aberrant phenotype and function of myeloid dendritic cells · aberrant phenotype and function of...
TRANSCRIPT

of June 11, 2018.This information is current as
ErythematosusDendritic Cells in Systemic Lupus Aberrant Phenotype and Function of Myeloid
Mariana J. KaplanDacheng Ding, Hemal Mehta, W. Joseph McCune and
http://www.jimmunol.org/content/177/9/5878doi: 10.4049/jimmunol.177.9.5878
2006; 177:5878-5889; ;J Immunol
Referenceshttp://www.jimmunol.org/content/177/9/5878.full#ref-list-1
, 32 of which you can access for free at: cites 101 articlesThis article
average*
4 weeks from acceptance to publicationFast Publication! •
Every submission reviewed by practicing scientistsNo Triage! •
from submission to initial decisionRapid Reviews! 30 days* •
Submit online. ?The JIWhy
Subscriptionhttp://jimmunol.org/subscription
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/About/Publications/JI/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/alertsReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists All rights reserved.Copyright © 2006 by The American Association of1451 Rockville Pike, Suite 650, Rockville, MD 20852The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on June 11, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
by guest on June 11, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

Aberrant Phenotype and Function of Myeloid Dendritic Cellsin Systemic Lupus Erythematosus1
Dacheng Ding, Hemal Mehta, W. Joseph McCune, and Mariana J. Kaplan2
Systemic lupus erythematosus (SLE) is characterized by a systemic autoimmune response with profound and diverse T cellchanges. Dendritic cells (DCs) are important orchestrators of immune responses and have an important role in the regulation ofT cell function. The objective of this study was to determine whether myeloid DCs from individuals with SLE display abnormal-ities in phenotype and promote abnormal T cell function. Monocyte-derived DCs and freshly isolated peripheral blood myeloidDCs from lupus patients displayed an abnormal phenotype characterized by accelerated differentiation, maturation, and secretionof proinflammatory cytokines. These abnormalities were characterized by higher expression of the DC differentiation markerCD1a, the maturation markers CD86, CD80, and HLA-DR, and the proinflammatory cytokine IL-8. In addition, SLE patientsdisplayed selective down-regulation of the maturation marker CD83 and had abnormal responses to maturation stimuli. Theseabnormalities have functional relevance, as SLE DCs were able to significantly increase proliferation and activation of allogeneicT cells when compared with control DCs. We conclude that myeloid DCs from SLE patients display significant changes inphenotype which promote aberrant T cell function and could contribute to the pathogenesis of SLE and organ damage. TheJournal of Immunology, 2006, 177: 5878–5889.
S ystemic lupus erythematosus is an autoimmune disease ofunclear etiology that affects multiple organs. Individualswith systemic lupus erythematosus (SLE)3 present with
numerous and diverse immune system abnormalities and display aprominent imbalance in different T cell subset functions (1–3).Indeed, individuals with SLE have autoreactive T cells that arespecific for ubiquitous self-peptides and provide pathogenic B cellhelp (4, 5). A pan T cell dysfunction in SLE is characterized byexaggerated CD4� T cell responses (6, 7) and diminished CD8� Tcell activities (8). Other alterations include changes in proliferativeT cell responses as well as increased expression of early and lateT cell activation markers (3, 7, 9–11). In addition, alterations inTh1 and/or Th2 lymphocyte function have been described (12–16),resulting in production of cytokines that up-regulate autoantibodyproduction by B cells, promote immune complex formation, andincrease the apoptotic load (17, 18). The exact role that accessorycells might have in inducing abnormal T cell responses in SLE isunclear; however, different groups have proposed that T cell dis-turbances in SLE could be induced or promoted, at least in part, byalterations in dendritic cell (DC) phenotype and function, becausethese are key regulators of the immune system (19–22). DCs com-
prise several subsets with different stages of maturation (20). Im-mature, Ag-capturing DCs sense diverse signals that induce themto undergo a maturation process, whose hallmarks are the up-reg-ulation of the molecule CD83, as well as of a number of costimu-latory and adhesion molecules, migration into lymphoid organs(23), and acquisition of the capacity to activate quiescent, naive,and memory lymphocytes (20, 24–30). DCs can induce either Tcell tolerance or strong innate and adaptive immunity to specificAg. In general, tolerance is initiated when DCs are immature,whereas the initiation of immunity requires an effective DC mat-uration signal. After reaching the lymph nodes, DCs present pro-cessed Ag to T cells via both classical (class I and II MHC) andnonclassical (CD1 family) pathways (24, 31, 32). In addition totheir maturation status, DCs can be subdivided into a CD11c�
(plasmacytoid DC) and a CD11c� (myeloid DC) subset (20, 33).The CD11c� subset follows a myeloid differentiation pathway inwhich monocytes serve as precursors. These DC subsets have dif-ferent migration programs and very likely have divergent roles inthe induction and regulation of the immune response (33, 34)
In the past few years, significant progress has been made inelucidating the important role that IFN-�-producing plasmacytoidDCs have in promoting autoimmune responses in SLE (22, 35, 36).Much less is known about the role that myeloid DCs play in con-tributing to the pathogenesis of SLE. Indeed, it is unclear whethermyeloid DCs present phenotypic and functional changes that maypromote the autoimmune features of lupus and whether such dif-ferences might potentiate aberrant cognate interactions with Tcells. We now report that myeloid DCs of individuals with SLEdisplay evidence of aberrant phenotype and function which maycontribute to the T cell abnormalities described in this disease.
Materials and MethodsPatient selection
Patients with SLE and rheumatoid arthritis (RA) fulfilled the AmericanCollege of Rheumatology criteria for these diseases (37–39) and were re-cruited from the outpatient rheumatology clinic and inpatient services atthe University of Michigan, and from the Michigan Lupus Cohort (AnnArbor, MI). Healthy controls were obtained by advertisement. SLE activity
Department of Internal Medicine, Division of Rheumatology, University of Michigan,Ann Arbor, MI 48109
Received for publication June 15, 2005. Accepted for publication August 12, 2006.
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby marked advertisement in accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.1 This work was supported by Public Health Service Grants AR050554 andAR048235 and by the Anthony S. Gramer Fund in Inflammation Research (all toM.J.K.). D.D. was supported by Training Grant T32 AR 07080. W.J.M. was sup-ported by the Herb and Carol Amster Lupus Research Fund and the Klein LupusResearch Fund. This research was also supported (in part) by the National Institutesof Health through University of Michigan’s Cancer Center Support Grant (P30CA46592) and the Rheumatic Diseases Core Center Grant (P30 AR48310).2 Address correspondence and reprint requests to Dr. Mariana J. Kaplan, Division ofRheumatology, University of Michigan, 5520 MSRBI, 1150 West Medical CenterDrive, Ann Arbor, MI 48109-0680. E-mail address: [email protected] Abbreviations used in this paper: SLE, systemic lupus erythematosus; DC, dendriticcell; RA, rheumatoid arthritis; SLEDAI, SLE disease activity index; 6-MP, 6-mer-captopurine; MMF, mycophenolate-mofetil.
The Journal of Immunology
Copyright © 2006 by The American Association of Immunologists, Inc. 0022-1767/06/$02.00
by guest on June 11, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

was assessed by the SLE disease activity index (SLEDAI) (40). Patient andcontrol cells were paired and studied in parallel. Overall, 146 SLE patients, 76healthy controls and 35 RA patients were studied. Information regarding thedemographics, disease activity, and use of medications is provided in Table I.
Reagents
Human rIL-4, TNF-�, and IL-2 were purchased from PeproTech. HumanGM-CSF was a gift from Berlex. rIFN-�2b was obtained from Schering.X-vivo 10 serum-free medium was from BioWhittaker. LPS and PHA werepurchased from Sigma-Aldrich. The following anti-human mAbs conju-gated to FITC, PE, CyChrome, and allophycocyanin were used: anti-CD1a,CD3, CD4, CD8, CD11c, CD14, CD25, CD40L, CD69, CD80, CD83,CD86, HLA-DR, and isotype controls (all obtained from BD Biosciences).The pan T cell isolation kit was obtained from Miltenyi Biotec. CFSE wasobtained from Molecular Probes.
Generation of monocyte-derived DCs
Myeloid DCs were generated from human peripheral monocytes as previ-ously described (41). In brief, human PBMC were separated by standarddensity gradient centrifugation on Ficoll-Hypaque Plus (Amersham Bio-sciences) and resuspended at 6 � 106 cells/ml in RPMI 1640 with L-glutamine and 10% FBS. The cells were transferred to tissue culture platesand allowed to adhere to the plastic surface for 1 h at 37°C. Nonadherentcells were then removed by washing with PBS. Monocyte recovery rateafter adherence was �89% in both healthy controls and SLE patients. Theadhered monocytes were further cultured for 5–7 days in DC inductionmedium (serum-free X-vivo-10 containing 20 ng/ml GM-CSF and 5–10ng/ml IL-4). In some experiments, cells were further purified using metri-zamide gradient (Sigma-Aldrich). The purity of myeloid DCs obtainedunder our experimental conditions was �90%, as confirmed by flow cy-tometric analysis (data not shown). At days 5–7, cells were harvested foranalysis or stimulated with DC maturation stimuli (41) (0.5–2 �g/ml LPSand/or 10–100 ng/ml TNF-�) for 48 h. In additional experiments, freshlyisolated monocytes were cultured for 7 days with GM-CSF, with or withoutIL-4, in the presence or absence of 100 or 1000 U of rIFN-�.
Immunofluorescence staining, FACS, and fluorescent microscopyanalysis
DCs were washed with PBS/0.2% BSA and FcRs were blocked by incu-bating cells for 20 min in PBS with 40% control human sera or with anti-FcAb (Miltenyi Biotec). Cells were then incubated for 30 min at 4°C with0.06–0.15 �g/ml fluorochrome-conjugated mAb following the manufac-turer’s directions. Cells were washed three times with PBS/0.2% BSA,fixed in 1% paraformaldehyde and analyzed in a FACSCalibur flow cy-tometer (BD Biosciences), using previously described protocols. Data anal-ysis was performed using Lysys II software (BD Biosciences) and Win-MDI 2.8 software (�http://facs.scripps.edu�). Stained cells were gated byside scatter and forward scatter characteristics and further identified bysurface markers. The results were expressed as the percentage of cellsstaining positive for different markers as well as by mean channel fluores-cence. The cutoff point for positive staining was above the level of thecontrol isotype Abs.
For fluorescent microscopy, DCs were derived from monocytes as statedabove. At day 7, cells were harvested, washed, and stained with the specificmAbs mentioned above to characterize DC phenotype and morphology.Cells were then fixed in 1% formalin in PBS for 30 min, and transferred toa microscope slide using ProLong Antifade kit (Molecular Probes). Slideswere analyzed using a Zeiss LSM 510 META Laser Scanning Microscope(Carl Zeiss Advanced Imaging Microscopy).
Isolation and characterization of myeloid DCs from peripheralblood
PBMCs were isolated from peripheral blood of SLE patients and controlsby Ficoll gradient as described above. PBMCs were incubated with twodifferent mixtures of Abs: 1) CD11c-FITC, CD1a-allophycocyanin, CD83-PE, and CD86-PE/Cy5, and 2) CD11c-FITC, CD14-allophycocyanin/Cy5,CD86-PE/Cy5 and HLA-DR-allophycocyanin. After incubation for 30 minwith these Abs at 4°C, cells were washed, fixed in PBS/1% paraformal-dehyde, and analyzed by FACS by gating the CD11c� population andexcluding CD14� cells.
