a peptide corresponding to gpiib, 300-312, a presumptive
TRANSCRIPT

THE JOURNAL OF BIOLOGICAL CHEMISTRY (0 1992 hy The American Society for Biochemistry and Molecular Biology, Inc
Vol. 267, No. 17, Issue of June 15, pp. 11729-11733,1992 Printed in U. S. A .
A Peptide Corresponding to GPIIb, 300-312, a Presumptive Fibrinogen y-Chain Binding Site on the Platelet Integrin GPIIb/IIIa, Inhibits the Adhesion of Platelets to at Least Four Adhesive Ligands*
(Received for publication, March 17, 1992)
Donald B. Taylor$ and T. Kent Gartner From the Department of Biology, Memphis State University, Memphis, Tennessee 38152
The platelet fibrinogen (Fg) receptor (GPIIbpIIa) is an integrin which plays a critical role in hemostasis by recognizing at least the four adhesive ligands: Fg, fi- bronectin (Fn), vitronectin (Vn), and von Willebrand factor (vWf). We reported that residues 309-312 of GPIIb, appear to comprise at least part of a Fg binding site on the Fg receptor (Gartner, T. K., and Taylor, D. B. (1990) Thrornb. Res. 60,291-309). Here we report that the peptide GPIIb, 300-312 (G13) inhibits plate- let aggregation and binds Fg and Vn. Significantly, this peptide inhibits the adhesion of stimulated plate- lets to Fg, Fn, Vn, and vWf, but not the adhesion of resting platelets to Fn. Thus, GPIIb 300-312 may constitute a specific but common recognition site on GPIIb/IIIa for both LGGAKQAGDV- and RGD-con- taining ligands.
In response to blood vessel injury, platelets mediate hemo- stasis by adhering, spreading, and aggregating on exposed extracellular matrix components of the damaged subendothe- lium (1, 2). These cellular adhesion events are critical for the maintenance of normal vascular integrity. These platelet functions involve the recognition of large adhesive glycopro- teins such as Fg,’ Fn, Vn, and vWf by the platelet integrin GPIIb/IIIa (3-7). Integrins are heterodimeric complexes con- sisting of distinct a and /3 subunits which serve as cell surface receptors for adhesive ligands (8-13). They participate in a wide variety of cellular functions including cell adhesion and migration, development, hemostasis, the immune response, and wound healing (8-13). Many of the integrins recognize a common amino acid sequence (RGD) as at least part of their minimum ligand recognition site (8). Synthetic peptides cor- responding to the individual RGDX sequences present in Fg, Fn, Vn, and vWf each can inhibit these ligand/receptor inter- actions (14-18). Consistent with this ability, RGD-containing peptides can interact directly with a& (aIIb = GPIIb; = GPIIIa) (19-21).
The binding of Fg to GPIIb/IIIa is more complex than the
* This work was supported by National Institutes of Health Grants HL42523 and HL46152. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
$Recipient of a Van Vleet Foundation research fellowship. To whom correspondence should be addressed Dept. of Biology, Mem- phis State University, Memphis, TN 38152. Tel.: 901-678-2977; Fax:
The abbreviations used are: Fg, fibrinogen; BSA, bovine serum
tor; G13, the peptide GDGRHDLLVGAPL; L10, the peptide albumin; Fn, fibronectin; Vn, vitronectin; vWf, von Willebrand fac-
LGGAKQAGDV; PBS, phosphate-buffered saline.
901-678-3299.
binding of Fn, Vn, and vWf to this receptor. This complexity exists because Fg contains a non-RGD-containing GPIIb/IIIa recognition sequence (HHLGGAKQAGDV), which is present in the carboxyl terminus of the y-chain of Fg (22, 23). The other GPIIb/IIIa ligands, Fn, Vn, and vWf, have the common RGD sequence but lack the HHLGGAKQAGDV site. A syn- thetic peptide derivative of the y-chain recognition sequence cross-links selectively to residues 294-314 of GPIIb (24). We reported that a peptide with the sequence Gly-Ala-Pro-Leu (GAPL) found as residues 309-312 in GPIIb appears to com- prise at least part of a Fg binding site on GPIIb (25). In collaboration with Amrani and Kirschbaum (26-28), we re- cently demonstrated that GAPL-containing peptides inhibit the binding of the Fg y-chain to platelets. To further char- acterize this platelet-specific LY subunit in platelet function, a peptide corresponding to residues 300-312 of GPIIb (GDGRHDLLVGAPL (G13)) was synthesized. Because G13 is present in the region of GPIIb that was cross-linked by a derivative of the y-chain of Fg, which is thought to represent a unique recognition site for Fg, we were interested in deter- mining if G13 could serve as a common recognition site for the binding of the other RGD adhesive ligands (which lack the HHLGGAKQAGDV sequence: Vn, vWf, and Fn) to GPIIb/IIIa.
EXPERIMENTAL PROCEDURES
Synthetic peptides were synthesized, purified, and characterized as previously described (25). The purified peak 1 Fg and the initial batch of the EIIYPAGLV peptide were generous gifts from Dr. David Amrani, University of Wisconsin; the a-thrombin was provided by Dr. John Fenton 11, New York State Dept. of Health; the purified vitronectin was kindly provided by Dr. Deane Mosher and Steve Bittorf, University of Wisconsin; the von Willebrand factor was generously donated by Dr. Zaverio Ruggeri, Scripps Research Clinic; the thrombospondin was a generous gift of Dr. Dan Walz, Wayne State University; ADP, L-epinephrine, ovalbumin, and BSA (bovine serum albumin) were from Sigma; collagen (calf skin, grade A) was purchased from Calbiochem; sodium chromate (51Cr, 250-500 mCi/ mg Cr) was from Du Pont-New England Nuclear; t-butoxycarbonyl amino acids and Merrifield resins for solid-phase peptide synthesis were from Bachem Inc. (Torrance, CA) and Peninsula Labs (Belmont, CA).
