an agency of the european union presented by: melanie leivers head of veterinary regulatory and...
TRANSCRIPT

An agency of the European Union
Presented by: Melanie LeiversHead of Veterinary Regulatory and Organisational Support, European Medicines Agency
TAIEX Workshop on EU legislation for
Veterinary Medicinal Products – Istanbul 18 - 19 April 2011
Marketing Authorisation Procedures for Veterinary Medicinal Products
in the European Union

2
Aim of this presentation is:
• To describe the different marketing authorisation procedures in the EU for veterinary medicinal products
• To show their differences and similarities
• To describe when a route can be used by an applicant
Marketing Authorisation Procedures for Veterinary Medicinal
Products in the European Union

3
Why is a marketing authorisation necessary?
• Any veterinary medicinal product placed on the market in a Member State of the
European Union must have a marketing authorisation (sometimes called in a
Member State a 'Product Licence') and a marketing authorisation holder who is
responsible for the product
• Reference is Directive 2001/82/EC of the European Parliament and of the Council on
the Community code relating to veterinary medicinal products
(http://ec.europa.eu/health/files/eudralex/vol-5/consol_2004/dir_2001_02-dir_2004_28-cons_en.pdf
)
Marketing Authorisation Procedures for Veterinary Medicinal
Products in the European Union

4
What products need to be authorised before they are placed on
the market?
The scope of the Directive states:
“The provisions of this Directive shall apply to veterinary medicinal
products intended to be placed on the market inter alia in the form
of medicinal products, ready-made veterinary medicinal products or
pre-mixes for medicated feedingstuffs”
Marketing Authorisation Procedures for Veterinary Medicinal
Products in the European Union

5
All authorisation procedures have the same basic requirements:
• Quality, Safety and Efficacy must be demonstrated;
• Same technical requirements for all procedures as defined in Annex I to Directive 2001/82/EC;
• Directly related to the technical requirements are European Guidelines – applicable for all European procedures
Authorisation procedures

6
In addition, the European Commission publishes guidance in consultation with the
competent authorities of the Member States and the European Medicines Agency
on the regulatory principles for marketing authorisations in the form of a
“Notice to Applicants”– NTA
http://ec.europa.eu/health/documents/eudralex/vol-6/index_en.htm
Volume 6 gives the regulatory guidelines related to procedural and regulatory requirements
such as for example:
How to present the application dossier;
Renewal procedures;
Dossier requirements for Variations;
Requirements for the summary of product characteristics;
Labelling and package leaflet requirements
Authorisation procedures

7
How are products authorised?
There are 4 possible routes to authorisation
• Centralised procedure
• Decentralised procedure
• Mutual Recognition procedure
• National procedure
Authorisation Procedures

8
Centralised Procedure
• Regulation (EC) No. 726/2004 of the European Parliament and of the Council http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2004:136:0001:0033:en:PDF
• Only for products not already authorised in the EU
• Compulsory for products based on biotechnology, genetically modified organisms (GMO) and growth promoters (Annex to the Regulation)
• Optional for other products ……………> > > >

9
Centralised Procedure
> > > ……“Any medicinal product not appearing in the Annex may be granted a
marketing authorisation by the Community in accordance with the provisions
of this
Regulation, if:
(a) the medicinal product contains a new active substance which, on the date of
entry into force of this Regulation, was not authorised in the Community; or
(b) the applicant shows that the medicinal product constitutes a significant
therapeutic, scientific or technical innovation or that the granting of
authorisation in accordance with this Regulation is in the interests of patients
or animal health at Community level.
Immunological veterinary medicinal products for the treatment of animal diseasesthat are subject to Community prophylactic measures may also be granted suchauthorisation”

10
Centralised Procedure
• Assessment of the application is coordinated by the European Medicines Agency through the Committee for Medicinal Products for Veterinary Use (CVMP) using the expertise of the Committee members
• One CVMP member is appointed as a “Rapporteur” for the assessment of the product application dossier and a second CVMP member as “Co-Rapporteur”
• Assessment reports of the Rapporteur and Co-Rapporteur are available around Day 85 and are sent to the Applicant for information
• A list of questions is adopted by the CVMP around Day 120, the clock is stopped and questions sent to the Applicant who is then given 6 months to respond

11
Centralised Procedure
• The clock is re-started and the responses of the Applicant are assessed - the Applicant is given the right to present their view to the CVMP (oral explanation)
• CVMP decides to give a positive or negative opinion at Day 210
• Applicant can request a re-examination of the opinion if they disagree with it
• The resulting Authorisation is issued by European Commission in the form of a Commission Decision which is valid throughout the EU (Iceland, Liechtenstein and Norway issue their own authorisations based on the Commission Decision)
• European Public Assessment Report (EPAR) published on EMEA website
http://www.ema.europa.eu/ema/index.jsp?curl=/pages/medicines/landing/vet_epar_search.jsp&murl=menus/medicines/medicines.jsp&mid=WC0b01ac058001fa1c