Drug treatment
Monocytes were cultured as stated above to induce DC differentiation, inthe presence or absence of graded concentrations of indomethacin (0.01–1�g/ml), hydroxychloroquine (0.02–2 �g/ml), hydrocortisone (0.01–1 �M),6-mercaptopurine (6-MP) (0.01–1 �M) and mycophenolate-mofetil(MMF) (0.04–4 �g/ml; all obtained from Sigma-Aldrich) or vehicle (42–46). A stock solution of 6-MP was prepared in dimethylformamide at aconcentration of 10 mg/ml. The stock solution was diluted in assay diluent(80% culture medium/20% ethanol) to yield a 6-MP working solution of 80�g/ml or less as indicated. The working solution of 6-MP as well as theother materials prepared in assay diluent were then further diluted 1/40 intothe cell cultures for the tests. The final concentrations of ethanol (0.1%) ordimethylformamide (�0.02%) do not yield significant effects on the cellcultures (11, 47). Indomethacin was prepared in a concentration of 500 mMin absolute ethanol, then diluted to final concentrations in the cell culturemedium (11). Control cells were treated with an equal volume of the sol-vent. MMF was prepared as previously described (43). At day 7, DCs werewashed and analyzed by flow cytometry for expression of differentiation(CD1a) and maturation (CD83, CD86, HLA-DR) markers.
RNA extraction and quantitative real-time RT-PCR
Total RNA was isolated from myeloid DCs using the RNeasy kit withDNase I digestion (Qiagen) to remove possible genomic DNA contamina-tion, and reverse transcribed to cDNA using the SuperScript III first-strandsynthesis system (Invitrogen Life Technologies) with Oligo(dT)30 primer.For real-time detection of target and reference gene expression, eight pairprimers and probes were designed as follows: 83 forward (F): 5�-CTGCTCCTGAGCTGCGCCTACA; CD83 reverse (R): 5�-CACCACCCTCCAATAACTTGAC; CD83 probe: 5�-ATCCGCAGGTTCCCTACACGGTCTCC; CD80 F: 5�-CTTCAACTGGAATACAACCAAGCA; CD80 R: 5�-TGCATCTTGGGGCAAAGCAGTA; CD80 probe: 5�-CTCCCATCCTGGGCCATTACCTTAATC; CD1a F: 5�-GTCCTCTACTGGGAGCATCACA; CD1a R: 5�-GTCTTAACAGAAACAGCGTTTCC; CD1a probe:
Table I. Demographic and clinical characteristics of human subjects studied
Variable Lupus Control RA
Number studied 146 76 35Age(mean range) 44.5 11.5 37 13.5 51 14.4Females n (%) 130 (89) 55 (72.3) 18 (51.5)Males n (%) 16 (11) 21 (27.6) 17 (48.5)
Lupus disease activity (mean SEM) 4.5 3SLEDAI � 2 (%) 77SLEDAI � 2 (%) 69
MedicationsAntimalarials (%) 68.5 40.5Azathioprine (%) 5.47 0Cyclophosphamide (%) 1.36 0Methotrexate (%) 1.36 51.4Mycophenolate mofetil (%) 24.65 0Prednisone (�0.5 mg/kg/day) (%) 34.2 51.4Prednisone (0.5–1 mg/kg/day) (%) 9.5 5.7Prednisone (�1 mg/kg/day) (%) 4.1 8.5
5879The Journal of Immunology
by guest on June 11, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

5�-CTTGGCGGTGATAGTGCCTTTACTTCTT; CD86 F: 5�-GACAGGCATTTGTGACAGCACTA; CD86 R: 5�-TCT GCA GTC TCA TTGAAA TAA GC; CD86 probe: 5�-TTCCTGCTCTCTGGTGCTGCTCCTCT;CD14 F: 5�-GGTGCCGCTGTGTAGAAAGAAGC; CD14 R: 5�-GGTTCTGGCGTGGTCGCAGAGAC; CD14 probe: 5�-TTATCGACCATGGAGCGCGCGT; DRa F: 5�-TCAAGGTGCATTGGCCAACATAG; DRaR: 5�-CTCTCAGTTCCACAGGGCTGTTC; DRa probe: 5�-CGATCACCAATGTACCTCCAGAG; PBGD-F 5�-GGCAATGCGGCTGCAA-3�;PBGD-R 5�-GGGTACCCACGCGAATCAC-3�; PBGD probe 5�-CTCATCTTTGGGCTGTTTTCTTCCGCC; �-actin F: 5�-AGCCTTCCTTCCTGGGCATGGA; �-actin R: 5�-CTCAGGAGGAGCAATGATCTTGA;�-actin probe: 5�-ACATCCGCAAAGACCTGTACGCCAACA.
Probes were labeled with 5�-6-FAM/3�-TAMRA (Integrated DNATechnologies). HotStar Taq polymerase (Qiagen) was used. PCR was per-formed using MyCycler Thermal Cycler (Bio-Rad) in a total reaction mix-ture of 20 �l containing 50 ng of cDNA, 1� HotStar TaqPCR buffer, and400 nM probe/primers mixture. After denaturation at 95°C for 15 min, 55cycles were performed at 95°C for 15 s, followed by 60°C for 1 min.Comparative cycle threshold method with PCR efficiency correction, alsoknown as Pfaffl’s method, was used for quantification, as previously de-scribed (48). The expression levels of the target genes were adjusted to theexpression levels of the housekeeping genes PBGD and �-actin.
Cytokine determination
Cytokines were measured using two different methods. The human cyto-kines IL-1�, IL-2, IL-4, IL-5, IL-6, IL-7, IL-8, IL-10, IL-12p70, IL-13,IFN-�, GM-CSF, and TNF-� were simultaneously quantified in duplicateusing the human cytokine multiplex kit (Linco Research). Supernatantsobtained from day 7 (unstimulated DCs) or day 9 (DCs treated with mat-uration stimuli for 48 h) cultures were aliquoted and stored at �80°C untilused. Sera from the same patients were also stored. Cytokine concentra-tions were determined following manufacturer’s instructions. In brief, stan-dards and samples were added into the appropriate wells. Mixed beadswere added to each well and the plate was incubated with agitation for 1 hat room temperature. Plate was washed and a detection Ab mixture wasadded to each well and incubated for 30 min at room temperature. Strepta-vidin-PE was added to each well containing the detection Ab mixture. Theplate was then incubated with agitation on a plate shaker for 30 min atroom temperature, washed, and sheath fluid was added for 5 min. Plate wasread on Luminex 100 (Luminex) and the concentration reported as pico-grams per milliliter.
In addition, the BD CBA Human Inflammation kit (BD Biosciences)was used to quantitatively measure IL-8, IL-1, IL-6, IL-10, TNF-�, andIL-12p70 protein levels in sera and supernatant samples, following man-ufacturer’s instructions. In brief, mixed capture beads were added to assaytubes. Human inflammation standard dilutions and test samples were addedto the assay tubes. Samples were incubated for 1.5 h at room temperatureand protected from direct exposure to light. Wash buffer was added andtubes were centrifuged at 200 � g for 5 min. Supernatants were discardedleaving 100 �l of liquid in each assay tube. Human inflammation PE de-tection reagent was added to the tubes and samples were incubated for 1.5 hat room temperature, washed, and analyzed by FACS using BD CytometricBead Array Software.
T cell proliferation and determination of T cell activation
T cells were isolated by negative selection using magnetic beads and in-structions provided by the manufacturer (pan T cell isolation kit; MiltenyiBiotec). Purity was �95%, as assessed by CD3 expression. T cell prolif-eration was analyzed using the intracellular fluorescent dye CFSE. Witheach cell division, the CFSE fluorescence intensity of the cells is reducedby half (49–51). T cells from healthy controls were labeled with 2 �MCFSE in RPMI 1640/10% FBS for 2 min at room temperature. After wash-ing to remove unbound fluorescent dye, 4 � 106 T cells were coculturedwith allogeneic DCs from SLE or healthy controls. Conditions includedunstimulated DCs or DCs stimulated with LPS and TNF-� for 48 h, asdescribed above. Cells were cocultured at a T cell:DC ratio of 5:1 to 10:1.After 1 and 5 days, cells were harvested and stained with mouse anti-human CD3-allophycocyanin/Cy7, CD11c-PE, and CD25-CyChrome, thenfixed with 2% formaldehyde/PBS and analyzed by FACSCalibur, usingCellQuest software (BD Biosciences) and WinMDI. In additional experi-ments, expression of additional activation markers (CD69 and CD40L) wasmeasured on these T cells. Expression was measured 24 h and 5 days aftercocultures were started. Proliferation of T cells was analyzed by the log-arithmic reduction of CFSE staining of CD3-positive cells. The percentageof T cells that had undergone specific numbers of cell divisions (G1–G5) orno cell divisions (G0) was calculated (52, 53). Controls included PHA-stimulated T cells and unstimulated T cells.
Statistical analysis
The difference between means was analyzed using paired t test or one-wayANOVA with post hoc analysis and Bonferroni correction. Spearman andPearson’s correlation were used to assess correlation between differentvariables. Analyses were performed with SPSS version 11.5. A value ofp � 0.05 was considered to be statistically significant.
ResultsMyeloid lupus DCs display abnormal levels of differentiationand maturation markers and have abnormal responses tomaturation stimuli
Confirming a previous study (54), no morphologic differences inthe capacity of monocytes to differentiate into DCs were foundbetween SLE and controls, using light and fluorescent microscopy(Fig. 1). After 5–7 days of culture, DCs from SLE patients andhealthy controls were found to be increased in size and developedtypical dendrites. To assess whether DCs from SLE individualsand controls differ in their differentiation and maturation poten-tials, monocyte-derived DCs from 31 individuals with SLE, 8 pa-tients with RA, and 20 healthy controls obtained at day 7 werestained with anti-human Abs to CD1a (to evaluate DC differenti-ation) (31) and CD83 (a marker of DC maturation) (55), and ex-pression of these markers was measured by FACS. As shown inFig. 2, A–C, SLE patients, have increased numbers of cells ex-pressing the differentiation marker CD1a, and decreased numbersof cells expressing the maturation marker CD83 ( p � 0.05). No
FIGURE 1. Monocyte-derived DCs from SLE patients display normalmorphology. A, Images represent different magnifications of cells from oneindividual with SLE, after culture in X-vivo medium with IL-4 and GM-CSF for 7 days. Cells display the characteristic membrane and nuclearfeatures of DCs. Arrows point at the cells that are displayed at highermagnification on the left panels. B, Fluorescent microscopy image of onecell from a patient with SLE that displays the classic morphology DCs, atday 7 in culture. The cell expresses both CD1a and CD83.
5880 ABNORMAL PHENOTYPE AND FUNCTION OF LUPUS DCs
by guest on June 11, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

significant differences were observed in mean fluorescence inten-sity (data not shown). No significant differences were detected be-tween RA patients and controls in CD1a expression (Fig. 2A) orCD83 expression (Fig. 2B). The differences between SLE and con-trols were confirmed at the mRNA level by real-time RT-PCR(Fig. 2D). Indeed, lupus patients (but not RA patients) displayedsignificantly decreased mRNA levels of the monocyte markerCD14 and higher levels of CD1a mRNA at day 7, suggesting an
acceleration of the differentiation from the monocyte stage to themyeloid DC stage (Fig. 2D). Baseline CD14 levels in monocytesdid no differ between controls and lupus patients (data not shown),suggesting that the differences seen on day 7 DCs were the con-sequence of accelerated differentiation from the monocyte to theDC stage and not due to baseline down-regulation of CD14 onlupus monocytes. Interestingly, while CD83 protein and mRNAlevels were significantly lower in lupus DCs at day 7 (Fig. 2, B–D),
FIGURE 2. Aberrant expression of differentiation and maturation markers in monocyte-derived DCs in SLE. Bar graph represents A, CD1a, and B, CD83percent expression, in 20 healthy controls, 8 RA patients, and 31 SLE patients. �, p � 0.05, results represent mean SEM of independent experiments.C, A representative density plot of CD83 and CD1a expression from one control and one SLE patient. D, Bar graphs represent CD1a, CD14, and CD83mRNA expression by real-time RT-PCR relative to housekeeping genes in day 7 DCs from 7 healthy controls, 22 SLE, and 7 RA patients. Results arereported as the fold change compared with healthy controls. �, p � 0.05, results represent mean SEM of independent experiments.
