a framework for identification of actionable cancer genome
TRANSCRIPT

A framework for identification of actionable cancergenome dependencies in small cell lung cancerMartin L. Sosa,b,c,d,1,2, Felix Dietleina,b,1, Martin Peifera,b, Jakob Schöttlea,b, Hyatt Balke-Wanta,b, Christian Müllera,b,Mirjam Kokera,b, André Richterse,f, Stefanie Heyncka,b, Florian Malchersa,b, Johannes M. Heuckmanna,b, Danila Seidela,b,Patrick A. Eyersg, Roland T. Ullrichb, Andrey P. Antonchickh, Viktor V. Vintonyakh, Peter M. Schneideri,Takashi Ninomiyaj, Herbert Waldmanne,h, Reinhard Büttnerk, Daniel Rauhe,f, Lukas C. Heukampk,and Roman K. Thomasa,b,k,2
aDepartment of Translational Genomics, University of Cologne, 50931 Cologne, Germany; bMax Planck Institute for Neurological Research, 50931 Cologne,Germany; cHoward Hughes Medical Institute, dDepartment of Cellular and Molecular Pharmacology, University of California, San Francisco, CA 94158;eChemical Genomics Center of the Max Planck Society, 44227 Dortmund, Germany; fTechnical University Dortmund, D-44221 Dortmund, Germany; gYorkshireCancer Research (YCR) Institute for Cancer Studies, Cancer Research United Kingdom (CR-UK)/YCR Sheffield Cancer Research Centre, Department of Oncology,University of Sheffield, Sheffield S10 2RX, United Kingdom; hMax Planck Institute of Molecular Physiology, D-44227 Dortmund, Germany; iInstitute of ForensicMedicine, University of Cologne, 50823 Cologne, Germany; jDepartment of Hematology, Oncology, and Respiratory Medicine, Okayama University GraduateSchool of Medicine, Dentistry, and Pharmaceutical Sciences, 700-8558 Okayama, Japan; and kInstitute of Pathology, University of Cologne, 50924 Cologne,Germany
Edited by Peter K. Vogt, The Scripps Research Institute, La Jolla, CA, and approved September 11, 2012 (received for review April 30, 2012)
Small cell lung cancer (SCLC) accounts for about 15% of all lungcancers. The prognosis of SCLC patients is devastating and no bi-ologically targeted therapeutics are active in this tumor type. Todevelop a framework for development of specific SCLC-targeteddrugs we conducted a combined genomic and pharmacological vul-nerability screen in SCLC cell lines. We show that SCLC cell linescapture the genomic landscape of primary SCLC tumors and pro-vide genetic predictors for activity of clinically relevant inhibitorsby screening 267 compounds across 44 of these cell lines. We showAurora kinase inhibitors are effective in SCLC cell lines bearingMYCamplification, which occur in 3–7% of SCLC patients. In MYC-ampli-fied SCLC cells Aurora kinase inhibition associates with G2/M-ar-rest, inactivation of PI3-kinase (PI3K) signaling, and induction ofapoptosis. Aurora dependency in SCLC primarily involved AuroraB, required its kinase activity, and was independent of depletion ofcytoplasmic levels of MYC. Our study suggests that a fraction ofSCLC patients may benefit from therapeutic inhibition of Aurora B.Thus, thorough chemical and genomic exploration of SCLC cell linesmay provide starting points for further development of rationaltargeted therapeutic intervention in this deadly tumor type.
Over the past years the development of targeted therapies hasdramatically affected clinical treatment of lung cancer (1–3).
This development was sparked by the identification of mutations inEGFR (4–6) that confer exquisite sensitivity to EGFR inhibitors (2,7) andEML4-ALK fusions (8) thatmake tumors susceptible toALKinhibition (3). The recent identification ofFGFR1 amplification andDDR2mutations in squamous cell lung cancer (SQLC) patients hasfueled hopes that not only lung tumors of never-smokers beartherapeutically amenable genetic alterations (9, 10). However, insmall cell lung cancer (SCLC) the lack of specimens suitable fordeep genomic characterization has so far hampered similar effortsto identify novel therapeutically relevant genome alterations.Among the genes recurrently affected by genomic alterations in
SCLCareTP53,RB1, as well as theMYC family genes such asMYC,MYCL1, and MYCN, which are frequently amplified in a mutuallyexclusive manner (11, 12). The PI3-kinase (PI3K) pathway has beenproposed to be a therapeutically actionable signaling cascade that isactivated in SCLC (11) but the frequency of genetic alterationsdriving PI3-kinase activation is currently unclear (13). Furthermore,the Hedgehog (HH) pathway has been identified as a potentiallydruggable target in SCLC mouse models (14) but it is presentlyunclear whether HH signaling dependency segregates with partic-ular genetic alterations.Given the inherent difficulties in the rational design of potent
inhibitors of MYC and other transcription factors, alternative ther-apeutic strategies such as inhibition of MYC-MAX dimerization
and “synthetic lethality” have emerged (15–19). As a complementaryapproach, screening of libraries of small molecules across genomi-cally characterized cell line panels has revealed direct oncogenedependencies as well as synthetic lethal dependencies (20–23).The use of small molecules offers the advantage of immediatelyaddressing the question of whether a given vulnerability can bechemically attacked.To identify therapeutically relevant genome alterations in
SCLC, we performed a combined genomic and chemical vul-nerability analysis in a panel of 60 SCLC cell lines. This studyinvolved the screening of a library of 267 compounds across 44SCLC cell lines coupled to genomic characterization of theseand additional cell lines.
