applications for t-cell epitope queries and tools in the immune epitope database and analysis...
TRANSCRIPT

Journal of Immunological Methods 374 (2011) 62–69
Contents lists available at ScienceDirect
Journal of Immunological Methods
j ourna l homepage: www.e lsev ie r.com/ locate / j im
Research paper
Applications for T-cell epitope queries and tools in the Immune EpitopeDatabase and Analysis Resource
Yohan Kim, Alessandro Sette, Bjoern Peters⁎La Jolla Institute for Allergy & Immunology, 9420 Athena Circle, La Jolla, CA 92037, USA
a r t i c l e i n f o
⁎ Corresponding author. Tel.: +1 858 752 6914; faxE-mail address: [email protected] (B. Peters).
0022-1759/$ – see front matter © 2010 Elsevier B.V.doi:10.1016/j.jim.2010.10.010
a b s t r a c t
Article history:Received 8 September 2010Received in revised form 23 October 2010Accepted 27 October 2010Available online 31 October 2010
The Immune Epitope Database and Analysis Resource (IEDB, http://www.iedb.org) hosts acontinuously growing set of immune epitope data curated from the literature, as well as datasubmitted directly by experimental scientists. In addition, the IEDB hosts a collection ofprediction tools for both MHC class I and II restricted T-cell epitopes that are regularly updated.In this review, we provide an overview of T-cell epitope data and prediction tools provided bythe IEDB. We then illustrate effective use of these resources to support experimental studies.We focus on two applications, namely identification of conserved epitopes in novel strains of apreviously studied pathogen, and prediction of novel T-cell epitopes to facilitate vaccine design.We address common questions and concerns faced by users, and identify patterns of usage thathave proven successful.
© 2010 Elsevier B.V. All rights reserved.
Keywords:Epitope conservationEpitope predictionsVaccine designMajor Histocompatibility Complex
1. Introduction
The goal of the IEDB (Peters et al., 2005; Peters and Sette,2007) is to catalog and organize information related to T- andB-cell epitopes, as well as to provide tools to predict novelepitopes and to analyze known epitopes to gain newinformation about them. The IEDB website has been updatedcontinuously based on user feedback, resulting in fundamen-tally revised versions of both the database itself (Vita et al.,2010) and the tools hosted in the associated AnalysisResource (Zhang et al., 2008). The continued use of thedatabase and tools has broadened our understanding of whatapplications are possible and refined the methodology toachieve optimal results.
While a similar suite of data and tools exists for B-cell/antibody epitopes, they are outside of the scope for thepresent article. Moreover, details of curation process used tocapture data from the literature (Vita et al., 2008), develop-ment of a formal ontology to represent the immune epitopedata (Sathiamurthy et al., 2005), all available prediction andanalysis tools (Zhang et al., 2008), and recent updates to the
: +1 858 752 6987.
All rights reserved.
database structure (Vita et al., 2010) are described elsewhere.We here present a summary of the currently availableresources in the IEDB that are related to T-cell epitopes.
Multiple applications can benefit from identifying T-cellepitopes, including the design of prophylactic vaccines(Kaech et al., 2002; Sette and Fikes, 2003; Purcell et al.,2007), therapeutics (Bousquet et al., 1998; Banchereau andPalucka, 2005), diagnostics (Brock et al., 2004; Pai et al.,2008), reagents for research (Altman et al., 1996), and de-immunization of biological drugs (Bryson et al., 2010).Designing vaccines against infectious agents is the mostfrequent application that can benefit from knowing T-cellepitopes. Basically, for a vaccine to induce the creation of amemory T-cell population capable of recognizing a pathogen,the vaccine has to contain T-cell epitopes from that pathogen.This does not mean that the vaccine itself has to be comprisedof individual epitopes, but minimally that antigens thatharbor the epitopes are being included. In the case oftherapeutics (sometimes also called therapeutic vaccines),the goal is to modulate an existing immune response thateither needs to be enhanced (cancer, chronic infection), orreduced (autoimmune diseases, allergy). Again, designingsuch a therapeutic requires knowing the epitope targets ofthe T-cell responses that are intended to be modulated. For

able 1ain curation categories of peptidic entries in the IEDB.
Main Category Total number journal articles % Processed
Infectious diseases 8616 99Allergens 741 97.7Autoimmune 3924 84.1Transplants/Alloantigens 631 20.4
or each category, a broad PubMed query was carried out to retrieve allurnal articles of potential relevance. These article abstracts were thenanned using a combination of automated and manual text classification tolter out false positives, resulting in the numbers shown in the secondlumn. Any epitopes from these articles were then manually curated intoe IEDB. The percentage completion of the manual curation process in eachtegory to date is shown in the last column.
63Y. Kim et al. / Journal of Immunological Methods 374 (2011) 62–69
diagnostics, the goal is to identify if a patient has been incontact with certain antigens in order to help identify apatient's disease status. The use of epitopes as diagnostics hasthe potential to distinguish infections with related pathogens,which can be difficult when relying on whole organism orantigen preparations, which can often be cross-recognized.Finally, a major application of T-cell epitopes is their directuse as reagents for basic research, as they allow trackingspecific T-cell populations, notably in the form of MHCtetramers.