5881The Journal of Immunology
by guest on June 11, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
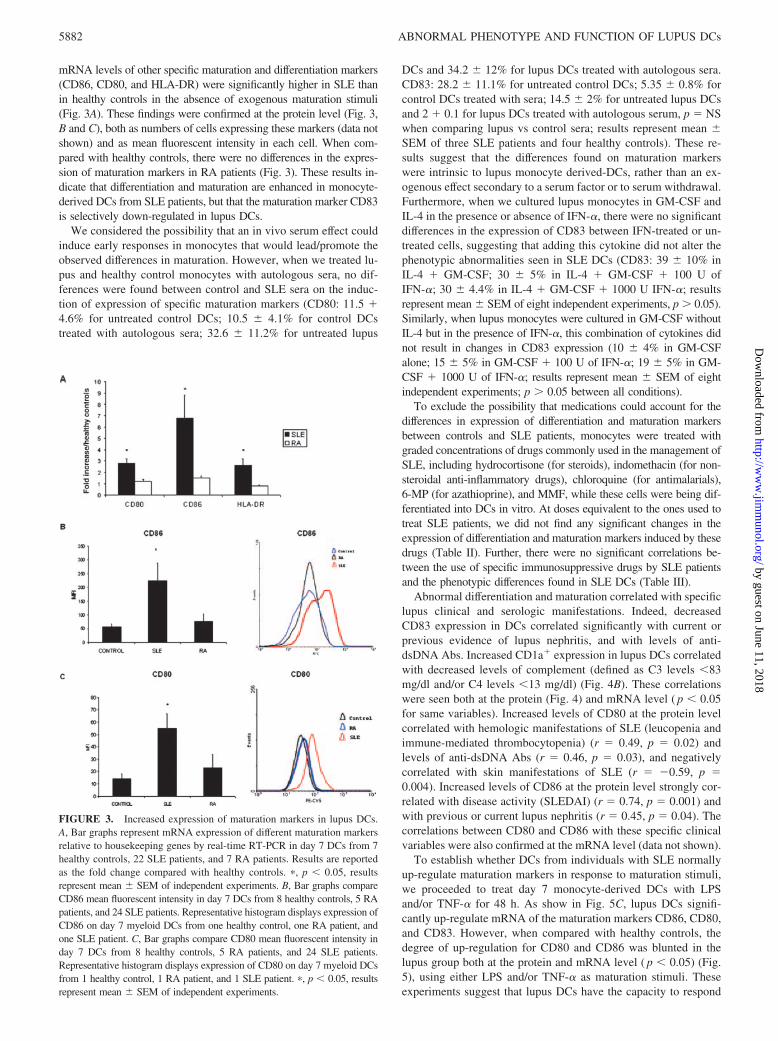
mRNA levels of other specific maturation and differentiation markers(CD86, CD80, and HLA-DR) were significantly higher in SLE thanin healthy controls in the absence of exogenous maturation stimuli(Fig. 3A). These findings were confirmed at the protein level (Fig. 3,B and C), both as numbers of cells expressing these markers (data notshown) and as mean fluorescent intensity in each cell. When com-pared with healthy controls, there were no differences in the expres-sion of maturation markers in RA patients (Fig. 3). These results in-dicate that differentiation and maturation are enhanced in monocyte-derived DCs from SLE patients, but that the maturation marker CD83is selectively down-regulated in lupus DCs.
We considered the possibility that an in vivo serum effect couldinduce early responses in monocytes that would lead/promote theobserved differences in maturation. However, when we treated lu-pus and healthy control monocytes with autologous sera, no dif-ferences were found between control and SLE sera on the induc-tion of expression of specific maturation markers (CD80: 11.5 �4.6% for untreated control DCs; 10.5 4.1% for control DCstreated with autologous sera; 32.6 11.2% for untreated lupus
DCs and 34.2 12% for lupus DCs treated with autologous sera.CD83: 28.2 11.1% for untreated control DCs; 5.35 0.8% forcontrol DCs treated with sera; 14.5 2% for untreated lupus DCsand 2 � 0.1 for lupus DCs treated with autologous serum, p NSwhen comparing lupus vs control sera; results represent mean SEM of three SLE patients and four healthy controls). These re-sults suggest that the differences found on maturation markerswere intrinsic to lupus monocyte derived-DCs, rather than an ex-ogenous effect secondary to a serum factor or to serum withdrawal.Furthermore, when we cultured lupus monocytes in GM-CSF andIL-4 in the presence or absence of IFN-�, there were no significantdifferences in the expression of CD83 between IFN-treated or un-treated cells, suggesting that adding this cytokine did not alter thephenotypic abnormalities seen in SLE DCs (CD83: 39 10% inIL-4 � GM-CSF; 30 5% in IL-4 � GM-CSF � 100 U ofIFN-�; 30 4.4% in IL-4 � GM-CSF � 1000 U IFN-�; resultsrepresent mean SEM of eight independent experiments, p � 0.05).Similarly, when lupus monocytes were cultured in GM-CSF withoutIL-4 but in the presence of IFN-�, this combination of cytokines didnot result in changes in CD83 expression (10 4% in GM-CSFalone; 15 5% in GM-CSF � 100 U of IFN-�; 19 5% in GM-CSF � 1000 U of IFN-�; results represent mean SEM of eightindependent experiments; p � 0.05 between all conditions).
To exclude the possibility that medications could account for thedifferences in expression of differentiation and maturation markersbetween controls and SLE patients, monocytes were treated withgraded concentrations of drugs commonly used in the management ofSLE, including hydrocortisone (for steroids), indomethacin (for non-steroidal anti-inflammatory drugs), chloroquine (for antimalarials),6-MP (for azathioprine), and MMF, while these cells were being dif-ferentiated into DCs in vitro. At doses equivalent to the ones used totreat SLE patients, we did not find any significant changes in theexpression of differentiation and maturation markers induced by thesedrugs (Table II). Further, there were no significant correlations be-tween the use of specific immunosuppressive drugs by SLE patientsand the phenotypic differences found in SLE DCs (Table III).
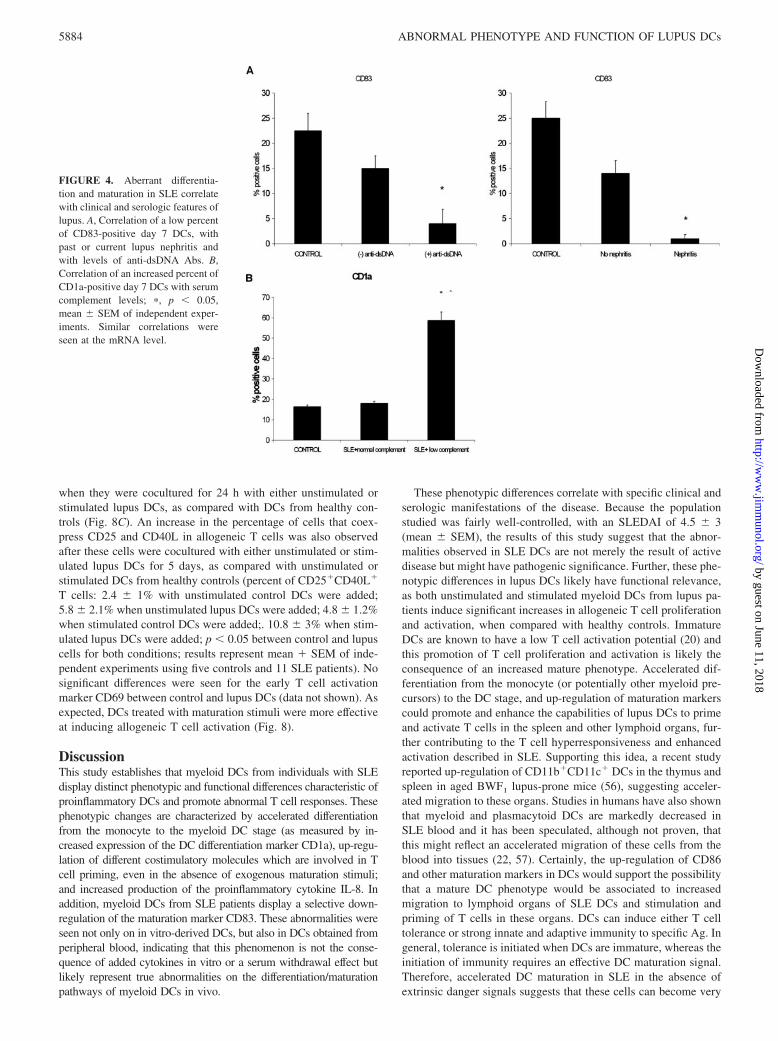
Abnormal differentiation and maturation correlated with specificlupus clinical and serologic manifestations. Indeed, decreasedCD83 expression in DCs correlated significantly with current orprevious evidence of lupus nephritis, and with levels of anti-dsDNA Abs. Increased CD1a� expression in lupus DCs correlatedwith decreased levels of complement (defined as C3 levels �83mg/dl and/or C4 levels �13 mg/dl) (Fig. 4B). These correlationswere seen both at the protein (Fig. 4) and mRNA level ( p � 0.05for same variables). Increased levels of CD80 at the protein levelcorrelated with hemologic manifestations of SLE (leucopenia andimmune-mediated thrombocytopenia) (r 0.49, p 0.02) andlevels of anti-dsDNA Abs (r 0.46, p 0.03), and negativelycorrelated with skin manifestations of SLE (r �0.59, p 0.004). Increased levels of CD86 at the protein level strongly cor-related with disease activity (SLEDAI) (r 0.74, p 0.001) andwith previous or current lupus nephritis (r 0.45, p 0.04). Thecorrelations between CD80 and CD86 with these specific clinicalvariables were also confirmed at the mRNA level (data not shown).
To establish whether DCs from individuals with SLE normallyup-regulate maturation markers in response to maturation stimuli,we proceeded to treat day 7 monocyte-derived DCs with LPSand/or TNF-� for 48 h. As show in Fig. 5C, lupus DCs signifi-cantly up-regulate mRNA of the maturation markers CD86, CD80,and CD83. However, when compared with healthy controls, thedegree of up-regulation for CD80 and CD86 was blunted in thelupus group both at the protein and mRNA level ( p � 0.05) (Fig.5), using either LPS and/or TNF-� as maturation stimuli. Theseexperiments suggest that lupus DCs have the capacity to respond
FIGURE 3. Increased expression of maturation markers in lupus DCs.A, Bar graphs represent mRNA expression of different maturation markersrelative to housekeeping genes by real-time RT-PCR in day 7 DCs from 7healthy controls, 22 SLE patients, and 7 RA patients. Results are reportedas the fold change compared with healthy controls. �, p � 0.05, resultsrepresent mean SEM of independent experiments. B, Bar graphs compareCD86 mean fluorescent intensity in day 7 DCs from 8 healthy controls, 5 RApatients, and 24 SLE patients. Representative histogram displays expression ofCD86 on day 7 myeloid DCs from one healthy control, one RA patient, andone SLE patient. C, Bar graphs compare CD80 mean fluorescent intensity inday 7 DCs from 8 healthy controls, 5 RA patients, and 24 SLE patients.Representative histogram displays expression of CD80 on day 7 myeloid DCsfrom 1 healthy control, 1 RA patient, and 1 SLE patient. �, p � 0.05, resultsrepresent mean SEM of independent experiments.
5882 ABNORMAL PHENOTYPE AND FUNCTION OF LUPUS DCs
by guest on June 11, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
to maturation stimuli, as shown by up-regulation of maturationmarkers; however, the level of up-regulation of these markers isdecreased when compared with healthy controls. When comparedwith healthy controls, there were no statistical differences in thecapacity of RA DCs to up-regulate maturation markers after ex-posure to exogenous maturation stimuli (Fig. 5, A and B).
To exclude the possibility that abnormal differentiation and mat-uration observed in SLE DCs were consequences of the in vitroconditions used, the expression of maturation and differentiationmarkers in myeloid DCs directly obtained from peripheral bloodwas examined. As shown in Fig. 6A, the myeloid DC populationobtained directly from lupus blood is also characterized by highernumbers of CD1a� cells, lower numbers of CD83� cells andhigher expression of CD86� cells when compared with healthycontrols, further confirming our in vitro findings. In addition, highlevels of CD1a� cells significantly correlated with levels of anti-dsDNA Abs (Fig. 6B) and with hypocomplementemia ( p 0.004). Furthermore, similar to what we found in DCs generated invitro, there were no significant correlations between the use ofspecific immunosuppressive drugs and phenotypic abnormalitiesseen in lupus DCs isolated from peripheral blood (data not shown).
