aiche 2011 - automatic reaction mechanism generation with group additive kinetics
DESCRIPTION
Automatic Reaction Mechanism Generation with Group Additive KineticsTalk 551d at the 2011 AIChE Annual Meeting in Minneapolis, MN. Abstract at http://aiche.confex.com/aiche/2011/webprogram/Paper236982.htmlTRANSCRIPT

Automatic Reaction Mechanism Generation with Group Additive KineticsRichard H. West
Joshua W. Allen
William H. Green
Massachusetts Institute of Technology

Combustion chemistry is complex
1250 715
0.1
10
Initial temperature (K)
Igni
tion
del
ay (m
s)
900
1
2

Modern kinetic models are large
1 80
1000
C atoms in largest alkane
Sp
ecie
s in
kin
etic
mo
del
3

Detailed kinetic modeling is complex
For each chemical reaction
we need:
•forward rate coefficient
•equilibrium constantk f = A exp
�−EaRT
�
∆G = ∆H − T∆S
k f
kr= Keq = exp
�−∆G
RT
�
A + B � C + D
r = k f [A][B]
4

Detailed kinetic modeling is complex
Estimating all the reactions is tedious and error prone.
What do we do?What do we do?
5

Detailed kinetic modeling is complex
Estimating all the reactions is tedious and error prone.
12/03/2009 12:17PM_rp_svg2.html
Page 1 of 1file:///Users/rwest/XCodeProjects/RMG/software/python/_rp_svg2.html
Scale = 0.1
-
!0.2
0
0.2
0.4
0.6
0.8
1
500 Tp (K)
1!(N/
N 0)
SimulationRaghavan
1000 1500 2000
12/03/2009 12:17PM_rp_svg2.html
Page 1 of 1file:///Users/rwest/XCodeProjects/RMG/software/python/_rp_svg2.html
Scale = 0.1
-
Recent advances allow complicated chemical reacting systems...
quantum calculations can provide data for unknown species and reactions...
...and software can automatically build kinetic models based on chemistry rules
many important systems have very complicated chemistry
it is common to make simple chemistry models...
and industrial reactors are at such different conditions...
...and fit the parameters to laboratory reactor data
...that the models may be invalid
I work on a modern approach to this problem, that you can use today!
but these models offer little insight into the underlying chemistry
These first-principles models offer more insight than simple linear models...
...to be modelled from first principles!
...and are better for extrapolating to new temperatures and pressures
the methods are now fast and accurate enough to interest the wider chemical engineering community
you can do better than that!
12/03/2009 12:17PM_rp_svg2.html
Page 1 of 1file:///Users/rwest/XCodeProjects/RMG/software/python/_rp_svg2.html
Scale = 0.1
-
!0.2
0
0.2
0.4
0.6
0.8
1
500 Tp (K)
1!(N/
N 0)
SimulationRaghavan
1000 1500 2000
12/03/2009 12:17PM_rp_svg2.html
Page 1 of 1file:///Users/rwest/XCodeProjects/RMG/software/python/_rp_svg2.html
Scale = 0.1
-
Recent advances allow complicated chemical reacting systems...
quantum calculations can provide data for unknown species and reactions...
...and software can automatically build kinetic models based on chemistry rules
many important systems have very complicated chemistry
it is common to make simple chemistry models...
and industrial reactors are at such different conditions...
...and fit the parameters to laboratory reactor data
...that the models may be invalid
I work on a modern approach to this problem, that you can use today!
but these models offer little insight into the underlying chemistry
These first-principles models offer more insight than simple linear models...
...to be modelled from first principles!
...and are better for extrapolating to new temperatures and pressures
the methods are now fast and accurate enough to interest the wider chemical engineering community
you can do better than that!
5