Platelet Aggregation-Whole blood obtained from aspirin-free hu- man donors was drawn into acid citrate dextrose (39 mM citric acid, 75 mM sodium citrate, 135 mM glucose, pH 4.5). Washed platelets were prepared as previously described (25). Aggregation studies were performed at 37 ‘C using a single-channel aggregometer (Chrono-log Corp., Havertown, PA). Briefly, 0.1 ml of a 1 X 109/ml platelet suspension was diluted with 0.35 ml of Ca2+-free Tyrode’s solution (1 g of glucose/liter, 8 g of NaCl/liter, 1 g of NaHCOa/liter, 0.2 g of KCl/liter, 0.1 g of MgC12. 6H20/liter, 0.05 g of NaH2P04. H20/liter, pH 7.4) and the indicated amounts of peptides were added 30 s prior to initiation of aggregation by 0.05 units of thrombin.
Dot Blot Assays-Briefly, purifiedpeak 1 Fg (5 pg) or other proteins were applied to the center of nitrocellulose squares, allowed to dry,
11729
11730 GPIIb 300-312 Peptide Acts as Common Ligand Binding Site and post-coated with 1% BSA for 1 h at 37 "C. Blots were incubated with 100 pg of peptide/0.3 ml of PBS at 37 "C for 2 h. Blots were washed 3 times with blot buffer (PBS, 0.05% Tween 20, and 0.5% BSA) and subsequently incubated with purified monospecific anti- G13 polyclonal Ig (5 pg/ml) or preimmune IgG (first antibody) for 90 min at ambient temperature. Following washes (3 times), the blots were incubated with a 1:lOOO goat anti-rabbit IgG horseradish per- oxidase conjugate (second antibody) for 1 h. After washing, blots were developed using H202 and 4-chloro-1-naphthol. Rabbits were immu- nized with GI3 derivatized to keyhole limpet hemocyanin (Sigma) according to a previously reported immunization protocol (29). Anti- serum raised against GI3 was preadsorbed against nonderivatized Sepharose 4B and precipitated with 40% (NH4)2S04, and IgGs were isolated by affinity chromatography using a protein G column (Boeh- ringer Mannheim). Anti-G13 IgG fractions were made monospecific by affinity chromatography with a GDGRHDLLVGAPL-Sepharose 4B column. IgGs were isolated from preimmune sera obtained from the same rabbit prior to immunization with the G13.
Platelet Adhesion Assays to Fg-The platelet adhesion assays were performed as previously described (25, 31). Chromium labeled plate- lets were stimulated sequentially with epinephrine (5 p ~ ) for 5 min and then with ADP (30 p ~ ) for 5 min prior to use (25,31). Adhesion was allowed to proceed for 30 min prior to termination of the assay. Two types of controls were used as indicators of nonspecific binding in these assays: 1) platelet adhesion was measured on BSA-coated wells, and 2) platelets were incubated at 37 "C at pH 7.8 in the presence of 5 mM EDTA for 15 min to irreversibly inactivate GPIIb/ IIIa complexes thereby minimizing adhesion to Fg mediated by its receptor (25). Typically 5-8% of the platelets in the platelet suspen- sion adhered.
Platelet Adhesion to Fn, Vn, and uWf-Platelet adhesion assays were performed as described (25,31) with the following modifications. Fn and Vn were dissolved in 0.1 ml of adhesion solution (25) at a final concentration of 5 pg/ml and incubated in wells of Immulon-1 microtiter plates (Dynatech) overnight at 4 "C. vWf was dissolved in 0.05 ml of adhesion solution at a final concentration of 10 pg/ml. For assays of platelet adhesion to Fn and Vn, labeled platelets were stimulated, first with epinephrine (5 pM for Vn, 20 pM for Fn) for 5 min and then with ADP (30 p ~ ) for 5 min, prior to use (25). For assays of platelet adhesion to vWf, platelets were stimulated with cy- thrombin (1.0 unit/ml). Adhesion was allowed to proceed for 30 min except for the Vn experiments, which were allowed to proceed for 60 min prior to termination of the assays. Adhesion of stimulated plate- lets to all three ligands was saturable and could be inhibited with GRGDSP or LGGAKQAGDV peptides and by 5 mM EDTA.
Adhesion of Resting Platelets to Fn-Assays of adhesion of stimu- lated platelelets to Fn were performed as described previously. Ex- periments for adhesion of resting platelets to Fn were performed exactly as described (33) except the platelets were allowed to incubate in the Fn-coated wells for 45 min instead of 30 prior to aspiration. Fn was purified essentially as reported (33). Briefly, fibronectin was isolated from fresh human citrated plasma (pH 6.4) by affinity chromatography on gelatin-Sepharose (Pharmacia). Plasma (100-150 ml) was applied to the column (50-ml beads) which had been equili- brated with 0.05 M Tris-HC1 buffer (pH 7.6), 0.15 M NaCl, 0.025 M c- aminocaproic acid. After all of the plasma had entered the beads, the column was washed extensively with equilibration buffer until the absorbance reached baseline. Prior to elution of bound fibronectin, the column was washed sequentially with 1 M NaCl and 1 M arginine in equilibration buffer (50 ml each). Fibronectin was eluted with 1 M NaBr in 0.05 Tris-HCI, 0.025 M c-aminocaproic acid (pH 5.3). The protein-containing fractions were pooled and extensively dialyzed against 0.039 M Tris/phosphate buffer, pH 8.6, and concentrated via ultrafiltration. The isolated fibronectin yielded a single band on sodium dodecyl sulfate-polyacrylamide gels under nonreducing con- ditions and two closely spaced bands of high molecular weight (215,000-230,000) under reducing conditions.