Lupus DCs secrete higher levels of IL-8
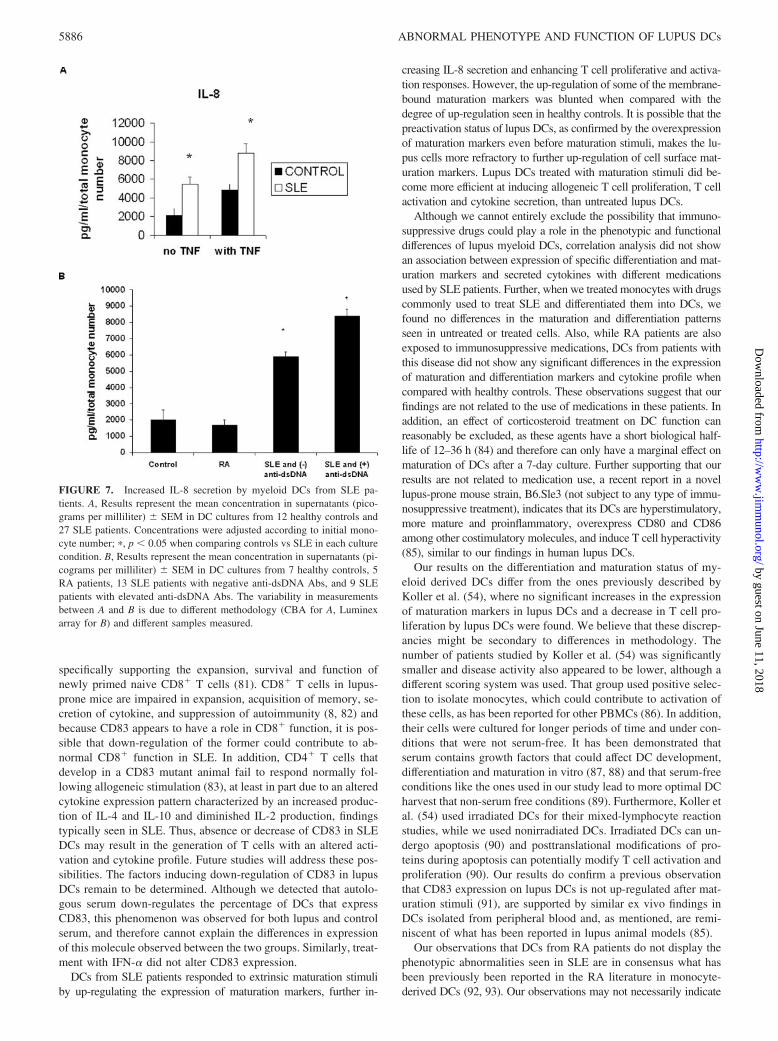
To evaluate whether SLE DCs show differential secretion of cy-tokines, we measured the secretion of a number of cytokines usingcytometric bead array and a cytokine multiplex kit array. Control,RA, and SLE DCs secrete detectable amounts of IL-6, IL-7, IL-8,IL-10, IL-13, IFN-�, and TNF-�. The cytokine secreted at thehighest levels was the proinflammatory cytokine IL-8, and lupusDCs secreted significantly higher levels of IL-8 than control DCsboth before (5459.2 813.7� vs 2137.9 759.3 pg/ml, respec-tively, mean SEM, p � 0.05) and after TNF-� stimulation
(8760.6 1048.6 vs 4817.2 689.4, respectively, mean SEM,p � 0.05) (Fig. 7A). Levels of IL-8 secreted by lupus DCs corre-lated with expression of the maturation marker CD80 (r 0.56,p 0.01, Pearson’s correlation), and patients with higher produc-tion of IL-8 had higher anti-dsDNA (Fig. 7B) and anti-cardiolipinlevels in sera (r 0.53, p 0.007 and r 0.45, p 0.02,respectively, Pearson’s correlation). When compared with healthycontrols, there were no significant increases in IL-8 secretion byDCs from RA patients (Fig. 7B). No significant differences be-tween control, SLE, and RA patients were observed when the othercytokines were measured in DC supernatants (data not shown). Ofinterest, IL-8 was also the most abundant cytokine detected inplasma from SLE patients and serum levels were higher than inhealthy controls, although no statistical significance was found(57.9 vs 36.3 pg/ml, P NS).
Lupus DCs up-regulate T cell proliferation and activation
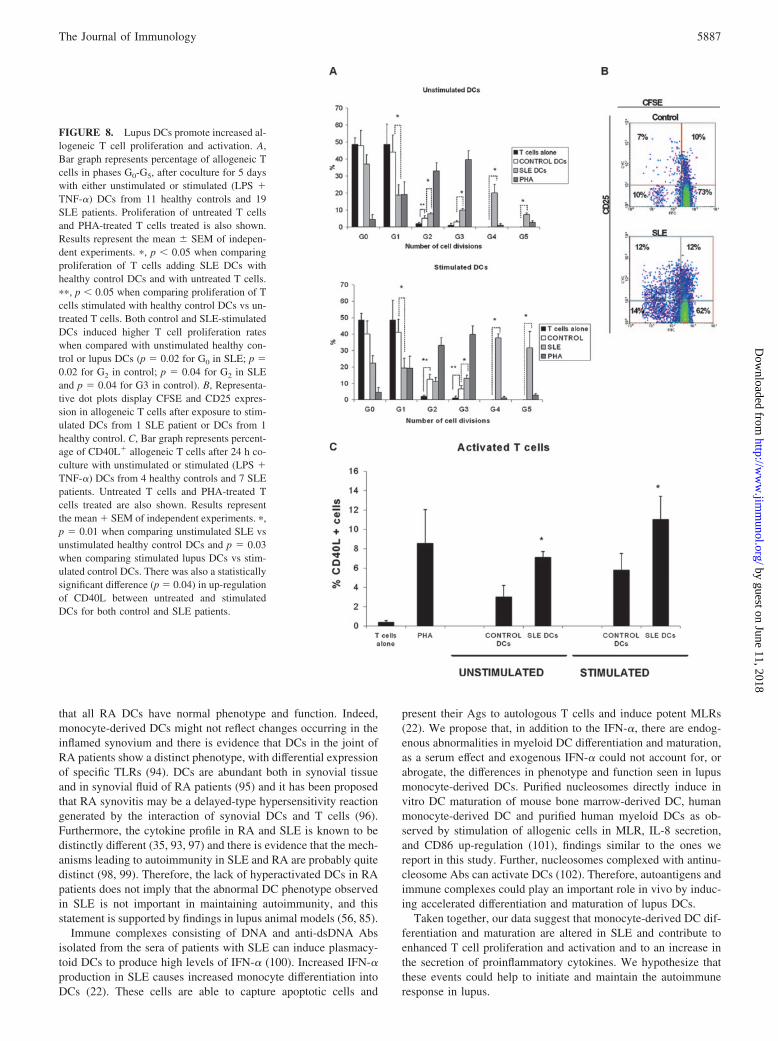
Unstimulated and stimulated DCs from lupus patients induced asignificant increase in allogeneic T cell proliferation when com-pared with DCs from healthy controls. As expected, unstimulatedDCs from healthy controls were poor stimulators of allogeneic Tcell proliferation and, after stimulation with maturation stimuli,their capacity to induce allogeneic T cell proliferation increasedsignificantly. When cocultured with lupus DCs, allogeneic T cellsshowed a significantly higher percentage of proliferating cells thanwhat was observed with control allogeneic DCs, and the effect wasmore pronounced in DCs treated with maturation stimuli (LPS andTNF-�) (Fig. 8, A and B). This increase in proliferation requiredcell-cell interactions, as supernatants from lupus DCs did not in-duce similar increases in T cell proliferation (data not shown).Similarly, there was a statistically significant increase in the ex-pression of the activation marker CD40L in allogeneic T cells
Table III. Correlation analysis of specific immunosuppressive/immunomodulator use and expression of differentiation and maturation markers inlupus DCsa
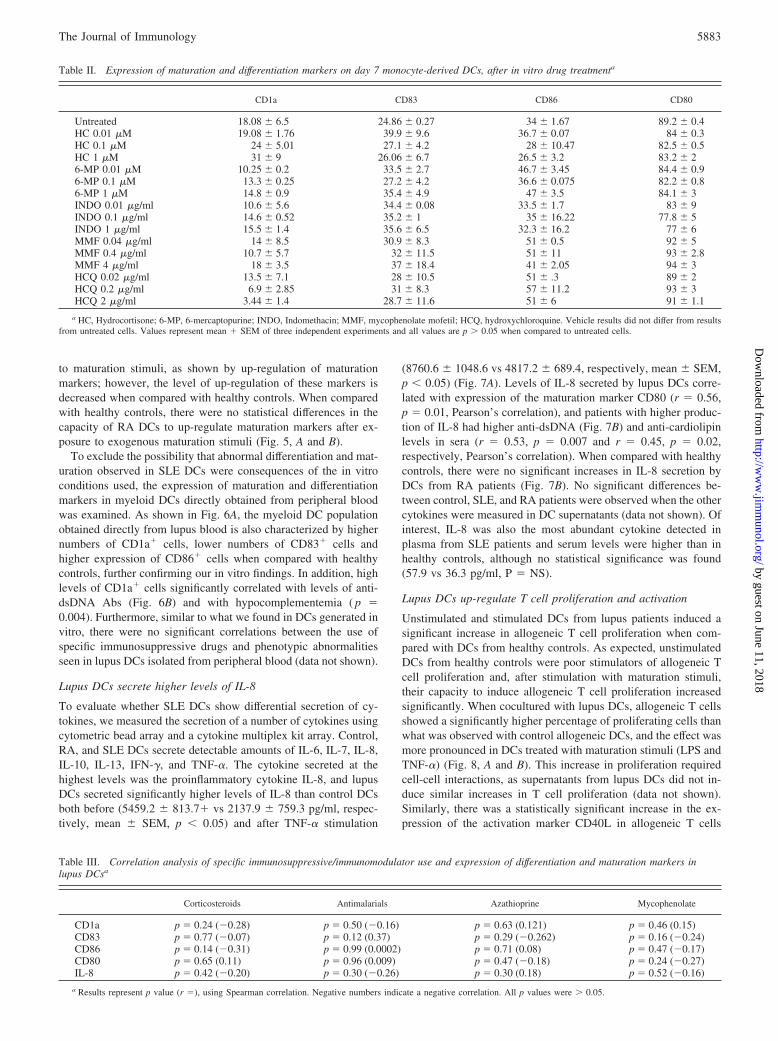
Corticosteroids Antimalarials Azathioprine Mycophenolate
CD1a p 0.24 (�0.28) p 0.50 (�0.16) p 0.63 (0.121) p 0.46 (0.15)CD83 p 0.77 (�0.07) p 0.12 (0.37) p 0.29 (�0.262) p 0.16 (�0.24)CD86 p 0.14 (�0.31) p 0.99 (0.0002) p 0.71 (0.08) p 0.47 (�0.17)CD80 p 0.65 (0.11) p 0.96 (0.009) p 0.47 (�0.18) p 0.24 (�0.27)IL-8 p 0.42 (�0.20) p 0.30 (�0.26) p 0.30 (0.18) p 0.52 (�0.16)
a Results represent p value (r ), using Spearman correlation. Negative numbers indicate a negative correlation. All p values were � 0.05.
