alignments
TRANSCRIPT

Multiple Sequence Alignment
James McInerneybioinf4biologists Feb 2009
Alignment can be easy or difficult
Easy
Difficult due to insertions or deletions
(indels)
GCGGCCCA TCAGGTAGTT GGTGG
GCGGCCCA TCAGGTAGTT GGTGG GCGTTCCA TCAGCTGGTT GGTGG
GCGTCCCA TCAGCTAGTT GGTGG
GCGGCGCA TTAGCTAGTT GGTGA
TTGACATG CCGGGG--- A AACCG
TTGACATG CCGGTG-- GT AAGCC TTGACATG - CTAGG--- A ACGCG
TTGACATG - CTAGGGAAC ACGCG
TTGACATC - CTCTG--- A ACGCG
Homology Definitionbull Homology similarity that is the result of inheritance
from a common ancestor - identification and analysis of homologies is central to phylogenetic systematics
bull An Alignment is an hypothesis of positional homology between basesAmino Acids
Multiple Sequence Alignment- Goals
bull To generate a concise information-rich summary of sequence data
bull Sometimes used to illustrate the dissimilarity between a group of sequences
bull Alignments can be treated as models that can be used to test hypotheses
bull Used to identify homologous residues within sequences
Multiple sequence alignments - problems
bull All sequences show some similarity (even random sequences)
bull Similarity levels might be high in some parts of the sequence and low in other parts
bull Sequences might show substantial length variation and presenceabsence of various domains
SSU rRNA
bull Structural RNA (not translated)bull Found in the small ribosomal subunitbull Widely-used for phylogeny reconstruction
(found in every species)bull Contains stem and loop structuresbull Stem structures usually conform to
watson-crick base pairing
Alignment of 16S rRNA can be guided by secondary structure
lt---------------(--------------------HELIX 19---------------------)lt---------------(22222222-000000-111111-00000-111111-0000-22222222Thermus ruber UCCGAUGC-UAAAGA-CCGAAG=CUCAA=CUUCGG=GGGU=GCGUUGGATh thermophilus UCCCAUGU-GAAAGA-CCACGG=CUCAA=CCGUGG=GGGA=GCGUGGGAEcoli UCAGAUGU-GAAAUC-CCCGGG=CUCAA=CCUGGG=AACU=GCAUCUGAAncystnidulans UCUGUUGU-CAAAGC-GUGGGG=CUCAA=CCUCAU=ACAG=GCAAUGGABsubtilis UCUGAUGU-GAAAGC-CCCCGG=CUCAA=CCGGGG=AGGG=UCAUUGGAChlaurantiacus UCGGCGCU-GAAAGC-GCCCCG=CUUAA=CGGGGC=GAGG=CGCGCCGAmatch
Alignment of 16S rRNA sequences from different bacteria
Protein Alignment may be guided Protein Alignment may be guided by Tertiary Structure Interactionsby Tertiary Structure Interactions
Homo sapiens DjlA protein
Escherichia coli DjlA protein
Multiple Sequence Alignment- Methods
ndash3 main methods of alignment
bull Manual (using custom-built text editors)bull Automatic (using custom-built alignment
software)bull Combined
Manual Alignment - reasonsbull Might be carried out because
ndash Alignment is easyndash There is some extraneous information (structural)
ndash Automated alignment methods have encountered the local minimum problem
ndash An automated alignment method can be ldquoimprovedrdquo
Local minimum
GARFIELDTHEFAT---CATGARFIELDTHEFATFATCAT
bull The dotplot provides a way of quickly visualizing the similarities between all parts of two sequences simultaneously
bull Lets consider a dotplot between sperm whale and human myoglobins
Dotplots
Sperm whale myoglobin
GLSDGEWQLV LNVWGKVEAD IPGHGQEVLI RLFKGHPETL EKFDKFKHLK SEDEMKASED LKKHGATVLT ALGGILKKKG HHEAEIKPLA QSHATKHKIP VKYLEFISEC IIQVLQSKHP GDFGADAQGA MNKALELFRK DMASNYKELG FQG
human myoglobin
VLSEGEWQLV LHVWAKVEAD VAGHGQDILI RLFKSHPETL EKFDRFKHLK TEAEMKASED LKKHGVTVLT ALGAILKKKG HHEAELKPLA QSHATKHKIP IKYLEFISEA IIHVLHSRHP GDFGADAQGA MNKALELFRK DIAAKYKELG YQG
bull Put one sequence on top
bull the other on the side
bull where residues are identical put a dot
bull Diagonal lines of dots show similarities
Dotplot example sperm whale vs human myg
Sperm whale myoglobin
G L S D G E W Q L V V L S E G E W Q L V
Human myoglobin
bullJust do the first 10 amino acids of eachbullMake a table with
ndashwhale sequence on top ndashhuman sequence on the side
bull This is the result for the whole sequence
bull It is easy to see that the diagonal is a line of dots
bull So sperm whale and human myoglobin are very similar
bull But the picture is noisy can smooth using a sliding window which considers neighbouring residues as well
Dotplot example sperm whale vs human myg
16
Sperm whale myoglobin
G L S D G E W Q L V V L S E G E W Q L V
Human myoglobin
bull can smooth noise using a sliding window which considers neighbouring residues as well
bull Have done this here can see the diagonal is highly similar
bull Also instead of using using a simple identity use a scoring matrix
Dotplot example sperm whale vs human myg
Dotplots in practicebull The best tool is an applet called dotlet
bull wwwisrecisb-sibchjavadotletDotlethtmlbull wwwbipbhamacukdotletDotlethtml
bull an applet is a program that runs in a web browser This means that you can produce dotplots within a netscapeIE window
bull Dotplots are often useful to identify things like repeated domains or duplications in big proteins
Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
bull Protein has many repeats bull SLIT_DROME (P24014)
MAAPSRTTLMPPPFRLQLRLLILPILLLLRHDAVHAEPYSGGFGSSAVSSGGLGSVGIHIPGGGVGVITEARCPRVCSCT GLNVDCSHRGLTSVPRKISADVERLELQGNNLTVIYETDFQRLTKLRMLQLTDNQIHTIERNSFQDLVSLERLDISNNVI TTVGRRVFKGAQSLRSLQLDNNQITCLDEHAFKGLVELEILTLNNNNLTSLPHNIFGGLGRLRALRLSDNPFACDCHLSW LSRFLRSATRLAPYTRCQSPSQLKGQNVADLHDQEFKCSGLTEHAPMECGAENSCPHPCRCADGIVDCREKSLTSVPVTL PDDTTDVRLEQNFITELPPKSFSSFRRLRRIDLSNNNISRIAHDALSGLKQLTTLVLYGNKIKDLPSGVFKGLGSLRLLL LNANEISCIRKDAFRDLHSLSLLSLYDNNIQSLANGTFDAMKSMKTVHLAKNPFICDCNLRWLADYLHKNPIETSGARCE SPKRMHRRRIESLREEKFKCSWGELRMKLSGECRMDSDCPAMCHCEGTTVDCTGRRLKEIPRDIPLHTTELLLNDNELGR ISSDGLFGRLPHLVKLELKRNQLTGIEPNAFEGASHIQELQLGENKIKEISNKMFLGLHQLKTLNLYDNQISCVMPGSFE HLNSLTSLNLASNPFNCNCHLAWFAECVRKKSLNGGAARCGAPSKVRDVQIKDLPHSEFKCSSENSEGCLGDGYCPPSCT CTGTVVACSRNQLKEIPRGIPAETSELYLESNEIEQIHYERIRHLRSLTRLDLSNNQITILSNYTFANLTKLSTLIISYN KLQCLQRHALSGLNNLRVVSLHGNRISMLPEGSFEDLKSLTHIALGSNPLYCDCGLKWFSDWIKLDYVEPGIARCAEPEQ MKDKLILSTPSSSFVCRGRVRNDILAKCNACFEQPCQNQAQCVALPQREYQCLCQPGYHGKHCEFMIDACYGNPCRNNAT CTVLEEGRFSCQCAPGYTGARCETNIDDCLGEIKCQNNATCIDGVESYKCECQPGFSGEFCDTKIQFCSPEFNPCANGAK CMDHFTHYSCDCQAGFHGTNCTDNIDDCQNHMCQNGGTCVDGINDYQCRCPDDYTGKYCEGHNMISMMYPQTSPCQNHEC KHGVCFQPNAQGSDYLCRCHPGYTGKWCEYLTSISFVHNNSFVELEPLRTRPEANVTIVFSSAEQNGILMYDGQDAHLAV ELFNGRIRVSYDVGNHPVSTMYSFEMVADGKYHAVELLAIKKNFTLRVDRGLARSIINEGSNDYLKLTTPMFLGGLPVDP AQQAYKNWQIRNLTSFKGCMKEVWINHKLVDFGNAQRQQKITPGCALLEGEQQEEEDDEQDFMDETPHIKEEPVDPCLEN KCRRGSRCVPNSNARDGYQCKCKHGQRGRYCDQGEGSTEPPTVTAASTCRKEQVREYYTENDCRSRQPLKYAKCVGGCGN QCCAAKIVRRRKVRMVCSNNRKYIKNLDIVRKCGCTKKCY
bull Perform a dotplot of the SLIT protein against itself wwwbiobhamacukdotletDotlethtml
Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
20Swiss-prot entry
For further discussion of dotplot see Attwood and Parry-Smith p116-8
Dynamic programming2 methodsbull Dynamic programming
ndash Consider 2 protein sequences of 100 amino acids in lengthndash If it takes 1002 seconds to exhaustively align these sequences
then it will take 1003 seconds to align 3 sequences 1004 to align 4 sequencesetc
ndash More time than the universe has existed to align 20 sequences exhaustively
bull Progressive alignment
Progressive Alignmentbull Devised by Feng and Doolittle in 1987bull Essentially a heuristic method and as such
is not guaranteed to find the lsquooptimalrsquo alignment
bull Requires n-1+n-2+n-3n-n+1 pairwise alignments as a starting point
bull Most successful implementation is Clustal (Des Higgins) This software is cited 3000 times per year in the scientific literature
Overview of ClustalW Procedure
1 PEEKSAVTALWGKVN--VDEVGG2 GEEKAAVLALWDKVN--EEEVGG3 PADKTNVKAAWGKVGAHAGEYGA4 AADKTNVKAAWSKVGGHAGEYGA5 EHEWQLVLHVWAKVEADVAGHGQ
Hbb_Human 1 -Hbb_Horse 2 17 -Hba_Human 3 59 60 -Hba_Horse 4 59 59 13 -Myg_Whale 5 77 77 75 75 -
Hbb_Human
Hbb_Horse
Hba_Horse
Hba_Human
Myg_Whale
2
1
3 4
2
1
3 4
alpha-helices
Quick pairwise alignment calculate distance matrix
Neighbor-joining tree(guide tree)
Progressive alignment following guide tree
CLUSTAL W
ClustalW- Pairwise Alignments
bull First perform all possible pairwise alignments between each pair of sequences There are (n-1)+(n-2)(n-n+1) possibilities
bull Calculate the lsquodistancersquo between each pair of sequences based on these isolated pairwise alignments
bull Generate a distance matrix
Path Graph for aligning two sequences
Possible alignment
1
1
0
1
0
-1
Scoring SchemebullMatch +1bullMismatch 0bullIndel -1
Score for this path= 2
Alignment using this path
GATTC-GAATTC
1
1
0
1
0
-1
Optimal Alignment 1
1
1
-1
1
1
1
Alignment score 4Alignment score 4
Alignment using this path
GA-TTCGA-TTCGAATTCGAATTC
Optimal Alignment 2
1
-1
1
1
1
1
Alignment score 4Alignment score 4
Alignment using this path
G-ATTCG-ATTCGAATTCGAATTC
Alignment of 3 sequences
ClustalW- Guide Tree
bull Generate a Neighbor-Joining lsquoguide treersquo from these pairwise distances
bull This guide tree gives the order in which the progressive alignment will be carried out
Neighbor joining method
bullThe neighbor-joining method is a greedy heuristic which joins at each step the two closest sub-trees that are not already joinedbullIt is based on the minimum evolution principlebullneighbors are defined as two taxa that are connected by a single node in an unrooted tree
A B
Node 1
PAM Spinach Rice Mosquito Monkey HumanSpinach 00 849 1056 908 863Rice 849 00 1178 1224 1226Mosquito 1056 1178 00 847 808Monkey 908 1224 847 00 33Human 863 1226 808 33 00
What is required for the Neighbour joining method
Distance matrixDistance Matrix
PAM distance 33 (Human - Monkey) is the minimum So well join Human and Monkey to MonHum and well calculate the new distances
Mon-Hum
MonkeyHumanSpinachMosquito Rice
First Step
After we have joined two species in a subtree we have to compute the distances from every other node to the new subtree We do this with a simple average of distancesDist[Spinach MonHum]
= (Dist[Spinach Monkey] + Dist[Spinach Human])2 = (908 + 863)2 = 8855
Mon-Hum
MonkeyHumanSpinach
Calculation of New Distances
PAM Spinach Rice Mosquito MonHumSpinach 00 849 1056 886Rice 849 00 1178 1225Mosquito 1056 1178 00 828MonHum 886 1225 828 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Next Cycle
PAM Spinach Rice MosMonHumSpinach 00 849 971Rice 849 00 1202MosMonHum 971 1202 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
Penultimate Cycle
PAM SpinRice MosMonHumSpinach 00 1087MosMonHum 1087 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
(Spin-Rice)-(Mos-(Mon-Hum))
Last Joining
Human
Monkey
MosquitoRice
Spinach
Unrooted Neighbor-Joining Tree
Multiple Alignment- First pairbull Align the two most closely-related sequences first
bull This alignment is then lsquofixedrsquo and will never change If a gap is to be introduced subsequently then it will be introduced in the same place in both sequences but their relative alignment remains unchanged
ClustalW- Decision timebull Consult the guide tree to see what alignment is performed next
ndash Align a third sequence to the first twoOrndash Align two entirely different sequences to each other
Option 1Option 1 Option 2Option 2
ClustalW- Alternative 1If the situation arises where a third sequence is aligned to the first two then when a gap has to be introduced to improve the alignment each of these two entities are treated as two single sequences
+
ClustalW- Alternative 2bull If on the other hand two separate
sequences have to be aligned together then the first pairwise alignment is placed to one side and the pairwise alignment of the other two is carried out
+
ClustalW- Progression
bull The alignment is progressively built up in this way with each step being treated as a pairwise alignment sometimes with each member of a lsquopairrsquo having more than one sequence
Progressive alignment - step 11 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
12345
Progressive alignment - step 21 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
12345
Progressive alignment - step 31 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
+
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
12345
Progressive alignment - final step1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
+
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
5 -ctcga-acgatacgatgactagct-
12345
ClustalW-Good pointsBad points
bull Advantagesndash Speed
bull Disadvantagesndash No objective functionndash No way of quantifying whether or not the alignment is good
ndash No way of knowing if the alignment is lsquocorrectrsquo
ClustalW-Local Minimumbull Potential problems
ndash Local minimum problem If an error is introduced early in the alignment process it is impossible to correct this later in the procedure
ndash Arbitrary alignment
Increasing the sophistication of the alignment process
bull Should we treat all the sequences in the same way - even though some sequences are closely-related and some sequences are distant relatives
bull Should we treat all positions in the sequences as though they were the same - even though they might have different functions and different locations in the 3-dimensional structure
ClustalW- Caveatsbull Sequence weightingbull Varying substitution matricesbull Residue-specific gap penalties and reduced penalties
in hydrophilic regions (external regions of protein sequences) encourage gaps in loops rather than in core regions
bull Positions in early alignments where gaps have been opened receive locally reduced gap penalties to encourage openings in subsequent alignments
ClustalW- User-supplied values
bull Two penalties are set by the user (there are default values but you should know that it is possible to change these)
bull GOP- Gap Opening Penalty is the cost of opening a gap in an alignment
bull GEP- Gap Extension Penalty is the cost of extending this gap
Position-Specific gap penaltiesbull Before any pair of (groups of) sequences are
aligned a table of GOPs are generated for each position in the two (sets of) sequences
bull The GOP is manipulated in a position-specific manner so that it can vary over the sequences
bull If there is a gap at a position the GOP and GEP penalties are lowered the other rules do not apply
bull This makes gaps more likely at positions where gaps already exist
Discouraging too many gaps bull If there is no gap opened then the GOP is increased if the
position is within 8 residues of an existing gapbull This discourages gaps that are too close togetherbull At any position within a run of hydrophilic residues the GOP is
decreasedbull These runs usually indicate loop regions in protein structuresbull A run of 5 hydrophilic residues is considered to be a hydrophilic
stretchbull The default hydrophilic residues are
ndash D E G K N Q P R Sndash But this can be changed by the user
Divergent Sequencesbull The most divergent sequences (most different on average
from all of the other sequences) are usually the most difficult to align
bull It is sometimes better to delay their aligment until later (when the easier sequences have already been aligned)
bull The user has the choice of setting a cutoff (default is 40 identity)
bull This will delay the alignment until the others have been aligned
Advice on progressive alignmentbull Progressive alignment is a mathematical process that is completely
independent of biological realitybull Can be a very good estimatebull Can be an impossibly poor estimatebull Requires user input and skillbull Treat cautiouslybull Can be improved by eye (usually)bull Often helps to have colour-codingbull Depending on the use the user should be able to make a judgement
on those regions that are reliable or notbull For phylogeny reconstruction only use those positions whose
hypothesis of positional homology is unimpeachable
Alignment of protein-coding DNA sequences
bull It is not very sensible to align the DNA sequences of protein-coding genes
ATGCTGTTAGGGATGACTCTGTTAGGG
ATG-CT--GTTAGGGATGACTCTGTTAGGG
The result might be highly-implausible and might not reflect what is known about biological processesIt is much more sensible to translate the sequences to their corresponding amino acid sequences align these protein sequences and then put the gaps in the DNA sequences according to where they are found in the amino acid alignment
Manual Alignment- softwareGDE- The Genetic Data Environment (UNIX)CINEMA- Java applet available from
ndash httpwwwbiochemuclacuk
SeqappSeqpup- MacPCUNIX available fromndash httpiubiobioindianaedu
SeAl for Macintosh available fromndash httpevolvezoooxacukSe-AlSe-Alhtml
BioEdit for PC available fromndash httpwwwmbioncsueduRNasePinfoprogramsBIOEDITbio
edithtml
- Multiple Sequence Alignment James McInerney bioinf4biologists Feb 2009
- Alignment can be easy or difficult
- Homology Definition
- Multiple Sequence Alignment- Goals
- Multiple sequence alignments - problems
- Slide 6
- Slide 7
- SSU rRNA
- Alignment of 16S rRNA can be guided by secondary structure
- Protein Alignment may be guided by Tertiary Structure Interactions
- Multiple Sequence Alignment- Methods
- Manual Alignment - reasons
- Local minimum
- Dotplots
- Dotplot example sperm whale vs human myg
- Slide 16
- Slide 17
- Dotplots in practice
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Dynamic programming
- Progressive Alignment
- Slide 23
- ClustalW- Pairwise Alignments
- Path Graph for aligning two sequences
- Possible alignment
- Alignment using this path
- Optimal Alignment 1
- Optimal Alignment 2
- Alignment of 3 sequences
- ClustalW- Guide Tree
- Neighbor joining method
- Distance Matrix
- First Step
- Calculation of New Distances
- Next Cycle
- Penultimate Cycle
- Last Joining
- Unrooted Neighbor-Joining Tree
- Multiple Alignment- First pair
- ClustalW- Decision time
- ClustalW- Alternative 1
- ClustalW- Progression
- Progressive alignment - step 1
- Progressive alignment - step 2
- Progressive alignment - step 3
- Progressive alignment - final step
- ClustalW-Good pointsBad points
- ClustalW-Local Minimum
- Increasing the sophistication of the alignment process
- Slide 51
- ClustalW- Caveats
- ClustalW- User-supplied values
- Position-Specific gap penalties
- Discouraging too many gaps
- Divergent Sequences
- Advice on progressive alignment
- Alignment of protein-coding DNA sequences
- Manual Alignment- software
-

Alignment can be easy or difficult
Easy
Difficult due to insertions or deletions
(indels)
GCGGCCCA TCAGGTAGTT GGTGG
GCGGCCCA TCAGGTAGTT GGTGG GCGTTCCA TCAGCTGGTT GGTGG
GCGTCCCA TCAGCTAGTT GGTGG
GCGGCGCA TTAGCTAGTT GGTGA
TTGACATG CCGGGG--- A AACCG
TTGACATG CCGGTG-- GT AAGCC TTGACATG - CTAGG--- A ACGCG
TTGACATG - CTAGGGAAC ACGCG
TTGACATC - CTCTG--- A ACGCG
Homology Definitionbull Homology similarity that is the result of inheritance
from a common ancestor - identification and analysis of homologies is central to phylogenetic systematics
bull An Alignment is an hypothesis of positional homology between basesAmino Acids
Multiple Sequence Alignment- Goals
bull To generate a concise information-rich summary of sequence data
bull Sometimes used to illustrate the dissimilarity between a group of sequences
bull Alignments can be treated as models that can be used to test hypotheses
bull Used to identify homologous residues within sequences
Multiple sequence alignments - problems
bull All sequences show some similarity (even random sequences)
bull Similarity levels might be high in some parts of the sequence and low in other parts
bull Sequences might show substantial length variation and presenceabsence of various domains
SSU rRNA
bull Structural RNA (not translated)bull Found in the small ribosomal subunitbull Widely-used for phylogeny reconstruction
(found in every species)bull Contains stem and loop structuresbull Stem structures usually conform to
watson-crick base pairing
Alignment of 16S rRNA can be guided by secondary structure
lt---------------(--------------------HELIX 19---------------------)lt---------------(22222222-000000-111111-00000-111111-0000-22222222Thermus ruber UCCGAUGC-UAAAGA-CCGAAG=CUCAA=CUUCGG=GGGU=GCGUUGGATh thermophilus UCCCAUGU-GAAAGA-CCACGG=CUCAA=CCGUGG=GGGA=GCGUGGGAEcoli UCAGAUGU-GAAAUC-CCCGGG=CUCAA=CCUGGG=AACU=GCAUCUGAAncystnidulans UCUGUUGU-CAAAGC-GUGGGG=CUCAA=CCUCAU=ACAG=GCAAUGGABsubtilis UCUGAUGU-GAAAGC-CCCCGG=CUCAA=CCGGGG=AGGG=UCAUUGGAChlaurantiacus UCGGCGCU-GAAAGC-GCCCCG=CUUAA=CGGGGC=GAGG=CGCGCCGAmatch
Alignment of 16S rRNA sequences from different bacteria
Protein Alignment may be guided Protein Alignment may be guided by Tertiary Structure Interactionsby Tertiary Structure Interactions
Homo sapiens DjlA protein
Escherichia coli DjlA protein
Multiple Sequence Alignment- Methods
ndash3 main methods of alignment
bull Manual (using custom-built text editors)bull Automatic (using custom-built alignment
software)bull Combined
Manual Alignment - reasonsbull Might be carried out because
ndash Alignment is easyndash There is some extraneous information (structural)
ndash Automated alignment methods have encountered the local minimum problem
ndash An automated alignment method can be ldquoimprovedrdquo
Local minimum
GARFIELDTHEFAT---CATGARFIELDTHEFATFATCAT
bull The dotplot provides a way of quickly visualizing the similarities between all parts of two sequences simultaneously
bull Lets consider a dotplot between sperm whale and human myoglobins
Dotplots
Sperm whale myoglobin
GLSDGEWQLV LNVWGKVEAD IPGHGQEVLI RLFKGHPETL EKFDKFKHLK SEDEMKASED LKKHGATVLT ALGGILKKKG HHEAEIKPLA QSHATKHKIP VKYLEFISEC IIQVLQSKHP GDFGADAQGA MNKALELFRK DMASNYKELG FQG
human myoglobin
VLSEGEWQLV LHVWAKVEAD VAGHGQDILI RLFKSHPETL EKFDRFKHLK TEAEMKASED LKKHGVTVLT ALGAILKKKG HHEAELKPLA QSHATKHKIP IKYLEFISEA IIHVLHSRHP GDFGADAQGA MNKALELFRK DIAAKYKELG YQG
bull Put one sequence on top
bull the other on the side
bull where residues are identical put a dot
bull Diagonal lines of dots show similarities
Dotplot example sperm whale vs human myg
Sperm whale myoglobin
G L S D G E W Q L V V L S E G E W Q L V
Human myoglobin
bullJust do the first 10 amino acids of eachbullMake a table with
ndashwhale sequence on top ndashhuman sequence on the side
bull This is the result for the whole sequence
bull It is easy to see that the diagonal is a line of dots
bull So sperm whale and human myoglobin are very similar
bull But the picture is noisy can smooth using a sliding window which considers neighbouring residues as well
Dotplot example sperm whale vs human myg
16
Sperm whale myoglobin
G L S D G E W Q L V V L S E G E W Q L V
Human myoglobin
bull can smooth noise using a sliding window which considers neighbouring residues as well
bull Have done this here can see the diagonal is highly similar
bull Also instead of using using a simple identity use a scoring matrix
Dotplot example sperm whale vs human myg
Dotplots in practicebull The best tool is an applet called dotlet
bull wwwisrecisb-sibchjavadotletDotlethtmlbull wwwbipbhamacukdotletDotlethtml
bull an applet is a program that runs in a web browser This means that you can produce dotplots within a netscapeIE window
bull Dotplots are often useful to identify things like repeated domains or duplications in big proteins
Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
bull Protein has many repeats bull SLIT_DROME (P24014)
MAAPSRTTLMPPPFRLQLRLLILPILLLLRHDAVHAEPYSGGFGSSAVSSGGLGSVGIHIPGGGVGVITEARCPRVCSCT GLNVDCSHRGLTSVPRKISADVERLELQGNNLTVIYETDFQRLTKLRMLQLTDNQIHTIERNSFQDLVSLERLDISNNVI TTVGRRVFKGAQSLRSLQLDNNQITCLDEHAFKGLVELEILTLNNNNLTSLPHNIFGGLGRLRALRLSDNPFACDCHLSW LSRFLRSATRLAPYTRCQSPSQLKGQNVADLHDQEFKCSGLTEHAPMECGAENSCPHPCRCADGIVDCREKSLTSVPVTL PDDTTDVRLEQNFITELPPKSFSSFRRLRRIDLSNNNISRIAHDALSGLKQLTTLVLYGNKIKDLPSGVFKGLGSLRLLL LNANEISCIRKDAFRDLHSLSLLSLYDNNIQSLANGTFDAMKSMKTVHLAKNPFICDCNLRWLADYLHKNPIETSGARCE SPKRMHRRRIESLREEKFKCSWGELRMKLSGECRMDSDCPAMCHCEGTTVDCTGRRLKEIPRDIPLHTTELLLNDNELGR ISSDGLFGRLPHLVKLELKRNQLTGIEPNAFEGASHIQELQLGENKIKEISNKMFLGLHQLKTLNLYDNQISCVMPGSFE HLNSLTSLNLASNPFNCNCHLAWFAECVRKKSLNGGAARCGAPSKVRDVQIKDLPHSEFKCSSENSEGCLGDGYCPPSCT CTGTVVACSRNQLKEIPRGIPAETSELYLESNEIEQIHYERIRHLRSLTRLDLSNNQITILSNYTFANLTKLSTLIISYN KLQCLQRHALSGLNNLRVVSLHGNRISMLPEGSFEDLKSLTHIALGSNPLYCDCGLKWFSDWIKLDYVEPGIARCAEPEQ MKDKLILSTPSSSFVCRGRVRNDILAKCNACFEQPCQNQAQCVALPQREYQCLCQPGYHGKHCEFMIDACYGNPCRNNAT CTVLEEGRFSCQCAPGYTGARCETNIDDCLGEIKCQNNATCIDGVESYKCECQPGFSGEFCDTKIQFCSPEFNPCANGAK CMDHFTHYSCDCQAGFHGTNCTDNIDDCQNHMCQNGGTCVDGINDYQCRCPDDYTGKYCEGHNMISMMYPQTSPCQNHEC KHGVCFQPNAQGSDYLCRCHPGYTGKWCEYLTSISFVHNNSFVELEPLRTRPEANVTIVFSSAEQNGILMYDGQDAHLAV ELFNGRIRVSYDVGNHPVSTMYSFEMVADGKYHAVELLAIKKNFTLRVDRGLARSIINEGSNDYLKLTTPMFLGGLPVDP AQQAYKNWQIRNLTSFKGCMKEVWINHKLVDFGNAQRQQKITPGCALLEGEQQEEEDDEQDFMDETPHIKEEPVDPCLEN KCRRGSRCVPNSNARDGYQCKCKHGQRGRYCDQGEGSTEPPTVTAASTCRKEQVREYYTENDCRSRQPLKYAKCVGGCGN QCCAAKIVRRRKVRMVCSNNRKYIKNLDIVRKCGCTKKCY
bull Perform a dotplot of the SLIT protein against itself wwwbiobhamacukdotletDotlethtml
Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
20Swiss-prot entry
For further discussion of dotplot see Attwood and Parry-Smith p116-8
Dynamic programming2 methodsbull Dynamic programming
ndash Consider 2 protein sequences of 100 amino acids in lengthndash If it takes 1002 seconds to exhaustively align these sequences
then it will take 1003 seconds to align 3 sequences 1004 to align 4 sequencesetc
ndash More time than the universe has existed to align 20 sequences exhaustively
bull Progressive alignment
Progressive Alignmentbull Devised by Feng and Doolittle in 1987bull Essentially a heuristic method and as such
is not guaranteed to find the lsquooptimalrsquo alignment
bull Requires n-1+n-2+n-3n-n+1 pairwise alignments as a starting point
bull Most successful implementation is Clustal (Des Higgins) This software is cited 3000 times per year in the scientific literature
Overview of ClustalW Procedure
1 PEEKSAVTALWGKVN--VDEVGG2 GEEKAAVLALWDKVN--EEEVGG3 PADKTNVKAAWGKVGAHAGEYGA4 AADKTNVKAAWSKVGGHAGEYGA5 EHEWQLVLHVWAKVEADVAGHGQ
Hbb_Human 1 -Hbb_Horse 2 17 -Hba_Human 3 59 60 -Hba_Horse 4 59 59 13 -Myg_Whale 5 77 77 75 75 -
Hbb_Human
Hbb_Horse
Hba_Horse
Hba_Human
Myg_Whale
2
1
3 4
2
1
3 4
alpha-helices
Quick pairwise alignment calculate distance matrix
Neighbor-joining tree(guide tree)
Progressive alignment following guide tree
CLUSTAL W
ClustalW- Pairwise Alignments
bull First perform all possible pairwise alignments between each pair of sequences There are (n-1)+(n-2)(n-n+1) possibilities
bull Calculate the lsquodistancersquo between each pair of sequences based on these isolated pairwise alignments
bull Generate a distance matrix
Path Graph for aligning two sequences
Possible alignment
1
1
0
1
0
-1
Scoring SchemebullMatch +1bullMismatch 0bullIndel -1
Score for this path= 2
Alignment using this path
GATTC-GAATTC
1
1
0
1
0
-1
Optimal Alignment 1
1
1
-1
1
1
1
Alignment score 4Alignment score 4
Alignment using this path
GA-TTCGA-TTCGAATTCGAATTC
Optimal Alignment 2
1
-1
1
1
1
1
Alignment score 4Alignment score 4
Alignment using this path
G-ATTCG-ATTCGAATTCGAATTC
Alignment of 3 sequences
ClustalW- Guide Tree
bull Generate a Neighbor-Joining lsquoguide treersquo from these pairwise distances
bull This guide tree gives the order in which the progressive alignment will be carried out
Neighbor joining method
bullThe neighbor-joining method is a greedy heuristic which joins at each step the two closest sub-trees that are not already joinedbullIt is based on the minimum evolution principlebullneighbors are defined as two taxa that are connected by a single node in an unrooted tree
A B
Node 1
PAM Spinach Rice Mosquito Monkey HumanSpinach 00 849 1056 908 863Rice 849 00 1178 1224 1226Mosquito 1056 1178 00 847 808Monkey 908 1224 847 00 33Human 863 1226 808 33 00
What is required for the Neighbour joining method
Distance matrixDistance Matrix
PAM distance 33 (Human - Monkey) is the minimum So well join Human and Monkey to MonHum and well calculate the new distances
Mon-Hum
MonkeyHumanSpinachMosquito Rice
First Step
After we have joined two species in a subtree we have to compute the distances from every other node to the new subtree We do this with a simple average of distancesDist[Spinach MonHum]
= (Dist[Spinach Monkey] + Dist[Spinach Human])2 = (908 + 863)2 = 8855
Mon-Hum
MonkeyHumanSpinach
Calculation of New Distances
PAM Spinach Rice Mosquito MonHumSpinach 00 849 1056 886Rice 849 00 1178 1225Mosquito 1056 1178 00 828MonHum 886 1225 828 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Next Cycle
PAM Spinach Rice MosMonHumSpinach 00 849 971Rice 849 00 1202MosMonHum 971 1202 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
Penultimate Cycle
PAM SpinRice MosMonHumSpinach 00 1087MosMonHum 1087 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
(Spin-Rice)-(Mos-(Mon-Hum))
Last Joining
Human
Monkey
MosquitoRice
Spinach
Unrooted Neighbor-Joining Tree
Multiple Alignment- First pairbull Align the two most closely-related sequences first
bull This alignment is then lsquofixedrsquo and will never change If a gap is to be introduced subsequently then it will be introduced in the same place in both sequences but their relative alignment remains unchanged
ClustalW- Decision timebull Consult the guide tree to see what alignment is performed next
ndash Align a third sequence to the first twoOrndash Align two entirely different sequences to each other
Option 1Option 1 Option 2Option 2
ClustalW- Alternative 1If the situation arises where a third sequence is aligned to the first two then when a gap has to be introduced to improve the alignment each of these two entities are treated as two single sequences
+
ClustalW- Alternative 2bull If on the other hand two separate
sequences have to be aligned together then the first pairwise alignment is placed to one side and the pairwise alignment of the other two is carried out
+
ClustalW- Progression
bull The alignment is progressively built up in this way with each step being treated as a pairwise alignment sometimes with each member of a lsquopairrsquo having more than one sequence
Progressive alignment - step 11 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
12345
Progressive alignment - step 21 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
12345
Progressive alignment - step 31 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
+
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
12345
Progressive alignment - final step1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
+
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
5 -ctcga-acgatacgatgactagct-
12345
ClustalW-Good pointsBad points
bull Advantagesndash Speed
bull Disadvantagesndash No objective functionndash No way of quantifying whether or not the alignment is good
ndash No way of knowing if the alignment is lsquocorrectrsquo
ClustalW-Local Minimumbull Potential problems
ndash Local minimum problem If an error is introduced early in the alignment process it is impossible to correct this later in the procedure
ndash Arbitrary alignment
Increasing the sophistication of the alignment process
bull Should we treat all the sequences in the same way - even though some sequences are closely-related and some sequences are distant relatives
bull Should we treat all positions in the sequences as though they were the same - even though they might have different functions and different locations in the 3-dimensional structure
ClustalW- Caveatsbull Sequence weightingbull Varying substitution matricesbull Residue-specific gap penalties and reduced penalties
in hydrophilic regions (external regions of protein sequences) encourage gaps in loops rather than in core regions
bull Positions in early alignments where gaps have been opened receive locally reduced gap penalties to encourage openings in subsequent alignments
ClustalW- User-supplied values
bull Two penalties are set by the user (there are default values but you should know that it is possible to change these)
bull GOP- Gap Opening Penalty is the cost of opening a gap in an alignment
bull GEP- Gap Extension Penalty is the cost of extending this gap
Position-Specific gap penaltiesbull Before any pair of (groups of) sequences are
aligned a table of GOPs are generated for each position in the two (sets of) sequences
bull The GOP is manipulated in a position-specific manner so that it can vary over the sequences
bull If there is a gap at a position the GOP and GEP penalties are lowered the other rules do not apply
bull This makes gaps more likely at positions where gaps already exist
Discouraging too many gaps bull If there is no gap opened then the GOP is increased if the
position is within 8 residues of an existing gapbull This discourages gaps that are too close togetherbull At any position within a run of hydrophilic residues the GOP is
decreasedbull These runs usually indicate loop regions in protein structuresbull A run of 5 hydrophilic residues is considered to be a hydrophilic
stretchbull The default hydrophilic residues are
ndash D E G K N Q P R Sndash But this can be changed by the user
Divergent Sequencesbull The most divergent sequences (most different on average
from all of the other sequences) are usually the most difficult to align
bull It is sometimes better to delay their aligment until later (when the easier sequences have already been aligned)
bull The user has the choice of setting a cutoff (default is 40 identity)
bull This will delay the alignment until the others have been aligned
Advice on progressive alignmentbull Progressive alignment is a mathematical process that is completely
independent of biological realitybull Can be a very good estimatebull Can be an impossibly poor estimatebull Requires user input and skillbull Treat cautiouslybull Can be improved by eye (usually)bull Often helps to have colour-codingbull Depending on the use the user should be able to make a judgement
on those regions that are reliable or notbull For phylogeny reconstruction only use those positions whose
hypothesis of positional homology is unimpeachable
Alignment of protein-coding DNA sequences
bull It is not very sensible to align the DNA sequences of protein-coding genes
ATGCTGTTAGGGATGACTCTGTTAGGG
ATG-CT--GTTAGGGATGACTCTGTTAGGG
The result might be highly-implausible and might not reflect what is known about biological processesIt is much more sensible to translate the sequences to their corresponding amino acid sequences align these protein sequences and then put the gaps in the DNA sequences according to where they are found in the amino acid alignment
Manual Alignment- softwareGDE- The Genetic Data Environment (UNIX)CINEMA- Java applet available from
ndash httpwwwbiochemuclacuk
SeqappSeqpup- MacPCUNIX available fromndash httpiubiobioindianaedu
SeAl for Macintosh available fromndash httpevolvezoooxacukSe-AlSe-Alhtml
BioEdit for PC available fromndash httpwwwmbioncsueduRNasePinfoprogramsBIOEDITbio
edithtml
- Multiple Sequence Alignment James McInerney bioinf4biologists Feb 2009
- Alignment can be easy or difficult
- Homology Definition
- Multiple Sequence Alignment- Goals
- Multiple sequence alignments - problems
- Slide 6
- Slide 7
- SSU rRNA
- Alignment of 16S rRNA can be guided by secondary structure
- Protein Alignment may be guided by Tertiary Structure Interactions
- Multiple Sequence Alignment- Methods
- Manual Alignment - reasons
- Local minimum
- Dotplots
- Dotplot example sperm whale vs human myg
- Slide 16
- Slide 17
- Dotplots in practice
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Dynamic programming
- Progressive Alignment
- Slide 23
- ClustalW- Pairwise Alignments
- Path Graph for aligning two sequences
- Possible alignment
- Alignment using this path
- Optimal Alignment 1
- Optimal Alignment 2
- Alignment of 3 sequences
- ClustalW- Guide Tree
- Neighbor joining method
- Distance Matrix
- First Step
- Calculation of New Distances
- Next Cycle
- Penultimate Cycle
- Last Joining
- Unrooted Neighbor-Joining Tree
- Multiple Alignment- First pair
- ClustalW- Decision time
- ClustalW- Alternative 1
- ClustalW- Progression
- Progressive alignment - step 1
- Progressive alignment - step 2
- Progressive alignment - step 3
- Progressive alignment - final step
- ClustalW-Good pointsBad points
- ClustalW-Local Minimum
- Increasing the sophistication of the alignment process
- Slide 51
- ClustalW- Caveats
- ClustalW- User-supplied values
- Position-Specific gap penalties
- Discouraging too many gaps
- Divergent Sequences
- Advice on progressive alignment
- Alignment of protein-coding DNA sequences
- Manual Alignment- software
-

Homology Definitionbull Homology similarity that is the result of inheritance
from a common ancestor - identification and analysis of homologies is central to phylogenetic systematics
bull An Alignment is an hypothesis of positional homology between basesAmino Acids
Multiple Sequence Alignment- Goals
bull To generate a concise information-rich summary of sequence data
bull Sometimes used to illustrate the dissimilarity between a group of sequences
bull Alignments can be treated as models that can be used to test hypotheses
bull Used to identify homologous residues within sequences
Multiple sequence alignments - problems
bull All sequences show some similarity (even random sequences)
bull Similarity levels might be high in some parts of the sequence and low in other parts
bull Sequences might show substantial length variation and presenceabsence of various domains
SSU rRNA
bull Structural RNA (not translated)bull Found in the small ribosomal subunitbull Widely-used for phylogeny reconstruction
(found in every species)bull Contains stem and loop structuresbull Stem structures usually conform to
watson-crick base pairing
Alignment of 16S rRNA can be guided by secondary structure
lt---------------(--------------------HELIX 19---------------------)lt---------------(22222222-000000-111111-00000-111111-0000-22222222Thermus ruber UCCGAUGC-UAAAGA-CCGAAG=CUCAA=CUUCGG=GGGU=GCGUUGGATh thermophilus UCCCAUGU-GAAAGA-CCACGG=CUCAA=CCGUGG=GGGA=GCGUGGGAEcoli UCAGAUGU-GAAAUC-CCCGGG=CUCAA=CCUGGG=AACU=GCAUCUGAAncystnidulans UCUGUUGU-CAAAGC-GUGGGG=CUCAA=CCUCAU=ACAG=GCAAUGGABsubtilis UCUGAUGU-GAAAGC-CCCCGG=CUCAA=CCGGGG=AGGG=UCAUUGGAChlaurantiacus UCGGCGCU-GAAAGC-GCCCCG=CUUAA=CGGGGC=GAGG=CGCGCCGAmatch
Alignment of 16S rRNA sequences from different bacteria
Protein Alignment may be guided Protein Alignment may be guided by Tertiary Structure Interactionsby Tertiary Structure Interactions
Homo sapiens DjlA protein
Escherichia coli DjlA protein
Multiple Sequence Alignment- Methods
ndash3 main methods of alignment
bull Manual (using custom-built text editors)bull Automatic (using custom-built alignment
software)bull Combined
Manual Alignment - reasonsbull Might be carried out because
ndash Alignment is easyndash There is some extraneous information (structural)
ndash Automated alignment methods have encountered the local minimum problem
ndash An automated alignment method can be ldquoimprovedrdquo
Local minimum
GARFIELDTHEFAT---CATGARFIELDTHEFATFATCAT
bull The dotplot provides a way of quickly visualizing the similarities between all parts of two sequences simultaneously
bull Lets consider a dotplot between sperm whale and human myoglobins
Dotplots
Sperm whale myoglobin
GLSDGEWQLV LNVWGKVEAD IPGHGQEVLI RLFKGHPETL EKFDKFKHLK SEDEMKASED LKKHGATVLT ALGGILKKKG HHEAEIKPLA QSHATKHKIP VKYLEFISEC IIQVLQSKHP GDFGADAQGA MNKALELFRK DMASNYKELG FQG
human myoglobin
VLSEGEWQLV LHVWAKVEAD VAGHGQDILI RLFKSHPETL EKFDRFKHLK TEAEMKASED LKKHGVTVLT ALGAILKKKG HHEAELKPLA QSHATKHKIP IKYLEFISEA IIHVLHSRHP GDFGADAQGA MNKALELFRK DIAAKYKELG YQG
bull Put one sequence on top
bull the other on the side
bull where residues are identical put a dot
bull Diagonal lines of dots show similarities
Dotplot example sperm whale vs human myg
Sperm whale myoglobin
G L S D G E W Q L V V L S E G E W Q L V
Human myoglobin
bullJust do the first 10 amino acids of eachbullMake a table with
ndashwhale sequence on top ndashhuman sequence on the side
bull This is the result for the whole sequence
bull It is easy to see that the diagonal is a line of dots
bull So sperm whale and human myoglobin are very similar
bull But the picture is noisy can smooth using a sliding window which considers neighbouring residues as well
Dotplot example sperm whale vs human myg
16
Sperm whale myoglobin
G L S D G E W Q L V V L S E G E W Q L V
Human myoglobin
bull can smooth noise using a sliding window which considers neighbouring residues as well
bull Have done this here can see the diagonal is highly similar
bull Also instead of using using a simple identity use a scoring matrix
Dotplot example sperm whale vs human myg
Dotplots in practicebull The best tool is an applet called dotlet
bull wwwisrecisb-sibchjavadotletDotlethtmlbull wwwbipbhamacukdotletDotlethtml
bull an applet is a program that runs in a web browser This means that you can produce dotplots within a netscapeIE window
bull Dotplots are often useful to identify things like repeated domains or duplications in big proteins
Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
bull Protein has many repeats bull SLIT_DROME (P24014)
MAAPSRTTLMPPPFRLQLRLLILPILLLLRHDAVHAEPYSGGFGSSAVSSGGLGSVGIHIPGGGVGVITEARCPRVCSCT GLNVDCSHRGLTSVPRKISADVERLELQGNNLTVIYETDFQRLTKLRMLQLTDNQIHTIERNSFQDLVSLERLDISNNVI TTVGRRVFKGAQSLRSLQLDNNQITCLDEHAFKGLVELEILTLNNNNLTSLPHNIFGGLGRLRALRLSDNPFACDCHLSW LSRFLRSATRLAPYTRCQSPSQLKGQNVADLHDQEFKCSGLTEHAPMECGAENSCPHPCRCADGIVDCREKSLTSVPVTL PDDTTDVRLEQNFITELPPKSFSSFRRLRRIDLSNNNISRIAHDALSGLKQLTTLVLYGNKIKDLPSGVFKGLGSLRLLL LNANEISCIRKDAFRDLHSLSLLSLYDNNIQSLANGTFDAMKSMKTVHLAKNPFICDCNLRWLADYLHKNPIETSGARCE SPKRMHRRRIESLREEKFKCSWGELRMKLSGECRMDSDCPAMCHCEGTTVDCTGRRLKEIPRDIPLHTTELLLNDNELGR ISSDGLFGRLPHLVKLELKRNQLTGIEPNAFEGASHIQELQLGENKIKEISNKMFLGLHQLKTLNLYDNQISCVMPGSFE HLNSLTSLNLASNPFNCNCHLAWFAECVRKKSLNGGAARCGAPSKVRDVQIKDLPHSEFKCSSENSEGCLGDGYCPPSCT CTGTVVACSRNQLKEIPRGIPAETSELYLESNEIEQIHYERIRHLRSLTRLDLSNNQITILSNYTFANLTKLSTLIISYN KLQCLQRHALSGLNNLRVVSLHGNRISMLPEGSFEDLKSLTHIALGSNPLYCDCGLKWFSDWIKLDYVEPGIARCAEPEQ MKDKLILSTPSSSFVCRGRVRNDILAKCNACFEQPCQNQAQCVALPQREYQCLCQPGYHGKHCEFMIDACYGNPCRNNAT CTVLEEGRFSCQCAPGYTGARCETNIDDCLGEIKCQNNATCIDGVESYKCECQPGFSGEFCDTKIQFCSPEFNPCANGAK CMDHFTHYSCDCQAGFHGTNCTDNIDDCQNHMCQNGGTCVDGINDYQCRCPDDYTGKYCEGHNMISMMYPQTSPCQNHEC KHGVCFQPNAQGSDYLCRCHPGYTGKWCEYLTSISFVHNNSFVELEPLRTRPEANVTIVFSSAEQNGILMYDGQDAHLAV ELFNGRIRVSYDVGNHPVSTMYSFEMVADGKYHAVELLAIKKNFTLRVDRGLARSIINEGSNDYLKLTTPMFLGGLPVDP AQQAYKNWQIRNLTSFKGCMKEVWINHKLVDFGNAQRQQKITPGCALLEGEQQEEEDDEQDFMDETPHIKEEPVDPCLEN KCRRGSRCVPNSNARDGYQCKCKHGQRGRYCDQGEGSTEPPTVTAASTCRKEQVREYYTENDCRSRQPLKYAKCVGGCGN QCCAAKIVRRRKVRMVCSNNRKYIKNLDIVRKCGCTKKCY
bull Perform a dotplot of the SLIT protein against itself wwwbiobhamacukdotletDotlethtml
Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
20Swiss-prot entry
For further discussion of dotplot see Attwood and Parry-Smith p116-8
Dynamic programming2 methodsbull Dynamic programming
ndash Consider 2 protein sequences of 100 amino acids in lengthndash If it takes 1002 seconds to exhaustively align these sequences
then it will take 1003 seconds to align 3 sequences 1004 to align 4 sequencesetc
ndash More time than the universe has existed to align 20 sequences exhaustively
bull Progressive alignment
Progressive Alignmentbull Devised by Feng and Doolittle in 1987bull Essentially a heuristic method and as such
is not guaranteed to find the lsquooptimalrsquo alignment
bull Requires n-1+n-2+n-3n-n+1 pairwise alignments as a starting point
bull Most successful implementation is Clustal (Des Higgins) This software is cited 3000 times per year in the scientific literature
Overview of ClustalW Procedure
1 PEEKSAVTALWGKVN--VDEVGG2 GEEKAAVLALWDKVN--EEEVGG3 PADKTNVKAAWGKVGAHAGEYGA4 AADKTNVKAAWSKVGGHAGEYGA5 EHEWQLVLHVWAKVEADVAGHGQ
Hbb_Human 1 -Hbb_Horse 2 17 -Hba_Human 3 59 60 -Hba_Horse 4 59 59 13 -Myg_Whale 5 77 77 75 75 -
Hbb_Human
Hbb_Horse
Hba_Horse
Hba_Human
Myg_Whale
2
1
3 4
2
1
3 4
alpha-helices
Quick pairwise alignment calculate distance matrix
Neighbor-joining tree(guide tree)
Progressive alignment following guide tree
CLUSTAL W
ClustalW- Pairwise Alignments
bull First perform all possible pairwise alignments between each pair of sequences There are (n-1)+(n-2)(n-n+1) possibilities
bull Calculate the lsquodistancersquo between each pair of sequences based on these isolated pairwise alignments
bull Generate a distance matrix
Path Graph for aligning two sequences
Possible alignment
1
1
0
1
0
-1
Scoring SchemebullMatch +1bullMismatch 0bullIndel -1
Score for this path= 2
Alignment using this path
GATTC-GAATTC
1
1
0
1
0
-1
Optimal Alignment 1
1
1
-1
1
1
1
Alignment score 4Alignment score 4
Alignment using this path
GA-TTCGA-TTCGAATTCGAATTC
Optimal Alignment 2
1
-1
1
1
1
1
Alignment score 4Alignment score 4
Alignment using this path
G-ATTCG-ATTCGAATTCGAATTC
Alignment of 3 sequences
ClustalW- Guide Tree
bull Generate a Neighbor-Joining lsquoguide treersquo from these pairwise distances
bull This guide tree gives the order in which the progressive alignment will be carried out
Neighbor joining method
bullThe neighbor-joining method is a greedy heuristic which joins at each step the two closest sub-trees that are not already joinedbullIt is based on the minimum evolution principlebullneighbors are defined as two taxa that are connected by a single node in an unrooted tree
A B
Node 1
PAM Spinach Rice Mosquito Monkey HumanSpinach 00 849 1056 908 863Rice 849 00 1178 1224 1226Mosquito 1056 1178 00 847 808Monkey 908 1224 847 00 33Human 863 1226 808 33 00
What is required for the Neighbour joining method
Distance matrixDistance Matrix
PAM distance 33 (Human - Monkey) is the minimum So well join Human and Monkey to MonHum and well calculate the new distances
Mon-Hum
MonkeyHumanSpinachMosquito Rice
First Step
After we have joined two species in a subtree we have to compute the distances from every other node to the new subtree We do this with a simple average of distancesDist[Spinach MonHum]
= (Dist[Spinach Monkey] + Dist[Spinach Human])2 = (908 + 863)2 = 8855
Mon-Hum
MonkeyHumanSpinach
Calculation of New Distances
PAM Spinach Rice Mosquito MonHumSpinach 00 849 1056 886Rice 849 00 1178 1225Mosquito 1056 1178 00 828MonHum 886 1225 828 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Next Cycle
PAM Spinach Rice MosMonHumSpinach 00 849 971Rice 849 00 1202MosMonHum 971 1202 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
Penultimate Cycle
PAM SpinRice MosMonHumSpinach 00 1087MosMonHum 1087 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
(Spin-Rice)-(Mos-(Mon-Hum))
Last Joining
Human
Monkey
MosquitoRice
Spinach
Unrooted Neighbor-Joining Tree
Multiple Alignment- First pairbull Align the two most closely-related sequences first
bull This alignment is then lsquofixedrsquo and will never change If a gap is to be introduced subsequently then it will be introduced in the same place in both sequences but their relative alignment remains unchanged
ClustalW- Decision timebull Consult the guide tree to see what alignment is performed next
ndash Align a third sequence to the first twoOrndash Align two entirely different sequences to each other
Option 1Option 1 Option 2Option 2
ClustalW- Alternative 1If the situation arises where a third sequence is aligned to the first two then when a gap has to be introduced to improve the alignment each of these two entities are treated as two single sequences
+
ClustalW- Alternative 2bull If on the other hand two separate
sequences have to be aligned together then the first pairwise alignment is placed to one side and the pairwise alignment of the other two is carried out
+
ClustalW- Progression
bull The alignment is progressively built up in this way with each step being treated as a pairwise alignment sometimes with each member of a lsquopairrsquo having more than one sequence
Progressive alignment - step 11 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
12345
Progressive alignment - step 21 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
12345
Progressive alignment - step 31 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
+
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
12345
Progressive alignment - final step1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
+
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
5 -ctcga-acgatacgatgactagct-
12345
ClustalW-Good pointsBad points
bull Advantagesndash Speed
bull Disadvantagesndash No objective functionndash No way of quantifying whether or not the alignment is good
ndash No way of knowing if the alignment is lsquocorrectrsquo
ClustalW-Local Minimumbull Potential problems
ndash Local minimum problem If an error is introduced early in the alignment process it is impossible to correct this later in the procedure
ndash Arbitrary alignment
Increasing the sophistication of the alignment process
bull Should we treat all the sequences in the same way - even though some sequences are closely-related and some sequences are distant relatives
bull Should we treat all positions in the sequences as though they were the same - even though they might have different functions and different locations in the 3-dimensional structure
ClustalW- Caveatsbull Sequence weightingbull Varying substitution matricesbull Residue-specific gap penalties and reduced penalties
in hydrophilic regions (external regions of protein sequences) encourage gaps in loops rather than in core regions
bull Positions in early alignments where gaps have been opened receive locally reduced gap penalties to encourage openings in subsequent alignments
ClustalW- User-supplied values
bull Two penalties are set by the user (there are default values but you should know that it is possible to change these)
bull GOP- Gap Opening Penalty is the cost of opening a gap in an alignment
bull GEP- Gap Extension Penalty is the cost of extending this gap
Position-Specific gap penaltiesbull Before any pair of (groups of) sequences are
aligned a table of GOPs are generated for each position in the two (sets of) sequences
bull The GOP is manipulated in a position-specific manner so that it can vary over the sequences
bull If there is a gap at a position the GOP and GEP penalties are lowered the other rules do not apply
bull This makes gaps more likely at positions where gaps already exist
Discouraging too many gaps bull If there is no gap opened then the GOP is increased if the
position is within 8 residues of an existing gapbull This discourages gaps that are too close togetherbull At any position within a run of hydrophilic residues the GOP is
decreasedbull These runs usually indicate loop regions in protein structuresbull A run of 5 hydrophilic residues is considered to be a hydrophilic
stretchbull The default hydrophilic residues are
ndash D E G K N Q P R Sndash But this can be changed by the user
Divergent Sequencesbull The most divergent sequences (most different on average
from all of the other sequences) are usually the most difficult to align
bull It is sometimes better to delay their aligment until later (when the easier sequences have already been aligned)
bull The user has the choice of setting a cutoff (default is 40 identity)
bull This will delay the alignment until the others have been aligned
Advice on progressive alignmentbull Progressive alignment is a mathematical process that is completely
independent of biological realitybull Can be a very good estimatebull Can be an impossibly poor estimatebull Requires user input and skillbull Treat cautiouslybull Can be improved by eye (usually)bull Often helps to have colour-codingbull Depending on the use the user should be able to make a judgement
on those regions that are reliable or notbull For phylogeny reconstruction only use those positions whose
hypothesis of positional homology is unimpeachable
Alignment of protein-coding DNA sequences
bull It is not very sensible to align the DNA sequences of protein-coding genes
ATGCTGTTAGGGATGACTCTGTTAGGG
ATG-CT--GTTAGGGATGACTCTGTTAGGG
The result might be highly-implausible and might not reflect what is known about biological processesIt is much more sensible to translate the sequences to their corresponding amino acid sequences align these protein sequences and then put the gaps in the DNA sequences according to where they are found in the amino acid alignment
Manual Alignment- softwareGDE- The Genetic Data Environment (UNIX)CINEMA- Java applet available from
ndash httpwwwbiochemuclacuk
SeqappSeqpup- MacPCUNIX available fromndash httpiubiobioindianaedu
SeAl for Macintosh available fromndash httpevolvezoooxacukSe-AlSe-Alhtml
BioEdit for PC available fromndash httpwwwmbioncsueduRNasePinfoprogramsBIOEDITbio
edithtml
- Multiple Sequence Alignment James McInerney bioinf4biologists Feb 2009
- Alignment can be easy or difficult
- Homology Definition
- Multiple Sequence Alignment- Goals
- Multiple sequence alignments - problems
- Slide 6
- Slide 7
- SSU rRNA
- Alignment of 16S rRNA can be guided by secondary structure
- Protein Alignment may be guided by Tertiary Structure Interactions
- Multiple Sequence Alignment- Methods
- Manual Alignment - reasons
- Local minimum
- Dotplots
- Dotplot example sperm whale vs human myg
- Slide 16
- Slide 17
- Dotplots in practice
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Dynamic programming
- Progressive Alignment
- Slide 23
- ClustalW- Pairwise Alignments
- Path Graph for aligning two sequences
- Possible alignment
- Alignment using this path
- Optimal Alignment 1
- Optimal Alignment 2
- Alignment of 3 sequences
- ClustalW- Guide Tree
- Neighbor joining method
- Distance Matrix
- First Step
- Calculation of New Distances
- Next Cycle
- Penultimate Cycle
- Last Joining
- Unrooted Neighbor-Joining Tree
- Multiple Alignment- First pair
- ClustalW- Decision time
- ClustalW- Alternative 1
- ClustalW- Progression
- Progressive alignment - step 1
- Progressive alignment - step 2
- Progressive alignment - step 3
- Progressive alignment - final step
- ClustalW-Good pointsBad points
- ClustalW-Local Minimum
- Increasing the sophistication of the alignment process
- Slide 51
- ClustalW- Caveats
- ClustalW- User-supplied values
- Position-Specific gap penalties
- Discouraging too many gaps
- Divergent Sequences
- Advice on progressive alignment
- Alignment of protein-coding DNA sequences
- Manual Alignment- software
-

Multiple Sequence Alignment- Goals
bull To generate a concise information-rich summary of sequence data
bull Sometimes used to illustrate the dissimilarity between a group of sequences
bull Alignments can be treated as models that can be used to test hypotheses
bull Used to identify homologous residues within sequences
Multiple sequence alignments - problems
bull All sequences show some similarity (even random sequences)
bull Similarity levels might be high in some parts of the sequence and low in other parts
bull Sequences might show substantial length variation and presenceabsence of various domains
SSU rRNA
bull Structural RNA (not translated)bull Found in the small ribosomal subunitbull Widely-used for phylogeny reconstruction
(found in every species)bull Contains stem and loop structuresbull Stem structures usually conform to
watson-crick base pairing
Alignment of 16S rRNA can be guided by secondary structure
lt---------------(--------------------HELIX 19---------------------)lt---------------(22222222-000000-111111-00000-111111-0000-22222222Thermus ruber UCCGAUGC-UAAAGA-CCGAAG=CUCAA=CUUCGG=GGGU=GCGUUGGATh thermophilus UCCCAUGU-GAAAGA-CCACGG=CUCAA=CCGUGG=GGGA=GCGUGGGAEcoli UCAGAUGU-GAAAUC-CCCGGG=CUCAA=CCUGGG=AACU=GCAUCUGAAncystnidulans UCUGUUGU-CAAAGC-GUGGGG=CUCAA=CCUCAU=ACAG=GCAAUGGABsubtilis UCUGAUGU-GAAAGC-CCCCGG=CUCAA=CCGGGG=AGGG=UCAUUGGAChlaurantiacus UCGGCGCU-GAAAGC-GCCCCG=CUUAA=CGGGGC=GAGG=CGCGCCGAmatch
Alignment of 16S rRNA sequences from different bacteria
Protein Alignment may be guided Protein Alignment may be guided by Tertiary Structure Interactionsby Tertiary Structure Interactions
Homo sapiens DjlA protein
Escherichia coli DjlA protein
Multiple Sequence Alignment- Methods
ndash3 main methods of alignment
bull Manual (using custom-built text editors)bull Automatic (using custom-built alignment
software)bull Combined
Manual Alignment - reasonsbull Might be carried out because
ndash Alignment is easyndash There is some extraneous information (structural)
ndash Automated alignment methods have encountered the local minimum problem
ndash An automated alignment method can be ldquoimprovedrdquo
Local minimum
GARFIELDTHEFAT---CATGARFIELDTHEFATFATCAT
bull The dotplot provides a way of quickly visualizing the similarities between all parts of two sequences simultaneously
bull Lets consider a dotplot between sperm whale and human myoglobins
Dotplots
Sperm whale myoglobin
GLSDGEWQLV LNVWGKVEAD IPGHGQEVLI RLFKGHPETL EKFDKFKHLK SEDEMKASED LKKHGATVLT ALGGILKKKG HHEAEIKPLA QSHATKHKIP VKYLEFISEC IIQVLQSKHP GDFGADAQGA MNKALELFRK DMASNYKELG FQG
human myoglobin
VLSEGEWQLV LHVWAKVEAD VAGHGQDILI RLFKSHPETL EKFDRFKHLK TEAEMKASED LKKHGVTVLT ALGAILKKKG HHEAELKPLA QSHATKHKIP IKYLEFISEA IIHVLHSRHP GDFGADAQGA MNKALELFRK DIAAKYKELG YQG
bull Put one sequence on top
bull the other on the side
bull where residues are identical put a dot
bull Diagonal lines of dots show similarities
Dotplot example sperm whale vs human myg
Sperm whale myoglobin
G L S D G E W Q L V V L S E G E W Q L V
Human myoglobin
bullJust do the first 10 amino acids of eachbullMake a table with
ndashwhale sequence on top ndashhuman sequence on the side
bull This is the result for the whole sequence
bull It is easy to see that the diagonal is a line of dots
bull So sperm whale and human myoglobin are very similar
bull But the picture is noisy can smooth using a sliding window which considers neighbouring residues as well
Dotplot example sperm whale vs human myg
16
Sperm whale myoglobin
G L S D G E W Q L V V L S E G E W Q L V
Human myoglobin
bull can smooth noise using a sliding window which considers neighbouring residues as well
bull Have done this here can see the diagonal is highly similar
bull Also instead of using using a simple identity use a scoring matrix
Dotplot example sperm whale vs human myg
Dotplots in practicebull The best tool is an applet called dotlet
bull wwwisrecisb-sibchjavadotletDotlethtmlbull wwwbipbhamacukdotletDotlethtml
bull an applet is a program that runs in a web browser This means that you can produce dotplots within a netscapeIE window
bull Dotplots are often useful to identify things like repeated domains or duplications in big proteins
Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
bull Protein has many repeats bull SLIT_DROME (P24014)
MAAPSRTTLMPPPFRLQLRLLILPILLLLRHDAVHAEPYSGGFGSSAVSSGGLGSVGIHIPGGGVGVITEARCPRVCSCT GLNVDCSHRGLTSVPRKISADVERLELQGNNLTVIYETDFQRLTKLRMLQLTDNQIHTIERNSFQDLVSLERLDISNNVI TTVGRRVFKGAQSLRSLQLDNNQITCLDEHAFKGLVELEILTLNNNNLTSLPHNIFGGLGRLRALRLSDNPFACDCHLSW LSRFLRSATRLAPYTRCQSPSQLKGQNVADLHDQEFKCSGLTEHAPMECGAENSCPHPCRCADGIVDCREKSLTSVPVTL PDDTTDVRLEQNFITELPPKSFSSFRRLRRIDLSNNNISRIAHDALSGLKQLTTLVLYGNKIKDLPSGVFKGLGSLRLLL LNANEISCIRKDAFRDLHSLSLLSLYDNNIQSLANGTFDAMKSMKTVHLAKNPFICDCNLRWLADYLHKNPIETSGARCE SPKRMHRRRIESLREEKFKCSWGELRMKLSGECRMDSDCPAMCHCEGTTVDCTGRRLKEIPRDIPLHTTELLLNDNELGR ISSDGLFGRLPHLVKLELKRNQLTGIEPNAFEGASHIQELQLGENKIKEISNKMFLGLHQLKTLNLYDNQISCVMPGSFE HLNSLTSLNLASNPFNCNCHLAWFAECVRKKSLNGGAARCGAPSKVRDVQIKDLPHSEFKCSSENSEGCLGDGYCPPSCT CTGTVVACSRNQLKEIPRGIPAETSELYLESNEIEQIHYERIRHLRSLTRLDLSNNQITILSNYTFANLTKLSTLIISYN KLQCLQRHALSGLNNLRVVSLHGNRISMLPEGSFEDLKSLTHIALGSNPLYCDCGLKWFSDWIKLDYVEPGIARCAEPEQ MKDKLILSTPSSSFVCRGRVRNDILAKCNACFEQPCQNQAQCVALPQREYQCLCQPGYHGKHCEFMIDACYGNPCRNNAT CTVLEEGRFSCQCAPGYTGARCETNIDDCLGEIKCQNNATCIDGVESYKCECQPGFSGEFCDTKIQFCSPEFNPCANGAK CMDHFTHYSCDCQAGFHGTNCTDNIDDCQNHMCQNGGTCVDGINDYQCRCPDDYTGKYCEGHNMISMMYPQTSPCQNHEC KHGVCFQPNAQGSDYLCRCHPGYTGKWCEYLTSISFVHNNSFVELEPLRTRPEANVTIVFSSAEQNGILMYDGQDAHLAV ELFNGRIRVSYDVGNHPVSTMYSFEMVADGKYHAVELLAIKKNFTLRVDRGLARSIINEGSNDYLKLTTPMFLGGLPVDP AQQAYKNWQIRNLTSFKGCMKEVWINHKLVDFGNAQRQQKITPGCALLEGEQQEEEDDEQDFMDETPHIKEEPVDPCLEN KCRRGSRCVPNSNARDGYQCKCKHGQRGRYCDQGEGSTEPPTVTAASTCRKEQVREYYTENDCRSRQPLKYAKCVGGCGN QCCAAKIVRRRKVRMVCSNNRKYIKNLDIVRKCGCTKKCY
bull Perform a dotplot of the SLIT protein against itself wwwbiobhamacukdotletDotlethtml
Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
20Swiss-prot entry
For further discussion of dotplot see Attwood and Parry-Smith p116-8
Dynamic programming2 methodsbull Dynamic programming
ndash Consider 2 protein sequences of 100 amino acids in lengthndash If it takes 1002 seconds to exhaustively align these sequences
then it will take 1003 seconds to align 3 sequences 1004 to align 4 sequencesetc
ndash More time than the universe has existed to align 20 sequences exhaustively
bull Progressive alignment
Progressive Alignmentbull Devised by Feng and Doolittle in 1987bull Essentially a heuristic method and as such
is not guaranteed to find the lsquooptimalrsquo alignment
bull Requires n-1+n-2+n-3n-n+1 pairwise alignments as a starting point
bull Most successful implementation is Clustal (Des Higgins) This software is cited 3000 times per year in the scientific literature
Overview of ClustalW Procedure
1 PEEKSAVTALWGKVN--VDEVGG2 GEEKAAVLALWDKVN--EEEVGG3 PADKTNVKAAWGKVGAHAGEYGA4 AADKTNVKAAWSKVGGHAGEYGA5 EHEWQLVLHVWAKVEADVAGHGQ
Hbb_Human 1 -Hbb_Horse 2 17 -Hba_Human 3 59 60 -Hba_Horse 4 59 59 13 -Myg_Whale 5 77 77 75 75 -
Hbb_Human
Hbb_Horse
Hba_Horse
Hba_Human
Myg_Whale
2
1
3 4
2
1
3 4
alpha-helices
Quick pairwise alignment calculate distance matrix
Neighbor-joining tree(guide tree)
Progressive alignment following guide tree
CLUSTAL W
ClustalW- Pairwise Alignments
bull First perform all possible pairwise alignments between each pair of sequences There are (n-1)+(n-2)(n-n+1) possibilities
bull Calculate the lsquodistancersquo between each pair of sequences based on these isolated pairwise alignments
bull Generate a distance matrix
Path Graph for aligning two sequences
Possible alignment
1
1
0
1
0
-1
Scoring SchemebullMatch +1bullMismatch 0bullIndel -1
Score for this path= 2
Alignment using this path
GATTC-GAATTC
1
1
0
1
0
-1
Optimal Alignment 1
1
1
-1
1
1
1
Alignment score 4Alignment score 4
Alignment using this path
GA-TTCGA-TTCGAATTCGAATTC
Optimal Alignment 2
1
-1
1
1
1
1
Alignment score 4Alignment score 4
Alignment using this path
G-ATTCG-ATTCGAATTCGAATTC
Alignment of 3 sequences
ClustalW- Guide Tree
bull Generate a Neighbor-Joining lsquoguide treersquo from these pairwise distances
bull This guide tree gives the order in which the progressive alignment will be carried out
Neighbor joining method
bullThe neighbor-joining method is a greedy heuristic which joins at each step the two closest sub-trees that are not already joinedbullIt is based on the minimum evolution principlebullneighbors are defined as two taxa that are connected by a single node in an unrooted tree
A B
Node 1
PAM Spinach Rice Mosquito Monkey HumanSpinach 00 849 1056 908 863Rice 849 00 1178 1224 1226Mosquito 1056 1178 00 847 808Monkey 908 1224 847 00 33Human 863 1226 808 33 00
What is required for the Neighbour joining method
Distance matrixDistance Matrix
PAM distance 33 (Human - Monkey) is the minimum So well join Human and Monkey to MonHum and well calculate the new distances
Mon-Hum
MonkeyHumanSpinachMosquito Rice
First Step
After we have joined two species in a subtree we have to compute the distances from every other node to the new subtree We do this with a simple average of distancesDist[Spinach MonHum]
= (Dist[Spinach Monkey] + Dist[Spinach Human])2 = (908 + 863)2 = 8855
Mon-Hum
MonkeyHumanSpinach
Calculation of New Distances
PAM Spinach Rice Mosquito MonHumSpinach 00 849 1056 886Rice 849 00 1178 1225Mosquito 1056 1178 00 828MonHum 886 1225 828 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Next Cycle
PAM Spinach Rice MosMonHumSpinach 00 849 971Rice 849 00 1202MosMonHum 971 1202 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
Penultimate Cycle
PAM SpinRice MosMonHumSpinach 00 1087MosMonHum 1087 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
(Spin-Rice)-(Mos-(Mon-Hum))
Last Joining
Human
Monkey
MosquitoRice
Spinach
Unrooted Neighbor-Joining Tree
Multiple Alignment- First pairbull Align the two most closely-related sequences first
bull This alignment is then lsquofixedrsquo and will never change If a gap is to be introduced subsequently then it will be introduced in the same place in both sequences but their relative alignment remains unchanged
ClustalW- Decision timebull Consult the guide tree to see what alignment is performed next
ndash Align a third sequence to the first twoOrndash Align two entirely different sequences to each other
Option 1Option 1 Option 2Option 2
ClustalW- Alternative 1If the situation arises where a third sequence is aligned to the first two then when a gap has to be introduced to improve the alignment each of these two entities are treated as two single sequences
+
ClustalW- Alternative 2bull If on the other hand two separate
sequences have to be aligned together then the first pairwise alignment is placed to one side and the pairwise alignment of the other two is carried out
+
ClustalW- Progression
bull The alignment is progressively built up in this way with each step being treated as a pairwise alignment sometimes with each member of a lsquopairrsquo having more than one sequence
Progressive alignment - step 11 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
12345
Progressive alignment - step 21 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
12345
Progressive alignment - step 31 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
+
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
12345
Progressive alignment - final step1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
+
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
5 -ctcga-acgatacgatgactagct-
12345
ClustalW-Good pointsBad points
bull Advantagesndash Speed
bull Disadvantagesndash No objective functionndash No way of quantifying whether or not the alignment is good
ndash No way of knowing if the alignment is lsquocorrectrsquo
ClustalW-Local Minimumbull Potential problems
ndash Local minimum problem If an error is introduced early in the alignment process it is impossible to correct this later in the procedure
ndash Arbitrary alignment
Increasing the sophistication of the alignment process
bull Should we treat all the sequences in the same way - even though some sequences are closely-related and some sequences are distant relatives
bull Should we treat all positions in the sequences as though they were the same - even though they might have different functions and different locations in the 3-dimensional structure
ClustalW- Caveatsbull Sequence weightingbull Varying substitution matricesbull Residue-specific gap penalties and reduced penalties
in hydrophilic regions (external regions of protein sequences) encourage gaps in loops rather than in core regions
bull Positions in early alignments where gaps have been opened receive locally reduced gap penalties to encourage openings in subsequent alignments
ClustalW- User-supplied values
bull Two penalties are set by the user (there are default values but you should know that it is possible to change these)
bull GOP- Gap Opening Penalty is the cost of opening a gap in an alignment
bull GEP- Gap Extension Penalty is the cost of extending this gap
Position-Specific gap penaltiesbull Before any pair of (groups of) sequences are
aligned a table of GOPs are generated for each position in the two (sets of) sequences
bull The GOP is manipulated in a position-specific manner so that it can vary over the sequences
bull If there is a gap at a position the GOP and GEP penalties are lowered the other rules do not apply
bull This makes gaps more likely at positions where gaps already exist
Discouraging too many gaps bull If there is no gap opened then the GOP is increased if the
position is within 8 residues of an existing gapbull This discourages gaps that are too close togetherbull At any position within a run of hydrophilic residues the GOP is
decreasedbull These runs usually indicate loop regions in protein structuresbull A run of 5 hydrophilic residues is considered to be a hydrophilic
stretchbull The default hydrophilic residues are
ndash D E G K N Q P R Sndash But this can be changed by the user
Divergent Sequencesbull The most divergent sequences (most different on average
from all of the other sequences) are usually the most difficult to align
bull It is sometimes better to delay their aligment until later (when the easier sequences have already been aligned)
bull The user has the choice of setting a cutoff (default is 40 identity)
bull This will delay the alignment until the others have been aligned
Advice on progressive alignmentbull Progressive alignment is a mathematical process that is completely
independent of biological realitybull Can be a very good estimatebull Can be an impossibly poor estimatebull Requires user input and skillbull Treat cautiouslybull Can be improved by eye (usually)bull Often helps to have colour-codingbull Depending on the use the user should be able to make a judgement
on those regions that are reliable or notbull For phylogeny reconstruction only use those positions whose
hypothesis of positional homology is unimpeachable
Alignment of protein-coding DNA sequences
bull It is not very sensible to align the DNA sequences of protein-coding genes
ATGCTGTTAGGGATGACTCTGTTAGGG
ATG-CT--GTTAGGGATGACTCTGTTAGGG
The result might be highly-implausible and might not reflect what is known about biological processesIt is much more sensible to translate the sequences to their corresponding amino acid sequences align these protein sequences and then put the gaps in the DNA sequences according to where they are found in the amino acid alignment
Manual Alignment- softwareGDE- The Genetic Data Environment (UNIX)CINEMA- Java applet available from
ndash httpwwwbiochemuclacuk
SeqappSeqpup- MacPCUNIX available fromndash httpiubiobioindianaedu
SeAl for Macintosh available fromndash httpevolvezoooxacukSe-AlSe-Alhtml
BioEdit for PC available fromndash httpwwwmbioncsueduRNasePinfoprogramsBIOEDITbio
edithtml
- Multiple Sequence Alignment James McInerney bioinf4biologists Feb 2009
- Alignment can be easy or difficult
- Homology Definition
- Multiple Sequence Alignment- Goals
- Multiple sequence alignments - problems
- Slide 6
- Slide 7
- SSU rRNA
- Alignment of 16S rRNA can be guided by secondary structure
- Protein Alignment may be guided by Tertiary Structure Interactions
- Multiple Sequence Alignment- Methods
- Manual Alignment - reasons
- Local minimum
- Dotplots
- Dotplot example sperm whale vs human myg
- Slide 16
- Slide 17
- Dotplots in practice
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Dynamic programming
- Progressive Alignment
- Slide 23
- ClustalW- Pairwise Alignments
- Path Graph for aligning two sequences
- Possible alignment
- Alignment using this path
- Optimal Alignment 1
- Optimal Alignment 2
- Alignment of 3 sequences
- ClustalW- Guide Tree
- Neighbor joining method
- Distance Matrix
- First Step
- Calculation of New Distances
- Next Cycle
- Penultimate Cycle
- Last Joining
- Unrooted Neighbor-Joining Tree
- Multiple Alignment- First pair
- ClustalW- Decision time
- ClustalW- Alternative 1
- ClustalW- Progression
- Progressive alignment - step 1
- Progressive alignment - step 2
- Progressive alignment - step 3
- Progressive alignment - final step
- ClustalW-Good pointsBad points
- ClustalW-Local Minimum
- Increasing the sophistication of the alignment process
- Slide 51
- ClustalW- Caveats
- ClustalW- User-supplied values
- Position-Specific gap penalties
- Discouraging too many gaps
- Divergent Sequences
- Advice on progressive alignment
- Alignment of protein-coding DNA sequences
- Manual Alignment- software
-