RESULTS
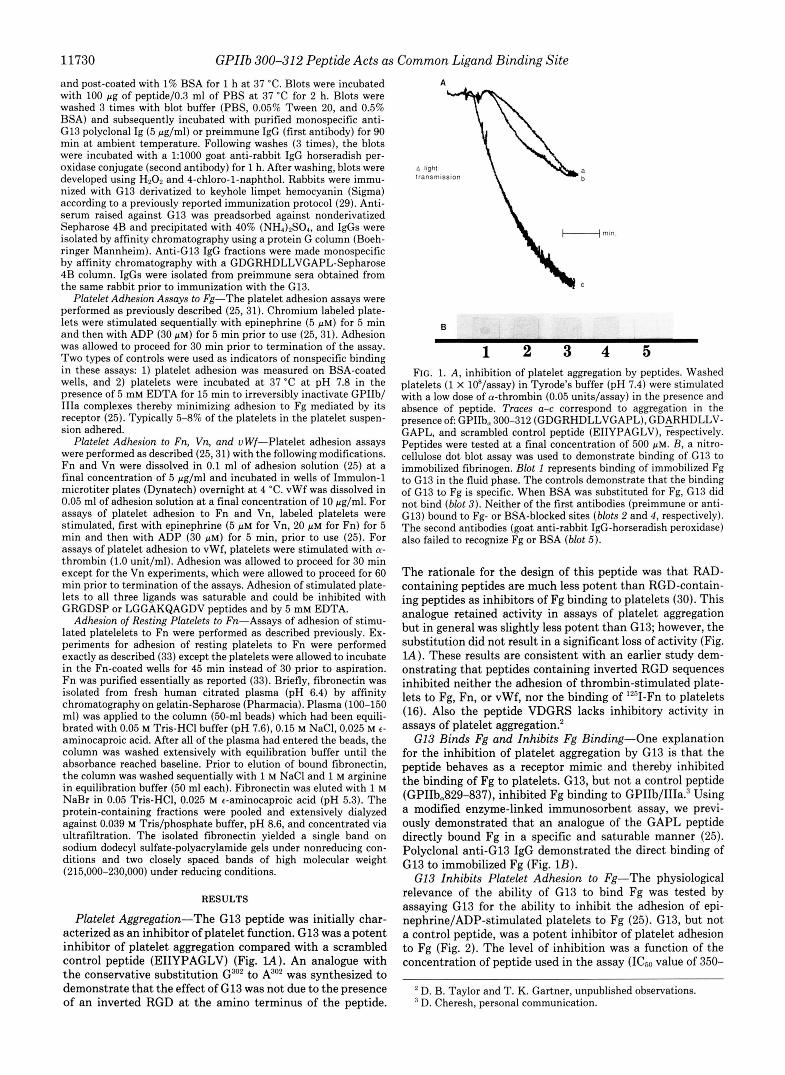
Platelet Aggregation-The G13 peptide was initially char- acterized as an inhibitor of platelet function. G13 was a potent inhibitor of platelet aggregation compared with a scrambled control peptide (EIIYPAGLV) (Fig. lA). An analogue with the conservative substitution G302 to A302 was synthesized to demonstrate that the effect of G13 was not due to the presence of an inverted RGD at the amino terminus of the peptide.
A
B
1 2 3 4 5 FIG. 1. A, inhibition of platelet aggregation by peptides. Washed
platelets (1 X IO*/assay) in Tyrode's buffer (pH 7.4) were stimulated with a low dose of cy-thrombin (0.05 units/assay) in the presence and
presence oE GPIIb, 300-312 (GDGRHDLLVGAPL), GDARHDLLV- absence of peptide. Traces a-c correspond to aggregation in the
GAPL, and scrambled control peptide (EIIYPAGLV), respectively. Peptides were tested at a final concentration of 500 p ~ . B, a nitro- cellulose dot blot assay was used to demonstrate binding of G13 to immobilized fibrinogen. Blot 1 represents binding of immobilized Fg to GI3 in the fluid phase. The controls demonstrate that the binding of G13 to Fg is specific. When BSA was substituted for Fg, G13 did not bind (blot 3). Neither of the first antibodies (preimmune or anti- G13) bound to Fg- or BSA-blocked sites (blots 2 and 4, respectively). The second antibodies (goat anti-rabbit IgG-horseradish peroxidase) also failed to recognize Fg or BSA (blot 5).
The rationale for the design of this peptide was that RAD- containing peptides are much less potent than RGD-contain- ing peptides as inhibitors of Fg binding to platelets (30). This analogue retained activity in assays of platelet aggregation but in general was slightly less potent than G13; however, the substitution did not result in a significant loss of activity (Fig. IA). These results are consistent with an earlier study dem- onstrating that peptides containing inverted RGD sequences inhibited neither the adhesion of thrombin-stimulated plate- lets to Fg, Fn, or vWf, nor the binding of lZ5I-Fn to platelets (16). Also the peptide VDGRS lacks inhibitory activity in assays of platelet aggregation?
G13 Binds Fg and Inhibits Fg Binding-One explanation for the inhibition of platelet aggregation by G13 is that the peptide behaves as a receptor mimic and thereby inhibited the binding of Fg to platelets. G13, but not a control peptide (GPIIb,829-837), inhibited Fg binding to GPIk1/111a.~ Using a modified enzyme-linked immunosorbent assay, we previ- ously demonstrated that an analogue of the GAPL peptide directly bound Fg in a specific and saturable manner (25). Polyclonal anti-G13 IgG demonstrated the direct binding of G13 to immobilized Fg (Fig. 1B).
