anti-kit monoclonal antibody inhibits imatinib-resistant
TRANSCRIPT

Anti-KIT monoclonal antibody inhibitsimatinib-resistant gastrointestinal stromaltumor growthBadreddin Edrisa,b,1, Stephen B. Willinghamc,1, Kipp Weiskopfc,1, Anne K. Volkmerc,d, Jens-Peter Volkmerc,Thomas Mühlenberge, Kelli D. Montgomerya, Humberto Contreras-Trujilloc, Agnieszka Czechowiczc,Jonathan A. Fletcherf, Robert B. Westa, Irving L. Weissmana,c,1,2, and Matt van de Rijna,1,2
aDepartment of Pathology, Stanford University Medical Center, Stanford, CA 94305; bDepartment of Genetics, Stanford University School of Medicine,Stanford, CA 94305; cInstitute for Stem Cell Biology and Regenerative Medicine and the Ludwig Cancer Institute, Stanford University School of Medicine,Stanford, CA 94305; dDepartment of Obstetrics and Gynecology, University of Dusseldorf, 40225 Dusseldorf, Germany; eSarcoma Center, West German CancerCenter, University of Duisburg-Essen Medical School, 45147 Essen, Germany; fDepartment of Pathology, Brigham and Women’s Hospital, Boston, MA 02115
Contributed by Irving L. Weissman, January 3, 2013 (sent for review August 2, 2012)
Gastrointestinal stromal tumor (GIST) is the most common sarcomaof the gastrointestinal tract and arises from the interstitial cells ofCajal. It is characterized by expression of the receptor tyrosine kinaseCD117 (KIT). In 70–80% of GIST cases, oncogenic mutations in KIT arepresent, leading to constitutive activation of the receptor, whichdrives the proliferation of these tumors. Treatment of GIST withimatinib, a small-molecule tyrosine kinase inhibitor, inhibits KIT-me-diated signaling and initially results in disease control in 70–85%of patients with KIT-positive GIST. However, the vast majority ofpatients eventually develop resistance to imatinib treatment, lead-ing to disease progression and posing a significant challenge in theclinical management of these tumors. Here, we show that an anti-KIT monoclonal antibody (mAb), SR1, is able to slow the growth ofthree human GIST cell lines in vitro. Importantly, these reductions incell growth were equivalent between imatinib-resistant and imati-nib-sensitive GIST cell lines. Treatment of GIST cell lines with SR1reduces cell-surface KIT expression, suggesting that mAb-inducedKIT down-regulation may be a mechanism by which SR1 inhibitsGIST growth. Furthermore, we also show that SR1 treatment en-hances phagocytosis of GIST cells by macrophages, indicating thattreatment with SR1 may enhance immune cell-mediated tumorclearance. Finally, using two xenotransplantation models of imatinib-sensitive and imatinib-resistant GIST, we demonstrate that SR1 isable to strongly inhibit tumor growth in vivo. These results sug-gest that treatment with mAbs targeting KIT may represent analternative, or complementary, approach for treating GIST.
cancer | immunotherapy | Gleevec | leiomyosarcoma
Gastrointestinal stromal tumor (GIST) is a neoplasm of mes-enchymal cells with an annual incidence of 10–20 cases per
million people. GIST frequently recurs locally after initial surgicalresection, and advanced disease is characterized by distant me-tastasis to the liver, lungs, and bone (1). Approximately 20 y ago,the interstitial cells of Cajal (ICCs) were first identified as the cell-of-origin forGIST. Found in themuscle wall of the gastrointestinaltract, ICCs are a population of cells that control peristaltic con-tractions and are characterized by their expression of the tyrosinekinase receptorKIT (CD117), a receptor for stem cell factor (SCF)(2). SCF binding to KIT leads to receptor homodimerization andintracellular kinase activation, setting off a signaling cascade thatpositively regulates both the MAPK and the PI3K-AKT pathways(3). The vast majority (95%) of GISTs are positive for KIT proteinexpression, and 70–80% of GISTs contain activating mutations inKIT, which result in constitutive activation via auto-phosphoryla-tion and SCF-independent signaling and cellular proliferation (4).Imatinib is a small-molecule tyrosine kinase inhibitor (TKI) that
was initially developed to inhibit the BCR-ABL fusion protein thatdrives the oncogenesis of chronic myelogenous leukemia. It waslater discovered that imatinib could also inhibit oncogenic KIT
signaling by stabilizing KIT in the inactivated form and preventingits constitutive auto-phosphorylation, leading to the rationale forits clinical use in the treatment of GIST (5–7). Before the use ofimatinib, less than 5% of GIST cases responded to conventionalchemotherapy, and patients with advanced disease showed a me-dian survival of 18 mo. In contrast, treatment of patients with KIT-mutation–positive GIST with imatinib resulted in disease controlin 70–85% of cases and in an increased median survival of 5 y (8).As such, GIST became one of the earliest examples of a solid tu-mor reacting to targeted therapy. Unfortunately, most patientseventually develop resistance to the drug, owing to mutations inKIT that render GIST cells imatinib-resistant and allow KIT sig-naling to remain constitutively active (1).Imatinib resistance poses a significant challenge in the clinical
management of GIST. The vast majority of approaches to datehave involved the use of alternative small-molecule TKIs targetingKIT or other receptor tyrosine kinases, such as PDGFRA andVEGFR (1). In the present work, we evaluate an alternative, andperhaps complementary, approach that involves targeting KITwith a monoclonal antibody (mAb), SR1 (9, 10). We show thatSR1 is able to potently inhibit the growth of GIST cells in vitroand in vivo. Significantly, the SR1-mediated effects on GISTgrowth were seen in both imatinib-sensitive and imatinib-resistanthuman GIST cell lines, suggesting that treatment with SR1 mayrepresent a fruitful approach to overcoming the significant clinicalchallenge of imatinib resistance in GIST.
