and levels of soluble - ulb bonnhss.ulb.uni-bonn.de/2009/1697/1697.pdf · role of pathogenic...
TRANSCRIPT

i
Role of Pathogenic Mediators in murine Arthritis and Levels of Serum Soluble CD21 and CD23 in autoimmune patients Dissertation
Zur Erlangung des Doktorgrades (Dr.rer.nat.)
der
Mathematisch‐Naturwissenschaftliche Fakultät
der
Rheinischen Friedrich‐Wilhelms‐Universität Bonn
Vorgelegt von
ANJANA SINGH
aus
(Delhi, INDIA)
Bonn (2009)
Rheinische Friedrich‐Wilhelms‐Universität Bonn
Mathematisch‐Naturwissenschaftliche Fakultät Wegelerstr. 10 53115 Bonn

ii
GUTACHTER‐I
Prof. Dr. Harald Illges
Professor of Immunology and Cell Biology
University of Applied Sciences
Bonn‐Rhein‐Sieg
Von‐Liebig Str. 20
53359 Rheinbach
Germany
GUTACHTER‐II
Prof. Dr. Waldemar Kolanus
Professor of Cellular Biochemistry
Institute of Molecular Physiology
and Developmental Biology
Division of Cellular Biochemistry
University of Bonn
Karlrobert‐Kreiten 13, D‐53115 Bonn
Germany
Date of Examination: 16th March 2009

iii
Dedicated to My Parents

iv
ACKNOWLEDGEMENT
This thesis work was carried out at Rheumatology unit, Department of Immunology and Cell Biology,
Rheinbach. I would like to express my gratitude to all colleagues and friends who have helped me
during the PhD studies.
My first, and most earnest, acknowledgement must go to my advisor Prof. Harald Illges. Nearly three
years ago, a telephone conversation with Prof. Illges started me on the path I traveled at Germany.
Prof. Illges has been instrumental in ensuring my academic, professional, financial, and moral well
being ever since. His support and guidance made my thesis work possible. He has been actively
interested in my work and has always been available to advise me. I am very grateful for his
patience, motivation, enthusiasm, and immense knowledge in B cell biology and Rheumatoid
Arthritis that, taken together, make him a great mentor. In every sense, none of this work would
have been possible without him.
Prof. Waldemar Kulanus for accepting to be my examiner.
I want to thank present and past members of the lab: Dr. Narendrian Rajashekaran, Hui Zhi Low,
Stephan Gütgemann, Danile Ferrando, sebastian Howe and Mahwish Iftikhar. Dr. Narendrian
Rajashekara gave me an introduction into the lab and for sharing his knowledge. I also thank
Sebastian Howe for helping me write the German version of thesis summary.
Far too many people to mention individually have assisted in so many ways during my work at
Germany. They all have my sincere gratitude. In particular, I would like to thank Mrs. Robert Karin
and Olaf Stock for timely help, Prof. Rolf Baüer for all the histology in this study, Prof. Miri Blank for
APS sera samples, Prof. Richard Buccala for MIF‐/‐ mice, Prof. Peter Angel for MMP 13‐/‐ mice, Dr.
Bettina Hartenstein‐Füssel and Mrs. Sibylle Teurich for the reagents and for help with the Semi‐
quantitative PCR experiments and also his valuable discussion for MMP13 paper, Prof. Zimmer for
SP‐/‐ and several anonymous reviewers for their help and comments that improved various published
papers.
I am also indebted to the Department of Immunology and Biological sciences at Rheinbach, for direct
financial aid through fellowships, awards, and travel grants.
For the non‐scientific side of my thesis, I particularly want to thank Mr. and Mrs. Baüer (House
owner) for sharing their apartment with me for 3 years for undertaking neat and adventurous
excursions in Rheinbach, and for being my eternal sunshine. Their support and care means a lot to
me.
A penultimate thank‐you goes to my wonderful parents, Anal Dharamshi, Jalpa Vasani and Thaniga
Subramaniam. For always being there when I needed them most, and never once complaining about
how infrequently I visit, they deserve far more credit than I can ever give them.

v
PUBLICATIONS
(1) Anjana Singh, Miri Blank, Yehuda Shoenfeld, Harald Illges
Antiphospholipid Syndrome patients display reduced titers of soluble CD21 in their plasma
irrespective of the presence of immune complex generating autoantibodies. Rheumatol Int 2008 Jan
3
(2) Madhan Masilamani, Narendiran Rajasekaran , Anjana Singh, Hui Zhi Low, Kerstin Albu, Swentje
Anders, Frank Behne, Peter Eiermann, Kathraina Konig, Clarissa Mindnich, Terdora Ribaraka, Harald
Illges.
Systemic reduction of soluble complement receptor II/CD21 during pregnancy reminiscent of
autoimmune disease. Rheumatol Int 2008 May 24
Manuscripts in preparation
(1) Anjana Singh, Bas Vestert, Harald Illges
Decreased levels of sCD21 and sCD23 in blood of patients with Systemic‐JA, Polyarticular‐JA,
Pauciarticular‐JA
(2) Anjana Singh, Narendiran Rajasekaran, Sibylle Teurich, Bettina Hartenstein‐Füssel, Gaida, Peter
Angel, Rolf Brauer and Harald Illges
Increased expression of collagenase‐3 (MMP‐13) deficiency protects from antibody‐induced arthritis
(3) Anjana Singh, Gaida , Rolf Brauer, Günter Fingerle‐Rowson, Richard Bucala, and Harald Illges
Inhibition of joint inflammation and destruction induced by K/BxN serum‐induced arthritis in mice
due to depletion of macrophage secreted macrophage inhibitory factor (MIF)
(4) Anjana Singh, A.Pfutzner, Harald Illges.
Decreased levels of sCD21 and sCD23 in patients with Insulin dependent Diabetes Mellitus
ABSTRACTS AND CONFERENCES
(1) Poster presentation in 16th European Congress of Immunology, Paris, 6th Sep‐9th Sep 2006.
(2) Oral and poster presentation in 13th International Congress of Immunology, Rio de Janeiro, Brazil,
21st Aug‐25th Aug 2007
(3) Poster Presentation 37th Annual meeting of German Society, Heidelberg, 5th Sep‐8th Sep 2007

vi
ABBREVATIONS
6‐OHDA 6‐hydroxydopamine Ag Antigen Ab Antibody APC Antigen presenting cell APS ALM 633 ALPS APS
Ammoniumpersulfate Alexa Fluor 633 C5 maleimide Autoimmune lymphoproliferative syndrome Antiphospholipid Syndrome
BCR B cell Receptor BSA CIA
Bovine Serum Albumin Collagen Induced Arthritis
CR Carboxy‐H2DCFDA CD CL‐3
Complement Receptor 5‐(and‐6)‐carboxy‐2´,7´‐dichlorodihydrofluorescein acetate Cluster of Differentiation collagenase‐3
DAF Decay Acceletrating Factor DC DMSO
Dendritic Cell Dimethyl Sulfoxide
EDTA Ethylene Diamine Tetraacetic Acid ELISA Enzyme‐linked Immunosorbant Assay FACS FBS
Fluorescence Activated Cell Sorter Fetal Bovine Serum
FDC Follicular Dendritic Cell FGF Fibroblast Growth Factor FITC Fluorescein Isothiocyanate FLS Fibroblast like Synoviocytes FC FCA
Fluorescence Channel Freund’s complete adjuvant
FSC Forward Scatter GC Germinal Center GM‐CSF Granulocyte‐Macrophage Colony Stimulating
Factor GPI Glucose‐6‐Phosphate‐Isomerase HCV Hepatitis‐C Virus HRP Horse Radish Peroxidase i.p. Intraperitoneal IC Immune Complex Ig Immunoglobin IL JA JRA
Interleukin Juvenile Arthritis Juvenile Rheumatoid Arthritis
K Da Kilo Dalton L, ml, µl Liter, millilitre, microlitter M, Mm Molar, Millimolar MCA Membrane Attack Complex MBL Mannose Binding Lectin

vii
MCP MIF MFI
Monocyte Chemoattractant Protein Macrophage inhibitory factor Median Fluorescence Intensity
MHC Major Histocompatibility Complex MMP Matrix Metalloproteinase NOD α7nAChR
Nonobese Diabetic Nicotinic Acetylcholine Receptor‐α7
O.D. PIA
Optical Density Pristane Induced Arthritis
PAGE Polyacryamide Gel Electrophoresis PBL Preipheral Blood Lymphocytes PBS Phosphate‐Buffered Saline PE PerCP P‐JA
Phycoerythrin Peridinin Chlorophyll Protein Pauciarticular Juvenile Arthritis
PVDF Polyvinylidene Fluoride RA Rheumatoid Arthritis RF Rheumatoid Factor ROS Reactive Oxygen Species RPE R‐Phycoerythrin SCR Short Consensus Repeat SDS Sodium Dodecyl Sulphate SLE SH S‐JA
Systemic Lupus Erythematosus Sulfhydryl Systemic Juvenile Arthritis
SP Substance p SS Sjoegren’s Syndrome SSC Side Scatter TEMED N, N,N, N’‐tetramethylethylenediamine TGF Transforming Growth Factor TMAO Trimethylamine N‐Oxide TNF Tumor Necrosis Factor v/v, w/w Volume/volume, weight/weight VCAM Vascular Cell Adhesion Molecule V Volt

viii
CONTENTS
1. CHAPTER I INTRODUCTION
1. Human Immune System……………………………………………………………………….1 1.1 Hematopoiesis……………………………………………………………………………………………………… 2 1.2 Innate immunity vs. adaptive immunity 1.1.1. Innate immunity……………………………………………………………………………………………. 4 1.1.2. Adaptive immunity………………………………………………………………………………………… 4 1.3. Complement pathway…………………………………………………………………………………………. 5 1.3.1. Activation of classical pathway………………………………………………………………………..6 1.3.1.1. C1 activation…………………………………………………………………………………………….. 6 1.3.1.2. C4 and C2 activation (generation of C3 convertase)…………………………………. 6 1.3.1.3. C3 activation (generation of C5 convertase)…………………………………………….. 6 1.3.2. Activation of alternative pathway…………………………………………………………………. 6 1.3.3. Initiation of Lectin pathway…………………………………………………………………………… 7 1.3.4. Membrane attack complex (MAC) formation ……………………………………………….. 7 1.3.5. Regulation of complement activation ………………………………………………………..... 7 1.3.5.1. Complement receptor (CR1 or CD35)………………………………………………………..8 1.3.5.2. Complement receptor 2 (CR2)…………………………………………………………………. 8 1.4. Autoimmunity…………………………………………………………………………………………………….. 9 1.4.1. Dimensions of autoimmune diseases…………………………………………………………… 13 1.5. Rheumatoid arthritis…………………………………………………………………………………………. 14 1.6. Autoimmune diseases 1.6.1. Animal models of rheumatoid arthritis 1.6.1.1. Type II collagen‐induced arthritis…………………………………………………………. 16 1.6.1.2. Adjuvant arthritis…………………………………………………………………………………. 17 1.6.1.3. Pristane‐induced arthritis………………………………………………………………….…. 17 1.6.1.4. Streptococcal cell wall‐induced arthritis………………………………………….…… 17 1.6.1.5. K/BxN murine model of RA……………………………………………………………….…..18 1.7. Juvenile arthritis………………………………………………………………………………………………. 21 1.8. Diabetes…………………………………………………………………………………………………………… 21 1.9. Antiphospholipd syndrome (APS)……………………………………………………………………. 22 1.10. Autoimmune lymphoproliferative syndrome (ALPS)…………………………………….. 23 1.11. Cellular abnormalties 1.11.1. Antigen presenting cells………………………………………………………………………….. 24 1.11.2. Macrophages…………………………………………………………………………………………… 24 1.11.3. Role of cytokines in RA………………………………………………………………………….... 25 1.12. Contributors to the inflammatory process 1.12.1. Neuropeptide connection in arthritis…………………………………………………….… 27 1.12.1.1. Role of Substance P in RA…………………………………………………………………. 28 1.12.2. Role of MMP 13 in RA……………………………………………………………………………... 29 1.12.3. Role of macrophage inhibitory factor (MIF) in RA………………………………….… 30 1.12.4. Role of redox‐reactions in RA………………………………………………………………….. 31

ix
1.13. Role of serum soluble CD21 and CD23 in autoimmune diseases 1.13.1. Soluble CD21…………………………………………………………………………………………. 32 1.13.1. Soluble CD23…………………………………………………………………………………………. 33 2. CHAPTER II AIMS OF THE STUDY…………………………………………………………………….. 34 3. CHAPTER III MATERIALS AND METHODS 3.1.1 Mouse strains
3.1.1.1 KRN transgenic mouse strain…………………………………………………………………….. 35 3.1.1.2. NOD/Lt mouse strain………………………………………………………………………………….35 3.1.1.3. K/BxN mouse strain………………………………………………………………………………….. 35 3.2. Knockout mouse 3.2.1. MMP 13‐/‐ mouse strain……………………………………………………………………………….. 36 3.2.2. Macrophage inhibitory factor‐/‐ mouse strain……………………………………………….. 36 3.2.3. Substance‐P‐/‐ mouse strain………………………………………………………………………..... 36 3.2.4. DBA mt FvB mouse strain……………………………………………………………………………… 36 3.2.5. B6.129 S7‐chrna 7tm1Bay/J mouse strain……………………………………………………….... 36 3.2.6. Balb/c and C57BL/6 mouse strain…………………………………………………………………. 36 3.3. Breeding experiments…………………………………………………………………........................ 36 3.4. Induction arthritis using K/BxN sera transfer 3.4.1. Production of K/BxN sera……………………………………………………………………………… 37 3.4.2. Establishment of antibody‐induced arthritis in mice…………………………………….. 37 3.4.3. Assessment of antibody‐induced arthritis by ankle thickness (mm) and clinical index (U)…………………………………………………………………………………………… 37
3.5. Histology 3.5.1. Decalcification…………………………………………………………………………………………….... 38 3.6. Preparation of adherent peritoneal macrophages and reconstitution of macrophages in MIF‐/‐ mice………………………………………………………………………………. 38 3.7. Immunological methods 3.7.1. Fluorescence activated cell sorter (FACS) Ficoll density gradient preparation of lymphocytes………………………………………. 38 3.7.2. RBC lysis………………………………………………………………………………………………………. 39 3.8. Quantification of sCD21 from human serum/plasma samples by ELISA…………… 39 3.9. Quantification of sCD23 from human serum/plasma samples by ELISA…………… 40 3.10. Immunoloblot 3.10.1. Western blot……………………………………………………………………………………………… 40 3.10.2. Dot blot……………………………………………………………………………………………………... 41 3.11.1. Transient sympathectomy …………………………………………………………………………… 42 3.11.2. Permanent sympathectomy…………………………………………………………………………. 42 3.12. Statistics 3.12.1. P‐value………………………………………………………………………………………………………. 43 3.12.2. Mann‐whitney test & Kruskal‐wallis test……………………………………………………. 43

x
4. CHAPTER IV RESULTS
4.1 Neurological experiments
4.1.1. Transient sympathectomy partially protects Balb/c mice from antibody‐induced arthritis…………………………………………………………………………………………………………… 44 4.1.2. Permanent symathectomy protects Balb/c mice from antibody‐induced arthritis……………………………………………………………………………………………………..…… 47 4.1.3. Nicotinic acetylcholine receptor‐α7 (α7nAChR) homozygous knockout mice were completely resistant to antibody‐induced arthritis…………………………………………….. 48 4.1.4. Capsaicin does not provide permanent sympathectomy in Balb/c mice…………… 50 4.1.5. Substance P knockout mice were partially protected from antibody‐induced arthritis……………………………………………………………………………………………………........... 52 4.2 Role of macrophage inhibitory factor in K/BxN serum‐induce arthritis 4.2.1. MIF‐/‐mice are protected from antibody induced arthritis…………………………………. 53 4.2.2. MIF deficiency reduced joint inflammation and cartilage destruction………………. 55 4.2.3. Reconstitution of MIF‐/‐ mice with naive macrophages restored susceptibility to arthritis………………………………………………………………………………………………………………. 57 4.3 Collagenase‐3 (MMP13) deficiency protects from antibody‐induced arthritis 4.3.1. An Essential role for MMP13 in K/BxN antibody‐induced arthritis…………………….. 59 4.3.2. Joint inflammation and cartilage destruction in MMP13‐/‐ mice………………………… 60 4.3.3. MMP13‐/‐ mice display reduced numbers of infiltrating neutrophil and increased number of monocytes in the joints………………………………………………………………………. 64 4.4. Role of redox reactions in RA antibody‐induced arthritis 4.4.1. In the K/BxN model phytol‐treated mice show an early onset of arthritis…………. 65 4.4.2. Increasing the number of R–SH groups on control neutrophils increased the arthritis………………………………………………………………………………………………………………. 66 4.4.3. No change of surface thiol levels on monocytes………………………………………………… 67 4.4.4. Measurement of intracellular ROS in neutrophils……………………………………………… 68 4.4.5. Measurement of intracellular ROS in monocytes………………………………………………. 69 4.4.6. Number of neutrohils increased after phytol treatment……………………………………. 70 4.4.7. No change in the levels of surface thiol in young and old KBN and BN mice……… 70 4.5. MCP‐1 antagonist treatment does not protect mice from antibody‐induced arthritis…………………………………………………………………………………………………………………… 71 4.6. DBA mt FvB mice are not protected from antibody‐induced arthritis…………………… 72 4.7. Soluble CD21 and CD23 concentration in autoimmune diseases 4.7.1. Antiphospholipid syndrome patients display reduced titers of soluble CD21 in their sera irrespective of circulating anti‐beta‐2‐glycoprotein‐ Autoantibodies…………………………………………………………………………………………………....75 4.7.2. Soluble CD21 levels in Autoimmune Lymphoproliferative Syndrome …………….. 77 4.7.3. Soluble CD21 levels in Vaccinated patients sera samples………………………………… 78 4.7.4. Decreased levels of sCD21 and sCD23 in patients with insulin dependent Diabetes Mellitus

xi
4.7.4.1. Levels of serum soluble CD21 are decreased in sera of patients with diabetes type I but not with diabetes type II…………………………………………….…….….…… 79 4.7.4.2. Levels of Serum soluble CD23 levels in sera of patients with Diabetes type I are decreased but not with diabetes type II…………………………………….…….…. 80 4.7.4.3. Decreased levels of plasma sCD21 after proinsulin treatment in diabetic obese patients……………………………………………………………………………………….… 81 4.7.4.4. Increased levels of plasma sCD23 after proinsulin treatment in diabetic obese patients…………………………………………………………………………………………….………82 4.7.5. Decreased levels of sCD21 and sCD23 in blood of patients with Systemic Juvenile Arthritis, Polyarticular Juvenile Arthritis and Pauciarticular Juvenile Arthritis 4.7.5.1. Low levels of soluble CD21 in plasma of patients with JA………………………... 84 4.7.5.2. Low levels of soluble CD23 in plasma of patients with JA…………………………. 85 4.7.5.3. Plasma sCD23 but not sCD21 levels correlate with age in healthy donors… 86 4.7.5.4. Plasma sCD21and sCD23 levels decline with age in JA……………………………… 87 4.7.5.6. Blood granulocytes were significantly increased in S‐JA patients compared with control and O‐JA and P‐JA patients……………………………………………………. 89 4.7.5.7. JA patients in remission showed increased sCD23 levels…………………………. 90 5. CHAPTER V DISCUSSION
5.1 The sympathetic nervous system and antibody‐induced arthritis……………………… 93 5.2. Macrophage inhibitory factor in antibody‐induced arthritis…………………………….. 96 5.3. MMP 13 (collagenase‐3) deficiency protects from antibody‐induced arthritis…. 97 5.4. Role of redox‐regulations in antibody‐induced arthritis …………………………………… 99 5.5. An antagonist of monocyte chemoattractant protein 1 (MCP‐1) did not inhibit arthritis in the K/BxN model……………………………………………………………………………… 100 5.6. DBA mt FvB mice are not protected from antibody‐induced arthritis………………. 101 5.7. Role of CD21 and CD23 in autoimmune disorders 5.7.1. Antiphospholipid syndrome……………………………………………………………………….103 5.7.2. Soluble CD21 levels in autoimmune lymphoproliferative syndrome………… 104 5.7.3. Diabetes……………………………………………………………………………………………………. 105 5.7.4. Juvenile arthritis………………………………………………………………………………………...106
6. CHAPTER VI SUMMARY………………………………………………………………………… 109
7. Zusammenfassung……………………………………………………………………………….. 113
8. References………………………………………………………………………………………………………….. 117
9. Lebenslauf……………………………………………………………………………………………. 133

- 1 -
CHAPTER‐I
INTRODUCTION
1. Human Immune system
An immune system can be divided into two related activities‐recognition and response. Once a
foreign organism has been recognized, the immune system enlists the participation of a variety of
cells (T cells, B cells, NK cells, helper cells, suppressor cells, macrophages, etc) and molecules to an
appropriate response, to eliminate or neutralize the invading organism.
The state of protection from infectious disease has both specific (adaptive immune system) and non
specific (innate immune system) components. The nonspecific components, innate immunity, are a
set of disease resistance mechanisms that are not specific to the particular pathogen. Phagocytic
cells such as macrophages, play an important role in many aspects of innate immunity. In contrast,
the specific components, adaptive immunity, display a high degree of specificity as well as the
remarkable property of ‘memory’.
There is an adaptive immune response against an antigen within five or six days after the initial
exposure to that antigen. Exposure to the same antigen sometime in the future results in a memory
response, the immune response to the second challenge occurs more quickly than the first, is
stronger, and often is more effective in neutralizing and clearing the pathogen. The major cellular
agents of adaptive immunity are lymphocytes and the antibodies.
Adaptive and innate immunity do not operate independently of each other; they function as a highly
interactive and cooperative system, producing a total response more effective than either could
alone. There is evidence that both the arms of immunity are highly inter‐connected and an efficient
immune response against a particular antigen requires both the arms of immunity.
The organs of the immune system are positioned throughout the body. They are called lymphoid
organ because they are home to lymphocytes, small while blood cells that are the key players in the
immune system. The lymphoid organs are boldly classified as primary (bone marrow and thymus)

- 2 -
and secondary lymphoid organs (spleen, lymph nodes, and lymphatic vessels). Primary lymphoid
organs provide appropriate microenvironments for the development and maturation of lymphocytes
while secondary lymphoid organs trap the antigen from the defined tissues or from vascular spaces
and are site where mature lymphocytes can interact effectively with antigen.
1.1 Hematopoiesis
Hematopoietic stem cells (HSCs) reside in the bone marrow and have the unique ability to give rise
to all of the different mature blood cell types. The stem cells produce hemocytoblasts and
differentiate into the preccursors of all the other types of cells.
The hematopoeitic system can also be divided into lymphoid and myeloid cells. Lymphoid progenitor
cells gives rise to the lymphocytes (B and T cells). B lymphocytes, which when activated differentiate
into plasma cells that secrete antibodies. There are two main classes of T cells Cytotoxic T cells killing
cells infected with viruses and T cells that activate other cells such as B cells and macrophages. While
the myeloid progenitor is the precursor of the granulocytes, macrophages and mast cells.
Hemocytoblasts mature into three types of blood cells: erythrocytes (RBCs), leukocytes (WBCs) and
thrombocytes (platelets). The leukocytes are further subdivided into granulocytes with large
granules in the cytoplasm and agranulocytes without granules. The granulocytes consists of
neutrophils, eosinophils and basophils. The agarulocytes are the lymphocytes (B and T cells) and
monocytes.

- 3 -
Hematopoietic stem cells produce cells in blood and lymph
Adapted from Biology of the Immune System, JAMA 278 (22)

- 4 -
1.2. Innate Immunity vs Adaptive Immunity
The immune system is typically divided into two categories‐innate and adaptive‐although these
distinctions are not mutually exclusive.
1.2.1 Innate immunity
Innate immunity refers to nonspecific defense mechanisms that come into play immediately or
within hours of an antigen's appearance in the body. These mechanisms include physical barriers
such as the skin, chemicals in the blood, and immune system cells that attack foreign cells in the
body. The innate immune response is activated by chemical properties of the antigen.
1.2.2 Adaptive immunity
Adaptive immunity refers to antigen‐specific immune response. The adaptive immune response is
more complex than the innate. The antigen first must be processed and recognized. Once an antigen
has been recognized, the adaptive immune system creates an army of immune cells specifically
designed to attack that antigen. Adaptive immunity also includes a "memory" that makes future
responses against a specific antigen more efficient.
Cells that make up the adaptive (specific) immune system include the B and T lymphocytes. After
exposure to antigen, B cells differentiate into plasma cells whose primary function is the production
of antibodies. Similarly, T cells can differentiate into either T cytotoxic (Tc) or T helper (Th) cells of
which there are two types Th1 and Th2 cells. Specificity on the adaptive immune response resides in
the antigen receptors on T and B cells, the TCR and BCR, respectively.
There are two major types of adaptive immune responses:
1. humoral immunity: humoral immunity involves the production of antibody molecules in
response to an antigen and is mediated by B‐lymphocytes.
2. Cell‐mediated immunity: the second class of adaptive immune response, activated T cells
react directly against a foreign antigen that is presented to them on the surface of a host
cell. The T cell, for example, might kill a virus‐infected host cell that has viral antigens on its

- 5 -
surface, thereby eliminating the infected cell before the virus has had a chance to replicate.
In other cases, the T cell produces signal molecules that activate macrophages to destroy the
invading microbes that they have phagocytosed.
Overview of the Immune System
1.3. Complement System
Complement is involved in the eradication of microorganisms and immune complexes, as well as in
inflammation and immunoregulation. The complement system comprises a series of 20 serum
glycoproteins that are activated in a cascade sequence, with pro enzymes that undergo sequential
proteolytic cleavage.
Two main pathways of complement activation exists, termed the classical pathway, and the
alternative pathway. These converge in the activation of C3, both forming individual C3 convertase.

- 6 -
This leads to the final common pathway with the assembly of C5‐C9 into the membrane attack
complex (MAC) which forms a ‘doughnut like’ transmembrane channel leading to cell lysis by
osmotic shock.
1.3.1. Activation of the Classical Pathway
1.3.1.1. C1 activation
C1, a multi‐subunit protein containing three different proteins (C1q, C1r and C1s), binds to the Fc
region of IgG and IgM antibody molecules that have bound to antigen. C1 binding does not occur to
antibodies that have not bound to antigen and binding requires calcium and magnesium ions. The
binding of C1 to antibodies is via C1q and C1q must cross link at least two antibody molecules before
it is firmly fixed. The binding of C1q results in the activation of C1r which in turns activate C1s. The
result is the formation of an activated ‘C1qrs’, which is an enzyme that cleaves C4 into fragments
C4a and C4b.
1.3.1.2 C4 and C2 activation (generation of C3 convertase)
The C4b fragment binds to the membrane and the C4a fragment is released into the environment.
Activated C1qrs also cleaves C2 into C2a and C2b. C2a binds to the membrane‐ associated C4b, and
C2b is released into the microenvironment. The resulting C4bC2a complex is a C3 convertase, which
cleaves C3 into C3a and C3b.
1.3.1.3. C3 activation (generation of C5 convertase)
C3b binds to the membrane in association with C4b and C2a, and C3a is released into the
microenvironment. The resulting C4bC2aC3b complex is a C5 convertase. The generation of C5
convertase is the end product of the classical pathway.
1.3.2. Activation of alternative pathway

- 7 -
The alternative pathway is initiated by the low‐grade activation of C3 by hydrolysed C3 (C3 (H2O))
and activated factor B (Bb). The activated C3b binds factor B (B), which is then cleaved into Bb by
factor D (D) to form the alternative pathway C3 convertase, C3bBb. Once C3b is attached to the cell
surface, the amplification loop consisting of the alternative‐pathway components is activated, and
the C3‐convertase enzymes cleave many molecules of C3 to C3b, which bind covalently around the
site of complement activation.
1.3.3. Initiation of Lectin pathway
This pathway is similar to the classical pathway but gets activated by mannose binding lectin (MBLs).
The lectin pathway is a subdivision of the classical pathways and it actiavtes classical pathway
without using the C1q complex.
1.3.4 Membrane attack complex formation
C5 convertase from the classical or lectin pathway (C4b2a3b) or alternative pathway (C3bBb3b)
cleaves C5 into C5a and C5b. C5a remains in the fluid phase and the C5b rapidly associates with C6
and C7 and inserts into the membrane. Subsequently C8 binds, followed by several molecules of C9.
The C9 molecules form a pore in the membrane through which the cellular content leaks and lysis
occurs.
Lysis is not an enzymatic process, it acts via physical damage to the membrane. The complex
consisting of C5bC6C7C8C9 is referred to as the membrane attack complex (MAC). C5a generated in
the lectin pathway has several potent biological activities. It is the most potent anaphylotoxin. In
addition, it is a chemotactic factor for neutrophils and stimulates the respiratory burst in them and it
stimulates inflammatory cytokine production by macrophages.
1.3.5. Regulation of complement activation
1.3.5.1 Complement Receptor 1 (CR1 or CD35)

- 8 -
Complement receptor type 1 (CR1, CD35, C3b/C4b receptor) is a 190 kDa transmembrane
glycoprotein expressed on several circulating cells, including erythrocytes, neutrophils,
monocytes/macrophages, B lymphocytes, and some T lymphocytes, as well as specific epithelial
cells. The CR1 is encoded along with CR2 at the single locus Cr2 on chromosome 1 in mice. The CR1
and CR2 consist of multiple repeating structures referred as short consensus repeats (SCR). The SCR
consists of conserved units of 60‐70 amino acids. CR2 consists of 16 SCR, a transmembrane region
and a 35 amino acid cytoplasmic tail, whereas CR1 includes all of CR2 and an additional 6 SCR on its
N‐terminal region. Just like factor H, CR1 acts as a cofactor and promotes cleavage of C3b and iC3by
factor I (Kinoshita et al., 1985; Molina et al., 1994). CR1 has moderate affinity for C3b but low
affinity for iC3b. It does not bind to the further metabolites of iC3b. CR1 also bind to the C4b. It helps
the phagocytes to generate opsonized particles, enhances antigen specific B cells endocytic
responses and clearance of antigen‐antibody complex by erythrocytes.
1.3.5.2 Complement Receptor 2
Human CD21 (complement receptor type 2, CR2) is a 145‐kDa type 1 transmembrane glycoprotein
that binds the surface‐fixed cleavage fragments of C3, iC3b/ C3d/ C3dg and serves as the receptor
for Epstein–Barr virus (EBV) on B lymphocytes (Fingeroth et al., 1984; Weis et al., 1984). CD21 is
comprised of a 954 aa extracellular domain, a 24 aa transmembrane domain and a 34 aa cytoplasmic
domain. The extracellular domain is composed of 15 or 16 tandemly repeated short consensus
repeats (SCR) sequences which are homologous to those described in other C3/C4‐binding proteins
(Ahearn and Fearon, 1989).
CD21 has initially been described as the receptor for the C3dg fragment of C3. The binding site is less
accessible in the native C3 molecule and in the C3b fragment so that the latter molecules exhibit a
lower affinity for CD21 than C3bi, C3dg and C3d (Kalli et al., 1991). CD21 also serves as the receptor
for EBV which binds to CD21 through its envelope glycoprotein gp350/220. Kaufman‐Paterson et al
demonstrate that human T cell lines and human thymocytes may be infected with EBV in vitro,
which relates to the expression of CD21 by these cells (Kelleher et al., 1995)(Kaufman‐Paterson et

- 9 -
al., 1995). An additional ligand for CD21 is CD23, a type II transmembrane protein expressed on a
variety of hemopoietic cell types that serves as the low‐affinity receptor for IgE. Cleavage of the
membrane receptor generates soluble forms of CD23 that are endowed with ‘cytokine‐ like’ activity
(Delespesse et al., 1991). Fluorescent liposomes carrying CD23 were shown to interact specifically
with CD21 on B cells, some T cells, follicular dendritic cells (Aubry et al., 1992; Pochon et al., 1992).
Human CD21 is expressed by B lymphocytes and B lymphoblastoid cell lines, human thymocytes
(Tsoukas and Lambris, 1988) a fraction of human T lymphocytes (Fischer et al., 1991), certain
leukemia T cell lines, the pharyngeal and cervical epithelium and by follicular dendritic cells. The
level of expression of CD21 is developmentally regulated: it is highest on mature B lymphocytes and
on a subpopulation of immature blastic thymocytes. A major function of CD21 is to contribute to
human B cell activation and proliferation. On the surface of B lymphocytes, CD21 was found to form
a complex with the membrane proteins CD19, TAPA‐1 and Leu‐13 (Bradbury et al., 1992).
Bonnefoy et al observed secondary cellular responses mediated through interaction of CD21 with
membrane and soluble CD23 had been demonstrated (Bonnefoy et al., 1995a). CD21/CD23 pairing
occurred between membrane forms of the receptors and were shown to be involved in homotypic
adhesion of B cells. Certain biological properties of soluble CD23 had been attributed to interaction
with CD21 on B cells. Thus, a subset of anti‐CD21 monoclonal antibodies has been shown to behave
in a similar fashion as soluble CD23 in decreasing the occurrence of apoptosis in germinal centre B
cells and in enhancing IL‐4‐induced IgE production by blood mononuclear cells.

