 22.17 0.38 0.082
n(O3) *(O1–C2) 4.26 0.94 0.058
n(O3) *(C4–C5) 2.33 1.26 0.049
n(O3) *(C2–O6) 39.57 0.35 0.107
n(O3) *(C4–C5) 22.17 0.38 0.082
n(O6) *(O1–C2) 2.50 1.03 0.046

272
aStabilisation (Delocalisation) energy. bEnergy difference between i (donor) and j (acceptor) NBO orbitals. eFock matrix element i and j NBO orbitals.
n(O6) *(C2–O3) 2.50 1.03 0.046
n(O6) *(O1–C2) 32.48 0.58 0.126
n(O6) *(C2–O3) 32.48 0.58 0.126
273
Table 7.8. The observed FTIR, FT–Raman and calculated frequencies determined by B3LYP method with 6–31G(d,p) , 6–311++G(d,p) and
cc–pVTZ basis sets along with their relative intensities, probable assignments and potential energy distribution (PED) of
4,5–dimethyl–1,3–dioxol–2–one.
Spec
ies
Observed
wavenumber (cm–1)
B3LYP/6–31G(d,p)
Calculated wavenumber
B3LYP/6–311++G(d,p)
Calculated wavenumber
B3LYP/cc–pVTZ
Calculated
wavenumber
Dep
olar
isat
ion
ratio
Ass
ignm
ent
%PE
D
FTIR
FTR
Uns
cale
d
(cm
–1)
Scal
ed
(cm
–1)
IR in
tens
ity
Uns
cale
d
(cm
–1)
Scal
ed
(cm
–1)
IR in
tens
ity
Ram
an
inte
nsity
Uns
cale
d
(cm
–1)
Scal
ed
(cm
–1)
IR in
tens
ity
A1 3003 w 3008 m 3148 2975 11.01 3125 2984 11.27 102.68 3126 2985 10.29 0.72 νaCH3 87CH
B2 2974 w 2977 m 3144 2971 10.39 3121 2981 9.52 12.16 3122 2982 9.64 0.75 νaCH3 85CH
A2 2939 w 2941 vs 3105 2934 19.91 3081 2942 0.00 200.68 3078 2939 18.77 0.75 νaCH3 82CH
B1 3105 2934 0.00 3081 2942 17.30 4.78 3078 2939 0.00 0.75 νaCH3 90CH
A1 2884 vw 2877 vw 3048 2880 9.08 3032 2896 9.00 495.42 3032 2896 8.55 0.03 νsCH3 92CH
B2 2868 vw 2869 vw 3047 2879 27.63 3031 2895 27.72 19.20 3031 2895 28.87 0.75 νsCH3 89CH
A1 1806 vs 1799 m 1938 1831 660.24 1902 1816 798.28 17.62 1908 1822 725.49 0.66 νC=O 95CO
A1 1745 s 1742 s 1799 1745 16.80 1785 1749 31.04 65.44 1789 1753 26.75 0.07 νC=C 85CC
B2 1460 m 1457 m 1505 1460 0.39 1493 1463 1.39 0.38 1494 1464 0.92 0.75 δaCH3 83CH

274
A1 1504 1459 5.68 1491 1461 6.35 44.64 1492 1462 5.70 0.55 δaCH3 82CH
B1 1442 s 1437 m 1482 1438 13.21 1472 1443 16.22 14.06 1472 1443 13.99 0.75 δaCH3 80CH
A2 1481 1437 0.00 1470 1441 0.00 1.95 1471 1442 0.00 0.75 δaCH3 84CH
A1 1392 s 1406 s 1439 1396 14.25 1428 1399 17.07 11.94 1431 1402 14.82 0.23 δsCH3 89CH
A2 1437 1394 0.66 1427 1398 0.00 13.49 1428 1399 0.01 0.75 ωCH3 55ωCH3+25tCH3
B2 1293 s 1287 vw 1322 1282 20.23 1310 1284 13.88 1.79 1312 1286 14.41 0.75 δsCH3 87CH
