a regulatory element in the ,b2-microglobulin promoter identified
TRANSCRIPT

Vol. 13, No. 11MOLECULAR AND CELLULAR BIOLOGY, Nov. 1993, p. 6629-66390270-7306/93/116629-11$02.00/0Copyright ©) 1993, American Society for Microbiology
A Regulatory Element in the ,B2-Microglobulin PromoterIdentified by In Vivo Footprinting
MAiTHEW LONERGAN,1 ANUP DEY,2 KEVIN G. BECKER,2 PAUL D. DREW,3AND KEIKO OZATO2*
Laboratory ofMolecular Growth Regulation, National Institute of Child Health and Human Development,2Howard Hughes Research Scholars Program, Howard Hughes Medical Institute,' and Neuroimmunology
Branch, National Institute ofNeurological Diseases and Stroke,3 Bethesda, Maryland 20892
Received 1 March 1993/Returned for modification 22 April 1993/Accepted 23 July 1993
Expression of the 12-microglobulin (132-m) and major histocompatibility complex (MHC) class I genes iscoordinately regulated. By ligation-mediated polymerase chain reaction, we have analyzed in vivo factor bindingto the promoter region of the murine ,12-m gene. In adult spleen, in which ,82-m is expressed, strong protectionwas found in three elements. Two of these elements, the ,82-m NF-KB binding site and the interferon consensussequence, are homologous to the regulatory elements of the MHC class I genes and were also found to beprotected in spleen. A third protected element, PAM, identified in this work, is unique to the 132-m gene. Noneof the elements showed protection in brain tissue, in which neither the 132-m nor the MHC class I gene isexpressed. In vivo footprinting was also performed with F9 embryonal carcinoma cells, in which expression of the132-m and MHC class I genes is induced at a low level only upon stimulation with retinoic acid (RA). No in vivoprotection was detected before and after RA treatment of F9 cells, indicating that RA induction of 132-m (andMHC class I) expression occurs without detectable in vivo factor occupancy, whereas EL4 T lymphocytesexpressing 132-m at a high level exhibited strong protection similar to that in spleen. Despite the lack of in vivooccupancy, the nuclear factors specific for each of the three elements were present in brain tissue and F9 cells aswell as in spleen tissue and EL4 cells. We show that PAM, an element identified by its in vivo protection, bindsnuclear factors ranging from 40 to 50 kDa in size and is capable of enhancing transcription of a reporter in F9and other cells. Taken together, these results indicate that in vivo factor occupancy for the ,82-m and MHC classI promoters is coordinated and occurs through a mechanism other than mere expression of relevant factors.
,2-Microglobulin (02-m) is a 12-kDa glycoprotein thatassociates with and directs the intracellular transport of themajor histocompatibility complex (MHC) class I molecule(57). Mice deficient in 02-m expression are deficient in MHCclass I expression and are defective in developing cytotoxicT cells (33, 66). ,2-m has additional functions, includingbinding to the Fc receptor in the neonatal intestine (59) andcontrolling migration of hemopoietic cells from bone marrow(8).Although the 12-m gene locus is not linked to the MHC,
expression of the 12-m and MHC class I genes is coordi-nately regulated. Both genes are expressed at high levels inlymphoid tissues (9), while the expression is virtually absentin the central nervous system (5, 33). Drezen et al. reportedthat 12-m and MHC mRNA levels correlate with the levelsof transcription of the two genes in various adult andembryonic tissues in mice (13). Both genes are inducible byinterferon (IFN) and tumor necrosis factor (12). Moreover,the two genes show a similar pattern of developmentalregulation: expression of both genes in mice begins to bedetected in a 9- to 10-day-old embryo at mid-somite stage(28, 47), although some level of asynchrony is noted (43).Expression of MHC class I and 12-m genes has been studiedin embryonal carcinoma (EC) cells, including F9 cells, as amodel to study developmental regulation (6, 7, 17, 40, 42, 46,58). In undifferentiated F9 cells, neither MHC class I nor12-m gene is expressed. However, treatment with retinoicacid (RA) leads to the induction of both genes (6, 40).
Coordinated expression of the two genes is thought to be
* Corresponding author.
controlled by shared regulatory elements in the conservedupstream region (see Fig. 1) (27, 32). The shared elementsinclude (i) an NF-KB binding site (KB) (1, 27, 50) and (ii) theIFN consensus sequence (ICS) (18). In MHC class I genes,the NF-KB binding site (also called enhancer A [32] or regionI [10]) serves as a constitutive but moderate enhancer, whilethe ICS is an IFN-inducible enhancer that is homologous tothe IFN-stimulated response elements (ISRE or IRE) ofmany other IFN-inducible genes (35, 64) (Fig. 1). The twoelements are closely juxtaposed in both genes, although inthe opposite orientation. The same juxtaposition of the twoelements (KB and the PRDI) is found in the INF-3 gene (19,63) (Fig. 1). The KB and related motifs bind proteins of theRel family (1, 4, 22, 31) expressed variably in different cells(2, 41), presumably the most prevalent form being thepSO-p65 heterodimer (1). The ICS and related sequences(ISRE or IRE) bind to the proteins of the IFN regulatoryfactor (IRF) family (14, 25, 61) that includes IRF-1, IRF-2,ICSBP, and the DNA-binding subunit of ISGF3. Some ofthese elements bind a C2H2 zinc finger protein, PRDIBF1(30).
In a previous genomic footprinting of MHC class I genes,we demonstrated that region I (KB-like), ICS, and an addi-tional element site, a, are occupied in vivo in a tissue-specific manner that closely parallels expression of MHCclass I genes in mice (10).
In the present study, we analyzed in vivo footprinting ofthe 12-m gene and addressed whether in vivo factor bindingof the 132-m gene is coordinated with that of MHC class Igenes, with respect to their tissue-specific expression anddevelopmental regulation. Secondly, we asked whether thepresence or absence of an in vivo footprint correlates with
6629
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 16
Dec
embe
r 20
21 b
y 18
0.24
1.80
.102
.

6630 LONERGAN ET AL.
-162 -110
13-2 m ATCACGGGACTTTATAAGAACATGAAACTGAAAATGGGAAAGTCCC1TTGTAAPAM ICS / IRE RI / NFkB
-127 -177H -2Kb CCAGGTTAGGTGCAGAAGTGAAACTGTGGAGATGGGGAATCCCCAGCCCTG
ICS / IRE RI / NFkB
-94 -44
,B- INF ATAGGMAACTGAAAGGGAGAAGTGAAAGTGGGAMATCCTCTGAATAGAG
PRDI / IRE PRDII / NFkB
FIG. 1. Comparison of the ,2-m with MHC class I and IFN-3promoters. The shared cis elements (ICS/IRE/PRDI) and NF-KBbinding sites (KB/RI/PRDII) are underlined. Sequences are fromreferences 19, 32, and 63. PAM, an element unique to P2-midentified in this work, is also underlined.
expression of specific nuclear factors in the cell. We showthat in vivo protection of the 02-m promoter is tissue specificand correlates with the expression of 02-m and MHC class Igenes. Furthermore, factors capable of binding to the regu-latory elements are found to be present in the cells andtissues, irrespective of in vivo protection. We also presentevidence that these elements are not occupied in F9 cellseven after the induction of ,2-m mRNA following RAtreatment. These results are discussed in the context ofpossible mechanisms conferring in vivo occupancy of thef2-m and MHC class I genes.
MATERIALS AND METHODS
Mice. Adult female C57BL/6 mice were used to prepareDNA for in vivo footprinting and nuclear extracts for elec-trophoretic mobility shift and UV cross-linking experiments.
Cells. F9 Tk- cells (40) and F9 clone ACc19 (6) werecultured in Dulbecco's modified Eagle's medium supple-mented with gentamicin, glutamine, and 10% fetal bovineserum at 37°C in 7% CO2 in humidified air. Cells were treatedwith RA (all-trans, S x 107 M; Sigma) and dibuturyl cyclicAMP (dcAMP; 0.5 mM; Sigma). ETA cells were cultured inRPMI 1640 supplemented with gentamicin, glutamine, and10% fetal bovine serum.