- 10 -
Overview of complement pathway

- 11 -
1.4. Autoimmunity
The concept of autoimmunity was first predicted by Nobel Laureate Paul Ehrlich at the start of the
twentieth century, and he described it as 'horror autotoxicus'. His experiments led him to conclude
that the immune system is normally focused on responding to foreign materials and has an inbuilt
tendency to avoid attacking self tissues. But when this process goes wrong, the immune system can
attack self tissues resulting in autoimmune disease. The perplexing issue of what allows the immune
system to attack self tissues is a continuing focus of research.
In the past, autoimmune diseases have been studied on the basis of the organ affected, but in recent
years the focus has switched to a more cross‐disciplinary approach with a view to providing a better
understanding of the common mechanisms underlying the pathogenesis of these diseases.
Autoimmunity is caused by an adaptive immune response against "self" antigen. The random
generation of many diverse TCR and BCR makes autoimmunity possible. Clonal deletion and anergy
of self‐specific lymphocytes greatly reduces but does not eliminate the possibility of low affinity self‐
specific responses. Transient autoimmune responses are common but usually cause no lasting
damage. Because self antigens are continually present in the body, when autoimmune responses are
prolonged the resulting tissue damage can be life‐threatening. Risk factors for autoimmune disease
include the presence of certain HLA alleles, sex hormone levels, infection, and other environmental
factors. Autoimmune diseases can be caused by antibodies or T cells and may be organ‐specific or
systemic.
Autoimmune diseases are initiated by activation of antigen‐specific T cells. Th2 cells activate B cells
to make autoantibodies, which (by activating complement) damage tissues directly or initiate
prolonged inflammation. CTL and macrophages activated by Th1 cells are directly cytotoxic and also
promote inflammation. The damage done by some autoimmune responses is limited to a single

- 12 -
organ, while other diseases cause systemic damage. The events that initiate specific autoimmune
diseases are not known.
Autoimmunity can be classified into two categories: organ specific autoimmune disease and
systemic autoimmune disease. Organ‐specific autoimmune diseases involve specific organs of the
body in which the target auto‐antigen is found. Examples of organ‐specific autoimmune diseases
include type 1 or insulin‐dependent diabetes (insulin secreting cells of the pancreas), thyroiditis
(thyroid), multiple sclerosis (myelin sheath of the nervous system; brain) and autoimmune gastritis;
which is the underlying cause of pernicious anaemia (acid secreting cells of the stomach). In systemic
autoimmune disease tissue injury and inflammation occur in multiple sites in organs without relation
to their antigenic makeup and are usually intitiated by vascular leakage and tissue deposition of
circulating autologous immune complexes. Sytemic autoimmune diseases include diseases such as
systemic lupus erythematosus (SLE), rheumatoid arthritis, scleroderma and polymyositis.(Table)
Organ specific autoimmune disease
Disease Self Antigen Immune response
Addision disease Adrenal cells Autoantibodies
Autoimmune hemolytic disease RBC membrane proteins Autoantibodies
Grave’s Disease Thyroid stimulating hormone
receptor
Autoantibodies
Hashimoto’s thyroiditis Thyroid proteins and cells TDTH cells, autoantibodies
Insuline dependent diabetes Pancreatic beta cells Autoantibodies
Myasthenia gravis Acetylcholine receptor Autoantibodies (blocking)
Myocardial infarction Heart Autoantibodies
Systemic autoimmune disease
Disease Self Antigen Immune response
SLE DNA, nuclear protein, RBC and
platelet membrane
Autoantibodies, mmune
complex
Multiple sclerosis Brain or while matter TDTH and Tc cells, autoantibodies
Rheumatoid arthritis Connective tissue, IgG Autoantibodies, immune
complexes

- 13 -
Scleroderma Heart, lungs, kidney,
gastrointestinal tract
Autoantibodies
Sjogren’s syndrome Salivery gland, liver, kidney,
thyroid
Autoantibody
1.4.1. The dimensions of the autoimmune diseases
Autoimmune diseases are a major threat to the health of worldwide. Understating, preventing and
managing chronic conditions are important steps for maintaining satisfactory health. Many chronic
autoimmune diseases affect women more than the men.
Female‐Male ratios in autoimmune diseases.
Hashimoto’s syndrome/hypothyroiditis 10:1
System lupus erythematosus 9:1
Sjoegren’s Syndrome 9:1
Antiphospholipid syndrome 9:1
Autoimmune hepaititis 8:1
Primary biliary cirrhosis 9:1
Grave’s disease 7:1
Scleroderma 3:1
Rheumatoid arthritis 2:5
Antiphospholipd syndrome‐primary 2:1
Autoimmune thromobocytopenia purpura 2:1
Multiple sclerosis 2:1
Myasthenia gravis 2:1

- 14 -
1.5. Rheumatoid Arthritis
Rheumatoid arthritis (RA) which affect approximately 1% of the population worldwide and
disproportionately affects woman, comprise a heterogenous group of poorly understood disorders
(Davidson and Diamond, 2001; Marrack et al., 2001). It is a chronic systemic inflammatory disease,
which predominately affects the joints. The symptoms include pain, swelling, stiffness and loss of
function of the joints. The disease often affects the writs and fingers joints closest to the hand and
occurs in a symmetrical pattern involving both the limbs. It can also affect other part of the body
besides the joints with symptoms of fatigue, occasional fever and malaise.
Courtesy of J. Cush 2002
The etiology of RA is still unknown. Even a basic question whether it is primarily an autoimmune
disease, or an inflammatory response/ to infection, is yet to be resolved. Genetic predisposition has
been implicated, while failure to demonstrate Mendelian inheritance means multiple genetic factors
may be involved. In human the MHC class II HLA‐DR4 allele is shown to be associated with
development and severity of RA. As the only known function of DR is to present peptide to CD4 T
cells this genetic association clearly indicates a role for T cells in the disease. It could be in defining
the repertoire of antigen receptors or in presentation of autoantigens. However, the autoantigen
recognized by autoreactive T cells is not known. The role of T‐lymphocytes in the pathology of RA is
always controversial. Firestein and Zvaifler, (Firestein and Zvaifler, 1990) suggested that T cells may

- 15 -
not be important in perpetuating the disease at late stages based on the lack of nay any evidence of
T cells proliferation in the synovium and low levels of T cells derived cytokines in the inflamed joint.
This is further supported by failed therapeutic trials targeting T cells functions.
Then, new animal models have shifted the focus to B cell‐secreted autoantibodies and innate
immunity mediators (Benoist and Mathis, 2000). The current concept being that while T and B cells
are important in the initiation of RA the pathogenesity is mostly mediated by autoantibodies and
innate immune mediators. The role of proinflammatory cytokines such as IL‐1 and TNF α are well
known and are a current target in RA therapy.
Normal Joint

- 16 -
1.6. Autoimmune diseases
1.6.1. Animal models of rheumatoid arthritis
Experimental animal models of arthritis, including type II collagen‐induced arthritis, adjuvant
arthritis, pristane‐induced arthritis, streptococcal cell wall‐induced arthritis and the K/BxN model of
arthritis have contributed to recent advances in the understanding of the immunopathology of
arthritis.
1.6.1.1. Type II collagen‐induced arthritis
The autoimmune targets in RA are not known but autoantibodies against various joint‐related
epitopes are detected in sera. Antibodies against epitopes modified by citrullination show the
highest specificity for RA and can be detected very early in the disease course (Masson‐Bessiere et
al., 2001; Rantapaa‐Dahlqvist et al., 2003; Schellekens et al., 1998). Antibodies against type II
collagen (CII) occur in a subset of RA, and CII‐specific B and T‐cells have been identified in
rheumatoid synovium and synovial fluid (Burkhardt et al., 2002; Cook et al., 1999; Kim et al., 1999;
Londei et al., 1989; Rudolphi et al., 1997; Tarkowski et al., 1989).
Immunization of mice with CII leads to the development of arthritis, the collagen‐induced arthritis
(CIA) model for RA. CII‐specific activation of both T and B cells is critical for the development of
arthritis, and the transfer of both rodent (Stuart and Dixon, 1983) and human (Wooley et al., 1984)
serum with CII‐specific antibodies induces arthritis in mice. Monoclonal CII‐specific autoantibodies
bind cartilage in vivo and induce arthritis (Holmdahl et al., 1986); the injection of large amounts of
several of such mAbs in cocktails induces severe arthritis (Nandakumar et al., 2003b; Terato et al.,
1992). Collagen‐antibody‐induced arthritis (CAIA) is an inflammation that is dependent on Fc
receptor and complement, involving the infiltration of both neutrophils and macrophages (Hietala et
al., 2004; Nandakumar et al., 2003a; Nandakumar et al., 2003b; Watson et al., 1987).

- 17 -
1.6.1.2. Adjuvant arthritis
It was initially demonstrated in suceptibale strains of rats, that the injection of Freund’s complete
adjuvant (FCA), a water‐in‐oil emulsion containing mycobacteria, causes a chronic polyarthritis
(Pearson, 1956). A single intrademal injection of adjuvant into the footpad or tail of rat induces a
severe arthropathy begining within 2 weeks involving the wrist, ankle, paws and caudal part of the
spine and tail. The arthropathy ultimately consists of synovitis with villus formation, pannus eroding
cartilage and bone, periostitis with new bone formation, and an accompanying inflammation and
fibrosis of the periarticular tissues.
1.6.1.3 Pristane‐induced arthritis
Two weeks after a single intradermal injection of 150 microliters of pristane, rats develop severe and
chronic arthritis. The inflammation is restricted to the joints and involves pannus formation, major
histocompatibility complex (MHC) class II expression, and T lymphocyte infiltration. The initial
development as well as the chronic stage of pristane‐induced arthritis is ameliorated by treatment
with antibodies to the alpha beta‐T‐cell receptor showing that the disease is T cell‐dependent.
Increased levels of interleukins in serum was seen after pristane injection but not during the chronic
stage of arthritis. Joint erosions were accompanied by elevated serum levels of cartilage oligomeric
matrix protein.
1.6.1.4. Streptococcal cell wall‐induced arthritis
Several groups have described the arthritogenic properties of bacterial preparations. The best
studied are the peptidogylcan‐carbohydrate polymers of group A streptococcal cell wall. This
fragment induces a peripheral arthritis, particularly in Lewis female rats. Histologically, it is
characterized by a synovial thickening, with surface fibrin, thickening of the synovial lining layer,
polymorphonuclear leucocytes exudation into the joint fluid, and predominately CD4+ T cells
mononuclear cell infiltrate. Ultimately, there is erosion of cartilage and bone, so that the clinical

- 18 -
features of this model do give resonable approximation to the histopathological appearance of
human RA.
1.6.1.5. K/BxN murine model of RA
A spontaneous murine disease with most of the characteristics of human RA was fortuitously
discovered by breeding the KRN TCR transgenic, mouse specific for bovine RNase (42‐56)/ I‐Ak, to the
nonbese diabetic (NOD) mouse strain (Kouskoff, V et al., 1996). The F1 generation between KRN and
NOD (K/BxN) spontaneously develops a progressive joint‐specific autoimmune disease between 3
and 5 weeks of age, characterized by the rapid symmetrical onset of peripheral joint inflammation
that is restricted primarily to the joints of the front and rear limbs. Pathology of disease in K/BxN
mice is similar to human RA with:
1) Pannus formation
2) Synovial hyperplasia
3) Increased synovial fluid volume
4) Cellular infiltration
5) Chronic remodeling of cartilage and bone
6) Elevated expression of proinflammatory cytokines
7) Autoreactive antibody production
One difference between the the K/BxN model and human RA is the absence of detectable
rheumatoid factor in the K/BxN mice (Kouskoff et al., 1996), although 20‐30% of human RA patients
are also negative for serum rheumatoid factor (Chen et al., 1987). Immune complexes of
rheumatoid factor‐IgG and potentially other Ab‐Ag immune complexes can be found in the joints of
human RA patients (Winchester et al., 1970), but their role in joint pathology and disease
progression has yet to be ascertained.

- 19 -
In the K/BxN model, KRN transgenic cells recognize a mouse (self derived) peptide bound to I‐Ak,
presented by B cells and other MHC class II‐positive APCs (Ji et al., 1999). The autoantigen for both
K/BxN arthritogenic Ig and KRN T‐cells was identified as glucose‐6‐phosphate isomerase (GPI), an
ubiquitous cytoplasmic enzyme that catalyzes the interconversion of fructose‐6‐phosphate and
glucose‐6‐phosphate during glycolysis. KRN TCR has dual ability to recognize RNase (42‐56)/ I‐Ak and
GPI (282‐294)/ I‐Ag7 (Basu et al., 2000; Basu et al., 2001). A working model of K/BxN mice postulates
that GPI‐specific B cells endocytose GPI via surface Ig receptors and present the I‐Ag7 restricted
epitope to incompletely tolerized CD4+ KRN T cells, which in turns provide specific help in maturation
and Ig isotype switching leading to production of autoantibodies (Ji et al., 1999; Kouskoff et al.,
1996; Matsumoto et al., 1999) and subsequently induction of joint inflammation.
Transfer of serum or purified Ig from arthritic K/BxN mice induces a synchronized joint‐specific
inflammatory reaction that mimic the K/BxN disease (Ji et al., 1999), indicating that arthritogenic Ig
can induce synovitis and rheumatoid‐like arthritic disease. The disease induced by single
administration of serum eventually resolves, unless arthritogenic Ig is reputedly transferred into
recipients (Ji et al., 1999). The development of an easily inducible model of RA with a rapid,
synchronized onset facilitates the study of the pathogenic mechanisms involved in the initiation of
joint specific autoimmunity.

- 20 -
K/BxN Mice Model
BN mice K/BxN mice

- 21 -
1.7. Juvenile Arthritis
Juvenile arthritis (JA) is also known as juvenile rheumatoid arthritis (JRA) and occurs with a
prevalence of 100‐400/100,000 (Gare, 1998). The characteristic symptoms of JA are chronic pain and
stiffness due to inflammation of the joints, sometime resulting in irreversible joint damage, growth
retardation and functional disability. The disease activity is generally considered to be T‐ cell
mediated (Panayi et al., 2001) although the contribution of B‐cells is debated (Edwards and
Cambridge, 2001; Edwards et al., 1999). Complement activation in JA seems to be mediated by the
alternative pathway (Aggarwal et al., 2000) although activation through the classical pathway is
reported as well (Miller et al., 1986).
Juvenile arthritis (JA) is a broad term encompassing several disorders starting before the age of
16. It is characterized by arthritis lasting more than 6 weeks, of unknown etiology and usually
persisting for six months initially. Of the various distinguishable clinical forms, oligoarticular JA is
the most frequent. It is characterised by an involvement of up to 4 joints during the first 6 months
and is mostly observed in females. Polyarticular JA involves more than 4 joints from the beginning
of the disease and can be subclassified into rheumatoid factor positive or negative disease.
Systemic onset JA (also called Still disease) usually involves a polyarticular arthritis accompanied
by systemic symptoms like a spiking fever for more than 14 days, a skin rash,
hepatosplenomegaly, serositis (or pericarditis) and lymphadenopathy. The prognosis of JA may be
complicated by the presence of uveitis, associated with an insidious progression. In systemic JA as
well as in some polyarticular forms, inflammation may continue into adulthood.
1.8 Diabetes
Diabetes is an epidemic health problem, affecting more than 150 million people worldwide. This
number is expected to double in the first decades of the third millennium. Insulin‐dependent
diabetes mellitus (type 1) diabetes is generally accepted to be an organ specific autoimmune disease
characterized by a mononuclear infiltrate within the Langerhans islet, immunocytes activation and

- 22 -
autoantibody production (Bach, 1994), but the mechanism of T cell‐dependent pancreatic B‐cell
destruction is still not exactly understood. Antigen presenting cells (APCs), which include dendritic
cells, macrophages, and B‐cells, play essential roles not only in the initiation of a protective immune
response against pathogen but also in the prevention of autoimmunity. While Type II diabetes
mellitus is strongly associated with various changes in the lymphocytes distribution in peripheral
blood; for example, T cell lymphocytosis (Chang and Shaio, 1995), T cell lymphopenia (Kimura et al.,
1998) and unchanged T cell subpopulation (Spooren et al., 1993). Some authors also suggest that, in
diabetic patients, metabolic control influences lymphocytes distribution.
Recent studies have demonstrated that immune markers on B‐cells like CD40, CD86 and T‐cells like
CD4 and CD8, it is possible to predict the disease even years before the clinical onset (Honeyman et
al., 1997). In the last few years, intervention trials have also begun to prevent the development of
type I diabetes at the stage of asymptomatic autoimmunity (Honeyman et al., 1997). However 5‐10
years will be required before the effect of preventive procedures, using humoral and/or metabolic
parameters as marker of progression to overt diabetes, are fully determined. This situation therefore
creates a strong need for identification of surrogate immune markers to show‐term effects of
manipulation of the autoimmune process, leading to the prevention of disease.
1.9 Antiphospholipid syndrome
The antiphospholipid syndrome (APS) is characterized by the presence of anti‐phospholipid
autoantibodies which bind target molecules mainly via ß2‐GPI (beta‐2‐glycoprotein‐I), and/or lupus
anticoagulants, associated with recurrent fetal loss, thromboembolic phenomena and
thrombocytopenia (Galli et al., 1990; Sammaritano and Gharavi, 1992; Shoenfeld, 2003). The
pathogenicity of anti ß2‐GPI antibodies was demonstrated by transfer into naive mice and rabbits as
well as preventing the ß2‐GPI antibodies generation and disease by oral tolerance with ß2‐GPI (Blank
et al., 1994; George et al., 1998). It has been postulated that anti ß2‐GPI antibodies exert a direct
pathogenic effect by interfering with homeostatic reactions occurring on the surface of platelets or
vascular endothelial cells as well as placenta (Blank et al., 1991; Blank et al., 1998; Blank et al., 2002;

- 23 -
Blank et al., 1999; Meroni et al., 2005). The factors causing production of anti ß2‐GPI remain
unidentified, but one of the causes were shown to be infectious agents by molecular mimicry.
1.10. Autoimmune lymphoproliferative syndrome (ALPS) ALPS was first described by Canale and Smith in 1967. In most cases disease is ravelled early in the
life, usually before 5 years of age. ALPS is characterized by the accumulation of a polyclonal
population of T cells called double‐negative T cells. These lymphocytes display the marker common
to mature T cells, CD3, and /β T‐cell–antigen receptors, but neither the CD4 nor the CD8
coreceptors (CD3+ T‐cell receptor /β+ CD4–CD8–). They normally account for less than 2 percent of
peripheral /β+ T cells and are distinct from the double‐negative thymocytes in the cortex of the
thymus, which lack CD3 and T‐cell receptors for antigen. The double‐negative T cells in patients with
ALPS are poorly responsive to mitogens and antigens and fail to produce growth and survival factors
such as interleukin‐2 (Fuss et al., 1997). In Fas‐deficient MRL lpr/lpr mice, the large population of
double‐negative T cells appears to originate from chronically activated CD8+ T cells that down‐
regulate the expression of CD8 and fail to undergo apoptosis (Nagata and Suda, 1995). In humans,
double‐negative T cells also seem to be antigen‐exposed T cells that have escaped apoptosis.
ALPS is classified according to the underlying genetic defect (Rieux‐Laucat et al., 2003). In type 0
disease, homozygous Fas mutations usually cause a complete deficiency of the Fas protein and a
severe form of the disease (Kasahara et al., 1998). In ALPS type I, heterozygous Fas mutations (ALPS
type Ia) (Jackson et al., 1999; Vaishnaw et al., 1999) or, more rarely, heterozygous mutations in the
gene for Fas ligand (ALPS type Ib) (Wu et al., 1996) are usually associated with a partial defect in
apoptosis mediated by Fas and its ligand. ALPS type II, which is characterized by resistance to Fas‐
mediated apoptosis despite the presence of normal Fas ligand and Fas, has been found in two
patients with caspase 10 mutations (Wang et al., 1999). In ALPS type III, Fas‐mediated apoptosis is
also normal in vitro, (Dianzani et al., 1997) and the genetic defect is unclear. Patients with ALPS type
III may not have all four classic features of the syndrome‐lymphoproliferation, excessive numbers of

- 24 -
double‐negative T cells, hypergammaglobulinemia, and autoimmune manifestations. (Fisher et al.,
1995; Rieux‐Laucat et al., 1995).
1.11. Cellular abnormalities
1.11.1. Antigen presenting cells
The concept that exogenous antigen, possibly from an infectious agent(s), is responsible for initiating
disease in RA, is an area of controversy. An exogenous arthritogenic antigen(s) has not been
recognized and it may be possible that endogenous antigen is capable of initiating and driving
disease processes. Dendritic cells (DC) are termed ‘professional APCs’ and have been shown to
accumulate in rheumatoid synovial tissue (Zvaifler et al., 1985), where they differentiate under the
influence of abundant monocytes and fibroblast derived cytokines and express high levels of MHC
class I and II and costimulatory molecules. Presentation of endogenous DC‐derived antigen,
including MHC‐derived peptides, to low affinity, self reactive CD4+T cells under the influence of TNF‐
α and GM‐CSF has been demonstrated (Thomas et al., 1994). This event provides a model by which
RA could be initiated and also accounts for the strong association between RA and MHC class II
alleles (Thomas and Lipsky, 1996).
1.11.2. Macrophages
Macrophages play a central role in the amplification of stimulatory signals nad tissue destruction in
RA. Activated macrophages are found in the synovium, destructive pannus tissue and rheumatoid
nodule (Highton et al., 1995). Both circulating and synovial macrophages produce large quantities of
prostanoids and proinflammatory cytokines like IL1, IL‐6, IL‐10, 1L‐13, IL‐15, IL‐18 and TNFα and they
are professional APCs. In the RA synovial environment, they are induced to upregulate Fc gamma RI
and Fc gamma RII, important in the capture of immune complexes (Burmester et al., 1997).
The other critical role of synovial macrophages is the induction and perpetuation of local
inflammatory changes in the joints. These occur in the cartilage pannus junction and are known to
mediate cartilage and bone destruction through secretion of matrixmetalloproteases.

- 25 -
1.11.3. Role of Cytokines in RA
Cytokines are protein mediators, now known to be involved in almost all important biological
processes, including cell growth and activation, inflammation, immunity, and differentiation. Thus, it
is not surprising that they also have a role in an autoimmune disease such as rheumatoid arthritis
(RA), in which there is chronic inflammation, with fibrosis and the eventual destruction of cartilage
and bone.
Analysis of cytokine‐mRNA and‐protein in rheumatoid arthritis tissue revealed that many
proinflammatory cytokines such as TNFα, IL‐1, IL‐6, GM‐CSF, and chemokines such as IL‐8 are
abundant in all patients regardless of therapy. This is compensated to some degree by the increased
production of anti‐inflammatory cytokines such as IL‐10 and TGFα and cytokine inhibitors such as IL‐
1ra and soluble TNF‐R. However, this upregulation in homeostatic regulatory mechanisms is not
sufficient as these are unable to neutralize all the TNFα and IL‐1 produced.
In rheumatoid arthritis, joint cell cultures that spontaneously produce IL‐1, TNFα was the major
dominant regulator of IL‐1. Subsequently, other proinflammatory cytokines were also inhibited if
TNFα was neutralized. This led to the hypothesis that TNFα was of major importance in rheumatoid
arthritis and became therapeutic target. A balance of proinflammatory and inflammatory cytokines
exists in the synovium and disturbance in this balance contributes to the pathogenesis in the
synovium.
Finally two theories are suggested regarding the pathogenesis of rheumatoid arthritis:
1) T cell centric theory of RA
According to this theory activation of CD4+ cells would trigger and maintain the
inflammatory process in the rheumatoid joint. Large numbers of CD4+ cells persist in the
synovium throughout the disease course, they appear to be inactive in the chronic phase of
the disease.
2) macrophage‐fibroblast theory

- 26 -
Cytokines known to be produced by effector cells (macrophages) and connective tissue cells
(fibroblasts) are expressed in abundance in RA synovium and synovial fluid . These cytokines
include IL1, IL6, TNF, IL8 and GM‐CSF. According to this theory T cells may be important in
inhitiating the disease, chronic inflammmation is self‐perpetuated by macrophages and
fibroblats in a T cell independent manner. This theory in based upon the relative absence of
activated T cell phenotypes in the chronic RA and the preponderance of activated
macrophages and fibroblats phenotype.
There is also a controversy as to whether the process of joint destruction is directly driven by the
inflammatory processor, the basic process of joint destruction with fibroblast proliferation and
activation of other cells is driven independently of the inflammatory process.
The central role of cytokines is confirmed by the success of cytokine‐directed immunotherapy. The
use of Anti –TNF‐α monoclonal antibodies gives therapeutic value to RA.

- 27 -
PMN –polymorphonuclear leukocytes
1.12. Contributors to the inflammatory process
1.12.1 Neuropeptide connection in arthritis
Many studies have shown that modulation of cytokine function is effective in ameliorating
symptoms of rheumatoid arthritis. Neuropeptides have recently been shown to have powerful
effects on the production and release of cytokines and have also been shown to exert potent
proinflammatory and anti‐inflammatory effects in animal models of inflammatory diseases. A lack of
a comprehensive understanding of the pathogenesis of rheumatoid arthritis (RA) has hampered the
development of novel therapeutic approaches to the treatment of this disease. Grimsholm and
colleagues (Grimsholm et al., 2005) demonstrated many different cell types, such as macrophages
and synoviocytes, have long been known to be involved in RA, it has been realized that the
peripheral nervous system has a key role in modulating the severity of RA (Eskandari et al., 2003;
Levine et al., 1985; Straub and Cutolo, 2001). Synovial tissue is richly innervated with neuropeptide‐

- 28 -
containing primary afferent and sympathetic neurons (Grigg, 2001; Mapp et al., 1990; Miller et al.,
2000) and there is evidence that the release of these neuropeptides powerfully influences the
severity of chronic inflammatory diseases, including RA (Cerinic et al., 1998; Niissalo et al., 2002).
Grimsholm and colleagues (Grimsholm et al., 2005) evaluated the relationship between synovial
fluid levels of neuropeptides, inflammatory cytokines and duration of disease in patients with RA.
The evidence for the involvement of neuropeptides in inflammatory diseases has focused
predominantly on a proinflammatory role for SP (O'Connor et al., 2004), as well as an immuno‐
modulatory role for neuropeptide Y (O'Connor et al., 2004) and an anti‐inflammatory role for VIP
(Niissalo et al., 2002).
1.12.1.1. Role of Substance‐P in RA
SP is a bio active undecapeptide found in high levels in primary afferent nociceptor, the central
nervous system and gastrointestinal tract. Since its initial characterization by Leeman and Carraway
the involvement of this peptide in a variety of physiological processes has been well documented.
Some of the functions attributed to SP include its potential roles as a sensory neurotransmitter,
regulator of cell proliferation, a modulator of vascular perfusion and permeability and a mediator of
immunological and inflammatory processes. Evidence supporting the interaction of SP in
immunological phenomena include its ability to bind to neutrophils and cause endogenous mediator
release, to activate lymphocytes and macrophages, as well as to cause degranulation of mast cells. In
addition, SP has been shown to enhance monocyte secretion of interleukin‐1 and tumor necrosis
factor, and can stimulate synovial cells to sectete postglandin E2and collagenase.
Levine at el, 1984 found that substance P concentration increased in RA patients. . They found that,
in the rat, joints that developed more severe arthritis (ankles) were more densely innervated by
substance P containing primary afferent neurons than were joints that developed less severe
arthritis (knees).

- 29 -
1.12.2. Role of MMP‐13 in arthritis
There is a growing body of evidence that implicates matrix metalloproteinases (MMPs) as major
players in numerous diseased conditions. The articular cartilage degradation that is characteristic of
rheumatoid arthritis (RA) is believed to be mediated by the collagenase subfamily of matrix
metalloproteinases. About 20 members of this family have been identified. Most MMPs are secreted
into the extracellular space in a latent proform, and require proteolytic cleavage for enzymatic
activity. A few MMPs, however, are activated intracellularly by a furin‐like mechanism and therefore,
these enzymes are fully active when they reach the extracellular space.
The articular cartilage degradation that is characteristic of rheumatoid arthritis (RA) is believed to be
mediated by the collagenases subfamily of MMPs. Collagenase cleave fibrillar collagens at neural pH
and play an important role in matrix remodeling. Collagens are the major structural proteins of all
connective tissues. The most abundant collagens are the type I, II and III, named interstitial
collagens. The expression of type I and type II collagen genes is tightly regulated during embryonic
development with each type of collagen being made in specific tissue at specific times (Miller and
Gay, 1987). Type I collagen is widely distributed, being present in bone, skin, tendons, and ligament
whereas type II collagen is located almost exclusively in hyline cartilage. One of the newest members
of the collagenase subfamily is collagenase‐3 (CL‐3), which was originally isolated from breast
carcinoma (Freije et al., 1994). In addition to being detected in brest tumers. CL‐3 mRNA has been
found in articular cartilage (Stahle‐Backdahl et al., 1997) and in the synovial tissue from patients with
osteoarthritis or rheumatoid arthritis (Wernicke et al., 1996). CL‐3 has been shown to degrade
collagen types I, II, III as well as being able to degrade the cartilage proteoglycan aggrecans (Fosang
et al., 1996). Biochemical characterization of CL‐3 has a broad spectrum of activity against
connective tissue components (Knauper et al., 1996a; Knauper et al., 1996b). The preference of
collagenase 3 (CL‐3) for collagen type II makes it a likely candidate in the turnover of articular
cartilage and a potential target for drug development. Improper management of MMP expression

- 30 -
and activation has been suggested to play major role diseases such as osteoarthritis, rheumatoid
arthritis, tumor invasion and metastasis and Alzheimer’s disease.
Most cells in the body express MMPs, even though some enzymes are often associated with a
particular cell type. For example, the principle substrate of MMP‐2 (also known as gelatinase A) and
MMP‐9 (also known as gelatinase B) is the type IV collagen in basement membrane and thus, these
enzymes are usually expressed by endothelial cells, although other cells (e.g. stromal fibroblasts,
macrophages, tumor cells) also express them (Borden and Heller, 1997; Nagase and Woessner, 1999;
Stetler‐Stevenson and Yu, 2001; Vincenti, 2001).
MMP‐3 (also known as stromelysin) activates MMP‐1 (also known as collagenase‐1) and cleaves a
broad range of matrix proteins; MMP‐1, which is an interstitial collagenase, and MMP‐3 are among
the most ubiquitously expressed MMPs. In contrast, MMP‐13 (also known as collagenase‐3) has a
more restricted pattern of expression within connective tissue, and is usually produced only by
cartilage and bone during development, and by chondrocytes in osteoarthritis (OA)(Borden et al.,
1996; Mengshol et al., 2000; Vincenti et al., 1998). The preference of collagenase‐3 (CL‐3) for
collagen type II makes it a likely candidate in the turnover of articular cartilage and a potential target
for drug development.
1.12.3. Role of macrophage inhibitory factor (MIF) in RA
Macrophage inhibitory factor is a cytokine with increasingly well recognized importance in the
regulation of immune and inflammatory responses. Its original description focused on its ability to
prevent the random migration of macrophages in the culture, but since its cloning in the mid‐1990
evidence of a much broad range of proinflammatory actions has emerged (Bacher et al., 1996;
Calandra et al., 1994; Churchill et al., 1975). MIF is released by activated T‐ lymphocytes and
macrophages and up regulates the proinflammatory activity of these cells. MIF induces macrophage
secretion of TNF‐alpha and promotes interferone‐gamma induced production of nitric oxide by

- 31 -
mouse macrophages (Attur et al., 1997; Bernhagen et al., 1994; Liew, 1994). Macrophage
intracellular killing and phagocytic function is upregulated in the presence of MIF (Juttner et al.,
1998; Onodera et al., 1997). MIF has also been demonstrated to be a crucial cofactor in T cell
activation (Bacher et al., 1996). The key proinflammatory actions exerted by MIF in vitro are
confirmed by the demonstration of its critical role in the development of both endotoxic shock and
delayed‐type hypersensitivity in vivo in mice (Bernhagen et al., 1996; Calandra and Bucala, 1995). Its
potential role in the development of autoimmune disease is highlighted by previous studies in which
immunoneutralization of MIF resulted in profound inhibition of adjuvant arthritis in the rat (Leech et
al., 1998). MIF antagonist has additionally been shown to delay the onset and lower the frequency
arthritis of collagen‐induced arthritis in the rat (Leech et al., 1998).
RA is characterized by synovial hyperplasia and a cellular infiltrate comprised predominately of
macrophage and T‐cells (Burmester et al., 1997). Macrophage‐derived cytokines, in particularly TNF‐
alpha, are thought to be crucial to the inflammatory process in RA (Elliott et al., 1993).
1.12.4. Role of redox‐reactions in RA
Reactive oxygen species (ROS) are generally thought to be harmful and to play a disease‐enhancing
role in autoimmune disease such as RA (Babior, 2000; Hitchon and El‐Gabalawy, 2004). However, we
have found that decrease capacity of intracellular ROS and surface thiol helps to increase arthritis is
K/BxN serum transfer model. The association between ROS and autoimmunity is interesting,
because it opens up the possibilities for new insight into the pathogenesis of complex autoimmune
disorders. NADPH oxidase complex that catalyzes the transfer of single electron from NADPH to
oxygen, generating ROS. The release of ROS and its downstream products from phagocytic cells is
known as a respiratory burst and is regarded as a part the protection against invading pathogen
(Babior, 2000). In healthy individuals, ROS and associated oxidative stresses are kept in check by
combination of antioxidant activates (Droge, 2002; Winyard et al., 2005). Human cells have
developed a formidable antioxidant defence against oxidant reactions. In particularly, they possess
enzymatic and non enzymatic antioxidant molecules, including thiols mainly glutathione, for

- 32 -
defence. One key chemical barrier against stress induced damage is the redox equilibrium of
sulfhydryl (SH)/ disulphides, by which low molecular weight thiol can be oxidized reversibly to
disulphide and/ or protein mixed disulphide in response to an oxidative stress (Cuozzo and Kaiser,
1999; Deneke, 2000; Dickinson and Forman, 2002). There is increasing evidence that ROS and the
resulting pro‐oxidant/antioxidant imbalance play a major role in the pathogenesis of RA (Brown‐
Galatola and Hall, 1992; Lawrence et al., 1996; Remans et al., 2005).
1.13. Role of Serum soluble CD21 in autoimmune diseases
1.13.1. Soluble CD21
Several lymphocyte surface proteins of are released into the extracellular environment by enzymatic
cleavage. The cleavage may occur close to the extracellular face of the membrane releasing
physiologically active proteins. We have characterized a soluble form of CD21 in human serum and
in culture supernatants of human lymphoblastoid B and T cell lines (Fremeaux‐Bacchi et al., 1996).
Soluble CD21 is a 135‐kDa protein corresponding to the extracytoplasmic portion of the molecule.
The CD21 protein has also been found in culture supernatants of tonsillar B cells and normal human
thymocytes. The release of soluble CD21(sCD21), from lymphocytes correlated with a parallel
decrease in the expression of the membrane‐associated molecule, indicating that sCD21 is
generated by cleavage and shedding of the membrane receptor. Furthermore, analysis of CD21
transcription excluded that alternative splicing is another mechanism leading to production of sCD21
(Illges et al., 1997). Soluble CD21 retains the ability to bind the ligands of the membrane form
(Fremeaux‐Bacchi et al., 1996). Analysis of sCD21 purified from normal human plasma indicated that
a part of sCD21 circulates in association with cleavage fragments of C3 and a trimeric form of sCD23
(Fremeaux‐Bacchi et al., 1998b). sCD21 levels are often altered in pathologic conditions including
lymphoproliferative leukemias, such as B‐CLL (chronic B‐lymphocytic leukemia) (Lowe et al., 1989),
acute EBV‐infection and other virus‐associated diseases (Delibrias et al., 1992; Moore et al., 1989)
and autoimmune diseases like RA, SLE, Sjoegren’s syndrome, Antiphospholipd syndrome, Juvenile
arthritis and diabetes. Therefore sCD21 can be regarded as a marker of chronic inflammatory

- 33 -
autoimmune disease. The levels of sCD21 in the serum may modulate the immunity as sCD21 levels
are correlated with several disorders like SLE, RA and Sjoegren’s syndrome, antiphospholipid
syndrom and diabetes.
1.13.2. Soluble CD23
sCD21 activates monocytes through binding to membrane CD23 and leads to degranulation of
basophils upon crosslinking (Fremeaux‐Bacchi et al., 1998b). CD23, the low affinity receptor for
the Fc portion of IgE (FcγRII), is expressed on a variety of haematopoietic cells including B cells
and monocytes (Delespesse et al., 1991). It is a 45 kDa membrane type II glycoprotein whose
proteolytic cleavage gives rise to unstable soluble fragments subsequently transformed into a
stable 25 kDa product referred to as soluble CD23 (sCD23). Cell surface CD23 expression and
sCD23 release are up regulated by IL‐4 in all CD23 expressing cell types, and inhibited by
interferon–gamma (IFN‐γ), transforming growth factor beta (TGF‐β) and glucocorticoids on B‐cells
(Delespesse et al., 1992). While sCD23 retains the capacity to bind IgE, it has many activities that
are IgE‐independent (Delespesse et al., 1992; Sarfati et al., 1992), including inhibition of apoptosis
(Liu et al., 1991). The amount of sCD21 and sCD23 in serum may modulate immunity as sCD21 and
sCD23 levels are correlated with several clinical conditions. In particularly reduced levels of sCD21
seems to be a marker of chronic inflammation

- 34 -
CHAPTER‐II
AIMS OF THE STUDY
The study presented here is an investigation of cellular and molecular factors involved in the
rheumatoid arthritis using the antibody induced arthritis model.
The first part of the study investigates the role of neuronal pathways in the antibody induced
arthritis. We determined the role of sympathetic, parasympathetic and peripheral nervous system
contribution to antibody induced‐arthritis.
The articular cartilage degradation that is characteristic of rheumatoid arthritis (RA) is believed to
be mediated by the collagenase subfamily of matrix metalloproteinases. MMP13 has been shown
to degrade collagen types I, II, III as well as being able to degrade the cartilage proteoglycan
aggrecans. The K/BxN model was used to determined the role of MMP13 in arthritis.
MIF is released by activated T‐ lymphocytes and macrophages and up regulates the
proinflammatory activity of these cells. It has been shown that macrophages play an important
role in K/BxN model (Solomon et al., 2005). Therefore we determined MIF role in K/BxN model.
Chnages in the redox state determine biochemical activities of variety of molecules and affect
immunity (Gelderman et al., 2006). Phytol, an oxidizing substance, has influence on the capacity
of T cells to mediate immunity (Hultqvist et al., 2006). Moreover, we analyzed the possible role of
ROS in K/BxN model.
2) The second aspect of the study was to determine the amounts of sCD21 and sCD23 in various
autoimmune disorders and healthy donor’s serum /plasma samples.