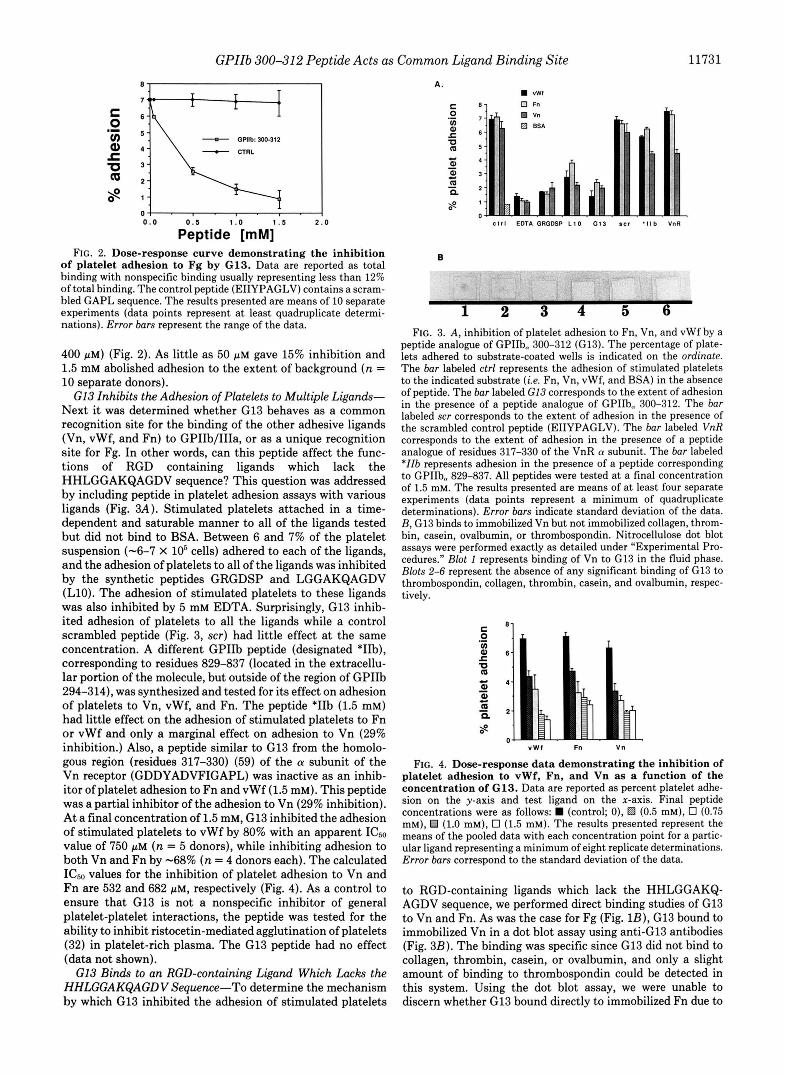
G13 Inhibits Platelet Adhesion to Fg-The physiological relevance of the ability of G13 to bind Fg was tested by assaying G13 for the ability to inhibit the adhesion of epi- nephrine/ADP-stimulated platelets to Fg (25). G13, but not a control peptide, was a potent inhibitor of platelet adhesion to Fg (Fig. 2). The level of inhibition was a function of the concentration of peptide used in the assay (ICso value of 350-
D. B. Taylor and T. K. Gartner, unpublished observations. D. Cheresh, personal communication.
GPIIb 300-312 Peptide Acts as Common Ligand Binding Site 11731
"b GPllb300-312
-t CTRL
O l . , . , . , . \ 0 . 0 0 . 5 1 . o 1 . 5 2 . 0
Peptide [mM] FIG. 2. Dose-response curve demonstrating the inhibition
of platelet adhesion to Fg by G13. Data are reported as total binding with nonspecific binding usually representing less than 12% of total binding. The control peptide (EIIYPAGLV) contains a scram- bled GAPL sequence. The results presented are means of 10 separate experiments (data points represent a t least quadruplicate determi- nations). Error bars represent the range of the data.
400 pM) (Fig. 2). As little as 50 p~ gave 15% inhibition and 1.5 mM abolished adhesion to the extent of background ( n = 10 separate donors).
G13 Inhibits the Adhesion of Platelets to Multiple Ligands- Next it was determined whether G13 behaves as a common recognition site for the binding of the other adhesive ligands (Vn, vWf, and Fn) to GPIIb/IIIa, or as a unique recognition site for Fg. In other words, can this peptide affect the func- tions of RGD containing ligands which lack the HHLGGAKQAGDV sequence? This question was addressed by including peptide in platelet adhesion assays with various ligands (Fig. 3A). Stimulated platelets attached in a time- dependent and saturable manner to all of the ligands tested but did not bind to BSA. Between 6 and 7% of the platelet suspension (-6-7 x lo5 cells) adhered to each of the ligands, and the adhesion of platelets to all of the ligands was inhibited by the synthetic peptides GRGDSP and LGGAKQAGDV (L10). The adhesion of stimulated platelets to these ligands was also inhibited by 5 mM EDTA. Surprisingly, G13 inhib- ited adhesion of platelets to all the ligands while a control scrambled peptide (Fig. 3, scr) had little effect at the same concentration. A different GPIIb peptide (designated *I%), corresponding to residues 829-837 (located in the extracellu- lar portion of the molecule, but outside of the region of GPIIb 294-314), was synthesized and tested for its effect on adhesion of platelets to Vn, vWf, and Fn. The peptide *IIb (1.5 mM) had little effect on the adhesion of stimulated platelets to Fn or vWf and only a marginal effect on adhesion to Vn (29% inhibition.) Also, a peptide similar to G13 from the homolo- gous region (residues 317-330) (59) of the a subunit of the Vn receptor (GDDYADVFIGAPL) was inactive as an inhib- itor of platelet adhesion to Fn and vWf (1.5 mM). This peptide was a partial inhibitor of the adhesion to Vn (29% inhibition). At a final concentration of 1.5 mM, G13 inhibited the adhesion of stimulated platelets to vWf by 80% with an apparent ICso value of 750 p~ ( n = 5 donors), while inhibiting adhesion to both Vn and Fn by -68% ( n = 4 donors each). The calculated ICs0 values for the inhibition of platelet adhesion to Vn and Fn are 532 and 682 p ~ , respectively (Fig. 4). As a control to ensure that G13 is not a nonspecific inhibitor of general platelet-platelet interactions, the peptide was tested for the ability to inhibit ristocetin-mediated agglutination of platelets (32) in platelet-rich plasma. The G13 peptide had no effect (data not shown).