In vivo footprinting. In vivo footprinting of adult mousetissues (brain and spleen) was performed by ligation-medi-ated polymerase chain reaction (PCR) (45) by using Seque-nase for first-strand extension and Taq polymerase foramplification. Vent polymerase was used for in vivo foot-printing of cultured cells for both extension and amplification(20). Briefly, brain and spleen tissues were disrupted at 4°Cin a Stomachar (Tekmar) for 30 to 90 s, producing a uniformcell suspension with greater than 90% cell viability (10). Thecells were treated with 0.1% dimethyl sulfate (DMS; Kodak)for 2 min at room temperature. F9 and ETA cells weretreated with 0.1% DMS for 70 s at room temperature.Genomic DNA was isolated and cleaved subsequently withpiperidine at 93°C (45). DNA for control G ladders wasprepared from spleen and F9 DNA treated with DMS invitro. An 8% denaturing polyacrylamide gel was used toresolve the reaction mixtures. Primers were synthesized inan Applied Biosystem 380B DNA synthesizer and purified ina 20% polyacrylamide gel. For the noncoding strand, threeprimers, 5' CTG AGT GGG ATA TTG TCA GC 3' (-288 to-269), 5' ATG AGG CGG TCC CAG GCT GAA CGA C 3'
(-258 to -235), and 5' ATG AGG CGG TCC CAG GCTGAA CGA CCA GAT ACA CCA AAC TC 3' (-258 to-218), were used. For the coding strand, 5' GCT CTT ATATAG TTC CTG 3' (-22 to -39), 5' TAG TTC CTG AACTCA CAG CCA ATC CG 3' (-31 to -56), and 5' CAG ATCCCG AGG TCG TCA CCA GTC TCC 3' (-61 to -87) wereused.RNA blot analysis. Total RNA was isolated by the guani-
dinium thiocyanate method (54). Fifteen micrograms of totalRNA was electrophoresed through 1.2% formaldehyde-aga-rose gels and electroblotted onto a nylon membrane (MicronSeparations). The blot was then hybridized with 106 cpm of32P-labeled probes per ml in 50% formamide. A 237-bp PstIfragment of the ,B2-m cDNA (48) and a 331-bp NcoI-EcoRIfragment of the chicken 1-actin cDNA (49) were labeled with32P by the random priming method by using Prime a Gene(Promega).EMSA. Nuclear extracts were prepared from the spleen
and brain tissues of adult C57BL/6 mice (10). Extracts fromF9 and ELA cells were prepared (11) with the followingmodifications. A combination of protease inhibitors, phenyl-methylsulfonyl fluoride (0.5 mM), Na-p-tosyl-L-lysine chlo-romethyl ketone (0.1 mM), N-tosyl-L-phenylalanine chlo-romethyl ketone (0.1 mM), leupeptin (10 ,ug/ml), aprotinin(33 ug/ml), and bestatin (10 ,ug/ml), and dithiothreitol (0.5mM) were used during the preparation of nuclear extracts.Undialyzed extracts (7.5 ,g) were incubated with approxi-mately 0.01 pmol of 3"P-labeled oligonucleotide (=10Ocpm)in electrophoretic mobility shift assay (EMSA) bindingbuffer for 30 min on ice. For competition experiments,nuclear extracts were preincubated with a 250-fold molarexcess of oligonucleotides for 15 min prior to the addition ofthe probe. Probes used are (i) PAM, 5' TGA AATATC ACGGGA CTIT TAT 3'; (ii) 32-m ICS, 5' TAA GAA CAT GAAACT GAA AAT GGG AAA G 3'; and (iii) 132-m KB, 5' ACAAAG GGA CTT TCC CAT TT 3'. Electrophoresis wasperformed as previously described (10). Rabbit polyclonalanti-peptide antibody against the NF-KB subunit proteinp50 (NL5; Santa Cruz Biotechnology) or control antibody,rabbit polyclonal anti-peptide antibody directed to mousePRDIIBF1 (2 RI), was added to the binding reaction mixture30 min prior to addition of labeled 12-m KB probe.UV cross-linking. The methods of Molitor et al. (41) or
Gray et al. (23) were used for cross-linking with 132-m ICSand PAM or with 132-m KB, respectively. Briefly, 30 pMtemplate and 300 pM primer oligonucleotides were annealedin TE buffer containing 50 mM NaCl. The DNA probe wasprepared by incorporation of 5-bromo-2'-deoxyuridine 5'triphosphate (BrdU) (Pharmacia), [a-32P]dCTP, [a-32P]dGTP (Amersham), and dATP (Pharmacia) with the Klenowfragment (U.S. Biochemical) at 16°C for 2 h in a 75-pIreaction mixture and subsequent centrifugation through aSephadex G-25 column twice. For PAM, a template oli-gomer, 5' TGA AAT ATC ACG GGA CTIT TAT 3', and aprimer, 5' ATAAAGTCC 3', were used, while the template5' TAA GAA CAT GAA ACT GAA AAT GGG AAA G 3'and primer 5' C''ITCCCA 3' were used for ICS. Fifteenmicrograms of nuclear extracts was incubated with approx-imately 0.03 pM primer (3 x 105 cpm) at 4°C for 30 min. Forcompetition assay, nuclear extracts were preincubated witha 250-fold molar excess of oligomers for 15 min prior to theaddition of labeled probe. For one-dimensional resolution,reaction mixtures were subjected to UV light at 312 nm for30 min and then run on a sodium dodecyl sulfate (SDS)-10.5% polyacrylamide gel. For two-dimensional resolution,reactions were first run on a 6% nondenaturing EMSA gel
MOL. CELL. BIOL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 16
Dec
embe
r 20
21 b
y 18
0.24
1.80
.102
.

IN VIVO FOOTPRINTING OF THE 02-m GENE 6631
and then subjected to UV light as described above. Laneswere cut, equilibrated in SDS loading buffer for 20 min, andthen resolved in the second dimension as described above.
Reporter constructs, transfection, and CAT assays. A wild-type monomeric oligomer or mutant PAM oligomer wasinserted into the BamHI site of the basal Tk-CAT reporter,PBL-CAT8+ (38). Correct insertions were confirmed bydideoxy sequencing. F9 cells (1.5 x 105 cells per plate) weretransfected with 1 ,ug of reporter constructs for 12 h by thecalcium phosphate precipitation method (46). Cells wereincubated for an additional 36 h and harvested. EL4 cells(2.5 x 107 cells in 250 ,ul) were transfected with 15 ,ug ofreporter constructs and 4 ,ug of pCMV-LacZ (37) by electro-poration according to the manufacturer's recommendations(Bethesda Research Laboratories Cell Porater, model 1600).Cells were incubated with normal medium for an additional48 h. Chloramphenicol acetyltransferase (CAT) activitieswere normalized relative to ,-galactosidase activity.
RESULTS
In vivo footprinting of the P2-m promoter in adult mousetissues. Like MHC class I genes, the j2-m gene is expressedat high levels in lymphoid tissues, whereas the expression isvirtually undetectable in brain tissues (5, 12, 34). Thisdifference in expression is largely accounted for by differenttranscriptional activities of the two genes. Drezen et al.showed that transcriptional activity of 32-m and MHC classI genes in lymphoid organs is at least 1 order of magnitudehigher than in brain tissue (13). To study promoter occu-pancy of the 32-m gene, we performed in vivo footprintinganalysis (45) with spleen and brain DNAs prepared fromadult mice. An upstream region spanning from -235 to -60was investigated. This region includes the NF-KB bindingsite (KB) and the ICS (IRE), which elicit constitutive andIFN-inducible enhancer activities, respectively (Fig. 1) (12,27, 32). Results of in vivo footprinting obtained by using theTaq polymerase are shown in Fig. 2A. The level of protec-tion in spleen was quantitated for each G residue by densi-tometric scanning (Fig. 2B). As seen in Fig. 2A (left panel),marked protection was detected in the coding strand ofspleen DNA over a long stretch of sequence from -167 to-121. This protected region includes the NF-KB site (-116to -127) and the ICS (IRE) (-130 to -139). Most G residuesin these elements showed greater than 50% protection(marked with closed arrows in Fig. 2B). The protectionextended farther upstream to a site designated PAM, inwhich two G residues at -157 and at -167 showed >40%protection. In the noncoding strand (Fig. 2A, right panel)protection was less prominent than in the coding strand; onlythe NF-KB site (-117 and -118) showed approximately 30%protection. The sequence in and near the NF-KB site mayform a secondary structure, since even the control G ladderproduced only two weak visible bands, rather than the threebands expected of the nucleotide sequence. No significantprotection was detected in the ICS and PAM in the noncod-ing strand. However, intense methylation hypersensitivitywas noted at -153 and -193 in the spleen DNA (Fig. 2A;open arrows in Fig. 2B). Methylation hypersensitivity hasbeen observed in other genes and seems to occur in residuesnear or within a factor binding region of an active gene (10,29, 45). No other sites in the region showed protection orhypersensitivity.