Detailed kinetic modeling is complex
Estimating all the reactions is tedious and error prone.
12/03/2009 12:17PM_rp_svg2.html
Page 1 of 1file:///Users/rwest/XCodeProjects/RMG/software/python/_rp_svg2.html
Scale = 0.1
-
!0.2
0
0.2
0.4
0.6
0.8
1
500 Tp (K)
1!(N/
N 0)
SimulationRaghavan
1000 1500 2000
12/03/2009 12:17PM_rp_svg2.html
Page 1 of 1file:///Users/rwest/XCodeProjects/RMG/software/python/_rp_svg2.html
Scale = 0.1
-
Recent advances allow complicated chemical reacting systems...
quantum calculations can provide data for unknown species and reactions...
...and software can automatically build kinetic models based on chemistry rules
many important systems have very complicated chemistry
it is common to make simple chemistry models...
and industrial reactors are at such different conditions...
...and fit the parameters to laboratory reactor data
...that the models may be invalid
I work on a modern approach to this problem, that you can use today!
but these models offer little insight into the underlying chemistry
These first-principles models offer more insight than simple linear models...
...to be modelled from first principles!
...and are better for extrapolating to new temperatures and pressures
the methods are now fast and accurate enough to interest the wider chemical engineering community
you can do better than that!
12/03/2009 12:17PM_rp_svg2.html
Page 1 of 1file:///Users/rwest/XCodeProjects/RMG/software/python/_rp_svg2.html
Scale = 0.1
-
!0.2
0
0.2
0.4
0.6
0.8
1
500 Tp (K)
1!(N/
N 0)
SimulationRaghavan
1000 1500 2000
12/03/2009 12:17PM_rp_svg2.html
Page 1 of 1file:///Users/rwest/XCodeProjects/RMG/software/python/_rp_svg2.html
Scale = 0.1
-
Recent advances allow complicated chemical reacting systems...
quantum calculations can provide data for unknown species and reactions...
...and software can automatically build kinetic models based on chemistry rules
many important systems have very complicated chemistry
it is common to make simple chemistry models...
and industrial reactors are at such different conditions...
...and fit the parameters to laboratory reactor data
...that the models may be invalid
I work on a modern approach to this problem, that you can use today!
but these models offer little insight into the underlying chemistry
These first-principles models offer more insight than simple linear models...
...to be modelled from first principles!
...and are better for extrapolating to new temperatures and pressures
the methods are now fast and accurate enough to interest the wider chemical engineering community
you can do better than that!
5

Detailed kinetic modeling is complex
Estimating all the reactions is tedious and error prone.
Teach the chemistry to a computer!
⇌facebook.com/rmg.mitrmg . sou rce f o rge . ne t
Reaction Mechanism Generator
•free and open source software
Teach the chemistry to a computer!
⇌facebook.com/rmg.mitrmg . sou rce f o rge . ne t
Reaction Mechanism Generator
•free and open source software
5
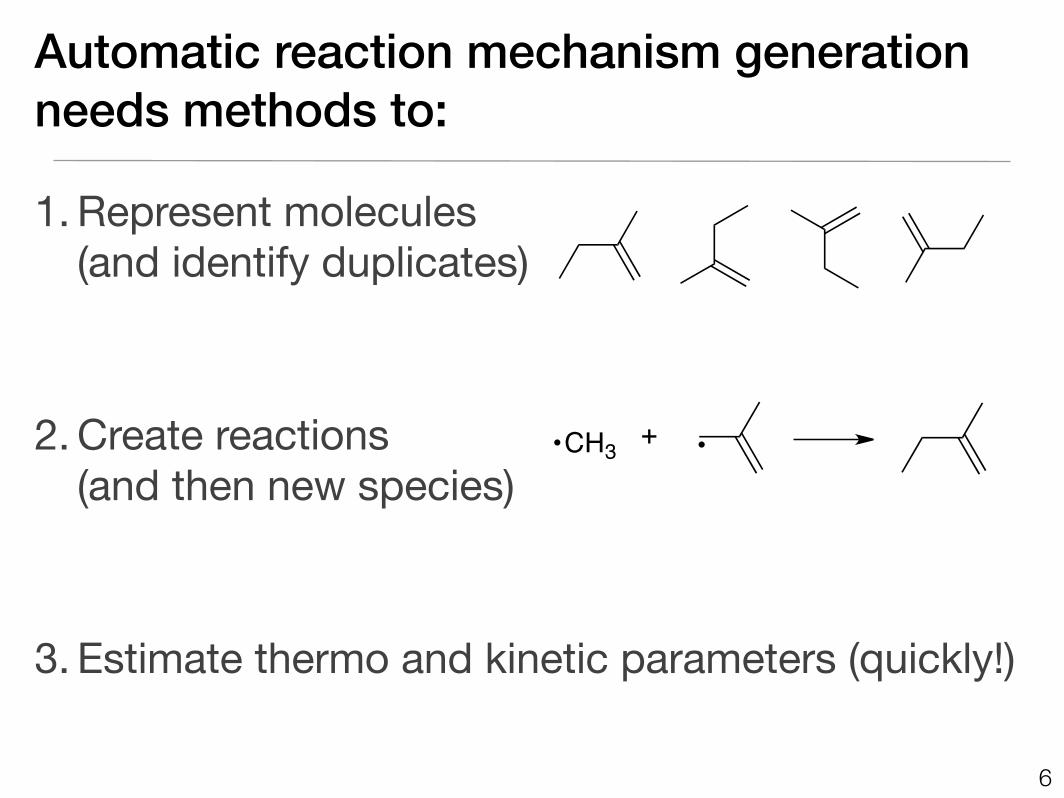
Automatic reaction mechanism generationneeds methods to:
1. Represent molecules (and identify duplicates)
2. Create reactions (and then new species)
3. Estimate thermo and kinetic parameters (quickly!)
CH3 +
6