ResultsSR1 Inhibits GIST Cell-Line Growth in Vitro and Reduces KIT Cell-Surface Expression. KIT protein expression was determined by im-munohistochemistry (IHC) on the human GIST cell lines GIST48,GIST430, and GIST882 and as a negative control on a humanleiomyosarcoma (LMS) cell line, LMS05 (Fig. 1A).GIST48 andGIST430 were derived from clinical specimens that
had developed resistance to imatinib treatment; GIST48 harborsa primary KIT exon 11 missense mutation and a secondary het-erozygous KIT exon 17 missense mutation, and GIST430 harbors
Author contributions: B.E., S.B.W., K.W., A.K.V., J.-P.V., A.C., I.L.W., and M.v.d.R. designedresearch; B.E., S.B.W., K.W., A.K.V., T.M., K.D.M., and H.C.-T. performed research; J.A.F.contributed new reagents/analytic tools; B.E., S.B.W., K.W., A.K.V., J.-P.V., A.C., R.B.W.,I.L.W., and M.v.d.R. analyzed data; and B.E., S.B.W., K.W., J.-P.V., I.L.W., and M.v.d.R.wrote the paper.
The authors declare no conflict of interest.
Freely available online through the PNAS open access option.1B.E., S.B.W., K.W., I.L.W., and M.v.d.R. contributed equally to this work.2To whom correspondence may be addressed. E-mail: [email protected] or [email protected].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1222893110/-/DCSupplemental.
www.pnas.org/cgi/doi/10.1073/pnas.1222893110 PNAS | February 26, 2013 | vol. 110 | no. 9 | 3501–3506
MED
ICALSC
IENCE
S

a primary heterozygous KIT exon 11 in-frame deletion and a sec-ondary heterozygous KIT exon 13 missense mutation (11). Incontrast, GIST882 was derived from a patient before treatmentwith imatinib and is sensitive to treatment with imatinib in vitro;GIST882 harbors a homozygous missense mutation in KIT exon13 (6). Consistent with their clinical origins, GIST882 cells treatedwith increasing doses of imatinib strongly inhibited cell viability,whereas GIST48 and GIST430 cells were significantly less sensi-tive to treatment in vitro; KIT-negative LMS05 cells showed noresponse to increasing doses of imatinib (Fig. 1B).To determine whether KIT-induced cell growth could be af-
fected using an anti-KIT mAb, GIST48, GIST430, GIST882, andLMS05 cells were plated and allowed to adhere overnight beforegrown in the presence of anti-KITmAb (clone SR1) or IgG controlantibody for 9 d. SR1 was equally effective in slowing the growthrate of both imatinib-resistant cell lines (GIST48 and GIST430)and an imatinib-sensitive cell line (GIST882), whereas the KIT-negative LMS05 cells showed no significant alteration in cell via-bility as a result of long-term culture with SR1 (Fig. 1C). Together,these results demonstrate that anti-KIT mAb possesses the po-tential to inhibit KIT-mediated cell growth in GIST cells in-dependently of their resistance or sensitivity to imatinib treatment.We observed no obvious decreases by Western blot in levels of
phosphorylated or total KIT upon treatment with SR1 (Fig. S1)and hypothesized that SR1 treatment of GIST cells could down-regulate levels ofKIT expression specifically on the cell surface. Todetermine levels of KIT cell-surface expression, we used a secondanti-KIT mAb (104D2) that binds a KIT epitope different fromSR1 (12). We found no decrease in the ability of 104D2 to bindKIT in the presence of SR1 at 4 °C, a condition where receptorinternalization or shedding does not occur (Fig. S2). Bortezomib,a small-molecule proteasome inhibitor that has been shown toinduce down-regulation, internalization, and degradation of cell-surface KIT, was used as a positive control (13). GIST48, GIST430,GIST882, and LMS05 cells were treated with IgG, SR1, DMSO,or bortezomib for 12 h, and cell-surface KIT expression was an-alyzed by flow cytometry. We found that, like bortezomib, SR1
was able to induce a significant decrease in cell-surface KIT ex-pression (Fig. 1D).
