arsenic in the soil environment: a...
TRANSCRIPT

ARSENIC IN THE SOIL ENVIRONMENT: A REVIEW
E. Smith,'?' R. Naidu,'y3* and A. M. Alston2
'CRC for Soil and Land Management Glen Osmond, South Australia 5064
Australia
*Department of Soil Science University of Adelaide
Glen Osmond, South Australia 5064 Australia
'CSIRO Division of Soils Glen Osmond, South Australia 5064
Australia
I. Introduction 11. Position in the Periodic Table
111. Background Sources
n! Anthropogenic Sources A. Background Concentrations of As in Soils
A. Industry B. Mining C. Other Sources D. Agriculture
A. Accumulation in Biota B. Human Exposure to As
A. Inorganic As Compounds B. Organic As Compounds C. The Soil Solution D. Adsorption-Desorption Processes E. Kinetics of As Adsorption-Desorption
A. Soil As and Plant Uptake VIII. Soil As and Microorganisms
A. Biotransforination of As in Soils
References
V. AsToxicity
VI. Physiochemical Behavior of As in Soil
VII. Soil As and Vegetation
IX. Conclusions
*Corresponding author.
149
.4hmrc.r in /lgronotnq, Vaiumr 64 Copyright 0 1098 by Academic Press. All rights of rrproducnon in m y form reserved.
1106s-zi im m o o

150 E. SMITH ET AL.
I. INTRODUCTION
Indiscriminate use of arsenical pesticides during the early to mid- 1900s has led to extensive contamination of soils worldwide. Contamination in excess of 1000 mg As kg-' has been recorded at many sites throughout Australia. Similar con- taminated sites also exist in the United States, Africa, and other parts of the world. The persistence of As residues in soil and toxicity to both plants and animals is of concern. Consequently, many investigators have studied the chemistry of As at contaminated sites. Arsenic shows no indication of being an essential element in biological processes, although some organic As compounds have been used in low concentrations as food additives to poultry and swine supplements (Leonard, 1991). The toxicity of As compounds depends on a number of factors, including the chemical form present: inorganic As forms are more toxic than organic, and arsenite (As"') is more toxic than arsenate (As") (World Health Organization, 1981).
In humans, the LD,, has been estimated from the incidents of poisoning to range from 1 to 5 mg As kg-' (Fowle 111 et al., 1991). Evidence of As poisoning of hu- mans has been reported in India (Das et al., 1996). Thousands of humans in Ben- gal have developed symptoms of As toxicity either through consumption of As- contaminated groundwater or through ingestion of As-containing food crops. The latter pathway seems unlikely given the low soil-plant transfer of As. Much re- search needs to be done on the dynamics of As in the soil-plant system under field conditions that assess plant uptake of As.
The presence of As in the environment may be due both to background and to anthropogenic sources. The soil environment is an important sink for As com- pounds. Arsenic deposited in the soil may accumulate rapidly since it is only slow- ly depleted through plant uptake, leaching, methylation, or erosion. Because of the known toxicity of As to human and animal systems and the presence of contami- nated sites throughout the world, there has been renewed interest in studying the dynamics of As with a view to developing management strategies. For this reason, numerous reviews have been published in recent years describing the behavior of As in the soil environment (Nriagu, 1994). Although these reviews consider As in the soil system, they lack details on the major sources of As, the rates of inputs, and the chemistry of As in the soil environment. This review addresses some of these issues.
11. POSITION IN THE PERIODIC TABLE
Arsenic (atomic number 33; atomic mass 74.9216) has an outer electronic con- figuration of 4s2 4p3 and belongs to subgroup V of the periodic table. The decrease

ARSENIC IN THE SOIL ENVIRONMENT: A REVIEW 1.51
in electronegativity that is found on descending this group is not sufficient to give As a metallic character, and it is often described as a metalloid. In soils, the chem- ical behavior of As is in many ways similar to that of phosphorus (P), especially in aerated systems, where the As" ion generally resembles orthophosphate ion closely (Walsh et al., 1977). However, under conditions normally encountered in soils, As is more mobile than P and unlike P can undergo changes in its valence state.
III. BACKGROUND SOURCES
The main source of As in soils is the parent materials from which the soil is de- rived (Yan-Chu, 1994). The native As content may vary considerably within an area and is often determined by the geological history of the region (Wild, 1993). Arsenate and As"' are the dominant As species in soils (Deuel and Swoboda, 1972; Walsh and Keeney, 1975), and anthropogenic sources of As pollution have en- hanced the background concentrations of these species.
The As content of rocks depends on the rock type, with the sedimentary rocks containing much higher concentrations than the igneous rocks (Bhumbla and Keefer, 1994). Although discernible differences exist between rock groups, the range of As concentrations within a rock type may vary considerably. Generally, the mean As concentrations in igneous rocks range from 1.5 to 3.0 mg As kg-I, whereas the mean As concentrations in sedimentary rocks range from 1.7 to 400 mg As kg-I.
Atmospheric deposition contributes significantly to the geochemical cycle of As (O'Neill, 1990). Chilvers and Peterson (1987) estimated a global atmospheric flux value of 73,540 t year-', with a 6 M O split between natural and anthropogenic sources. This compares with Nriagu and Pacyna (1988), who estimated a ratio of 70:30, with an anthropogenic As input of 18,800 t year-'. About 60% of the at- mospheric As flux has been estimated to be due to low-temperature volatilization, with volcanic activity the next most important natural source (Chilvers and Peter- son, 1987). However, on a localized scale, volcanic activity may be the dominant source of atmospheric deposition (O'Neill, 1990).
A. BACKGROUND CONCENTRATIONS OF As IN SOILS
The distribution of As in soils may vary with soil type, depending on the nature of the parent material. Background concentrations do not generally exceed 15 mg As kg-l (National Research Council of Canada [NRCC], 1978), although con- centrations ranging from 0.2 to 40 mg As kg-l soil have been reported (Walsh et

152 E. SMITH ETAL.
Table I
Arsenic Concentrations from Noncontaminated and Contaminated Soils in North Americaa ~~~~ ~~
Total As content
Noncontaminated Contaminated soil Sampling site soil (mg kg-') (mg kg-') Crop
Colorado 1.3-2.3 13-69 orchard Florida 8 18-28 potato Idaho 0-10 138-204 orchard Indiana 2-4 56-250 orchard Maine 9 10-40 bluebeny Maryland 19-41 21-238 orchard New Jersey 10 92-270 orchard New York 3-12 9 M 2 5 orchard North Carolina 4 1-5 tobacco Nova Scotia 0-7.9 10-124 orchard Ontario 1.1-8.6 10-121 orchard Oregon 2.5-14 17-439 orchard
3-32 4-103 orchard Washington 6 1 3 106-830 orchard
8-80 106-2553 orchard 4-13 48 orchard
Wisconsin 2.2 6-26 potato
"Reprinted with permission from Walsh and Keeney, 1975, 0 1975 American Chemical Society.
al., 1977). Dudas and Pawluk (1980) reported background As concentrations that averaged 5 mg As kg-' in 78 chernonzemic and luvisolic soil samples in Alberta. Much higher As concentrations have been reported in acid sulphate soils devel- oped on pyritic parent material. For instance, Dudas (1987) attributed elevated As concentrations that ranged from 8 to 40 mg As kg-I in Canadian acid sulphate soils to the weathering of pyrites in the parent material.
Other studies have reported a similar variability among soils from various re- gions. Reviewing literature on As concentrations in nonpolluted and polluted soils, Walsh and Keeney (1975) concluded that nonpolluted soils in North America (Table I) rarely contain more than 10 mg As kg-' soil. Similarly, the NRCC (1978) report on the effects of As in the Canadian environment concluded that background As concentrations in soils rarely exceed 15 mg As kg-' soil.
A limited number of similar studies have been reported in Australia. Merry et al. (1983) studied 15 surface (0-150 mrn) soils from South Australia and 6 from Tasmania that were considered unlikely to have received anthropogenic sources of As. The median As concentrations in South Australian and Tasmanian soils were 3.9 mg As kg-' (+2.0) and 0.6 mg As kg-' (+0.55), respectively. Tiller (1992)