Multiple sequence alignments - problems
bull All sequences show some similarity (even random sequences)
bull Similarity levels might be high in some parts of the sequence and low in other parts
bull Sequences might show substantial length variation and presenceabsence of various domains
SSU rRNA
bull Structural RNA (not translated)bull Found in the small ribosomal subunitbull Widely-used for phylogeny reconstruction
(found in every species)bull Contains stem and loop structuresbull Stem structures usually conform to
watson-crick base pairing
Alignment of 16S rRNA can be guided by secondary structure
lt---------------(--------------------HELIX 19---------------------)lt---------------(22222222-000000-111111-00000-111111-0000-22222222Thermus ruber UCCGAUGC-UAAAGA-CCGAAG=CUCAA=CUUCGG=GGGU=GCGUUGGATh thermophilus UCCCAUGU-GAAAGA-CCACGG=CUCAA=CCGUGG=GGGA=GCGUGGGAEcoli UCAGAUGU-GAAAUC-CCCGGG=CUCAA=CCUGGG=AACU=GCAUCUGAAncystnidulans UCUGUUGU-CAAAGC-GUGGGG=CUCAA=CCUCAU=ACAG=GCAAUGGABsubtilis UCUGAUGU-GAAAGC-CCCCGG=CUCAA=CCGGGG=AGGG=UCAUUGGAChlaurantiacus UCGGCGCU-GAAAGC-GCCCCG=CUUAA=CGGGGC=GAGG=CGCGCCGAmatch
Alignment of 16S rRNA sequences from different bacteria
Protein Alignment may be guided Protein Alignment may be guided by Tertiary Structure Interactionsby Tertiary Structure Interactions
Homo sapiens DjlA protein
Escherichia coli DjlA protein
Multiple Sequence Alignment- Methods
ndash3 main methods of alignment
bull Manual (using custom-built text editors)bull Automatic (using custom-built alignment
software)bull Combined
Manual Alignment - reasonsbull Might be carried out because
ndash Alignment is easyndash There is some extraneous information (structural)
ndash Automated alignment methods have encountered the local minimum problem
ndash An automated alignment method can be ldquoimprovedrdquo
Local minimum
GARFIELDTHEFAT---CATGARFIELDTHEFATFATCAT
bull The dotplot provides a way of quickly visualizing the similarities between all parts of two sequences simultaneously
bull Lets consider a dotplot between sperm whale and human myoglobins
Dotplots
Sperm whale myoglobin
GLSDGEWQLV LNVWGKVEAD IPGHGQEVLI RLFKGHPETL EKFDKFKHLK SEDEMKASED LKKHGATVLT ALGGILKKKG HHEAEIKPLA QSHATKHKIP VKYLEFISEC IIQVLQSKHP GDFGADAQGA MNKALELFRK DMASNYKELG FQG
human myoglobin
VLSEGEWQLV LHVWAKVEAD VAGHGQDILI RLFKSHPETL EKFDRFKHLK TEAEMKASED LKKHGVTVLT ALGAILKKKG HHEAELKPLA QSHATKHKIP IKYLEFISEA IIHVLHSRHP GDFGADAQGA MNKALELFRK DIAAKYKELG YQG
bull Put one sequence on top
bull the other on the side
bull where residues are identical put a dot
bull Diagonal lines of dots show similarities
Dotplot example sperm whale vs human myg
Sperm whale myoglobin
G L S D G E W Q L V V L S E G E W Q L V
Human myoglobin
bullJust do the first 10 amino acids of eachbullMake a table with
ndashwhale sequence on top ndashhuman sequence on the side
bull This is the result for the whole sequence
bull It is easy to see that the diagonal is a line of dots
bull So sperm whale and human myoglobin are very similar
bull But the picture is noisy can smooth using a sliding window which considers neighbouring residues as well
Dotplot example sperm whale vs human myg
16
Sperm whale myoglobin
G L S D G E W Q L V V L S E G E W Q L V
Human myoglobin
bull can smooth noise using a sliding window which considers neighbouring residues as well
bull Have done this here can see the diagonal is highly similar
bull Also instead of using using a simple identity use a scoring matrix
Dotplot example sperm whale vs human myg
Dotplots in practicebull The best tool is an applet called dotlet
bull wwwisrecisb-sibchjavadotletDotlethtmlbull wwwbipbhamacukdotletDotlethtml
bull an applet is a program that runs in a web browser This means that you can produce dotplots within a netscapeIE window
bull Dotplots are often useful to identify things like repeated domains or duplications in big proteins
Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
bull Protein has many repeats bull SLIT_DROME (P24014)
MAAPSRTTLMPPPFRLQLRLLILPILLLLRHDAVHAEPYSGGFGSSAVSSGGLGSVGIHIPGGGVGVITEARCPRVCSCT GLNVDCSHRGLTSVPRKISADVERLELQGNNLTVIYETDFQRLTKLRMLQLTDNQIHTIERNSFQDLVSLERLDISNNVI TTVGRRVFKGAQSLRSLQLDNNQITCLDEHAFKGLVELEILTLNNNNLTSLPHNIFGGLGRLRALRLSDNPFACDCHLSW LSRFLRSATRLAPYTRCQSPSQLKGQNVADLHDQEFKCSGLTEHAPMECGAENSCPHPCRCADGIVDCREKSLTSVPVTL PDDTTDVRLEQNFITELPPKSFSSFRRLRRIDLSNNNISRIAHDALSGLKQLTTLVLYGNKIKDLPSGVFKGLGSLRLLL LNANEISCIRKDAFRDLHSLSLLSLYDNNIQSLANGTFDAMKSMKTVHLAKNPFICDCNLRWLADYLHKNPIETSGARCE SPKRMHRRRIESLREEKFKCSWGELRMKLSGECRMDSDCPAMCHCEGTTVDCTGRRLKEIPRDIPLHTTELLLNDNELGR ISSDGLFGRLPHLVKLELKRNQLTGIEPNAFEGASHIQELQLGENKIKEISNKMFLGLHQLKTLNLYDNQISCVMPGSFE HLNSLTSLNLASNPFNCNCHLAWFAECVRKKSLNGGAARCGAPSKVRDVQIKDLPHSEFKCSSENSEGCLGDGYCPPSCT CTGTVVACSRNQLKEIPRGIPAETSELYLESNEIEQIHYERIRHLRSLTRLDLSNNQITILSNYTFANLTKLSTLIISYN KLQCLQRHALSGLNNLRVVSLHGNRISMLPEGSFEDLKSLTHIALGSNPLYCDCGLKWFSDWIKLDYVEPGIARCAEPEQ MKDKLILSTPSSSFVCRGRVRNDILAKCNACFEQPCQNQAQCVALPQREYQCLCQPGYHGKHCEFMIDACYGNPCRNNAT CTVLEEGRFSCQCAPGYTGARCETNIDDCLGEIKCQNNATCIDGVESYKCECQPGFSGEFCDTKIQFCSPEFNPCANGAK CMDHFTHYSCDCQAGFHGTNCTDNIDDCQNHMCQNGGTCVDGINDYQCRCPDDYTGKYCEGHNMISMMYPQTSPCQNHEC KHGVCFQPNAQGSDYLCRCHPGYTGKWCEYLTSISFVHNNSFVELEPLRTRPEANVTIVFSSAEQNGILMYDGQDAHLAV ELFNGRIRVSYDVGNHPVSTMYSFEMVADGKYHAVELLAIKKNFTLRVDRGLARSIINEGSNDYLKLTTPMFLGGLPVDP AQQAYKNWQIRNLTSFKGCMKEVWINHKLVDFGNAQRQQKITPGCALLEGEQQEEEDDEQDFMDETPHIKEEPVDPCLEN KCRRGSRCVPNSNARDGYQCKCKHGQRGRYCDQGEGSTEPPTVTAASTCRKEQVREYYTENDCRSRQPLKYAKCVGGCGN QCCAAKIVRRRKVRMVCSNNRKYIKNLDIVRKCGCTKKCY
bull Perform a dotplot of the SLIT protein against itself wwwbiobhamacukdotletDotlethtml
Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
20Swiss-prot entry
For further discussion of dotplot see Attwood and Parry-Smith p116-8
Dynamic programming2 methodsbull Dynamic programming
ndash Consider 2 protein sequences of 100 amino acids in lengthndash If it takes 1002 seconds to exhaustively align these sequences
then it will take 1003 seconds to align 3 sequences 1004 to align 4 sequencesetc
ndash More time than the universe has existed to align 20 sequences exhaustively
bull Progressive alignment
Progressive Alignmentbull Devised by Feng and Doolittle in 1987bull Essentially a heuristic method and as such
is not guaranteed to find the lsquooptimalrsquo alignment
bull Requires n-1+n-2+n-3n-n+1 pairwise alignments as a starting point
bull Most successful implementation is Clustal (Des Higgins) This software is cited 3000 times per year in the scientific literature
Overview of ClustalW Procedure
1 PEEKSAVTALWGKVN--VDEVGG2 GEEKAAVLALWDKVN--EEEVGG3 PADKTNVKAAWGKVGAHAGEYGA4 AADKTNVKAAWSKVGGHAGEYGA5 EHEWQLVLHVWAKVEADVAGHGQ
Hbb_Human 1 -Hbb_Horse 2 17 -Hba_Human 3 59 60 -Hba_Horse 4 59 59 13 -Myg_Whale 5 77 77 75 75 -
Hbb_Human
Hbb_Horse
Hba_Horse
Hba_Human
Myg_Whale
2
1
3 4
2
1
3 4
alpha-helices
Quick pairwise alignment calculate distance matrix
Neighbor-joining tree(guide tree)
Progressive alignment following guide tree
CLUSTAL W
ClustalW- Pairwise Alignments
bull First perform all possible pairwise alignments between each pair of sequences There are (n-1)+(n-2)(n-n+1) possibilities
bull Calculate the lsquodistancersquo between each pair of sequences based on these isolated pairwise alignments
bull Generate a distance matrix
Path Graph for aligning two sequences
Possible alignment
1
1
0
1
0
-1
Scoring SchemebullMatch +1bullMismatch 0bullIndel -1
Score for this path= 2
Alignment using this path
GATTC-GAATTC
1
1
0
1
0
-1
Optimal Alignment 1
1
1
-1
1
1
1
Alignment score 4Alignment score 4
Alignment using this path
GA-TTCGA-TTCGAATTCGAATTC
Optimal Alignment 2
1
-1
1
1
1
1
Alignment score 4Alignment score 4
Alignment using this path
G-ATTCG-ATTCGAATTCGAATTC
Alignment of 3 sequences
ClustalW- Guide Tree
bull Generate a Neighbor-Joining lsquoguide treersquo from these pairwise distances
bull This guide tree gives the order in which the progressive alignment will be carried out
Neighbor joining method
bullThe neighbor-joining method is a greedy heuristic which joins at each step the two closest sub-trees that are not already joinedbullIt is based on the minimum evolution principlebullneighbors are defined as two taxa that are connected by a single node in an unrooted tree
A B
Node 1
PAM Spinach Rice Mosquito Monkey HumanSpinach 00 849 1056 908 863Rice 849 00 1178 1224 1226Mosquito 1056 1178 00 847 808Monkey 908 1224 847 00 33Human 863 1226 808 33 00
What is required for the Neighbour joining method
Distance matrixDistance Matrix
PAM distance 33 (Human - Monkey) is the minimum So well join Human and Monkey to MonHum and well calculate the new distances
Mon-Hum
MonkeyHumanSpinachMosquito Rice
First Step
After we have joined two species in a subtree we have to compute the distances from every other node to the new subtree We do this with a simple average of distancesDist[Spinach MonHum]
= (Dist[Spinach Monkey] + Dist[Spinach Human])2 = (908 + 863)2 = 8855
Mon-Hum
MonkeyHumanSpinach
Calculation of New Distances
PAM Spinach Rice Mosquito MonHumSpinach 00 849 1056 886Rice 849 00 1178 1225Mosquito 1056 1178 00 828MonHum 886 1225 828 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Next Cycle
PAM Spinach Rice MosMonHumSpinach 00 849 971Rice 849 00 1202MosMonHum 971 1202 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
Penultimate Cycle
PAM SpinRice MosMonHumSpinach 00 1087MosMonHum 1087 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
(Spin-Rice)-(Mos-(Mon-Hum))
Last Joining
Human
Monkey
MosquitoRice
Spinach
Unrooted Neighbor-Joining Tree
Multiple Alignment- First pairbull Align the two most closely-related sequences first
bull This alignment is then lsquofixedrsquo and will never change If a gap is to be introduced subsequently then it will be introduced in the same place in both sequences but their relative alignment remains unchanged
ClustalW- Decision timebull Consult the guide tree to see what alignment is performed next
ndash Align a third sequence to the first twoOrndash Align two entirely different sequences to each other
Option 1Option 1 Option 2Option 2
ClustalW- Alternative 1If the situation arises where a third sequence is aligned to the first two then when a gap has to be introduced to improve the alignment each of these two entities are treated as two single sequences
+
ClustalW- Alternative 2bull If on the other hand two separate
sequences have to be aligned together then the first pairwise alignment is placed to one side and the pairwise alignment of the other two is carried out
+
ClustalW- Progression
bull The alignment is progressively built up in this way with each step being treated as a pairwise alignment sometimes with each member of a lsquopairrsquo having more than one sequence
Progressive alignment - step 11 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
12345
Progressive alignment - step 21 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
12345
Progressive alignment - step 31 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
+
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
12345
Progressive alignment - final step1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
+
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
5 -ctcga-acgatacgatgactagct-
12345
ClustalW-Good pointsBad points
bull Advantagesndash Speed
bull Disadvantagesndash No objective functionndash No way of quantifying whether or not the alignment is good
ndash No way of knowing if the alignment is lsquocorrectrsquo
ClustalW-Local Minimumbull Potential problems
ndash Local minimum problem If an error is introduced early in the alignment process it is impossible to correct this later in the procedure
ndash Arbitrary alignment
Increasing the sophistication of the alignment process
bull Should we treat all the sequences in the same way - even though some sequences are closely-related and some sequences are distant relatives
bull Should we treat all positions in the sequences as though they were the same - even though they might have different functions and different locations in the 3-dimensional structure
ClustalW- Caveatsbull Sequence weightingbull Varying substitution matricesbull Residue-specific gap penalties and reduced penalties
in hydrophilic regions (external regions of protein sequences) encourage gaps in loops rather than in core regions
bull Positions in early alignments where gaps have been opened receive locally reduced gap penalties to encourage openings in subsequent alignments
ClustalW- User-supplied values
bull Two penalties are set by the user (there are default values but you should know that it is possible to change these)
bull GOP- Gap Opening Penalty is the cost of opening a gap in an alignment
bull GEP- Gap Extension Penalty is the cost of extending this gap
Position-Specific gap penaltiesbull Before any pair of (groups of) sequences are
aligned a table of GOPs are generated for each position in the two (sets of) sequences
bull The GOP is manipulated in a position-specific manner so that it can vary over the sequences
bull If there is a gap at a position the GOP and GEP penalties are lowered the other rules do not apply
bull This makes gaps more likely at positions where gaps already exist
Discouraging too many gaps bull If there is no gap opened then the GOP is increased if the
position is within 8 residues of an existing gapbull This discourages gaps that are too close togetherbull At any position within a run of hydrophilic residues the GOP is
decreasedbull These runs usually indicate loop regions in protein structuresbull A run of 5 hydrophilic residues is considered to be a hydrophilic
stretchbull The default hydrophilic residues are
ndash D E G K N Q P R Sndash But this can be changed by the user
Divergent Sequencesbull The most divergent sequences (most different on average
from all of the other sequences) are usually the most difficult to align
bull It is sometimes better to delay their aligment until later (when the easier sequences have already been aligned)
bull The user has the choice of setting a cutoff (default is 40 identity)
bull This will delay the alignment until the others have been aligned
Advice on progressive alignmentbull Progressive alignment is a mathematical process that is completely
independent of biological realitybull Can be a very good estimatebull Can be an impossibly poor estimatebull Requires user input and skillbull Treat cautiouslybull Can be improved by eye (usually)bull Often helps to have colour-codingbull Depending on the use the user should be able to make a judgement
on those regions that are reliable or notbull For phylogeny reconstruction only use those positions whose
hypothesis of positional homology is unimpeachable
Alignment of protein-coding DNA sequences
bull It is not very sensible to align the DNA sequences of protein-coding genes
ATGCTGTTAGGGATGACTCTGTTAGGG
ATG-CT--GTTAGGGATGACTCTGTTAGGG
The result might be highly-implausible and might not reflect what is known about biological processesIt is much more sensible to translate the sequences to their corresponding amino acid sequences align these protein sequences and then put the gaps in the DNA sequences according to where they are found in the amino acid alignment
Manual Alignment- softwareGDE- The Genetic Data Environment (UNIX)CINEMA- Java applet available from
ndash httpwwwbiochemuclacuk
SeqappSeqpup- MacPCUNIX available fromndash httpiubiobioindianaedu
SeAl for Macintosh available fromndash httpevolvezoooxacukSe-AlSe-Alhtml
BioEdit for PC available fromndash httpwwwmbioncsueduRNasePinfoprogramsBIOEDITbio
edithtml
- Multiple Sequence Alignment James McInerney bioinf4biologists Feb 2009
- Alignment can be easy or difficult
- Homology Definition
- Multiple Sequence Alignment- Goals
- Multiple sequence alignments - problems
- Slide 6
- Slide 7
- SSU rRNA
- Alignment of 16S rRNA can be guided by secondary structure
- Protein Alignment may be guided by Tertiary Structure Interactions
- Multiple Sequence Alignment- Methods
- Manual Alignment - reasons
- Local minimum
- Dotplots
- Dotplot example sperm whale vs human myg
- Slide 16
- Slide 17
- Dotplots in practice
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Dynamic programming
- Progressive Alignment
- Slide 23
- ClustalW- Pairwise Alignments
- Path Graph for aligning two sequences
- Possible alignment
- Alignment using this path
- Optimal Alignment 1
- Optimal Alignment 2
- Alignment of 3 sequences
- ClustalW- Guide Tree
- Neighbor joining method
- Distance Matrix
- First Step
- Calculation of New Distances
- Next Cycle
- Penultimate Cycle
- Last Joining
- Unrooted Neighbor-Joining Tree
- Multiple Alignment- First pair
- ClustalW- Decision time
- ClustalW- Alternative 1
- ClustalW- Progression
- Progressive alignment - step 1
- Progressive alignment - step 2
- Progressive alignment - step 3
- Progressive alignment - final step
- ClustalW-Good pointsBad points
- ClustalW-Local Minimum
- Increasing the sophistication of the alignment process
- Slide 51
- ClustalW- Caveats
- ClustalW- User-supplied values
- Position-Specific gap penalties
- Discouraging too many gaps
- Divergent Sequences
- Advice on progressive alignment
- Alignment of protein-coding DNA sequences
- Manual Alignment- software
-
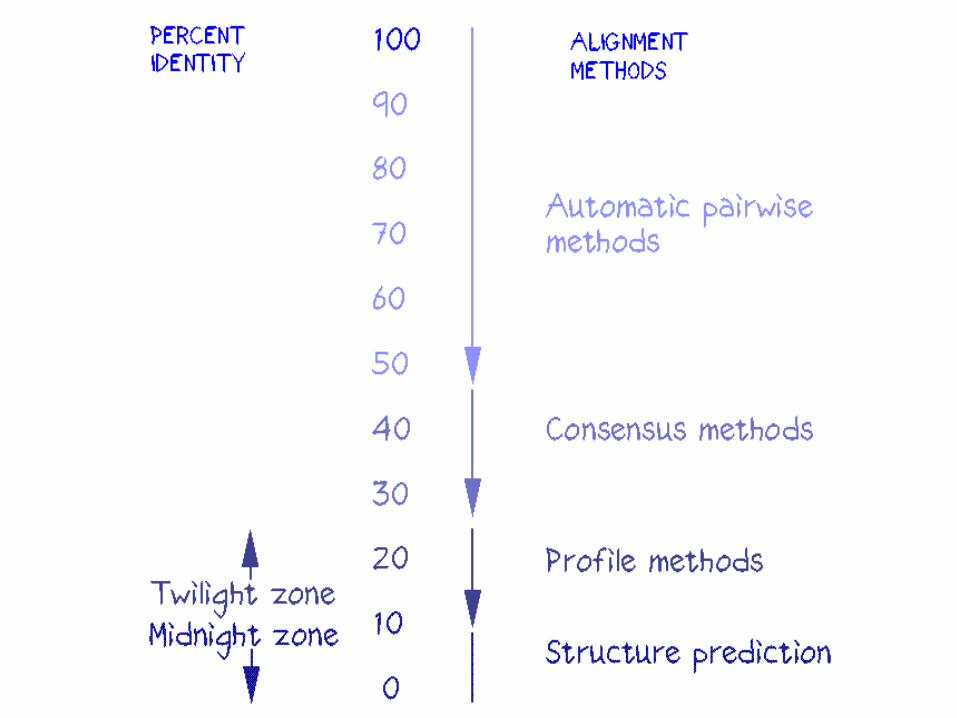
SSU rRNA
bull Structural RNA (not translated)bull Found in the small ribosomal subunitbull Widely-used for phylogeny reconstruction
(found in every species)bull Contains stem and loop structuresbull Stem structures usually conform to
watson-crick base pairing
Alignment of 16S rRNA can be guided by secondary structure
lt---------------(--------------------HELIX 19---------------------)lt---------------(22222222-000000-111111-00000-111111-0000-22222222Thermus ruber UCCGAUGC-UAAAGA-CCGAAG=CUCAA=CUUCGG=GGGU=GCGUUGGATh thermophilus UCCCAUGU-GAAAGA-CCACGG=CUCAA=CCGUGG=GGGA=GCGUGGGAEcoli UCAGAUGU-GAAAUC-CCCGGG=CUCAA=CCUGGG=AACU=GCAUCUGAAncystnidulans UCUGUUGU-CAAAGC-GUGGGG=CUCAA=CCUCAU=ACAG=GCAAUGGABsubtilis UCUGAUGU-GAAAGC-CCCCGG=CUCAA=CCGGGG=AGGG=UCAUUGGAChlaurantiacus UCGGCGCU-GAAAGC-GCCCCG=CUUAA=CGGGGC=GAGG=CGCGCCGAmatch
Alignment of 16S rRNA sequences from different bacteria
Protein Alignment may be guided Protein Alignment may be guided by Tertiary Structure Interactionsby Tertiary Structure Interactions
Homo sapiens DjlA protein
Escherichia coli DjlA protein
Multiple Sequence Alignment- Methods
ndash3 main methods of alignment
bull Manual (using custom-built text editors)bull Automatic (using custom-built alignment
software)bull Combined
Manual Alignment - reasonsbull Might be carried out because
ndash Alignment is easyndash There is some extraneous information (structural)
ndash Automated alignment methods have encountered the local minimum problem
ndash An automated alignment method can be ldquoimprovedrdquo
Local minimum
GARFIELDTHEFAT---CATGARFIELDTHEFATFATCAT
bull The dotplot provides a way of quickly visualizing the similarities between all parts of two sequences simultaneously
bull Lets consider a dotplot between sperm whale and human myoglobins
Dotplots
Sperm whale myoglobin
GLSDGEWQLV LNVWGKVEAD IPGHGQEVLI RLFKGHPETL EKFDKFKHLK SEDEMKASED LKKHGATVLT ALGGILKKKG HHEAEIKPLA QSHATKHKIP VKYLEFISEC IIQVLQSKHP GDFGADAQGA MNKALELFRK DMASNYKELG FQG
human myoglobin
VLSEGEWQLV LHVWAKVEAD VAGHGQDILI RLFKSHPETL EKFDRFKHLK TEAEMKASED LKKHGVTVLT ALGAILKKKG HHEAELKPLA QSHATKHKIP IKYLEFISEA IIHVLHSRHP GDFGADAQGA MNKALELFRK DIAAKYKELG YQG
bull Put one sequence on top
bull the other on the side
bull where residues are identical put a dot
bull Diagonal lines of dots show similarities
Dotplot example sperm whale vs human myg
Sperm whale myoglobin
G L S D G E W Q L V V L S E G E W Q L V
Human myoglobin
bullJust do the first 10 amino acids of eachbullMake a table with
ndashwhale sequence on top ndashhuman sequence on the side
bull This is the result for the whole sequence
bull It is easy to see that the diagonal is a line of dots
bull So sperm whale and human myoglobin are very similar
bull But the picture is noisy can smooth using a sliding window which considers neighbouring residues as well
Dotplot example sperm whale vs human myg
16
Sperm whale myoglobin
G L S D G E W Q L V V L S E G E W Q L V
Human myoglobin
bull can smooth noise using a sliding window which considers neighbouring residues as well
bull Have done this here can see the diagonal is highly similar
bull Also instead of using using a simple identity use a scoring matrix
Dotplot example sperm whale vs human myg
Dotplots in practicebull The best tool is an applet called dotlet
bull wwwisrecisb-sibchjavadotletDotlethtmlbull wwwbipbhamacukdotletDotlethtml
bull an applet is a program that runs in a web browser This means that you can produce dotplots within a netscapeIE window
bull Dotplots are often useful to identify things like repeated domains or duplications in big proteins
Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
bull Protein has many repeats bull SLIT_DROME (P24014)
MAAPSRTTLMPPPFRLQLRLLILPILLLLRHDAVHAEPYSGGFGSSAVSSGGLGSVGIHIPGGGVGVITEARCPRVCSCT GLNVDCSHRGLTSVPRKISADVERLELQGNNLTVIYETDFQRLTKLRMLQLTDNQIHTIERNSFQDLVSLERLDISNNVI TTVGRRVFKGAQSLRSLQLDNNQITCLDEHAFKGLVELEILTLNNNNLTSLPHNIFGGLGRLRALRLSDNPFACDCHLSW LSRFLRSATRLAPYTRCQSPSQLKGQNVADLHDQEFKCSGLTEHAPMECGAENSCPHPCRCADGIVDCREKSLTSVPVTL PDDTTDVRLEQNFITELPPKSFSSFRRLRRIDLSNNNISRIAHDALSGLKQLTTLVLYGNKIKDLPSGVFKGLGSLRLLL LNANEISCIRKDAFRDLHSLSLLSLYDNNIQSLANGTFDAMKSMKTVHLAKNPFICDCNLRWLADYLHKNPIETSGARCE SPKRMHRRRIESLREEKFKCSWGELRMKLSGECRMDSDCPAMCHCEGTTVDCTGRRLKEIPRDIPLHTTELLLNDNELGR ISSDGLFGRLPHLVKLELKRNQLTGIEPNAFEGASHIQELQLGENKIKEISNKMFLGLHQLKTLNLYDNQISCVMPGSFE HLNSLTSLNLASNPFNCNCHLAWFAECVRKKSLNGGAARCGAPSKVRDVQIKDLPHSEFKCSSENSEGCLGDGYCPPSCT CTGTVVACSRNQLKEIPRGIPAETSELYLESNEIEQIHYERIRHLRSLTRLDLSNNQITILSNYTFANLTKLSTLIISYN KLQCLQRHALSGLNNLRVVSLHGNRISMLPEGSFEDLKSLTHIALGSNPLYCDCGLKWFSDWIKLDYVEPGIARCAEPEQ MKDKLILSTPSSSFVCRGRVRNDILAKCNACFEQPCQNQAQCVALPQREYQCLCQPGYHGKHCEFMIDACYGNPCRNNAT CTVLEEGRFSCQCAPGYTGARCETNIDDCLGEIKCQNNATCIDGVESYKCECQPGFSGEFCDTKIQFCSPEFNPCANGAK CMDHFTHYSCDCQAGFHGTNCTDNIDDCQNHMCQNGGTCVDGINDYQCRCPDDYTGKYCEGHNMISMMYPQTSPCQNHEC KHGVCFQPNAQGSDYLCRCHPGYTGKWCEYLTSISFVHNNSFVELEPLRTRPEANVTIVFSSAEQNGILMYDGQDAHLAV ELFNGRIRVSYDVGNHPVSTMYSFEMVADGKYHAVELLAIKKNFTLRVDRGLARSIINEGSNDYLKLTTPMFLGGLPVDP AQQAYKNWQIRNLTSFKGCMKEVWINHKLVDFGNAQRQQKITPGCALLEGEQQEEEDDEQDFMDETPHIKEEPVDPCLEN KCRRGSRCVPNSNARDGYQCKCKHGQRGRYCDQGEGSTEPPTVTAASTCRKEQVREYYTENDCRSRQPLKYAKCVGGCGN QCCAAKIVRRRKVRMVCSNNRKYIKNLDIVRKCGCTKKCY
bull Perform a dotplot of the SLIT protein against itself wwwbiobhamacukdotletDotlethtml
Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
20Swiss-prot entry
For further discussion of dotplot see Attwood and Parry-Smith p116-8
Dynamic programming2 methodsbull Dynamic programming
ndash Consider 2 protein sequences of 100 amino acids in lengthndash If it takes 1002 seconds to exhaustively align these sequences
then it will take 1003 seconds to align 3 sequences 1004 to align 4 sequencesetc
ndash More time than the universe has existed to align 20 sequences exhaustively
bull Progressive alignment
Progressive Alignmentbull Devised by Feng and Doolittle in 1987bull Essentially a heuristic method and as such
is not guaranteed to find the lsquooptimalrsquo alignment
bull Requires n-1+n-2+n-3n-n+1 pairwise alignments as a starting point
bull Most successful implementation is Clustal (Des Higgins) This software is cited 3000 times per year in the scientific literature
Overview of ClustalW Procedure
1 PEEKSAVTALWGKVN--VDEVGG2 GEEKAAVLALWDKVN--EEEVGG3 PADKTNVKAAWGKVGAHAGEYGA4 AADKTNVKAAWSKVGGHAGEYGA5 EHEWQLVLHVWAKVEADVAGHGQ
Hbb_Human 1 -Hbb_Horse 2 17 -Hba_Human 3 59 60 -Hba_Horse 4 59 59 13 -Myg_Whale 5 77 77 75 75 -
Hbb_Human
Hbb_Horse
Hba_Horse
Hba_Human
Myg_Whale
2
1
3 4
2
1
3 4
alpha-helices
Quick pairwise alignment calculate distance matrix
Neighbor-joining tree(guide tree)
Progressive alignment following guide tree
CLUSTAL W
ClustalW- Pairwise Alignments
bull First perform all possible pairwise alignments between each pair of sequences There are (n-1)+(n-2)(n-n+1) possibilities
bull Calculate the lsquodistancersquo between each pair of sequences based on these isolated pairwise alignments
bull Generate a distance matrix
Path Graph for aligning two sequences
Possible alignment
1
1
0
1
0
-1
Scoring SchemebullMatch +1bullMismatch 0bullIndel -1
Score for this path= 2
Alignment using this path
GATTC-GAATTC
1
1
0
1
0
-1
Optimal Alignment 1
1
1
-1
1
1
1
Alignment score 4Alignment score 4
Alignment using this path
GA-TTCGA-TTCGAATTCGAATTC
Optimal Alignment 2
1
-1
1
1
1
1
Alignment score 4Alignment score 4
Alignment using this path
G-ATTCG-ATTCGAATTCGAATTC
Alignment of 3 sequences
ClustalW- Guide Tree
bull Generate a Neighbor-Joining lsquoguide treersquo from these pairwise distances
bull This guide tree gives the order in which the progressive alignment will be carried out
Neighbor joining method
bullThe neighbor-joining method is a greedy heuristic which joins at each step the two closest sub-trees that are not already joinedbullIt is based on the minimum evolution principlebullneighbors are defined as two taxa that are connected by a single node in an unrooted tree
A B
Node 1
PAM Spinach Rice Mosquito Monkey HumanSpinach 00 849 1056 908 863Rice 849 00 1178 1224 1226Mosquito 1056 1178 00 847 808Monkey 908 1224 847 00 33Human 863 1226 808 33 00
What is required for the Neighbour joining method
Distance matrixDistance Matrix
PAM distance 33 (Human - Monkey) is the minimum So well join Human and Monkey to MonHum and well calculate the new distances
Mon-Hum
MonkeyHumanSpinachMosquito Rice
First Step
After we have joined two species in a subtree we have to compute the distances from every other node to the new subtree We do this with a simple average of distancesDist[Spinach MonHum]
= (Dist[Spinach Monkey] + Dist[Spinach Human])2 = (908 + 863)2 = 8855
Mon-Hum
MonkeyHumanSpinach
Calculation of New Distances
PAM Spinach Rice Mosquito MonHumSpinach 00 849 1056 886Rice 849 00 1178 1225Mosquito 1056 1178 00 828MonHum 886 1225 828 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Next Cycle
PAM Spinach Rice MosMonHumSpinach 00 849 971Rice 849 00 1202MosMonHum 971 1202 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
Penultimate Cycle
PAM SpinRice MosMonHumSpinach 00 1087MosMonHum 1087 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
(Spin-Rice)-(Mos-(Mon-Hum))
Last Joining
Human
Monkey
MosquitoRice
Spinach
Unrooted Neighbor-Joining Tree
Multiple Alignment- First pairbull Align the two most closely-related sequences first
bull This alignment is then lsquofixedrsquo and will never change If a gap is to be introduced subsequently then it will be introduced in the same place in both sequences but their relative alignment remains unchanged
ClustalW- Decision timebull Consult the guide tree to see what alignment is performed next
ndash Align a third sequence to the first twoOrndash Align two entirely different sequences to each other
Option 1Option 1 Option 2Option 2
ClustalW- Alternative 1If the situation arises where a third sequence is aligned to the first two then when a gap has to be introduced to improve the alignment each of these two entities are treated as two single sequences
+
ClustalW- Alternative 2bull If on the other hand two separate
sequences have to be aligned together then the first pairwise alignment is placed to one side and the pairwise alignment of the other two is carried out
+
ClustalW- Progression
bull The alignment is progressively built up in this way with each step being treated as a pairwise alignment sometimes with each member of a lsquopairrsquo having more than one sequence
Progressive alignment - step 11 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
12345
Progressive alignment - step 21 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
12345
Progressive alignment - step 31 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
+
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
12345
Progressive alignment - final step1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
+
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
5 -ctcga-acgatacgatgactagct-
12345
ClustalW-Good pointsBad points
bull Advantagesndash Speed
bull Disadvantagesndash No objective functionndash No way of quantifying whether or not the alignment is good
ndash No way of knowing if the alignment is lsquocorrectrsquo
ClustalW-Local Minimumbull Potential problems
ndash Local minimum problem If an error is introduced early in the alignment process it is impossible to correct this later in the procedure
ndash Arbitrary alignment
Increasing the sophistication of the alignment process
bull Should we treat all the sequences in the same way - even though some sequences are closely-related and some sequences are distant relatives
bull Should we treat all positions in the sequences as though they were the same - even though they might have different functions and different locations in the 3-dimensional structure
ClustalW- Caveatsbull Sequence weightingbull Varying substitution matricesbull Residue-specific gap penalties and reduced penalties
in hydrophilic regions (external regions of protein sequences) encourage gaps in loops rather than in core regions
bull Positions in early alignments where gaps have been opened receive locally reduced gap penalties to encourage openings in subsequent alignments
ClustalW- User-supplied values
bull Two penalties are set by the user (there are default values but you should know that it is possible to change these)
bull GOP- Gap Opening Penalty is the cost of opening a gap in an alignment
bull GEP- Gap Extension Penalty is the cost of extending this gap
Position-Specific gap penaltiesbull Before any pair of (groups of) sequences are
aligned a table of GOPs are generated for each position in the two (sets of) sequences
bull The GOP is manipulated in a position-specific manner so that it can vary over the sequences
bull If there is a gap at a position the GOP and GEP penalties are lowered the other rules do not apply
bull This makes gaps more likely at positions where gaps already exist
Discouraging too many gaps bull If there is no gap opened then the GOP is increased if the
position is within 8 residues of an existing gapbull This discourages gaps that are too close togetherbull At any position within a run of hydrophilic residues the GOP is
decreasedbull These runs usually indicate loop regions in protein structuresbull A run of 5 hydrophilic residues is considered to be a hydrophilic
stretchbull The default hydrophilic residues are
ndash D E G K N Q P R Sndash But this can be changed by the user
Divergent Sequencesbull The most divergent sequences (most different on average
from all of the other sequences) are usually the most difficult to align
bull It is sometimes better to delay their aligment until later (when the easier sequences have already been aligned)
bull The user has the choice of setting a cutoff (default is 40 identity)
bull This will delay the alignment until the others have been aligned
Advice on progressive alignmentbull Progressive alignment is a mathematical process that is completely
independent of biological realitybull Can be a very good estimatebull Can be an impossibly poor estimatebull Requires user input and skillbull Treat cautiouslybull Can be improved by eye (usually)bull Often helps to have colour-codingbull Depending on the use the user should be able to make a judgement
on those regions that are reliable or notbull For phylogeny reconstruction only use those positions whose
hypothesis of positional homology is unimpeachable
Alignment of protein-coding DNA sequences
bull It is not very sensible to align the DNA sequences of protein-coding genes
ATGCTGTTAGGGATGACTCTGTTAGGG
ATG-CT--GTTAGGGATGACTCTGTTAGGG
The result might be highly-implausible and might not reflect what is known about biological processesIt is much more sensible to translate the sequences to their corresponding amino acid sequences align these protein sequences and then put the gaps in the DNA sequences according to where they are found in the amino acid alignment
Manual Alignment- softwareGDE- The Genetic Data Environment (UNIX)CINEMA- Java applet available from
ndash httpwwwbiochemuclacuk
SeqappSeqpup- MacPCUNIX available fromndash httpiubiobioindianaedu
SeAl for Macintosh available fromndash httpevolvezoooxacukSe-AlSe-Alhtml
BioEdit for PC available fromndash httpwwwmbioncsueduRNasePinfoprogramsBIOEDITbio
edithtml
- Multiple Sequence Alignment James McInerney bioinf4biologists Feb 2009
- Alignment can be easy or difficult
- Homology Definition
- Multiple Sequence Alignment- Goals
- Multiple sequence alignments - problems
- Slide 6
- Slide 7
- SSU rRNA
- Alignment of 16S rRNA can be guided by secondary structure
- Protein Alignment may be guided by Tertiary Structure Interactions
- Multiple Sequence Alignment- Methods
- Manual Alignment - reasons
- Local minimum
- Dotplots
- Dotplot example sperm whale vs human myg
- Slide 16
- Slide 17
- Dotplots in practice
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Dynamic programming
- Progressive Alignment
- Slide 23
- ClustalW- Pairwise Alignments
- Path Graph for aligning two sequences
- Possible alignment
- Alignment using this path
- Optimal Alignment 1
- Optimal Alignment 2
- Alignment of 3 sequences
- ClustalW- Guide Tree
- Neighbor joining method
- Distance Matrix
- First Step
- Calculation of New Distances
- Next Cycle
- Penultimate Cycle
- Last Joining
- Unrooted Neighbor-Joining Tree
- Multiple Alignment- First pair
- ClustalW- Decision time
- ClustalW- Alternative 1
- ClustalW- Progression
- Progressive alignment - step 1
- Progressive alignment - step 2
- Progressive alignment - step 3
- Progressive alignment - final step
- ClustalW-Good pointsBad points
- ClustalW-Local Minimum
- Increasing the sophistication of the alignment process
- Slide 51
- ClustalW- Caveats
- ClustalW- User-supplied values
- Position-Specific gap penalties
- Discouraging too many gaps
- Divergent Sequences
- Advice on progressive alignment
- Alignment of protein-coding DNA sequences
- Manual Alignment- software
-

Alignment of 16S rRNA can be guided by secondary structure
lt---------------(--------------------HELIX 19---------------------)lt---------------(22222222-000000-111111-00000-111111-0000-22222222Thermus ruber UCCGAUGC-UAAAGA-CCGAAG=CUCAA=CUUCGG=GGGU=GCGUUGGATh thermophilus UCCCAUGU-GAAAGA-CCACGG=CUCAA=CCGUGG=GGGA=GCGUGGGAEcoli UCAGAUGU-GAAAUC-CCCGGG=CUCAA=CCUGGG=AACU=GCAUCUGAAncystnidulans UCUGUUGU-CAAAGC-GUGGGG=CUCAA=CCUCAU=ACAG=GCAAUGGABsubtilis UCUGAUGU-GAAAGC-CCCCGG=CUCAA=CCGGGG=AGGG=UCAUUGGAChlaurantiacus UCGGCGCU-GAAAGC-GCCCCG=CUUAA=CGGGGC=GAGG=CGCGCCGAmatch
Alignment of 16S rRNA sequences from different bacteria
Protein Alignment may be guided Protein Alignment may be guided by Tertiary Structure Interactionsby Tertiary Structure Interactions
Homo sapiens DjlA protein
Escherichia coli DjlA protein
Multiple Sequence Alignment- Methods
ndash3 main methods of alignment
bull Manual (using custom-built text editors)bull Automatic (using custom-built alignment
software)bull Combined
Manual Alignment - reasonsbull Might be carried out because
ndash Alignment is easyndash There is some extraneous information (structural)
ndash Automated alignment methods have encountered the local minimum problem
ndash An automated alignment method can be ldquoimprovedrdquo
Local minimum
GARFIELDTHEFAT---CATGARFIELDTHEFATFATCAT
bull The dotplot provides a way of quickly visualizing the similarities between all parts of two sequences simultaneously
bull Lets consider a dotplot between sperm whale and human myoglobins
Dotplots
Sperm whale myoglobin
GLSDGEWQLV LNVWGKVEAD IPGHGQEVLI RLFKGHPETL EKFDKFKHLK SEDEMKASED LKKHGATVLT ALGGILKKKG HHEAEIKPLA QSHATKHKIP VKYLEFISEC IIQVLQSKHP GDFGADAQGA MNKALELFRK DMASNYKELG FQG
human myoglobin
VLSEGEWQLV LHVWAKVEAD VAGHGQDILI RLFKSHPETL EKFDRFKHLK TEAEMKASED LKKHGVTVLT ALGAILKKKG HHEAELKPLA QSHATKHKIP IKYLEFISEA IIHVLHSRHP GDFGADAQGA MNKALELFRK DIAAKYKELG YQG
bull Put one sequence on top
bull the other on the side
bull where residues are identical put a dot
bull Diagonal lines of dots show similarities
Dotplot example sperm whale vs human myg
Sperm whale myoglobin
G L S D G E W Q L V V L S E G E W Q L V
Human myoglobin
bullJust do the first 10 amino acids of eachbullMake a table with
ndashwhale sequence on top ndashhuman sequence on the side
bull This is the result for the whole sequence
bull It is easy to see that the diagonal is a line of dots
bull So sperm whale and human myoglobin are very similar
bull But the picture is noisy can smooth using a sliding window which considers neighbouring residues as well
Dotplot example sperm whale vs human myg
16
Sperm whale myoglobin
G L S D G E W Q L V V L S E G E W Q L V
Human myoglobin
bull can smooth noise using a sliding window which considers neighbouring residues as well
bull Have done this here can see the diagonal is highly similar
bull Also instead of using using a simple identity use a scoring matrix
Dotplot example sperm whale vs human myg
Dotplots in practicebull The best tool is an applet called dotlet
bull wwwisrecisb-sibchjavadotletDotlethtmlbull wwwbipbhamacukdotletDotlethtml
bull an applet is a program that runs in a web browser This means that you can produce dotplots within a netscapeIE window
bull Dotplots are often useful to identify things like repeated domains or duplications in big proteins
Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
bull Protein has many repeats bull SLIT_DROME (P24014)
MAAPSRTTLMPPPFRLQLRLLILPILLLLRHDAVHAEPYSGGFGSSAVSSGGLGSVGIHIPGGGVGVITEARCPRVCSCT GLNVDCSHRGLTSVPRKISADVERLELQGNNLTVIYETDFQRLTKLRMLQLTDNQIHTIERNSFQDLVSLERLDISNNVI TTVGRRVFKGAQSLRSLQLDNNQITCLDEHAFKGLVELEILTLNNNNLTSLPHNIFGGLGRLRALRLSDNPFACDCHLSW LSRFLRSATRLAPYTRCQSPSQLKGQNVADLHDQEFKCSGLTEHAPMECGAENSCPHPCRCADGIVDCREKSLTSVPVTL PDDTTDVRLEQNFITELPPKSFSSFRRLRRIDLSNNNISRIAHDALSGLKQLTTLVLYGNKIKDLPSGVFKGLGSLRLLL LNANEISCIRKDAFRDLHSLSLLSLYDNNIQSLANGTFDAMKSMKTVHLAKNPFICDCNLRWLADYLHKNPIETSGARCE SPKRMHRRRIESLREEKFKCSWGELRMKLSGECRMDSDCPAMCHCEGTTVDCTGRRLKEIPRDIPLHTTELLLNDNELGR ISSDGLFGRLPHLVKLELKRNQLTGIEPNAFEGASHIQELQLGENKIKEISNKMFLGLHQLKTLNLYDNQISCVMPGSFE HLNSLTSLNLASNPFNCNCHLAWFAECVRKKSLNGGAARCGAPSKVRDVQIKDLPHSEFKCSSENSEGCLGDGYCPPSCT CTGTVVACSRNQLKEIPRGIPAETSELYLESNEIEQIHYERIRHLRSLTRLDLSNNQITILSNYTFANLTKLSTLIISYN KLQCLQRHALSGLNNLRVVSLHGNRISMLPEGSFEDLKSLTHIALGSNPLYCDCGLKWFSDWIKLDYVEPGIARCAEPEQ MKDKLILSTPSSSFVCRGRVRNDILAKCNACFEQPCQNQAQCVALPQREYQCLCQPGYHGKHCEFMIDACYGNPCRNNAT CTVLEEGRFSCQCAPGYTGARCETNIDDCLGEIKCQNNATCIDGVESYKCECQPGFSGEFCDTKIQFCSPEFNPCANGAK CMDHFTHYSCDCQAGFHGTNCTDNIDDCQNHMCQNGGTCVDGINDYQCRCPDDYTGKYCEGHNMISMMYPQTSPCQNHEC KHGVCFQPNAQGSDYLCRCHPGYTGKWCEYLTSISFVHNNSFVELEPLRTRPEANVTIVFSSAEQNGILMYDGQDAHLAV ELFNGRIRVSYDVGNHPVSTMYSFEMVADGKYHAVELLAIKKNFTLRVDRGLARSIINEGSNDYLKLTTPMFLGGLPVDP AQQAYKNWQIRNLTSFKGCMKEVWINHKLVDFGNAQRQQKITPGCALLEGEQQEEEDDEQDFMDETPHIKEEPVDPCLEN KCRRGSRCVPNSNARDGYQCKCKHGQRGRYCDQGEGSTEPPTVTAASTCRKEQVREYYTENDCRSRQPLKYAKCVGGCGN QCCAAKIVRRRKVRMVCSNNRKYIKNLDIVRKCGCTKKCY
bull Perform a dotplot of the SLIT protein against itself wwwbiobhamacukdotletDotlethtml
Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
20Swiss-prot entry
For further discussion of dotplot see Attwood and Parry-Smith p116-8
Dynamic programming2 methodsbull Dynamic programming
ndash Consider 2 protein sequences of 100 amino acids in lengthndash If it takes 1002 seconds to exhaustively align these sequences
then it will take 1003 seconds to align 3 sequences 1004 to align 4 sequencesetc
ndash More time than the universe has existed to align 20 sequences exhaustively
bull Progressive alignment
Progressive Alignmentbull Devised by Feng and Doolittle in 1987bull Essentially a heuristic method and as such
is not guaranteed to find the lsquooptimalrsquo alignment
bull Requires n-1+n-2+n-3n-n+1 pairwise alignments as a starting point
bull Most successful implementation is Clustal (Des Higgins) This software is cited 3000 times per year in the scientific literature
Overview of ClustalW Procedure
1 PEEKSAVTALWGKVN--VDEVGG2 GEEKAAVLALWDKVN--EEEVGG3 PADKTNVKAAWGKVGAHAGEYGA4 AADKTNVKAAWSKVGGHAGEYGA5 EHEWQLVLHVWAKVEADVAGHGQ
Hbb_Human 1 -Hbb_Horse 2 17 -Hba_Human 3 59 60 -Hba_Horse 4 59 59 13 -Myg_Whale 5 77 77 75 75 -
Hbb_Human
Hbb_Horse
Hba_Horse
Hba_Human
Myg_Whale
2
1
3 4
2
1
3 4
alpha-helices
Quick pairwise alignment calculate distance matrix
Neighbor-joining tree(guide tree)
Progressive alignment following guide tree
CLUSTAL W
ClustalW- Pairwise Alignments
bull First perform all possible pairwise alignments between each pair of sequences There are (n-1)+(n-2)(n-n+1) possibilities
bull Calculate the lsquodistancersquo between each pair of sequences based on these isolated pairwise alignments
bull Generate a distance matrix
Path Graph for aligning two sequences
Possible alignment
1
1
0
1
0
-1
Scoring SchemebullMatch +1bullMismatch 0bullIndel -1
Score for this path= 2
Alignment using this path
GATTC-GAATTC
1
1
0
1
0
-1
Optimal Alignment 1
1
1
-1
1
1
1
Alignment score 4Alignment score 4
Alignment using this path
GA-TTCGA-TTCGAATTCGAATTC
Optimal Alignment 2
1
-1
1
1
1
1
Alignment score 4Alignment score 4
Alignment using this path
G-ATTCG-ATTCGAATTCGAATTC
Alignment of 3 sequences
ClustalW- Guide Tree
bull Generate a Neighbor-Joining lsquoguide treersquo from these pairwise distances
bull This guide tree gives the order in which the progressive alignment will be carried out
Neighbor joining method
bullThe neighbor-joining method is a greedy heuristic which joins at each step the two closest sub-trees that are not already joinedbullIt is based on the minimum evolution principlebullneighbors are defined as two taxa that are connected by a single node in an unrooted tree
A B
Node 1
PAM Spinach Rice Mosquito Monkey HumanSpinach 00 849 1056 908 863Rice 849 00 1178 1224 1226Mosquito 1056 1178 00 847 808Monkey 908 1224 847 00 33Human 863 1226 808 33 00
What is required for the Neighbour joining method
Distance matrixDistance Matrix
PAM distance 33 (Human - Monkey) is the minimum So well join Human and Monkey to MonHum and well calculate the new distances
Mon-Hum
MonkeyHumanSpinachMosquito Rice
First Step
After we have joined two species in a subtree we have to compute the distances from every other node to the new subtree We do this with a simple average of distancesDist[Spinach MonHum]
= (Dist[Spinach Monkey] + Dist[Spinach Human])2 = (908 + 863)2 = 8855
Mon-Hum
MonkeyHumanSpinach
Calculation of New Distances
PAM Spinach Rice Mosquito MonHumSpinach 00 849 1056 886Rice 849 00 1178 1225Mosquito 1056 1178 00 828MonHum 886 1225 828 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Next Cycle
PAM Spinach Rice MosMonHumSpinach 00 849 971Rice 849 00 1202MosMonHum 971 1202 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
Penultimate Cycle
PAM SpinRice MosMonHumSpinach 00 1087MosMonHum 1087 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
(Spin-Rice)-(Mos-(Mon-Hum))
Last Joining
Human
Monkey
MosquitoRice
Spinach
Unrooted Neighbor-Joining Tree
Multiple Alignment- First pairbull Align the two most closely-related sequences first
bull This alignment is then lsquofixedrsquo and will never change If a gap is to be introduced subsequently then it will be introduced in the same place in both sequences but their relative alignment remains unchanged
ClustalW- Decision timebull Consult the guide tree to see what alignment is performed next
ndash Align a third sequence to the first twoOrndash Align two entirely different sequences to each other
Option 1Option 1 Option 2Option 2
ClustalW- Alternative 1If the situation arises where a third sequence is aligned to the first two then when a gap has to be introduced to improve the alignment each of these two entities are treated as two single sequences
+
ClustalW- Alternative 2bull If on the other hand two separate
sequences have to be aligned together then the first pairwise alignment is placed to one side and the pairwise alignment of the other two is carried out
+
ClustalW- Progression
bull The alignment is progressively built up in this way with each step being treated as a pairwise alignment sometimes with each member of a lsquopairrsquo having more than one sequence
Progressive alignment - step 11 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
12345
Progressive alignment - step 21 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
12345
Progressive alignment - step 31 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
+
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
12345
Progressive alignment - final step1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
+
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
5 -ctcga-acgatacgatgactagct-
12345
ClustalW-Good pointsBad points
bull Advantagesndash Speed
bull Disadvantagesndash No objective functionndash No way of quantifying whether or not the alignment is good
ndash No way of knowing if the alignment is lsquocorrectrsquo
ClustalW-Local Minimumbull Potential problems
ndash Local minimum problem If an error is introduced early in the alignment process it is impossible to correct this later in the procedure
ndash Arbitrary alignment
Increasing the sophistication of the alignment process
bull Should we treat all the sequences in the same way - even though some sequences are closely-related and some sequences are distant relatives
bull Should we treat all positions in the sequences as though they were the same - even though they might have different functions and different locations in the 3-dimensional structure
ClustalW- Caveatsbull Sequence weightingbull Varying substitution matricesbull Residue-specific gap penalties and reduced penalties
in hydrophilic regions (external regions of protein sequences) encourage gaps in loops rather than in core regions
bull Positions in early alignments where gaps have been opened receive locally reduced gap penalties to encourage openings in subsequent alignments
ClustalW- User-supplied values
bull Two penalties are set by the user (there are default values but you should know that it is possible to change these)
bull GOP- Gap Opening Penalty is the cost of opening a gap in an alignment
bull GEP- Gap Extension Penalty is the cost of extending this gap
Position-Specific gap penaltiesbull Before any pair of (groups of) sequences are
aligned a table of GOPs are generated for each position in the two (sets of) sequences
bull The GOP is manipulated in a position-specific manner so that it can vary over the sequences
bull If there is a gap at a position the GOP and GEP penalties are lowered the other rules do not apply
bull This makes gaps more likely at positions where gaps already exist
Discouraging too many gaps bull If there is no gap opened then the GOP is increased if the
position is within 8 residues of an existing gapbull This discourages gaps that are too close togetherbull At any position within a run of hydrophilic residues the GOP is
decreasedbull These runs usually indicate loop regions in protein structuresbull A run of 5 hydrophilic residues is considered to be a hydrophilic
stretchbull The default hydrophilic residues are
ndash D E G K N Q P R Sndash But this can be changed by the user
Divergent Sequencesbull The most divergent sequences (most different on average
from all of the other sequences) are usually the most difficult to align
bull It is sometimes better to delay their aligment until later (when the easier sequences have already been aligned)
bull The user has the choice of setting a cutoff (default is 40 identity)
bull This will delay the alignment until the others have been aligned
Advice on progressive alignmentbull Progressive alignment is a mathematical process that is completely
independent of biological realitybull Can be a very good estimatebull Can be an impossibly poor estimatebull Requires user input and skillbull Treat cautiouslybull Can be improved by eye (usually)bull Often helps to have colour-codingbull Depending on the use the user should be able to make a judgement
on those regions that are reliable or notbull For phylogeny reconstruction only use those positions whose
hypothesis of positional homology is unimpeachable
Alignment of protein-coding DNA sequences
bull It is not very sensible to align the DNA sequences of protein-coding genes
ATGCTGTTAGGGATGACTCTGTTAGGG
ATG-CT--GTTAGGGATGACTCTGTTAGGG
The result might be highly-implausible and might not reflect what is known about biological processesIt is much more sensible to translate the sequences to their corresponding amino acid sequences align these protein sequences and then put the gaps in the DNA sequences according to where they are found in the amino acid alignment
Manual Alignment- softwareGDE- The Genetic Data Environment (UNIX)CINEMA- Java applet available from
ndash httpwwwbiochemuclacuk
SeqappSeqpup- MacPCUNIX available fromndash httpiubiobioindianaedu
SeAl for Macintosh available fromndash httpevolvezoooxacukSe-AlSe-Alhtml
BioEdit for PC available fromndash httpwwwmbioncsueduRNasePinfoprogramsBIOEDITbio
edithtml
- Multiple Sequence Alignment James McInerney bioinf4biologists Feb 2009
- Alignment can be easy or difficult
- Homology Definition
- Multiple Sequence Alignment- Goals
- Multiple sequence alignments - problems
- Slide 6
- Slide 7
- SSU rRNA
- Alignment of 16S rRNA can be guided by secondary structure
- Protein Alignment may be guided by Tertiary Structure Interactions
- Multiple Sequence Alignment- Methods
- Manual Alignment - reasons
- Local minimum
- Dotplots
- Dotplot example sperm whale vs human myg
- Slide 16
- Slide 17
- Dotplots in practice
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Dynamic programming
- Progressive Alignment
- Slide 23
- ClustalW- Pairwise Alignments
- Path Graph for aligning two sequences
- Possible alignment
- Alignment using this path
- Optimal Alignment 1
- Optimal Alignment 2
- Alignment of 3 sequences
- ClustalW- Guide Tree
- Neighbor joining method
- Distance Matrix
- First Step
- Calculation of New Distances
- Next Cycle
- Penultimate Cycle
- Last Joining
- Unrooted Neighbor-Joining Tree
- Multiple Alignment- First pair
- ClustalW- Decision time
- ClustalW- Alternative 1
- ClustalW- Progression
- Progressive alignment - step 1
- Progressive alignment - step 2
- Progressive alignment - step 3
- Progressive alignment - final step
- ClustalW-Good pointsBad points
- ClustalW-Local Minimum
- Increasing the sophistication of the alignment process
- Slide 51
- ClustalW- Caveats
- ClustalW- User-supplied values
- Position-Specific gap penalties
- Discouraging too many gaps
- Divergent Sequences
- Advice on progressive alignment
- Alignment of protein-coding DNA sequences
- Manual Alignment- software
-