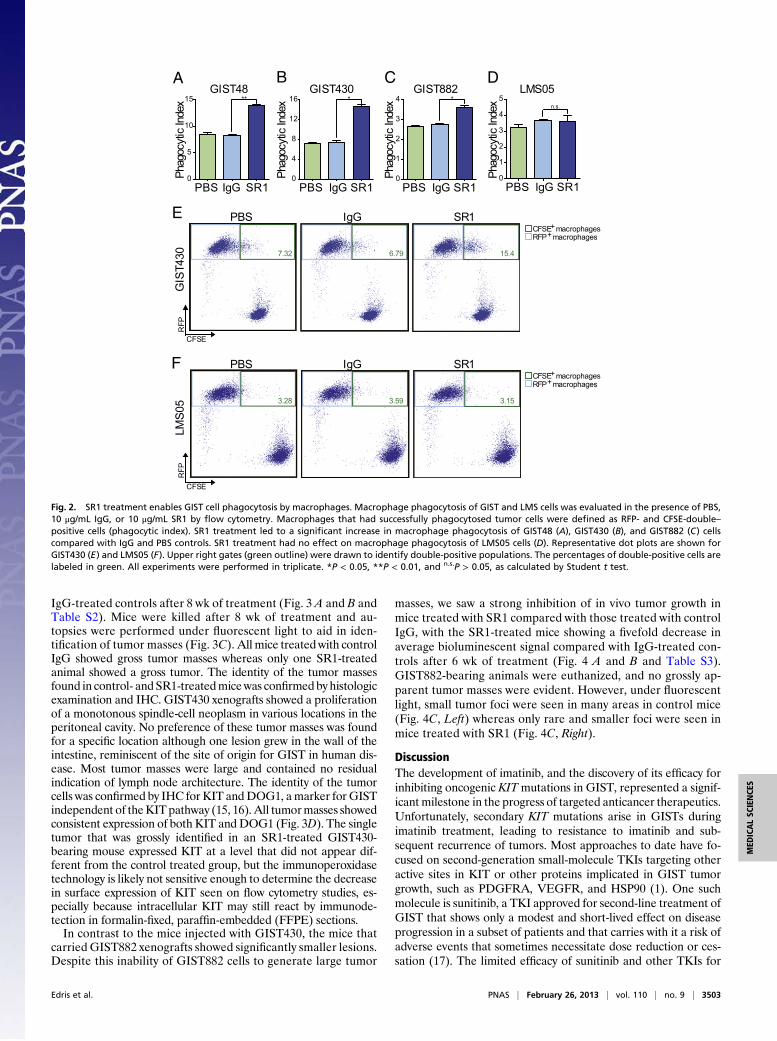
SR1 Allows for Phagocytosis of GIST Cells by Macrophages. As mAbscan also mediate immune cell-mediated tumor clearance, wenext performed coculture assays with macrophages to ascertainwhether SR1 treatment could induce phagocytosis of GIST tumors.GIST48, GIST430, GIST882, and LMS05 cells were green fluo-rescently labeled and then incubated for 2 h with bone marrow-derivedmacrophages from red fluorescent protein (RFP)-positivemice in the presence of PBS, control IgG, or SR1. The cells werethen analyzed by flow cytometry to determine the level of tumorcell phagocytosis by the macrophages. We found that in all threeGIST cell lines, SR1 treatment led to a statistically significantincrease in macrophage phagocytosis (Fig. 2 A–C and E and Fig.S3). In contrast, KIT-negative LMS05 cells showed no differencein their potential to be phagocytosed regardless of PBS, IgG, orSR1 treatment (Fig. 2 D and F and Fig. S3).