ResultsSimilarity of SCLC Cell Lines and Primary Tumors. We analyzedchromosomal gene copy number alterations in 60 patient-derivedSCLC cell lines (Dataset S1) using Affymetrix 6.0 SNP arrays anddetermined significant copy number alterations using the pre-viously described GISTIC algorithm (Dataset S2) (24, 25). Next,we compared the significant alterations present in the cell linecollection to the genetic alterations of a previously describedcollection of 63 primary SCLC specimens (Fig. 1A) (13). Con-firming an overall high similarity of SCLC cell lines and primarytumors, this analysis revealed a significant (r = 0.83) correlation
Author contributions: M.L.S., F.D., M.P., and R.K.T. designed research; M.L.S., F.D., J.S.,H.B.-W., C.M., M.K., S.H., F.M., J.M.H., P.M.S., and L.C.H. performed research; A.R., P.A.E.,R.T.U., A.P.A., V.V.V., T.N., H.W., and D.R. contributed new reagents/analytic tools; M.L.S.,F.D., M.P., J.S., H.B.-W., C.M., M.K., A.R., S.H., F.M., J.M.H., D.S., P.A.E., R.T.U., A.P.A., V.V.V.,P.M.S., T.N., H.W., R.B., D.R., L.C.H., and R.K.T. analyzed data; and M.L.S., F.D., and R.K.T.wrote the paper.
Conflict of interest statement: R.K.T. received consulting and lecture fees from Sanofi-Aventis, Merck KGaA, Bayer, Lilly, Roche, Boehringer Ingelheim, Johnson & Johnson,AstraZeneca, Atlas-Biolabs, Daiichi-Sankyo, and Blackfield as well as research supportfrom AstraZeneca, Merck, and EOS. R.K.T. is a founder and shareholder of Blackfield,a company involved in cancer genome services and cancer genomics-baseddrug discovery.
This article is a PNAS Direct Submission.
Freely available online through the PNAS open access option.
Data deposition: The SNP array data reported in this paper have been deposited in theGene Expression Omnibus (GEO) database, www.ncbi.nlm.nih.gov/geo (accession no.GSE40142).1M.L.S. and F.D. contributed equally to this work.2To whom correspondence may be addressed. E-mail: [email protected] or [email protected].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1207310109/-/DCSupplemental.
www.pnas.org/cgi/doi/10.1073/pnas.1207310109 PNAS Early Edition | 1 of 6
MED
ICALSC
IENCE
S

of copy number alterations in both datasets (Fig. 1A and SI Ap-pendix, Fig. S1A), similar to findings in other tumor types (20–22).Our cell line collection captures hallmark events of SCLC such asrecurrent deletions of RB1 and PTEN (13, 26) but also amplifica-tion of genes such as FGFR1 (11, 13). Furthermore, in bothdatasets we identified recurrent and focal amplification ofMYCL1,MYCN, and MYC (13). High-level MYCN amplification (inferredcopy number > 4) occurred in about 4–6% of cases in both data-sets, whereas MYCL1 (primary samples, 8% and cell lines, 22%)(Dataset S3) and MYC amplification (primary samples, 3% andcell lines, 15%)was detected with a higher prevalence in SCLC celllines (Fig. 1A–C andDataset S3) (27). Althoughmajor events suchas MYC amplification are found in both datasets, overall the sig-nificant copy number changes of SCLC differ from those found innon-small cell lung cancer (NSCLC) (r = 0.57) (SI Appendix, Fig.S1 B and C). Given the high prevalence of limited stage disease(∼68%) in the cohort of patient samples and the high prevalence ofadvanced stage disease in the case of the cell lines (∼95%) thefrequency ofMYCL1 (Dataset S3) andMYC amplification is likely
associated with the stage of the tumor as seen previously forMYC(Fig. 1 A–C and Dataset S3) (28, 29). However, cell line artifactsand a treatment bias might contribute to this association andcannot be formally excluded.To confirm our findings of significant copy number changes in
SCLC, we analyzed an independent cohort of 55 primary SCLCtissues for the presence ofMYC amplification using FISH (Fig. 1D).In accordance with published data (25), we identified high-levelamplification of the MYC gene in about 5.5% of primary SCLCsamples (Fig. 1 D and E). Thus, our data suggest that our cell linecollection captures major copy number alterations of SCLC.