Molecules are represented as graphs
CH3CH2. C C*
H
H
H H
H
=
7

Thermochemistry is estimated by Benson group contributions
C-(C)(H)3
C-(C)2(H)2
Cb-(H)
C-(C)(Cb)(O)(H)
8

Reaction families propose all possible reactions with given chemical species
•Template for recognizing reactive sites
•Recipe for changing the bonding at the site
•Rules for estimating the rate, based on local chemical structure
bond breaking and hydrogen abstraction
intramolecularH-abstraction
9
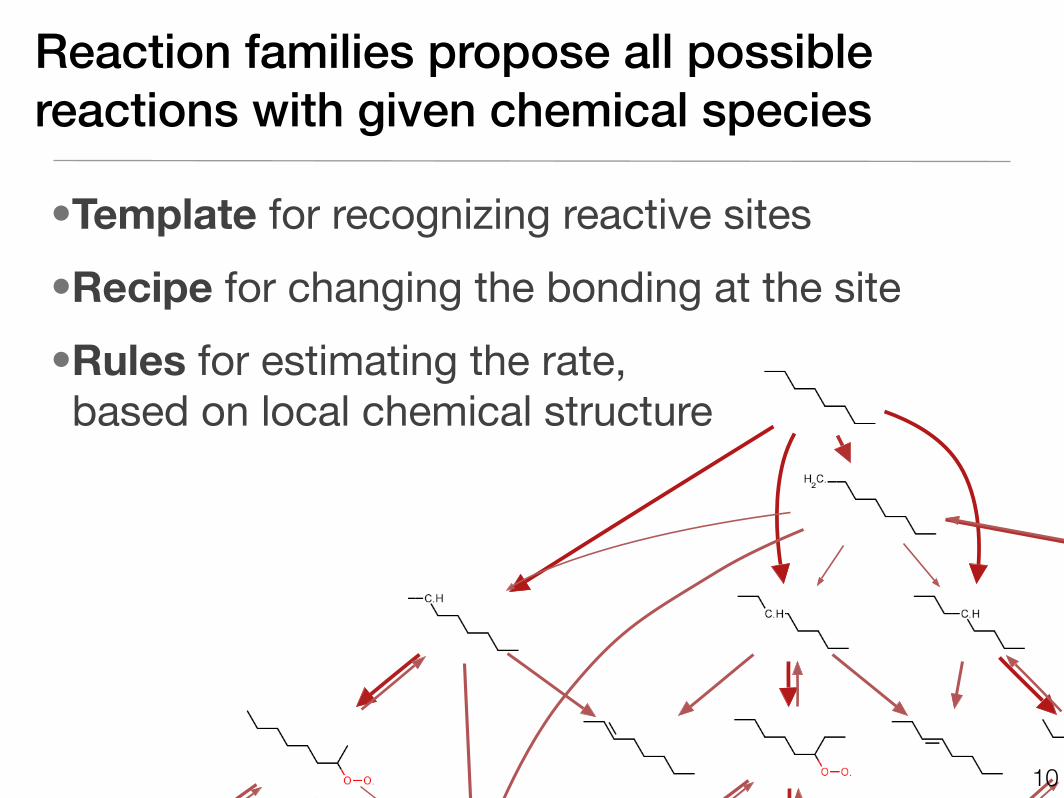
Reaction families propose all possible reactions with given chemical species
•Template for recognizing reactive sites
•Recipe for changing the bonding at the site
•Rules for estimating the rate, based on local chemical structure
10