Protein Alignment may be guided Protein Alignment may be guided by Tertiary Structure Interactionsby Tertiary Structure Interactions
Homo sapiens DjlA protein
Escherichia coli DjlA protein
Multiple Sequence Alignment- Methods
ndash3 main methods of alignment
bull Manual (using custom-built text editors)bull Automatic (using custom-built alignment
software)bull Combined
Manual Alignment - reasonsbull Might be carried out because
ndash Alignment is easyndash There is some extraneous information (structural)
ndash Automated alignment methods have encountered the local minimum problem
ndash An automated alignment method can be ldquoimprovedrdquo
Local minimum
GARFIELDTHEFAT---CATGARFIELDTHEFATFATCAT
bull The dotplot provides a way of quickly visualizing the similarities between all parts of two sequences simultaneously
bull Lets consider a dotplot between sperm whale and human myoglobins
Dotplots
Sperm whale myoglobin
GLSDGEWQLV LNVWGKVEAD IPGHGQEVLI RLFKGHPETL EKFDKFKHLK SEDEMKASED LKKHGATVLT ALGGILKKKG HHEAEIKPLA QSHATKHKIP VKYLEFISEC IIQVLQSKHP GDFGADAQGA MNKALELFRK DMASNYKELG FQG
human myoglobin
VLSEGEWQLV LHVWAKVEAD VAGHGQDILI RLFKSHPETL EKFDRFKHLK TEAEMKASED LKKHGVTVLT ALGAILKKKG HHEAELKPLA QSHATKHKIP IKYLEFISEA IIHVLHSRHP GDFGADAQGA MNKALELFRK DIAAKYKELG YQG
bull Put one sequence on top
bull the other on the side
bull where residues are identical put a dot
bull Diagonal lines of dots show similarities
Dotplot example sperm whale vs human myg
Sperm whale myoglobin
G L S D G E W Q L V V L S E G E W Q L V
Human myoglobin
bullJust do the first 10 amino acids of eachbullMake a table with
ndashwhale sequence on top ndashhuman sequence on the side
bull This is the result for the whole sequence
bull It is easy to see that the diagonal is a line of dots
bull So sperm whale and human myoglobin are very similar
bull But the picture is noisy can smooth using a sliding window which considers neighbouring residues as well
Dotplot example sperm whale vs human myg
16
Sperm whale myoglobin
G L S D G E W Q L V V L S E G E W Q L V
Human myoglobin
bull can smooth noise using a sliding window which considers neighbouring residues as well
bull Have done this here can see the diagonal is highly similar
bull Also instead of using using a simple identity use a scoring matrix
Dotplot example sperm whale vs human myg
Dotplots in practicebull The best tool is an applet called dotlet
bull wwwisrecisb-sibchjavadotletDotlethtmlbull wwwbipbhamacukdotletDotlethtml
bull an applet is a program that runs in a web browser This means that you can produce dotplots within a netscapeIE window
bull Dotplots are often useful to identify things like repeated domains or duplications in big proteins
Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
bull Protein has many repeats bull SLIT_DROME (P24014)
MAAPSRTTLMPPPFRLQLRLLILPILLLLRHDAVHAEPYSGGFGSSAVSSGGLGSVGIHIPGGGVGVITEARCPRVCSCT GLNVDCSHRGLTSVPRKISADVERLELQGNNLTVIYETDFQRLTKLRMLQLTDNQIHTIERNSFQDLVSLERLDISNNVI TTVGRRVFKGAQSLRSLQLDNNQITCLDEHAFKGLVELEILTLNNNNLTSLPHNIFGGLGRLRALRLSDNPFACDCHLSW LSRFLRSATRLAPYTRCQSPSQLKGQNVADLHDQEFKCSGLTEHAPMECGAENSCPHPCRCADGIVDCREKSLTSVPVTL PDDTTDVRLEQNFITELPPKSFSSFRRLRRIDLSNNNISRIAHDALSGLKQLTTLVLYGNKIKDLPSGVFKGLGSLRLLL LNANEISCIRKDAFRDLHSLSLLSLYDNNIQSLANGTFDAMKSMKTVHLAKNPFICDCNLRWLADYLHKNPIETSGARCE SPKRMHRRRIESLREEKFKCSWGELRMKLSGECRMDSDCPAMCHCEGTTVDCTGRRLKEIPRDIPLHTTELLLNDNELGR ISSDGLFGRLPHLVKLELKRNQLTGIEPNAFEGASHIQELQLGENKIKEISNKMFLGLHQLKTLNLYDNQISCVMPGSFE HLNSLTSLNLASNPFNCNCHLAWFAECVRKKSLNGGAARCGAPSKVRDVQIKDLPHSEFKCSSENSEGCLGDGYCPPSCT CTGTVVACSRNQLKEIPRGIPAETSELYLESNEIEQIHYERIRHLRSLTRLDLSNNQITILSNYTFANLTKLSTLIISYN KLQCLQRHALSGLNNLRVVSLHGNRISMLPEGSFEDLKSLTHIALGSNPLYCDCGLKWFSDWIKLDYVEPGIARCAEPEQ MKDKLILSTPSSSFVCRGRVRNDILAKCNACFEQPCQNQAQCVALPQREYQCLCQPGYHGKHCEFMIDACYGNPCRNNAT CTVLEEGRFSCQCAPGYTGARCETNIDDCLGEIKCQNNATCIDGVESYKCECQPGFSGEFCDTKIQFCSPEFNPCANGAK CMDHFTHYSCDCQAGFHGTNCTDNIDDCQNHMCQNGGTCVDGINDYQCRCPDDYTGKYCEGHNMISMMYPQTSPCQNHEC KHGVCFQPNAQGSDYLCRCHPGYTGKWCEYLTSISFVHNNSFVELEPLRTRPEANVTIVFSSAEQNGILMYDGQDAHLAV ELFNGRIRVSYDVGNHPVSTMYSFEMVADGKYHAVELLAIKKNFTLRVDRGLARSIINEGSNDYLKLTTPMFLGGLPVDP AQQAYKNWQIRNLTSFKGCMKEVWINHKLVDFGNAQRQQKITPGCALLEGEQQEEEDDEQDFMDETPHIKEEPVDPCLEN KCRRGSRCVPNSNARDGYQCKCKHGQRGRYCDQGEGSTEPPTVTAASTCRKEQVREYYTENDCRSRQPLKYAKCVGGCGN QCCAAKIVRRRKVRMVCSNNRKYIKNLDIVRKCGCTKKCY
bull Perform a dotplot of the SLIT protein against itself wwwbiobhamacukdotletDotlethtml
Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
20Swiss-prot entry
For further discussion of dotplot see Attwood and Parry-Smith p116-8
Dynamic programming2 methodsbull Dynamic programming
ndash Consider 2 protein sequences of 100 amino acids in lengthndash If it takes 1002 seconds to exhaustively align these sequences
then it will take 1003 seconds to align 3 sequences 1004 to align 4 sequencesetc
ndash More time than the universe has existed to align 20 sequences exhaustively
bull Progressive alignment
Progressive Alignmentbull Devised by Feng and Doolittle in 1987bull Essentially a heuristic method and as such
is not guaranteed to find the lsquooptimalrsquo alignment
bull Requires n-1+n-2+n-3n-n+1 pairwise alignments as a starting point
bull Most successful implementation is Clustal (Des Higgins) This software is cited 3000 times per year in the scientific literature
Overview of ClustalW Procedure
1 PEEKSAVTALWGKVN--VDEVGG2 GEEKAAVLALWDKVN--EEEVGG3 PADKTNVKAAWGKVGAHAGEYGA4 AADKTNVKAAWSKVGGHAGEYGA5 EHEWQLVLHVWAKVEADVAGHGQ
Hbb_Human 1 -Hbb_Horse 2 17 -Hba_Human 3 59 60 -Hba_Horse 4 59 59 13 -Myg_Whale 5 77 77 75 75 -
Hbb_Human
Hbb_Horse
Hba_Horse
Hba_Human
Myg_Whale
2
1
3 4
2
1
3 4
alpha-helices
Quick pairwise alignment calculate distance matrix
Neighbor-joining tree(guide tree)
Progressive alignment following guide tree
CLUSTAL W
ClustalW- Pairwise Alignments
bull First perform all possible pairwise alignments between each pair of sequences There are (n-1)+(n-2)(n-n+1) possibilities
bull Calculate the lsquodistancersquo between each pair of sequences based on these isolated pairwise alignments
bull Generate a distance matrix
Path Graph for aligning two sequences
Possible alignment
1
1
0
1
0
-1
Scoring SchemebullMatch +1bullMismatch 0bullIndel -1
Score for this path= 2
Alignment using this path
GATTC-GAATTC
1
1
0
1
0
-1
Optimal Alignment 1
1
1
-1
1
1
1
Alignment score 4Alignment score 4
Alignment using this path
GA-TTCGA-TTCGAATTCGAATTC
Optimal Alignment 2
1
-1
1
1
1
1
Alignment score 4Alignment score 4
Alignment using this path
G-ATTCG-ATTCGAATTCGAATTC
Alignment of 3 sequences
ClustalW- Guide Tree
bull Generate a Neighbor-Joining lsquoguide treersquo from these pairwise distances
bull This guide tree gives the order in which the progressive alignment will be carried out
Neighbor joining method
bullThe neighbor-joining method is a greedy heuristic which joins at each step the two closest sub-trees that are not already joinedbullIt is based on the minimum evolution principlebullneighbors are defined as two taxa that are connected by a single node in an unrooted tree
A B
Node 1
PAM Spinach Rice Mosquito Monkey HumanSpinach 00 849 1056 908 863Rice 849 00 1178 1224 1226Mosquito 1056 1178 00 847 808Monkey 908 1224 847 00 33Human 863 1226 808 33 00
What is required for the Neighbour joining method
Distance matrixDistance Matrix
PAM distance 33 (Human - Monkey) is the minimum So well join Human and Monkey to MonHum and well calculate the new distances
Mon-Hum
MonkeyHumanSpinachMosquito Rice
First Step
After we have joined two species in a subtree we have to compute the distances from every other node to the new subtree We do this with a simple average of distancesDist[Spinach MonHum]
= (Dist[Spinach Monkey] + Dist[Spinach Human])2 = (908 + 863)2 = 8855
Mon-Hum
MonkeyHumanSpinach
Calculation of New Distances
PAM Spinach Rice Mosquito MonHumSpinach 00 849 1056 886Rice 849 00 1178 1225Mosquito 1056 1178 00 828MonHum 886 1225 828 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Next Cycle
PAM Spinach Rice MosMonHumSpinach 00 849 971Rice 849 00 1202MosMonHum 971 1202 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
Penultimate Cycle
PAM SpinRice MosMonHumSpinach 00 1087MosMonHum 1087 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
(Spin-Rice)-(Mos-(Mon-Hum))
Last Joining
Human
Monkey
MosquitoRice
Spinach
Unrooted Neighbor-Joining Tree
Multiple Alignment- First pairbull Align the two most closely-related sequences first
bull This alignment is then lsquofixedrsquo and will never change If a gap is to be introduced subsequently then it will be introduced in the same place in both sequences but their relative alignment remains unchanged
ClustalW- Decision timebull Consult the guide tree to see what alignment is performed next
ndash Align a third sequence to the first twoOrndash Align two entirely different sequences to each other
Option 1Option 1 Option 2Option 2
ClustalW- Alternative 1If the situation arises where a third sequence is aligned to the first two then when a gap has to be introduced to improve the alignment each of these two entities are treated as two single sequences
+
ClustalW- Alternative 2bull If on the other hand two separate
sequences have to be aligned together then the first pairwise alignment is placed to one side and the pairwise alignment of the other two is carried out
+
ClustalW- Progression
bull The alignment is progressively built up in this way with each step being treated as a pairwise alignment sometimes with each member of a lsquopairrsquo having more than one sequence
Progressive alignment - step 11 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
12345
Progressive alignment - step 21 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
12345
Progressive alignment - step 31 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
+
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
12345
Progressive alignment - final step1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
+
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
5 -ctcga-acgatacgatgactagct-
12345
ClustalW-Good pointsBad points
bull Advantagesndash Speed
bull Disadvantagesndash No objective functionndash No way of quantifying whether or not the alignment is good
ndash No way of knowing if the alignment is lsquocorrectrsquo
ClustalW-Local Minimumbull Potential problems
ndash Local minimum problem If an error is introduced early in the alignment process it is impossible to correct this later in the procedure
ndash Arbitrary alignment
Increasing the sophistication of the alignment process
bull Should we treat all the sequences in the same way - even though some sequences are closely-related and some sequences are distant relatives
bull Should we treat all positions in the sequences as though they were the same - even though they might have different functions and different locations in the 3-dimensional structure
ClustalW- Caveatsbull Sequence weightingbull Varying substitution matricesbull Residue-specific gap penalties and reduced penalties
in hydrophilic regions (external regions of protein sequences) encourage gaps in loops rather than in core regions
bull Positions in early alignments where gaps have been opened receive locally reduced gap penalties to encourage openings in subsequent alignments
ClustalW- User-supplied values
bull Two penalties are set by the user (there are default values but you should know that it is possible to change these)
bull GOP- Gap Opening Penalty is the cost of opening a gap in an alignment
bull GEP- Gap Extension Penalty is the cost of extending this gap
Position-Specific gap penaltiesbull Before any pair of (groups of) sequences are
aligned a table of GOPs are generated for each position in the two (sets of) sequences
bull The GOP is manipulated in a position-specific manner so that it can vary over the sequences
bull If there is a gap at a position the GOP and GEP penalties are lowered the other rules do not apply
bull This makes gaps more likely at positions where gaps already exist
Discouraging too many gaps bull If there is no gap opened then the GOP is increased if the
position is within 8 residues of an existing gapbull This discourages gaps that are too close togetherbull At any position within a run of hydrophilic residues the GOP is
decreasedbull These runs usually indicate loop regions in protein structuresbull A run of 5 hydrophilic residues is considered to be a hydrophilic
stretchbull The default hydrophilic residues are
ndash D E G K N Q P R Sndash But this can be changed by the user
Divergent Sequencesbull The most divergent sequences (most different on average
from all of the other sequences) are usually the most difficult to align
bull It is sometimes better to delay their aligment until later (when the easier sequences have already been aligned)
bull The user has the choice of setting a cutoff (default is 40 identity)
bull This will delay the alignment until the others have been aligned
Advice on progressive alignmentbull Progressive alignment is a mathematical process that is completely
independent of biological realitybull Can be a very good estimatebull Can be an impossibly poor estimatebull Requires user input and skillbull Treat cautiouslybull Can be improved by eye (usually)bull Often helps to have colour-codingbull Depending on the use the user should be able to make a judgement
on those regions that are reliable or notbull For phylogeny reconstruction only use those positions whose
hypothesis of positional homology is unimpeachable
Alignment of protein-coding DNA sequences
bull It is not very sensible to align the DNA sequences of protein-coding genes
ATGCTGTTAGGGATGACTCTGTTAGGG
ATG-CT--GTTAGGGATGACTCTGTTAGGG
The result might be highly-implausible and might not reflect what is known about biological processesIt is much more sensible to translate the sequences to their corresponding amino acid sequences align these protein sequences and then put the gaps in the DNA sequences according to where they are found in the amino acid alignment
Manual Alignment- softwareGDE- The Genetic Data Environment (UNIX)CINEMA- Java applet available from
ndash httpwwwbiochemuclacuk
SeqappSeqpup- MacPCUNIX available fromndash httpiubiobioindianaedu
SeAl for Macintosh available fromndash httpevolvezoooxacukSe-AlSe-Alhtml
BioEdit for PC available fromndash httpwwwmbioncsueduRNasePinfoprogramsBIOEDITbio
edithtml
- Multiple Sequence Alignment James McInerney bioinf4biologists Feb 2009
- Alignment can be easy or difficult
- Homology Definition
- Multiple Sequence Alignment- Goals
- Multiple sequence alignments - problems
- Slide 6
- Slide 7
- SSU rRNA
- Alignment of 16S rRNA can be guided by secondary structure
- Protein Alignment may be guided by Tertiary Structure Interactions
- Multiple Sequence Alignment- Methods
- Manual Alignment - reasons
- Local minimum
- Dotplots
- Dotplot example sperm whale vs human myg
- Slide 16
- Slide 17
- Dotplots in practice
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Dynamic programming
- Progressive Alignment
- Slide 23
- ClustalW- Pairwise Alignments
- Path Graph for aligning two sequences
- Possible alignment
- Alignment using this path
- Optimal Alignment 1
- Optimal Alignment 2
- Alignment of 3 sequences
- ClustalW- Guide Tree
- Neighbor joining method
- Distance Matrix
- First Step
- Calculation of New Distances
- Next Cycle
- Penultimate Cycle
- Last Joining
- Unrooted Neighbor-Joining Tree
- Multiple Alignment- First pair
- ClustalW- Decision time
- ClustalW- Alternative 1
- ClustalW- Progression
- Progressive alignment - step 1
- Progressive alignment - step 2
- Progressive alignment - step 3
- Progressive alignment - final step
- ClustalW-Good pointsBad points
- ClustalW-Local Minimum
- Increasing the sophistication of the alignment process
- Slide 51
- ClustalW- Caveats
- ClustalW- User-supplied values
- Position-Specific gap penalties
- Discouraging too many gaps
- Divergent Sequences
- Advice on progressive alignment
- Alignment of protein-coding DNA sequences
- Manual Alignment- software
-

Multiple Sequence Alignment- Methods
ndash3 main methods of alignment
bull Manual (using custom-built text editors)bull Automatic (using custom-built alignment
software)bull Combined
Manual Alignment - reasonsbull Might be carried out because
ndash Alignment is easyndash There is some extraneous information (structural)
ndash Automated alignment methods have encountered the local minimum problem
ndash An automated alignment method can be ldquoimprovedrdquo
Local minimum
GARFIELDTHEFAT---CATGARFIELDTHEFATFATCAT
bull The dotplot provides a way of quickly visualizing the similarities between all parts of two sequences simultaneously
bull Lets consider a dotplot between sperm whale and human myoglobins
Dotplots
Sperm whale myoglobin
GLSDGEWQLV LNVWGKVEAD IPGHGQEVLI RLFKGHPETL EKFDKFKHLK SEDEMKASED LKKHGATVLT ALGGILKKKG HHEAEIKPLA QSHATKHKIP VKYLEFISEC IIQVLQSKHP GDFGADAQGA MNKALELFRK DMASNYKELG FQG
human myoglobin
VLSEGEWQLV LHVWAKVEAD VAGHGQDILI RLFKSHPETL EKFDRFKHLK TEAEMKASED LKKHGVTVLT ALGAILKKKG HHEAELKPLA QSHATKHKIP IKYLEFISEA IIHVLHSRHP GDFGADAQGA MNKALELFRK DIAAKYKELG YQG
bull Put one sequence on top
bull the other on the side
bull where residues are identical put a dot
bull Diagonal lines of dots show similarities
Dotplot example sperm whale vs human myg
Sperm whale myoglobin
G L S D G E W Q L V V L S E G E W Q L V
Human myoglobin
bullJust do the first 10 amino acids of eachbullMake a table with
ndashwhale sequence on top ndashhuman sequence on the side
bull This is the result for the whole sequence
bull It is easy to see that the diagonal is a line of dots
bull So sperm whale and human myoglobin are very similar
bull But the picture is noisy can smooth using a sliding window which considers neighbouring residues as well
Dotplot example sperm whale vs human myg
16
Sperm whale myoglobin
G L S D G E W Q L V V L S E G E W Q L V
Human myoglobin
bull can smooth noise using a sliding window which considers neighbouring residues as well
bull Have done this here can see the diagonal is highly similar
bull Also instead of using using a simple identity use a scoring matrix
Dotplot example sperm whale vs human myg
Dotplots in practicebull The best tool is an applet called dotlet
bull wwwisrecisb-sibchjavadotletDotlethtmlbull wwwbipbhamacukdotletDotlethtml
bull an applet is a program that runs in a web browser This means that you can produce dotplots within a netscapeIE window
bull Dotplots are often useful to identify things like repeated domains or duplications in big proteins
Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
bull Protein has many repeats bull SLIT_DROME (P24014)
MAAPSRTTLMPPPFRLQLRLLILPILLLLRHDAVHAEPYSGGFGSSAVSSGGLGSVGIHIPGGGVGVITEARCPRVCSCT GLNVDCSHRGLTSVPRKISADVERLELQGNNLTVIYETDFQRLTKLRMLQLTDNQIHTIERNSFQDLVSLERLDISNNVI TTVGRRVFKGAQSLRSLQLDNNQITCLDEHAFKGLVELEILTLNNNNLTSLPHNIFGGLGRLRALRLSDNPFACDCHLSW LSRFLRSATRLAPYTRCQSPSQLKGQNVADLHDQEFKCSGLTEHAPMECGAENSCPHPCRCADGIVDCREKSLTSVPVTL PDDTTDVRLEQNFITELPPKSFSSFRRLRRIDLSNNNISRIAHDALSGLKQLTTLVLYGNKIKDLPSGVFKGLGSLRLLL LNANEISCIRKDAFRDLHSLSLLSLYDNNIQSLANGTFDAMKSMKTVHLAKNPFICDCNLRWLADYLHKNPIETSGARCE SPKRMHRRRIESLREEKFKCSWGELRMKLSGECRMDSDCPAMCHCEGTTVDCTGRRLKEIPRDIPLHTTELLLNDNELGR ISSDGLFGRLPHLVKLELKRNQLTGIEPNAFEGASHIQELQLGENKIKEISNKMFLGLHQLKTLNLYDNQISCVMPGSFE HLNSLTSLNLASNPFNCNCHLAWFAECVRKKSLNGGAARCGAPSKVRDVQIKDLPHSEFKCSSENSEGCLGDGYCPPSCT CTGTVVACSRNQLKEIPRGIPAETSELYLESNEIEQIHYERIRHLRSLTRLDLSNNQITILSNYTFANLTKLSTLIISYN KLQCLQRHALSGLNNLRVVSLHGNRISMLPEGSFEDLKSLTHIALGSNPLYCDCGLKWFSDWIKLDYVEPGIARCAEPEQ MKDKLILSTPSSSFVCRGRVRNDILAKCNACFEQPCQNQAQCVALPQREYQCLCQPGYHGKHCEFMIDACYGNPCRNNAT CTVLEEGRFSCQCAPGYTGARCETNIDDCLGEIKCQNNATCIDGVESYKCECQPGFSGEFCDTKIQFCSPEFNPCANGAK CMDHFTHYSCDCQAGFHGTNCTDNIDDCQNHMCQNGGTCVDGINDYQCRCPDDYTGKYCEGHNMISMMYPQTSPCQNHEC KHGVCFQPNAQGSDYLCRCHPGYTGKWCEYLTSISFVHNNSFVELEPLRTRPEANVTIVFSSAEQNGILMYDGQDAHLAV ELFNGRIRVSYDVGNHPVSTMYSFEMVADGKYHAVELLAIKKNFTLRVDRGLARSIINEGSNDYLKLTTPMFLGGLPVDP AQQAYKNWQIRNLTSFKGCMKEVWINHKLVDFGNAQRQQKITPGCALLEGEQQEEEDDEQDFMDETPHIKEEPVDPCLEN KCRRGSRCVPNSNARDGYQCKCKHGQRGRYCDQGEGSTEPPTVTAASTCRKEQVREYYTENDCRSRQPLKYAKCVGGCGN QCCAAKIVRRRKVRMVCSNNRKYIKNLDIVRKCGCTKKCY
bull Perform a dotplot of the SLIT protein against itself wwwbiobhamacukdotletDotlethtml
Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
20Swiss-prot entry
For further discussion of dotplot see Attwood and Parry-Smith p116-8
Dynamic programming2 methodsbull Dynamic programming
ndash Consider 2 protein sequences of 100 amino acids in lengthndash If it takes 1002 seconds to exhaustively align these sequences
then it will take 1003 seconds to align 3 sequences 1004 to align 4 sequencesetc
ndash More time than the universe has existed to align 20 sequences exhaustively
bull Progressive alignment
Progressive Alignmentbull Devised by Feng and Doolittle in 1987bull Essentially a heuristic method and as such
is not guaranteed to find the lsquooptimalrsquo alignment
bull Requires n-1+n-2+n-3n-n+1 pairwise alignments as a starting point
bull Most successful implementation is Clustal (Des Higgins) This software is cited 3000 times per year in the scientific literature
Overview of ClustalW Procedure
1 PEEKSAVTALWGKVN--VDEVGG2 GEEKAAVLALWDKVN--EEEVGG3 PADKTNVKAAWGKVGAHAGEYGA4 AADKTNVKAAWSKVGGHAGEYGA5 EHEWQLVLHVWAKVEADVAGHGQ
Hbb_Human 1 -Hbb_Horse 2 17 -Hba_Human 3 59 60 -Hba_Horse 4 59 59 13 -Myg_Whale 5 77 77 75 75 -
Hbb_Human
Hbb_Horse
Hba_Horse
Hba_Human
Myg_Whale
2
1
3 4
2
1
3 4
alpha-helices
Quick pairwise alignment calculate distance matrix
Neighbor-joining tree(guide tree)
Progressive alignment following guide tree
CLUSTAL W
ClustalW- Pairwise Alignments
bull First perform all possible pairwise alignments between each pair of sequences There are (n-1)+(n-2)(n-n+1) possibilities
bull Calculate the lsquodistancersquo between each pair of sequences based on these isolated pairwise alignments
bull Generate a distance matrix
Path Graph for aligning two sequences
Possible alignment
1
1
0
1
0
-1
Scoring SchemebullMatch +1bullMismatch 0bullIndel -1
Score for this path= 2
Alignment using this path
GATTC-GAATTC
1
1
0
1
0
-1
Optimal Alignment 1
1
1
-1
1
1
1
Alignment score 4Alignment score 4
Alignment using this path
GA-TTCGA-TTCGAATTCGAATTC
Optimal Alignment 2
1
-1
1
1
1
1
Alignment score 4Alignment score 4
Alignment using this path
G-ATTCG-ATTCGAATTCGAATTC
Alignment of 3 sequences
ClustalW- Guide Tree
bull Generate a Neighbor-Joining lsquoguide treersquo from these pairwise distances
bull This guide tree gives the order in which the progressive alignment will be carried out
Neighbor joining method
bullThe neighbor-joining method is a greedy heuristic which joins at each step the two closest sub-trees that are not already joinedbullIt is based on the minimum evolution principlebullneighbors are defined as two taxa that are connected by a single node in an unrooted tree
A B
Node 1
PAM Spinach Rice Mosquito Monkey HumanSpinach 00 849 1056 908 863Rice 849 00 1178 1224 1226Mosquito 1056 1178 00 847 808Monkey 908 1224 847 00 33Human 863 1226 808 33 00
What is required for the Neighbour joining method
Distance matrixDistance Matrix
PAM distance 33 (Human - Monkey) is the minimum So well join Human and Monkey to MonHum and well calculate the new distances
Mon-Hum
MonkeyHumanSpinachMosquito Rice
First Step
After we have joined two species in a subtree we have to compute the distances from every other node to the new subtree We do this with a simple average of distancesDist[Spinach MonHum]
= (Dist[Spinach Monkey] + Dist[Spinach Human])2 = (908 + 863)2 = 8855
Mon-Hum
MonkeyHumanSpinach
Calculation of New Distances
PAM Spinach Rice Mosquito MonHumSpinach 00 849 1056 886Rice 849 00 1178 1225Mosquito 1056 1178 00 828MonHum 886 1225 828 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Next Cycle
PAM Spinach Rice MosMonHumSpinach 00 849 971Rice 849 00 1202MosMonHum 971 1202 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
Penultimate Cycle
PAM SpinRice MosMonHumSpinach 00 1087MosMonHum 1087 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
(Spin-Rice)-(Mos-(Mon-Hum))
Last Joining
Human
Monkey
MosquitoRice
Spinach
Unrooted Neighbor-Joining Tree
Multiple Alignment- First pairbull Align the two most closely-related sequences first
bull This alignment is then lsquofixedrsquo and will never change If a gap is to be introduced subsequently then it will be introduced in the same place in both sequences but their relative alignment remains unchanged
ClustalW- Decision timebull Consult the guide tree to see what alignment is performed next
ndash Align a third sequence to the first twoOrndash Align two entirely different sequences to each other
Option 1Option 1 Option 2Option 2
ClustalW- Alternative 1If the situation arises where a third sequence is aligned to the first two then when a gap has to be introduced to improve the alignment each of these two entities are treated as two single sequences
+
ClustalW- Alternative 2bull If on the other hand two separate
sequences have to be aligned together then the first pairwise alignment is placed to one side and the pairwise alignment of the other two is carried out
+
ClustalW- Progression
bull The alignment is progressively built up in this way with each step being treated as a pairwise alignment sometimes with each member of a lsquopairrsquo having more than one sequence
Progressive alignment - step 11 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
12345
Progressive alignment - step 21 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
12345
Progressive alignment - step 31 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
+
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
12345
Progressive alignment - final step1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
+
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
5 -ctcga-acgatacgatgactagct-
12345
ClustalW-Good pointsBad points
bull Advantagesndash Speed
bull Disadvantagesndash No objective functionndash No way of quantifying whether or not the alignment is good
ndash No way of knowing if the alignment is lsquocorrectrsquo
ClustalW-Local Minimumbull Potential problems
ndash Local minimum problem If an error is introduced early in the alignment process it is impossible to correct this later in the procedure
ndash Arbitrary alignment
Increasing the sophistication of the alignment process
bull Should we treat all the sequences in the same way - even though some sequences are closely-related and some sequences are distant relatives
bull Should we treat all positions in the sequences as though they were the same - even though they might have different functions and different locations in the 3-dimensional structure
ClustalW- Caveatsbull Sequence weightingbull Varying substitution matricesbull Residue-specific gap penalties and reduced penalties
in hydrophilic regions (external regions of protein sequences) encourage gaps in loops rather than in core regions
bull Positions in early alignments where gaps have been opened receive locally reduced gap penalties to encourage openings in subsequent alignments
ClustalW- User-supplied values
bull Two penalties are set by the user (there are default values but you should know that it is possible to change these)
bull GOP- Gap Opening Penalty is the cost of opening a gap in an alignment
bull GEP- Gap Extension Penalty is the cost of extending this gap
Position-Specific gap penaltiesbull Before any pair of (groups of) sequences are
aligned a table of GOPs are generated for each position in the two (sets of) sequences
bull The GOP is manipulated in a position-specific manner so that it can vary over the sequences
bull If there is a gap at a position the GOP and GEP penalties are lowered the other rules do not apply
bull This makes gaps more likely at positions where gaps already exist
Discouraging too many gaps bull If there is no gap opened then the GOP is increased if the
position is within 8 residues of an existing gapbull This discourages gaps that are too close togetherbull At any position within a run of hydrophilic residues the GOP is
decreasedbull These runs usually indicate loop regions in protein structuresbull A run of 5 hydrophilic residues is considered to be a hydrophilic
stretchbull The default hydrophilic residues are
ndash D E G K N Q P R Sndash But this can be changed by the user
Divergent Sequencesbull The most divergent sequences (most different on average
from all of the other sequences) are usually the most difficult to align
bull It is sometimes better to delay their aligment until later (when the easier sequences have already been aligned)
bull The user has the choice of setting a cutoff (default is 40 identity)
bull This will delay the alignment until the others have been aligned
Advice on progressive alignmentbull Progressive alignment is a mathematical process that is completely
independent of biological realitybull Can be a very good estimatebull Can be an impossibly poor estimatebull Requires user input and skillbull Treat cautiouslybull Can be improved by eye (usually)bull Often helps to have colour-codingbull Depending on the use the user should be able to make a judgement
on those regions that are reliable or notbull For phylogeny reconstruction only use those positions whose
hypothesis of positional homology is unimpeachable
Alignment of protein-coding DNA sequences
bull It is not very sensible to align the DNA sequences of protein-coding genes
ATGCTGTTAGGGATGACTCTGTTAGGG
ATG-CT--GTTAGGGATGACTCTGTTAGGG
The result might be highly-implausible and might not reflect what is known about biological processesIt is much more sensible to translate the sequences to their corresponding amino acid sequences align these protein sequences and then put the gaps in the DNA sequences according to where they are found in the amino acid alignment
Manual Alignment- softwareGDE- The Genetic Data Environment (UNIX)CINEMA- Java applet available from
ndash httpwwwbiochemuclacuk
SeqappSeqpup- MacPCUNIX available fromndash httpiubiobioindianaedu
SeAl for Macintosh available fromndash httpevolvezoooxacukSe-AlSe-Alhtml
BioEdit for PC available fromndash httpwwwmbioncsueduRNasePinfoprogramsBIOEDITbio
edithtml
- Multiple Sequence Alignment James McInerney bioinf4biologists Feb 2009
- Alignment can be easy or difficult
- Homology Definition
- Multiple Sequence Alignment- Goals
- Multiple sequence alignments - problems
- Slide 6
- Slide 7
- SSU rRNA
- Alignment of 16S rRNA can be guided by secondary structure
- Protein Alignment may be guided by Tertiary Structure Interactions
- Multiple Sequence Alignment- Methods
- Manual Alignment - reasons
- Local minimum
- Dotplots
- Dotplot example sperm whale vs human myg
- Slide 16
- Slide 17
- Dotplots in practice
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Dynamic programming
- Progressive Alignment
- Slide 23
- ClustalW- Pairwise Alignments
- Path Graph for aligning two sequences
- Possible alignment
- Alignment using this path
- Optimal Alignment 1
- Optimal Alignment 2
- Alignment of 3 sequences
- ClustalW- Guide Tree
- Neighbor joining method
- Distance Matrix
- First Step
- Calculation of New Distances
- Next Cycle
- Penultimate Cycle
- Last Joining
- Unrooted Neighbor-Joining Tree
- Multiple Alignment- First pair
- ClustalW- Decision time
- ClustalW- Alternative 1
- ClustalW- Progression
- Progressive alignment - step 1
- Progressive alignment - step 2
- Progressive alignment - step 3
- Progressive alignment - final step
- ClustalW-Good pointsBad points
- ClustalW-Local Minimum
- Increasing the sophistication of the alignment process
- Slide 51
- ClustalW- Caveats
- ClustalW- User-supplied values
- Position-Specific gap penalties
- Discouraging too many gaps
- Divergent Sequences
- Advice on progressive alignment
- Alignment of protein-coding DNA sequences
- Manual Alignment- software
-
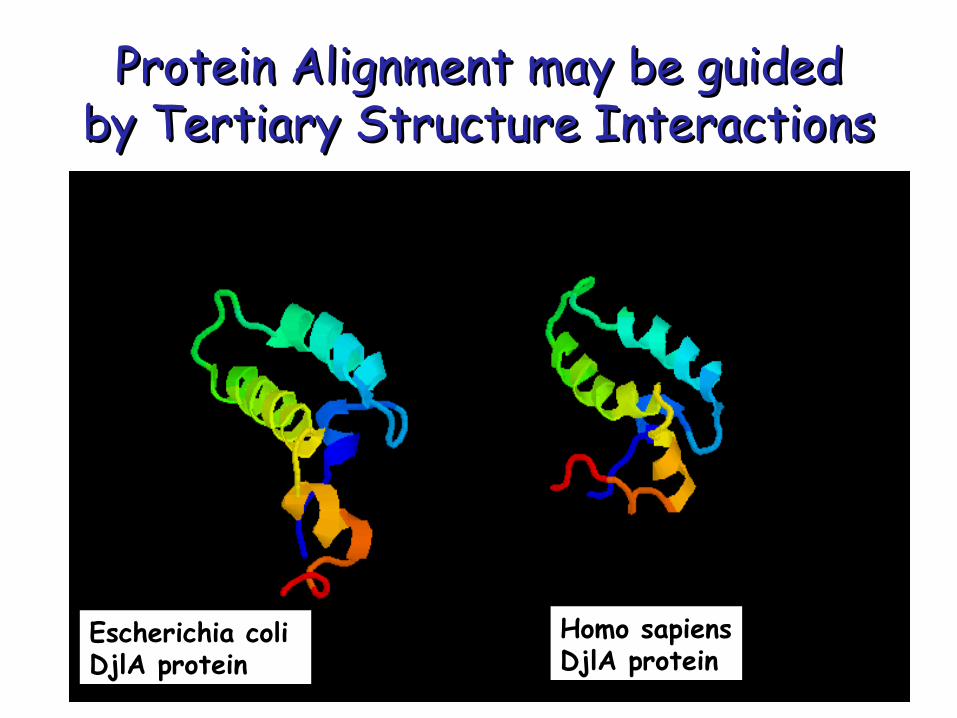
Manual Alignment - reasonsbull Might be carried out because
ndash Alignment is easyndash There is some extraneous information (structural)
ndash Automated alignment methods have encountered the local minimum problem
ndash An automated alignment method can be ldquoimprovedrdquo
Local minimum
GARFIELDTHEFAT---CATGARFIELDTHEFATFATCAT
bull The dotplot provides a way of quickly visualizing the similarities between all parts of two sequences simultaneously
bull Lets consider a dotplot between sperm whale and human myoglobins
Dotplots
Sperm whale myoglobin
GLSDGEWQLV LNVWGKVEAD IPGHGQEVLI RLFKGHPETL EKFDKFKHLK SEDEMKASED LKKHGATVLT ALGGILKKKG HHEAEIKPLA QSHATKHKIP VKYLEFISEC IIQVLQSKHP GDFGADAQGA MNKALELFRK DMASNYKELG FQG
human myoglobin
VLSEGEWQLV LHVWAKVEAD VAGHGQDILI RLFKSHPETL EKFDRFKHLK TEAEMKASED LKKHGVTVLT ALGAILKKKG HHEAELKPLA QSHATKHKIP IKYLEFISEA IIHVLHSRHP GDFGADAQGA MNKALELFRK DIAAKYKELG YQG
bull Put one sequence on top
bull the other on the side
bull where residues are identical put a dot
bull Diagonal lines of dots show similarities
Dotplot example sperm whale vs human myg
Sperm whale myoglobin
G L S D G E W Q L V V L S E G E W Q L V
Human myoglobin
bullJust do the first 10 amino acids of eachbullMake a table with
ndashwhale sequence on top ndashhuman sequence on the side
bull This is the result for the whole sequence
bull It is easy to see that the diagonal is a line of dots
bull So sperm whale and human myoglobin are very similar
bull But the picture is noisy can smooth using a sliding window which considers neighbouring residues as well
Dotplot example sperm whale vs human myg
16
Sperm whale myoglobin
G L S D G E W Q L V V L S E G E W Q L V
Human myoglobin
bull can smooth noise using a sliding window which considers neighbouring residues as well
bull Have done this here can see the diagonal is highly similar
bull Also instead of using using a simple identity use a scoring matrix
Dotplot example sperm whale vs human myg
Dotplots in practicebull The best tool is an applet called dotlet
bull wwwisrecisb-sibchjavadotletDotlethtmlbull wwwbipbhamacukdotletDotlethtml
bull an applet is a program that runs in a web browser This means that you can produce dotplots within a netscapeIE window
bull Dotplots are often useful to identify things like repeated domains or duplications in big proteins
Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
bull Protein has many repeats bull SLIT_DROME (P24014)
MAAPSRTTLMPPPFRLQLRLLILPILLLLRHDAVHAEPYSGGFGSSAVSSGGLGSVGIHIPGGGVGVITEARCPRVCSCT GLNVDCSHRGLTSVPRKISADVERLELQGNNLTVIYETDFQRLTKLRMLQLTDNQIHTIERNSFQDLVSLERLDISNNVI TTVGRRVFKGAQSLRSLQLDNNQITCLDEHAFKGLVELEILTLNNNNLTSLPHNIFGGLGRLRALRLSDNPFACDCHLSW LSRFLRSATRLAPYTRCQSPSQLKGQNVADLHDQEFKCSGLTEHAPMECGAENSCPHPCRCADGIVDCREKSLTSVPVTL PDDTTDVRLEQNFITELPPKSFSSFRRLRRIDLSNNNISRIAHDALSGLKQLTTLVLYGNKIKDLPSGVFKGLGSLRLLL LNANEISCIRKDAFRDLHSLSLLSLYDNNIQSLANGTFDAMKSMKTVHLAKNPFICDCNLRWLADYLHKNPIETSGARCE SPKRMHRRRIESLREEKFKCSWGELRMKLSGECRMDSDCPAMCHCEGTTVDCTGRRLKEIPRDIPLHTTELLLNDNELGR ISSDGLFGRLPHLVKLELKRNQLTGIEPNAFEGASHIQELQLGENKIKEISNKMFLGLHQLKTLNLYDNQISCVMPGSFE HLNSLTSLNLASNPFNCNCHLAWFAECVRKKSLNGGAARCGAPSKVRDVQIKDLPHSEFKCSSENSEGCLGDGYCPPSCT CTGTVVACSRNQLKEIPRGIPAETSELYLESNEIEQIHYERIRHLRSLTRLDLSNNQITILSNYTFANLTKLSTLIISYN KLQCLQRHALSGLNNLRVVSLHGNRISMLPEGSFEDLKSLTHIALGSNPLYCDCGLKWFSDWIKLDYVEPGIARCAEPEQ MKDKLILSTPSSSFVCRGRVRNDILAKCNACFEQPCQNQAQCVALPQREYQCLCQPGYHGKHCEFMIDACYGNPCRNNAT CTVLEEGRFSCQCAPGYTGARCETNIDDCLGEIKCQNNATCIDGVESYKCECQPGFSGEFCDTKIQFCSPEFNPCANGAK CMDHFTHYSCDCQAGFHGTNCTDNIDDCQNHMCQNGGTCVDGINDYQCRCPDDYTGKYCEGHNMISMMYPQTSPCQNHEC KHGVCFQPNAQGSDYLCRCHPGYTGKWCEYLTSISFVHNNSFVELEPLRTRPEANVTIVFSSAEQNGILMYDGQDAHLAV ELFNGRIRVSYDVGNHPVSTMYSFEMVADGKYHAVELLAIKKNFTLRVDRGLARSIINEGSNDYLKLTTPMFLGGLPVDP AQQAYKNWQIRNLTSFKGCMKEVWINHKLVDFGNAQRQQKITPGCALLEGEQQEEEDDEQDFMDETPHIKEEPVDPCLEN KCRRGSRCVPNSNARDGYQCKCKHGQRGRYCDQGEGSTEPPTVTAASTCRKEQVREYYTENDCRSRQPLKYAKCVGGCGN QCCAAKIVRRRKVRMVCSNNRKYIKNLDIVRKCGCTKKCY
bull Perform a dotplot of the SLIT protein against itself wwwbiobhamacukdotletDotlethtml
Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
20Swiss-prot entry
For further discussion of dotplot see Attwood and Parry-Smith p116-8
Dynamic programming2 methodsbull Dynamic programming
ndash Consider 2 protein sequences of 100 amino acids in lengthndash If it takes 1002 seconds to exhaustively align these sequences
then it will take 1003 seconds to align 3 sequences 1004 to align 4 sequencesetc
ndash More time than the universe has existed to align 20 sequences exhaustively
bull Progressive alignment
Progressive Alignmentbull Devised by Feng and Doolittle in 1987bull Essentially a heuristic method and as such
is not guaranteed to find the lsquooptimalrsquo alignment
bull Requires n-1+n-2+n-3n-n+1 pairwise alignments as a starting point
bull Most successful implementation is Clustal (Des Higgins) This software is cited 3000 times per year in the scientific literature
Overview of ClustalW Procedure
1 PEEKSAVTALWGKVN--VDEVGG2 GEEKAAVLALWDKVN--EEEVGG3 PADKTNVKAAWGKVGAHAGEYGA4 AADKTNVKAAWSKVGGHAGEYGA5 EHEWQLVLHVWAKVEADVAGHGQ
Hbb_Human 1 -Hbb_Horse 2 17 -Hba_Human 3 59 60 -Hba_Horse 4 59 59 13 -Myg_Whale 5 77 77 75 75 -
Hbb_Human
Hbb_Horse
Hba_Horse
Hba_Human
Myg_Whale
2
1
3 4
2
1
3 4
alpha-helices
Quick pairwise alignment calculate distance matrix
Neighbor-joining tree(guide tree)
Progressive alignment following guide tree
CLUSTAL W
ClustalW- Pairwise Alignments
bull First perform all possible pairwise alignments between each pair of sequences There are (n-1)+(n-2)(n-n+1) possibilities
bull Calculate the lsquodistancersquo between each pair of sequences based on these isolated pairwise alignments
bull Generate a distance matrix
Path Graph for aligning two sequences
Possible alignment
1
1
0
1
0
-1
Scoring SchemebullMatch +1bullMismatch 0bullIndel -1
Score for this path= 2
Alignment using this path
GATTC-GAATTC
1
1
0
1
0
-1
Optimal Alignment 1
1
1
-1
1
1
1
Alignment score 4Alignment score 4
Alignment using this path
GA-TTCGA-TTCGAATTCGAATTC
Optimal Alignment 2
1
-1
1
1
1
1
Alignment score 4Alignment score 4
Alignment using this path
G-ATTCG-ATTCGAATTCGAATTC
Alignment of 3 sequences
ClustalW- Guide Tree
bull Generate a Neighbor-Joining lsquoguide treersquo from these pairwise distances
bull This guide tree gives the order in which the progressive alignment will be carried out
Neighbor joining method
bullThe neighbor-joining method is a greedy heuristic which joins at each step the two closest sub-trees that are not already joinedbullIt is based on the minimum evolution principlebullneighbors are defined as two taxa that are connected by a single node in an unrooted tree
A B
Node 1
PAM Spinach Rice Mosquito Monkey HumanSpinach 00 849 1056 908 863Rice 849 00 1178 1224 1226Mosquito 1056 1178 00 847 808Monkey 908 1224 847 00 33Human 863 1226 808 33 00
What is required for the Neighbour joining method
Distance matrixDistance Matrix
PAM distance 33 (Human - Monkey) is the minimum So well join Human and Monkey to MonHum and well calculate the new distances
Mon-Hum
MonkeyHumanSpinachMosquito Rice
First Step
After we have joined two species in a subtree we have to compute the distances from every other node to the new subtree We do this with a simple average of distancesDist[Spinach MonHum]
= (Dist[Spinach Monkey] + Dist[Spinach Human])2 = (908 + 863)2 = 8855
Mon-Hum
MonkeyHumanSpinach
Calculation of New Distances
PAM Spinach Rice Mosquito MonHumSpinach 00 849 1056 886Rice 849 00 1178 1225Mosquito 1056 1178 00 828MonHum 886 1225 828 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Next Cycle
PAM Spinach Rice MosMonHumSpinach 00 849 971Rice 849 00 1202MosMonHum 971 1202 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
Penultimate Cycle
PAM SpinRice MosMonHumSpinach 00 1087MosMonHum 1087 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
(Spin-Rice)-(Mos-(Mon-Hum))
Last Joining
Human
Monkey
MosquitoRice
Spinach
Unrooted Neighbor-Joining Tree
Multiple Alignment- First pairbull Align the two most closely-related sequences first
bull This alignment is then lsquofixedrsquo and will never change If a gap is to be introduced subsequently then it will be introduced in the same place in both sequences but their relative alignment remains unchanged
ClustalW- Decision timebull Consult the guide tree to see what alignment is performed next
ndash Align a third sequence to the first twoOrndash Align two entirely different sequences to each other
Option 1Option 1 Option 2Option 2
ClustalW- Alternative 1If the situation arises where a third sequence is aligned to the first two then when a gap has to be introduced to improve the alignment each of these two entities are treated as two single sequences
+
ClustalW- Alternative 2bull If on the other hand two separate
sequences have to be aligned together then the first pairwise alignment is placed to one side and the pairwise alignment of the other two is carried out
+
ClustalW- Progression
bull The alignment is progressively built up in this way with each step being treated as a pairwise alignment sometimes with each member of a lsquopairrsquo having more than one sequence
Progressive alignment - step 11 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
12345
Progressive alignment - step 21 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
12345
Progressive alignment - step 31 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
+
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
12345
Progressive alignment - final step1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
+
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
5 -ctcga-acgatacgatgactagct-
12345
ClustalW-Good pointsBad points
bull Advantagesndash Speed
bull Disadvantagesndash No objective functionndash No way of quantifying whether or not the alignment is good
ndash No way of knowing if the alignment is lsquocorrectrsquo
ClustalW-Local Minimumbull Potential problems
ndash Local minimum problem If an error is introduced early in the alignment process it is impossible to correct this later in the procedure
ndash Arbitrary alignment
Increasing the sophistication of the alignment process
bull Should we treat all the sequences in the same way - even though some sequences are closely-related and some sequences are distant relatives
bull Should we treat all positions in the sequences as though they were the same - even though they might have different functions and different locations in the 3-dimensional structure
ClustalW- Caveatsbull Sequence weightingbull Varying substitution matricesbull Residue-specific gap penalties and reduced penalties
in hydrophilic regions (external regions of protein sequences) encourage gaps in loops rather than in core regions
bull Positions in early alignments where gaps have been opened receive locally reduced gap penalties to encourage openings in subsequent alignments
ClustalW- User-supplied values
bull Two penalties are set by the user (there are default values but you should know that it is possible to change these)
bull GOP- Gap Opening Penalty is the cost of opening a gap in an alignment
bull GEP- Gap Extension Penalty is the cost of extending this gap
Position-Specific gap penaltiesbull Before any pair of (groups of) sequences are
aligned a table of GOPs are generated for each position in the two (sets of) sequences
bull The GOP is manipulated in a position-specific manner so that it can vary over the sequences
bull If there is a gap at a position the GOP and GEP penalties are lowered the other rules do not apply
bull This makes gaps more likely at positions where gaps already exist
Discouraging too many gaps bull If there is no gap opened then the GOP is increased if the
position is within 8 residues of an existing gapbull This discourages gaps that are too close togetherbull At any position within a run of hydrophilic residues the GOP is
decreasedbull These runs usually indicate loop regions in protein structuresbull A run of 5 hydrophilic residues is considered to be a hydrophilic
stretchbull The default hydrophilic residues are
ndash D E G K N Q P R Sndash But this can be changed by the user
Divergent Sequencesbull The most divergent sequences (most different on average
from all of the other sequences) are usually the most difficult to align
bull It is sometimes better to delay their aligment until later (when the easier sequences have already been aligned)
bull The user has the choice of setting a cutoff (default is 40 identity)
bull This will delay the alignment until the others have been aligned
Advice on progressive alignmentbull Progressive alignment is a mathematical process that is completely
independent of biological realitybull Can be a very good estimatebull Can be an impossibly poor estimatebull Requires user input and skillbull Treat cautiouslybull Can be improved by eye (usually)bull Often helps to have colour-codingbull Depending on the use the user should be able to make a judgement
on those regions that are reliable or notbull For phylogeny reconstruction only use those positions whose
hypothesis of positional homology is unimpeachable
Alignment of protein-coding DNA sequences
bull It is not very sensible to align the DNA sequences of protein-coding genes
ATGCTGTTAGGGATGACTCTGTTAGGG
ATG-CT--GTTAGGGATGACTCTGTTAGGG
The result might be highly-implausible and might not reflect what is known about biological processesIt is much more sensible to translate the sequences to their corresponding amino acid sequences align these protein sequences and then put the gaps in the DNA sequences according to where they are found in the amino acid alignment
Manual Alignment- softwareGDE- The Genetic Data Environment (UNIX)CINEMA- Java applet available from
ndash httpwwwbiochemuclacuk
SeqappSeqpup- MacPCUNIX available fromndash httpiubiobioindianaedu
SeAl for Macintosh available fromndash httpevolvezoooxacukSe-AlSe-Alhtml
BioEdit for PC available fromndash httpwwwmbioncsueduRNasePinfoprogramsBIOEDITbio
edithtml
- Multiple Sequence Alignment James McInerney bioinf4biologists Feb 2009
- Alignment can be easy or difficult
- Homology Definition
- Multiple Sequence Alignment- Goals
- Multiple sequence alignments - problems
- Slide 6
- Slide 7
- SSU rRNA
- Alignment of 16S rRNA can be guided by secondary structure
- Protein Alignment may be guided by Tertiary Structure Interactions
- Multiple Sequence Alignment- Methods
- Manual Alignment - reasons
- Local minimum
- Dotplots
- Dotplot example sperm whale vs human myg
- Slide 16
- Slide 17
- Dotplots in practice
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Dynamic programming
- Progressive Alignment
- Slide 23
- ClustalW- Pairwise Alignments
- Path Graph for aligning two sequences
- Possible alignment
- Alignment using this path
- Optimal Alignment 1
- Optimal Alignment 2
- Alignment of 3 sequences
- ClustalW- Guide Tree
- Neighbor joining method
- Distance Matrix
- First Step
- Calculation of New Distances
- Next Cycle
- Penultimate Cycle
- Last Joining
- Unrooted Neighbor-Joining Tree
- Multiple Alignment- First pair
- ClustalW- Decision time
- ClustalW- Alternative 1
- ClustalW- Progression
- Progressive alignment - step 1
- Progressive alignment - step 2
- Progressive alignment - step 3
- Progressive alignment - final step
- ClustalW-Good pointsBad points
- ClustalW-Local Minimum
- Increasing the sophistication of the alignment process
- Slide 51
- ClustalW- Caveats
- ClustalW- User-supplied values
- Position-Specific gap penalties
- Discouraging too many gaps
- Divergent Sequences
- Advice on progressive alignment
- Alignment of protein-coding DNA sequences
- Manual Alignment- software
-

Local minimum
GARFIELDTHEFAT---CATGARFIELDTHEFATFATCAT
bull The dotplot provides a way of quickly visualizing the similarities between all parts of two sequences simultaneously
bull Lets consider a dotplot between sperm whale and human myoglobins
Dotplots
Sperm whale myoglobin
GLSDGEWQLV LNVWGKVEAD IPGHGQEVLI RLFKGHPETL EKFDKFKHLK SEDEMKASED LKKHGATVLT ALGGILKKKG HHEAEIKPLA QSHATKHKIP VKYLEFISEC IIQVLQSKHP GDFGADAQGA MNKALELFRK DMASNYKELG FQG
human myoglobin
VLSEGEWQLV LHVWAKVEAD VAGHGQDILI RLFKSHPETL EKFDRFKHLK TEAEMKASED LKKHGVTVLT ALGAILKKKG HHEAELKPLA QSHATKHKIP IKYLEFISEA IIHVLHSRHP GDFGADAQGA MNKALELFRK DIAAKYKELG YQG
bull Put one sequence on top
bull the other on the side
bull where residues are identical put a dot
bull Diagonal lines of dots show similarities
Dotplot example sperm whale vs human myg
Sperm whale myoglobin
G L S D G E W Q L V V L S E G E W Q L V
Human myoglobin
bullJust do the first 10 amino acids of eachbullMake a table with
ndashwhale sequence on top ndashhuman sequence on the side
bull This is the result for the whole sequence
bull It is easy to see that the diagonal is a line of dots
bull So sperm whale and human myoglobin are very similar
bull But the picture is noisy can smooth using a sliding window which considers neighbouring residues as well
Dotplot example sperm whale vs human myg
16
Sperm whale myoglobin
G L S D G E W Q L V V L S E G E W Q L V
Human myoglobin
bull can smooth noise using a sliding window which considers neighbouring residues as well
bull Have done this here can see the diagonal is highly similar
bull Also instead of using using a simple identity use a scoring matrix
Dotplot example sperm whale vs human myg
Dotplots in practicebull The best tool is an applet called dotlet
bull wwwisrecisb-sibchjavadotletDotlethtmlbull wwwbipbhamacukdotletDotlethtml
bull an applet is a program that runs in a web browser This means that you can produce dotplots within a netscapeIE window
bull Dotplots are often useful to identify things like repeated domains or duplications in big proteins
Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
bull Protein has many repeats bull SLIT_DROME (P24014)
MAAPSRTTLMPPPFRLQLRLLILPILLLLRHDAVHAEPYSGGFGSSAVSSGGLGSVGIHIPGGGVGVITEARCPRVCSCT GLNVDCSHRGLTSVPRKISADVERLELQGNNLTVIYETDFQRLTKLRMLQLTDNQIHTIERNSFQDLVSLERLDISNNVI TTVGRRVFKGAQSLRSLQLDNNQITCLDEHAFKGLVELEILTLNNNNLTSLPHNIFGGLGRLRALRLSDNPFACDCHLSW LSRFLRSATRLAPYTRCQSPSQLKGQNVADLHDQEFKCSGLTEHAPMECGAENSCPHPCRCADGIVDCREKSLTSVPVTL PDDTTDVRLEQNFITELPPKSFSSFRRLRRIDLSNNNISRIAHDALSGLKQLTTLVLYGNKIKDLPSGVFKGLGSLRLLL LNANEISCIRKDAFRDLHSLSLLSLYDNNIQSLANGTFDAMKSMKTVHLAKNPFICDCNLRWLADYLHKNPIETSGARCE SPKRMHRRRIESLREEKFKCSWGELRMKLSGECRMDSDCPAMCHCEGTTVDCTGRRLKEIPRDIPLHTTELLLNDNELGR ISSDGLFGRLPHLVKLELKRNQLTGIEPNAFEGASHIQELQLGENKIKEISNKMFLGLHQLKTLNLYDNQISCVMPGSFE HLNSLTSLNLASNPFNCNCHLAWFAECVRKKSLNGGAARCGAPSKVRDVQIKDLPHSEFKCSSENSEGCLGDGYCPPSCT CTGTVVACSRNQLKEIPRGIPAETSELYLESNEIEQIHYERIRHLRSLTRLDLSNNQITILSNYTFANLTKLSTLIISYN KLQCLQRHALSGLNNLRVVSLHGNRISMLPEGSFEDLKSLTHIALGSNPLYCDCGLKWFSDWIKLDYVEPGIARCAEPEQ MKDKLILSTPSSSFVCRGRVRNDILAKCNACFEQPCQNQAQCVALPQREYQCLCQPGYHGKHCEFMIDACYGNPCRNNAT CTVLEEGRFSCQCAPGYTGARCETNIDDCLGEIKCQNNATCIDGVESYKCECQPGFSGEFCDTKIQFCSPEFNPCANGAK CMDHFTHYSCDCQAGFHGTNCTDNIDDCQNHMCQNGGTCVDGINDYQCRCPDDYTGKYCEGHNMISMMYPQTSPCQNHEC KHGVCFQPNAQGSDYLCRCHPGYTGKWCEYLTSISFVHNNSFVELEPLRTRPEANVTIVFSSAEQNGILMYDGQDAHLAV ELFNGRIRVSYDVGNHPVSTMYSFEMVADGKYHAVELLAIKKNFTLRVDRGLARSIINEGSNDYLKLTTPMFLGGLPVDP AQQAYKNWQIRNLTSFKGCMKEVWINHKLVDFGNAQRQQKITPGCALLEGEQQEEEDDEQDFMDETPHIKEEPVDPCLEN KCRRGSRCVPNSNARDGYQCKCKHGQRGRYCDQGEGSTEPPTVTAASTCRKEQVREYYTENDCRSRQPLKYAKCVGGCGN QCCAAKIVRRRKVRMVCSNNRKYIKNLDIVRKCGCTKKCY
bull Perform a dotplot of the SLIT protein against itself wwwbiobhamacukdotletDotlethtml
Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
20Swiss-prot entry
For further discussion of dotplot see Attwood and Parry-Smith p116-8
Dynamic programming2 methodsbull Dynamic programming
ndash Consider 2 protein sequences of 100 amino acids in lengthndash If it takes 1002 seconds to exhaustively align these sequences
then it will take 1003 seconds to align 3 sequences 1004 to align 4 sequencesetc
ndash More time than the universe has existed to align 20 sequences exhaustively
bull Progressive alignment
Progressive Alignmentbull Devised by Feng and Doolittle in 1987bull Essentially a heuristic method and as such
is not guaranteed to find the lsquooptimalrsquo alignment
bull Requires n-1+n-2+n-3n-n+1 pairwise alignments as a starting point
bull Most successful implementation is Clustal (Des Higgins) This software is cited 3000 times per year in the scientific literature
Overview of ClustalW Procedure
1 PEEKSAVTALWGKVN--VDEVGG2 GEEKAAVLALWDKVN--EEEVGG3 PADKTNVKAAWGKVGAHAGEYGA4 AADKTNVKAAWSKVGGHAGEYGA5 EHEWQLVLHVWAKVEADVAGHGQ
Hbb_Human 1 -Hbb_Horse 2 17 -Hba_Human 3 59 60 -Hba_Horse 4 59 59 13 -Myg_Whale 5 77 77 75 75 -
Hbb_Human
Hbb_Horse
Hba_Horse
Hba_Human
Myg_Whale
2
1
3 4
2
1
3 4
alpha-helices
Quick pairwise alignment calculate distance matrix
Neighbor-joining tree(guide tree)
Progressive alignment following guide tree
CLUSTAL W
ClustalW- Pairwise Alignments
bull First perform all possible pairwise alignments between each pair of sequences There are (n-1)+(n-2)(n-n+1) possibilities
bull Calculate the lsquodistancersquo between each pair of sequences based on these isolated pairwise alignments
bull Generate a distance matrix
Path Graph for aligning two sequences
Possible alignment
1
1
0
1
0
-1
Scoring SchemebullMatch +1bullMismatch 0bullIndel -1
Score for this path= 2
Alignment using this path
GATTC-GAATTC
1
1
0
1
0
-1
Optimal Alignment 1
1
1
-1
1
1
1
Alignment score 4Alignment score 4
Alignment using this path
GA-TTCGA-TTCGAATTCGAATTC
Optimal Alignment 2
1
-1
1
1
1
1
Alignment score 4Alignment score 4
Alignment using this path
G-ATTCG-ATTCGAATTCGAATTC
Alignment of 3 sequences
ClustalW- Guide Tree
bull Generate a Neighbor-Joining lsquoguide treersquo from these pairwise distances
bull This guide tree gives the order in which the progressive alignment will be carried out
Neighbor joining method
bullThe neighbor-joining method is a greedy heuristic which joins at each step the two closest sub-trees that are not already joinedbullIt is based on the minimum evolution principlebullneighbors are defined as two taxa that are connected by a single node in an unrooted tree
A B
Node 1
PAM Spinach Rice Mosquito Monkey HumanSpinach 00 849 1056 908 863Rice 849 00 1178 1224 1226Mosquito 1056 1178 00 847 808Monkey 908 1224 847 00 33Human 863 1226 808 33 00
What is required for the Neighbour joining method
Distance matrixDistance Matrix
PAM distance 33 (Human - Monkey) is the minimum So well join Human and Monkey to MonHum and well calculate the new distances
Mon-Hum
MonkeyHumanSpinachMosquito Rice
First Step
After we have joined two species in a subtree we have to compute the distances from every other node to the new subtree We do this with a simple average of distancesDist[Spinach MonHum]
= (Dist[Spinach Monkey] + Dist[Spinach Human])2 = (908 + 863)2 = 8855
Mon-Hum
MonkeyHumanSpinach
Calculation of New Distances
PAM Spinach Rice Mosquito MonHumSpinach 00 849 1056 886Rice 849 00 1178 1225Mosquito 1056 1178 00 828MonHum 886 1225 828 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Next Cycle
PAM Spinach Rice MosMonHumSpinach 00 849 971Rice 849 00 1202MosMonHum 971 1202 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
Penultimate Cycle
PAM SpinRice MosMonHumSpinach 00 1087MosMonHum 1087 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
(Spin-Rice)-(Mos-(Mon-Hum))
Last Joining
Human
Monkey
MosquitoRice
Spinach
Unrooted Neighbor-Joining Tree
Multiple Alignment- First pairbull Align the two most closely-related sequences first
bull This alignment is then lsquofixedrsquo and will never change If a gap is to be introduced subsequently then it will be introduced in the same place in both sequences but their relative alignment remains unchanged
ClustalW- Decision timebull Consult the guide tree to see what alignment is performed next
ndash Align a third sequence to the first twoOrndash Align two entirely different sequences to each other
Option 1Option 1 Option 2Option 2
ClustalW- Alternative 1If the situation arises where a third sequence is aligned to the first two then when a gap has to be introduced to improve the alignment each of these two entities are treated as two single sequences
+
ClustalW- Alternative 2bull If on the other hand two separate
sequences have to be aligned together then the first pairwise alignment is placed to one side and the pairwise alignment of the other two is carried out
+
ClustalW- Progression
bull The alignment is progressively built up in this way with each step being treated as a pairwise alignment sometimes with each member of a lsquopairrsquo having more than one sequence
Progressive alignment - step 11 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
12345
Progressive alignment - step 21 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
12345
Progressive alignment - step 31 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
+
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
12345
Progressive alignment - final step1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
+
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
5 -ctcga-acgatacgatgactagct-
12345
ClustalW-Good pointsBad points
bull Advantagesndash Speed
bull Disadvantagesndash No objective functionndash No way of quantifying whether or not the alignment is good
ndash No way of knowing if the alignment is lsquocorrectrsquo
ClustalW-Local Minimumbull Potential problems
ndash Local minimum problem If an error is introduced early in the alignment process it is impossible to correct this later in the procedure
ndash Arbitrary alignment
Increasing the sophistication of the alignment process
bull Should we treat all the sequences in the same way - even though some sequences are closely-related and some sequences are distant relatives
bull Should we treat all positions in the sequences as though they were the same - even though they might have different functions and different locations in the 3-dimensional structure
ClustalW- Caveatsbull Sequence weightingbull Varying substitution matricesbull Residue-specific gap penalties and reduced penalties
in hydrophilic regions (external regions of protein sequences) encourage gaps in loops rather than in core regions
bull Positions in early alignments where gaps have been opened receive locally reduced gap penalties to encourage openings in subsequent alignments
ClustalW- User-supplied values
bull Two penalties are set by the user (there are default values but you should know that it is possible to change these)
bull GOP- Gap Opening Penalty is the cost of opening a gap in an alignment
bull GEP- Gap Extension Penalty is the cost of extending this gap
Position-Specific gap penaltiesbull Before any pair of (groups of) sequences are
aligned a table of GOPs are generated for each position in the two (sets of) sequences
bull The GOP is manipulated in a position-specific manner so that it can vary over the sequences
bull If there is a gap at a position the GOP and GEP penalties are lowered the other rules do not apply
bull This makes gaps more likely at positions where gaps already exist
Discouraging too many gaps bull If there is no gap opened then the GOP is increased if the
position is within 8 residues of an existing gapbull This discourages gaps that are too close togetherbull At any position within a run of hydrophilic residues the GOP is
decreasedbull These runs usually indicate loop regions in protein structuresbull A run of 5 hydrophilic residues is considered to be a hydrophilic
stretchbull The default hydrophilic residues are
ndash D E G K N Q P R Sndash But this can be changed by the user
Divergent Sequencesbull The most divergent sequences (most different on average
from all of the other sequences) are usually the most difficult to align
bull It is sometimes better to delay their aligment until later (when the easier sequences have already been aligned)
bull The user has the choice of setting a cutoff (default is 40 identity)
bull This will delay the alignment until the others have been aligned
Advice on progressive alignmentbull Progressive alignment is a mathematical process that is completely
independent of biological realitybull Can be a very good estimatebull Can be an impossibly poor estimatebull Requires user input and skillbull Treat cautiouslybull Can be improved by eye (usually)bull Often helps to have colour-codingbull Depending on the use the user should be able to make a judgement
on those regions that are reliable or notbull For phylogeny reconstruction only use those positions whose
hypothesis of positional homology is unimpeachable
Alignment of protein-coding DNA sequences
bull It is not very sensible to align the DNA sequences of protein-coding genes
ATGCTGTTAGGGATGACTCTGTTAGGG
ATG-CT--GTTAGGGATGACTCTGTTAGGG
The result might be highly-implausible and might not reflect what is known about biological processesIt is much more sensible to translate the sequences to their corresponding amino acid sequences align these protein sequences and then put the gaps in the DNA sequences according to where they are found in the amino acid alignment
Manual Alignment- softwareGDE- The Genetic Data Environment (UNIX)CINEMA- Java applet available from
ndash httpwwwbiochemuclacuk
SeqappSeqpup- MacPCUNIX available fromndash httpiubiobioindianaedu
SeAl for Macintosh available fromndash httpevolvezoooxacukSe-AlSe-Alhtml
BioEdit for PC available fromndash httpwwwmbioncsueduRNasePinfoprogramsBIOEDITbio
edithtml
- Multiple Sequence Alignment James McInerney bioinf4biologists Feb 2009
- Alignment can be easy or difficult
- Homology Definition
- Multiple Sequence Alignment- Goals
- Multiple sequence alignments - problems
- Slide 6
- Slide 7
- SSU rRNA
- Alignment of 16S rRNA can be guided by secondary structure
- Protein Alignment may be guided by Tertiary Structure Interactions
- Multiple Sequence Alignment- Methods
- Manual Alignment - reasons
- Local minimum
- Dotplots
- Dotplot example sperm whale vs human myg
- Slide 16
- Slide 17
- Dotplots in practice
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Dynamic programming
- Progressive Alignment
- Slide 23
- ClustalW- Pairwise Alignments
- Path Graph for aligning two sequences
- Possible alignment
- Alignment using this path
- Optimal Alignment 1
- Optimal Alignment 2
- Alignment of 3 sequences
- ClustalW- Guide Tree
- Neighbor joining method
- Distance Matrix
- First Step
- Calculation of New Distances
- Next Cycle
- Penultimate Cycle
- Last Joining
- Unrooted Neighbor-Joining Tree
- Multiple Alignment- First pair
- ClustalW- Decision time
- ClustalW- Alternative 1
- ClustalW- Progression
- Progressive alignment - step 1
- Progressive alignment - step 2
- Progressive alignment - step 3
- Progressive alignment - final step
- ClustalW-Good pointsBad points
- ClustalW-Local Minimum
- Increasing the sophistication of the alignment process
- Slide 51
- ClustalW- Caveats
- ClustalW- User-supplied values
- Position-Specific gap penalties
- Discouraging too many gaps
- Divergent Sequences
- Advice on progressive alignment
- Alignment of protein-coding DNA sequences
- Manual Alignment- software
-