SR1 Inhibits Growth of Both Imatinib-Sensitive and Imatinib-ResistantGIST Xenografts in Mice.We next evaluated whether anti-KIT mAbscould inhibit the growth of xenotransplanted GIST tumors in mice.Imatinib-resistant GIST48 and GIST430 cells, and imatinib-sen-sitive GIST882 cells, were first transduced in vitro with a lentivirusdesigned to express GFP and luciferase, enabling the use of bio-luminescent imaging to monitor tumor engraftment and growth invivo. For all cell lines, 100,000 cells were injected into the perito-neal cavity of 4- to 8-wk-old NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ(NSG) immunodeficient mice, which lack functional T-cells andB-cells but retain macrophages capable of phagocytosis (14). Twoweeks after transplantation, engraftment was evaluated usingbioluminescent imaging; GIST48 tumors failed to engraft, but tu-mor cell engraftment was confirmed in GIST430 and GIST882mice. The animals were then randomized according to baselinetumor luminescence and treatment commenced (full treatmentprotocols are available in Table S1).In imatinib-resistant GIST430 xenografts, treatment with SR1
potently inhibited tumor growth, as evidenced by an ∼10-folddecrease in average bioluminescent signal compared with the
H&
EK
IT
GIST48 GIST430 GIST882 LMS05A B
.00 .04 .08 .16 .32 .63 1.25 2.5 5.0
100
75
50
25
0
125
150 GIST48 GIST430 GIST882 LMS05
C
GIST48 GIST430 GIST882 LMS050
25
50
75
100
125 SR1IgG* ** **
Cell
viab
ility
( % Ig
G)
Abs o
rban
ce
Imatinib concentration (µM)
GIST48 GIST430 GIST882
150
100
50
0
IgG SR1 DMSO Bortezomib
******
MFI
(%
IgG
)
D
Fig. 1. SR1 treatment slows in vitro GIST cell growth and reduces cell-surface KIT expression. KIT protein expression was analyzed by IHC on paraffin-em-bedded pellets of GIST48, GIST430, GIST882, and LMS05 cell lines (A). Cell viability assays were carried out to ascertain sensitivity to imatinib treatment for 72 hwith GIST and LMS cell lines (B). Viable cell number, as measured by WST-1 absorbance, after 9 d in the presence of 10 μg/mL IgG control or SR1 was evaluatedin GIST and LMS cells (C). Cell-surface KIT expression was evaluated in GIST cells by flow cytometry after 12 h of incubation with 10 μg/mL IgG, 10 μg/mL SR1,DMSO (1:1,000), or 100 nM bortezomib (D). All experiments were performed in triplicate. *P < 0.05 and **P < 0.01, as calculated by Student t test.
3502 | www.pnas.org/cgi/doi/10.1073/pnas.1222893110 Edris et al.
IgG-treated controls after 8 wk of treatment (Fig. 3 A and B andTable S2). Mice were killed after 8 wk of treatment and au-topsies were performed under fluorescent light to aid in iden-tification of tumor masses (Fig. 3C). All mice treatedwith controlIgG showed gross tumor masses whereas only one SR1-treatedanimal showed a gross tumor. The identity of the tumor massesfound in control- and SR1-treatedmicewas confirmed by histologicexamination and IHC. GIST430 xenografts showed a proliferationof a monotonous spindle-cell neoplasm in various locations in theperitoneal cavity. No preference of these tumor masses was foundfor a specific location although one lesion grew in the wall of theintestine, reminiscent of the site of origin for GIST in human dis-ease. Most tumor masses were large and contained no residualindication of lymph node architecture. The identity of the tumorcells was confirmed by IHC for KIT andDOG1, amarker for GISTindependent of theKIT pathway (15, 16). All tumormasses showedconsistent expression of both KIT and DOG1 (Fig. 3D). The singletumor that was grossly identified in an SR1-treated GIST430-bearing mouse expressed KIT at a level that did not appear dif-ferent from the control treated group, but the immunoperoxidasetechnology is likely not sensitive enough to determine the decreasein surface expression of KIT seen on flow cytometry studies, es-pecially because intracellular KIT may still react by immunode-tection in formalin-fixed, paraffin-embedded (FFPE) sections.In contrast to the mice injected with GIST430, the mice that
carried GIST882 xenografts showed significantly smaller lesions.Despite this inability of GIST882 cells to generate large tumor
masses, we saw a strong inhibition of in vivo tumor growth inmice treated with SR1 compared with those treated with controlIgG, with the SR1-treated mice showing a fivefold decrease inaverage bioluminescent signal compared with IgG-treated con-trols after 6 wk of treatment (Fig. 4 A and B and Table S3).GIST882-bearing animals were euthanized, and no grossly ap-parent tumor masses were evident. However, under fluorescentlight, small tumor foci were seen in many areas in control mice(Fig. 4C, Left) whereas only rare and smaller foci were seen inmice treated with SR1 (Fig. 4C, Right).
DiscussionThe development of imatinib, and the discovery of its efficacy forinhibiting oncogenic KITmutations in GIST, represented a signif-icant milestone in the progress of targeted anticancer therapeutics.Unfortunately, secondary KIT mutations arise in GISTs duringimatinib treatment, leading to resistance to imatinib and sub-sequent recurrence of tumors. Most approaches to date have fo-cused on second-generation small-molecule TKIs targeting otheractive sites in KIT or other proteins implicated in GIST tumorgrowth, such as PDGFRA, VEGFR, and HSP90 (1). One suchmolecule is sunitinib, a TKI approved for second-line treatment ofGIST that shows only a modest and short-lived effect on diseaseprogression in a subset of patients and that carries with it a risk ofadverse events that sometimes necessitate dose reduction or ces-sation (17). The limited efficacy of sunitinib and other TKIs for
PBS IgG SR10
5
10
15 **
Phag
ocyt
i c In
dex
GIST48A
Phag
o cy t
ic In
dex
GIST430B
Phag
ocyt
ic In
dex
GIST882C
Phag
ocyt
ic In
dex
LMS05D
PBS IgG SR10
4
8
12
16 *
E
PBS IgG SR10
1
2
3
4 *
PBS IgG SR10
1
2
3
4
5n.s.