In contrast to spleen, neither protection nor hypersensi-tivity was detected with brain DNA either in the coding ornoncoding strand (Fig. 2A). The presence of protection (and
hypersensitivity) in spleen DNA and their absence in brainDNA were consistently observed with separate preparationsof DNA tested (not shown). It should be mentioned thatprotection in spleen DNA was partial for all three sites,although reproducibly observed for the same G residues.These results indicate that in vivo occupancy of the 12-mpromoter correlates with the expression of the gene, as hasbeen noted for MHC class I genes (10).The lack of in vivo protection in F9 EC cells before and after
RA-induced 132-m expression. Neither the ,B2-m nor the MHCclass I gene is expressed in undifferentiated EC cells (6, 7,42). However, RA treatment induces transcription of bothgenes (6) that leads to a low level of the surface expressionof H-2 molecules. This induction mimics expression of thetwo genes during mouse embryogenesis in vivo (28, 47). Tostudy whether in vivo protection of the 12-m gene is devel-opmentally controlled, we performed in vivo footprinting ofF9 cells before and after RA treatment, with or withoutdcAMP. Treatment with RA alone has been shown topromote differentiation into visceral endodermal cells, whilea combination of RA and dcAMP stimulates differentiationinto parietal endodermal cells (60). In vivo footprinting wasalso performed for EML murine T lymphocytes as a repre-sentative of a high 12-m expressor. To confirm RA inductionof 132-m gene in F9 cells, RNA blot analysis was performed.Data in Fig. 3A show that untreated F9 cells do not expressan appreciable level of 12-m RNA, while the mRNA isinduced following treatment with RA and dcAMP for 5 days.This induction was most likely due to transcriptional activa-tion (13, 56). The level of ,B2-mmRNA expressed in RA- anddcAMP-treated F9 cells was, however, more than 20 timesless than that expressed constitutively in ELM lymphocytes.A similar low level of 12-m RNA was induced followingtreatment with RA alone (not shown).
Results of in vivo footprinting obtained for F9 and EL4cells by using the Vent polymerase are shown in Fig. 3B.Although the Vent polymerase allowed for clearer visualiza-tion of protected bands than the Taq polymerase, it generallyproduced more extraneous bands of low intensity that werederived from non-G residues. Such extraneous bands weremore abundant in cell or tissue DNA than in purified DNAused for G ladders and were seen for other genes as well (10).These extraneous bands were not a product of degradation,since they were uniformly distributed over a long stretch ofDNA, with a pattern identical among different preparationsof DNA. These bands may be produced by a nonspecificcleaving agent(s) present in cells or tissue preparations thatmay be active during DMS treatment. G ladders observedwith the Taq and Vent polymerases showed a slight differ-ence in band intensity for some G residues, the basis ofwhich has not been investigated. For EML cells, prominentprotection was observed over the NF-KB binding site, ICS,and PAM of the coding strand (Fig. 3B), with the protectedG residues being identical to those seen in spleen tissue (Fig.2A). In the noncoding strand, the NF-KB site showed partialprotection, as was seen in spleen tissue (as for the tissueDNA, G residues in this area were compressed). Methyla-tion hypersensitivity was seen at -153 and -193 in EML, asseen in splenic DNA (Fig. 2A). In contrast, no detectableprotection (and hypersensitivity) was demonstrated in any ofthe elements in either untreated F9 cells or F9 cells treatedwith RA and dcAMP for 5 days (Fig. 3B). F9 cells treatedwith RA alone for 3 or 5 days were also negative for in vivoprotection (not shown). Likewise, shorter periods of RAtreatment (6 to 24 h) did not produce in vivo footprinting (notshown). Furthermore, clone ACc19, established following
VOL. 13, 1993
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 16
Dec
embe
r 20
21 b
y 18
0.24
1.80
.102
.

6632 LONERGAN ET AL.
TissuesA Coding Noncoding
-117-119 : " " RI/NFKB
FIG. 2. In vivo protection and methylation hypersensitivity ofthe 32-m promoter in mouse tissues. (A) Comparison between adultspleen and brain tissues. In vivo footprinting was performed byligation-mediated PCR with the Taq polymerase. Lanes G-ladder,positions of G residues revealed from spleen DNA treated withDMS in vitro and cleaved by piperidine. Numbers represent nucle-otide positions relative to the transcription initiation site. (B) Quan-titation of splenic protection and methylation hypersensitivity.Percent protection was quantitated by densitometric scanning of theintensity of each G residue and normalized to that of the brain Gresidue. Levels of protection greater than 30% are marked asprotected residues (closed arrows). Sites of hypersensitivity to DMSare marked with open arrows (values represent percent increase inband intensity). The core sequences of the three protected sites,PAM, ICS (IRE), and NF-KB (RI), are shown by a shaded box.
-135IRE/ICS
-1l42 _>sx
-153 --158 :: PAM
1233
Joh. .
1 2] 3 .
B
AGTAGGGCAC CAAGGGTCCA
TCATCCC TG GTTCCCAGGT-200 -190
4M%o
42% 51%
GCCCAGGCTG TTTGAAATAT CACGGGACTT TATAAGAACAPAM
CGGGTCCGAC AAACTTTATA GTGCCCT AA ATATTCTTGT-180 -170 -160 -150
80%
52% 62% 57% 42%
;; ;;-1X~~~~~~~12TGAAACTGAA AATGGGAAAG TCCCTTTGTA
t /IRE PJ/NFKB
ACTTTGACTT TTACCCTTTC AGGGAAACAT-140 -130
31%
prolonged RA treatment of F9 cells (6) exhibiting propertiesof differentiated cells, also failed to produce detectable invivo footprinting (not shown). Thus, the regulatory region ofthe 12-m gene is not protected in F9 cells before and afterRA treatment, even when 32-m mRNA is expressed at a lowlevel. These results indicate that the absence of in vivoprotection does not necessarily signify the absence of geneexpression. As will be evident below, the lack of in vivoprotection in F9 cells (and in brain tissue) was not attribut-able to the lack of factors that bind to the relevant elements.To correlate these results with in vivo occupancy of the
MHC class I regulatory region, we performed in vivo foot-printing of an MHC class I gene in F9 and ELA cells. Sincethese cells are of the H-2" haplotype, we were able to useprimers for the H-2K! gene described in our previous work(10). In agreement with results with the 12-m gene, no
protection was detected for the H-2K" gene before and after
RA treatment of F9 cells (with and without dcAMP). How-ever, EIA cells exhibited strong in vivo protection overregion I (KB), ICS, and site a that was very similar to thatseen in spleen tissue (10) (not shown).PAM binds a specific nuclear factor and enhances transcrip-
tion from a heterologous promoter. Both in spleen and in ETAcells, the PAM sequence (-167 to -154) consistentlyshowed strong protection and hypersensitivity. PAM in-cludes the hexanucleotide ATCACG that is conserved in thehuman and mouse 132-m genes (24). To our knowledge,neither in vitro factor binding nor the functional significanceof this sequence has been elucidated so far.To study factor binding activity specific for the PAM site,
EMSAs were performed with nuclear extracts from the cellsand tissues tested above. As seen in Fig. 4A, extracts fromall cells and tissues produced an intense band (Fig. 4A,PAM) which was inhibited by excess unlabeled PAM but not
-167
-157
-139
-133
-1 25-127
-121
-113
PAM*0*
IIRE/ICS
iRI/NFKB
I 2 3
-110 -101
ACCTAGTTCA
TGGATCAAGT
MOL. CELL. BIOL.
ci73 c':. Q Zft .Id -;.
r. ;-.; U) =
r.v r-W -!.CL I
ur) =
0,0
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 16
Dec
embe
r 20
21 b
y 18
0.24
1.80
.102
.

IN VIVO FOOTPRINTING OF THE ,B2-m GENE 6633
A
- 28S
-18S
--2m- 0
B-Cactin- lw
1 2 3
CellsB Coding
-s -
-157 PAM
-139
-133
-125-127-
-121
1 2 3 4
Noncoding
-117-119 .-
-135
-142
IRE/ICS -153-
-158-160 ---
RI/NFKB
-193
FIG. 3. 12-m mRNA expression and in vivo protection in F9cells and ELA T lymphocytes. (A) RNA blot. Total RNA (15 ,ug)from untreated F9 cells, F9 cells treated with RA and dcAMP (5days), or ELA cells were hybridized with 32P-labeled 132-m probe orchicken 1-actin probe. The positions of rRNA (28S and 18S) areshown. (B) In vivo footprinting of untreated F9 cells, F9 cellstreated with RA and dcAMP (5 days), or ELA cells. In vivofootprinting was performed as for Fig. 2, except that the Ventpolymerase was used in these experiments.
