aristacat - information services division · 16. reference list ... concentration than that...
TRANSCRIPT


ARISTACAT
Chief Investigator Professor David Cameron
Edinburgh Cancer Centre, Western General Hospital Crewe Road South Edinburgh, EH4 2XU Tel: 0131 777 3512 Fax: 0131 777 3520 Email: [email protected]
Co-Investigator
Professor Peter Barrett-Lee Velindre Cancer Centre Velindre Road, Whitchurch Cardiff, CF14 2TL Tel: 029 2061 5888 Fax: 029 2019 6835 Email: [email protected]
Clinical Coordinator
Dr Stefan Symeonides Edinburgh Cancer Centre, Western General Hospital Crewe Road South Edinburgh, EH4 2XU Tel: 0131 537 1000 Fax: 0131 537 1029 Email: [email protected]
Sponsor
The Common Services Agency Gyle Square, 1 South Gyle Crescent, Edinburgh, EH12 9EB
Funder (partial funding)
AstraZeneca UK Limited 2, Kingdom Street London, W2 6BD
Senior Trial Coordinator
Eve Macdonald Gyle Square, 1 South Gyle Crescent, Edinburgh, EH12 9EB Tel: 0131 275 7058 Fax: 0131 275 7512 Email: [email protected]
Trial Statistician Haiying Nan Gyle Square, 1 South Gyle Crescent, Edinburgh, EH12 9EB Tel: 0131 275 6226 Fax: 0131 275 7512 Email: [email protected]
__________________________________________________________________________
INFORMATION SERVICES
rdVersion 1.1; Date: 23 January 2012
Page 2 of 45

ARISTACAT
__________________________________________________________________________
INFORMATION SERVICES
Version 1.1; Date: 23rd January 2012
Page 3 of 45
Synopsis Protocol ID: ARISTACAT Protocol Title: ARomatase Inhibition plus/minus SaracaTinib as Advanced breast
CAncer Therapy (ARISTACAT): a randomised phase II study of aromatase inhibition plus/minus the Src-inhibitor, saracatinib (AZD0530) in post-menopausal women with advanced breast cancer.
Development Phase: Randomised phase II Primary Endpoint: Comparison of progression free survival between cohort receiving
aromatase inhibition plus saracatinib, versus those receiving aromatase inhibition plus placebo.
Secondary Endpoints: Toxicity, response rate and overall survival. Translational sub-studies
also planned. Study Design: Multi-centre, placebo-controlled, double-blind, randomised phase II.
Post-menopausal women with advanced, incurable breast cancer, due to start first or second line hormonal treatment, will be randomised to receive aromatase inhibitor plus either saracatinib or placebo.
Patient Accrual: 140 patients will be recruited over a 2 year period at around 20 sites
in the UK. Final Analysis: The final analysis will be performed after 110 PFS events have
occurred or at minimum follow up of 6 months, whichever is later.

ARISTACAT
TABLE OF CONTENTS
1. INTRODUCTION..................................................................................................................7
1.1. BACKGROUND............................................................................................................................... 7 1.2. INVESTIGATIONAL MEDICINAL PRODUCT .................................................................................... 7 1.3. PRE-CLINICAL DATA .................................................................................................................... 7 1.4. CLINICAL DATA............................................................................................................................ 8 1.5. TRIAL RATIONALE ........................................................................................................................ 9
2. TRIAL OBJECTIVES .........................................................................................................10
3. TRIAL DESIGN ..................................................................................................................10
3.1. GENERAL DESIGN ....................................................................................................................... 10 3.2. INCLUSION CRITERIA.................................................................................................................. 10 3.3. EXCLUSION CRITERIA................................................................................................................. 11 3.4. RANDOMISATION CODES ............................................................................................................ 11 3.5. TRANSLATIONAL RESEARCH ...................................................................................................... 11 3.6. WITHDRAWAL OF SUBJECTS....................................................................................................... 12 3.7. ENDPOINTS ................................................................................................................................. 12
4. TREATMENT .....................................................................................................................12
4.1. TREATMENT SCHEDULE.............................................................................................................. 12 4.2. DOSE REDUCTION OR TOXICITY MODIFICATIONS...................................................................... 13 4.3. CONCOMITANT THERAPY ........................................................................................................... 14 4.4. COMPLIANCE WITH TREATMENT ................................................................................................ 18 4.5. DRUG SUPPLIES AND LABELLING............................................................................................... 18 4.6. DRUG STORAGE AND ACCOUNTABILITY .................................................................................... 19
5. ASSESSMENT OF EFFICACY..........................................................................................21
5.1. TREATMENT AND EXAMINATION SCHEDULE ............................................................................. 21 5.2. SCHEDULE OF ASSESSMENTS ...................................................................................................... 23
6. SUB-STUDIES ...................................................................................................................28
6.1. TRANSLATIONAL ........................................................................................................................ 28 6.2. BONE-ONLY METASTASES .......................................................................................................... 28
7. PHARMACOVIGILANCE...................................................................................................28
7.1. DEFINITIONS ............................................................................................................................... 28 7.2. EMERGING SAFETY PROFILE ....................................................................................................... 29 7.3. RECORDING AND REPORTING OF ADVERSE REACTIONS ............................................................ 29 7.4. RECORDING AND REPORTING OF SERIOUS ADVERSE EVENTS ................................................... 30 7.5. NON-REPORTABLE EVENTS........................................................................................................ 31 7.6. CODE BREAKING PROCEDURE.................................................................................................... 31 7.7. DEVELOPMENTAL SAFETY UPDATE REPORT.............................................................................. 31 7.8. PREGNANCIES ............................................................................................................................. 31
8. DATA MANAGEMENT ......................................................................................................32
8.1. DATA COLLECTION..................................................................................................................... 32 8.2. RECORD KEEPING AND ARCHIVING............................................................................................ 32
9. STATISTICS ......................................................................................................................32 __________________________________________________________________________
INFORMATION SERVICES
rdVersion 1.1; Date: 23 January 2012
Page 4 of 45

ARISTACAT
__________________________________________________________________________
INFORMATION SERVICES
Version 1.1; Date: 23rd January 2012
Page 5 of 45
9.1. SAMPLE SIZE............................................................................................................................... 32 9.2. RANDOMISATION AND STRATIFICATION .................................................................................... 33 9.3. ANALYSIS PLAN.......................................................................................................................... 33 9.4. END OF STUDY............................................................................................................................ 34
10. ACCESS TO SOURCE DATA/ DOCUMENTS ................................................................34
11. QUALITY CONTROL AND QUALITY ASSURANCE......................................................34
11.1. MONITORING VISITS................................................................................................................. 34 11.2. DATA MONITORING AND ETHICS COMMITTEE......................................................................... 34 11.3. TRIAL STEERING COMMITTEE .................................................................................................. 34
12. ETHICAL CONSIDERATIONS ........................................................................................34
12.1. PATIENT CONFIDENTIALITY ..................................................................................................... 35 12.2. INFORMED CONSENT ................................................................................................................ 35
13. RESEARCH GOVERNANCE...........................................................................................36
13.1. TRIAL ORGANISATION .............................................................................................................. 36
14. FINANCING AND INSURANCE ......................................................................................37
15. PUBLICATION POLICY ..................................................................................................37
16. REFERENCE LIST ..........................................................................................................38
Appendix 1 – Causality ............................................................................................................. 40 Appendix 2 – Efficacy Evaluation – RECIST Criteria ............................................................. 41 Appendix 3 – Investigator Statement (CSA Copy) ................................................................... 41 Appendix 4 – Investigator Statement (Investigator Copy)........................................................ 43 Please refer to attached documents: .......................................................................................... 44 Appendix 5 – Patient Information Sheet & Informed Consent Form (Main Trial & Bone Resorption sub-study) ............................................................................................................... 44 Appendix 6 – Patient Information Sheet & Informed Consent Form (Changes in the tumour sub-study) .................................................................................................................................. 44 Appendix 7 – The Principles of ICH Good Clinical Practice.................................................... 45

ARISTACAT
__________________________________________________________________________
INFORMATION SERVICES
Version 1.1; Date: 23rd January 2012
Page 6 of 45
GLOSSARY OF ABREVIATIONS AE Adverse Event AI Aromatase Inhibitor ALP Alkaline phosphatase ALT Alanine Amino Transferase AST Aspartate Amino Transferase AR Adverse Reaction CACTUS Cancer Clinical Trials Unit, Scotland CI Chief Investigator CRF Case Report Form CRP C-Reactive Protein CT Computed Tomography CTCAE Common Toxicity Criteria for Adverse Events CTAAC Clinical Trials Awards and Advisory Committee CTC Common Toxicity Criteria CTU Clinical Trials Unit CSA Common Services Agency DSMC Data Safety Monitoring Committee ECOG Eastern Cooperative Oncology Group EU European ECG Electrocardiogram FBC Full Blood Count GCP Good Clinical Practice GP General Practitioner GMP Good Medical Practice HER2 Human Epidermal growth factor Receptor 2 HRCT High resolution Computed Tomography IB Investigator Brochure ICH International Council for Harmonization ISD Information Services Division IMP Investigational Medicinal Product ISD Information Services Division IRB Institutional Review Board IEC Independent Ethics Committee IWRS Interactive Web Response System MHRA Medicines and Healthcare products Regulatory Agency MREC Main Research Ethics Committee NOAEL No Observed Adverse Effect Level PI Principal Investigator PK Pharmacokinetic RCC Renal Cell Carcinoma SAE Serious Adverse Event SDV Source Data Verification SOP Standard Operating Procedure SUSAR Suspected Unexpected Serious Adverse Reaction TMG Trial Management Group TSC Trial Steering Committee ULN Upper Limit of Normal

ARISTACAT
__________________________________________________________________________
INFORMATION SERVICES
Version 1.1; Date: 23rd January 2012
Page 7 of 45
1. INTRODUCTION
1.1. Background
Hormone receptor positive breast cancer in post-menopausal women is commonly treated with an aromatase inhibitor, both in the adjuvant and metastatic settings. However, most patients with metastatic disease, and a significant minority of patients with early breast cancer, develop resistance to this well-tolerated therapy. Ultimately, this leads to disease progression and, in many, premature death. There are many mechanisms by which this resistance develops but one route may well be via activation of Src, a non-receptor tyrosine kinase with multiple roles in malignant cell behaviour. Src activation can occur in up to 40% of ER+ breast cancers, and is involved in endocrine resistance, likely via accelerated proteolysis of p27 (1,2). Since p27 is a mediator of endocrine therapy-induced cell cycle arrest, loss of p27 reduces or abrogates the efficacy of endocrine therapies. 1.2. Investigational Medicinal Product
Saracatinib (AZD0530) is a potent, oral, inhibitor of the Src protein tyrosine kinase. Src itself was the first proto-oncogene to be described (3,4) and is now known to regulate multiple key signalling pathways involved in cell proliferation, differentiation, adhesion, migration, survival and angiogenesis. As a key regulator protein, Src is expressed ubiquitously. Platelets, osteoclasts and neural cells contain higher levels, with the essential role of Src in osteoclast function best described amongst these (5). In many malignant and premalignant tissues, including breast cancers (6,7), Src expression and activation is increased and has been linked with poor prognosis (8). Its interaction with multiple oncogenic pathways has implications for disease activity and in resistance to other therapies (9). 1.3. Pre-Clinical Data
Saracatinib is a potent and selective inhibitor of Src family kinases, with an IC50 of 0.0027 μM for Src and 0.004-0.011 μM for other Src family kinases (10). The only other kinases affected in the <1 μM range are v-Abl, EGFR, c-Kit & EphA2. Cell culture assays with single-agent Saracatinib have demonstrated growth inhibition at an IC50 of <1 μM in a number of cell lines. Other Src-regulated characteristics are also inhibited including adhesion, migration and invasion (11) and these may occur at a lower concentration than that required to inhibit proliferation. Similar results have been seen in exograft and explant models, with dose-dependent activity at doses up to 25mg/kg/day in one mouse model, and evidence that metastasis formation was reduced at lower doses (11, 12, 13, 14). Cell culture assays of saracatinib in combination with various chemotherapeutics have shown an increased anti-proliferative effect and provided evidence of restoring chemosensitivity in models of resistance. Similarly, and of particular interest, saracatinib has been found to enhance the anti-proliferative effect of tamoxifen, fulvestrant and anastrazole on breast cancer cell lines (15, 16). It prevented the development of a tamoxifen-resistant phenotype in MCF7 cells and restored the sensitivity of resistant cell lines. Again, similar results were seen in xenografts (16, 19). Osteoclasts natively express high levels of Src and 0.1-5 μM saracatinib had a dose-dependent effect on bone resorption in an in vitro assay of rabbit osteoclasts. Similarly, it