As more protein based drugs are developed, minimizingimmunogenicity of these drugs becomes another importantapplication of T-cell epitope identification. The term ‘de-immunization’ has been coined to refer to a set of technol-ogies that reduce immunogenicity by removing T-cellepitopes from protein therapeutics (Jones et al., 2009). Theprocedure entails first identifying epitopes and their anchorresidues for HLA binding, applying point mutations to theseanchor residues to neutral ones such that mutations do notinterfere with folding and function of proteins, and finallycarrying out validations to confirm absence of immunoge-nicity of themutated proteins (Scott and De Groot, 2010). Thegeneral T-cell epitope identification tools in the IEDB can beapplied to this problem, but currently no dedicated tools existin the IEDB to e.g. suggest what mutations in a protein drugare recommended.
Following the overview of resources in the IEDB, wepresent examples of applications how the IEDB can be used to1) discover conserved epitopes in a novel antigen, and 2)predict novel epitopes to facilitate vaccine design. Overall,this article provides examples on how the IEDB data and toolscan be used effectively for practical experimental applica-tions, and addresses commonly encountered issues.
2. Overview of T-cell epitope related resources available inthe IEDB
2.1. Data contained in the IEDB
The IEDB provides a catalog of experimentally character-ized T-cell epitopes, as well as data on Major Histocompat-ibility Complex (MHC) binding and MHC ligand elutionexperiments. These three types of experimental data differin what they convey: While MHC binding data shows amolecular association between a ligand (typically a peptide)and an MHC molecule, MHC ligand elution data shows thatthe eluted ligand is not only capable of binding an MHCmolecule, but can also be created by the cellular antigenprocessing machinery. T-cell epitope mapping experiments,on the other hand, test the ability to engage a T-cell receptor(TCR), which requires binding to an MHC molecule andactivation of the TCR. The IEDB represents the molecularstructures tested for MHC binding, MHC ligand elution or T-cell recognition, and the details of the experimental contextsin which these molecules were tested. Only peptides that testpositive in a T-cell recognition assays are truly ‘T-cellepitopes’, while those that are only shown to be positive ina binding assay should be referred to as ‘MHC binders’ andthose that can be eluted from MHC on the cells surface are‘naturally processed ligands’. Epitopes recognized in humans,nonhuman primates, rodents, pigs, cats and all other tested
host species are included in the IEDB. Moreover, both positiveand negative experimental results are captured. Lastly, thescope of the database to date includes data relating to epitopederived from all infectious diseases, including NIAID CategoryA, B and C priority pathogens (http://www3.niaid.nih.gov/topics/BiodefenseRelated/Biodefense/research/CatA.html),NIAID Emerging and Reemerging infectious diseases (http://www3.niaid.nih.gov/topics/emerging/), allergens, and auto-antigens involved in autoimune disorders. In particular, HIVepitopes are explicitly excluded, which can instead be foundin the Los Alamos HIV Molecular Immunology Database(http://www.hiv.lanl.gov) (Korber et al., 2006).
Since the IEDB was initiated 4 years ago, the data from158,067 T-cell, 198,877 MHC binding, and 2007 MHC ligandelution assays have been collected through either manualcuration or direct submissions. Curated data from theliterature covers 99% of all publicly available journal articleson peptidic epitopes mapped in infectious agents (excludingHIV) and 97.7% of those mapped in allergens. In addition, thecuration of epitopes related to autoimmunity is expected tobe completed by the end of 2010. In terms of directsubmissions, 194,862 assays were submitted by investigatorsto the IEDB that would otherwise be unavailable to the public.Table 1 provides an overview of the journal articles availablein each of themain categories of peptidic epitopes included inthe IEDB and their level of completion.
2.2. Query interfaces
The query interface of the IEDB has been completelyredesigned in early 2009 for the IEDB 2.0 release (Vita et al.,2010). For instance, more frequently used query features areprominently displayed in the left panel of the home page(Fig. 1). The query interface is divided into epitope structure(to query by e.g. peptide sequence), epitope source (to queryby the antigen or organism in which an epitope is contained)and immunological contexts (to query by the type of assay,host organism of the response, or MHC restriction). The usercan for example search for any epitope contained in InfluenzaA viruses that is recognized in a T-cell assay by humaneffector cells and restricted by MHC class II molecule. Theresults of this query are presented in Fig. 2. For users with theneed to perform more detailed queries, an advanced querymode is provided through the ‘Search’ menu item, allowingone to search any field in the database. In addition, epitopeentries can be ‘browsed’ using features such as MHC alleles or
TM
Fjoscfi
cothca

Fig. 1. Main IEDB query interface for retrieving epitopes. Using the interface, one can specify epitope structure type (Epitope Structure), organism from which icame from (Epitope Source), and contexts in which the epitope was determined (Immune Recognition Context). An example query for retrieving all T-celepitopes derived from Influenza A virus and restricted to human host and MHC class II is shown.