Table II. Expression of maturation and differentiation markers on day 7 monocyte-derived DCs, after in vitro drug treatmenta
CD1a CD83 CD86 CD80
Untreated 18.08 6.5 24.86 0.27 34 1.67 89.2 0.4HC 0.01 �M 19.08 1.76 39.9 9.6 36.7 0.07 84 0.3HC 0.1 �M 24 5.01 27.1 4.2 28 10.47 82.5 0.5HC 1 �M 31 9 26.06 6.7 26.5 3.2 83.2 26-MP 0.01 �M 10.25 0.2 33.5 2.7 46.7 3.45 84.4 0.96-MP 0.1 �M 13.3 0.25 27.2 4.2 36.6 0.075 82.2 0.86-MP 1 �M 14.8 0.9 35.4 4.9 47 3.5 84.1 3INDO 0.01 �g/ml 10.6 5.6 34.4 0.08 33.5 1.7 83 9INDO 0.1 �g/ml 14.6 0.52 35.2 1 35 16.22 77.8 5INDO 1 �g/ml 15.5 1.4 35.6 6.5 32.3 16.2 77 6MMF 0.04 �g/ml 14 8.5 30.9 8.3 51 0.5 92 5MMF 0.4 �g/ml 10.7 5.7 32 11.5 51 11 93 2.8MMF 4 �g/ml 18 3.5 37 18.4 41 2.05 94 3HCQ 0.02 �g/ml 13.5 7.1 28 10.5 51 .3 89 2HCQ 0.2 �g/ml 6.9 2.85 31 8.3 57 11.2 93 3HCQ 2 �g/ml 3.44 1.4 28.7 11.6 51 6 91 1.1
a HC, Hydrocortisone; 6-MP, 6-mercaptopurine; INDO, Indomethacin; MMF, mycophenolate mofetil; HCQ, hydroxychloroquine. Vehicle results did not differ from resultsfrom untreated cells. Values represent mean � SEM of three independent experiments and all values are p � 0.05 when compared to untreated cells.
5883The Journal of Immunology
by guest on June 11, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
when they were cocultured for 24 h with either unstimulated orstimulated lupus DCs, as compared with DCs from healthy con-trols (Fig. 8C). An increase in the percentage of cells that coex-press CD25 and CD40L in allogeneic T cells was also observedafter these cells were cocultured with either unstimulated or stim-ulated lupus DCs for 5 days, as compared with unstimulated orstimulated DCs from healthy controls (percent of CD25�CD40L�
T cells: 2.4 1% with unstimulated control DCs were added;5.8 2.1% when unstimulated lupus DCs were added; 4.8 1.2%when stimulated control DCs were added;. 10.8 3% when stim-ulated lupus DCs were added; p � 0.05 between control and lupuscells for both conditions; results represent mean � SEM of inde-pendent experiments using five controls and 11 SLE patients). Nosignificant differences were seen for the early T cell activationmarker CD69 between control and lupus DCs (data not shown). Asexpected, DCs treated with maturation stimuli were more effectiveat inducing allogeneic T cell activation (Fig. 8).
DiscussionThis study establishes that myeloid DCs from individuals with SLEdisplay distinct phenotypic and functional differences characteristic ofproinflammatory DCs and promote abnormal T cell responses. Thesephenotypic changes are characterized by accelerated differentiationfrom the monocyte to the myeloid DC stage (as measured by in-creased expression of the DC differentiation marker CD1a), up-regu-lation of different costimulatory molecules which are involved in Tcell priming, even in the absence of exogenous maturation stimuli;and increased production of the proinflammatory cytokine IL-8. Inaddition, myeloid DCs from SLE patients display a selective down-regulation of the maturation marker CD83. These abnormalities wereseen not only on in vitro-derived DCs, but also in DCs obtained fromperipheral blood, indicating that this phenomenon is not the conse-quence of added cytokines in vitro or a serum withdrawal effect butlikely represent true abnormalities on the differentiation/maturationpathways of myeloid DCs in vivo.
These phenotypic differences correlate with specific clinical andserologic manifestations of the disease. Because the populationstudied was fairly well-controlled, with an SLEDAI of 4.5 3(mean SEM), the results of this study suggest that the abnor-malities observed in SLE DCs are not merely the result of activedisease but might have pathogenic significance. Further, these phe-notypic differences in lupus DCs likely have functional relevance,as both unstimulated and stimulated myeloid DCs from lupus pa-tients induce significant increases in allogeneic T cell proliferationand activation, when compared with healthy controls. ImmatureDCs are known to have a low T cell activation potential (20) andthis promotion of T cell proliferation and activation is likely theconsequence of an increased mature phenotype. Accelerated dif-ferentiation from the monocyte (or potentially other myeloid pre-cursors) to the DC stage, and up-regulation of maturation markerscould promote and enhance the capabilities of lupus DCs to primeand activate T cells in the spleen and other lymphoid organs, fur-ther contributing to the T cell hyperresponsiveness and enhancedactivation described in SLE. Supporting this idea, a recent studyreported up-regulation of CD11b�CD11c� DCs in the thymus andspleen in aged BWF1 lupus-prone mice (56), suggesting acceler-ated migration to these organs. Studies in humans have also shownthat myeloid and plasmacytoid DCs are markedly decreased inSLE blood and it has been speculated, although not proven, thatthis might reflect an accelerated migration of these cells from theblood into tissues (22, 57). Certainly, the up-regulation of CD86and other maturation markers in DCs would support the possibilitythat a mature DC phenotype would be associated to increasedmigration to lymphoid organs of SLE DCs and stimulation andpriming of T cells in these organs. DCs can induce either T celltolerance or strong innate and adaptive immunity to specific Ag. Ingeneral, tolerance is initiated when DCs are immature, whereas theinitiation of immunity requires an effective DC maturation signal.Therefore, accelerated DC maturation in SLE in the absence ofextrinsic danger signals suggests that these cells can become very
FIGURE 4. Aberrant differentia-tion and maturation in SLE correlatewith clinical and serologic features oflupus. A, Correlation of a low percentof CD83-positive day 7 DCs, withpast or current lupus nephritis andwith levels of anti-dsDNA Abs. B,Correlation of an increased percent ofCD1a-positive day 7 DCs with serumcomplement levels; �, p � 0.05,mean SEM of independent exper-iments. Similar correlations wereseen at the mRNA level.
5884 ABNORMAL PHENOTYPE AND FUNCTION OF LUPUS DCs
by guest on June 11, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

efficient autoantigen-presenting cells and drive autoimmune re-sponses. In addition, the role of costimulatory molecules is welldocumented in murine models of lupus, including the observationthat treatment with a combination of blocking mAbs to CD80 andCD86, before the onset of murine lupus, significantly improvessurvival and severity of the disease (58, 59).
Given that mature myeloid DCs are able to break tolerance andinduce lupus autoantibodies in normal hosts (60), the increase in DCmaturation in SLE suggests that these abnormalities might be veryrelevant in the induction and perpetuation of autoimmunity in SLE.
Increased secretion of IL-8 by lupus DCs could potentially con-tribute to tissue damage, as this cytokine has been proposed to playan important role in the development of lupus nephritis (61–64)
and has been found to be elevated in lupus patients with CNS in-volvement (65). IL-8 is produced by numerous cell types, includingmonocytes/macrophages and DCs (64, 66, 67). IL-8 is mainly activeon neutrophils, promoting their recruitment and also their strong ac-tivation which triggers the leukotriene pathway, induces the release oftheir granular content, elastase and lactoferrin and increases their ad-herence to endothelial cells (68–71). In fact, migration of neutrophilsis influenced by DCs primarily by IL-8 (64). IL-8 is also a chemoat-tractant for other cell types including T cells (72). In addition, wefound a significant association between IL-8 secretion and serum lev-els of anti-dsDNA. Anti-dsDNA can enhance the release of proin-flammatory cytokines, including IL-8 and TNF-� from mononuclearcells to augment inflammatory reactions and can polarize the immunereaction toward the Th2 pathway (72–74). Therefore, an increase inthe production of this cytokine by DCs might enhance the ability torecruit cells in the glomerulus and enhance neutrophil adhesion tovascular endothelium which in turn may contribute to renal and vas-cular damage. Interestingly, a recent study has shown that the IL-8gene and its receptor CXCR-2 are up-regulated in PBMCs from SLEpatients using microarrays (75).
The functional relevance of decreased levels of CD83 in SLE isunclear at this point. CD83 is a cell surface membrane glycopro-tein whose surface expression is largely restricted to DCs (55). Theprecise functions of this molecule remain unknown (76, 77), butCD83 may serve important roles during intercellular interactions(77–79), as membrane-bound CD83 increases the stimulatory ca-pacity of DCs (79). Further, previous studies suggest that CD83mediates adhesion to monocytes and CD8� T cells (80). CD83-Igenhances T cell proliferation and increases the proportion of CD8�
T cells (80), and engagement of CD83 delivers a significant signal
FIGURE 5. Responses of monocyte-derived DCs to maturation stimuliare abnormal in lupus DCs. A, Bar graphs display mean fluorescence in-tensity of CD86 in DCs from 10 healthy controls, 5 RA patients, and 20SLE patients before and after stimulation with TNF-�. B, Representativehistograms of CD86 expression on DCs from 1 healthy control, 1 patientwith SLE, and 1 patient with RA before and after stimulation with LPS andTNF-�. C, Bar graphs display mRNA expression by real-time RT-PCR ofdifferentiation and maturation markers normalized to housekeeping genesin DCs, after stimulation with LPS and TNF-� in 6 healthy controls and 17SLE patients. Results are reported as the fold change when compared withunstimulated day 7 DCs. �, p � 0.05 when compared before and afterstimulation; results are presented as mean SEM.
FIGURE 6. Aberrant expression of differentiation and maturation mark-ers in lupus myeloid DCs obtained from peripheral blood. A, Bar graphsdisplay expression of DC differentiation and maturation markers in freshlyisolated CD11c� cells from peripheral blood, in 11 healthy controls and 24SLE patients. B, Bar graphs display correlation of anti-dsDNA Abs withCD1a� DCs in peripheral blood. �, p � 0.05, results represent the mean SEM of independent experiments.
5885The Journal of Immunology
by guest on June 11, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
specifically supporting the expansion, survival and function ofnewly primed naive CD8� T cells (81). CD8� T cells in lupus-prone mice are impaired in expansion, acquisition of memory, se-cretion of cytokine, and suppression of autoimmunity (8, 82) andbecause CD83 appears to have a role in CD8� function, it is pos-sible that down-regulation of the former could contribute to ab-normal CD8� function in SLE. In addition, CD4� T cells thatdevelop in a CD83 mutant animal fail to respond normally fol-lowing allogeneic stimulation (83), at least in part due to an alteredcytokine expression pattern characterized by an increased produc-tion of IL-4 and IL-10 and diminished IL-2 production, findingstypically seen in SLE. Thus, absence or decrease of CD83 in SLEDCs may result in the generation of T cells with an altered acti-vation and cytokine profile. Future studies will address these pos-sibilities. The factors inducing down-regulation of CD83 in lupusDCs remain to be determined. Although we detected that autolo-gous serum down-regulates the percentage of DCs that expressCD83, this phenomenon was observed for both lupus and controlserum, and therefore cannot explain the differences in expressionof this molecule observed between the two groups. Similarly, treat-ment with IFN-� did not alter CD83 expression.
DCs from SLE patients responded to extrinsic maturation stimuliby up-regulating the expression of maturation markers, further in-
creasing IL-8 secretion and enhancing T cell proliferative and activa-tion responses. However, the up-regulation of some of the membrane-bound maturation markers was blunted when compared with thedegree of up-regulation seen in healthy controls. It is possible that thepreactivation status of lupus DCs, as confirmed by the overexpressionof maturation markers even before maturation stimuli, makes the lu-pus cells more refractory to further up-regulation of cell surface mat-uration markers. Lupus DCs treated with maturation stimuli did be-come more efficient at inducing allogeneic T cell proliferation, T cellactivation and cytokine secretion, than untreated lupus DCs.