ARISTACAT
__________________________________________________________________________
INFORMATION SERVICES
Version 1.1; Date: 23rd January 2012
Page 8 of 45
reversibly inhibited the formation and activation of human osteoclasts (17). In a mouse model of prostate bony metastases, saracatinib inhibited osteoclast activity, reducing bone resorption and the development and progression of bone lesions (18). Pharmacokinetics and safety profiling were carried out in rats and dogs in preclinical tests. Pharmacokinetics were comparable to the subsequent human data. Safety testing showed that most toxicities were mild and reversible and included: anaemia (seemingly secondary to GI tract effects), lymphopenia, thrombocytopenia or thrombocytosis, histiocytosis, GI hypertrophy or degenerative changes (with effects on smooth muscle contractility implicated), mild renal impairment, transaminitis and bone changes. However sustained high doses caused dose-limiting toxicities including GI toxicity, bone changes and reversible multi-organ damage. The no observed adverse effect level (NOAEL) for 6 months treatment in rats was 2.5 mg/kg/day, with 20 mg/kg/day not tolerated (bone changes). The NOAEL for 6 months treatment in dogs was 0.5 mg/kg/day, with 10 mg/kg/day not tolerated (GI toxicity). Very high concentrations were required for cardiac electrophysiological disturbance and there was no evidence of mutagenic, clastogenic or phototoxicity potential. However, there was evidence of foetal toxicity. 1.4. Clinical Data
1.4.1. Safety Data
187 healthy volunteers and 81 patients have received saracatinib during the course of Phase I pharmacokinetic and safety studies completed to date. 12 further patients are receiving saracatinib in an ongoing Phase I study in a Japanese population. By the 20th of December 2010, 608 subjects (187 healthy volunteers & 421 patients) have received single or multiple once-daily oral doses of saracatinib in AstraZeneca-sponsored trials.
Human pharmacokinetics showed slow absorption and clearance, with 90% protein-bound in plasma and a large volume of distribution. Food did not affect bioavailability but there was considerable inter-patient variability in plasma levels for a given fixed daily dose. Bioavailability increased with regular once-daily dosage, leading to higher steady state levels when steady state is reach at around Day 10 - Day 12. Hepatic metabolism produces the predominant metabolite, N-desmethyl saracatinib, which retains some activity. However this reaches just 20% of the dose at steady state, while most saracatinib is excreted unmetabolised via biliary elimination (with 9-16% renally excreted). Other minor metabolites are also produced. Both saracatinib and its major N-desmethyl metabolite caused moderate CYP3A4 inhibition (as well as some CYP2D6 inhibition), with once-daily saracatinib 125mg increasing concentrations of midazolam to around 2-fold. There was no evidence of CYP3A4 induction. Both are also inhibitors of the OCT2 influx pump and BRCP. In healthy volunteers, 1000mg was found to be the maximum tolerated single dose, 250mg the maximum tolerated regular once-daily dose. For patients, 175mg was found to be the maximum tolerated once-daily dose, with grade 3 neutropenia a clear dose-limiting toxicity as well as other events which can be less confidently ascribed to the drug. The maximum tolerated dose in the ongoing Japanese trial is 125mg, with dose-limiting transaminitis. Reported toxicities were generally mild and included headache, diarrhoea, nausea, vomiting, papular rash, anaemia, transaminitis and flu-like symptoms (fever, myalgia, upper respiratory tract symptoms, raised CRP). However some of these, including the flu-like symptoms, were

ARISTACAT
__________________________________________________________________________
INFORMATION SERVICES
Version 1.1; Date: 23rd January 2012
Page 9 of 45
also seen in placebo cohorts. Specific effects included an increase in CTC grade 1 bleeding events (predominantly epistaxis) and positive urine dipstick protein or blood. No clinically relevant changes in blood pressure, heart rate, ECGs, pulmonary function tests, adrenal function, platelet aggregation or bleeding times were observed. A small number of serious pneumonitis-type events have been reported in patients with advanced cancer; however there were confounding factors in each case and the link is uncertain as yet. Analysis of blood tests revealed a trend to a mild dose-dependent reduction in neutrophil and platelet counts. There was also a mild, but reversible, dose-dependent rise in serum creatinine (by 5-30%), without a change in insulin clearance. This was due to reduced tubular creatinine secretion, with renal function unaffected (20). There was a trend to decreased serum oestradiol in male volunteers and a dose-dependent reduction in T-cell IL-2 production (presumably an effect on immune-cell restricted Src family kinases). The effects on osteoclast activity lead to a reduction in bone resorption markers (21), as well as a trend to mild hypocalcaemia or hypophosphataemia. Trials combining saracatinib with other agents have mainly identified additional toxicities that are amongst the generally recognised side-effects of the other agents. However, the addition of saracatinib may exacerbate hypokalaemia or hyponatraemia in patients receiving platinum agents. 1.4.2. Efficacy Data
To date, over 600 patients with solid malignancies have received saracatinib monotherapy or combination therapy in early phase clinical trials. Reported data suggests that there have been some clinically meaningful durations of stabilised disease with monotherapy, while some partial responses have been observed with combination therapies. Study D8180C00034 is currently ongoing, comparing saracatinib to Zoledronate in 139 bisphosphonate-naïve patients with bone metastases from breast or prostate cancer. While concomitant hormonal therapy was allowed, chemotherapy was not and a subset of these patients would be similar to the patient group being considered for the trial described in this protocol. Encouragingly, treatment is reported to have been well tolerated, with pharmacokinetics and toxicities similar to other studies. Furthermore, the pattern of bone turnover markers in the saracatinib group compared favourably with the Zoledronate group. This is particularly interesting in the light of newly-presented adjuvant data from the AZURE (Adjuvant Zoledronic Acid to Reduce Recurrence) trial which has shown a reduction in breast cancer recurrence rates in post-menopausal women given adjuvant Zoledronate (22). 1.5. Trial Rationale
Recent data suggest that Src-inhibition may help prevent the development of resistance to endocrine treatment in breast cancer. Despite the progress made in treating breast cancer by the introduction of both AIs and other endocrine agents such as fulvestrant, many women every year develop resistance to this endocrine therapy. Other than HER2-blockade, there are no other agents in the clinic that reverse endocrine resistance, nor prevent its occurrence. Chemotherapy is used to deliver additional clinical benefits for patients with ER+ breast cancer but comes with its own set of risks and eventual relapse with chemotherapy-resistant disease as inevitable. New approaches are required if survival rates are to improve. Prolonging the effectiveness of endocrine treatment could yield significant benefits for this group of patients.

ARISTACAT
__________________________________________________________________________
INFORMATION SERVICES
Version 1.1; Date: 23rd January 2012
Page 10 of 45
2. TRIAL OBJECTIVES
The aim of this study is to seek evidence that the addition of a potent Src-inhibitor, AZD0530, to conventional aromatase inhibition, delays the onset of endocrine resistance in post-menopausal women with advanced, incurable breast cancer. 3. TRIAL DESIGN
3.1. General Design
This is a double blind randomised phase II study. Patients will be randomised to receive aromatase inhibitor and saracatinib (AZD0530) or aromatase inhibitor and placebo. Patients will be assessed at the same frequency in both arms and progression will be assessed against RECIST 1.1 criteria. Please refer to Appendix 2 for RECIST 1.1 guidelines. Patients will be enrolled into one of two strata: an “AI-sensitive/naïve” group of patients with potentially AI-sensitive tumours, and a second “prior-AI” group of women whose cancers have already progressed on an AI, but for whom there is likely to still be some endocrine sensitivity. 3.2. Inclusion Criteria
1. Females who are clearly post menopausal with ER positive (Allred score ≥ 3) advanced, incurable breast cancer with at least one lesion which is measurable. They may also have additional evaluable but non-measurable lesions. (Patient is post-menopausal as per the indications for aromatase inhibition. Where there is doubt about postmenopausal status clinically, this should be determined biochemically (luteinizing-hormone [LH] follicle stimulating hormone [FSH], and/or oestradiol levels).
2. Patients must be performance status 0 – 2. 3. Suitable for treatment with an aromatase inhibitor. 4. Life expectancy > 3 months. 5. Cancer must be HER2- (by FISH and/or IHC as appropriate), OR if the cancer is HER2+
the patient must not be a candidate for anti-HER2 therapy. 6. All patients will need to also meet inclusion criteria for one of the two main strata:
a. “AI-sensitive/naive” group – either never previously treated with an aromatase inhibitor, but if treated with tamoxifen must not have rapid progression on tamoxifen (i.e. treated for at least 24 months adjuvant or ≥ 6 months in metastatic setting); or, if previously treated with an AI, only in the adjuvant or neo-adjuvant setting AND have remained free of progression for at least 12 months whilst not being treated with an AI.
b. “prior AI” group – patients NOT meeting the criteria in 6 (a) above, but
previously treated with a non-steroidal AI without progression for at least 24 months in the (neo-) adjuvant setting or for at least 6 months for advanced disease.
7. Patients who have had two lines of prior AI therapy will not be eligible UNLESS they were switched from one AI to another ONLY for reasons of toxicity, and ONLY during (neo-) adjuvant therapy AND in the absence of any evidence of progression/relapse.
8. Single site of bone disease must be histologically confirmed and known not to be ER negative.
9. Palliative radiotherapy can be given to bone lesions within 4 weeks of trial entry provided not more than 20% of the bone marrow is irradiated, AND there is at least one other measurable bone lesion which has clearly progressed since any prior irradiation.