64 Y. Kim et al. / Journal of Immunological Methods 374 (2011) 62–69
source organisms using NCBI taxonomy tree (See ‘Browse’ tabat the top).
2.3. T-cell epitope prediction tools
Complementing the experimentally determined immuneepitope data contained in the database, the Analysis Resourcecomponent of the IEDB provides tools for predicting andanalyzing epitope data (http://tools.immuneepitope.org).Table 2 gives an overview of different T-cell relevantcategories of tools available. The analysis tools provide theability to map epitopes onto 3D protein structure of thesource antigen or its homologue (Beaver et al., 2007), clusterepitopes into groups with similar sequences, calculate thedegree of conservation of epitopes in a given set of proteins(Bui et al., 2007), or predict the population coverage of a set ofepitopes based on the frequency of the HLA alleles they arerestricted by (Bui et al., 2006).
The T-cell epitope prediction tools are divided into MHCbinding and processing prediction tools. The former categorycontains tools that predict binding affinities of peptides toeither MHC class I or class II molecules. Based on pastbenchmarks of MHC-I binding prediction methods, NetMHC-3.0 (called ANN on the IEDB website) and SMM methods areamong the best performing individual methods (Peters et al.,2006; Lin et al., 2008a). For MHC-II, Consensus and NN-alignhave been shown to be best performing (Lin et al., 2008b;Wang et al., 2008). Consensus prediction methods, which
tl
combine outputs of different prediction approaches are alsoprovided. Such methods have proven to be a successfulstrategy in many bioinformatics applications (Cuff et al.,1998; Ginalski et al., 2003; Arai et al., 2004).
The MHC class I processing predictions combine thebinding predictions described above with predictions ofproteasomal cleavage and TAP transport to identify peptidesthat are ‘naturally processed’. Some studies have shown thatthe top predicted peptides aremore likely to be recognized byT-cells than those selected by MHC binding predictions alone(Peters et al., 2003; Tenzer et al., 2005; Kessler and Melief,2007). For example, in (Kessler and Melief, 2007), a numberof integrated CD8+ epitope prediction methods available onthe web were tested against a list of 64 nonamer peptidesderived from a known tumor associated antigen, PRAME, thatwere previously studied. Out of 5 methods tested against thislist, 4 methods identified 2 out of 3 known epitopes as amongtop 10 predicted peptides. The prediction method from theIEDB identified all 3.
However, no large-scale evaluation has been performedthat clearly quantified the advantage of using the processingpredictions, and determined how many epitopes are errone-ously predicted not to be processed. Based on our experienceto date, the advantage of using processing predictions overbinding predictions – if any – seems to be relatively small.Also, any advantage has to be weighed against the increasedcomplexity of interpreting the results of processing predic-tions, specifically when predicting epitopes in pathogens that
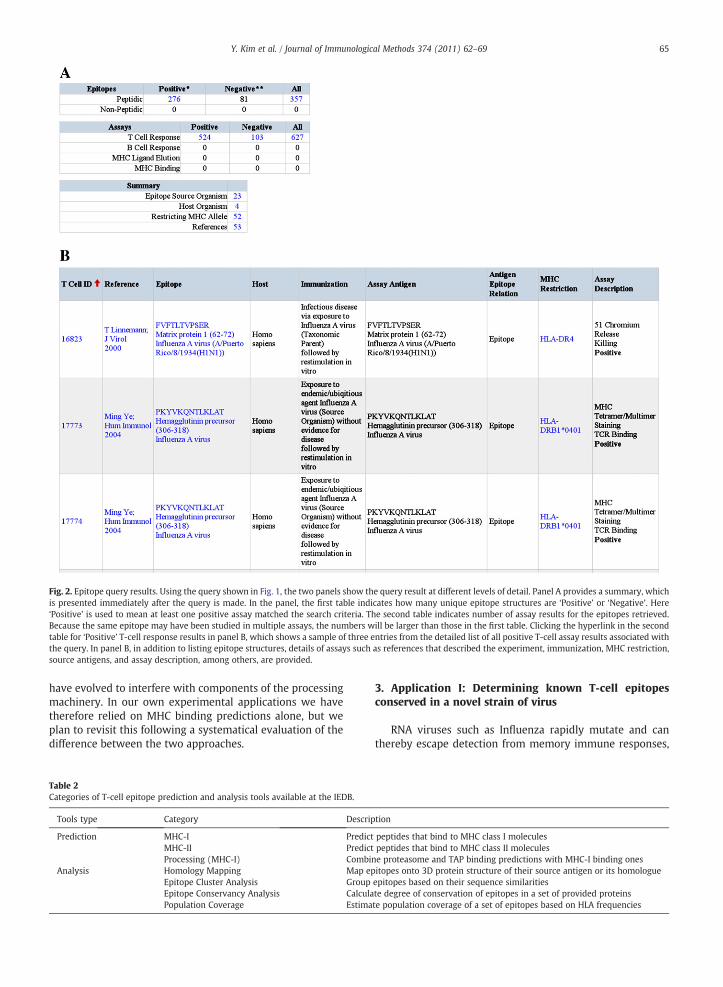
Fig. 2. Epitope query results. Using the query shown in Fig. 1, the two panels show the query result at different levels of detail. Panel A provides a summary, whichis presented immediately after the query is made. In the panel, the first table indicates how many unique epitope structures are ‘Positive’ or ‘Negative’. Here‘Positive’ is used to mean at least one positive assay matched the search criteria. The second table indicates number of assay results for the epitopes retrieved.Because the same epitope may have been studied in multiple assays, the numbers will be larger than those in the first table. Clicking the hyperlink in the secondtable for ‘Positive’ T-cell response results in panel B, which shows a sample of three entries from the detailed list of all positive T-cell assay results associated withthe query. In panel B, in addition to listing epitope structures, details of assays such as references that described the experiment, immunization, MHC restriction,source antigens, and assay description, among others, are provided.