bull The dotplot provides a way of quickly visualizing the similarities between all parts of two sequences simultaneously
bull Lets consider a dotplot between sperm whale and human myoglobins
Dotplots
Sperm whale myoglobin
GLSDGEWQLV LNVWGKVEAD IPGHGQEVLI RLFKGHPETL EKFDKFKHLK SEDEMKASED LKKHGATVLT ALGGILKKKG HHEAEIKPLA QSHATKHKIP VKYLEFISEC IIQVLQSKHP GDFGADAQGA MNKALELFRK DMASNYKELG FQG
human myoglobin
VLSEGEWQLV LHVWAKVEAD VAGHGQDILI RLFKSHPETL EKFDRFKHLK TEAEMKASED LKKHGVTVLT ALGAILKKKG HHEAELKPLA QSHATKHKIP IKYLEFISEA IIHVLHSRHP GDFGADAQGA MNKALELFRK DIAAKYKELG YQG
bull Put one sequence on top
bull the other on the side
bull where residues are identical put a dot
bull Diagonal lines of dots show similarities
Dotplot example sperm whale vs human myg
Sperm whale myoglobin
G L S D G E W Q L V V L S E G E W Q L V
Human myoglobin
bullJust do the first 10 amino acids of eachbullMake a table with
ndashwhale sequence on top ndashhuman sequence on the side
bull This is the result for the whole sequence
bull It is easy to see that the diagonal is a line of dots
bull So sperm whale and human myoglobin are very similar
bull But the picture is noisy can smooth using a sliding window which considers neighbouring residues as well
Dotplot example sperm whale vs human myg
16
Sperm whale myoglobin
G L S D G E W Q L V V L S E G E W Q L V
Human myoglobin
bull can smooth noise using a sliding window which considers neighbouring residues as well
bull Have done this here can see the diagonal is highly similar
bull Also instead of using using a simple identity use a scoring matrix
Dotplot example sperm whale vs human myg
Dotplots in practicebull The best tool is an applet called dotlet
bull wwwisrecisb-sibchjavadotletDotlethtmlbull wwwbipbhamacukdotletDotlethtml
bull an applet is a program that runs in a web browser This means that you can produce dotplots within a netscapeIE window
bull Dotplots are often useful to identify things like repeated domains or duplications in big proteins
Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
bull Protein has many repeats bull SLIT_DROME (P24014)
MAAPSRTTLMPPPFRLQLRLLILPILLLLRHDAVHAEPYSGGFGSSAVSSGGLGSVGIHIPGGGVGVITEARCPRVCSCT GLNVDCSHRGLTSVPRKISADVERLELQGNNLTVIYETDFQRLTKLRMLQLTDNQIHTIERNSFQDLVSLERLDISNNVI TTVGRRVFKGAQSLRSLQLDNNQITCLDEHAFKGLVELEILTLNNNNLTSLPHNIFGGLGRLRALRLSDNPFACDCHLSW LSRFLRSATRLAPYTRCQSPSQLKGQNVADLHDQEFKCSGLTEHAPMECGAENSCPHPCRCADGIVDCREKSLTSVPVTL PDDTTDVRLEQNFITELPPKSFSSFRRLRRIDLSNNNISRIAHDALSGLKQLTTLVLYGNKIKDLPSGVFKGLGSLRLLL LNANEISCIRKDAFRDLHSLSLLSLYDNNIQSLANGTFDAMKSMKTVHLAKNPFICDCNLRWLADYLHKNPIETSGARCE SPKRMHRRRIESLREEKFKCSWGELRMKLSGECRMDSDCPAMCHCEGTTVDCTGRRLKEIPRDIPLHTTELLLNDNELGR ISSDGLFGRLPHLVKLELKRNQLTGIEPNAFEGASHIQELQLGENKIKEISNKMFLGLHQLKTLNLYDNQISCVMPGSFE HLNSLTSLNLASNPFNCNCHLAWFAECVRKKSLNGGAARCGAPSKVRDVQIKDLPHSEFKCSSENSEGCLGDGYCPPSCT CTGTVVACSRNQLKEIPRGIPAETSELYLESNEIEQIHYERIRHLRSLTRLDLSNNQITILSNYTFANLTKLSTLIISYN KLQCLQRHALSGLNNLRVVSLHGNRISMLPEGSFEDLKSLTHIALGSNPLYCDCGLKWFSDWIKLDYVEPGIARCAEPEQ MKDKLILSTPSSSFVCRGRVRNDILAKCNACFEQPCQNQAQCVALPQREYQCLCQPGYHGKHCEFMIDACYGNPCRNNAT CTVLEEGRFSCQCAPGYTGARCETNIDDCLGEIKCQNNATCIDGVESYKCECQPGFSGEFCDTKIQFCSPEFNPCANGAK CMDHFTHYSCDCQAGFHGTNCTDNIDDCQNHMCQNGGTCVDGINDYQCRCPDDYTGKYCEGHNMISMMYPQTSPCQNHEC KHGVCFQPNAQGSDYLCRCHPGYTGKWCEYLTSISFVHNNSFVELEPLRTRPEANVTIVFSSAEQNGILMYDGQDAHLAV ELFNGRIRVSYDVGNHPVSTMYSFEMVADGKYHAVELLAIKKNFTLRVDRGLARSIINEGSNDYLKLTTPMFLGGLPVDP AQQAYKNWQIRNLTSFKGCMKEVWINHKLVDFGNAQRQQKITPGCALLEGEQQEEEDDEQDFMDETPHIKEEPVDPCLEN KCRRGSRCVPNSNARDGYQCKCKHGQRGRYCDQGEGSTEPPTVTAASTCRKEQVREYYTENDCRSRQPLKYAKCVGGCGN QCCAAKIVRRRKVRMVCSNNRKYIKNLDIVRKCGCTKKCY
bull Perform a dotplot of the SLIT protein against itself wwwbiobhamacukdotletDotlethtml
Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
20Swiss-prot entry
For further discussion of dotplot see Attwood and Parry-Smith p116-8
Dynamic programming2 methodsbull Dynamic programming
ndash Consider 2 protein sequences of 100 amino acids in lengthndash If it takes 1002 seconds to exhaustively align these sequences
then it will take 1003 seconds to align 3 sequences 1004 to align 4 sequencesetc
ndash More time than the universe has existed to align 20 sequences exhaustively
bull Progressive alignment
Progressive Alignmentbull Devised by Feng and Doolittle in 1987bull Essentially a heuristic method and as such
is not guaranteed to find the lsquooptimalrsquo alignment
bull Requires n-1+n-2+n-3n-n+1 pairwise alignments as a starting point
bull Most successful implementation is Clustal (Des Higgins) This software is cited 3000 times per year in the scientific literature
Overview of ClustalW Procedure
1 PEEKSAVTALWGKVN--VDEVGG2 GEEKAAVLALWDKVN--EEEVGG3 PADKTNVKAAWGKVGAHAGEYGA4 AADKTNVKAAWSKVGGHAGEYGA5 EHEWQLVLHVWAKVEADVAGHGQ
Hbb_Human 1 -Hbb_Horse 2 17 -Hba_Human 3 59 60 -Hba_Horse 4 59 59 13 -Myg_Whale 5 77 77 75 75 -
Hbb_Human
Hbb_Horse
Hba_Horse
Hba_Human
Myg_Whale
2
1
3 4
2
1
3 4
alpha-helices
Quick pairwise alignment calculate distance matrix
Neighbor-joining tree(guide tree)
Progressive alignment following guide tree
CLUSTAL W
ClustalW- Pairwise Alignments
bull First perform all possible pairwise alignments between each pair of sequences There are (n-1)+(n-2)(n-n+1) possibilities
bull Calculate the lsquodistancersquo between each pair of sequences based on these isolated pairwise alignments
bull Generate a distance matrix
Path Graph for aligning two sequences
Possible alignment
1
1
0
1
0
-1
Scoring SchemebullMatch +1bullMismatch 0bullIndel -1
Score for this path= 2
Alignment using this path
GATTC-GAATTC
1
1
0
1
0
-1
Optimal Alignment 1
1
1
-1
1
1
1
Alignment score 4Alignment score 4
Alignment using this path
GA-TTCGA-TTCGAATTCGAATTC
Optimal Alignment 2
1
-1
1
1
1
1
Alignment score 4Alignment score 4
Alignment using this path
G-ATTCG-ATTCGAATTCGAATTC
Alignment of 3 sequences
ClustalW- Guide Tree
bull Generate a Neighbor-Joining lsquoguide treersquo from these pairwise distances
bull This guide tree gives the order in which the progressive alignment will be carried out
Neighbor joining method
bullThe neighbor-joining method is a greedy heuristic which joins at each step the two closest sub-trees that are not already joinedbullIt is based on the minimum evolution principlebullneighbors are defined as two taxa that are connected by a single node in an unrooted tree
A B
Node 1
PAM Spinach Rice Mosquito Monkey HumanSpinach 00 849 1056 908 863Rice 849 00 1178 1224 1226Mosquito 1056 1178 00 847 808Monkey 908 1224 847 00 33Human 863 1226 808 33 00
What is required for the Neighbour joining method
Distance matrixDistance Matrix
PAM distance 33 (Human - Monkey) is the minimum So well join Human and Monkey to MonHum and well calculate the new distances
Mon-Hum
MonkeyHumanSpinachMosquito Rice
First Step
After we have joined two species in a subtree we have to compute the distances from every other node to the new subtree We do this with a simple average of distancesDist[Spinach MonHum]
= (Dist[Spinach Monkey] + Dist[Spinach Human])2 = (908 + 863)2 = 8855
Mon-Hum
MonkeyHumanSpinach
Calculation of New Distances
PAM Spinach Rice Mosquito MonHumSpinach 00 849 1056 886Rice 849 00 1178 1225Mosquito 1056 1178 00 828MonHum 886 1225 828 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Next Cycle
PAM Spinach Rice MosMonHumSpinach 00 849 971Rice 849 00 1202MosMonHum 971 1202 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
Penultimate Cycle
PAM SpinRice MosMonHumSpinach 00 1087MosMonHum 1087 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
(Spin-Rice)-(Mos-(Mon-Hum))
Last Joining
Human
Monkey
MosquitoRice
Spinach
Unrooted Neighbor-Joining Tree
Multiple Alignment- First pairbull Align the two most closely-related sequences first
bull This alignment is then lsquofixedrsquo and will never change If a gap is to be introduced subsequently then it will be introduced in the same place in both sequences but their relative alignment remains unchanged
ClustalW- Decision timebull Consult the guide tree to see what alignment is performed next
ndash Align a third sequence to the first twoOrndash Align two entirely different sequences to each other
Option 1Option 1 Option 2Option 2
ClustalW- Alternative 1If the situation arises where a third sequence is aligned to the first two then when a gap has to be introduced to improve the alignment each of these two entities are treated as two single sequences
+
ClustalW- Alternative 2bull If on the other hand two separate
sequences have to be aligned together then the first pairwise alignment is placed to one side and the pairwise alignment of the other two is carried out
+
ClustalW- Progression
bull The alignment is progressively built up in this way with each step being treated as a pairwise alignment sometimes with each member of a lsquopairrsquo having more than one sequence
Progressive alignment - step 11 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
12345
Progressive alignment - step 21 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
12345
Progressive alignment - step 31 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
+
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
12345
Progressive alignment - final step1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
+
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
5 -ctcga-acgatacgatgactagct-
12345
ClustalW-Good pointsBad points
bull Advantagesndash Speed
bull Disadvantagesndash No objective functionndash No way of quantifying whether or not the alignment is good
ndash No way of knowing if the alignment is lsquocorrectrsquo
ClustalW-Local Minimumbull Potential problems
ndash Local minimum problem If an error is introduced early in the alignment process it is impossible to correct this later in the procedure
ndash Arbitrary alignment
Increasing the sophistication of the alignment process
bull Should we treat all the sequences in the same way - even though some sequences are closely-related and some sequences are distant relatives
bull Should we treat all positions in the sequences as though they were the same - even though they might have different functions and different locations in the 3-dimensional structure
ClustalW- Caveatsbull Sequence weightingbull Varying substitution matricesbull Residue-specific gap penalties and reduced penalties
in hydrophilic regions (external regions of protein sequences) encourage gaps in loops rather than in core regions
bull Positions in early alignments where gaps have been opened receive locally reduced gap penalties to encourage openings in subsequent alignments
ClustalW- User-supplied values
bull Two penalties are set by the user (there are default values but you should know that it is possible to change these)
bull GOP- Gap Opening Penalty is the cost of opening a gap in an alignment
bull GEP- Gap Extension Penalty is the cost of extending this gap
Position-Specific gap penaltiesbull Before any pair of (groups of) sequences are
aligned a table of GOPs are generated for each position in the two (sets of) sequences
bull The GOP is manipulated in a position-specific manner so that it can vary over the sequences
bull If there is a gap at a position the GOP and GEP penalties are lowered the other rules do not apply
bull This makes gaps more likely at positions where gaps already exist
Discouraging too many gaps bull If there is no gap opened then the GOP is increased if the
position is within 8 residues of an existing gapbull This discourages gaps that are too close togetherbull At any position within a run of hydrophilic residues the GOP is
decreasedbull These runs usually indicate loop regions in protein structuresbull A run of 5 hydrophilic residues is considered to be a hydrophilic
stretchbull The default hydrophilic residues are
ndash D E G K N Q P R Sndash But this can be changed by the user
Divergent Sequencesbull The most divergent sequences (most different on average
from all of the other sequences) are usually the most difficult to align
bull It is sometimes better to delay their aligment until later (when the easier sequences have already been aligned)
bull The user has the choice of setting a cutoff (default is 40 identity)
bull This will delay the alignment until the others have been aligned
Advice on progressive alignmentbull Progressive alignment is a mathematical process that is completely
independent of biological realitybull Can be a very good estimatebull Can be an impossibly poor estimatebull Requires user input and skillbull Treat cautiouslybull Can be improved by eye (usually)bull Often helps to have colour-codingbull Depending on the use the user should be able to make a judgement
on those regions that are reliable or notbull For phylogeny reconstruction only use those positions whose
hypothesis of positional homology is unimpeachable
Alignment of protein-coding DNA sequences
bull It is not very sensible to align the DNA sequences of protein-coding genes
ATGCTGTTAGGGATGACTCTGTTAGGG
ATG-CT--GTTAGGGATGACTCTGTTAGGG
The result might be highly-implausible and might not reflect what is known about biological processesIt is much more sensible to translate the sequences to their corresponding amino acid sequences align these protein sequences and then put the gaps in the DNA sequences according to where they are found in the amino acid alignment
Manual Alignment- softwareGDE- The Genetic Data Environment (UNIX)CINEMA- Java applet available from
ndash httpwwwbiochemuclacuk
SeqappSeqpup- MacPCUNIX available fromndash httpiubiobioindianaedu
SeAl for Macintosh available fromndash httpevolvezoooxacukSe-AlSe-Alhtml
BioEdit for PC available fromndash httpwwwmbioncsueduRNasePinfoprogramsBIOEDITbio
edithtml
- Multiple Sequence Alignment James McInerney bioinf4biologists Feb 2009
- Alignment can be easy or difficult
- Homology Definition
- Multiple Sequence Alignment- Goals
- Multiple sequence alignments - problems
- Slide 6
- Slide 7
- SSU rRNA
- Alignment of 16S rRNA can be guided by secondary structure
- Protein Alignment may be guided by Tertiary Structure Interactions
- Multiple Sequence Alignment- Methods
- Manual Alignment - reasons
- Local minimum
- Dotplots
- Dotplot example sperm whale vs human myg
- Slide 16
- Slide 17
- Dotplots in practice
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Dynamic programming
- Progressive Alignment
- Slide 23
- ClustalW- Pairwise Alignments
- Path Graph for aligning two sequences
- Possible alignment
- Alignment using this path
- Optimal Alignment 1
- Optimal Alignment 2
- Alignment of 3 sequences
- ClustalW- Guide Tree
- Neighbor joining method
- Distance Matrix
- First Step
- Calculation of New Distances
- Next Cycle
- Penultimate Cycle
- Last Joining
- Unrooted Neighbor-Joining Tree
- Multiple Alignment- First pair
- ClustalW- Decision time
- ClustalW- Alternative 1
- ClustalW- Progression
- Progressive alignment - step 1
- Progressive alignment - step 2
- Progressive alignment - step 3
- Progressive alignment - final step
- ClustalW-Good pointsBad points
- ClustalW-Local Minimum
- Increasing the sophistication of the alignment process
- Slide 51
- ClustalW- Caveats
- ClustalW- User-supplied values
- Position-Specific gap penalties
- Discouraging too many gaps
- Divergent Sequences
- Advice on progressive alignment
- Alignment of protein-coding DNA sequences
- Manual Alignment- software
-

bull Put one sequence on top
bull the other on the side
bull where residues are identical put a dot
bull Diagonal lines of dots show similarities
Dotplot example sperm whale vs human myg
Sperm whale myoglobin
G L S D G E W Q L V V L S E G E W Q L V
Human myoglobin
bullJust do the first 10 amino acids of eachbullMake a table with
ndashwhale sequence on top ndashhuman sequence on the side
bull This is the result for the whole sequence
bull It is easy to see that the diagonal is a line of dots
bull So sperm whale and human myoglobin are very similar
bull But the picture is noisy can smooth using a sliding window which considers neighbouring residues as well
Dotplot example sperm whale vs human myg
16
Sperm whale myoglobin
G L S D G E W Q L V V L S E G E W Q L V
Human myoglobin
bull can smooth noise using a sliding window which considers neighbouring residues as well
bull Have done this here can see the diagonal is highly similar
bull Also instead of using using a simple identity use a scoring matrix
Dotplot example sperm whale vs human myg
Dotplots in practicebull The best tool is an applet called dotlet
bull wwwisrecisb-sibchjavadotletDotlethtmlbull wwwbipbhamacukdotletDotlethtml
bull an applet is a program that runs in a web browser This means that you can produce dotplots within a netscapeIE window
bull Dotplots are often useful to identify things like repeated domains or duplications in big proteins
Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
bull Protein has many repeats bull SLIT_DROME (P24014)
MAAPSRTTLMPPPFRLQLRLLILPILLLLRHDAVHAEPYSGGFGSSAVSSGGLGSVGIHIPGGGVGVITEARCPRVCSCT GLNVDCSHRGLTSVPRKISADVERLELQGNNLTVIYETDFQRLTKLRMLQLTDNQIHTIERNSFQDLVSLERLDISNNVI TTVGRRVFKGAQSLRSLQLDNNQITCLDEHAFKGLVELEILTLNNNNLTSLPHNIFGGLGRLRALRLSDNPFACDCHLSW LSRFLRSATRLAPYTRCQSPSQLKGQNVADLHDQEFKCSGLTEHAPMECGAENSCPHPCRCADGIVDCREKSLTSVPVTL PDDTTDVRLEQNFITELPPKSFSSFRRLRRIDLSNNNISRIAHDALSGLKQLTTLVLYGNKIKDLPSGVFKGLGSLRLLL LNANEISCIRKDAFRDLHSLSLLSLYDNNIQSLANGTFDAMKSMKTVHLAKNPFICDCNLRWLADYLHKNPIETSGARCE SPKRMHRRRIESLREEKFKCSWGELRMKLSGECRMDSDCPAMCHCEGTTVDCTGRRLKEIPRDIPLHTTELLLNDNELGR ISSDGLFGRLPHLVKLELKRNQLTGIEPNAFEGASHIQELQLGENKIKEISNKMFLGLHQLKTLNLYDNQISCVMPGSFE HLNSLTSLNLASNPFNCNCHLAWFAECVRKKSLNGGAARCGAPSKVRDVQIKDLPHSEFKCSSENSEGCLGDGYCPPSCT CTGTVVACSRNQLKEIPRGIPAETSELYLESNEIEQIHYERIRHLRSLTRLDLSNNQITILSNYTFANLTKLSTLIISYN KLQCLQRHALSGLNNLRVVSLHGNRISMLPEGSFEDLKSLTHIALGSNPLYCDCGLKWFSDWIKLDYVEPGIARCAEPEQ MKDKLILSTPSSSFVCRGRVRNDILAKCNACFEQPCQNQAQCVALPQREYQCLCQPGYHGKHCEFMIDACYGNPCRNNAT CTVLEEGRFSCQCAPGYTGARCETNIDDCLGEIKCQNNATCIDGVESYKCECQPGFSGEFCDTKIQFCSPEFNPCANGAK CMDHFTHYSCDCQAGFHGTNCTDNIDDCQNHMCQNGGTCVDGINDYQCRCPDDYTGKYCEGHNMISMMYPQTSPCQNHEC KHGVCFQPNAQGSDYLCRCHPGYTGKWCEYLTSISFVHNNSFVELEPLRTRPEANVTIVFSSAEQNGILMYDGQDAHLAV ELFNGRIRVSYDVGNHPVSTMYSFEMVADGKYHAVELLAIKKNFTLRVDRGLARSIINEGSNDYLKLTTPMFLGGLPVDP AQQAYKNWQIRNLTSFKGCMKEVWINHKLVDFGNAQRQQKITPGCALLEGEQQEEEDDEQDFMDETPHIKEEPVDPCLEN KCRRGSRCVPNSNARDGYQCKCKHGQRGRYCDQGEGSTEPPTVTAASTCRKEQVREYYTENDCRSRQPLKYAKCVGGCGN QCCAAKIVRRRKVRMVCSNNRKYIKNLDIVRKCGCTKKCY
bull Perform a dotplot of the SLIT protein against itself wwwbiobhamacukdotletDotlethtml
Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
20Swiss-prot entry
For further discussion of dotplot see Attwood and Parry-Smith p116-8
Dynamic programming2 methodsbull Dynamic programming
ndash Consider 2 protein sequences of 100 amino acids in lengthndash If it takes 1002 seconds to exhaustively align these sequences
then it will take 1003 seconds to align 3 sequences 1004 to align 4 sequencesetc
ndash More time than the universe has existed to align 20 sequences exhaustively
bull Progressive alignment
Progressive Alignmentbull Devised by Feng and Doolittle in 1987bull Essentially a heuristic method and as such
is not guaranteed to find the lsquooptimalrsquo alignment
bull Requires n-1+n-2+n-3n-n+1 pairwise alignments as a starting point
bull Most successful implementation is Clustal (Des Higgins) This software is cited 3000 times per year in the scientific literature
Overview of ClustalW Procedure
1 PEEKSAVTALWGKVN--VDEVGG2 GEEKAAVLALWDKVN--EEEVGG3 PADKTNVKAAWGKVGAHAGEYGA4 AADKTNVKAAWSKVGGHAGEYGA5 EHEWQLVLHVWAKVEADVAGHGQ
Hbb_Human 1 -Hbb_Horse 2 17 -Hba_Human 3 59 60 -Hba_Horse 4 59 59 13 -Myg_Whale 5 77 77 75 75 -
Hbb_Human
Hbb_Horse
Hba_Horse
Hba_Human
Myg_Whale
2
1
3 4
2
1
3 4
alpha-helices
Quick pairwise alignment calculate distance matrix
Neighbor-joining tree(guide tree)
Progressive alignment following guide tree
CLUSTAL W
ClustalW- Pairwise Alignments
bull First perform all possible pairwise alignments between each pair of sequences There are (n-1)+(n-2)(n-n+1) possibilities
bull Calculate the lsquodistancersquo between each pair of sequences based on these isolated pairwise alignments
bull Generate a distance matrix
Path Graph for aligning two sequences
Possible alignment
1
1
0
1
0
-1
Scoring SchemebullMatch +1bullMismatch 0bullIndel -1
Score for this path= 2
Alignment using this path
GATTC-GAATTC
1
1
0
1
0
-1
Optimal Alignment 1
1
1
-1
1
1
1
Alignment score 4Alignment score 4
Alignment using this path
GA-TTCGA-TTCGAATTCGAATTC
Optimal Alignment 2
1
-1
1
1
1
1
Alignment score 4Alignment score 4
Alignment using this path
G-ATTCG-ATTCGAATTCGAATTC
Alignment of 3 sequences
ClustalW- Guide Tree
bull Generate a Neighbor-Joining lsquoguide treersquo from these pairwise distances
bull This guide tree gives the order in which the progressive alignment will be carried out
Neighbor joining method
bullThe neighbor-joining method is a greedy heuristic which joins at each step the two closest sub-trees that are not already joinedbullIt is based on the minimum evolution principlebullneighbors are defined as two taxa that are connected by a single node in an unrooted tree
A B
Node 1
PAM Spinach Rice Mosquito Monkey HumanSpinach 00 849 1056 908 863Rice 849 00 1178 1224 1226Mosquito 1056 1178 00 847 808Monkey 908 1224 847 00 33Human 863 1226 808 33 00
What is required for the Neighbour joining method
Distance matrixDistance Matrix
PAM distance 33 (Human - Monkey) is the minimum So well join Human and Monkey to MonHum and well calculate the new distances
Mon-Hum
MonkeyHumanSpinachMosquito Rice
First Step
After we have joined two species in a subtree we have to compute the distances from every other node to the new subtree We do this with a simple average of distancesDist[Spinach MonHum]
= (Dist[Spinach Monkey] + Dist[Spinach Human])2 = (908 + 863)2 = 8855
Mon-Hum
MonkeyHumanSpinach
Calculation of New Distances
PAM Spinach Rice Mosquito MonHumSpinach 00 849 1056 886Rice 849 00 1178 1225Mosquito 1056 1178 00 828MonHum 886 1225 828 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Next Cycle
PAM Spinach Rice MosMonHumSpinach 00 849 971Rice 849 00 1202MosMonHum 971 1202 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
Penultimate Cycle
PAM SpinRice MosMonHumSpinach 00 1087MosMonHum 1087 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
(Spin-Rice)-(Mos-(Mon-Hum))
Last Joining
Human
Monkey
MosquitoRice
Spinach
Unrooted Neighbor-Joining Tree
Multiple Alignment- First pairbull Align the two most closely-related sequences first
bull This alignment is then lsquofixedrsquo and will never change If a gap is to be introduced subsequently then it will be introduced in the same place in both sequences but their relative alignment remains unchanged
ClustalW- Decision timebull Consult the guide tree to see what alignment is performed next
ndash Align a third sequence to the first twoOrndash Align two entirely different sequences to each other
Option 1Option 1 Option 2Option 2
ClustalW- Alternative 1If the situation arises where a third sequence is aligned to the first two then when a gap has to be introduced to improve the alignment each of these two entities are treated as two single sequences
+
ClustalW- Alternative 2bull If on the other hand two separate
sequences have to be aligned together then the first pairwise alignment is placed to one side and the pairwise alignment of the other two is carried out
+
ClustalW- Progression
bull The alignment is progressively built up in this way with each step being treated as a pairwise alignment sometimes with each member of a lsquopairrsquo having more than one sequence
Progressive alignment - step 11 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
12345
Progressive alignment - step 21 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
12345
Progressive alignment - step 31 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
+
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
12345
Progressive alignment - final step1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
+
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
5 -ctcga-acgatacgatgactagct-
12345
ClustalW-Good pointsBad points
bull Advantagesndash Speed
bull Disadvantagesndash No objective functionndash No way of quantifying whether or not the alignment is good
ndash No way of knowing if the alignment is lsquocorrectrsquo
ClustalW-Local Minimumbull Potential problems
ndash Local minimum problem If an error is introduced early in the alignment process it is impossible to correct this later in the procedure
ndash Arbitrary alignment
Increasing the sophistication of the alignment process
bull Should we treat all the sequences in the same way - even though some sequences are closely-related and some sequences are distant relatives
bull Should we treat all positions in the sequences as though they were the same - even though they might have different functions and different locations in the 3-dimensional structure
ClustalW- Caveatsbull Sequence weightingbull Varying substitution matricesbull Residue-specific gap penalties and reduced penalties
in hydrophilic regions (external regions of protein sequences) encourage gaps in loops rather than in core regions
bull Positions in early alignments where gaps have been opened receive locally reduced gap penalties to encourage openings in subsequent alignments
ClustalW- User-supplied values
bull Two penalties are set by the user (there are default values but you should know that it is possible to change these)
bull GOP- Gap Opening Penalty is the cost of opening a gap in an alignment
bull GEP- Gap Extension Penalty is the cost of extending this gap
Position-Specific gap penaltiesbull Before any pair of (groups of) sequences are
aligned a table of GOPs are generated for each position in the two (sets of) sequences
bull The GOP is manipulated in a position-specific manner so that it can vary over the sequences
bull If there is a gap at a position the GOP and GEP penalties are lowered the other rules do not apply
bull This makes gaps more likely at positions where gaps already exist
Discouraging too many gaps bull If there is no gap opened then the GOP is increased if the
position is within 8 residues of an existing gapbull This discourages gaps that are too close togetherbull At any position within a run of hydrophilic residues the GOP is
decreasedbull These runs usually indicate loop regions in protein structuresbull A run of 5 hydrophilic residues is considered to be a hydrophilic
stretchbull The default hydrophilic residues are
ndash D E G K N Q P R Sndash But this can be changed by the user
Divergent Sequencesbull The most divergent sequences (most different on average
from all of the other sequences) are usually the most difficult to align
bull It is sometimes better to delay their aligment until later (when the easier sequences have already been aligned)
bull The user has the choice of setting a cutoff (default is 40 identity)
bull This will delay the alignment until the others have been aligned
Advice on progressive alignmentbull Progressive alignment is a mathematical process that is completely
independent of biological realitybull Can be a very good estimatebull Can be an impossibly poor estimatebull Requires user input and skillbull Treat cautiouslybull Can be improved by eye (usually)bull Often helps to have colour-codingbull Depending on the use the user should be able to make a judgement
on those regions that are reliable or notbull For phylogeny reconstruction only use those positions whose
hypothesis of positional homology is unimpeachable
Alignment of protein-coding DNA sequences
bull It is not very sensible to align the DNA sequences of protein-coding genes
ATGCTGTTAGGGATGACTCTGTTAGGG
ATG-CT--GTTAGGGATGACTCTGTTAGGG
The result might be highly-implausible and might not reflect what is known about biological processesIt is much more sensible to translate the sequences to their corresponding amino acid sequences align these protein sequences and then put the gaps in the DNA sequences according to where they are found in the amino acid alignment
Manual Alignment- softwareGDE- The Genetic Data Environment (UNIX)CINEMA- Java applet available from
ndash httpwwwbiochemuclacuk
SeqappSeqpup- MacPCUNIX available fromndash httpiubiobioindianaedu
SeAl for Macintosh available fromndash httpevolvezoooxacukSe-AlSe-Alhtml
BioEdit for PC available fromndash httpwwwmbioncsueduRNasePinfoprogramsBIOEDITbio
edithtml
- Multiple Sequence Alignment James McInerney bioinf4biologists Feb 2009
- Alignment can be easy or difficult
- Homology Definition
- Multiple Sequence Alignment- Goals
- Multiple sequence alignments - problems
- Slide 6
- Slide 7
- SSU rRNA
- Alignment of 16S rRNA can be guided by secondary structure
- Protein Alignment may be guided by Tertiary Structure Interactions
- Multiple Sequence Alignment- Methods
- Manual Alignment - reasons
- Local minimum
- Dotplots
- Dotplot example sperm whale vs human myg
- Slide 16
- Slide 17
- Dotplots in practice
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Dynamic programming
- Progressive Alignment
- Slide 23
- ClustalW- Pairwise Alignments
- Path Graph for aligning two sequences
- Possible alignment
- Alignment using this path
- Optimal Alignment 1
- Optimal Alignment 2
- Alignment of 3 sequences
- ClustalW- Guide Tree
- Neighbor joining method
- Distance Matrix
- First Step
- Calculation of New Distances
- Next Cycle
- Penultimate Cycle
- Last Joining
- Unrooted Neighbor-Joining Tree
- Multiple Alignment- First pair
- ClustalW- Decision time
- ClustalW- Alternative 1
- ClustalW- Progression
- Progressive alignment - step 1
- Progressive alignment - step 2
- Progressive alignment - step 3
- Progressive alignment - final step
- ClustalW-Good pointsBad points
- ClustalW-Local Minimum
- Increasing the sophistication of the alignment process
- Slide 51
- ClustalW- Caveats
- ClustalW- User-supplied values
- Position-Specific gap penalties
- Discouraging too many gaps
- Divergent Sequences
- Advice on progressive alignment
- Alignment of protein-coding DNA sequences
- Manual Alignment- software
-

bull This is the result for the whole sequence
bull It is easy to see that the diagonal is a line of dots
bull So sperm whale and human myoglobin are very similar
bull But the picture is noisy can smooth using a sliding window which considers neighbouring residues as well
Dotplot example sperm whale vs human myg
16
Sperm whale myoglobin
G L S D G E W Q L V V L S E G E W Q L V
Human myoglobin
bull can smooth noise using a sliding window which considers neighbouring residues as well
bull Have done this here can see the diagonal is highly similar
bull Also instead of using using a simple identity use a scoring matrix
Dotplot example sperm whale vs human myg
Dotplots in practicebull The best tool is an applet called dotlet
bull wwwisrecisb-sibchjavadotletDotlethtmlbull wwwbipbhamacukdotletDotlethtml
bull an applet is a program that runs in a web browser This means that you can produce dotplots within a netscapeIE window
bull Dotplots are often useful to identify things like repeated domains or duplications in big proteins
Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
bull Protein has many repeats bull SLIT_DROME (P24014)
MAAPSRTTLMPPPFRLQLRLLILPILLLLRHDAVHAEPYSGGFGSSAVSSGGLGSVGIHIPGGGVGVITEARCPRVCSCT GLNVDCSHRGLTSVPRKISADVERLELQGNNLTVIYETDFQRLTKLRMLQLTDNQIHTIERNSFQDLVSLERLDISNNVI TTVGRRVFKGAQSLRSLQLDNNQITCLDEHAFKGLVELEILTLNNNNLTSLPHNIFGGLGRLRALRLSDNPFACDCHLSW LSRFLRSATRLAPYTRCQSPSQLKGQNVADLHDQEFKCSGLTEHAPMECGAENSCPHPCRCADGIVDCREKSLTSVPVTL PDDTTDVRLEQNFITELPPKSFSSFRRLRRIDLSNNNISRIAHDALSGLKQLTTLVLYGNKIKDLPSGVFKGLGSLRLLL LNANEISCIRKDAFRDLHSLSLLSLYDNNIQSLANGTFDAMKSMKTVHLAKNPFICDCNLRWLADYLHKNPIETSGARCE SPKRMHRRRIESLREEKFKCSWGELRMKLSGECRMDSDCPAMCHCEGTTVDCTGRRLKEIPRDIPLHTTELLLNDNELGR ISSDGLFGRLPHLVKLELKRNQLTGIEPNAFEGASHIQELQLGENKIKEISNKMFLGLHQLKTLNLYDNQISCVMPGSFE HLNSLTSLNLASNPFNCNCHLAWFAECVRKKSLNGGAARCGAPSKVRDVQIKDLPHSEFKCSSENSEGCLGDGYCPPSCT CTGTVVACSRNQLKEIPRGIPAETSELYLESNEIEQIHYERIRHLRSLTRLDLSNNQITILSNYTFANLTKLSTLIISYN KLQCLQRHALSGLNNLRVVSLHGNRISMLPEGSFEDLKSLTHIALGSNPLYCDCGLKWFSDWIKLDYVEPGIARCAEPEQ MKDKLILSTPSSSFVCRGRVRNDILAKCNACFEQPCQNQAQCVALPQREYQCLCQPGYHGKHCEFMIDACYGNPCRNNAT CTVLEEGRFSCQCAPGYTGARCETNIDDCLGEIKCQNNATCIDGVESYKCECQPGFSGEFCDTKIQFCSPEFNPCANGAK CMDHFTHYSCDCQAGFHGTNCTDNIDDCQNHMCQNGGTCVDGINDYQCRCPDDYTGKYCEGHNMISMMYPQTSPCQNHEC KHGVCFQPNAQGSDYLCRCHPGYTGKWCEYLTSISFVHNNSFVELEPLRTRPEANVTIVFSSAEQNGILMYDGQDAHLAV ELFNGRIRVSYDVGNHPVSTMYSFEMVADGKYHAVELLAIKKNFTLRVDRGLARSIINEGSNDYLKLTTPMFLGGLPVDP AQQAYKNWQIRNLTSFKGCMKEVWINHKLVDFGNAQRQQKITPGCALLEGEQQEEEDDEQDFMDETPHIKEEPVDPCLEN KCRRGSRCVPNSNARDGYQCKCKHGQRGRYCDQGEGSTEPPTVTAASTCRKEQVREYYTENDCRSRQPLKYAKCVGGCGN QCCAAKIVRRRKVRMVCSNNRKYIKNLDIVRKCGCTKKCY
bull Perform a dotplot of the SLIT protein against itself wwwbiobhamacukdotletDotlethtml
Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
20Swiss-prot entry
For further discussion of dotplot see Attwood and Parry-Smith p116-8
Dynamic programming2 methodsbull Dynamic programming
ndash Consider 2 protein sequences of 100 amino acids in lengthndash If it takes 1002 seconds to exhaustively align these sequences
then it will take 1003 seconds to align 3 sequences 1004 to align 4 sequencesetc
ndash More time than the universe has existed to align 20 sequences exhaustively
bull Progressive alignment
Progressive Alignmentbull Devised by Feng and Doolittle in 1987bull Essentially a heuristic method and as such
is not guaranteed to find the lsquooptimalrsquo alignment
bull Requires n-1+n-2+n-3n-n+1 pairwise alignments as a starting point
bull Most successful implementation is Clustal (Des Higgins) This software is cited 3000 times per year in the scientific literature
Overview of ClustalW Procedure
1 PEEKSAVTALWGKVN--VDEVGG2 GEEKAAVLALWDKVN--EEEVGG3 PADKTNVKAAWGKVGAHAGEYGA4 AADKTNVKAAWSKVGGHAGEYGA5 EHEWQLVLHVWAKVEADVAGHGQ
Hbb_Human 1 -Hbb_Horse 2 17 -Hba_Human 3 59 60 -Hba_Horse 4 59 59 13 -Myg_Whale 5 77 77 75 75 -
Hbb_Human
Hbb_Horse
Hba_Horse
Hba_Human
Myg_Whale
2
1
3 4
2
1
3 4
alpha-helices
Quick pairwise alignment calculate distance matrix
Neighbor-joining tree(guide tree)
Progressive alignment following guide tree
CLUSTAL W
ClustalW- Pairwise Alignments
bull First perform all possible pairwise alignments between each pair of sequences There are (n-1)+(n-2)(n-n+1) possibilities
bull Calculate the lsquodistancersquo between each pair of sequences based on these isolated pairwise alignments
bull Generate a distance matrix
Path Graph for aligning two sequences
Possible alignment
1
1
0
1
0
-1
Scoring SchemebullMatch +1bullMismatch 0bullIndel -1
Score for this path= 2
Alignment using this path
GATTC-GAATTC
1
1
0
1
0
-1
Optimal Alignment 1
1
1
-1
1
1
1
Alignment score 4Alignment score 4
Alignment using this path
GA-TTCGA-TTCGAATTCGAATTC
Optimal Alignment 2
1
-1
1
1
1
1
Alignment score 4Alignment score 4
Alignment using this path
G-ATTCG-ATTCGAATTCGAATTC
Alignment of 3 sequences
ClustalW- Guide Tree
bull Generate a Neighbor-Joining lsquoguide treersquo from these pairwise distances
bull This guide tree gives the order in which the progressive alignment will be carried out
Neighbor joining method
bullThe neighbor-joining method is a greedy heuristic which joins at each step the two closest sub-trees that are not already joinedbullIt is based on the minimum evolution principlebullneighbors are defined as two taxa that are connected by a single node in an unrooted tree
A B
Node 1
PAM Spinach Rice Mosquito Monkey HumanSpinach 00 849 1056 908 863Rice 849 00 1178 1224 1226Mosquito 1056 1178 00 847 808Monkey 908 1224 847 00 33Human 863 1226 808 33 00
What is required for the Neighbour joining method
Distance matrixDistance Matrix
PAM distance 33 (Human - Monkey) is the minimum So well join Human and Monkey to MonHum and well calculate the new distances
Mon-Hum
MonkeyHumanSpinachMosquito Rice
First Step
After we have joined two species in a subtree we have to compute the distances from every other node to the new subtree We do this with a simple average of distancesDist[Spinach MonHum]
= (Dist[Spinach Monkey] + Dist[Spinach Human])2 = (908 + 863)2 = 8855
Mon-Hum
MonkeyHumanSpinach
Calculation of New Distances
PAM Spinach Rice Mosquito MonHumSpinach 00 849 1056 886Rice 849 00 1178 1225Mosquito 1056 1178 00 828MonHum 886 1225 828 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Next Cycle
PAM Spinach Rice MosMonHumSpinach 00 849 971Rice 849 00 1202MosMonHum 971 1202 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
Penultimate Cycle
PAM SpinRice MosMonHumSpinach 00 1087MosMonHum 1087 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
(Spin-Rice)-(Mos-(Mon-Hum))
Last Joining
Human
Monkey
MosquitoRice
Spinach
Unrooted Neighbor-Joining Tree
Multiple Alignment- First pairbull Align the two most closely-related sequences first
bull This alignment is then lsquofixedrsquo and will never change If a gap is to be introduced subsequently then it will be introduced in the same place in both sequences but their relative alignment remains unchanged
ClustalW- Decision timebull Consult the guide tree to see what alignment is performed next
ndash Align a third sequence to the first twoOrndash Align two entirely different sequences to each other
Option 1Option 1 Option 2Option 2
ClustalW- Alternative 1If the situation arises where a third sequence is aligned to the first two then when a gap has to be introduced to improve the alignment each of these two entities are treated as two single sequences
+
ClustalW- Alternative 2bull If on the other hand two separate
sequences have to be aligned together then the first pairwise alignment is placed to one side and the pairwise alignment of the other two is carried out
+
ClustalW- Progression
bull The alignment is progressively built up in this way with each step being treated as a pairwise alignment sometimes with each member of a lsquopairrsquo having more than one sequence
Progressive alignment - step 11 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
12345
Progressive alignment - step 21 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
12345
Progressive alignment - step 31 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
+
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
12345
Progressive alignment - final step1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
+
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
5 -ctcga-acgatacgatgactagct-
12345
ClustalW-Good pointsBad points
bull Advantagesndash Speed
bull Disadvantagesndash No objective functionndash No way of quantifying whether or not the alignment is good
ndash No way of knowing if the alignment is lsquocorrectrsquo
ClustalW-Local Minimumbull Potential problems
ndash Local minimum problem If an error is introduced early in the alignment process it is impossible to correct this later in the procedure
ndash Arbitrary alignment
Increasing the sophistication of the alignment process
bull Should we treat all the sequences in the same way - even though some sequences are closely-related and some sequences are distant relatives
bull Should we treat all positions in the sequences as though they were the same - even though they might have different functions and different locations in the 3-dimensional structure
ClustalW- Caveatsbull Sequence weightingbull Varying substitution matricesbull Residue-specific gap penalties and reduced penalties
in hydrophilic regions (external regions of protein sequences) encourage gaps in loops rather than in core regions
bull Positions in early alignments where gaps have been opened receive locally reduced gap penalties to encourage openings in subsequent alignments
ClustalW- User-supplied values
bull Two penalties are set by the user (there are default values but you should know that it is possible to change these)
bull GOP- Gap Opening Penalty is the cost of opening a gap in an alignment
bull GEP- Gap Extension Penalty is the cost of extending this gap
Position-Specific gap penaltiesbull Before any pair of (groups of) sequences are
aligned a table of GOPs are generated for each position in the two (sets of) sequences
bull The GOP is manipulated in a position-specific manner so that it can vary over the sequences
bull If there is a gap at a position the GOP and GEP penalties are lowered the other rules do not apply
bull This makes gaps more likely at positions where gaps already exist
Discouraging too many gaps bull If there is no gap opened then the GOP is increased if the
position is within 8 residues of an existing gapbull This discourages gaps that are too close togetherbull At any position within a run of hydrophilic residues the GOP is
decreasedbull These runs usually indicate loop regions in protein structuresbull A run of 5 hydrophilic residues is considered to be a hydrophilic
stretchbull The default hydrophilic residues are
ndash D E G K N Q P R Sndash But this can be changed by the user
Divergent Sequencesbull The most divergent sequences (most different on average
from all of the other sequences) are usually the most difficult to align
bull It is sometimes better to delay their aligment until later (when the easier sequences have already been aligned)
bull The user has the choice of setting a cutoff (default is 40 identity)
bull This will delay the alignment until the others have been aligned
Advice on progressive alignmentbull Progressive alignment is a mathematical process that is completely
independent of biological realitybull Can be a very good estimatebull Can be an impossibly poor estimatebull Requires user input and skillbull Treat cautiouslybull Can be improved by eye (usually)bull Often helps to have colour-codingbull Depending on the use the user should be able to make a judgement
on those regions that are reliable or notbull For phylogeny reconstruction only use those positions whose
hypothesis of positional homology is unimpeachable
Alignment of protein-coding DNA sequences
bull It is not very sensible to align the DNA sequences of protein-coding genes
ATGCTGTTAGGGATGACTCTGTTAGGG
ATG-CT--GTTAGGGATGACTCTGTTAGGG
The result might be highly-implausible and might not reflect what is known about biological processesIt is much more sensible to translate the sequences to their corresponding amino acid sequences align these protein sequences and then put the gaps in the DNA sequences according to where they are found in the amino acid alignment
Manual Alignment- softwareGDE- The Genetic Data Environment (UNIX)CINEMA- Java applet available from
ndash httpwwwbiochemuclacuk
SeqappSeqpup- MacPCUNIX available fromndash httpiubiobioindianaedu
SeAl for Macintosh available fromndash httpevolvezoooxacukSe-AlSe-Alhtml
BioEdit for PC available fromndash httpwwwmbioncsueduRNasePinfoprogramsBIOEDITbio
edithtml
- Multiple Sequence Alignment James McInerney bioinf4biologists Feb 2009
- Alignment can be easy or difficult
- Homology Definition
- Multiple Sequence Alignment- Goals
- Multiple sequence alignments - problems
- Slide 6
- Slide 7
- SSU rRNA
- Alignment of 16S rRNA can be guided by secondary structure
- Protein Alignment may be guided by Tertiary Structure Interactions
- Multiple Sequence Alignment- Methods
- Manual Alignment - reasons
- Local minimum
- Dotplots
- Dotplot example sperm whale vs human myg
- Slide 16
- Slide 17
- Dotplots in practice
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Dynamic programming
- Progressive Alignment
- Slide 23
- ClustalW- Pairwise Alignments
- Path Graph for aligning two sequences
- Possible alignment
- Alignment using this path
- Optimal Alignment 1
- Optimal Alignment 2
- Alignment of 3 sequences
- ClustalW- Guide Tree
- Neighbor joining method
- Distance Matrix
- First Step
- Calculation of New Distances
- Next Cycle
- Penultimate Cycle
- Last Joining
- Unrooted Neighbor-Joining Tree
- Multiple Alignment- First pair
- ClustalW- Decision time
- ClustalW- Alternative 1
- ClustalW- Progression
- Progressive alignment - step 1
- Progressive alignment - step 2
- Progressive alignment - step 3
- Progressive alignment - final step
- ClustalW-Good pointsBad points
- ClustalW-Local Minimum
- Increasing the sophistication of the alignment process
- Slide 51
- ClustalW- Caveats
- ClustalW- User-supplied values
- Position-Specific gap penalties
- Discouraging too many gaps
- Divergent Sequences
- Advice on progressive alignment
- Alignment of protein-coding DNA sequences
- Manual Alignment- software
-

bull can smooth noise using a sliding window which considers neighbouring residues as well
bull Have done this here can see the diagonal is highly similar
bull Also instead of using using a simple identity use a scoring matrix
Dotplot example sperm whale vs human myg
Dotplots in practicebull The best tool is an applet called dotlet
bull wwwisrecisb-sibchjavadotletDotlethtmlbull wwwbipbhamacukdotletDotlethtml
bull an applet is a program that runs in a web browser This means that you can produce dotplots within a netscapeIE window
bull Dotplots are often useful to identify things like repeated domains or duplications in big proteins
Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
bull Protein has many repeats bull SLIT_DROME (P24014)
MAAPSRTTLMPPPFRLQLRLLILPILLLLRHDAVHAEPYSGGFGSSAVSSGGLGSVGIHIPGGGVGVITEARCPRVCSCT GLNVDCSHRGLTSVPRKISADVERLELQGNNLTVIYETDFQRLTKLRMLQLTDNQIHTIERNSFQDLVSLERLDISNNVI TTVGRRVFKGAQSLRSLQLDNNQITCLDEHAFKGLVELEILTLNNNNLTSLPHNIFGGLGRLRALRLSDNPFACDCHLSW LSRFLRSATRLAPYTRCQSPSQLKGQNVADLHDQEFKCSGLTEHAPMECGAENSCPHPCRCADGIVDCREKSLTSVPVTL PDDTTDVRLEQNFITELPPKSFSSFRRLRRIDLSNNNISRIAHDALSGLKQLTTLVLYGNKIKDLPSGVFKGLGSLRLLL LNANEISCIRKDAFRDLHSLSLLSLYDNNIQSLANGTFDAMKSMKTVHLAKNPFICDCNLRWLADYLHKNPIETSGARCE SPKRMHRRRIESLREEKFKCSWGELRMKLSGECRMDSDCPAMCHCEGTTVDCTGRRLKEIPRDIPLHTTELLLNDNELGR ISSDGLFGRLPHLVKLELKRNQLTGIEPNAFEGASHIQELQLGENKIKEISNKMFLGLHQLKTLNLYDNQISCVMPGSFE HLNSLTSLNLASNPFNCNCHLAWFAECVRKKSLNGGAARCGAPSKVRDVQIKDLPHSEFKCSSENSEGCLGDGYCPPSCT CTGTVVACSRNQLKEIPRGIPAETSELYLESNEIEQIHYERIRHLRSLTRLDLSNNQITILSNYTFANLTKLSTLIISYN KLQCLQRHALSGLNNLRVVSLHGNRISMLPEGSFEDLKSLTHIALGSNPLYCDCGLKWFSDWIKLDYVEPGIARCAEPEQ MKDKLILSTPSSSFVCRGRVRNDILAKCNACFEQPCQNQAQCVALPQREYQCLCQPGYHGKHCEFMIDACYGNPCRNNAT CTVLEEGRFSCQCAPGYTGARCETNIDDCLGEIKCQNNATCIDGVESYKCECQPGFSGEFCDTKIQFCSPEFNPCANGAK CMDHFTHYSCDCQAGFHGTNCTDNIDDCQNHMCQNGGTCVDGINDYQCRCPDDYTGKYCEGHNMISMMYPQTSPCQNHEC KHGVCFQPNAQGSDYLCRCHPGYTGKWCEYLTSISFVHNNSFVELEPLRTRPEANVTIVFSSAEQNGILMYDGQDAHLAV ELFNGRIRVSYDVGNHPVSTMYSFEMVADGKYHAVELLAIKKNFTLRVDRGLARSIINEGSNDYLKLTTPMFLGGLPVDP AQQAYKNWQIRNLTSFKGCMKEVWINHKLVDFGNAQRQQKITPGCALLEGEQQEEEDDEQDFMDETPHIKEEPVDPCLEN KCRRGSRCVPNSNARDGYQCKCKHGQRGRYCDQGEGSTEPPTVTAASTCRKEQVREYYTENDCRSRQPLKYAKCVGGCGN QCCAAKIVRRRKVRMVCSNNRKYIKNLDIVRKCGCTKKCY
bull Perform a dotplot of the SLIT protein against itself wwwbiobhamacukdotletDotlethtml
Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
20Swiss-prot entry
For further discussion of dotplot see Attwood and Parry-Smith p116-8
Dynamic programming2 methodsbull Dynamic programming
ndash Consider 2 protein sequences of 100 amino acids in lengthndash If it takes 1002 seconds to exhaustively align these sequences
then it will take 1003 seconds to align 3 sequences 1004 to align 4 sequencesetc
ndash More time than the universe has existed to align 20 sequences exhaustively
bull Progressive alignment
Progressive Alignmentbull Devised by Feng and Doolittle in 1987bull Essentially a heuristic method and as such
is not guaranteed to find the lsquooptimalrsquo alignment
bull Requires n-1+n-2+n-3n-n+1 pairwise alignments as a starting point
bull Most successful implementation is Clustal (Des Higgins) This software is cited 3000 times per year in the scientific literature
Overview of ClustalW Procedure
1 PEEKSAVTALWGKVN--VDEVGG2 GEEKAAVLALWDKVN--EEEVGG3 PADKTNVKAAWGKVGAHAGEYGA4 AADKTNVKAAWSKVGGHAGEYGA5 EHEWQLVLHVWAKVEADVAGHGQ
Hbb_Human 1 -Hbb_Horse 2 17 -Hba_Human 3 59 60 -Hba_Horse 4 59 59 13 -Myg_Whale 5 77 77 75 75 -
Hbb_Human
Hbb_Horse
Hba_Horse
Hba_Human
Myg_Whale
2
1
3 4
2
1
3 4
alpha-helices
Quick pairwise alignment calculate distance matrix
Neighbor-joining tree(guide tree)
Progressive alignment following guide tree
CLUSTAL W
ClustalW- Pairwise Alignments
bull First perform all possible pairwise alignments between each pair of sequences There are (n-1)+(n-2)(n-n+1) possibilities
bull Calculate the lsquodistancersquo between each pair of sequences based on these isolated pairwise alignments
bull Generate a distance matrix
Path Graph for aligning two sequences
Possible alignment
1
1
0
1
0
-1
Scoring SchemebullMatch +1bullMismatch 0bullIndel -1
Score for this path= 2
Alignment using this path
GATTC-GAATTC
1
1
0
1
0
-1
Optimal Alignment 1
1
1
-1
1
1
1
Alignment score 4Alignment score 4
Alignment using this path
GA-TTCGA-TTCGAATTCGAATTC
Optimal Alignment 2
1
-1
1
1
1
1
Alignment score 4Alignment score 4
Alignment using this path
G-ATTCG-ATTCGAATTCGAATTC
Alignment of 3 sequences
ClustalW- Guide Tree
bull Generate a Neighbor-Joining lsquoguide treersquo from these pairwise distances
bull This guide tree gives the order in which the progressive alignment will be carried out
Neighbor joining method
bullThe neighbor-joining method is a greedy heuristic which joins at each step the two closest sub-trees that are not already joinedbullIt is based on the minimum evolution principlebullneighbors are defined as two taxa that are connected by a single node in an unrooted tree
A B
Node 1
PAM Spinach Rice Mosquito Monkey HumanSpinach 00 849 1056 908 863Rice 849 00 1178 1224 1226Mosquito 1056 1178 00 847 808Monkey 908 1224 847 00 33Human 863 1226 808 33 00
What is required for the Neighbour joining method
Distance matrixDistance Matrix
PAM distance 33 (Human - Monkey) is the minimum So well join Human and Monkey to MonHum and well calculate the new distances
Mon-Hum
MonkeyHumanSpinachMosquito Rice
First Step
After we have joined two species in a subtree we have to compute the distances from every other node to the new subtree We do this with a simple average of distancesDist[Spinach MonHum]
= (Dist[Spinach Monkey] + Dist[Spinach Human])2 = (908 + 863)2 = 8855
Mon-Hum
MonkeyHumanSpinach
Calculation of New Distances
PAM Spinach Rice Mosquito MonHumSpinach 00 849 1056 886Rice 849 00 1178 1225Mosquito 1056 1178 00 828MonHum 886 1225 828 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Next Cycle
PAM Spinach Rice MosMonHumSpinach 00 849 971Rice 849 00 1202MosMonHum 971 1202 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
Penultimate Cycle
PAM SpinRice MosMonHumSpinach 00 1087MosMonHum 1087 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
(Spin-Rice)-(Mos-(Mon-Hum))
Last Joining
Human
Monkey
MosquitoRice
Spinach
Unrooted Neighbor-Joining Tree
Multiple Alignment- First pairbull Align the two most closely-related sequences first
bull This alignment is then lsquofixedrsquo and will never change If a gap is to be introduced subsequently then it will be introduced in the same place in both sequences but their relative alignment remains unchanged
ClustalW- Decision timebull Consult the guide tree to see what alignment is performed next
ndash Align a third sequence to the first twoOrndash Align two entirely different sequences to each other
Option 1Option 1 Option 2Option 2
ClustalW- Alternative 1If the situation arises where a third sequence is aligned to the first two then when a gap has to be introduced to improve the alignment each of these two entities are treated as two single sequences
+
ClustalW- Alternative 2bull If on the other hand two separate
sequences have to be aligned together then the first pairwise alignment is placed to one side and the pairwise alignment of the other two is carried out
+
ClustalW- Progression
bull The alignment is progressively built up in this way with each step being treated as a pairwise alignment sometimes with each member of a lsquopairrsquo having more than one sequence
Progressive alignment - step 11 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
12345
Progressive alignment - step 21 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
12345
Progressive alignment - step 31 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
+
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
12345
Progressive alignment - final step1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
+
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
5 -ctcga-acgatacgatgactagct-
12345
ClustalW-Good pointsBad points
bull Advantagesndash Speed
bull Disadvantagesndash No objective functionndash No way of quantifying whether or not the alignment is good
ndash No way of knowing if the alignment is lsquocorrectrsquo
ClustalW-Local Minimumbull Potential problems
ndash Local minimum problem If an error is introduced early in the alignment process it is impossible to correct this later in the procedure
ndash Arbitrary alignment
Increasing the sophistication of the alignment process
bull Should we treat all the sequences in the same way - even though some sequences are closely-related and some sequences are distant relatives
bull Should we treat all positions in the sequences as though they were the same - even though they might have different functions and different locations in the 3-dimensional structure
ClustalW- Caveatsbull Sequence weightingbull Varying substitution matricesbull Residue-specific gap penalties and reduced penalties
in hydrophilic regions (external regions of protein sequences) encourage gaps in loops rather than in core regions
bull Positions in early alignments where gaps have been opened receive locally reduced gap penalties to encourage openings in subsequent alignments
ClustalW- User-supplied values
bull Two penalties are set by the user (there are default values but you should know that it is possible to change these)
bull GOP- Gap Opening Penalty is the cost of opening a gap in an alignment
bull GEP- Gap Extension Penalty is the cost of extending this gap
Position-Specific gap penaltiesbull Before any pair of (groups of) sequences are
aligned a table of GOPs are generated for each position in the two (sets of) sequences
bull The GOP is manipulated in a position-specific manner so that it can vary over the sequences
bull If there is a gap at a position the GOP and GEP penalties are lowered the other rules do not apply
bull This makes gaps more likely at positions where gaps already exist
Discouraging too many gaps bull If there is no gap opened then the GOP is increased if the
position is within 8 residues of an existing gapbull This discourages gaps that are too close togetherbull At any position within a run of hydrophilic residues the GOP is
decreasedbull These runs usually indicate loop regions in protein structuresbull A run of 5 hydrophilic residues is considered to be a hydrophilic
stretchbull The default hydrophilic residues are
ndash D E G K N Q P R Sndash But this can be changed by the user
Divergent Sequencesbull The most divergent sequences (most different on average
from all of the other sequences) are usually the most difficult to align
bull It is sometimes better to delay their aligment until later (when the easier sequences have already been aligned)
bull The user has the choice of setting a cutoff (default is 40 identity)
bull This will delay the alignment until the others have been aligned
Advice on progressive alignmentbull Progressive alignment is a mathematical process that is completely
independent of biological realitybull Can be a very good estimatebull Can be an impossibly poor estimatebull Requires user input and skillbull Treat cautiouslybull Can be improved by eye (usually)bull Often helps to have colour-codingbull Depending on the use the user should be able to make a judgement
on those regions that are reliable or notbull For phylogeny reconstruction only use those positions whose
hypothesis of positional homology is unimpeachable
Alignment of protein-coding DNA sequences
bull It is not very sensible to align the DNA sequences of protein-coding genes
ATGCTGTTAGGGATGACTCTGTTAGGG
ATG-CT--GTTAGGGATGACTCTGTTAGGG
The result might be highly-implausible and might not reflect what is known about biological processesIt is much more sensible to translate the sequences to their corresponding amino acid sequences align these protein sequences and then put the gaps in the DNA sequences according to where they are found in the amino acid alignment
Manual Alignment- softwareGDE- The Genetic Data Environment (UNIX)CINEMA- Java applet available from
ndash httpwwwbiochemuclacuk
SeqappSeqpup- MacPCUNIX available fromndash httpiubiobioindianaedu
SeAl for Macintosh available fromndash httpevolvezoooxacukSe-AlSe-Alhtml
BioEdit for PC available fromndash httpwwwmbioncsueduRNasePinfoprogramsBIOEDITbio
edithtml
- Multiple Sequence Alignment James McInerney bioinf4biologists Feb 2009
- Alignment can be easy or difficult
- Homology Definition
- Multiple Sequence Alignment- Goals
- Multiple sequence alignments - problems
- Slide 6
- Slide 7
- SSU rRNA
- Alignment of 16S rRNA can be guided by secondary structure
- Protein Alignment may be guided by Tertiary Structure Interactions
- Multiple Sequence Alignment- Methods
- Manual Alignment - reasons
- Local minimum
- Dotplots
- Dotplot example sperm whale vs human myg
- Slide 16
- Slide 17
- Dotplots in practice
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Dynamic programming
- Progressive Alignment
- Slide 23
- ClustalW- Pairwise Alignments
- Path Graph for aligning two sequences
- Possible alignment
- Alignment using this path
- Optimal Alignment 1
- Optimal Alignment 2
- Alignment of 3 sequences
- ClustalW- Guide Tree
- Neighbor joining method
- Distance Matrix
- First Step
- Calculation of New Distances
- Next Cycle
- Penultimate Cycle
- Last Joining
- Unrooted Neighbor-Joining Tree
- Multiple Alignment- First pair
- ClustalW- Decision time
- ClustalW- Alternative 1
- ClustalW- Progression
- Progressive alignment - step 1
- Progressive alignment - step 2
- Progressive alignment - step 3
- Progressive alignment - final step
- ClustalW-Good pointsBad points
- ClustalW-Local Minimum
- Increasing the sophistication of the alignment process
- Slide 51
- ClustalW- Caveats
- ClustalW- User-supplied values
- Position-Specific gap penalties
- Discouraging too many gaps
- Divergent Sequences
- Advice on progressive alignment
- Alignment of protein-coding DNA sequences
- Manual Alignment- software
-

Dotplots in practicebull The best tool is an applet called dotlet
bull wwwisrecisb-sibchjavadotletDotlethtmlbull wwwbipbhamacukdotletDotlethtml
bull an applet is a program that runs in a web browser This means that you can produce dotplots within a netscapeIE window
bull Dotplots are often useful to identify things like repeated domains or duplications in big proteins
Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
bull Protein has many repeats bull SLIT_DROME (P24014)
MAAPSRTTLMPPPFRLQLRLLILPILLLLRHDAVHAEPYSGGFGSSAVSSGGLGSVGIHIPGGGVGVITEARCPRVCSCT GLNVDCSHRGLTSVPRKISADVERLELQGNNLTVIYETDFQRLTKLRMLQLTDNQIHTIERNSFQDLVSLERLDISNNVI TTVGRRVFKGAQSLRSLQLDNNQITCLDEHAFKGLVELEILTLNNNNLTSLPHNIFGGLGRLRALRLSDNPFACDCHLSW LSRFLRSATRLAPYTRCQSPSQLKGQNVADLHDQEFKCSGLTEHAPMECGAENSCPHPCRCADGIVDCREKSLTSVPVTL PDDTTDVRLEQNFITELPPKSFSSFRRLRRIDLSNNNISRIAHDALSGLKQLTTLVLYGNKIKDLPSGVFKGLGSLRLLL LNANEISCIRKDAFRDLHSLSLLSLYDNNIQSLANGTFDAMKSMKTVHLAKNPFICDCNLRWLADYLHKNPIETSGARCE SPKRMHRRRIESLREEKFKCSWGELRMKLSGECRMDSDCPAMCHCEGTTVDCTGRRLKEIPRDIPLHTTELLLNDNELGR ISSDGLFGRLPHLVKLELKRNQLTGIEPNAFEGASHIQELQLGENKIKEISNKMFLGLHQLKTLNLYDNQISCVMPGSFE HLNSLTSLNLASNPFNCNCHLAWFAECVRKKSLNGGAARCGAPSKVRDVQIKDLPHSEFKCSSENSEGCLGDGYCPPSCT CTGTVVACSRNQLKEIPRGIPAETSELYLESNEIEQIHYERIRHLRSLTRLDLSNNQITILSNYTFANLTKLSTLIISYN KLQCLQRHALSGLNNLRVVSLHGNRISMLPEGSFEDLKSLTHIALGSNPLYCDCGLKWFSDWIKLDYVEPGIARCAEPEQ MKDKLILSTPSSSFVCRGRVRNDILAKCNACFEQPCQNQAQCVALPQREYQCLCQPGYHGKHCEFMIDACYGNPCRNNAT CTVLEEGRFSCQCAPGYTGARCETNIDDCLGEIKCQNNATCIDGVESYKCECQPGFSGEFCDTKIQFCSPEFNPCANGAK CMDHFTHYSCDCQAGFHGTNCTDNIDDCQNHMCQNGGTCVDGINDYQCRCPDDYTGKYCEGHNMISMMYPQTSPCQNHEC KHGVCFQPNAQGSDYLCRCHPGYTGKWCEYLTSISFVHNNSFVELEPLRTRPEANVTIVFSSAEQNGILMYDGQDAHLAV ELFNGRIRVSYDVGNHPVSTMYSFEMVADGKYHAVELLAIKKNFTLRVDRGLARSIINEGSNDYLKLTTPMFLGGLPVDP AQQAYKNWQIRNLTSFKGCMKEVWINHKLVDFGNAQRQQKITPGCALLEGEQQEEEDDEQDFMDETPHIKEEPVDPCLEN KCRRGSRCVPNSNARDGYQCKCKHGQRGRYCDQGEGSTEPPTVTAASTCRKEQVREYYTENDCRSRQPLKYAKCVGGCGN QCCAAKIVRRRKVRMVCSNNRKYIKNLDIVRKCGCTKKCY
bull Perform a dotplot of the SLIT protein against itself wwwbiobhamacukdotletDotlethtml
Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
20Swiss-prot entry
For further discussion of dotplot see Attwood and Parry-Smith p116-8
Dynamic programming2 methodsbull Dynamic programming
ndash Consider 2 protein sequences of 100 amino acids in lengthndash If it takes 1002 seconds to exhaustively align these sequences
then it will take 1003 seconds to align 3 sequences 1004 to align 4 sequencesetc
ndash More time than the universe has existed to align 20 sequences exhaustively
bull Progressive alignment
Progressive Alignmentbull Devised by Feng and Doolittle in 1987bull Essentially a heuristic method and as such
is not guaranteed to find the lsquooptimalrsquo alignment
bull Requires n-1+n-2+n-3n-n+1 pairwise alignments as a starting point
bull Most successful implementation is Clustal (Des Higgins) This software is cited 3000 times per year in the scientific literature
Overview of ClustalW Procedure
1 PEEKSAVTALWGKVN--VDEVGG2 GEEKAAVLALWDKVN--EEEVGG3 PADKTNVKAAWGKVGAHAGEYGA4 AADKTNVKAAWSKVGGHAGEYGA5 EHEWQLVLHVWAKVEADVAGHGQ
Hbb_Human 1 -Hbb_Horse 2 17 -Hba_Human 3 59 60 -Hba_Horse 4 59 59 13 -Myg_Whale 5 77 77 75 75 -
Hbb_Human
Hbb_Horse
Hba_Horse
Hba_Human
Myg_Whale
2
1
3 4
2
1
3 4
alpha-helices
Quick pairwise alignment calculate distance matrix
Neighbor-joining tree(guide tree)
Progressive alignment following guide tree
CLUSTAL W
ClustalW- Pairwise Alignments
bull First perform all possible pairwise alignments between each pair of sequences There are (n-1)+(n-2)(n-n+1) possibilities
bull Calculate the lsquodistancersquo between each pair of sequences based on these isolated pairwise alignments
bull Generate a distance matrix
Path Graph for aligning two sequences
Possible alignment
1
1
0
1
0
-1
Scoring SchemebullMatch +1bullMismatch 0bullIndel -1
Score for this path= 2
Alignment using this path
GATTC-GAATTC
1
1
0
1
0
-1
Optimal Alignment 1
1
1
-1
1
1
1
Alignment score 4Alignment score 4
Alignment using this path
GA-TTCGA-TTCGAATTCGAATTC
Optimal Alignment 2
1
-1
1
1
1
1
Alignment score 4Alignment score 4
Alignment using this path
G-ATTCG-ATTCGAATTCGAATTC
Alignment of 3 sequences
ClustalW- Guide Tree
bull Generate a Neighbor-Joining lsquoguide treersquo from these pairwise distances
bull This guide tree gives the order in which the progressive alignment will be carried out
Neighbor joining method
bullThe neighbor-joining method is a greedy heuristic which joins at each step the two closest sub-trees that are not already joinedbullIt is based on the minimum evolution principlebullneighbors are defined as two taxa that are connected by a single node in an unrooted tree
A B
Node 1
PAM Spinach Rice Mosquito Monkey HumanSpinach 00 849 1056 908 863Rice 849 00 1178 1224 1226Mosquito 1056 1178 00 847 808Monkey 908 1224 847 00 33Human 863 1226 808 33 00
What is required for the Neighbour joining method
Distance matrixDistance Matrix
PAM distance 33 (Human - Monkey) is the minimum So well join Human and Monkey to MonHum and well calculate the new distances
Mon-Hum
MonkeyHumanSpinachMosquito Rice
First Step
After we have joined two species in a subtree we have to compute the distances from every other node to the new subtree We do this with a simple average of distancesDist[Spinach MonHum]
= (Dist[Spinach Monkey] + Dist[Spinach Human])2 = (908 + 863)2 = 8855
Mon-Hum
MonkeyHumanSpinach
Calculation of New Distances
PAM Spinach Rice Mosquito MonHumSpinach 00 849 1056 886Rice 849 00 1178 1225Mosquito 1056 1178 00 828MonHum 886 1225 828 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Next Cycle
PAM Spinach Rice MosMonHumSpinach 00 849 971Rice 849 00 1202MosMonHum 971 1202 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
Penultimate Cycle
PAM SpinRice MosMonHumSpinach 00 1087MosMonHum 1087 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
(Spin-Rice)-(Mos-(Mon-Hum))
Last Joining
Human
Monkey
MosquitoRice
Spinach
Unrooted Neighbor-Joining Tree
Multiple Alignment- First pairbull Align the two most closely-related sequences first
bull This alignment is then lsquofixedrsquo and will never change If a gap is to be introduced subsequently then it will be introduced in the same place in both sequences but their relative alignment remains unchanged
ClustalW- Decision timebull Consult the guide tree to see what alignment is performed next
ndash Align a third sequence to the first twoOrndash Align two entirely different sequences to each other
Option 1Option 1 Option 2Option 2
ClustalW- Alternative 1If the situation arises where a third sequence is aligned to the first two then when a gap has to be introduced to improve the alignment each of these two entities are treated as two single sequences
+
ClustalW- Alternative 2bull If on the other hand two separate
sequences have to be aligned together then the first pairwise alignment is placed to one side and the pairwise alignment of the other two is carried out
+
ClustalW- Progression
bull The alignment is progressively built up in this way with each step being treated as a pairwise alignment sometimes with each member of a lsquopairrsquo having more than one sequence
Progressive alignment - step 11 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
12345
Progressive alignment - step 21 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
12345
Progressive alignment - step 31 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
+
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
12345
Progressive alignment - final step1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
+
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
5 -ctcga-acgatacgatgactagct-
12345
ClustalW-Good pointsBad points
bull Advantagesndash Speed
bull Disadvantagesndash No objective functionndash No way of quantifying whether or not the alignment is good
ndash No way of knowing if the alignment is lsquocorrectrsquo
ClustalW-Local Minimumbull Potential problems
ndash Local minimum problem If an error is introduced early in the alignment process it is impossible to correct this later in the procedure
ndash Arbitrary alignment
Increasing the sophistication of the alignment process
bull Should we treat all the sequences in the same way - even though some sequences are closely-related and some sequences are distant relatives
bull Should we treat all positions in the sequences as though they were the same - even though they might have different functions and different locations in the 3-dimensional structure
ClustalW- Caveatsbull Sequence weightingbull Varying substitution matricesbull Residue-specific gap penalties and reduced penalties
in hydrophilic regions (external regions of protein sequences) encourage gaps in loops rather than in core regions
bull Positions in early alignments where gaps have been opened receive locally reduced gap penalties to encourage openings in subsequent alignments
ClustalW- User-supplied values
bull Two penalties are set by the user (there are default values but you should know that it is possible to change these)
bull GOP- Gap Opening Penalty is the cost of opening a gap in an alignment
bull GEP- Gap Extension Penalty is the cost of extending this gap
Position-Specific gap penaltiesbull Before any pair of (groups of) sequences are
aligned a table of GOPs are generated for each position in the two (sets of) sequences
bull The GOP is manipulated in a position-specific manner so that it can vary over the sequences
bull If there is a gap at a position the GOP and GEP penalties are lowered the other rules do not apply
bull This makes gaps more likely at positions where gaps already exist
Discouraging too many gaps bull If there is no gap opened then the GOP is increased if the
position is within 8 residues of an existing gapbull This discourages gaps that are too close togetherbull At any position within a run of hydrophilic residues the GOP is
decreasedbull These runs usually indicate loop regions in protein structuresbull A run of 5 hydrophilic residues is considered to be a hydrophilic
stretchbull The default hydrophilic residues are
ndash D E G K N Q P R Sndash But this can be changed by the user
Divergent Sequencesbull The most divergent sequences (most different on average
from all of the other sequences) are usually the most difficult to align
bull It is sometimes better to delay their aligment until later (when the easier sequences have already been aligned)
bull The user has the choice of setting a cutoff (default is 40 identity)
bull This will delay the alignment until the others have been aligned
Advice on progressive alignmentbull Progressive alignment is a mathematical process that is completely
independent of biological realitybull Can be a very good estimatebull Can be an impossibly poor estimatebull Requires user input and skillbull Treat cautiouslybull Can be improved by eye (usually)bull Often helps to have colour-codingbull Depending on the use the user should be able to make a judgement
on those regions that are reliable or notbull For phylogeny reconstruction only use those positions whose
hypothesis of positional homology is unimpeachable
Alignment of protein-coding DNA sequences
bull It is not very sensible to align the DNA sequences of protein-coding genes
ATGCTGTTAGGGATGACTCTGTTAGGG
ATG-CT--GTTAGGGATGACTCTGTTAGGG
The result might be highly-implausible and might not reflect what is known about biological processesIt is much more sensible to translate the sequences to their corresponding amino acid sequences align these protein sequences and then put the gaps in the DNA sequences according to where they are found in the amino acid alignment
Manual Alignment- softwareGDE- The Genetic Data Environment (UNIX)CINEMA- Java applet available from
ndash httpwwwbiochemuclacuk
SeqappSeqpup- MacPCUNIX available fromndash httpiubiobioindianaedu
SeAl for Macintosh available fromndash httpevolvezoooxacukSe-AlSe-Alhtml
BioEdit for PC available fromndash httpwwwmbioncsueduRNasePinfoprogramsBIOEDITbio
edithtml
- Multiple Sequence Alignment James McInerney bioinf4biologists Feb 2009
- Alignment can be easy or difficult
- Homology Definition
- Multiple Sequence Alignment- Goals
- Multiple sequence alignments - problems
- Slide 6
- Slide 7
- SSU rRNA
- Alignment of 16S rRNA can be guided by secondary structure
- Protein Alignment may be guided by Tertiary Structure Interactions
- Multiple Sequence Alignment- Methods
- Manual Alignment - reasons
- Local minimum
- Dotplots
- Dotplot example sperm whale vs human myg
- Slide 16
- Slide 17
- Dotplots in practice
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Dynamic programming
- Progressive Alignment
- Slide 23
- ClustalW- Pairwise Alignments
- Path Graph for aligning two sequences
- Possible alignment
- Alignment using this path
- Optimal Alignment 1
- Optimal Alignment 2
- Alignment of 3 sequences
- ClustalW- Guide Tree
- Neighbor joining method
- Distance Matrix
- First Step
- Calculation of New Distances
- Next Cycle
- Penultimate Cycle
- Last Joining
- Unrooted Neighbor-Joining Tree
- Multiple Alignment- First pair
- ClustalW- Decision time
- ClustalW- Alternative 1
- ClustalW- Progression
- Progressive alignment - step 1
- Progressive alignment - step 2
- Progressive alignment - step 3
- Progressive alignment - final step
- ClustalW-Good pointsBad points
- ClustalW-Local Minimum
- Increasing the sophistication of the alignment process
- Slide 51
- ClustalW- Caveats
- ClustalW- User-supplied values
- Position-Specific gap penalties
- Discouraging too many gaps
- Divergent Sequences
- Advice on progressive alignment
- Alignment of protein-coding DNA sequences
- Manual Alignment- software
-

Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
bull Protein has many repeats bull SLIT_DROME (P24014)
MAAPSRTTLMPPPFRLQLRLLILPILLLLRHDAVHAEPYSGGFGSSAVSSGGLGSVGIHIPGGGVGVITEARCPRVCSCT GLNVDCSHRGLTSVPRKISADVERLELQGNNLTVIYETDFQRLTKLRMLQLTDNQIHTIERNSFQDLVSLERLDISNNVI TTVGRRVFKGAQSLRSLQLDNNQITCLDEHAFKGLVELEILTLNNNNLTSLPHNIFGGLGRLRALRLSDNPFACDCHLSW LSRFLRSATRLAPYTRCQSPSQLKGQNVADLHDQEFKCSGLTEHAPMECGAENSCPHPCRCADGIVDCREKSLTSVPVTL PDDTTDVRLEQNFITELPPKSFSSFRRLRRIDLSNNNISRIAHDALSGLKQLTTLVLYGNKIKDLPSGVFKGLGSLRLLL LNANEISCIRKDAFRDLHSLSLLSLYDNNIQSLANGTFDAMKSMKTVHLAKNPFICDCNLRWLADYLHKNPIETSGARCE SPKRMHRRRIESLREEKFKCSWGELRMKLSGECRMDSDCPAMCHCEGTTVDCTGRRLKEIPRDIPLHTTELLLNDNELGR ISSDGLFGRLPHLVKLELKRNQLTGIEPNAFEGASHIQELQLGENKIKEISNKMFLGLHQLKTLNLYDNQISCVMPGSFE HLNSLTSLNLASNPFNCNCHLAWFAECVRKKSLNGGAARCGAPSKVRDVQIKDLPHSEFKCSSENSEGCLGDGYCPPSCT CTGTVVACSRNQLKEIPRGIPAETSELYLESNEIEQIHYERIRHLRSLTRLDLSNNQITILSNYTFANLTKLSTLIISYN KLQCLQRHALSGLNNLRVVSLHGNRISMLPEGSFEDLKSLTHIALGSNPLYCDCGLKWFSDWIKLDYVEPGIARCAEPEQ MKDKLILSTPSSSFVCRGRVRNDILAKCNACFEQPCQNQAQCVALPQREYQCLCQPGYHGKHCEFMIDACYGNPCRNNAT CTVLEEGRFSCQCAPGYTGARCETNIDDCLGEIKCQNNATCIDGVESYKCECQPGFSGEFCDTKIQFCSPEFNPCANGAK CMDHFTHYSCDCQAGFHGTNCTDNIDDCQNHMCQNGGTCVDGINDYQCRCPDDYTGKYCEGHNMISMMYPQTSPCQNHEC KHGVCFQPNAQGSDYLCRCHPGYTGKWCEYLTSISFVHNNSFVELEPLRTRPEANVTIVFSSAEQNGILMYDGQDAHLAV ELFNGRIRVSYDVGNHPVSTMYSFEMVADGKYHAVELLAIKKNFTLRVDRGLARSIINEGSNDYLKLTTPMFLGGLPVDP AQQAYKNWQIRNLTSFKGCMKEVWINHKLVDFGNAQRQQKITPGCALLEGEQQEEEDDEQDFMDETPHIKEEPVDPCLEN KCRRGSRCVPNSNARDGYQCKCKHGQRGRYCDQGEGSTEPPTVTAASTCRKEQVREYYTENDCRSRQPLKYAKCVGGCGN QCCAAKIVRRRKVRMVCSNNRKYIKNLDIVRKCGCTKKCY
bull Perform a dotplot of the SLIT protein against itself wwwbiobhamacukdotletDotlethtml
Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
20Swiss-prot entry
For further discussion of dotplot see Attwood and Parry-Smith p116-8
Dynamic programming2 methodsbull Dynamic programming
ndash Consider 2 protein sequences of 100 amino acids in lengthndash If it takes 1002 seconds to exhaustively align these sequences
then it will take 1003 seconds to align 3 sequences 1004 to align 4 sequencesetc
ndash More time than the universe has existed to align 20 sequences exhaustively
bull Progressive alignment
Progressive Alignmentbull Devised by Feng and Doolittle in 1987bull Essentially a heuristic method and as such
is not guaranteed to find the lsquooptimalrsquo alignment
bull Requires n-1+n-2+n-3n-n+1 pairwise alignments as a starting point
bull Most successful implementation is Clustal (Des Higgins) This software is cited 3000 times per year in the scientific literature
Overview of ClustalW Procedure
1 PEEKSAVTALWGKVN--VDEVGG2 GEEKAAVLALWDKVN--EEEVGG3 PADKTNVKAAWGKVGAHAGEYGA4 AADKTNVKAAWSKVGGHAGEYGA5 EHEWQLVLHVWAKVEADVAGHGQ
Hbb_Human 1 -Hbb_Horse 2 17 -Hba_Human 3 59 60 -Hba_Horse 4 59 59 13 -Myg_Whale 5 77 77 75 75 -
Hbb_Human
Hbb_Horse
Hba_Horse
Hba_Human
Myg_Whale
2
1
3 4
2
1
3 4
alpha-helices
Quick pairwise alignment calculate distance matrix
Neighbor-joining tree(guide tree)
Progressive alignment following guide tree
CLUSTAL W
ClustalW- Pairwise Alignments
bull First perform all possible pairwise alignments between each pair of sequences There are (n-1)+(n-2)(n-n+1) possibilities
bull Calculate the lsquodistancersquo between each pair of sequences based on these isolated pairwise alignments
bull Generate a distance matrix
Path Graph for aligning two sequences
Possible alignment
1
1
0
1
0
-1
Scoring SchemebullMatch +1bullMismatch 0bullIndel -1
Score for this path= 2
Alignment using this path
GATTC-GAATTC
1
1
0
1
0
-1
Optimal Alignment 1
1
1
-1
1
1
1
Alignment score 4Alignment score 4
Alignment using this path
GA-TTCGA-TTCGAATTCGAATTC
Optimal Alignment 2
1
-1
1
1
1
1
Alignment score 4Alignment score 4
Alignment using this path
G-ATTCG-ATTCGAATTCGAATTC
Alignment of 3 sequences
ClustalW- Guide Tree
bull Generate a Neighbor-Joining lsquoguide treersquo from these pairwise distances
bull This guide tree gives the order in which the progressive alignment will be carried out
Neighbor joining method
bullThe neighbor-joining method is a greedy heuristic which joins at each step the two closest sub-trees that are not already joinedbullIt is based on the minimum evolution principlebullneighbors are defined as two taxa that are connected by a single node in an unrooted tree
A B
Node 1
PAM Spinach Rice Mosquito Monkey HumanSpinach 00 849 1056 908 863Rice 849 00 1178 1224 1226Mosquito 1056 1178 00 847 808Monkey 908 1224 847 00 33Human 863 1226 808 33 00
What is required for the Neighbour joining method
Distance matrixDistance Matrix
PAM distance 33 (Human - Monkey) is the minimum So well join Human and Monkey to MonHum and well calculate the new distances
Mon-Hum
MonkeyHumanSpinachMosquito Rice
First Step
After we have joined two species in a subtree we have to compute the distances from every other node to the new subtree We do this with a simple average of distancesDist[Spinach MonHum]
= (Dist[Spinach Monkey] + Dist[Spinach Human])2 = (908 + 863)2 = 8855
Mon-Hum
MonkeyHumanSpinach
Calculation of New Distances
PAM Spinach Rice Mosquito MonHumSpinach 00 849 1056 886Rice 849 00 1178 1225Mosquito 1056 1178 00 828MonHum 886 1225 828 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Next Cycle
PAM Spinach Rice MosMonHumSpinach 00 849 971Rice 849 00 1202MosMonHum 971 1202 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
Penultimate Cycle
PAM SpinRice MosMonHumSpinach 00 1087MosMonHum 1087 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
(Spin-Rice)-(Mos-(Mon-Hum))
Last Joining
Human
Monkey
MosquitoRice
Spinach
Unrooted Neighbor-Joining Tree
Multiple Alignment- First pairbull Align the two most closely-related sequences first
bull This alignment is then lsquofixedrsquo and will never change If a gap is to be introduced subsequently then it will be introduced in the same place in both sequences but their relative alignment remains unchanged
ClustalW- Decision timebull Consult the guide tree to see what alignment is performed next
ndash Align a third sequence to the first twoOrndash Align two entirely different sequences to each other
Option 1Option 1 Option 2Option 2
ClustalW- Alternative 1If the situation arises where a third sequence is aligned to the first two then when a gap has to be introduced to improve the alignment each of these two entities are treated as two single sequences
+
ClustalW- Alternative 2bull If on the other hand two separate
sequences have to be aligned together then the first pairwise alignment is placed to one side and the pairwise alignment of the other two is carried out
+
ClustalW- Progression
bull The alignment is progressively built up in this way with each step being treated as a pairwise alignment sometimes with each member of a lsquopairrsquo having more than one sequence
Progressive alignment - step 11 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
12345
Progressive alignment - step 21 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
12345
Progressive alignment - step 31 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
+
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
12345
Progressive alignment - final step1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
+
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
5 -ctcga-acgatacgatgactagct-
12345
ClustalW-Good pointsBad points
bull Advantagesndash Speed
bull Disadvantagesndash No objective functionndash No way of quantifying whether or not the alignment is good
ndash No way of knowing if the alignment is lsquocorrectrsquo
ClustalW-Local Minimumbull Potential problems
ndash Local minimum problem If an error is introduced early in the alignment process it is impossible to correct this later in the procedure
ndash Arbitrary alignment
Increasing the sophistication of the alignment process
bull Should we treat all the sequences in the same way - even though some sequences are closely-related and some sequences are distant relatives
bull Should we treat all positions in the sequences as though they were the same - even though they might have different functions and different locations in the 3-dimensional structure
ClustalW- Caveatsbull Sequence weightingbull Varying substitution matricesbull Residue-specific gap penalties and reduced penalties
in hydrophilic regions (external regions of protein sequences) encourage gaps in loops rather than in core regions
bull Positions in early alignments where gaps have been opened receive locally reduced gap penalties to encourage openings in subsequent alignments
ClustalW- User-supplied values
bull Two penalties are set by the user (there are default values but you should know that it is possible to change these)
bull GOP- Gap Opening Penalty is the cost of opening a gap in an alignment
bull GEP- Gap Extension Penalty is the cost of extending this gap
Position-Specific gap penaltiesbull Before any pair of (groups of) sequences are
aligned a table of GOPs are generated for each position in the two (sets of) sequences
bull The GOP is manipulated in a position-specific manner so that it can vary over the sequences
bull If there is a gap at a position the GOP and GEP penalties are lowered the other rules do not apply
bull This makes gaps more likely at positions where gaps already exist
Discouraging too many gaps bull If there is no gap opened then the GOP is increased if the
position is within 8 residues of an existing gapbull This discourages gaps that are too close togetherbull At any position within a run of hydrophilic residues the GOP is
decreasedbull These runs usually indicate loop regions in protein structuresbull A run of 5 hydrophilic residues is considered to be a hydrophilic
stretchbull The default hydrophilic residues are
ndash D E G K N Q P R Sndash But this can be changed by the user
Divergent Sequencesbull The most divergent sequences (most different on average
from all of the other sequences) are usually the most difficult to align
bull It is sometimes better to delay their aligment until later (when the easier sequences have already been aligned)
bull The user has the choice of setting a cutoff (default is 40 identity)
bull This will delay the alignment until the others have been aligned
Advice on progressive alignmentbull Progressive alignment is a mathematical process that is completely
independent of biological realitybull Can be a very good estimatebull Can be an impossibly poor estimatebull Requires user input and skillbull Treat cautiouslybull Can be improved by eye (usually)bull Often helps to have colour-codingbull Depending on the use the user should be able to make a judgement
on those regions that are reliable or notbull For phylogeny reconstruction only use those positions whose
hypothesis of positional homology is unimpeachable
Alignment of protein-coding DNA sequences
bull It is not very sensible to align the DNA sequences of protein-coding genes
ATGCTGTTAGGGATGACTCTGTTAGGG
ATG-CT--GTTAGGGATGACTCTGTTAGGG
The result might be highly-implausible and might not reflect what is known about biological processesIt is much more sensible to translate the sequences to their corresponding amino acid sequences align these protein sequences and then put the gaps in the DNA sequences according to where they are found in the amino acid alignment
Manual Alignment- softwareGDE- The Genetic Data Environment (UNIX)CINEMA- Java applet available from
ndash httpwwwbiochemuclacuk
SeqappSeqpup- MacPCUNIX available fromndash httpiubiobioindianaedu
SeAl for Macintosh available fromndash httpevolvezoooxacukSe-AlSe-Alhtml
BioEdit for PC available fromndash httpwwwmbioncsueduRNasePinfoprogramsBIOEDITbio
edithtml
- Multiple Sequence Alignment James McInerney bioinf4biologists Feb 2009
- Alignment can be easy or difficult
- Homology Definition
- Multiple Sequence Alignment- Goals
- Multiple sequence alignments - problems
- Slide 6
- Slide 7
- SSU rRNA
- Alignment of 16S rRNA can be guided by secondary structure
- Protein Alignment may be guided by Tertiary Structure Interactions
- Multiple Sequence Alignment- Methods
- Manual Alignment - reasons
- Local minimum
- Dotplots
- Dotplot example sperm whale vs human myg
- Slide 16
- Slide 17
- Dotplots in practice
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Dynamic programming
- Progressive Alignment
- Slide 23
- ClustalW- Pairwise Alignments
- Path Graph for aligning two sequences
- Possible alignment
- Alignment using this path
- Optimal Alignment 1
- Optimal Alignment 2
- Alignment of 3 sequences
- ClustalW- Guide Tree
- Neighbor joining method
- Distance Matrix
- First Step
- Calculation of New Distances
- Next Cycle
- Penultimate Cycle
- Last Joining
- Unrooted Neighbor-Joining Tree
- Multiple Alignment- First pair
- ClustalW- Decision time
- ClustalW- Alternative 1
- ClustalW- Progression
- Progressive alignment - step 1
- Progressive alignment - step 2
- Progressive alignment - step 3
- Progressive alignment - final step
- ClustalW-Good pointsBad points
- ClustalW-Local Minimum
- Increasing the sophistication of the alignment process
- Slide 51
- ClustalW- Caveats
- ClustalW- User-supplied values
- Position-Specific gap penalties
- Discouraging too many gaps
- Divergent Sequences
- Advice on progressive alignment
- Alignment of protein-coding DNA sequences
- Manual Alignment- software
-

Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
20Swiss-prot entry
For further discussion of dotplot see Attwood and Parry-Smith p116-8
Dynamic programming2 methodsbull Dynamic programming
ndash Consider 2 protein sequences of 100 amino acids in lengthndash If it takes 1002 seconds to exhaustively align these sequences
then it will take 1003 seconds to align 3 sequences 1004 to align 4 sequencesetc
ndash More time than the universe has existed to align 20 sequences exhaustively
bull Progressive alignment
Progressive Alignmentbull Devised by Feng and Doolittle in 1987bull Essentially a heuristic method and as such
is not guaranteed to find the lsquooptimalrsquo alignment
bull Requires n-1+n-2+n-3n-n+1 pairwise alignments as a starting point
bull Most successful implementation is Clustal (Des Higgins) This software is cited 3000 times per year in the scientific literature
Overview of ClustalW Procedure
1 PEEKSAVTALWGKVN--VDEVGG2 GEEKAAVLALWDKVN--EEEVGG3 PADKTNVKAAWGKVGAHAGEYGA4 AADKTNVKAAWSKVGGHAGEYGA5 EHEWQLVLHVWAKVEADVAGHGQ
Hbb_Human 1 -Hbb_Horse 2 17 -Hba_Human 3 59 60 -Hba_Horse 4 59 59 13 -Myg_Whale 5 77 77 75 75 -
Hbb_Human
Hbb_Horse
Hba_Horse
Hba_Human
Myg_Whale
2
1
3 4
2
1
3 4
alpha-helices
Quick pairwise alignment calculate distance matrix
Neighbor-joining tree(guide tree)
Progressive alignment following guide tree
CLUSTAL W
ClustalW- Pairwise Alignments
bull First perform all possible pairwise alignments between each pair of sequences There are (n-1)+(n-2)(n-n+1) possibilities
bull Calculate the lsquodistancersquo between each pair of sequences based on these isolated pairwise alignments
bull Generate a distance matrix
Path Graph for aligning two sequences
Possible alignment
1
1
0
1
0
-1
Scoring SchemebullMatch +1bullMismatch 0bullIndel -1
Score for this path= 2
Alignment using this path
GATTC-GAATTC
1
1
0
1
0
-1
Optimal Alignment 1
1
1
-1
1
1
1
Alignment score 4Alignment score 4
Alignment using this path
GA-TTCGA-TTCGAATTCGAATTC
Optimal Alignment 2
1
-1
1
1
1
1
Alignment score 4Alignment score 4
Alignment using this path
G-ATTCG-ATTCGAATTCGAATTC
Alignment of 3 sequences
ClustalW- Guide Tree
bull Generate a Neighbor-Joining lsquoguide treersquo from these pairwise distances
bull This guide tree gives the order in which the progressive alignment will be carried out
Neighbor joining method
bullThe neighbor-joining method is a greedy heuristic which joins at each step the two closest sub-trees that are not already joinedbullIt is based on the minimum evolution principlebullneighbors are defined as two taxa that are connected by a single node in an unrooted tree
A B
Node 1
PAM Spinach Rice Mosquito Monkey HumanSpinach 00 849 1056 908 863Rice 849 00 1178 1224 1226Mosquito 1056 1178 00 847 808Monkey 908 1224 847 00 33Human 863 1226 808 33 00
What is required for the Neighbour joining method
Distance matrixDistance Matrix
PAM distance 33 (Human - Monkey) is the minimum So well join Human and Monkey to MonHum and well calculate the new distances
Mon-Hum
MonkeyHumanSpinachMosquito Rice
First Step
After we have joined two species in a subtree we have to compute the distances from every other node to the new subtree We do this with a simple average of distancesDist[Spinach MonHum]
= (Dist[Spinach Monkey] + Dist[Spinach Human])2 = (908 + 863)2 = 8855
Mon-Hum
MonkeyHumanSpinach
Calculation of New Distances
PAM Spinach Rice Mosquito MonHumSpinach 00 849 1056 886Rice 849 00 1178 1225Mosquito 1056 1178 00 828MonHum 886 1225 828 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Next Cycle
PAM Spinach Rice MosMonHumSpinach 00 849 971Rice 849 00 1202MosMonHum 971 1202 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
Penultimate Cycle
PAM SpinRice MosMonHumSpinach 00 1087MosMonHum 1087 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
(Spin-Rice)-(Mos-(Mon-Hum))
Last Joining
Human
Monkey
MosquitoRice
Spinach
Unrooted Neighbor-Joining Tree
Multiple Alignment- First pairbull Align the two most closely-related sequences first
bull This alignment is then lsquofixedrsquo and will never change If a gap is to be introduced subsequently then it will be introduced in the same place in both sequences but their relative alignment remains unchanged
ClustalW- Decision timebull Consult the guide tree to see what alignment is performed next
ndash Align a third sequence to the first twoOrndash Align two entirely different sequences to each other
Option 1Option 1 Option 2Option 2
ClustalW- Alternative 1If the situation arises where a third sequence is aligned to the first two then when a gap has to be introduced to improve the alignment each of these two entities are treated as two single sequences
+
ClustalW- Alternative 2bull If on the other hand two separate
sequences have to be aligned together then the first pairwise alignment is placed to one side and the pairwise alignment of the other two is carried out
+
ClustalW- Progression
bull The alignment is progressively built up in this way with each step being treated as a pairwise alignment sometimes with each member of a lsquopairrsquo having more than one sequence
Progressive alignment - step 11 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
12345
Progressive alignment - step 21 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
12345
Progressive alignment - step 31 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
+
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
12345
Progressive alignment - final step1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
+
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
5 -ctcga-acgatacgatgactagct-
12345
ClustalW-Good pointsBad points
bull Advantagesndash Speed
bull Disadvantagesndash No objective functionndash No way of quantifying whether or not the alignment is good
ndash No way of knowing if the alignment is lsquocorrectrsquo
ClustalW-Local Minimumbull Potential problems
ndash Local minimum problem If an error is introduced early in the alignment process it is impossible to correct this later in the procedure
ndash Arbitrary alignment
Increasing the sophistication of the alignment process
bull Should we treat all the sequences in the same way - even though some sequences are closely-related and some sequences are distant relatives
bull Should we treat all positions in the sequences as though they were the same - even though they might have different functions and different locations in the 3-dimensional structure
ClustalW- Caveatsbull Sequence weightingbull Varying substitution matricesbull Residue-specific gap penalties and reduced penalties
in hydrophilic regions (external regions of protein sequences) encourage gaps in loops rather than in core regions
bull Positions in early alignments where gaps have been opened receive locally reduced gap penalties to encourage openings in subsequent alignments
ClustalW- User-supplied values
bull Two penalties are set by the user (there are default values but you should know that it is possible to change these)
bull GOP- Gap Opening Penalty is the cost of opening a gap in an alignment
bull GEP- Gap Extension Penalty is the cost of extending this gap
Position-Specific gap penaltiesbull Before any pair of (groups of) sequences are
aligned a table of GOPs are generated for each position in the two (sets of) sequences
bull The GOP is manipulated in a position-specific manner so that it can vary over the sequences
bull If there is a gap at a position the GOP and GEP penalties are lowered the other rules do not apply
bull This makes gaps more likely at positions where gaps already exist
Discouraging too many gaps bull If there is no gap opened then the GOP is increased if the
position is within 8 residues of an existing gapbull This discourages gaps that are too close togetherbull At any position within a run of hydrophilic residues the GOP is
decreasedbull These runs usually indicate loop regions in protein structuresbull A run of 5 hydrophilic residues is considered to be a hydrophilic
stretchbull The default hydrophilic residues are
ndash D E G K N Q P R Sndash But this can be changed by the user
Divergent Sequencesbull The most divergent sequences (most different on average
from all of the other sequences) are usually the most difficult to align
bull It is sometimes better to delay their aligment until later (when the easier sequences have already been aligned)
bull The user has the choice of setting a cutoff (default is 40 identity)
bull This will delay the alignment until the others have been aligned
Advice on progressive alignmentbull Progressive alignment is a mathematical process that is completely
independent of biological realitybull Can be a very good estimatebull Can be an impossibly poor estimatebull Requires user input and skillbull Treat cautiouslybull Can be improved by eye (usually)bull Often helps to have colour-codingbull Depending on the use the user should be able to make a judgement
on those regions that are reliable or notbull For phylogeny reconstruction only use those positions whose
hypothesis of positional homology is unimpeachable
Alignment of protein-coding DNA sequences
bull It is not very sensible to align the DNA sequences of protein-coding genes
ATGCTGTTAGGGATGACTCTGTTAGGG
ATG-CT--GTTAGGGATGACTCTGTTAGGG
The result might be highly-implausible and might not reflect what is known about biological processesIt is much more sensible to translate the sequences to their corresponding amino acid sequences align these protein sequences and then put the gaps in the DNA sequences according to where they are found in the amino acid alignment
Manual Alignment- softwareGDE- The Genetic Data Environment (UNIX)CINEMA- Java applet available from
ndash httpwwwbiochemuclacuk
SeqappSeqpup- MacPCUNIX available fromndash httpiubiobioindianaedu
SeAl for Macintosh available fromndash httpevolvezoooxacukSe-AlSe-Alhtml
BioEdit for PC available fromndash httpwwwmbioncsueduRNasePinfoprogramsBIOEDITbio
edithtml
- Multiple Sequence Alignment James McInerney bioinf4biologists Feb 2009
- Alignment can be easy or difficult
- Homology Definition
- Multiple Sequence Alignment- Goals
- Multiple sequence alignments - problems
- Slide 6
- Slide 7
- SSU rRNA
- Alignment of 16S rRNA can be guided by secondary structure
- Protein Alignment may be guided by Tertiary Structure Interactions
- Multiple Sequence Alignment- Methods
- Manual Alignment - reasons
- Local minimum
- Dotplots
- Dotplot example sperm whale vs human myg
- Slide 16
- Slide 17
- Dotplots in practice
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Dynamic programming
- Progressive Alignment
- Slide 23
- ClustalW- Pairwise Alignments
- Path Graph for aligning two sequences
- Possible alignment
- Alignment using this path
- Optimal Alignment 1
- Optimal Alignment 2
- Alignment of 3 sequences
- ClustalW- Guide Tree
- Neighbor joining method
- Distance Matrix
- First Step
- Calculation of New Distances
- Next Cycle
- Penultimate Cycle
- Last Joining
- Unrooted Neighbor-Joining Tree
- Multiple Alignment- First pair
- ClustalW- Decision time
- ClustalW- Alternative 1
- ClustalW- Progression
- Progressive alignment - step 1
- Progressive alignment - step 2
- Progressive alignment - step 3
- Progressive alignment - final step
- ClustalW-Good pointsBad points
- ClustalW-Local Minimum
- Increasing the sophistication of the alignment process
- Slide 51
- ClustalW- Caveats
- ClustalW- User-supplied values
- Position-Specific gap penalties
- Discouraging too many gaps
- Divergent Sequences
- Advice on progressive alignment
- Alignment of protein-coding DNA sequences
- Manual Alignment- software
-

Dynamic programming2 methodsbull Dynamic programming
ndash Consider 2 protein sequences of 100 amino acids in lengthndash If it takes 1002 seconds to exhaustively align these sequences
then it will take 1003 seconds to align 3 sequences 1004 to align 4 sequencesetc
ndash More time than the universe has existed to align 20 sequences exhaustively
bull Progressive alignment
Progressive Alignmentbull Devised by Feng and Doolittle in 1987bull Essentially a heuristic method and as such
is not guaranteed to find the lsquooptimalrsquo alignment
bull Requires n-1+n-2+n-3n-n+1 pairwise alignments as a starting point
bull Most successful implementation is Clustal (Des Higgins) This software is cited 3000 times per year in the scientific literature
Overview of ClustalW Procedure
1 PEEKSAVTALWGKVN--VDEVGG2 GEEKAAVLALWDKVN--EEEVGG3 PADKTNVKAAWGKVGAHAGEYGA4 AADKTNVKAAWSKVGGHAGEYGA5 EHEWQLVLHVWAKVEADVAGHGQ
Hbb_Human 1 -Hbb_Horse 2 17 -Hba_Human 3 59 60 -Hba_Horse 4 59 59 13 -Myg_Whale 5 77 77 75 75 -
Hbb_Human
Hbb_Horse
Hba_Horse
Hba_Human
Myg_Whale
2
1
3 4
2
1
3 4
alpha-helices
Quick pairwise alignment calculate distance matrix
Neighbor-joining tree(guide tree)
Progressive alignment following guide tree
CLUSTAL W
ClustalW- Pairwise Alignments
bull First perform all possible pairwise alignments between each pair of sequences There are (n-1)+(n-2)(n-n+1) possibilities
bull Calculate the lsquodistancersquo between each pair of sequences based on these isolated pairwise alignments
bull Generate a distance matrix
Path Graph for aligning two sequences
Possible alignment
1
1
0
1
0
-1
Scoring SchemebullMatch +1bullMismatch 0bullIndel -1
Score for this path= 2
Alignment using this path
GATTC-GAATTC
1
1
0
1
0
-1
Optimal Alignment 1
1
1
-1
1
1
1
Alignment score 4Alignment score 4
Alignment using this path
GA-TTCGA-TTCGAATTCGAATTC
Optimal Alignment 2
1
-1
1
1
1
1
Alignment score 4Alignment score 4
Alignment using this path
G-ATTCG-ATTCGAATTCGAATTC
Alignment of 3 sequences
ClustalW- Guide Tree
bull Generate a Neighbor-Joining lsquoguide treersquo from these pairwise distances
bull This guide tree gives the order in which the progressive alignment will be carried out
Neighbor joining method
bullThe neighbor-joining method is a greedy heuristic which joins at each step the two closest sub-trees that are not already joinedbullIt is based on the minimum evolution principlebullneighbors are defined as two taxa that are connected by a single node in an unrooted tree
A B
Node 1
PAM Spinach Rice Mosquito Monkey HumanSpinach 00 849 1056 908 863Rice 849 00 1178 1224 1226Mosquito 1056 1178 00 847 808Monkey 908 1224 847 00 33Human 863 1226 808 33 00
What is required for the Neighbour joining method
Distance matrixDistance Matrix
PAM distance 33 (Human - Monkey) is the minimum So well join Human and Monkey to MonHum and well calculate the new distances
Mon-Hum
MonkeyHumanSpinachMosquito Rice
First Step
After we have joined two species in a subtree we have to compute the distances from every other node to the new subtree We do this with a simple average of distancesDist[Spinach MonHum]
= (Dist[Spinach Monkey] + Dist[Spinach Human])2 = (908 + 863)2 = 8855
Mon-Hum
MonkeyHumanSpinach
Calculation of New Distances
PAM Spinach Rice Mosquito MonHumSpinach 00 849 1056 886Rice 849 00 1178 1225Mosquito 1056 1178 00 828MonHum 886 1225 828 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Next Cycle
PAM Spinach Rice MosMonHumSpinach 00 849 971Rice 849 00 1202MosMonHum 971 1202 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
Penultimate Cycle
PAM SpinRice MosMonHumSpinach 00 1087MosMonHum 1087 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
(Spin-Rice)-(Mos-(Mon-Hum))
Last Joining
Human
Monkey
MosquitoRice
Spinach
Unrooted Neighbor-Joining Tree
Multiple Alignment- First pairbull Align the two most closely-related sequences first
bull This alignment is then lsquofixedrsquo and will never change If a gap is to be introduced subsequently then it will be introduced in the same place in both sequences but their relative alignment remains unchanged
ClustalW- Decision timebull Consult the guide tree to see what alignment is performed next
ndash Align a third sequence to the first twoOrndash Align two entirely different sequences to each other
Option 1Option 1 Option 2Option 2
ClustalW- Alternative 1If the situation arises where a third sequence is aligned to the first two then when a gap has to be introduced to improve the alignment each of these two entities are treated as two single sequences
+
ClustalW- Alternative 2bull If on the other hand two separate
sequences have to be aligned together then the first pairwise alignment is placed to one side and the pairwise alignment of the other two is carried out
+
ClustalW- Progression
bull The alignment is progressively built up in this way with each step being treated as a pairwise alignment sometimes with each member of a lsquopairrsquo having more than one sequence
Progressive alignment - step 11 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
12345
Progressive alignment - step 21 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
12345
Progressive alignment - step 31 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
+
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
12345
Progressive alignment - final step1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
+
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
5 -ctcga-acgatacgatgactagct-
12345
ClustalW-Good pointsBad points
bull Advantagesndash Speed
bull Disadvantagesndash No objective functionndash No way of quantifying whether or not the alignment is good
ndash No way of knowing if the alignment is lsquocorrectrsquo
ClustalW-Local Minimumbull Potential problems
ndash Local minimum problem If an error is introduced early in the alignment process it is impossible to correct this later in the procedure
ndash Arbitrary alignment
Increasing the sophistication of the alignment process
bull Should we treat all the sequences in the same way - even though some sequences are closely-related and some sequences are distant relatives
bull Should we treat all positions in the sequences as though they were the same - even though they might have different functions and different locations in the 3-dimensional structure
ClustalW- Caveatsbull Sequence weightingbull Varying substitution matricesbull Residue-specific gap penalties and reduced penalties
in hydrophilic regions (external regions of protein sequences) encourage gaps in loops rather than in core regions
bull Positions in early alignments where gaps have been opened receive locally reduced gap penalties to encourage openings in subsequent alignments
ClustalW- User-supplied values
bull Two penalties are set by the user (there are default values but you should know that it is possible to change these)
bull GOP- Gap Opening Penalty is the cost of opening a gap in an alignment
bull GEP- Gap Extension Penalty is the cost of extending this gap
Position-Specific gap penaltiesbull Before any pair of (groups of) sequences are
aligned a table of GOPs are generated for each position in the two (sets of) sequences
bull The GOP is manipulated in a position-specific manner so that it can vary over the sequences
bull If there is a gap at a position the GOP and GEP penalties are lowered the other rules do not apply
bull This makes gaps more likely at positions where gaps already exist
Discouraging too many gaps bull If there is no gap opened then the GOP is increased if the
position is within 8 residues of an existing gapbull This discourages gaps that are too close togetherbull At any position within a run of hydrophilic residues the GOP is
decreasedbull These runs usually indicate loop regions in protein structuresbull A run of 5 hydrophilic residues is considered to be a hydrophilic
stretchbull The default hydrophilic residues are
ndash D E G K N Q P R Sndash But this can be changed by the user
Divergent Sequencesbull The most divergent sequences (most different on average
from all of the other sequences) are usually the most difficult to align
bull It is sometimes better to delay their aligment until later (when the easier sequences have already been aligned)
bull The user has the choice of setting a cutoff (default is 40 identity)
bull This will delay the alignment until the others have been aligned
Advice on progressive alignmentbull Progressive alignment is a mathematical process that is completely
independent of biological realitybull Can be a very good estimatebull Can be an impossibly poor estimatebull Requires user input and skillbull Treat cautiouslybull Can be improved by eye (usually)bull Often helps to have colour-codingbull Depending on the use the user should be able to make a judgement
on those regions that are reliable or notbull For phylogeny reconstruction only use those positions whose
hypothesis of positional homology is unimpeachable
Alignment of protein-coding DNA sequences
bull It is not very sensible to align the DNA sequences of protein-coding genes
ATGCTGTTAGGGATGACTCTGTTAGGG
ATG-CT--GTTAGGGATGACTCTGTTAGGG
The result might be highly-implausible and might not reflect what is known about biological processesIt is much more sensible to translate the sequences to their corresponding amino acid sequences align these protein sequences and then put the gaps in the DNA sequences according to where they are found in the amino acid alignment
Manual Alignment- softwareGDE- The Genetic Data Environment (UNIX)CINEMA- Java applet available from
ndash httpwwwbiochemuclacuk
SeqappSeqpup- MacPCUNIX available fromndash httpiubiobioindianaedu
SeAl for Macintosh available fromndash httpevolvezoooxacukSe-AlSe-Alhtml
BioEdit for PC available fromndash httpwwwmbioncsueduRNasePinfoprogramsBIOEDITbio
edithtml
- Multiple Sequence Alignment James McInerney bioinf4biologists Feb 2009
- Alignment can be easy or difficult
- Homology Definition
- Multiple Sequence Alignment- Goals
- Multiple sequence alignments - problems
- Slide 6
- Slide 7
- SSU rRNA
- Alignment of 16S rRNA can be guided by secondary structure
- Protein Alignment may be guided by Tertiary Structure Interactions
- Multiple Sequence Alignment- Methods
- Manual Alignment - reasons
- Local minimum
- Dotplots
- Dotplot example sperm whale vs human myg
- Slide 16
- Slide 17
- Dotplots in practice
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Dynamic programming
- Progressive Alignment
- Slide 23
- ClustalW- Pairwise Alignments
- Path Graph for aligning two sequences
- Possible alignment
- Alignment using this path
- Optimal Alignment 1
- Optimal Alignment 2
- Alignment of 3 sequences
- ClustalW- Guide Tree
- Neighbor joining method
- Distance Matrix
- First Step
- Calculation of New Distances
- Next Cycle
- Penultimate Cycle
- Last Joining
- Unrooted Neighbor-Joining Tree
- Multiple Alignment- First pair
- ClustalW- Decision time
- ClustalW- Alternative 1
- ClustalW- Progression
- Progressive alignment - step 1
- Progressive alignment - step 2
- Progressive alignment - step 3
- Progressive alignment - final step
- ClustalW-Good pointsBad points
- ClustalW-Local Minimum
- Increasing the sophistication of the alignment process
- Slide 51
- ClustalW- Caveats
- ClustalW- User-supplied values
- Position-Specific gap penalties
- Discouraging too many gaps
- Divergent Sequences
- Advice on progressive alignment
- Alignment of protein-coding DNA sequences
- Manual Alignment- software
-

Progressive Alignmentbull Devised by Feng and Doolittle in 1987bull Essentially a heuristic method and as such
is not guaranteed to find the lsquooptimalrsquo alignment
bull Requires n-1+n-2+n-3n-n+1 pairwise alignments as a starting point
bull Most successful implementation is Clustal (Des Higgins) This software is cited 3000 times per year in the scientific literature
Overview of ClustalW Procedure
1 PEEKSAVTALWGKVN--VDEVGG2 GEEKAAVLALWDKVN--EEEVGG3 PADKTNVKAAWGKVGAHAGEYGA4 AADKTNVKAAWSKVGGHAGEYGA5 EHEWQLVLHVWAKVEADVAGHGQ
Hbb_Human 1 -Hbb_Horse 2 17 -Hba_Human 3 59 60 -Hba_Horse 4 59 59 13 -Myg_Whale 5 77 77 75 75 -
Hbb_Human
Hbb_Horse
Hba_Horse
Hba_Human
Myg_Whale
2
1
3 4
2
1
3 4
alpha-helices
Quick pairwise alignment calculate distance matrix
Neighbor-joining tree(guide tree)
Progressive alignment following guide tree
CLUSTAL W
ClustalW- Pairwise Alignments
bull First perform all possible pairwise alignments between each pair of sequences There are (n-1)+(n-2)(n-n+1) possibilities
bull Calculate the lsquodistancersquo between each pair of sequences based on these isolated pairwise alignments
bull Generate a distance matrix
Path Graph for aligning two sequences
Possible alignment
1
1
0
1
0
-1
Scoring SchemebullMatch +1bullMismatch 0bullIndel -1
Score for this path= 2
Alignment using this path
GATTC-GAATTC
1
1
0
1
0
-1
Optimal Alignment 1
1
1
-1
1
1
1
Alignment score 4Alignment score 4
Alignment using this path
GA-TTCGA-TTCGAATTCGAATTC
Optimal Alignment 2
1
-1
1
1
1
1
Alignment score 4Alignment score 4
Alignment using this path
G-ATTCG-ATTCGAATTCGAATTC
Alignment of 3 sequences
ClustalW- Guide Tree
bull Generate a Neighbor-Joining lsquoguide treersquo from these pairwise distances
bull This guide tree gives the order in which the progressive alignment will be carried out
Neighbor joining method
bullThe neighbor-joining method is a greedy heuristic which joins at each step the two closest sub-trees that are not already joinedbullIt is based on the minimum evolution principlebullneighbors are defined as two taxa that are connected by a single node in an unrooted tree
A B
Node 1
PAM Spinach Rice Mosquito Monkey HumanSpinach 00 849 1056 908 863Rice 849 00 1178 1224 1226Mosquito 1056 1178 00 847 808Monkey 908 1224 847 00 33Human 863 1226 808 33 00
What is required for the Neighbour joining method
Distance matrixDistance Matrix
PAM distance 33 (Human - Monkey) is the minimum So well join Human and Monkey to MonHum and well calculate the new distances
Mon-Hum
MonkeyHumanSpinachMosquito Rice
First Step
After we have joined two species in a subtree we have to compute the distances from every other node to the new subtree We do this with a simple average of distancesDist[Spinach MonHum]
= (Dist[Spinach Monkey] + Dist[Spinach Human])2 = (908 + 863)2 = 8855
Mon-Hum
MonkeyHumanSpinach
Calculation of New Distances
PAM Spinach Rice Mosquito MonHumSpinach 00 849 1056 886Rice 849 00 1178 1225Mosquito 1056 1178 00 828MonHum 886 1225 828 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Next Cycle
PAM Spinach Rice MosMonHumSpinach 00 849 971Rice 849 00 1202MosMonHum 971 1202 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
Penultimate Cycle
PAM SpinRice MosMonHumSpinach 00 1087MosMonHum 1087 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
(Spin-Rice)-(Mos-(Mon-Hum))
Last Joining
Human
Monkey
MosquitoRice
Spinach
Unrooted Neighbor-Joining Tree
Multiple Alignment- First pairbull Align the two most closely-related sequences first
bull This alignment is then lsquofixedrsquo and will never change If a gap is to be introduced subsequently then it will be introduced in the same place in both sequences but their relative alignment remains unchanged
ClustalW- Decision timebull Consult the guide tree to see what alignment is performed next
ndash Align a third sequence to the first twoOrndash Align two entirely different sequences to each other
Option 1Option 1 Option 2Option 2
ClustalW- Alternative 1If the situation arises where a third sequence is aligned to the first two then when a gap has to be introduced to improve the alignment each of these two entities are treated as two single sequences
+
ClustalW- Alternative 2bull If on the other hand two separate
sequences have to be aligned together then the first pairwise alignment is placed to one side and the pairwise alignment of the other two is carried out
+
ClustalW- Progression
bull The alignment is progressively built up in this way with each step being treated as a pairwise alignment sometimes with each member of a lsquopairrsquo having more than one sequence
Progressive alignment - step 11 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
12345
Progressive alignment - step 21 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
12345
Progressive alignment - step 31 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
+
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
12345
Progressive alignment - final step1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
+
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
5 -ctcga-acgatacgatgactagct-
12345
ClustalW-Good pointsBad points
bull Advantagesndash Speed
bull Disadvantagesndash No objective functionndash No way of quantifying whether or not the alignment is good
ndash No way of knowing if the alignment is lsquocorrectrsquo
ClustalW-Local Minimumbull Potential problems
ndash Local minimum problem If an error is introduced early in the alignment process it is impossible to correct this later in the procedure
ndash Arbitrary alignment
Increasing the sophistication of the alignment process
bull Should we treat all the sequences in the same way - even though some sequences are closely-related and some sequences are distant relatives
bull Should we treat all positions in the sequences as though they were the same - even though they might have different functions and different locations in the 3-dimensional structure
ClustalW- Caveatsbull Sequence weightingbull Varying substitution matricesbull Residue-specific gap penalties and reduced penalties
in hydrophilic regions (external regions of protein sequences) encourage gaps in loops rather than in core regions
bull Positions in early alignments where gaps have been opened receive locally reduced gap penalties to encourage openings in subsequent alignments
ClustalW- User-supplied values
bull Two penalties are set by the user (there are default values but you should know that it is possible to change these)
bull GOP- Gap Opening Penalty is the cost of opening a gap in an alignment
bull GEP- Gap Extension Penalty is the cost of extending this gap
Position-Specific gap penaltiesbull Before any pair of (groups of) sequences are
aligned a table of GOPs are generated for each position in the two (sets of) sequences
bull The GOP is manipulated in a position-specific manner so that it can vary over the sequences
bull If there is a gap at a position the GOP and GEP penalties are lowered the other rules do not apply
bull This makes gaps more likely at positions where gaps already exist
Discouraging too many gaps bull If there is no gap opened then the GOP is increased if the
position is within 8 residues of an existing gapbull This discourages gaps that are too close togetherbull At any position within a run of hydrophilic residues the GOP is
decreasedbull These runs usually indicate loop regions in protein structuresbull A run of 5 hydrophilic residues is considered to be a hydrophilic
stretchbull The default hydrophilic residues are
ndash D E G K N Q P R Sndash But this can be changed by the user
Divergent Sequencesbull The most divergent sequences (most different on average
from all of the other sequences) are usually the most difficult to align
bull It is sometimes better to delay their aligment until later (when the easier sequences have already been aligned)
bull The user has the choice of setting a cutoff (default is 40 identity)
bull This will delay the alignment until the others have been aligned
Advice on progressive alignmentbull Progressive alignment is a mathematical process that is completely
independent of biological realitybull Can be a very good estimatebull Can be an impossibly poor estimatebull Requires user input and skillbull Treat cautiouslybull Can be improved by eye (usually)bull Often helps to have colour-codingbull Depending on the use the user should be able to make a judgement
on those regions that are reliable or notbull For phylogeny reconstruction only use those positions whose
hypothesis of positional homology is unimpeachable
Alignment of protein-coding DNA sequences
bull It is not very sensible to align the DNA sequences of protein-coding genes
ATGCTGTTAGGGATGACTCTGTTAGGG
ATG-CT--GTTAGGGATGACTCTGTTAGGG
The result might be highly-implausible and might not reflect what is known about biological processesIt is much more sensible to translate the sequences to their corresponding amino acid sequences align these protein sequences and then put the gaps in the DNA sequences according to where they are found in the amino acid alignment
Manual Alignment- softwareGDE- The Genetic Data Environment (UNIX)CINEMA- Java applet available from
ndash httpwwwbiochemuclacuk
SeqappSeqpup- MacPCUNIX available fromndash httpiubiobioindianaedu
SeAl for Macintosh available fromndash httpevolvezoooxacukSe-AlSe-Alhtml
BioEdit for PC available fromndash httpwwwmbioncsueduRNasePinfoprogramsBIOEDITbio
edithtml
- Multiple Sequence Alignment James McInerney bioinf4biologists Feb 2009
- Alignment can be easy or difficult
- Homology Definition
- Multiple Sequence Alignment- Goals
- Multiple sequence alignments - problems
- Slide 6
- Slide 7
- SSU rRNA
- Alignment of 16S rRNA can be guided by secondary structure
- Protein Alignment may be guided by Tertiary Structure Interactions
- Multiple Sequence Alignment- Methods
- Manual Alignment - reasons
- Local minimum
- Dotplots
- Dotplot example sperm whale vs human myg
- Slide 16
- Slide 17
- Dotplots in practice
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Dynamic programming
- Progressive Alignment
- Slide 23
- ClustalW- Pairwise Alignments
- Path Graph for aligning two sequences
- Possible alignment
- Alignment using this path
- Optimal Alignment 1
- Optimal Alignment 2
- Alignment of 3 sequences
- ClustalW- Guide Tree
- Neighbor joining method
- Distance Matrix
- First Step
- Calculation of New Distances
- Next Cycle
- Penultimate Cycle
- Last Joining
- Unrooted Neighbor-Joining Tree
- Multiple Alignment- First pair
- ClustalW- Decision time
- ClustalW- Alternative 1
- ClustalW- Progression
- Progressive alignment - step 1
- Progressive alignment - step 2
- Progressive alignment - step 3
- Progressive alignment - final step
- ClustalW-Good pointsBad points
- ClustalW-Local Minimum
- Increasing the sophistication of the alignment process
- Slide 51
- ClustalW- Caveats
- ClustalW- User-supplied values
- Position-Specific gap penalties
- Discouraging too many gaps
- Divergent Sequences
- Advice on progressive alignment
- Alignment of protein-coding DNA sequences
- Manual Alignment- software
-

Overview of ClustalW Procedure
1 PEEKSAVTALWGKVN--VDEVGG2 GEEKAAVLALWDKVN--EEEVGG3 PADKTNVKAAWGKVGAHAGEYGA4 AADKTNVKAAWSKVGGHAGEYGA5 EHEWQLVLHVWAKVEADVAGHGQ
Hbb_Human 1 -Hbb_Horse 2 17 -Hba_Human 3 59 60 -Hba_Horse 4 59 59 13 -Myg_Whale 5 77 77 75 75 -
Hbb_Human
Hbb_Horse
Hba_Horse
Hba_Human
Myg_Whale
2
1
3 4
2
1
3 4
alpha-helices
Quick pairwise alignment calculate distance matrix
Neighbor-joining tree(guide tree)
Progressive alignment following guide tree
CLUSTAL W
ClustalW- Pairwise Alignments
bull First perform all possible pairwise alignments between each pair of sequences There are (n-1)+(n-2)(n-n+1) possibilities
bull Calculate the lsquodistancersquo between each pair of sequences based on these isolated pairwise alignments
bull Generate a distance matrix
Path Graph for aligning two sequences
Possible alignment
1
1
0
1
0
-1
Scoring SchemebullMatch +1bullMismatch 0bullIndel -1
Score for this path= 2
Alignment using this path
GATTC-GAATTC
1
1
0
1
0
-1
Optimal Alignment 1
1
1
-1
1
1
1
Alignment score 4Alignment score 4
Alignment using this path
GA-TTCGA-TTCGAATTCGAATTC
Optimal Alignment 2
1
-1
1
1
1
1
Alignment score 4Alignment score 4
Alignment using this path
G-ATTCG-ATTCGAATTCGAATTC
Alignment of 3 sequences
ClustalW- Guide Tree
bull Generate a Neighbor-Joining lsquoguide treersquo from these pairwise distances
bull This guide tree gives the order in which the progressive alignment will be carried out
Neighbor joining method
bullThe neighbor-joining method is a greedy heuristic which joins at each step the two closest sub-trees that are not already joinedbullIt is based on the minimum evolution principlebullneighbors are defined as two taxa that are connected by a single node in an unrooted tree
A B
Node 1
PAM Spinach Rice Mosquito Monkey HumanSpinach 00 849 1056 908 863Rice 849 00 1178 1224 1226Mosquito 1056 1178 00 847 808Monkey 908 1224 847 00 33Human 863 1226 808 33 00
What is required for the Neighbour joining method
Distance matrixDistance Matrix
PAM distance 33 (Human - Monkey) is the minimum So well join Human and Monkey to MonHum and well calculate the new distances
Mon-Hum
MonkeyHumanSpinachMosquito Rice
First Step
After we have joined two species in a subtree we have to compute the distances from every other node to the new subtree We do this with a simple average of distancesDist[Spinach MonHum]
= (Dist[Spinach Monkey] + Dist[Spinach Human])2 = (908 + 863)2 = 8855
Mon-Hum
MonkeyHumanSpinach
Calculation of New Distances
PAM Spinach Rice Mosquito MonHumSpinach 00 849 1056 886Rice 849 00 1178 1225Mosquito 1056 1178 00 828MonHum 886 1225 828 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Next Cycle
PAM Spinach Rice MosMonHumSpinach 00 849 971Rice 849 00 1202MosMonHum 971 1202 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
Penultimate Cycle
PAM SpinRice MosMonHumSpinach 00 1087MosMonHum 1087 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
(Spin-Rice)-(Mos-(Mon-Hum))
Last Joining
Human
Monkey
MosquitoRice
Spinach
Unrooted Neighbor-Joining Tree
Multiple Alignment- First pairbull Align the two most closely-related sequences first
bull This alignment is then lsquofixedrsquo and will never change If a gap is to be introduced subsequently then it will be introduced in the same place in both sequences but their relative alignment remains unchanged
ClustalW- Decision timebull Consult the guide tree to see what alignment is performed next
ndash Align a third sequence to the first twoOrndash Align two entirely different sequences to each other
Option 1Option 1 Option 2Option 2
ClustalW- Alternative 1If the situation arises where a third sequence is aligned to the first two then when a gap has to be introduced to improve the alignment each of these two entities are treated as two single sequences
+
ClustalW- Alternative 2bull If on the other hand two separate
sequences have to be aligned together then the first pairwise alignment is placed to one side and the pairwise alignment of the other two is carried out
+
ClustalW- Progression
bull The alignment is progressively built up in this way with each step being treated as a pairwise alignment sometimes with each member of a lsquopairrsquo having more than one sequence
Progressive alignment - step 11 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
12345
Progressive alignment - step 21 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
12345
Progressive alignment - step 31 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
+
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
12345
Progressive alignment - final step1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
+
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
5 -ctcga-acgatacgatgactagct-
12345
ClustalW-Good pointsBad points
bull Advantagesndash Speed
bull Disadvantagesndash No objective functionndash No way of quantifying whether or not the alignment is good
ndash No way of knowing if the alignment is lsquocorrectrsquo
ClustalW-Local Minimumbull Potential problems
ndash Local minimum problem If an error is introduced early in the alignment process it is impossible to correct this later in the procedure
ndash Arbitrary alignment
Increasing the sophistication of the alignment process
bull Should we treat all the sequences in the same way - even though some sequences are closely-related and some sequences are distant relatives
bull Should we treat all positions in the sequences as though they were the same - even though they might have different functions and different locations in the 3-dimensional structure
ClustalW- Caveatsbull Sequence weightingbull Varying substitution matricesbull Residue-specific gap penalties and reduced penalties
in hydrophilic regions (external regions of protein sequences) encourage gaps in loops rather than in core regions
bull Positions in early alignments where gaps have been opened receive locally reduced gap penalties to encourage openings in subsequent alignments
ClustalW- User-supplied values
bull Two penalties are set by the user (there are default values but you should know that it is possible to change these)
bull GOP- Gap Opening Penalty is the cost of opening a gap in an alignment
bull GEP- Gap Extension Penalty is the cost of extending this gap
Position-Specific gap penaltiesbull Before any pair of (groups of) sequences are
aligned a table of GOPs are generated for each position in the two (sets of) sequences
bull The GOP is manipulated in a position-specific manner so that it can vary over the sequences
bull If there is a gap at a position the GOP and GEP penalties are lowered the other rules do not apply
bull This makes gaps more likely at positions where gaps already exist
Discouraging too many gaps bull If there is no gap opened then the GOP is increased if the
position is within 8 residues of an existing gapbull This discourages gaps that are too close togetherbull At any position within a run of hydrophilic residues the GOP is
decreasedbull These runs usually indicate loop regions in protein structuresbull A run of 5 hydrophilic residues is considered to be a hydrophilic
stretchbull The default hydrophilic residues are
ndash D E G K N Q P R Sndash But this can be changed by the user
Divergent Sequencesbull The most divergent sequences (most different on average
from all of the other sequences) are usually the most difficult to align
bull It is sometimes better to delay their aligment until later (when the easier sequences have already been aligned)
bull The user has the choice of setting a cutoff (default is 40 identity)
bull This will delay the alignment until the others have been aligned
Advice on progressive alignmentbull Progressive alignment is a mathematical process that is completely
independent of biological realitybull Can be a very good estimatebull Can be an impossibly poor estimatebull Requires user input and skillbull Treat cautiouslybull Can be improved by eye (usually)bull Often helps to have colour-codingbull Depending on the use the user should be able to make a judgement
on those regions that are reliable or notbull For phylogeny reconstruction only use those positions whose
hypothesis of positional homology is unimpeachable
Alignment of protein-coding DNA sequences
bull It is not very sensible to align the DNA sequences of protein-coding genes
ATGCTGTTAGGGATGACTCTGTTAGGG
ATG-CT--GTTAGGGATGACTCTGTTAGGG
The result might be highly-implausible and might not reflect what is known about biological processesIt is much more sensible to translate the sequences to their corresponding amino acid sequences align these protein sequences and then put the gaps in the DNA sequences according to where they are found in the amino acid alignment
Manual Alignment- softwareGDE- The Genetic Data Environment (UNIX)CINEMA- Java applet available from
ndash httpwwwbiochemuclacuk
SeqappSeqpup- MacPCUNIX available fromndash httpiubiobioindianaedu
SeAl for Macintosh available fromndash httpevolvezoooxacukSe-AlSe-Alhtml
BioEdit for PC available fromndash httpwwwmbioncsueduRNasePinfoprogramsBIOEDITbio
edithtml
- Multiple Sequence Alignment James McInerney bioinf4biologists Feb 2009
- Alignment can be easy or difficult
- Homology Definition
- Multiple Sequence Alignment- Goals
- Multiple sequence alignments - problems
- Slide 6
- Slide 7
- SSU rRNA
- Alignment of 16S rRNA can be guided by secondary structure
- Protein Alignment may be guided by Tertiary Structure Interactions
- Multiple Sequence Alignment- Methods
- Manual Alignment - reasons
- Local minimum
- Dotplots
- Dotplot example sperm whale vs human myg
- Slide 16
- Slide 17
- Dotplots in practice
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Dynamic programming
- Progressive Alignment
- Slide 23
- ClustalW- Pairwise Alignments
- Path Graph for aligning two sequences
- Possible alignment
- Alignment using this path
- Optimal Alignment 1
- Optimal Alignment 2
- Alignment of 3 sequences
- ClustalW- Guide Tree
- Neighbor joining method
- Distance Matrix
- First Step
- Calculation of New Distances
- Next Cycle
- Penultimate Cycle
- Last Joining
- Unrooted Neighbor-Joining Tree
- Multiple Alignment- First pair
- ClustalW- Decision time
- ClustalW- Alternative 1
- ClustalW- Progression
- Progressive alignment - step 1
- Progressive alignment - step 2
- Progressive alignment - step 3
- Progressive alignment - final step
- ClustalW-Good pointsBad points
- ClustalW-Local Minimum
- Increasing the sophistication of the alignment process
- Slide 51
- ClustalW- Caveats
- ClustalW- User-supplied values
- Position-Specific gap penalties
- Discouraging too many gaps
- Divergent Sequences
- Advice on progressive alignment
- Alignment of protein-coding DNA sequences
- Manual Alignment- software
-

ClustalW- Pairwise Alignments
bull First perform all possible pairwise alignments between each pair of sequences There are (n-1)+(n-2)(n-n+1) possibilities
bull Calculate the lsquodistancersquo between each pair of sequences based on these isolated pairwise alignments
bull Generate a distance matrix
Path Graph for aligning two sequences
Possible alignment
1
1
0
1
0
-1
Scoring SchemebullMatch +1bullMismatch 0bullIndel -1
Score for this path= 2
Alignment using this path
GATTC-GAATTC
1
1
0
1
0
-1
Optimal Alignment 1
1
1
-1
1
1
1
Alignment score 4Alignment score 4
Alignment using this path
GA-TTCGA-TTCGAATTCGAATTC
Optimal Alignment 2
1
-1
1
1
1
1
Alignment score 4Alignment score 4
Alignment using this path
G-ATTCG-ATTCGAATTCGAATTC
Alignment of 3 sequences
ClustalW- Guide Tree
bull Generate a Neighbor-Joining lsquoguide treersquo from these pairwise distances
bull This guide tree gives the order in which the progressive alignment will be carried out
Neighbor joining method
bullThe neighbor-joining method is a greedy heuristic which joins at each step the two closest sub-trees that are not already joinedbullIt is based on the minimum evolution principlebullneighbors are defined as two taxa that are connected by a single node in an unrooted tree
A B
Node 1
PAM Spinach Rice Mosquito Monkey HumanSpinach 00 849 1056 908 863Rice 849 00 1178 1224 1226Mosquito 1056 1178 00 847 808Monkey 908 1224 847 00 33Human 863 1226 808 33 00
What is required for the Neighbour joining method
Distance matrixDistance Matrix
PAM distance 33 (Human - Monkey) is the minimum So well join Human and Monkey to MonHum and well calculate the new distances
Mon-Hum
MonkeyHumanSpinachMosquito Rice
First Step
After we have joined two species in a subtree we have to compute the distances from every other node to the new subtree We do this with a simple average of distancesDist[Spinach MonHum]
= (Dist[Spinach Monkey] + Dist[Spinach Human])2 = (908 + 863)2 = 8855
Mon-Hum
MonkeyHumanSpinach
Calculation of New Distances
PAM Spinach Rice Mosquito MonHumSpinach 00 849 1056 886Rice 849 00 1178 1225Mosquito 1056 1178 00 828MonHum 886 1225 828 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Next Cycle
PAM Spinach Rice MosMonHumSpinach 00 849 971Rice 849 00 1202MosMonHum 971 1202 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
Penultimate Cycle
PAM SpinRice MosMonHumSpinach 00 1087MosMonHum 1087 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
(Spin-Rice)-(Mos-(Mon-Hum))
Last Joining
Human
Monkey
MosquitoRice
Spinach
Unrooted Neighbor-Joining Tree
Multiple Alignment- First pairbull Align the two most closely-related sequences first
bull This alignment is then lsquofixedrsquo and will never change If a gap is to be introduced subsequently then it will be introduced in the same place in both sequences but their relative alignment remains unchanged
ClustalW- Decision timebull Consult the guide tree to see what alignment is performed next
ndash Align a third sequence to the first twoOrndash Align two entirely different sequences to each other
Option 1Option 1 Option 2Option 2
ClustalW- Alternative 1If the situation arises where a third sequence is aligned to the first two then when a gap has to be introduced to improve the alignment each of these two entities are treated as two single sequences
+
ClustalW- Alternative 2bull If on the other hand two separate
sequences have to be aligned together then the first pairwise alignment is placed to one side and the pairwise alignment of the other two is carried out
+
ClustalW- Progression
bull The alignment is progressively built up in this way with each step being treated as a pairwise alignment sometimes with each member of a lsquopairrsquo having more than one sequence
Progressive alignment - step 11 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
12345
Progressive alignment - step 21 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
12345
Progressive alignment - step 31 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
+
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
12345
Progressive alignment - final step1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
+
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
5 -ctcga-acgatacgatgactagct-
12345
ClustalW-Good pointsBad points
bull Advantagesndash Speed
bull Disadvantagesndash No objective functionndash No way of quantifying whether or not the alignment is good
ndash No way of knowing if the alignment is lsquocorrectrsquo
ClustalW-Local Minimumbull Potential problems
ndash Local minimum problem If an error is introduced early in the alignment process it is impossible to correct this later in the procedure
ndash Arbitrary alignment
Increasing the sophistication of the alignment process
bull Should we treat all the sequences in the same way - even though some sequences are closely-related and some sequences are distant relatives
bull Should we treat all positions in the sequences as though they were the same - even though they might have different functions and different locations in the 3-dimensional structure
ClustalW- Caveatsbull Sequence weightingbull Varying substitution matricesbull Residue-specific gap penalties and reduced penalties
in hydrophilic regions (external regions of protein sequences) encourage gaps in loops rather than in core regions
bull Positions in early alignments where gaps have been opened receive locally reduced gap penalties to encourage openings in subsequent alignments
ClustalW- User-supplied values
bull Two penalties are set by the user (there are default values but you should know that it is possible to change these)
bull GOP- Gap Opening Penalty is the cost of opening a gap in an alignment
bull GEP- Gap Extension Penalty is the cost of extending this gap
Position-Specific gap penaltiesbull Before any pair of (groups of) sequences are
aligned a table of GOPs are generated for each position in the two (sets of) sequences
bull The GOP is manipulated in a position-specific manner so that it can vary over the sequences
bull If there is a gap at a position the GOP and GEP penalties are lowered the other rules do not apply
bull This makes gaps more likely at positions where gaps already exist
Discouraging too many gaps bull If there is no gap opened then the GOP is increased if the
position is within 8 residues of an existing gapbull This discourages gaps that are too close togetherbull At any position within a run of hydrophilic residues the GOP is
decreasedbull These runs usually indicate loop regions in protein structuresbull A run of 5 hydrophilic residues is considered to be a hydrophilic
stretchbull The default hydrophilic residues are
ndash D E G K N Q P R Sndash But this can be changed by the user
Divergent Sequencesbull The most divergent sequences (most different on average
from all of the other sequences) are usually the most difficult to align
bull It is sometimes better to delay their aligment until later (when the easier sequences have already been aligned)
bull The user has the choice of setting a cutoff (default is 40 identity)
bull This will delay the alignment until the others have been aligned
Advice on progressive alignmentbull Progressive alignment is a mathematical process that is completely
independent of biological realitybull Can be a very good estimatebull Can be an impossibly poor estimatebull Requires user input and skillbull Treat cautiouslybull Can be improved by eye (usually)bull Often helps to have colour-codingbull Depending on the use the user should be able to make a judgement
on those regions that are reliable or notbull For phylogeny reconstruction only use those positions whose
hypothesis of positional homology is unimpeachable
Alignment of protein-coding DNA sequences
bull It is not very sensible to align the DNA sequences of protein-coding genes
ATGCTGTTAGGGATGACTCTGTTAGGG
ATG-CT--GTTAGGGATGACTCTGTTAGGG
The result might be highly-implausible and might not reflect what is known about biological processesIt is much more sensible to translate the sequences to their corresponding amino acid sequences align these protein sequences and then put the gaps in the DNA sequences according to where they are found in the amino acid alignment
Manual Alignment- softwareGDE- The Genetic Data Environment (UNIX)CINEMA- Java applet available from
ndash httpwwwbiochemuclacuk
SeqappSeqpup- MacPCUNIX available fromndash httpiubiobioindianaedu
SeAl for Macintosh available fromndash httpevolvezoooxacukSe-AlSe-Alhtml
BioEdit for PC available fromndash httpwwwmbioncsueduRNasePinfoprogramsBIOEDITbio
edithtml
- Multiple Sequence Alignment James McInerney bioinf4biologists Feb 2009
- Alignment can be easy or difficult
- Homology Definition
- Multiple Sequence Alignment- Goals
- Multiple sequence alignments - problems
- Slide 6
- Slide 7
- SSU rRNA
- Alignment of 16S rRNA can be guided by secondary structure
- Protein Alignment may be guided by Tertiary Structure Interactions
- Multiple Sequence Alignment- Methods
- Manual Alignment - reasons
- Local minimum
- Dotplots
- Dotplot example sperm whale vs human myg
- Slide 16
- Slide 17
- Dotplots in practice
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Dynamic programming
- Progressive Alignment
- Slide 23
- ClustalW- Pairwise Alignments
- Path Graph for aligning two sequences
- Possible alignment
- Alignment using this path
- Optimal Alignment 1
- Optimal Alignment 2
- Alignment of 3 sequences
- ClustalW- Guide Tree
- Neighbor joining method
- Distance Matrix
- First Step
- Calculation of New Distances
- Next Cycle
- Penultimate Cycle
- Last Joining
- Unrooted Neighbor-Joining Tree
- Multiple Alignment- First pair
- ClustalW- Decision time
- ClustalW- Alternative 1
- ClustalW- Progression
- Progressive alignment - step 1
- Progressive alignment - step 2
- Progressive alignment - step 3
- Progressive alignment - final step
- ClustalW-Good pointsBad points
- ClustalW-Local Minimum
- Increasing the sophistication of the alignment process
- Slide 51
- ClustalW- Caveats
- ClustalW- User-supplied values
- Position-Specific gap penalties
- Discouraging too many gaps
- Divergent Sequences
- Advice on progressive alignment
- Alignment of protein-coding DNA sequences
- Manual Alignment- software
-

Path Graph for aligning two sequences
Possible alignment
1
1
0
1
0
-1
Scoring SchemebullMatch +1bullMismatch 0bullIndel -1
Score for this path= 2
Alignment using this path
GATTC-GAATTC
1
1
0
1
0
-1
Optimal Alignment 1
1
1
-1
1
1
1
Alignment score 4Alignment score 4
Alignment using this path
GA-TTCGA-TTCGAATTCGAATTC
Optimal Alignment 2
1
-1
1
1
1
1
Alignment score 4Alignment score 4
Alignment using this path
G-ATTCG-ATTCGAATTCGAATTC
Alignment of 3 sequences
ClustalW- Guide Tree
bull Generate a Neighbor-Joining lsquoguide treersquo from these pairwise distances
bull This guide tree gives the order in which the progressive alignment will be carried out
Neighbor joining method
bullThe neighbor-joining method is a greedy heuristic which joins at each step the two closest sub-trees that are not already joinedbullIt is based on the minimum evolution principlebullneighbors are defined as two taxa that are connected by a single node in an unrooted tree
A B
Node 1
PAM Spinach Rice Mosquito Monkey HumanSpinach 00 849 1056 908 863Rice 849 00 1178 1224 1226Mosquito 1056 1178 00 847 808Monkey 908 1224 847 00 33Human 863 1226 808 33 00
What is required for the Neighbour joining method
Distance matrixDistance Matrix
PAM distance 33 (Human - Monkey) is the minimum So well join Human and Monkey to MonHum and well calculate the new distances
Mon-Hum
MonkeyHumanSpinachMosquito Rice
First Step
After we have joined two species in a subtree we have to compute the distances from every other node to the new subtree We do this with a simple average of distancesDist[Spinach MonHum]
= (Dist[Spinach Monkey] + Dist[Spinach Human])2 = (908 + 863)2 = 8855
Mon-Hum
MonkeyHumanSpinach
Calculation of New Distances
PAM Spinach Rice Mosquito MonHumSpinach 00 849 1056 886Rice 849 00 1178 1225Mosquito 1056 1178 00 828MonHum 886 1225 828 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Next Cycle
PAM Spinach Rice MosMonHumSpinach 00 849 971Rice 849 00 1202MosMonHum 971 1202 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
Penultimate Cycle
PAM SpinRice MosMonHumSpinach 00 1087MosMonHum 1087 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
(Spin-Rice)-(Mos-(Mon-Hum))
Last Joining
Human
Monkey
MosquitoRice
Spinach
Unrooted Neighbor-Joining Tree
Multiple Alignment- First pairbull Align the two most closely-related sequences first
bull This alignment is then lsquofixedrsquo and will never change If a gap is to be introduced subsequently then it will be introduced in the same place in both sequences but their relative alignment remains unchanged
ClustalW- Decision timebull Consult the guide tree to see what alignment is performed next
ndash Align a third sequence to the first twoOrndash Align two entirely different sequences to each other
Option 1Option 1 Option 2Option 2
ClustalW- Alternative 1If the situation arises where a third sequence is aligned to the first two then when a gap has to be introduced to improve the alignment each of these two entities are treated as two single sequences
+
ClustalW- Alternative 2bull If on the other hand two separate
sequences have to be aligned together then the first pairwise alignment is placed to one side and the pairwise alignment of the other two is carried out
+
ClustalW- Progression
bull The alignment is progressively built up in this way with each step being treated as a pairwise alignment sometimes with each member of a lsquopairrsquo having more than one sequence
Progressive alignment - step 11 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
12345
Progressive alignment - step 21 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
12345
Progressive alignment - step 31 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
+
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
12345
Progressive alignment - final step1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
+
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
5 -ctcga-acgatacgatgactagct-
12345
ClustalW-Good pointsBad points
bull Advantagesndash Speed
bull Disadvantagesndash No objective functionndash No way of quantifying whether or not the alignment is good
ndash No way of knowing if the alignment is lsquocorrectrsquo
ClustalW-Local Minimumbull Potential problems
ndash Local minimum problem If an error is introduced early in the alignment process it is impossible to correct this later in the procedure
ndash Arbitrary alignment
Increasing the sophistication of the alignment process
bull Should we treat all the sequences in the same way - even though some sequences are closely-related and some sequences are distant relatives
bull Should we treat all positions in the sequences as though they were the same - even though they might have different functions and different locations in the 3-dimensional structure
ClustalW- Caveatsbull Sequence weightingbull Varying substitution matricesbull Residue-specific gap penalties and reduced penalties
in hydrophilic regions (external regions of protein sequences) encourage gaps in loops rather than in core regions
bull Positions in early alignments where gaps have been opened receive locally reduced gap penalties to encourage openings in subsequent alignments
ClustalW- User-supplied values
bull Two penalties are set by the user (there are default values but you should know that it is possible to change these)
bull GOP- Gap Opening Penalty is the cost of opening a gap in an alignment
bull GEP- Gap Extension Penalty is the cost of extending this gap
Position-Specific gap penaltiesbull Before any pair of (groups of) sequences are
aligned a table of GOPs are generated for each position in the two (sets of) sequences
bull The GOP is manipulated in a position-specific manner so that it can vary over the sequences
bull If there is a gap at a position the GOP and GEP penalties are lowered the other rules do not apply
bull This makes gaps more likely at positions where gaps already exist
Discouraging too many gaps bull If there is no gap opened then the GOP is increased if the
position is within 8 residues of an existing gapbull This discourages gaps that are too close togetherbull At any position within a run of hydrophilic residues the GOP is
decreasedbull These runs usually indicate loop regions in protein structuresbull A run of 5 hydrophilic residues is considered to be a hydrophilic
stretchbull The default hydrophilic residues are
ndash D E G K N Q P R Sndash But this can be changed by the user
Divergent Sequencesbull The most divergent sequences (most different on average
from all of the other sequences) are usually the most difficult to align
bull It is sometimes better to delay their aligment until later (when the easier sequences have already been aligned)
bull The user has the choice of setting a cutoff (default is 40 identity)
bull This will delay the alignment until the others have been aligned
Advice on progressive alignmentbull Progressive alignment is a mathematical process that is completely
independent of biological realitybull Can be a very good estimatebull Can be an impossibly poor estimatebull Requires user input and skillbull Treat cautiouslybull Can be improved by eye (usually)bull Often helps to have colour-codingbull Depending on the use the user should be able to make a judgement
on those regions that are reliable or notbull For phylogeny reconstruction only use those positions whose
hypothesis of positional homology is unimpeachable
Alignment of protein-coding DNA sequences
bull It is not very sensible to align the DNA sequences of protein-coding genes
ATGCTGTTAGGGATGACTCTGTTAGGG
ATG-CT--GTTAGGGATGACTCTGTTAGGG
The result might be highly-implausible and might not reflect what is known about biological processesIt is much more sensible to translate the sequences to their corresponding amino acid sequences align these protein sequences and then put the gaps in the DNA sequences according to where they are found in the amino acid alignment
Manual Alignment- softwareGDE- The Genetic Data Environment (UNIX)CINEMA- Java applet available from
ndash httpwwwbiochemuclacuk
SeqappSeqpup- MacPCUNIX available fromndash httpiubiobioindianaedu
SeAl for Macintosh available fromndash httpevolvezoooxacukSe-AlSe-Alhtml
BioEdit for PC available fromndash httpwwwmbioncsueduRNasePinfoprogramsBIOEDITbio
edithtml
- Multiple Sequence Alignment James McInerney bioinf4biologists Feb 2009
- Alignment can be easy or difficult
- Homology Definition
- Multiple Sequence Alignment- Goals
- Multiple sequence alignments - problems
- Slide 6
- Slide 7
- SSU rRNA
- Alignment of 16S rRNA can be guided by secondary structure
- Protein Alignment may be guided by Tertiary Structure Interactions
- Multiple Sequence Alignment- Methods
- Manual Alignment - reasons
- Local minimum
- Dotplots
- Dotplot example sperm whale vs human myg
- Slide 16
- Slide 17
- Dotplots in practice
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Dynamic programming
- Progressive Alignment
- Slide 23
- ClustalW- Pairwise Alignments
- Path Graph for aligning two sequences
- Possible alignment
- Alignment using this path
- Optimal Alignment 1
- Optimal Alignment 2
- Alignment of 3 sequences
- ClustalW- Guide Tree
- Neighbor joining method
- Distance Matrix
- First Step
- Calculation of New Distances
- Next Cycle
- Penultimate Cycle
- Last Joining
- Unrooted Neighbor-Joining Tree
- Multiple Alignment- First pair
- ClustalW- Decision time
- ClustalW- Alternative 1
- ClustalW- Progression
- Progressive alignment - step 1
- Progressive alignment - step 2
- Progressive alignment - step 3
- Progressive alignment - final step
- ClustalW-Good pointsBad points
- ClustalW-Local Minimum
- Increasing the sophistication of the alignment process
- Slide 51
- ClustalW- Caveats
- ClustalW- User-supplied values
- Position-Specific gap penalties
- Discouraging too many gaps
- Divergent Sequences
- Advice on progressive alignment
- Alignment of protein-coding DNA sequences
- Manual Alignment- software
-