ARSENIC IN THE SOIL ENVIRONMENT: A REVIEW 153
~~
Total As (rng kg-I)
Melbourne Hobart Sydney Adelaide
< 0.2-8.1 2-45
0.6-1 1 0.2-16
“‘Iiller, 1m; reprinted by permission of CSIRO Aushlia
has also compared background As concentrations from several studies of urban soils (Table 11) demonstrating a wide range in soil-As concentrations. In contrast to studies by Merry e? al. (1983) and Tiller ( 1992), Fergus ( I 955) reported elevat- ed As concentrations in soils derived from weathered quartzite (70-100 mg As kg-’ at 0-75 mm) that resulted in restricted growth and toxicity symptoms on the leaves of banana palms in Queensland. These regional variations in As concentra- tions in soils highlight the wide variability in soil As.
IV. ANTHROPOGEMC SOURCES
Anthropogenic activities that contribute As to the soil environment originate from primary and secondary industries. These varying sources add As that differs widely in nature and composition. Such variations in the composition and nature of As have implications for biological availability as well as the mobility of As in soils. The following sections briefly consider the sources and forms of As enter- ing the soil environment through anthropogenic activities.
A. INDUSTRY
Arsenic trioxide (As,O,) is the major form of As that is produced for industry. Industrial uses include the manufacture of ceramics and glass, electronics, pig- ments and antifouling agents, cosmetics, and fireworks (Leonard, 1991). Arsenic is also added as a minor constituent to Cu and Cu-based alloys to raise the corro- sion resistance of the metal(s) (Nriagu, 1994).
Arsenic trioxide is recovered from the smelting or roasting of nonferrous met- al ores or concentrates (Loebenstein, 1993). From the limited data available (Fig. 1 ), the world production of As,O, appears to have remained relatively constant
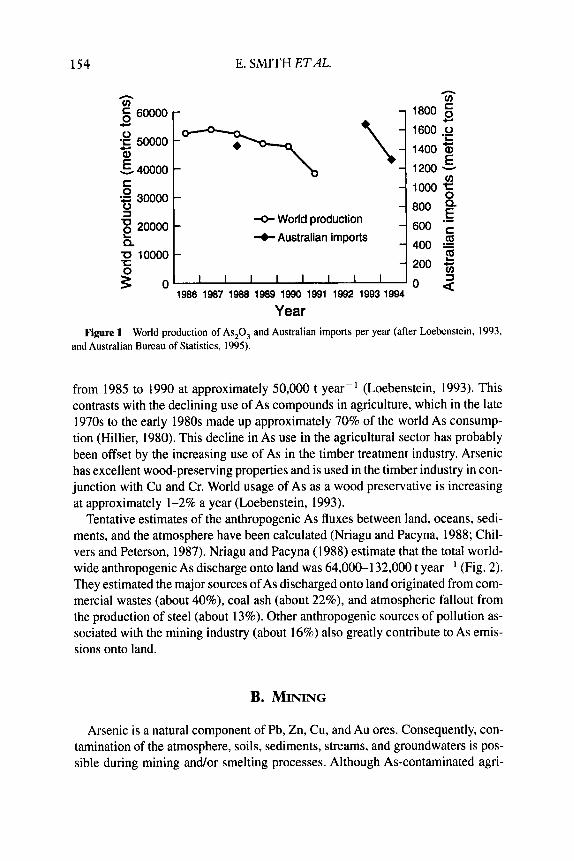
154 E. SMITH E T A .
- 7
:\ - L: -
- +World production - 4- Australian imports -
- - I I I I I I 1 I
n v) g 60000 mg 50000 c.
+
l! v 40000 C
0 3
-s 30000 3 20000 Q 10000
8 s o
n v)
1800 g CI
1600 -4 1400 5 1200 -
L
E
1000
800
600 'c a 400 = 2
200 'j
0 2
Year Figure 1 World production of As,O, and Australian imports per year (after Loebenstein, 1993,
and Australian Bureau of Statistics, 1995).
from 1985 to 1990 at approximately 50,000 t year-' (Loebenstein, 1993). This contrasts with the declining use of As compounds in agriculture, which in the late 1970s to the early 1980s made up approximately 70% of the world As consump- tion (Hillier, 1980). This decline in As use in the agricultural sector has probably been offset by the increasing use of As in the timber treatment industry. Arsenic has excellent wood-preserving properties and is used in the timber industry in con- junction with Cu and Cr. World usage of As as a wood preservative is increasing at approximately I-2% a year (Loebenstein, 1993).
Tentative estimates of the anthropogenic As fluxes between land, oceans, sedi- ments, and the atmosphere have been calculated (Nriagu and Pacyna, 1988; Chil- vers and Peterson, 1987). Nriagu and Pacyna (1988) estimate that the total world- wide anthropogenic As discharge onto land was 64,000-132,000 t year-' (Fig. 2). They estimated the major sources of As discharged onto land originated from com- mercial wastes (about 40%), coal ash (about 22%), and atmospheric fallout from the production of steel (about 13%). Other anthropogenic sources of pollution as- sociated with the mining industry (about 16%) also greatly contribute to As emis- sions onto land.
B. MINING
Arsenic is a natural component of Pb, Zn, Cu, and Au ores. Consequently, con- tamination of the atmosphere, soils, sediments, streams, and groundwaters is pos- sible during mining and/or smelting processes. Although As-contaminated agri-

ARSENIC IN THE SOIL ENVIRONMENT: A REVIEW 15s
Figure 2 Worldwide As discharges onto soils (after Nriagu and Pacyna, 1988).
cultural soils have been the subject of numerous detailed investigations, limited studies have been carried out on the detailed nature and dynamics of As in mine spoils.
Peterson et al. (1979) investigated the total As concentrations and nature of As in grossly contaminated mine spoils of southwest England. Total As concentra- tions in the spoils were greater than 20,000 mg As kg- ', with the maximum con- centration at a depth of 20-40 mm, and very high As concentrations being detect- ed to the lowest depth sampled (33,750 mg As kg-' at 250-300 mm). These investigators also assessed bioavailability of As with deionized water. They found that water-soluble As concentrations (0.3 1 mg As kg- ' at 0-20 mm) were gener- ally less than 1 % of total As. Arsenate and As"' were the major As forms present in the water-soluble soil extracts, although dimethylarsine was also detected in sur- face samples. Although As" was the predominant form throughout the soil profile, similar concentrations of As"' were reported in the surface samples. Even though these soils had been relatively undisturbed for 70 years, the contaminated sites were largely barren and supported only a limited number of plant species that gen- erally covered less than 1 % of the contaminated area. Grasses were the predomi- nant species present (Agrostis stolonifera and A. tenuis), with As concentrations in leaves greater than I000 mg As kg- ' (dry weight). Wild ( 1 973-74) reported that 72 plant species were found on 15 Rhodesian arsenical mine dumps with total As concentrations ranging from 200 to 30,000 mg As kg-'. As may be expected, the number of plant species and their density was found to increase as As content de- creased. Of the mine dumps surveyed, the Banshee mine dump (30,000 mg As

156 E. SMITH ETAL.
kg-l) was affected the worst and was incapable of supporting vegetation. Gener- ally, weed species were found to be the most important species in terms of plant numbers on the mine dumps. Ffaveria trinervia (Gaika Weed) was often the dom- inant or most important weed species present on the mine dumps. Of the grass species, Cynodon dactylon was the most important species, being present in soils with As concentrations ranging from 200 to 30,000 mg As kg- *. While As and oth- er heavy metal concentrations at some mine dumps may inhibit stabilizing soil vegetation from establishing, some plants, such as Cydon dactylon, can tolerate high As concentrations and may be useful for stabilizing mine dump soils. They may also offer a long-term, low-cost solution for remediating mine dumps when other remediation techniques are impractical.
There have been a number of reported incidences of atmospheric As release dur- ing the smelting of Pb, Zn, Au, and Cu ores (Crecelius et al., 1974; Ragaini et al., 1977; Li andThornton, 1993). Crecelius et aE. (1974) reported that a large Cu mine near Tacoma, Washington, released approximately 300 t of particulate matter into the atmosphere per year. Dust containing approximately 20-30% As contaminat- ed the soil (0-30 mm) within a 5-km radius of the smelter, with up to 380 mg As kg-' occurring at some of the sites sampled. Li and Thornton (1 993) studied the As contamination of soil from three ore smelting areas in England-Derbyshire, Cornwall, and Somerset. They reported that As concentrations in the topsoil (0-1 50 mm) were elevated above background measurements (7.69-8.97 mg As kg-l) and ranged between 16 and 925 mg As kg-', depending on the sampling area. Although most mining and smelting in these regions ceased at the end of the 19th century, As contamination in some areas is still particularly high. This em- phasizes the general long-term problem posed by the recalcitrant nature of com- pounds associated with soil contamination from industrial sources.
Unlike many heavy metals such as Cr, Cd, and Hg, As has been detected in groundwaters especially at sites contaminated by mill tailings. Bernard (1 983) in- vestigated the contamination of groundwater and the subsequent contamination of Lake Moira, Canada, and found that haphazard disposal of mill tailings and other slag wastes resulted in considerable leaching of As from these sites. Water sam- ples collected from around the tailings and As storage areas in a hydrological in- vestigation of groundwater had As concentrations ranging from 600 to 2200 mg As liter-'. Extensive mitigation methods have been required to alleviate the high As concentrations in the lake. Similarly, Leblanc et a f . (1996) reported that the dis- solved As content of an acidic stream (pH 2.2-4) originating from a waste mine dump of the Camoulbs Pb-(Zn) mine in Gard, France, was extremely high (aver- age 250 mg As liter-'). Leblanc et al. (1996) also observed that As was precipi- tated and concentrated in Fe-As bacterial stromatolites. It was proposed that the accumulation of As was through direct or induced microbial action. Rittle el al. (1995) also reported that the immobilization of As"' into a Fe-As-S solid phase was also linked to microbial activity.

ARSENIC IN THE SOIL ENVIRONMENT: A REVIEW 157
The mitigation of polluted As sites is an important aspect of the remediation process and is a research area that is receiving considerable attention. However, discussion of these remediation processes is beyond the scope of this review.
c. OTHERSOURCES
1. coal
Fly ash presents an increasing waste problem worldwide because of the contin- uing demand for coal-fired power stations. In addition to directly releasing As into the atmosphere, coal combustion produces fly and bottom ash containing As. The physical and chemical properties of fly ash restrict the general utilization of fly ash, resulting in a large proportion of the ash being used as land fill (Beretka and Nel- son, 1994).
Generally, As concentrations in coal vary from 2 to 82 mg As kg-I, depending on geological origin (Adriano et al., 1980). However, very high concentrations of As (1500 mg As kg- I ) have been recorded in brown coal from the former Czecho- slovakia (Bencko and Symon, 1977).
An important feature of fly ash is the variation of elemental concentration with particle size. Natush er al. (1975) have observed that the concentrations of As and other metals in fly ash tend to increase as particle size decreases. Smaller particles of fly ash may escape emission-control devices and therefore may have greater im- pacts on biological systems in the vicinity of the emission source.
The oxidation state of As in coal ash and its leachate are of concern, since As"' is considerably more toxic than As". However, there is a lack of information on the dominant redox state of As present in fly ash. Turner (1981) reported that pond effluent from 12 ash disposal systems contained quantities of total dissolved As of less than 0.5-1 50 pg As literp1. Arsenate was the dominant As species pre- sent, but the more mobile and toxic As"' accounted for between 3 and 40% of the dissolved As. This highlights the fact that, although As" may be the dominant As species present, conditions may be favorable for the presence of As"' as well. In contrast to the relatively low As"' concentrations in the ash porewater, interstitial As"' concentrations collected from two wells ranged from 1.2 to 550 pg As liter-' at4-6 m and43-1480 pg As liter-' at 10-12 mdepth, respectively (Turner, 1981). These relatively high concentrations of As"' may be of environmental and human and animal health concerns if contamination of surface andor groundwater were to occur.
The disposal of fly ash in Australia is typical of that found in many Western countries. Australia currently produces approximately 7.7 X lo6 t (1992) of fly ash annually from major power stations (Beretka and Nelson, 1994). Of this, approx- imately 7.7 X lo5 t were used in the production of cement, sand replacement in