Although we cannot entirely exclude the possibility that immuno-suppressive drugs could play a role in the phenotypic and functionaldifferences of lupus myeloid DCs, correlation analysis did not showan association between expression of specific differentiation and mat-uration markers and secreted cytokines with different medicationsused by SLE patients. Further, when we treated monocytes with drugscommonly used to treat SLE and differentiated them into DCs, wefound no differences in the maturation and differentiation patternsseen in untreated or treated cells. Also, while RA patients are alsoexposed to immunosuppressive medications, DCs from patients withthis disease did not show any significant differences in the expressionof maturation and differentiation markers and cytokine profile whencompared with healthy controls. These observations suggest that ourfindings are not related to the use of medications in these patients. Inaddition, an effect of corticosteroid treatment on DC function canreasonably be excluded, as these agents have a short biological half-life of 12–36 h (84) and therefore can only have a marginal effect onmaturation of DCs after a 7-day culture. Further supporting that ourresults are not related to medication use, a recent report in a novellupus-prone mouse strain, B6.Sle3 (not subject to any type of immu-nosuppressive treatment), indicates that its DCs are hyperstimulatory,more mature and proinflammatory, overexpress CD80 and CD86among other costimulatory molecules, and induce T cell hyperactivity(85), similar to our findings in human lupus DCs.
Our results on the differentiation and maturation status of my-eloid derived DCs differ from the ones previously described byKoller et al. (54), where no significant increases in the expressionof maturation markers in lupus DCs and a decrease in T cell pro-liferation by lupus DCs were found. We believe that these discrep-ancies might be secondary to differences in methodology. Thenumber of patients studied by Koller et al. (54) was significantlysmaller and disease activity also appeared to be lower, although adifferent scoring system was used. That group used positive selec-tion to isolate monocytes, which could contribute to activation ofthese cells, as has been reported for other PBMCs (86). In addition,their cells were cultured for longer periods of time and under con-ditions that were not serum-free. It has been demonstrated thatserum contains growth factors that could affect DC development,differentiation and maturation in vitro (87, 88) and that serum-freeconditions like the ones used in our study lead to more optimal DCharvest that non-serum free conditions (89). Furthermore, Koller etal. (54) used irradiated DCs for their mixed-lymphocyte reactionstudies, while we used nonirradiated DCs. Irradiated DCs can un-dergo apoptosis (90) and posttranslational modifications of pro-teins during apoptosis can potentially modify T cell activation andproliferation (90). Our results do confirm a previous observationthat CD83 expression on lupus DCs is not up-regulated after mat-uration stimuli (91), are supported by similar ex vivo findings inDCs isolated from peripheral blood and, as mentioned, are remi-niscent of what has been reported in lupus animal models (85).
Our observations that DCs from RA patients do not display thephenotypic abnormalities seen in SLE are in consensus what hasbeen previously been reported in the RA literature in monocyte-derived DCs (92, 93). Our observations may not necessarily indicate
FIGURE 7. Increased IL-8 secretion by myeloid DCs from SLE pa-tients. A, Results represent the mean concentration in supernatants (pico-grams per milliliter) SEM in DC cultures from 12 healthy controls and27 SLE patients. Concentrations were adjusted according to initial mono-cyte number; �, p � 0.05 when comparing controls vs SLE in each culturecondition. B, Results represent the mean concentration in supernatants (pi-cograms per milliliter) SEM in DC cultures from 7 healthy controls, 5RA patients, 13 SLE patients with negative anti-dsDNA Abs, and 9 SLEpatients with elevated anti-dsDNA Abs. The variability in measurementsbetween A and B is due to different methodology (CBA for A, Luminexarray for B) and different samples measured.
5886 ABNORMAL PHENOTYPE AND FUNCTION OF LUPUS DCs
by guest on June 11, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
that all RA DCs have normal phenotype and function. Indeed,monocyte-derived DCs might not reflect changes occurring in theinflamed synovium and there is evidence that DCs in the joint ofRA patients show a distinct phenotype, with differential expressionof specific TLRs (94). DCs are abundant both in synovial tissueand in synovial fluid of RA patients (95) and it has been proposedthat RA synovitis may be a delayed-type hypersensitivity reactiongenerated by the interaction of synovial DCs and T cells (96).Furthermore, the cytokine profile in RA and SLE is known to bedistinctly different (35, 93, 97) and there is evidence that the mech-anisms leading to autoimmunity in SLE and RA are probably quitedistinct (98, 99). Therefore, the lack of hyperactivated DCs in RApatients does not imply that the abnormal DC phenotype observedin SLE is not important in maintaining autoimmunity, and thisstatement is supported by findings in lupus animal models (56, 85).
Immune complexes consisting of DNA and anti-dsDNA Absisolated from the sera of patients with SLE can induce plasmacy-toid DCs to produce high levels of IFN-� (100). Increased IFN-�production in SLE causes increased monocyte differentiation intoDCs (22). These cells are able to capture apoptotic cells and
present their Ags to autologous T cells and induce potent MLRs(22). We propose that, in addition to the IFN-�, there are endog-enous abnormalities in myeloid DC differentiation and maturation,as a serum effect and exogenous IFN-� could not account for, orabrogate, the differences in phenotype and function seen in lupusmonocyte-derived DCs. Purified nucleosomes directly induce invitro DC maturation of mouse bone marrow-derived DC, humanmonocyte-derived DC and purified human myeloid DCs as ob-served by stimulation of allogenic cells in MLR, IL-8 secretion,and CD86 up-regulation (101), findings similar to the ones wereport in this study. Further, nucleosomes complexed with antinu-cleosome Abs can activate DCs (102). Therefore, autoantigens andimmune complexes could play an important role in vivo by induc-ing accelerated differentiation and maturation of lupus DCs.
Taken together, our data suggest that monocyte-derived DC dif-ferentiation and maturation are altered in SLE and contribute toenhanced T cell proliferation and activation and to an increase inthe secretion of proinflammatory cytokines. We hypothesize thatthese events could help to initiate and maintain the autoimmuneresponse in lupus.
FIGURE 8. Lupus DCs promote increased al-logeneic T cell proliferation and activation. A,Bar graph represents percentage of allogeneic Tcells in phases G0-G5, after coculture for 5 dayswith either unstimulated or stimulated (LPS �TNF-�) DCs from 11 healthy controls and 19SLE patients. Proliferation of untreated T cellsand PHA-treated T cells treated is also shown.Results represent the mean SEM of indepen-dent experiments. �, p � 0.05 when comparingproliferation of T cells adding SLE DCs withhealthy control DCs and with untreated T cells.��, p � 0.05 when comparing proliferation of Tcells stimulated with healthy control DCs vs un-treated T cells. Both control and SLE-stimulatedDCs induced higher T cell proliferation rateswhen compared with unstimulated healthy con-trol or lupus DCs (p 0.02 for G0 in SLE; p 0.02 for G2 in control; p 0.04 for G2 in SLEand p 0.04 for G3 in control). B, Representa-tive dot plots display CFSE and CD25 expres-sion in allogeneic T cells after exposure to stim-ulated DCs from 1 SLE patient or DCs from 1healthy control. C, Bar graph represents percent-age of CD40L� allogeneic T cells after 24 h co-culture with unstimulated or stimulated (LPS �TNF-�) DCs from 4 healthy controls and 7 SLEpatients. Untreated T cells and PHA-treated Tcells treated are also shown. Results representthe mean � SEM of independent experiments. �,p 0.01 when comparing unstimulated SLE vsunstimulated healthy control DCs and p 0.03when comparing stimulated lupus DCs vs stim-ulated control DCs. There was also a statisticallysignificant difference (p 0.04) in up-regulationof CD40L between untreated and stimulatedDCs for both control and SLE patients.
5887The Journal of Immunology
by guest on June 11, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

AcknowledgmentsWe thank Drs. David Fox, Bruce Richardson, and Michael Denny for crit-ical review of the manuscript; Joel Joshua for help in patient recruitment,and Berlex for providing human GM-CSF.
DisclosuresThe authors have no financial conflict of interest.
References1. Tsokos, G. C., H. Wong, E. Enyedi, and M. Nambiar. 2000. Immune cell sig-
naling in lupus. Curr. Opin. Rheumatol. 12: 355–363.2. Kammer, G., A. Perl, B. Richardson, and G. Tsokos. 2002. Abnormal T cell
signal transduction in systemic lupus erythematosus. Arthritis Rheum. 46:1139–1154.
3. Tsokos, G. C., J. Mitchell, and Y. Juang. 2003. T cell abnormalities in human andmouse lupus: intrinsic and extrinsic. Curr. Opin. Rheumatol. 15: 542–547.
4. Busser, B. W., B. Adair, J. Erikson, and T. Laufer. 2003. Activation of diverserepertoires of autoreactive T cells enhances the loss of anti-dsDNA B cell toler-ance. J. Clin. Invest. 112: 1361–1371.
5. Riemekasten, G., D. Langnickel, F. Ebling, G. Karpouzas, J. Kalsi, G. Herberth,B. Tsao, P. Henklein, S. Langer, G. Burmester, et al. 2003. Identification andcharacterization of SmD183–119-reactive T cells that provide T cell help forpathogenic anti-double-stranded DNA antibodies. Arthritis Rheum 48: 475–485.
6. Kaplan, M. J., Q. Lu, A. Wu, J. Attwood, and B. Richardson. 2004. Demethyl-ation of promoter regulatory elements contributes to perforin overexpression inCD4� lupus T cells. J. Immunol. 172: 3652–3661.
7. Tsai, H. F., J. Lai, A. Chou, T. Wang, C. Wu, and P. Hsu. 2004. Induction ofcostimulation of human CD4 T cells by tumor necrosis factor-related apoptosis-inducing ligand: possible role in T cell activation in systemic lupus erythemato-sus. Arthritis Rheum 50: 629–639.
8. Karpouzas, G. A., A. La Cava, F. M. Ebling, R. R. Singh, and B. H. Hahn. 2004.Differences between CD8� T cells in lupus-prone (NZB � NZW)F1 mice andhealthy (BALB/c � NZW)F1 mice may influence autoimmunity in the lupusmodel. Eur. J. Immunol. 34: 2489–2499.
9. Wenzel, J., S. Henze, S. Brahler, T. Bieber, and T. Tuting. 2005. The expressionof human leukocyte antigen-DR and CD25 on circulating T cells in cutaneouslupus erythematosus and correlation with disease activity. Exp. Dermatol. 14:454–459.
10. Zielinski, C. E., S. Jacob, F. Bouzahzah, B. Ehrlich, and J. Craft. 2005. NaiveCD4� T cells from lupus-prone Fas-intact MRL mice display TCR-mediatedhyperproliferation due to intrinsic threshold defects in activation. J. Immunol.174: 5100–5109.
11. Kaplan, M. J., E. Lewis, E. Shelden, E. Somers, R. Pavlic, W. McCune, andB. Richardson. 2002. The apoptotic ligands TRAIL, TWEAK, and Fas ligandmediate monocyte death induced by autologous lupus T cells. J. Immunol. 169:6020–6029.
12. Calvani, N., H. Richards, M. Tucci, G. Pannarale, and F. Silvestris. 2004. Up-regulation of IL-18 and predominance of a Th1 immune response is a hallmarkof lupus nephritis. Clin. Exp. Immunol. 138: 171–178.
13. Gomez, D., P. Correa, L. Gomez, J. Cadena, J. Molina, and J. Anaya. 2004.Th1/Th2 cytokines in patients with systemic lupus erythematosus: is tumor ne-crosis factor � protective? Semin. Arthritis Rheum. 33: 404–413.
14. Zeng, D., Y. Liu, S. Sidobre, M. Kronenberg, and S. Strober. 2003. Activation ofnatural killer T cells in NZB/W mice induces Th1-type immune responses ex-acerbating lupus. J. Clin. Invest. 112: 1211–1222.
15. Singh, R. 2003. IL-4 and many roads to lupus like autoimmunity. Clin. Immunol.108: 73–79.
16. Nakajima, A., S. Hirose, H. Yagita, and K. Okumura. 1997. Roles of IL-4 andIL-12 in the development of lupus in NZB/W F1 mice. J. Immunol. 158:1466–1472.
17. Tyrrell-Price, J., P. Lydyard, and D. Isenberg. 2001. The effect of interleukin-10and of interleukin-12 on the in vitro production of anti-double-stranded DNAantibodies from patients with systemic lupus erythematosus. Clin. Exp. Immunol.124: 118–125.