65Y. Kim et al. / Journal of Immunological Methods 374 (2011) 62–69
have evolved to interfere with components of the processingmachinery. In our own experimental applications we havetherefore relied on MHC binding predictions alone, but weplan to revisit this following a systematical evaluation of thedifference between the two approaches.
Table 2Categories of T-cell epitope prediction and analysis tools available at the IEDB.
Tools type Category Descrip
Prediction MHC-I PredictMHC-II PredictProcessing (MHC-I) Combi
Analysis Homology Mapping Map epEpitope Cluster Analysis GroupEpitope Conservancy Analysis CalculaPopulation Coverage Estima
3. Application I: Determining known T-cell epitopesconserved in a novel strain of virus
RNA viruses such as Influenza rapidly mutate and canthereby escape detection from memory immune responses,
tion
peptides that bind to MHC class I moleculespeptides that bind to MHC class II molecules
ne proteasome and TAP binding predictions with MHC-I binding onesitopes onto 3D protein structure of their source antigen or its homologueepitopes based on their sequence similaritieste degree of conservation of epitopes in a set of provided proteinste population coverage of a set of epitopes based on HLA frequencies

66 Y. Kim et al. / Journal of Immunological Methods 374 (2011) 62–69
allowing recurring infections of the same host by differentstrains from the same viral species. However, in every RNAvirus there are some parts that mutate much less frequently,presumably because those parts are of high functionalrelevance, and most mutations result in a significant of lossof fitness. The identification of epitopes that are conservedbetween different strains of a viral species is of particularinterest. For both Dengue (Khan et al., 2008) and Influenzaviruses (Heiny et al., 2007), known and predicted T-cellepitopes have been analysed for their conservation acrossmultiple viral strains. A similar analysis was performed forthe pandemic H1N1 strain in 2009 (Greenbaum et al., 2009),which was specifically probed for epitopes that were alsopresent in recently circulating seasonal Influenza strains. Alarge fraction of T-cell epitopes was indeed found to beconserved in the pandemic strain, and these were experi-mentally confirmed to be targets of pre-existing T cellimmunity.
The basic technical execution of identifying conserved T-cell epitopes from the IEDB in all of the above applications issimilar. First, the IEDB is queried for epitopes from the viralspecies of interest using the ‘source organism’ field. Forexample, in Fig. 1, ‘Influenza A virus’ is used as the ‘sourceorganism’. In addition, restrictions can be posed depending
Fig. 3. Epitope conservancy tool. The tool allows the user to determine to what extentext input boxes allow the user to enter epitopes and protein sequences of interediscontinuous) and sequence identity threshold and submitting, results are presenthreshold. Additional analysis results such as alignments of epitopes and proteins a
on the application, such as only T-cell epitopes could be ofinterest, or only those defined in humans. Further refinementof the query, such as restricting epitopes to those mappedfollowing infection with virus (in contrast to immunizationwith individual antigens), requires using the advanced searchoption. Once the set of epitopes has been retrieved from theIEDB, the sequences can be exported as an Excel spreadsheet,and analyzed for conservation offline. This can either be donemanually, or with custom computer code, or using the IEDBepitope conservancy tool (Bui et al., 2007). Using the epitopeconservancy tool, the retrieved epitopes can be checked forconservation in different antigen sequences. For instance, byentering the protein sequences of the 2009 pandemicInfluenza strain (see Fig. 3), and selecting a sequence identitythreshold of 100%, all previously known Influenza epitopesthat are 100% conserved in the pandemic strain can beidentified.
For individual antigens, a simple alternative to search forall known epitopes that are conserved in it is to enter theantigen protein sequence in the ‘Linear Peptide’ field (seeFig. 1), and selecting the ‘Blast 90%’ option rather than ‘ExactMatches’ in the pull down menu to retrieve partiallymatching epitopes meeting the similarity threshold. Thisquery can again be combined with further restrictions, such
t user-provided epitopes are conserved in a list of protein sequences. The twost, respectively. After specifying the type of epitope structure (i.e. linear vsted that indicate how many proteins contain each epitope at the specifiedre also provided.