158 E. SMITH ETAL.
concrete, and the manufacture of blended Portland fly-ash cement. Small quanti- ties are also used as a mineral filler in asphalt, and the remaining fly-ash (about 90%) is disposed of either as mine fill or as a codisposal material in waste dumps (Beretka and Nelson, 1994). This cheap and therefore very attractive form of disposal may be of environmental concern given the relatively high concentrations of potentially toxic and mobile As"' in interstitial porewaters that have been re- ported.
2. Tannery Wastes
Arsenic was historically used as a pesticide in the treatment of animal hides (Sadler et al., 1994). Sadler et al. (1994) investigated the As status of surface and subsurface soil contamination by long-term disposal of tannery wastes in Queens- land, Australia. For over 81 years, liquid wastes from a tannery were pumped or transported by tanker to a site in Brisbane. The liquid waste was disposed of either by burial or spray irrigation methods. Surface-soil contamination (0- 125 mm) dis- played considerable variation, ranging from less than 1 to 435 mg As kg- soil across the site. Similarly, subsurface soil (250-375 mm) contamination also var- ied considerably, ranging from less than 1 to 1010 mg As kg-' soil. Although the amount of As contamination of the soil is not as high compared with mining and other reported sites, contamination at this site extended to considerable depth (600-725 mm) in the soil. This may be due both to the nature of As in the wastes and to the soil characteristics. Sodium arsenite was the active ingredient in the pes- ticide formulation used extensively to treat animal hides (Sadler ef al., 1994). Pres- ence of As at considerable depth in sandy soils may be attributed to the high mo- bility of As"' in such soils ( T a m e s and de Lint, 1969).
3. Forestry
The wood preservative industry is the major market for As in the United States (Loebenstein, 1993), and in 1990 this industry accounted for approximately 70% of the domestic As demand in the United States (Loebenstein, 1993). Although the wood preservative industry is a major end user of imported As,O,, there are few reported incidences of contamination. Nevertheless, Lund and Fobian ( 199 1 ) re- ported elevated As concentrations in two soil types (typic haplorthod and typic hapludalf) due to spillage of chemicals used in impregnating wood. Arsenic con- centrations in the haplorthod were highest in the surface soil (3290 mg As kg-' soil) and showed a general decline with increasing soil depth. Similar trends were evident for the hapludalf (surface sample approximately 380 mg As kg- ' soil), but there were large variations in the profile that could not be explained by the com- position of the soil horizons (Lund and Fobian, 1991). Generally, As was retained in the A and B horizons of both profiles. In the A horizon, the retention of As was

ARSEMC IN THE SOIL ENVIRONMENT: A REVIEW 159
associated with high organic-matter content, whereas retention in the B horizon may be associated with adsorption by Mn, Fe, and A1 oxides (Lund and Fobian, 1991). The mechanism of As adsorption and the role of oxidic materials is further discussed in the section on adsorption mechanisms.
McLaren et al. ( 1 994) investigated the leaching of Cu, Cr, and As (CCA) solu- tion through free-draining, coarse-textured surface and subsurface soils (typic ustipsamment and udic ustochrept) using undisturbed soil lysimeters. Cumulative amounts of As leached through the lysimeters ranged from 4 to 30% of the total CCA solution applied (90 mg Cu, 157 mg Cr, and 130 mg As). The large amount of As leached is probably due to As being present as a simple salt (H,AsO,) in the CCA solution and therefore represents an increased leaching potential in compar- ison with metals in sewage sludge, which are in relatively immobile forms (McLaren et af., 1994).
D. AGRICULTURE
Agricultural inputs such as pesticides, desiccants, and fertilizers are the major sources of As in soils (Jiang and Singh, 1994). Numerous cases of As contamina- tion of agricultural soils have been recorded (Bishop and Chisholm, 1961; Wool- son et al., 1971a; Hess and Blanchar, 1976; Merry et al., 1983).
I. Pesticides
From the late 1800s and until the introduction of dichlorodiphenyltrichlo- roethane (DDT), Pb arsenate (PbAsO,), calcium arsenate (CaAsO,), magnesium arsenate (MgAsO,), zinc arsenate (ZnAsO,), zinc arsenite (Zn(AsO,),), and Paris green [C~(CH,COO);~CU(A~O~)~] were used extensively as pesticides in or- chards (Anastasia and Kender, 1973; Merry et al., 1983). The resultant pollution of orchard soils by inorganic Pb and As pesticides has been extensively reported in the literature(BishopandChisholm, 1961;Franketal., 1976;Merryetal., 1983; Peryea and Creger, 1994).
Bishop and Chisholm (1 96 1) investigated As soil pollution on 25 Annapolis Val- ley orchards (mostly sandy loams; pH 6.2-6.7). It was found that the use of of ar- senical pesticides had resulted in the accumulation of 9.8-124 mg As kg-' in the topsoil (0-1 SO mm) (Table 111). The considerable variations in As concentrations were attributed to different spraying practices at each orchard. Frank et al. ( 1976) reported similar findings in apple orchards to which PbAsO, sprays were applied for periods ranging from S to 70 years. The mean As concentrations were 54.2 2 25.8 mg As kg-' and 20.9 ? 13.6 mg As kg-*, respectively, in the 0-150-mm and 150-300-mm layers of soil. A comparison of the age of orchards versus As con- centration in the surface soil showed an increase of 7-121 mg As kg-' after 70

160 E. SMITH ETAL.
Table 111
Arsenic Contamination of Orchard Sites
Total soil As (&I50 mm)
Background Contaminated concentration concentration
Apple orchards (mg kg-') (mg kg-') Source
Torbrook 4.2 124.4 Bishop and Chisholm, 1961 Kentville 4.5 15.0 Ibid. Woodville -0 53.8 bid. Morristown 1.0 30.4 Ibid. Cornwallis trace 9.8 bid. Mean of 3 1 orchards 6.21 54.2 Frank et al., 1976 Mean of 98 orchardsb 3.9'; 0.6d 29' Merry et al., 1983
~ ~ ~ _ _ _ _ _ _ _ _ ~ ~ ~ _ _ _ _ _ _ _ _ ~
"No data available. bTen apple and pear orchards from South Australia and 60 from Tasmania. CMean of 15 soils from South Australia. %lean of 6 soils from Tasmania. 'Samples from depth of 0-100 mm.
years of pesticide applications. Increases in the As concentration were also evident in the 150-300-mm layer, although the concentrations were much lower. Howev- er, comparison of the Pb-As ratio between the untreated and treated topsoils indi- cates that there may have been considerable loss of As from the surface soil (Frank et af., 1976). This loss is reflected to some extent in the accumulation of As in the 150-300-mm horizon. Merry et af. (1983) reported that although there was con- siderable accumulation of As in 98 surface soils of apple and pear orchards in South Australia and Tasmania (Table III), there was evidence of loss of As from the surface soil at some sites. Translocation by leaching in soil solution or colloids in suspension were suggested as possible mechanisms for losses (Merry et al., 1983), although losses ofAs compounds through volatilization may also be an im- portant but difficult pathway to quantify. Barrow (1974) and Davenport and Peryea ( 1 99 1) have reported that P amendments to soil may contribute to the displacement of As in soils, and leaching may be accentuated in sandy soils (Tammes and de Lint, 1969). Peryea and Creger (1994) found that the movement of Pb and As was greater in soils with low clay and organic contents, high irrigation rates, and high application rates of Pb arsenate pesticide. This study highlights that As mobility is a result of complex interactions between soil and solution factors that influence the leaching of As from the surface soil.
Arsenical pesticides were also widely used in livestock dips to control ticks, fleas, and lice (Vaughan, 1993). In Australia, As-based pesticide solutions were widely used in Queensland and northern New South Wales from the early 1900s

ARSENIC IN THE SOIL ENVIRONMENT: A REVIEW 161
Table N
Average As Residues in Aggregated Soil in and around Cattle-Dip Sites"
As (mg kg-' soil)
Location within dip site Mean Range
Adjacent to dip bath 290 0-1636 Draining pen 436 2-870 Scooping mound 720 15-3000 Disposal pit 467 (L2600
"After DIPMAC Report, 1992.
to 1955 (DIPMAC Report, 1992) to control ticks in cattle. Investigations of pos- sible polluted sites have identified 1607 known cattle dip sites, of which 1041 sites were still in operation in 1990 (DIPMAC Report, 1992). High concentrations of As residues have been identified at some dip sites (Table IV), especially in the im- mediate area around the dip bath and draining pen (DIPMAC Report, 1992; Barzi et al., 1996).
Arsenic residues (<50 to >3000 mg As kg- l ) have also been identified in sub- surface layers at depths exceeding 500 mm (Naidu et al., 1995). Some of the As concentrations present in the contaminated soil around livestock dips are compa- rable to As concentrations present in mine spoils but are perhaps more toxic be- cause of the soluble nature of the As compounds present. Residential development of the contaminated sites may pose a considerable risk to human health. Similar problems also exist at former sheep dip sites due to the use of As-based pesticides by pastoralists in Australian Capital Temtory, Australia. Similar contaminated sites, resulting from animal dips, exist in Africa and many parts of the United States.
2. Herbicides
Since the late 19th century, inorganic arsenical compounds were used as non- selective soil sterilants and weed killers (Vaughan, 1993). Because of their persis- tence in the soil and toxicity to humans and stock they were superseded by organoarsenical herbicides (McMillan, 1988).
Monosodium methanearsonate (MSMA) and disodium methanearsonate (DSMA) have been used extensively as preemergence and postemergence herbi- cides in cotton and turf grasses (Sachs and Michael, 1971). Although both MSMA and DSMA are effective as selective grass suppressors, MSMA has been used al- most exclusively in Australia (McMillan, 1988). The use of MSMA and DSMA in

162 E. SMITH ETAL.
the cotton industry has declined since the development of more effective contact herbicides.
Despite the low mammalian toxicity of the methanearsonates, there is some con- cern with the ultimate fate of these As compounds, because they may persist in the soil environment andor be phyto-available (Hiltbold et al., 1974). Hiltbold et al. ( 1974) investigated the distribution of MSMA after repeated application to three soil types. After 6 years of applying MSMAat various rates (0-40 kg ha- year- I ) ,
the cumulative As concentration in the soils (0-900 mm) ranged from 10 to 3 1 mg As kg-' in a Decatur silt loam (rhodic paleudult), 2.4 to 23.6 mg As kg-' in a Hartsells fine sandy loam (typic hapludult), and 5.2 to 2 1.2 mg As kg- ' in a Dothan loamy sand (plinthic paleudult). There was no apparent decline in the cotton yield over the 6-year period, and only low concentration of As were detected in the cot- ton seed (<0.2 mg As kg-'). Gilmor and Wells (l980), in contrast, reported that the residual effects of MSMA increased the sterility of rice cultivars grown in a Crowley silt loam soil (typic albaqualf). It was observed that the occurrence of straighthead in the rice cultivars increased with increasing application rates of MSMA (1.1-11.2 kg As ha-'). No sterility or yield decrease was found with ei- ther midseason draining and drying treatment, which is a common management practice to prevent straighthead, or at low rates of As application to the rice culti- vars (Gilmor and Wells, 1980).
Although many soils have repeatedly received applications of As-based herbi- cides, the highest reported concentrations of As residues have been observed in soils that have been treated with arsenical-based pesticides. This is mainly due to the differences between herbicide and pesticide application rates, with herbicides being applied at significantly lower rates (Vaughan, 1993).
3. Fertilizers
Information on the effect of P fertilizers on the As content of soils is limited. Goodroad and Caldwell(l979) reported that there was no increase in As concen- trations in a Nicollet clay loam soil (aquic hapludoll) and Port Byron silt loam soil (typic halpudoll) after receiving various P-fertilizer treatments. Phosphate fertil- izer (concentrated superphosphate; about 20% P) was applied in two parts at var- ious rates with a cumulative total of 0-8,888 kg ha-' of superphosphate applied to the Nicollet soil, while the Port Byron soil received five annual treatments of no P, 99 kg ha-' of concentrated superphosphate, 73 kg ha-' of calcium metaphos- phate, 82 kg ha- * of phosphoric acid, and 352 kg ha-' of southern rock phosphate. Soil samples were collected from the Ap horizon (0-250 mm, Nicollet clay loam; and 0-225 mm, Port Byron silt loam).
Charter et al. (1995) analyzed commercial phosphorus fertilizers marketed in Iowa and phosphate rocks used in the production of P fertilizers worldwide for