by the unrelated oligomer UCR (16). The intensity of thePAM band produced from F9 cell extracts was comparableto that in ELA cells. The positions of retarded bands ap-peared to differ slightly depending on cells or tissues thatexpress or do not express 32-m. Extracts from RA-treatedF9 cells and from spleen tissue expressing P2-m revealedanother faster-migrating band (Fig. 4A, *). The faster-mi-grating band was, however, not detected in another 12-m-expressing cell, ELA (Fig. 4A, lanes 10 to 12). These resultsshow that PAM binds to nuclear factors from a variety ofcells and tissues, irrespective of in vivo factor occupancy.To examine sequence specificity of PAM binding, mutantoligomers were tested for their ability to compete for factorbinding. Results from spleen extracts are shown in Fig. 4B.Mutants M2 and M3, which had mutations in the conservedhexanucleotide, failed to compete for binding, while muta-tions in either side of the core had no effect. The samecompetition pattern was observed with extracts from braintissue and F9 cells (not shown). In addition, factor binding toPAM was not inhibited by the 32-m KB oligomer, nor was itinhibited by MHC class I region I oligomer (Fig. 4B, H-2RI).Partial competition was noted by MHC region II (Fig. 4B,H-2RII), to which the nuclear hormone receptor retinoid Xreceptor ,B binds (46). The basis of this partial competitionhas not been studied in detail. The PAM core hexanucleotidedid not match with any of the cis regulatory elements listedin the transcription factor data base to date (21), suggestingthat PAM is a unique cis element.To assess the nature of the factor(s) that binds to PAM,
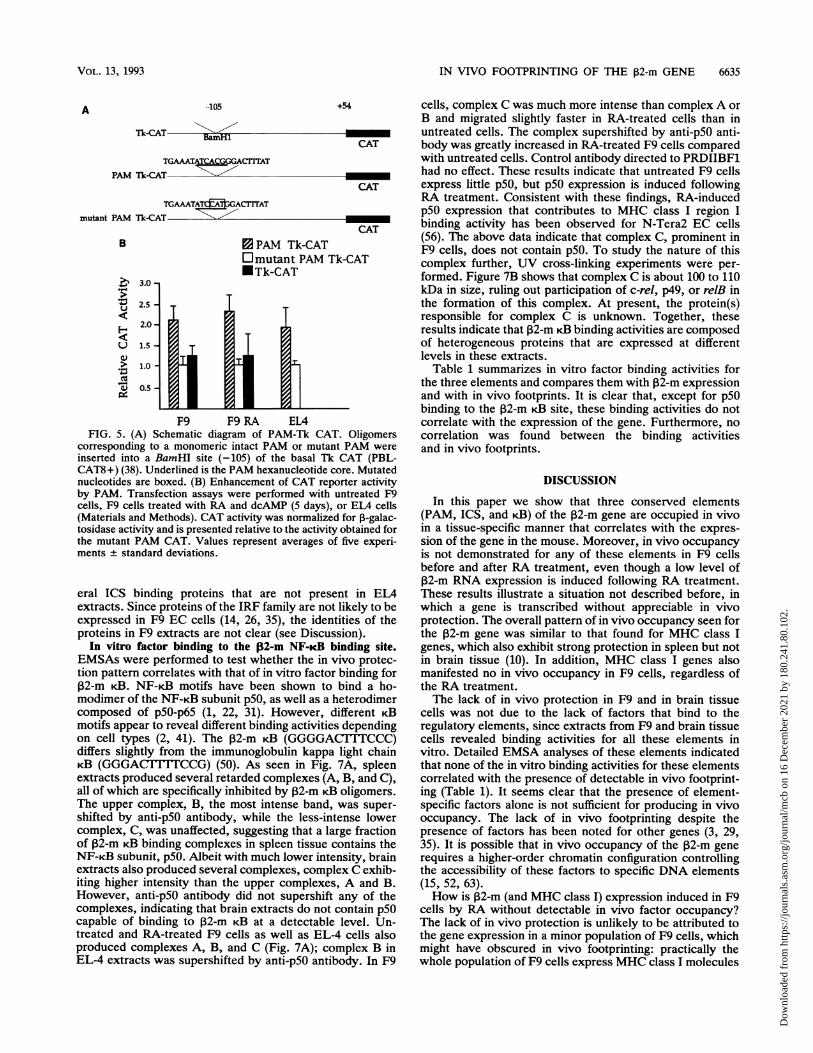
UV cross-linking experiments were performed by using aBrdU-substituted PAM probe (Materials and Methods).Cross-linked complexes were resolved by SDS-polyacryl-amide gel electrophoresis (PAGE) (Fig. 4C). Multiple bandsthat clustered around 40 to 55 kDa were detected in bothspleen and brain extracts; the size of the cross-linked pro-teins was estimated on the basis of the apparent mobility ofthe free probe (roughly 3.4 kDa). These cross-linked bandsrepresent specific DNA-protein complexes, since they wereeliminated by unlabeled PAM competitor (Fig. 4C, lanes 2and 5), but not by the control competitor (Fig. 4C, lanes 3and 6). These results suggest that PAM binds multipleproteins that are somewhat heterogeneous in size and tissuedistribution. It is, however, possible that the multiple bandsare produced by partial proteolysis of a protein.To address whether PAM is transcriptionally functional,
chimeric reporters in which a monomeric PAM oligomer ora mutant PAM oligomer was placed upstream of the Tk CATgene were constructed (see Fig. 5A for scheme). The mutantPAM used here corresponds to M3 in Fig. 4B, which failedto compete for PAM binding. Results of transient transfec-tion assays are shown in Fig. SB. In F9 cells, CAT activityproduced by the PAM-Tk reporter was about twice that bythe mutant PAM-Tk reporter. CAT activity by this mutantwas very similar to that seen by the basal Tk CAT reporter.Essentially the same, roughly twofold, increase was ob-served when transfection was performed with RA-treated F9cells. Similarly, in EL4 cells, PAM-Tk CAT elicited about atwofold-greater reporter activity than the mutant PAM (Fig.SB). CAT enhancement by PAM was highly reproducibleand was also observed in mouse L fibroblasts (not shown).Thus, PAM is capable of enhancing transcription from aheterologous promoter in a variety of cells, without clear celltype specificity.
In vitro factor binding to the 132-m ICS. To test whether invivo footprint of the ICS correlates with the presence of ICSbinding factors, EMSAs were performed by using 132-m ICS
VOL. 13, 1993
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 16
Dec
embe
r 20
21 b
y 18
0.24
1.80
.102
.

6634 LONERGAN ET AL.
WY N- / F1j w!Fk4 4Mh1r RA -ld RI;A 14 SpIleel Brain
B
i. - -.4 -
,z - -~-
PAM * 4
*s 4 it
P9AM-i81Xi1|1 "w. id tw ~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
1~~~~~~~~~~~S~:;i...
,
_ '?':
'I 2 4 m 6 7 8 10 !I 12 13 14 15 1h 1717 2 5 4 6 7 8 9 10 11
PAM TGAAATATCACGGGACTTTATMl - --CCG-M2 CGA--
M3 - CAT--
M4 - TTC------
M5 ---AGG--
FIG. 4. In vitro characterization of PAM binding factors. (A) Tissue and cell line distribution of PAM binding factors. EMSAs were
performed with 7.5 pLg of nuclear extract protein by using a 32P-labeled PAM oligomer as a probe. Lanes 1, 4, 7, 10, 13, and 16, no competitor;lanes 2, 5, 8, 11, 14, and 17, 250-fold molar excess of unlabeled PAM; lanes 3, 6, 9, 12, 15, and 18, 250-fold molar excess of control competitor(UCR). (B) Sequence specificity of PAM binding. EMSAs were performed with spleen nuclear extracts as described above, and mutantcompetitors (sequence in the bottom panel) were added at a 250-fold molar excess. Only mutated nucleotides are shown. Nucleotides identicalto the native PAM are shown by dashes. The PAM hexanucleotide is underlined. Also added at a 250-fold molar excess are MHC class I regionII (H-2RII) and mutant region II (H-2RIIM) (46), and control competitors immunoglobulin KB (50) and MHC class I region I (H-2RI) (10, 31).(C) UV cross-linking of PAM-protein complexes. Nuclear extracts (15 pg) from brain and spleen tissues were incubated with a 32P-labeledBrdU-substituted PAM probe, irradiated with UV, and then resolved by SDS-10.5% PAGE (Materials and Methods). Lanes 1 and 4, no
competitor added; lanes 2 and 5, PAM (unsubstituted, 250-fold molar excess) added; lanes 3 and 6, control UCR (250-fold molar excess)added. Numbers on the right represent approximate molecular masses (in kilodaltons).