.

67Y. Kim et al. / Journal of Immunological Methods 374 (2011) 62–69
as limiting the results to epitopes recognized by T-cells,defined in humans and derived from Influenza A virus.
4. Application II: Identifying novel T-cell epitope targets tofacilitate vaccine design
The purpose of identifying T-cell epitopes for vaccinedesign can be multifold. Sets of epitopes can be used directlyas a vaccine (Sette and Fikes, 2003; Kessler and Melief, 2007;Purcell et al., 2007), or epitope mapping can be used toidentify antigens that should be included in a subunit vaccine(Scarselli et al., 2005; Zvi et al., 2008), or epitopes can be usedas a correlate of protective efficacy to evaluate differentvaccine constructs. The task at hand in each of these cases isto identify peptides in a given antigen that elicit a T-cellresponse. In contrast to the applications above, we hereconsider the identification of novel epitopes in antigens thathave not been studied exhaustively. For vaccine designapplications that involve well-studied pathogens, the querystrategies described in the previous section should be applied.To identify novel epitopes, a multi-step approach has beenshown to be successful, in which prediction tools are used toidentify epitope candidates that are then tested in validationexperiments. The goal of predictions is therefore to identify asmall enough set of peptide candidates that make experi-mental validation feasible.
Whenmaking peptide:MHCbinding predictions, there are anumber of important things to consider to achieve optimalresults. First, the length of peptides that are considered to bepotential ligands are different for MHC class I and II molecules.Most MHC class I restricted T-cell epitopes are 9 residues long,but depending on theMHC allele, epitopes of lengths 8, 10 and11 are also found. That means, for a given antigen, all peptidesof these lengths are potential candidate epitopes. In contrast,MHC class II restricted T-cell epitopes are of more variablelengths between 13 and 25 residues, as they are bound in anopen ended groove and ends of a bound peptide can be outsideof the core binding region. The core region of six to nine epitoperesidues makes direct contact with the binding pocket of theMHC class II molecule. Peptides containing the same coreregion with flanking residues of different length can activatethe same T-cells, arguably making them variants of the sameepitope. However, the core binding region by itself is a poorepitope, aspeptides of length9or shorter donot bindMHCclassII well. A peptide overhang of 2–3 residues at each end isrequired to stabilize the peptides, largely independent of whatresidues are involved. Thismeans 13-mers or 15-mers aremoreappropriate to test as MHC class II epitope candidates. In ourexperimental applications, we have historically used 15-merpeptides.
Second, MHC alleles for which peptide binding predictionswill be made need to be carefully chosen. If the goal of thestudy is to identify T-cell epitopes for use in an animal model,the selected MHC alleles should match those of the speciesand strain of animals used. If the goal is to design a vaccine fora human population, the frequency of MHC alleles in thepopulation of interest should be considered to maximize thevaccine's population coverage. As every human expressesmultiple MHC class I and II molecules, it is not required that avaccine covers all of them, but at least one MHC allele in themajority of individuals should be included. There are different
approaches for achieving this goal. For instance, it has beennoted that binding specificities of MHC-I molecules overlapand can be grouped into 9 supertypes (Sidney et al., 2008). Byselecting alleles representing several or all of these super-types, one can achieve a wide coverage. Alternatively, acollection of MHC alleles can be chosen and the populationcoverage of a set of epitopes reactive for each of these allelescan be projected using the Population coverage tool (Bui et al.,2006).
As an example application, we will consider identificationof T-cell epitopes in Sabia virus restricted by two prototypesupertype alleles, HLA A*0201 and HLA A*1101. This is asimplified version of the strategy in (Kotturi et al., 2009) inwhich a crossreactive vaccine for multiple Arenavirus specieswas designed. First, one needs to obtain protein sequencesfrom Sabia virus, which can be done by searching the NCBIdatabase or a specialist resource such as the NIAID Bioinfor-matics Resource Centers (Greene et al., 2007) for full genomicsequences of Sabia. The virus has two genomic segmentsencoding four proteins with a total of 3362 residues (in thisexample, the proteins with genbank identifiers gi|52627080,gi|52627081, gi|52627077 and gi|52627078 were used).These sequences are then pasted in FASTA format into theMHC class I binding prediction tool. Selecting ‘HLA A*0201’ asthe allele to make binding predictions for, and ‘all lengths’ tomake predictions for and ‘submit’ results in 6656 predictionsof affinities for HLA A*0201 for all 9 and 10-mer peptidescontained in the proteins. Multiple prediction methods areavailable, but by default the method considered best is used,which currently is the ‘ann’ method (NetMHC, (Lundegaardet al., 2008)). The peptides can be sorted by IC50 value withlow values corresponding to high affinity binders. The samecan now be done for HLA A*1101, resulting in a second list ofpeptide candidates sorted by their predicted affinity.