ARSENIC IN THE SOIL ENVIRONMENT: A REVIEW 163
trace metal contamination. The concentrations of As and Mo were greater and more variable than other trace metals analyzed for in concentrated superphosphate, monoammonium phosphate (MAP), diammonium phosphate (DAP) and phos- phate rocks (PRs). Arsenic concentrations in the samples were in the range 2.4-18.5 mg As kg-' (TSP), 8.1-17.8 mg As kg-I (MAP), 6.8-12.4 mg As kg-I (DAP), and 3.2-32.1 mg As kg-I (PRs).
V. ASTOXICITY
For many soil pollutants, regulatory criteria for remediation or health are usu- ally based on the effects on human health as an endpoint, but certain pollutants are toxic to plants and soil biota at concentrations that do not affect animal or human health. Arsenic is a good example of one such pollutant.
The speciation of As in the environment is of critical importance because or- ganic and inorganic compounds differ largely in their toxicity (Leonard, 1991). The toxicity of As is also related to the rate that it is metabolized from the body and the degree to which it accumulates in the tissues. The general pattern of tox- icity is ASH, > As"' > As" > RAs-X (Fowler, 1977). Therefore, as a rule, inor- ganic arsenicals are more toxic than organic arsenicals, and the trivalent oxidation state is more toxic than the pentavalent oxidation state (NRCC, 1978).
Although the pharmacokinetic aspects of As toxicology are important, they lie largely outside the scope of this review, and no attempt has been made to assess the vast amount of literature on this topic.
A. ACCUMULATION IN BIOTA
Arsenic is present in many plants and plant products (Fowler, 1977) but typi- cally does not exceed 1 mg As kg-' (Kiss et al., 1992). MacLean and Langille (1981) studied the uptake of As in apples grown on orchard sites and found that the As concentration in the peel and pulp of the fruit did not exceed 0.36 and 0.30 mg As kg-I, respectively. The uptake of As by radishes and silverbeet has been studied by Merry et al. (1986) in a glasshouse experiment with eight soils (3 X typic rhodoxeralf, 2 X ultic palexeralf, typic pelloxeralf, dystic xeropsamment, lithic xeropsamment). They found that with soil-As concentrations ranging be- tween 26 and 260 mg As kg- ', none of the plants grown in these experiments con- tained As that exceeded currently accepted health limits for human consumption of 1 .O mg As kg-' (dry weight) (National Food Authority, 1993). The general as- sumption, therefore, is that in many situations the soil-plant transfer of As is low.

164 E. SMITH E T A .
Although the biochemical role of As in animals has been studied extensively (WHO, 1981; Petito and Beck, 1990), little is known about the biochemical role of As in plants (Kabata-Pendias and Pendias, 1992). Arsenic induces phytotoxic- ity that effectively protects humans from As poisoning. Phytotoxicity also results in restricted plant growth, which is undesirable (Sheppard, 1992). The effects of phytotoxicity have been reported to vary with soil type, with As being more toxic in sandy soils than in clay soils (Sheppard, 1992), which may be attributable to the greater As bioavailability in sandy soils. However, Jacobs et al. (1 970) reported that As may stimulate plant yields at low concentrations in the soil. Similarly, mi- croorganisms have been shown to display a range of sensitivities to As compounds, with the responses being dependent on soil type, the nature of the As species, and the concentration of As in the soil (Maliszewska et al., 1985).
B. HUMAN EXPOSURE TO As
Humans may be exposed to As from a variety of environmental sources, but food constitutes the largest source of As intake, with smaller contributions from air and drinking water (Chen and Lin, 1994).
“Normal” As concentrations in human whole blood and urine have been re- ported to be about 100 and 15 pg As liter-’ (Fowler, 1977), respectively, but this may vary widely depending on environmental exposure. Approximately 5-1 5% of As ingested by humans is absorbed (NRCC, 1978), and As compounds are dis- tributed in the liver, kidney, lungs, spleen, and the wall of the gastrointestinal tract within 24 hours of absorption. Some As may also be deposited in the bones, hair, nails, and skin (Leonard, 199 1). Children may be exposed to higher amounts of As through the direct ingestion of soil.
Effects of acute and chronic As poisoning in humans vary depending on the sex, age, dose, and duration of exposure (Fowler, 1977) and the chemical form and oxidation state of the As compound (NRCC, 1978). The acute effects caused by the ingestion of inorganic As compounds, mainly As,O,, are well document- ed in the literature. The fatal human dose for ingested As,O, ranges between 70 and 180 mg (WHO, 1981). Induction of cancer appears to be the most common long-term effect of chronic exposure to inorganic As. However, most animal ex- periments have not been able to demonstrate a direct relationship between As and carcinogenicity (Leonard, 199 l), although epidemiological studies have demon- strated a causal relationship between environmental, occupational, and medicinal exposure of humans to inorganic As and cancer of the skin and lungs (National Academy of Sciences, 1977). Organoarsenic compounds that accumulate in ma- rine seafood appear to pose little health risk to animals and humans, because the As compounds ingested are rapidly excreted in unchanged forms (Tamaki and Frankenberger, 1992). There are many clinical manifestations of As poisoning,

ARSENIC IN THE SOIL ENVIRONMENT: A REVIEW 165
but the most commonly observed symptoms are conjunctivitis, melanosis, and hyperkeratosis (Das et al., 1996). Chronic As poisoning has been intermittently reported in the literature and is most commonly associated with As groundwater contamination (Lu, 1990). The most recent reported incident of chronic As poi- soning induced by groundwater was in six districts of West Bengal, India. More than 800,000 people in this region are drinking As-contaminated water (total As range: 0.05-3.7 kg As liter-l), and at least 175,000 people show skin lesions caused by As poisoning (Das et al., 1996). The high As concentration in ground- water is geological in origin, and the water demands in the region are met main- ly by groundwater resources.
The main concern with contaminated soils is that the presence of As may pose an immediate or, more likely, a potential long-term hazard to the health of plants and animals, including humans. Various guidelines have been developed to pro- vide a framework for the prevention, assessment, cleanup, and management of ex- isting and future contaminated sites. For example, the Australian and New Zealand Environment and Conservation Council (ANZECC) have recommended maxi- mum As concentrations in soils for the assessment and management of contami- nated sites. These guidelines are generally followed by most Australian states. 0th- er countries have developed their own soil-contamination criteria; however, many countries have not, using accepted soil contamination criteria from other countries (Tiller, 1992). The soil-contamination criteria of the U.S. Environmental Protec- tion Agency, Britain, or the Netherlands are often quoted. All regulatory measures are based on total As concentration regardless of the well-accepted fact that the most toxic fraction is that which is bioavailable. Indeed, the term "bioavailabili- ty" is abused and shows lack of clarity by consultants, regulatory bodies, and re- searchers. Toxic substances such as As need special attention to delineate between total and bioavailable fractions.
VI. PWSIOCHEMICAL BEHAVIOR OF As IN SOIL
Arsenic forms a variety of inorganic and organic compounds in soils (Vaughan, 1993 ) and is present mainly as inorganic species, either AsV or As"' (Massche- leyn et al., 1991). Under oxic soil conditions (Eh > 200 mV; pH 5-8), As is com- monly present in the +5 oxidation state. However, As"' is the predominant form under reducing conditions (Masscheleyn et al., 199 1 ; Marin el al., 1993). Both AsV and As"' species have been reported to be subject to chemical and/or microbial ox- idation-reduction and methylation reactions in soils and sediments (Braman and Foreback, 1973; Brannon and Patrick, 1987). Many different As compounds have been identified in the soil environment (Table V), and they may be classified into two major groups: inorganic As compounds and organic As compounds.

166 E. SMITH ETAL.
Table V
Arsenic Compounds of Environmental Importance
Name Synonym Formula
Inorganic As, trivalent As trioxide
Arsenenous acid Arsenite As chloride As sulfide Arsine
As oxide As acid Arsenenic acid Arsenate
Organic As Methylarsonic acid
Inorganic As, pentavalent
Dimethylarsinic acid Trimethylarsine oxide Methy larsine Dimethy larsine Trimethylarsine Arsenobetaine Arsenoc holine Arsanilic acid Cu acetoarsenite
As trioxide, arsenous oxide, white oxide
Arsenious acid Salts of arsenous acid As trichloride As trisulfide, orpiment -
As pentoxide Orthoarsenic acid Metaarsenic acid Salts of arsenic acid
Methanearsonic acid,
Cacodylic acid or monomethylarsonic acid
-
- 4-aminophenylarsonic acid Paris green
As203
HAsO, H,AsO,-. HASO,,-, or As0,'- AsCI, As2S3 ASH,
As,O,
HAsO, H,AsO,-, or HAsOd2-, AsOd3-
CH,AsO(OH),
(CH,),AsO(OH) (CH,),AsO CH,AsH, (CH,),AsH (CH,),As (CH,),As+CH,COOH (CH,),As + CH,CH,OH H,NC,H,AsO(OH), Cu(CH,CO0),~3Cu(AsO2),
H,AsO,
"After Vaughan, 1993.
A. INORGANIC As COMPOUNDS
Among the As species found in the soil environment, compounds of AsV and As"' are the most important inorganic As species in the soil, because their com- pounds are highly soluble in water (Vaughan, 1993) and may change valency states depending on the pH (Masscheleyn et al., 1991) and redox conditions (Marin et al., 1993). The equilibria for arsenic acid (As") and arsenous acid (As"') in aque- ous solutions are given in Eqs. 1-6 (O'Neill, 1990).
Arsenic acid
H,AsO, + H,O H,AsO, + H,O+ pKa 2.20