ARISTACAT
__________________________________________________________________________
INFORMATION SERVICES
Version 1.1; Date: 23rd January 2012
Page 11 of 45
10. Haematology – commensurate with a phase II hormonal therapy study: Neutrophils > 1.5 * 109/l, Hb> 10.0 g/dl and Platelets > 100 * 109/l
11. Biochemistry – similar: albumin normal, ALT/AST < 2.5 ULN, Alk Phos < 5 * ULN unless of bone origin, e-GFR > 50ml/min. Normal urea & electrolytes. (If e-GFR is not routinely used at site then please contact the Aristacat trial team who can provide a formula to calculate e-GFR).
12. Patients receiving bisphosphonates are eligible, provided they are commenced before, or at, trial entry. Patients will be stratified by use of, or stated intention to give, bisphosphonate at randomisation. Patients ideally should have been on therapy for at least 1 week before starting trial therapy, but must start within 1 week after starting trial therapy.
13. Aged ≥18 years (no upper age limit). 3.3. Exclusion Criteria
1. Patients with short life expectancy or significant co-morbidity from other severe or uncontrolled systemic conditions (e.g. interstitial lung disease [bilateral, diffuse, parenchymal change])
2. Rapidly progressive visceral disease (lymphangitis, diffuse liver disease, uncontrolled CNS disease).
3. Resting ECG with a measureable QTc >480 msec. 4. Life expectancy < 3 months. 5. Contra-indication to either AZD0530 (or excipients) or aromatase inhibition. 6. Concomitant chemotherapy or anti-HER2 therapy. 7. Patients requiring medications that are contra-indicated in combination with AZD0530
due to significant CYP3A4 interaction (Section 4.3.) 8. Unable or unwilling to give informed consent. 9. Pregnant and/or lactating. 3.4. Randomisation Codes
After ensuring that the patient meets all eligibility criteria and has consented to participate in the study, please complete the randomisation form and phone the Information Services Division (ISD) Cancer Clinical Trials team using the randomisation line below.
ISD CANCER CLINICAL TRIALS TEAM
RANDOMISATION LINE: 0131 275 7276 OR: 0131 316 4278
RANDOMISATION FAX: 0131 275 7512
Subjects will be randomised and ISD Cancer Clinical Trials Team will appoint a patient identification number which should be used in all correspondence. 3.5. Translational Research
There are two translational aspects associated with the study. The first optional study relates to changes in the tumour. In patients for whom accessible tumour material is available, samples will be taken at Screening and their Week 6 visit. These samples will be stored and subsequently compared with response to the AI and AI+ saracatinib therapies in order to

ARISTACAT
__________________________________________________________________________
INFORMATION SERVICES
Version 1.1; Date: 23rd January 2012
Page 12 of 45
determine if any of the preclinical changes that are induced by Src-inhibition can (a) be identified in human tumours and (b) correlate with/ predict for benefit. The second mandatory translational study will look at pre-and on-treatment markers of bone resorption to test the hypothesis that Src-inhibition in bone can additionally reduce bone turnover in patients with metastatic bone disease. For all patients, irrespective of the presence of bone metastases, baseline blood samples will be collected to measure markers of bone turnover (synthesis and resorption). Sampling will be repeated at Week 6, Week 24 and at discontinuation of study IMP/placebo. 3.6. Withdrawal of Subjects
Treatment with study drug should be discontinued if it is considered to be in the best interest of the patient. Reasons for treatment discontinuation include:
Disease progression Occurrence of intolerable side effects Patient withdrawal of consent or non-compliance
Discontinuation Patients discontinued from the study for reasons such as safety, non-compliance or withdrawal of consent, etc. These patients will continue to be followed up for efficacy and safety as per protocol, unless they are lost to follow up, deceased or withdrew consent to the study. In addition all patients randomised are to be included in the ITT analysis set and are evaluable for efficacy (per the ITT principle). All patients who received at least 1 dose of study drug/placebo are to be included in the safety analysis set and are evaluable for safety. Patients may withdraw from study at any point, with no further data collection will take place, and will be censored at the point of withdrawal. The reason for patient withdrawal should be included in the CRFs. The “Intent to Treat” analysis will include all patients, regardless of whether they have received study drug or not. 3.7. Endpoints
Primary: Progression free survival. Time to progression will be measured through standard,
regular, clinical assessment. Secondary:
Toxicity Change in tumour size analysed using a Waterfall plot in the two strata separately Overall survival Response rate
4. TREATMENT
4.1. Treatment Schedule
Patients will be enrolled into one of two strata: an “AI-sensitive/naïve” group of patients with potentially AI-sensitive tumours, and a second “prior-AI” group of women whose cancers

ARISTACAT
__________________________________________________________________________
INFORMATION SERVICES
Version 1.1; Date: 23rd January 2012
Page 13 of 45
have already progressed on an AI, but for whom there is likely to still be some endocrine sensitivity. For women in the “AI-sensitive/naïve” group, treatment would be with anastrazole 1mg daily ± saracatinib 175 mg daily. For women in the “prior AI” group, treatment would be exemestane 25mg daily ± saracatinib 175 mg daily. Saracatinib (AZD0530) is an oral Src inhibitor and can be administered with or without food. Allocation to treatment with either aromatase inhibitor plus placebo, or aromatase inhibitor plus saracatinib (AZD0530), will be performed using a minimisation algorithm. Stratification factors will be: AI sensitivity strata, disease site (bone metastases alone v any other sites), bisphosphonate use, performance status (0 v 1 v 2) and treatment centre. Randomisations will be obtained by contacting CSA, Edinburgh. Treatment will then continue until one of the following occurs: progression of disease, toxicity (requiring either dose reduction or termination of study drug) or patient choice. It is estimated that at the 3 year point there will be fewer than 10% of patients still on treatment. Patients will be followed up whilst on therapy for efficacy and toxicity until the later of either progression or 30 days after cessation of therapy. Thus patients who progress on therapy will be followed up for a further 30 days – patients who come off therapy because of toxicity before progression will be followed up only to the date of progression. Beyond that point, patients will be tracked for overall survival only. If a patient forgets to take a tablet, and it is within 6 hours of the scheduled time then the patient should be advised to take them as soon as possible. If it is more than 6 hours after the scheduled time, then study medication should not be taken for that day. Study medication should continue as scheduled previously on the subsequent day. A patient should not take more than a single day’s dose of tablets, within a day. In the event that the patient cannot hold the tablet(s) down (if the patient vomits) within 30 minutes from taking the tablet(s) or if can identify the tablet (s) in the vomit content, the patient can re-take new tablet(s) from the bottle(s). 4.2. Dose Reduction or Toxicity Modifications
Adverse events most commonly associated with single agent saracatinib (≥20% of the study population irrespective of reported causality) were: anaemia (37%), nausea (37%), anorexia (36%), asthenia (32%), pyrexia (27%), vomiting (27%), and diarrhoea (26%). Management of toxicity and overview of dose reductions The following general guidance should be followed for management of toxicities and dose reduction.
Treat each of the toxicities with maximum supportive care (including withholding the agent suspected of causing the toxicity where required), according to local protocols.
If the symptoms promptly resolve with supportive care, consideration should be given to continuing the same dose of study medications along with appropriate continuing supportive care. If medically appropriate, dose reductions are permitted (see below).
All dose changes should be documented with clear reasoning and documentation of the approach taken.

ARISTACAT
__________________________________________________________________________
INFORMATION SERVICES
Version 1.1; Date: 23rd January 2012
Page 14 of 45
An interruption in saracatinib / placebo treatment is recommended in the presence of any CTCAE grade 3 toxicity, or any grade 2 toxicity that persists for greater than 1 week despite symptomatic management. Treatment should then be restarted at a dose reduction (Table 1). Treatment should be ceased after any grade 4 toxicity.
However, if the investigator concludes that toxicity is clearly related to the concomitant endocrine therapy (typical arthralgia, flushing, etc), saracatinib / placebo treatment need not be adjusted.
Table 1 Dose Reduction Information
Dose Step
Saracatinib
Starting dose 175 mg
1st reduction 125 mg
2nd reduction 50 mg
Study drug dosing may be withheld for up to 21 days for management of toxicity. If a longer interruption is required due to unresolved toxicity, study drug should be discontinued and the patient comes off study, with subsequent endocrine therapy to be continued as the physician and patient decide. Study drug should also be withheld if the patient requires a contraindicated concomitant medication (see 4.3) or if they are to undergo anything other than very minor surgery. Interstitial lung disease
A small number of pneumonitis-type events (including fatal outcomes) have been reported in patients with advanced cancer during treatment with saracatinib. There were confounding features for each event, including pulmonary metastatic disease, co-morbidities and/or use of concomitants medications with known association with pneumonitis; however saracatinib cannot be excluded as a cause for these pneumonitis-type events. Therefore all patients are required to have a high-resolution CT image of their thorax at baseline. In the event of any new or worsening respiratory symptoms (e.g. dyspnoea and/or cough) and/or demonstration of new pulmonary radiological finding consistent with interstitial lung disease a high resolution CT should be performed along with respiratory function tests. Study drug should be stopped and contact with the clinical trials centre and PI should take place. 4.3. Concomitant Therapy
In vitro and in vivo studies indicate that saracatinib is metabolised mainly by CYP3A4 and produces slight competitive inhibition of this enzyme at concentrations of 30μg/mL and above, in vitro. It is advised to exercise caution with concomitant medications that are significantly metabolised by CYP3A4 or which significantly affect CYP3A4 activity themselves. Table 2 lists drugs that are potent inhibitors of CYP3A4 and should not be given while on saracatinib / placebo. Similarly Table 3 lists potent inducers of CYP3A4 and Table 5 lists drugs that are

ARISTACAT
__________________________________________________________________________
INFORMATION SERVICES
Version 1.1; Date: 23rd January 2012
Page 15 of 45
significantly metabolised by CYP3A4. These drugs should also not be given while on saracatinib / placebo. A record will be made at baseline to document if the patient is currently taking any of the medications from Tables 2 to 6 and this should be updated at each follow-up visit. If it is imperative that a patient needs to take a drug from Tables 2, 3 or 5, then the study drug should be withheld during this period.
Drugs affecting CYP3A4 metabolism which should not be combined with study drug
Table 2 Potent CYP3A4 inhibitors may increase exposure to study drug more
than 3-fold
Ketoconazole
Ritonavir
Saquinavir
Indanavir
Nefazodone
Minimum of 48 hours washout prior to study drug
administration and must avoid for the duration of
the study and for 14 days following discontinuation
of study drug
Itraconazole
Clarithromycin (250mg or 500mg bd)
Erythromycin
Fluconazole 400mg
Minimum of 7 days washout prior to study drug
administration and must avoid for the duration of
the study and for 14 days following discontinuation
of study drug
Diltiazem
Minimum of 14 days washout prior to study drug
administration and must avoid for the duration of
the study and for 14 days following discontinuation
of study drug
Table 3 Potent inducers of CYP3A4 may reduce exposure to study drug by
more than 3-fold
Barbiturates
Carbamazepine
Phenytoin
Rifampicin, Rifabutin
St John’s Wort
Minimum of 14 days washout prior to study drug
administration and must avoid for the duration of
the study and for 14 days following discontinuation
of study drug

ARISTACAT
__________________________________________________________________________
INFORMATION SERVICES
Version 1.1; Date: 23rd January 2012
Page 16 of 45
There are currently no data confirming that there is a pharmacokinetic (PK) interaction between these agents and saracatinib; however, a potential interaction is considered on the basis of the current knowledge of the PK profile of saracatinib. This list is not intended to be exhaustive, and a similar restriction will apply to other agents that are known to strongly modulate CYP3A4 activity. Appropriate medical judgment is required. Please contact AstraZeneca with any queries you have on this issue.
Drugs affecting CYP3A4 metabolism that may allowed with caution
Table 4 Moderate Inhibitors of CYP3A4 may increase exposure to saracatinib
Warning of possible interaction:
Verapamil
Nelfinavir
Fluconazole <400mg
Drugs are permitted but caution should be
exercised and patients monitored closely for
possible drug interactions. Please refer to full
prescribing information for all drugs prior to co-
administration with study drug
Grapefruit juice
Seville oranges (and other products
containing Seville oranges)
Patients should abstain from eating large amounts
of grapefruit and Seville oranges (and other
products containing these fruits e.g., grapefruit
juice or marmalade) during the study (e.g., no
more than a small glass of grapefruit juice (120
mL) or half a grapefruit or 1-2 teaspoons (15 g) of
Seville orange marmalade daily).