Once the sorted lists of peptide candidates are available, themost promising candidates for validation experiments can bechosen using a number of strategies. One strategy is to choosethe top0.5%–3%scoringpeptides for eachallele, therebyequallyrepresenting all alleles. Another strategy is to use the same IC50cutoff for all alleles, which will result in different number ofpeptides for alleles, because of different sizes of bindingpeptiderepertoires. What strategy is preferred depends on theapplication: If it is more important to identify one or moreepitopes per allele, one should represent the alleles equally, anduse a percentile cutoff. If it is more important to maximize thechances of getting a response per peptide, independent of therestricting allele, we believe it is better to choose an absoluteIC50 value cutoff. Still another is based on the use of apromiscuity score, which is defined as the number of allelesthat a peptide binds at a given binding affinity score threshold.Using this strategy, peptides with higher promiscuity scoreswould be selected. We have been primarily using the firststrategy in order to have unbiased representation of each allelein the candidate set. We are planning to perform a metaanalysis of the results from these studies to determine ifdifferent strategies of peptide selection would have resulted inhigher success rates.
The top 67 peptides (=1%) of all predicted binders shouldinclude more than 80% of all epitopes restricted by the allele ofinterest in a given virus (Moutaftsi et al., 2006; Kotturi et al.,2007). This number of peptides is also easily testable in

68 Y. Kim et al. / Journal of Immunological Methods 374 (2011) 62–69
validation experiments. Validation experiments should typi-cally be carried out in a staggered fashion, where highthroughput experiments are carried out first to further narrowthe set of candidates to be tested in more resource intensiveexperiments. In (Kotturi et al., 2009), predicted bindingpeptides from Arenaviruses were first tested for recognitionby T-cells from HLA transgenic mice that were infected withrecombinant vaccinia virus (rVACV) expressing Arenavirusantigens. The use of HLA transgenic mice rather than samplesfrom previously infected humans was necessary as the latterare very hard to obtain. Similarly, the use of rVACV wasnecessary as using live Arenavirus infections poses substantialbiosafety concerns. Peptides that were targeted by strongresponses were selected for further validation, carried out byimmunizing HLA transgenic mice with the peptide, and thenchallenging them with a lethal dose of recombinant rVACV.Ultimately, a set of epitopes was assembled that resulted inprotection against different strains of Arenavirus, demonstrat-ing cross-protectiveness of the vaccine. The HLA-A*0201restricted Sabia virus peptides included in the vaccine were#2 and #37 highest predicted binders (GLLEWIFRA andLLPDALLFTL) in the candidate list generated as describedabove. This illustrates that a significant fraction of peptidecandidates will not be able to induce effective protection, eventhough they had similar or better predicted binding affinities tothe ones that did. This always has to be expected, as amultitudeof additional factors influence the ability of an epitope toprotect, such as availability of a T cell repertoire, or kinetics ofantigen expression.
5. Conclusion
The IEDB (http://www.iedb.org) contains useful resourcesfor those studying the immune system and developingimmune-based vaccines, therapeutics and diagnostics. Thisreview has shown examples demonstrating how T-cell im-muneepitopedata andepitopeprediction tools providedby theIEDB can be effectively used. We expect that, as epitope basedapplicationsare increasingly used in ahigh throughputmanner,new tools will become available that better predict whatepitopes are most appropriate for different applications. Also,we are continuously working on improving the usefulness ofthe IEDB resource, in particular in further automating theworkflow of novel epitope identification. We are grateful forthe feedback we have received from the IEDB user community,and want to further encourage constructive criticism, asultimately the success of the IEDB is measured by the value itprovides to its user base.
Acknowledgment
This work was supported by the National Institutes ofHealth's (National Institute of Allergy and Infectious Disease)Contract HHSN26620040006C.
References
Altman, J.D., Moss, P.A.H., Goulder, P.J.R., Barouch, D.H., McHeyzer-Williams,M.G., Bell, J.I., McMichael, A.J., Davis, M.M., 1996. Phenotypic analysis ofantigen-specific T lymphocytes. Science 274, 94.
Arai, M., Mitsuke, H., Ikeda, M., Xia, J.-X., Kikuchi, T., Satake, M., Shimizu, T.,2004. ConPred II: a consensus prediction method for obtaining
transmembrane topology models with high reliability. Nucl. Acids Res.32, W390.
Banchereau, J., Palucka, A.K., 2005. Dendritic cells as therapeutic vaccinesagainst cancer. Nat. Rev. Immunol. 5, 296.
Beaver, J., Bourne, P. and Ponomarenko, J. (2007) EpitopeViewer: a Javaapplication for the visualization and analysis of immune epitopes in theImmune Epitope Database and Analysis Resource (IEDB). ImmunomeResearch 3, 3.
Bousquet, J., Lockey, R., Malling, H.-J., 1998. Allergen immunotherapy:therapeutic vaccines for allergic diseases A WHO position paper. J. AllergyClin. Immunol. 102, 558.