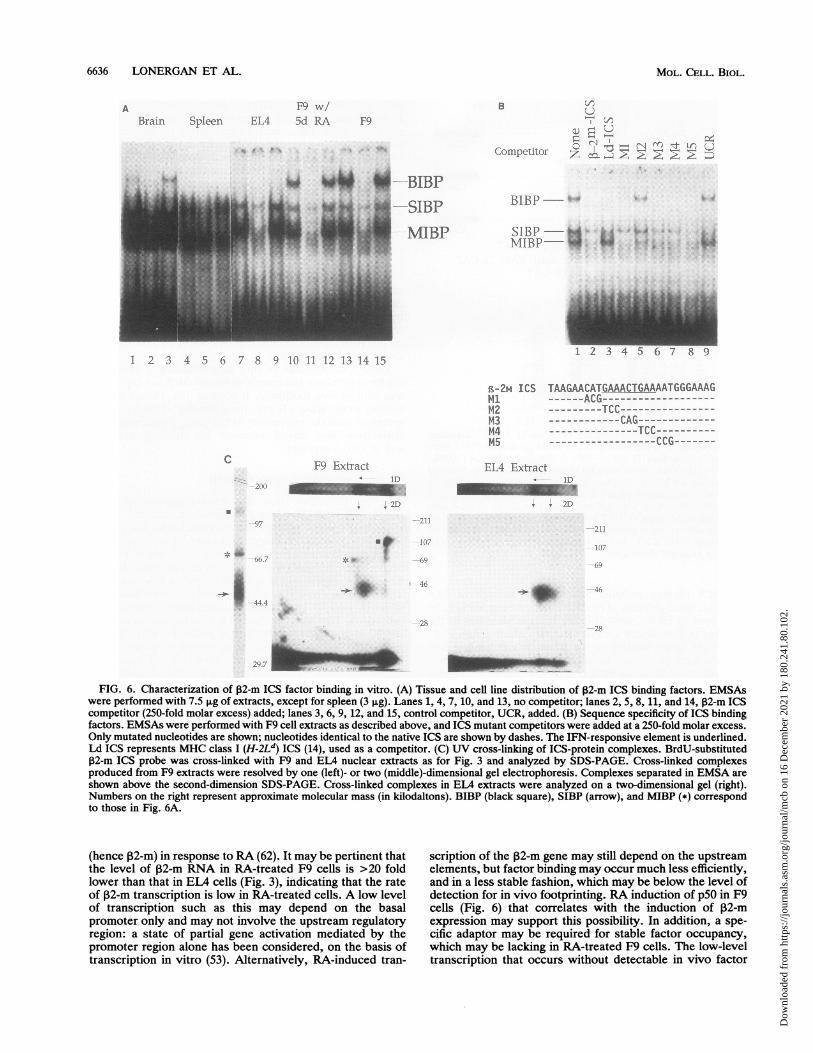
as a probe and the same set of extracts as for Fig. 4. Resultsare shown in Fig. 6A. All of these extracts generatedICS-specific bands, although a large amount of nonspecificbinding was noted with extracts from tissues. Spleen andEL4 cell extracts produced two faster-migrating bands (Fig.6A, MIBP and SIBP), while brain and F9 cell extractsproduced an additional slowly migrating band (Fig. 6A,BIBP), indicating that 132-m ICS binds a set of heterogeneousnuclear factors present in these extracts. None of the bandsprecisely correlated with the 32-m expression or with in vivofootprint: SIBP and MIBP were found in all samples regard-less of 132-m expression, while BIBP was found in thosewhere in vivo footprint was absent. Thus, the lack of in vivofootprint in the ICS in brain and F9 cells is not due to thelack of ICS binding factors. Sequence specificity of 132-mICS binding was then tested with F9 extracts, since theupper band (BIBP) present in F9 cells (and brain tissue) wasabsent in EL4 and spleen extracts and did not correspond tothe previously observed bands with other cells (12, 14). Asseen in Fig. 6B, M2 that carries a mutation in the regionhomologous to the ISRE (IRE) failed to compete for both theupper (BIBP) and the lower band that contained SIBP andMIBP. This mutation resides within the minimum IFN-responsive motif (36) found in many IFN-responsive genes
(35, 65). All other mutants competed for both upper andlower bands. It is significant that M3 and M4 that havesubstitutions within the homologous ISRE competed forthese bands, suggesting that they are distinct from theknown ISRE binding factors (14, 21, 39, 61). The middle,presumably nonspecific, band present in these experimentsdid not show significant competition. Although 32-m ICScompeted for both upper and lower bands, the MHC class IICS competed only for the upper band, indicating that thereare multiple proteins that bind to various ICS sequences withvarious affinities. To assess the molecular sizes of ICSbinding factors, UV cross-linking with extracts from F9 andEL4 cells was performed. As seen in Fig. 6C, EMSA gelsrevealing retarded complexes (top) were resolved by SDS-PAGE (bottom). The uppermost band (BIBP) produced byF9 extracts was resolved into proteins with molecularmasses ranging from 70 to 110 kDa (estimating the molecularmass of the ICS probe to be roughly about 5 kDa). The lowertwo bands (MIBP and SIBP) produced two discrete spots ofabout 70 and 40 to 45 kDa, respectively. ETA extractsproduced only one large spot of 40 to 45 kDa. Resolution ofF9 extracts in a one-dimensional gel (Fig. 6C, left) producedthree bands consistent with the three spots (Fig. 6C, middlepanel). These results indicate that F9 extracts contain sev-
A
MOL. CELL. BIOL.
C 1,-)pIcen Braill
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 16
Dec
embe
r 20
21 b
y 18
0.24
1.80
.102
.
IN VIVO FOOTPRINTING OF THE 1B2-m GENE 6635
+54105
Tk-CAT BamHlCAT
TGAAATATCACGGGACTEAT
PAM Tk-CATCAT
TGAAATA;GACJTAT
mutant PAM Tk-CAT \CAT
B
2>3.0.-I
2.5
1.5
> 2.0
0.5 -
S PAM Tk-CATEJmutant PAM Tk-CAT*Tk-CAT
F9 F9 RA EL4FIG. 5. (A) Schematic diagram of PAM-Tk CAT. Oligomers
corresponding to a monomeric intact PAM or mutant PAM wereinserted into a BamHI site (-105) of the basal Tk CAT (PBL-CAT8+) (38). Underlined is the PAM hexanucleotide core. Mutatednucleotides are boxed. (B) Enhancement of CAT reporter activityby PAM. Transfection assays were performed with untreated F9cells, F9 cells treated with RA and dcAMP (5 days), or ELA cells(Materials and Methods). CAT activity was normalized for 1-galac-tosidase activity and is presented relative to the activity obtained forthe mutant PAM CAT. Values represent averages of five experi-ments standard deviations.
eral ICS binding proteins that are not present in EL4extracts. Since proteins of the IRF family are not likely to beexpressed in F9 EC cells (14, 26, 35), the identities of theproteins in F9 extracts are not clear (see Discussion).
In vitro factor binding to the 132-m NF-KB binding site.EMSAs were performed to test whether the in vivo protec-tion pattern correlates with that of in vitro factor binding for,B2-m KB. NF-KB motifs have been shown to bind a ho-modimer of the NF-KB subunit p50, as well as a heterodimercomposed of pSO-p65 (1, 22, 31). However, different KBmotifs appear to reveal different binding activities dependingon cell types (2, 41). The 12-m KB (GGGGACTTrCCC)differs slightly from the immunoglobulin kappa light chainKB (GGGACT-FITCCG) (50). As seen in Fig. 7A, spleenextracts produced several retarded complexes (A, B, and C),all of which are specifically inhibited by 12-m KB oligomers.The upper complex, B, the most intense band, was super-shifted by anti-p50 antibody, while the less-intense lowercomplex, C, was unaffected, suggesting that a large fractionof 12-m KB binding complexes in spleen tissue contains theNF-KB subunit, p50. Albeit with much lower intensity, brainextracts also produced several complexes, complex C exhib-iting higher intensity than the upper complexes, A and B.However, anti-p50 antibody did not supershift any of thecomplexes, indicating that brain extracts do not contain p50capable of binding to 12-m KB at a detectable level. Un-treated and RA-treated F9 cells as well as EL-4 cells alsoproduced complexes A, B, and C (Fig. 7A); complex B inEL-4 extracts was supershifted by anti-p50 antibody. In F9
cells, complex C was much more intense than complex A orB and migrated slightly faster in RA-treated cells than inuntreated cells. The complex supershifted by anti-p50 anti-body was greatly increased in RA-treated F9 cells comparedwith untreated cells. Control antibody directed to PRDIIBF1had no effect. These results indicate that untreated F9 cellsexpress little p50, but p50 expression is induced followingRA treatment. Consistent with these findings, RA-inducedp50 expression that contributes to MHC class I region Ibinding activity has been observed for N-Tera2 EC cells(56). The above data indicate that complex C, prominent inF9 cells, does not contain p50. To study the nature of thiscomplex further, UV cross-linking experiments were per-formed. Figure 7B shows that complex C is about 100 to 110kDa in size, ruling out participation of c-rel, p49, or relB inthe formation of this complex. At present, the protein(s)responsible for complex C is unknown. Together, theseresults indicate that 32-m KB binding activities are composedof heterogeneous proteins that are expressed at differentlevels in these extracts.Table 1 summarizes in vitro factor binding activities for
the three elements and compares them with 32-m expressionand with in vivo footprints. It is clear that, except for p50binding to the 02-m KB site, these binding activities do notcorrelate with the expression of the gene. Furthermore, nocorrelation was found between the binding activitiesand in vivo footprints.