G13 Binds to an RGD-containing Ligand Which Lacks the HHLGGAKQAGDV Sequence-To determine the mechanism by which G13 inhibited the adhesion of stimulated platelets
A. m "WI
0 Fn
c l r l EDTA GRGDSP L 1 0 G 1 3 9 c 1 ' l l b VnR
1 2 3 4 5 6 FIG. 3. A, inhibition of platelet adhesion to Fn, Vn, and vWf by a
peptide analogue of GPIIb- 300-312 (G13). The percentage of plate- lets adhered to substrate-coated wells is indicated on the ordinate. The bar labeled ctrl represents the adhesion of stimulated platelets to the indicated substrate ( i e . Fn, Vn, vWf, and BSA) in the absence of peptide. The bar labeled C13 corresponds to the extent of adhesion in the presence of a peptide analogue of GPIIb, 300-312. The bar
the scrambled control peptide (EIIYPAGLV). The bar labeled VnR labeled scr corresponds to the extent of adhesion in the presence of
corresponds to the extent of adhesion in the presence of a peptide analogue of residues 317-330 of the VnR a subunit. The bar labeled *IIb represents adhesion in the presence of a peptide corresponding to GPIIbm 829-837. All peptides were tested at a final concentration of 1.5 mM. The results presented are means of a t least four separate experiments (data points represent a minimum of quadruplicate determinations). Error bars indicate standard deviation of the data. B , G13 binds to immobilized Vn but not immobilized collagen, throm- bin, casein, ovalbumin, or thrombospondin. Nitrocellulose dot blot assays were performed exactly as detailed under "Experimental Pro- cedures." Blot 1 represents binding of Vn to G13 in the fluid phase. Blots 2-6 represent the absence of any significant binding of G13 to thrombospondin, collagen, thrombin, casein, and ovalbumin, respec- tively.
" W I Fn Vn
FIG. 4. Dose-response data demonstrating the inhibition of platelet adhesion to vWf, Fn, and Vn as a function of the concentration of G13. Data are reported as percent platelet adhe- sion on the y-axis and test ligand on the x-axis. Final peptide concentrations were as follows: (control; o), f?d (0.5 mM), 0 (0.75 mM), €4 (1.0 mM), 0 (1.5 mM). The results presented represent the means of the pooled data with each concentration point for a partic- ular ligand representing a minimum of eight replicate determinations. Error bars correspond to the standard deviation of the data.
to RGD-containing ligands which lack the HHLGGAKQ- AGDV sequence, we performed direct binding studies of G13 to Vn and Fn. As was the case for Fg (Fig. lB), G13 bound to immobilized Vn in a dot blot assay using anti-G13 antibodies (Fig. 3B). The binding was specific since G13 did not bind to collagen, thrombin, casein, or ovalbumin, and only a slight amount of binding to thrombospondin could be detected in this system. Using the dot blot assay, we were unable to discern whether G13 bound directly to immobilized Fn due to
11732 GPIIb 300-312 Peptide Acts as Common Ligand Binding Site
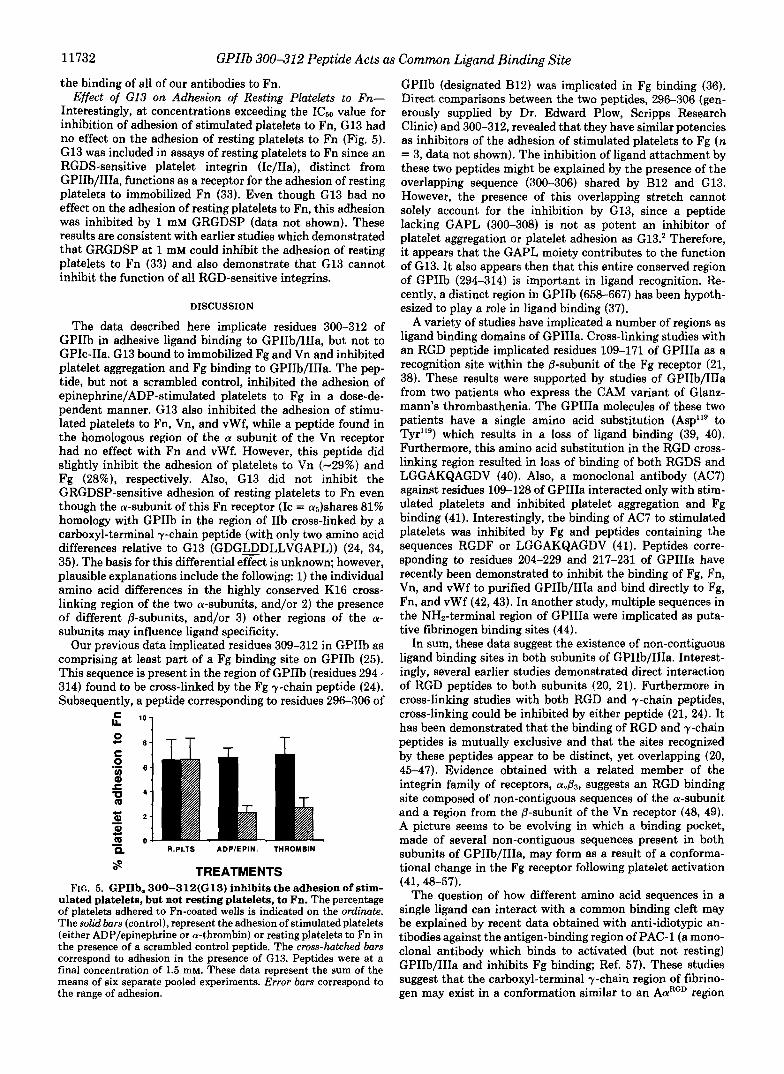
the binding of all of our antibodies to Fn. Effect of GI3 on Adhesion of Resting Platelets to Fn-
Interestingly, at concentrations exceeding the ICso value for inhibition of adhesion of stimulated platelets to Fn, G13 had no effect on the adhesion of resting platelets to Fn (Fig. 5). G13 was included in assays of resting platelets to Fn since an RGDS-sensitive platelet integrin (Ic/IIa), distinct from GPIIb/IIIa, functions as a receptor for the adhesion of resting platelets to immobilized Fn (33). Even though G13 had no effect on the adhesion of resting platelets to Fn, this adhesion was inhibited by 1 mM GRGDSP (data not shown). These results are consistent with earlier studies which demonstrated that GRGDSP at 1 mM could inhibit the adhesion of resting platelets to Fn (33) and also demonstrate that G13 cannot inhibit the function of all RGD-sensitive integrins.