- 35 -
CHAPTER‐III MATERIALS AND METHODS 3. Mouse strains
3.1.1. KRN transgenic mouse strain
The transgenic mouse strain was propagated in the animal facility, Rheinbach. The strain harbours
the transgenes encoding the αβ T cell receptor capable of recognizing Glucose‐6‐phosphate
isomerase in the context of IAg7 molecule (Kouskoff et al., 1996). The strain was maintained by serial
crossing the heterozygotes positive for the KRN T cell receptor against the C57BL/6 background.
Inbred strain of C57BL/6 mice were also maintained in the same animal facility, Rheinbach, Germany
and were maintained under pathogen free conditions. This mouse line was used for breeding KRN
mouse strain and also for various experiments.
3.1.2. NOD/Lt mouse strain
The nonobese diabetic (NOD) mouse strain was also propagated in animal facility of University of
Applied Sciences, Bonn‐Rhein‐Sieg, Rheinbach. The mice were originally derived from out bred JCL‐
ICR mice. This strain serves as an excellent model for the autoimmune disease, insulin dependent
diabetes mellitus.
3.1.3. K/BxN mouse stain (KRNxNOD)
The K/BxN mouse strain is obtained through a genetic crossing between K/B (KRN) transgenic mice
with C57BL/6 background) and the NOD strain. The KRN transgeneic mice being heterozygous, the
F1 offspring were either carrier or non carrier of the KRN transgene. The carrier were designated as
K/BxN and these develop arthritis within one monthafter birth. The non carrier were designated as
BxN.

- 36 -
3.2 Knockout mice
3.2.1 MMP13 knockout mouse strain
MMP 13‐/‐ mice were obtained from Prof. Peter Angel, Heidelberg, Germany (Angel P et al. 2006)
3.2.2. Macrophage inhibitory factor (MIF) knockout mice
MIF ‐/‐ were obtained from Prof. Bucala, Tke Picower Institute for Medical Research, Manhasset, New
York. USA.
3.2.3. Substance P knockout mouse strain
SP ‐/‐ mice were obtained from Prof. Dr. Zimmer, University of Bonn, Germany (Bilkei‐ Gorzo et al.,
2002) and propagated in the animal facility, University of Applied Sciences, Bonn‐Rhein‐Sieg,
Rheinbach.
3.2.4. DBA mt FvB mouse strain
DBA mt FvB (ATP deficient) mouse strain and wild type DBA/1J were a kind gift from Dr. Ibrahim,
University of Rostock, Germany.
3.2.5. B6.129 S7‐Chrna7 tm1Bay/J mouse strain
B6.129 S7‐Chrna7 tm1Bay/J mice were obtained from Jackson laboratory.
All knockout mice were C57BL/6 background, maintained and breed under pathogen free condition,
University of Applied Sciences, Bonn‐Rhein‐Sieg, Rheinbach.
3.2.6. BALB/c mice and CD57BL/6 mice
BALB/c and C57BL/6 mice were obtained from the Charles Liver Laboratory.
3.3. Breeding Experiments
All mouse strains were kept in a pathogen free environment in the University of applied Sciences,
University of Applied Sciences, Bonn‐Rhein‐Sieg, Rheinbach. The KRN strain was bred with the
C57BL/6 strain to maintain the KRN mouse line in a heterozygous condition on C57BL/6 background.

- 37 -
The mice were screened by FACS analysis of the peripheral blood lymphocytes using CD4 PE and
TCR‐Vβ6‐ FITC antibodies. The double positive phenotype were selected for breeding.
3.4 Induction of arthritis using K/BxN sera transfer
3.4.1. Production of K/BxN sera
The K/BxN mice were the F1 generation of KRN transgenic mice crossed with the NOD strain and,
develop arthritis around day 30 after birth.
K/BxN mice produce anti‐GPI antibodies and when sera from theses arthritic mice are transferred to
susceptible stains like BALB/c and C57BL/6 mice they develop arthritis. For all experiments involving
sera transfer K/BxN mice around 2‐5 months old were bled by their tail and collected blood was
allowed to clot at 4°C overnight. The samples were then centrifuged at 2000 rpm at 4°C and clot was
removed. The samples were again centrifuged at 2,000 rpm to remove the residual blood cells. The
sera were pooled and stored at ‐200C and used for arthritis induction.
3.4.2. Establishment of antibody‐induced arthritis in mice
Arthritis was induced into the recipient mice by an intraperitoneal injection of 200µl of pooled
K/BxN sera on day 0.
3.4.3 Assessment of antibody‐induced arthritis by ankle thickness (mm) and clinical score
(U)
Increase in ankle thickness was measured in all experiments using a calliper from Hann &Kolb (model
no 33185). Ankle thickness was measured till day 14 and it was expressed in millimetres as summed
average of ankle thickness of the four limbs of each mouse. Clinical index (I) were also measured
along with the ankle thickness and I a scale varying from 0‐4. The lowest score of 0 was given for a
leg when ankle is not affected; 1 if redening and highly swollen ankle was observed. The error was
expressed as standard error of mean (SEM).

- 38 -
3.5 Histology
Ankle and knee joints of the mice were carefully dissected out without damage to the joints. Skin,
hair and soft tissue were removed surgically and the complete joints were fixed overnight in 4%
formaldehyde solution.
3.5.1 Decalcification
Fixed joints were decalcified by treatment with ethylenediaminetetraacetic acid (EDTA) pH 7.2
solution for two weeks with gentle stirring with the help of scalpel and daily replacement of solution.
Soon after decalcification the joints were washed with PBS and stored in 50% ethanol. The treated
joints were then, embedded in paraffin for sectioning. About 5‐10µm thick sagittal serial section
were cut. The sections were placed on glass slides and quality of sections were tested
microscopically.
5.6. Preparation of adherent peritoneal macrophages and reconstitution of macrophages
in MIF knockout Mice
Adherent peritoneal macrophages were isolated from 6–8 weeks old C57BL/6 mice. 5‐8 ml ice‐cold
PBS, was intraperitoneally injected into mice, killed by cervical dislocation and the cells in the
peritoneal fluid were collected. After centrifugation from the peritoneal extract, the cells were
resuspended in PBS. The macrophages were purified by adherence on 90‐mm culture dishes
(Greiner, Frickenhausen, Germany) for 1 h in 5% CO2 at 37°C. Non‐adherent cells, which are mainly
lymphocytes, were removed by washing two times with PBS. Adherent cells, which were mainly
macrophages by morphology, were collected and resuspended in PBS. To reconstitute macrophages,
1.5×107 cells in a total volume of 0.2 ml PBS were injected intraperitoneally into MIF knockout mice.
3.7 Immunological Methods
3.7.1 Fluorescence Activated Cell Sorting (FACS)
Ficoll density gradient preparation of lymphocytes

- 39 -
Mice were bled from the tail vein and blood was directly collected into the 1.5ml centrifuge tubes
containing 400 µl of Alsever’s solution (0.42g NaCl, 0.8g tri‐Na‐Citrate‐2H2O, 2.05g D‐Glucose in
100ml of water, pH6.1 adjusted with 10% citric acid). Samples were mixed properly in order to avoid
blood coagulation and kept at 4oC. Ficoll gradient purification of samples were performed at 2500
rpm for 10 min at 4oC in 1.5 ml centrifuge tubes. The lymphocytes were carefully removed from the
interface of the two layers and transferred into 96 well plates. Then, the cells washed with 1x PBS.
The supernatant was discarded and the pellet resuspended in 50µl of antibody mixture.
All the antibodies were diluted in 1x PBS. The anti‐CD4 and anti‐Vß6 antibody mixture was used to
check for the KRN TCR transgene.
Cells in the antibody solution were incubated in the dark for 30 min on ice. Then, cells were washed
with 100‐200µl of 1x PBS and centrifuged at 1200 rpm for 10 min at 4oC. Finally, the pellet was
resuspended in 100µl of 1xPBS and transfered into the FACS tubes (BD Pharmingen, Heidelberg,
Germany). Data were collected in a FACS Calibur using the ‘Cell Quest’ software (BD‐ Pharmingen,
Heidelberg, Germany).
3.7.2. RBC Lysis
Mice were bled from the tail and blood was collected into 1.5ml centrifuge tube containing 400µl
Alsever’s solution. The whole blood sample was transferred into 15ml falcon tube and 10‐12 ml RBC
lysis buffer was added [8.29 grams NH4Cl (0.15M), 1 gram KHCO3 (10mM), 37.2 mg Na2EDTA
(0.1mM), added 800ml H2O and adjusted pH 7.2‐7.4 with HCl ].The cells were incubated at room
temperature for 10 min and then, cells were centrifuged at 1200 rpm for 10 min. The supernatant
was discarded and the cells washed with 1xPBS at 1200rpm for 10 min. After centrifugation the
pellet was resuspended in 100µl of 1x PBS and stained the cells with different antibody mixtures.
3.8. Quantification of sCD21 from human serum/plasma by ELISA
The monoclonal antibodies THB5 and biotinylated BU32 were used as capture and detection
antibody respectively. Titration was performed with purified sCD21 and a standard curve was
plotted.THB5 was coated onto an ELISA plate (NUNC, Germany) at a concentration of 5µg/ml in

- 40 -
coating buffer (phosphate buffer, pH 7.2) for overnight at 4°C. Then the plate was empted to get rid
of residual buffer and tapped on an adsorbent paper. Subsequently the plate was blocked with
5%BSA in PBS for 2 h. The serum samples at a dilution of 1:50, were added into the plate along with
the standard in triplicates. The plates were incubated overnight at 4°C. Then, the plate were washed
two times with PBS‐T and incubated with biotinylated BU32 for 1h at RT. Then, after three washes,
streptavidin coupled to horse‐radish peroxidase was added and TMB as substrate/coloring agent.
The enzyme reaction was stopped by adding 1M H2SO4 and was quantified by taking the optical
density at 450nm and reference wavelength at 690nm in an ELISA reader (Anthos Micro System,
Germany). The sCD21 concentration was calculated by extrapolating from the standard graph.
3.9. Quantification of sCD23 from human serum/plasma by ELISA
sCD23 levels in serum were determined using a commercial sCD23 ELISA in accordance with the
manufacturer's instructions (Bender Medsystems, Vienna, Austria).
Briefly, plates were coated with 5µg/ml coating antibody over night at 4°C. Then, plates were
washed with wash buffer (0.05% Tween 20 in PBS), blocked with assay buffer (5%BSA, 0.05% Tween
20 in PBS) for two hours. After washing, sera samples were added at a dilution of 1:10 into the plates
along with the standard and biotin‐conjugate in duplicates and the plates were incubated for 2 hour
at room temperature. Then, after incubation plates were washed with wash buffer and streptavidin‐
HRP added in all the wells. Finally, TMB (Sigma) was added as a substrats. The enzyme reaction was
stopped by addition of 1M H2SO4 and the optical density measured at 450nm, reference wavelength
at 690nm, in an ELISA reader Anthos Microsystem, Germany). The sCD23 concentrations were
calculated by extrapolating from the standard graph.
3.10. Immunoblot
3.10.1. Western blot
Immunoblotting is a technique used for the detection of the macromolecular antigens (usually
proteis) using specific antibody (Towbin et al., 1979). Proteins bind to the nitrocellulose chiefly by
hydrophobic bonds (van Oss et al., 1987) and PVDF membranes ensure much stronger hydrophobic

- 41 -
interactions . Polyacrylamide gels were, carefully removed from the electrophoresis chambers and
placed on the top of an equal size nitrocellulose or PVDF membrane soaked in transfer buffer
(50mM Tris; pH 8.8, 250mM Glycine; pH 8.3, 20% Methanol). Six pieces of Whatmann 3mm paper of
equal size were also socked in the transfer buffer to saturation. Three piece Whatmann 3MM paper
saturated with transfer buffer were placed on the bottom of the gel‐membrane sandwich and three
on the top. The complete sandwich was placed on the bed of the transfer apparatus in such a way
that the membrane faces the anode. Transfer was carried out for 1 h at 20 V and 200mA. Soon after
the transfer the membrane was carefully taken out from the sandwich and stained with Ponceau S
solution for the detection of the transferred polypeptide bands. The band pattern was scanned
electronically.
The membrane containing the polypeptide bands were placed in the blocking buffer (5% BSA in PBS)
for overnight at 4oC. After blocking, the membrane was incubated in primary antibody solution
(dilution in blocking buffer according to the manufacture’s instructions) for 1 h at room temperature
on a shaker. The unbound antibodies were removed by washing the membrane three times for 10
minute with PBS‐T. After the last wash the membrane was incubated with Horse radish peroxidase
(HRP)‐conjugated secondary antibody for 1 hour at room temperature on a shaker. After that, the
membrane was washed three times with wash buffer.
Super Signal chemiluminescent substrate (Pierce, Rockford, USA) was prepared by mixing equal
volumes of Solution A and solution B. The cocktail was added to the membrane and incubated for
five minute at room temperature. Pictures were taken by Chemidoc (Biorad, Germany).
3.10.2. Dot Blot
Instead of running a SDS PAGE, protein samples were spotted directly onto a nitrocellulose
membrane. The membrane was blocked with 5% BSA in PBS at 4°C overnight. After blocking, the
membrane was incubated in primary antibody solution (dilution in blocking buffer according to the
manufacture’s instructions) for 1 h at room temperature on a shaker. The unbound antibodies were
removed by washing the membrane three times for 10 minute with PBS‐T. After the last wash the

- 42 -
membrane was incubated with Horse radish peroxidase (HRP)‐conjugated secondary antibody for 1
hour at room temperature on a shaker. After that, the membrane was washed three times with
wash buffer.
Super Signal chemiluminescent substrate (Pierce, Rockford,USA) was prepared by mixing equal
volumes of Solution A and solution B. The cocktail was added to the membrane and incubated for
five minute at room temperature. Pictures were taken by Chemidoc (Biorad, Germany).
3.11 Chemical Sympathectomy
BALB/c mice 8 weeks old were obtained from the Charles Liver laboratories, Germany. Chemical
Sympathectomy was performed by injecting 6‐OHDA (6‐Hydroxydopamine hydrochloride, Sigma
Aldrich Chemie GmbH, Germany). 6‐ OHDA was dissolved in 0.8% NaCl containing 1x10‐3 M
Ascorbate as antioxidant. The solution was stored in the dark at ‐20°C.
3.11.1. Transient sympathectomy
In this method eight weeks old BALB/c mice were injected weekly once with 6‐OHDA (150mg/kg of
body weight) for seven weeks consecutively. The control mice were injected with vehicle alone
(0.8% NaCl containing 1x10‐3 M Ascorbate as antioxidant). Mice were maintained in pathogen free
environment. At the end of the last injection mice were taken for the arthritis induction by serum
transfer.
3.11.2. Permanent sympathectomy
For permanent sympathectomy, pubs were injected with 6‐OHDA (50mg/kg of body weight) at
postnatal day 1. The control pubs were treated with vehicle alone (0.8% NaCl containing 1x10‐3 M
Ascorbate as antioxidant). At 8 weeks of the age the mice in this group were taken for the serum
transfer experiments for arthritis induction.

- 43 -
3.12. Statistics
Statistical calculations and graphical illustrations were performed using Instat/Prism software.
Kruskal Wallis test for nonparametric analysis of variance (ANOVA) and Mann‐Whitney test to obtain
nonparametric two‐tail P value were performed.
3.12.1. P value
Having collected data from several samples and means, it is possible that the populations have the
same mean and the difference observed is a coincidence of random sampling. There is no way we
can ever be sure whether the difference observed reflects a true difference or a coincidence of
random sampling. All we can do is to calculate the probabilities. P‐value answers this question: If
the populations really did have the same mean, what is the probability of observing such a large
difference (or larger) between sample means in an experiment of this size? The P‐value is a
probability with a value ranging from zero to one. If the P value is small, we can conclude that the
difference is quite unlikely to be caused by random sampling. Instead, the populations have
different means. The lower the P‐value is, the higher is the significance of the observed differences.
3.12.2. Mann‐Whitney test & Kruskal‐Wallis test
The Mann‐Whitney test is a nonparametric test to compare two unpaired groups. The Kruskal‐Wallis
test is a nonparametric test to compare three or more unpaired groups answering the same
question as above. The key result is a P‐value that answers this question: If the populations really
have the same median, what is the chance that random sampling would result in medians as far
apart (or more so) as observed in this experiment? If the P‐value is small, the difference is not a
coincidence; instead the populations have different medians. If the P‐value is large, the data do not
give any reason to conclude that the overall medians differ.

- 44 -
CHAPTER‐IV RESULTS 4.1 Neurological Experiments:
4.1.1 Transient sympathectomy partially protects Balb/c mice from antibody induced
arthritis
Rheumatoid arthritis is a chronic, painful, inflammatory disease that causes debilitation of distal
joints. The disease is also characterised by spontaneous ‘flare ups’, causing an exacerbation of the
pain and inflammation. Inflammatory events that take place within the joints include oedema
formation, vascular remodelling and a significant amount of cellular infiltration, amongst other. For
many years it has been considered that sensory nerves, which mediate neurogenic inflammation in a
variety of disease states, may play a role in the onset and progression of disease. Neurogenic
inflammation is defined as the oedema formation, increased blood flow and inflammatory cells
recruitment observed after stimulation of sensory nerves (usually C and Aδ fibers) and release of
neuropeptides. The symmetrical inflammation of joints observed in patients with rheumatoid
arthritis suggests that the progression of the disease has a neural influence. Sensory nerves are
involved in the development of joint disease, however no immunological explanation is given so far
for this fact. In order to evaluate the role of the sympathectic nervous system in RA pathogenesis,
we performed chemical sympathectomy followed by induction of arthritis by K/BxN sera induction.
Sympathectomy was done by administration of 6‐OH Dopamine and depletion of the peripheral
sympathetic nervous system was checked by western blot. Since lymphoid organs are rich with
sympathetic nerve fibres, we selected the spleen for western blot analysis of tyrosine hyroxylase
(TH), a marker for sympathetic nerve fibers innervating lymphoid organs. TH is expressed in basal
and stress conditions in spleen. After 6‐OH Dopamine injection, we observed 60% denervation of

- 45 -
sympathetic nerve fibers in the spleen of adult mice and 99% denervation in neonatal compared
with vehicle treated mice Fig. 1.1.
Figure 1.1: Effect of 6‐OH Dopamine induced denervation in murine spleens.
Western blot analysis of Tyrosine Hydroxylase (TH) in spleens of adult and neonatal 6‐OH Dopamine
treated mice and brain used as control. The 60 KDa TH bands were stronger in the mice which were
treated four times with 6‐OHDA at 8 weeks of age compared to the neonatal treated mice.
Quantification of TH content was done by chemiluminescence scanning.
Figure 1.2: Densitometric analysis of the Western blot.
Balb/c mice were taken for neurological experiments because of their highest susceptibility for
K/BxN sera‐induced arthritis (Maccioni et al., 2002; Solomon et al., 2002). For transient
sympathectomy Balb/c mice were treated with 6‐OH Dopamine weekly once for four weeks. Then,
arthritis was induced by intraperitoneal injection of 200 µl of K/BxN sera. The control group were
60 KDa
Brain Control 6‐OHDA Untreated6‐OHDA Untreated
Neonatal Adult

- 46 -
treated with vehicle only followed by 200 µl of K/BxN sera. Clinical index and ankle measurement
were observed at days 3, 6, 9, 12, 15, 18 and 21 post injection of K/BxN sera. Administration of
K/BxN serum into transient sympathectomized mice resulted in significantly decreased ankle
thickness and clinical index from day 6 to 15. In transient sympathectomized mice ankle thickness
reached a maximum value of 2 mm on day 6 and swelling decreased slowly from day 9 on and finally
reached an average ankle thickness of about 0.5 mm by day 15. Similarly, in vehicle treated mice
ankle thickness reached a maximum value of 3.4 mm on day 6 and disease decreased slowly from
day 9 and finally reached an average ankle thickness of about 1.0 mm by day 15 (Fig.1.3 a).
Clinical Index
A decrease in the clinical index of the disease was also observed, with the maximum clinical index 4
in vehicle treated mice at day 9 while in transient sympathectomized mice maximum clinical index
reached up to 3 at day 9. Decreased clinical index was significant at days 3, 6 and 9 (Fig 1.3 b).
(a) (b)
Figure 1.3: Transient Sympathectomy results in significantly reduced arthritis.

- 47 -
Transient sypathectomized Balb/c mice (n=10) and control mice (n=10) were treated with 200µl
K/BxN sera and arthritis evaluated by measuring (a) ankle thickness and (b) clinical index. There was
significant difference in ankle thickness and clinical index between the two groups. The values
represent the mean ± SEM of ankles/ time points, which were compared with student t‐test to the
wild type mice.
4.1.2. Permanent sympathectomy protects Balb/c mice from antibody induced arthritis.
Permanent sympathectomy was done by injection of 6‐OH dopamine on postnatal day one. The
control mice were treated with vehicle only. Arthritis was induced in the permanent
sympathectomized mice at 8 week of age. In vehicle treated mice maximum ankle thickness reached
3 mm on day 9 while in permanent sympathectomised mice maximum ankle thickness reached up to
2.5mm at day 9 and the swelling decreased slowly from day 9 on and finally reached 2 mm and 1.5
mm for vehicle treated mice and permanent sympathectomized mice respectively. A significantly
reduced arthritis was observed at day 3 and 9 in the permanent sympathectomized mice as
compared to vehicle treated mice.
There has been considerable debate regarding the role of the sympathetic nervous system in the
inflammatory diseases (Fitzgerald, 1989; Kidd et al., 1992; Levine et al., 1988). Usually pain is
associated with stress, which increases the sympathetic activity. Sympathetic terminals terminate
near blood vessels and mast cells in the synovium and therefore it is conceivable that
neurotransmitters released from sympathetic nerves would influence vascular permeability and
mast cells activity in joints. ATP released from the adrenergic terminals influence mast cells to
degranulate (Wilhelm et al., 2005).
Clinical index
The clinical index for vehicle treated mice reached a maximum of 3.7 mm and for permanent
sympathectomized mice of 3 mm. The difference was significant at days 3, 6 and 9 (Fig. 1.4 b).

- 48 -
(a) (b)
Figure 1.4 : Permanent Sympathectomy
(a) Average Ankle thickness and (b) clinical index of permanently sympathectomized Balb/c mice
(n=6) and vehicle treated mice (n=7). Control mice and permanently sympathectomized
Balb/c mice were treated with 200µl K/BxN sera.
4.1.3. Nicotinic acetylcholine receptor‐α7 (α7nAChR) homozygous knockout mice were
completely resistant to K/BxN antibody‐induced arthritis
The nicotinic acetylcholine receptor‐α7 (α7nAChR) subunit is an essential regulator of inflammation.
Activation of parasympathetic nervous system results in the activation of cholinergic nerve fibers of
the efferent nerve and release of acetylcholine at the synapses. Acetylcholine attenuate the release
of proinflammatory cytokines (TNF, IL‐1β, IL‐6 and IL‐18) but not the anti‐inflammatory cytokine IL‐
10 in LPS stimulated human macrophage culture.
Acetrylcholine acts through 2 types of receptors: muscarinic and nicotine, the α7subunit of the
nicotine acetylcholine receptor is expressed on macrophages. Because of the immunosuppressive
effects of acetylcholine in vitro, we studied the possible immunomodulatory role of the
parasympathetic nervous system in K/BxN antibody‐induced arthritis model.

- 49 -
The α7nAChR homozygous‐/‐, heterozygous‐/+ α7nAChR mice and genotype matched C57BL/6 control
mice were injected with 200 µl K/BxN serum. The mouse cohorts were followed up for signs of
arthritis development by caliper measurement of ankle thickness and by visual examination of ankle
swelling to obtain the clinical score on days 0, 3, 5, 7, 9, 11 and 13. Ankle swelling in the control
cohorts was evident beginning at day 2 after K/BxN sera transfer and progressing up to day 12/ 13
and peak ankle thickness occurred between days 5 to 7. On the contrary the α7nAChR‐/‐ mice were
found to be completely resistant to arthritis, not showing any symptom of joint inflammation and
clinical index while α7nAChR‐/+ mice were showing almost the same clinical index, and ankle
measurements just like control mice. The average ankle thickness of diseased control C57BL/6 mice
reached its maximum increase of 2.2 mm on day 7, in α7nAChR‐/‐ mice was 0.2 mm on day 3 and in
α7nAChR‐/+ mice the value was 1.5 on day 7. In control and α7nAChR heterozygous‐/+ mice the
disease decreased from day 9 and reached normal values on day 11. The reduction in ankle thickness
was significant when we compared control groups with the α7nAChR‐/‐ experimental groups (Fig 1.5
a).
Clinical index
A significant decrease in clinical index was observed between control C57BL/6 mice and α7nAChR‐/‐
mice after K/BxN sera injection. The difference was not significant when we compared α7nAChR‐/+
and control mice (Fig 1.5 b).
Wild type (440bp)
α7nAChR Homozygous‐/‐ (770bp) α7nAChR Heterozygous+/‐ (440 and 750bp)
DNA Ladder

- 50 -
Figure 1.4: The genotype of α7nAChR ‐/‐ and ‐/+ mice were checked by PCR analysis. DNA was taken
from the mice tail, was described in materials and methods. C57BL/6 mice DNA was used as control
with 440bp α7nAChR‐specific band, α7nAChR ‐/‐ 750bp band and α7nAChR ‐/+ mice shared with 440
and 750bp bands.
(a) (b)
Figure 1.5 : α7nAChR ‐/‐ deficient mice are resistant to arthritis. On day 0, 200 µl K/BxN serum was
injected intraperitoneally in α7nAChR homozygous deficient mice, α7nAChR ‐/+ and wild type control
C57BL/6 mice. The mice were assessed for arthritis development by (a) ankle thickness and (b)
clinical index on days 0, 3, 5, 7, 9 and 11. α7nAChR ‐/‐ mice were completely resistant to arthritis while
α7nAChR ‐/+ mice showed disease just like the wild type control mice. Data are expressed as mean ±
SEM; n=3.
4.1.4. Capsaicin does not provide permanent sympathectomy in Balb/c mice
Pups were treated with capsaicin (50mg/kg of body weight) at postnatal day 1. After 8 weeks,
arthritis was induced by injection of 200 µl K/BxN sera. Average ankle thickness was not significantly

- 51 -
decreased between the control mice (n=5) and capsaicin treated mice (n=5). The values represent
the mean ± SEM of ankles/ time points, which were compared with student t‐test to the wild type
mice (Fig 1.6 a). The experiment was repeated 3 times with same result.
Clinical Index
The mice were assessed for arthritis development by clinical index score on days 0, 2, 4, 6, 8, 10 and
12. There was no significant difference between control and capsacine treated mice (Fig 1.6 b)
(a) (b)
Figure 1.6: Permanent Sympathectomy showed no significant difference between the control and
capsaicin treated pups.
The effect of capsaicin, an ingredient of hot pepper, on rheumatoid arthritis. Pups were treated with
capsaicin (50mg/kg of body weight) at postnatal day 1. After 8 weeks, arthritis was induced by
injection of 200 µl K/BxN sera. Average ankle thickness (a) and clinical index (b) was not significantly
decreased comparing the control mice (n=5) and capsaicin treated mice (n=5). The values represent

- 52 -
the mean ± SEM of ankles/ time points, which were compared with student t‐test to the wild type
mice.
4.1.5. Substance P knockout mice were partially protected from antibody induced arthritis
SP is known to be widely distributed throughout the central and peripheral nervous system and the
intestinal tract of many species including humans (Hokfelt et al., 1975; Jiang et al., 1982; Pernow,
1983). In order to specifically observe the influence of the SP in arthritis, we used mice deficient in
the peptide neurotransmitter SP. SP is released from unmyelinated C fibers innervating ankle and
knee joints. The role of SP has been studied in adjuvant‐induced arthritis (Levine et al., 1984). Levine
et al found in the rat, joints that developed more severe arthritis were more densely innervated by
SP containing primary afferent neurons than were joints that developed less severe arthritis. The
concentration of SP increased in the joints affected by arthritis (Lotz et al., 1987). Joints having dense
innervations display more severe inflammation and tissue destruction in adjuvant and collagen
induced arthritis (Mitchell and Fries, 1982).
The Substance P‐/‐ mice and genotype matched C57BL/6 control mice were injected with 200 µl
K/BxN serum. Ankle thickness and clinical index were recorded as given in the materials and
methods section. A reduced arthritis was observed in SP‐/‐ mice. Clinical manifestation of the disease
was evident on day 3 as in control C57/BL/6 mice and SP‐/‐mice. The average ankle thickness of
diseased C57BL/6 mice reached its maximum value with 2.5 mm on day 9 and in SP‐/‐ mice was 2 mm
on day 9. In both the groups the disease was decreased from day 10 and reached normal values on
day 21. The reduction in ankle thickness was significant at day 3, 9, 12 and 15 when comparing
control groups with SP knockout groups (Fig 1.7 a).
Clinical index
A significant decrease in the clinical index was observed comparing control C57BL/6 mice and SP‐/‐
mice after K/BxN sera injection. The difference was significant at days 3, 6, 9 and 15 (Fig 1.7 b).