RFP+macrophagesCFSE+macrophages
PBS IgG SR1
7.32 6.79 15.4
F
3.28 3.59 3.15
RFP
CFSE
GIS
T430
RFP+macrophagesCFSE+macrophages
PBS IgG SR1
RFP
CFSE
LMS0
5
Fig. 2. SR1 treatment enables GIST cell phagocytosis by macrophages. Macrophage phagocytosis of GIST and LMS cells was evaluated in the presence of PBS,10 μg/mL IgG, or 10 μg/mL SR1 by flow cytometry. Macrophages that had successfully phagocytosed tumor cells were defined as RFP- and CFSE-double–positive cells (phagocytic index). SR1 treatment led to a significant increase in macrophage phagocytosis of GIST48 (A), GIST430 (B), and GIST882 (C) cellscompared with IgG and PBS controls. SR1 treatment had no effect on macrophage phagocytosis of LMS05 cells (D). Representative dot plots are shown forGIST430 (E) and LMS05 (F). Upper right gates (green outline) were drawn to identify double-positive populations. The percentages of double-positive cells arelabeled in green. All experiments were performed in triplicate. *P < 0.05, **P < 0.01, and n.s.P > 0.05, as calculated by Student t test.
Edris et al. PNAS | February 26, 2013 | vol. 110 | no. 9 | 3503
MED
ICALSC
IENCE
S

imatinib-resistant GIST highlights the need for alternative ap-proaches to treat these tumors.In parallel with efforts focusing on improving small-molecule
drug candidates for oncology indications, the development ofmAbs as therapeutic molecules for the treatment of cancer hasoffered a large number of treatment options across a broad range oftumor types. mAbs are capable of exerting multiple, and often si-multaneous, effects on cancer cells via their interaction with theirtargets. When successful, these mAb-induced interactions can ul-timately combine to produce antitumor effects in vitro and in vivothrough various modes of action, including inhibition of the target’sability to activate downstream signaling targets, internalization anddegradation of a cell-surface target, phagocytosis, and/or antibody-dependent cell-mediated cytotoxicity (ADCC), a process throughwhich target cells are lysed by cytotoxic granules released by naturalkiller (NK) cells, granulocytes, and other leukocytes (18).In the present work, we show that treatment of GIST cell lines
with the anti-KITmAbSR1 resulted in a significant decrease in cellgrowth and in cell-surfaceKITexpression, suggesting that the SR1-mediated growth inhibition in GIST cells may be occurring due tointernalization and degradation of KIT. We also show that SR1treatment increased phagocytosis of GIST cells by macrophages.Further study is needed, however, to fully elucidate additionalconsequences of SR1 treatment on GIST cells. The impact of SR1treatment on GIST cell interactions with additional immune ef-fector cells, such as T-cells, B-cells, and NK cells, which are notpresent in themice used for our xenotransplantation studies, needsto be investigated. In the future, SR1, or other KIT-specific mAbs,could be modified to enhance affinity to KIT and/or to potentiateone or more mAb-mediated antitumor cell functions, such as re-ceptor internalization, receptor homodimerization inhibition,macrophage phagocytosis, or ADCC. Also, although this reportfocused on treatment of GIST cells, SR1 or other anti-KIT mAbtreatment may prove to be useful in other KIT-positive tumors,such as pancreatic adenocarcinoma, testicular seminoma, mela-noma, neuroblastoma, and breast cancer (19–23).
In conclusion, we show that treatment of human GIST cell lineswith the anti-KIT mAb SR1 can inhibit tumor growth in vitro andin vivo and that decreasing cell-surface KIT expression and im-mune cell-mediated tumor clearance are two mechanisms thatlikely contribute to SR1’s efficacy. Importantly, SR1-mediated in-hibition in tumor growth was independent of imatinib sensitivity orresistance in human GIST cell lines, suggesting that anti-KIT mAbtherapy may effectively address the significant clinical problemof imatinib resistance in GIST and thereby form the rationale forevaluating the clinical efficacy of mAb therapy in GIST patients.