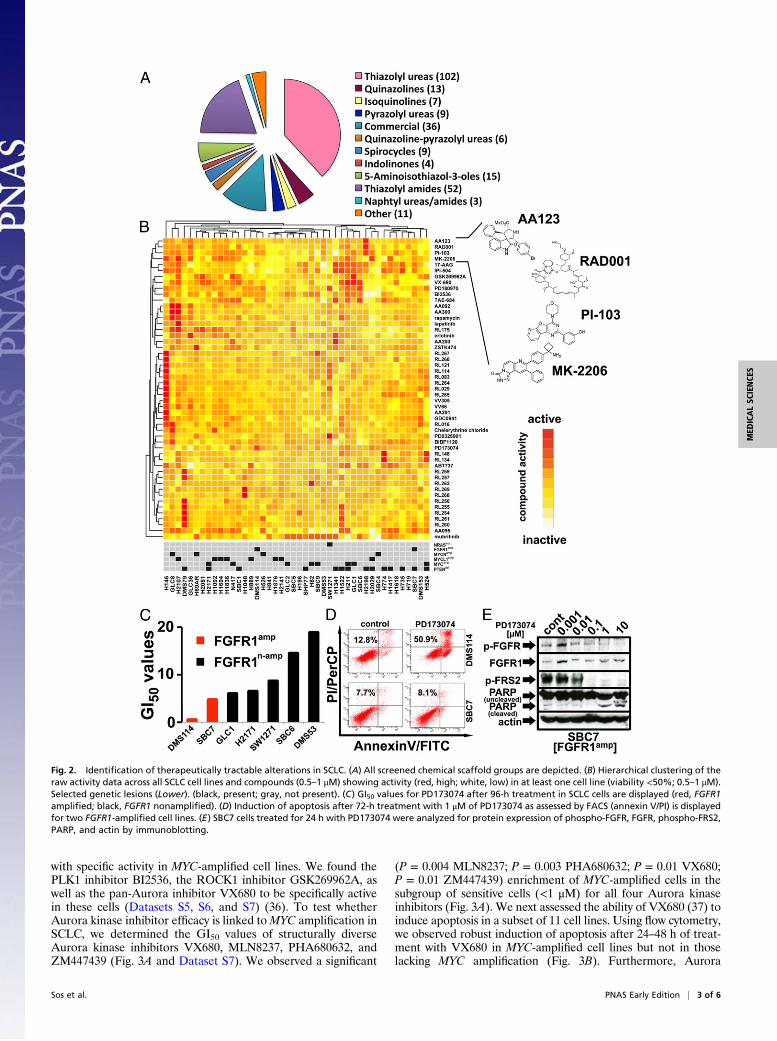
Activity Profiles of Clinically Relevant Targeted Compounds AcrossSCLC Cell Lines. We performed a systematic cell-based screen (44SCLC cell lines) against a library of 267 compounds with diversescaffolds (Fig. 2A), targeting a wide range of cellular proteins(Dataset S4 and SI Appendix, Fig. S2A) (30–34). Compound ac-tivity was assessed across cell lines as the remaining cellular via-bility at two different concentrations (Dataset S5). The resultingactivity profiles ranged from compounds with no activity (n = 97)at high concentrations (5–10 μM) across all cell lines to com-pounds with high activity at low concentrations (0.5–1 μM) acrossthe majority of cells (e.g., IPI-504) to highly selective compounds(e.g., PD173074 and PD0325904) (SI Appendix, Fig. S2B andDataset S5) showing activity in only a few cell lines.Using hierarchical clustering of the raw inhibitor activity data,
we identified compound groups of different scaffolds indicatingcommon targets (Fig. 2B). For example, the mTOR inhibitoreverolimus shared a cluster with the AKT inhibitor MK-2206, thePI3K inhibitor PI-103, and the spirooxindole derivative AA123,previously described to induce mitotic arrest in cellular assays(Fig. 2B and Dataset S4) (30). Our data therefore suggest thatAA123 might be a scaffold that inhibits the PI3K-signaling path-way. This analysis supports the robustness of our screening ap-proach and affords identification of unexpected cellular targetsfor unique compounds.To identify genetic predictors for the activity of the screened
compounds, we used signal-to-noise–based feature selectioncombined with the K-nearest-neighbor (KNN) algorithm (22)(Dataset S6). This analysis revealed that PTEN loss predicts cy-totoxic activity of the HSP90 inhibitor IPI-504 and its close ho-molog 17-AAG (P = 0.02 and P = 0.01; Fisher’s exact) (DatasetS6). Surprisingly, PTEN loss did not predict efficacy of PI3Kinhibitors (Dataset S6). Overall, these results suggest that in theclinical setting PTEN loss may be a genetic marker for the efficacyof HSP90 inhibitors but not PI3K inhibitors in SCLC.Next, we identified FGFR1 amplification as a predictor for the
activity (P = 0.05) of the FGFR inhibitor PD173074 (Dataset S6).FGFR1 amplification is a recurrent genome alteration in SQLC,associated with FGFR dependency in some lung cancer cell lines(9, 11, 35). To test whether FGFR1 amplification is also linked withcytotoxic activity of FGFR inhibition in SCLC, we determined theGI50 values for a subset of seven SCLC cells (Fig. 2C). One of thetwo FGFR1-amplified cell lines, DMS114, was previously shown tobe sensitive to PD173074 (GI50 = 0.46 µM) (9) (Fig. 2C). Bycontrast, the other FGFR1-amplified cell line, SBC7, was resistantto PD173074 and no apoptosis was induced upon treatment withthe FGFR inhibitor (Fig. 2 C and D and SI Appendix, Fig. S3A).These data suggest that not all FGFR1-amplified SCLC tumorsmay be dependent on FGFR1 activity. Because these cells lackexpression of PTEN (SI Appendix, Fig. S3B), we speculate thatPTEN loss may contribute to modulation of FGFR inhibitor sen-sitivity. Consequently, despite inducing dephosphorylation of theadaptor protein FRS2 in SBC7 cells (Fig. 2E), PD173074 inducedapoptosis in DMS114 cells but not in SBC7 cells (Fig. 2D).