ARSENIC IN THE SOIL ENVIRONMENT: A REVIEW 167
H,AsO; + H,O - w HASO:- + H,O+ pKa 6.97 (2)
HASO:- + H,O - w AsO2- + H,O+ pKa 11.53 (3)
Arsenous acid
H,AsO, + H,O H H,AsO; + H,O+ pKa 9.22 (4) H,AsO; + H,O w As0;- + H,O+ (5 )
HAsOS- + H,O w As0:- + H,O+ pKa 13.4 (6)
pKa 12.13
Geochemical systems are commonly interpreted in terms of their response to re- dox potential (Eh) and pH. The most thermodynamically stable species over the normal soil pH range 4-8 are H,AsO, (As"'), H,AsO;, and HASO:- (As").
B. ORGANIC As COMPOUNDS
Organic As compounds (Table V) exist in both the trivalent and pentavalent states in soils (Vaughan, 1993). Microbial methylation of the As oxyanions may occur, forming methylarsenic compounds such as monomethylarsonics and di- and trimethylarsines (O'Neill, 1990; Vaughan, 1993), and ultimately may lead to the formation of arsine gas (NRCC, 1978).
Different microorganisms vary in their ability to methylate inorganic As com- pounds present in the soil (NRCC, 1978). The methylation pathway for bacteria and fungi differ, with biomethylation of As by bacteria proceeding only to di- methylarsine, which is stable in the absence of oxygen. In comparison, fungi are able to transform inorganic and organic As compounds into volatile methylarsines (Cullen and Reimer, 1989; Tamaki and Frankenberger, 1992). Some microorgan- isms can methylate As compounds over a wide range of soil conditions, whereas other microorganisms are limited by the As substrates they can methylate and the degree of methylation of those substrates (NRCC, 1978).
The equilibria for methylarsonic acid and dimethylarsinic acid in aqueous so- lution are given in Eqs. 7-9 (O'Neill, 1990). Few studies have reported the pres- ence of organoarsenical compounds in soil systems, but this is probably due to the analytical difficulties of determining trace levels of organoarsenic species.
Monnrneth.vlar.ronic acid
(CH,)AsO(OH), - + H,O w (CH,)AsO,(OH)- + H,O+ pKa 4.19 (7)
(CH,)AsO,(OH)- + H,O H (CH,)AsO:- + H,O+ pKa, 8.77 (8)
Dimethylarsinic acid
(CH,),AsO(OH) + H,O w (CH,),AsO; + H,O' pKa 6.27 (9)

168 E. SMITH E T A .
C. THE SOIL SOLUTION
Limited information is available on the concentration and nature of As in soil solutions under field conditions. However, considerable information is available on the solubility and nature of As species in soils under known reducing condi- tions simulated in the laboratory (Deuel and Swoboda, 1972; Masscheleyn et al., 1991; Marin et al., 1993; Onken and Hossner, 1995). These studies reveal that un- der moderately reducing conditions As"' is the predominant species in the soil so- lution. Deuel and Swoboda (1972) found that there was an increase of As"' in soil solution over time under flooded soil conditions, which they attributed to the re- lease of As during dissolution of iron oxyhydroxide minerals that have a strong affinity for AsV under aerobic conditions. Thus, minerals such as FeAsO, and oth- er forms of Fe"' are reduced to the soluble Fe", and sorbed As" is released into solution (Takamatsu et al., 1982). These reactions are in general agreement with processes that have been observed in groundwaters. Intermittent incidents of As contamination of groundwater and the consequential As poisoning of people have been reported (Lu, 1990; Das et al., 1996). In a recently reported case of As poi- soning in six districts encompassing an area of 34,000 km2 in West Bengal (Das et al., 1996), As concentrations were found to be above the maximum permissible limit established by the World Health Organization of 0.05 mg As liter-'. It has been proposed that As enters the groundwater through changes in the geochemi- cal environment produced by the high withdrawal rate of groundwater. It is likely that the major mechanism of As release is through the decomposition of ar- senopyrite according to Eq. 10 (Rimstidt et al., 1994).
FeAsS + 13Fe3+ + 8H20 (10) Onken and Hossner ( 1995) identified the concentration and nature of As species
present in the solution from two flooded soils (entic pelludert and typic ochraqualf) treated with sodium arsenate or sodium arsenite (rates of 0 4 5 mg As kg-I) in a glasshouse study. In soils treated with sodium arsenite (25 mg As kg- I), As"' was the major As species present in aqueous solution at day 0, but by day 10 conver- sion to As" had occurred (Midland silt loam, about 50% of total As present as AsV; Beaumont clay, about 20% of total As present as As") due to the relatively high redox potential of the soils. Similarly, a Midland silt loam treated with sodium ar- senate (25 mg As kg-') contained no As"' at day 0, but by day 10 about 80% of total As was present as As"' in solution. The increase in As"' in aqueous solution resulted from the conversion of As" to As"' as the redox potential in the flooded soils declined. However, complete reduction of As" to As"' was not observed.
Masscheleyn et al. (199 I ) studied the influence of redox potential and pH on As speciation and solubility in a contaminated soil (aeric ochraqualf). Changes in the redox potential and pH greatly affected the As species present in the soil solution. At higher soil redox potential (500-200 mV), As solubility was low, and AsV was
14Fe2+ + SO$- + 13H+ + H,AsO,(aq)

ARSENIC IN THE SOIL ENVIRONMENT: A REVIEW 169
the predominant species in solution. Increasing pH or reducing AsV to As"' in- creased the concentration of As species in the solution. Based on these studies they concluded that the solubility of As under moderately reduced conditions was con- trolled by the dissolution of Fe hydroxides, which is consistent with the observa- tions of Deuel and Swoboda (1972). More recent studies by Marin et al. (1993) support the earlier findings.
The transition of As" to As"' is not surprising given that As"' is thermodynam- ically more stable than AsV under reducing soil conditions; i.e., where free elec- tron activity (pE) + pH < 8, and pH < 6 (Sadiq et al., 1983). Despite the ther- modynamic stability of As"' relative to As", numerous investigators (Masscheleyn et al., 199 1; Marin et al., 1993) have reported the presence of As" in aqueous so- lution under reducing conditions. Masscheleyn et al. (199 l ) suggested two possi- ble reasons for this: ( 1 ) competition of Fe"' as a terminal electron acceptor in mi- crobial respiration (Eq. l l ) , or (2) presence of manganeseIv oxides (Eq. I2), which have been shown to be effective oxidants of As"' (Oscarson et al., 1981).
Fe20, + 4H+ + AsOi- 2Fe2+ + As0;- + 2H,O E" = 0.21 V (1 1)
MnO, + 2H+ + AsOi- e Mn2+ + As0;- + H,O E" = 0.67 V (12)
The redox potential of soil depends on the half-cell potentials (E") of all the re- ducing and oxidizing systems in the soil, and due to the heterogenetic nature of soils, these relationships are very complex.
D. ADSORPTION-DESORPTION PROCESSES
As with many other contaminants, the concentration of As in the soil solution concentration is controlled both by soil physical and soil chemical properties that influence adsorption-desorption processes. Compared to the large volume of lit- erature on metal adsorption by pure silicate and oxidic mineral systems, little in- formation is available on As adsorption and transport in soils. Studies on pure sys- tems suggest that As has a high affinity for oxidic surfaces, although reactivity of oxides may vary considerably, depending on pH, charge density, and soil solution composition. Soil texture (Wauchope, 1975; Frost and Griffin, 1977), nature of constituent minerals (Walsh er al., 1977; Pierce and Moore, 1980), pH, and the na- ture of competing ions have all been shown to influence adsorption processes.
Few researchers have investigated the mechanisms involved in As sorption. The studies that have been conducted have generated considerable evidence for the for- mation of inner sphere complexes (specific adsorption) with soil components (Hingston et al., 197 1 ; Anderson and Malotky, 1979). Direct evidence for the for- mation of AsV inner sphere complexes have been obtained using extended X-ray absorption fine structure (EXAFS) spectroscopy (Waychunas et al., 1993) and wide-angle X-ray scattering (Waychunas et al., 1996) on the ferrihydrite, infrared

170 E, SMITH ETAL.
spectroscopy by Lumsdon et al. (1984) on goethite, and on other hydrous Fe ox- ides by Harrison and Berkheiser (1982). Waychunas and co-workers (1993,1996) have postulated that AsV adsorbs onto ferrihydrite by forming binuclear, inner sphere complexes. However, monodentate complexes were also observed and ac- counted for approximately 30% of all As-Fe correlations (Waychunas et al., 1993), and monodentate AsV-Fe complexes were comparable to the number of bidentate AsV-Fe complexes at low total As concentrations. Similarly, Fendorf et al. (1997) investigated the surface structure of AsV and chromate sorption on goethitc. They concluded, from EXAFS spectroscopy examination of the surface, that AsV formed three different complexes on goethite. A monodentate complex was fa- vored at low surface coverage, whereas the bidentate complexes were favored at higher surface coverages. Indirect methods have also been used to study sorption mechanisms. Specific anion adsorption produces a shift in the zero-point charge (pH ) of the adsorbent. Pierce and Moore (1980) investigated the sorption of AsIfiZnto amorphous Fe hydroxide and observed that the pHzF decreased with in- creasing addition of As"'. This was assumed to be indicative of As"' being specif- ically sorbed to the hydrous Fe hydroxide surface.
Theoretically, all the adsorbed metal may be desorbed from the soil constituents. However, investigations to date report that substantial proportions of trace metals sorbed by soil constituents are not readily released into the soil solution. Few stud- ies to date have investigated desorption of As from soil constituents.
Phosphate has been reported to displace adsorbed As from soils (Woolson et al., 1973; Peryea, 1991). Heavy additions of P to As-polluted soils have been report- ed to displace approximately 77% of the total As in the soil, with the water-solu- ble As fraction being redistributed to lower depths in the soil profile (Woolson et al., 1973). Peryea (1991) observed that although P increased As solubility, de- sorption of As was dependent on the soil type, with the As concentration in soil so- lution from a volcanic soil not altering after the addition of P. These volcanic soils have high anion-fixing and pH-buffering capacities due to the presence of allo- phanic minerals; this implies that only large additions of P to high anion-fixing soils may affect As solubility. In leaching experiments with columns of repacked soils collected from PbAs0,-contaminated apple orchards, the addition of P in the form of MAP or monocalcium P (MCP) significantly increased the amount of As leached from the columns (Davenport and Peryea, 1991).
Although As"' has been recognized as being more mobile and toxic than As", there have been few reported desorption studies of As"' from the soil. Tammes and de Lint (1969) found that even after extremely high concentrations of As"' appli- cations, symptoms of phytotoxicity in potatoes gradually disappeared over time, which probably indicates the leaching of As"' from the soil root zone. Elkhatib et al. (1984), in contrast, reported that As"' sorption was not reversible, since only a small amount of the sorbed As"' was released after five desorption steps.
Considering the importance of desorption processes in controlling As concen-