B2 1256 s 1253 1253 125.30 1242 1242 144.90 0.01 1246 1246 131.97 0.75 νaOCO 88OCO
A1 1241 vs 1251 1251 152.92 1228 1228 170.44 1.26 1232 1232 161.49 0.46 νsOCO 87OCO
A2 1083 vs 1074 1074 0.00 1067 1067 0.00 0.34 1076 1076 0.00 0.75 ωCH3 52ωCH3+24tCH3
B1 1059 m 1051 vw 1070 1070 3.24 1062 1062 1.49 0.27 1067 1067 1.78 0.75 tCH3 56tCH3+22ωCH3
A1 1040 m 1039 1039 14.46 1027 1027 22.26 1.88 1033 1033 17.66 0.75 νCO 79CO
B2 1020 vs 1022 s 1034 1034 66.02 1027 1027 85.68 8.56 1029 1029 81.73 0.16 νCO 80CO
A1 970 970 0.25 948 948 0.59 1.40 952 952 0.04 0.75 ρCH3 62ρCH3+18CC
A1 936 s 937 m 935 935 26.99 931 931 31.66 2.42 932 932 29.53 0.17 νC–CH3 728CC + 18δCH3
B2 808 m 813 s 817 817 0.00 817 817 0.12 5.80 816 816 0.07 0.47 νC–CH3 70CC + 14δCH3
A1 770 vs 760 760 21.36 769 769 21.57 0.29 777 777 19.55 0.75 βOCO 65OCO+12CCO
B1 626 vs 632 vs 635 635 8.92 631 631 9.86 16.14 634 634 8.83 0.17 γC=O 79γCO+12OCO
A1 629 629 4.61 630 630 3.16 2.41 629 629 4.01 0.75 βCOC 67COC+14CO

275
A2 597 m 593 593 0.00 586 586 0.00 3.08 620 620 0.00 0.75 γOCC 62OCC+16CO
B2 587 m 579 579 2.03 583 583 1.77 0.96 585 585 1.42 0.75 βC=O 65CO+18OCO
B1 339 339 2.58 335 335 4.46 0.15 340 340 3.59 0.75 γOCO 60γOCO+18γCCO
A2 320 m 312 312 0.00 314 314 0.00 3.14 315 315 0.00 0.75 γCOC 56γCOC+15γCO
A2 266 m 249 249 0.05 250 250 0.06 1.41 251 251 0.05 0.63 γOCC 52γOCC+20γCCC
A2 219 219 0.00 218 218 0.00 0.10 221 221 0.00 0.75 γOCC 55γCH+18γCO
A2 174 174 0.00 176 176 0.00 0.32 178 178 0.00 0.75 γC–CH3 51γCC+22γCCO
B1 108 w 158 158 0.03 156 156 0.18 0.64 159 159 0.04 0.75 γC–CH3 53γCC+24γCCO
B1 153 153 0.24 155 155 0.20 0.32 156 156 0.22 0.75 tCH3 52tCH2+25ωCH2 aν–stretching; β–in–plane bending; δ–deformation; ρ–rocking; γ–out of plane bending; ω–wagging and τ–twisting/torsion, wavenumbers (cm–1); IR
intensities (km/mol) and Raman intensities (Å)4/(a.m.u).

276
References
[1] I. Shoji, T. Yasushi, H. Ryoichi, S. Fumio, I. Koji, T. Goro, Chem. Pharm. Bull. 36 (1988) 394.
[2] T. Hiyama, S. Fujita, H. Nozaki, Bull. Chem. Soc. Japan, 45 (1972) 2797.
[3] S. Ikeda, F. Sakamoto, H. Kondo, M. Moriyama, G. Tsukamoto, Chem. Pharm. Bull. 32 (1984) 4316.
[4] F. Sakamoto, S. Ikeda, G. Tsukamoto, Chem. Pharm. Bull. 32 (1984) 2241.
[5] J.L. Alonso, R. Cervellati, A.D. Esposti, D.G. Lister, P. PaImieri, J. Chem. Soc. Faraday Trans. II, 82 (1986) 337.
[6] J.E. Boggs, F.R. Cordell, J. Mol. Struct. 76 (1981) 329.