DISCUSSION
In this paper we show that three conserved elements(PAM, ICS, and KB) of the 32-m gene are occupied in vivoin a tissue-specific manner that correlates with the expres-sion of the gene in the mouse. Moreover, in vivo occupancyis not demonstrated for any of these elements in F9 cellsbefore and after RA treatment, even though a low level of12-m RNA expression is induced following RA treatment.These results illustrate a situation not described before, inwhich a gene is transcribed without appreciable in vivoprotection. The overall pattern of in vivo occupancy seen forthe 12-m gene was similar to that found for MHC class Igenes, which also exhibit strong protection in spleen but notin brain tissue (10). In addition, MHC class I genes alsomanifested no in vivo occupancy in F9 cells, regardless ofthe RA treatment.The lack of in vivo protection in F9 and in brain tissue
cells was not due to the lack of factors that bind to theregulatory elements, since extracts from F9 and brain tissuecells revealed binding activities for all these elements invitro. Detailed EMSA analyses of these elements indicatedthat none of the in vitro binding activities for these elementscorrelated with the presence of detectable in vivo footprint-ing (Table 1). It seems clear that the presence of element-specific factors alone is not sufficient for producing in vivooccupancy. The lack of in vivo footprinting despite thepresence of factors has been noted for other genes (3, 29,35). It is possible that in vivo occupancy of the 12-m generequires a higher-order chromatin configuration controllingthe accessibility of these factors to specific DNA elements(15, 52, 63).How is 132-m (and MHC class I) expression induced in F9
cells by RA without detectable in vivo factor occupancy?The lack of in vivo protection is unlikely to be attributed tothe gene expression in a minor population of F9 cells, whichmight have obscured in vivo footprinting: practically thewhole population of F9 cells express MHC class I molecules
A
VOL. 13, 1993
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 16
Dec
embe
r 20
21 b
y 18
0.24
1.80
.102
.
6636 LONERGAN ET AL.
Brain SpleenF9 w/
EL4 5d RA F9B r .
-
Cpr, a,
Co.mpeltitr z IMT co
t --BIBP's
| .._Li MIBPBIIBP --
SIBP- - 'fiMNIBP- Q: 14
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15
s-2M ICSMlM2M3M4M5
F9 Extract
2((~~ ~ ~~~2
67 107__ _ ~~~~~~~~~~~~. 46
,191
*14.4
28
TAAGAACATGAAACTGAAAATGGGAAAG--ACG-------TCC--------CAG------------ ----TCC-------------------CCG--
EL4 Extract
v v 2D
211
107
29.7
FIG. 6. Characterization of 12-m ICS factor binding in vitro. (A) Tissue and cell line distribution of 32-m ICS binding factors. EMSAswere performed with 7.5 pug of extracts, except for spleen (3 ,ug). Lanes 1, 4, 7, 10, and 13, no competitor; lanes 2, 5, 8, 11, and 14, 12-m ICScompetitor (250-fold molar excess) added; lanes 3, 6, 9, 12, and 15, control competitor, UCR, added. (B) Sequence specificity of ICS bindingfactors. EMSAs were performed with F9 cell extracts as described above, and ICS mutant competitors were added at a 250-fold molar excess.
Only mutated nucleotides are shown; nucleotides identical to the native ICS are shown by dashes. The IFN-responsive element is underlined.Ld ICS represents MHC class I (H-2Ld) ICS (14), used as a competitor. (C) UV cross-linking of ICS-protein complexes. BrdU-substituted12-m ICS probe was cross-linked with F9 and EL4 nuclear extracts as for Fig. 3 and analyzed by SDS-PAGE. Cross-linked complexesproduced from F9 extracts were resolved by one (left)- or two (middle)-dimensional gel electrophoresis. Complexes separated in EMSA are
shown above the second-dimension SDS-PAGE. Cross-linked complexes in EL4 extracts were analyzed on a two-dimensional gel (right).Numbers on the right represent approximate molecular mass (in kilodaltons). BIBP (black square), SIBP (arrow), and MIBP (*) correspondto those in Fig. 6A.
(hence 132-m) in response to RA (62). It may be pertinent thatthe level of 12-m RNA in RA-treated F9 cells is >20 foldlower than that in ELA cells (Fig. 3), indicating that the rateof 32-m transcription is low in RA-treated cells. A low levelof transcription such as this may depend on the basalpromoter only and may not involve the upstream regulatoryregion: a state of partial gene activation mediated by thepromoter region alone has been considered, on the basis oftranscription in vitro (53). Alternatively, RA-induced tran-
scription of the 132-m gene may still depend on the upstreamelements, but factor binding may occur much less efficiently,and in a less stable fashion, which may be below the level ofdetection for in vivo footprinting. RA induction of p50 in F9cells (Fig. 6) that correlates with the induction of 12-mexpression may support this possibility. In addition, a spe-cific adaptor may be required for stable factor occupancy,which may be lacking in RA-treated F9 cells. The low-leveltranscription that occurs without detectable in vivo factor
A
N-. 1-t 'r r 1
C
411,4 .
MOL. CELL. BIOL.
1D
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 16
Dec
embe
r 20
21 b
y 18
0.24
1.80
.102
.

IN VIVO FOOTPRINTING OF THE 12-m GENE 6637
F9 w1F9 5dl RA
B
EL4
-A-B
-(C
F9 Extract
I 1) -+.: X
2 1)
21 I--
16Q-46-
Prh-Proxbe
1 2 3 4 5 6 7 8 9 110 12 13 1415 16 17 18 1920 21 22 23 24 25 26
FIG. 7. Factor binding to the 12-m KB site in vitro. (A) EMSA performed with extracts as for Fig. 6A. Lane 1, probe only; lanes 2, 7, 12,17, and 22, no competitor; lanes 3, 8, 13, 18, and 23, 12-m KB competitor (250-fold molar excess); lanes 4, 9, 14, 19, and 24, control competitorUCR; lanes 5, 10, 15, 20, and 25, anti-NF-KB p50 antibody; lanes 6, 11, 21, and 26, control anti-PRDIIBF1 antibody. A supershifted band ismarked with asterisks. (B) UV cross-linking of the 12-m KB-protein complex C from untreated F9 extracts. UV cross-linking was performed(23), and reactions were resolved by SDS-PAGE as for Fig. 6C. The small arrow indicates a major spot produced by complex C.
occupancy appears to represent a distinct state of geneexpression and appears to be maintained in a stable fashion:an F9 clone, ACc19, that exhibits properties of differentiatedcells likewise showed no discernible in vivo occupancy.
In vivo footprinting allowed us to identify a new ciselement, PAM. We show that DAM has a conserved hexa-nucleotide core to which multiple factors of 40 to 50 kDabind. A G residue in the core sequence required for factorbinding in vitro coincided with the site of in vivo protection,suggesting that the proteins that bind PAM in vitro also bindPAM in vivo. However, the precise molecular nature of thefactors binding to PAM is not clear: since the PAM core
TABLE 1. Summary of ,B2-m expression, in vivo footprinting,and in vitro factor binding activities
Tissue or In vivo In vitro DNA bindingecell line Expression footprinting pAMb IcsC KW
TissuesBrain - - - +Spleen + + + + + _ ++
CellsEIA ++ ++ - - ++F9 - - - + -/+F9 (RA) + + + +
a Binding activity not found in every cell and tissue is examined for acorrelation with the 32-m expression or with in vivo footprinting.
b No discernible correlation between the PAM binding activity and the,B2-m expression or in vivo footprinting. The lower band marked with anasterisk in Fig. 4 is found in spleen tissue and RA-treated F9 cells but not inEL4 cells that express 12-m.
c No discernible correlation is found, although the presence of BIBP in Fig.5 correlates with the lack of in vivo footprinting (but not with ,B2-mexpression).
d Complex B containing p5O (Fig. 7) correlates with 132-m expression (butnot in vivo footprinting).
sequence is not related to other well known factor bindingsites, they may represent a series of new DNA-bindingproteins. We note that a G residue farther upstream at -167,outside the PAM core, is also protected. It is possible that anadditional factor(s) is involved in the formation of the PAMcomplex in vivo. Furthermore, the PAM sequence, whenplaced on a heterologous promoter, enhanced reporter ac-tivity in several cells (Fig. 5B), indicating that it is atranscriptionally functional element. The level of PAM en-hancement seen with this reporter was modest (generallyabout twice that of the control), although PAM may elicit ahigher level of enhancement in a native promoter. It isinteresting to note that the level of PAM enhancement seenin F9 cells was essentially the same as that in ELA cells, eventhough the endogenous PAM is presumably nonfunctional inF9 cells.
Lastly, the property of ICS binding factors found in F9cells warrants attention. The ICS of the 32-m and MHC classI genes is homologous to the ISRE of other genes induced byIFNs (36). It has been documented that members of the IRFfamily, IRF-1, IRF-2, ICSBP, and ISGF3-y, that bind to theICS/ISRE (14, 26, 39, 61) range from 40 to 50 kDa in size,matching with the broad spot seen in both ELA and F9 cells(Fig. 6). However, none of the factors revealed in F9 cells islikely to be a known member of the IRF family, sincemutations that changed the conserved ISRE did not changetheir competition for binding in EMSA. Furthermore, it hasbeen shown that in F9 cells, IRF-1, IRF-2, and ICSBP arenot expressed (14, 26). On the basis of molecular mass,PRDIBF1 (30) is also an unlikely candidate for these factors.There may be a series of unidentified ICS binding factorsexpressed in F9 cells.
ACKNOWLEDGMENTS
We thank D. Harter, Director, HHMI Research Scholar's Pro-gram, for his support, J. Segars for the TkCAT plasmid, and K.