MYC Amplification Predicts Efficacy of Aurora Kinase Inhibitors. Us-ing the KNN algorithm, we next sought to identify compounds
Fig. 1. SCLC cell line collection reflects major genetic lesions of SCLCpatients. (A) Significant copy number changes (amplifications are in thelower panel and deletions are in the upper panel) as defined by GISTIC (qvalues) in SCLC primary samples (green and brown) and in SCLC cells (red andblue). Selected genes are annotated. (B) Significant amplifications (exten-sive, red; limited, black) as defined by the GISTIC algorithm (g score) iden-tified in SCLC tumors. (C) Copy number changes at the MYC locus aredisplayed for the top 10 MYC amplified of extensive- (Left) and limited(Right)-stage samples. (D) FISH analysis of a sample showing amplification ofMYC (Left) and a sample with no MYC amplification (Right) (MYC, green;control, red). (E) Frequency of MYC amplification in primary samples asdetermined by SNP arrays in a published dataset (25) and by FISH in theindependent SCLC cohort.
2 of 6 | www.pnas.org/cgi/doi/10.1073/pnas.1207310109 Sos et al.
with specific activity in MYC-amplified cell lines. We found thePLK1 inhibitor BI2536, the ROCK1 inhibitor GSK269962A, aswell as the pan-Aurora inhibitor VX680 to be specifically activein these cells (Datasets S5, S6, and S7) (36). To test whetherAurora kinase inhibitor efficacy is linked toMYC amplification inSCLC, we determined the GI50 values of structurally diverseAurora kinase inhibitors VX680, MLN8237, PHA680632, andZM447439 (Fig. 3A and Dataset S7). We observed a significant
(P = 0.004 MLN8237; P = 0.003 PHA680632; P = 0.01 VX680;P = 0.01 ZM447439) enrichment of MYC-amplified cells in thesubgroup of sensitive cells (<1 μM) for all four Aurora kinaseinhibitors (Fig. 3A). We next assessed the ability of VX680 (37) toinduce apoptosis in a subset of 11 cell lines. Using flow cytometry,we observed robust induction of apoptosis after 24–48 h of treat-ment with VX680 in MYC-amplified cell lines but not in thoselacking MYC amplification (Fig. 3B). Furthermore, Aurora
Fig. 2. Identification of therapeutically tractable alterations in SCLC. (A) All screened chemical scaffold groups are depicted. (B) Hierarchical clustering of theraw activity data across all SCLC cell lines and compounds (0.5–1 μM) showing activity (red, high; white, low) in at least one cell line (viability <50%; 0.5–1 μM).Selected genetic lesions (Lower). (black, present; gray, not present). (C) GI50 values for PD173074 after 96-h treatment in SCLC cells are displayed (red, FGFR1amplified; black, FGFR1 nonamplified). (D) Induction of apoptosis after 72-h treatment with 1 μM of PD173074 as assessed by FACS (annexin V/PI) is displayedfor two FGFR1-amplified cell lines. (E) SBC7 cells treated for 24 h with PD173074 were analyzed for protein expression of phospho-FGFR, FGFR, phospho-FRS2,PARP, and actin by immunoblotting.
Sos et al. PNAS Early Edition | 3 of 6
MED
ICALSC
IENCE
S

inhibition induced a collapse of the mitochondrial membrane po-tential, specifically in MYC-amplified SCLC cells (SI Appendix,Fig. S4A). Inhibition of Aurora kinases also led to significantlyfaster G2/M-arrest inMYC-amplified SCLC cell lines, compatiblewith a generally increased proliferation rate in these cells (Fig. 3C).To test whether MYC-amplified SCLC cells are generally vulner-able to induction of a G2/M-arrest, we measured apoptosis aftertreatment with the prometaphase-arresting agent nocodazole. Nodifference between the induction of apoptosis with nocodazole orVX680was observed inMYC-nonamplified SCLC cells, whereas inMYC-amplified cells, we measured a significant difference (Wil-coxon test; P = 0.025) between nocodazole and VX680 (SI Ap-pendix, Fig. S4B), suggesting that VX680-induced cell cycle arrestis not the main driver of cytotoxicity in MYC-amplified cells.VX680 treatment led to dephosphorylation of Aurora A/B/C as
well as of histone H3 (HH3), a surrogate marker of Aurora Bsignaling, in all cell lines at concentrations in the range of the de-termined GI50 values (Fig. 3 A and D). Aurora inhibition wasparalleled by poly(ADP-ribose)-polymerase (PARP) cleavage anda reduction of phosphorylation of AKT in MYC-amplified cells(GLC1 and N417) but not in MYC-nonamplified SBC6 cells (Fig.