DISCUSSION
The data described here implicate. residues 300-312 of GPIIb in adhesive ligand binding to GPIIb/IIIa, but not to GPIc-IIa. G13 bound to immobilized Fg and Vn and inhibited platelet aggregation and Fg binding to GPIIb/IIIa. The pep- tide, but not a scrambled control, inhibited the adhesion of epinephrine/ADP-stimulated platelets to Fg in a dose-de- pendent manner. G13 also inhibited the adhesion of stimu- lated platelets to Fn, Vn, and vWf, while a peptide found in the homologous region of the a subunit of the Vn receptor had no effect with Fn and vWf. However, this peptide did slightly inhibit the adhesion of platelets to Vn (-29%) and Fg (28%), respectively. Also, G13 did not inhibit the GRGDSP-sensitive adhesion of resting platelets to Fn even though the a-subunit of this Fn receptor (IC = a6)shares 81% homology with GPIIb in the region of IIb cross-linked by a carboxyl-terminal y-chain peptide (with only two amino acid differences relative to G13 (GDGaDLLVGAPL)) (24, 34, 35). The basis for this differential effect is unknown; however, plausible explanations include the following: 1) the individual amino acid differences in the highly conserved K16 cross- linking region of the two a-subunits, and/or 2) the presence of different @-subunits, and/or 3) other regions of the a- subunits may influence ligand specificity.
Our previous data implicated residues 309-312 in GPIIb as comprising at least part of a Fg binding site on GPIIb (25). This sequence is present in the region of GPIIb (residues 294- 314) found to be cross-linked by the Fg y-chain peptide (24). Subsequently, a peptide corresponding to residues 296-306 of
T
- n R.PLTS ADPIEPIN. THROMBIN
8 TREATMENTS FIG. 5. GPIIb, 300-312(G13) inhibits the adhesion of stim-
ulated platelets, but not resting platelets, to Fn. The percentage of platelets adhered to Fn-coated wells is indicated on the ordinate. The solid bars (control), represent the adhesion of stimulatedplatelets (either ADP/epinephrine or a-thrombin) or resting platelets to Fn in the presence of a scrambled control peptide. The cross-hatched bars correspond to adhesion in the presence of G13. Peptides were at a final concentration of 1.5 mM. These data represent the sum of the means of six separate pooled experiments. Error bars correspond to the range of adhesion.
GPIIb (designated B12) was implicated in Fg binding (36). Direct comparisons between the two peptides, 296-306 (gen- erously supplied by Dr. Edward Plow, Scripps Research Clinic) and 300-312, revealed that they have similar potencies as inhibitors of the adhesion of stimulated platelets to Fg (n = 3, data not shown). The inhibition of ligand attachment by these two peptides might be explained by the presence of the overlapping sequence (300-306) shared by B12 and G13. However, the presence of this overlapping stretch cannot solely account for the inhibition by G13, since a peptide lacking GAPL (300-308) is not as potent an inhibitor of platelet aggregation or platelet adhesion as G13.' Therefore, it appears that the GAPL moiety contributes to the function of G13. It also appears then that this entire conserved region of GPIIb (294-314) is important in ligand recognition. Re- cently, a distinct region in GPIIb (658-667) has been hypoth- esized to play a role in ligand binding (37).
A variety of studies have implicated a number of regions as ligand binding domains of GPIIIa. Cross-linking studies with an RGD peptide implicated residues 109-171 of GPIIIa as a recognition site within the @-subunit of the Fg receptor (21, 38). These results were supported by studies of GPIIb/IIIa from two patients who express the CAM variant of Glanz- mann's thrombasthenia. The GPIIIa molecules of these two patients have a single amino acid substitution (Asp"' to Tyr119) which results in a loss of ligand binding (39, 40). Furthermore, this amino acid substitution in the RGD cross- linking region resulted in loss of binding of both RGDS and LGGAKQAGDV (40). Also, a monoclonal antibody (AC7) against residues 109-128 of GPIIIa interacted only with stim- ulated platelets and inhibited platelet aggregation and Fg binding (41). Interestingly, the binding of AC7 to stimulated platelets was inhibited by Fg and peptides containing the sequences RGDF or LGGAKQAGDV (41). Peptides corre- sponding to residues 204-229 and 217-231 of GPIIIa have recently been demonstrated to inhibit the binding of Fg, Fn, Vn, and vWf to purified GPIIb/IIIa and bind directly to Fg, Fn, and vWf (42,43). In another study, multiple sequences in the NHz-terminal region of GPIIIa were implicated as puta- tive fibrinogen binding sites (44).