- 53 -
These results suggest a significant physiological difference between joints that develop mild and
severe arthritis and indicate that release of intraneuronal substance P in joints contributes to the
severity of the arthritis.
(a) (b)
Figure 1.7: Mice deficient in the neurotransmitter Substance P (n=5) showed reduced inflammation
after the injection of 200µl K/BxN sera. There was a significant decrease in (a) average ankle
thickness (mm) (b) Clinical index between SP knockout mice and C57BL/6 mice. The values represent
the mean ± SEM of ankles/ time points.
4.2. Role of macrophage inhibitory factor in K/BxN serum‐induced arthritis
4.2.1. MIF knockout mice are protected from antibody‐induced arthritis
Macrophage migration inhibitory factor (MIF) is increasingly recognized as an important regulatory
cytokine in immune and inflammatory responses. MIF is a product of activated macrophages, T‐cells
and endothelial cells, and up‐regulates the proinflammatory activity of these cells. Here we have

- 54 -
shown that murine K/BxN serum‐transfer model is MIF‐dependent because MIF ‐/‐ mice are
protected from antibody‐induced arthritis.
Figure 1.8: The genotype of MIF‐/‐ mice was checked analysing genomic DNA by PCR. DNA was
isolated from the mice tail. PCR of C57BL/6 mice DNA was used as control with a 544bp MIF wildtype
band and MIF‐/‐ mice DNA with a 383bp band.
MIF ‐/‐ mice with C57BL/6 genetic background were treated with 200 µl K/BxN sera to induce arthritis
at day 0. Control mice were treated in parallel with 200 µl K/BxN sera. These cohorts were assessed
for arthritis development, measuring ankle thickness and clinical index scores on days 0, 2, 4, 6, 8, 10
and 12. Ankle thickness from MIF‐/‐ mice showed significant decrease throughout the entire
experiment as compared with wild type controls.
Clinical manifestation of the disease was evident on day 2 both in control C57BL/6 mice and MIF ‐/‐
mice. The average ankle thickness of diseased C57BL/6 mice reached its maximum value of 2 mm
and in MIF ‐/‐ mice with 0.8 mm on day 6 and 8. In both the groups the disease decreased from day 8
on and reached normal values on day 14 (Fig 1.9 a).
Clinical Index
A significant decrease in clinical index was observed between control C57BL/6 mice and MIF‐/‐ mice
after K/BxN sera injection. The difference was significant at days 2, 4, 6, 8 and 10 (Fig 1.9 b).
DNA Ladder
Wild type (544bp)
MIF‐/‐ (388bp)

- 55 -
(a) (b)
Figure 1.9: MIF‐/‐ mice (n=8) and control C57BL/6 mice (n=8) were treated with intraperitoneal
injection of 200µl of K/BxN sera and arthritis evaluated by measuring ankle thickness (a) and clinical
index (b). There was a significant difference in ankle thickness and clinical index scores between the
two groups. The values represent the mean ± SEM of ankles/ time points, which were compared
student t‐test.
4.2.2. MIF deficiency reduced joint inflammation and cartilage destruction
To accurately assess the degree of inflammation and destruction of bone and cartilage, ankle
sections were examined using a histopathologic scoring system and found joint swelling and clinical
severity of arthritis were higher in wild type mice compared with MIF ‐/‐ (day 8 mean ±SD
inflammation score 4±0.5 in wild type mice, 1±0.5 in MIF‐/‐ mice; P < 0.0004 and cartilage destruction
score 0.9±0.5 in wild type mice, 0.2±0.5 in MIF‐/‐ mice; P < 0.0043) (Fig 1.10). Histologic evaluation of
synovial inflammation revealed high degrees of infiltration by polymorphonuclear leukocytes,
exudation of granulocytes and hyperplasia of joints in wild type compared to MIF‐/‐ mice.
Examination of the histologic features of arthritis revealed significant differences in the severity of
arthritis, including less inflammation, pannus formation, and bone and cartilage destruction in MIF‐/‐
mice joints as compared with wild type mice joints at day 8 post serum transfer (Fig 1.10).

- 56 -
(a)
(b)
P < 0.0004
P < 0.0043

- 57 -
Figure 1.10: Arthritis scores of 8 weeks old wild type C57BL/6 and MIF‐/‐ mice after administration of
K/BxN serum (n=3 per genotype per time point) (a) Inflammation (Infiltration of the synovial
membrane by polymorphonuclear leukocytes and exudation of granulocytes) and (b) cartilage
damage in mice was induced with K/BxN sera in wild type and MIF‐/‐ mice to evaluate the
contribution of MIF. The histology scores for synovial inflammation, bone and cartilage erosion are
significantly less MIF‐/‐ mice compared to the wild type C57BL/6 (P < 0.0004, 0.0043 respectively).
Bars show the mean and SEM. P values were determined by student t‐test.
4.2.3. Reconstitution of MIF‐/‐ mice with naive macrophages restored susceptibility to
arthritis
The activities of MIF in vivo and in vitro are strongly suggestive of a role for MIF in the pathogenesis
of many inflammatory diseases, including RA. However, it has not been known which cells contribute
to the MIF dependent pathology in RA. We hypothesized that macrophage were key to the
pathology of arthritis. Therefore, macrophages from C57BL/6 mice were adoptively transferred into
MIF‐/‐ mice. For this, 1.5 x 107 peritoneal macrophages suspended in 200 µl of PBS injected
intraperitoneally into MIF‐/‐ mice, and this was followed immediately by 200 µl K/BxN serum to
induce arthritis. Wild type cohorts were treated in parallel, with PBS only injections followed by
arthritis induction by K/BxN serum. These mice were assessed for arthritis development, measuring
ankle thickness (mm) and clinical index scores on days 0, 3, 5, 7, 9 and 12 (Fig 1.11).
The macrophage reconstituted MIF‐/‐ mice were found to be susceptible to arthritis induction upon
K/BxN serum transfer, as evident from the ankle thickness and clinical index scores. Typical K/BxN
serum‐induced arthritis manifested with a peak swelling around day 5‐6 and inflammation
progressed up to day 8‐12. Also, there was no difference in the inflammation (P < 0.6445) and
cartilage score (P < 0.4710) when we compared wild type with MIF‐/‐ mice reconstituted with
peritoneal macrophages (Fig 1.11c). Thus, reconstitution of MIF‐/‐ mice with peritoneal macrophages
showed that these are indeed one of the key cellular players in arthritis pathogenesis.

- 58 -
(a) (b)
(c)
Figure 1.11: Macrophage‐reconstituted MIF‐/‐ mice are susceptible to arthritis. Peritoneal
macrophages were injected intraperitoneally into the MIF‐/‐ mice, followed by 200 µl K/BxN sera. The
mice were assessed for arthritis development by ankle thickness and clinical index. MIF‐/‐ mice
reconstituted with wildtype peritoneal macrophages showed arthritis just like control C57BL/6 mice.
Data are expressed as mean ± SEM; n=5.
(c)There was no difference in inflammation and cartilage scores between wild type C57BL/6 and MIF‐
/‐ mice reconstituted with peritoneal macrophages.

- 59 -
4.3 Collagenase‐3 (MMP13) deficiency protects from antibody‐induced
arthritis
4.3.1. An Essential role for MMP13 in K/BxN antibody‐induced arthritis
Transfer of K/BxN serum into normal mice induced rapid and synchronous development of arthritis,
the first signs of joint inflammation appearing with in 24 hrs in fully susceptible strains. The
degradation of articular cartilage is a characteristic of rheumatoid arthritis. MMP 13 has been
suggested to play a role in this degradation. Expression of MMP 13 is low in normal cells, and these
low levels allow for healthy connective tissue remodeling (Uitto et al., 1998). In the pathogenic
conditions, however the level of MMP 13 expression increases considerably, resulting in connective
tissue destruction. Excess MMP‐production is associated with the pathology of many diseases,
including atherosclerosis tumour invasion/metastasis and arthritic diseases.
To elucidate the role of MMP 13 in the antibody induced arthritis, MMP 13‐/‐ and wild type mice
were injected with 200µl of K/BxN sera intra‐peritonealy at day 0. After induction of arthritis, the
control mice showed typical signs and symptoms of arthritis with increasing ankle thickness and
clinical index while MMP 13‐/‐ mice showed 50% reduction of disease. Arthritis was monitored every
2 days by measuring ankle thickness and clinical index. The onset of arthritis and ankle thickening
started during 24 h to 48 h after injection of sera in both MMP13‐/‐ and control C57BL/6 mice
(Figure1.11 a). The ankle thickness increased to a maximum value around day 10 after which the
diseases resolved until day 18 (Figure 1.11 a). The average ankle thickness of the MMP13‐/‐ mice was
decreased by 1.5 fold (P<0.0001) when compared to the wild type. The MMP13‐/‐ mice showed
significant difference in the ankle thickness values when compared to the wild type C57BL/6 mice.
Inflammation and joint destruction was assessed histologically from ankle sections of mice sacrificed
on days 0, 2, 6, 8 and 14. As expected severe cellular infiltration and synovial proliferation was
observed in the arthritic wild type mice on day 8. Cartilage and bone destruction was observed on
histological sections of mice sacrificed on day 6 and day 8 (Fig 1.12). There was a difference in

- 60 -
synovial cells infiltration and pannus tissue formation between MMP13‐/‐ and control mice. These
clear cut results indicate that in this serum‐transfer model mediated by arthritogenic Igs, MMP13
plays an essential role, critically required for disease progression.
Clinical Index
A significant decrease in clinical index was observed between control C57BL/6 mice and MMP 13‐/‐
mice after K/BxN sera injection. The difference was significant at days 10, 12 and 14 (Fig 11.1 b).
(a) (b)
Figure 1.11: MMP13 ‐/‐ mice were protected from antibody‐induced arthritis.
MMP13‐/‐ mice (n=21) and control C57BL/6 (n=20) mice were treated with 200µl of K/BxN sera and
arthritis evaluated by measuring ankle thickness (a) and clinical index (b). There was a significant
difference in ankle thickness and clinical index scores between the two groups. The values represent
the mean ± SEM of ankles/ time points, which were compared using the student t‐test.
Mice Incidence Day of Onset Max AT Max CI Histo. Score
C57BL/6 20 1, 2, 3, 4 2.9 4.0 6, 1.5
MMP13 -/- 21 1, 2, -, - 1.5 2.0 2, 0.8

- 61 -
Tabulation of results for 21 MMP 13 ‐/‐ mice and age/gender matched 20 control mice (C57/BL6);
Max AT=maximum increase in ankle thickness in millimeter. Max CI=maximum clinical index (for each
limb: 0, no disease; 0.5, mild swelling of paw; 1, clear joint inflammation in ankle). Histological score,
inflammatory score and cartilage destruction.
4.3.2. Joint inflammation and cartilage destruction in MMP13‐/‐ mice
Inflammation and joint destruction was assessed histologically from ankle sections of mice sacrificed
on days 0, 2, 6, 8 and 14. K/BxN sera was injected in wild type and MMP13‐/‐ to evaluate the
contribution of MMP13 in arthritis development and progression. Joint swelling and clinical severity
of arthritis were higher in wild type mice compared with MMP 13‐/‐ (day 8 mean ±SD inflammation
score 6±0.5 in wild type mice, 1.8±0.5 in MMP13‐/‐ mice; P < 0.05 and cartilage destruction score
1.5±0.5 in wild type mice, 0.6±0.5 in MMP13‐/‐ mice; P < 0.0043) (Fig 1.12). Histologic evaluation of
synovial inflammation revealed high degrees of infiltration by polymorphonuclear leukocytes,
exudation of granulocytes and hyperplasia of joints in wild type compared to MMP13‐/‐ mice.
Examination of histologic features of arthritis revealed that there was a significant difference in the
severity of arthritis, including the extent of inflammation, bone and cartilage destruction in MMP13 ‐
/‐ mice joints as compared to the wild type joints at days 4 and 8 post serum transfer (Fig 1.12 b).
Histological analysis of joints on day 8 from wild type arthritic mice demonstrated a characteristic
inflammation, synovial hypertrophy and joint erosion, findings that were completely absent or
minimally present in MMP13‐/‐ mice (Fig1.13). On day 8 post serum transfer wild type mice (Fig 1.13
b) showed, the proliferating inflamed synovial tissue (pannus) expanded over the surface of the
articular cartilage, and at places of contact of the pannus tissue with the cartilage matrix and/or
bone degradation of the cartilage and erosion of the bone surface, whereas in MMP13‐/‐ mice there
was no cartilage destruction and pannus formation.

- 62 -
(a)
( b)
Figure 1.12: Arthritis scores of 8 weeks old wild type C57BL/6 and MMP13‐/‐ mice after
administration of K/BxN serum (n=3 per genotype per time point) (a) Inflammation (infiltration of the
synovial membrane by polymorphonuclear leukocytes and exudation of granulocytes) and (b)
cartilage damage in mice was induced with K/BxN sera in wild type and MMP13‐/‐ mice to evaluate
the contribution of MMP13 in arthritis. The histology scores for synovial inflammation, bone and
cartilage erosion were significantly less in MMP13‐/‐ mice compared to the wild type C57BL/6 mice
(P<0.05). Bars show the mean and ± SEM. P values were determined using student t‐test.

- 63 -
(a)
(b)
(c)
Figure (1.13): Representative histology of ankle joints from wildtype C57BL/6 and MMP13‐/‐ mice
during (a) day 0 (before sera transfer) and (b,c)day 8 (post sera transfer.(b) wildtype joints display
synovial inflammation, cartilage destruction, pannus tissue formation, bone erosion and synovial

- 64 -
hypertrophy whereas (c) MMP13‐/‐ joints were free of inflammation and the synovium retained its
relatively acellular composition.
4.3.3. MMP13‐/‐ mice display reduced numbers of infiltrating neutrophil and increased
number of monocytes in the joints.
The K/BxN antibody‐induced arthritis is characterized by high neutrophil infiltration during onset of
disease. The infiltrating cells in wild type and MMP13‐/‐ ankle mainly consist of lymphocytes,
granulocytes and macrophages. However, MMP13‐/‐ mice had less number of neutrophils compared
to wild type C57BL/6 mice. There was an increase in the numbers of neutrophils (Fig 1.14 a)and
surprisingly a decrease in the numbers of monocytes (Fig 1.14 b) found in wild type C57BL/6 mice at
day 7 after serum transfer as compared to MMP13‐/‐ mice. FACS analysis showed that the Ly6G and
CD11b populations were decreased and increased in MMP13‐/‐ mice respectively. The number of
neutrophils in wild type joints correlated with the levels of inflammation and ankle thickness. These
data suggested that less inflammation and joint destruction as seen in the MMP13‐/‐ following K/BxN
serum transfer may be due to lower number of blood neutophils.
(a) (b)

- 65 -
Figure 1.14: (a) Less neutrophils and (b) more monocytes were found in MMP13‐/‐ peripheral blood
following injection of K/BxN serum
Immunophenotying of peripheral blood cells showed an increase of neutrophils and a decrease of
monocytes in the blood of MMP13 ‐/‐ mice compared to wild type C57BL/6 mice at day 7. Peripheral
blood was isolated from the tail vein of mice. Red blood cells were lysed with lysis buffer followed by
staining with Ly6G and CD11b antibodies for 30 minutes.
4.4. Role of redox reactions in RA antibody‐induced arthritis
4.4.1. In the K/BxN model phytol‐treated mice show an early onset of arthritis
To elucidate the role of changes in the redox state, phytol (3, 7, 11, 15‐tetramethyl‐2‐hexadecene‐
1ol) was applied in the antibody‐induced arthritis model. For this Balb/c mice were injected with 200
µl of phytol. After five days, arthritis was induced in the phytol‐ treated and control Balb/c mice.
Onset and progression of arthritis was observed through the next two weeks. Arthritis was
monitored every two days by measuring ankle thickness and clinical index. The onset of ankle
thickness and clinical index occurred within a 24 hrs in phytol‐treated mice. The phytol‐treated mice
showed enhanced arthritis during the first autoimmune phase of the K/BxN model. After day 4, the
disease started resolving and on day 8 the disease was resolved completely. The average ankle
thickness of the phytol‐treated mice reached 2.5 mm on day 4 and the disease decreased from day 6
and reached normal values on day 10 as opposed to control Balb/c mice (Fig 1.15).
Immunohistological examination of normal mouse joint tissue showed the presence of GPI in the
articular cavity, along the cartilage surface; these GPI deposits were amplified in arthritic mice
(Matsumoto et al., 2002). During autoimmune phase arthritogenic K/BxN sera containing anti‐GPI
antibodies bind directly to pre‐existing extracellular GPI in normal healthy mouse joints, followed by
the formation of immune complexs on the articular surface. And in the chronic phase mice showed
histological features like leukocyte invasion, synovitis, pannus formation, cartilage, and bone
destruction. In control mice the arthritis appeared on day 2 and the average ankle thickness reached
its maximum value with 3.0 mm on day 8 and disease was resolved by day 14. Comparing phytol‐

- 66 -
treated and control mice, difference in the reduction of ankle thickness was significant at days 6, 8,
10 and 12 (Fig 1.15 a).
Clinical Index
A significant decrease in the clinical index was observed between control Balb/c mice and phytol‐
treated mice after K/BxN sera injection. The difference was significant at days 6, 8, 10 and 12 (Fig
1.15 b).
(a) (b)
Figure 1.15: Effect of phytol in K/BxN sera‐induced arthritis. Control Balb/c mice (n=21) and
Phytol‐treated mice (n=21) were injected with 200µl of K/BxN sera i.p. on day 0 and arthritis
evaluated by measuring ankle thickness (a) and clinical index (b).There was a significant
difference in ankle thickness and clinical index scores between the two groups. The values
represent the mean ± SEM of ankles/time points, using student t‐test to the wild type
mice.***‐P<0.0001, **‐P<0.001, *‐P<0.05.
4.4.2. Increasing the number of R–SH groups on control neutrophils increased the arthritis
To measure the overall level of cell surface R‐SH, we took advantage of the readily available
AlexaFluor‐maleimide reagents. The nucleophilic maleimide covalently couples to an available R‐SH
moiety. Maleimide is effective in its ability to react with thiols and is considered to be highly specific

- 67 -
for thiol groups. The Alexa dyes that are coupled to maleimide do not interfere with the maleimide‐
thiol interaction and as they are charged molecules, do not enter the cell, thus enabling a FACS‐
detectable, overall cell surface‐thiol determination. For assessment of surface thiol levels in phytol‐
treated mice cells were stained as described in the material and method section (Chapter III). After
K/BxN sera transfer surface thiol levels on neutrophils were significantly decreased on day 6
compared with control mice. The decreased surface thiol levels parallel the resolving disease on day
6. During the autoimmune phase the surface thiol levels were increased and in pathogenic phase
surface thiol levels decreased.
Figure 1.16: Surface thiol levels on neutrophils. Phytol treated mice (n=15) have significantly lower
levels of surface thiol on neutrophils than the control mice (n=15) at day 6. Cells were stained with
Alexa maleimide (10µM) followed by staining with Ly6G antibodies. The levels of thiol on the surface
of neutrophils are presented as a units of mean fluorescence intensity (MFI). The data are mean
±SEM,*, Significant difference (P<0.05).
4.4.3. No change of surface thiol levels on monocytes
Surface thiol levels on monocytes were determined in the peripheral blood collected from phytol‐
treated and control mice Balb/c. After lysisof red blood cells the leukocytes were stained with
*

- 68 -
antibodies of R‐SH group (Alexa maleimide dye) and CD11b (monocyte marker). After FACS analysis,
we found there was no significant change in the monocytes population as compared with control
mice.
Figure 1.17: Surface thiol levels on monocytes. Surface thiol levels on monocytes were determined by
FACS. Peripheral blood was isolated from mice tail vein. Blood cells were lysed with lysis buffer and
then leukocytes were stained with Alexa maleimide dye (10µM) followed by staining with CD11b
antibodies. The difference was not significant comparing phytol‐treated (n=15) and control mice
(n=15), P< 0.9619.
4.4.4. Measurement of intracellular ROS in neutrophils
Polymorphonuclear neutrophils (PMNs) are crucial in host defence against invading microorganisms
through reactive oxygen species (ROS) production. However, generated ROS released in excess can
damage the host tissue. Intracellular PMN ROS‐production was measured using 2′‐7′‐
dichlorofluorescen diacetate (DCFH‐DA) and flow cytometry. The dyes is nonfluorescent until
oxidized by ROS and an increase in fluorescence of DCFDA indicates oxidation by peroxides,
peroxynitrite, and/or hydroxyl radicals. PMN were incubated with DCFDA before Ly6G staining, and
the median fluorescence intensity was determined. Fig 1.19 demonstrates that there were no

- 69 -
differences in intracellular ROS in PMN comparing control and phytol‐treated mice after K/BxN sera
transfer.
Figure 1.19: Intracellular ROS levels in neutophils. There was no significant difference when we
compared control mice with phytol‐treated mice, P < 0.9432.
4.5.5. Measurement of intracellular ROS in monocytes
Comparisons between the control and phytol treated monocytes revealed that the generation of
ROS by phytol‐treated cells was insignificantly higher than in the controls. There was no difference in
intracellular ROS generation in monocytes after phytol‐treatment when compared with the MFI of
control mice.

- 70 -
Figure 1.20: Intracellular ROS levels in monocytes. The levels were insignificantly increased in phytol
treated mice (n=15) on day 6 as compared to control mice (n=15), P < 0.8607.
4.4.6. Number of neutrohils increased after phytol treatment
The absolute number of neutrophils after phytol‐treatment were measured using true count beads.
The number of neutrophils were calculated per microliter. We found that before sera induction the
numbers of neutrophils were significantly higher in phytol treated mice as compared to control mice
but after sera transfer there was no significant difference between the groups.
Figure 1.21: Neutrophils were counted by BD true count beads. At day 0, phytol‐treated mice (n=8)
neutrophil counts were significantly higher than in control mice (n=8), P < 0.008.
4.4.6. No change in the levels of surface thiol in young and old KBN and BN mice
Surface redox levels on monocytes and neutrophils from young and old KBN mice and young and
old BN mice were determined by FACS after staining the cells for reduced thiol (‐SH) groups and
leukocytes specific markers. No difference in the levels of surface thiols on monocytes and
neutrophils was observed when we compared young KBN with young BN and old KBN with an old BN
mice. There were increased surface thiol levels on monocytes and neutrophils in old KBN and old BN
than the young KBN and BN mice, although the difference was not significant.
**
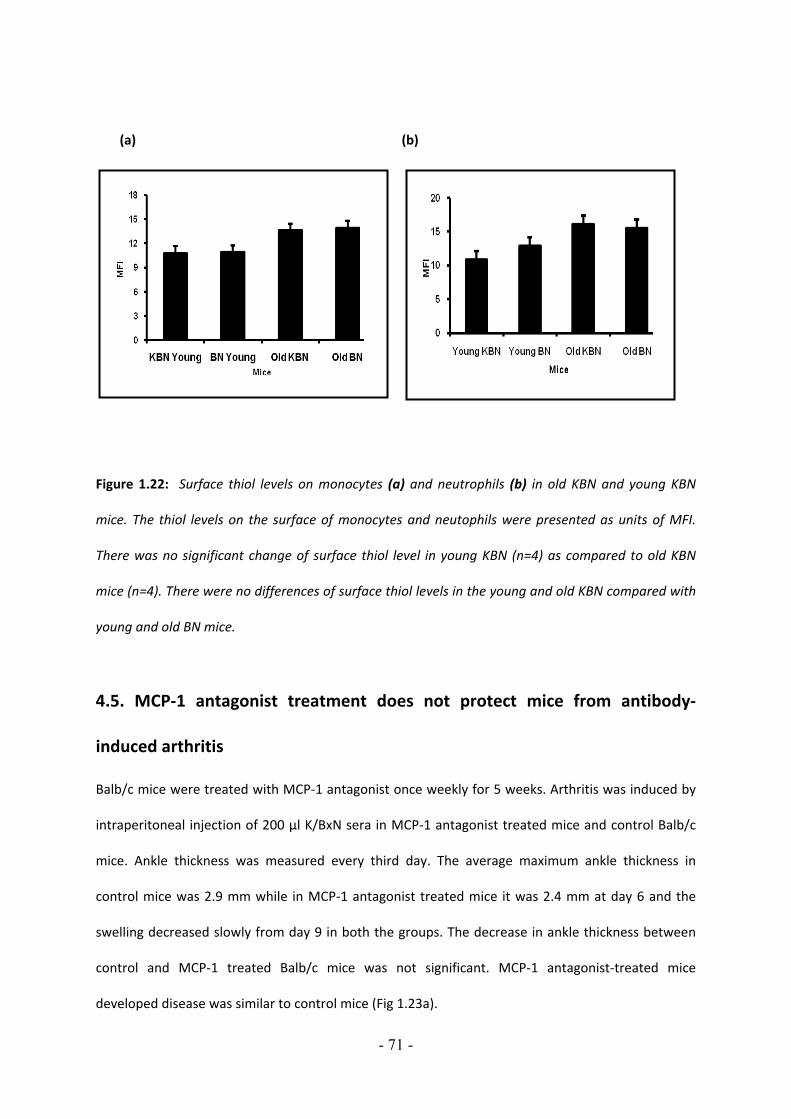
- 71 -
(a) (b)
Figure 1.22: Surface thiol levels on monocytes (a) and neutrophils (b) in old KBN and young KBN
mice. The thiol levels on the surface of monocytes and neutophils were presented as units of MFI.
There was no significant change of surface thiol level in young KBN (n=4) as compared to old KBN
mice (n=4). There were no differences of surface thiol levels in the young and old KBN compared with
young and old BN mice.
4.5. MCP‐1 antagonist treatment does not protect mice from antibody‐
induced arthritis
Balb/c mice were treated with MCP‐1 antagonist once weekly for 5 weeks. Arthritis was induced by
intraperitoneal injection of 200 µl K/BxN sera in MCP‐1 antagonist treated mice and control Balb/c
mice. Ankle thickness was measured every third day. The average maximum ankle thickness in
control mice was 2.9 mm while in MCP‐1 antagonist treated mice it was 2.4 mm at day 6 and the
swelling decreased slowly from day 9 in both the groups. The decrease in ankle thickness between
control and MCP‐1 treated Balb/c mice was not significant. MCP‐1 antagonist‐treated mice
developed disease was similar to control mice (Fig 1.23a).

- 72 -
Clinical Index
The clinical index was measured every third day in both the groups. There was no decrease in the
clinical index comparing control mice and MCP‐1 antagonist‐treated mice (Fig 1.23b).
(a) (b)
Figure 1.23: MCP‐1 antagonist‐treated mice were not protected from serum‐induced arthritis.
Balb/c mice were treated with MCP antagonist (2mg/kg body weight) and control mice were treated
with vehicle only. Arthritis was induced injecting 200 µl K/BxN sera. The difference was not significant
in ankle thickness (a) and clinical index (b) between MCP‐1 antagonist treated mice (n=10) and
control mice (n=10).The values represent the mean ± SEM of ankles/ time points, using student t‐test.
4.6. DBA mt FvB mice are not protected from antibody induced arthritis
Rheumatoid arthritis (RA) is a complex, multifactorial polygenic disease that is influenced by age and
sex as well as hormonal and environmental factors (Silman, 1994). Susceptibility to RA is controlled
by genetic factors. Quantitative traits such as complex diseases are controlled by many genes with
small‐effect that are difficult to identify. The genetic contribution to RA susceptibility is estimated to
be as much as 60%, of which the HLA– DRB1 locus is thought to account for 30–50% (MacGregor et
al., 2000; Wordsworth et al., 1989). For arthritis severity, recently a new QTL, Cia2 and Cia4 was

- 73 -
identified (Bauer et al., 2004). Cia2 control the severity and Cia4 control the onset of disease.
However, Cia2 contributed only to 16% to the phenotypic variations, indicating that there should be
some small‐effect QTL.
Here, we used (DBA/1J × FVB/N) F2 progeny for the sera‐induced arthritis and DBA/1 used as wild
type control. We found that DBA mt FvB mice were as highly sensitive to arthritis as were wildtype
DBA/1 mice.
Arthritis was evaluated by measuring ankle thickness every two days. The average maximum ankle
thickness in control DBA/1 mice it was 3.8 mm while in DBA mt FvB mice was 3.4 mm at day 6 and
the swelling decreased slowly from day 9 in both the groups. The decrease in ankle thickness
between DBA/1 and DBA mt FvB mice was not significant. DBA mt FvB sera treated mice developed
arthritis and progress of the disease was similar as DBA/1 mice (Fig 1.24 a).
Clinical Index: The clinical index was measured every third day in both the groups. There was no
decrease in clinical index comparing DBA/1 mice and DBA mt FvB sera‐treated mice (Fig 1.24 b).
(a) (b)

- 74 -
Figure 1.25: DBA mt FvB mice were not protected from serum induced arthritis. DBA mt FvB (n=5)
and wild type DBA/1 (n=5) were injected with 200µl of pooled serum from arthritic K/BxN mice on
day 0. Arthritis was evaluated by measuring ankle thickness (a) and clinical index (b) every 2 days.
The values represent the mean ± SEM of ankles/ time points, using student t‐test.

- 75 -
4.7. Soluble CD21 and CD23 concentration in autoimmune diseases
4.7.1. Antiphospholipid syndrome patients display reduced titers of soluble CD21 in their
sera irrespective of circulating anti‐beta‐2‐glycoprotein‐I autoantibodies.
Sera from 49 APS patients and 24 controls (table 1) were analyzed for the presence of sCD21 using a
sandwich ELISA. The sera were diluted 1:50 and applied to the ELISA assay. For control standard sera
with known sCD21 titer was used in serial dilution. Comparing standard, patients and healthy control
sera we determined the concentration of sCD21 in ng/ml. We used the Mann‐Whitney test for
statistical analysis. The level of sCD21 in healthy control ranged from 100‐500 ng/ml (median 330
ng/ml) while in APS it was 10‐250 ng/ml (Median 121.95 ng/ml) (Fig 2.1). Thus sCD21 is significantly
reduced in APS patients sera compared to healthy controls.
p<0.0001
Healthy Doner APS0
250
500
750Healthy DonerAPS
Figure2.1. Serum soluble CD21 (sCD21) levels in Antiphospholipid Syndrome (APS). Serum sCD21
concentrations of APS patients (n=49) were compared to healthy donors (n=24). Serum sCD21 levels
are significantly decreased in APS patient samples (P<0.0001). Horizontal lines in the graph represent
the median value. Statistical significance was calculated using nonparametric Mann‐Whitney test.