3D). The observed induction of apoptosis translated into inhibitionof tumor growth of VX680-treated (60mg/kg) mice engrafted withMYC-amplified cells (GLC1) (SI Appendix, Fig. S5). By contrast,no growth inhibition was observed in nude mice engrafted withMYC-nonamplified SW1271 cells (SI Appendix, Fig. S5). Of note,increasing concentrations of VX680 led to enhanced levels of p-AKT in the control cell line, indicating that inMYC-amplified cellsa lack of feedback loops that activate the PI3K pathway maycontribute to the Aurora dependency (Fig. 3D). Depletion ofAurora A was recently shown to destabilize MYCN protein inMYCN-amplified neuroblastoma; the catalytical activity of AuroraA was, however, not required (15). By contrast, inMYC-amplifiedSCLC, inhibition of Aurora kinase activity did not affect MYCprotein levels (Fig. 3D). Overall, these data indicate that Aurorakinase activity is specifically required for the survival of MYC-amplified SCLC cells and that the mechanism differs from Auroradependency in MYCN-amplified neuroblastoma.
Dependency on AURKB Activity in MYC-Amplified SCLC Cells. MYC isknown to be involved in the pathogenesis of diverse cancer types(38). To test the relevance of MYC amplification in SCLC, we
Fig. 3. Inhibition of Aurora kinases leads to celldeath and apoptosis in MYC-amplified cells. (A) GI50values (Dataset S7) for the Aurora kinase inhibitorsMLN8237, PHA680632, VX680, and ZM447439 (96-htreatment) in a subset of 34 SCLC cell lines (*MYCamplified). P values (Fisher’s exact test) are dis-played. (B) Induction of apoptosis after treatmentwith 1 μM of VX680 (12–48 h) as assessed by FACS(annexin V/PI) is displayed for six MYC-amplified andfive MYC-nonamplified cell lines. Inset shows repre-sentative pictures of GLC1 and SBC6 cells treatedwith either control or VX680. (C) Depicted is thefraction of cells in the G2/M-phase as measured byflow cytometry (PI signal) in six MYC-amplified(black) and five MYC-nonamplified cell lines (white).Error bars represent SD. (D) GLC1, N417, and SBC6cells treated with VX680 (24 h) were analyzed forprotein expression of Aurora A, phospho-HH3, PARP,phospho-AKT, AKT, MYC, and actin by immuno-blotting. Lysates for detection of phospho-AURKA/B/C were sonificated.
4 of 6 | www.pnas.org/cgi/doi/10.1073/pnas.1207310109 Sos et al.

silenced expression ofMYC (SI Appendix, Fig. S6A) in a subset ofSCLC cell lines and assessed their viability (Fig. 4A). We ob-served a strong association betweenMYC amplification and MYCdependency resulting in induction of cell death in MYC-amplifiedbut not in MYC-nonamplified cells (Fig. 4A). The MYC de-pendency ofMYC-amplified cells (SI Appendix, Fig. S6B) was alsolinked to gene expression of MYC but not AURKA and AURKB(SI Appendix, Fig. S6C) and independent of MYC, Aurora A, andAurora B protein expression (SI Appendix, Fig. S6D). Knockdownof MYC led to induction of apoptosis in MYC-amplified cells asassessed by PARP cleavage in immunoblotting assays (Fig. 4B).To validate our findings, we silenced expression of AURKA and
AURKB in a panel of SCLC cell lines (Fig. 4C). We had observedpreferential but subtle dephosphorylation of Aurora B over Au-rora A in two of the cell lines treated with VX680 (Fig. 3D, Centerand Left), compatible with requirement of Aurora B in the contextofMYC amplification in SCLC. Supporting this notion, knockdown
of AURKB but not of AURKA induced PARP cleavage in MYC-amplified SCLC cell lines (Fig. 4C). Note that the AURKA-tar-geted hairpin 3 also has off-target effects against AURKB (Fig. 4C,Left). The observed dependency on AURKB expression was cor-related with MYC dependency (r = 0.96) (SI Appendix, Fig. S6E).As a consequence, knockdown of AURKB (Fig. 4D) but notAURKA (Fig. 4E) resulted in a reduction of cell viability in MYC-amplified SCLC cells. As a further confirmation of preferentialdependency of MYC-amplified SCLC cells on Aurora B, we nexttested barasertib-hQPA (AZD1152), a compound with ∼1,000-fold selectivity against Aurora B compared with Aurora A (39).AZD1152 induced marked dephosphorylation of Aurora B andpartially Aurora C but not AuroraA at concentrations of 0.1 μM inall cell lines (Fig. 4F). However, AZD1152 treatment led to a re-duction of cell viability at lownanomolar concentrations only in thetwo MYC-amplified but not in MYC-nonamplified cells (Fig. 4G).Thus, further extending results obtained in genetically engineeredcells (36), the observed activity of Aurora kinase inhibition ispredominantly mediated by inhibition of Aurora B in MYC-dependent cells.