ARSENIC IN THE SOIL ENVIRONMENT: A REVIEW 171
trations in soil solution, further studies in this area are urgently needed for a more complete understanding of desorption processes.
I . Soil Properties
Among the factors that influence sorption of As, soil properties have been most extensively studied. These investigations show that both the amount of clay and the nature of constituent clay minerals control As adsorption in soils. Johnson and Hiltbold ( 1969) reported that approximately 90% of As present in the soil was as- sociated with the clay fraction after 4 years of repeated applications of MSMA, monoammonium methanearsonate (MAMA), and DSMA to turf. Livesey and Huang (198 1 ) studied retention of As" in four Saskatchewan surface soils (orthic dark gray Carrot River, orthic black Melfort, orthic black Oxbow, low-humic elu- viated gleysol Oxbow) and reported that at dilute As concentrations adsorp- tion-desorption processes controlled retention of As. They found that sorption was linearly related to ammonium oxalate-extractable Al and, to a lesser extent, to clay and ammonium oxalate-extractable Fe. Wauchope ( 1975) investigated the ad- sorption of As", P, DSMA, and the sodium salt of dimethylarsinic acid by 16 Mis- sissippi River alluvial flood-plain soils. Sorption of the two organoarsenical her- bicides was strongly correlated with As" and P sorption, and all four As species were found to be correlated @ < 0.01) with the clay and Fe oxide contents of the soils. This suggests that Fe and Fe coatings on clay surfaces may be important in controlling As adsorption-desorption processes in soils. Gustafsson and Jacks (1995) examined As solid-phase relationships in forest soil profiles. They report- ed that As" was the dominant As species present in the soil (entic haplocryod, typ- ic halpocryod, typic cryorthent). On addition of As" to the soil, it was found that adsorption of As by imogolite-type materials and fenihydnte were the key prop- erties that determined As" concentration in solution.
Clays may often be coated with Fe and Al oxides (Shuman, 1976; Schutless and Huang, 1990; Naidu et al., 1990; Naidu etaf., 1994), and this may modify As-clay interactions. Fordham and Nomsh (1979) reported that clay minerals were rela- tively unimportant in comparison with Fe oxides and, to a lesser extent, titanium oxides in the adsorption of As" in several acidic soils. Fordham and Norrish ( 1983), in a later study, reported that the adsorption of As" by a lateritic podzol surface soil (palexeralf) was controlled mainly by Fe oxides of approximately 50 nm diameter. Iron oxides were associated with other soil components, forming sur- face deposits on larger kaolin flakes or microaggregates with smaller flakes. Tita- nium oxides competed with Fe oxides for As" and were able to dominate As" ad- sorption when Fe oxides were chemically removed. Similarly, Elkhatib et al. (1984) examined As"' sorption in the A and B horizons of five major West Vir- ginian soils (typic hapludults, typic haplualf, and fluventic dystrocrept). Iron OX-
ide and pH were the soil properties most related to As"' sorption.

172 E. SMITH ETAL.
I I I I I I I I I I
I I I I I I ,OH
I I I I
I I I I I I
I I I I I I
-Mn- 0 -Mn -OH
0 OH
-Mn- 0- Mn -OH -Mn - 0 -Mn -OH
0 I 0
I
I
0 I
I
0 0
-Mn- 0 -Mn -As= 0 -Mn- 0 -Mn - As
0 OH
-Mn-O- Mn-OH
-Mn-0 -Mn-OH
0 I 'OH 0 0 0 0
-Mn- 0- Mn -OH - Mn - 0 - Mn -OH
D Release of Mnll
E surfaces
I I I I I I
I I I
I I I I I I I I
-Mn- 0- Mn -OH -Mn - 0-Mn-OH
OH OH
Mn2+ -Mn-0-Mn-OH 0-Mn=O
OH
I I I I
I I
0 I
OH 0
-Mn-OH
OH OH 0 0
-Mn- 0- Mn -OH -Mn - 0 -Mn-OH
Figure 3 Proposed schematic representation of the cross-section of the surface layer of a Mn'" oxide (birnessite) and the proposed As"' adsorption and subsequent As" oxidation and release (reprint- ed with permission from Scott and Morgan, 1995.0 1995 American Chemical Society).
Oscarson et al. ( I 983a) reported that Mn oxides may play an important role in the adsorption of As"' and As" from soil solution as well as the oxidation of the more toxic and mobile As"' to As" (Oscarson et al., 1981). Sorption of As by Mn oxides after the addition of As"' to soil solution (pH 7) was reported to be in the order: cryptomelane (a-MnO,) > birnessite (6-Mn0,) > pyrolusite (p-MnO,) (Oscarson et al., 1983a). The amount of As adsorbed by Mn oxides appears to be related to the pHZ and the surface area of the oxides, as well as to the oxidation of As"' to As". TLs implies that in some environments that have been contami- nated with As"', the presence of Mn oxides such as cryptomelane or birnessite in the system may decrease the potential toxicity of As"' by converting As"' to the less toxic As" and the subsequent adsorption of this species. Scott and Morgan (1995) proposed that the surface redox reactions between As"' and a Mn'" oxide (6-Mn0,) occurs through a multiprocess mechanism (Fig. 3 ) . Oscarson et al. (1983b), in a later study, reported that Fe and Al oxides and CaCO, coatings de- posited on Mn oxides affected the adsorption of As from solution. The coatings

ARSENIC IN THE SOIL ENVIRONMENT: A REVIEW 173
evidently masked the electron-accepting sites on Mn dioxides for converting As"' to AsV (Oscarson et al., 1983b).
2. Effect of pH
The effect of pH on As sorption has been studied widely using both pure min- eral systems and soils (Frost and Griffin, 1977; Pierce and Moore, 1980; Xu er d., 1988, 1991). These investigations showed that the pH of the soil solution has a large influence on adsorption of As. Generally, the effect of pH on sorption varies with the As species. Frost and Griffin ( 1977) reported that As" sorption by the lay- er silicate minerals kaolinite and montmorillonite exhibited a maximum pH of 4-6. Arsenite, in contrast, was adsorbed steadily from pH 4 to 9 on kaolinite and peaked at pH 7 on montmorillonite. Goldberg and Glaubig (1988) also investigated sorp- tion of AsV on montmorillonite, kaolinite, and calcite. The shape of the sorption curves closely agreed with those found by Frost and Griffin (1977). However, in contrast to Frost and Griffin (1977), similar amounts of As" were sorbed onto both kaolinite and montmorillonite. This may be attributable to the similar surface ar- eas of the kaolinite and montmorillonite clays (Table VI) used by Goldberg and Glaubig (1 988), compared with those used by Frost and Griffin (1 977). Perhaps importantly, Goldberg and Glaubig (1988) found that carbonates play an impor- tant role in As" sorption in the pH 9-12 range.
Xu and his co-workers (Xu et al., 1988, 1991) studied the adsorption of AsV, monomethylarsonic acid (MMAA), dimethylarsinic acid (DMAA), and As"' on alumina, hematite, and quartz. The adsorption of all four forms of As was strong- ly influenced by pH (Fig. 4), and this was attributed to the pH-dependent charge and the distribution of As species in soil solution. Based on stability constants, H,AsO; and HASO:- are the main AsV species, H,AsO, the main As"' species, CH,AsO,OH- the main MMAA species, and either (CH,),AsO(OH) or (CH,),AsO; the main DMAA (pKa 6.2) species present in the pH range (pH 4-9).
The pHzpc for alumina and haematite is approximately pH 6.5-7, and the solid surfaces are therefore negatively charged at a pH above this, which may explain why the adsorption of AsV (in deprotonated form) rapidly decreases above pH 7.
Table VI
Surface Area of Kaolinite and Montmorillonite Clays
Surface area (rn2g-1)
Kaolinite Montrnorillonite Source
34.2 86.0 Frost and Griffin, 1977 20.5 18.6 Goldberg and Glaubig, 1988

174 E. SMITH ETAL.
-0- As' -0- As"' -M- MMAA 0 DMAA
PH Figure 4 Adsorption of As". As"'. MMAA, and DMAA on alumina as a function of pH (As",
As"') = 10-hM: MMAA, DMAA = 10-8M; adsorbent-solution = 25 g liter-') (Xu et al., 1991; reprinted with kind permission from Kluwer Academic Publishers).
The P H : ~ of quartz is approximately 2, and surfaces are negatively charged, thus depressing the adsorption of As". Similar reasoning may be used to explain the adsorption of As"' and organoarsenic species to the solids. However, Xu er al. (1991) noted adsorption discrepancies in the sorption maxima of DMAA and MMAA for both compounds and suggested that other factors may also affect the sorption of these ions.
3. Effect of Competing Ions
Appreciable quantities of both inorganic and organic ligand ions are present in many soils and aquatic systems. This is especially true for Australian soils where over 30% of the soils are affected by salt (Naidu et al., 1993) and the ligand ions generally include C1-, SO:-, PO:- ions (Naidu and Rengasamy, 1993). In addi- tion, soils contain organic ligands arising from both plant root exudates and de- composing plant residues (Harter and Naidu, 1995). Competition for adsorption sites between some of these ligand ions and As can appreciably affect the amounts of As sorbed.
Phosphate is known to displace sorbed As from soils (Woolson et al., 1973). Ap- plications of relatively high rates of P fertilizers (about 8-12 mmol P kg-' soil [Peryea, 19911 and 0-48.6 mmol P kg-' soil [Melamed el al., 19951) have been shown to enhance As mobility in laboratory columns (Melamed er al., 1995) and As solution concentration in laboratory batch studies (Peryea, 199 1).
The presence of P in the equilibrating solution has been reported to suppress the adsorption of As, whereas the addition of CI-, NO;, and SO:- to the equilibrat- ing solution had little significant effect on As adsorption (Livesey and Huang,

