apo-sys workshop on data analysis and pathway charting igor ulitsky ron shamir ’ s computational...
Post on 19-Dec-2015
220 views
TRANSCRIPT

APO-SYS workshop on data APO-SYS workshop on data analysis and pathway analysis and pathway
charting charting
Igor UlitskyRon Shamir’s Computational Genomics Group

Part I: PresentationsPart I: PresentationsEXPANDER AMADEUSSPIKEMATISSE

Part II: Hands-on Part II: Hands-on SessionSession
EXPANDERMATISSESPIKE

EXPEXPression ression ANANalyzer and alyzer and DDisplayisplayERER
Adi Maron-KatzAdi Maron-KatzChaim LinhartChaim LinhartAmos TanayAmos TanayRani ElkonRani ElkonIsrael SteinfeldIsrael Steinfeld
Seagull ShavitSeagull ShavitIgor UlitskyIgor UlitskyRoded SharanRoded SharanYossi ShilohYossi ShilohRon ShamirRon Shamir
http://acgt.cs.tau.ac.il/http://acgt.cs.tau.ac.il/expanderexpander

EXPANDER
– Low level analysis:• Missing data estimation (KNN or manual)• Normalization: quantile, loess• Filtering: fold change, variation, t-test• Standardization: mean 0 std 1, take log, fixed norm
– High level gene partition analysis:• Clustering• Biclustering
– Ascribing biological meaning to patterns:• Enriched functional categories (Gene Ontology)• Identify transcriptional regulators – promoter analysis
• Built-in support for 9 organisms:– human, mouse, rat, chicken, zebrafish, fly, worm, arabidopsis, yeast

Clustering(CLICK, SOM,
K-means, Hierarchical)
Input data
Biclustering(SAMBA)
Functional enrichment(TANGO)
Normalization/Filtering
Promoter signals (PRIMA)
Lin
ks to
pu
blic
ann
ota
tion
da
tab
ase
s V
isua
lizatio
n utilitie
s

EXPANDER - Preprocessing• Input data:
Expression matrix (probe-row; condition-column)• One-channel data (e.g., Affymetrix)• Dual-channel data (cDNA microarrays, data are (log)
ratios between the Red and Green channels)• ‘.cel’ files
ID conversion file: map probes to genesGene sets data
Data definitions: Defining condition subsets Data type & scale (log)

EXPANDER – Preprocessing (II)
Data Adjustments: Missing value estimation (KNN or arbitrary)Merging conditions
Normalization: removal of systematic biases from the analyzed chips Implemented methods: quantile, lowess Visualization: box plots, scatter plots (simple,
M vs. A)

EXPANDER – Preprocessing (III) Filtering: Focus downstream analysis on the set
of “responding genes” Fold-Change Variation Statistical tests (T-test)
Standardization : Create a common scale For each probe Mean=0, STD=1 Log data (base 2) Fixed Norm (divide by norm of probe vector)

Clustering(CLICK, SOM,
K-means, Hierarchical)
Input data
Biclustering(SAMBA)
Functional enrichment(TANGO)
Normalization/Filtering
Promoter signals (PRIMA)
Lin
ks to
pu
blic
ann
ota
tion
da
tab
ase
sV
isua
lizatio
n utilitie
s

Cluster Analysis
• Partition the responding genes into distinct sets, each with a particular expression pattern Identify major patterns in the data: reduce the
dimensionality of the problem co-expression → co-function co-expression → co-regulation
• Partition the genes to achieve: Homogeneity: genes inside a cluster show
highly similar expression pattern. Separation: genes from different clusters have
different expression patterns.

Cluster Analysis (II)• Implemented algorithms:
– CLICK, K-means, SOM, Hierarchical
• Visualization: – Mean expression patterns
– Heat-maps
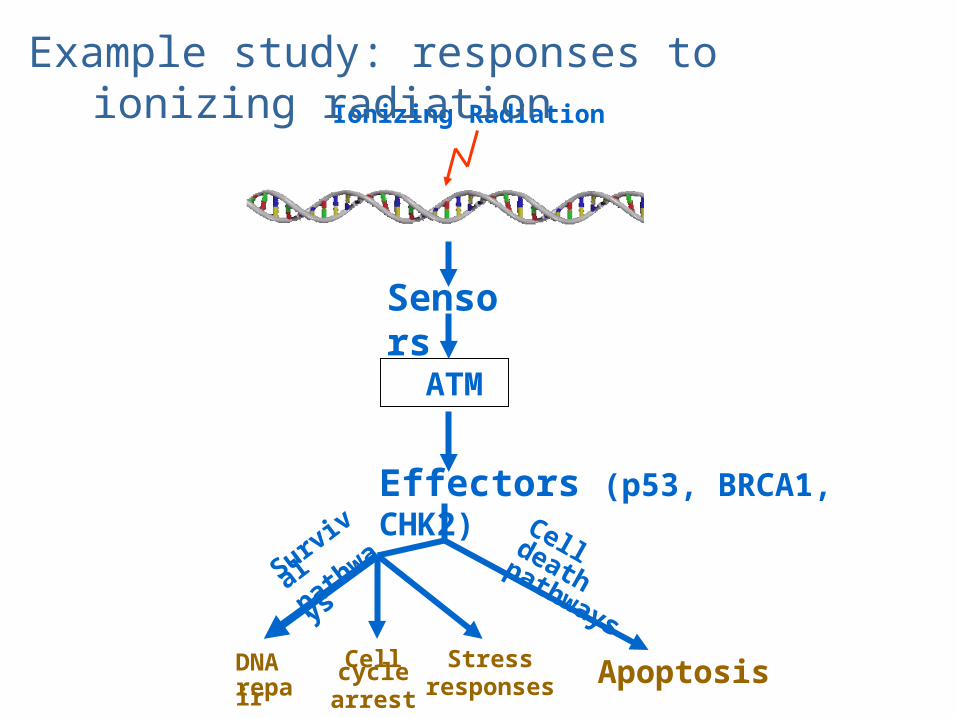
Ionizing Radiation
Effectors (p53, BRCA1, CHK2)
DNArepair
Cell cyclearrest
Stressresponses
Survival
pathways
Apoptosis
Cell death pathways
Sensors
ATM
Double Strand Breaks
Example study: responses to ionizing radiation