DiscussionThe findings of this combined genomic and chemical vulnerabilityscreen support the use of this approach to develop strategies forgenetically tailored therapies against SCLC. We demonstrate thatour cell line panel captures the major copy number alterations ofprimary SCLC tumors, thereby allowing extrapolating to actual pa-tient populations. Furthermore, the diversity of the screened inhib-itors provides a first broad assessment of pathway dependenciesacross representative SCLC genotypes. Building on previous studies(11, 40), the scaling of both the number of cell lines and the numberof compounds afforded identification of vulnerabilities associatedwith infrequent genome alterations, such as amplifications ofFGFR1andMYC. In the case ofFGFR1 amplification, further studies will berequired to clarify the frequency of FGFR1 dependency in SCLC,as other genetic lesions may play a role in the responsiveness toFGFR inhibition.Furthermore, we describe and functionally characterize the
dependency of MYC-amplified SCLC tumors on Aurora B (41).We find thatMYC-amplified tumors depend on the kinase activityof Aurora B for their survival (36). Currently a series of Aurorakinase inhibitors (e.g., MLN8237 and PHA739358) including theAurora B-selective inhibitor barasertib (AZD1152) are un-dergoing clinical evaluation in phase I/II studies (42, 43). Ourdata provide a rationale for the testing of these compounds ingenetically defined SCLC patient groups.Previous studies did not support a role for Aurora dependency in
NSCLC cell lines of different genotpyes (22) or implied differentMYC family genes in amixed lung cancer panel (40), thus indicatingthat the role of amplifiedMYC and its dependency onAURKB (36)may differ between different cancer subtypes. Thus, our data pro-vide a lineage-specific extension of previous data generated inlymphoma mouse models driven by an active transgene of MYC(36). Compatible with this notion is the recent finding that BRAFmutations associate with BRAF inhibitor sensitivity in melanomabut not colorectal cancer (44, 45). Our data not only point out thedifferences in the biology of distinct subtypes of lung cancer but alsounderscore the differences in oncogenic signaling of MYC genefamily members: although amplifications of MYCL1 and MYCNamplifications occur in a mutually exclusive fashion with MYCamplification (suggesting genetic epistasis), they do not segregatewith vulnerability to Aurora B inhibition in SCLC.Although MYC-amplified SCLC represents only a small subset
of lung cancer patients, recent experience with the successful in-troduction of ALK inhibitors for the treatment of about 2–3%ALK fusion positive adenocarcinomas suggest that genetic strati-fication is feasible and beneficial, even in small subgroups (3). Ourstudy provides a framework for preclinical testing of genetically
Fig. 4. MYC-amplified SCLC cells depend on MYC and Aurora B proteinexpression. (A) Viability of SCLC cells after transduction with MYC-shRNAselected from a set of four shRNA constructs (SI Appendix, Fig. S6A) com-pared with controls is displayed. Error bars represent SD. (B) GLC1, H211,N417, and SBC6 cells after transduction with either control or MYC-shRNAwere analyzed for MYC and PARP protein by immunoblotting. (C) N417(Left) and SW1271 (Right) cells after transduction with either control orAURKA/B-shRNA constructs were analyzed for protein expression of AuroraA/B, PARP, and MYC by immunoblotting. (D) Viability of SCLC cells aftertransduction with AURKB-shRNA or AURKA-shRNA (E) compared withcontrols was assessed. (F) N417, GLC1, SW1271, and SBC4 cells treated withAZD1152 (24 h) were analyzed for protein expression of phospho-Aurora A/B/C, Aurora A/B, and actin by immunoblotting. Lysates for detection ofphospho-AURKA/B/C were sonificated. (G) Viability of N417, GLC1, (MYC-amp, red) SW1271, and SBC4 (MYC-nonamp, black) cells after 96-h treat-ment with barasertib (AZD1152) was determined with celltiter-glo (CTG)assays. Error bars represent SD.
Sos et al. PNAS Early Edition | 5 of 6
MED
ICALSC
IENCE
S

encrypted vulnerabilities in SCLC. In support of this notion, ourinitial screen has already yielded actionable targets for furtherpreclinical validation and possible clinical testing. We thereforehope that—in the longer term—our approach might help to im-prove the disappointing survival rates of these patients.