ARSENIC IN THE SOIL ENVIRONMENT: A REVIEW 175
4 -
v
b 3 - 0
5 6 2 - Y
P i - 0
1981). Roy et ul. ( I 986) reported P and molybdate (Mo) suppressed the adsorp- tion of As on a Cecil clay (typic hapludult), with P being more effective than Mo at suppressing AsV adsorption. Competitive adsorption equations of the Freundlich type developed by DiGiano et al. (1978) and Shiendorf et al. (198 1) for the adsorption of dilute organic compounds by activated carbon appear to be po- tentially useful for describing competitive interactions of AsV with other ions on clay (Roy et al., 1986). Both equations appeared to apply only in situations where the equilibrium concentration of the competing P or Mo anion was much less than that of AsV (Roy et al., 1986), which is not the case in many soil solutions.
Recent studies of competitive adsorption interactions of anions on pure miner- al systems (Xu et al., 1988) suggests that at pH < 7, the SO:- (20 mg liter-') an- ion decreased the adsorption of AsV on alumina (Fig. 5). Increasing the SO:- con- centration (40 mg liter-') has little effect on AsV adsorption, indicating the sorption mechanisms of AsV and SO:- are not identical (Xu et al., 1988).
The presence of fulvic acid greatly affected the adsorption of AsV on alumina at a pH between 3 and 7.5 (Xu et al., 1988). Fulvic acid may be adsorbed on alu- mina by coulombic attraction (Xu et al., 1988), or fulvic acid may react directly with As (Thanabalasingam and Pickering, 1986), which tends to decrease the ad- sorption of the corresponding As complex (Xu et al., 1988). Few studies have in- vestigated the adsorption of As by organic matter. Thanabalasingam and Pick- ering (1986) reported that adsorption of AsV and As"' by humic acid was pH-de- pendent. This trend was more apparent when a high-ash-containing humic acid was used. The maximum adsorption of As" occurred at approximately pH 5.5, whereas As"' maxima occurred at a much higher pH of 8.5. Adsorption of As"' was less than AsV, which is a trend that has been noted by other authors (Frost and
-0 so:- = 0.0 mg liter-' -0- SO:- = 20.0 mg liter-'
-D SO:- = 40.0 mg liter-' -H- SO:- = 80.0 mg liter-'

176 E. SMITH ETAL.
Griffin, 1977). The general behavior of the As species was largely attributed to hu- mic acids becoming more soluble as pH increased (more alkaline), which de- creased their ability to remove As from solution. Alternatively, the observed pH effect could reflect the changes in the protonation of both the adsorbent and ab- sorbate (Thanabalasingam and Picketing, 1986).
4. Other Effects on As Adsorption
Although numerous studies have investigated the effects of pH and ion compe- tition on adsorption behavior of As in soil systems, few studies have considered other factors, such as ionic strength and index cations (e.g., Na and Ca). Many of these other factors, however, have been studied with other anions such as P and S . There is considerable data showing that when P is adsorbed by soil, or a soil con- stituent, adsorption varies with the concentration and nature of the background so- lution, although the underlying mechanisms are open to debate. The effects of dif- ferent cations have been attributed to a number of mechanisms, including the formation of surface P complexes with divalent cations (Heylar et af., 1976), the formation of insoluble Ca-P compounds (Freeman and Rowell, 198 l), and differ- ences in surface electrostatic potential (Barrow, 1983; Curtin et al., 1992).
Similarly, differences in ionic strength have been shown to affect P (Barrow, 1984; Bolan et af., 1986), S (Bolan et al., 1986; Ajwa and Tabatabai, 1995), and other anions in soil solution. In the case of P, increasing ionic strength has been shown to decrease the adsorption of P below the zpc and to promote the adsorp- tion of P above the zpc on variable-charged surfaces (Barrow et al., 1980; Bolan et al., 1986). Researchers suggest that the effects of ionic strength operate through its effect on the electrostatic potential in the plane of adsorption (Bolan et al., 1986). Therefore, at a pH above the zpc of a variable-charge surface, increasing ionic strength decreases the negative potential in the plane of adsorption, where- as at pH less than zpc it decreases the positive potential in the plane of adsorption (Barrow et al., 1980). Although this type of adsorption behavior for P is displayed over a wide range of ionic strengths, other anions, such as S , behave differently on variable-charge surfaces. Although little information is available about these af- fects of soil solution composition on the adsorption of As, studies (Woolson et al., 1973; Barrow, 1974; Peryea, 1991) have shown that P and AsV behave very sim- ilarly in soils. This suggests that the affects of ionic strength and different index cations on the behavior of As" adsorption are similar to the adsorption behavior of P. Current unpublished data in our laboratory confirm this conclusion.
E. KINETICS OF As ADSORPTION-DESORPTION
Adsorption and desorption processes are the principal factors affecting the transport, degradation, and biological availability of compounds in soils. Numer-

ARSENIC IN THE SOIL ENVIRONMENT: A REVIEW 177
ous studies have accumulated a large amount of data from anion adsorption and interpreted these results with adsorption models. Application of these models to soil systems is not simple, because a number of soil processes complicate the mea- surement and interpretation of the results. However, it is generally agreed that in most cases anionic adsorption is bimodal, since it occurs in fast and slow stages. The adsorption of both AsV and As'" on pure minerals and soils has been well stud- ied (refer to earlier sections), and researchers have found that the initial adsorption of both As species is rapid (Anderson ef al., 1976; Pierce and Moore, 1980; Elkhat- ib et al., 1984; Scott and Morgan, 1995). Anderson et al. (1976) reported that the rate of AsV adsorption on aluminium hydroxide was initially rapid, with over 90% of the adsorption reaction (75 mg As liter-' added) taking place before the sam- ple could be collected. After 1 hour the rate of AsV adsorption slowed consider- ably, but As" adsorption continued at a slow rate for the duration of the observa- tional period (70 hours). Similarly, Elkhatib et al. (1984) reported that the initial reaction of As"' with five surface (A) and subsurface (B) soils having a range of chemical and physical properties (coarse-loamy, mixed, mesic typic hapludults; fine-loamy, mixed, mesic typic hapludults; coarse-loamy, mixed, mesic fluventic dystrochrepts; fine, mixed, mesic typic hapludalfs) was rapid, with more than 50% (5-500 mg As"' liter-') of the original As"' present being adsorbed in the first 30 min. Conventional methods (batch and flow methods) are too slow to observe the kinetics of most surface chemical reactions. Pressure-jump @-jump) relaxation technique allows the determination of extremely rapid surface chemical reactions. Grossl ef al. (1997) investigated the rapid adsorption-desorption of AsV and chro- mate on goethite using the p-jump technique. From information elucidated using this technique, Grossl et al. (1997) proposed that the rapid adsorption of AsV on goethite was a two-step process that resulted in the formation of an inner sphere bidentate complex. This was in general agreement with EXAFS spectroscopy data obtained by Fendorf et al. (1997).
For many of these batch studies, the apparent equilibrium between the solution and the solid was assumed to be reached in a few days. However, few studies have investigated the long-term adsorption behavior of As in soils, and the magnitude of this slow fraction is unknown. Examples from long-term studies with other el- ements indicate that the apparent adsorption distribution coefficient ( Kd) can in- crease as much as 10-fold between short contact times (1-3 days) and long times (Pignatello and Xing, 1996).
In contrast to adsorption studies, little information is available on the desorp- tion of As or other elements from soils. Elkhatib et al. (1984) reported that as As"' desorption was quite hysteric and only slowly desorbed from five soils where As"' had been in contact with the soils for 24 hours. Carbonell Barrachina et al. (1996) have also studied the desorption behaviour of As"' and found that As"' sorption was a reversible process from three freshly saturated soils (aridisol gyp- siorthid torriothent, inceptisol haplumbret dystochrepts, and entisol torriorthent haplargid calciorthid). Carbonell Barrachina et al. (1996) have suggested that the

178 E. SMITH ETAL.
difference between their results and Elkhatib et al. (1984) may be explained through the different sorption capacities of the soils, since the soils used by Elkhat- ib et al. (1984) had a greater sorption capacity than those used by Carbonell Bar- rachina et d. (1996). Other ions, such as P, exhibit desorption behavior similar to that ofAs. Many desorption studies often reveal a fast desorbable fraction followed by a slow fraction (Garcia-Rodeja and Gil-Sortes, 1995; Lookman et al., 1995). Garcia-Rodeja and Gil-Sortes (1995) studied the desorption of P from 3 1 surface- soil samples collected from northwest Spain that had been spiked with varying amounts of P (200-2000 mg P kg-') and maintained at 75% field capacity for 1 year. They reported that Pdesorption was initially very rapid but over time became progressively slower. Lookman et al. (1995) also studied the desorption of P from 44 soil samples in long-term spiking trials. They reported that the desorption of P could be described by considering that P occupies two discrete pools-in one pool P was readily available, and in the other pool P was strongly fixed and desorbed slowly. The desorption of P from the fast and slow pools could be described by a two-component first-order model (Eq. 13), where Ql,o and Q2.0 are the amounts of P initially present in the labile pool, k , and k , are the rate of desorption from each descrete pool, and t is the time.
(13) However, equilibrium and thermodynamic considerations make it difficult to vi- sualize the presence of such discrete pools of P. Mathematical equations of the na- ture described here assist us in explaining the trends in desorption, but they often fall short of the mechanism of interactions.
Kinetic studies are becoming increasingly important in clarifying adsorption- desorption processes (Pignatello and Xing, 1996). Adsorption data may be fitted to any number of equations, ranging from zero- and second-order equations to the parabolic diffusion law, the Elovich equation, and the modified Freundlich equa- tion (Table VII).
The particular equation used to describe adsorption-desorption rates is the one that best fits the data. Few researchers have investigated the adsorptiondesorp- tion kinetics of As, but Elkhatib et al. ( I 984) found that the Elovich and modified Freundlich equations described the sorption kinetics of As"' by 10 surface and sub- surface soils, whereas the modified Freundlich equation described the desorption kinetics of As"'. The fact that the Elovich equation did not describe the desorption rate of As"' may be indicative of the premises on which the equation is based and may limit the application of the equation across a broad spectrum of soils.
A number of studies have investigated the adsorption of AsV and As"' by pure minerals and soils. Many of these studies employed the Freundlich or Langmuir equations, but these equations do not adequately describe the adsorption of As to surfaces. Surface-complexation models are chemical models that give a general molecular description of adsorption using an equilibrium approach (Goldberg,
Q,,,(t) = Q,,o(l - ePkl .7 + QJl - e-k2.t)

ARSENIC IN THE SOIL ENVIRONMENT: A REVIEW 179
Table VII
Summary of Equations Used to Describe Various Kinetic Models"
Equation Formulah
Zero order First order
Parabolic diffusion Two-constant rate Elovich-type equation Modified Freundlich
C, = C, + Kr InC, = InC,, + Kt
C, = C,, + Kdt InC, = InC,, + K In t C, = C,, + K In t
InC, = InC, + p, In t + p2C + &T
Second order VC, = K,, - Kt
"Garcia-Rodeja and Gil-Sortes, 1995: reprinted by permission of the publisher.
"C, = final concentration of adsorbate as a function of contact time; C, = initial concentration of adsorbate; K = rate constant; t = time; p and Tare independent variables.
1992). Only a few studies (Goldberg, 1986; Goldberg and Glaubig, 1988; Belzile and Tessier, 1990; Manning and Goldberg, 1996) have investigated surface com- plexation modeling as a means of quantifying the adsorption of AsV by different surfaces. The constant-capacitance model (CCM) has been used to describe AsV adsorption on pure systems (Goldberg, 1986; Manning and Goldberg, 1996) and soils (Goldberg and Glaubig, 1988). Goldberg and Glaubig (1988) showed that the CCM adequately described AsV adsorption on an Imperial soil series (fine, mont- morillonitic, hyperthermic vertic torrifluvent) up to pH 9 but was unable to de- scribe As" adsorption in the pH 9-12 range. Similarly, Manning and Goldberg ( 1 996) reported that the CCM adequately described adsorption envelopes of AsV, P, and Mo by goethite and gibbsite. However, the authors applied the CCM mod- el using both the one-site (monodentate) and two-site (monodentate + bidentate) conceptualization of the oxide surface. The CCM, using both these approaches, gave similar descriptions of the experimental data, indicating that the present un- derstanding of anion adsorption on mineral surfaces is not complete (Manning and Goldberg, 1996).
VII. SOIL As AND VEGETATION
Arsenical compounds have been widely used as pesticides and herbicides in agriculture. Attention has therefore focused on the accumulation of As in agricul- tural soils and the possible toxic effects on plant production. Extensive research on the effects of As on plant production are well documented (Jacobs ef al., 1970;