18. Llorente, L., W. Zou, Y. Levy, Y. Richaud-Patin, J. Wijdenes, J. Alcocer-Varela,B. Morel-Fourrier, J. Brouet, D. Alarcon-Segovia, P. Galanaud, and D. Emilie.1995. Role of interleukin 10 in the B lymphocyte hyperactivity and autoantibodyproduction of human systemic lupus erythematosus. J. Exp. Med. 181: 839–844.
19. Palucka, A. K., J. Banchereau, P. Blanco, and V. Pascual. 2002. The interplay ofdendritic cell subsets in systemic lupus erythematosus. 80: 484–488.
20. Banchereau, J., and R. M. Steinman. 1998. Dendritic cells and the control ofimmunity. Nature 392: 245–252.
21. Kono, D. H., R. Baccala, and A. Theofilopoulos. 2003. Inhibition of lupus bygenetic alteration of the interferon-�/� receptor. Autoimmunity 36: 503–510.
22. Blanco, P., A. Palucka, M. Gill, V. Pascual, and J. Banchereau. 2001. Inductionof dendritic cell differentiation by IFN-� in systemic lupus erythematosus. Sci-ence 294: 1540–1543.
23. Stoll, S., J. Delon, T. M. Brotz, and R. N. Germain. 2002. Dynamic imaging ofT cell-dendritic cell interactions in lymph nodes. Science 296: 1873–1876.
24. Mailliard, R. B., S. Egawa, Q. Cai, A. Kalinska, S. N. Bykovskaya, M. T. Lotze,M. L. Kapsenberg, W. J. Storkus, and P. Kalinski. 2002. Complementary den-dritic cell-activating function of CD8� and CD4� T cells: helper role of CD8�
T cells in the development of T helper type 1 responses. J. Exp. Med. 195:473–483.
25. Angelini, G., S. Gardella, M. Ardy, M. R. Ciriolo, G. Filomeni, G. Di Trapani,F. Clarke, R. Sitia, and A. Rubartelli. 2002. Antigen-presenting dendritic cellsprovide the reducing extracellular microenvironment required for T lymphocyteactivation. Proc. Natl. Acad. Sci. USA 99: 1491–1496.
26. Cella, M., D. Scheidegger, K. Palmer-Lehmann, P. Lane, A. Lanzavecchia, andG. Alber. 1996. Ligation of CD40 on dendritic cells triggers production of highlevels of interleukin-12 and enhances T cell stimulatory capacity: T-T help viaAPC activation. J. Exp. Med. 184: 747–752.
27. Daniel, P. T., C. Scholz, J. Westermann, B. Dorken, and A. Pezzutto. 1998.Dendritic cells prevent CD95 mediated T lymphocyte death through costimula-tory signals. Adv. Exp. Med. Biol. 451: 173–177.
28. Ingulli, E., D. R. Ulman, M. M. Lucido, and M. K. Jenkins. 2002. In situ analysisreveals physical interactions between CD11b� dendritic cells and antigen-specificCD4 T cells after subcutaneous injection of antigen. J. Immunol. 169: 2247–2252.
29. Maldonado-Lopez, R., T. De Smedt, P. Michel, J. Godfroid, B. Pajak,C. Heirman, K. Thielemans, O. Leo, J. Urbain, and M. Moser. 1999. CD8�� andCD8�� subclasses of dendritic cells direct the development of distinct T helpercells in vivo. J. Exp. Med. 189: 587–592.
30. Sallusto, F., and A. Lanzavecchia. 2002. The instructive role of dendritic cells onT-cell responses. Arthritis Res. 4(Suppl. 3): S127–S132.
31. Brigl, M., and M. Brenner. 2004. CD1: antigen presentation and T cell function.Annu. Rev. Immunol. 22: 817–890.
32. Langenkamp, A., G. Casorati, C. Garavaglia, P. Dellabona, A. Lanzavecchia, andF. Sallusto. 2002. T cell priming by dendritic cells: thresholds for proliferation,differentiation and death and intraclonal functional diversification. Eur. J. Immu-nol. 32: 2046–2054.
33. Penna, G., M. Vulcano, A. Roncari, F. Facchetti, S. Sozzani, and L. Adorini.2002. Differential chemokine production by myeloid and plasmacytoid dendriticcells. J. Immunol. 169: 6673–6676.
34. Pulendran, B., J. L. Smith, G. Caspary, K. Brasel, D. Pettit, E. Maraskovsky, andC. R. Maliszewski. 1999. Distinct dendritic cell subsets differentially regulate theclass of immune response in vivo. Proc. Natl. Acad. Sci. USA 96: 1036–1041.
35. Banchereau, J., V. Pascual, and A. Palucka. 2004. Autoimmunity through cyto-kine-induced dendritic cell activation. Immunity 20: 539–550.
36. Santiago-Raber, M. L., R. Baccala, K. Haraldsson, D. Choubey, T. Stewart,D. Kono, and A. Theofilopoulos. 2003. Type-I interferon receptor deficiencyreduces lupus-like disease in NZB mice. J. Exp. Med. 197: 777–788.
37. Tan, E. M., A. Cohen, J. Fries, A. Masi, D. McShane, N. Rothfield, J. Schaller,N. Talal, and R. Winchester. 1982. The 1982 revised criteria for the classificationof systemic lupus erythematosus. Arthritis Rheum. 25: 1271–1277.
38. Arnett, F. C., S. Edworthy, D. Bloch, D. McShane, J. Fries, N. Cooper, L. Healey,S. Kaplan, M. Liang, H. Luthra, et al. 1988. The American Rheumatism Asso-ciation 1987 revised criteria for the classification of rheumatoid arthritis. ArthritisRheum. 31: 315–324.
39. Hochberg, M. C., for the Diagnostic and Therapeutic Criteria Committee of theAmerican College of Rheumatology. 1997. Updating the American College ofRheumatology revised criteria for the classification of systemic lupus erythem-atosus [letter]. Arthritis Rheum. 40: 1725.
40. Bombardier, C., D. Gladman, M. Urowitz, D. Caron, and C. Chang. 1992. Der-ivation of the SLEDAI: a disease activity index for lupus patients. The Commit-tee on Prognosis Studies in SLE. Arthritis Rheum. 35: 630–640.
41. Scandella, E., Y. Men, S. Gillessen, R. Forster, and M. Groettrup. 2002. Prosta-glandin E2 is a key factor for CCR7 surface expression and migration of mono-cyte-derived dendritic cells. Blood 100: 1354–1361.
42. Munster, T., J. P. Gibbs, D. Shen, B. A. Baethge, G. R. Botstein, J. Caldwell,F. Dietz, R. Ettlinger, H. E. Golden, H. Lindsley, et al. 2002. Hydroxychloro-quine concentration-response relationships in patients with rheumatoid arthritis.Arthritis Rheum. 46: 1460–1469.
43. Ensom, M. H., N. Partovi, D. Decarie, A. P. Ignaszewski, G. J. Fradet, andR. D. Levy. 2003. Mycophenolate pharmacokinetics in early period followinglung or heart transplantation. Ann. Pharmacother. 37: 1761–1767.
44. Doria, A., M. Cutolo, A. Ghirardello, S. Zampieri, F. Vescovi, A. Sulli,M. Giusti, A. Piccoli, P. Grella, and P. F. Gambari. 2002. Steroid hormones anddisease activity during pregnancy in systemic lupus erythematosus. ArthritisRheum. 47: 202–209.
45. Iqbal, M. P., J. A. Baig, A. A. Ali, S. K. Niazi, N. Mehboobali, andM. A. Hussain. 1998. The effects of non-steroidal anti-inflammatory drugs on thedisposition of methotrexate in patients with rheumatoid arthritis. Biopharm. DrugDispos. 19: 163–167.
46. Mawatari, H., K. Unei, S. Nishimura, N. Sakura, and K. Ueda. 2001. Compar-ative pharmacokinetics of oral 6-mercaptopurine and intravenous 6-mercaptopu-rine riboside in children. Pediatr. Int. 43: 673–677.
47. Lieberman, M. M., G. M. Patterson, and R. E. Moore. 2001. In vitro bioassays foranticancer drug screening: effects of cell concentration and other assay parame-ters on growth inhibitory activity. Cancer Lett. 173: 21–29.
48. Tichopad, A., M. Dilger, G. Schwarz, and M. W. Pfaffl. 2003. Standardizeddetermination of real-time PCR efficiency from a single reaction set-up. NucleicAcids Res. 31: e122.
49. Green, K. J. 2002. Improving understanding of exercise effects on in vitro T-lymphocyte function—the role of fluorescent cell division tracking. Exerc. Im-munol. Rev. 8: 101–115.
50. Mannering, S. I., J. Morris, K. Jensen, A. Purcell, M. Honeyman, P. van Endert,and L. Harrison. 2003. A sensitive method for detecting proliferation of rareautoantigen-specific human T cells. J. Immunol. Methods 283: 173–183.
51. Thompson, B. S., and T. C. Mitchell. 2004. Measurement of daughter cell accu-mulation during lymphocyte proliferation in vivo. J. Immunol. Methods 295:79–87.
5888 ABNORMAL PHENOTYPE AND FUNCTION OF LUPUS DCs
by guest on June 11, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

52. Chen, J. C., M. L. Chang, and M. O. Muench. 2003. A kinetic study of the murinemixed lymphocyte reaction by 5,6-carboxyfluorescein diacetate succinimidyl es-ter labeling. J. Immunol. Methods 279: 123–133.
53. Renno, T., A. Attinger, S. Locatelli, T. Bakker, S. Vacheron, andH. R. MacDonald. 1999. Cutting edge: apoptosis of superantigen-activated Tcells occurs preferentially after a discrete number of cell divisions in vivo. J. Im-munol. 162: 6312–6315.
54. Koller, M., B. Zwolfer, G. Steiner, J. Smolen, and C. Scheinecker. 2004. Phe-notypic and functional deficiencies of monocyte-derived dendritic cells in sys-temic lupus erythematosus (SLE) patients. Int. Immunol. 16: 1595–1604.
55. Cao, W., S. Lee, and J. Lu. 2004. CD83 is preformed inside monocytes, macro-phages and dendritic cells but it is only stably expressed on activated dendriticcells. Biochem. J. 385: 85–93.
56. Adachi, Y., S. Taketani, J. Toki, K. Ikebukuro, K. Sugiura, H. Oyaizu,R. Yasumizu, M. Tomita, H. Kaneda, Y. Amoh, et al. 2002. Marked increase innumber of dendritic cells in autoimmune-prone (NZW � BXSB)F1 mice withage. Stem Cells 20: 61–72.
57. Scheinecker, C., B. Zwolfer, M. Koller, G. Manner, and J. Smolen. 2001. Alter-ations of dendritic cells in systemic lupus erythematosus: phenotypic and func-tional deficiencies. Arthritis Rheum. 44: 856–865.
58. Nakajima, A., M. Azuma, S. Kodera, S. Nuriya, A. Terashi, M. Abe, S. Hirose,T. Shirai, H. Yagita, and K. Okumura. 1995. Preferential dependence of autoan-tibody production in murine lupus on CD86 costimulatory molecule. Eur. J. Im-munol. 25: 3060–3069.
59. Kinoshita, K., G. Tesch, A. Schwarting, R. Maron, A. Sharpe, and V. Kelley.2000. Costimulation by B7-1 and B7-2 is required for autoimmune disease inMRL-Faslpr mice. J. Immunol. 164: 6046–6056.
60. Georgiev, M., L. Agle, J. Chu, K. Elkon, and D. Ashany. 2005. Mature dendriticcells readily break tolerance in normal mice but do not lead to disease expression.Arthritis Rheum. 52: 225–238.
61. Holcombe, R. F., B. Baethge, R. Wolf, K. Betzing, R. Stewart, V. Hall, andM. Fukuda. 1994. Correlation of serum interleukin-8 and cell surface lysosome-associated membrane protein expression with clinical disease activity in systemiclupus erythematosus. Lupus 3: 97–102.