In sum, these data suggest the existence of non-contiguous ligand binding sites in both subunits of GPIIb/IIIa. Interest- ingly, several earlier studies demonstrated direct interaction of RGD peptides to both subunits (20, 21). Furthermore in cross-linking studies with both RGD and y-chain peptides, cross-linking could be inhibited by either peptide (21, 24). It has been demonstrated that the binding of RGD and y-chain peptides is mutually exclusive and that the sites recognized by these peptides appear to be distinct, yet overlapping (20, 45-47). Evidence obtained with a related member of the integrin family of receptors, a,& suggests an RGD binding site composed of non-contiguous sequences of the a-subunit and a region from the P-subunit of the Vn receptor (48, 49). A picture seems to be evolving in which a binding pocket, made of several non-contiguous sequences present in both subunits of GPIIb/IIIa, may form as a result of a conforma- tional change in the Fg receptor following platelet activation
The question of how different amino acid sequences in a single ligand can interact with a common binding cleft may be explained by recent data obtained with anti-idiotypic an- tibodies against the antigen-binding region of PAC-1 (a mono- clonal antibody which binds to activated (but not resting) GPIIb/IIIa and inhibits Fg binding; Ref. 57). These studies suggest that the carboxyl-terminal y-chain region of fibrino- gen may exist in a conformation similar to an AaRGD region
(41, 48-57).

GPIIb 300-312 Peptide Acts as Common Ligand Binding Site 11733
of the molecule (58). If this were the case, it would provide a rationale to explain the inhibition of binding of multiple adhesive RGD ligands to the Fg receptor by short linear peptides (such as G13) which represent a presumptive Fg y- chain binding domain of GPIIb/IIIa. Our data demonstrating direct binding of G13 to Vn (an RGD containing ligand which lacks the HHLGGAKQAGDV sequence) are consistent with this hypothesis. While our data do not exclude an effect of the peptide on platelets, our data which directly demonstrate that G13 binds to at least Fg and Vn and the distinct inhibi- tion responses of platelet adhesion to the different ligands make this explanation unlikely.
Acknowledgments-We thank Dr. David Amrani for the kind gift of peak 1 Fg and the initial batch of the EIIYPAGLV peptide, Dr. John Fenton I1 for the a-thrombin, Dr. Deane Mosher and Steve Bittorf for the vitronectin, Dr. Zaverio Ruggeri for the Von Wille- brand factor, and Dr. Dan Walz for the thrombospondin. We espe- cially thank Drs. Charles A. Lessman and David F. Smith for com- ments concerning the manuscript. We thank Robert Loudon and Jerry Derrick for technical assistance.
REFERENCES 1. George, J. N., Nurden, A. T., and Phillips, D. R. (1984) N. Engl. J. Med.
2. Harker, L. A. (1990) in Hematology (Williams, J. W., Beutler, E., Erslev, 3 11, 1084-1098
A. J., and Lichtman, M. A., eds) 4th Ed., pp. 1559-1569, McGraw-Hill, New York
3. Phillips, D. R., Charo, I. F., Parise, L. V., and Fitzgerald, L. A. (1988) Blood 71,831-843
4. Ginsberg, M. H., Loftus, J. C., and Plow, E. F. (1988) Thromb. Haemost.
5. Bennett, J. S. (1990) Sem. Hematol. 2 7 , 186-204 6. Kieffer, N.. and Phillius. D. R. (1990) Annu. Reu. Cell Biol. 6.. 329-357
6 9 , 1-6
7. Bennett, J.. S. (1991) Ann. N. Y.. Acad. Sci. 614,214-228 ”
8. Ruoslahti, E., and Pierschbacher, M. D. (1987) Science 238,491-497 9. Tamkun, J. W., DeSimone, D. W., Fonda. D.. Patel. R. $3.. and Buck. C.
(1986) Cell 46,271-282
2421-2428 10. Buck, C., Shea, E., Duggan, K., and Horwitz, A. F. (1986) J. Cell Biol. 1 0 3 ,
11. Hynes, R. 0. (1987) Cell 48,549-554 12. Hemler, M. E., Huang, C., and Schwartz, L. (1987) J. Biol. Chem 262 ,
13. Anderson, D. C., and Springer, T. A. (1987) Annu. Reu. Med. 38,175-194 14. Gartner, T. K., and Bennett, J. S. (1985) J. Biol. Chem. 260,11891-11894 15. Plow, E. F., Pierschbacher, M. D., Ruoslahti, E., Marguerie, G., and
Ginsberg, M. H. (1985) Proc. Natl. Acad. Sei. U. S. A . 82,8057-8061 16. Haverstick, D. M., Cowan, J. F., Yamada, K. M., and Santoro, S. A. (1985)
Blood 66,946-952 17. Ginsberg, M. H., Pierschbacher, M. D., Ruoslahti, E., Marguerie, G., and
Plow, E. F. (1985) J. Biol. Chem. 260,3931-3936 18. Thiagarajan, P., and Kelly, K. L. (1987) J. Biol. Chem. 263,3035-3038 19. Gardner, J. M., and Hynes, R. 0. (1985) Cell 42,439-448
21. D’Souza, S. E., Ginsberg, M. H., Lam, S. C.-T., and Plow, E. F. (1988) J. 20. Santoro, S. A., and Lawing, W. J. (1987) Cell 48,867-873
22. Kloczewiak, M., Timmons, S., and Hawiger, J. (1983) Thromb. Res. 2 9 ,
3300-3309
Biol. Chem. 263,3943-3951
249-255
23.
24.
25. 26. 27.
28.
30. 29.
31.
32.
33.
34.
35.
36.
37.
38.
39.
40.
41.
42.
43.
44.
45.
46.
47.
49. 48.
50.
51.
52.
53.
54.
55.
56.
57.
58.
59.
Kloczewiak. M.. Timmons. S.. Lukas. T. J.. and Hawker. J. (1984) Bio- chemist6 23; 1767-1774 ’
Biol. Chem. 266.3440-3446
I , - ~ , . ~~~ ,~
D’Souza, S. E., Ginsberg, M. H., Burke, T. A., and Plow, E. F. (1990) J.