Possible alignment
1
1
0
1
0
-1
Scoring SchemebullMatch +1bullMismatch 0bullIndel -1
Score for this path= 2
Alignment using this path
GATTC-GAATTC
1
1
0
1
0
-1
Optimal Alignment 1
1
1
-1
1
1
1
Alignment score 4Alignment score 4
Alignment using this path
GA-TTCGA-TTCGAATTCGAATTC
Optimal Alignment 2
1
-1
1
1
1
1
Alignment score 4Alignment score 4
Alignment using this path
G-ATTCG-ATTCGAATTCGAATTC
Alignment of 3 sequences
ClustalW- Guide Tree
bull Generate a Neighbor-Joining lsquoguide treersquo from these pairwise distances
bull This guide tree gives the order in which the progressive alignment will be carried out
Neighbor joining method
bullThe neighbor-joining method is a greedy heuristic which joins at each step the two closest sub-trees that are not already joinedbullIt is based on the minimum evolution principlebullneighbors are defined as two taxa that are connected by a single node in an unrooted tree
A B
Node 1
PAM Spinach Rice Mosquito Monkey HumanSpinach 00 849 1056 908 863Rice 849 00 1178 1224 1226Mosquito 1056 1178 00 847 808Monkey 908 1224 847 00 33Human 863 1226 808 33 00
What is required for the Neighbour joining method
Distance matrixDistance Matrix
PAM distance 33 (Human - Monkey) is the minimum So well join Human and Monkey to MonHum and well calculate the new distances
Mon-Hum
MonkeyHumanSpinachMosquito Rice
First Step
After we have joined two species in a subtree we have to compute the distances from every other node to the new subtree We do this with a simple average of distancesDist[Spinach MonHum]
= (Dist[Spinach Monkey] + Dist[Spinach Human])2 = (908 + 863)2 = 8855
Mon-Hum
MonkeyHumanSpinach
Calculation of New Distances
PAM Spinach Rice Mosquito MonHumSpinach 00 849 1056 886Rice 849 00 1178 1225Mosquito 1056 1178 00 828MonHum 886 1225 828 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Next Cycle
PAM Spinach Rice MosMonHumSpinach 00 849 971Rice 849 00 1202MosMonHum 971 1202 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
Penultimate Cycle
PAM SpinRice MosMonHumSpinach 00 1087MosMonHum 1087 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
(Spin-Rice)-(Mos-(Mon-Hum))
Last Joining
Human
Monkey
MosquitoRice
Spinach
Unrooted Neighbor-Joining Tree
Multiple Alignment- First pairbull Align the two most closely-related sequences first
bull This alignment is then lsquofixedrsquo and will never change If a gap is to be introduced subsequently then it will be introduced in the same place in both sequences but their relative alignment remains unchanged
ClustalW- Decision timebull Consult the guide tree to see what alignment is performed next
ndash Align a third sequence to the first twoOrndash Align two entirely different sequences to each other
Option 1Option 1 Option 2Option 2
ClustalW- Alternative 1If the situation arises where a third sequence is aligned to the first two then when a gap has to be introduced to improve the alignment each of these two entities are treated as two single sequences
+
ClustalW- Alternative 2bull If on the other hand two separate
sequences have to be aligned together then the first pairwise alignment is placed to one side and the pairwise alignment of the other two is carried out
+
ClustalW- Progression
bull The alignment is progressively built up in this way with each step being treated as a pairwise alignment sometimes with each member of a lsquopairrsquo having more than one sequence
Progressive alignment - step 11 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
12345
Progressive alignment - step 21 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
12345
Progressive alignment - step 31 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
+
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
12345
Progressive alignment - final step1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
+
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
5 -ctcga-acgatacgatgactagct-
12345
ClustalW-Good pointsBad points
bull Advantagesndash Speed
bull Disadvantagesndash No objective functionndash No way of quantifying whether or not the alignment is good
ndash No way of knowing if the alignment is lsquocorrectrsquo
ClustalW-Local Minimumbull Potential problems
ndash Local minimum problem If an error is introduced early in the alignment process it is impossible to correct this later in the procedure
ndash Arbitrary alignment
Increasing the sophistication of the alignment process
bull Should we treat all the sequences in the same way - even though some sequences are closely-related and some sequences are distant relatives
bull Should we treat all positions in the sequences as though they were the same - even though they might have different functions and different locations in the 3-dimensional structure
ClustalW- Caveatsbull Sequence weightingbull Varying substitution matricesbull Residue-specific gap penalties and reduced penalties
in hydrophilic regions (external regions of protein sequences) encourage gaps in loops rather than in core regions
bull Positions in early alignments where gaps have been opened receive locally reduced gap penalties to encourage openings in subsequent alignments
ClustalW- User-supplied values
bull Two penalties are set by the user (there are default values but you should know that it is possible to change these)
bull GOP- Gap Opening Penalty is the cost of opening a gap in an alignment
bull GEP- Gap Extension Penalty is the cost of extending this gap
Position-Specific gap penaltiesbull Before any pair of (groups of) sequences are
aligned a table of GOPs are generated for each position in the two (sets of) sequences
bull The GOP is manipulated in a position-specific manner so that it can vary over the sequences
bull If there is a gap at a position the GOP and GEP penalties are lowered the other rules do not apply
bull This makes gaps more likely at positions where gaps already exist
Discouraging too many gaps bull If there is no gap opened then the GOP is increased if the
position is within 8 residues of an existing gapbull This discourages gaps that are too close togetherbull At any position within a run of hydrophilic residues the GOP is
decreasedbull These runs usually indicate loop regions in protein structuresbull A run of 5 hydrophilic residues is considered to be a hydrophilic
stretchbull The default hydrophilic residues are
ndash D E G K N Q P R Sndash But this can be changed by the user
Divergent Sequencesbull The most divergent sequences (most different on average
from all of the other sequences) are usually the most difficult to align
bull It is sometimes better to delay their aligment until later (when the easier sequences have already been aligned)
bull The user has the choice of setting a cutoff (default is 40 identity)
bull This will delay the alignment until the others have been aligned
Advice on progressive alignmentbull Progressive alignment is a mathematical process that is completely
independent of biological realitybull Can be a very good estimatebull Can be an impossibly poor estimatebull Requires user input and skillbull Treat cautiouslybull Can be improved by eye (usually)bull Often helps to have colour-codingbull Depending on the use the user should be able to make a judgement
on those regions that are reliable or notbull For phylogeny reconstruction only use those positions whose
hypothesis of positional homology is unimpeachable
Alignment of protein-coding DNA sequences
bull It is not very sensible to align the DNA sequences of protein-coding genes
ATGCTGTTAGGGATGACTCTGTTAGGG
ATG-CT--GTTAGGGATGACTCTGTTAGGG
The result might be highly-implausible and might not reflect what is known about biological processesIt is much more sensible to translate the sequences to their corresponding amino acid sequences align these protein sequences and then put the gaps in the DNA sequences according to where they are found in the amino acid alignment
Manual Alignment- softwareGDE- The Genetic Data Environment (UNIX)CINEMA- Java applet available from
ndash httpwwwbiochemuclacuk
SeqappSeqpup- MacPCUNIX available fromndash httpiubiobioindianaedu
SeAl for Macintosh available fromndash httpevolvezoooxacukSe-AlSe-Alhtml
BioEdit for PC available fromndash httpwwwmbioncsueduRNasePinfoprogramsBIOEDITbio
edithtml
- Multiple Sequence Alignment James McInerney bioinf4biologists Feb 2009
- Alignment can be easy or difficult
- Homology Definition
- Multiple Sequence Alignment- Goals
- Multiple sequence alignments - problems
- Slide 6
- Slide 7
- SSU rRNA
- Alignment of 16S rRNA can be guided by secondary structure
- Protein Alignment may be guided by Tertiary Structure Interactions
- Multiple Sequence Alignment- Methods
- Manual Alignment - reasons
- Local minimum
- Dotplots
- Dotplot example sperm whale vs human myg
- Slide 16
- Slide 17
- Dotplots in practice
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Dynamic programming
- Progressive Alignment
- Slide 23
- ClustalW- Pairwise Alignments
- Path Graph for aligning two sequences
- Possible alignment
- Alignment using this path
- Optimal Alignment 1
- Optimal Alignment 2
- Alignment of 3 sequences
- ClustalW- Guide Tree
- Neighbor joining method
- Distance Matrix
- First Step
- Calculation of New Distances
- Next Cycle
- Penultimate Cycle
- Last Joining
- Unrooted Neighbor-Joining Tree
- Multiple Alignment- First pair
- ClustalW- Decision time
- ClustalW- Alternative 1
- ClustalW- Progression
- Progressive alignment - step 1
- Progressive alignment - step 2
- Progressive alignment - step 3
- Progressive alignment - final step
- ClustalW-Good pointsBad points
- ClustalW-Local Minimum
- Increasing the sophistication of the alignment process
- Slide 51
- ClustalW- Caveats
- ClustalW- User-supplied values
- Position-Specific gap penalties
- Discouraging too many gaps
- Divergent Sequences
- Advice on progressive alignment
- Alignment of protein-coding DNA sequences
- Manual Alignment- software
-

Alignment using this path
GATTC-GAATTC
1
1
0
1
0
-1
Optimal Alignment 1
1
1
-1
1
1
1
Alignment score 4Alignment score 4
Alignment using this path
GA-TTCGA-TTCGAATTCGAATTC
Optimal Alignment 2
1
-1
1
1
1
1
Alignment score 4Alignment score 4
Alignment using this path
G-ATTCG-ATTCGAATTCGAATTC
Alignment of 3 sequences
ClustalW- Guide Tree
bull Generate a Neighbor-Joining lsquoguide treersquo from these pairwise distances
bull This guide tree gives the order in which the progressive alignment will be carried out
Neighbor joining method
bullThe neighbor-joining method is a greedy heuristic which joins at each step the two closest sub-trees that are not already joinedbullIt is based on the minimum evolution principlebullneighbors are defined as two taxa that are connected by a single node in an unrooted tree
A B
Node 1
PAM Spinach Rice Mosquito Monkey HumanSpinach 00 849 1056 908 863Rice 849 00 1178 1224 1226Mosquito 1056 1178 00 847 808Monkey 908 1224 847 00 33Human 863 1226 808 33 00
What is required for the Neighbour joining method
Distance matrixDistance Matrix
PAM distance 33 (Human - Monkey) is the minimum So well join Human and Monkey to MonHum and well calculate the new distances
Mon-Hum
MonkeyHumanSpinachMosquito Rice
First Step
After we have joined two species in a subtree we have to compute the distances from every other node to the new subtree We do this with a simple average of distancesDist[Spinach MonHum]
= (Dist[Spinach Monkey] + Dist[Spinach Human])2 = (908 + 863)2 = 8855
Mon-Hum
MonkeyHumanSpinach
Calculation of New Distances
PAM Spinach Rice Mosquito MonHumSpinach 00 849 1056 886Rice 849 00 1178 1225Mosquito 1056 1178 00 828MonHum 886 1225 828 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Next Cycle
PAM Spinach Rice MosMonHumSpinach 00 849 971Rice 849 00 1202MosMonHum 971 1202 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
Penultimate Cycle
PAM SpinRice MosMonHumSpinach 00 1087MosMonHum 1087 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
(Spin-Rice)-(Mos-(Mon-Hum))
Last Joining
Human
Monkey
MosquitoRice
Spinach
Unrooted Neighbor-Joining Tree
Multiple Alignment- First pairbull Align the two most closely-related sequences first
bull This alignment is then lsquofixedrsquo and will never change If a gap is to be introduced subsequently then it will be introduced in the same place in both sequences but their relative alignment remains unchanged
ClustalW- Decision timebull Consult the guide tree to see what alignment is performed next
ndash Align a third sequence to the first twoOrndash Align two entirely different sequences to each other
Option 1Option 1 Option 2Option 2
ClustalW- Alternative 1If the situation arises where a third sequence is aligned to the first two then when a gap has to be introduced to improve the alignment each of these two entities are treated as two single sequences
+
ClustalW- Alternative 2bull If on the other hand two separate
sequences have to be aligned together then the first pairwise alignment is placed to one side and the pairwise alignment of the other two is carried out
+
ClustalW- Progression
bull The alignment is progressively built up in this way with each step being treated as a pairwise alignment sometimes with each member of a lsquopairrsquo having more than one sequence
Progressive alignment - step 11 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
12345
Progressive alignment - step 21 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
12345
Progressive alignment - step 31 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
+
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
12345
Progressive alignment - final step1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
+
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
5 -ctcga-acgatacgatgactagct-
12345
ClustalW-Good pointsBad points
bull Advantagesndash Speed
bull Disadvantagesndash No objective functionndash No way of quantifying whether or not the alignment is good
ndash No way of knowing if the alignment is lsquocorrectrsquo
ClustalW-Local Minimumbull Potential problems
ndash Local minimum problem If an error is introduced early in the alignment process it is impossible to correct this later in the procedure
ndash Arbitrary alignment
Increasing the sophistication of the alignment process
bull Should we treat all the sequences in the same way - even though some sequences are closely-related and some sequences are distant relatives
bull Should we treat all positions in the sequences as though they were the same - even though they might have different functions and different locations in the 3-dimensional structure
ClustalW- Caveatsbull Sequence weightingbull Varying substitution matricesbull Residue-specific gap penalties and reduced penalties
in hydrophilic regions (external regions of protein sequences) encourage gaps in loops rather than in core regions
bull Positions in early alignments where gaps have been opened receive locally reduced gap penalties to encourage openings in subsequent alignments
ClustalW- User-supplied values
bull Two penalties are set by the user (there are default values but you should know that it is possible to change these)
bull GOP- Gap Opening Penalty is the cost of opening a gap in an alignment
bull GEP- Gap Extension Penalty is the cost of extending this gap
Position-Specific gap penaltiesbull Before any pair of (groups of) sequences are
aligned a table of GOPs are generated for each position in the two (sets of) sequences
bull The GOP is manipulated in a position-specific manner so that it can vary over the sequences
bull If there is a gap at a position the GOP and GEP penalties are lowered the other rules do not apply
bull This makes gaps more likely at positions where gaps already exist
Discouraging too many gaps bull If there is no gap opened then the GOP is increased if the
position is within 8 residues of an existing gapbull This discourages gaps that are too close togetherbull At any position within a run of hydrophilic residues the GOP is
decreasedbull These runs usually indicate loop regions in protein structuresbull A run of 5 hydrophilic residues is considered to be a hydrophilic
stretchbull The default hydrophilic residues are
ndash D E G K N Q P R Sndash But this can be changed by the user
Divergent Sequencesbull The most divergent sequences (most different on average
from all of the other sequences) are usually the most difficult to align
bull It is sometimes better to delay their aligment until later (when the easier sequences have already been aligned)
bull The user has the choice of setting a cutoff (default is 40 identity)
bull This will delay the alignment until the others have been aligned
Advice on progressive alignmentbull Progressive alignment is a mathematical process that is completely
independent of biological realitybull Can be a very good estimatebull Can be an impossibly poor estimatebull Requires user input and skillbull Treat cautiouslybull Can be improved by eye (usually)bull Often helps to have colour-codingbull Depending on the use the user should be able to make a judgement
on those regions that are reliable or notbull For phylogeny reconstruction only use those positions whose
hypothesis of positional homology is unimpeachable
Alignment of protein-coding DNA sequences
bull It is not very sensible to align the DNA sequences of protein-coding genes
ATGCTGTTAGGGATGACTCTGTTAGGG
ATG-CT--GTTAGGGATGACTCTGTTAGGG
The result might be highly-implausible and might not reflect what is known about biological processesIt is much more sensible to translate the sequences to their corresponding amino acid sequences align these protein sequences and then put the gaps in the DNA sequences according to where they are found in the amino acid alignment
Manual Alignment- softwareGDE- The Genetic Data Environment (UNIX)CINEMA- Java applet available from
ndash httpwwwbiochemuclacuk
SeqappSeqpup- MacPCUNIX available fromndash httpiubiobioindianaedu
SeAl for Macintosh available fromndash httpevolvezoooxacukSe-AlSe-Alhtml
BioEdit for PC available fromndash httpwwwmbioncsueduRNasePinfoprogramsBIOEDITbio
edithtml
- Multiple Sequence Alignment James McInerney bioinf4biologists Feb 2009
- Alignment can be easy or difficult
- Homology Definition
- Multiple Sequence Alignment- Goals
- Multiple sequence alignments - problems
- Slide 6
- Slide 7
- SSU rRNA
- Alignment of 16S rRNA can be guided by secondary structure
- Protein Alignment may be guided by Tertiary Structure Interactions
- Multiple Sequence Alignment- Methods
- Manual Alignment - reasons
- Local minimum
- Dotplots
- Dotplot example sperm whale vs human myg
- Slide 16
- Slide 17
- Dotplots in practice
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Dynamic programming
- Progressive Alignment
- Slide 23
- ClustalW- Pairwise Alignments
- Path Graph for aligning two sequences
- Possible alignment
- Alignment using this path
- Optimal Alignment 1
- Optimal Alignment 2
- Alignment of 3 sequences
- ClustalW- Guide Tree
- Neighbor joining method
- Distance Matrix
- First Step
- Calculation of New Distances
- Next Cycle
- Penultimate Cycle
- Last Joining
- Unrooted Neighbor-Joining Tree
- Multiple Alignment- First pair
- ClustalW- Decision time
- ClustalW- Alternative 1
- ClustalW- Progression
- Progressive alignment - step 1
- Progressive alignment - step 2
- Progressive alignment - step 3
- Progressive alignment - final step
- ClustalW-Good pointsBad points
- ClustalW-Local Minimum
- Increasing the sophistication of the alignment process
- Slide 51
- ClustalW- Caveats
- ClustalW- User-supplied values
- Position-Specific gap penalties
- Discouraging too many gaps
- Divergent Sequences
- Advice on progressive alignment
- Alignment of protein-coding DNA sequences
- Manual Alignment- software
-

Optimal Alignment 1
1
1
-1
1
1
1
Alignment score 4Alignment score 4
Alignment using this path
GA-TTCGA-TTCGAATTCGAATTC
Optimal Alignment 2
1
-1
1
1
1
1
Alignment score 4Alignment score 4
Alignment using this path
G-ATTCG-ATTCGAATTCGAATTC
Alignment of 3 sequences
ClustalW- Guide Tree
bull Generate a Neighbor-Joining lsquoguide treersquo from these pairwise distances
bull This guide tree gives the order in which the progressive alignment will be carried out
Neighbor joining method
bullThe neighbor-joining method is a greedy heuristic which joins at each step the two closest sub-trees that are not already joinedbullIt is based on the minimum evolution principlebullneighbors are defined as two taxa that are connected by a single node in an unrooted tree
A B
Node 1
PAM Spinach Rice Mosquito Monkey HumanSpinach 00 849 1056 908 863Rice 849 00 1178 1224 1226Mosquito 1056 1178 00 847 808Monkey 908 1224 847 00 33Human 863 1226 808 33 00
What is required for the Neighbour joining method
Distance matrixDistance Matrix
PAM distance 33 (Human - Monkey) is the minimum So well join Human and Monkey to MonHum and well calculate the new distances
Mon-Hum
MonkeyHumanSpinachMosquito Rice
First Step
After we have joined two species in a subtree we have to compute the distances from every other node to the new subtree We do this with a simple average of distancesDist[Spinach MonHum]
= (Dist[Spinach Monkey] + Dist[Spinach Human])2 = (908 + 863)2 = 8855
Mon-Hum
MonkeyHumanSpinach
Calculation of New Distances
PAM Spinach Rice Mosquito MonHumSpinach 00 849 1056 886Rice 849 00 1178 1225Mosquito 1056 1178 00 828MonHum 886 1225 828 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Next Cycle
PAM Spinach Rice MosMonHumSpinach 00 849 971Rice 849 00 1202MosMonHum 971 1202 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
Penultimate Cycle
PAM SpinRice MosMonHumSpinach 00 1087MosMonHum 1087 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
(Spin-Rice)-(Mos-(Mon-Hum))
Last Joining
Human
Monkey
MosquitoRice
Spinach
Unrooted Neighbor-Joining Tree
Multiple Alignment- First pairbull Align the two most closely-related sequences first
bull This alignment is then lsquofixedrsquo and will never change If a gap is to be introduced subsequently then it will be introduced in the same place in both sequences but their relative alignment remains unchanged
ClustalW- Decision timebull Consult the guide tree to see what alignment is performed next
ndash Align a third sequence to the first twoOrndash Align two entirely different sequences to each other
Option 1Option 1 Option 2Option 2
ClustalW- Alternative 1If the situation arises where a third sequence is aligned to the first two then when a gap has to be introduced to improve the alignment each of these two entities are treated as two single sequences
+
ClustalW- Alternative 2bull If on the other hand two separate
sequences have to be aligned together then the first pairwise alignment is placed to one side and the pairwise alignment of the other two is carried out
+
ClustalW- Progression
bull The alignment is progressively built up in this way with each step being treated as a pairwise alignment sometimes with each member of a lsquopairrsquo having more than one sequence
Progressive alignment - step 11 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
12345
Progressive alignment - step 21 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
12345
Progressive alignment - step 31 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
+
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
12345
Progressive alignment - final step1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
+
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
5 -ctcga-acgatacgatgactagct-
12345
ClustalW-Good pointsBad points
bull Advantagesndash Speed
bull Disadvantagesndash No objective functionndash No way of quantifying whether or not the alignment is good
ndash No way of knowing if the alignment is lsquocorrectrsquo
ClustalW-Local Minimumbull Potential problems
ndash Local minimum problem If an error is introduced early in the alignment process it is impossible to correct this later in the procedure
ndash Arbitrary alignment
Increasing the sophistication of the alignment process
bull Should we treat all the sequences in the same way - even though some sequences are closely-related and some sequences are distant relatives
bull Should we treat all positions in the sequences as though they were the same - even though they might have different functions and different locations in the 3-dimensional structure
ClustalW- Caveatsbull Sequence weightingbull Varying substitution matricesbull Residue-specific gap penalties and reduced penalties
in hydrophilic regions (external regions of protein sequences) encourage gaps in loops rather than in core regions
bull Positions in early alignments where gaps have been opened receive locally reduced gap penalties to encourage openings in subsequent alignments
ClustalW- User-supplied values
bull Two penalties are set by the user (there are default values but you should know that it is possible to change these)
bull GOP- Gap Opening Penalty is the cost of opening a gap in an alignment
bull GEP- Gap Extension Penalty is the cost of extending this gap
Position-Specific gap penaltiesbull Before any pair of (groups of) sequences are
aligned a table of GOPs are generated for each position in the two (sets of) sequences
bull The GOP is manipulated in a position-specific manner so that it can vary over the sequences
bull If there is a gap at a position the GOP and GEP penalties are lowered the other rules do not apply
bull This makes gaps more likely at positions where gaps already exist
Discouraging too many gaps bull If there is no gap opened then the GOP is increased if the
position is within 8 residues of an existing gapbull This discourages gaps that are too close togetherbull At any position within a run of hydrophilic residues the GOP is
decreasedbull These runs usually indicate loop regions in protein structuresbull A run of 5 hydrophilic residues is considered to be a hydrophilic
stretchbull The default hydrophilic residues are
ndash D E G K N Q P R Sndash But this can be changed by the user
Divergent Sequencesbull The most divergent sequences (most different on average
from all of the other sequences) are usually the most difficult to align
bull It is sometimes better to delay their aligment until later (when the easier sequences have already been aligned)
bull The user has the choice of setting a cutoff (default is 40 identity)
bull This will delay the alignment until the others have been aligned
Advice on progressive alignmentbull Progressive alignment is a mathematical process that is completely
independent of biological realitybull Can be a very good estimatebull Can be an impossibly poor estimatebull Requires user input and skillbull Treat cautiouslybull Can be improved by eye (usually)bull Often helps to have colour-codingbull Depending on the use the user should be able to make a judgement
on those regions that are reliable or notbull For phylogeny reconstruction only use those positions whose
hypothesis of positional homology is unimpeachable
Alignment of protein-coding DNA sequences
bull It is not very sensible to align the DNA sequences of protein-coding genes
ATGCTGTTAGGGATGACTCTGTTAGGG
ATG-CT--GTTAGGGATGACTCTGTTAGGG
The result might be highly-implausible and might not reflect what is known about biological processesIt is much more sensible to translate the sequences to their corresponding amino acid sequences align these protein sequences and then put the gaps in the DNA sequences according to where they are found in the amino acid alignment
Manual Alignment- softwareGDE- The Genetic Data Environment (UNIX)CINEMA- Java applet available from
ndash httpwwwbiochemuclacuk
SeqappSeqpup- MacPCUNIX available fromndash httpiubiobioindianaedu
SeAl for Macintosh available fromndash httpevolvezoooxacukSe-AlSe-Alhtml
BioEdit for PC available fromndash httpwwwmbioncsueduRNasePinfoprogramsBIOEDITbio
edithtml
- Multiple Sequence Alignment James McInerney bioinf4biologists Feb 2009
- Alignment can be easy or difficult
- Homology Definition
- Multiple Sequence Alignment- Goals
- Multiple sequence alignments - problems
- Slide 6
- Slide 7
- SSU rRNA
- Alignment of 16S rRNA can be guided by secondary structure
- Protein Alignment may be guided by Tertiary Structure Interactions
- Multiple Sequence Alignment- Methods
- Manual Alignment - reasons
- Local minimum
- Dotplots
- Dotplot example sperm whale vs human myg
- Slide 16
- Slide 17
- Dotplots in practice
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Dynamic programming
- Progressive Alignment
- Slide 23
- ClustalW- Pairwise Alignments
- Path Graph for aligning two sequences
- Possible alignment
- Alignment using this path
- Optimal Alignment 1
- Optimal Alignment 2
- Alignment of 3 sequences
- ClustalW- Guide Tree
- Neighbor joining method
- Distance Matrix
- First Step
- Calculation of New Distances
- Next Cycle
- Penultimate Cycle
- Last Joining
- Unrooted Neighbor-Joining Tree
- Multiple Alignment- First pair
- ClustalW- Decision time
- ClustalW- Alternative 1
- ClustalW- Progression
- Progressive alignment - step 1
- Progressive alignment - step 2
- Progressive alignment - step 3
- Progressive alignment - final step
- ClustalW-Good pointsBad points
- ClustalW-Local Minimum
- Increasing the sophistication of the alignment process
- Slide 51
- ClustalW- Caveats
- ClustalW- User-supplied values
- Position-Specific gap penalties
- Discouraging too many gaps
- Divergent Sequences
- Advice on progressive alignment
- Alignment of protein-coding DNA sequences
- Manual Alignment- software
-

Optimal Alignment 2
1
-1
1
1
1
1
Alignment score 4Alignment score 4
Alignment using this path
G-ATTCG-ATTCGAATTCGAATTC
Alignment of 3 sequences
ClustalW- Guide Tree
bull Generate a Neighbor-Joining lsquoguide treersquo from these pairwise distances
bull This guide tree gives the order in which the progressive alignment will be carried out
Neighbor joining method
bullThe neighbor-joining method is a greedy heuristic which joins at each step the two closest sub-trees that are not already joinedbullIt is based on the minimum evolution principlebullneighbors are defined as two taxa that are connected by a single node in an unrooted tree
A B
Node 1
PAM Spinach Rice Mosquito Monkey HumanSpinach 00 849 1056 908 863Rice 849 00 1178 1224 1226Mosquito 1056 1178 00 847 808Monkey 908 1224 847 00 33Human 863 1226 808 33 00
What is required for the Neighbour joining method
Distance matrixDistance Matrix
PAM distance 33 (Human - Monkey) is the minimum So well join Human and Monkey to MonHum and well calculate the new distances
Mon-Hum
MonkeyHumanSpinachMosquito Rice
First Step
After we have joined two species in a subtree we have to compute the distances from every other node to the new subtree We do this with a simple average of distancesDist[Spinach MonHum]
= (Dist[Spinach Monkey] + Dist[Spinach Human])2 = (908 + 863)2 = 8855
Mon-Hum
MonkeyHumanSpinach
Calculation of New Distances
PAM Spinach Rice Mosquito MonHumSpinach 00 849 1056 886Rice 849 00 1178 1225Mosquito 1056 1178 00 828MonHum 886 1225 828 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Next Cycle
PAM Spinach Rice MosMonHumSpinach 00 849 971Rice 849 00 1202MosMonHum 971 1202 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
Penultimate Cycle
PAM SpinRice MosMonHumSpinach 00 1087MosMonHum 1087 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
(Spin-Rice)-(Mos-(Mon-Hum))
Last Joining
Human
Monkey
MosquitoRice
Spinach
Unrooted Neighbor-Joining Tree
Multiple Alignment- First pairbull Align the two most closely-related sequences first
bull This alignment is then lsquofixedrsquo and will never change If a gap is to be introduced subsequently then it will be introduced in the same place in both sequences but their relative alignment remains unchanged
ClustalW- Decision timebull Consult the guide tree to see what alignment is performed next
ndash Align a third sequence to the first twoOrndash Align two entirely different sequences to each other
Option 1Option 1 Option 2Option 2
ClustalW- Alternative 1If the situation arises where a third sequence is aligned to the first two then when a gap has to be introduced to improve the alignment each of these two entities are treated as two single sequences
+
ClustalW- Alternative 2bull If on the other hand two separate
sequences have to be aligned together then the first pairwise alignment is placed to one side and the pairwise alignment of the other two is carried out
+
ClustalW- Progression
bull The alignment is progressively built up in this way with each step being treated as a pairwise alignment sometimes with each member of a lsquopairrsquo having more than one sequence
Progressive alignment - step 11 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
12345
Progressive alignment - step 21 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
12345
Progressive alignment - step 31 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
+
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
12345
Progressive alignment - final step1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
+
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
5 -ctcga-acgatacgatgactagct-
12345
ClustalW-Good pointsBad points
bull Advantagesndash Speed
bull Disadvantagesndash No objective functionndash No way of quantifying whether or not the alignment is good
ndash No way of knowing if the alignment is lsquocorrectrsquo
ClustalW-Local Minimumbull Potential problems
ndash Local minimum problem If an error is introduced early in the alignment process it is impossible to correct this later in the procedure
ndash Arbitrary alignment
Increasing the sophistication of the alignment process
bull Should we treat all the sequences in the same way - even though some sequences are closely-related and some sequences are distant relatives
bull Should we treat all positions in the sequences as though they were the same - even though they might have different functions and different locations in the 3-dimensional structure
ClustalW- Caveatsbull Sequence weightingbull Varying substitution matricesbull Residue-specific gap penalties and reduced penalties
in hydrophilic regions (external regions of protein sequences) encourage gaps in loops rather than in core regions
bull Positions in early alignments where gaps have been opened receive locally reduced gap penalties to encourage openings in subsequent alignments
ClustalW- User-supplied values
bull Two penalties are set by the user (there are default values but you should know that it is possible to change these)
bull GOP- Gap Opening Penalty is the cost of opening a gap in an alignment
bull GEP- Gap Extension Penalty is the cost of extending this gap
Position-Specific gap penaltiesbull Before any pair of (groups of) sequences are
aligned a table of GOPs are generated for each position in the two (sets of) sequences
bull The GOP is manipulated in a position-specific manner so that it can vary over the sequences
bull If there is a gap at a position the GOP and GEP penalties are lowered the other rules do not apply
bull This makes gaps more likely at positions where gaps already exist
Discouraging too many gaps bull If there is no gap opened then the GOP is increased if the
position is within 8 residues of an existing gapbull This discourages gaps that are too close togetherbull At any position within a run of hydrophilic residues the GOP is
decreasedbull These runs usually indicate loop regions in protein structuresbull A run of 5 hydrophilic residues is considered to be a hydrophilic
stretchbull The default hydrophilic residues are
ndash D E G K N Q P R Sndash But this can be changed by the user
Divergent Sequencesbull The most divergent sequences (most different on average
from all of the other sequences) are usually the most difficult to align
bull It is sometimes better to delay their aligment until later (when the easier sequences have already been aligned)
bull The user has the choice of setting a cutoff (default is 40 identity)
bull This will delay the alignment until the others have been aligned
Advice on progressive alignmentbull Progressive alignment is a mathematical process that is completely
independent of biological realitybull Can be a very good estimatebull Can be an impossibly poor estimatebull Requires user input and skillbull Treat cautiouslybull Can be improved by eye (usually)bull Often helps to have colour-codingbull Depending on the use the user should be able to make a judgement
on those regions that are reliable or notbull For phylogeny reconstruction only use those positions whose
hypothesis of positional homology is unimpeachable
Alignment of protein-coding DNA sequences
bull It is not very sensible to align the DNA sequences of protein-coding genes
ATGCTGTTAGGGATGACTCTGTTAGGG
ATG-CT--GTTAGGGATGACTCTGTTAGGG
The result might be highly-implausible and might not reflect what is known about biological processesIt is much more sensible to translate the sequences to their corresponding amino acid sequences align these protein sequences and then put the gaps in the DNA sequences according to where they are found in the amino acid alignment
Manual Alignment- softwareGDE- The Genetic Data Environment (UNIX)CINEMA- Java applet available from
ndash httpwwwbiochemuclacuk
SeqappSeqpup- MacPCUNIX available fromndash httpiubiobioindianaedu
SeAl for Macintosh available fromndash httpevolvezoooxacukSe-AlSe-Alhtml
BioEdit for PC available fromndash httpwwwmbioncsueduRNasePinfoprogramsBIOEDITbio
edithtml
- Multiple Sequence Alignment James McInerney bioinf4biologists Feb 2009
- Alignment can be easy or difficult
- Homology Definition
- Multiple Sequence Alignment- Goals
- Multiple sequence alignments - problems
- Slide 6
- Slide 7
- SSU rRNA
- Alignment of 16S rRNA can be guided by secondary structure
- Protein Alignment may be guided by Tertiary Structure Interactions
- Multiple Sequence Alignment- Methods
- Manual Alignment - reasons
- Local minimum
- Dotplots
- Dotplot example sperm whale vs human myg
- Slide 16
- Slide 17
- Dotplots in practice
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Dynamic programming
- Progressive Alignment
- Slide 23
- ClustalW- Pairwise Alignments
- Path Graph for aligning two sequences
- Possible alignment
- Alignment using this path
- Optimal Alignment 1
- Optimal Alignment 2
- Alignment of 3 sequences
- ClustalW- Guide Tree
- Neighbor joining method
- Distance Matrix
- First Step
- Calculation of New Distances
- Next Cycle
- Penultimate Cycle
- Last Joining
- Unrooted Neighbor-Joining Tree
- Multiple Alignment- First pair
- ClustalW- Decision time
- ClustalW- Alternative 1
- ClustalW- Progression
- Progressive alignment - step 1
- Progressive alignment - step 2
- Progressive alignment - step 3
- Progressive alignment - final step
- ClustalW-Good pointsBad points
- ClustalW-Local Minimum
- Increasing the sophistication of the alignment process
- Slide 51
- ClustalW- Caveats
- ClustalW- User-supplied values
- Position-Specific gap penalties
- Discouraging too many gaps
- Divergent Sequences
- Advice on progressive alignment
- Alignment of protein-coding DNA sequences
- Manual Alignment- software
-

Alignment of 3 sequences
ClustalW- Guide Tree
bull Generate a Neighbor-Joining lsquoguide treersquo from these pairwise distances
bull This guide tree gives the order in which the progressive alignment will be carried out
Neighbor joining method
bullThe neighbor-joining method is a greedy heuristic which joins at each step the two closest sub-trees that are not already joinedbullIt is based on the minimum evolution principlebullneighbors are defined as two taxa that are connected by a single node in an unrooted tree
A B
Node 1
PAM Spinach Rice Mosquito Monkey HumanSpinach 00 849 1056 908 863Rice 849 00 1178 1224 1226Mosquito 1056 1178 00 847 808Monkey 908 1224 847 00 33Human 863 1226 808 33 00
What is required for the Neighbour joining method
Distance matrixDistance Matrix
PAM distance 33 (Human - Monkey) is the minimum So well join Human and Monkey to MonHum and well calculate the new distances
Mon-Hum
MonkeyHumanSpinachMosquito Rice
First Step
After we have joined two species in a subtree we have to compute the distances from every other node to the new subtree We do this with a simple average of distancesDist[Spinach MonHum]
= (Dist[Spinach Monkey] + Dist[Spinach Human])2 = (908 + 863)2 = 8855
Mon-Hum
MonkeyHumanSpinach
Calculation of New Distances
PAM Spinach Rice Mosquito MonHumSpinach 00 849 1056 886Rice 849 00 1178 1225Mosquito 1056 1178 00 828MonHum 886 1225 828 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Next Cycle
PAM Spinach Rice MosMonHumSpinach 00 849 971Rice 849 00 1202MosMonHum 971 1202 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
Penultimate Cycle
PAM SpinRice MosMonHumSpinach 00 1087MosMonHum 1087 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
(Spin-Rice)-(Mos-(Mon-Hum))
Last Joining
Human
Monkey
MosquitoRice
Spinach
Unrooted Neighbor-Joining Tree
Multiple Alignment- First pairbull Align the two most closely-related sequences first
bull This alignment is then lsquofixedrsquo and will never change If a gap is to be introduced subsequently then it will be introduced in the same place in both sequences but their relative alignment remains unchanged
ClustalW- Decision timebull Consult the guide tree to see what alignment is performed next
ndash Align a third sequence to the first twoOrndash Align two entirely different sequences to each other
Option 1Option 1 Option 2Option 2
ClustalW- Alternative 1If the situation arises where a third sequence is aligned to the first two then when a gap has to be introduced to improve the alignment each of these two entities are treated as two single sequences
+
ClustalW- Alternative 2bull If on the other hand two separate
sequences have to be aligned together then the first pairwise alignment is placed to one side and the pairwise alignment of the other two is carried out
+
ClustalW- Progression
bull The alignment is progressively built up in this way with each step being treated as a pairwise alignment sometimes with each member of a lsquopairrsquo having more than one sequence
Progressive alignment - step 11 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
12345
Progressive alignment - step 21 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
12345
Progressive alignment - step 31 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
+
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
12345
Progressive alignment - final step1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
+
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
5 -ctcga-acgatacgatgactagct-
12345
ClustalW-Good pointsBad points
bull Advantagesndash Speed
bull Disadvantagesndash No objective functionndash No way of quantifying whether or not the alignment is good
ndash No way of knowing if the alignment is lsquocorrectrsquo
ClustalW-Local Minimumbull Potential problems
ndash Local minimum problem If an error is introduced early in the alignment process it is impossible to correct this later in the procedure
ndash Arbitrary alignment
Increasing the sophistication of the alignment process
bull Should we treat all the sequences in the same way - even though some sequences are closely-related and some sequences are distant relatives
bull Should we treat all positions in the sequences as though they were the same - even though they might have different functions and different locations in the 3-dimensional structure
ClustalW- Caveatsbull Sequence weightingbull Varying substitution matricesbull Residue-specific gap penalties and reduced penalties
in hydrophilic regions (external regions of protein sequences) encourage gaps in loops rather than in core regions
bull Positions in early alignments where gaps have been opened receive locally reduced gap penalties to encourage openings in subsequent alignments
ClustalW- User-supplied values
bull Two penalties are set by the user (there are default values but you should know that it is possible to change these)
bull GOP- Gap Opening Penalty is the cost of opening a gap in an alignment
bull GEP- Gap Extension Penalty is the cost of extending this gap
Position-Specific gap penaltiesbull Before any pair of (groups of) sequences are
aligned a table of GOPs are generated for each position in the two (sets of) sequences
bull The GOP is manipulated in a position-specific manner so that it can vary over the sequences
bull If there is a gap at a position the GOP and GEP penalties are lowered the other rules do not apply
bull This makes gaps more likely at positions where gaps already exist
Discouraging too many gaps bull If there is no gap opened then the GOP is increased if the
position is within 8 residues of an existing gapbull This discourages gaps that are too close togetherbull At any position within a run of hydrophilic residues the GOP is
decreasedbull These runs usually indicate loop regions in protein structuresbull A run of 5 hydrophilic residues is considered to be a hydrophilic
stretchbull The default hydrophilic residues are
ndash D E G K N Q P R Sndash But this can be changed by the user
Divergent Sequencesbull The most divergent sequences (most different on average
from all of the other sequences) are usually the most difficult to align
bull It is sometimes better to delay their aligment until later (when the easier sequences have already been aligned)
bull The user has the choice of setting a cutoff (default is 40 identity)
bull This will delay the alignment until the others have been aligned
Advice on progressive alignmentbull Progressive alignment is a mathematical process that is completely
independent of biological realitybull Can be a very good estimatebull Can be an impossibly poor estimatebull Requires user input and skillbull Treat cautiouslybull Can be improved by eye (usually)bull Often helps to have colour-codingbull Depending on the use the user should be able to make a judgement
on those regions that are reliable or notbull For phylogeny reconstruction only use those positions whose
hypothesis of positional homology is unimpeachable
Alignment of protein-coding DNA sequences
bull It is not very sensible to align the DNA sequences of protein-coding genes
ATGCTGTTAGGGATGACTCTGTTAGGG
ATG-CT--GTTAGGGATGACTCTGTTAGGG
The result might be highly-implausible and might not reflect what is known about biological processesIt is much more sensible to translate the sequences to their corresponding amino acid sequences align these protein sequences and then put the gaps in the DNA sequences according to where they are found in the amino acid alignment
Manual Alignment- softwareGDE- The Genetic Data Environment (UNIX)CINEMA- Java applet available from
ndash httpwwwbiochemuclacuk
SeqappSeqpup- MacPCUNIX available fromndash httpiubiobioindianaedu
SeAl for Macintosh available fromndash httpevolvezoooxacukSe-AlSe-Alhtml
BioEdit for PC available fromndash httpwwwmbioncsueduRNasePinfoprogramsBIOEDITbio
edithtml
- Multiple Sequence Alignment James McInerney bioinf4biologists Feb 2009
- Alignment can be easy or difficult
- Homology Definition
- Multiple Sequence Alignment- Goals
- Multiple sequence alignments - problems
- Slide 6
- Slide 7
- SSU rRNA
- Alignment of 16S rRNA can be guided by secondary structure
- Protein Alignment may be guided by Tertiary Structure Interactions
- Multiple Sequence Alignment- Methods
- Manual Alignment - reasons
- Local minimum
- Dotplots
- Dotplot example sperm whale vs human myg
- Slide 16
- Slide 17
- Dotplots in practice
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Dynamic programming
- Progressive Alignment
- Slide 23
- ClustalW- Pairwise Alignments
- Path Graph for aligning two sequences
- Possible alignment
- Alignment using this path
- Optimal Alignment 1
- Optimal Alignment 2
- Alignment of 3 sequences
- ClustalW- Guide Tree
- Neighbor joining method
- Distance Matrix
- First Step
- Calculation of New Distances
- Next Cycle
- Penultimate Cycle
- Last Joining
- Unrooted Neighbor-Joining Tree
- Multiple Alignment- First pair
- ClustalW- Decision time
- ClustalW- Alternative 1
- ClustalW- Progression
- Progressive alignment - step 1
- Progressive alignment - step 2
- Progressive alignment - step 3
- Progressive alignment - final step
- ClustalW-Good pointsBad points
- ClustalW-Local Minimum
- Increasing the sophistication of the alignment process
- Slide 51
- ClustalW- Caveats
- ClustalW- User-supplied values
- Position-Specific gap penalties
- Discouraging too many gaps
- Divergent Sequences
- Advice on progressive alignment
- Alignment of protein-coding DNA sequences
- Manual Alignment- software
-

ClustalW- Guide Tree
bull Generate a Neighbor-Joining lsquoguide treersquo from these pairwise distances
bull This guide tree gives the order in which the progressive alignment will be carried out
Neighbor joining method
bullThe neighbor-joining method is a greedy heuristic which joins at each step the two closest sub-trees that are not already joinedbullIt is based on the minimum evolution principlebullneighbors are defined as two taxa that are connected by a single node in an unrooted tree
A B
Node 1
PAM Spinach Rice Mosquito Monkey HumanSpinach 00 849 1056 908 863Rice 849 00 1178 1224 1226Mosquito 1056 1178 00 847 808Monkey 908 1224 847 00 33Human 863 1226 808 33 00
What is required for the Neighbour joining method
Distance matrixDistance Matrix
PAM distance 33 (Human - Monkey) is the minimum So well join Human and Monkey to MonHum and well calculate the new distances
Mon-Hum
MonkeyHumanSpinachMosquito Rice
First Step
After we have joined two species in a subtree we have to compute the distances from every other node to the new subtree We do this with a simple average of distancesDist[Spinach MonHum]
= (Dist[Spinach Monkey] + Dist[Spinach Human])2 = (908 + 863)2 = 8855
Mon-Hum
MonkeyHumanSpinach
Calculation of New Distances
PAM Spinach Rice Mosquito MonHumSpinach 00 849 1056 886Rice 849 00 1178 1225Mosquito 1056 1178 00 828MonHum 886 1225 828 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Next Cycle
PAM Spinach Rice MosMonHumSpinach 00 849 971Rice 849 00 1202MosMonHum 971 1202 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
Penultimate Cycle
PAM SpinRice MosMonHumSpinach 00 1087MosMonHum 1087 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
(Spin-Rice)-(Mos-(Mon-Hum))
Last Joining
Human
Monkey
MosquitoRice
Spinach
Unrooted Neighbor-Joining Tree
Multiple Alignment- First pairbull Align the two most closely-related sequences first
bull This alignment is then lsquofixedrsquo and will never change If a gap is to be introduced subsequently then it will be introduced in the same place in both sequences but their relative alignment remains unchanged
ClustalW- Decision timebull Consult the guide tree to see what alignment is performed next
ndash Align a third sequence to the first twoOrndash Align two entirely different sequences to each other
Option 1Option 1 Option 2Option 2
ClustalW- Alternative 1If the situation arises where a third sequence is aligned to the first two then when a gap has to be introduced to improve the alignment each of these two entities are treated as two single sequences
+
ClustalW- Alternative 2bull If on the other hand two separate
sequences have to be aligned together then the first pairwise alignment is placed to one side and the pairwise alignment of the other two is carried out
+
ClustalW- Progression
bull The alignment is progressively built up in this way with each step being treated as a pairwise alignment sometimes with each member of a lsquopairrsquo having more than one sequence
Progressive alignment - step 11 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
12345
Progressive alignment - step 21 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
12345
Progressive alignment - step 31 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
+
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
12345
Progressive alignment - final step1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
+
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
5 -ctcga-acgatacgatgactagct-
12345
ClustalW-Good pointsBad points
bull Advantagesndash Speed
bull Disadvantagesndash No objective functionndash No way of quantifying whether or not the alignment is good
ndash No way of knowing if the alignment is lsquocorrectrsquo
ClustalW-Local Minimumbull Potential problems
ndash Local minimum problem If an error is introduced early in the alignment process it is impossible to correct this later in the procedure
ndash Arbitrary alignment
Increasing the sophistication of the alignment process
bull Should we treat all the sequences in the same way - even though some sequences are closely-related and some sequences are distant relatives
bull Should we treat all positions in the sequences as though they were the same - even though they might have different functions and different locations in the 3-dimensional structure
ClustalW- Caveatsbull Sequence weightingbull Varying substitution matricesbull Residue-specific gap penalties and reduced penalties
in hydrophilic regions (external regions of protein sequences) encourage gaps in loops rather than in core regions
bull Positions in early alignments where gaps have been opened receive locally reduced gap penalties to encourage openings in subsequent alignments
ClustalW- User-supplied values
bull Two penalties are set by the user (there are default values but you should know that it is possible to change these)
bull GOP- Gap Opening Penalty is the cost of opening a gap in an alignment
bull GEP- Gap Extension Penalty is the cost of extending this gap
Position-Specific gap penaltiesbull Before any pair of (groups of) sequences are
aligned a table of GOPs are generated for each position in the two (sets of) sequences
bull The GOP is manipulated in a position-specific manner so that it can vary over the sequences
bull If there is a gap at a position the GOP and GEP penalties are lowered the other rules do not apply
bull This makes gaps more likely at positions where gaps already exist
Discouraging too many gaps bull If there is no gap opened then the GOP is increased if the
position is within 8 residues of an existing gapbull This discourages gaps that are too close togetherbull At any position within a run of hydrophilic residues the GOP is
decreasedbull These runs usually indicate loop regions in protein structuresbull A run of 5 hydrophilic residues is considered to be a hydrophilic
stretchbull The default hydrophilic residues are
ndash D E G K N Q P R Sndash But this can be changed by the user
Divergent Sequencesbull The most divergent sequences (most different on average
from all of the other sequences) are usually the most difficult to align
bull It is sometimes better to delay their aligment until later (when the easier sequences have already been aligned)
bull The user has the choice of setting a cutoff (default is 40 identity)
bull This will delay the alignment until the others have been aligned
Advice on progressive alignmentbull Progressive alignment is a mathematical process that is completely
independent of biological realitybull Can be a very good estimatebull Can be an impossibly poor estimatebull Requires user input and skillbull Treat cautiouslybull Can be improved by eye (usually)bull Often helps to have colour-codingbull Depending on the use the user should be able to make a judgement
on those regions that are reliable or notbull For phylogeny reconstruction only use those positions whose
hypothesis of positional homology is unimpeachable
Alignment of protein-coding DNA sequences
bull It is not very sensible to align the DNA sequences of protein-coding genes
ATGCTGTTAGGGATGACTCTGTTAGGG
ATG-CT--GTTAGGGATGACTCTGTTAGGG
The result might be highly-implausible and might not reflect what is known about biological processesIt is much more sensible to translate the sequences to their corresponding amino acid sequences align these protein sequences and then put the gaps in the DNA sequences according to where they are found in the amino acid alignment
Manual Alignment- softwareGDE- The Genetic Data Environment (UNIX)CINEMA- Java applet available from
ndash httpwwwbiochemuclacuk
SeqappSeqpup- MacPCUNIX available fromndash httpiubiobioindianaedu
SeAl for Macintosh available fromndash httpevolvezoooxacukSe-AlSe-Alhtml
BioEdit for PC available fromndash httpwwwmbioncsueduRNasePinfoprogramsBIOEDITbio
edithtml
- Multiple Sequence Alignment James McInerney bioinf4biologists Feb 2009
- Alignment can be easy or difficult
- Homology Definition
- Multiple Sequence Alignment- Goals
- Multiple sequence alignments - problems
- Slide 6
- Slide 7
- SSU rRNA
- Alignment of 16S rRNA can be guided by secondary structure
- Protein Alignment may be guided by Tertiary Structure Interactions
- Multiple Sequence Alignment- Methods
- Manual Alignment - reasons
- Local minimum
- Dotplots
- Dotplot example sperm whale vs human myg
- Slide 16
- Slide 17
- Dotplots in practice
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Dynamic programming
- Progressive Alignment
- Slide 23
- ClustalW- Pairwise Alignments
- Path Graph for aligning two sequences
- Possible alignment
- Alignment using this path
- Optimal Alignment 1
- Optimal Alignment 2
- Alignment of 3 sequences
- ClustalW- Guide Tree
- Neighbor joining method
- Distance Matrix
- First Step
- Calculation of New Distances
- Next Cycle
- Penultimate Cycle
- Last Joining
- Unrooted Neighbor-Joining Tree
- Multiple Alignment- First pair
- ClustalW- Decision time
- ClustalW- Alternative 1
- ClustalW- Progression
- Progressive alignment - step 1
- Progressive alignment - step 2
- Progressive alignment - step 3
- Progressive alignment - final step
- ClustalW-Good pointsBad points
- ClustalW-Local Minimum
- Increasing the sophistication of the alignment process
- Slide 51
- ClustalW- Caveats
- ClustalW- User-supplied values
- Position-Specific gap penalties
- Discouraging too many gaps
- Divergent Sequences
- Advice on progressive alignment
- Alignment of protein-coding DNA sequences
- Manual Alignment- software
-
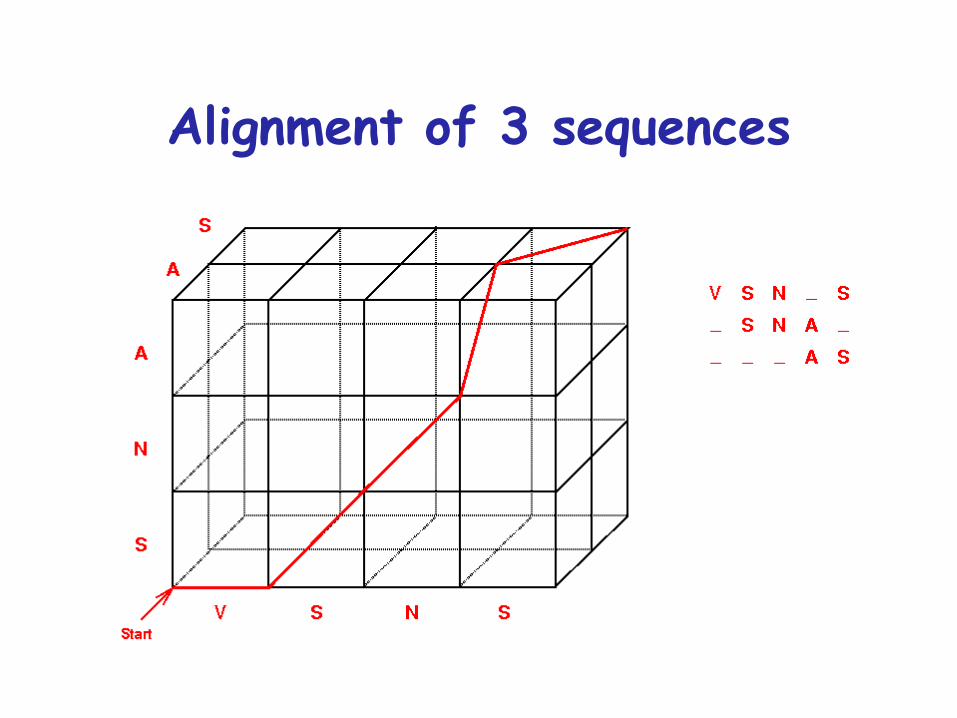
Neighbor joining method
bullThe neighbor-joining method is a greedy heuristic which joins at each step the two closest sub-trees that are not already joinedbullIt is based on the minimum evolution principlebullneighbors are defined as two taxa that are connected by a single node in an unrooted tree
A B
Node 1
PAM Spinach Rice Mosquito Monkey HumanSpinach 00 849 1056 908 863Rice 849 00 1178 1224 1226Mosquito 1056 1178 00 847 808Monkey 908 1224 847 00 33Human 863 1226 808 33 00
What is required for the Neighbour joining method
Distance matrixDistance Matrix
PAM distance 33 (Human - Monkey) is the minimum So well join Human and Monkey to MonHum and well calculate the new distances
Mon-Hum
MonkeyHumanSpinachMosquito Rice
First Step
After we have joined two species in a subtree we have to compute the distances from every other node to the new subtree We do this with a simple average of distancesDist[Spinach MonHum]
= (Dist[Spinach Monkey] + Dist[Spinach Human])2 = (908 + 863)2 = 8855
Mon-Hum
MonkeyHumanSpinach
Calculation of New Distances
PAM Spinach Rice Mosquito MonHumSpinach 00 849 1056 886Rice 849 00 1178 1225Mosquito 1056 1178 00 828MonHum 886 1225 828 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Next Cycle
PAM Spinach Rice MosMonHumSpinach 00 849 971Rice 849 00 1202MosMonHum 971 1202 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
Penultimate Cycle
PAM SpinRice MosMonHumSpinach 00 1087MosMonHum 1087 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
(Spin-Rice)-(Mos-(Mon-Hum))
Last Joining
Human
Monkey
MosquitoRice
Spinach
Unrooted Neighbor-Joining Tree
Multiple Alignment- First pairbull Align the two most closely-related sequences first
bull This alignment is then lsquofixedrsquo and will never change If a gap is to be introduced subsequently then it will be introduced in the same place in both sequences but their relative alignment remains unchanged
ClustalW- Decision timebull Consult the guide tree to see what alignment is performed next
ndash Align a third sequence to the first twoOrndash Align two entirely different sequences to each other
Option 1Option 1 Option 2Option 2
ClustalW- Alternative 1If the situation arises where a third sequence is aligned to the first two then when a gap has to be introduced to improve the alignment each of these two entities are treated as two single sequences
+
ClustalW- Alternative 2bull If on the other hand two separate
sequences have to be aligned together then the first pairwise alignment is placed to one side and the pairwise alignment of the other two is carried out
+
ClustalW- Progression
bull The alignment is progressively built up in this way with each step being treated as a pairwise alignment sometimes with each member of a lsquopairrsquo having more than one sequence
Progressive alignment - step 11 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
12345
Progressive alignment - step 21 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
12345
Progressive alignment - step 31 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
+
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
12345
Progressive alignment - final step1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
+
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
5 -ctcga-acgatacgatgactagct-
12345
ClustalW-Good pointsBad points
bull Advantagesndash Speed
bull Disadvantagesndash No objective functionndash No way of quantifying whether or not the alignment is good
ndash No way of knowing if the alignment is lsquocorrectrsquo
ClustalW-Local Minimumbull Potential problems
ndash Local minimum problem If an error is introduced early in the alignment process it is impossible to correct this later in the procedure
ndash Arbitrary alignment
Increasing the sophistication of the alignment process
bull Should we treat all the sequences in the same way - even though some sequences are closely-related and some sequences are distant relatives
bull Should we treat all positions in the sequences as though they were the same - even though they might have different functions and different locations in the 3-dimensional structure
ClustalW- Caveatsbull Sequence weightingbull Varying substitution matricesbull Residue-specific gap penalties and reduced penalties
in hydrophilic regions (external regions of protein sequences) encourage gaps in loops rather than in core regions
bull Positions in early alignments where gaps have been opened receive locally reduced gap penalties to encourage openings in subsequent alignments
ClustalW- User-supplied values
bull Two penalties are set by the user (there are default values but you should know that it is possible to change these)
bull GOP- Gap Opening Penalty is the cost of opening a gap in an alignment
bull GEP- Gap Extension Penalty is the cost of extending this gap
Position-Specific gap penaltiesbull Before any pair of (groups of) sequences are
aligned a table of GOPs are generated for each position in the two (sets of) sequences
bull The GOP is manipulated in a position-specific manner so that it can vary over the sequences
bull If there is a gap at a position the GOP and GEP penalties are lowered the other rules do not apply
bull This makes gaps more likely at positions where gaps already exist
Discouraging too many gaps bull If there is no gap opened then the GOP is increased if the
position is within 8 residues of an existing gapbull This discourages gaps that are too close togetherbull At any position within a run of hydrophilic residues the GOP is
decreasedbull These runs usually indicate loop regions in protein structuresbull A run of 5 hydrophilic residues is considered to be a hydrophilic
stretchbull The default hydrophilic residues are
ndash D E G K N Q P R Sndash But this can be changed by the user
Divergent Sequencesbull The most divergent sequences (most different on average
from all of the other sequences) are usually the most difficult to align
bull It is sometimes better to delay their aligment until later (when the easier sequences have already been aligned)
bull The user has the choice of setting a cutoff (default is 40 identity)
bull This will delay the alignment until the others have been aligned
Advice on progressive alignmentbull Progressive alignment is a mathematical process that is completely
independent of biological realitybull Can be a very good estimatebull Can be an impossibly poor estimatebull Requires user input and skillbull Treat cautiouslybull Can be improved by eye (usually)bull Often helps to have colour-codingbull Depending on the use the user should be able to make a judgement
on those regions that are reliable or notbull For phylogeny reconstruction only use those positions whose
hypothesis of positional homology is unimpeachable
Alignment of protein-coding DNA sequences
bull It is not very sensible to align the DNA sequences of protein-coding genes
ATGCTGTTAGGGATGACTCTGTTAGGG
ATG-CT--GTTAGGGATGACTCTGTTAGGG
The result might be highly-implausible and might not reflect what is known about biological processesIt is much more sensible to translate the sequences to their corresponding amino acid sequences align these protein sequences and then put the gaps in the DNA sequences according to where they are found in the amino acid alignment
Manual Alignment- softwareGDE- The Genetic Data Environment (UNIX)CINEMA- Java applet available from
ndash httpwwwbiochemuclacuk
SeqappSeqpup- MacPCUNIX available fromndash httpiubiobioindianaedu
SeAl for Macintosh available fromndash httpevolvezoooxacukSe-AlSe-Alhtml
BioEdit for PC available fromndash httpwwwmbioncsueduRNasePinfoprogramsBIOEDITbio
edithtml
- Multiple Sequence Alignment James McInerney bioinf4biologists Feb 2009
- Alignment can be easy or difficult
- Homology Definition
- Multiple Sequence Alignment- Goals
- Multiple sequence alignments - problems
- Slide 6
- Slide 7
- SSU rRNA
- Alignment of 16S rRNA can be guided by secondary structure
- Protein Alignment may be guided by Tertiary Structure Interactions
- Multiple Sequence Alignment- Methods
- Manual Alignment - reasons
- Local minimum
- Dotplots
- Dotplot example sperm whale vs human myg
- Slide 16
- Slide 17
- Dotplots in practice
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Dynamic programming
- Progressive Alignment
- Slide 23
- ClustalW- Pairwise Alignments
- Path Graph for aligning two sequences
- Possible alignment
- Alignment using this path
- Optimal Alignment 1
- Optimal Alignment 2
- Alignment of 3 sequences
- ClustalW- Guide Tree
- Neighbor joining method
- Distance Matrix
- First Step
- Calculation of New Distances
- Next Cycle
- Penultimate Cycle
- Last Joining
- Unrooted Neighbor-Joining Tree
- Multiple Alignment- First pair
- ClustalW- Decision time
- ClustalW- Alternative 1
- ClustalW- Progression
- Progressive alignment - step 1
- Progressive alignment - step 2
- Progressive alignment - step 3
- Progressive alignment - final step
- ClustalW-Good pointsBad points
- ClustalW-Local Minimum
- Increasing the sophistication of the alignment process
- Slide 51
- ClustalW- Caveats
- ClustalW- User-supplied values
- Position-Specific gap penalties
- Discouraging too many gaps
- Divergent Sequences
- Advice on progressive alignment
- Alignment of protein-coding DNA sequences
- Manual Alignment- software
-

PAM Spinach Rice Mosquito Monkey HumanSpinach 00 849 1056 908 863Rice 849 00 1178 1224 1226Mosquito 1056 1178 00 847 808Monkey 908 1224 847 00 33Human 863 1226 808 33 00
What is required for the Neighbour joining method
Distance matrixDistance Matrix
PAM distance 33 (Human - Monkey) is the minimum So well join Human and Monkey to MonHum and well calculate the new distances
Mon-Hum
MonkeyHumanSpinachMosquito Rice
First Step
After we have joined two species in a subtree we have to compute the distances from every other node to the new subtree We do this with a simple average of distancesDist[Spinach MonHum]
= (Dist[Spinach Monkey] + Dist[Spinach Human])2 = (908 + 863)2 = 8855
Mon-Hum
MonkeyHumanSpinach
Calculation of New Distances
PAM Spinach Rice Mosquito MonHumSpinach 00 849 1056 886Rice 849 00 1178 1225Mosquito 1056 1178 00 828MonHum 886 1225 828 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Next Cycle
PAM Spinach Rice MosMonHumSpinach 00 849 971Rice 849 00 1202MosMonHum 971 1202 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
Penultimate Cycle
PAM SpinRice MosMonHumSpinach 00 1087MosMonHum 1087 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
(Spin-Rice)-(Mos-(Mon-Hum))
Last Joining
Human
Monkey
MosquitoRice
Spinach
Unrooted Neighbor-Joining Tree
Multiple Alignment- First pairbull Align the two most closely-related sequences first
bull This alignment is then lsquofixedrsquo and will never change If a gap is to be introduced subsequently then it will be introduced in the same place in both sequences but their relative alignment remains unchanged
ClustalW- Decision timebull Consult the guide tree to see what alignment is performed next
ndash Align a third sequence to the first twoOrndash Align two entirely different sequences to each other
Option 1Option 1 Option 2Option 2
ClustalW- Alternative 1If the situation arises where a third sequence is aligned to the first two then when a gap has to be introduced to improve the alignment each of these two entities are treated as two single sequences
+
ClustalW- Alternative 2bull If on the other hand two separate
sequences have to be aligned together then the first pairwise alignment is placed to one side and the pairwise alignment of the other two is carried out
+
ClustalW- Progression
bull The alignment is progressively built up in this way with each step being treated as a pairwise alignment sometimes with each member of a lsquopairrsquo having more than one sequence
Progressive alignment - step 11 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
12345
Progressive alignment - step 21 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
12345
Progressive alignment - step 31 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
+
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
12345
Progressive alignment - final step1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
+
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
5 -ctcga-acgatacgatgactagct-
12345
ClustalW-Good pointsBad points
bull Advantagesndash Speed
bull Disadvantagesndash No objective functionndash No way of quantifying whether or not the alignment is good
ndash No way of knowing if the alignment is lsquocorrectrsquo
ClustalW-Local Minimumbull Potential problems
ndash Local minimum problem If an error is introduced early in the alignment process it is impossible to correct this later in the procedure
ndash Arbitrary alignment
Increasing the sophistication of the alignment process
bull Should we treat all the sequences in the same way - even though some sequences are closely-related and some sequences are distant relatives
bull Should we treat all positions in the sequences as though they were the same - even though they might have different functions and different locations in the 3-dimensional structure
ClustalW- Caveatsbull Sequence weightingbull Varying substitution matricesbull Residue-specific gap penalties and reduced penalties
in hydrophilic regions (external regions of protein sequences) encourage gaps in loops rather than in core regions
bull Positions in early alignments where gaps have been opened receive locally reduced gap penalties to encourage openings in subsequent alignments
ClustalW- User-supplied values
bull Two penalties are set by the user (there are default values but you should know that it is possible to change these)
bull GOP- Gap Opening Penalty is the cost of opening a gap in an alignment
bull GEP- Gap Extension Penalty is the cost of extending this gap
Position-Specific gap penaltiesbull Before any pair of (groups of) sequences are
aligned a table of GOPs are generated for each position in the two (sets of) sequences
bull The GOP is manipulated in a position-specific manner so that it can vary over the sequences
bull If there is a gap at a position the GOP and GEP penalties are lowered the other rules do not apply
bull This makes gaps more likely at positions where gaps already exist
Discouraging too many gaps bull If there is no gap opened then the GOP is increased if the
position is within 8 residues of an existing gapbull This discourages gaps that are too close togetherbull At any position within a run of hydrophilic residues the GOP is
decreasedbull These runs usually indicate loop regions in protein structuresbull A run of 5 hydrophilic residues is considered to be a hydrophilic
stretchbull The default hydrophilic residues are
ndash D E G K N Q P R Sndash But this can be changed by the user
Divergent Sequencesbull The most divergent sequences (most different on average
from all of the other sequences) are usually the most difficult to align
bull It is sometimes better to delay their aligment until later (when the easier sequences have already been aligned)
bull The user has the choice of setting a cutoff (default is 40 identity)
bull This will delay the alignment until the others have been aligned
Advice on progressive alignmentbull Progressive alignment is a mathematical process that is completely
independent of biological realitybull Can be a very good estimatebull Can be an impossibly poor estimatebull Requires user input and skillbull Treat cautiouslybull Can be improved by eye (usually)bull Often helps to have colour-codingbull Depending on the use the user should be able to make a judgement
on those regions that are reliable or notbull For phylogeny reconstruction only use those positions whose
hypothesis of positional homology is unimpeachable
Alignment of protein-coding DNA sequences
bull It is not very sensible to align the DNA sequences of protein-coding genes
ATGCTGTTAGGGATGACTCTGTTAGGG
ATG-CT--GTTAGGGATGACTCTGTTAGGG
The result might be highly-implausible and might not reflect what is known about biological processesIt is much more sensible to translate the sequences to their corresponding amino acid sequences align these protein sequences and then put the gaps in the DNA sequences according to where they are found in the amino acid alignment
Manual Alignment- softwareGDE- The Genetic Data Environment (UNIX)CINEMA- Java applet available from
ndash httpwwwbiochemuclacuk
SeqappSeqpup- MacPCUNIX available fromndash httpiubiobioindianaedu
SeAl for Macintosh available fromndash httpevolvezoooxacukSe-AlSe-Alhtml
BioEdit for PC available fromndash httpwwwmbioncsueduRNasePinfoprogramsBIOEDITbio
edithtml
- Multiple Sequence Alignment James McInerney bioinf4biologists Feb 2009
- Alignment can be easy or difficult
- Homology Definition
- Multiple Sequence Alignment- Goals
- Multiple sequence alignments - problems
- Slide 6
- Slide 7
- SSU rRNA
- Alignment of 16S rRNA can be guided by secondary structure
- Protein Alignment may be guided by Tertiary Structure Interactions
- Multiple Sequence Alignment- Methods
- Manual Alignment - reasons
- Local minimum
- Dotplots
- Dotplot example sperm whale vs human myg
- Slide 16
- Slide 17
- Dotplots in practice
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Dynamic programming
- Progressive Alignment
- Slide 23
- ClustalW- Pairwise Alignments
- Path Graph for aligning two sequences
- Possible alignment
- Alignment using this path
- Optimal Alignment 1
- Optimal Alignment 2
- Alignment of 3 sequences
- ClustalW- Guide Tree
- Neighbor joining method
- Distance Matrix
- First Step
- Calculation of New Distances
- Next Cycle
- Penultimate Cycle
- Last Joining
- Unrooted Neighbor-Joining Tree
- Multiple Alignment- First pair
- ClustalW- Decision time
- ClustalW- Alternative 1
- ClustalW- Progression
- Progressive alignment - step 1
- Progressive alignment - step 2
- Progressive alignment - step 3
- Progressive alignment - final step
- ClustalW-Good pointsBad points
- ClustalW-Local Minimum
- Increasing the sophistication of the alignment process
- Slide 51
- ClustalW- Caveats
- ClustalW- User-supplied values
- Position-Specific gap penalties
- Discouraging too many gaps
- Divergent Sequences
- Advice on progressive alignment
- Alignment of protein-coding DNA sequences
- Manual Alignment- software
-