Brock, I., Weldingh, K., Leyten, E.M.S., Arend, S.M., Ravn, P., Andersen, P.,2004. Specific T-cell epitopes for immunoassay-based diagnosis ofMycobacterium tuberculosis infection. J. Clin. Microbiol. 42, 2379.
Bryson, C.J., Jones, T.D. and Baker, M.P. (2010) Prediction of Immunogenicityof Therapeutic Proteins: Validity of Computational Tools. BioDrugs 24, -.
Bui, H.-H., Sidney, J., Dinh, K., Southwood, S., Newman, M. and Sette, A.(2006) Predicting population coverage of T-cell epitope-based diagnos-tics and vaccines. BMC Bioinformatics 7, 153.
Bui, H.-H., Sidney, J., Li, W., Fusseder, N. and Sette, A. (2007) Development ofan epitope conservancy analysis tool to facilitate the design of epitope-based diagnostics and vaccines. BMC Bioinformatics 8, 361.
Cuff, J., Clamp, M., Siddiqui, A., Finlay, M., Barton, G., 1998. JPred: a consensussecondary structure prediction server. Bioinformatics 14, 892.
Ginalski, K., Elofsson, A., Fischer, D., Rychlewski, L., 2003. 3D-Jury: a simpleapproach to improve protein structure predictions. Bioinformatics 19,1015.
Greenbaum, J.A., Kotturi, M.F., Kim, Y., Oseroff, C., Vaughan, K., Salimi, N., Vita,R., Ponomarenko, J., Scheuermann, R.H., Sette, A., Peters, B., 2009. Pre-existing immunity against swine-origin H1N1 influenza viruses in thegeneral human population. Proc. Natl. Acad. Sci. 106, 20365.
Greene, J.M., Collins, F., Lefkowitz, E.J., Roos, D., Scheuermann, R.H., Sobral, B.,Stevens, R., White, O., Di Francesco, V., 2007. National Institute of Allergyand Infectious Diseases Bioinformatics Resource Centers: new assets forpathogen informatics. Infect. Immun. 75, 3212.
Heiny, A.T., Miotto, O., Srinivasan, K.N., Khan, A.M., Zhang, G.L., Brusic, V., Tan,T.W., August, J.T., 2007. Evolutionarily conserved protein sequences ofinfluenza A viruses, avian and human, as vaccine targets. PLoS ONE 2,e1190.
Jones, T., Crompton, L., Carr, F., Baker, M., 2009. Deimmunization ofmonoclonal antibodies. Meth. Mol. Biol. 525, 405.
Kaech, S.M., Wherry, E.J., Ahmed, R., 2002. Effector and memory T-celldifferentiation: implications for vaccine development. Nat. Rev. Immu-nol. 2, 251.
Kessler, J.H., Melief, C.J.M., 2007. Identification of T-cell epitopes for cancerimmunotherapy. Leukemia 21, 1859.
Khan, A.M., Miotto, O., Nascimento, E.J.M., Srinivasan, K.N., Heiny, A.T., Zhang,G.L., Marques, E.T., Tan, T.W., Brusic, V., Salmon, J., August, J.T., 2008.Conservation and variability of dengue virus proteins: implications forvaccine design. PLoS Negl.Trop. Dis. 2, e272.
Korber, B.T.M., Brander, C., Haynes, B.F., Koup, R., Moore, J.P., Walker, B.D. andWatkins, D.I. HIV Molecular Immunology 2006/2007. In. Los AlamosNational Laboratory, Theoretical Biology and Biophysics, Los Alamos,New Mexico. LA-UR 07-4752.
Kotturi, M.F., Botten, J., Sidney, J., Bui, H.-H., Giancola, L., Maybeno, M., Babin,J., Oseroff, C., Pasquetto, V., Greenbaum, J.A., Peters, B., Ting, J., Do, D.,Vang, L., Alexander, J., Grey, H., Buchmeier, M.J. and Sette, A. (2009) AMultivalent and Cross-Protective Vaccine Strategy against ArenavirusesAssociated with Human Disease. PLoS Pathog 5, e1000695.
Kotturi, M.F., Peters, B., Buendia-Laysa Jr., F., Sidney, J., Oseroff, C., Botten, J.,Grey, H., Buchmeier, M.J., Sette, A., 2007. The CD8+ T-cell response tolymphocytic choriomeningitis virus involves the L antigen: uncoveringnew tricks for an old virus. J. Virol. 81, 4928.
Lin, H., Ray, S., Tongchusak, S., Reinherz, E. and Brusic, V. (2008a) Evaluationof MHC class I peptide binding prediction servers: Applications forvaccine research. BMC Immunology 9, 8.
Lin, H., Zhang, G., Tongchusak, S., Reinherz, E. and Brusic, V. (2008b)Evaluation of MHC-II peptide binding prediction servers: applications forvaccine research. BMC Bioinformatics 9, S22.
Lundegaard, C., Lamberth, K., Harndahl, M., Buus, S., Lund, O., Nielsen, M.,2008. NetMHC-3.0: accurate web accessible predictions of human,mouse and monkey MHC class I affinities for peptides of length 8–11.Nucl. Acids Res. 36, W509.