Materials and MethodsCell Culture. GIST cell lines were developed in the Fletcher laboratory atBrigham and Women’s Hospital, Boston, using a protocol approved by theBrigham and Women’s Hospital institutional research board (6, 11). Theirderivations and culture conditions have been described previously (6, 11, 24).SR-1 (mouse IgG2A) binds to human c-kit and was purified by Protein G fromsupernatant obtained from hybridoma cells cultured under standard conditions.
Cell Viability Assays. For imatinib sensitivity assays, cells were seeded at adensity of 4,000 cells per well in 96-well tissue culture plates and allowedto adhere overnight. Imatinib mesylate (Santa Cruz Biotechnology) wasdissolved in DMSO to a stock concentration of 10 mM, and cells were thentreated with a range of concentrations from 0 to 5 μM; total DMSO was heldconstant. After 72 h, cell numbers were assessed using WST-1 reagent(Roche); viable cell numbers are directly related to WST-1 absorbance. ForSR1 sensitivity assays, cells were seeded at a density of 4,000 cells per well in96-well plates and allowed to adhere overnight. Cells were then cultured inthe presence of SR1 or mouse IgG control antibody at a concentration of 10μg/mL. The antibody-containing media was refreshed every 3 d, and cellviability was assessed using WST-1 reagent (Roche) after 9 d of antibodytreatment. Purified SR1 mAb was provided by the laboratory of Judith Shi-zuru (Stanford University School of Medicine). Control mouse IgG antibodywas purchased from Innovative Research.
Cell-Surface Protein Expression Assays. A total of 500,000 tumor cells wereplated in each well of a six-well tissue culture plate (BD Biosciences), allowedto adhere overnight, and treated in triplicate with 10 μg/mL SR1, 10 μg/mLIgG control, DMSO (1:1,000), or 100 nM bortezomib (Selleck Chemicals) for12 h. The cells were then dissociated using TrypLE (Life Technologies), washed
3.0
2.5
2.0
1.5
1.0
0.5
x10 9
p/sec/cm^2/sr
Color BarMin - 1.25e8Max - 3.00e9a
A B C
Brig
htfie
ldG
FP
IgG SR1
D H&E KIT DOG1
p=0.0009
p=0.0206p=0.0042
p<0.0001
Fold
Cha
nge
1900
1700
1500400300200
150100500
-50 2 4 6 8
Week
IgGSR1
Fig. 3. SR1 treatment inhibits tumor growth in a xenotransplantation model of imatinib-resistant GIST. Eight weeks of treatment with SR1 significantlydecreased GIST430 xenograft growth compared with IgG control treatment (A). Representative images of SR1 and IgG control-treated mice are shown (B).Fluorescent microscopy was used to evaluate the presence of GFP-positive growths after animal sacrifice (C). The identity of GIST430 xenotransplanted tumorswas confirmed by H&E staining and by IHC for GIST markers KIT and DOG1 (D).
3504 | www.pnas.org/cgi/doi/10.1073/pnas.1222893110 Edris et al.

with PBS, and stained with anti-KIT mAb (clone 104D2) directly conjugatedto phycoerythrin (STEMCELL Technologies), which can bind to KIT in thepresence of SR1 (Fig. S2). The cells were then washed twice, stained withDAPI, and analyzed on an LSRFortessa cell analyzer (BD Biosciences).
Western Blotting. Protein lysates were prepared from GIST430 and GIST882cell monolayers using RIPA buffer (Thermo Scientific) supplemented withprotease and phosphatase inhibitors (Roche) and PMSF (Sigma-Aldrich). Pro-tein concentrations were determined with the Bio-Rad Protein Assay (Bio-Rad Laboratories). Electrophoresis and immunoblotting for KIT, phospho-KITY703, phospho-KIT Y719, and β-actin were carried out as previously de-scribed (25). Protein expression and phosphorylation changes were visual-ized by chemiluminescence, captured using a GelDoc system (Bio-Rad), andprocessed using GIMP and Inkscape software.
Macrophage Phagocytosis Assays. C57BL/Ka Rosa26-mRFP1 transgenic micewere used for red-fluorescent macrophage derivation (26), carried out asdescribed previously (27). Tumor cells were green fluorescently labeledwith 1 μM carboxyfluorescein succinimidyl ester (CFSE) (Invitrogen) for10 m at 37 °C, washed twice with HBSS (Invitrogen), and incubated for 30 m
in Iscove’s modified Dulbecco’s media (IMDM) (Invitrogen) with mouse IgG orSR-1. A total of 50,000 macrophages were then incubated with 100,000 tumorcells for 2 h in Ultra-low Cluster 96-well plates (BD Biosciences), washed twice,stained with DAPI, and analyzed on an LSRFortessa cell analyzer (BD Bio-sciences). The phagocytic index was defined as the percentage of macrophagesthat had successfully phagocytosed tumor cells (i.e., RFP-positive and CFSE-highcells) (Fig. 2 E and F and Fig. S3), and differences in phagocytosis betweentreatment groups were evaluated using Student t tests.