- 76 -
Samples n p<0.05 Rank
Control/APS 24/49 *** S
APS‐SLE‐ve/+ve 14/34 0.7832 Ns
APS‐DVT‐ve/+ve 16/32 0.0716 Ns
APS‐B2GPI‐ve/+ve 24/24 0.1539 Ns
APS‐P‐APS‐ve/+ve 21/27 0.8835 Ns
APS‐Art‐Th‐ve/+ve 20/28 0.3953 Ns
APS‐TH‐ve/+ve 31/16 0.7448 Ns
APS‐FETAL‐ve/+ve 35/13 0.1635 Ns
APS‐Joints‐ve/+ve 34/12 0.3656 Ns
Table 1. Serum soluble CD21 levels in other disorders of Antiphospholipid Syndrome. There were no
significant changes in serum sCD21 levels. Statistical significance was calculated using nonparametric
Mann‐Whitney test.
Table 2
APS Patients Characteristic (n=49) %*
Deep Vein Thrombosis(DVT) 16.5
Beta 2GPI (IgM) 12.5
Beta 2GPI (IgG) 25
Primary antiphosholipid Syndrome (P‐APS) 14
Systemic Lupus Erythematosus (SLE) 7.5
Thrombosis 22
Fetal Loss 6.5
Venous Thrombosis 17
Arterial Thrombosis (Leg or hands) 2.5
Transient Ischmemic Attcak (TIA) 2.5
Stroke 3.5
Multiinfarct dementia 7.5
Pulmonary embolism and infarction 6.5
Digital gangrene 0.5
Retinal artey Thrombosis 0

- 77 -
Arterial Thrombosis 10.5
Epilepsy 1.5
Migraine 2
Vegetations 0.5
Avascular necrosis of bone 0.5
Joints 6.5
Livedo reticularis 2
Autoimmune hemolytic anemia 1
Thrombocytopenia 8.5
*The values given represent percentage of patients which were positive for the symptom.
4.7.2. Soluble CD21 levels in Autoimmune Lymphoproliferative Syndrome (ALPS).
Autoimmune Lymphoproliferative Syndrome (ALPS) is an inherited disorder of the immune system
that affects both children and adults. Autoimmune lymphoproliferative syndrome is caused by
genetic mutations that interfere with apoptosis or programmed cell death. Normally, all of the
body’s cells have a programmed cell death or survival time known as apoptosis. And normally,
apoptosis prevents the accumulation of mature lymphocytes. symptoms in autoimmune
lymphoproliferative syndrome include moderate to massive splenomegaly (enlarged spleen), skin
rashes, frequent nosebleeds, enlarged lymph nodes especially in the neck, enlarged liver, petechia
(small bruises), edema, evidence of autoimmunity expressed usually as autoimmune hemolytic
anemia, increased levels of gamma immunoglobulins (hypergammaglobulinemia), B‐cell
lymphocytosis (increased number of B‐lymphocytes), thrombocytopenia (decreased platelets),
autoimmune neutropenia (decreased number of segmented granulocytes), and the expansion of an
unusual population of CD4‐CD8‐ T cells (lymphocytes with markers for T‐helper and T‐suppressor
functions) that express the alpha/beta T‐cell receptor (TCR).
The soluble CD21 concentration were estimated in the sera from 17 ALPS patients and compared
with healthy volunteers. The range of sCD21 in healthy individuals was 50‐300 ng/ml with a median
of 200±5 ng/ml while the levels of sCD21 in ALPS patients were 50‐1300 ng/ml with a median 650±5
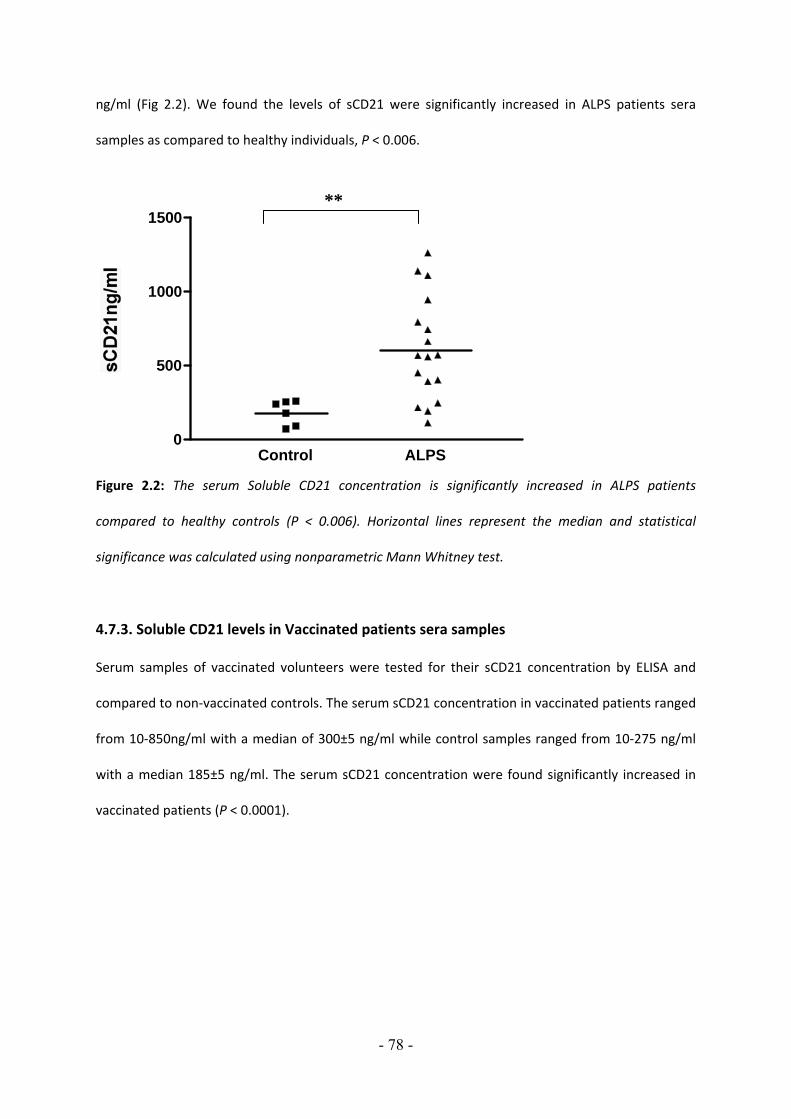
- 78 -
ng/ml (Fig 2.2). We found the levels of sCD21 were significantly increased in ALPS patients sera
samples as compared to healthy individuals, P < 0.006.
Control ALPS 0
500
1000
1500
Figure 2.2: The serum Soluble CD21 concentration is significantly increased in ALPS patients
compared to healthy controls (P < 0.006). Horizontal lines represent the median and statistical
significance was calculated using nonparametric Mann Whitney test.
4.7.3. Soluble CD21 levels in Vaccinated patients sera samples
Serum samples of vaccinated volunteers were tested for their sCD21 concentration by ELISA and
compared to non‐vaccinated controls. The serum sCD21 concentration in vaccinated patients ranged
from 10‐850ng/ml with a median of 300±5 ng/ml while control samples ranged from 10‐275 ng/ml
with a median 185±5 ng/ml. The serum sCD21 concentration were found significantly increased in
vaccinated patients (P < 0.0001).
**

- 79 -
Heathy Donor Vaccinated Donor0
250
500
750
Figure 2.3: Serum sCD21 concentration of vaccinated volunteers (n=40) were estimated and
compared with non‐vaccinated controls (n=25). Serum sCD21 was significantly increased in
vaccinated patients (P < 0.0001). Statistical significance was calculated using nonparametric Mann
Whitney test.
4.7.4 Decreased levels of sCD21 and sCD23 in patients with autoimmune diabetes type I
not with type II
4.7.4.1. Levels of serum soluble CD21 are decreased in sera of patients with Diabetes type I
but not with Diabetes type II
Sera from 30 age matched healthy donors and 30 each diabetes type I and II patients were analyzed
for the presence of sCD21 using a sandwich ELISA. The sera were diluted 1:50 and applied to the
ELISA assay. For control standard sera with known sCD21 titer was used in serial dilution.
Comparing standard, patients and healthy control sera we determined the concentration of sCD21 in
ng/ml. We compared sCD21 concentration in age matched healthy controls with those in diabetic
type I and II patients sera samples and found a statistically significant reduction with diabetes type I
(P < 0.0067) and no statistically significant reduction with diabetes type II patients sera samples (P <
***

- 80 -
0.8603). The levels of sCD21 in healthy control ranged from 30 to 400 ng/ml (Median 204 ng/ml),
diabetes type I and II were 50‐400ng/ml (Median 166 ng/ml) and 100‐400ng/ml (Median 216 ng/ml)
respectively (Fig 2.4.1). Mann‐ Whitney test was used for statistical comparison.
Control Diabetes I Diabetes II 0
100
200
300
400
Figure 2.4.1: The serum sCD21 concentration was significantly decreased in diabetes I (n=30) (P <
0.0067) but not in diabetes II (n=30) patients as compared to healthy controls (n=30). Statistical
significance was calculated using the nonparametric Mann Whitney test.
4.7.4.2. Levels of Serum soluble CD23 levels in sera of patients with Diabetes type I are
decreased but not with diabetes type II
Serum sCD23 concentration of diabetic I and II patients (n=30 each) were estimated and compared
to age‐matched healthy donors (n=30). The sera samples were diluted 1:5 and applied to the sCD23
ELISA assay. The median values of sCD23 in healthy donor were 72 U/ml while in diabetes type I and
II were 42 U/ml and 63 U/ml respectively (Fig 2.4.2). A significant decrease of sCD23 in sera of
diabetic type I patients (P <0.0001) and non significant increase in diabetes type II (P < 0.5692)
compared to age matched healthy controls. The Mann‐Whitney test was used for statistical analysis.
**

- 81 -
Control Diabetes I DiabetesII0
20
40
60
80
100
Figure 2.4.2: The serum sCD23 concentration of diabetes I (n=30) patients were estimated and
compared with healthy controls (n=30). Serum sCD23 levels were significantly decreased in diabetes I
(P < 0.0001) samples but not in diabetes II. Horizontal lines represent the median and statistical
significance was calculated using nonparametric Mann Whitney test.
4.8.4.3. Decreased levels of plasma sCD21 after pro insulin treatment in diabetic obese
patients
Next we measured sCD21 concentration in plasma of obese diabetic patients after (n=87) and before
(n=87) proinsulin treatment. The median values of sCD21 in the pre treatment group were 200
ng/ml and in after treatment were 174 ng/ml. We found sCD21 levels were significantly reduced in
patients after treatment (n=87) with pro insulin as compared with before treatment (n=87) (P <
0.001).We used the Mann‐Whitney test for statistical analysis (Fig 2.4.3).
***

- 82 -
Before Treatment After Treatment0
250
500
750
Figure 2.4.3: The serum soluble CD21 concentration was significantly decreased in the diabetic
patients with BMI < 90 treated with proinsulin (n=87) compared to untreated group (n=87) (P<0.001).
Horizontal lines in the graph represent the median value. Statistical significance was calculated using
the nonparametric Mann Whitney test.
4.8.4.4. Increased levels of plasma sCD23 after pro insulin treatment in diabetic obese
patients
40 Plasma samples from obese diabetic patients before and after treatment with pro insulin were
analyzed for the presence of sCD23 using a sandwich ELISA. The patient plasma samples were
diluted 1:5 and applied to the sCD23 ELISA assay. The levels of sCD23 in pre treatment group with
median 101 U/ml and in after treatment group were 132 U/ml. Soluble. We found sCD23 levels were
significantly increased in patient’s after treatment (n=40) with proinsulin treatment as compared
with before (n=40) treatment (P < 0.0001).We used the Mann‐Whitney test for statistical analysis.
**

- 83 -
Before Treatment After Treatment0
100
200
300
Figure 2.4.4: The serum soluble CD23 concentration was significantly increased in the diabetic
patients with BMI < 90 (n=40) of treated group compared to untreated persons (n=40) (P<0.0001).
Horizontal lines in the graph represent the median value. Statistical significance was calculated using
the nonparametric Mann Whitney test.
Samples P‐Value Significance
sCD21
Diabetes I (n=30) 0.0067 **
Diabetes II (n=30) 0.8603 ns
sCD23
Diabetes I (n=30) 0.0001 ***
Diabetes II(n=30) 0.5692 ns
Table 1: P‐values of sCD21 and sCD23 of diabetic I and II samples. Statistical significance was
calculated by nonparametric student t‐test of graph pad prism software.
***

- 84 -
4.7.5. Decreased levels of sCD21 and sCD23 in blood of patients with
Systemic Juvenile Arthritis, Polyarticular Juvenile Arthritis and Pauciarticular
Juvenile Arthritis
4.7.5.1. Low levels of soluble CD21 in plasma of patients with JA
Plasma from 20 age‐matched healthy donors and 20 of each subtype of JA patients were analyzed
for the presence of sCD21 using a sandwich ELISA. The plasma were diluted 1:50 and applied to
the ELISA assay. For control standard sera with known sCD21 titer were used in serial dilution.
Comparing standard, patients and healthy control plasma, we determined the concentration of
sCD21 in ng/ml. The median levels of sCD21 in healthy controls were 700ng/ml, in O‐JA 200 ng/ml,
in P‐JA 400 ng/ml and in S‐JA 225ng/ml (Fig 2.5.1). When we applied the Mann‐Whitney test
comparing data from all subtypes of JA with controls we found that sCD21 levels were significantly
reduced in all subtypes of JA as compared with healthy donors (P < 0.0001 for O‐JA and S‐JA and P
< 0.0068 for P‐JA).
We also compared the sCD21 levels among the JA subtypes. Comparing P‐JA with O‐JA and S‐JA
showed significant decrease in sCD21 (P < 0.0001 and 0.0036 respectively).

- 85 -
Control O-JA P-JA S-JA0
200
400
600
800
1000
1200
Figure 2.5.1: Plasma sCD21 levels in patients suffering from different forms of Juvenile Arthritis.
Plasma sCD21 concentration of O‐JA (n=20), P‐JA (n=20) and S‐ JA (n=20) were determined and
compared to healthy controls. Plasma sCD21 levels were significantly decreased in all subtypes of
JA samples (P < 0.0001). Horizontal lines represent the median. Statistical significance was
calculated using nonparametric student t‐test.
4.7.5.2. Low levels of soluble CD23 in plasma of patients with JA
Next we measured sCD23 levels in the plasma of patients representing the 3 subtypes of JA by
ELISA. Median sCD23 levels in healthy controls were 45 U/ml, in O‐JA 20 U/ml, 30 U/ml in P‐JA and
35 U/ml in S‐JA (Fig 2.5.2). The levels of sCD23 were significantly reduced in P‐JA (P < 0.0001) and
S‐JA (P < 0.0184) while the reduction of sCD23 in O‐JA was not significant as compared with
healthy controls.
Unlike sCD21 levels comparing the subtypes of JA, sCD23 levels were significantly increased when
we compared P‐JA with S‐JA (P < 0.009) but we found non‐significant decreased levels when we
compared P‐JA with O‐JA (P < 0.4181).
***
*** **
*** **

- 86 -
Control O-JA P-JA S-JA0
50
100
150
200
Figure 2.5.2: Plasma sCD23 levels in patients suffering from different forms of Juvenile arthritis.
The sCD23 concentrations were measured in plasma from healthy controls (n=20) and compared
with JA patient samples. Plasma sCD23 were significantly decrease in P‐JA (P < 0.0001) and S‐JA
samples (P < 0.0184). Horizontal lines represent the median. Statistical significance was calculated
using the nonparametric student t‐test.
4.7.5.3. Plasma sCD23 but not sCD21 levels correlate with age in healthy donors
In the previous study (Masilamani et al., 2003) we found that sCD21 levels decline significantly
with age, comparing groups spanning 0‐20, 21‐40 years of life and so on. The decline was stronger
in the younger age, therefore we here analyzed children grouped in cohorts spanning 5 years of
age (0‐5, 6‐10 and 11‐15 years). Analyzing healthy donors, we found that levels of sCD21
significantly decrease with age comparing the 0‐5 years group with the 11‐15 group (P < 0.0077)
(Fig 2.5.3a). The levels of sCD23 decreased with age comparing the 0‐5 years group with the 6‐10
and the 11‐15 years group (P < 0.0068, 0.0021 respectively, Fig 2.5.3b).
**
*
***

- 87 -
(a) (b)
Figure 2.5.3: Plasma sCD23 not sCD21 concentration decreases with age. The data were grouped
0‐5, 6‐10, 11‐15 years age group. The Mann‐Whitney test was done to obtain non‐parametric two‐
tail P values (a) The sCD21 levels was significant decreased in 11‐15 years age group when
compared with 0‐5 years age group (P < 0.0077) whereas (b) sCD23 values were significant
decreased in 6‐10 and 11‐15 years age group when compared with 0‐5 years age group( P <
0.0068 and 0.0021 respectively ).
4.7.5.4. Plasma sCD21and sCD23 levels decline with age in JA
sCD21 and sCD23 levels were determined in all age groups as above comparing the JA subtypes
with healthy controls. sCD21 levels in S‐JA in the 0‐5 year group were 300 ng/ml average
compared to 800 ng/ml in the control group (difference statistically not significant). However, the
difference was significant in the 6‐10 (median 257 ng/ml) and 11‐15 years age group (median 230
ng/ml) of S‐JA when compared with the control groups (6‐10 years median 609 ng/ml; 11‐15 years
median 500 ng/ml (P < 0.0039, 0.0057 respectively) (Fig 2.5.4a).
Like in S‐JA, sCD21 levels were also significantly decreased with age in the 6‐10 years group
(median 255 ng/ml) and the 11‐15 years group (median 195 ng/ml) in O‐JA when we compared
**
**
0-5 6-10 11-150
500
1000
1500
2000
Age (Years)0-5 6-10 11-15
0
100
200
300
400
500
Age (Years)
**
***

- 88 -
with their respective controls (group 6‐10 years median 609 ng/ml; group 11‐15 years median 500
ng/ml (P < 0.0038, 0.0004 respectively) (Fig 2.5.4b). In contrast there was no significant difference
in the P‐JA age groups (data not shown).
Analysis of sCD23 levels in S‐JA showed a statistically significant difference when compared to the
levels of healthy controls only in the 6‐10 years group (P < 0.01) (Fig 2.5.5a).
Similarly, in P‐JA, the sCD23 levels in the 6‐10 year age group (median 25 U/ml) were significantly
lower compared with the controls (median 35 U/ml) (P < 0.0001) (Fig 2.5.5b).
In O‐JA, sCD23 levels were significantly lower in the 6‐10 years age group (median 25 U/ml) and
11‐15 years age group (median 20 U/ml) compared with the respective control 6‐10 years
(median 35 U/ml) and 11‐15 years age group (median 35 U/ml) (P < 0.005) (Fig 5c). Thus the
reduction of sCD23 and sCD21 in JA is independent of age.
(a) (b)
S-JA
6-10 6-10 11-15 11-150
250
500
750
1000
Age (Years)
Figure 2.5.4: Levels of sCD21 in JA subtypes patients are low irrespective of age. The control and JA
subtypes sCD21 data were grouped according to age. sCD21 levels from healthy volunteers were
compared with the corresponding age group of (a) S‐JA and (b) O‐JA.
*****
** ***
O-JA
6-10 6-10 11-15 11-150
250
500
750
1000
Age (Years)

- 89 -
(a) (b)
S-JA
6-10 6-10 11-15 11-150
50
100
150
200
Age (Years)
(c)
O-JA
6-10 6-10 11-15 11-150
50
100
150
200
Age (Years)
Figure 2.5.5: Levels of sCD23 in control and JA subtypes grouped according to age. Control sCD23
levels compared with their respective age group of (a) S‐JA, (b) P‐JA and (c) O‐JA.
4.7.5.6. Blood granulocytes were significantly increased in S‐JA patients compared with
control and O‐JA and P‐JA patients
****
**
**
P-JA
6-10 6-10 11-15 11-150
50
100
150
200
Age (Years)
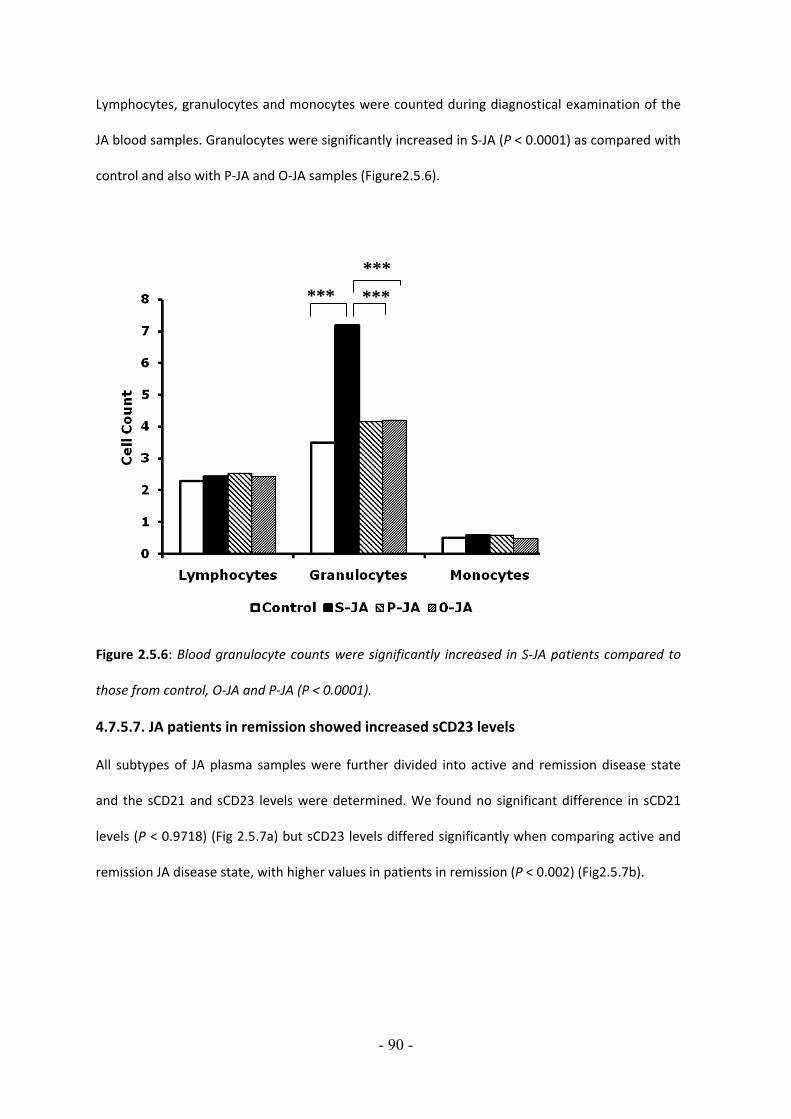
- 90 -
Lymphocytes, granulocytes and monocytes were counted during diagnostical examination of the
JA blood samples. Granulocytes were significantly increased in S‐JA (P < 0.0001) as compared with
control and also with P‐JA and O‐JA samples (Figure2.5.6).
Figure 2.5.6: Blood granulocyte counts were significantly increased in S‐JA patients compared to
those from control, O‐JA and P‐JA (P < 0.0001).
4.7.5.7. JA patients in remission showed increased sCD23 levels
All subtypes of JA plasma samples were further divided into active and remission disease state
and the sCD21 and sCD23 levels were determined. We found no significant difference in sCD21
levels (P < 0.9718) (Fig 2.5.7a) but sCD23 levels differed significantly when comparing active and
remission JA disease state, with higher values in patients in remission (P < 0.002) (Fig2.5.7b).
******
***

- 91 -
(a) (b)
Active Remission0
250
500
750
1000
Figure 2.5.7: (a) sCD21 and (b) sCD23 levels in active and remission disease state. Plasma sCD21
levels were insignificant comparing active and remission disease state while difference in sCD23
levels were significant (P<0.0022), using the nonparametric student t‐test.
Table 2.5.1: P‐ values of sCD21 and sCD23 of JA samples. Statistical significance was calculated by
nonparametric student t‐test of graphpad prism software.
Samples P Value Significance sCD21 O‐JA 0.0001 *** P‐JA 0.0068 ** S‐JA 0.0001 ***sCD23 O‐JA 0.3843 ns P‐JA 0.0001 *** S‐JA 0.0184 * Table 5.2.2: P‐values of sCD21 and sCD23 in active and remission disease state.
Active and Remission P Value Significance
sCD21 0.9718 ns
sCD23 0.0022 **
**
Active Remission0
20
40
60
80
100

- 92 -
Table 5.2.3: Median and interqartile range of (a) sCD21 and (b) sCD23 in healthy Controls and
subtypes of JA.
(a) Patient Group (sCD21) Median Range Interquartile Range Control (n=20) 600 ng/ml±10 180‐1100 ng/ml JA‐O (n=20) 200 ng/ml±10 10‐400 ng/ml JA‐P (n=20) 400 ng/ml±10 150‐800 ng/ml JA‐S (n=20) 220 ng/ml±10 20‐500 ng/ml JA‐All (n=20) 250 ng/ml±10 0‐800 ng/ml (b) Patient Group (sCD23) Median Range Interquartile Range Control (n=20) 45 U/ml ±10 0‐170 U/ml O‐JA (n=20) 25 U/ml ±10 0‐150 U/ml P‐JA (n=20) 30 U/ml ±10 0‐90 U/ml S‐JA (n=20) 35 U/ml ±10 0‐100 U/ml All‐JA (n=20) 25 U/ml ±10 0‐150 U/ml

- 93 -
CHAPTER‐V DISCUSSION 5.1. The sympathetic nervous system and antibody‐induced arthritis
Importance of the nervous system in immune regulation is clearly suggested by innervations of
lymphoid tissues by sympathetic nerve fibers. In a disease like arthritis, there is a persistent
inflammation characterized by micro vascular permeability and neutrophil influx into the joints. In
the antibody‐induced arthritis model, the onset of the disease is marked by joint swelling within 24
hours of pathogenic antibody injection in Balb/c mice. This strain is highly susceptible to antibody
induced‐arthritis (Maccioni et al., 2002; Solomon et al., 2002). In this model of arthritis, the
alternative pathway of the complement system is involved (Ji et al., 1999; Solomon et al., 2002).
Development of disease requires FcγRIII and C5aR as well as neutrophils (Wipke and Allen, 2001),
mast cells (Kneilling et al., 2007; Lee et al., 2002), macrophages (Solomon et al., 2002) and
inflammatory cytokines like TNF‐α and IL‐1β.
Here, we have provided evidence that neural pathways contribute to the clinical manifestation of
antibody‐induced arthritis. There has been considerable debate regarding the role of sympathetic
nerves in chronic inflammatory diseases. Joints are supplied with nerves contacting myelinated and
unmyelinated primary afferent fibers. Neurogenic inflammation is defined as the oedema formation,
increased blood flow and involvement of inflammatory cells observed after stimulation of sensory
nerve fibers and release of neuropeptides, such as substance P (SP), calcitonin gene related peptide,
and neuropeptide Y.
The role of substance P has been studied in adjuvant‐induced arthritis (Levine et al., 1984). The
concentration of substance P is increased in the joints affected by arthritis. Joints having dense
innervations display more severe inflammation and tissue destruction in adjuvant or collagen‐
induced arthritis (Levine et al., 1984).

- 94 -
Substance P has a potent effect on the cellular components of inflammation, such as neutrophil
accumulation (Cao et al., 2000). Moreover, Substance P is a powerful mediator of vascular
permeability. It is released from the unmyelinated C‐fibers in the joints. Besides joints and mucosal
epithelium immunoreactive Substance P, Substance P mRNA and NK‐1R are found in the
macrophages and mast cells where they stimulate the release of TNF‐α (Ansel et al., 1993; Luber‐
Narod et al., 1994; Pascual and Bost, 1990). The Substance P receptor NK‐1R is also present on the
post capillary venular endothelium (Baluk et al., 1996) and NK‐1R is expressed on lymphocytes,
macrophages, endothelial cells, neurons and other cell types. The release of Substance P is an early
event in immune complex mediated injury and acts as an amplifier of the subsequent inflammatory
changes. Substance P acts on the vascular endothelium to cause leakage of plasma proteins and
thereby increasing the concentration of more and more complement components at the site of
inflammation. Complement components are fixed by the immune complexes and ultimately release
C5a (Sylvestre and Ravetch, 1996).
Macrophages/monocytes and the proinflammatory mediators, such as TNF‐α, prostaglandin E2
(PGE2), macrophage inflammatory protein (MIP)‐1α and MIP‐1α, play a critical role in the
progression of immunological disorders including rheumatoid arthritis, Behçets disease and Crohns
disease.
It has been known that if adult and neonatal pups are treated with 6‐OH Dopamine they denervate
the sympathetic nerve fibers (Taxt et al., 1983). Considering the role of the sympathetic nervous
system in the K/BxN sera‐induced arthritis model, we sympathectomised adult mice and neonatal
pups with 6‐OH Dopamine to denervate the sympathetic nervous system and we observed
protection in transiently and permanently sympathectomised mice.
Next we determined the role of capsaicin in the neonatal pups. Capsaicin, the parent compound of a
group of vanillyl fatty acid amides isolated from capsicum, is a distinct, single chemical entity (Buck
and Burks, 1986; Cordell and Araujo, 1993), which has been used as an experimental tool to
investigate the functional and neurochemical characteristics of peptide containing primary sensory

- 95 -
neurones (Gamse et al., 1982). Biologically, when a sensory neuron is exposed to capsaicin the
neuron will release its supply of Substance P (Martine et al., 1991). Capsaicin evoked Substance P
release is due to its ability to induce calcium influx into nerve terminals (Buck and Burks, 1986;
Cordell and Araujo, 1993). In adjuvant arthritis, capsaicin pre‐treated rats were not protected
(Ahmed et al., 1995). We demonstrated that neonatal pups treated with capsaicin are not protected
from the serum induced arthritis. Because capsaicin stimulates Substance release, we found disease
after K/BxN serum transfer while SP‐/‐ mice were partially protected.
To understand the role of the cholinergic anti‐inflammatory pathway in K/BxN antibody‐induced
arthritis model we used α7nAChR‐/‐ mice. This pathway plays a critical role in controlling the
inflammatory response through interaction with peripheral α7 subunit‐containing nicotine
acetylcholine receptors which are expressed on macrophages. Nicotine acetylcholine receptor‐α7 is
an essential candidate in the inflammatory cascades. We found that α7nAChR‐/‐ homozygous mice
were completely resistant to inflammation induced by transfer of K/BxN serum. The main function of
this receptor family is to transmit signals for the neurotransmitter acetylcholine at neuromuscular
junctions and in the central and peripheral nervous systems (Le Novere and Changeux, 1995;
Leonard and Bertrand, 2001; Marubio and Changeux, 2000; Steinlein, 1998). It has been shown that
acetylcholine inhibits the release of TNF‐α and other cytokines through a post‐transcriptional
mechanism that is dependent on α‐ nicotinic receptors on primary human macrophages (Borovikova
et al., 2000). Macrophages are an important source of TNF‐α and play a crucial role for macrophages
in the pathology of K/BxN serum induced arthritis (Solomon et al., 2005).
Particular focus was placed on TNF‐α due to the importance of this cytokine in human disease, as
suggested by the successful treatment of RA patients with antibodies or fusion proteins capable of
blocking the TNF‐α/TNF‐α receptor interaction. (Brennan, 2001; Feldmann et al., 1996; Feldmann
and Maini, 2001). Both macrophages and TNF‐α are the important target to prevent the disease.
α7nAChR deficiency in mice might prevent the release of TNF‐α from macrophages, which could be
the reason why α7nAChR‐/‐ mice were completely resistant to disease after K/BxN sera transfer.

- 96 -
Taken together, this study adds further support to link between the neural pathways and
inflammatory diseases which may exemplify the emerging intimate connection between
neuropeptides and the immune system.
5.2. Macrophage inhibitory factor in antibody‐induced arthritis
The cytokine profile of RA synovium is an area of intense research interest. Pro‐inflammatory
cytokines such as TNF α and IL‐1β are known to induce, in vitro, many of the mediators of
inflammation and joint injury operative in RA. Moreover, the discovery of the expression of these
cytokines in RA synovium has led to the development of specific therapeutic strategies targeted at
these molecules. To date, however no such strategies have proved curative, and the search for other
therapeutic targets in RA continues.
Many aspects of the function of MIF suggest it as an important cytokine in RA. Recombinant MIF is
an inducer of monocyte/macrophage TNF α and MIF also induces or enhances other aspects of
monocytes/macrophage activity, including interferon‐γ‐induced nitric oxide production, intracellular
killing and phagocytic function (Bernhagen et al., 1994). MIF also up‐regulates MMPs and
cyclooxygenase 2 mRNA in rheumatoid synovial fibroblast (Leech et al., 1998). In addition, the recent
report of the expression of MIF by endothelial cells suggest it has a uniquely broad range of activates
within the immune/inflammatory response involved in RA pathology.
Macrophages play a crucial role in the K/BxN serum‐induced arthritis model (Solomon et al., 2005),
and are present in high numbers in inflamed tissue and at the cartilage‐pannus interface in RA. The
data presented here demonstrate the role of MIF in antibody‐induced arthritis. Age‐matched
C57BL/6 wild type mice developed arthritis within a day after the arthritic K/BxN serum transfer
while MIF ‐/‐ mice were disease‐free by measureable clinical parameters at that time. On day 6 and 8
MIF ‐/‐ mice showed a significant reduction in the ankle thickness and clinical index compared with
wild type mice.
Eleveated MIF levels have been reported in RA serum, synovial fluid and synovial tissues, with the
later correlating with disease activity (Leech et al., 1999; Morand et al., 2002). Cellular sourch of MIF

- 97 -
in RA include monocytes/macrophages, fibroblasts like synoviocytes (Leech et al., 1999), endothelial
cells and to a lesser extent T cells (Bacher et al., 1996).
MIF is known to promote TNF α secretion from macrophage cell lines and peritoneal mouse
macrophages (Bernhagen et al., 1994; Calandra et al., 1995). To determine whether macrophages
are responsible for the MIF‐production relevant to the RA pathogenesis we reconstituted the MIF‐/‐
mice with naive macrophages and found that adoptive transfer completely restores the
susceptibility to arthritis in MIF ‐/‐ with measurable ankle thickness and clinical index just like wild
type mice.
In conclusion, we have shown that a deficiency in the MIF gene inhibits joint inflammation and
destruction induced by antibody‐induced arthritis. We show for the first time that macrophage
derived MIF is the key to arthritis. These results support the hypothesis that MIF is an important
factor in the inflammation and cartilage degradation of RA. These data further support the view that
MIF is a potential target in RA.
5.3. MMP 13 (collagenase‐3) deficiency protects from antibody‐induced arthritis
The degradation of articular cartilage is a characteristic of rheumatoid arthritis. The collagenases
(MMP‐1, MMP‐8 and MMP‐13) have the unique ability to cleave the triple helix of collagen, thereby
allowing the chains to unwind, and are believed to play a major role in the collagen degradation
associated with arthritic conditions. MMP‐13 was originally isolated from breast tumor cells (Freije
et al., 1994).
Expression of MMPs is low in normal cells, and this low levels allow for healthy connective tissue
remodeling. In pathologic conditions however, the level of MMP expression increases considerably,
resulting in tissue destruction. Excess MMP production is associated with the pathology of many
diseases, including artherosclerosis, tumor invasion/metastasis and RA. (Borden and Heller, 1997;
Brinckerhoff et al., 2000; Vincenti, 2001).
Among all MMPs, MMP‐13 has been suggested to play a major role in the pathogenesis and tissue
destruction in rheumatoid arthritis because MMP‐13 mRNA has been found in the synovial tissue

- 98 -
from patients with rheumatoid arthritis (Wernicke et al., 1996). MMP‐13 has been shown to degrade
collagen type I, II, III as well as being able to degrade cartilage proteoglycan aggrecan (Fosang et al.,
1996). Unlike MMP‐1, which has a substrate preference for collagen type III, MMP‐13 prefers
collagen type II which is the primary collagen found in articular cartilage (Knauper et al., 1996b).
Also, MMP 13 mRNA and protein have been found in synoviocytes of the pannus‐hard tissue
junction of RA patients (Konttinen et al., 1999). TNF‐α expression by monocyte/macrophages has
been demonstrated both in synovial tissue and at the cartilage‐pannus junction. IL‐1ß and TNF‐α can
induce expression of MMP‐13 in the synovium. Therefore we have chosen to analyze the role of
MMP‐13 in antibody induced arthritis.
The data presented in this study show an important role of MMP13 in the progression of arthritis in
the K/BxN antibody induced arthritis. We tested whether mice lacking MMP13 would be capable of
developing arthritis. Although age‐matched C57Bl/6 wt controls developed arthritis within 2 days of
K/BxN serum injection, MMP13‐/‐ mice remained disease free by measurable clinical parameter (Fig
1.11). In MMP13‐/‐ mice, arthritis was significantly reduced in the progressive stage of the disease on
days 4, 6 and 8 as shown by the ankle measurement and clinical index when compared to wild type
mice. In wild type mice the arthritis was significantly evident on day 6. On day 8, significant
difference between wild type and MMP13 ‐/‐ was also confirmed by histological examination. The
wild type showed chronic inflammation with infiltration of mononuclear cells of the synovial
membrane, hyperplasia of synovial lining cells and fibrosis of synovial membrane and inflammation
was accompanied by pannus tissue formation and cartilage and bone destruction while ankle joint
sections of MMP13 ‐/‐ showed complete absence of pannus tissue, bone erosion and cartilage
destruction (Fig. 1.13).
Our data indicate that MMP‐13 is involved in triggering the inflammation in antibody‐induced
arthritis and MMP13 is required to prevent a sustained inflammatory response that occurs in the
effecter phase of arthritis. Therefore a drug aiming to inhibit MMP‐13 function might be candidate
for RA therapy.