ARISTACAT
__________________________________________________________________________
INFORMATION SERVICES
Version 1.1; Date: 23rd January 2012
Page 17 of 45
Medicines that are significantly metabolised by CYP3A4 that should not be used with study drug
Table 5 Exposure, pharmacological action and toxicity may be increased by
inhibition of CYP3A4 by study drug
Alfentanil
Cyclosporin
Tacrolimus
Atorvastatin
Lovastatin
Simvastatin
Minimum of 7 days washout prior to study drug
administration and must avoid for the duration of
the study and for 14 days following discontinuation
of study drug
Carbamazepine
Bepridil
Lidocaine
Minimum of 14 days washout prior to study drug
administration and must avoid for the duration of
the study and for 14 days following discontinuation
of study drug
Amiodarone Minimum of 6 month washout prior to study drug
administration and must avoid for the duration of
the study and for 14 days following discontinuation
of study drug.
There are currently no data confirming that there is a pharmacokinetic (PK) interaction between these agents and saracatinib; however a potential interaction is considered on the basis of the current knowledge of the PK profile of saracatinib. This list is not intended to be exhaustive, and a similar restriction will apply to other agents with narrow therapeutic windows that are known to depend on CYP3A4 for metabolism. Appropriate medical judgment is required. Please contact AstraZeneca with any queries you have on this issue.
Medicines that are significantly metabolised by CYP3A4 that may be allowed with caution

ARISTACAT
__________________________________________________________________________
INFORMATION SERVICES
Version 1.1; Date: 23rd January 2012
Page 18 of 45
Table 6 Exposure, pharmacological action and toxicity may be increased by
inhibition of CYP3A4 by study drug
Warning of possible interaction:
Alprazolam
Midazolam
Triazolam
Felodipine
Isradipine
Nifedipine
Amlodipine
and possibly other calcium antagonists
Methylprednisolone
Pimozide
Quinidine
Drugs are permitted but caution should be
exercised and patients monitored closely for
possible drug interactions. Please refer to full
prescribing information for all drugs prior to co-
administration with saracatinib.
Caution should also be exercised in concomitant use of any medication that may affect hepatic cytochrome P450 drug metabolising activity by way of enzyme induction (e.g., phenytoin) or inhibition (e.g., ketoconazole, ritonavir, erythromycin) within 2 weeks of the first dose of saracatanib and throughout the study period. 4.4. Compliance with Treatment
Patients will be required to return all bottles of study medication at their Week 2, 6, 12, 18, 24 (then 3 monthly to 18 months & 6 monthly thereafter) and when they discontinue from taking the study drug/placebo. The number of missed doses should be recorded on the CRF and the number of tablets remaining in each bottle should be documented in the pharmacy log. Date of treatment, dose, delays and reasons for delays of all study treatments will be recorded. Non-compliance Patients who interrupt study drug intake for greater than 21 days, without direction from the doctors, will be considered as non-compliant and will be discontinued from the study. These patients will be included in the safety and efficacy assessments. 4.5. Drug Supplies and Labelling
All study drug supplied by AstraZeneca will be packaged and labelled in accordance with local regulations and Good Manufacturing Practice, stating that the drug is for clinical use only and should be kept out of the reach and sight of children. The drugs will be labelled, QP released and distributed by Fisher Clinical Services. Supply will be managed by the IWRS. The packaging and tablets will appear identical for both active and matching placebo treatments. The label attached to each package of blinded study material will have a unique treatment kit number that is linked to the randomisation scheme.

ARISTACAT
__________________________________________________________________________
INFORMATION SERVICES
Version 1.1; Date: 23rd January 2012
Page 19 of 45
Each centre will be provided with kits of packaged drugs. The IWRS will allocate the appropriate kit number from those available at the centre. Patients enrolled in the study will be dispensed bottles saracatinib/placebo tablets as determined by the IWRS and randomisation scheme. A detachable tear-off label will be affixed to each container and will contain space for centre number, patient number and date of dispensing to be completed and attached to the patient drug dispensing log located in the pharmacy pack at the time of dispensing. Patients will be supplied with sufficient medication for each visit. There will be sufficient tablets in the bottle to cover the visit window. 4.6. Drug Storage and Accountability
Storage All investigational products must be kept in a secure place under appropriate storage conditions. A description of the appropriate storage and shipment conditions are specified on the investigational labels. Study treatment must be kept out of the reach and sight of children. Accountability The investigator or a delegated individual (e.g. pharmacist) must ensure that the study drug is stored and dispensed in accordance with hospital standard operating procedures and applicable regulatory requirements. The medication provided for this study is for use only as directed in the protocol. Drug distribution and accountability logs will be provided to the site in a pharmacy pack. It is the investigator’s responsibility to establish a system for handling the investigational product to ensure that:
Deliveries of investigational products from AstraZeneca via Fisher Clinical Services are correctly received by a responsible person (e.g., pharmacist or suitable pharmacy designee) and are handled and stored correctly and safely
Investigational products are dispensed only to study participants, and in accordance with the protocol
Participants return any unused investigational product and all empty containers to the investigator
A dispensing record (which will include the identification of the participant to whom the investigational product was dispensed, the date of dispensing, the quantity of investigational product dispensed, and the date and quantity of any unused investigational product returned to the pharmacy) is accurately maintained. Any discrepancies must be accounted for on the appropriate form.
In the case that any study drug is damaged, please contact the CTU for reconciliation and replacement. At the termination of the study or at the request of the sponsor, all unused drugs will be accounted for and destroyed locally at the study sites. Certificates of delivery and destruction or return must be signed and copies retained in the Investigator Site File.

ARISTACAT
__________________________________________________________________________
INFORMATION SERVICES
Version 1.1; Date: 23rd January 2012
Page 20 of 45
Accountability records must be completed and any study drug remaining at the end of the trial must be destroyed according to the sites local standard procedures.
ARISTACAT
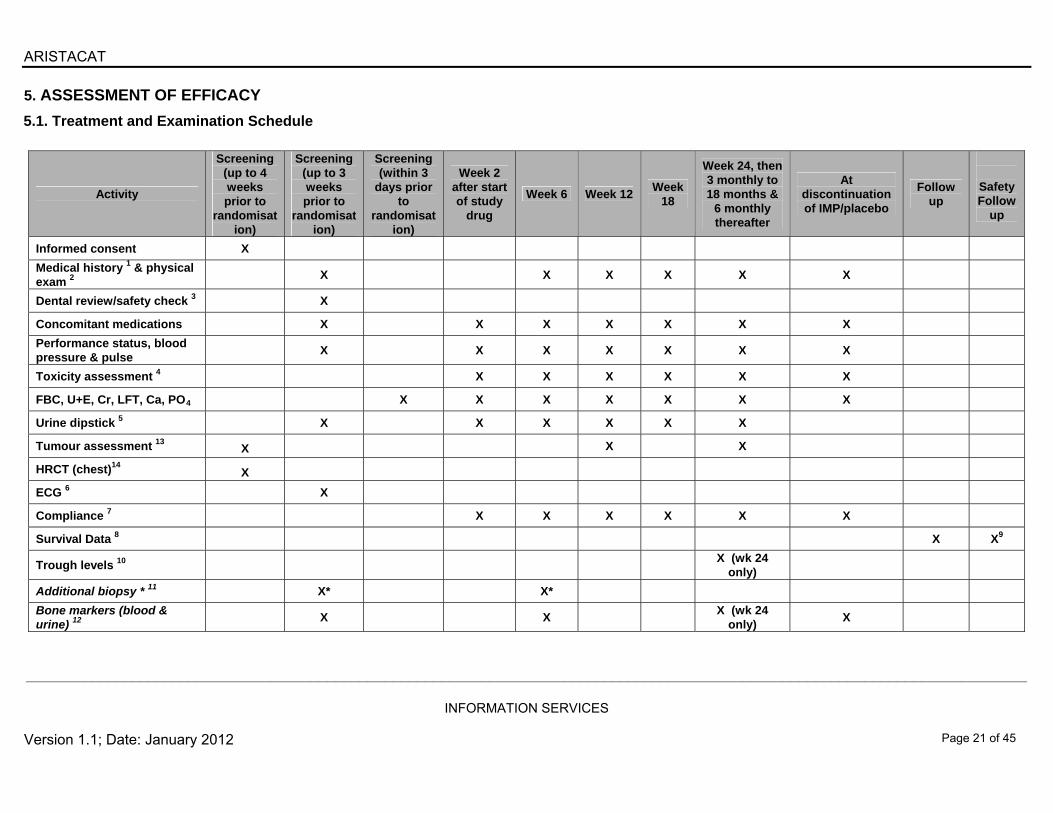
5. ASSESSMENT OF EFFICACY
5.1. Treatment and Examination Schedule
Activity
Screening (up to 4 weeks prior to
randomisation)
Screening (up to 3 weeks prior to
randomisation)
Screening (within 3
days prior to
randomisation)
Week 2 after start of study
drug
Week 6 Week 12 Week
18
Week 24, then 3 monthly to 18 months &
6 monthly thereafter
At discontinuation of IMP/placebo
Follow up
Safety Follow
up
Informed consent X
Medical history 1 & physical exam 2
X X X X X X
Dental review/safety check 3 X
Concomitant medications X X X X X X X
Performance status, blood pressure & pulse
X X X X X X X
Toxicity assessment 4 X X X X X X
FBC, U+E, Cr, LFT, Ca, PO4 X X X X X X X
Urine dipstick 5 X X X X X X
Tumour assessment 13
X X X
HRCT (chest)14
X
ECG 6 X
Compliance 7 X X X X X X
Survival Data 8 X X9
Trough levels 10 X (wk 24
only)
Additional biopsy * 11 X* X*
Bone markers (blood & urine) 12
X X X (wk 24
only) X
___________________________________________________________________________________________________________________________
INFORMATION SERVICES
Version 1.1; Date: January 2012
Page 21 of 45

ARISTACAT
___________________________________________________________________________________________________________________________
INFORMATION SERVICES
Version 1.1; Date: January 2012
Page 22 of 45
1. Full medical history, including history of breast cancer diagnosis & treatment, other diseases (active or resolved) concomitant illnesses and existing signs and symptoms. 2. Physical examination to include record of height & weight. 3. Routine dental review as a baseline safety check (ideally by patient’s own dentist), unless patient has already had a satisfactory dental review in the 6 months prior to randomisation. 4. CTCAE version 4.0 to be used when grading toxicities. 5. If a patient has a change of two plus (++) from baseline on two consecutive urine protein dipstick measurements, please measure urine protein : creatinine ratio or collect a 24-hour
urine for total protein. 6. ECG: Only at screening 7. Tablet bottle(s) including any unused tablets must be returned to the clinic for drug accountability. 8. Follow-up survival information will be collected for all patients by clinic visit or telephone contact every 3 months. 9. An additional safety visit will be performed 28 days after receiving final study medication/placebo. 10. Trough levels to be performed at specific named sites only. Weight to be repeated. 11. This biopsy would be purely for research purposes. The additional biopsy can be taken from consenting patients if the tissue is easily accessible without requiring CT (for example
lymph nodes). A formalin-fixed paraffin embedded block is required: additional snap frozen material to be stored (locally) at -80C and to be used for future research. Patients entering the main study are under no obligation to enter this part of the study.
12. These samples are for all patients, to look at pre- and on-treatment markers of bone resorption. 13. Measurable lesions are to be assessed 3-monthly for the first 18-months and 6-monthly thereafter. THE SAME technique must be used, which can be CT/MRI/plain
radiology/ultrasound or clinical examination as appropriate 14. Baseline High resolution assessment of the thorax is required in all patients for safety purposes. This can be done by reconstruction from a standard CT chest/abdo/pelvis, and this
same scan can be used as the baseline for subsequent tumour assessments if CT is an appropriate form of assessment. T* Represents optional translational research.