Octane autoxidation has many pathways
11

•Some pathways go further than others.
12

Need reasonable rate estimates,even of unlikely reactions
•Faster pathways are explored
•Slower pathways are ignored
•Exploration continues until tolerance satisfied.
AB
CD
E
FG
H
AB
CD
E
F
13
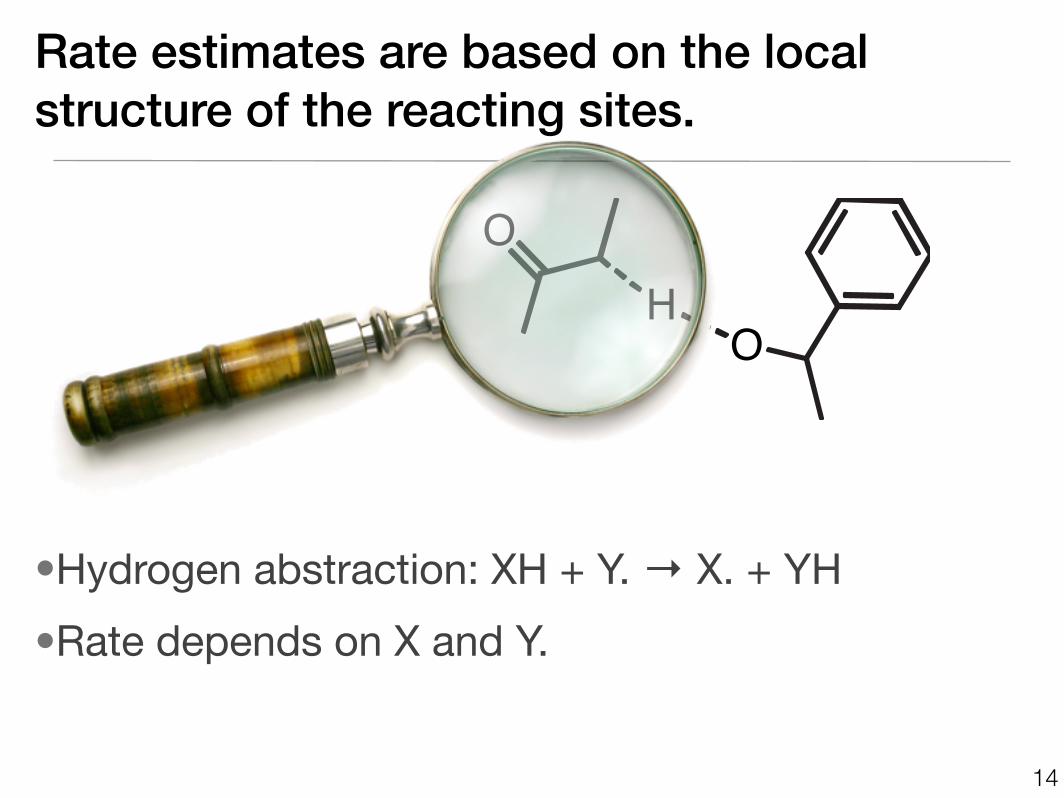
Rate estimates are based on the local structure of the reacting sites.
•Hydrogen abstraction: XH + Y. → X. + YH
•Rate depends on X and Y.
14
OH
O

Rate estimation rules are organized in a tree
•Most general structure at top
•More specific structures are children
15