Example study: experimental design
• Genotypes: Atm-/- and control w.t. mice
• Tissue: Lymph node
• Treatment: Ionizing radiation
• Time points: 0, 30 min, 120 min
• Microarrays: Affymetrix U74Av2 (12k probesets)

Test case - Data Analysis • Dataset: six conditions (2 genotypes, 3 time
points)• Normalization• Filtering step – define the ‘responding genes’ set
• genes whose expression level is changed by at least 1.75 fold
• Over 700 genes met this criterion
• The set contains genes with various response patterns – we applied CLICK to this set of genes
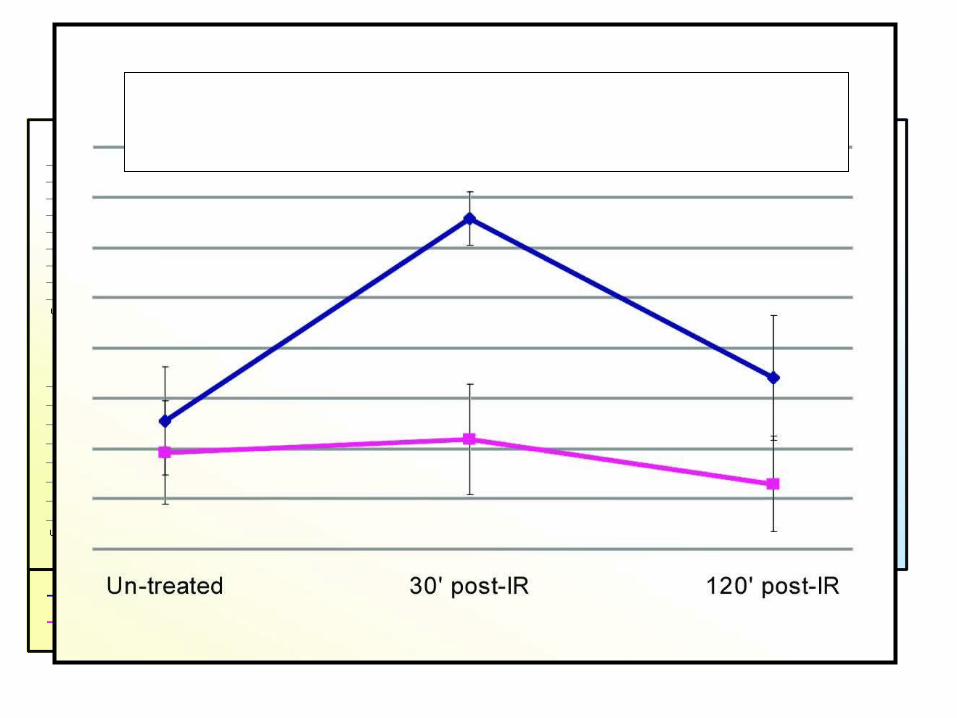
Major Gene Clusters – Irradiated Lymph nodeAtm-dependent early responding genes

Major Gene Clusters – Irradiated Lymph nodeAtm-dependent 2nd wave of responding genes
Clustering(CLICK, SOM,
K-means, Hierarchical)
Input data
Biclustering(SAMBA)
Functional enrichment(TANGOTANGO)
Normalization/Filtering
Promoter signals (PRIMA)
Lin
ks to
pu
blic
ann
ota
tion
da
tab
ase
sV
isua
lizatio
n utilitie
s

Ascribe Functional Meaning to the Clusters
• Gene Ontology (GO) annotations for human, mouse, rat, chicken, fly, worm, Arabidopsis, Zebrafish and yeast.
• TANGO: Apply statistical tests that seek over-represented GO functional categories in the clusters.