ARISTACAT
5.2. Schedule of assessments
5.2.1. Screening procedures
Screening procedures within 4 weeks prior to randomisation
Clinical assessments o Signed informed consent
Radiological assessment o CT chest, abdomen & pelvis, including HRCT chest
Screening procedures within 3 weeks prior to randomisation
Clinical assessments o Complete medical history, including breast cancer diagnosis and previous
treatment, history of other diseases (active or resolved), concomitant illnesses and medications (especially those interacting via CYP3A4)
o Toxicity/symptoms evaluation (CTCAE grading version 4) o Physical examination including blood pressure & pulse o ECOG performance status o Dental review and safety check (ideally by patient’s own dentist), unless
patient has already had a satisfactory dental review in the 6 months prior to randomisation.
Laboratory determinations
o FBC, U+E, Cr, LFT, Ca, PO4 o Urine dipstick. If a patient has a change of two plus (++) from baseline on two
consecutive urine protein dipstick measurements, please measure urine protein : creatinine ratio or collect a 24-hour urine for total protein.
Cardiac assessment
o 12 lead ECG
Translational aspects – Additional biopsy (optional) o Signed informed consent. o Additional tissue collection. This additional biopsy would be purely for
research purposes. The additional biopsy can be taken from consenting patients if the tissue is easily accessible without requiring CT (for example lymph nodes). This tissue will be formalin fixed paraffin embedded, and if the site has local approved storage (ECMC/HTA etc.) then additional snap frozen material to be stored at -80C can also be taken and will be used for future research. Patients entering the main study are under no obligation to enter this tissue collection part of the study.
Translational aspects – Bone markers (mandatory)
o Signed informed consent (there is an additional section to sign in the main trial informed consent form for this sub-study)
o Blood samples for bone resorption markers in all patients. Screening procedures within 72 hours prior to randomisation
__________________________________________________________________________
INFORMATION SERVICES
rdVersion 1.1 ; Date: 23 January 2012
Page 23 of 45

ARISTACAT
Laboratory Determinations o FBC, U+E, Cr, LFT, Ca, PO4
5.2.2. Randomisation
The participant’s research nurse and/or doctor will screen the participant to ensure that they meet the trial eligibility criteria, after obtaining participant consent for any additional trial screening procedures. The research nurse will then contact ISD-CCTT using the randomisation telephone number provided in section 3.4. ISD-CCTT will randomise the patient through the Sealed Envelope randomisation system. The Sealed Envelope system will require confirmation of the eligibility criteria before allocating a Drug Code and Patient Identifier. The research nurse will then log onto the Interactive Web Response System (IWRS) and complete the Patient Randomisation Confirmation Form using the Patient Identifier and Drug Code. Each user that requires access to the IWRS will receive their login details and a user guide once the site has been activated in the system Randomisation can only be performed once the participant has signed the consent form. The packaging and tablets will appear identical for both active and matching placebo treatments. The label attached to each package of blinded study material will have a unique treatment kit number that is linked to the randomisation scheme. Each centre will be provided with kits of packaged drugs. The IWRS will allocate the appropriate kit number from those available at the centre. The randomisation is centralised, and the assigned randomisation number and associated kit numbers will not be sequential within a centre. If a patient is given the incorrect kit, the patient should continue to take the medication they have been allocated. The site must inform the IWRS immediately after the error is identified. At randomisation, the participant will be given a unique participant trial number which should be recorded on the participant randomisation form and the top copy returned to the CTU within four weeks. The centre will inform the participant’s General Practitioner (GP) of the participant’s enrolment, if the participant gives consent to do so. It may be possible for participants to be recruited into other clinical trials, but this should be discussed with the CI via the CTU before this is considered. A minimum of 4 weeks is required following last study drug administration before a patient may be recruited into another trial. Patients should start study drug within 72 hours of randomisation 5.2.3. Assessment at week 2 (+/-3 days)
Clinical assessment o Toxicity assessment including ECOG performance status, blood pressure and
pulse. o History of any changes to concomitant medications
__________________________________________________________________________
INFORMATION SERVICES
rdVersion 1.1 ; Date: 23 January 2012
Page 24 of 45

ARISTACAT
Laboratory determinations
o Haematology and biochemistry (FBC, LFT, U+E, Cr, Ca, PO4) o Urine dipstick. If a patient has a change of two plus (++) from baseline on two
consecutive urine protein dipstick measurements, please measure urine protein : creatinine ratio or collect a 24-hour urine for total protein.
5.2.4. Assessment at week 6 (+/-1 week)
Clinical assessment o Physical examination including blood pressure & pulse o Toxicity assessment including ECOG performance status, blood pressure and
pulse. o History of any changes to concomitant medications o Compliance evaluation (tablet bottle(s) including any unused tablets must be
returned to the clinic for drug accountability at the end of each cycle).
Laboratory determinations o Haematology and biochemistry (FBC, U&E, LFT, Ca, PO4) o Urine dipstick. If a patient has a change of two plus (++) from baseline on two
consecutive urine protein dipstick measurements, please measure urine protein : creatinine ratio or collect a 24-hour urine for total protein.
Translational research (+/-7 days)
o Blood & urine samples for bone resorption markers in all patients o Additional tissue collection. This additional optional biopsy would be purely for
research purposes. The additional biopsy can be taken from consenting patients if the tissue is easily accessible without requiring CT (for example lymph nodes). One part will be formalin-fixed paraffin embedded, but if there are locally approved storage facilities (ECMC/ HTA approved etc.) then additional tissue will taken and snapfrozen at -80C and will be used for future research. Patients entering the translational component of this study are under no obligation to enter this tissue collection part of the study.
5.2.5. Assessment at week 12 (+/- 2 weeks)
Clinical assessment o Physical examination including blood pressure & pulse o Toxicity assessment including ECOG performance status, blood pressure and
pulse. o History of any changes to concomitant medications o Compliance evaluation (tablet bottle(s) including any unused tablets must be
returned to the clinic for drug accountability at the end of each cycle).
Laboratory determinations o Haematology and biochemistry (FBC, U&E, LFT, Ca, PO4) o Urine dipstick. If a patient has a change of two plus (++) from baseline on two
consecutive urine protein dipstick measurements, please measure urine protein : creatinine ratio or collect a 24-hour urine for total protein.
Tumour assessment
__________________________________________________________________________
INFORMATION SERVICES
rdVersion 1.1 ; Date: 23 January 2012
Page 25 of 45

ARISTACAT
o CT chest, abdomen & pelvis or other appropriate imaging/clinical measurements to assess the measurable lesions
5.2.6. Assessment at week 18 (+/- 2 weeks)
Clinical assessment o Physical examination including blood pressure & pulse o Toxicity assessment including ECOG performance status, blood pressure and
pulse. o History of any changes to concomitant medications o Compliance evaluation (tablet bottle(s) including any unused tablets must be
returned to the clinic for drug accountability at the end of each cycle).
Laboratory determinations o Haematology and biochemistry (FBC, U&E, LFT, Ca, PO4) o Urine dipstick. If a patient has a change of two plus (++) from baseline on two
consecutive urine protein dipstick measurements, please measure urine protein : creatinine ratio or collect a 24-hour urine for total protein.
5.2.7. Assessment at week 24 (and subsequent 3 month intervals until month 18, then 6 monthly to progression) (+/- 4 weeks)
Clinical assessment o Physical examination including blood pressure & pulse o Toxicity assessment including ECOG performance status, blood pressure and
pulse. o History of any changes to concomitant medications o Compliance evaluation (tablet bottle(s) including any unused tablets must be
returned to the clinic for drug accountability at the end of each cycle).
Laboratory determinations o Haematology and biochemistry (FBC, U&E, LFT, Ca, PO4). o Urine dipstick. If a patient has a change of two plus (++) from baseline on two
consecutive urine protein dipstick measurements, please measure urine protein : creatinine ratio or collect a 24-hour urine for total protein.
Tumour assessment
o CT chest, abdomen & pelvis or other appropriate imaging/clinical measurements to assess the measurable lesions
Translational research (week 24 only) (+/- 4 weeks)
o Blood & urine samples for bone resorption markers in all patients. o Blood for trough saracatinib levels. Repeat weight.
5.2.8. At discontinuation of IMP/placebo
Clinical assessment o Physical examination including blood pressure & pulse o Toxicity assessment including ECOG performance status, blood pressure and
pulse. o History of any changes to concomitant medications.
__________________________________________________________________________
INFORMATION SERVICES
rdVersion 1.1 ; Date: 23 January 2012
Page 26 of 45

ARISTACAT
__________________________________________________________________________
INFORMATION SERVICES
Version 1.1 ; Date: 23rd January 2012
Page 27 of 45
o Compliance evaluation (tablet bottle(s) including any unused tablets must be returned to the clinic for drug accountability at the end of each cycle).
Laboratory determinations
o Haematology and biochemistry (FBC, U&E, LFT, Ca, PO4). o Urine dipstick. If a patient has a change of two plus (++) from baseline on two
consecutive urine protein dipstick measurements, please measure urine protein : creatinine ratio or collect a 24-hour urine for total protein.
Tumour assessment
o CT chest, abdomen & pelvis or other appropriate imaging/clinical measurements to assess the measurable lesions
o Sites of progression should be noted. 5.2.9. Follow-up (After progressive disease or patients have come off IMP/placebo)
Survival data (follow-up survival information will be collected for all patients by clinic visit or telephone contact every 3 months. This will include further systemic therapy).
5.2.10. Safety Follow-up
For all patients receiving study drug an additional safety visit will be performed 28 days after receiving final study medication.