12
Decentralised Procedure
• For products not already authorised in the EU
• Coordinated through the Coordination Group for Mutual Recognition and Decentralised Procedures – Veterinary (CMDv)
• One Reference Member State assesses the application dossier and circulates an assessment report
• Between 1 and 26 Concerned Member States plus EEA countries (Norway, Iceland and Lichtenstein)
13
Decentralised Procedure
Scope:
• 210 days (120 days for the Reference Member State to prepare an assessment report and a 90 day procedure for Member States to agree)
• A 60 day referral period to CMDv if Member States cannot agree
• CMDv Best Practice Guide on the Decentralised Procedure:
http://www.hma.eu/uploads/media/CMDv_BPG-002-04_DC_EMA-CMDv-63793-2006_Final.pdf
• Authorisation is granted nationally in the Member States who were part of the procedure – if no agreement after CMDv referral, then referral to CVMP for final opinion (known as an Article 33.4 referral)

14
Mutual Recognition Procedure
• Based on 'mutual recognition' of an authorised product
• National authority of original Member State where the product received a marketing authorisation acts as the 'Reference Member State' for the procedure - Preparation/Updating of the original assessment report within 90 days
• Between 1 and 26 'Concerned Member States' plus EEA countries (Norway, Iceland and Lichtenstein)
• Coordinated through the Coordination Group for Mutual Recognition and Decentralised Procedures – Veterinary (CMDv)

15
Mutual Recognition Procedure
• Mutual Recognition Procedure of 90 days with 60 more days for referral at CMDv if no agreement
• CMDv Best Practice Guide on the Mutual Recognition Procedure
http://www.hma.eu/uploads/media/BPG-001-03_FINAL_-_MRP_EMEA-CMDv-83618-2006.pdf
• Authorisation is granted nationally in the Member States who were part of the procedure If no agreement after CMDv referral, then referral to CVMP for final opinion (known as an Article 33.4 referral)

16
National Marketing Authorisation
• Issued by one national competent authority only
• Permits marketing in one Member State only
• Assessment procedure lasts a maximum of 210 days
• May form the basis for a mutual recognition procedure in the future
17
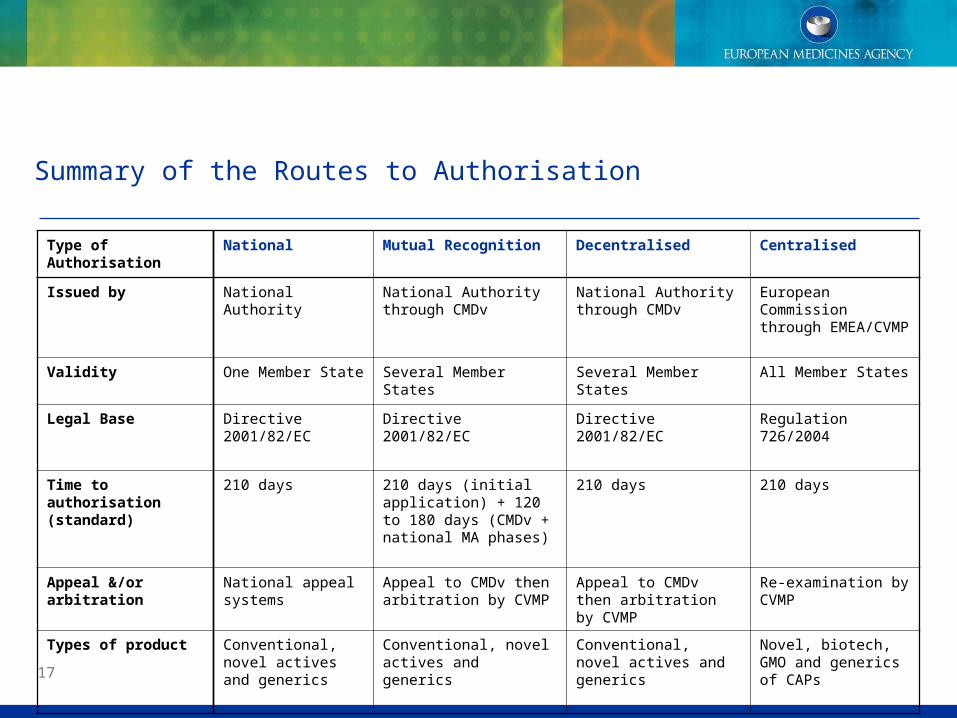
Summary of the Routes to Authorisation
Type of Authorisation
National Mutual Recognition Decentralised Centralised
Issued by National Authority National Authority through CMDv
National Authority through CMDv
European Commission through EMEA/CVMP
Validity One Member State Several Member States Several Member States
All Member States
Legal Base Directive 2001/82/EC
Directive 2001/82/EC Directive 2001/82/EC Regulation 726/2004
Time to authorisation (standard)
210 days 210 days (initial application) + 120 to 180 days (CMDv + national MA phases)
210 days 210 days
Appeal &/or arbitration
National appeal systems
Appeal to CMDv then arbitration by CVMP
Appeal to CMDv then arbitration by CVMP
Re-examination by CVMP
Types of product Conventional, novel actives and generics
Conventional, novel actives and generics
Conventional, novel actives and generics
Novel, biotech, GMO and generics of CAPs