MethodsAll patients gave written informed consent sample analysis. The tumor speci-mens have been collected under local institutional review board approval(University of Cologne, Cologne, Germany) and genomic analyses were per-formed as described elsewhere (13). GISTIC analyses of a group of 60 SCLC celllines were performed as described previously (24, 25). For a subgroup of 36 celllines, previously published copy number data (www.sanger.ac.uk/genetics/CGP/CellLines) were used. The human genome build hg18 was used. All rawcopy number data have been deposited in the Gene Expression Omnibus da-tabase (accession no. GSE40142). The cell line collection of 44 unique small celllung cancer patient-derived cells was used for cell-based screening against 267inhibitors using CellTiterGlo as a growth inhibition assay. Detailed analysis wasperformedona subset of 51 compounds that showedactivity in at least one cellline at concentrations of 0.5–1 μM. Annexin V and propidium iodide (PI)staining was used to measure apoptosis in flow cytometry assays. For thesubgroup of cell lines where the raw copy number datawere not generated inhouse, genotyping of a panel of 11 SNPs was performed to control the re-spective annotation. Cell viability was measured at two concentrations in
triplicates and compared with DMSO controls. Calculation of the P values wasperformed using a Wilcoxon rank sum test, a two-tailed t test, or a Fisher’sexact test implemented in “R”. Pharmacodynamic response of signaling wasmeasured by immunoblotting of cellular lysates of treated cells using phospho-specific antibodies. Not all bands were detected at the samemembrane due tooverlapping protein sizes. RT-PCR assayswere performedwith SYBRGreen andprimers for the respective genes and GAPDH as control. For gene silencing,lentiviruses were produced with pLKO.1-Puro–based vectors. After trans-duction, cell viability was measured by measuring cell numbers of quad-ruplicates and normalized to viability of cell lines transduced with controlconstructs. All animal procedures were performed in agreement with the an-imal protection committee and the local authorities and treatment was per-formed as described previously (22).
ACKNOWLEDGMENTS. We thank Drs. Christian Reinhardt and HamidKashkar for support and AstraZeneca for providing barasertib-hQPA(AZD1152). This work was supported by the European Union-FrameworkProgramme CURELUNG (HEALTH-F2-2010-258677 to R.K.T.); by the Deut-sche Forschungsgemeinschaft through TH1386/3-1 (to R.K.T. and M.L.S.)and through SFB832 (TP6 to R.K.T. and TP5 to L.C.H.); by the German Min-istry of Science and Education as part of the National Network for the Studyof the Genome (NGFN) plus program (Grant 01GS08100 to R.K.T.), by theMax Planck Society and by the Behrensweise Foundation (M.I.F.A.NEUR8061 to R.K.T.); and by an anonymous foundation (R.K.T.). M.L.S. isa fellow of the International Association for the Study of Lung Cancer.
1. Pao W, Chmielecki J (2010) Rational, biologically based treatment of EGFR-mutantnon-small-cell lung cancer. Nat Rev Cancer 10:760–774.
2. Rosell R, et al.; Spanish Lung Cancer Group (2009) Screening for epidermal growthfactor receptor mutations in lung cancer. N Engl J Med 361:958–967.
3. Kwak EL, et al. (2010) Anaplastic lymphoma kinase inhibition in non-small-cell lungcancer. N Engl J Med 363:1693–1703.
4. Lynch TJ, et al. (2004) Activating mutations in the epidermal growth factor receptorunderlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med350:2129–2139.
5. Paez JG, et al. (2004) EGFR mutations in lung cancer: correlation with clinical responseto gefitinib therapy. Science 304:1497–1500.
6. Pao W, et al. (2004) EGF receptor gene mutations are common in lung cancers from“never smokers” and are associated with sensitivity of tumors to gefitinib and erlo-tinib. Proc Natl Acad Sci USA 101:13306–13311.
7. Mok TS, et al. (2009) Gefitinib or carboplatin-paclitaxel in pulmonary adenocarci-noma. N Engl J Med 361:947–957.
8. Soda M, et al. (2007) Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 448:561–566.
9. Weiss J, et al.; Frequent and Focal FGFR1 Amplification Associates with Therapeuti-cally Tractable FGFR1 Dependency in Squamous Cell Lung Cancer (2010) Frequent andfocal FGFR1 amplification associates with therapeutically tractable FGFR1 dependencyin squamous cell lung cancer. Sci Transl Med 2:62ra93.
10. Hammerman PS, et al. (2011) Mutations in the DDR2 kinase gene identify a noveltherapeutic target in squamous cell lung cancer. Cancer Discov 1:78–89.
11. Voortman J, et al. (2010) Array comparative genomic hybridization-based charac-terization of genetic alterations in pulmonary neuroendocrine tumors. Proc Natl AcadSci USA 107:13040–13045.
12. Wistuba II, Gazdar AF, Minna JD (2001) Molecular genetics of small cell lung carci-noma. Semin Oncol 28(2, Suppl 4):3–13.
13. Peifer M, et al. (2012) Integrative genome analyses identify key somatic driver mu-tations of small-cell lung cancer. Nat Genet, 10.1038/ng.2396.
14. Park KS, et al. (2011) A crucial requirement for Hedgehog signaling in small cell lungcancer. Nat Med 17:1504–1508.
15. Otto T, et al. (2009) Stabilization of N-Myc is a critical function of Aurora A in humanneuroblastoma. Cancer Cell 15:67–78.
16. Prochownik EV, Vogt PK (2010) Therapeutic targeting of Myc. Genes Cancer 1(6):650–659.
17. Kessler JD, et al. (2012) A SUMOylation-dependent transcriptional subprogram isrequired for Myc-driven tumorigenesis. Science 335:348–353.
18. Toyoshima M, et al. (2012) Functional genomics identifies therapeutic targets forMYC-driven cancer. Proc Natl Acad Sci USA 109:9545–9550.