ARISTACAT
6. Sub-Studies
6.1. Translational
There are two translational aspects associated with the study. The first optional study relates to changes in the tumour. In patients for whom accessible tumour material is available, samples will be taken at Screening and their Week 6 visit. These samples will be stored and subsequently compared with response to the AI and AI+ saracatinib therapies in order to determine if any of the preclinical changes that are induced by Src-inhibition can (a) be identified in human tumours and (b) correlate with/ predict for benefit. The second mandatory translational study will look at pre-and on-treatment markers of bone resorption to test the hypothesis that Src-inhibition in bone can additionally reduce bone turnover in patients with metastatic bone disease. For all patients, baseline blood samples will be collected to measure markers of bone turnover (synthesis and resorption). Sampling will be repeated at Week 6, Week 24 and at discontinuation of study IMP/placebo. Please refer to section 6.2 for further information. 6.2. Bone-only metastases
Those patients with bone-only metastases have the potential to benefit from the effect of saracatinib on a specific group of non-tumour cells and hence these patients form an important sub-group in this study. Given the role of Src in osteoclast function (5), the activity of saracatinib in preclinical (17,18) and clinical (D8180C00034) studies would support a direct effect on bone turnover by inhibiting osteoclast activity. This study is not designed to look at skeletal events and patients at clear risk of this would be expected to be on standard bisphosphonate treatment. However, there is emerging data in the adjuvant setting that bone agents may affect the natural history of breast cancer, with a reduction in breast cancer recurrence rates in post-menopausal women given adjuvant Zoledronate (22). Biochemical markers of bone turnover will be investigated in one of the translational sub-studies (Section 6.1.), but clinical endpoints will also form an important sub-study. The primary endpoint will be progression-free survival and this will be assessed separately for patients receiving, or not receiving, concomitant bisphosphonate. Any effect is expected to be most apparent in the latter group. As with the main study, secondary endpoints are toxicity, response rate & overall survival. It is therefore important that thought is given to the regular on-study assessment of patients entered with measureable bone lesions in order that accurate data can be obtained on time to progression. 7. Pharmacovigilance
7.1. Definitions
Adverse Event (AE): An adverse event (AE) is any untoward medical occurrence in a patient which does not necessarily have a causal relationship with the study treatments or procedures. An adverse event can therefore be any unfavourable and unintended sign (including an abnormal laboratory finding), symptom, or disease temporally associated with a treatment or procedure, whether or not considered related.
Adverse Reaction (AR): All noxious and unintended responses related to a study treatment or procedure should be considered adverse drug reactions.
__________________________________________________________________________
INFORMATION SERVICES
rdVersion 1.1 ; Date: 23 January 2012
Page 28 of 45

ARISTACAT
Serious Adverse Event (SAE): Any untoward medical occurrence in a patient that
a) Results in death b) Is life-threatening c) Requires hospitalisation or prolongation of existing hospitalisation d) Results in persistent or significant disability or incapacity e) Consists of a congenital anomaly or birth defect f) Is otherwise considered to be medically significant by the investigator (e.g.
intensive treatment in an emergency room or at home for allergic bronchospasm, blood dyscrasias or convulsions that do not result in hospitalisation).
The term “life-threatening” refers to an event in which the patient was at risk of death at the time of the event; it does not refer to an event which hypothetically might have caused death if it were more severe.
Suspected Unexpected Serious Adverse Reaction (SUSAR): A suspected unexpected serious adverse reaction (SUSAR) is an adverse reaction that is classified as serious and it is thought to be caused by a study treatment or procedure. Expected events are detailed within the Investigator Brochure (IB). The nature, severity or outcome of this adverse reaction must not be consistent with IB for the treatment or procedure.
7.2. Emerging safety profile
This section lists those events that are to be regarded as expected for regulatory reporting purposes (as at April 2011, please refer to the IB for any updates).
Gastrointestinal disorders: nausea, vomiting, diarrhoea, anorexia. General disorders: flu-like syndromes (e.g., musculoskeletal pains, fever, headache,
fatigue, elevations in serum C-reactive protein), asthenia. Haematologic disorders: neutropenia, lymphopenia, thrombocytopenia, bleeding
(generally mild, e.g. epistaxis), bruising (generally mild, e.g. venipuncture site bruising).
Investigations: increases in serum creatinine, positive findings on urine dipstick testing for protein and/or blood (generally occurring in isolation of one another, occasionally concomitantly), mild elevations in serum transaminases, mild reductions in serum calcium, mild reductions in serum phosphate.
Respiratory disorders: pneumonitis Skin and subcutaneous tissue disorders: rash (generally papular). In combination with chemotherapy:
o Febrile neutropenia has been observed in patients receiving saracatinib in combination with carboplatin plus paclitaxel.
o Hypokalaemia has been observed in patients receiving saracatinib in combination with carboplatin plus paclitaxel.
o Mild reductions in serum sodium levels have been observed in patients receiving saracatinib in combination with carboplatin-containing chemotherapy.
7.3. Recording and Reporting of Adverse Reactions
All related Adverse Reactions will be recorded in the Case Report Form. Any Adverse Reaction considered unrelated will not be recorded.
__________________________________________________________________________
INFORMATION SERVICES
rdVersion 1.1 ; Date: 23 January 2012
Page 29 of 45

ARISTACAT
All adverse reactions that occur after the signing of written informed consent and within 30 days after the final study treatment will be recorded on the appropriate CRF page. The exception to this would be any event occurring after signing the informed consent and prior to commencing study treatment that is considered unrelated to trial procedures. In addition any events occurring more than 30 days after final study treatment that are deemed to be related to the study drug should be notified to the CSA as detailed in section 7.4. Please refer to Appendix 1 for details on the Causality. 7.4. Recording and Reporting of Serious Adverse Events
Contact Details for Reporting SAEs
CSA Fax: +44 131 275 7512 (preferred method) CSA Telephone: +44 131 275 7276 (Mon – Fri 9am-5pm) Or: +44 131 316 4278 (Mon – Fri 9am-5pm) All serious adverse events that occur after the signing of written informed consent and within 30 days after the final study treatment will be recorded on the SAE report form. The exception to this would be any event occurring after signing the informed consent and prior to commencing study treatment that is considered unrelated to trial procedures. In addition any events occurring more than 30 days after final study treatment that are deemed to be related to the study drug should be notified to the CSA as above. The SAE report form must be signed by the Principal Investigator of the centre involved and faxed to CSA within 24 hours of first becoming aware of the event. All initial SAE reports should contain the following minimum information:
Reporter information At least one subject identifier (trial number/patient initials) Event term Assessment of relatedness Serious criteria
A fax receipt will be sent to the relevant centre by CSA to acknowledge receipt of the SAE report form, and CSA will notify the Chief Investigator (CI). All SAEs will be forwarded to the CI by CSA for assessment of expectedness against the IB. Any SAE that is deemed to be both related and unexpected (i.e. a SUSAR) will be notified to the appropriate Competent Authorities and Research Ethics Committees within 7 days of becoming aware of the event for fatal or life threatening events and 15 days for all other serious events. SUSARs should be reported to the MREC accompanied by the ‘safety reports to the MREC covering form’. The coordinator of the MREC should acknowledge receipt of the safety report within 30 days by signing and returning a copy of the covering form.
__________________________________________________________________________
INFORMATION SERVICES
rdVersion 1.1 ; Date: 23 January 2012
Page 30 of 45

ARISTACAT
CSA will then notify the CI and the PI’s at all of the participating centres of the occurrence of all SUSARs. Hospitalisations planned prior to enrolment in the trial or for social reasons should not normally be considered as SAEs unless the hospitalisation has to be prolonged. Treatment in an emergency room of less than 24 hours or on an out-patient basis that does not meet any other serious criteria should not be considered as an SAE. Please refer to Appendix 1 for details on the Causality. 7.5. Non-Reportable Events
Disease progression or death as a result of disease progression are not considered to be SAEs however they will need to be reported as adverse events on the CRF. Due to the nature and stage of the disease in this study, the following situations that fulfil the definition of an SAE are excluded from recording/reporting on an SAE form however they should be recorded on the CRF and in the medical records.
Elective hospitalisation and surgery for treatment of breast cancer or its complications.
Elective hospitalisation to make treatment or procedures easier. Elective hospitalisation for pre-existing conditions that have not been exacerbated by
trial treatment. 7.6. Code Breaking Procedure
A patient unblinding form will be available to Investigators in the Cenduit IWR system. Directions to use this form can be found in the Cenduit Study Investigator and Site User Guide. The Investigator and trials pharmacist (if applicable) will have access to unblind through this system in case of emergency. In the event that the Investigator (or pharmacist) is not available to unblind the following Cenduit 24 hour helpline is provided 00 80010121960. Cenduit will contact the Chief Investigator who will advise on whether Cenduit can unblind the patient. If a site has provided 24 hour pharmacy unblinding contact details, Cenduit may contact the pharmacist instead of the Chief Investigator. The treatment code must not be broken except in medical emergencies when the appropriate management of the patient necessitates knowledge of the treatment randomisation. If the treatment code is broken then the investigator(s) must document this and report it to the Cancer Clinical Trials Team within 24 hours of the occurrence. Once a patient has been unblinded in the IWRS no further material will be allowed to be dispensed. 7.7. Developmental Safety Update Report
A developmental safety update report will be submitted to the appropriate Competent Authorities and Ethics Committees, once a year for the duration of the trial. The time frame for the report starts with the date of first authorisation by a competent authority in an EU member state and the report should be submitted within 60 days of the anniversary of first authorisation. 7.8. Pregnancies
Any pregnancy in a trial participant that occurs during study participation should be reported to CSA within 24 hours of becoming aware of its occurrence, using the contact details in
__________________________________________________________________________
INFORMATION SERVICES
rdVersion 1.1 ; Date: 23 January 2012
Page 31 of 45

ARISTACAT
Section 7.3. The pregnancy should be followed up to determine outcome, including spontaneous or voluntary abortion, details of birth and presence or absence of any birth defects, congenital abnormalities or maternal or newborn complications. Any birth defects or congenital abnormalities must be reported as SAEs. In addition, since pregnancy in a patient indicates that the patient no longer meets inclusion criteria (they are not post-menopausal), they must come off the study drug (if appropriate) as soon as any pregnancy is identified. 8. DATA MANAGEMENT
All data will be handled, computerised and stored in accordance with the Data Protection Act 1998 and NHS National Services Scotland Confidentiality Guidelines. 8.1. Data Collection
Data generated will be collected by CSA, who will be responsible for checking the data, entering it on the trial database and validating it. The data collected will include:
initial clinical details at randomisation drug administration concomitant medications adverse events survival/ recurrence details
8.2. Record Keeping and Archiving
CSA will store study documentation until the end of patient follow up. The documentation will then be sent back to the Sponsor for archiving. 9. STATISTICS
9.1. Sample Size
The study is designed to detect a 50% improvement (from 8 months to 12 months) in median PFS by adding Src-inhibitor AZD0530 to aromatase inhibition with 80% power at the 10% 1-sided level of statistical significance (or equivalently with 90% power at the 20% level of statistical significance). This decision making process follows a 3-outcome type design (Shengyan Hong and Yanping Wang A three-outcome design for randomized comparative phase II clinical Statist. Med. 2007; 26:3525–3534). This requires 140 patients recruited over 2 years with 1 year subsequent follow-up. As all patients will have ongoing residual advanced cancer at the end of study, it is highly unlikely that any patients will be lost to follow up prior to capturing the primary endpoint-related event(s). At the end of the study if the observed PFS difference in favour of AZD0530 is
statistically significant at 10% this will be a clear signal that a subsequent phase III study is warranted.
A result that is statistically significant at the 20% level (but not 10%) will proceed to phase III only if significant change in response as assessed by waterfall plot.
__________________________________________________________________________
INFORMATION SERVICES
rdVersion 1.1 ; Date: 23 January 2012
Page 32 of 45