Xenotransplantation Studies. All animal procedures were approved by theAdministrative Panel on Laboratory Animal Care at Stanford University.Lentiviral production of a pCDH-CMV-EF1-puro construct (Systems Bio-science) containing a ubiquitin promoter driving the expression of a fusionprotein containing the Luc2 (pgl4) luciferase gene (Promega) and the eGFPgene (Becton Dickinson) was carried out using standard protocols. NSGmicewere used for xenotransplantation studies (14). GIST48, GIST430, andGIST882 cells were transduced with lentivirus, and 100,000 GFP+ cells wereinjected intraperitoneally into 4- to 8-wk-old NSG mice as described pre-viously (28). Bioluminescent activity was visualized in vivo after D-luciferininjection (Biosynth) on an IVIS Spectrum (Caliper Life Sciences) instrument
A
B
C IgG Treatment SR1 Treatment
GFP GFP
IgG Treatment SR1 Treatment
0
50
100
150
Week 2 Week 4 Week 6
Fold
Cha
nge
in T
otal
Flu
x
p=0.0210
p<0.0001IgG
SR1
Fig. 4. SR1 treatment inhibits tumor growth in a xenotransplantation model of imatinib-sensitive GIST. Six weeks of treatment with SR1 significantly de-creased GIST882 xenograft growth compared with IgG control treatment (A). Representative images of SR1 and IgG control-treated mice are shown (B).Fluorescent microscopy was used to evaluate the presence of GFP-positive growths after animal sacrifice. White arrows denote tumor nodules (C).
Edris et al. PNAS | February 26, 2013 | vol. 110 | no. 9 | 3505
MED
ICALSC
IENCE
S

and quantified using Image 4.0 software, as described previously (28). Totalflux (photons/second) values were obtained from mice. Mice were matchedbased on total flux 2 wk after cell engraftment and subsequently treatedweekly via i.p. injections with SR1 (500 μg) or mouse IgG. Mice were imagedevery 2 wk, and differences in tumor growth were assessed using Student ttest. At autopsy, tumor masses were visualized using a fluorescent dissectingmicroscope (Leica). Each symbol in Figs. 3A and 4A (GIST430 and GIST882,respectively) represent an individual mouse. All treatment cohorts consist offive mice, with the exception of the SR1 cohort in the GIST430 treatment (n =4) due to the death of one animal while performing bioluminescent imagingbefore initiation of treatment.
Histology and Immunohistochemistry. KIT and DOG1 protein expression wasevaluated on FFPE cell pellets or on full cross-sections of FFPE xenografttumors, which were dissected after animal sacrifice, as described previously
(24). Samples were then stained with primary antibodies against KIT (Dako;1:200) and DOG1 (Leica; clone K9, 1:100) on a Benchmark autostainer (VentanaMedical Systems). The IHC reactions were visualized using mouse versionsof the EnVision + system (Dako) with diaminobenzidine.
ACKNOWLEDGMENTS. We thank A. Logan, A. M. Ring, G. Krampitz, J. Oak,R. Li, X. Guo, S. K. Gupta, E. Gilbert, and members of the StanfordImmunodiagnosis Laboratory for technical assistance and helpful discus-sions. Grant support came from National Institutes of Health Grants CA112270 and CA 139490, the National Cancer Institute (F30 CA168059 toK.W.), Deutsche Forschungsgemeinschaft (Grant VO 1976/1 to A.K.V.), theLife Raft Group, the GIST Cancer Research Fund, the Lacob Program ofExcellence in Gynecologic–Ovarian Cancer Research and Treatment, andthe Ludwig Institute for Cancer Research. B.E. is a recipient of the NationalScience Foundation Graduate Research Fellowship. Dedicated to thememory of our colleague Angela Lee Riepel, who lost her life to GIST.
1. Corless CL, Barnett CM, Heinrich MC (2011) Gastrointestinal stromal tumours: Originand molecular oncology. Nat Rev Cancer 11(12):865–878.
2. Kindblom LG, Remotti HE, Aldenborg F, Meis-Kindblom JM (1998) Gastrointestinalpacemaker cell tumor (GIPACT): Gastrointestinal stromal tumors show phenotypiccharacteristics of the interstitial cells of Cajal. Am J Pathol 152(5):1259–1269.
3. Yuzawa S, et al. (2007) Structural basis for activation of the receptor tyrosine kinaseKIT by stem cell factor. Cell 130(2):323–334.
4. Hirota S, et al. (1998) Gain-of-function mutations of c-kit in human gastrointestinalstromal tumors. Science 279(5350):577–580.
5. Heinrich MC, et al. (2000) Inhibition of c-kit receptor tyrosine kinase activity by STI571, a selective tyrosine kinase inhibitor. Blood 96(3):925–932.