- 99 -
5.4. Role of redox‐regulations in antibody‐induced arthritis
Our results showed that increased arthritis in Phytol‐treated mice is mediated by increased oxidative
burst in vivo the disease was resolved soon as compared to wild type mice.
Generally, ROS is thought to play a major role in defence against invading pathogens. ROS
production is maintained by the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase
complex which functions as an electron donor in the oxidative burst process via reduction of oxygen
[22] to superoxide that in turn could yield various reactive oxygen species. The most important
component of the NADPH complex is the Ncf1 (Neutrophil cytosolic factor) protein. It has been
shown that complete lack of ROS due to Ncf 1 deficiency results in chronic granulomatous disease in
both mice (Jackson et al., 1995) and human. Ncf1 is heavily phosphorylated and it is responsible for
transporting the cytosolic complex to the membrane during the activation.
Before sera injection into the phytol‐treated mice, we found significantly increased numbers of
neutrophils as compared to control mice. Neurophils play a key role in host defence against invading
pathogens and have a major role in inflammation in general (Babior, 2000; Klebanoff, 2005; Segal,
2005). In response to a variety of agents, they release large quantities of superoxide anions (O2.‐)
(Chanock et al., 1994; Groemping and Rittinger, 2005; Quinn and Gauss, 2004). O2.‐ production by
neutrophils is dependent on activation of the NADPH oxidase complex. Increased numbers of
neutrophils in phytol‐treated mice could be responsible for an increase the O2.‐ concentration which
quenches the ROS, and decreased ROS might be responsible to increase the disease in phytol‐
treated mice at days 2 and 4 as compared to control mice (Fig 1.15). It has been shown that lower
oxidative burst led to a reduction in the cell membrane proteins and activation of autorecative and
arthritogenic T cells (Gelderman et al., 2006).
To determine relative amounts of surface R‐SH on neutrophils and monocytes we used ALM. This
assay takes advantage of the fluorescent Alexa dye (FL‐4) covalently bound to maleimide, a well
known neutrophilic molecule widely used to react with‐SH groups on proteins and other molecules.
ALM reagents are commonly used to make Alexa‐ labeled mAb for use in FACS. However, here we

- 100 -
used ALM for labeling cell surface thiols. We observed on day 6, the surface R‐SH levels on
neutrophils were significantly decreased in the phytol‐treated mice compared with control mice. The
reason for this could be that in the phytol‐treated mice on day 4 the diseases starts resolving. In
K/BxN mice model, it has been shown that neutrophils play an important role in the early stage of
the disease (Wipke and Allen, 2001). We also measured the absolute numbers of neutrophils after
phytol‐treatment and found that on day 0 neutrophil numbers were significantly higher as compared
with controls. This increased numbers of neutrophils in the phytol‐treated mice could be responsible
for onset of disease within a 24 hrs. Along with the surface thiol levels on neutrophils we also
determined the surface thiol levels on monocytes. Unlike thiol levels on neutrophils, we did not find
any change on surface R‐SH levels on monocytes.
Along with the surface R‐SH we also determined the intracellular ROS levels in neutrophils and in
monocytes and found there was no significant correlation between phytol‐ treated and control mice.
5.5. An antagonist of monocyte chemoattractant protein 1 (MCP‐1) did not inhibits
arthritis in the K/BxN model
Monocyte chemoattractant protein (MCP)‐1 is a chemoattractant cytokine (chemokine) (Baggiolini
et al., 1994) that promotes the migration and activation of monocytes (Van Damme et al., 1992;
Zachariae et al., 1990). It has been associated with several inflammatory diseases (Furie and
Randolph, 1995), but a causal relationship has been difficult to prove. Monocyte infiltrates are
prominent in rheumatoid arthritis (RA) and their products, such as cytokines that amplify the
inflammatory response and enzymes that destroy connective tissue (Furie and Randolph, 1995), are
readily detected in diseased joints. MCP‐1 is produced by both synovial cells and infiltrated
monocytes in RA (Harigai et al., 1993; Koch et al., 1992; Kunkel et al., 1996).
To test the hypothesis that, by inhibiting endogenous MCP‐1, the inflammatory reaction was
reduced, we examined its effect in the K/BxN mouse model of arthritis. Daily injections of the
antagonist did not prevent the onset of arthritis as monitored by measuring joint swelling and by
clinical score of the joints. The MCP‐1 antagonist‐treated mice spontaneously developed a chronic

- 101 -
inflammatory arthritis that was similar to control Balb/c mice. MCP‐1 accelerated the onset and
enhanced the symptoms of joint inflammation (Fig 1.23). In vitro, MCP‐1 results in migration only if a
gradient is formed, high concentrations, MCP‐1 inhibits migration by collapsing the gradient
(Randolph and Furie, 1995). It is unlikely that the enhancement we have observed in vivo with MCP‐
1 antagonist is simply nonspecific. Rather, it is more likely that the systemic levels of MCP‐1 in these
mice are insufficient to cause non‐responsiveness of the target cells to MCP‐1 antagonist. Our
interpretation for the enhancement of the onset and swelling is that the injected MCP‐1
accumulates in the tissues and causes activation of monocytes and other target cells, whereas the
MCP‐1 that remains in the vascular compartment is eliminated rapidly.
5.6. DBA mt FvB mice are not protected from antibody‐induced arthritis
Antibody‐induced arthritis in mice is one of the most widely used autoimmune experimental models,
with many features similar to humsn rheumatoid arthritis. Five of the eight small‐effect QTL in the
DBA/1 strain are arthritis‐enhancing alleles, while in the FVB/N strain are the arthritis‐enhancing
alleles are the other three QTL, indicating that some susceptibility genes could come from the
resistant strain. Several studies have been carried out to detect gene expression profile responsible
for disease in CIA model, all of which used joints as the investigated tissue. The susceptibility and
resistance of DBA/1J and FVB/N mouse strains respectively, to the induction of CIA is well
documented. Previously it has been shown that the (DBA1JxFVB/N) F1 progeny was susceptible to
CIA. A comparison and arthritis indexes between the F2 progeny and parental DBA/1J showed
significant difference suggesting the existence of protective FvB/N loci (Ibrahim et al., 2002;
Thornton et al., 2002).
In this study, we used (DBA/1J×FVB/N) F2 mice and found after arthritic sera transfer mice were
susceptible to disease and showed severity to antibody‐induced arthritis similar to DBA/1 wild type
mice. The reason for this could be differential expression of a gene could result not only from allele
difference between two strains.

- 102 -
5.7. Role of CD21 and CD23 in autoimmune disorders
Autoimmune diseases are often characterized by high levels of circulating autoantibodies and/or the
presence, of T cells, in affected organs with reactivity against self‐antigens. It is well established that
complement plays a crucial role in the acute inflammatory response following attack on self tissue
by autoantibodies and deposition of complement activating immune complexes in glomeruli and
small vessels (Sheerin et al., 1997; Wang et al., 2000).
Complement facilitates the clearance of immune complexes (IC) from circulation (Schifferli et al.,
1986). In human, erythrocytes bear clusters of CD35 (Chevalier and Kazatchkine, 1989; Fearon,
1980) that mediate multivalent binding of C3 and/or C4 fragments deposited on IC in the periphery
and transport the complexes to the liver and spleen where transfer to phagocytic macrophages
takes place (Bogers et al., 1991; Davies et al., 1993). But, in autoimmune diseases, disposal of IC to
the liver and spleen could fail, resulting in the deposition of IC in glomeruli and small vessels
followed by inflammatory reactions.
The role of B cells in various autoimmune diseases, like diabetes (Falcone et al., 1998; Serreze et al.,
1996) in mice, rheumatoid arthritis in human (Edwards and Cambridge, 2001; Takemura et al., 2001)
and systemic lupus erythematosus (SLE)is shown. SLE is a systemic, autoimmune disease with clinical
features, including glomerulonephritis, haemolytic anaemia, thrombocytopenia and central nervous
system involvement (Robson and Walport, 2001). SLE is characterized by polyclonal B cell activation
and (Gross et al., 2000; Levinson et al., 1981) excessive production of autoantibodies.
CD21 is expressed by B lymphocytes and B lymphoblastoid cell lines, human thymocytes (Tsoukas
and Lambris, 1988) a fraction of human T lymphocytes (Fischer et al., 1991) certain leukemia T cell
lines, the pharyngeal and cervical epithelium and by follicular dendritic cells. The level of CD21
expression is developmentally regulated: it is highest on mature B lymphocytes and on a
subpopulation of immature blastic thymocytes. The soluble form of CD21 (sCD21) is found in human
plasma and in cell culture supernatants of lymphoid cells (Huemer et al., 1993; Ling et al., 1991;
Myones and Ross, 1987).

- 103 -
The cellular response of CD21 is mediated through interaction of CD21 with membrane and soluble
CD23 (Bonnefoy et al., 1995.) CD21/CD23 pairing occurred between membrane forms of the
receptors and is involved in homotypic adhesion of B cells. Certain biological properties of soluble
CD23 had been attributed to interaction with CD21 on B cells.
CD23 is a ligand for CD21, a type II transmembrane protein expressed on a variety of hemopoietic
cell types that serves as the low‐affinity receptor for IgE. Cleavage of the membrane receptor
generates soluble forms of CD23 that are endowed with ‘cytokine‐like’ activity (Delespesse et al.,
1991).
CD23, is expressed on a variety of hematopoietic cells including B cells and monocytes (Delespesse
et al., 1991). It is a 45 kDa membrane type II glycoprotein whose proteolytic cleavage gives rise to
unstable soluble fragments subsequently transformed into a stable 25 kDa product referred to as
soluble CD23 (sCD23). Cell surface CD23 expression and sCD23 release are up regulated by IL‐4 in all
CD23 expressing cell types, and inhibited by interferon‐gamma (IFN‐�), transforming growth factor
beta (TGF‐�) and glucocorticoids on B‐cells (Delespesse et al., 1992). While sCD23 retains the
capacity to bind IgE, it has many activities that are IgE‐independent (Delespesse et al., 1992; Sarfati
et al., 1992), including inhibition of apoptosis (Liu et al., 1991). The amount of sCD21 and sCD23 in
serum may modulate immunity as sCD21 and sCD23 levels are correlated with several clinical
conditions.
sCD21 and sCD23 levels differ in various autoimmune diseases from normal values and can be
regarded as diagnostical markers for autoimmne diseases. That is why we were interested to
determine their levels in autoimmune sera/plasma samples.
5.7.1. Antiphospholipid syndrome
sCD21 is significantly reduced in APS patients sera compared to healthy controls. Because sCD21
binds to C3d on immune complexes (Carel et al., 1990; Lyubchenko et al., 2005; Szakonyi et al.,
2001) we reasoned that the presence of autoantibodies forming immune complexes might
influences the amounts of sCD21 due to phagocytic clearance of theses complexes in the liver. The

- 104 -
presence of anti‐ ß2GPI autoantibodies and the occurrence of the respective antigen (ß2‐GPI) in sera
in quantities of 150‐300µg/ml exceeds the amounts of sCD21 in healthy donors and autoimmune
patients by about 1000 fold. Because the complement activation is an enzymatic amplification
cascade, one would expect a further molar increase in complement activation products like C3d in
these immune complexes. Therefore one would expect to observe a reduction of sCD21 due to
binding to immune complexes formed by autoantibodies, complement and autoantigens. However,
when we compared sCD21 titers from patients having anti‐ß2GPI autoantibodies with those lacking
these antibodies, we could not find significant differences. This finding is astonishing because it
sheds some doubts on the presence of classical/textbook style immune complexes in the blood of
these patients. We analyzed previously a large cohort of rheumatoid arthritis patients for the
presence of sCD21 and found no difference between patients with and without rheumatoid factors
(Masilamani et al., 2004b). Because IgM is the strongest complement activator among the
immunoglobulin isotypes, we expected in particular the IgM anti IgG rheumatoid factors to bind
sCD21 via activated complement C3d and finally be cleared via the liver. In rheumatoid arthritis as
well as in APS the autoantibodies and their ligands outnumber by far sCD21 and thus should
significantly reduce amounts of circulating sCD21 in the blood. This is obviously not the case.
5.7.2. Soluble CD21 levels in autoimmune lymphoproliferative syndrome (ALPS) Autoimmune lymphoproliferative syndrome (ALPS) is a rare disorder in which there is chronic non‐
malignant lymphoproliferation and autoimmunity, most often involving cells of the haematopoietic
system. It can present in children or adults but usually presents in early childhood (Deutsch et al.,
2004). It is caused by a genetic defect in the mechanism of programmed cell death (apoptosis) and is
characterized by the presence of double‐negative (TCR alpha/beta CD4‐ CD8‐) T lymphocytes (DNT).
Our study indicates sCD21 levels significantly increased in ALPS patient’s sera as compared with age
matched healthy controls. During the first decade of life, T cells are generated from the thymus and
double negative thymocytes express high levels of CD21 (Fischer et al., 1999). The increased sCD21

- 105 -
concentration in ALPS patients could be a result of thehigh expression of CD21 on double negative T
cells in the periphery.
5.7.3. Diabetes
Diabetes is occurring in two forms type I and type II, where diabetes type I is an result of an
autoimmune reaction while type II has occur for non immunological reasons. Thus diabetes is an
interesting model to address the question whether changes in sCD21 and sCD23 levels are realted to
autoimmunity. Clinically altered levels of sCD21 and sCD23 have been shown in various autoimmune
diseases. Therefore we were interested to determine sCD21 and sCD23 levels in diabetes patients
samples.
In the present study we demonstrated that the decreased sCD21 and sCD23 levels in autoimmune
diabetes type I (P < 0.0067 and 0.0001 respectively) while there was no change in the levels of sCD21
and sCD23 in diabetes type II (P < 0.8603, 0.5692 respectively) as compared to age‐matched
controls. The lower levels of sCD21 and sCD23 in autoimmune diabetes type I could be because of
decreased activation and insufficient autoproteolysis of surface CD21 and CD23 antigen on immune‐
competent cells while no change in diabetes type II because it is not an autoimmune disease.
The lower levels of sCD23 were found in subjects with the lowest insulin secretion, who represent
the group with the highest risk of progression to autoimmune type I diabetes. These findings could
be explained by Gladstone & Nepom’s hypothesis that insulin can substitute IL‐4 function and locally
enhance the Th2 subset in pancreatic β‐cells at an early stage of pancreatic β‐cell destruction.
Honeyman et al. suggested that the imbalance between Th1 cells secreting interleukin (IL‐1) and
Interferon‐γ and Th2 lymphocytes producing mainly IL‐4 and IL‐5, plays a key role in the autoimmune
process leading to pancreatic β‐ cells destruction (Honeyman et al., 1997; Hussain et al., 1998). It
was also shown that surface expression of CD23 lymphocytes and sCD23 release are up regulated by
IL‐4, but down regulated by IFN‐γ (Corominas et al., 1998). The high expression of CD23 on T cells
would favour the interaction between T and B cells by means of CD23 expressed on T cells and its
receptor CD21 on B cells. This would lead to IgE production.

- 106 -
Secondly, we have found that after proinsulin treatment the sCD21 levels were significantly
decreased in diabetic patients with BMI < 90 when compared with without treatment (P < 0.001).
But in contrast surprisingly, sCD23 levels significantly increased after treatment (P < 0.0001). It has
been found that anti‐CD3 and nasal proinsulin combination therapy enhances remission from
autoimmune type I diabetes by inducing Tregs (Bresson et al., 2006). The anti‐CD3 would create a
systemic immune modulatory milieu to facilitate the islet‐antigen‐specific induction of Tregs and
also induce a shift in the cytokine profile (mainly Th1 to Th2). The levels of CD4+ lymphocytes
significantly increased after proinsulin treatment. CD23 like CD21 is considered to be an activation
marker of B cells. The expression of CD4+ cells after proinsulin treatment could be one of the reasons
for the increased sCD23 levels, whereas numbers of B cells remain constant after proinsulin
treatment. It has been known that B cells are involved in diabetes (Charlton et al., 2001) but it is not
clear at which stage they play a role: early, late or both while destruction of pancreatic β cells is
dependent on T cells.
In conclusion our study suggests that sCD21 and sCD23 titers have the potential to be used as an
additional markers for type I diabetes and could play a role in determining the efficacy of prevention
trials.
5.7.4. Juvenile arthritis
JA involves joint pain and inflammation and is the most common chronic arthritis in children.
Shedding of CD21 seems to be correlated with that of CD23. Both are ligands of each other
(Bonnefoy et al., 1995b) and both are expressed and/or released by B‐cells, possibly allowing
homotypic interactions to take place (Cherukuri et al., 2001). Such interaction may contribute to
B‐cell hyperactivity and autoantibody production. Analysis of sCD21 purified from normal human
plasma indicated that a part of sCD21 circulates in association with a cleavage fragment of C3 and
a trimeric form of sCD23 (Fremeaux‐Bacchi et al., 1998a). Therefore we were interested to
analyze both sCD21 and sCD23 in JA.

- 107 -
Reduction of sCD21 levels have been reported in SLE, RA, anti‐phospholipid syndrome and Sjogren’s
syndrome (Illges et al., 2000; Masilamani et al., 2003; Masilamani et al., 2004a; Masilamani et al.,
2004b; Singh et al., 2008) and here in JA subtypes. Unlike sCD21, sCD23 levels were found elevated
in various autoimmune disorder like localized scleroderma (Sato et al., 1996), rheumatoid arthritis
and systemic lupus erythematosus (Bansal et al., 1992). Therefore, sCD21 and sCD23 appear to be
markers for autoimmunity and/or chronic inflammation in human. Supporting this, in a mouse
model, genetic ablation of CD21 results in enhanced autoimmunity (Del Nagro et al., 2005). A further
support of our hypothesis that sCD21 is modulated in immunity comes from our analysis of sCD21
levels during pregnancy. In pregnancy immunity is changed to prevent immune attack to the fetus.
During pregnancy sCD21 declines to levels reminiscent of autoimmune diseases and it takes month
after delivery to reach normal levels again (Masilamani et al., 2008). Levels of sCD23 also changed
during pregnancy (Wegmann et al., 1993).
In a previous study we analyzed a cohort of JA sera, which was undifferentiated for the subtypes,
and concluded that JA displayed a just not significant decline in sCD21 levels (Masilamani et al.,
2004a). The difference to our present study may relate to the following: the controls of our
previous study were age related less stringently and as we have shown here there are differences
in the JA subtypes with respect to sCD21 levels, which may have biased our previous results.
Our study shows sCD23 levels decreased in plasma of patients with JA while in RA, the percentage
of B cells expressing CD23 and serum sCD23 levels were elevated (Chomarat et al., 1993). In
contrast to our findings, Kutukculer et al. (Kutukculer and Caglayan, 1998) found an increased
sCD23 levels in patients with chronic juvenile arthritis. Their observation could, however, reflect
the later stage of the autoimmune process than observed in present study. On the basis of
present results we could suggest that the changes in the levels of sCD23 on peripheral
lymphocytes could serve as a marker of different stages of autoimmune process.
The study presented here demonstrates that the significant decline of sCD21 and sCD23 in healthy
donors was independent on gender (Fig 2.5.4 and Fig 2.5.5). Sällfors et. al., 2003 (Sallfors et al.,

- 108 -
2003) found age differences in pain, coping and health status among children and chronic
arthritis. As found in RA, the reduction of sCD21 in JA is independent of age and this indicates that
pathological mechanisms are responsible for lowering systemic sCD21 (and sCD23).
JA is also associated with effusion and swelling of soft tissue. This inflammatory process is
generated by a series of events, including the migration of leucocytes (Pitzalis, 1993), lymphocytes
(Silverman et al., 1993), granulocytes and monocytes (Kashiwagi et al., 1998) from the blood
stream into the tissue, their activation to become effector cells and, finally, their local retention
which faciliates the continuing immune reaction. We found that in circulating blood of S‐JA
patients granulocytes significantly increased as compared with those of healthy subjects and
other subtypes of JA (Fig 2.5.6). It has been shown that granulocytes and monocytes apheresis
suppress the symptoms of rheumatoid arthritis (Nagashima et al., 1998). Therefore, the increase
in granulocytes in S‐JA can be of diagnostical value and our data suggest that apheresis of
granulocytes could be useful particularly in S‐JA but not in O‐JA and P‐JA.
Active JA stage is associated with higher levels of soluble E‐selectin (sE‐selectin) and soluble
intracellular adhesion molecules‐1 (sICAM) while during remission state their levels goes down
(Chen et al., 2002). C‐Y Chen et. al found that sE‐selectin and sICAM levels were significantly
higher in S‐JA. Difference in the levels of sCD21 were not significant when we compared active
state with remission state while the difference was significant in sCD23 (P < 0.0022) (Fig 2.5.7b). In
pregnancy we observed that the sCD21 levels need months to be in the normal range again. This
may be the same for sCD21 here with respect to the remission stage. In contrast to sCD21, sCD23
levels increased again in remission state, which suggest that the mechanism of shedding though
related has different aspects at least in regulation.

- 109 -
CHAPTER VI
SUMMARY
The K/BxN sera induced arthritis is a unique model used to study the inflammatory process taking
place in the joints independent of the autoimmune mechanisms that are required for autoimmune
production. The K/BxN mouse model shows many features common to human RA and other forms
of inflammatory arthritis, including leukocyte invasion, synoviocyte proliferation, pannus formation,
synovitis and cartilage and bone erosion. Intraperitoneal injection of sera from arthritic K/BxN mice
containing antibodies against Glucose‐6‐phosphate isomerase (GPI) is capable of inducing arthritis in
the susceptible mice like Balb/c and C57Bl/6. This disease is independent of T and B cells. The
inflammatory mediators (cellular and molecular) involved in the K/BxN sera‐induced arthritis model
are examined in this research work.
Many studies have shown that modulation of cytokine function is effective in ameliorating
symptoms of rheumatoid arthritis. Neuropeptides have recently been shown to have powerful
effects on the production and release of cytokines and have also been shown to exert potent
proinflammatory and anti‐inflammatory effects in animal models of inflammatory diseases.
Importance of the nervous system in immune regulation is clearly suggested by innervations of
lymphoid tissues by sympathetic nerve fibers. Substance‐P, Nicotine Acetylcholine receptor
(α7nAChR) is a powerful mediator of vascular permeability and is released from unmyelinated C
fibers and macrophages respectively. The release of substance P is an early event in immune‐
complex‐mediated injury and could act as an amplifier of the subsequent inflammatory changes.
Vagus nerve releases acetylcholine (ACh) in the vicinity of macrophages. ACh can interact specifically
with macrophage α7 subunits of nicotinic ACh receptors, leading to cellular deactivation and
inhibition of cytokine release mainly TNF‐α. TNF‐α was identified in the synovial membrane, and
particularly at the cartilage‐pannus junction (Chu et al., 1991), promoted cartilage and bone

- 110 -
resorption (Bertolini et al., 1986; Saklatvala, 1986) and induced the release of prostaglandin E2 and
collagenase (Dayer et al., 1985) by synovial cells of patients with RA.
In antibody‐induced arthritis, it was observed that transiently and permanently sympathectomised
mice showed partial protection from disease. Similarly substance‐P knockout mice showed partial
protection whereas α7nAChR ‐/‐ mice showed completely resistant from antibody induced arthritis.
The matrix metalloproteinase (MMP) family members are the major enzymes that degrade the
components of the extracellular matrix (Nagase and Woessner, 1999; Vincenti, 2001). We focused
mainly on MMP13 because it may have a particular role in cartilage degradation because it is
expressed by chondrocytes, and because it hydrolyzes type‐II collagen more efficiently than the
other collagenases (Mitchell et al., 1996). We found that MMP13 deficient mice showed protection
from arthritis.
Cytokines play important roles in the pathology of rheumatoid arthritis. Macropahge migration
inhibitory factor (MIF) is a cytokine with a broad spectrum of actions including induction of
monocyte TNF‐α in monocytes. MIF is a cytokine with well recognised importance in the regulation
of immune and inflammatory responses. Our results demonstrate that MIF plays an important role
in the antibody induced arthritis. MIF knockout mice showed significant protection compared with
wild type mice. In addition we reconstituted the MIF deficient mice with peritoneal macrophages
and found the MIF knockout mice were susceptible to arthritis induction after K/BxN sera transfer
just like control mice. Our data indicate for the first time that macrophage devoid MIF is responsible
for the MIF related pathology of arthritis.
Inflammation is not always abnormal, but in fact plays an important part in the body's defense
against infection. As part of their activity against disease‐causing bacteria, the white blood cells
known as granulocytes generate reactive oxygen species (ROS), sometimes known as “free radicals.”
To elucidate the role of redox reactions we determined the importance of phytol in antibody
induced arthritis. Phytol (3,7,11,15‐tetramethyl‐2‐hexadecene‐1‐ol) increased oxidative burst in vivo.
We found that onset of disease within 24 hours in phytol treated mice while in control mice disease

- 111 -
took 48 or 72 hours. Surprisingly the disease resolved faster in phytol‐treated mice as compared
with control mice. We also found in phytol treated mice increased numbers of neutrophil before
induction of arthritis as compared to control mice. Increased neutrophils in phytol treated mice
could be responsible increasing the O2.‐ which quenches the ROS, and decreased ROS might be
responsible to increase the disease in phytol treated mice at days 2 and 4 as compared with control
mice (Fig 1,15), because it has been shown that lower oxidative burst led to a reduction in the cell
membrane proteins and activation of autorecative and arthritogenic T cells (Gelderman et al., 2006).
Monocyte chemoattractant protein (MCP)‐1 is a chemoattractant cytokine (chemokine) (Baggiolini
et al., 1994) that promotes the migration and activation of monocytes (Van Damme et al., 1992;
Zachariae et al., 1990). It has been associated with several inflammatory diseases. To test the
hypothesis that, by inhibiting endogenous MCP‐1, the antagonist has antiinflammatory activity in
vivo, we examined its effect in the K/BxN mouse model of arthritis. Daily injection of the MCP‐1
antagonist did not prevent the onset of arthritis as monitored by measuring joint swelling and by
clinical score of the joints.
sCD21 and sCD23 levels in autoimmune disorders
CD21 and CD23 are released as a soluble form from the cell surface by proteolytic activity known as
‘Shedding’. sCD21 retains the capacity to bind iC3b and CD23, the known ligands of membrane
CD21. In a similar fashion to IgE complexes, another ligand of CD23, the sCD21 was shown to
efficiently trigger CD23‐signalling pathways in human monocytes. The clinical significance of sCD21
and sCD23 has been shown by changes in their concentrations in various autoimmune disorders.
We estimated the sCD21 and sCD23 concentration in antiphospholipid syndrome (APS), juvenile
arthritis (JA), diabetes and autoimmune lymphoproliferative syndrome (ALPS) patients sera/plasma
samples.
sCD21 levels were significantly reduced in APS patients, in comparison with healthy donors (P <
0.0001). The presence of specific ß2‐GPI autoantibodies did not correlate with sCD21 titers.

- 112 -
However, the levels of sCD21 in these patients are not further influenced by additional autoimmune
SLE, vasculitis, DVT (Deep vein thrombosis) fetal loss or thrombosis.
In case of JA, we investigated the levels of plasma sCD21 and sCD23 in subtypes of JA patients.
Our data demonstrate that levels of sCD21 were decreased in all subtypes of JA patients while
plasma sCD23 levels were significantly reduced in P‐JA and S‐JA samples (P < 0.0001, 0.018), but
not in O‐JA (P < 0.3843).
As the CD21 and CD23 antigens were shown to be involved in the pathogenesis of various
autoimmune diseases, we evaluated the levels of sCD21 and sCD23 in sera of healthy and diabetes
type I and II patients. Our data demonstrate that levels of sCD21 and sCD23 in diabetes type I
individuals significantly decrease (P < 0.0067 and P < 0.0001 respectively) while there is no change in
diabetes type II (P < 0.8603 and 0.5692 respectively) as compared to age matched healthy controls.
In case of ALPS patient’s sera, the soluble CD21 concentration were estimated in the sera from 17
ALPS patients and compared with healthy volunteers. We found the levels of sCD21 were
significantly increased in ALPS patient’s sera samples as compared to healthy individuals, P < 0.006.
This disease is found in children or adults but usually presents in early childhood. The increased
sCD21 in ALPS patients sera, could be related to the presence of double‐negative (TCR alpha/beta
CD4‐ CD8‐) T lymphocytes (DNT). During the first decade of life, T cells are generated from the
thymus and double negative thymocytes express higher levels of CD21.
Our study indicates that sCD21 and sCD23 are additional markers for autoimmune diseases.

- 113 -
Zusammenfassung
Die K/BxN Serum induzierende Arthritis ist ein einzigartiges Modell zur Untersuchung von
Entzündungsprozessen, die in den Gelenken stattfinden. Das K/BxN Mausmodell zeigt viele
Merkmale der menschlichen rheumatischen Arthritis und anderen Arthritisformen, einschließlich
Leukozyteneinfall, Synoviozytenproliferation, Pannusformation, Synovitis sowie Knorpel‐ und
Knochenerosion. Peritonale Injektion des Serums von arthritischen K/BxN Mäusen Glukose‐6‐
Phosphat Isomerase (GPI) enthält in anfälligen Mäusen stämme wie Balb/c und C57BI/6, welches
Antikörper gegen, verursacht Arthritis. Diese Krankheit ist unabhängig von T‐ und B‐Zellen. Die
Entzündungsmediatoren (zelluläre und molekulare), die im K/BxN Serum induzierten Arthritismodell
eine Rolle spielen, werden in dieser Forschungsarbeit untersucht.
Viele Studien haben gezeigt, dass die Modulierung der Zytokinfunktion sehr effektiv ist um die
Symptome von rheumatischer Arthritis zu verbessern. Neuropeptide haben starke Effekte auf die
Produktion und Abgabe von Zytokinen . Außerdem üben sie starke entzündungshemmende und
entzündungsverursachende Effekte in Tiermodellen mit Entzündungskrankheiten aus. Die Bedeutung
des Nervensystems für das Immunesystem liegt nahe, da Lymphgewebe von sympathischen
Nervensträngen angeregt werder. Die Substanz P, ist ein starker Mediator der vaskulären
Permeabilität und wird von unmyelinierten C Nervenfasern und Makrophagen abgegeben. Die
Abgabe von Substanz P ist ein frühes Ereignis bei immun‐komplex verursachten Verletzungen und
könnte als Verstärker von nachfolgenden entzündlichen Veränderungen agieren. Der Vagusnerv gibt
Acetylcholine (Ach) in die Umgebung von Makrophagen ab. ACh kann spezifisch mit der
Makrophagen α7 Untereinheit des nikotinischen Acetylcholinrezeptors interagieren, welches zu
zellulärer Deaktivierung und Unterdrückung der Zytokinabgabe, hauptsächlich TNF‐α, führt. TNF‐α
wurde in der Synovialmembran identifiziert, insbesondere an der Knorpel‐Pannus‐Grenze (Chu et al.,
1991) bei begünstigter Knorpel‐ und Knochenresorption (Bertolini et al., 1986; Saklatvala, 1986) und
induzierter Abgabe von Prostaglandin E2 und Collagenase (Dayer et al., 1985) von Synovialzellen
rheumatischer Arthritis Patienten.