[7] F. Cser, Acta Chim. Acad. Sci. Hung. 80 (1974) 49.
[8] Y. Takebe, K. Iuchi, G. Tsukamoto, U.S. Patent No. 4554358 (1985).
[9] K. Xu, Chem. Rev. 104 (2004) 4303.
[10] PNGV Battery Test Manual, Revision 2, August 1999, DOE/ID-10597.
[11] E. Peled, J. Electrochem. Soc. 126 (1979) 2047.
[12] E. Peled, D. Golodnitsky, G. Ardel, J. Electrochem. Soc. 144 (1997) L208.
[13] M. Xu, W. Li, B.L. Lucht, J. Power Sources 193 (2009) 804.
[14] L.E. Ouatani, R. Dedryvere, C. Siret, P. Biensan, S. Reynaud, P. Iratcabal, D. Gonbeau, J. Electrochem. Soc. 156 (2009) A103.
[15] D. Aurbach, K. Gamolsky, B. Markovsky, Y. Gofer, M. Schmidt, U. Heider, Electrochim. Acta 47 (2002) 1423.
[16] H. Ota, Y. Sakata, A. Inoue, S. Yamaguchi, J. Electrochem. Soc. 151 (2004) A1659.
[17] H. Ota, K. Shima, M. Ue, J.-I. Yamaki, Electrochim. Acta 49 (2004) 565.
[18] S.S. Zhang, K. Xu, T.R. Jow, Electrochim. Acta 51 (2006) 1636.
[19] M. Lu, H. Cheng, Y. Yang, Electrochim. Acta 53 (2008) 3539.
[20] M.Q. Xu, W.S. Li, X.X. Zuo, J.S. Liu, X. Xu, J. Power Sources 174 (2007) 705.
[21] X. Zuo, M. Xu, W. Li, D. Su, J. Liu, Electrochem. Solid-State Lett. 9 (2006) A196.
[22] A. Xiao, L. Yang, B.L. Lucht, S.-H. Kang, D.P. Abraham, J. Electrochem. Soc. 156 (2009) A318.
[23] K. Xu, U. Lee, S.S. Zhang, T.R. Jow, J. Electrochem. Soc. 151 (2004) A2106.
[24] W. Xu, C.A. Angell, Electrochem. Solid-State Lett. 4 (2001) E1.
[25] S.S. Zhang, Electrochem. Commun. 8 (2006) 1423.

277
[26] J. Li, W.H. Yao, Y.S. Meng, Y. Yang, J. Phys. Chem. C 112 (2008) 12550.
[27] Y.S. Hu, W.H. Kong, H. Li, X.J. Huang, L.Q. Chen, Electrochem. Commun. 6 (2004) 126.
[28] Mengqing Xu, Liu Zhou, Lidan Xing, Weishan Li, Brett L. Lucht, Electrochim. Acta 55 (2010) 6743.
[29] I. Ohno, K. Hirata, C. Ishida, M. Ihara, K. Matsudab, S. Kagabua, Bioorg. & Med. Chem. Lett. 17 (2007) 4500.
[30] W. Kosmus, K. Kalcher, J. Cryst. Spectrosc. Res. 13 (1983) 143.
[31] K. Kalcher, W. Kosmus, J. Cryst. Spectrosc. Res. 13 (1983) 135.
[32] G. Naray−Szabo, M.R. Peterson, J. Mol. Struct. (Theochem.) 85 (1981) 249.
[33] P. Hohenberg, W. Kohn, Phys. Rev. B 136 (1964) 864.
[34] A.D. Becke, J. Chem. Phys. 98 (1993) 5648.
[35] C. Lee, W. Yang, R.G. Parr, Phys. Rev. B 37 (1988) 785.