Part of the tree for X
16

Part of the tree for Y

Ideal tree: lots of data
18

Typical tree: sparse data
19

RMG averages obscure the source of data
•The pair (O_pri, Ct_rad) is not in the database.
•It is estimated by averaging pairs that are:• H_Abstraction estimate: (Average of: (Average of: (Average of: (O_pri O2b) && Average of: (O/H/NonDeC O2b) && O_pri
H_rad && Average of: (O/H/NonDeC H_rad && O/H/OneDe H_rad) && Average of: (O_pri C_methyl && Average of: (O_pri C_rad/H2/Cs)) && Average of: (O/H/NonDeC C_methyl && Average of: (O/H/NonDeC C_rad/H2/Cs) && Average of: (O/H/NonDeC C_rad/H/NonDeC) && Average of: (Average of: (O/H/NonDeC C_rad/Cs3)) && Average of: (Average of: (H2O2 C4H9O/c12345 && H2O2 C4H9O/c134(2)5 && H2O2 C4H9O/c134(2)5 && H2O2 C4H9O/c14(2,3)5) && Average of: (H2O2 C3H5/c132)) && Average of: (Average of: (H2O2 C4H9O/c12345 && H2O2 C4H9O/c12345 && H2O2 C4H9O/c134(2)5) && Average of: (Average of: (H2O2 C4H9O/c12345))) && Average of: (Average of: (Average of: (H2O2 C4H9O/c134(2)5))) && O/H/OneDe C_methyl) && Average of: (O_pri Cd_pri_rad) && Average of: (O/H/NonDeC Cd_pri_rad && Average of: (H2O2 C4H7/c1342) && Average of: (H2O2 Cd_rad/NonDeC)) && Average of: (O/H/NonDeC Ct_rad) && Average of: (O_pri CO_pri_rad) && Average of: (O_pri O_pri_rad && Average of: (O_pri O_rad/NonDeC)) && Average of: (O/H/NonDeC O_pri_rad && Average of: (H2O2 O_rad/NonDeO && H2O2 O_rad/OneDe)))))
20
O_priCt_rad

New approach: group additive log(k)
•O_pri is in the database and contributes -2.35 to log(k@1000K)
•Ct_rad is in the databaseand contributes +2.53 to log(k@1000K)
•Add these to a base rate, to get rate estimate.
21
O_priCt_rad

Group Additive Kinetics through the years
•Reference reaction + thermodynamic corrections
•Willems and Froment (1988)
•Reference reaction + generalized corrective factors
•Truong (2000)
•Estimate thermodynamics of transition state
•Sumathi et al. (2001)
•Direct estimation of Arrhenius parameters
•Saeys et al (2004-)22

23
How to generate kinetics group additivity values
Hierarchy offunc.onal groups
Database of reac.ons
Assign groups for each reac.on
Solve op.miza.onproblem
Validate withtest set Group addi.vity
values
Check tree forwell-‐formedness

24
The ideal training set…
...would use real reactant and product species
...would only have one k(T) for each reaction
...would only have well-known k(T) values
...would be large