Moutaftsi, M., Peters, B., Pasquetto, V., Tscharke, D.C., Sidney, J., Bui, H.-H.,Grey, H., Sette, A., 2006. A consensus epitope prediction approachidentifies the breadth of murine TCD8+-cell responses to vaccinia virus.Nat. Biotech. 24, 817.
Pai, M., Zwerling, A., Menzies, D., 2008. Systematic review: T-cell basedassays for the diagnosis of latent tuberculosis infection: an update. Ann.Intern. Med. 149, 177.

69Y. Kim et al. / Journal of Immunological Methods 374 (2011) 62–69
Peters, B., Bui, H.-H., Frankild, S., Nielsen, M., Lundegaard, C., Kostem, E.,Basch, D., Lamberth, K., Harndahl, M., Fleri, W., Wilson, S.S., Sidney, J.,Lund, O., Buus, S. and Sette, A. (2006) A Community ResourceBenchmarking Predictions of Peptide Binding to MHC-I Molecules.PLoS Comput Biol 2, e65.
Peters, B., Bulik, S., Tampe, R., van Endert, P.M., Holzhutter, H.-G., 2003.Identifying MHC Class I epitopes by predicting the TAP transportefficiency of epitope precursors. J. Immunol. 171, 1741.
Peters, B., Sette, A., 2007. Integrating epitope data into the emerging web ofbiomedical knowledge resources. Nat. Rev. Immunol. 7, 485.
Peters, B., Sidney, J., Bourne, P., Bui, H.-H., Buus, S., Doh,G., Fleri,W., Kronenberg,M., Kubo, R., Lund, O., Nemazee, D., Ponomarenko, J.V., Sathiamurthy, M.,Schoenberger, S., Stewart, S., Surko, P., Way, S., Wilson, S. and Sette, A.(2005) The Immune Epitope Database and Analysis Resource: From Visionto Blueprint. PLoS Biol 3, e91.
Purcell, A.W., McCluskey, J., Rossjohn, J., 2007. More than one reason torethink the use of peptides in vaccine design. Nat. Rev. Drug Discov. 6,404.
Sathiamurthy, M., Peters, B., Bui, H.-H., Sidney, J., Mokili, J., Wilson, S., Fleri,W., McGuinness, D., Bourne, P. and Sette, A. (2005) An ontology forimmune epitopes: application to the design of a broad scope database ofimmune reactivities. Immunome Research 1, 2.
Scarselli, M., Giuliani, M.M., Adu-Bobie, J., Pizza, M., Rappuoli, R., 2005. Theimpact of genomics on vaccine design. Trends Biotechnol. 23, 84.
Scott, D.W., De Groot, A.S., 2010. Can we prevent immunogenicity of humanprotein drugs?, p. i72.
Sette, A., Fikes, J., 2003. Epitope-based vaccines: an update on epitopeidentification, vaccine design and delivery. Curr. Opin. Immunol. 15, 461.
Sidney, J., Peters, B., Frahm, N., Brander, C. and Sette, A. (2008) HLA class Isupertypes: a revised and updated classification. BMC Immunology 9, 1.
Tenzer, S., Peters, B., Bulik, S., Schoor,O., Lemmel, C., Schatz,M.M., Kloetzel, P.-M.,Rammensee, H.-G., Schild, H. and Holzhütter, H.-G. (2005) Modeling theMHC class I pathway by combiningpredictionsof proteasomal cleavage,TAPtransport and MHC class I binding. In, p. 1025.
Vita, R., Peters, B., Sette, A., 2008. The curation guidelines of the immuneepitope database and analysis resource. Cytometry 73A, 1066.
Vita, R., Zarebski, L., Greenbaum, J.A., Emami, H., Hoof, I., Salimi, N., Damle, R.,Sette, A., Peters, B., 2010. The Immune Epitope Database 2.0. Nucl. AcidsRes. 38, D854.
Wang, P., Sidney, J., Dow, C., Mothé, B., Sette, A. and Peters, B. (2008) ASystematic Assessment of MHC Class II Peptide Binding Predictions andEvaluation of a Consensus Approach. PLoS Comput Biol 4, e1000048.
Zhang, Q.,Wang, P., Kim, Y.,Haste-Andersen, P., Beaver, J., Bourne, P.E., Bui, H.-H.,Buus, S., Frankild, S., Greenbaum, J., Lund, O., Lundegaard, C., Nielsen, M.,Ponomarenko, J., Sette, A., Zhu, Z., Peters, B., 2008. Immune epitopedatabaseanalysis resource (IEDB-AR). Nucl. Acids Res. 36, W513.
Zvi, A., Ariel, N., Fulkerson, J., Sadoff, J., Shafferman, A., 2008. Whole genomeidentification of Mycobacterium tuberculosis vaccine candidates bycomprehensive data mining and bioinformatic analyses. BMC Med. Genet.1, 18.