A Brain KSpleen
*4~~~ v
i.
VOL. 13, 1993
I
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 16
Dec
embe
r 20
21 b
y 18
0.24
1.80
.102
.

6638 LONERGAN ET AL.
Rubin and T. Timmerman for expert secretarial help. We areindebted to members of the Ozato laboratory for critical reading ofthe manuscript.
REFERENCES1. Baeuerle, P. A. 1991. The inducible transcription activator
NF-KB: regulation by distinct protein subunit. Biochim. Bio-phys. Acta 1072:63-80.
2. Ballard, D. W., W. H. Walker, S. Doerre, P. Sista, J. A. Molitor,E. P. Dixon, N. J. Peffer, M. Hannink, and W. C. Greene. 1990.The v-rel oncogene encodes a KB enhancer binding protein thatinhibits NF-KB function. Cell 63:803-814.
3. Becker, P. B., S. Ruppert, and G. Schutz. 1987. Genomicfootprinting reveals cell type specific DNA binding of ubiqui-tous factors. Cell 51:435-443.
4. Bours, V., J. Villalobos, P. R. Burd, K. Kelly, and U. Siebenlist.1990. Cloning of a mitogen-inducible gene encoding a KBDNA-binding protein with homology to the rel oncogene and tocell-cycle motifs. Nature (London) 348:76-80.
5. Chamberlain, J. W., J. A. Nolan, P. J. Conrad, H. A. Vasavada,H. H. Vasavada, S. Ganguly, C. A. Janeway, Jr., and S. M.Weissman. 1988. Tissue-specific and cell surface expression ofhuman major histocompatibility complex class I heavy (HLA-B7) and light (032-microglobulin) chain genes in transgenic mice.Proc. Natl. Acad. Sci. USA 85:7690-7694.
6. Croce, C. M., A. Linnenbach, K. Huebner, J. R. Parnes, D. H.Margulies, E. Appella, and J. G. Seidman. 1981. Control ofexpression of histocompatibility antigens (H-2) and 32-microglo-bulin in F9 teratocarcinoma stem cells. Proc. Natl. Acad. Sci.USA 78:5754-5758.
7. Daniel, F., D. Morello, 0. Le Bail, P. Chambon, Y. Cayre, andP. Kourilsky. 1983. Structure and expression of the mouse02-microglobulin gene isolated from somatic and non-express-ing teratocarcinoma cells. EMBO J. 2:1061-1065.
8. Dargemont, C., D. Dunon, M.-A. Deugnier, M. Denoyelle, J.-M.Girault, F. Lederer, K. H. D. U, F. Godeau, J. P. Thiery, andB. A. Imhof. 1989. Thymotaxin, a chemotactic protein, is iden-tical to P2-microglobulin. Science 246:803-806.
9. David-Watine, B., A. Israel, and P. Kourilsky. 1990. The regu-lation and expression of MHC class I genes. Immunol. Today11:286-292.
10. Dey, A., A. M. Thornton, M. Lonergan, S. M. Weissman, J. W.Chamberlain, and K. Ozato. 1992. Occupancy of upstreamregulatory sites in vivo coincides with major histocompatibilitycomplex class I gene expression in mouse tissues. Mol. Cell.Biol. 12:3590-3599.
11. Dignam, J. D., R. M. Lebowitz, and R. G. Roeder. 1983.Accurate transcription initiation by RNA polymerase II in asoluble extract from isolated mammalian nuclei. Nucleic AcidsRes. 11:1475-1489.
12. Drew, P. D., M. Lonergan, M. D. Goldstein, L. A. Lampson, K.Ozato, and D. E. McFarlin. 1993. Regulation of MHC class I and32-microglobulin gene expression in human neuronal cells:
factor binding to conserved cis-acting regulatory sequencescorrelates with expression of the genes. J. Immunol. 150:3300-3310.
13. Drezen, J. M., C. Babinet, and D. Morello. 1993. Transcriptionalcontrol of MHC class I and 02-microglobulin genes in vivo. J.Immunol. 150:2805-2813.
14. Driggers, P. H., D. L. Ennist, S. L. Gleason, W. Mak, M. S.Marks, B. Levi, J. R. Flanagan, E. Appella, and K Ozato. 1990.An interferon gamma-regulated protein that binds the interfer-on-inducible enhancer element of major histocompatibility com-plex class I genes. Proc. Natl. Acad. Sci. USA 87:3743-3747.
15. Evans, T., G. Felsenfeld, and M. Reitman. 1990. Control ofglobin gene transcription. Annu. Rev. Cell Biol. 6:95-124.
16. Flanagan, J. R., K. G. Becker, D. L. Ennist, S. L. Gleason, P. H.Driggers, B.-Z. Levi, E. Appella, and K. Ozato. 1992. Cloning ofa negative transcription factor that binds to the upstreamconserved region of Moloney murine leukemia virus. Mol. Cell.Biol. 12:38-44.
17. Flanagan, J. R., M. Murata, P. A. Burke, Y. Shirayoshi, E.Appella, P. A. Sharp, and K. Ozato. 1991. Negative regulation of
the major histocompatibility complex class I promoter in em-bryonal carcinoma cells. Proc. Natl. Acad. Sci. USA 88:8555-8559.
18. Friedman, R., and G. Stark. 1985. a-Interferon induced tran-scription of HLA and metallothionein genes containing homol-ogous upstream sequences. Nature (London) 314:637-639.
19. Fujita, T., M. Miyamoto, Y. Kimura, J. Hammer, and T.Taniguchi. 1989. Involvement of a cis element that binds onH2TF-IINFKB-like factor(s) in the virus-induced interferon-1gene expression. Nucleic Acids Res. 17:3335-3346.
20. Garrity, P. A., and B. J. Wold. 1992. Effects of different DNApolymerases in ligation-mediated PCR: enhanced genomic se-quencing and in vivo footprinting. Proc. Natl. Acad. Sci. USA89:1021-1025.
21. Ghosh, D. 1990. A relational database of transcription factors.Nucleic Acids Res. 18:1749-1756.
22. Ghosh, S., A. M. Gifford, L. Riviere, P. Tempst, G. P. Nolan,and D. Baltimore. 1990. Cloning of the p50 DNA binding subunitof NF-KB: homology to rel and dorsal. Cell 62:1019-1029.
23. Gray, T. A., D. L. Gumucio, and F. S. Collins. 1990. Atwo-dimensional electrophoresis system for DNA-binding pro-teins. Technique J. Methods Cell Mol. Biol. 2:147-154.
24. Gfissow, D., R. Rein, I. Ginjaar, F. Hochstenbach, G. Seeman,A. Kottmann, and H. Ploegh. 1987. The human f32-microglobulingene. Primary structure and definition of the transcriptionalunit. J. Immunol. 139:3132-3138.
25. Harada, H., T. Fujita, M. Miyamoto, Y. Kimura, M. Maruyama,A. Furia, T. Miyata, and T. Taniguchi. 1989. Structurally similarbut functionally distinct factors, IRF-1 and IRF-2, bind to thesame regulatory elements of IFN and IFN-inducible genes. Cell58:729-739.
26. Harada, H., K. Willison, J. Sakakibara, M. Miyamoto, T. Fujita,and T. Taniguchi. 1990. Absence of the type I IFN system in ECcells: transcriptional activator (IRF-1) and repressor (IRF-2)genes are developmentally regulated. Cell 63:303-312.
27. Israel, A., A. Kimura, M. Kieran, 0. Yano, J. Kanellopoulos, 0.Le Bail, and P. Kourilsky. 1987. A common positive trans-actingfactor binds to enhancer sequences in the promoters of mouseH-2 and 32-microglobulin genes. Proc. Natl. Acad. Sci. USA84:2653-2657.
28. Jaffe, L., L. Jeannotte, E. K. Bikoff, and E. J. Robertson. 1990.Analysis of 02-microglobulin gene expression in the developingmouse embryo and placenta. J. Immunol. 145:3474-3482.
29. Kara, C. J., and L. H. Glimcher. 1991. In vivo footprinting ofMHC class II genes: bare promoters in the bare lymphocytesyndrome. Science 252:709-712.
30. Keller, A. D., and T. Maniatis. 1991. Identification and charac-terization of a novel repressor of beta-interferon gene expres-sion. Genes Dev. 5:868-879.
31. Kieran, M., V. Blank, F. Logeat, J. Vandekerckhove, F. Lott-speich, 0. Le Bail, M. B. Urban, P. Kourilsky, P. A. Baeuerie,and A. Israel. 1990. The DNA binding subunit of NF-KB isidentical to factor KBF1 and homologous to the rel oncogeneproduct. Cell 62:1007-1018.
32. Kimura, A., A. Israel, 0. Le Bail, and P. Kourilsky. 1986.Detailed analysis of the mouse H-2Kb promoter: enhancer-likesequences and their role in the regulation of class I geneexpression. Cell 44:261-272.