The ideal training set does not exist.
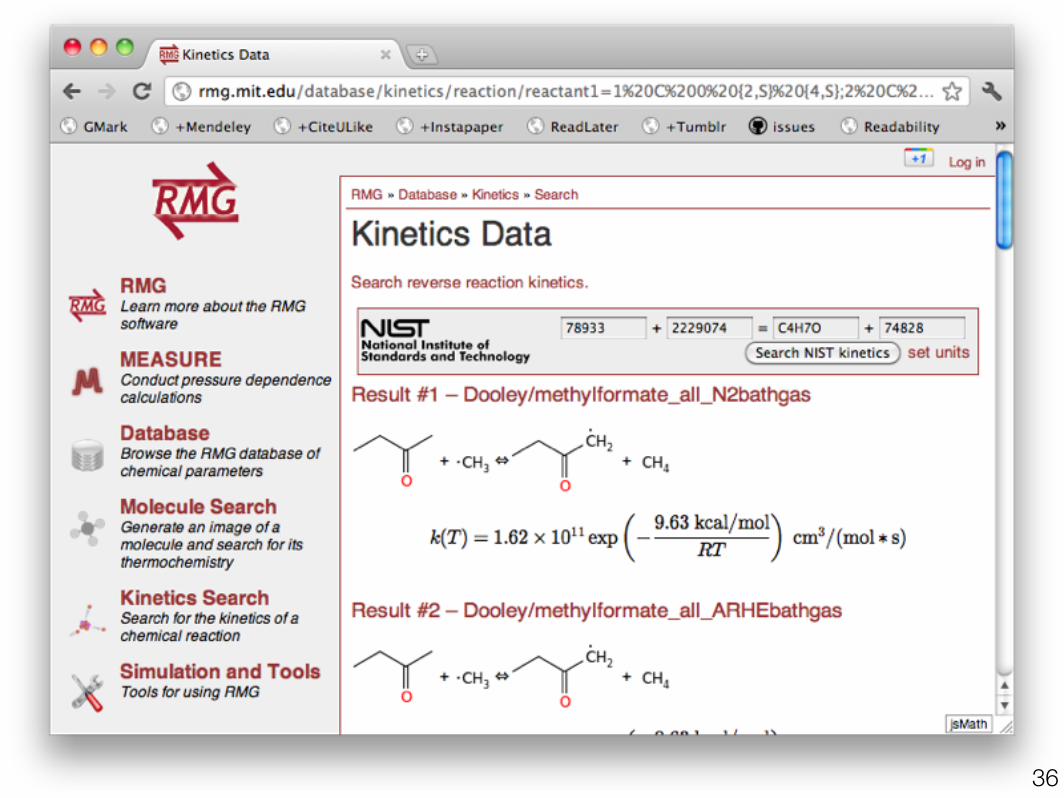
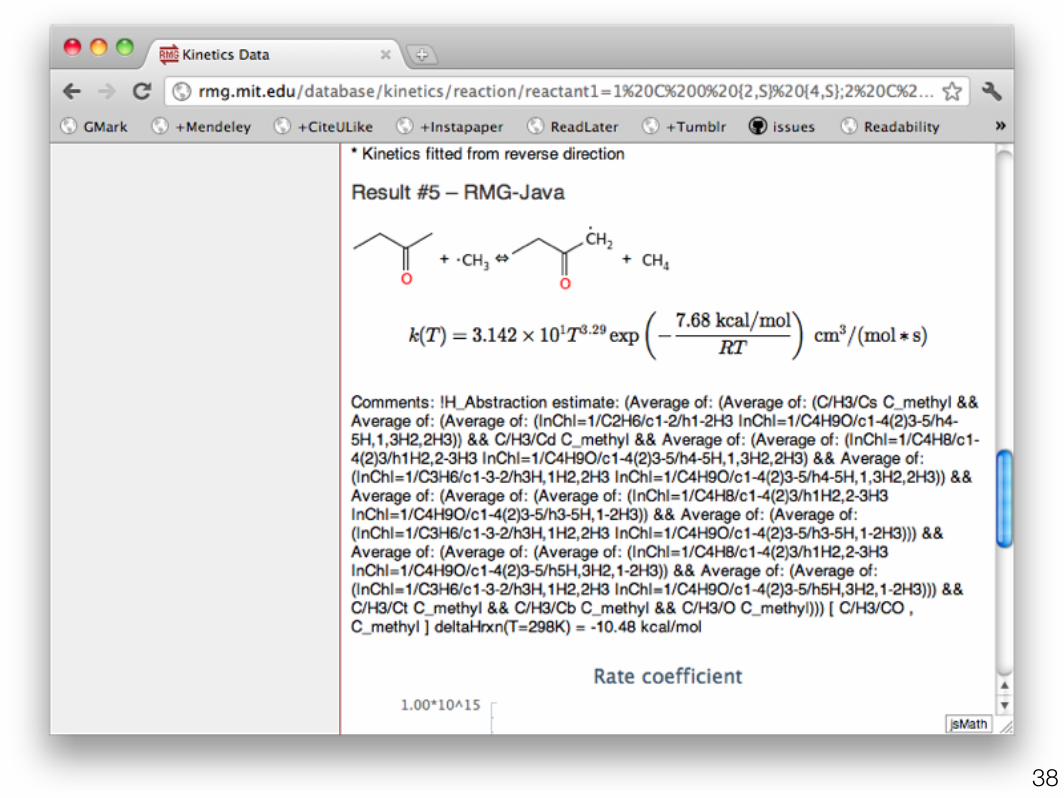
•PrIMe (primekinetics.org)•Transcription errors•No temperature ranges
•NIST (kinetics.nist.gov)•duplicates•estimates•no API
•Current RMG rules (rmg.mit.edu)•functional groups not molecules•current choice
25
⇌