6. Tuveson DA, et al. (2001) STI571 inactivation of the gastrointestinal stromal tumor c-KIT oncoprotein: Biological and clinical implications. Oncogene 20(36):5054–5058.
7. Mol CD, et al. (2004) Structural basis for the autoinhibition and STI-571 inhibition of c-Kit tyrosine kinase. J Biol Chem 279(30):31655–31663.
8. Dematteo RP, Heinrich MC, El-Rifai WM, Demetri G (2002) Clinical management ofgastrointestinal stromal tumors: Before and after STI-571. Hum Pathol 33(5):466–477.
9. Broudy VC, et al. (1992) Isolation and characterization of a monoclonal antibody thatrecognizes the human c-kit receptor. Blood 79(2):338–346.
10. Ashman LK, Bühring HJ, Aylett GW, Broudy VC, Müller C (1994) Epitope mapping andfunctional studies with three monoclonal antibodies to the c-kit receptor tyrosinekinase, YB5.B8, 17F11, and SR-1. J Cell Physiol 158(3):545–554.
11. Bauer S, Yu LK, Demetri GD, Fletcher JA (2006) Heat shock protein 90 inhibition inimatinib-resistant gastrointestinal stromal tumor. Cancer Res 66(18):9153–9161.
12. Bühring HJ, et al. (1993) Modulation of p145c-kit function in cells of patients withacute myeloblastic leukemia. Cancer Res 53(18):4424–4431.
13. Fang HT, et al. (2012) Bortezomib interferes with C-KIT processing and transforms thet(8;21)-generated fusion proteins into tumor-suppressing fragments in leukemia cells.Proc Natl Acad Sci USA 109(7):2521–2526.
14. Ito M, et al. (2002) NOD/SCID/gamma(c)(null) mouse: An excellent recipient mousemodel for engraftment of human cells. Blood 100(9):3175–3182.
15. West RB, et al. (2004) The novel marker, DOG1, is expressed ubiquitously in gastro-intestinal stromal tumors irrespective of KIT or PDGFRA mutation status. Am J Pathol165(1):107–113.
16. Espinosa I, et al. (2008) A novel monoclonal antibody against DOG1 is a sensitive andspecific marker for gastrointestinal stromal tumors. Am J Surg Pathol 32(2):210–218.
17. Matsumoto K, et al. (2011) Clinical efficacy and safety of sunitinib after imatinibfailure in Japanese patients with gastrointestinal stromal tumor. Jpn J Clin Oncol41(1):57–62.
18. Weiner LM, Surana R, Wang S (2010) Monoclonal antibodies: Versatile platforms forcancer immunotherapy. Nat Rev Immunol 10(5):317–327.
19. Esposito I, et al. (2002) The stem cell factor-c-kit system and mast cells in humanpancreatic cancer. Lab Invest 82(11):1481–1492.
20. Kemmer K, et al. (2004) KIT mutations are common in testicular seminomas. Am JPathol 164(1):305–313.
21. Woodman SE, Davies MA (2010) Targeting KIT in melanoma: A paradigm of molec-ular medicine and targeted therapeutics. Biochem Pharmacol 80(5):568–574.
22. Cohen PS, Chan JP, Lipkunskaya M, Biedler JL, Seeger RC; The Children’s Cancer Group(1994) Expression of stem cell factor and c-kit in human neuroblastoma. Blood 84(10):3465–3472.
23. Johansson I, et al. (2012) Increased gene copy number of KIT and VEGFR2 at 4q12 inprimary breast cancer is related to an aggressive phenotype and impaired prognosis.Genes Chromosomes Cancer 51(4):375–383.
24. Edris B, et al. (2012) ROR2 is a novel prognostic biomarker and a potential thera-peutic target in leiomyosarcoma and gastrointestinal stromal tumour. J Pathol 227(2):223–233.
25. Mühlenberg T, et al. (2009) Inhibitors of deacetylases suppress oncogenic KIT sig-naling, acetylate HSP90, and induce apoptosis in gastrointestinal stromal tumors.Cancer Res 69(17):6941–6950.
26. Ueno H, Weissman IL (2006) Clonal analysis of mouse development reveals a poly-clonal origin for yolk sac blood islands. Dev Cell 11(4):519–533.
27. Edris B, et al. (2012) Antibody therapy targeting the CD47 protein is effective ina model of aggressive metastatic leiomyosarcoma. Proc Natl Acad Sci USA 109(17):6656–6661.
28. Willingham SB, et al. (2012) The CD47-signal regulatory protein alpha (SIRPa) in-teraction is a therapeutic target for human solid tumors. Proc Natl Acad Sci USA 109(17):6662–6667.
3506 | www.pnas.org/cgi/doi/10.1073/pnas.1222893110 Edris et al.