Gartner, T. K., and Taylor, D. B. (1990) Thromb. Res. 6 0 , 291-309 Amrani, D. L., and Kirschbaum, N. E. (1991) Thromb. Haemost. 66,1069a Kirschbaum. N. E.. Mosesson. M. W.. and Amrani. D. L. (1992) Blood. in
nrew T&%;: D. B., Amrani, D. L., Kirschbaum, N. E., Matsueda, G. L., and
Knutson, V. P. (1988) J. Biol. Chem., 263 , 14146-14151 Gartner, T. K., Bennett, J. S., and Ogilvie, M. L. (1987) Blood 70,351a Gartner, T. K., and Taylor, D. B. (1991) Proc. SOC. Erp. Biol. Med. 198,
Gartner, T. K. (1991) J. Cell Biol. 116 , 1669a
649-657 Muranog, G., and Bick, R. L. (1980) in Plotelet Disorders (Triplett, D., ed)
pp. 101-112, CRC Press, Boca Raton, FL Piotrowicz, R. S., Orchekowski, R. P., Nugent, D. J., Yamada, K. M., and
Kunicki, T. J. (1988) J. Cell Biol. 106,1359-1364 Poncz, M., Eisman, R., Heidenreich, R., Silver, S. M., Vilaire, G., Surrey,
S., Schwartz, E., and,Bennett, J. S.(1987) J. Biol. Chem. 262,8476-8482 Argraves, W. S., Suzukl, S., Arai, H., Thompson, K., Pierschbacher, M. D.,
and Ruoslahti, E. (1987) J. Cell Biol. 106,1183-1190 D’Souza, S. E., Ginsberg, M. H., Matsueda, G. R., and Plow, E. F. (1991)
Nature 360,66-68 Calvete, J. J., Arias, J., Alvarez, M. V., Lopez, M. M., Henschen, A,,
Gonzalez-Rodriguez, J. (1991) Biochem. J. 273,767-775 D’Souza, S. E., Ginsberg, M. H., Burke, T. A., Lam, S. C.-T., and Plow, E.
F. (1988) Science 242,91-93 Loftus, J. C., O’Toole, T. E., Plow, E. F. Glass, A., Frelinger, A. L.,
Ginsberg, M. H. (1990) Science 249,915-bl8 Ginsberg, M. H., Loftus, J. C D’Souza, S. E. O’Toole, T. E., Frelinger, A.
L., 111, and Plow, E. F. (19;O) J. Cell Bioehkm. Su 1 14, 1398 Andrieux, A., Rabiet, M. J., Chapel, A., Concord, f?,.and Marguerie, G.
(1991) J. Biol. Chem. 266,14202-14207 Charo, I. F., Nannizzi, L., Phillips, D. R., Hsu, M. A., and Scarborough, R.
M. (1991) J. Biol. Chem. 266,1415-1421 Cook, J. J., Trybulec, M., Lasz, E. C., Khan, S., and Niewiarowski, S.
(1992) Biochrm. B~ophys. Acta 11 19,346-354 Calvete, J. J., Arias, J., Alvarez, M. V., Lopez, M. M., Henschen, A., and
Gonzalez-Rodriguez, J. (1991) Biochem. J. 274,457-463 Lam, S. C.-T., Plow, E. F., Smith, M. A., Andrieux, A., Ryckwaert, J. J.,
Marguerie, G., and Ginsberg, M. H. (1987) J. Biol. Chem. 262,947-950 Bennett, J. S. Shattil, S. S. Power, J. W., and Gartner, T. K. (1988) J.
Bwl. Chem. 263,12948-1i593 Andrieux, A., Hudry-Clergeon, G., Ryckewaert, J. J., Chapel, A., Ginsberg,
M. H., Plow, E. F., and Marguerie, G. (1989) J. Biol. Chem. 264,9258- 9265
Smith, J. W., and Cheresh D. A. (1988) J. Biol. Chem. 263, 18726-18731 Smith, J. W., and Cheresh: D. A. (1990) J. Biol. Chem. 266,2168- 2172 Parise, L. V., Helgerson, L. L., Steiner, B., Nannizzi, L., and Phillips, D.
Frelinger A. L., 111, Lam S. C-T. Plow, E. F., Smith, M. A,, Loftus, J. C., R. (1987) J. Biol. Chem. 262,12597-12602
Frelinger, A. L., 111, Cohen I., Plow, E. F., Smith M. A. Roberts, J., Lam, and Ginsberg, M. H. (i988) J. Biol. Chem. 263,12397-12402
Kouns, W. C., Wall, C. D., White, M. M., Fox, C. F., and Jennings, L. K. S. C-T., and Ginsberg, M . H. (1990) J. Biol. C&m. 266,6346-6352
O’Toole, T. E., Loftus, J. C., Du, X., Glass, A. A. Ruggeri, Z. M. Shattil, (1990) J. Biol. Chem. 265,20594-20601
Du, X., Plow, E. F., Frelin er A. L. I11 O’Toole, T. E., Loftus, J. C., and S. J., Plow, E. F., and Ginsberg, M. H. (1990) dell Regul. 1,883”893
Frelinger, A. L., 111, Du, X., Plow, E. F., and Ginsberg, M. H. (1991) J. Ginsberg, M. H. (1991) &li66,4b9-i16
Shattil, S. J., Hoxie, J. A., Cunningham, M., and Brass, L. F. (1985) J. Biol. Chem. 266,17106-17111
Biol. Chem. 260. 11107-11114 Abrams, C. S., Ruggeri, 2. M., Taub, R., Hoxie, J. A., Nagaswami, C.,
Welsel, J. W., and Shattil, S. J. (1992) J. B~ol. Chem. 2 6 7 , 2775-2785 Suzuki, S. Argraves, W. S Arai, H., Languino L. R., Pierschbacher, M.
D., Ruohahti, E. (1987) j. Biol. Chem. 262,1b080-14085