Group values trained using old RMG rules,then tested against PrIMe database.
•Take PrIMe Database
•Filter only Hydrogen Abstraction reactions
•Correct obvious errors (eg. Avogadro number)
•Try to predict with RMG
26
warehouse.primekine.cs.org
3118 C/H/O reac.ons
348 C/H/O hydrogen abstrac.on reac.ons
13654 reac.ons
1075 C/H/O template reac.ons

27
Good agreement when we havean exact match of a rate rule
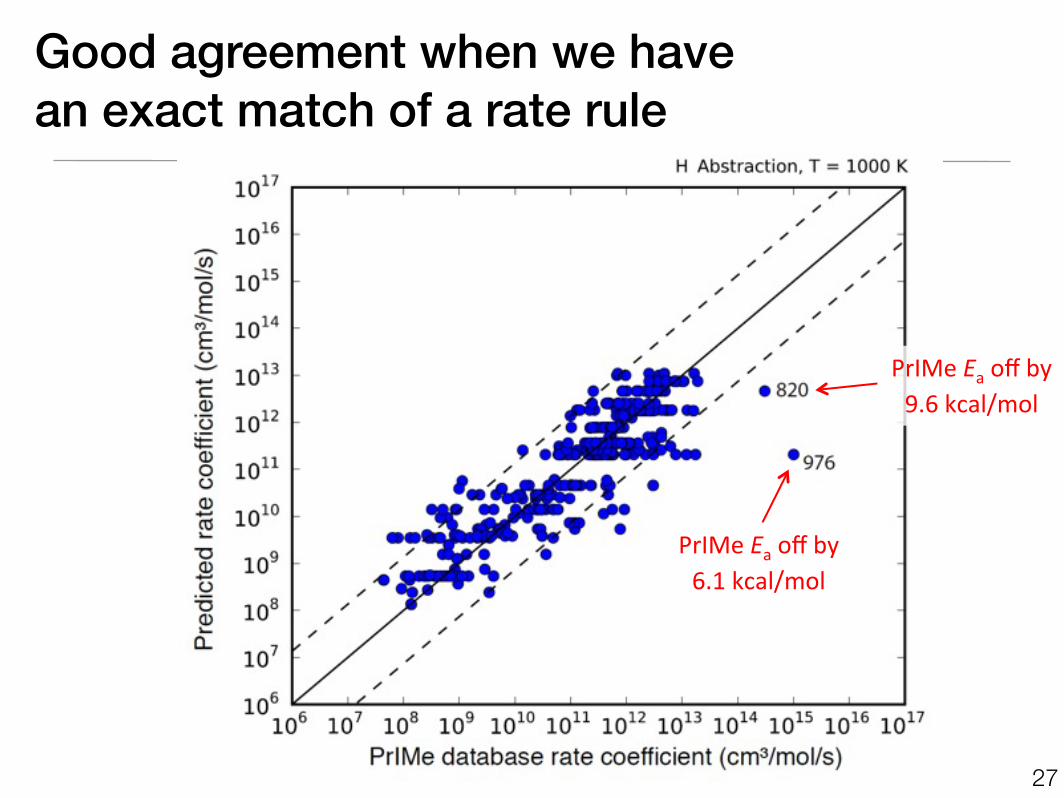
27
Good agreement when we havean exact match of a rate rule
PrIMe Ea off by9.6 kcal/mol
PrIMe Ea off by6.1 kcal/mol