[36] M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb, J.R. Cheeseman, J.A. Montgomery, Jr., T. Vreven, K.N. Kudin, J.C. Burant, J.M. Millam, S.S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G.A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J.E. Knox, H.P. Hratchian, J.B. Cross, C. Adamo, J. Jaramillo, R. Gomperts, R.E. Stratmann, O. Yazyev, A.J. Austin, R. Cammi, C. Pomelli, J.W. Ochterski, P.Y. Ayala, K. Morokuma, A. Voth, P. Salvador, J.J. Dannenberg, V.G. Zakrzewski, S. Dapprich, A.D. Daniels, M.C. Strain, O. Farkas, D.K. Malick, A.D. Rabuck, K. Raghavachari, J.B. Foresman, J.V. Ortiz, Q. Cui, A.G. Baboul, S. Clifford, J. Cioslowski, B.B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R.L. Martin, D.J. Fox, T. Keith, M.A. Al−Laham, C.Y. Peng, A. Nanayakkara, M. Challacombe, P.M.W. Gill, B. Johnson,W. Chen, M.W. Wong, C. Gonzalez, J.A. Pople, Gaussian Inc., Wallingford, CT, 2004.
[37] E.B. Wilson Jr, J. Chem. Phys. 7 (1939) 1047.
[38] E.B. Wilson Jr, J. Chem. Phys. 9 (1941) 76.
[39] E.B. Wilson Jr, J.C. Decius, P.C. Cross, Molecular Vibrations, McGraw Hill, New York, 1955.
[40] H. Fuhrer, V.B. Kartha, K.L. Kidd, P.J. Kruger, H.H. Mantsch, Computer Program for Infrared and Spectrometry, Normal Coordinate Analysis, Vol. 5, National Research Council, Ottawa, Canada, 1976.
[41] P. M. Matias, G.A. Jeffrey, L.M. Wingert, J.R. Ruble, J. Mol. Struct. (Theochem.) 184 (1989) 247.

278
[42] V. Arjunan, I. saravanan, P. Ravindran, S. Mohan, Spectrochim. Acta 77A (2010) 28.
[43] A.E. Reed, F. Weinhold, J. Chem. Phys. 83 (1985) 1736.
[44] J.M. Seminario, Recent Developments and Applications of Modern Density Functional Theory, Vol.4, Elsevier,1996, pp. 800–806 .
[45] A.E. Frisch, H.P. Hratchian, R.D. Dennington II, et al., GaussView, Version 5.0.8, Gaussian Inc., 235 Wallingford CT, 2009.
[46] J.R. Durig, J.W. Clark, J.M. Casper, J. Mol Struct. 5 (1970) 67.
[47] R.A. Pethrick, A.D. Wilson, Spectrochim. Acta 30A (1974) 1073.
[48] V. Arjunan, S. Mohan, J. Mol. Struct. 892 (2008) 289.
[49] V. Arjunan, S. Mohan, Spectrochim. Acta 72A (2009) 436.
[51] J.A. Pople, H.B. Schlegel, R. Krishnan, D.J. Defrees, J.S Binkley, M.J. Frish, R.A. Whitside, R.H. Hout, W.J. Hehre, Int. J. Quantum Chem. Quantum Chem. Symp. 15 (1981) 269.
[52] V. Arjunan, S. Mohan, J. Mol. Struct. 892 (2008) 289.
[53] V. Arjunan, S. Mohan, Spectrochim. Acta 72A (2009) 436.
[54] H.F. Hameka, J.O. Jensen, J. Mol. Struct. (Theochem.) 362 (1996) 325.
[55] J.O. Jensen, A. Banerjee, C.N. Merrow, D. Zeroka, J.M. Lochner, J. Mol. Struct. (Theochem.) 531 (2000) 323.
[56] A.P. Scott, L. Radom, J. Phys. Chem. 100 (1996) 16502.
[57] M.P. Andersson, P. Uvdal, J. Chem. Phys. A 109 (2005) 2937.
[58] M. Alcolea Palafox, M. Gill, N.J. Nunez, V.K. Rastogi, Lalit Mittal, Rekha Sharma, Int. J. Quant. Chem. 103 (2005) 394.
[59] M. Alcolea Palafox, Int. J. Quant. Chem. 77 (2000) 661.
[60] V. Arjunan, P. Ravindran, T. Rani, S. Mohan, J. Mol. Struct. 988 (2011) 91.