- 114 -
In Antikörper induzierter Arthritis wurde beobachtet, das vorübergehende und dauerhafte
sympathektomierte Mäuse teilweise vor der Krankheit geschützt sind. Substanz P Knockout Mäuse
zeigten einen ähnlichen Schutz. α7nAChR‐/‐ Mäuse zeigten eine komplette Resistenz gegen
Antikörper induzierte Arthritis. Mitglieder der Matrix Metalloproteinase (MMP) Familie sind die
Hauptenzyme, die Komponenten der extrazellulären Matrix abbauen (Nagase and Woessner, 1999;
Vincenti, 2001).Wir konzentrierten uns hauptsächlich auf MMP13 weil es eine entscheidende Rolle
im Knorpelabbau haben könnte. Es wird von Chondrozyten exprimiert und hydrolysiert Typ‐II
Collagen effizienter als andere Collagenasen (Mitchell et al., 1996). Wir fanden heraus, dass MMP13
defiziente Mäuse Schutz vor Arthritis besitzen.
Zytokine spielen eine wichtige Rolle in der Pathologie von rheumatischer Arthritis. Makrophagen
migration inhibitory factor (MIF) ist ein Zytokin mit einem breiten Wirkspektrum einschließlich der
Induktion von TNF‐α in Monozyten. Unsere Ergebnisse zeigen das MIF eine wichtige Rolle in
Antikörper induzierter Arthritis spielt. MIF defiziente Mäuse zeigten einen erheblichen Schutz vor
Arthritis im Vergleich mit Wildtyp Mäusen. Außerdem haben wir die MIF Knockoutmäuse mit
peritonalen Makrophagen rekonstituiert und herausgefunden, dass die MIF Knockout‐Mäuse
anfällig für Arthritis Induktion nach K/BxN Serum Transfer wurden, ebenso wie die Kontrollmäuse.
Unsere Daten zeigen zum ersten Mal, das MIF von Makrophagen verantwortlich für die Pathologie
von Arthritis im Zusammenhang mit MIF ist. Entzündungen sind im eigentlichen Sinne ein wichtiger
Teil der körpereigenen Abwehr gegen Infektionen. Als Teil der Aktivität gegen krankheitserregende
Bakterien, generieren weiße Blutkörperchen bekannt als Granulozyten, reaktive Sauerstoff Formen
(ROS), auch bekannt als „freie Radikale“. Um die Rolle von Redox Reaktionen zu untersuchen haben
wir die Bedeutung von Phytol in Antikörper induzierter Arthritis bestimmt. Phytol (3,7,11,15‐
tetramethyl‐2‐hexadecene‐1‐ol) erhöht die Radikalfreisetzung (oxidative burst) in vivo. Wir fanden,
das der Beginn der Arthritis innerhalb von 24 Stunden in Phytol behandelten Mäuse stattfand
während es in Kontrollmäusen 48 bis 72 Stunden dauerte. Überraschenderweise wurde die
Krankheit in Phytol behandelten Mäusen schneller beendet im Vergleich zu den Kontrollmäusen. Wir

- 115 -
fanden außerdem eine erhöhte Zahl Neutrophile in Phytol behandelten Mäusen vor der Arthritis
Induktion im Vergleich zu Kontrollmäusen. Eine erhöhte Zahl von Neutrophilen in Phytol
behandelten Mäusen könnte verantwortlich für erhöhtes O2 sein, welches die ROS unterdrückt.
Verringerte ROS könnten verantwortlich dafür sein die Krankheit in Phytol behandelten Mäusen an
Tag 2 und 4 im Vergleich zu den Kontrollmäusen, zu verschlimmern (Fig 1,15), weil gezeigt wurde,
dass eine verringerte Radikalfreisetzung (oxidative burst) zu einer Reduktion von Zellmembran
proteinen und der Aktivierung von selbstreaktiven und arthritischen T‐Zellen führt (Gelderman et al.,
2006).
Monocyte chemoattractant protein (MCP)‐1 ist ein chemotaktisches Zytokin (Chemokin) (Baggiolini
et al., 1994) welches die Migration und Aktivierung von Monozyten fördert (Van Damme et al., 1992;
Zachariae et al., 1990). Es wurde mit mehreren Entzündungskrankheiten in Verbindung gebracht.
Um die Hypothese, mit der Hemmung von endogenem MCP‐1 habe der Antagonist
entzündungshemmende Wirkung in vivo, zu testen, haben wir den Effekt im K/BxN Arthritis
Mausmodell untersucht. Tägliche Injektionen des MCP‐1 Antagonisten hat den Beginn der Arthritis
nicht verhindert,.
sCD21 und sCD23 Titer in Autoimmunstörungen
CD21 und CD23 werden als lösliche Form von der Zelloberfläche mit Hilfe proteolytischer Aktivität,
bekannt als „Shedding“, abgegeben. sCD21 behält die Fähigkeit iC3b und CD23 zu binden, die
bekannten Liganden von membranem CD21. In ähnlicher Weise zu IgE, einem anderen Liganden von
CD23, wurde gezeigt, dass sCD21 wirkungsvoll CD23 Signalwege in menschlichen Monozyten auslöst.
Die klinische Bedeutung von sCD21 und sCD23 wurde mit Veränderungen ihrer Konzentrationen in
verschiedenen Autoimmunerkrankungen gezeigt.
Wir haben die Konzentrationen von sCD21 und sCD23 im Serum und Plasma von antiphospholipid
syndrome (APS), juvenile arthritis (JA), Diabetes und autoimmune lymphoproliferative syndrom
(ALPS) Patienten untersucht. sCD21 Titer waren in APS Patienten im Vergleich mit gesunden
Spendern (P < 0.0001) deutlich verringert. Die Anwesenheit von spezifischen �2‐GPI Autoantikörpern

- 116 -
stand nicht in Beziehung zu dem sCD21 Titer. Allerdings sind die sCD21 Level in diesen Patienten
nicht von zusätzlichen Autoimmunerkrankungen wie SLE, vasculitis, DVT (deep vein thrombosis),
Fehlgeburten oder Thrombosis beeinflusst.
Im Falle von JA haben wir die Titer des Plasma sCD21 und sCD23 in Untergruppen von JA untersucht.
Unsere Daten demonstrieren, dass die sCD21 Titer in allen Untergruppen der JA Patienten gesunken
sind, während die Plasma sCD23 Level nur bedeutend in P‐JA und S‐JA Proben (P < 0.0001, 0.018)
gesunken sind, aber nicht in O‐JA (P < 0.3843).
Da gezeigt wurde das CD21 und CD23 an mehreren Autoimmun krankheiten beteiligt sind, haben wir
die Level von sCD21 und sCD23 in den Seren von gesunden und Diabetes Typ I und II Patienten
untersucht. Unsere Daten zeigen dass die Titer von sCD21 und sCD23 in Diabetes Typ I verringert
sind (P < 0.0067 und P < 0.0001) während es keine Veränderungen in Diabetes Typ II im Vergleich zu
gesunden Kontrollen gleichen Alters gibt.
Im Falle von ALPS Patienten wurde die Konzentration des löslichen CD21 in Seren von 17 ALPS
Patienten untersucht und mit gesunden Freiwilligen verglichen. Wir fanden heraus, dass die Titer
von sCD21 in den ALPS Patientenproben stark erhöht waren im Vergleich zu den gesunden
Personen, P < 0.006. Diese Krankheit wird in Kindern oder Erwachsenen gefunden ist aber meist in
der frühen Kindheit präsent. Das erhöhte sCD21 in ALPS Patientenseren könnte mit der Anwesenheit
von doppel‐negativen (TCR alpha/beta CD4‐ CD8‐) T‐Lymphozyten (DNT) in der Peripherie
zusammenhängen. Während dem ersten Jahrzehnt werden die T‐Zellen vom Thymus erzeugt, wobei
doppel‐negative Thymozyten höhere Level von CD21 exprimieren.
Unsere Arbeten zeigen auf, dass sCD21 und sCD23 zusätzliche Marker für Autoimmunerkrankungen
sind.

- 117 -
REFERENCES Aggarwal, A., Bhardwaj, A., Alam, S. and Misra, R. (2000). Evidence for activation of the alternate complement pathway in patients with juvenile rheumatoid arthritis. Rheumatology (Oxford) 39, 189‐92. Ahearn, J. M. and Fearon, D. T. (1989). Structure and function of the complement receptors, CR1 (CD35) and CR2 (CD21). Adv Immunol 46, 183‐219. Ahmed, M., Bjurholm, A., Srinivasan, G. R., Lundeberg, T., Theodorsson, E., Schultzberg, M. and Kreicbergs, A. (1995). Capsaicin effects on substance P and CGRP in rat adjuvant arthritis. Regul Pept 55, 85‐102. Ansel, J. C., Brown, J. R., Payan, D. G. and Brown, M. A. (1993). Substance P selectively activates TNF‐alpha gene expression in murine mast cells. J Immunol 150, 4478‐85. Attur, M. G., Patel, R. N., Abramson, S. B. and Amin, A. R. (1997). Interleukin‐17 up‐regulation of nitric oxide production in human osteoarthritis cartilage. Arthritis Rheum 40, 1050‐3. Aubry, J. P., Pochon, S., Graber, P., Jansen, K. U. and Bonnefoy, J. Y. (1992). CD21 is a ligand for CD23 and regulates IgE production. Nature 358, 505‐7. Babior, B. M. (2000). Phagocytes and oxidative stress. Am J Med 109, 33‐44. Bach, J. F. (1994). Insulin‐dependent diabetes mellitus as an autoimmune disease. Endocr Rev 15, 516‐42. Bacher, M., Metz, C. N., Calandra, T., Mayer, K., Chesney, J., Lohoff, M., Gemsa, D., Donnelly, T. and Bucala, R. (1996). An essential regulatory role for macrophage migration inhibitory factor in T‐cell activation. Proc Natl Acad Sci U S A 93, 7849‐54. Baggiolini, M., Dewald, B. and Moser, B. (1994). Interleukin‐8 and related chemotactic cytokines‐‐CXC and CC chemokines. Adv Immunol 55, 97‐179. Baluk, P., Bertrand, C., Geppetti, P., McDonald, D. M. and Nadel, J. A. (1996). NK1 receptor antagonist CP‐99,994 inhibits cigarette smoke‐induced neutrophil and eosinophil adhesion in rat tracheal venules. Exp Lung Res 22, 409‐18. Bansal, A., Roberts, T., Hay, E. M., Kay, R., Pumphrey, R. S. and Wilson, P. B. (1992). Soluble CD23 levels are elevated in the serum of patients with primary Sjogren's syndrome and systemic lupus erythematosus. Clin Exp Immunol 89, 452‐5. Basu, D., Horvath, S., Matsumoto, I., Fremont, D. H. and Allen, P. M. (2000). Molecular basis for recognition of an arthritic peptide and a foreign epitope on distinct MHC molecules by a single TCR. J Immunol 164, 5788‐96. Basu, D., Horvath, S., O'Mara, L., Donermeyer, D. and Allen, P. M. (2001). Two MHC surface amino acid differences distinguish foreign peptide recognition from autoantigen specificity. J Immunol 166, 4005‐11. Bauer, K., Yu, X., Wernhoff, P., Koczan, D., Thiesen, H. J. and Ibrahim, S. M. (2004). Identification of new quantitative trait loci in mice with collagen‐induced arthritis. Arthritis Rheum 50, 3721‐8. Benoist, C. and Mathis, D. (2000). A revival of the B cell paradigm for rheumatoid arthritis pathogenesis? Arthritis Res 2, 90‐4. Bernhagen, J., Bacher, M., Calandra, T., Metz, C. N., Doty, S. B., Donnelly, T. and Bucala, R. (1996). An essential role for macrophage migration inhibitory factor in the tuberculin delayed‐type hypersensitivity reaction. J Exp Med 183, 277‐82.

- 118 -
Bernhagen, J., Mitchell, R. A., Calandra, T., Voelter, W., Cerami, A. and Bucala, R. (1994). Purification, bioactivity, and secondary structure analysis of mouse and human macrophage migration inhibitory factor (MIF). Biochemistry 33, 14144‐55. Bertolini, D. R., Nedwin, G. E., Bringman, T. S., Smith, D. D. and Mundy, G. R. (1986). Stimulation of bone resorption and inhibition of bone formation in vitro by human tumour necrosis factors. Nature 319, 516‐8. Blank, M., Cohen, J., Toder, V. and Shoenfeld, Y. (1991). Induction of anti‐phospholipid syndrome in naive mice with mouse lupus monoclonal and human polyclonal anti‐cardiolipin antibodies. Proc Natl Acad Sci U S A 88, 3069‐73. Blank, M., George, J., Barak, V., Tincani, A., Koike, T. and Shoenfeld, Y. (1998). Oral tolerance to low dose beta 2‐glycoprotein I: immunomodulation of experimental antiphospholipid syndrome. J Immunol 161, 5303‐12. Blank, M., Krause, I., Fridkin, M., Keller, N., Kopolovic, J., Goldberg, I., Tobar, A. and Shoenfeld, Y. (2002). Bacterial induction of autoantibodies to beta2‐glycoprotein‐I accounts for the infectious etiology of antiphospholipid syndrome. J Clin Invest 109, 797‐804. Blank, M., Shoenfeld, Y., Cabilly, S., Heldman, Y., Fridkin, M. and Katchalski‐Katzir, E. (1999). Prevention of experimental antiphospholipid syndrome and endothelial cell activation by synthetic peptides. Proc Natl Acad Sci U S A 96, 5164‐8. Blank, M., Tincani, A. and Shoenfeld, Y. (1994). Induction of experimental antiphospholipid syndrome in naive mice with purified IgG antiphosphatidylserine antibodies. J Rheumatol 21, 100‐4. Bogers, W. M., Stad, R. K., Janssen, D. J., van Rooijen, N., van Es, L. A. and Daha, M. R. (1991). Kupffer cell depletion in vivo results in preferential elimination of IgG aggregates and immune complexes via specific Fc receptors on rat liver endothelial cells. Clin Exp Immunol 86, 328‐33. Bonnefoy, J. Y., Gauchat, J. F., Life, P., Graber, P., Aubry, J. P. and Lecoanet‐Henchoz, S. (1995a). Regulation of IgE synthesis by CD23/CD21 interaction. Int Arch Allergy Immunol 107, 40‐2. Bonnefoy, J. Y., Lecoanet‐Henchoz, S., Aubry, J. P., Gauchat, J. F. and Graber, P. (1995b). CD23 and B‐cell activation. Curr Opin Immunol 7, 355‐9. Borden, P. and Heller, R. A. (1997). Transcriptional control of matrix metalloproteinases and the tissue inhibitors of matrix metalloproteinases. Crit Rev Eukaryot Gene Expr 7, 159‐78. Borden, P., Solymar, D., Sucharczuk, A., Lindman, B., Cannon, P. and Heller, R. A. (1996). Cytokine control of interstitial collagenase and collagenase‐3 gene expression in human chondrocytes. J Biol Chem 271, 23577‐81. Borovikova, L. V., Ivanova, S., Zhang, M., Yang, H., Botchkina, G. I., Watkins, L. R., Wang, H., Abumrad, N., Eaton, J. W. and Tracey, K. J. (2000). Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 405, 458‐62. Bradbury, L. E., Kansas, G. S., Levy, S., Evans, R. L. and Tedder, T. F. (1992). The CD19/CD21 signal transducing complex of human B lymphocytes includes the target of antiproliferative antibody‐1 and Leu‐13 molecules. J Immunol 149, 2841‐50. Brennan, F. M. (2001). A follow‐up to "Anti‐cytokine therapy in chronic destructive arthritis" by Wim B van den Berg. Arthritis Res 3, 211‐3; discussion 214‐5.

- 119 -
Bresson, M., Cirimele, V., Villain, M. and Kintz, P. (2006). Doping control for metandienone using hair analyzed by gas chromatography‐tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 836, 124‐8. Brinckerhoff, C. E., Rutter, J. L. and Benbow, U. (2000). Interstitial collagenases as markers of tumor progression. Clin Cancer Res 6, 4823‐30. Brown‐Galatola, C. H. and Hall, N. D. (1992). Impaired suppressor cell activity due to surface sulphydryl oxidation in rheumatoid arthritis. Br J Rheumatol 31, 599‐603. Buck, S. H. and Burks, T. F. (1986). The neuropharmacology of capsaicin: review of some recent observations. Pharmacol Rev 38, 179‐226. Burkhardt, H., Koller, T., Engstrom, A., Nandakumar, K. S., Turnay, J., Kraetsch, H. G., Kalden, J. R. and Holmdahl, R. (2002). Epitope‐specific recognition of type II collagen by rheumatoid arthritis antibodies is shared with recognition by antibodies that are arthritogenic in collagen‐induced arthritis in the mouse. Arthritis Rheum 46, 2339‐48. Burmester, G. R., Stuhlmuller, B., Keyszer, G. and Kinne, R. W. (1997). Mononuclear phagocytes and rheumatoid synovitis. Mastermind or workhorse in arthritis? Arthritis Rheum 40, 5‐18. Calandra, T., Bernhagen, J., Metz, C. N., Spiegel, L. A., Bacher, M., Donnelly, T., Cerami, A. and Bucala, R. (1995). MIF as a glucocorticoid‐induced modulator of cytokine production. Nature 377, 68‐71. Calandra, T., Bernhagen, J., Mitchell, R. A. and Bucala, R. (1994). The macrophage is an important and previously unrecognized source of macrophage migration inhibitory factor. J Exp Med 179, 1895‐902. Calandra, T. and Bucala, R. (1995). Macrophage migration inhibitory factor: a counter‐regulator of glucocorticoid action and critical mediator of septic shock. J Inflamm 47, 39‐51. Cao, T., Pinter, E., Al‐Rashed, S., Gerard, N., Hoult, J. R. and Brain, S. D. (2000). Neurokinin‐1 receptor agonists are involved in mediating neutrophil accumulation in the inflamed, but not normal, cutaneous microvasculature: an in vivo study using neurokinin‐1 receptor knockout mice. J Immunol 164, 5424‐9. Carel, J. C., Myones, B. L., Frazier, B. and Holers, V. M. (1990). Structural requirements for C3d,g/Epstein‐Barr virus receptor (CR2/CD21) ligand binding, internalization, and viral infection. In J Biol Chem, vol. 265, pp. 12293‐9. Cerinic, M. M., Konttinen, Y., Generini, S. and Cutolo, M. (1998). Neuropeptides and steroid hormones in arthritis. Curr Opin Rheumatol 10, 220‐35. Chang, F. Y. and Shaio, M. F. (1995). Decreased cell‐mediated immunity in patients with non‐insulin‐dependent diabetes mellitus. Diabetes Res Clin Pract 28, 137‐46. Chanock, S. J., el Benna, J., Smith, R. M. and Babior, B. M. (1994). The respiratory burst oxidase. J Biol Chem 269, 24519‐22. Charlton, B., Zhang, M. D. and Slattery, R. M. (2001). B lymphocytes not required for progression from insulitis to diabetes in non‐obese diabetic mice. Immunol Cell Biol 79, 597‐601. Chen, C. Y., Tsao, C. H., Ou, L. S., Yang, M. H., Kuo, M. L. and Huang, J. L. (2002). Comparison of soluble adhesion molecules in juvenile idiopathic arthritis between the active and remission stages. Ann Rheum Dis 61, 167‐70. Chen, P. P., Fong, S. and Carson, D. A. (1987). Rheumatoid factor. Rheum Dis Clin North Am 13, 545‐68.

- 120 -
Cherukuri, A., Cheng, P. C. and Pierce, S. K. (2001). The role of the CD19/CD21 complex in B cell processing and presentation of complement‐tagged antigens. J Immunol 167, 163‐72. Chevalier, J. and Kazatchkine, M. D. (1989). Distribution in clusters of complement receptor type one (CR1) on human erythrocytes. J Immunol 142, 2031‐6. Chomarat, P., Briolay, J., Banchereau, J. and Miossec, P. (1993). Increased production of soluble CD23 in rheumatoid arthritis, and its regulation by interleukin‐4. Arthritis Rheum 36, 234‐42. Chu, C. Q., Field, M., Feldmann, M. and Maini, R. N. (1991). Localization of tumor necrosis factor alpha in synovial tissues and at the cartilage‐pannus junction in patients with rheumatoid arthritis. Arthritis Rheum 34, 1125‐32. Churchill, W. H., Jr., Piessens, W. F., Sulis, C. A. and David, J. R. (1975). Macrophages activated as suspension cultures with lymphocyte mediators devoid of antigen become cytotoxic for tumor cells. J Immunol 115, 781‐6. Cook, A. D., Stockman, A., Brand, C. A., Tait, B. D., Mackay, I. R., Muirden, K. D., Bernard, C. C. and Rowley, M. J. (1999). Antibodies to type II collagen and HLA disease susceptibility markers in rheumatoid arthritis. Arthritis Rheum 42, 2569‐76. Cordell, G. A. and Araujo, O. E. (1993). Capsaicin: identification, nomenclature, and pharmacotherapy. Ann Pharmacother 27, 330‐6. Corominas, M., Mestre, M., Bas, J. and Buendia, E. (1998). Distinct modulation by interferon‐gamma (IFN‐gamma) of CD23 expression on B and T lymphocytes of atopic subjects. Clin Exp Immunol 112, 276‐80. Cuozzo, J. W. and Kaiser, C. A. (1999). Competition between glutathione and protein thiols for disulphide‐bond formation. Nat Cell Biol 1, 130‐5. Davidson, A. and Diamond, B. (2001). Autoimmune diseases. N Engl J Med 345, 340‐50. Davies, K. A., Erlendsson, K., Beynon, H. L., Peters, A. M., Steinsson, K., Valdimarsson, H. and Walport, M. J. (1993). Splenic uptake of immune complexes in man is complement‐dependent. J Immunol 151, 3866‐73. Dayer, J. M., Beutler, B. and Cerami, A. (1985). Cachectin/tumor necrosis factor stimulates collagenase and prostaglandin E2 production by human synovial cells and dermal fibroblasts. J Exp Med 162, 2163‐8. Del Nagro, C. J., Kolla, R. V. and Rickert, R. C. (2005). A critical role for complement C3d and the B cell coreceptor (CD19/CD21) complex in the initiation of inflammatory arthritis. J Immunol 175, 5379‐89. Delespesse, G., Sarfati, M., Wu, C. Y., Fournier, S. and Letellier, M. (1992). The low‐affinity receptor for IgE. Immunol Rev 125, 77‐97. Delespesse, G., Suter, U., Mossalayi, D., Bettler, B., Sarfati, M., Hofstetter, H., Kilcherr, E., Debre, P. and Dalloul, A. (1991). Expression, structure, and function of the CD23 antigen. Adv Immunol 49, 149‐91. Delibrias, C. C., Fischer, E., Bismuth, G. and Kazatchkine, M. D. (1992). Expression, molecular association, and functions of C3 complement receptors CR1 (CD35) and CR2 (CD21) on the human T cell line HPB‐ALL. J Immunol 149, 768‐74. Deneke, S. M. (2000). Thiol‐based antioxidants. Curr Top Cell Regul 36, 151‐80. Deutsch, M., Tsopanou, E. and Dourakis, S. P. (2004). The autoimmune lymphoproliferative syndrome (Canale‐Smith) in adulthood. Clin Rheumatol 23, 43‐4.

- 121 -
Dianzani, U., Bragardo, M., DiFranco, D., Alliaudi, C., Scagni, P., Buonfiglio, D., Redoglia, V., Bonissoni, S., Correra, A., Dianzani, I. et al. (1997). Deficiency of the Fas apoptosis pathway without Fas gene mutations in pediatric patients with autoimmunity/lymphoproliferation. Blood 89, 2871‐9. Dickinson, D. A. and Forman, H. J. (2002). Cellular glutathione and thiols metabolism. Biochem Pharmacol 64, 1019‐26. Droge, W. (2002). Free radicals in the physiological control of cell function. Physiol Rev 82, 47‐95. Edwards, J. C. and Cambridge, G. (2001). Sustained improvement in rheumatoid arthritis following a protocol designed to deplete B lymphocytes. Rheumatology (Oxford) 40, 205‐11. Edwards, J. C., Cambridge, G. and Abrahams, V. M. (1999). Do self‐perpetuating B lymphocytes drive human autoimmune disease? Immunology 97, 188‐96. Elliott, M. J., Maini, R. N., Feldmann, M., Long‐Fox, A., Charles, P., Katsikis, P., Brennan, F. M., Walker, J., Bijl, H., Ghrayeb, J. et al. (1993). Treatment of rheumatoid arthritis with chimeric monoclonal antibodies to tumor necrosis factor alpha. Arthritis Rheum 36, 1681‐90. Eskandari, F., Webster, J. I. and Sternberg, E. M. (2003). Neural immune pathways and their connection to inflammatory diseases. Arthritis Res Ther 5, 251‐65. Falcone, M., Lee, J., Patstone, G., Yeung, B. and Sarvetnick, N. (1998). B lymphocytes are crucial antigen‐presenting cells in the pathogenic autoimmune response to GAD65 antigen in nonobese diabetic mice. J Immunol 161, 1163‐8. Fearon, D. T. (1980). Identification of the membrane glycoprotein that is the C3b receptor of the human erythrocyte, polymorphonuclear leukocyte, B lymphocyte, and monocyte. J Exp Med 152, 20‐30. Feldmann, M., Brennan, F. M. and Maini, R. N. (1996). Role of cytokines in rheumatoid arthritis. Annu Rev Immunol 14, 397‐440. Feldmann, M. and Maini, R. N. (2001). Anti‐TNF alpha therapy of rheumatoid arthritis: what have we learned? Annu Rev Immunol 19, 163‐96. Fingeroth, J. D., Weis, J. J., Tedder, T. F., Strominger, J. L., Biro, P. A. and Fearon, D. T. (1984). Epstein‐Barr virus receptor of human B lymphocytes is the C3d receptor CR2. Proc Natl Acad Sci U S A 81, 4510‐4. Firestein, G. S. and Zvaifler, N. J. (1990). How important are T cells in chronic rheumatoid synovitis? Arthritis Rheum 33, 768‐73. Fischer, E., Delibrias, C. and Kazatchkine, M. D. (1991). Expression of CR2 (the C3dg/EBV receptor, CD21) on normal human peripheral blood T lymphocytes. J Immunol 146, 865‐9. Fischer, E. M., Mouhoub, A., Maillet, F., Fremeaux‐Bacchi, V., Krief, C., Gould, H., Berrih‐Aknin, S. and Kazatchkine, M. D. (1999). Expression of CD21 is developmentally regulated during thymic maturation of human T lymphocytes. Int Immunol 11, 1841‐9. Fisher, G. H., Rosenberg, F. J., Straus, S. E., Dale, J. K., Middleton, L. A., Lin, A. Y., Strober, W., Lenardo, M. J. and Puck, J. M. (1995). Dominant interfering Fas gene mutations impair apoptosis in a human autoimmune lymphoproliferative syndrome. Cell 81, 935‐46. Fitzgerald, M. (1989). Arthritis and the nervous system. Trends Neurosci 12, 86‐7. Fosang, A. J., Last, K., Knauper, V., Murphy, G. and Neame, P. J. (1996). Degradation of cartilage aggrecan by collagenase‐3 (MMP‐13). FEBS Lett 380, 17‐20.

- 122 -
Freije, J. M., Diez‐Itza, I., Balbin, M., Sanchez, L. M., Blasco, R., Tolivia, J. and Lopez‐Otin, C. (1994). Molecular cloning and expression of collagenase‐3, a novel human matrix metalloproteinase produced by breast carcinomas. J Biol Chem 269, 16766‐73. Fremeaux‐Bacchi, V., Aubry, J. P., Bonnefoy, J. Y., Kazatchkine, M. D., Kolb, J. P. and Fischer, E. M. (1998a). Soluble CD21 induces activation and differentiation of human monocytes through binding to membrane CD23. Eur J Immunol 28, 4268‐74. Fremeaux‐Bacchi, V., Bernard, I., Maillet, F., Mani, J. C., Fontaine, M., Bonnefoy, J. Y., Kazatchkine, M. D. and Fischer, E. (1996). Human lymphocytes shed a soluble form of CD21 (the C3dg/Epstein‐Barr virus receptor, CR2) that binds iC3b and CD23. Eur J Immunol 26, 1497‐503. Fremeaux‐Bacchi, V., Fischer, E., Lecoanet‐Henchoz, S., Mani, J. C., Bonnefoy, J. Y. and Kazatchkine, M. D. (1998b). Soluble CD21 (sCD21) forms biologically active complexes with CD23: sCD21 is present in normal plasma as a complex with trimeric CD23 and inhibits soluble CD23‐induced IgE synthesis by B cells. Int Immunol 10, 1459‐66. Furie, M. B. and Randolph, G. J. (1995). Chemokines and tissue injury. Am J Pathol 146, 1287‐301. Fuss, I. J., Strober, W., Dale, J. K., Fritz, S., Pearlstein, G. R., Puck, J. M., Lenardo, M. J. and Straus, S. E. (1997). Characteristic T helper 2 T cell cytokine abnormalities in autoimmune lymphoproliferative syndrome, a syndrome marked by defective apoptosis and humoral autoimmunity. J Immunol 158, 1912‐8. Galli, M., Comfurius, P., Maassen, C., Hemker, H. C., de Baets, M. H., van Breda‐Vriesman, P. J., Barbui, T., Zwaal, R. F. and Bevers, E. M. (1990). Anticardiolipin antibodies (ACA) directed not to cardiolipin but to a plasma protein cofactor. Lancet 335, 1544‐7. Gamse, R., Petsche, U., Lembeck, F. and Jancso, G. (1982). Capsaicin applied to peripheral nerve inhibits axoplasmic transport of substance P and somatostatin. Brain Res 239, 447‐62. Gare, B. A. (1998). Epidemiology. Baillieres Clin Rheumatol 12, 191‐208. Gelderman, K. A., Hultqvist, M., Holmberg, J., Olofsson, P. and Holmdahl, R. (2006). T cell surface redox levels determine T cell reactivity and arthritis susceptibility. Proc Natl Acad Sci U S A 103, 12831‐6. George, J., Blank, M., Levy, Y., Meroni, P., Damianovich, M., Tincani, A. and Shoenfeld, Y. (1998). Differential effects of anti‐beta2‐glycoprotein I antibodies on endothelial cells and on the manifestations of experimental antiphospholipid syndrome. Circulation 97, 900‐6. Grigg, P. (2001). Properties of sensory neurons innervating synovial joints. Cells Tissues Organs 169, 218‐25. Grimsholm, O., Rantapaa‐Dahlqvist, S. and Forsgren, S. (2005). Levels of gastrin‐releasing peptide and substance P in synovial fluid and serum correlate with levels of cytokines in rheumatoid arthritis. Arthritis Res Ther 7, R416‐26. Groemping, Y. and Rittinger, K. (2005). Activation and assembly of the NADPH oxidase: a structural perspective. Biochem J 386, 401‐16. Gross, J. A., Johnston, J., Mudri, S., Enselman, R., Dillon, S. R., Madden, K., Xu, W., Parrish‐Novak, J., Foster, D., Lofton‐Day, C. et al. (2000). TACI and BCMA are receptors for a TNF homologue implicated in B‐cell autoimmune disease. Nature 404, 995‐9. Harigai, M., Hara, M., Yoshimura, T., Leonard, E. J., Inoue, K. and Kashiwazaki, S. (1993). Monocyte chemoattractant protein‐1 (MCP‐1) in inflammatory joint diseases and

- 123 -
its involvement in the cytokine network of rheumatoid synovium. Clin Immunol Immunopathol 69, 83‐91. Hietala, M. A., Nandakumar, K. S., Persson, L., Fahlen, S., Holmdahl, R. and Pekna, M. (2004). Complement activation by both classical and alternative pathways is critical for the effector phase of arthritis. Eur J Immunol 34, 1208‐16. Highton, J., Carlisle, B. and Palmer, D. G. (1995). Changes in the phenotype of monocytes/macrophages and expression of cytokine mRNA in peripheral blood and synovial fluid of patients with rheumatoid arthritis. Clin Exp Immunol 102, 541‐6. Hitchon, C. A. and El‐Gabalawy, H. S. (2004). Oxidation in rheumatoid arthritis. Arthritis Res Ther 6, 265‐78. Hokfelt, T., Kellerth, J. O., Nilsson, G. and Pernow, B. (1975). Substance p: localization in the central nervous system and in some primary sensory neurons. Science 190, 889‐90. Holmdahl, R., Rubin, K., Klareskog, L., Larsson, E. and Wigzell, H. (1986). Characterization of the antibody response in mice with type II collagen‐induced arthritis, using monoclonal anti‐type II collagen antibodies. Arthritis Rheum 29, 400‐10. Honeyman, M., Wasserfall, C., Nerup, J. and Rossini, A. (1997). Prediction and prevention of IDDM. Diabetologia 40 Suppl 3, B58‐61. Huemer, H. P., Larcher, C., Prodinger, W. M., Petzer, A. L., Mitterer, M. and Falser, N. (1993). Determination of soluble CD21 as a parameter of B cell activation. Clin Exp Immunol 93, 195‐9. Hultqvist, M., Olofsson, P., Gelderman, K. A., Holmberg, J. and Holmdahl, R. (2006). A new arthritis therapy with oxidative burst inducers. PLoS Med 3, e348. Hussain, M. J., Maher, J., Warnock, T., Vats, A., Peakman, M. and Vergani, D. (1998). Cytokine overproduction in healthy first degree relatives of patients with IDDM. Diabetologia 41, 343‐9. Ibrahim, S. M., Koczan, D. and Thiesen, H. J. (2002). Gene‐expression profile of collagen‐induced arthritis. J Autoimmun 18, 159‐67. Illges, H., Braun, M., Peter, H. H. and Melchers, I. (1997). Analysis of the human CD21 transcription unit reveals differential splicing of exon 11 in mature transcripts and excludes alternative splicing as the mechanism causing solubilization of CD21. Mol Immunol 34, 683‐93. Illges, H., Braun, M., Peter, H. H. and Melchers, I. (2000). Reduced expression of the complement receptor type 2 (CR2, CD21) by synovial fluid B and T lymphocytes. Clin Exp Immunol 122, 270‐6. Jackson, C. E., Fischer, R. E., Hsu, A. P., Anderson, S. M., Choi, Y., Wang, J., Dale, J. K., Fleisher, T. A., Middelton, L. A., Sneller, M. C. et al. (1999). Autoimmune lymphoproliferative syndrome with defective Fas: genotype influences penetrance. Am J Hum Genet 64, 1002‐14. Jackson, S. H., Gallin, J. I. and Holland, S. M. (1995). The p47phox mouse knock‐out model of chronic granulomatous disease. J Exp Med 182, 751‐8. Ji, H., Korganow, A. S., Mangialaio, S., Hoglund, P., Andre, I., Luhder, F., Gonzalez, A., Poirot, L., Benoist, C. and Mathis, D. (1999). Different modes of pathogenesis in T‐cell‐dependent autoimmunity: clues from two TCR transgenic systems. Immunol Rev 169, 139‐46. Jiang, Z., Dun, N. J. and Karczmar, A. G. (1982). Substance P: a putative sensory transmitter in mammalian autonomic ganglia. Science 217, 739‐41.