ARISTACAT
A result that is not statistically significant at 20% level will not be worthwhile proceeding to phase III.
If a statistically significant difference in terms of PFS in favour of AZD0530 is found in the whole patient group, then a further examination will be made of whether this difference is apparent in each of the two aromatase inhibition subgroups (those treated with exemestane and those treated with anstrazole). The requirement for this is that the observed hazard ratio in the subgroup favours the study AZD0530 (this is equivalent to using a 50% 1-sided level of statistical significance). There is a >85% probability that this will be observed in a subgroup if the 50% improvement in median PFS applies as long as we have at a least 35 patients/27 PFS events in that subgroup. 9.2. Randomisation and Stratification
This is a multi centre, double blind randomisation study and a centralised registration system will be used to assign patients to treatment groups (1:1 allocation; 70 patients in each group). Randomisations will be obtained by phoning the CaCTUS Cancer Clinical Trials Team in Edinburgh who will issue a patient identifier and study drug pack number. Patients will be allocated to treatments using minimisation algorithm. The factors used in the algorithm will be:
Al sensitivity Disease site (bone metastases alone vs any other sites) Bisphosphonate use Performance status (0 vs 1 vs 2) Treatment Centre
9.3. Analysis Plan
All analyses will be conducted on an intention-to-treat basis. Progression-free survival will be compared between the treatment groups in the context of a Cox model incorporating the baseline stratification factors. The p-value for the observed hazard ratio will be determined from this model. The progression-free survival associated with the addition of AZD0530 will be determined in the two aromitase inhibition subgroups in the same manner. The overall survival and PFS will be illustrated using Kaplan-Meier plots. RECIST1.1 (Please refer to Appendix 2 for RECIST 1.1 guidelines). Response rates will be compared using Pearson’s chi-square test. Waterfall plots will also be used to compare response; a non-parametric approach based on the log ratio of tumour shrinkage will be adopted (Karrison, TG; Maitland, ML; Stadler, WM, et al.Design of phase II cancer trials using a continuous endpoint of change in tumor size: application to a study of sorafenib and erlotinib in non-small-cell lung cancer JNCI 2007; 99(19); 1456-1461). The worst toxicity grades experienced during treatment will be compared between the treatment groups using the Mann-Whitney U test. Interim Analysis There is no formal stopping rule for efficacy or futility but the study data will be reviewed six months after the first patient has entered the study by the data safety committee, who will assess the data primarily from the stand point of safety and treatment deliverability. Further meetings are expected to be biannual, but may be frequent if requested by the DSMC.
__________________________________________________________________________
INFORMATION SERVICES
rdVersion 1.1 ; Date: 23 January 2012
Page 33 of 45

ARISTACAT
Final Analysis The final analysis will be performed after 110 PFS events have occurred or at minimum follow up of 6 months, whichever is later. 9.4. End of Study
This study will end at the earliest of either 31st March 2017 (beyond which time no patient can receive the sarcatinib/placebo) or the last patient comes off study medication. 10. ACCESS TO SOURCE DATA/ DOCUMENTS
The investigator, by accepting to participate to this protocol, agrees to co-operate fully with any quality assurance visit undertaken by third parties, including representatives from the Sponsor, CSA or the Coordinating Centre, national and/or foreign regulatory authorities or company supplying the product under investigation, as well as to allow direct access to documentation pertaining to the clinical trial (including CRFs, source documents, hospital patient charts and other study files) to these authorised individuals. 11. QUALITY CONTROL AND QUALITY ASSURANCE
Quality control will be maintained through adherence to GCP and the coordinating centre’s SOPs. The coordinating centre will monitor receipt of CRFs and evaluate incoming CRFs for compliance with the protocol, inconsistencies and missing data. 11.1. Monitoring Visits
We have allowed for site visits in the UK to enable monitoring by CSA to check patient consent forms, confirm compliance with the protocol and complete source data verification (SDV) on the patient data as defined in the Data Monitoring Plan. Higher levels of monitoring will be performed, if requested, by the Data Monitoring and Ethics Committee, or if the investigators, the Trial Management Group or Trial Steering Committee identify particular safety issues. 11.2. Data Monitoring and Ethics Committee
An independent Data Monitoring and Ethics Committee will be established and will meet 6 monthly in the first instance and annually thereafter (and at any other time at the committee’s discretion). There will be an extra meeting of the committee after 50% recruitment. None of the members of the committee will be involved in the trial. The committee will receive regular reports from the CSA. It will submit its comments and recommendations to the Trial Steering Committee and Trial Management Group. 11.3. Trial Steering Committee
A trial steering committee will be established to provide overall supervision of the trial, in particular: trial progress, adherence to protocol, patient safety, and consideration of new information. The committee will meet annually. 12. ETHICAL CONSIDERATIONS
Ethical approval by a Multi-Centre Research Ethics Committee and Local Research Ethics Committee will be needed before the trial can be started. The trial will be carried out according to guidelines of good clinical practice (GCP) as defined by paragraph 28 and
__________________________________________________________________________
INFORMATION SERVICES
rdVersion 1.1 ; Date: 23 January 2012
Page 34 of 45

ARISTACAT
Schedule 1 Part 2 of the Medicines for Human Use (Clinical Trials) Regulations, 2004, and the Clinical Trials Directive (2001/20/EC) elsewhere in the European Union and follow the principles of research governance. 12.1. Patient Confidentiality
The patient’s full name, date of birth, hospital number and NHS number (Community Health Index and/or hospital number in Scotland) will be collected to enable tracing through national records. The personal data recorded on all records will be regarded as confidential, and to preserve each patient’s anonymity, only their initials and age will be recorded on CRFs. The patients will be identified within the CRFs by the use of a unique trial number allocated to them upon entry into the study. The Principal Investigator (or delegate) at each site must keep a log of patients’ trial numbers, names, addresses and hospital numbers. The Principal Investigator must ensure that patient confidentiality is maintained and that all trial documents (e.g. consent forms) are maintained in strict confidence. CSA will maintain the confidentiality of all patient data and will not reproduce or disclose any information by which patients could be identified. Patients will only be referred to by Trial Number, Initials and Age in any essential trial related correspondence, including Case Report Forms and Serious Adverse Event Reports. All patient identifiable data will be handled, computerised and stored in accordance with the Data Protection Act 1998 and NHS National Services Scotland Confidentiality Guidelines. 12.2. Informed Consent
All patients will be informed of the aims of the study, the possible adverse events, the procedures and possible hazards to which they will be exposed, and the mechanism of treatment allocation. They will be informed as to the strict confidentiality of their patient data, but that their medical records may be reviewed for trial purposes by authorized individuals other than their treating physician. It will be emphasized that the participation is voluntary and that the patient is allowed to refuse further participation in the protocol whenever they want. This will not prejudice the patient’s subsequent care. A copy of the patient information sheet is given in Appendix 2. Documented informed consent must be obtained for all patients included in the study before they are enrolled or randomized. This must be done in accordance with the national and local regulatory requirements and must conform to guidelines on Good Clinical Practice. That is, “the written informed consent form should be signed and personally dated by the patient or by the patient’s legally acceptable representative”. A copy of the informed consent form is given in Appendix 3. All Patient Information Sheets & Informed Consent Forms will be version controlled and dated and this information will always be stated in any communication with ethics committees
__________________________________________________________________________
INFORMATION SERVICES
rdVersion 1.1 ; Date: 23 January 2012
Page 35 of 45

ARISTACAT
13. RESEARCH GOVERNANCE
13.1. Trial Organisation
Chief Investigator – The Chief Investigator will have overall responsibility for the design, co-ordination and management of the study. These include:
Trial authorization including responsibility for the protocol and obtaining approvals Ensuring that the trial is conducted according to Good Clinical Practice (GCP) Assessment of SAEs and providing a prompt response as to whether the SAE is a
SUSAR. Clinical Trials Unit – The Chief Investigator has delegated the responsibility for overall project management, data management and monitoring to ISD Cancer Clinical Trials Team. Responsibilities include:
Assistance with completion of the IRAS form and MREC communication Production of trial specific documentation (i.e. CRFs) Assistance with SSA procedures within centres Facilitating set up of trial centres Data management Monitoring Pharmacovigilance – Reporting of SARs / SUSARs
Statistical Analysis – Haiying Nan, based at CSA, Edinburgh will undertake the final analysis arising for this study. Local Project Teams – These will consist of Surgeons and/or Oncologists (responsible for introducing the patient to the study and ensuring eligibility and consent), Research Nurse (responsible for patient recruitment, obtaining consent and co-ordination of all aspects of data collection), Radiologists and Radiographers (responsible for completing CT scans to protocol). Centres are specifically responsible for conducting the trial in accordance with the protocol, Standard Operating Procedures (SOPs), the trial agreement and Good Clinical Practice. Trial Steering Committee and Data Monitoring and Ethics Committee – CSA will act as study sponsor. Central study co-ordination, data collection, monitoring and organisation of the data for the statistical analyses will be undertaken by ISD Cancer Clinical Trials Team, which has processes in place to ensure that the study will not open to recruitment until appropriate approvals and authorisations have been obtained from the independent research ethics committee, and NHS Research and Development departments. The trial steering committee (TSC), including members of the research team and an independent radiologist, oncologist, statistician, and lay member, will be responsible for the progress and conduct of the study and convene annually. A Trial Management Group will meet quarterly. A Data Monitoring and Ethics Committee will convene 6 monthly initially, and annually thereafter, to review all data including adverse events and develop a stopping policy for the trial, if necessary. Specific issues that will be looked at include: tolerability of initial dose, dose reductions, toxicities (including interaction of the mild increased bleeding risk), imaging & review schedules. There will be an extra meeting of the committee after 50% recruitment. Laboratories – Accredited local laboratories shall be used for all on-study tests. Samples from the two translational studies will be transferred to the Edinburgh Cancer Research UK
__________________________________________________________________________
INFORMATION SERVICES
rdVersion 1.1 ; Date: 23 January 2012
Page 36 of 45

ARISTACAT
Centre, Western General Hospital, Crewe Road South, EDINBURGH. The details of the sampling, storage and shipping protocols will be covered by the “Translational SOP” and will accord with all prevailing regulatory requirements. 14. FINANCING AND INSURANCE
This study is funded by AstraZeneca Pharmaceutical and endorsed by Cancer Research UK’s Clinical Trials Awards & Advisory Committee (CTAAC). Indemnity for participating hospitals is provided by the usual NHS indemnity arrangements. 15. PUBLICATION POLICY
All presentations and publications relating to the trial must be authorized by the Trial Management Group. The main trial results will be published in the name of the trial in a peer-reviewed journal, on behalf of all collaborators. The manuscript will be prepared by Trial Management Group, representatives from ISD Cancer Clinical Trials Team and high accruing clinicians. The trials offices and all participating Centers and clinicians will be acknowledged in this publication. Any data that might detrimentally affect the progress of the trial will not be released prior to the end of the trial. No investigator may present or attempt to publish data concerning their patients, which is directly relevant to the questions posed in the trial, until the main results have been published.
__________________________________________________________________________
INFORMATION SERVICES
rdVersion 1.1 ; Date: 23 January 2012
Page 37 of 45