18
Is that all?
No!
Once a marketing authorisation is issued (whichever route has been
used), the Marketing Authorisation Holder must keep the dossier
updated and correct:
After a marketing authorisation has been issued, the holder must take into
account scientific and technical progress and introduce any changes that may be
required to enable that veterinary medicinal product to be manufactured and
checked by means of generally accepted scientific methods (e.g. to submit to the
competent authority applications for changes to the authorisation via variation
applications)
Marketing Authorisation Procedures for Veterinary Medicinal
Products in the European Union

19
Is that all? No! A marketing authorisation is valid for 5 years and the Marketing
Authorisation Holder must also apply for the renewal of the authorisation
- a second renewal can only be requested by the competent authority on
pharmacovigilance grounds
In addition the Marketing Authorisation Holder must comply with the
requirements of Pharmacovigilance – about which more tomorrow!
Marketing Authorisation Procedures for Veterinary Medicinal
Products in the European Union

20
Thank you for your attention!

An agency of the European Union
Presented by: Melanie LeiversHead of Veterinary Regulatory and Organisational Support
TAIEX Workshop on EU legislation for
Veterinary Medicinal Products – Istanbul 18 - 19 April 2011
The Role and Tasks of the European Medicines Agency

22
The Role and tasks of the European Medicines Agency
• The Agency is headed by an Executive Director and has a secretariat of approximately 600 full-time staff (plus contractors e.g. in IT).
• The Management Board is the supervisory body of the Agency
• The Agency is responsible for the scientific evaluation of applications for both human and veterinary medicines submitted via the centralised procedure
• The Agency provides the secretariat for the CVMP and CMDv

23
The Role and tasks of the European Medicines Agency
• The Agency is involved in referral procedures relating to medicines that are approved or under consideration by Member States in non-centralised authorisation procedures
• The Agency constantly monitors the safety of medicines through a pharmacovigilance network, and takes appropriate actions if adverse drug reaction reports suggest that the benefit-risk balance of a medicine has changed since it was authorised
• For veterinary medicines, the Agency has the responsibility to establish safe limits for medicinal residues in food of animal origin (MRLs)

24
The Role and tasks of the European Medicines Agency
• A dedicated SME Office, established in 2005, provides special assistance to small and medium-sized enterprises
• Six scientific committees, composed of members of all EU and EEA-EFTA states, some including patients’ and doctors’ representatives, conduct the main scientific work of the Agency: the Committee for Medicinal Products for Human Use (CHMP), the Committee for Medicinal Products for Veterinary Use (CVMP), the Committee for Orphan Medicinal Products (COMP), the Committee on Herbal Medicinal Products (HMPC), the Paediatric Committee (PDCO) and the Committee for Advanced Therapies (CAT)

25
The Role and tasks of the European Medicines Agency
• The Agency works within a network of over 4,500 European experts (members of the Agency's scientific committees, working parties and product scientific assessment teams). These experts come from the national competent authorities of the EU and EFTA states
• We work closely with other partner organisations, including the World Health Organisation and the regulatory authorities of non-European nations e.g FDA
• The Agency is continually involved in a wide range of cooperation activities with its international partners, to help the timely exchange of regulatory and scientific expertise and development of best practices in the regulatory field

26
Summary of CVMP activities
• Establishment of Maximum Residue Limits (MRLs)• Scientific advice and other assistance to companies for the
development of new medicines• Centralised procedure applications• Referrals• Development of guidelines on quality safety and efficacy• Production of scientific strategy documents• Comment on legislative proposals• Post-authorisation activities (maintenance activities of centralised
authorisations – Pharmacovigilance -Sampling and testing• Co-ordination of Inspection of manufacturers (MSs)