- 124 -
Juttner, S., Bernhagen, J., Metz, C. N., Rollinghoff, M., Bucala, R. and Gessner, A. (1998). Migration inhibitory factor induces killing of Leishmania major by macrophages: dependence on reactive nitrogen intermediates and endogenous TNF‐alpha. J Immunol 161, 2383‐90. Kalli, K. R., Ahearn, J. M. and Fearon, D. T. (1991). Interaction of iC3b with recombinant isotypic and chimeric forms of CR2. J Immunol 147, 590‐4. Kasahara, Y., Wada, T., Niida, Y., Yachie, A., Seki, H., Ishida, Y., Sakai, T., Koizumi, F., Koizumi, S., Miyawaki, T. et al. (1998). Novel Fas (CD95/APO‐1) mutations in infants with a lymphoproliferative disorder. Int Immunol 10, 195‐202. Kashiwagi, N., Hirata, I. and Kasukawa, R. (1998). A role for granulocyte and monocyte apheresis in the treatment of rheumatoid arthritis. Ther Apher 2, 134‐41. Kelleher, C. A., Paterson, R. K., Dreyfus, D. H., Streib, J. E., Xu, J. W., Takase, K., Jones, J. F. and Gelfand, E. W. (1995). Epstein‐Barr virus replicative gene transcription during de novo infection of human thymocytes: simultaneous early expression of BZLF‐1 and its repressor RAZ. Virology 208, 685‐95. Kidd, B. L., Cruwys, S., Mapp, P. I. and Blake, D. R. (1992). Role of the sympathetic nervous system in chronic joint pain and inflammation. Ann Rheum Dis 51, 1188‐91. Kim, H. Y., Kim, W. U., Cho, M. L., Lee, S. K., Youn, J., Kim, S. I., Yoo, W. H., Park, J. H., Min, J. K., Lee, S. H. et al. (1999). Enhanced T cell proliferative response to type II collagen and synthetic peptide CII (255‐274) in patients with rheumatoid arthritis. Arthritis Rheum 42, 2085‐93. Kimura, M., Tanaka, S., Isoda, F., Sekigawa, K., Yamakawa, T. and Sekihara, H. (1998). T lymphopenia in obese diabetic (db/db) mice is non‐selective and thymus independent. Life Sci 62, 1243‐50. Kinoshita, T., Lavoie, S. and Nussenzweig, V. (1985). Regulatory proteins for the activated third and fourth components of complement (C3b and C4b) in mice. II. Identification and properties of complement receptor type 1 (CR1). J Immunol 134, 2564‐70. Klebanoff, S. J. (2005). Myeloperoxidase: friend and foe. J Leukoc Biol 77, 598‐625. Knauper, V., Lopez‐Otin, C., Smith, B., Knight, G. and Murphy, G. (1996a). Biochemical characterization of human collagenase‐3. J Biol Chem 271, 1544‐50. Knauper, V., Will, H., Lopez‐Otin, C., Smith, B., Atkinson, S. J., Stanton, H., Hembry, R. M. and Murphy, G. (1996b). Cellular mechanisms for human procollagenase‐3 (MMP‐13) activation. Evidence that MT1‐MMP (MMP‐14) and gelatinase a (MMP‐2) are able to generate active enzyme. J Biol Chem 271, 17124‐31. Kneilling, M., Hultner, L., Pichler, B. J., Mailhammer, R., Morawietz, L., Solomon, S., Eichner, M., Sabatino, J., Biedermann, T., Krenn, V. et al. (2007). Targeted mast cell silencing protects against joint destruction and angiogenesis in experimental arthritis in mice. Arthritis Rheum 56, 1806‐16. Koch, A. E., Kunkel, S. L., Harlow, L. A., Johnson, B., Evanoff, H. L., Haines, G. K., Burdick, M. D., Pope, R. M. and Strieter, R. M. (1992). Enhanced production of monocyte chemoattractant protein‐1 in rheumatoid arthritis. J Clin Invest 90, 772‐9. Konttinen, Y. T., Salo, T., Hanemaaijer, R., Valleala, H., Sorsa, T., Sutinen, M., Ceponis, A., Xu, J. W., Santavirta, S., Teronen, O. et al. (1999). Collagenase‐3 (MMP‐13) and its activators in rheumatoid arthritis: localization in the pannus‐hard tissue junction and inhibition by alendronate. Matrix Biol 18, 401‐12.

- 125 -
Kouskoff, V., Korganow, A. S., Duchatelle, V., Degott, C., Benoist, C. and Mathis, D. (1996). Organ‐specific disease provoked by systemic autoimmunity. Cell 87, 811‐22. Kunkel, S. L., Lukacs, N., Kasama, T. and Strieter, R. M. (1996). The role of chemokines in inflammatory joint disease. J Leukoc Biol 59, 6‐12. Kutukculer, N. and Caglayan, S. (1998). Plasma and synovial fluid soluble CD23 concentrations in children with juvenile chronic arthritis. Autoimmunity 27, 155‐8. Lander, E. S. and Botstein, D. (1989). Mapping mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics 121, 185‐99. Lawrence, D. A., Song, R. and Weber, P. (1996). Surface thiols of human lymphocytes and their changes after in vitro and in vivo activation. J Leukoc Biol 60, 611‐8. Le Novere, N. and Changeux, J. P. (1995). Molecular evolution of the nicotinic acetylcholine receptor: an example of multigene family in excitable cells. J Mol Evol 40, 155‐72. Lee, D. M., Friend, D. S., Gurish, M. F., Benoist, C., Mathis, D. and Brenner, M. B. (2002). Mast cells: a cellular link between autoantibodies and inflammatory arthritis. Science 297, 1689‐92. Leech, M., Metz, C., Hall, P., Hutchinson, P., Gianis, K., Smith, M., Weedon, H., Holdsworth, S. R., Bucala, R. and Morand, E. F. (1999). Macrophage migration inhibitory factor in rheumatoid arthritis: evidence of proinflammatory function and regulation by glucocorticoids. Arthritis Rheum 42, 1601‐8. Leech, M., Metz, C., Santos, L., Peng, T., Holdsworth, S. R., Bucala, R. and Morand, E. F. (1998). Involvement of macrophage migration inhibitory factor in the evolution of rat adjuvant arthritis. Arthritis Rheum 41, 910‐7. Leonard, S. and Bertrand, D. (2001). Neuronal nicotinic receptors: from structure to function. Nicotine Tob Res 3, 203‐23. Levine, J. D., Clark, R., Devor, M., Helms, C., Moskowitz, M. A. and Basbaum, A. I. (1984). Intraneuronal substance P contributes to the severity of experimental arthritis. Science 226, 547‐9. Levine, J. D., Coderre, T. J., Helms, C. and Basbaum, A. I. (1988). Beta 2‐adrenergic mechanisms in experimental arthritis. Proc Natl Acad Sci U S A 85, 4553‐6. Levine, J. D., Collier, D. H., Basbaum, A. I., Moskowitz, M. A. and Helms, C. A. (1985). Hypothesis: the nervous system may contribute to the pathophysiology of rheumatoid arthritis. J Rheumatol 12, 406‐11. Levinson, A. I., Dziarski, A., Pincus, T., deHoratius, R. J. and Zweiman, B. (1981). Heterogeneity of polyclonal B‐cell activity in systemic lupus erythematosus. J Clin Lab Immunol 5, 17‐22. Liew, F. Y. (1994). Regulation of nitric oxide synthesis in infectious and autoimmune diseases. Immunol Lett 43, 95‐8. Ling, N., Hansel, T., Richardson, P. and Brown, B. (1991). Cellular origins of serum complement receptor type 2 in normal individuals and in hypogammaglobulinaemia. Clin Exp Immunol 84, 16‐22. Liu, Y. J., Cairns, J. A., Holder, M. J., Abbot, S. D., Jansen, K. U., Bonnefoy, J. Y., Gordon, J. and MacLennan, I. C. (1991). Recombinant 25‐kDa CD23 and interleukin 1 alpha promote the survival of germinal center B cells: evidence for bifurcation in the development of centrocytes rescued from apoptosis. Eur J Immunol 21, 1107‐14. Londei, M., Savill, C. M., Verhoef, A., Brennan, F., Leech, Z. A., Duance, V., Maini, R. N. and Feldmann, M. (1989). Persistence of collagen type II‐specific T‐cell clones in the

- 126 -
synovial membrane of a patient with rheumatoid arthritis. Proc Natl Acad Sci U S A 86, 636‐40. Lotz, M., Carson, D. A. and Vaughan, J. H. (1987). Substance P activation of rheumatoid synoviocytes: neural pathway in pathogenesis of arthritis. Science 235, 893‐5. Lowe, J., Brown, B., Hardie, D., Richardson, P. and Ling, N. (1989). Soluble forms of CD21 and CD23 antigens in the serum in B cell chronic lymphocytic leukaemia. Immunol Lett 20, 103‐9. Luber‐Narod, J., Kage, R. and Leeman, S. E. (1994). Substance P enhances the secretion of tumor necrosis factor‐alpha from neuroglial cells stimulated with lipopolysaccharide. J Immunol 152, 819‐24. Lyubchenko, T., dal Porto, J., Cambier, J. C. and Holers, V. M. (2005). Coligation of the B cell receptor with complement receptor type 2 (CR2/CD21) using its natural ligand C3dg: activation without engagement of an inhibitory signaling pathway. J Immunol 174, 3264‐72. Maccioni, M., Zeder‐Lutz, G., Huang, H., Ebel, C., Gerber, P., Hergueux, J., Marchal, P., Duchatelle, V., Degott, C., van Regenmortel, M. et al. (2002). Arthritogenic monoclonal antibodies from K/BxN mice. J Exp Med 195, 1071‐7. MacGregor, A. J., Snieder, H., Rigby, A. S., Koskenvuo, M., Kaprio, J., Aho, K. and Silman, A. J. (2000). Characterizing the quantitative genetic contribution to rheumatoid arthritis using data from twins. Arthritis Rheum 43, 30‐7. Mapp, P. I., Kidd, B. L., Gibson, S. J., Terry, J. M., Revell, P. A., Ibrahim, N. B., Blake, D. R. and Polak, J. M. (1990). Substance P‐, calcitonin gene‐related peptide‐ and C‐flanking peptide of neuropeptide Y‐immunoreactive fibres are present in normal synovium but depleted in patients with rheumatoid arthritis. Neuroscience 37, 143‐53. Marrack, P., Kappler, J. and Kotzin, B. L. (2001). Autoimmune disease: why and where it occurs. Nat Med 7, 899‐905. Marubio, L. M. and Changeux, J. (2000). Nicotinic acetylcholine receptor knockout mice as animal models for studying receptor function. Eur J Pharmacol 393, 113‐21. Masilamani, M., Kassahn, D., Mikkat, S., Glocker, M. O. and Illges, H. (2003). B cell activation leads to shedding of complement receptor type II (CR2/CD21). Eur J Immunol 33, 2391‐7. Masilamani, M., Nowack, R., Witte, T., Schlesier, M., Warnatz, K., Glocker, M. O., Peter, H. H. and Illges, H. (2004a). Reduction of soluble complement receptor 2/CD21 in systemic lupus erythomatosus and Sjogren's syndrome but not juvenile arthritis. Scand J Immunol 60, 625‐30. Masilamani, M., Rajasekaran, N., Singh, A., Low, H. Z., Albus, K., Anders, S., Behne, F., Eiermann, P., Konig, K., Mindnich, C. et al. (2008). Systemic reduction of soluble complement receptor II/CD21 during pregnancy to levels reminiscent of autoimmune disease. Rheumatol Int 28, 1137‐41. Masilamani, M., von Kempis, J. and Illges, H. (2004b). Decreased levels of serum soluble complement receptor‐II (CR2/CD21) in patients with rheumatoid arthritis. Rheumatology (Oxford) 43, 186‐90. Masson‐Bessiere, C., Sebbag, M., Girbal‐Neuhauser, E., Nogueira, L., Vincent, C., Senshu, T. and Serre, G. (2001). The major synovial targets of the rheumatoid arthritis‐specific antifilaggrin autoantibodies are deiminated forms of the alpha‐ and beta‐chains of fibrin. J Immunol 166, 4177‐84.

- 127 -
Matsumoto, I., Maccioni, M., Lee, D. M., Maurice, M., Simmons, B., Brenner, M., Mathis, D. and Benoist, C. (2002). How antibodies to a ubiquitous cytoplasmic enzyme may provoke joint‐specific autoimmune disease. Nat Immunol 3, 360‐5. Matsumoto, I., Okada, S., Kuroda, K., Iwamoto, I., Saito, Y., Tokuhisa, T., Nishioka, K. and Sumida, T. (1999). Single cell analysis of T cells infiltrating labial salivary glands from patients with Sjogren's syndrome. Int J Mol Med 4, 519‐27. Mengshol, J. A., Vincenti, M. P., Coon, C. I., Barchowsky, A. and Brinckerhoff, C. E. (2000). Interleukin‐1 induction of collagenase 3 (matrix metalloproteinase 13) gene expression in chondrocytes requires p38, c‐Jun N‐terminal kinase, and nuclear factor kappaB: differential regulation of collagenase 1 and collagenase 3. Arthritis Rheum 43, 801‐11. Meroni, P., Ronda, N., Raschi, E. and Borghi, M. O. (2005). Humoral autoimmunity against endothelium: theory or reality? Trends Immunol 26, 275‐81. Miller, E. J. and Gay, S. (1987). The collagens: an overview and update. Methods Enzymol 144, 3‐41. Miller, J. J., 3rd, Olds, L. C. and Huene, D. B. (1986). Complement activation products and factors influencing phagocyte migration in synovial fluids from children with chronic arthritis. Clin Exp Rheumatol 4, 53‐6. Miller, L. E., Justen, H. P., Scholmerich, J. and Straub, R. H. (2000). The loss of sympathetic nerve fibers in the synovial tissue of patients with rheumatoid arthritis is accompanied by increased norepinephrine release from synovial macrophages. Faseb J 14, 2097‐107. Mitchell, P. G., Magna, H. A., Reeves, L. M., Lopresti‐Morrow, L. L., Yocum, S. A., Rosner, P. J., Geoghegan, K. F. and Hambor, J. E. (1996). Cloning, expression, and type II collagenolytic activity of matrix metalloproteinase‐13 from human osteoarthritic cartilage. J Clin Invest 97, 761‐8. Molina, H., Kinoshita, T., Webster, C. B. and Holers, V. M. (1994). Analysis of C3b/C3d binding sites and factor I cofactor regions within mouse complement receptors 1 and 2. J Immunol 153, 789‐95. Moore, M. D., DiScipio, R. G., Cooper, N. R. and Nemerow, G. R. (1989). Hydrodynamic, electron microscopic, and ligand‐binding analysis of the Epstein‐Barr virus/C3dg receptor (CR2). J Biol Chem 264, 20576‐82. Morand, E. F., Leech, M., Weedon, H., Metz, C., Bucala, R. and Smith, M. D. (2002). Macrophage migration inhibitory factor in rheumatoid arthritis: clinical correlations. Rheumatology (Oxford) 41, 558‐62. Myones, B. L. and Ross, G. D. (1987). Identification of a spontaneously shed fragment of B cell complement receptor type two (CR2) containing the C3d‐binding site. Complement 4, 87‐98. Nagase, H. and Woessner, J. F., Jr. (1999). Matrix metalloproteinases. J Biol Chem 274, 21491‐4. Nagashima, M., Yoshino, S., Tanaka, H., Yoshida, N., Kashiwagi, N. and Saniabadi, A. R. (1998). Granulocyte and monocyte apheresis suppresses symptoms of rheumatoid arthritis: a pilot study. Rheumatol Int 18, 113‐8. Nagata, S. and Suda, T. (1995). Fas and Fas ligand: lpr and gld mutations. Immunol Today 16, 39‐43. Nandakumar, K. S., Andren, M., Martinsson, P., Bajtner, E., Hellstrom, S., Holmdahl, R. and Kleinau, S. (2003a). Induction of arthritis by single monoclonal IgG anti‐

- 128 -
collagen type II antibodies and enhancement of arthritis in mice lacking inhibitory FcgammaRIIB. Eur J Immunol 33, 2269‐77. Nandakumar, K. S., Svensson, L. and Holmdahl, R. (2003b). Collagen type II‐specific monoclonal antibody‐induced arthritis in mice: description of the disease and the influence of age, sex, and genes. Am J Pathol 163, 1827‐37. Niissalo, S., Hukkanen, M., Imai, S., Tornwall, J. and Konttinen, Y. T. (2002). Neuropeptides in experimental and degenerative arthritis. Ann N Y Acad Sci 966, 384‐99. O'Connor, T. M., O'Connell, J., O'Brien, D. I., Goode, T., Bredin, C. P. and Shanahan, F. (2004). The role of substance P in inflammatory disease. J Cell Physiol 201, 167‐80. Onodera, S., Suzuki, K., Matsuno, T., Kaneda, K., Takagi, M. and Nishihira, J. (1997). Macrophage migration inhibitory factor induces phagocytosis of foreign particles by macrophages in autocrine and paracrine fashion. Immunology 92, 131‐7. Panayi, G. S., Corrigall, V. M. and Pitzalis, C. (2001). Pathogenesis of rheumatoid arthritis. The role of T cells and other beasts. Rheum Dis Clin North Am 27, 317‐34. Pascual, D. W. and Bost, K. L. (1990). Substance P production by P388D1 macrophages: a possible autocrine function for this neuropeptide. Immunology 71, 52‐6. Pearson, C. M. (1956). Development of arthritis, periarthritis and periostitis in rats given adjuvants. Proc Soc Exp Biol Med 91, 95‐101. Pernow, B. (1983). Substance P‐‐a putative mediator of antidromic vasodilation. Gen Pharmacol 14, 13‐6. Pitzalis, C. (1993). Adhesion and migration of inflammatory cells. Clin Exp Rheumatol 11 Suppl 8, S71‐6. Pochon, S., Graber, P., Yeager, M., Jansen, K., Bernard, A. R., Aubry, J. P. and Bonnefoy, J. Y. (1992). Demonstration of a second ligand for the low affinity receptor for immunoglobulin E (CD23) using recombinant CD23 reconstituted into fluorescent liposomes. J Exp Med 176, 389‐97. Quinn, M. T. and Gauss, K. A. (2004). Structure and regulation of the neutrophil respiratory burst oxidase: comparison with nonphagocyte oxidases. J Leukoc Biol 76, 760‐81. Randolph, G. J. and Furie, M. B. (1995). A soluble gradient of endogenous monocyte chemoattractant protein‐1 promotes the transendothelial migration of monocytes in vitro. J Immunol 155, 3610‐8. Rantapaa‐Dahlqvist, S., de Jong, B. A., Berglin, E., Hallmans, G., Wadell, G., Stenlund, H., Sundin, U. and van Venrooij, W. J. (2003). Antibodies against cyclic citrullinated peptide and IgA rheumatoid factor predict the development of rheumatoid arthritis. Arthritis Rheum 48, 2741‐9. Remans, P. H., van Oosterhout, M., Smeets, T. J., Sanders, M., Frederiks, W. M., Reedquist, K. A., Tak, P. P., Breedveld, F. C. and van Laar, J. M. (2005). Intracellular free radical production in synovial T lymphocytes from patients with rheumatoid arthritis. Arthritis Rheum 52, 2003‐9. Rieux‐Laucat, F., Fischer, A. and Deist, F. L. (2003). Cell‐death signaling and human disease. Curr Opin Immunol 15, 325‐31. Rieux‐Laucat, F., Le Deist, F., Hivroz, C., Roberts, I. A., Debatin, K. M., Fischer, A. and de Villartay, J. P. (1995). Mutations in Fas associated with human lymphoproliferative syndrome and autoimmunity. Science 268, 1347‐9. Robson, M. G. and Walport, M. J. (2001). Pathogenesis of systemic lupus erythematosus (SLE). Clin Exp Allergy 31, 678‐85.

- 129 -
Rudolphi, U., Rzepka, R., Batsford, S., Kaufmann, S. H., von der Mark, K., Peter, H. H. and Melchers, I. (1997). The B cell repertoire of patients with rheumatoid arthritis. II. Increased frequencies of IgG+ and IgA+ B cells specific for mycobacterial heat‐shock protein 60 or human type II collagen in synovial fluid and tissue. Arthritis Rheum 40, 1409‐19. Saklatvala, J. (1986). Tumour necrosis factor alpha stimulates resorption and inhibits synthesis of proteoglycan in cartilage. Nature 322, 547‐9. Sallfors, C., Hallberg, L. R. and Fasth, A. (2003). Gender and age differences in pain, coping and health status among children with chronic arthritis. Clin Exp Rheumatol 21, 785‐93. Sammaritano, L. R. and Gharavi, A. E. (1992). Antiphospholipid antibody syndrome. Clin Lab Med 12, 41‐59. Sarfati, M., Fournier, S., Wu, C. Y. and Delespesse, G. (1992). Expression, regulation and function of human Fc epsilon RII (CD23) antigen. Immunol Res 11, 260‐72. Sato, S., Fujimoto, M., Kikuchi, K., Ihn, H., Tamaki, K. and Takehara, K. (1996). Elevated soluble CD23 levels in the sera from patients with localized scleroderma. Arch Dermatol Res 288, 74‐8. Schellekens, G. A., de Jong, B. A., van den Hoogen, F. H., van de Putte, L. B. and van Venrooij, W. J. (1998). Citrulline is an essential constituent of antigenic determinants recognized by rheumatoid arthritis‐specific autoantibodies. J Clin Invest 101, 273‐81. Schifferli, J. A., Ng, Y. C. and Peters, D. K. (1986). The role of complement and its receptor in the elimination of immune complexes. N Engl J Med 315, 488‐95. Segal, A. W. (2005). How neutrophils kill microbes. Annu Rev Immunol 23, 197‐223. Serreze, D. V., Chapman, H. D., Varnum, D. S., Hanson, M. S., Reifsnyder, P. C., Richard, S. D., Fleming, S. A., Leiter, E. H. and Shultz, L. D. (1996). B lymphocytes are essential for the initiation of T cell‐mediated autoimmune diabetes: analysis of a new "speed congenic" stock of NOD.Ig mu null mice. J Exp Med 184, 2049‐53. Sheerin, N. S., Springall, T., Carroll, M. C., Hartley, B. and Sacks, S. H. (1997). Protection against anti‐glomerular basement membrane (GBM)‐mediated nephritis in C3‐ and C4‐deficient mice. Clin Exp Immunol 110, 403‐9. Shoenfeld, Y. (2003). Systemic antiphospholipid syndrome. Lupus 12, 497‐8. Silman, A. J. (1994). Epidemiology of rheumatoid arthritis. Apmis 102, 721‐8. Silverman, E. D., Isacovics, B., Petsche, D. and Laxer, R. M. (1993). Synovial fluid cells in juvenile arthritis: evidence of selective T cell migration to inflamed tissue. Clin Exp Immunol 91, 90‐5. Singh, A., Blank, M., Shoenfeld, Y. and Illges, H. (2008). Antiphospholipid syndrome patients display reduced titers of soluble CD21 in their sera irrespective of circulating anti‐beta2‐glycoprotein‐I autoantibodies. Rheumatol Int. Solomon, S., Kolb, C., Mohanty, S., Jeisy‐Walder, E., Preyer, R., Schollhorn, V. and Illges, H. (2002). Transmission of antibody‐induced arthritis is independent of complement component 4 (C4) and the complement receptors 1 and 2 (CD21/35). Eur J Immunol 32, 644‐51. Solomon, S., Rajasekaran, N., Jeisy‐Walder, E., Snapper, S. B. and Illges, H. (2005). A crucial role for macrophages in the pathology of K/B x N serum‐induced arthritis. Eur J Immunol 35, 3064‐73. Spooren, P. F., Vermes, I. and Soons, J. W. (1993). Similar alterations of lymphocyte subpopulations in type I and type II diabetes. Neth J Med 42, 163‐7.

- 130 -
Stahle‐Backdahl, M., Sandstedt, B., Bruce, K., Lindahl, A., Jimenez, M. G., Vega, J. A. and Lopez‐Otin, C. (1997). Collagenase‐3 (MMP‐13) is expressed during human fetal ossification and re‐expressed in postnatal bone remodeling and in rheumatoid arthritis. Lab Invest 76, 717‐28. Steinlein, O. (1998). New functions for nicotinic acetylcholine receptors? Behav Brain Res 95, 31‐5. Stetler‐Stevenson, W. G. and Yu, A. E. (2001). Proteases in invasion: matrix metalloproteinases. Semin Cancer Biol 11, 143‐52. Straub, R. H. and Cutolo, M. (2001). Involvement of the hypothalamic‐‐pituitary‐‐adrenal/gonadal axis and the peripheral nervous system in rheumatoid arthritis: viewpoint based on a systemic pathogenetic role. Arthritis Rheum 44, 493‐507. Stuart, J. M. and Dixon, F. J. (1983). Serum transfer of collagen‐induced arthritis in mice. J Exp Med 158, 378‐92. Sylvestre, D. L. and Ravetch, J. V. (1996). A dominant role for mast cell Fc receptors in the Arthus reaction. Immunity 5, 387‐90. Szakonyi, G., Guthridge, J. M., Li, D., Young, K., Holers, V. M. and Chen, X. S. (2001). Structure of complement receptor 2 in complex with its C3d ligand. In Science, vol. 292, pp. 1725‐8. Takemura, S., Klimiuk, P. A., Braun, A., Goronzy, J. J. and Weyand, C. M. (2001). T cell activation in rheumatoid synovium is B cell dependent. J Immunol 167, 4710‐8. Tarkowski, A., Klareskog, L., Carlsten, H., Herberts, P. and Koopman, W. J. (1989). Secretion of antibodies to types I and II collagen by synovial tissue cells in patients with rheumatoid arthritis. Arthritis Rheum 32, 1087‐92. Taxt, T., Tonnesen, H. and Jansen, J. K. (1983). Neonatal treatment with 6‐OH dopamine and the elimination of polyneuronal innervation of skeletal muscle in the rat. Acta Physiol Scand 119, 293‐7. Terato, K., Hasty, K. A., Reife, R. A., Cremer, M. A., Kang, A. H. and Stuart, J. M. (1992). Induction of arthritis with monoclonal antibodies to collagen. J Immunol 148, 2103‐8. Thomas, R., Davis, L. S. and Lipsky, P. E. (1994). Rheumatoid synovium is enriched in mature antigen‐presenting dendritic cells. J Immunol 152, 2613‐23. Thomas, R. and Lipsky, P. E. (1996). Could endogenous self‐peptides presented by dendritic cells initiate rheumatoid arthritis? Immunol Today 17, 559‐64. Thornton, S., Sowders, D., Aronow, B., Witte, D. P., Brunner, H. I., Giannini, E. H. and Hirsch, R. (2002). DNA microarray analysis reveals novel gene expression profiles in collagen‐induced arthritis. Clin Immunol 105, 155‐68. Towbin, H., Staehelin, T. and Gordon, J. (1979). Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci U S A 76, 4350‐4. Tsoukas, C. D. and Lambris, J. D. (1988). Expression of CR2/EBV receptors on human thymocytes detected by monoclonal antibodies. Eur J Immunol 18, 1299‐302. Uitto, V. J., Airola, K., Vaalamo, M., Johansson, N., Putnins, E. E., Firth, J. D., Salonen, J., Lopez‐Otin, C., Saarialho‐Kere, U. and Kahari, V. M. (1998). Collagenase‐3 (matrix metalloproteinase‐13) expression is induced in oral mucosal epithelium during chronic inflammation. Am J Pathol 152, 1489‐99.

- 131 -
Vaishnaw, A. K., Orlinick, J. R., Chu, J. L., Krammer, P. H., Chao, M. V. and Elkon, K. B. (1999). The molecular basis for apoptotic defects in patients with CD95 (Fas/Apo‐1) mutations. J Clin Invest 103, 355‐63. Van Damme, J., Proost, P., Lenaerts, J. P. and Opdenakker, G. (1992). Structural and functional identification of two human, tumor‐derived monocyte chemotactic proteins (MCP‐2 and MCP‐3) belonging to the chemokine family. J Exp Med 176, 59‐65. van Oss, C. J., Chaudhury, M. K. and Good, R. J. (1987). Monopolar surfaces. Adv Colloid Interface Sci 28, 35‐64. Vincenti, M. P. (2001). The matrix metalloproteinase (MMP) and tissue inhibitor of metalloproteinase (TIMP) genes. Transcriptional and posttranscriptional regulation, signal transduction and cell‐type‐specific expression. Methods Mol Biol 151, 121‐48. Vincenti, M. P., Coon, C. I., Mengshol, J. A., Yocum, S., Mitchell, P. and Brinckerhoff, C. E. (1998). Cloning of the gene for interstitial collagenase‐3 (matrix metalloproteinase‐13) from rabbit synovial fibroblasts: differential expression with collagenase‐1 (matrix metalloproteinase‐1). Biochem J 331 ( Pt 1), 341‐6. Wang, J., Zheng, L., Lobito, A., Chan, F. K., Dale, J., Sneller, M., Yao, X., Puck, J. M., Straus, S. E. and Lenardo, M. J. (1999). Inherited human Caspase 10 mutations underlie defective lymphocyte and dendritic cell apoptosis in autoimmune lymphoproliferative syndrome type II. Cell 98, 47‐58. Wang, Y., Kristan, J., Hao, L., Lenkoski, C. S., Shen, Y. and Matis, L. A. (2000). A role for complement in antibody‐mediated inflammation: C5‐deficient DBA/1 mice are resistant to collagen‐induced arthritis. J Immunol 164, 4340‐7. Watson, W. C., Brown, P. S., Pitcock, J. A. and Townes, A. S. (1987). Passive transfer studies with type II collagen antibody in B10.D2/old and new line and C57Bl/6 normal and beige (Chediak‐Higashi) strains: evidence of important roles for C5 and multiple inflammatory cell types in the development of erosive arthritis. Arthritis Rheum 30, 460‐5. Wegmann, T. G., Lin, H., Guilbert, L. and Mosmann, T. R. (1993). Bidirectional cytokine interactions in the maternal‐fetal relationship: is successful pregnancy a TH2 phenomenon? Immunol Today 14, 353‐6. Weis, J. J., Tedder, T. F. and Fearon, D. T. (1984). Identification of a 145,000 Mr membrane protein as the C3d receptor (CR2) of human B lymphocytes. Proc Natl Acad Sci U S A 81, 881‐5. Wernicke, D., Seyfert, C., Hinzmann, B. and Gromnica‐Ihle, E. (1996). Cloning of collagenase 3 from the synovial membrane and its expression in rheumatoid arthritis and osteoarthritis. J Rheumatol 23, 590‐5. Wilhelm, M., Silver, R. and Silverman, A. J. (2005). Central nervous system neurons acquire mast cell products via transgranulation. Eur J Neurosci 22, 2238‐48. Winchester, R. J., Agnello, V. and Kunkel, H. G. (1970). Gamma globulin complexes in synovial fluids of patients with rheumatoid arthritis. Partial characterization and relationship to lowered complement levels. Clin Exp Immunol 6, 689‐706. Winyard, P. G., Moody, C. J. and Jacob, C. (2005). Oxidative activation of antioxidant defence. Trends Biochem Sci 30, 453‐61. Wipke, B. T. and Allen, P. M. (2001). Essential role of neutrophils in the initiation and progression of a murine model of rheumatoid arthritis. J Immunol 167, 1601‐8. Wooley, P. H., Luthra, H. S., Singh, S. K., Huse, A. R., Stuart, J. M. and David, C. S. (1984). Passive transfer of arthritis to mice by injection of human anti‐type II collagen antibody. Mayo Clin Proc 59, 737‐43.

- 132 -
Wordsworth, B. P., Lanchbury, J. S., Sakkas, L. I., Welsh, K. I., Panayi, G. S. and Bell, J. I. (1989). HLA‐DR4 subtype frequencies in rheumatoid arthritis indicate that DRB1 is the major susceptibility locus within the HLA class II region. Proc Natl Acad Sci U S A 86, 10049‐53. Wu, J., Wilson, J., He, J., Xiang, L., Schur, P. H. and Mountz, J. D. (1996). Fas ligand mutation in a patient with systemic lupus erythematosus and lymphoproliferative disease. J Clin Invest 98, 1107‐13. Zachariae, C. O., Anderson, A. O., Thompson, H. L., Appella, E., Mantovani, A., Oppenheim, J. J. and Matsushima, K. (1990). Properties of monocyte chemotactic and activating factor (MCAF) purified from a human fibrosarcoma cell line. J Exp Med 171, 2177‐82. Zvaifler, N. J., Steinman, R. M., Kaplan, G., Lau, L. L. and Rivelis, M. (1985). Identification of immunostimulatory dendritic cells in the synovial effusions of patients with rheumatoid arthritis. J Clin Invest 76, 789‐800.