33. Koller, B. H., P. Marrack, J. W. Kappler, and 0. Smithies. 1990.Normal development of mice deficient in 132M, MHC class Iproteins, and CD8' T cells. Science 248:1227-1230.
34. Lampson, L. A., C. A. Fisher, and J. P. Whelan. 1983. Strikingpaucity of HLA-A,B,C, and 32-microglobulin on human neuro-blastoma cell lines. J. Immunol. 130:2471-2478.
35. Levy, D. E., D. S. Kessler, R. Pine, and J. E. Darnell, Jr. 1989.Cytoplasmic activation of ISGF3, the positive regulator ofinterferon-alpha-stimulated transcription, reconstituted in vitro.Genes Dev. 3:1362-1371.
36. MacDonald, N. J., D. Kuhl, D. Maguire, D. Naf, P. Gallant, A.Goswamy, H. Hug, H. Bueler, M. Chatuvedi, J. de la Fuente, H.Ruffher, F. Meyer, and C. Weismann. 1990. Different pathwaysmediate virus inducibility of the human IFN-alphal and IFN-beta genes. Cell 60:767-779.
MOL. CELL. BIOL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 16
Dec
embe
r 20
21 b
y 18
0.24
1.80
.102
.

IN VIVO FOOTPRINTING OF THE 02-m GENE 6639
37. MacGregor, G. R., and T. Caskey. 1989. Construction of plas-mids that express E. coli 0-galactosidase in mammalian cells.Nucleic Acids Res. 17:2365.
38. Martinez, E., F. Givel, and W. Wahli. 1987. The estrogen-responsive element as an inducible enhancer: DNA sequencerequirements and conversion to a glucocorticoid-responsiveelement. EMBO J. 6:3719-3727.
39. Miyamoto, M., T. Fujita, Y. Kimura, M. Maruyama, H. Harada,Y. Sudo, T. Miyata, and T. Taniguchi. 1988. Regulated expres-sion of a gene encoding a nuclear factor, IRF-1, that specificallybinds to IFNI3 gene regulatory elements. Cell 54:903-913.
40. Miyazaki, J.-I., E. Appella, and K. Ozato. 1986. Negativeregulation of the major histocompatibility class I gene in undif-ferentiated embryonal carcinoma cells. Proc. Natl. Acad. Sci.USA 83:9537-9541.
41. Molitor, J. A., W. H. Walker, S. Doerre, D. W. Ballard, andW. C. Greene. 1990. NF-KB: a family of inducible and differen-tially expressed enhancer-binding proteins in human T cells.Proc. Natl. Acad. Sci. USA 87:10028-10032.
42. Morello, D., F. Daniel, P. Baldacci, Y. Cayre, G. Gachelin, andP. Kourfisky. 1982. Absence of significant H-2 and P2-microglo-bulin mRNA expression by mouse embryonal carcinoma cells.Nature (London) 296:260-262.
43. Morello, D., P. Duprey, A. Israel, and C. Babinet. 1985. Asyn-chronous regulation of mouse H-2D and beta-2 microglobulinRNA transcripts. Immunogenetics 22:441-452.
44. Mueller, P. R., S. J. Salser, and B. Wold. 1988. Constitutive andmeta-inducible protein: DNA interactions at the mouse metal-lothionine I promoter examined by in vivo and in vitro footprint-ing. Genes Dev. 2:412-427.
45. Mueller, P. R., and B. Wold. 1989. In vivo footprinting of amuscle specific enhancer by ligation-mediated PCR. Science246:780-786.
46. Nagata, T., J. H. Segars, B.-Z. Levi, and K. Ozato. 1992.Retinoic acid-dependent transactivation of major histocompati-bility complex class I promoters by the nuclear hormone recep-tor H-2RIIBP in undifferentiated embryonal carcinoma cells.Proc. Natl. Acad. Sci. USA 89:937-941.
47. Ozato, K., Y.-J. Wan, and B. Orrison. 1985. Mouse majorhistocompatibility class I gene expression begins at midsomitestage and is inducible in earlier-stage embryos by interferon.Proc. Natl. Acad. Sci. USA 82:2427-2431.
48. Parnes, J. R., B. Velan, A. Felsenfeld, L. Ramanathan, U.Ferrini, E. Appella, and J. G. Seidman. 1981. Mouse 2-microglobulin cDNA clones: a screening procedure for cDNAclones corresponding to rare mRNAs. Proc. Natl. Acad. Sci.USA 78:2253-2257.
49. Paterson, B. M., and J. D. Eldridge. 1984. a-Cardiac actin is amajor sarcomeric isoform expressed in embryonic avian skeletalmuscle. Science 224:1436-1438.
50. Picard, D., and W. Schaftfer. 1984. A lymphocyte-specificenhancer in the mouse immunoglobulin K gene. Nature (Lon-don) 307:80-82.
51. Richard-Foy, H., and G. L. Hager. 1987. Sequence-specificpositioning of nucleosomes over the steroid-inducible MMTVpromoter. EMBO J. 6:2321-2328.
52. Riek, A., G. Schutz, and A. F. Stewart. 1991. Glucocorticoidsare required for establishment and maintenance of an alteration
in chromatin structure: induction leads to a reversible disruptionof nucleosomes over an enhancer. EMBO J. 10:2569-2576.
53. Roeder, R. G. 1991. The complexities of eukaryotic transcrip-tion initiation: regulation of preinitiation complex assembly.Trends Biochem. Sci. 16:402-408.
54. Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecularcloning: a laboratory manual, 2nd ed. Cold Spring HarborLaboratory Press, Cold Spring Harbor, N.Y.
55. Schindler, C., X.-Y. Fu, T. Improta, R. Aebersold, and J. E.Darnell, Jr. 1992. Proteins of transcription factor ISGF-3: onegene encodes the 91- and 84-kDa ISGF-3 proteins that areactivated by interferon a. Proc. Natl. Acad. Sci. USA 89:7836-7839.
56. Segars, J. H., T. Nagata, V. Bours, J. A. Medin, G. Franzoso,J. C. G. Blanco, P. Drew, K. G. Becker, J. An, T. Tang, D. A.Stephany, B. Neel, U. Siebenlist, and K. Ozato. 1993. Retinoicacid induction of major histocompatibility complex class I genesin N-Tera2 embryonal carcinoma cells involves coactivation ofNF-KB (pSO-p65) and retinoic acid receptor ,B-retinoid X recep-tor K heterodimers. Mol. Cell. Biol. 13:6157-6169.
57. Severinsson, L., and P. A. Peterson. 1984. 02-Microglobulininduces intracellular transcript of human class I transplantationantigen heavy chains in Xenopus laevis oocytes. J. Cell Biol.99:226-232.
58. Silverman, T., A. Rein, B. Orrison, J. Langloss, G. Bratthauer,J.-I. Miyazaki, and K. Ozato. 1988. Establishment of cell linesfrom somite stage mouse embryos and expression of majorhistocompatibility class I genes in these cells. J. Immunol.140:4378-4387.
59. Simister, N. E., and K. E. Mostov. 1989. An Fc receptorstructurally related to MHC class I antigens. Nature (London)337:184-187.
60. Strickland, S., K. K. Smith, and R. Marotti. 1980. Hormonalinduction of differentiation in teratocarcinoma stem cells: gen-eration of parietal endoderm by retinoic acid and dibutyrylcAMP. Cell 21:347-355.
61. Veals, S. A., C. Schindler, D. Leonard, X.-Y. Fu, R. Aebersold,J. E. Darnell, Jr., and D. E. Levy. 1992. Subunit of an alpha-interferon-responsive transcription factor is related to interferonregulatory factor and Myb families of DNA-binding proteins.Mol. Cell. Biol. 12:3315-3324.
62. Wan, Y.-J., B. M. Orrison, R. Lieberman, P. Lazarovici, and K.Ozato. 1987. Induction of major histocompatibility class I anti-gens by interferons in undifferentiated F9 cells. J. Cell. Physiol.130:276-283.
63. Weih, F., D. Nitsch, A. Reik, and P. B. Becker. 1991. Analysis ofCpG methylation and genomic footprinting at the tyrosineaminotransferase gene: DNA methylation alone is not sufficientto prevent protein binding in vivo. EMBO J. 10:2559-2568.
64. Whittemore, L.-A., and T. Maniatis. 1990. Postinduction repres-sion of the ,B-interferon gene is mediated through two positiveregulatory domains. Proc. Natl. Acad. Sci. USA 87:7799-7803.
65. Williams, B. R. G. 1991. Transcriptional regulation of interfer-on-stimulated genes. Eur. J. Biochem. 200:1-11.
66. ZUlstra, M., M. Bix, N. E. Simister, J. M. Loring, D. H. Raulet,and R. Jaenisch. 1990. 32-Microglobulin deficient mice lackCD4-8+ cytolytic T cells. Nature (London) 344:742-746.
VOL. 13, 1993
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 16
Dec
embe
r 20
21 b
y 18
0.24
1.80
.102
.