PAM distance 33 (Human - Monkey) is the minimum So well join Human and Monkey to MonHum and well calculate the new distances
Mon-Hum
MonkeyHumanSpinachMosquito Rice
First Step
After we have joined two species in a subtree we have to compute the distances from every other node to the new subtree We do this with a simple average of distancesDist[Spinach MonHum]
= (Dist[Spinach Monkey] + Dist[Spinach Human])2 = (908 + 863)2 = 8855
Mon-Hum
MonkeyHumanSpinach
Calculation of New Distances
PAM Spinach Rice Mosquito MonHumSpinach 00 849 1056 886Rice 849 00 1178 1225Mosquito 1056 1178 00 828MonHum 886 1225 828 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Next Cycle
PAM Spinach Rice MosMonHumSpinach 00 849 971Rice 849 00 1202MosMonHum 971 1202 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
Penultimate Cycle
PAM SpinRice MosMonHumSpinach 00 1087MosMonHum 1087 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
(Spin-Rice)-(Mos-(Mon-Hum))
Last Joining
Human
Monkey
MosquitoRice
Spinach
Unrooted Neighbor-Joining Tree
Multiple Alignment- First pairbull Align the two most closely-related sequences first
bull This alignment is then lsquofixedrsquo and will never change If a gap is to be introduced subsequently then it will be introduced in the same place in both sequences but their relative alignment remains unchanged
ClustalW- Decision timebull Consult the guide tree to see what alignment is performed next
ndash Align a third sequence to the first twoOrndash Align two entirely different sequences to each other
Option 1Option 1 Option 2Option 2
ClustalW- Alternative 1If the situation arises where a third sequence is aligned to the first two then when a gap has to be introduced to improve the alignment each of these two entities are treated as two single sequences
+
ClustalW- Alternative 2bull If on the other hand two separate
sequences have to be aligned together then the first pairwise alignment is placed to one side and the pairwise alignment of the other two is carried out
+
ClustalW- Progression
bull The alignment is progressively built up in this way with each step being treated as a pairwise alignment sometimes with each member of a lsquopairrsquo having more than one sequence
Progressive alignment - step 11 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
12345
Progressive alignment - step 21 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
12345
Progressive alignment - step 31 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
+
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
12345
Progressive alignment - final step1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
+
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
5 -ctcga-acgatacgatgactagct-
12345
ClustalW-Good pointsBad points
bull Advantagesndash Speed
bull Disadvantagesndash No objective functionndash No way of quantifying whether or not the alignment is good
ndash No way of knowing if the alignment is lsquocorrectrsquo
ClustalW-Local Minimumbull Potential problems
ndash Local minimum problem If an error is introduced early in the alignment process it is impossible to correct this later in the procedure
ndash Arbitrary alignment
Increasing the sophistication of the alignment process
bull Should we treat all the sequences in the same way - even though some sequences are closely-related and some sequences are distant relatives
bull Should we treat all positions in the sequences as though they were the same - even though they might have different functions and different locations in the 3-dimensional structure
ClustalW- Caveatsbull Sequence weightingbull Varying substitution matricesbull Residue-specific gap penalties and reduced penalties
in hydrophilic regions (external regions of protein sequences) encourage gaps in loops rather than in core regions
bull Positions in early alignments where gaps have been opened receive locally reduced gap penalties to encourage openings in subsequent alignments
ClustalW- User-supplied values
bull Two penalties are set by the user (there are default values but you should know that it is possible to change these)
bull GOP- Gap Opening Penalty is the cost of opening a gap in an alignment
bull GEP- Gap Extension Penalty is the cost of extending this gap
Position-Specific gap penaltiesbull Before any pair of (groups of) sequences are
aligned a table of GOPs are generated for each position in the two (sets of) sequences
bull The GOP is manipulated in a position-specific manner so that it can vary over the sequences
bull If there is a gap at a position the GOP and GEP penalties are lowered the other rules do not apply
bull This makes gaps more likely at positions where gaps already exist
Discouraging too many gaps bull If there is no gap opened then the GOP is increased if the
position is within 8 residues of an existing gapbull This discourages gaps that are too close togetherbull At any position within a run of hydrophilic residues the GOP is
decreasedbull These runs usually indicate loop regions in protein structuresbull A run of 5 hydrophilic residues is considered to be a hydrophilic
stretchbull The default hydrophilic residues are
ndash D E G K N Q P R Sndash But this can be changed by the user
Divergent Sequencesbull The most divergent sequences (most different on average
from all of the other sequences) are usually the most difficult to align
bull It is sometimes better to delay their aligment until later (when the easier sequences have already been aligned)
bull The user has the choice of setting a cutoff (default is 40 identity)
bull This will delay the alignment until the others have been aligned
Advice on progressive alignmentbull Progressive alignment is a mathematical process that is completely
independent of biological realitybull Can be a very good estimatebull Can be an impossibly poor estimatebull Requires user input and skillbull Treat cautiouslybull Can be improved by eye (usually)bull Often helps to have colour-codingbull Depending on the use the user should be able to make a judgement
on those regions that are reliable or notbull For phylogeny reconstruction only use those positions whose
hypothesis of positional homology is unimpeachable
Alignment of protein-coding DNA sequences
bull It is not very sensible to align the DNA sequences of protein-coding genes
ATGCTGTTAGGGATGACTCTGTTAGGG
ATG-CT--GTTAGGGATGACTCTGTTAGGG
The result might be highly-implausible and might not reflect what is known about biological processesIt is much more sensible to translate the sequences to their corresponding amino acid sequences align these protein sequences and then put the gaps in the DNA sequences according to where they are found in the amino acid alignment
Manual Alignment- softwareGDE- The Genetic Data Environment (UNIX)CINEMA- Java applet available from
ndash httpwwwbiochemuclacuk
SeqappSeqpup- MacPCUNIX available fromndash httpiubiobioindianaedu
SeAl for Macintosh available fromndash httpevolvezoooxacukSe-AlSe-Alhtml
BioEdit for PC available fromndash httpwwwmbioncsueduRNasePinfoprogramsBIOEDITbio
edithtml
- Multiple Sequence Alignment James McInerney bioinf4biologists Feb 2009
- Alignment can be easy or difficult
- Homology Definition
- Multiple Sequence Alignment- Goals
- Multiple sequence alignments - problems
- Slide 6
- Slide 7
- SSU rRNA
- Alignment of 16S rRNA can be guided by secondary structure
- Protein Alignment may be guided by Tertiary Structure Interactions
- Multiple Sequence Alignment- Methods
- Manual Alignment - reasons
- Local minimum
- Dotplots
- Dotplot example sperm whale vs human myg
- Slide 16
- Slide 17
- Dotplots in practice
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Dynamic programming
- Progressive Alignment
- Slide 23
- ClustalW- Pairwise Alignments
- Path Graph for aligning two sequences
- Possible alignment
- Alignment using this path
- Optimal Alignment 1
- Optimal Alignment 2
- Alignment of 3 sequences
- ClustalW- Guide Tree
- Neighbor joining method
- Distance Matrix
- First Step
- Calculation of New Distances
- Next Cycle
- Penultimate Cycle
- Last Joining
- Unrooted Neighbor-Joining Tree
- Multiple Alignment- First pair
- ClustalW- Decision time
- ClustalW- Alternative 1
- ClustalW- Progression
- Progressive alignment - step 1
- Progressive alignment - step 2
- Progressive alignment - step 3
- Progressive alignment - final step
- ClustalW-Good pointsBad points
- ClustalW-Local Minimum
- Increasing the sophistication of the alignment process
- Slide 51
- ClustalW- Caveats
- ClustalW- User-supplied values
- Position-Specific gap penalties
- Discouraging too many gaps
- Divergent Sequences
- Advice on progressive alignment
- Alignment of protein-coding DNA sequences
- Manual Alignment- software
-
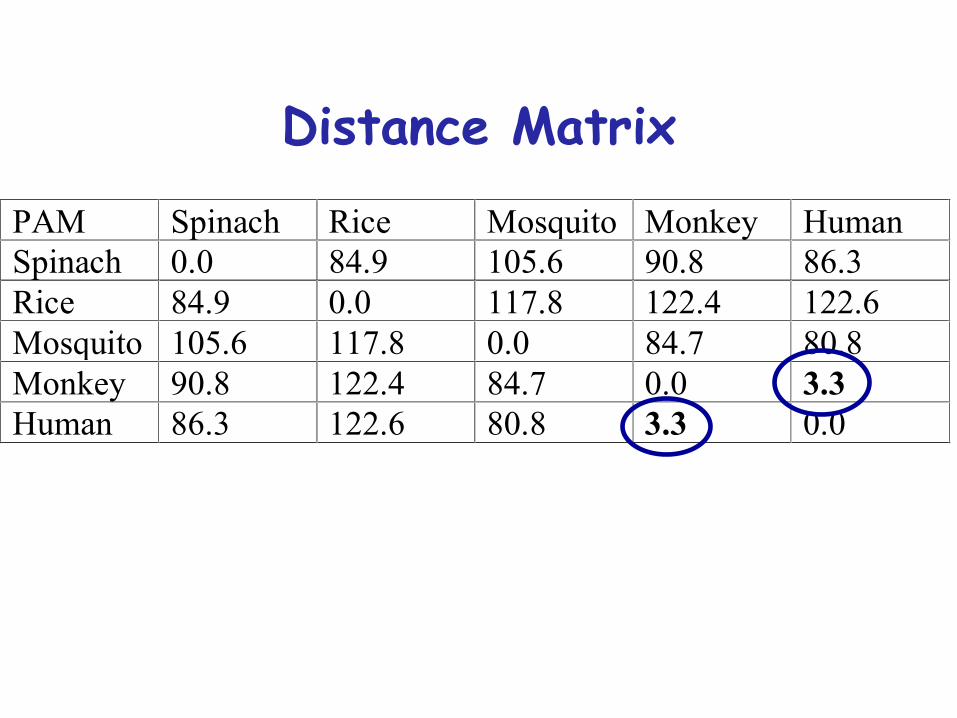
After we have joined two species in a subtree we have to compute the distances from every other node to the new subtree We do this with a simple average of distancesDist[Spinach MonHum]
= (Dist[Spinach Monkey] + Dist[Spinach Human])2 = (908 + 863)2 = 8855
Mon-Hum
MonkeyHumanSpinach
Calculation of New Distances
PAM Spinach Rice Mosquito MonHumSpinach 00 849 1056 886Rice 849 00 1178 1225Mosquito 1056 1178 00 828MonHum 886 1225 828 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Next Cycle
PAM Spinach Rice MosMonHumSpinach 00 849 971Rice 849 00 1202MosMonHum 971 1202 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
Penultimate Cycle
PAM SpinRice MosMonHumSpinach 00 1087MosMonHum 1087 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
(Spin-Rice)-(Mos-(Mon-Hum))
Last Joining
Human
Monkey
MosquitoRice
Spinach
Unrooted Neighbor-Joining Tree
Multiple Alignment- First pairbull Align the two most closely-related sequences first
bull This alignment is then lsquofixedrsquo and will never change If a gap is to be introduced subsequently then it will be introduced in the same place in both sequences but their relative alignment remains unchanged
ClustalW- Decision timebull Consult the guide tree to see what alignment is performed next
ndash Align a third sequence to the first twoOrndash Align two entirely different sequences to each other
Option 1Option 1 Option 2Option 2
ClustalW- Alternative 1If the situation arises where a third sequence is aligned to the first two then when a gap has to be introduced to improve the alignment each of these two entities are treated as two single sequences
+
ClustalW- Alternative 2bull If on the other hand two separate
sequences have to be aligned together then the first pairwise alignment is placed to one side and the pairwise alignment of the other two is carried out
+
ClustalW- Progression
bull The alignment is progressively built up in this way with each step being treated as a pairwise alignment sometimes with each member of a lsquopairrsquo having more than one sequence
Progressive alignment - step 11 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
12345
Progressive alignment - step 21 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
12345
Progressive alignment - step 31 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
+
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
12345
Progressive alignment - final step1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
+
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
5 -ctcga-acgatacgatgactagct-
12345
ClustalW-Good pointsBad points
bull Advantagesndash Speed
bull Disadvantagesndash No objective functionndash No way of quantifying whether or not the alignment is good
ndash No way of knowing if the alignment is lsquocorrectrsquo
ClustalW-Local Minimumbull Potential problems
ndash Local minimum problem If an error is introduced early in the alignment process it is impossible to correct this later in the procedure
ndash Arbitrary alignment
Increasing the sophistication of the alignment process
bull Should we treat all the sequences in the same way - even though some sequences are closely-related and some sequences are distant relatives
bull Should we treat all positions in the sequences as though they were the same - even though they might have different functions and different locations in the 3-dimensional structure
ClustalW- Caveatsbull Sequence weightingbull Varying substitution matricesbull Residue-specific gap penalties and reduced penalties
in hydrophilic regions (external regions of protein sequences) encourage gaps in loops rather than in core regions
bull Positions in early alignments where gaps have been opened receive locally reduced gap penalties to encourage openings in subsequent alignments
ClustalW- User-supplied values
bull Two penalties are set by the user (there are default values but you should know that it is possible to change these)
bull GOP- Gap Opening Penalty is the cost of opening a gap in an alignment
bull GEP- Gap Extension Penalty is the cost of extending this gap
Position-Specific gap penaltiesbull Before any pair of (groups of) sequences are
aligned a table of GOPs are generated for each position in the two (sets of) sequences
bull The GOP is manipulated in a position-specific manner so that it can vary over the sequences
bull If there is a gap at a position the GOP and GEP penalties are lowered the other rules do not apply
bull This makes gaps more likely at positions where gaps already exist
Discouraging too many gaps bull If there is no gap opened then the GOP is increased if the
position is within 8 residues of an existing gapbull This discourages gaps that are too close togetherbull At any position within a run of hydrophilic residues the GOP is
decreasedbull These runs usually indicate loop regions in protein structuresbull A run of 5 hydrophilic residues is considered to be a hydrophilic
stretchbull The default hydrophilic residues are
ndash D E G K N Q P R Sndash But this can be changed by the user
Divergent Sequencesbull The most divergent sequences (most different on average
from all of the other sequences) are usually the most difficult to align
bull It is sometimes better to delay their aligment until later (when the easier sequences have already been aligned)
bull The user has the choice of setting a cutoff (default is 40 identity)
bull This will delay the alignment until the others have been aligned
Advice on progressive alignmentbull Progressive alignment is a mathematical process that is completely
independent of biological realitybull Can be a very good estimatebull Can be an impossibly poor estimatebull Requires user input and skillbull Treat cautiouslybull Can be improved by eye (usually)bull Often helps to have colour-codingbull Depending on the use the user should be able to make a judgement
on those regions that are reliable or notbull For phylogeny reconstruction only use those positions whose
hypothesis of positional homology is unimpeachable
Alignment of protein-coding DNA sequences
bull It is not very sensible to align the DNA sequences of protein-coding genes
ATGCTGTTAGGGATGACTCTGTTAGGG
ATG-CT--GTTAGGGATGACTCTGTTAGGG
The result might be highly-implausible and might not reflect what is known about biological processesIt is much more sensible to translate the sequences to their corresponding amino acid sequences align these protein sequences and then put the gaps in the DNA sequences according to where they are found in the amino acid alignment
Manual Alignment- softwareGDE- The Genetic Data Environment (UNIX)CINEMA- Java applet available from
ndash httpwwwbiochemuclacuk
SeqappSeqpup- MacPCUNIX available fromndash httpiubiobioindianaedu
SeAl for Macintosh available fromndash httpevolvezoooxacukSe-AlSe-Alhtml
BioEdit for PC available fromndash httpwwwmbioncsueduRNasePinfoprogramsBIOEDITbio
edithtml
- Multiple Sequence Alignment James McInerney bioinf4biologists Feb 2009
- Alignment can be easy or difficult
- Homology Definition
- Multiple Sequence Alignment- Goals
- Multiple sequence alignments - problems
- Slide 6
- Slide 7
- SSU rRNA
- Alignment of 16S rRNA can be guided by secondary structure
- Protein Alignment may be guided by Tertiary Structure Interactions
- Multiple Sequence Alignment- Methods
- Manual Alignment - reasons
- Local minimum
- Dotplots
- Dotplot example sperm whale vs human myg
- Slide 16
- Slide 17
- Dotplots in practice
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Dynamic programming
- Progressive Alignment
- Slide 23
- ClustalW- Pairwise Alignments
- Path Graph for aligning two sequences
- Possible alignment
- Alignment using this path
- Optimal Alignment 1
- Optimal Alignment 2
- Alignment of 3 sequences
- ClustalW- Guide Tree
- Neighbor joining method
- Distance Matrix
- First Step
- Calculation of New Distances
- Next Cycle
- Penultimate Cycle
- Last Joining
- Unrooted Neighbor-Joining Tree
- Multiple Alignment- First pair
- ClustalW- Decision time
- ClustalW- Alternative 1
- ClustalW- Progression
- Progressive alignment - step 1
- Progressive alignment - step 2
- Progressive alignment - step 3
- Progressive alignment - final step
- ClustalW-Good pointsBad points
- ClustalW-Local Minimum
- Increasing the sophistication of the alignment process
- Slide 51
- ClustalW- Caveats
- ClustalW- User-supplied values
- Position-Specific gap penalties
- Discouraging too many gaps
- Divergent Sequences
- Advice on progressive alignment
- Alignment of protein-coding DNA sequences
- Manual Alignment- software
-

PAM Spinach Rice Mosquito MonHumSpinach 00 849 1056 886Rice 849 00 1178 1225Mosquito 1056 1178 00 828MonHum 886 1225 828 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Next Cycle
PAM Spinach Rice MosMonHumSpinach 00 849 971Rice 849 00 1202MosMonHum 971 1202 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
Penultimate Cycle
PAM SpinRice MosMonHumSpinach 00 1087MosMonHum 1087 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
(Spin-Rice)-(Mos-(Mon-Hum))
Last Joining
Human
Monkey
MosquitoRice
Spinach
Unrooted Neighbor-Joining Tree
Multiple Alignment- First pairbull Align the two most closely-related sequences first
bull This alignment is then lsquofixedrsquo and will never change If a gap is to be introduced subsequently then it will be introduced in the same place in both sequences but their relative alignment remains unchanged
ClustalW- Decision timebull Consult the guide tree to see what alignment is performed next
ndash Align a third sequence to the first twoOrndash Align two entirely different sequences to each other
Option 1Option 1 Option 2Option 2
ClustalW- Alternative 1If the situation arises where a third sequence is aligned to the first two then when a gap has to be introduced to improve the alignment each of these two entities are treated as two single sequences
+
ClustalW- Alternative 2bull If on the other hand two separate
sequences have to be aligned together then the first pairwise alignment is placed to one side and the pairwise alignment of the other two is carried out
+
ClustalW- Progression
bull The alignment is progressively built up in this way with each step being treated as a pairwise alignment sometimes with each member of a lsquopairrsquo having more than one sequence
Progressive alignment - step 11 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
12345
Progressive alignment - step 21 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
12345
Progressive alignment - step 31 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
+
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
12345
Progressive alignment - final step1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
+
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
5 -ctcga-acgatacgatgactagct-
12345
ClustalW-Good pointsBad points
bull Advantagesndash Speed
bull Disadvantagesndash No objective functionndash No way of quantifying whether or not the alignment is good
ndash No way of knowing if the alignment is lsquocorrectrsquo
ClustalW-Local Minimumbull Potential problems
ndash Local minimum problem If an error is introduced early in the alignment process it is impossible to correct this later in the procedure
ndash Arbitrary alignment
Increasing the sophistication of the alignment process
bull Should we treat all the sequences in the same way - even though some sequences are closely-related and some sequences are distant relatives
bull Should we treat all positions in the sequences as though they were the same - even though they might have different functions and different locations in the 3-dimensional structure
ClustalW- Caveatsbull Sequence weightingbull Varying substitution matricesbull Residue-specific gap penalties and reduced penalties
in hydrophilic regions (external regions of protein sequences) encourage gaps in loops rather than in core regions
bull Positions in early alignments where gaps have been opened receive locally reduced gap penalties to encourage openings in subsequent alignments
ClustalW- User-supplied values
bull Two penalties are set by the user (there are default values but you should know that it is possible to change these)
bull GOP- Gap Opening Penalty is the cost of opening a gap in an alignment
bull GEP- Gap Extension Penalty is the cost of extending this gap
Position-Specific gap penaltiesbull Before any pair of (groups of) sequences are
aligned a table of GOPs are generated for each position in the two (sets of) sequences
bull The GOP is manipulated in a position-specific manner so that it can vary over the sequences
bull If there is a gap at a position the GOP and GEP penalties are lowered the other rules do not apply
bull This makes gaps more likely at positions where gaps already exist
Discouraging too many gaps bull If there is no gap opened then the GOP is increased if the
position is within 8 residues of an existing gapbull This discourages gaps that are too close togetherbull At any position within a run of hydrophilic residues the GOP is
decreasedbull These runs usually indicate loop regions in protein structuresbull A run of 5 hydrophilic residues is considered to be a hydrophilic
stretchbull The default hydrophilic residues are
ndash D E G K N Q P R Sndash But this can be changed by the user
Divergent Sequencesbull The most divergent sequences (most different on average
from all of the other sequences) are usually the most difficult to align
bull It is sometimes better to delay their aligment until later (when the easier sequences have already been aligned)
bull The user has the choice of setting a cutoff (default is 40 identity)
bull This will delay the alignment until the others have been aligned
Advice on progressive alignmentbull Progressive alignment is a mathematical process that is completely
independent of biological realitybull Can be a very good estimatebull Can be an impossibly poor estimatebull Requires user input and skillbull Treat cautiouslybull Can be improved by eye (usually)bull Often helps to have colour-codingbull Depending on the use the user should be able to make a judgement
on those regions that are reliable or notbull For phylogeny reconstruction only use those positions whose
hypothesis of positional homology is unimpeachable
Alignment of protein-coding DNA sequences
bull It is not very sensible to align the DNA sequences of protein-coding genes
ATGCTGTTAGGGATGACTCTGTTAGGG
ATG-CT--GTTAGGGATGACTCTGTTAGGG
The result might be highly-implausible and might not reflect what is known about biological processesIt is much more sensible to translate the sequences to their corresponding amino acid sequences align these protein sequences and then put the gaps in the DNA sequences according to where they are found in the amino acid alignment
Manual Alignment- softwareGDE- The Genetic Data Environment (UNIX)CINEMA- Java applet available from
ndash httpwwwbiochemuclacuk
SeqappSeqpup- MacPCUNIX available fromndash httpiubiobioindianaedu
SeAl for Macintosh available fromndash httpevolvezoooxacukSe-AlSe-Alhtml
BioEdit for PC available fromndash httpwwwmbioncsueduRNasePinfoprogramsBIOEDITbio
edithtml
- Multiple Sequence Alignment James McInerney bioinf4biologists Feb 2009
- Alignment can be easy or difficult
- Homology Definition
- Multiple Sequence Alignment- Goals
- Multiple sequence alignments - problems
- Slide 6
- Slide 7
- SSU rRNA
- Alignment of 16S rRNA can be guided by secondary structure
- Protein Alignment may be guided by Tertiary Structure Interactions
- Multiple Sequence Alignment- Methods
- Manual Alignment - reasons
- Local minimum
- Dotplots
- Dotplot example sperm whale vs human myg
- Slide 16
- Slide 17
- Dotplots in practice
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Dynamic programming
- Progressive Alignment
- Slide 23
- ClustalW- Pairwise Alignments
- Path Graph for aligning two sequences
- Possible alignment
- Alignment using this path
- Optimal Alignment 1
- Optimal Alignment 2
- Alignment of 3 sequences
- ClustalW- Guide Tree
- Neighbor joining method
- Distance Matrix
- First Step
- Calculation of New Distances
- Next Cycle
- Penultimate Cycle
- Last Joining
- Unrooted Neighbor-Joining Tree
- Multiple Alignment- First pair
- ClustalW- Decision time
- ClustalW- Alternative 1
- ClustalW- Progression
- Progressive alignment - step 1
- Progressive alignment - step 2
- Progressive alignment - step 3
- Progressive alignment - final step
- ClustalW-Good pointsBad points
- ClustalW-Local Minimum
- Increasing the sophistication of the alignment process
- Slide 51
- ClustalW- Caveats
- ClustalW- User-supplied values
- Position-Specific gap penalties
- Discouraging too many gaps
- Divergent Sequences
- Advice on progressive alignment
- Alignment of protein-coding DNA sequences
- Manual Alignment- software
-

PAM Spinach Rice MosMonHumSpinach 00 849 971Rice 849 00 1202MosMonHum 971 1202 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
Penultimate Cycle
PAM SpinRice MosMonHumSpinach 00 1087MosMonHum 1087 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
(Spin-Rice)-(Mos-(Mon-Hum))
Last Joining
Human
Monkey
MosquitoRice
Spinach
Unrooted Neighbor-Joining Tree
Multiple Alignment- First pairbull Align the two most closely-related sequences first
bull This alignment is then lsquofixedrsquo and will never change If a gap is to be introduced subsequently then it will be introduced in the same place in both sequences but their relative alignment remains unchanged
ClustalW- Decision timebull Consult the guide tree to see what alignment is performed next
ndash Align a third sequence to the first twoOrndash Align two entirely different sequences to each other
Option 1Option 1 Option 2Option 2
ClustalW- Alternative 1If the situation arises where a third sequence is aligned to the first two then when a gap has to be introduced to improve the alignment each of these two entities are treated as two single sequences
+
ClustalW- Alternative 2bull If on the other hand two separate
sequences have to be aligned together then the first pairwise alignment is placed to one side and the pairwise alignment of the other two is carried out
+
ClustalW- Progression
bull The alignment is progressively built up in this way with each step being treated as a pairwise alignment sometimes with each member of a lsquopairrsquo having more than one sequence
Progressive alignment - step 11 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
12345
Progressive alignment - step 21 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
12345
Progressive alignment - step 31 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
+
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
12345
Progressive alignment - final step1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
+
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
5 -ctcga-acgatacgatgactagct-
12345
ClustalW-Good pointsBad points
bull Advantagesndash Speed
bull Disadvantagesndash No objective functionndash No way of quantifying whether or not the alignment is good
ndash No way of knowing if the alignment is lsquocorrectrsquo
ClustalW-Local Minimumbull Potential problems
ndash Local minimum problem If an error is introduced early in the alignment process it is impossible to correct this later in the procedure
ndash Arbitrary alignment
Increasing the sophistication of the alignment process
bull Should we treat all the sequences in the same way - even though some sequences are closely-related and some sequences are distant relatives
bull Should we treat all positions in the sequences as though they were the same - even though they might have different functions and different locations in the 3-dimensional structure
ClustalW- Caveatsbull Sequence weightingbull Varying substitution matricesbull Residue-specific gap penalties and reduced penalties
in hydrophilic regions (external regions of protein sequences) encourage gaps in loops rather than in core regions
bull Positions in early alignments where gaps have been opened receive locally reduced gap penalties to encourage openings in subsequent alignments
ClustalW- User-supplied values
bull Two penalties are set by the user (there are default values but you should know that it is possible to change these)
bull GOP- Gap Opening Penalty is the cost of opening a gap in an alignment
bull GEP- Gap Extension Penalty is the cost of extending this gap
Position-Specific gap penaltiesbull Before any pair of (groups of) sequences are
aligned a table of GOPs are generated for each position in the two (sets of) sequences
bull The GOP is manipulated in a position-specific manner so that it can vary over the sequences
bull If there is a gap at a position the GOP and GEP penalties are lowered the other rules do not apply
bull This makes gaps more likely at positions where gaps already exist
Discouraging too many gaps bull If there is no gap opened then the GOP is increased if the
position is within 8 residues of an existing gapbull This discourages gaps that are too close togetherbull At any position within a run of hydrophilic residues the GOP is
decreasedbull These runs usually indicate loop regions in protein structuresbull A run of 5 hydrophilic residues is considered to be a hydrophilic
stretchbull The default hydrophilic residues are
ndash D E G K N Q P R Sndash But this can be changed by the user
Divergent Sequencesbull The most divergent sequences (most different on average
from all of the other sequences) are usually the most difficult to align
bull It is sometimes better to delay their aligment until later (when the easier sequences have already been aligned)
bull The user has the choice of setting a cutoff (default is 40 identity)
bull This will delay the alignment until the others have been aligned
Advice on progressive alignmentbull Progressive alignment is a mathematical process that is completely
independent of biological realitybull Can be a very good estimatebull Can be an impossibly poor estimatebull Requires user input and skillbull Treat cautiouslybull Can be improved by eye (usually)bull Often helps to have colour-codingbull Depending on the use the user should be able to make a judgement
on those regions that are reliable or notbull For phylogeny reconstruction only use those positions whose
hypothesis of positional homology is unimpeachable
Alignment of protein-coding DNA sequences
bull It is not very sensible to align the DNA sequences of protein-coding genes
ATGCTGTTAGGGATGACTCTGTTAGGG
ATG-CT--GTTAGGGATGACTCTGTTAGGG
The result might be highly-implausible and might not reflect what is known about biological processesIt is much more sensible to translate the sequences to their corresponding amino acid sequences align these protein sequences and then put the gaps in the DNA sequences according to where they are found in the amino acid alignment
Manual Alignment- softwareGDE- The Genetic Data Environment (UNIX)CINEMA- Java applet available from
ndash httpwwwbiochemuclacuk
SeqappSeqpup- MacPCUNIX available fromndash httpiubiobioindianaedu
SeAl for Macintosh available fromndash httpevolvezoooxacukSe-AlSe-Alhtml
BioEdit for PC available fromndash httpwwwmbioncsueduRNasePinfoprogramsBIOEDITbio
edithtml
- Multiple Sequence Alignment James McInerney bioinf4biologists Feb 2009
- Alignment can be easy or difficult
- Homology Definition
- Multiple Sequence Alignment- Goals
- Multiple sequence alignments - problems
- Slide 6
- Slide 7
- SSU rRNA
- Alignment of 16S rRNA can be guided by secondary structure
- Protein Alignment may be guided by Tertiary Structure Interactions
- Multiple Sequence Alignment- Methods
- Manual Alignment - reasons
- Local minimum
- Dotplots
- Dotplot example sperm whale vs human myg
- Slide 16
- Slide 17
- Dotplots in practice
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Dynamic programming
- Progressive Alignment
- Slide 23
- ClustalW- Pairwise Alignments
- Path Graph for aligning two sequences
- Possible alignment
- Alignment using this path
- Optimal Alignment 1
- Optimal Alignment 2
- Alignment of 3 sequences
- ClustalW- Guide Tree
- Neighbor joining method
- Distance Matrix
- First Step
- Calculation of New Distances
- Next Cycle
- Penultimate Cycle
- Last Joining
- Unrooted Neighbor-Joining Tree
- Multiple Alignment- First pair
- ClustalW- Decision time
- ClustalW- Alternative 1
- ClustalW- Progression
- Progressive alignment - step 1
- Progressive alignment - step 2
- Progressive alignment - step 3
- Progressive alignment - final step
- ClustalW-Good pointsBad points
- ClustalW-Local Minimum
- Increasing the sophistication of the alignment process
- Slide 51
- ClustalW- Caveats
- ClustalW- User-supplied values
- Position-Specific gap penalties
- Discouraging too many gaps
- Divergent Sequences
- Advice on progressive alignment
- Alignment of protein-coding DNA sequences
- Manual Alignment- software
-

PAM SpinRice MosMonHumSpinach 00 1087MosMonHum 1087 00
HumanMosquito
Mon-Hum
MonkeySpinachRice
Mos-(Mon-Hum)
Spin-Rice
(Spin-Rice)-(Mos-(Mon-Hum))
Last Joining
Human
Monkey
MosquitoRice
Spinach
Unrooted Neighbor-Joining Tree
Multiple Alignment- First pairbull Align the two most closely-related sequences first
bull This alignment is then lsquofixedrsquo and will never change If a gap is to be introduced subsequently then it will be introduced in the same place in both sequences but their relative alignment remains unchanged
ClustalW- Decision timebull Consult the guide tree to see what alignment is performed next
ndash Align a third sequence to the first twoOrndash Align two entirely different sequences to each other
Option 1Option 1 Option 2Option 2
ClustalW- Alternative 1If the situation arises where a third sequence is aligned to the first two then when a gap has to be introduced to improve the alignment each of these two entities are treated as two single sequences
+
ClustalW- Alternative 2bull If on the other hand two separate
sequences have to be aligned together then the first pairwise alignment is placed to one side and the pairwise alignment of the other two is carried out
+
ClustalW- Progression
bull The alignment is progressively built up in this way with each step being treated as a pairwise alignment sometimes with each member of a lsquopairrsquo having more than one sequence
Progressive alignment - step 11 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
12345
Progressive alignment - step 21 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
12345
Progressive alignment - step 31 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
+
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
12345
Progressive alignment - final step1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
+
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
5 -ctcga-acgatacgatgactagct-
12345
ClustalW-Good pointsBad points
bull Advantagesndash Speed
bull Disadvantagesndash No objective functionndash No way of quantifying whether or not the alignment is good
ndash No way of knowing if the alignment is lsquocorrectrsquo
ClustalW-Local Minimumbull Potential problems
ndash Local minimum problem If an error is introduced early in the alignment process it is impossible to correct this later in the procedure
ndash Arbitrary alignment
Increasing the sophistication of the alignment process
bull Should we treat all the sequences in the same way - even though some sequences are closely-related and some sequences are distant relatives
bull Should we treat all positions in the sequences as though they were the same - even though they might have different functions and different locations in the 3-dimensional structure
ClustalW- Caveatsbull Sequence weightingbull Varying substitution matricesbull Residue-specific gap penalties and reduced penalties
in hydrophilic regions (external regions of protein sequences) encourage gaps in loops rather than in core regions
bull Positions in early alignments where gaps have been opened receive locally reduced gap penalties to encourage openings in subsequent alignments
ClustalW- User-supplied values
bull Two penalties are set by the user (there are default values but you should know that it is possible to change these)
bull GOP- Gap Opening Penalty is the cost of opening a gap in an alignment
bull GEP- Gap Extension Penalty is the cost of extending this gap
Position-Specific gap penaltiesbull Before any pair of (groups of) sequences are
aligned a table of GOPs are generated for each position in the two (sets of) sequences
bull The GOP is manipulated in a position-specific manner so that it can vary over the sequences
bull If there is a gap at a position the GOP and GEP penalties are lowered the other rules do not apply
bull This makes gaps more likely at positions where gaps already exist
Discouraging too many gaps bull If there is no gap opened then the GOP is increased if the
position is within 8 residues of an existing gapbull This discourages gaps that are too close togetherbull At any position within a run of hydrophilic residues the GOP is
decreasedbull These runs usually indicate loop regions in protein structuresbull A run of 5 hydrophilic residues is considered to be a hydrophilic
stretchbull The default hydrophilic residues are
ndash D E G K N Q P R Sndash But this can be changed by the user
Divergent Sequencesbull The most divergent sequences (most different on average
from all of the other sequences) are usually the most difficult to align
bull It is sometimes better to delay their aligment until later (when the easier sequences have already been aligned)
bull The user has the choice of setting a cutoff (default is 40 identity)
bull This will delay the alignment until the others have been aligned
Advice on progressive alignmentbull Progressive alignment is a mathematical process that is completely
independent of biological realitybull Can be a very good estimatebull Can be an impossibly poor estimatebull Requires user input and skillbull Treat cautiouslybull Can be improved by eye (usually)bull Often helps to have colour-codingbull Depending on the use the user should be able to make a judgement
on those regions that are reliable or notbull For phylogeny reconstruction only use those positions whose
hypothesis of positional homology is unimpeachable
Alignment of protein-coding DNA sequences
bull It is not very sensible to align the DNA sequences of protein-coding genes
ATGCTGTTAGGGATGACTCTGTTAGGG
ATG-CT--GTTAGGGATGACTCTGTTAGGG
The result might be highly-implausible and might not reflect what is known about biological processesIt is much more sensible to translate the sequences to their corresponding amino acid sequences align these protein sequences and then put the gaps in the DNA sequences according to where they are found in the amino acid alignment
Manual Alignment- softwareGDE- The Genetic Data Environment (UNIX)CINEMA- Java applet available from
ndash httpwwwbiochemuclacuk
SeqappSeqpup- MacPCUNIX available fromndash httpiubiobioindianaedu
SeAl for Macintosh available fromndash httpevolvezoooxacukSe-AlSe-Alhtml
BioEdit for PC available fromndash httpwwwmbioncsueduRNasePinfoprogramsBIOEDITbio
edithtml
- Multiple Sequence Alignment James McInerney bioinf4biologists Feb 2009
- Alignment can be easy or difficult
- Homology Definition
- Multiple Sequence Alignment- Goals
- Multiple sequence alignments - problems
- Slide 6
- Slide 7
- SSU rRNA
- Alignment of 16S rRNA can be guided by secondary structure
- Protein Alignment may be guided by Tertiary Structure Interactions
- Multiple Sequence Alignment- Methods
- Manual Alignment - reasons
- Local minimum
- Dotplots
- Dotplot example sperm whale vs human myg
- Slide 16
- Slide 17
- Dotplots in practice
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Dynamic programming
- Progressive Alignment
- Slide 23
- ClustalW- Pairwise Alignments
- Path Graph for aligning two sequences
- Possible alignment
- Alignment using this path
- Optimal Alignment 1
- Optimal Alignment 2
- Alignment of 3 sequences
- ClustalW- Guide Tree
- Neighbor joining method
- Distance Matrix
- First Step
- Calculation of New Distances
- Next Cycle
- Penultimate Cycle
- Last Joining
- Unrooted Neighbor-Joining Tree
- Multiple Alignment- First pair
- ClustalW- Decision time
- ClustalW- Alternative 1
- ClustalW- Progression
- Progressive alignment - step 1
- Progressive alignment - step 2
- Progressive alignment - step 3
- Progressive alignment - final step
- ClustalW-Good pointsBad points
- ClustalW-Local Minimum
- Increasing the sophistication of the alignment process
- Slide 51
- ClustalW- Caveats
- ClustalW- User-supplied values
- Position-Specific gap penalties
- Discouraging too many gaps
- Divergent Sequences
- Advice on progressive alignment
- Alignment of protein-coding DNA sequences
- Manual Alignment- software
-

Human
Monkey
MosquitoRice
Spinach
Unrooted Neighbor-Joining Tree
Multiple Alignment- First pairbull Align the two most closely-related sequences first
bull This alignment is then lsquofixedrsquo and will never change If a gap is to be introduced subsequently then it will be introduced in the same place in both sequences but their relative alignment remains unchanged
ClustalW- Decision timebull Consult the guide tree to see what alignment is performed next
ndash Align a third sequence to the first twoOrndash Align two entirely different sequences to each other
Option 1Option 1 Option 2Option 2
ClustalW- Alternative 1If the situation arises where a third sequence is aligned to the first two then when a gap has to be introduced to improve the alignment each of these two entities are treated as two single sequences
+
ClustalW- Alternative 2bull If on the other hand two separate
sequences have to be aligned together then the first pairwise alignment is placed to one side and the pairwise alignment of the other two is carried out
+
ClustalW- Progression
bull The alignment is progressively built up in this way with each step being treated as a pairwise alignment sometimes with each member of a lsquopairrsquo having more than one sequence
Progressive alignment - step 11 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
12345
Progressive alignment - step 21 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
12345
Progressive alignment - step 31 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
+
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
12345
Progressive alignment - final step1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
+
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
5 -ctcga-acgatacgatgactagct-
12345
ClustalW-Good pointsBad points
bull Advantagesndash Speed
bull Disadvantagesndash No objective functionndash No way of quantifying whether or not the alignment is good
ndash No way of knowing if the alignment is lsquocorrectrsquo
ClustalW-Local Minimumbull Potential problems
ndash Local minimum problem If an error is introduced early in the alignment process it is impossible to correct this later in the procedure
ndash Arbitrary alignment
Increasing the sophistication of the alignment process
bull Should we treat all the sequences in the same way - even though some sequences are closely-related and some sequences are distant relatives
bull Should we treat all positions in the sequences as though they were the same - even though they might have different functions and different locations in the 3-dimensional structure
ClustalW- Caveatsbull Sequence weightingbull Varying substitution matricesbull Residue-specific gap penalties and reduced penalties
in hydrophilic regions (external regions of protein sequences) encourage gaps in loops rather than in core regions
bull Positions in early alignments where gaps have been opened receive locally reduced gap penalties to encourage openings in subsequent alignments
ClustalW- User-supplied values
bull Two penalties are set by the user (there are default values but you should know that it is possible to change these)
bull GOP- Gap Opening Penalty is the cost of opening a gap in an alignment
bull GEP- Gap Extension Penalty is the cost of extending this gap
Position-Specific gap penaltiesbull Before any pair of (groups of) sequences are
aligned a table of GOPs are generated for each position in the two (sets of) sequences
bull The GOP is manipulated in a position-specific manner so that it can vary over the sequences
bull If there is a gap at a position the GOP and GEP penalties are lowered the other rules do not apply
bull This makes gaps more likely at positions where gaps already exist
Discouraging too many gaps bull If there is no gap opened then the GOP is increased if the
position is within 8 residues of an existing gapbull This discourages gaps that are too close togetherbull At any position within a run of hydrophilic residues the GOP is
decreasedbull These runs usually indicate loop regions in protein structuresbull A run of 5 hydrophilic residues is considered to be a hydrophilic
stretchbull The default hydrophilic residues are
ndash D E G K N Q P R Sndash But this can be changed by the user
Divergent Sequencesbull The most divergent sequences (most different on average
from all of the other sequences) are usually the most difficult to align
bull It is sometimes better to delay their aligment until later (when the easier sequences have already been aligned)
bull The user has the choice of setting a cutoff (default is 40 identity)
bull This will delay the alignment until the others have been aligned
Advice on progressive alignmentbull Progressive alignment is a mathematical process that is completely
independent of biological realitybull Can be a very good estimatebull Can be an impossibly poor estimatebull Requires user input and skillbull Treat cautiouslybull Can be improved by eye (usually)bull Often helps to have colour-codingbull Depending on the use the user should be able to make a judgement
on those regions that are reliable or notbull For phylogeny reconstruction only use those positions whose
hypothesis of positional homology is unimpeachable
Alignment of protein-coding DNA sequences
bull It is not very sensible to align the DNA sequences of protein-coding genes
ATGCTGTTAGGGATGACTCTGTTAGGG
ATG-CT--GTTAGGGATGACTCTGTTAGGG
The result might be highly-implausible and might not reflect what is known about biological processesIt is much more sensible to translate the sequences to their corresponding amino acid sequences align these protein sequences and then put the gaps in the DNA sequences according to where they are found in the amino acid alignment
Manual Alignment- softwareGDE- The Genetic Data Environment (UNIX)CINEMA- Java applet available from
ndash httpwwwbiochemuclacuk
SeqappSeqpup- MacPCUNIX available fromndash httpiubiobioindianaedu
SeAl for Macintosh available fromndash httpevolvezoooxacukSe-AlSe-Alhtml
BioEdit for PC available fromndash httpwwwmbioncsueduRNasePinfoprogramsBIOEDITbio
edithtml
- Multiple Sequence Alignment James McInerney bioinf4biologists Feb 2009
- Alignment can be easy or difficult
- Homology Definition
- Multiple Sequence Alignment- Goals
- Multiple sequence alignments - problems
- Slide 6
- Slide 7
- SSU rRNA
- Alignment of 16S rRNA can be guided by secondary structure
- Protein Alignment may be guided by Tertiary Structure Interactions
- Multiple Sequence Alignment- Methods
- Manual Alignment - reasons
- Local minimum
- Dotplots
- Dotplot example sperm whale vs human myg
- Slide 16
- Slide 17
- Dotplots in practice
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Dynamic programming
- Progressive Alignment
- Slide 23
- ClustalW- Pairwise Alignments
- Path Graph for aligning two sequences
- Possible alignment
- Alignment using this path
- Optimal Alignment 1
- Optimal Alignment 2
- Alignment of 3 sequences
- ClustalW- Guide Tree
- Neighbor joining method
- Distance Matrix
- First Step
- Calculation of New Distances
- Next Cycle
- Penultimate Cycle
- Last Joining
- Unrooted Neighbor-Joining Tree
- Multiple Alignment- First pair
- ClustalW- Decision time
- ClustalW- Alternative 1
- ClustalW- Progression
- Progressive alignment - step 1
- Progressive alignment - step 2
- Progressive alignment - step 3
- Progressive alignment - final step
- ClustalW-Good pointsBad points
- ClustalW-Local Minimum
- Increasing the sophistication of the alignment process
- Slide 51
- ClustalW- Caveats
- ClustalW- User-supplied values
- Position-Specific gap penalties
- Discouraging too many gaps
- Divergent Sequences
- Advice on progressive alignment
- Alignment of protein-coding DNA sequences
- Manual Alignment- software
-

Multiple Alignment- First pairbull Align the two most closely-related sequences first
bull This alignment is then lsquofixedrsquo and will never change If a gap is to be introduced subsequently then it will be introduced in the same place in both sequences but their relative alignment remains unchanged
ClustalW- Decision timebull Consult the guide tree to see what alignment is performed next
ndash Align a third sequence to the first twoOrndash Align two entirely different sequences to each other
Option 1Option 1 Option 2Option 2
ClustalW- Alternative 1If the situation arises where a third sequence is aligned to the first two then when a gap has to be introduced to improve the alignment each of these two entities are treated as two single sequences
+
ClustalW- Alternative 2bull If on the other hand two separate
sequences have to be aligned together then the first pairwise alignment is placed to one side and the pairwise alignment of the other two is carried out
+
ClustalW- Progression
bull The alignment is progressively built up in this way with each step being treated as a pairwise alignment sometimes with each member of a lsquopairrsquo having more than one sequence
Progressive alignment - step 11 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
12345
Progressive alignment - step 21 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
12345
Progressive alignment - step 31 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
+
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
12345
Progressive alignment - final step1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
+
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
5 -ctcga-acgatacgatgactagct-
12345
ClustalW-Good pointsBad points
bull Advantagesndash Speed
bull Disadvantagesndash No objective functionndash No way of quantifying whether or not the alignment is good
ndash No way of knowing if the alignment is lsquocorrectrsquo
ClustalW-Local Minimumbull Potential problems
ndash Local minimum problem If an error is introduced early in the alignment process it is impossible to correct this later in the procedure
ndash Arbitrary alignment
Increasing the sophistication of the alignment process
bull Should we treat all the sequences in the same way - even though some sequences are closely-related and some sequences are distant relatives
bull Should we treat all positions in the sequences as though they were the same - even though they might have different functions and different locations in the 3-dimensional structure
ClustalW- Caveatsbull Sequence weightingbull Varying substitution matricesbull Residue-specific gap penalties and reduced penalties
in hydrophilic regions (external regions of protein sequences) encourage gaps in loops rather than in core regions
bull Positions in early alignments where gaps have been opened receive locally reduced gap penalties to encourage openings in subsequent alignments
ClustalW- User-supplied values
bull Two penalties are set by the user (there are default values but you should know that it is possible to change these)
bull GOP- Gap Opening Penalty is the cost of opening a gap in an alignment
bull GEP- Gap Extension Penalty is the cost of extending this gap
Position-Specific gap penaltiesbull Before any pair of (groups of) sequences are
aligned a table of GOPs are generated for each position in the two (sets of) sequences
bull The GOP is manipulated in a position-specific manner so that it can vary over the sequences
bull If there is a gap at a position the GOP and GEP penalties are lowered the other rules do not apply
bull This makes gaps more likely at positions where gaps already exist
Discouraging too many gaps bull If there is no gap opened then the GOP is increased if the
position is within 8 residues of an existing gapbull This discourages gaps that are too close togetherbull At any position within a run of hydrophilic residues the GOP is
decreasedbull These runs usually indicate loop regions in protein structuresbull A run of 5 hydrophilic residues is considered to be a hydrophilic
stretchbull The default hydrophilic residues are
ndash D E G K N Q P R Sndash But this can be changed by the user
Divergent Sequencesbull The most divergent sequences (most different on average
from all of the other sequences) are usually the most difficult to align
bull It is sometimes better to delay their aligment until later (when the easier sequences have already been aligned)
bull The user has the choice of setting a cutoff (default is 40 identity)
bull This will delay the alignment until the others have been aligned
Advice on progressive alignmentbull Progressive alignment is a mathematical process that is completely
independent of biological realitybull Can be a very good estimatebull Can be an impossibly poor estimatebull Requires user input and skillbull Treat cautiouslybull Can be improved by eye (usually)bull Often helps to have colour-codingbull Depending on the use the user should be able to make a judgement
on those regions that are reliable or notbull For phylogeny reconstruction only use those positions whose
hypothesis of positional homology is unimpeachable
Alignment of protein-coding DNA sequences
bull It is not very sensible to align the DNA sequences of protein-coding genes
ATGCTGTTAGGGATGACTCTGTTAGGG
ATG-CT--GTTAGGGATGACTCTGTTAGGG
The result might be highly-implausible and might not reflect what is known about biological processesIt is much more sensible to translate the sequences to their corresponding amino acid sequences align these protein sequences and then put the gaps in the DNA sequences according to where they are found in the amino acid alignment
Manual Alignment- softwareGDE- The Genetic Data Environment (UNIX)CINEMA- Java applet available from
ndash httpwwwbiochemuclacuk
SeqappSeqpup- MacPCUNIX available fromndash httpiubiobioindianaedu
SeAl for Macintosh available fromndash httpevolvezoooxacukSe-AlSe-Alhtml
BioEdit for PC available fromndash httpwwwmbioncsueduRNasePinfoprogramsBIOEDITbio
edithtml
- Multiple Sequence Alignment James McInerney bioinf4biologists Feb 2009
- Alignment can be easy or difficult
- Homology Definition
- Multiple Sequence Alignment- Goals
- Multiple sequence alignments - problems
- Slide 6
- Slide 7
- SSU rRNA
- Alignment of 16S rRNA can be guided by secondary structure
- Protein Alignment may be guided by Tertiary Structure Interactions
- Multiple Sequence Alignment- Methods
- Manual Alignment - reasons
- Local minimum
- Dotplots
- Dotplot example sperm whale vs human myg
- Slide 16
- Slide 17
- Dotplots in practice
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Dynamic programming
- Progressive Alignment
- Slide 23
- ClustalW- Pairwise Alignments
- Path Graph for aligning two sequences
- Possible alignment
- Alignment using this path
- Optimal Alignment 1
- Optimal Alignment 2
- Alignment of 3 sequences
- ClustalW- Guide Tree
- Neighbor joining method
- Distance Matrix
- First Step
- Calculation of New Distances
- Next Cycle
- Penultimate Cycle
- Last Joining
- Unrooted Neighbor-Joining Tree
- Multiple Alignment- First pair
- ClustalW- Decision time
- ClustalW- Alternative 1
- ClustalW- Progression
- Progressive alignment - step 1
- Progressive alignment - step 2
- Progressive alignment - step 3
- Progressive alignment - final step
- ClustalW-Good pointsBad points
- ClustalW-Local Minimum
- Increasing the sophistication of the alignment process
- Slide 51
- ClustalW- Caveats
- ClustalW- User-supplied values
- Position-Specific gap penalties
- Discouraging too many gaps
- Divergent Sequences
- Advice on progressive alignment
- Alignment of protein-coding DNA sequences
- Manual Alignment- software
-

ClustalW- Decision timebull Consult the guide tree to see what alignment is performed next
ndash Align a third sequence to the first twoOrndash Align two entirely different sequences to each other
Option 1Option 1 Option 2Option 2
ClustalW- Alternative 1If the situation arises where a third sequence is aligned to the first two then when a gap has to be introduced to improve the alignment each of these two entities are treated as two single sequences
+
ClustalW- Alternative 2bull If on the other hand two separate
sequences have to be aligned together then the first pairwise alignment is placed to one side and the pairwise alignment of the other two is carried out
+
ClustalW- Progression
bull The alignment is progressively built up in this way with each step being treated as a pairwise alignment sometimes with each member of a lsquopairrsquo having more than one sequence
Progressive alignment - step 11 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
12345
Progressive alignment - step 21 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
12345
Progressive alignment - step 31 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
+
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
12345
Progressive alignment - final step1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
+
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
5 -ctcga-acgatacgatgactagct-
12345
ClustalW-Good pointsBad points
bull Advantagesndash Speed
bull Disadvantagesndash No objective functionndash No way of quantifying whether or not the alignment is good
ndash No way of knowing if the alignment is lsquocorrectrsquo
ClustalW-Local Minimumbull Potential problems
ndash Local minimum problem If an error is introduced early in the alignment process it is impossible to correct this later in the procedure
ndash Arbitrary alignment
Increasing the sophistication of the alignment process
bull Should we treat all the sequences in the same way - even though some sequences are closely-related and some sequences are distant relatives
bull Should we treat all positions in the sequences as though they were the same - even though they might have different functions and different locations in the 3-dimensional structure
ClustalW- Caveatsbull Sequence weightingbull Varying substitution matricesbull Residue-specific gap penalties and reduced penalties
in hydrophilic regions (external regions of protein sequences) encourage gaps in loops rather than in core regions
bull Positions in early alignments where gaps have been opened receive locally reduced gap penalties to encourage openings in subsequent alignments
ClustalW- User-supplied values
bull Two penalties are set by the user (there are default values but you should know that it is possible to change these)
bull GOP- Gap Opening Penalty is the cost of opening a gap in an alignment
bull GEP- Gap Extension Penalty is the cost of extending this gap
Position-Specific gap penaltiesbull Before any pair of (groups of) sequences are
aligned a table of GOPs are generated for each position in the two (sets of) sequences
bull The GOP is manipulated in a position-specific manner so that it can vary over the sequences
bull If there is a gap at a position the GOP and GEP penalties are lowered the other rules do not apply
bull This makes gaps more likely at positions where gaps already exist
Discouraging too many gaps bull If there is no gap opened then the GOP is increased if the
position is within 8 residues of an existing gapbull This discourages gaps that are too close togetherbull At any position within a run of hydrophilic residues the GOP is
decreasedbull These runs usually indicate loop regions in protein structuresbull A run of 5 hydrophilic residues is considered to be a hydrophilic
stretchbull The default hydrophilic residues are
ndash D E G K N Q P R Sndash But this can be changed by the user
Divergent Sequencesbull The most divergent sequences (most different on average
from all of the other sequences) are usually the most difficult to align
bull It is sometimes better to delay their aligment until later (when the easier sequences have already been aligned)
bull The user has the choice of setting a cutoff (default is 40 identity)
bull This will delay the alignment until the others have been aligned
Advice on progressive alignmentbull Progressive alignment is a mathematical process that is completely
independent of biological realitybull Can be a very good estimatebull Can be an impossibly poor estimatebull Requires user input and skillbull Treat cautiouslybull Can be improved by eye (usually)bull Often helps to have colour-codingbull Depending on the use the user should be able to make a judgement
on those regions that are reliable or notbull For phylogeny reconstruction only use those positions whose
hypothesis of positional homology is unimpeachable
Alignment of protein-coding DNA sequences
bull It is not very sensible to align the DNA sequences of protein-coding genes
ATGCTGTTAGGGATGACTCTGTTAGGG
ATG-CT--GTTAGGGATGACTCTGTTAGGG
The result might be highly-implausible and might not reflect what is known about biological processesIt is much more sensible to translate the sequences to their corresponding amino acid sequences align these protein sequences and then put the gaps in the DNA sequences according to where they are found in the amino acid alignment
Manual Alignment- softwareGDE- The Genetic Data Environment (UNIX)CINEMA- Java applet available from
ndash httpwwwbiochemuclacuk
SeqappSeqpup- MacPCUNIX available fromndash httpiubiobioindianaedu
SeAl for Macintosh available fromndash httpevolvezoooxacukSe-AlSe-Alhtml
BioEdit for PC available fromndash httpwwwmbioncsueduRNasePinfoprogramsBIOEDITbio
edithtml
- Multiple Sequence Alignment James McInerney bioinf4biologists Feb 2009
- Alignment can be easy or difficult
- Homology Definition
- Multiple Sequence Alignment- Goals
- Multiple sequence alignments - problems
- Slide 6
- Slide 7
- SSU rRNA
- Alignment of 16S rRNA can be guided by secondary structure
- Protein Alignment may be guided by Tertiary Structure Interactions
- Multiple Sequence Alignment- Methods
- Manual Alignment - reasons
- Local minimum
- Dotplots
- Dotplot example sperm whale vs human myg
- Slide 16
- Slide 17
- Dotplots in practice
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Dynamic programming
- Progressive Alignment
- Slide 23
- ClustalW- Pairwise Alignments
- Path Graph for aligning two sequences
- Possible alignment
- Alignment using this path
- Optimal Alignment 1
- Optimal Alignment 2
- Alignment of 3 sequences
- ClustalW- Guide Tree
- Neighbor joining method
- Distance Matrix
- First Step
- Calculation of New Distances
- Next Cycle
- Penultimate Cycle
- Last Joining
- Unrooted Neighbor-Joining Tree
- Multiple Alignment- First pair
- ClustalW- Decision time
- ClustalW- Alternative 1
- ClustalW- Progression
- Progressive alignment - step 1
- Progressive alignment - step 2
- Progressive alignment - step 3
- Progressive alignment - final step
- ClustalW-Good pointsBad points
- ClustalW-Local Minimum
- Increasing the sophistication of the alignment process
- Slide 51
- ClustalW- Caveats
- ClustalW- User-supplied values
- Position-Specific gap penalties
- Discouraging too many gaps
- Divergent Sequences
- Advice on progressive alignment
- Alignment of protein-coding DNA sequences
- Manual Alignment- software
-

ClustalW- Alternative 1If the situation arises where a third sequence is aligned to the first two then when a gap has to be introduced to improve the alignment each of these two entities are treated as two single sequences
+
ClustalW- Alternative 2bull If on the other hand two separate
sequences have to be aligned together then the first pairwise alignment is placed to one side and the pairwise alignment of the other two is carried out
+
ClustalW- Progression
bull The alignment is progressively built up in this way with each step being treated as a pairwise alignment sometimes with each member of a lsquopairrsquo having more than one sequence
Progressive alignment - step 11 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
12345
Progressive alignment - step 21 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
12345
Progressive alignment - step 31 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
+
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
12345
Progressive alignment - final step1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
+
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
5 -ctcga-acgatacgatgactagct-
12345
ClustalW-Good pointsBad points
bull Advantagesndash Speed
bull Disadvantagesndash No objective functionndash No way of quantifying whether or not the alignment is good
ndash No way of knowing if the alignment is lsquocorrectrsquo
ClustalW-Local Minimumbull Potential problems
ndash Local minimum problem If an error is introduced early in the alignment process it is impossible to correct this later in the procedure
ndash Arbitrary alignment
Increasing the sophistication of the alignment process
bull Should we treat all the sequences in the same way - even though some sequences are closely-related and some sequences are distant relatives
bull Should we treat all positions in the sequences as though they were the same - even though they might have different functions and different locations in the 3-dimensional structure
ClustalW- Caveatsbull Sequence weightingbull Varying substitution matricesbull Residue-specific gap penalties and reduced penalties
in hydrophilic regions (external regions of protein sequences) encourage gaps in loops rather than in core regions
bull Positions in early alignments where gaps have been opened receive locally reduced gap penalties to encourage openings in subsequent alignments
ClustalW- User-supplied values
bull Two penalties are set by the user (there are default values but you should know that it is possible to change these)
bull GOP- Gap Opening Penalty is the cost of opening a gap in an alignment
bull GEP- Gap Extension Penalty is the cost of extending this gap
Position-Specific gap penaltiesbull Before any pair of (groups of) sequences are
aligned a table of GOPs are generated for each position in the two (sets of) sequences
bull The GOP is manipulated in a position-specific manner so that it can vary over the sequences
bull If there is a gap at a position the GOP and GEP penalties are lowered the other rules do not apply
bull This makes gaps more likely at positions where gaps already exist
Discouraging too many gaps bull If there is no gap opened then the GOP is increased if the
position is within 8 residues of an existing gapbull This discourages gaps that are too close togetherbull At any position within a run of hydrophilic residues the GOP is
decreasedbull These runs usually indicate loop regions in protein structuresbull A run of 5 hydrophilic residues is considered to be a hydrophilic
stretchbull The default hydrophilic residues are
ndash D E G K N Q P R Sndash But this can be changed by the user
Divergent Sequencesbull The most divergent sequences (most different on average
from all of the other sequences) are usually the most difficult to align
bull It is sometimes better to delay their aligment until later (when the easier sequences have already been aligned)
bull The user has the choice of setting a cutoff (default is 40 identity)
bull This will delay the alignment until the others have been aligned
Advice on progressive alignmentbull Progressive alignment is a mathematical process that is completely
independent of biological realitybull Can be a very good estimatebull Can be an impossibly poor estimatebull Requires user input and skillbull Treat cautiouslybull Can be improved by eye (usually)bull Often helps to have colour-codingbull Depending on the use the user should be able to make a judgement
on those regions that are reliable or notbull For phylogeny reconstruction only use those positions whose
hypothesis of positional homology is unimpeachable
Alignment of protein-coding DNA sequences
bull It is not very sensible to align the DNA sequences of protein-coding genes
ATGCTGTTAGGGATGACTCTGTTAGGG
ATG-CT--GTTAGGGATGACTCTGTTAGGG
The result might be highly-implausible and might not reflect what is known about biological processesIt is much more sensible to translate the sequences to their corresponding amino acid sequences align these protein sequences and then put the gaps in the DNA sequences according to where they are found in the amino acid alignment
Manual Alignment- softwareGDE- The Genetic Data Environment (UNIX)CINEMA- Java applet available from
ndash httpwwwbiochemuclacuk
SeqappSeqpup- MacPCUNIX available fromndash httpiubiobioindianaedu
SeAl for Macintosh available fromndash httpevolvezoooxacukSe-AlSe-Alhtml
BioEdit for PC available fromndash httpwwwmbioncsueduRNasePinfoprogramsBIOEDITbio
edithtml
- Multiple Sequence Alignment James McInerney bioinf4biologists Feb 2009
- Alignment can be easy or difficult
- Homology Definition
- Multiple Sequence Alignment- Goals
- Multiple sequence alignments - problems
- Slide 6
- Slide 7
- SSU rRNA
- Alignment of 16S rRNA can be guided by secondary structure
- Protein Alignment may be guided by Tertiary Structure Interactions
- Multiple Sequence Alignment- Methods
- Manual Alignment - reasons
- Local minimum
- Dotplots
- Dotplot example sperm whale vs human myg
- Slide 16
- Slide 17
- Dotplots in practice
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Dynamic programming
- Progressive Alignment
- Slide 23
- ClustalW- Pairwise Alignments
- Path Graph for aligning two sequences
- Possible alignment
- Alignment using this path
- Optimal Alignment 1
- Optimal Alignment 2
- Alignment of 3 sequences
- ClustalW- Guide Tree
- Neighbor joining method
- Distance Matrix
- First Step
- Calculation of New Distances
- Next Cycle
- Penultimate Cycle
- Last Joining
- Unrooted Neighbor-Joining Tree
- Multiple Alignment- First pair
- ClustalW- Decision time
- ClustalW- Alternative 1
- ClustalW- Progression
- Progressive alignment - step 1
- Progressive alignment - step 2
- Progressive alignment - step 3
- Progressive alignment - final step
- ClustalW-Good pointsBad points
- ClustalW-Local Minimum
- Increasing the sophistication of the alignment process
- Slide 51
- ClustalW- Caveats
- ClustalW- User-supplied values
- Position-Specific gap penalties
- Discouraging too many gaps
- Divergent Sequences
- Advice on progressive alignment
- Alignment of protein-coding DNA sequences
- Manual Alignment- software
-

ClustalW- Progression
bull The alignment is progressively built up in this way with each step being treated as a pairwise alignment sometimes with each member of a lsquopairrsquo having more than one sequence
Progressive alignment - step 11 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
12345
Progressive alignment - step 21 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
12345
Progressive alignment - step 31 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
+
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
12345
Progressive alignment - final step1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
+
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
5 -ctcga-acgatacgatgactagct-
12345
ClustalW-Good pointsBad points
bull Advantagesndash Speed
bull Disadvantagesndash No objective functionndash No way of quantifying whether or not the alignment is good
ndash No way of knowing if the alignment is lsquocorrectrsquo
ClustalW-Local Minimumbull Potential problems
ndash Local minimum problem If an error is introduced early in the alignment process it is impossible to correct this later in the procedure
ndash Arbitrary alignment
Increasing the sophistication of the alignment process
bull Should we treat all the sequences in the same way - even though some sequences are closely-related and some sequences are distant relatives
bull Should we treat all positions in the sequences as though they were the same - even though they might have different functions and different locations in the 3-dimensional structure
ClustalW- Caveatsbull Sequence weightingbull Varying substitution matricesbull Residue-specific gap penalties and reduced penalties
in hydrophilic regions (external regions of protein sequences) encourage gaps in loops rather than in core regions
bull Positions in early alignments where gaps have been opened receive locally reduced gap penalties to encourage openings in subsequent alignments
ClustalW- User-supplied values
bull Two penalties are set by the user (there are default values but you should know that it is possible to change these)
bull GOP- Gap Opening Penalty is the cost of opening a gap in an alignment
bull GEP- Gap Extension Penalty is the cost of extending this gap
Position-Specific gap penaltiesbull Before any pair of (groups of) sequences are
aligned a table of GOPs are generated for each position in the two (sets of) sequences
bull The GOP is manipulated in a position-specific manner so that it can vary over the sequences
bull If there is a gap at a position the GOP and GEP penalties are lowered the other rules do not apply
bull This makes gaps more likely at positions where gaps already exist
Discouraging too many gaps bull If there is no gap opened then the GOP is increased if the
position is within 8 residues of an existing gapbull This discourages gaps that are too close togetherbull At any position within a run of hydrophilic residues the GOP is
decreasedbull These runs usually indicate loop regions in protein structuresbull A run of 5 hydrophilic residues is considered to be a hydrophilic
stretchbull The default hydrophilic residues are
ndash D E G K N Q P R Sndash But this can be changed by the user
Divergent Sequencesbull The most divergent sequences (most different on average
from all of the other sequences) are usually the most difficult to align
bull It is sometimes better to delay their aligment until later (when the easier sequences have already been aligned)
bull The user has the choice of setting a cutoff (default is 40 identity)
bull This will delay the alignment until the others have been aligned
Advice on progressive alignmentbull Progressive alignment is a mathematical process that is completely
independent of biological realitybull Can be a very good estimatebull Can be an impossibly poor estimatebull Requires user input and skillbull Treat cautiouslybull Can be improved by eye (usually)bull Often helps to have colour-codingbull Depending on the use the user should be able to make a judgement
on those regions that are reliable or notbull For phylogeny reconstruction only use those positions whose
hypothesis of positional homology is unimpeachable
Alignment of protein-coding DNA sequences
bull It is not very sensible to align the DNA sequences of protein-coding genes
ATGCTGTTAGGGATGACTCTGTTAGGG
ATG-CT--GTTAGGGATGACTCTGTTAGGG
The result might be highly-implausible and might not reflect what is known about biological processesIt is much more sensible to translate the sequences to their corresponding amino acid sequences align these protein sequences and then put the gaps in the DNA sequences according to where they are found in the amino acid alignment
Manual Alignment- softwareGDE- The Genetic Data Environment (UNIX)CINEMA- Java applet available from
ndash httpwwwbiochemuclacuk
SeqappSeqpup- MacPCUNIX available fromndash httpiubiobioindianaedu
SeAl for Macintosh available fromndash httpevolvezoooxacukSe-AlSe-Alhtml
BioEdit for PC available fromndash httpwwwmbioncsueduRNasePinfoprogramsBIOEDITbio
edithtml
- Multiple Sequence Alignment James McInerney bioinf4biologists Feb 2009
- Alignment can be easy or difficult
- Homology Definition
- Multiple Sequence Alignment- Goals
- Multiple sequence alignments - problems
- Slide 6
- Slide 7
- SSU rRNA
- Alignment of 16S rRNA can be guided by secondary structure
- Protein Alignment may be guided by Tertiary Structure Interactions
- Multiple Sequence Alignment- Methods
- Manual Alignment - reasons
- Local minimum
- Dotplots
- Dotplot example sperm whale vs human myg
- Slide 16
- Slide 17
- Dotplots in practice
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Dynamic programming
- Progressive Alignment
- Slide 23
- ClustalW- Pairwise Alignments
- Path Graph for aligning two sequences
- Possible alignment
- Alignment using this path
- Optimal Alignment 1
- Optimal Alignment 2
- Alignment of 3 sequences
- ClustalW- Guide Tree
- Neighbor joining method
- Distance Matrix
- First Step
- Calculation of New Distances
- Next Cycle
- Penultimate Cycle
- Last Joining
- Unrooted Neighbor-Joining Tree
- Multiple Alignment- First pair
- ClustalW- Decision time
- ClustalW- Alternative 1
- ClustalW- Progression
- Progressive alignment - step 1
- Progressive alignment - step 2
- Progressive alignment - step 3
- Progressive alignment - final step
- ClustalW-Good pointsBad points
- ClustalW-Local Minimum
- Increasing the sophistication of the alignment process
- Slide 51
- ClustalW- Caveats
- ClustalW- User-supplied values
- Position-Specific gap penalties
- Discouraging too many gaps
- Divergent Sequences
- Advice on progressive alignment
- Alignment of protein-coding DNA sequences
- Manual Alignment- software
-