28
Much larger uncertainty when we useaveraged rate rules

28
Much larger uncertainty when we useaveraged rate rules
Complex “average-‐of” es.mate
29
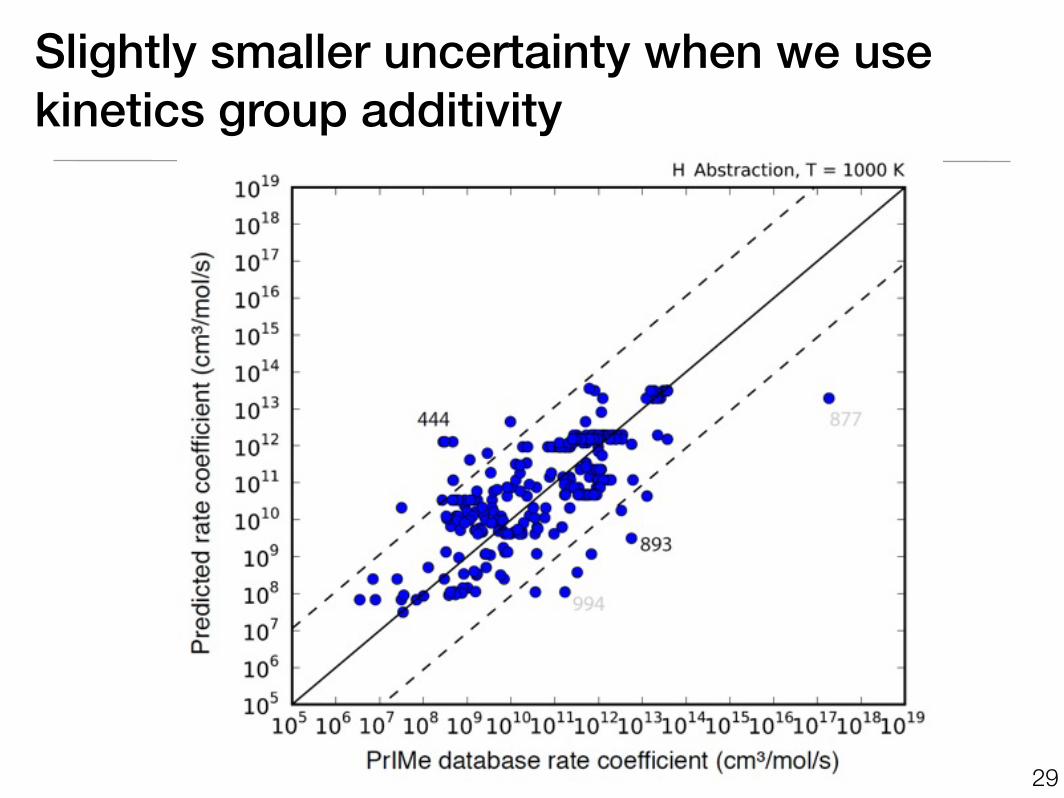
Slightly smaller uncertainty when we usekinetics group additivity

29
Slightly smaller uncertainty when we usekinetics group additivity
Trained on1 rule

30
We can use the group valuesto design a better tree
±0.0 (233)log kXH(1000 K) [cm3/mol*s] Number of entries trained against

30
We can use the group valuesto design a better tree
+0.18 (120) -‐0.55 (25) -‐2.31 (5) +0.79 (22) -‐0.05 (34)-‐0.59 (19)
+0.05 (23) +1.83 (2) +2.09 (1) +1.42 (3)-‐0.12 (17)
+0.16 (47) +0.18 (28) +0.56 (29)-‐0.45 (16)
±0.0 (233)log kXH(1000 K) [cm3/mol*s] Number of entries trained against

31
We can use the group valuesto design a better tree
±0.0 (233)log kY.(1000 K) [cm3/mol*s] Number of entries trained against

31
We can use the group valuesto design a better tree
±0.0 (233)
-‐0.09 (218) +1.68 (13)
+0.54 (23) -‐0.69 (34) -‐0.96 (23) -‐1.11 (17)-‐7.82 (12) +3.52 (1)
-‐0.56 (97) +1.02 (26) +2.62 (7)+2.21 (23)-‐7.01 (13) -‐1.13 (12) +0.77 (37)
log kY.(1000 K) [cm3/mol*s] Number of entries trained against

32

33

34

35
36

37
38

39

Benefits of group additive approach
•Easier to explain and justify than averaging method
•Possible to include uncertainty estimates
•Trained against real reactions
•Easy to modify trees and update rules
40
Next steps
•Collect reliable, clean, database of reaction rates.
•Formalize the estimation of uncertainties
•Extend to other reaction families
•cyclic transition states?
41
!
!
"
"
#
#

42
Acknowledgements
Prof William H. Green
Joshua W. Allen
Connie Gao
Dr. Michael Harper
Amrit Jalan
Gregory Magoon
Shamel Merchant
⇌rmg.mi t .edurmg.s f .ne t

43
Contributions
Developed framework for fitting kinetics group additivity parameters
Group additivity kinetics estimation shows promise for hydrogen abstraction reactions
Key challenge: Getting lots of data