19. Liu L, et al. (2012) Deregulated MYC expression induces dependence upon AMPK-related kinase 5. Nature 483:608–612.
20. Barretina J, et al. (2012) The Cancer Cell Line Encyclopedia enables predictive mod-elling of anticancer drug sensitivity. Nature 483:603–607.
21. Garnett MJ, et al. (2012) Systematic identification of genomic markers of drug sen-sitivity in cancer cells. Nature 483:570–575.
22. Sos ML, et al. (2009) Predicting drug susceptibility of non-small cell lung cancers basedon genetic lesions. J Clin Invest 119:1727–1740.
23. Solit DB, et al. (2006) BRAF mutation predicts sensitivity to MEK inhibition. Nature439:358–362.
24. Beroukhim R, et al. (2007) Assessing the significance of chromosomal aberrations incancer: Methodology and application to glioma. Proc Natl Acad Sci USA 104:20007–20012.
25. Beroukhim R, et al. (2010) The landscape of somatic copy-number alteration acrosshuman cancers. Nature 463:899–905.
26. Sutherland KD, et al. (2011) Cell of origin of small cell lung cancer: Inactivation ofTrp53 and Rb1 in distinct cell types of adult mouse lung. Cancer Cell 19:754–764.
27. Kim YH, et al. (2006) Combined microarray analysis of small cell lung cancer revealsaltered apoptotic balance and distinct expression signatures of MYC family geneamplification. Oncogene 25:130–138.
28. Johnson BE, et al. (1987) myc family oncogene amplification in tumor cell lines es-tablished from small cell lung cancer patients and its relationship to clinical status andcourse. J Clin Invest 79:1629–1634.
29. Gazzeri S, et al. (1991) Activation of myc gene family in human lung carcinomas andduring heterotransplantation into nude mice. Cancer Res 51:2566–2571.
30. Antonchick AP, et al. (2010) Highly enantioselective synthesis and cellular evaluationof spirooxindoles inspired by natural products. Nat Chem 2:735–740.
31. Getlik M, et al. (2009) Hybrid compound design to overcome the gatekeeper T338Mmutation in cSrc. J Med Chem 52:3915–3926.
32. Rode HB, et al. (2011) Synthesis and biological evaluation of 7-substituted-1-(3-bro-mophenylamino)isoquinoline-4-carbonitriles as inhibitors of myosin light chain kinaseand epidermal growth factor receptor. Bioorg Med Chem 19:429–439.
33. Simard JR, et al. (2009) A new screening assay for allosteric inhibitors of cSrc. NatChem Biol 5:394–396.
34. Vintonyak VV, et al. (2010) Identification of thiazolidinones spiro-fused to indolin-2-ones as potent and selective inhibitors of the Mycobacterium tuberculosis proteintyrosine phosphatase B. Angew Chem Int Ed Engl 49:5902–5905.
35. Dutt A, et al. (2011) Inhibitor-sensitive FGFR1 amplification in human non-small celllung cancer. PLoS ONE 6:e20351.
36. Yang D, et al. (2010) Therapeutic potential of a synthetic lethal interaction betweenthe MYC proto-oncogene and inhibition of aurora-B kinase. Proc Natl Acad Sci USA107:13836–13841.
37. Tyler RK, Shpiro N, Marquez R, Eyers PA (2007) VX-680 inhibits Aurora A and Aurora Bkinase activity in human cells. Cell Cycle 6:2846–2854.
38. Meyer N, Penn LZ (2008) Reflecting on 25 years with MYC. Nat Rev Cancer 8:976–990.39. Mortlock AA, et al. (2007) Discovery, synthesis, and in vivo activity of a new class of
pyrazoloquinazolines as selective inhibitors of aurora B kinase. J Med Chem 50:2213–2224.
40. Hook KE, et al. (2012) An integrated genomic approach to identify predictive bio-markers of response to the aurora kinase inhibitor PF-03814735. Mol Cancer Ther 11:710–719.
41. Carmena M, Earnshaw WC (2003) The cellular geography of aurora kinases. Nat RevMol Cell Biol 4:842–854.
42. Cohen RB, et al. (2009) A phase I dose-escalation study of danusertib (PHA-739358)administered as a 24-hour infusion with and without granulocyte colony-stimulatingfactor in a 14-day cycle in patients with advanced solid tumors. Clin Cancer Res 15:6694–6701.
43. Löwenberg B, et al. (2011) Phase 1/2 study to assess the safety, efficacy, and phar-macokinetics of barasertib (AZD1152) in patients with advanced acute myeloid leu-kemia. Blood 118:6030–6036.
44. Corcoran RB, et al. (2012) EGFR-mediated re-activation of MAPK signaling contributesto insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib.Cancer Discov 2:227–235.
45. Prahallad A, et al. (2012) Unresponsiveness of colon cancer to BRAF(V600E) inhibitionthrough feedback activation of EGFR. Nature 483:100–103.
6 of 6 | www.pnas.org/cgi/doi/10.1073/pnas.1207310109 Sos et al.