62. Rovin, B. H., L. Lu, and X. Zhang. 2002. A novel interleukin-8 polymorphism isassociated with severe systemic lupus erythematosus nephritis. Kidney Int. 62:261–265.
63. Tsai, C. Y., T. H. Wu, C. Yu, J. Lu, and Y. Tsai. 2000. Increased excretions of�2-microglobulin, IL-6, and IL-8 and decreased excretion of Tamm-Horsfall gly-coprotein in urine of patients with active lupus nephritis. Nephron 85: 207–214.
64. Scimone, M. L., V. P. Lutzky, S. Zittermann, P. Maffia, C. Jancic, F. Buzzola,A. Issekutz, and H. Chuluyan. 2005. Migration of polymorphonuclear leucocytesis influenced by dendritic cells. Immunology 114: 375–385.
65. Trysberg, E., H. Carlsten, and A. Tarkowski. 2000. Intrathecal cytokines in sys-temic lupus erythematosus with central nervous system involvement. Lupus 9:498–503.
66. Zhu, K., Q. Shen, M. Ulrich, and M. Zheng. 2000. Human monocyte-deriveddendritic cells expressing both chemotactic cytokines IL-8, MCP-1, RANTESand their receptors, and their selective migration to these chemokines. Chin. Med.J. 113: 1124–1128.
67. Mayer, G., K. Pohlmeyer, A. Caliebe, E. Heimueller, B. Behnke, G. Steimann,C. Lange, and J. Beuth. 2000. Low molecular thymic peptides stimulate humanblood dendritic cells. Anticancer Res. 20: 2873–2883.
68. Bickel, B. M. 1993. The role of interleukin-8 in inflammation and mechanisms ofregulation. J. Periodontol. 64(Suppl. 5): 456–460.
69. Huang, S., L. Mills, B. Mian, C. Tellez, M. McCarty, X. Yang, J. Gudas, andM. Bar-Eli. 2002. Fully humanized neutralizing antibodies to interleukin-8(ABX-IL8) inhibit angiogenesis, tumor growth, and metastasis of human mela-noma. Am. J. Pathol. 161: 125–134.
70. Mukaida, N. 2000. Interleukin-8: an expanding universe beyond neutrophil che-motaxis and activation. Am. J. Hematol. 72: 391–398.
71. Gerszten, T. E., E. Garcia-Zepeda, Y. Lim, M. Yushida, H. Ding, M. Gimbrone,A. Luster, F. Luscinskas, and A. Rosenzweig. 1999. MCP-1 and IL-8 trigger firmadhesion of monocytes to vascular endothelium under flow conditions. Nature398: 718–723.
72. Casilli, F., A. Bianchini, I. Gloaguen, L. Biordi, E. Alesse, C. Festuccia,B. Cavalieri, R. Strippoli, M. Cervellera, R. Di Bitondo, et al. 2005. Inhibition ofinterleukin-8 (CXCL8/IL-8) responses by repertaxin, a new inhibitor of the che-mokine receptors CXCR1 and CXCR2. Biochem. Pharmacol. 69: 385–394.
73. Lai, K., J. Leung, K. Lai, and C. Lai. 1997. Effect of anti-DNA autoantibodies onthe gene expression of interleukin 8, transforming growth factor-�, and nitricoxide synthase in cultured endothelial cells. Scand. J. Rheumatol. 26: 461–467.
74. Sun, K. H., C. Yu, S. Tang, and G. Sun. 2000. Monoclonal anti-double-strandedDNA autoantibody stimulates the expression and release of Il-1�, Il-6, IL-8 IL-10and TNF-� from normal human mononuclear cells involving in the lupus patho-genesis. Immunology 99: 352–360.
75. Rus, V., S. Atamas, V. Shustova, I. Luzina, F. Selaru, L. Madger, and C. Via.2002. Expression of cytokine- and chemokine-related genes in peripheral bloodmononuclear cels from lupus patients by cDNA array. Clin. Immunol. 102:283–290.
76. Scholler, N., M. Hayden-Ledbetter, K. Hellstrom, I. Hellstrom, and J. Ledbetter.2001. CD83 is a sialic acid-binding Ig-like lectin (Siglec) adhesion receptor that bindsmonocytes and a subset of activated CD8� T cells. J. Immunol. 166: 3865–3872
77. Kruse, M., O. Rosorius, F. Kratzer, D. Bevec, C. Kuhnt, A. Steinkasserer,G. Schuler, and J. Hauber. 2000. Inhibition of CD83 cell surface expressionduring dendritic cell maturation by interference with nuclear export of CD83mRNA. J. Exp. Med. 191: 1581–1590.
78. Lechmann, M., E. Zinser, A. Golka, and A. Steinkasserer. 2002. Role of CD83in the immunomodulation of dendritic cells. Int. Arch. Allergy Immunol. 129:113–118.
79. Lechmann, M., E. Kremmer, H. Sticht, and A. Steinkasserer. 2002. Overexpression,purification, and biochemical characterization of the extracellular human CD83 do-main and generation of monoclonal antibodies. Protein Expr. Purif. 24: 445–452.
80. Scholler, N., M. Hayden-Ledbetter, A. Dahlin, I. Hellstrom, K. Hellstrom, andJ. Ledbetter. 2002. Cutting edge: CD83 regulates the development of cellularimmunity. J. Immunol. 168: 2599–2602.
81. Hirano, N., M. O. Butler, Z. Xia, S. Ansen, M. S. von Bergwelt-Baildon,D. Neuberg, G. J. Freeman, and L. M. Nadler. 2005. Engagement of CD83ligand induces prolonged expansion of CD8� T cells and preferential enrich-ment for antigen specificity. Blood 107: 1528–1536.
82. Filaci, G., S. Bacilieri, M. Fravega, M. Monetti, P. Contini, M. Ghio, M. Setti,F. Puppo, and F. Indiveri. 2001. Impairment of CD8� T suppressor cell function inpatients with active systemic lupus erythematosus. J. Immunol. 166: 6452–6457.
83. Garcia-Martinez, L., M. Appleby, K. Staehling-Hampton, D. Andrews, Y. Chen,M. McEuen, P. Tang, R. Rhinehart, S. Proll, B. Paeper, et al. 2004. A novelmutation in CD83 results in the development of a unique population of CD4�
T cells. J. Immunol. 173: 2995–3001.84. Schwab, M., and U. Klotz. 2001. Pharmacokinetic considerations in the treat-
ment of inflammatory bowel disease. Clin. Pharmacokinet. 40: 723–751.85. Zhu, J., X. Liu, C. Xie, M. Yan, Y. Yu, E. S. Sobel, E. K. Wakeland, and
C. Mohan. 2005. T cell hyperactivity in lupus as a consequence of hyperstimu-latory antigen-presenting cells. J. Clin. Invest. 115: 1869–1878.
86. Oren, A., C. Husebo, A. C. Iversen, and R. Austgulen. 2005. A comparativestudy of immunomagnetic methods used for separation of human natural killercells from peripheral blood. J. Immunol. Methods 303: 1–10.
87. Tobiasova-Czetoova, Z., A. Palmborg, A. Lundqvist, G. Karlsson, L. Adamson,J. Bartunkova, G. Masucci, and P. Pisa. 2005. Effects of human plasma proteins onmaturation of monocyte-derived dendritic cells. Immunol. Lett. 100: 113–119.
88. Tkachenko, N., K. Wojas, J. Tabarkiewicz, and J. Rolinski. 2005. Generation ofdendritic cells from human peripheral blood monocytes—comparison of differ-ent culture medium. Folia Histochem. Cytobiol. 43: 25–30.
89. Kufner, S., H. Zitzelsberger, T. Kroell, R. Pelka-Fleischer, A. Salem, F. de Valle,C. Schweiger, V. Nuessler, C. Schmid, H. J. Kolb, and H. M. Schmetzer. 2005.Leukemia-derived dendritic cells can be generated from blood or bone marrow cellsfrom patients with acute myeloid leukaemia: a methodological approach under se-rum-free culture conditions. Scand. J. Immunol. 62: 86–98.
90. Chernysheva, A. D., K. A. Kirou, and M. K. Crow. 2002. T cell proliferationinduced by autologous non-T cells is a response to apoptotic cells processed bydendritic cells. J. Immunol. 169: 1241–1250.
91. Banki, Z., L. Kacani, B. Mullauer, D. Wilflingseder, G. Obermoser,H. Niederegger, H. Schennach, G. Sprinzl, N. Sepp, A. Erdei, et al. 2003. Cross-linking of CD32 induces maturation of human monocyte-derived dendritic cellsvia NF-�B signaling pathway. J. Immunol. 170: 3963–3970.
92. Radstake, T. R., K. C. Nabbe, M. H. Wenink, M. F. Roelofs, A. Oosterlaar,A. W. van Lieshout, P. Barrera, P. L. van Lent, and W. B. van den Berg. 2005.Dendritic cells from patients with rheumatoid arthritis lack the interleukin 13mediated increase of Fc�RII expression, which has clear functional conse-quences. Ann. Rheum. Dis. 64: 1737–1743.
93. Radstake, T. R., A. B. Blom, A. W. Sloetjes, E. O. van Gorselen, G. J. Pesman,L. Engelen, R. Torensma, W. B. van den Berg, C. G. Figdor, P. L. van Lent, etal. 2004. Increased Fc�RII expression and aberrant tumour necrosis factor �production by mature dendritic cells from patients with active rheumatoid ar-thritis. Ann. Rheum. Dis. 63: 1556–1563.
94. Roelofs, M. F., L. A. Joosten, S. Abdollahi-Roodsaz, A. W. van Lieshout,T. Sprong, F. H. van den Hoogen, W. B. van den Berg, and T. R. Radstake.2005. The expression of Toll-like receptors 3 and 7 in rheumatoid arthritissynovium is increased and costimulation of Toll-like receptors 3, 4, and 7/8results in synergistic cytokine production by dendritic cells. Arthritis Rheum. 52:2313–2322.
95. Tran, C. N., S. K. Lundy, and D. A. Fox. 2005. Synovial biology and T cells inrheumatoid arthritis. Pathophysiology 12: 183–189.
96. Klareskog, L., U. Forsum, A. Scheynius, D. Kabelitz, and H. Wigzell. 1982.Evidence in support of a self-perpetuating HLA-DR-dependent delayed-typecell reaction in rheumatoid arthritis. Proc. Natl. Acad. Sci. USA 79: 3632–3636.
97. Palucka, A. K., J. P. Blanck, L. Bennett, V. Pascual, and J. Banchereau. 2005.Cross-regulation of TNF and IFN-� in autoimmune diseases. Proc. Natl. Acad.Sci. USA 102: 3372–3377.
98. Kyttaris, V. C., Y. T. Juang, and G. C. Tsokos. 2005. Immune cells and cyto-kines in systemic lupus erythematosus: an update. Curr. Opin. Rheumatol. 17:518–522.
99. Lorenz, H. M., M. Herrmann, and J. R. Kalden. 2001. The pathogenesis ofautoimmune diseases. Scand. J. Clin. Lab. Invest. Suppl. 235: 16–26.
100. Vallin, H., A. Perers, G.V. Alm, and L. Ronnblom. 1999. Anti-double-strandedDNA antibodies and immunostimulatory plasmid DNA in combination mimicthe endogenous IFN-� inducer in systemic lupus erythematosus. J. Immunol.163: 6306–6313.
101. Boule, M., C. Broughton, F. Mackay, S. Akira, A. Marshak-Rothstein, and I. Rifkin.2004. Toll-like receptor 9-dependent and -independent dendritic cell activation bychromatin-immunoglobulin G complexes. J. Exp. Med. 199: 1631–1640.
102. Decker, P., H. Singh-Jasuja, S. Haager, I. Kotter, and H. Rammensee. 2005.Nucleosome, the main autoantigen in systemic lupus erythematosus, inducesdirect dendritic cell activation via a MyD88-independent pathway: conse-quences on inflammation. J. Immunol. 174: 3326–3334.
5889The Journal of Immunology
by guest on June 11, 2018http://w
ww
.jimm
unol.org/D
ownloaded from