27
CVMP
Secretarial support from EMEA
Efficacy WPSecretary: Barbara Cyrus CVMP
Secretary: B. Mustafov
Scientific AdviceWorking Party
Secretary: Karen Quigley
Safety WPSecretary: Nicholas Jarrett
Joint CHMP/CVMP Quality WP
Vet Secretary: Teresa Potter
Environmental Risk Assessment WP
Secretary: Jordi Torren
Immunologicals WP
Secretary: Nikolaus Križ
Pharmacovigilance Working Party
Secretary: Raquel Gopal Antimicrobial Scientific Advisory
GroupSecretary: Jordi Torren
CMDvSecretary: Emily Drury

28
European Medicines Network

29
The Role and tasks of the European Medicines Agency
Elements of the Medicines regulatory network• National Competent Authorities (around 42) coordinated by Heads of
Medicines Agencies (HMA)– Human/Joint Agencies/Authorities– Veterinary Agencies/Authorities– Inspection Agencies/Authorities– 42 agencies, including EEA
• European Medicines Agency• European Commission• European Directorate for the Quality of Medicines & Healthcare (EDQM)• Network of Official Medicines Control Laboratories• MRA partners

30
The European Medicines Regulatory Networking Model
Lessons learnt during the building of the European Regulatory Network:
• Sharing of resources can be effective and efficient
• Mutual trust and transparency are essential and increase with experience
• Gains to industry– Time– Resource– Equal treatment– Predictability

31
The European Medicines Regulatory Networking Model
Prospects for the future
• Increase in work-sharing?
• Emphasis on training and cooperation
• European Commission is currently reviewing the legislation to assess the need for
– simplification
– ensuring a proportionate regulatory burden
– adapting to the needs of the veterinary sector

32
Agency Roadmap
Roadmap to 2010 largely complete:
e.g.
• Delivery of activities in the area of availability– Input into MRL regulation– MUMS initiative
• Implementation of the CVMP Strategy on Antimicrobial Resistance

33
The Agency’s Road Map to 2015
• Continuation of work started in previous Roadmap
• Provides the Agency’s vision on how it should further develop as a public
and animal health Agency
• Encompasses the Agency’s longer term strategy for both human and
veterinary medicines
• Consistent with, and complementary to, strategies of the European
Commission and HMA
• Recognises the important contribution of the National Competent Authorities

34
The Agency’s Road Map to 2015
• New and emerging science
− Novel therapies and approaches in human and veterinary medicine
• Impact of increasing globalisation
− Global nature of medicines development and research (movement of clinical research and manufacturing to lower cost countries)
− Specific veterinary issues (harmonisation of regulatory requirements through VICH, increased activity by OIE, increased cooperation with Codex in setting acceptable residue limits for veterinary medicines in animal foodstuffs)

35
The Agency’s Road Map to 2015
• Ensuring safety (animal, user, environment)
− Veterinary pharmacovigilance legislation ‘lagging behind’ human sector in terms of legislative review
− Need to develop a framework and tools appropriate for the needs of the veterinary sector
• Demands for more transparency and openness
− Aim is to strengthen trust by stakeholders
− Focus has to be both on the tools applied and the content of the information -balance between earlier availability of information vs.the protection of commercially confidential information

36
The Agency’s Road Map to 2015
• Efficient operation of the Agency’s core business
− Roles and responsibilities have expanded
− Tasks have become more complex
− Drive for efficiency
• First priority for the next five years
FOCUS on QUALITY of output

37
The Agency’s Road Map to 2015
3 strategic areas identified:
• Addressing public and animal health needs
• Facilitating access to medicines
• Optimising the safe and rational use of medicines

38
The Agency’s Road Map to 2015
Addressing public and animal health needs:
• Focus will be on engagement with Community Animal Health Strategy
and the European Technology Platform for Global Animal Health -
DISCONTOOLS
• New and emerging science to what extent is new legislation for this area needed in the veterinary sector?
• Public health threats e.g. Antimicrobial resistance

39
The Agency’s Road Map to 2015
Medicines regulation − A complex concept !
− Encompasses various elements which are undergoing a review (requirements for medicine development, benefit/risk balance, point of decision-making, post-authorisation follow-up)
− To what extent should legislation for veterinary medicines be tailored to the specific requirements of the veterinary sector?

40
The Agency’s Road Map to 2015
Facilitating access to medicines:
• Medicine development process, early assessment and continuing
dialogue in Guidelines, Scientific advice and the Assessment process
• Benefit Risk (B/R) assessment process - reinforce the B/R
methodology in the assessment procedure and better communicate
to stakeholders for veterinary medicines

41
The Agency’s Road Map to 2015
Optimising the safe and rational use of medicines:
• Responding to proposals for pharmacovigilance legislation within the
proposed European Commission Impact Assessment
• Post-authorisation follow-up – opportunity to development of an appropriate
risk management framework for veterinary medicines
-------------------------------------------------------------------------
“From Vision to Reality” - implementing measures – coming soon!
----------------------------------------------------------------------------------

42
Thank you for your attention!