Progressive alignment - step 11 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
12345
Progressive alignment - step 21 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
12345
Progressive alignment - step 31 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
+
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
12345
Progressive alignment - final step1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
+
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
5 -ctcga-acgatacgatgactagct-
12345
ClustalW-Good pointsBad points
bull Advantagesndash Speed
bull Disadvantagesndash No objective functionndash No way of quantifying whether or not the alignment is good
ndash No way of knowing if the alignment is lsquocorrectrsquo
ClustalW-Local Minimumbull Potential problems
ndash Local minimum problem If an error is introduced early in the alignment process it is impossible to correct this later in the procedure
ndash Arbitrary alignment
Increasing the sophistication of the alignment process
bull Should we treat all the sequences in the same way - even though some sequences are closely-related and some sequences are distant relatives
bull Should we treat all positions in the sequences as though they were the same - even though they might have different functions and different locations in the 3-dimensional structure
ClustalW- Caveatsbull Sequence weightingbull Varying substitution matricesbull Residue-specific gap penalties and reduced penalties
in hydrophilic regions (external regions of protein sequences) encourage gaps in loops rather than in core regions
bull Positions in early alignments where gaps have been opened receive locally reduced gap penalties to encourage openings in subsequent alignments
ClustalW- User-supplied values
bull Two penalties are set by the user (there are default values but you should know that it is possible to change these)
bull GOP- Gap Opening Penalty is the cost of opening a gap in an alignment
bull GEP- Gap Extension Penalty is the cost of extending this gap
Position-Specific gap penaltiesbull Before any pair of (groups of) sequences are
aligned a table of GOPs are generated for each position in the two (sets of) sequences
bull The GOP is manipulated in a position-specific manner so that it can vary over the sequences
bull If there is a gap at a position the GOP and GEP penalties are lowered the other rules do not apply
bull This makes gaps more likely at positions where gaps already exist
Discouraging too many gaps bull If there is no gap opened then the GOP is increased if the
position is within 8 residues of an existing gapbull This discourages gaps that are too close togetherbull At any position within a run of hydrophilic residues the GOP is
decreasedbull These runs usually indicate loop regions in protein structuresbull A run of 5 hydrophilic residues is considered to be a hydrophilic
stretchbull The default hydrophilic residues are
ndash D E G K N Q P R Sndash But this can be changed by the user
Divergent Sequencesbull The most divergent sequences (most different on average
from all of the other sequences) are usually the most difficult to align
bull It is sometimes better to delay their aligment until later (when the easier sequences have already been aligned)
bull The user has the choice of setting a cutoff (default is 40 identity)
bull This will delay the alignment until the others have been aligned
Advice on progressive alignmentbull Progressive alignment is a mathematical process that is completely
independent of biological realitybull Can be a very good estimatebull Can be an impossibly poor estimatebull Requires user input and skillbull Treat cautiouslybull Can be improved by eye (usually)bull Often helps to have colour-codingbull Depending on the use the user should be able to make a judgement
on those regions that are reliable or notbull For phylogeny reconstruction only use those positions whose
hypothesis of positional homology is unimpeachable
Alignment of protein-coding DNA sequences
bull It is not very sensible to align the DNA sequences of protein-coding genes
ATGCTGTTAGGGATGACTCTGTTAGGG
ATG-CT--GTTAGGGATGACTCTGTTAGGG
The result might be highly-implausible and might not reflect what is known about biological processesIt is much more sensible to translate the sequences to their corresponding amino acid sequences align these protein sequences and then put the gaps in the DNA sequences according to where they are found in the amino acid alignment
Manual Alignment- softwareGDE- The Genetic Data Environment (UNIX)CINEMA- Java applet available from
ndash httpwwwbiochemuclacuk
SeqappSeqpup- MacPCUNIX available fromndash httpiubiobioindianaedu
SeAl for Macintosh available fromndash httpevolvezoooxacukSe-AlSe-Alhtml
BioEdit for PC available fromndash httpwwwmbioncsueduRNasePinfoprogramsBIOEDITbio
edithtml
- Multiple Sequence Alignment James McInerney bioinf4biologists Feb 2009
- Alignment can be easy or difficult
- Homology Definition
- Multiple Sequence Alignment- Goals
- Multiple sequence alignments - problems
- Slide 6
- Slide 7
- SSU rRNA
- Alignment of 16S rRNA can be guided by secondary structure
- Protein Alignment may be guided by Tertiary Structure Interactions
- Multiple Sequence Alignment- Methods
- Manual Alignment - reasons
- Local minimum
- Dotplots
- Dotplot example sperm whale vs human myg
- Slide 16
- Slide 17
- Dotplots in practice
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Dynamic programming
- Progressive Alignment
- Slide 23
- ClustalW- Pairwise Alignments
- Path Graph for aligning two sequences
- Possible alignment
- Alignment using this path
- Optimal Alignment 1
- Optimal Alignment 2
- Alignment of 3 sequences
- ClustalW- Guide Tree
- Neighbor joining method
- Distance Matrix
- First Step
- Calculation of New Distances
- Next Cycle
- Penultimate Cycle
- Last Joining
- Unrooted Neighbor-Joining Tree
- Multiple Alignment- First pair
- ClustalW- Decision time
- ClustalW- Alternative 1
- ClustalW- Progression
- Progressive alignment - step 1
- Progressive alignment - step 2
- Progressive alignment - step 3
- Progressive alignment - final step
- ClustalW-Good pointsBad points
- ClustalW-Local Minimum
- Increasing the sophistication of the alignment process
- Slide 51
- ClustalW- Caveats
- ClustalW- User-supplied values
- Position-Specific gap penalties
- Discouraging too many gaps
- Divergent Sequences
- Advice on progressive alignment
- Alignment of protein-coding DNA sequences
- Manual Alignment- software
-

Progressive alignment - step 21 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgacagcta
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
5 ctcgaacgatacgatgactagct
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
12345
Progressive alignment - step 31 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
+
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
12345
Progressive alignment - final step1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
+
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
5 -ctcga-acgatacgatgactagct-
12345
ClustalW-Good pointsBad points
bull Advantagesndash Speed
bull Disadvantagesndash No objective functionndash No way of quantifying whether or not the alignment is good
ndash No way of knowing if the alignment is lsquocorrectrsquo
ClustalW-Local Minimumbull Potential problems
ndash Local minimum problem If an error is introduced early in the alignment process it is impossible to correct this later in the procedure
ndash Arbitrary alignment
Increasing the sophistication of the alignment process
bull Should we treat all the sequences in the same way - even though some sequences are closely-related and some sequences are distant relatives
bull Should we treat all positions in the sequences as though they were the same - even though they might have different functions and different locations in the 3-dimensional structure
ClustalW- Caveatsbull Sequence weightingbull Varying substitution matricesbull Residue-specific gap penalties and reduced penalties
in hydrophilic regions (external regions of protein sequences) encourage gaps in loops rather than in core regions
bull Positions in early alignments where gaps have been opened receive locally reduced gap penalties to encourage openings in subsequent alignments
ClustalW- User-supplied values
bull Two penalties are set by the user (there are default values but you should know that it is possible to change these)
bull GOP- Gap Opening Penalty is the cost of opening a gap in an alignment
bull GEP- Gap Extension Penalty is the cost of extending this gap
Position-Specific gap penaltiesbull Before any pair of (groups of) sequences are
aligned a table of GOPs are generated for each position in the two (sets of) sequences
bull The GOP is manipulated in a position-specific manner so that it can vary over the sequences
bull If there is a gap at a position the GOP and GEP penalties are lowered the other rules do not apply
bull This makes gaps more likely at positions where gaps already exist
Discouraging too many gaps bull If there is no gap opened then the GOP is increased if the
position is within 8 residues of an existing gapbull This discourages gaps that are too close togetherbull At any position within a run of hydrophilic residues the GOP is
decreasedbull These runs usually indicate loop regions in protein structuresbull A run of 5 hydrophilic residues is considered to be a hydrophilic
stretchbull The default hydrophilic residues are
ndash D E G K N Q P R Sndash But this can be changed by the user
Divergent Sequencesbull The most divergent sequences (most different on average
from all of the other sequences) are usually the most difficult to align
bull It is sometimes better to delay their aligment until later (when the easier sequences have already been aligned)
bull The user has the choice of setting a cutoff (default is 40 identity)
bull This will delay the alignment until the others have been aligned
Advice on progressive alignmentbull Progressive alignment is a mathematical process that is completely
independent of biological realitybull Can be a very good estimatebull Can be an impossibly poor estimatebull Requires user input and skillbull Treat cautiouslybull Can be improved by eye (usually)bull Often helps to have colour-codingbull Depending on the use the user should be able to make a judgement
on those regions that are reliable or notbull For phylogeny reconstruction only use those positions whose
hypothesis of positional homology is unimpeachable
Alignment of protein-coding DNA sequences
bull It is not very sensible to align the DNA sequences of protein-coding genes
ATGCTGTTAGGGATGACTCTGTTAGGG
ATG-CT--GTTAGGGATGACTCTGTTAGGG
The result might be highly-implausible and might not reflect what is known about biological processesIt is much more sensible to translate the sequences to their corresponding amino acid sequences align these protein sequences and then put the gaps in the DNA sequences according to where they are found in the amino acid alignment
Manual Alignment- softwareGDE- The Genetic Data Environment (UNIX)CINEMA- Java applet available from
ndash httpwwwbiochemuclacuk
SeqappSeqpup- MacPCUNIX available fromndash httpiubiobioindianaedu
SeAl for Macintosh available fromndash httpevolvezoooxacukSe-AlSe-Alhtml
BioEdit for PC available fromndash httpwwwmbioncsueduRNasePinfoprogramsBIOEDITbio
edithtml
- Multiple Sequence Alignment James McInerney bioinf4biologists Feb 2009
- Alignment can be easy or difficult
- Homology Definition
- Multiple Sequence Alignment- Goals
- Multiple sequence alignments - problems
- Slide 6
- Slide 7
- SSU rRNA
- Alignment of 16S rRNA can be guided by secondary structure
- Protein Alignment may be guided by Tertiary Structure Interactions
- Multiple Sequence Alignment- Methods
- Manual Alignment - reasons
- Local minimum
- Dotplots
- Dotplot example sperm whale vs human myg
- Slide 16
- Slide 17
- Dotplots in practice
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Dynamic programming
- Progressive Alignment
- Slide 23
- ClustalW- Pairwise Alignments
- Path Graph for aligning two sequences
- Possible alignment
- Alignment using this path
- Optimal Alignment 1
- Optimal Alignment 2
- Alignment of 3 sequences
- ClustalW- Guide Tree
- Neighbor joining method
- Distance Matrix
- First Step
- Calculation of New Distances
- Next Cycle
- Penultimate Cycle
- Last Joining
- Unrooted Neighbor-Joining Tree
- Multiple Alignment- First pair
- ClustalW- Decision time
- ClustalW- Alternative 1
- ClustalW- Progression
- Progressive alignment - step 1
- Progressive alignment - step 2
- Progressive alignment - step 3
- Progressive alignment - final step
- ClustalW-Good pointsBad points
- ClustalW-Local Minimum
- Increasing the sophistication of the alignment process
- Slide 51
- ClustalW- Caveats
- ClustalW- User-supplied values
- Position-Specific gap penalties
- Discouraging too many gaps
- Divergent Sequences
- Advice on progressive alignment
- Alignment of protein-coding DNA sequences
- Manual Alignment- software
-

Progressive alignment - step 31 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
+
3 gctcgatacacgatgactagcta
4 gctcgatacacgatgacgagcga
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
12345
Progressive alignment - final step1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
+
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
5 -ctcga-acgatacgatgactagct-
12345
ClustalW-Good pointsBad points
bull Advantagesndash Speed
bull Disadvantagesndash No objective functionndash No way of quantifying whether or not the alignment is good
ndash No way of knowing if the alignment is lsquocorrectrsquo
ClustalW-Local Minimumbull Potential problems
ndash Local minimum problem If an error is introduced early in the alignment process it is impossible to correct this later in the procedure
ndash Arbitrary alignment
Increasing the sophistication of the alignment process
bull Should we treat all the sequences in the same way - even though some sequences are closely-related and some sequences are distant relatives
bull Should we treat all positions in the sequences as though they were the same - even though they might have different functions and different locations in the 3-dimensional structure
ClustalW- Caveatsbull Sequence weightingbull Varying substitution matricesbull Residue-specific gap penalties and reduced penalties
in hydrophilic regions (external regions of protein sequences) encourage gaps in loops rather than in core regions
bull Positions in early alignments where gaps have been opened receive locally reduced gap penalties to encourage openings in subsequent alignments
ClustalW- User-supplied values
bull Two penalties are set by the user (there are default values but you should know that it is possible to change these)
bull GOP- Gap Opening Penalty is the cost of opening a gap in an alignment
bull GEP- Gap Extension Penalty is the cost of extending this gap
Position-Specific gap penaltiesbull Before any pair of (groups of) sequences are
aligned a table of GOPs are generated for each position in the two (sets of) sequences
bull The GOP is manipulated in a position-specific manner so that it can vary over the sequences
bull If there is a gap at a position the GOP and GEP penalties are lowered the other rules do not apply
bull This makes gaps more likely at positions where gaps already exist
Discouraging too many gaps bull If there is no gap opened then the GOP is increased if the
position is within 8 residues of an existing gapbull This discourages gaps that are too close togetherbull At any position within a run of hydrophilic residues the GOP is
decreasedbull These runs usually indicate loop regions in protein structuresbull A run of 5 hydrophilic residues is considered to be a hydrophilic
stretchbull The default hydrophilic residues are
ndash D E G K N Q P R Sndash But this can be changed by the user
Divergent Sequencesbull The most divergent sequences (most different on average
from all of the other sequences) are usually the most difficult to align
bull It is sometimes better to delay their aligment until later (when the easier sequences have already been aligned)
bull The user has the choice of setting a cutoff (default is 40 identity)
bull This will delay the alignment until the others have been aligned
Advice on progressive alignmentbull Progressive alignment is a mathematical process that is completely
independent of biological realitybull Can be a very good estimatebull Can be an impossibly poor estimatebull Requires user input and skillbull Treat cautiouslybull Can be improved by eye (usually)bull Often helps to have colour-codingbull Depending on the use the user should be able to make a judgement
on those regions that are reliable or notbull For phylogeny reconstruction only use those positions whose
hypothesis of positional homology is unimpeachable
Alignment of protein-coding DNA sequences
bull It is not very sensible to align the DNA sequences of protein-coding genes
ATGCTGTTAGGGATGACTCTGTTAGGG
ATG-CT--GTTAGGGATGACTCTGTTAGGG
The result might be highly-implausible and might not reflect what is known about biological processesIt is much more sensible to translate the sequences to their corresponding amino acid sequences align these protein sequences and then put the gaps in the DNA sequences according to where they are found in the amino acid alignment
Manual Alignment- softwareGDE- The Genetic Data Environment (UNIX)CINEMA- Java applet available from
ndash httpwwwbiochemuclacuk
SeqappSeqpup- MacPCUNIX available fromndash httpiubiobioindianaedu
SeAl for Macintosh available fromndash httpevolvezoooxacukSe-AlSe-Alhtml
BioEdit for PC available fromndash httpwwwmbioncsueduRNasePinfoprogramsBIOEDITbio
edithtml
- Multiple Sequence Alignment James McInerney bioinf4biologists Feb 2009
- Alignment can be easy or difficult
- Homology Definition
- Multiple Sequence Alignment- Goals
- Multiple sequence alignments - problems
- Slide 6
- Slide 7
- SSU rRNA
- Alignment of 16S rRNA can be guided by secondary structure
- Protein Alignment may be guided by Tertiary Structure Interactions
- Multiple Sequence Alignment- Methods
- Manual Alignment - reasons
- Local minimum
- Dotplots
- Dotplot example sperm whale vs human myg
- Slide 16
- Slide 17
- Dotplots in practice
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Dynamic programming
- Progressive Alignment
- Slide 23
- ClustalW- Pairwise Alignments
- Path Graph for aligning two sequences
- Possible alignment
- Alignment using this path
- Optimal Alignment 1
- Optimal Alignment 2
- Alignment of 3 sequences
- ClustalW- Guide Tree
- Neighbor joining method
- Distance Matrix
- First Step
- Calculation of New Distances
- Next Cycle
- Penultimate Cycle
- Last Joining
- Unrooted Neighbor-Joining Tree
- Multiple Alignment- First pair
- ClustalW- Decision time
- ClustalW- Alternative 1
- ClustalW- Progression
- Progressive alignment - step 1
- Progressive alignment - step 2
- Progressive alignment - step 3
- Progressive alignment - final step
- ClustalW-Good pointsBad points
- ClustalW-Local Minimum
- Increasing the sophistication of the alignment process
- Slide 51
- ClustalW- Caveats
- ClustalW- User-supplied values
- Position-Specific gap penalties
- Discouraging too many gaps
- Divergent Sequences
- Advice on progressive alignment
- Alignment of protein-coding DNA sequences
- Manual Alignment- software
-

Progressive alignment - final step1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
+
5 ctcgaacgatacgatgactagct
1 gctcgatacgatacgatgactagcta
2 gctcgatacaagacgatgac-agcta
3 gctcgatacacga---tgactagcta
4 gctcgatacacga---tgacgagcga
5 -ctcga-acgatacgatgactagct-
12345
ClustalW-Good pointsBad points
bull Advantagesndash Speed
bull Disadvantagesndash No objective functionndash No way of quantifying whether or not the alignment is good
ndash No way of knowing if the alignment is lsquocorrectrsquo
ClustalW-Local Minimumbull Potential problems
ndash Local minimum problem If an error is introduced early in the alignment process it is impossible to correct this later in the procedure
ndash Arbitrary alignment
Increasing the sophistication of the alignment process
bull Should we treat all the sequences in the same way - even though some sequences are closely-related and some sequences are distant relatives
bull Should we treat all positions in the sequences as though they were the same - even though they might have different functions and different locations in the 3-dimensional structure
ClustalW- Caveatsbull Sequence weightingbull Varying substitution matricesbull Residue-specific gap penalties and reduced penalties
in hydrophilic regions (external regions of protein sequences) encourage gaps in loops rather than in core regions
bull Positions in early alignments where gaps have been opened receive locally reduced gap penalties to encourage openings in subsequent alignments
ClustalW- User-supplied values
bull Two penalties are set by the user (there are default values but you should know that it is possible to change these)
bull GOP- Gap Opening Penalty is the cost of opening a gap in an alignment
bull GEP- Gap Extension Penalty is the cost of extending this gap
Position-Specific gap penaltiesbull Before any pair of (groups of) sequences are
aligned a table of GOPs are generated for each position in the two (sets of) sequences
bull The GOP is manipulated in a position-specific manner so that it can vary over the sequences
bull If there is a gap at a position the GOP and GEP penalties are lowered the other rules do not apply
bull This makes gaps more likely at positions where gaps already exist
Discouraging too many gaps bull If there is no gap opened then the GOP is increased if the
position is within 8 residues of an existing gapbull This discourages gaps that are too close togetherbull At any position within a run of hydrophilic residues the GOP is
decreasedbull These runs usually indicate loop regions in protein structuresbull A run of 5 hydrophilic residues is considered to be a hydrophilic
stretchbull The default hydrophilic residues are
ndash D E G K N Q P R Sndash But this can be changed by the user
Divergent Sequencesbull The most divergent sequences (most different on average
from all of the other sequences) are usually the most difficult to align
bull It is sometimes better to delay their aligment until later (when the easier sequences have already been aligned)
bull The user has the choice of setting a cutoff (default is 40 identity)
bull This will delay the alignment until the others have been aligned
Advice on progressive alignmentbull Progressive alignment is a mathematical process that is completely
independent of biological realitybull Can be a very good estimatebull Can be an impossibly poor estimatebull Requires user input and skillbull Treat cautiouslybull Can be improved by eye (usually)bull Often helps to have colour-codingbull Depending on the use the user should be able to make a judgement
on those regions that are reliable or notbull For phylogeny reconstruction only use those positions whose
hypothesis of positional homology is unimpeachable
Alignment of protein-coding DNA sequences
bull It is not very sensible to align the DNA sequences of protein-coding genes
ATGCTGTTAGGGATGACTCTGTTAGGG
ATG-CT--GTTAGGGATGACTCTGTTAGGG
The result might be highly-implausible and might not reflect what is known about biological processesIt is much more sensible to translate the sequences to their corresponding amino acid sequences align these protein sequences and then put the gaps in the DNA sequences according to where they are found in the amino acid alignment
Manual Alignment- softwareGDE- The Genetic Data Environment (UNIX)CINEMA- Java applet available from
ndash httpwwwbiochemuclacuk
SeqappSeqpup- MacPCUNIX available fromndash httpiubiobioindianaedu
SeAl for Macintosh available fromndash httpevolvezoooxacukSe-AlSe-Alhtml
BioEdit for PC available fromndash httpwwwmbioncsueduRNasePinfoprogramsBIOEDITbio
edithtml
- Multiple Sequence Alignment James McInerney bioinf4biologists Feb 2009
- Alignment can be easy or difficult
- Homology Definition
- Multiple Sequence Alignment- Goals
- Multiple sequence alignments - problems
- Slide 6
- Slide 7
- SSU rRNA
- Alignment of 16S rRNA can be guided by secondary structure
- Protein Alignment may be guided by Tertiary Structure Interactions
- Multiple Sequence Alignment- Methods
- Manual Alignment - reasons
- Local minimum
- Dotplots
- Dotplot example sperm whale vs human myg
- Slide 16
- Slide 17
- Dotplots in practice
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Dynamic programming
- Progressive Alignment
- Slide 23
- ClustalW- Pairwise Alignments
- Path Graph for aligning two sequences
- Possible alignment
- Alignment using this path
- Optimal Alignment 1
- Optimal Alignment 2
- Alignment of 3 sequences
- ClustalW- Guide Tree
- Neighbor joining method
- Distance Matrix
- First Step
- Calculation of New Distances
- Next Cycle
- Penultimate Cycle
- Last Joining
- Unrooted Neighbor-Joining Tree
- Multiple Alignment- First pair
- ClustalW- Decision time
- ClustalW- Alternative 1
- ClustalW- Progression
- Progressive alignment - step 1
- Progressive alignment - step 2
- Progressive alignment - step 3
- Progressive alignment - final step
- ClustalW-Good pointsBad points
- ClustalW-Local Minimum
- Increasing the sophistication of the alignment process
- Slide 51
- ClustalW- Caveats
- ClustalW- User-supplied values
- Position-Specific gap penalties
- Discouraging too many gaps
- Divergent Sequences
- Advice on progressive alignment
- Alignment of protein-coding DNA sequences
- Manual Alignment- software
-

ClustalW-Good pointsBad points
bull Advantagesndash Speed
bull Disadvantagesndash No objective functionndash No way of quantifying whether or not the alignment is good
ndash No way of knowing if the alignment is lsquocorrectrsquo
ClustalW-Local Minimumbull Potential problems
ndash Local minimum problem If an error is introduced early in the alignment process it is impossible to correct this later in the procedure
ndash Arbitrary alignment
Increasing the sophistication of the alignment process
bull Should we treat all the sequences in the same way - even though some sequences are closely-related and some sequences are distant relatives
bull Should we treat all positions in the sequences as though they were the same - even though they might have different functions and different locations in the 3-dimensional structure
ClustalW- Caveatsbull Sequence weightingbull Varying substitution matricesbull Residue-specific gap penalties and reduced penalties
in hydrophilic regions (external regions of protein sequences) encourage gaps in loops rather than in core regions
bull Positions in early alignments where gaps have been opened receive locally reduced gap penalties to encourage openings in subsequent alignments
ClustalW- User-supplied values
bull Two penalties are set by the user (there are default values but you should know that it is possible to change these)
bull GOP- Gap Opening Penalty is the cost of opening a gap in an alignment
bull GEP- Gap Extension Penalty is the cost of extending this gap
Position-Specific gap penaltiesbull Before any pair of (groups of) sequences are
aligned a table of GOPs are generated for each position in the two (sets of) sequences
bull The GOP is manipulated in a position-specific manner so that it can vary over the sequences
bull If there is a gap at a position the GOP and GEP penalties are lowered the other rules do not apply
bull This makes gaps more likely at positions where gaps already exist
Discouraging too many gaps bull If there is no gap opened then the GOP is increased if the
position is within 8 residues of an existing gapbull This discourages gaps that are too close togetherbull At any position within a run of hydrophilic residues the GOP is
decreasedbull These runs usually indicate loop regions in protein structuresbull A run of 5 hydrophilic residues is considered to be a hydrophilic
stretchbull The default hydrophilic residues are
ndash D E G K N Q P R Sndash But this can be changed by the user
Divergent Sequencesbull The most divergent sequences (most different on average
from all of the other sequences) are usually the most difficult to align
bull It is sometimes better to delay their aligment until later (when the easier sequences have already been aligned)
bull The user has the choice of setting a cutoff (default is 40 identity)
bull This will delay the alignment until the others have been aligned
Advice on progressive alignmentbull Progressive alignment is a mathematical process that is completely
independent of biological realitybull Can be a very good estimatebull Can be an impossibly poor estimatebull Requires user input and skillbull Treat cautiouslybull Can be improved by eye (usually)bull Often helps to have colour-codingbull Depending on the use the user should be able to make a judgement
on those regions that are reliable or notbull For phylogeny reconstruction only use those positions whose
hypothesis of positional homology is unimpeachable
Alignment of protein-coding DNA sequences
bull It is not very sensible to align the DNA sequences of protein-coding genes
ATGCTGTTAGGGATGACTCTGTTAGGG
ATG-CT--GTTAGGGATGACTCTGTTAGGG
The result might be highly-implausible and might not reflect what is known about biological processesIt is much more sensible to translate the sequences to their corresponding amino acid sequences align these protein sequences and then put the gaps in the DNA sequences according to where they are found in the amino acid alignment
Manual Alignment- softwareGDE- The Genetic Data Environment (UNIX)CINEMA- Java applet available from
ndash httpwwwbiochemuclacuk
SeqappSeqpup- MacPCUNIX available fromndash httpiubiobioindianaedu
SeAl for Macintosh available fromndash httpevolvezoooxacukSe-AlSe-Alhtml
BioEdit for PC available fromndash httpwwwmbioncsueduRNasePinfoprogramsBIOEDITbio
edithtml
- Multiple Sequence Alignment James McInerney bioinf4biologists Feb 2009
- Alignment can be easy or difficult
- Homology Definition
- Multiple Sequence Alignment- Goals
- Multiple sequence alignments - problems
- Slide 6
- Slide 7
- SSU rRNA
- Alignment of 16S rRNA can be guided by secondary structure
- Protein Alignment may be guided by Tertiary Structure Interactions
- Multiple Sequence Alignment- Methods
- Manual Alignment - reasons
- Local minimum
- Dotplots
- Dotplot example sperm whale vs human myg
- Slide 16
- Slide 17
- Dotplots in practice
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Dynamic programming
- Progressive Alignment
- Slide 23
- ClustalW- Pairwise Alignments
- Path Graph for aligning two sequences
- Possible alignment
- Alignment using this path
- Optimal Alignment 1
- Optimal Alignment 2
- Alignment of 3 sequences
- ClustalW- Guide Tree
- Neighbor joining method
- Distance Matrix
- First Step
- Calculation of New Distances
- Next Cycle
- Penultimate Cycle
- Last Joining
- Unrooted Neighbor-Joining Tree
- Multiple Alignment- First pair
- ClustalW- Decision time
- ClustalW- Alternative 1
- ClustalW- Progression
- Progressive alignment - step 1
- Progressive alignment - step 2
- Progressive alignment - step 3
- Progressive alignment - final step
- ClustalW-Good pointsBad points
- ClustalW-Local Minimum
- Increasing the sophistication of the alignment process
- Slide 51
- ClustalW- Caveats
- ClustalW- User-supplied values
- Position-Specific gap penalties
- Discouraging too many gaps
- Divergent Sequences
- Advice on progressive alignment
- Alignment of protein-coding DNA sequences
- Manual Alignment- software
-
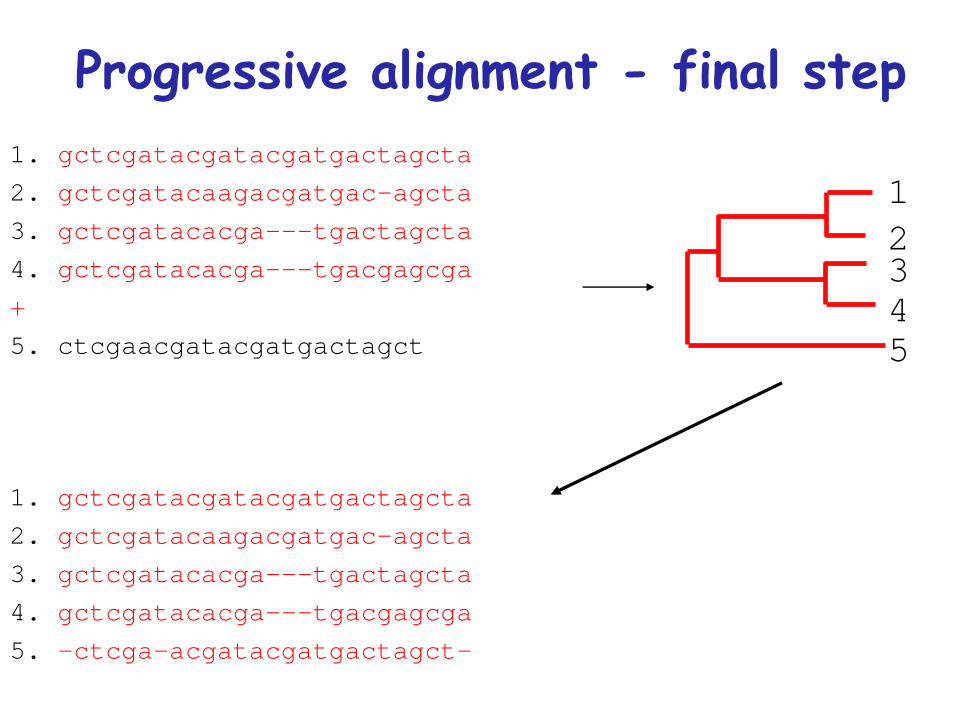
ClustalW-Local Minimumbull Potential problems
ndash Local minimum problem If an error is introduced early in the alignment process it is impossible to correct this later in the procedure
ndash Arbitrary alignment
Increasing the sophistication of the alignment process
bull Should we treat all the sequences in the same way - even though some sequences are closely-related and some sequences are distant relatives
bull Should we treat all positions in the sequences as though they were the same - even though they might have different functions and different locations in the 3-dimensional structure
ClustalW- Caveatsbull Sequence weightingbull Varying substitution matricesbull Residue-specific gap penalties and reduced penalties
in hydrophilic regions (external regions of protein sequences) encourage gaps in loops rather than in core regions
bull Positions in early alignments where gaps have been opened receive locally reduced gap penalties to encourage openings in subsequent alignments
ClustalW- User-supplied values
bull Two penalties are set by the user (there are default values but you should know that it is possible to change these)
bull GOP- Gap Opening Penalty is the cost of opening a gap in an alignment
bull GEP- Gap Extension Penalty is the cost of extending this gap
Position-Specific gap penaltiesbull Before any pair of (groups of) sequences are
aligned a table of GOPs are generated for each position in the two (sets of) sequences
bull The GOP is manipulated in a position-specific manner so that it can vary over the sequences
bull If there is a gap at a position the GOP and GEP penalties are lowered the other rules do not apply
bull This makes gaps more likely at positions where gaps already exist
Discouraging too many gaps bull If there is no gap opened then the GOP is increased if the
position is within 8 residues of an existing gapbull This discourages gaps that are too close togetherbull At any position within a run of hydrophilic residues the GOP is
decreasedbull These runs usually indicate loop regions in protein structuresbull A run of 5 hydrophilic residues is considered to be a hydrophilic
stretchbull The default hydrophilic residues are
ndash D E G K N Q P R Sndash But this can be changed by the user
Divergent Sequencesbull The most divergent sequences (most different on average
from all of the other sequences) are usually the most difficult to align
bull It is sometimes better to delay their aligment until later (when the easier sequences have already been aligned)
bull The user has the choice of setting a cutoff (default is 40 identity)
bull This will delay the alignment until the others have been aligned
Advice on progressive alignmentbull Progressive alignment is a mathematical process that is completely
independent of biological realitybull Can be a very good estimatebull Can be an impossibly poor estimatebull Requires user input and skillbull Treat cautiouslybull Can be improved by eye (usually)bull Often helps to have colour-codingbull Depending on the use the user should be able to make a judgement
on those regions that are reliable or notbull For phylogeny reconstruction only use those positions whose
hypothesis of positional homology is unimpeachable
Alignment of protein-coding DNA sequences
bull It is not very sensible to align the DNA sequences of protein-coding genes
ATGCTGTTAGGGATGACTCTGTTAGGG
ATG-CT--GTTAGGGATGACTCTGTTAGGG
The result might be highly-implausible and might not reflect what is known about biological processesIt is much more sensible to translate the sequences to their corresponding amino acid sequences align these protein sequences and then put the gaps in the DNA sequences according to where they are found in the amino acid alignment
Manual Alignment- softwareGDE- The Genetic Data Environment (UNIX)CINEMA- Java applet available from
ndash httpwwwbiochemuclacuk
SeqappSeqpup- MacPCUNIX available fromndash httpiubiobioindianaedu
SeAl for Macintosh available fromndash httpevolvezoooxacukSe-AlSe-Alhtml
BioEdit for PC available fromndash httpwwwmbioncsueduRNasePinfoprogramsBIOEDITbio
edithtml
- Multiple Sequence Alignment James McInerney bioinf4biologists Feb 2009
- Alignment can be easy or difficult
- Homology Definition
- Multiple Sequence Alignment- Goals
- Multiple sequence alignments - problems
- Slide 6
- Slide 7
- SSU rRNA
- Alignment of 16S rRNA can be guided by secondary structure
- Protein Alignment may be guided by Tertiary Structure Interactions
- Multiple Sequence Alignment- Methods
- Manual Alignment - reasons
- Local minimum
- Dotplots
- Dotplot example sperm whale vs human myg
- Slide 16
- Slide 17
- Dotplots in practice
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Dynamic programming
- Progressive Alignment
- Slide 23
- ClustalW- Pairwise Alignments
- Path Graph for aligning two sequences
- Possible alignment
- Alignment using this path
- Optimal Alignment 1
- Optimal Alignment 2
- Alignment of 3 sequences
- ClustalW- Guide Tree
- Neighbor joining method
- Distance Matrix
- First Step
- Calculation of New Distances
- Next Cycle
- Penultimate Cycle
- Last Joining
- Unrooted Neighbor-Joining Tree
- Multiple Alignment- First pair
- ClustalW- Decision time
- ClustalW- Alternative 1
- ClustalW- Progression
- Progressive alignment - step 1
- Progressive alignment - step 2
- Progressive alignment - step 3
- Progressive alignment - final step
- ClustalW-Good pointsBad points
- ClustalW-Local Minimum
- Increasing the sophistication of the alignment process
- Slide 51
- ClustalW- Caveats
- ClustalW- User-supplied values
- Position-Specific gap penalties
- Discouraging too many gaps
- Divergent Sequences
- Advice on progressive alignment
- Alignment of protein-coding DNA sequences
- Manual Alignment- software
-

Increasing the sophistication of the alignment process
bull Should we treat all the sequences in the same way - even though some sequences are closely-related and some sequences are distant relatives
bull Should we treat all positions in the sequences as though they were the same - even though they might have different functions and different locations in the 3-dimensional structure
ClustalW- Caveatsbull Sequence weightingbull Varying substitution matricesbull Residue-specific gap penalties and reduced penalties
in hydrophilic regions (external regions of protein sequences) encourage gaps in loops rather than in core regions
bull Positions in early alignments where gaps have been opened receive locally reduced gap penalties to encourage openings in subsequent alignments
ClustalW- User-supplied values
bull Two penalties are set by the user (there are default values but you should know that it is possible to change these)
bull GOP- Gap Opening Penalty is the cost of opening a gap in an alignment
bull GEP- Gap Extension Penalty is the cost of extending this gap
Position-Specific gap penaltiesbull Before any pair of (groups of) sequences are
aligned a table of GOPs are generated for each position in the two (sets of) sequences
bull The GOP is manipulated in a position-specific manner so that it can vary over the sequences
bull If there is a gap at a position the GOP and GEP penalties are lowered the other rules do not apply
bull This makes gaps more likely at positions where gaps already exist
Discouraging too many gaps bull If there is no gap opened then the GOP is increased if the
position is within 8 residues of an existing gapbull This discourages gaps that are too close togetherbull At any position within a run of hydrophilic residues the GOP is
decreasedbull These runs usually indicate loop regions in protein structuresbull A run of 5 hydrophilic residues is considered to be a hydrophilic
stretchbull The default hydrophilic residues are
ndash D E G K N Q P R Sndash But this can be changed by the user
Divergent Sequencesbull The most divergent sequences (most different on average
from all of the other sequences) are usually the most difficult to align
bull It is sometimes better to delay their aligment until later (when the easier sequences have already been aligned)
bull The user has the choice of setting a cutoff (default is 40 identity)
bull This will delay the alignment until the others have been aligned
Advice on progressive alignmentbull Progressive alignment is a mathematical process that is completely
independent of biological realitybull Can be a very good estimatebull Can be an impossibly poor estimatebull Requires user input and skillbull Treat cautiouslybull Can be improved by eye (usually)bull Often helps to have colour-codingbull Depending on the use the user should be able to make a judgement
on those regions that are reliable or notbull For phylogeny reconstruction only use those positions whose
hypothesis of positional homology is unimpeachable
Alignment of protein-coding DNA sequences
bull It is not very sensible to align the DNA sequences of protein-coding genes
ATGCTGTTAGGGATGACTCTGTTAGGG
ATG-CT--GTTAGGGATGACTCTGTTAGGG
The result might be highly-implausible and might not reflect what is known about biological processesIt is much more sensible to translate the sequences to their corresponding amino acid sequences align these protein sequences and then put the gaps in the DNA sequences according to where they are found in the amino acid alignment
Manual Alignment- softwareGDE- The Genetic Data Environment (UNIX)CINEMA- Java applet available from
ndash httpwwwbiochemuclacuk
SeqappSeqpup- MacPCUNIX available fromndash httpiubiobioindianaedu
SeAl for Macintosh available fromndash httpevolvezoooxacukSe-AlSe-Alhtml
BioEdit for PC available fromndash httpwwwmbioncsueduRNasePinfoprogramsBIOEDITbio
edithtml
- Multiple Sequence Alignment James McInerney bioinf4biologists Feb 2009
- Alignment can be easy or difficult
- Homology Definition
- Multiple Sequence Alignment- Goals
- Multiple sequence alignments - problems
- Slide 6
- Slide 7
- SSU rRNA
- Alignment of 16S rRNA can be guided by secondary structure
- Protein Alignment may be guided by Tertiary Structure Interactions
- Multiple Sequence Alignment- Methods
- Manual Alignment - reasons
- Local minimum
- Dotplots
- Dotplot example sperm whale vs human myg
- Slide 16
- Slide 17
- Dotplots in practice
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Dynamic programming
- Progressive Alignment
- Slide 23
- ClustalW- Pairwise Alignments
- Path Graph for aligning two sequences
- Possible alignment
- Alignment using this path
- Optimal Alignment 1
- Optimal Alignment 2
- Alignment of 3 sequences
- ClustalW- Guide Tree
- Neighbor joining method
- Distance Matrix
- First Step
- Calculation of New Distances
- Next Cycle
- Penultimate Cycle
- Last Joining
- Unrooted Neighbor-Joining Tree
- Multiple Alignment- First pair
- ClustalW- Decision time
- ClustalW- Alternative 1
- ClustalW- Progression
- Progressive alignment - step 1
- Progressive alignment - step 2
- Progressive alignment - step 3
- Progressive alignment - final step
- ClustalW-Good pointsBad points
- ClustalW-Local Minimum
- Increasing the sophistication of the alignment process
- Slide 51
- ClustalW- Caveats
- ClustalW- User-supplied values
- Position-Specific gap penalties
- Discouraging too many gaps
- Divergent Sequences
- Advice on progressive alignment
- Alignment of protein-coding DNA sequences
- Manual Alignment- software
-

ClustalW- Caveatsbull Sequence weightingbull Varying substitution matricesbull Residue-specific gap penalties and reduced penalties
in hydrophilic regions (external regions of protein sequences) encourage gaps in loops rather than in core regions
bull Positions in early alignments where gaps have been opened receive locally reduced gap penalties to encourage openings in subsequent alignments
ClustalW- User-supplied values
bull Two penalties are set by the user (there are default values but you should know that it is possible to change these)
bull GOP- Gap Opening Penalty is the cost of opening a gap in an alignment
bull GEP- Gap Extension Penalty is the cost of extending this gap
Position-Specific gap penaltiesbull Before any pair of (groups of) sequences are
aligned a table of GOPs are generated for each position in the two (sets of) sequences
bull The GOP is manipulated in a position-specific manner so that it can vary over the sequences
bull If there is a gap at a position the GOP and GEP penalties are lowered the other rules do not apply
bull This makes gaps more likely at positions where gaps already exist
Discouraging too many gaps bull If there is no gap opened then the GOP is increased if the
position is within 8 residues of an existing gapbull This discourages gaps that are too close togetherbull At any position within a run of hydrophilic residues the GOP is
decreasedbull These runs usually indicate loop regions in protein structuresbull A run of 5 hydrophilic residues is considered to be a hydrophilic
stretchbull The default hydrophilic residues are
ndash D E G K N Q P R Sndash But this can be changed by the user
Divergent Sequencesbull The most divergent sequences (most different on average
from all of the other sequences) are usually the most difficult to align
bull It is sometimes better to delay their aligment until later (when the easier sequences have already been aligned)
bull The user has the choice of setting a cutoff (default is 40 identity)
bull This will delay the alignment until the others have been aligned
Advice on progressive alignmentbull Progressive alignment is a mathematical process that is completely
independent of biological realitybull Can be a very good estimatebull Can be an impossibly poor estimatebull Requires user input and skillbull Treat cautiouslybull Can be improved by eye (usually)bull Often helps to have colour-codingbull Depending on the use the user should be able to make a judgement
on those regions that are reliable or notbull For phylogeny reconstruction only use those positions whose
hypothesis of positional homology is unimpeachable
Alignment of protein-coding DNA sequences
bull It is not very sensible to align the DNA sequences of protein-coding genes
ATGCTGTTAGGGATGACTCTGTTAGGG
ATG-CT--GTTAGGGATGACTCTGTTAGGG
The result might be highly-implausible and might not reflect what is known about biological processesIt is much more sensible to translate the sequences to their corresponding amino acid sequences align these protein sequences and then put the gaps in the DNA sequences according to where they are found in the amino acid alignment
Manual Alignment- softwareGDE- The Genetic Data Environment (UNIX)CINEMA- Java applet available from
ndash httpwwwbiochemuclacuk
SeqappSeqpup- MacPCUNIX available fromndash httpiubiobioindianaedu
SeAl for Macintosh available fromndash httpevolvezoooxacukSe-AlSe-Alhtml
BioEdit for PC available fromndash httpwwwmbioncsueduRNasePinfoprogramsBIOEDITbio
edithtml
- Multiple Sequence Alignment James McInerney bioinf4biologists Feb 2009
- Alignment can be easy or difficult
- Homology Definition
- Multiple Sequence Alignment- Goals
- Multiple sequence alignments - problems
- Slide 6
- Slide 7
- SSU rRNA
- Alignment of 16S rRNA can be guided by secondary structure
- Protein Alignment may be guided by Tertiary Structure Interactions
- Multiple Sequence Alignment- Methods
- Manual Alignment - reasons
- Local minimum
- Dotplots
- Dotplot example sperm whale vs human myg
- Slide 16
- Slide 17
- Dotplots in practice
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Dynamic programming
- Progressive Alignment
- Slide 23
- ClustalW- Pairwise Alignments
- Path Graph for aligning two sequences
- Possible alignment
- Alignment using this path
- Optimal Alignment 1
- Optimal Alignment 2
- Alignment of 3 sequences
- ClustalW- Guide Tree
- Neighbor joining method
- Distance Matrix
- First Step
- Calculation of New Distances
- Next Cycle
- Penultimate Cycle
- Last Joining
- Unrooted Neighbor-Joining Tree
- Multiple Alignment- First pair
- ClustalW- Decision time
- ClustalW- Alternative 1
- ClustalW- Progression
- Progressive alignment - step 1
- Progressive alignment - step 2
- Progressive alignment - step 3
- Progressive alignment - final step
- ClustalW-Good pointsBad points
- ClustalW-Local Minimum
- Increasing the sophistication of the alignment process
- Slide 51
- ClustalW- Caveats
- ClustalW- User-supplied values
- Position-Specific gap penalties
- Discouraging too many gaps
- Divergent Sequences
- Advice on progressive alignment
- Alignment of protein-coding DNA sequences
- Manual Alignment- software
-

ClustalW- User-supplied values
bull Two penalties are set by the user (there are default values but you should know that it is possible to change these)
bull GOP- Gap Opening Penalty is the cost of opening a gap in an alignment
bull GEP- Gap Extension Penalty is the cost of extending this gap
Position-Specific gap penaltiesbull Before any pair of (groups of) sequences are
aligned a table of GOPs are generated for each position in the two (sets of) sequences
bull The GOP is manipulated in a position-specific manner so that it can vary over the sequences
bull If there is a gap at a position the GOP and GEP penalties are lowered the other rules do not apply
bull This makes gaps more likely at positions where gaps already exist
Discouraging too many gaps bull If there is no gap opened then the GOP is increased if the
position is within 8 residues of an existing gapbull This discourages gaps that are too close togetherbull At any position within a run of hydrophilic residues the GOP is
decreasedbull These runs usually indicate loop regions in protein structuresbull A run of 5 hydrophilic residues is considered to be a hydrophilic
stretchbull The default hydrophilic residues are
ndash D E G K N Q P R Sndash But this can be changed by the user
Divergent Sequencesbull The most divergent sequences (most different on average
from all of the other sequences) are usually the most difficult to align
bull It is sometimes better to delay their aligment until later (when the easier sequences have already been aligned)
bull The user has the choice of setting a cutoff (default is 40 identity)
bull This will delay the alignment until the others have been aligned
Advice on progressive alignmentbull Progressive alignment is a mathematical process that is completely
independent of biological realitybull Can be a very good estimatebull Can be an impossibly poor estimatebull Requires user input and skillbull Treat cautiouslybull Can be improved by eye (usually)bull Often helps to have colour-codingbull Depending on the use the user should be able to make a judgement
on those regions that are reliable or notbull For phylogeny reconstruction only use those positions whose
hypothesis of positional homology is unimpeachable
Alignment of protein-coding DNA sequences
bull It is not very sensible to align the DNA sequences of protein-coding genes
ATGCTGTTAGGGATGACTCTGTTAGGG
ATG-CT--GTTAGGGATGACTCTGTTAGGG
The result might be highly-implausible and might not reflect what is known about biological processesIt is much more sensible to translate the sequences to their corresponding amino acid sequences align these protein sequences and then put the gaps in the DNA sequences according to where they are found in the amino acid alignment
Manual Alignment- softwareGDE- The Genetic Data Environment (UNIX)CINEMA- Java applet available from
ndash httpwwwbiochemuclacuk
SeqappSeqpup- MacPCUNIX available fromndash httpiubiobioindianaedu
SeAl for Macintosh available fromndash httpevolvezoooxacukSe-AlSe-Alhtml
BioEdit for PC available fromndash httpwwwmbioncsueduRNasePinfoprogramsBIOEDITbio
edithtml
- Multiple Sequence Alignment James McInerney bioinf4biologists Feb 2009
- Alignment can be easy or difficult
- Homology Definition
- Multiple Sequence Alignment- Goals
- Multiple sequence alignments - problems
- Slide 6
- Slide 7
- SSU rRNA
- Alignment of 16S rRNA can be guided by secondary structure
- Protein Alignment may be guided by Tertiary Structure Interactions
- Multiple Sequence Alignment- Methods
- Manual Alignment - reasons
- Local minimum
- Dotplots
- Dotplot example sperm whale vs human myg
- Slide 16
- Slide 17
- Dotplots in practice
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Dynamic programming
- Progressive Alignment
- Slide 23
- ClustalW- Pairwise Alignments
- Path Graph for aligning two sequences
- Possible alignment
- Alignment using this path
- Optimal Alignment 1
- Optimal Alignment 2
- Alignment of 3 sequences
- ClustalW- Guide Tree
- Neighbor joining method
- Distance Matrix
- First Step
- Calculation of New Distances
- Next Cycle
- Penultimate Cycle
- Last Joining
- Unrooted Neighbor-Joining Tree
- Multiple Alignment- First pair
- ClustalW- Decision time
- ClustalW- Alternative 1
- ClustalW- Progression
- Progressive alignment - step 1
- Progressive alignment - step 2
- Progressive alignment - step 3
- Progressive alignment - final step
- ClustalW-Good pointsBad points
- ClustalW-Local Minimum
- Increasing the sophistication of the alignment process
- Slide 51
- ClustalW- Caveats
- ClustalW- User-supplied values
- Position-Specific gap penalties
- Discouraging too many gaps
- Divergent Sequences
- Advice on progressive alignment
- Alignment of protein-coding DNA sequences
- Manual Alignment- software
-
Position-Specific gap penaltiesbull Before any pair of (groups of) sequences are
aligned a table of GOPs are generated for each position in the two (sets of) sequences
bull The GOP is manipulated in a position-specific manner so that it can vary over the sequences
bull If there is a gap at a position the GOP and GEP penalties are lowered the other rules do not apply
bull This makes gaps more likely at positions where gaps already exist
Discouraging too many gaps bull If there is no gap opened then the GOP is increased if the
position is within 8 residues of an existing gapbull This discourages gaps that are too close togetherbull At any position within a run of hydrophilic residues the GOP is
decreasedbull These runs usually indicate loop regions in protein structuresbull A run of 5 hydrophilic residues is considered to be a hydrophilic
stretchbull The default hydrophilic residues are
ndash D E G K N Q P R Sndash But this can be changed by the user
Divergent Sequencesbull The most divergent sequences (most different on average
from all of the other sequences) are usually the most difficult to align
bull It is sometimes better to delay their aligment until later (when the easier sequences have already been aligned)
bull The user has the choice of setting a cutoff (default is 40 identity)
bull This will delay the alignment until the others have been aligned
Advice on progressive alignmentbull Progressive alignment is a mathematical process that is completely
independent of biological realitybull Can be a very good estimatebull Can be an impossibly poor estimatebull Requires user input and skillbull Treat cautiouslybull Can be improved by eye (usually)bull Often helps to have colour-codingbull Depending on the use the user should be able to make a judgement
on those regions that are reliable or notbull For phylogeny reconstruction only use those positions whose
hypothesis of positional homology is unimpeachable
Alignment of protein-coding DNA sequences
bull It is not very sensible to align the DNA sequences of protein-coding genes
ATGCTGTTAGGGATGACTCTGTTAGGG
ATG-CT--GTTAGGGATGACTCTGTTAGGG
The result might be highly-implausible and might not reflect what is known about biological processesIt is much more sensible to translate the sequences to their corresponding amino acid sequences align these protein sequences and then put the gaps in the DNA sequences according to where they are found in the amino acid alignment
Manual Alignment- softwareGDE- The Genetic Data Environment (UNIX)CINEMA- Java applet available from
ndash httpwwwbiochemuclacuk
SeqappSeqpup- MacPCUNIX available fromndash httpiubiobioindianaedu
SeAl for Macintosh available fromndash httpevolvezoooxacukSe-AlSe-Alhtml
BioEdit for PC available fromndash httpwwwmbioncsueduRNasePinfoprogramsBIOEDITbio
edithtml
- Multiple Sequence Alignment James McInerney bioinf4biologists Feb 2009
- Alignment can be easy or difficult
- Homology Definition
- Multiple Sequence Alignment- Goals
- Multiple sequence alignments - problems
- Slide 6
- Slide 7
- SSU rRNA
- Alignment of 16S rRNA can be guided by secondary structure
- Protein Alignment may be guided by Tertiary Structure Interactions
- Multiple Sequence Alignment- Methods
- Manual Alignment - reasons
- Local minimum
- Dotplots
- Dotplot example sperm whale vs human myg
- Slide 16
- Slide 17
- Dotplots in practice
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Dynamic programming
- Progressive Alignment
- Slide 23
- ClustalW- Pairwise Alignments
- Path Graph for aligning two sequences
- Possible alignment
- Alignment using this path
- Optimal Alignment 1
- Optimal Alignment 2
- Alignment of 3 sequences
- ClustalW- Guide Tree
- Neighbor joining method
- Distance Matrix
- First Step
- Calculation of New Distances
- Next Cycle
- Penultimate Cycle
- Last Joining
- Unrooted Neighbor-Joining Tree
- Multiple Alignment- First pair
- ClustalW- Decision time
- ClustalW- Alternative 1
- ClustalW- Progression
- Progressive alignment - step 1
- Progressive alignment - step 2
- Progressive alignment - step 3
- Progressive alignment - final step
- ClustalW-Good pointsBad points
- ClustalW-Local Minimum
- Increasing the sophistication of the alignment process
- Slide 51
- ClustalW- Caveats
- ClustalW- User-supplied values
- Position-Specific gap penalties
- Discouraging too many gaps
- Divergent Sequences
- Advice on progressive alignment
- Alignment of protein-coding DNA sequences
- Manual Alignment- software
-

Discouraging too many gaps bull If there is no gap opened then the GOP is increased if the
position is within 8 residues of an existing gapbull This discourages gaps that are too close togetherbull At any position within a run of hydrophilic residues the GOP is
decreasedbull These runs usually indicate loop regions in protein structuresbull A run of 5 hydrophilic residues is considered to be a hydrophilic
stretchbull The default hydrophilic residues are
ndash D E G K N Q P R Sndash But this can be changed by the user
Divergent Sequencesbull The most divergent sequences (most different on average
from all of the other sequences) are usually the most difficult to align
bull It is sometimes better to delay their aligment until later (when the easier sequences have already been aligned)
bull The user has the choice of setting a cutoff (default is 40 identity)
bull This will delay the alignment until the others have been aligned
Advice on progressive alignmentbull Progressive alignment is a mathematical process that is completely
independent of biological realitybull Can be a very good estimatebull Can be an impossibly poor estimatebull Requires user input and skillbull Treat cautiouslybull Can be improved by eye (usually)bull Often helps to have colour-codingbull Depending on the use the user should be able to make a judgement
on those regions that are reliable or notbull For phylogeny reconstruction only use those positions whose
hypothesis of positional homology is unimpeachable
Alignment of protein-coding DNA sequences
bull It is not very sensible to align the DNA sequences of protein-coding genes
ATGCTGTTAGGGATGACTCTGTTAGGG
ATG-CT--GTTAGGGATGACTCTGTTAGGG
The result might be highly-implausible and might not reflect what is known about biological processesIt is much more sensible to translate the sequences to their corresponding amino acid sequences align these protein sequences and then put the gaps in the DNA sequences according to where they are found in the amino acid alignment
Manual Alignment- softwareGDE- The Genetic Data Environment (UNIX)CINEMA- Java applet available from
ndash httpwwwbiochemuclacuk
SeqappSeqpup- MacPCUNIX available fromndash httpiubiobioindianaedu
SeAl for Macintosh available fromndash httpevolvezoooxacukSe-AlSe-Alhtml
BioEdit for PC available fromndash httpwwwmbioncsueduRNasePinfoprogramsBIOEDITbio
edithtml
- Multiple Sequence Alignment James McInerney bioinf4biologists Feb 2009
- Alignment can be easy or difficult
- Homology Definition
- Multiple Sequence Alignment- Goals
- Multiple sequence alignments - problems
- Slide 6
- Slide 7
- SSU rRNA
- Alignment of 16S rRNA can be guided by secondary structure
- Protein Alignment may be guided by Tertiary Structure Interactions
- Multiple Sequence Alignment- Methods
- Manual Alignment - reasons
- Local minimum
- Dotplots
- Dotplot example sperm whale vs human myg
- Slide 16
- Slide 17
- Dotplots in practice
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Dynamic programming
- Progressive Alignment
- Slide 23
- ClustalW- Pairwise Alignments
- Path Graph for aligning two sequences
- Possible alignment
- Alignment using this path
- Optimal Alignment 1
- Optimal Alignment 2
- Alignment of 3 sequences
- ClustalW- Guide Tree
- Neighbor joining method
- Distance Matrix
- First Step
- Calculation of New Distances
- Next Cycle
- Penultimate Cycle
- Last Joining
- Unrooted Neighbor-Joining Tree
- Multiple Alignment- First pair
- ClustalW- Decision time
- ClustalW- Alternative 1
- ClustalW- Progression
- Progressive alignment - step 1
- Progressive alignment - step 2
- Progressive alignment - step 3
- Progressive alignment - final step
- ClustalW-Good pointsBad points
- ClustalW-Local Minimum
- Increasing the sophistication of the alignment process
- Slide 51
- ClustalW- Caveats
- ClustalW- User-supplied values
- Position-Specific gap penalties
- Discouraging too many gaps
- Divergent Sequences
- Advice on progressive alignment
- Alignment of protein-coding DNA sequences
- Manual Alignment- software
-

Divergent Sequencesbull The most divergent sequences (most different on average
from all of the other sequences) are usually the most difficult to align
bull It is sometimes better to delay their aligment until later (when the easier sequences have already been aligned)
bull The user has the choice of setting a cutoff (default is 40 identity)
bull This will delay the alignment until the others have been aligned
Advice on progressive alignmentbull Progressive alignment is a mathematical process that is completely
independent of biological realitybull Can be a very good estimatebull Can be an impossibly poor estimatebull Requires user input and skillbull Treat cautiouslybull Can be improved by eye (usually)bull Often helps to have colour-codingbull Depending on the use the user should be able to make a judgement
on those regions that are reliable or notbull For phylogeny reconstruction only use those positions whose
hypothesis of positional homology is unimpeachable
Alignment of protein-coding DNA sequences
bull It is not very sensible to align the DNA sequences of protein-coding genes
ATGCTGTTAGGGATGACTCTGTTAGGG
ATG-CT--GTTAGGGATGACTCTGTTAGGG
The result might be highly-implausible and might not reflect what is known about biological processesIt is much more sensible to translate the sequences to their corresponding amino acid sequences align these protein sequences and then put the gaps in the DNA sequences according to where they are found in the amino acid alignment
Manual Alignment- softwareGDE- The Genetic Data Environment (UNIX)CINEMA- Java applet available from
ndash httpwwwbiochemuclacuk
SeqappSeqpup- MacPCUNIX available fromndash httpiubiobioindianaedu
SeAl for Macintosh available fromndash httpevolvezoooxacukSe-AlSe-Alhtml
BioEdit for PC available fromndash httpwwwmbioncsueduRNasePinfoprogramsBIOEDITbio
edithtml
- Multiple Sequence Alignment James McInerney bioinf4biologists Feb 2009
- Alignment can be easy or difficult
- Homology Definition
- Multiple Sequence Alignment- Goals
- Multiple sequence alignments - problems
- Slide 6
- Slide 7
- SSU rRNA
- Alignment of 16S rRNA can be guided by secondary structure
- Protein Alignment may be guided by Tertiary Structure Interactions
- Multiple Sequence Alignment- Methods
- Manual Alignment - reasons
- Local minimum
- Dotplots
- Dotplot example sperm whale vs human myg
- Slide 16
- Slide 17
- Dotplots in practice
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Dynamic programming
- Progressive Alignment
- Slide 23
- ClustalW- Pairwise Alignments
- Path Graph for aligning two sequences
- Possible alignment
- Alignment using this path
- Optimal Alignment 1
- Optimal Alignment 2
- Alignment of 3 sequences
- ClustalW- Guide Tree
- Neighbor joining method
- Distance Matrix
- First Step
- Calculation of New Distances
- Next Cycle
- Penultimate Cycle
- Last Joining
- Unrooted Neighbor-Joining Tree
- Multiple Alignment- First pair
- ClustalW- Decision time
- ClustalW- Alternative 1
- ClustalW- Progression
- Progressive alignment - step 1
- Progressive alignment - step 2
- Progressive alignment - step 3
- Progressive alignment - final step
- ClustalW-Good pointsBad points
- ClustalW-Local Minimum
- Increasing the sophistication of the alignment process
- Slide 51
- ClustalW- Caveats
- ClustalW- User-supplied values
- Position-Specific gap penalties
- Discouraging too many gaps
- Divergent Sequences
- Advice on progressive alignment
- Alignment of protein-coding DNA sequences
- Manual Alignment- software
-

Advice on progressive alignmentbull Progressive alignment is a mathematical process that is completely
independent of biological realitybull Can be a very good estimatebull Can be an impossibly poor estimatebull Requires user input and skillbull Treat cautiouslybull Can be improved by eye (usually)bull Often helps to have colour-codingbull Depending on the use the user should be able to make a judgement
on those regions that are reliable or notbull For phylogeny reconstruction only use those positions whose
hypothesis of positional homology is unimpeachable
Alignment of protein-coding DNA sequences
bull It is not very sensible to align the DNA sequences of protein-coding genes
ATGCTGTTAGGGATGACTCTGTTAGGG
ATG-CT--GTTAGGGATGACTCTGTTAGGG
The result might be highly-implausible and might not reflect what is known about biological processesIt is much more sensible to translate the sequences to their corresponding amino acid sequences align these protein sequences and then put the gaps in the DNA sequences according to where they are found in the amino acid alignment
Manual Alignment- softwareGDE- The Genetic Data Environment (UNIX)CINEMA- Java applet available from
ndash httpwwwbiochemuclacuk
SeqappSeqpup- MacPCUNIX available fromndash httpiubiobioindianaedu
SeAl for Macintosh available fromndash httpevolvezoooxacukSe-AlSe-Alhtml
BioEdit for PC available fromndash httpwwwmbioncsueduRNasePinfoprogramsBIOEDITbio
edithtml
- Multiple Sequence Alignment James McInerney bioinf4biologists Feb 2009
- Alignment can be easy or difficult
- Homology Definition
- Multiple Sequence Alignment- Goals
- Multiple sequence alignments - problems
- Slide 6
- Slide 7
- SSU rRNA
- Alignment of 16S rRNA can be guided by secondary structure
- Protein Alignment may be guided by Tertiary Structure Interactions
- Multiple Sequence Alignment- Methods
- Manual Alignment - reasons
- Local minimum
- Dotplots
- Dotplot example sperm whale vs human myg
- Slide 16
- Slide 17
- Dotplots in practice
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Dynamic programming
- Progressive Alignment
- Slide 23
- ClustalW- Pairwise Alignments
- Path Graph for aligning two sequences
- Possible alignment
- Alignment using this path
- Optimal Alignment 1
- Optimal Alignment 2
- Alignment of 3 sequences
- ClustalW- Guide Tree
- Neighbor joining method
- Distance Matrix
- First Step
- Calculation of New Distances
- Next Cycle
- Penultimate Cycle
- Last Joining
- Unrooted Neighbor-Joining Tree
- Multiple Alignment- First pair
- ClustalW- Decision time
- ClustalW- Alternative 1
- ClustalW- Progression
- Progressive alignment - step 1
- Progressive alignment - step 2
- Progressive alignment - step 3
- Progressive alignment - final step
- ClustalW-Good pointsBad points
- ClustalW-Local Minimum
- Increasing the sophistication of the alignment process
- Slide 51
- ClustalW- Caveats
- ClustalW- User-supplied values
- Position-Specific gap penalties
- Discouraging too many gaps
- Divergent Sequences
- Advice on progressive alignment
- Alignment of protein-coding DNA sequences
- Manual Alignment- software
-

Alignment of protein-coding DNA sequences
bull It is not very sensible to align the DNA sequences of protein-coding genes
ATGCTGTTAGGGATGACTCTGTTAGGG
ATG-CT--GTTAGGGATGACTCTGTTAGGG
The result might be highly-implausible and might not reflect what is known about biological processesIt is much more sensible to translate the sequences to their corresponding amino acid sequences align these protein sequences and then put the gaps in the DNA sequences according to where they are found in the amino acid alignment
Manual Alignment- softwareGDE- The Genetic Data Environment (UNIX)CINEMA- Java applet available from
ndash httpwwwbiochemuclacuk
SeqappSeqpup- MacPCUNIX available fromndash httpiubiobioindianaedu
SeAl for Macintosh available fromndash httpevolvezoooxacukSe-AlSe-Alhtml
BioEdit for PC available fromndash httpwwwmbioncsueduRNasePinfoprogramsBIOEDITbio
edithtml
- Multiple Sequence Alignment James McInerney bioinf4biologists Feb 2009
- Alignment can be easy or difficult
- Homology Definition
- Multiple Sequence Alignment- Goals
- Multiple sequence alignments - problems
- Slide 6
- Slide 7
- SSU rRNA
- Alignment of 16S rRNA can be guided by secondary structure
- Protein Alignment may be guided by Tertiary Structure Interactions
- Multiple Sequence Alignment- Methods
- Manual Alignment - reasons
- Local minimum
- Dotplots
- Dotplot example sperm whale vs human myg
- Slide 16
- Slide 17
- Dotplots in practice
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Dynamic programming
- Progressive Alignment
- Slide 23
- ClustalW- Pairwise Alignments
- Path Graph for aligning two sequences
- Possible alignment
- Alignment using this path
- Optimal Alignment 1
- Optimal Alignment 2
- Alignment of 3 sequences
- ClustalW- Guide Tree
- Neighbor joining method
- Distance Matrix
- First Step
- Calculation of New Distances
- Next Cycle
- Penultimate Cycle
- Last Joining
- Unrooted Neighbor-Joining Tree
- Multiple Alignment- First pair
- ClustalW- Decision time
- ClustalW- Alternative 1
- ClustalW- Progression
- Progressive alignment - step 1
- Progressive alignment - step 2
- Progressive alignment - step 3
- Progressive alignment - final step
- ClustalW-Good pointsBad points
- ClustalW-Local Minimum
- Increasing the sophistication of the alignment process
- Slide 51
- ClustalW- Caveats
- ClustalW- User-supplied values
- Position-Specific gap penalties
- Discouraging too many gaps
- Divergent Sequences
- Advice on progressive alignment
- Alignment of protein-coding DNA sequences
- Manual Alignment- software
-

Manual Alignment- softwareGDE- The Genetic Data Environment (UNIX)CINEMA- Java applet available from
ndash httpwwwbiochemuclacuk
SeqappSeqpup- MacPCUNIX available fromndash httpiubiobioindianaedu
SeAl for Macintosh available fromndash httpevolvezoooxacukSe-AlSe-Alhtml
BioEdit for PC available fromndash httpwwwmbioncsueduRNasePinfoprogramsBIOEDITbio
edithtml
- Multiple Sequence Alignment James McInerney bioinf4biologists Feb 2009
- Alignment can be easy or difficult
- Homology Definition
- Multiple Sequence Alignment- Goals
- Multiple sequence alignments - problems
- Slide 6
- Slide 7
- SSU rRNA
- Alignment of 16S rRNA can be guided by secondary structure
- Protein Alignment may be guided by Tertiary Structure Interactions
- Multiple Sequence Alignment- Methods
- Manual Alignment - reasons
- Local minimum
- Dotplots
- Dotplot example sperm whale vs human myg
- Slide 16
- Slide 17
- Dotplots in practice
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Example dotplot - repeated domains in Drosophila melanogaster SLIT protein
- Dynamic programming
- Progressive Alignment
- Slide 23
- ClustalW- Pairwise Alignments
- Path Graph for aligning two sequences
- Possible alignment
- Alignment using this path
- Optimal Alignment 1
- Optimal Alignment 2
- Alignment of 3 sequences
- ClustalW- Guide Tree
- Neighbor joining method
- Distance Matrix
- First Step
- Calculation of New Distances
- Next Cycle
- Penultimate Cycle
- Last Joining
- Unrooted Neighbor-Joining Tree
- Multiple Alignment- First pair
- ClustalW- Decision time
- ClustalW- Alternative 1
- ClustalW- Progression
- Progressive alignment - step 1
- Progressive alignment - step 2
- Progressive alignment - step 3
- Progressive alignment - final step
- ClustalW-Good pointsBad points
- ClustalW-Local Minimum
- Increasing the sophistication of the alignment process
- Slide 51
- ClustalW- Caveats
- ClustalW- User-supplied values
- Position-Specific gap penalties
- Discouraging too many gaps
- Divergent Sequences
- Advice on progressive alignment
- Alignment of protein-coding DNA sequences
- Manual Alignment- software
-