Functional Enrichment - Visualization

Functional Categories
cell cycle control (p<1x10-6 )

Cell cycle control (p<5x10-6)Apoptosis (p=0.001)
Functional Categories

Clustering(CLICK, SOM,
K-means, Hierarchical)
Input data
Biclustering(SAMBA)
Functional enrichment(TANGO)
Normalization/Filtering
Promoter signals (PRIMA)
Lin
ks to
pu
blic
ann
ota
tion
da
tab
ase
sV
isua
lizatio
n utilitie
s

? ? ? ? ?p53TF-C TF-B TF-ANEW
ATM
g3g13 g12 g10 g9 g1g8 g7 g6 g5 g4g11 g2
Hidden layer
Observed layer
Clues are in the
promoters
Identify Transcriptional Regulators

‘Reverse engineering’ of transcriptional networks
• Infers regulatory mechanisms from gene expression data– Assumption:
co-expression → transcriptional co-regulation → common cis-regulatory promoter elements
• Step 1: Identification of co-expressed genes using microarray technology (clustering algs)
• Step 2: Computational identification of cis-regulatory elements that are over-represented in promoters of the co-expressed gene

PRIMA – general description
• Input: – Target set (e.g., co-expressed genes)– Background set (e.g., all genes on the chip)
• Analysis:– Identify transcription factors whose binding
site signatures are enriched in the ‘Target set’ with respect to the ‘Background set’.
• TF binding site models – TRANSFAC DB• Default: From -1000 bp to 200 bp relative
the TSS

Promoter Analysis - Visualization

PRIMA - Results

Transcription factor
Enrichment factor
P-value
Transcription factor
Enrichment factor
P-value
CREB2.66.0x10-5
PRIMA – Results
NF-B 5.1 3.8x10-8
p53 4.2 9.6x10-7
STAT-1 3.2 5.4x10-6
Sp-1 1.7 6.5x10-4
Clustering(CLICK, SOM,
K-means, Hierarchical)
Input data
Biclustering(SAMBA)
Functional enrichment(TANGO)
Normalization/Filtering
Promoter signals (PRIMA)
Lin
ks to
pu
blic
ann
ota
tion
da
tab
ase
sV
isua
lizatio
n utilitie
s

Biclustering
Clustering becomes too restrictive on large datasets: • Seeks global partition of
genes according to similarity in their expression across ALL conditions
Relevant knowledge can be revealed by identifying genes with common pattern across a subset of the conditions
• Biclustering algorithmic approach

* Bicluster (=module) : subset of genes with similar behavior in a subset of conditions
* Computationally challenging: has to consider many combinations of sub-conditions
Biclustering: SAMBAStatistical Algorithmic Method for Bicluster Analysis
A. Tanay, R. Sharan, R. Shamir RECOMB 02
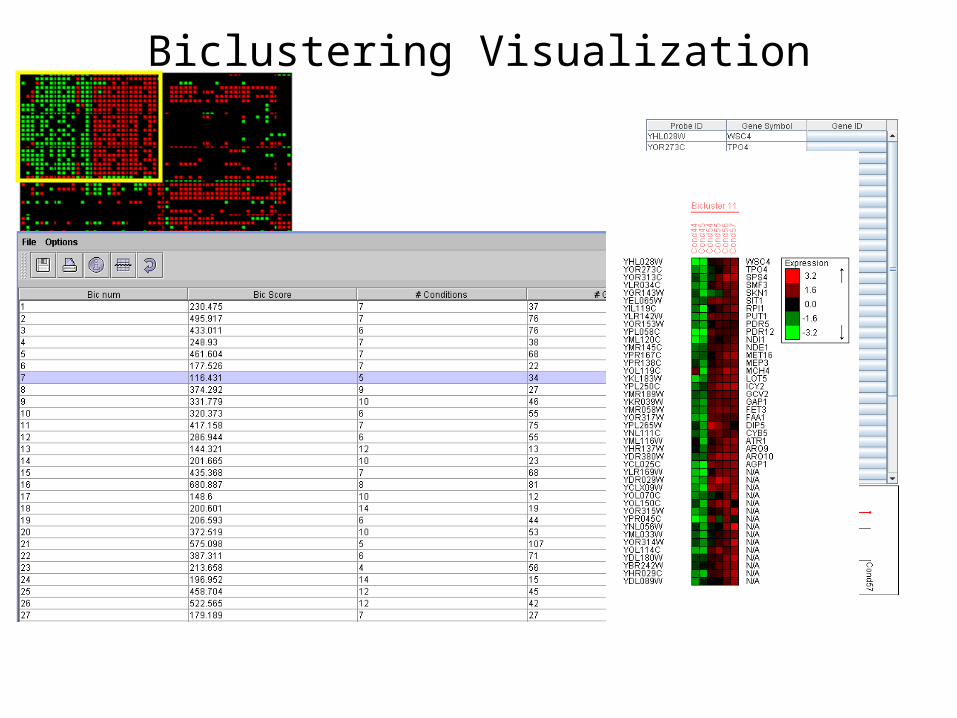
Biclustering Visualization