ARISTACAT
16. REFERENCE LIST
1. Chu I, Sun J, Arnaout A, Kahn H, Hanna W, Narod S, Sun P, Tan CK, Hengst L,
Slingerland J (2007) p27 phosphorylation by Src regulates inhibition of cyclin E-Cdk2. Cell 128(2):281–294
2. Grimmler M, Wang Y, Mund T, Cilensek Z, Keidel EM, Waddell MB, Jakel H, Kullmann M, Kriwacki RW, Hengst L (2007) Cdk inhibitory activity and stability of p27Kip1 are directly regulated oncogenic tyrosine kinases. Cell 128(2):269–280
3. Brugge JS, Erikson RL. Identification of a transformation-specific antigen induced by an avian sarcoma virus. Nature 1977;269:346-8.
4. Hunter T, Sefton BM. Transforming gene product of Rous sarcoma virus phosphorylates tyrosine. Proc Natl Acad Sci USA 1980;77:1311-5.
5. Horne WC, Sanjay A, Bruzzaniti A, Baron R. The role(s) of Src kinase and Cbl proteins in the regulation of osteoclast differentiation and function. Immunological Reviews 2005;208:106-25.
6. Verbeek BS, Vroom TM, Adriaansen-Slot SS, Ottenhoff-Kalff AE, Geertzema JG, Hennipman A, et al. c-Src protein expression is increased in human breast cancer. An immunohistochemical and biochemical analysis. J Pathol 1996;180:383-8.
7. Reissig D, Clement J, Sanger J, Berndt A, Kosmehl H, Bohmer FD. Elevated activity and expression of Src-family kinases in human breast carcinoma tissue versus matched non-tumor tissue. J Cancer Res Clin Oncol 2001;127:226-30.
8. Bild AH, Yao G, Chang JT, Wang Q, Potti A, Chasse D, et al. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature Letters 2006; 439:353-7.
9. Pengetzne Y, Steed M, Roby KF, Terranova PF, Taylor CC. Src tyrosine kinase promotes survival and resistance to chemotherapeutics in a mouse ovarian cancer cell line. Biochem Biophys Res Commun 2003;309:377-83.
10. Green TP, Fennell M, Whittaker R, Curwen J, Jacobs V, Allen J, et al. Preclinical anticancer activity of the potent, oral Src inhibitor AZD0530. Mol Oncol 2009;3:248-61.
11. Dong M, Rice L, Siemann DW. Impact of the small molecule Src inhibitor AZD0530 on the metastatic phenotype of a fibrosarcoma (KHT) tumour model. Anticancer Res 2010; 30: 4405-4413.
12. Boyer B, Bourgeois Y, Poupon M-F. Src kinase contributes to the metastatic spread of carcinoma cells. Oncogene 2002;21(15):2347-56.
13. Phillips KA, Parikh NU, Park SI, Kuwai T, Nokomura T, Green TP et al. Inhibition of colon tumour metastasis in orthotopic nude mouse models with the dual selective Src/Abl kinase inhibitor, AZD0530. AACR-NCI-EORTC International Conference:
__________________________________________________________________________
INFORMATION SERVICES
rdVersion 1.1 ; Date: 23 January 2012
Page 38 of 45

ARISTACAT
__________________________________________________________________________
INFORMATION SERVICES
Version 1.1 ; Date: 23rd January 2012
Page 39 of 45
Molecular Targets and Cancer Therapuetics Oct 22-26, 2007;San Francisco, California, USA. Abstract, Proffered Paper 11, page 64.
14. Yang JC, Ok J-H, Busby JE, Borowsky AD, Kung H-J, Evans CP. Aberrant activation of androgen receptor in a new neuropeptide-autocrine model of androgen-insensitive prostate cancer. Cancer Res 2009; 69:151-60.
15. Hiscox S, Jordon NJ, Smith C, James M, Morgan L,Taylor K et al. Dual targeting of Src and ER prevents acquired antihormone resistance in breast cancer cells. Breast Cancer Res Treat 2009:115:57–67
16. Chen Y, Guggisberg N, Jorda M, Gonzalez-Angulo A, Hennessy B, Mills GB, et al.
Combined Src and Aromatase inhibition impairs human breast cancer growth in vivo and bypass pathways are activated in AZD0530-resistant tumours. Clin Cancer Res 2009;15:3396-405.
17. deVries TJ, Mullender MG, vanDuin MA, Semeins CM, James N, Green TP, et al. The Src inhibitor AZD0530 reversibly inhibits the formation and activity of human osteoclasts. Mol Cancer Res 2009;7:476-88.
18. Yang JC, Bai L, Yap S, Gao AC, Kung H-J, Evans CP. Effect of the specifci Src family kinase inhibitor saracatinib on osteolytic lesions using the PC-3 bone model. Mol Cancer Ther 2010; 9: 1629-1637.
19. Chen Y, Alvarez EA, Azzam D, Wander SA, Guggisberg N, Jorda M, et al. Combined Src and ER blockade impairs human breast cancer proliferation in vitro and in vivo. Breast Cancer Res Treat 2011; 128; 69-78.
20. Dalton RN, Chetty R, Stuart M, Iacona RB, Swaisland A. Effects of the Src inhibitor saracatinib (AZD0530) on renal function in healthy subjects. Anticancer Res 2010;30(7):2935-2942.
21. Hannon RA, Clack G, Rimmer M, Swaisland A, Lockton AJ, Finkelman RD, et al. Effects of the Src Kinase Inhibitor Saracatinib (AZD0530) on Bone Turnover in Healthy Men: A Randomized, Double-Blind, Placebo-Controlled, Multiple Ascending Dose Phase I Trial. JBMR 2010; 25(3): 463-471.
22. Coleman R et al. "Discordant treatment effects according to menopausal status
following adjuvant zoledronic acid in stage II/III breast cancer -- The AZURE Trial (BIG 01/04)" ECCO-ESMO 2011; Abstract 5019.

ARISTACAT
Appendix 1 – Causality
The assignment of causality should be made by the investigator responsible for the care of the patient, using the definitions in the table below. If any doubt about the causality exists the investigator should inform the Trials Centre who will notify the Chief Investigator. The pharma companies and/or other clinicians may be asked to advice in difficult cases. In the case of discrepant views on causality between the investigator and others, the case will be discussed by all parties. In the event that no agreement is made, the MHRA will be informed of both points of view.
Table 7 Description of causality of adverse events
Unrelated There is no evidence of any causal relationship.
Unlikely There is little evidence to suggest there is a causal
relationship (e.g. the event did not occur within a
reasonable time after administration of the trial
medication). There is another reasonable
explanation for the event (e.g. the patient’s clinical
condition, other concomitant treatment).
Possible There is some evidence to suggest a causal
relationship (e.g. because the event occurs within
a reasonable time after administration of the trial
medication). However, the influence of other
factors may have contributed to the event (e.g. the
patient’s clinical condition, other concomitant
treatments).
Probable There is evidence to suggest a causal relationship
and the influence of other factors is unlikely.
Definitely There is clear evidence to suggest a causal
relationship and other possible contributing factors
can be ruled out.
__________________________________________________________________________
INFORMATION SERVICES
rdVersion 1.1 ; Date: 23 January 2012
Page 40 of 45

ARISTACAT
Appendix 2 – Efficacy Evaluation – RECIST Criteria Please refer to http://www.recist.com/ for RECIST 1.1 guidelines. Appendix 3 – Investigator Statement (CSA Copy)
__________________________________________________________________________
INFORMATION SERVICES
rdVersion 1.1 ; Date: 23 January 2012
Page 41 of 45

ARISTACAT
ARISTACAT
ARomatase Inhibition plus/minus SaracaTinib as Advanced breast CAncer Therapy (ARISTACAT): a randomised phase II study of aromatase inhibition
plus/minus the Src-inhibitor, saracatinib (AZD0530) in post-menopausal women with advanced breast cancer.
Principal Investigator Declaration
I acknowledge receipt of version <#> date <dd/mmm/yyyy> of the ARISTACAT trial protocol (MREC approved <dd/mmm/yyyy>) and I agree to perform this trial in accordance with this version of the protocol and Good Clinical Practice.
I understand that the safety of the patient is my first concern
Print Name: --------------------------------------------------------
Hospital: --------------------------------------------------------
Signed: --------------------------------------------------------
Date: --------------------------------------------------------
Please return this copy to: <Trial Coordinator name>
ISD Cancer Clinical Trials Team, (Partner in CSA - Cancer Clinical Trials Unit Scotland), Gyle Square, 1 South Gyle Crescent, Edinburgh, EH12 9EB
__________________________________________________________________________
INFORMATION SERVICES
rdVersion 1.1 ; Date: 23 January 2012
Page 42 of 45

ARISTACAT
__________________________________________________________________________
INFORMATION SERVICES
Version 1.1 ; Date: 23rd January 2012
Page 43 of 45
Appendix 4 – Investigator Statement (Investigator Copy)
ARISTACAT
ARomatase Inhibition plus/minus SaracaTinib as Advanced breast CAncer Therapy (ARISTACAT): a randomised phase II study of aromatase inhibition
plus/minus the Src-inhibitor, saracatinib (AZD0530) in post-menopausal women with advanced breast cancer.
Principal Investigator Declaration
I acknowledge receipt of version <#> date <dd/mmm/yyyy> of the ARISTACAT trial protocol (MREC approved <dd/mmm/yyyy>) and I agree to perform this trial in accordance with this version of the protocol and Good Clinical Practice.
I understand that the safety of the patient is my first concern
Print Name: --------------------------------------------------------
Hospital: --------------------------------------------------------
Signed: --------------------------------------------------------
Date: --------------------------------------------------------
Please retain this copy and file in Investigator Site File.

ARISTACAT
Please refer to attached documents: Appendix 5 – Patient Information Sheet & Informed Consent Form (Main Trial & Bone Resorption sub-study) Appendix 6 – Patient Information Sheet & Informed Consent Form (Changes in the tumour sub-study)
__________________________________________________________________________
INFORMATION SERVICES
rdVersion 1.1 ; Date: 23 January 2012
Page 44 of 45

ARISTACAT
__________________________________________________________________________
INFORMATION SERVICES
Version 1.1 ; Date: 23rd January 2012
Page 45 of 45
Appendix 7 – The Principles of ICH Good Clinical Practice
1. Clinical trials should be conducted in accordance with the ethical principles that have their origin in the Declaration of Helsinki, and that are consistent with GCP and the applicable regulatory requirement(s).
2. Before a trial is initiated, foreseeable risks and inconveniences should be weighed against the anticipated benefit for the individual trial subject and society. A trial should be initiated and continued only if the anticipated benefits justify the risks.
3. The rights, safety, and well-being of the trial subjects are the most important considerations and should prevail over interests of science and society.
4. The available non-clinical and clinical information on an investigational product should be adequate to support the proposed clinical trial.
5. Clinical trials should be scientifically sound, and described in a clear, detailed protocol.
6. A trial should be conducted in compliance with the protocol that has received prior institutional review board (IRB)/independent ethics committee (IEC) approval/ favorable opinion.
7. The medical care given to, and medical decisions made on behalf of, subjects should always be the responsibility of a qualified physician or, when appropriate, of a qualified dentist.
8. Each individual involved in conducting a trial should be qualified by education, training, and experience to perform his or her respective task(s).
9. Freely given informed consent should be obtained from every subject prior to clinical trial participation.
10. All clinical trial information should be recorded, handled, and stored in a way that allows its accurate reporting, interpretation and verification.
11. The confidentiality of records that could identify subjects should be protected, respecting the privacy and confidentiality rules in accordance with the applicable regulatory requirement(s).
12. Investigational products should be manufactured, handled, and stored in accordance with applicable good manufacturing practice (GMP). They should be used in accordance with the approved protocol.
13. Systems with procedures that assure the quality of every aspect of the trial should be implemented.