180 E. SMITH ETAL.
Steevens et al., 1972; Anastasia and Kender, 1973; MacLean and Leangille, 198 1 ; Jiang and Singh, 1994).
A. SOIL As AND PLANT UPTAKE
The accumulation of As in the edible parts of most plants is generally low (Vaughan, 1993; O'Neill, 1995). Plants seldom accumulate As at concentrations hazardous to human and animal health because phytotoxicity usually occurs be- fore such concentrations are reached (Walsh and Keeney, 1975). Thus, the major hazard for animal and human systems is ingesting As-contaminated soils or con- suming contaminated water.
Uptake of As by plants occurs primarily through the root system, and the high- est As concentrations are reported in plant roots and tubers (Anastasia and Kender, 1973; Marin et al., 1993). Therefore, tuber crops (e.g., potatoes) could be expect- ed to have higher As concentrations than other crop types when grown in pollut- ed soils. This appears not to be the case, since potatoes grown in a sandy soil that received As additions ranging from 45 to 720 kg As ha-' accumulated only 0.5 mg As kg-' in the potato tuber (Jacobs et al., 1970). In contrast, the external pota- to peels had As concentrations of up to 84 mg As kg- I . This was attributed to con- tamination from soil adhering to the surface peels.
1. Crop Tolerance to As
A considerable variation in plant sensitivity to As exists among plant species (Jacobs et al., 1970; Jiang and Singh, 1994). Vegetable crops grown in three soils (Lakeland loamy sand, Hagerstown clay loam, and Christiana clay loam) in a greenhouse trial exhibited a range of sensitivities to sodium arsenate (0-500 mg As kg- I ) . Plant sensitivity followed the order green beans > lima beans = spinach > radish > tomato > cabbage (Woolson, 1973).
The symptoms of phytotoxicity may vary between species. Tomato plants grown in soils with high As background concentrations (100-130 mg As kg- I )
showed leaf dieback from the tip and poor-quality fruit set (Fergus, 1955). Fruit trees grown on replanted orchard sites commonly exhibit retarded early growth, to which As toxicity may contribute (Davenport and Peryea, 1991). Similarly, rice grown on former cotton-producing soils that had a history of repeated MSMA ap- plications showed indications of susceptibility to straighthead disease (abnormal- ly developed or sterile flowers resulting in low grain yields) under flooded soils conditions (Wells and Gilmor, 1977). The range of soil-As concentrations that may be phytotoxic is summarized in Fig. 6. Although the data are not extensive, they highlight both the broad range of concentrations of soil As over which toxicity symptoms may occur and the narrow margin that exists between background con-

ARSENIC IN THE SOIL ENVIRONMENT: A REVIEW 181
A
, TF I E
I I I &
Figure 6 Range of As concentrations in soils at which crops may exhibit phytotoxic symptoms. (A) Woolson, 1973; ( B ) Jacobs ef a/., 1970; (C) Woolson ef a/., 1973; (D) Woolson et al., 1971b; (E) Steevens ef al., 1972; (F) Wells and Gilmor, 1977.
centrations of As (<40 mg As kg- in most soils) and phytotoxic concentrations. As discussed earlier, the total concentration of As in soil is a poor indicator of
its bioavailability. The concept of bioavailability generally refers to some fraction of the total amount of As present in the soil that best correlates with plant avail- ability (Pierynski et al., 1994). Woolson et al. (1971a) related plant-available As to the total soil As and found that the amount of water-soluble As was better cor- related with plant growth than with total soil As. Plants absorb nutrients and met- als from the soil solution, and the “bioavailable” As soil pool may be a better in- dicator than total As concentration of plant phytotoxicity in the soil (Woolson et al., 1971; O’Neill, 1995). Sadiq (1986) reported that As concentration in maize was correlated with the water-extractable As but not with the total As concentra- tions in calcareous soils. Jiang and Singh (1994) found a similar relationship be- tween the As concentrations of barley and ryegrass plants grown in greenhouse ex- periments and soils (typic udipsamment and humaquept) fortified with As.
Reviewing the literature on As, Sheppard (1992) concluded that soil type is the only significant variable when considering plant phytotoxicity for inorganic As. It was reported that inorganic As was five times more toxic to plants in sands (mean = 40 mg As k g ’ ) than in clay (mean = 200 mg As kg-’) soils. Arsenic phyto- toxicity is expected to be greater in sandy soils than in other soil types, since sandy soils generally contain low amounts of Fe and A1 oxides and clays. These soil con- stituents have been implicated in the adsorption of As from solution in soils.

182 E. SMITH ETAL.
The nature of As species in the soil solution may also determine the phytotox- icity. Although As is primarily present as AsV or As"' in the soil-water environ- ment (Bohn, 1976), MMAA and DMAA compounds may also be present. Marin et af. (1992) reported that both As']' and MMAA were found to be phytotoxic to rice plants grown in nutrient solution, whereas AsV did not affect plant growth at the same concentration (0.8 mg As liter-l). The degree of As uptake by rice fol- lowed the trend As"' > MMAA > AsV > DMAA. However, these observations were made after nutrient solutions were amended with different As compounds to produce concentrations (0.05,0.20, and 0.80 mg As literp1) that were much high- er than those encountered under normal field conditions.
Furthermore, the effect of soil As on plant growth may be complicated by the presence of competing anions, notably P. Phosphate and As exhibit similar phys- iochemical behavior in the soil, and both synergistic and antagonistic effects of P on uptake of As by plants have been reported. Davenport and Peryea (1991) have reported a reduction of As uptake by plants after the application of P. In other stud- ies, P has been found to increase As availability and therefore to increase the As concentration in plants (Woolson, 1973). The increased availability of As is par- ticularly noticeable in sandy soils, where fewer adsorption sites are present and added Pmay displace some of the bound As ions into soil solution (O'Neill, 1995). Apart from P, N fertilizers, lime, and SO:- have also been observed to alleviate or depress the availability of As to plants (Thomas, 1977; Merry et al., 1986).
VIII. SOIL As AND MICROORGANISMS
The effects of metal pollution on biotic communities have been extensively stud- ied, and many of these studies have focused on the adaptation of bacterial com- munities through the development of resistance or tolerance mechanisms.
Biological transformations are important in redistributing As in soils (Fig. 7). Arsenic may have a direct influence on the microbial populations present in the soil. Decreases have been reported in microbial populations in soils that have been polluted with As compounds (Malone, 1971; Bisessar, 1982; Maliszewska et al., 1985). Bisessar (1982) reported that the decrease in population counts of bacteria, actinomycetes, fungi, and nematodes was significantly correlated (p 5 0.05) with concentrations of As in soil collected from several sites near a secondary Pb smelter (Table VIII). Although As may have influenced a decline in the soil mi- croflora population, the decline of the soil biological population was probably due to the combined effects of all metals present. Speir et af. (1992), in contrast, re- ported that very few negative effects were attributable to Cu, Cr, and As in a pot trial conducted to assess the feasibility of using CCA and boric-treated sawdust as a soil amendment. The total concentrations of elements in the pot trial (45, 136,

ARSENIC IN THE SOIL ENVIRONMENT: A REVIEW 183
Oxygen present
HAs02 H2AS04-
(CH&&O(OH)
1 Arsenical Herbicides (CH&AsO and Pesticldes
Industrial Sources Local
Dust As406 I I Consumptton
Oxygen absent
H 2 A ~ 0 4 - H A s 0 2
T
I iH
Leaching
Figure 7 Soil-air cycle (WHO, 198 1; reprinted by permission of the publisher).
63, and 32 mg kg- for Cu, Cr, As, and B, respectively) were considerably lower than those present in Bisessar's (1982) work.
Considerable variation in tolerance to As compounds applied to the soil has been shown by the soil microflora. Maliszewska et al. (1985) reported that As"' com- pounds were more toxic than As" compounds to microorganisms important to maintaining soil fertility. Arsenite compounds were applied at 500, 1500, and 3000 mg As kg- I and As" compounds at 1000,5000, and 10,000 mg As kg- to sandy and alluvial soils. The influence of As"' and As" compounds on the soil microflo- ra differed, depending on the microflora and the soil type. In sandy soil, As"' de- pressed the growth of bacteria, whereas As" stimulated their proliferation. Over- all, both As compounds had little effect on the development of actinomycetes and fungi flora and suppressed the growth of Azotobacter sp.
Maliszewska et al. (1985) also measured a decrease of approximately 30% in dehydrogenase activity in the soils. The decrease in activity was greatest in the sandy soil and with the application of As"'. Dehydrogenase is an unspecific en- zyme for assessing the effect of As compounds on the soil microflora; other au- thors have found that enzymes involved in more specific biochemical reactions are inhibited by the addition of As to the soil environment. Tabatabai (1977) reported that As"' greatly inhibited the urease activity of some soils at a concentration of 375 mg As"' kg- I , but the addition of As" at a similar concentration to the soil had no effect on activity. Bardgett er al. (1 994) investigated microbial properties

Table VIII
Total As, Pb, Cd, and Cu Concentration and Counts of Sod Microflora Near a Secondary Pb SmelteP
Total heavy metal concentration (mg kg-I) Total no. of colonies in log soil
Location (eastofsource) Pb As Cd Cu Bacteria Actinomyces Fungi Nematodes Earthworms
~ _ _ _ _ _ _ _ _ _ _ _ ~ ~
15 m 28,000 972 151 599 10.5 1 2.5 16 0 90 m 8333 554 102 398 12 1.4 3.2 30.9 0 150 m 4800 230 33 287 12.6 1.6 3.5 26.6 0 180 m 3564 163 26 333 15.1 2.1 4.3 58 1.3 Control (1000 m 703 57 5 73 17.2 2.4 4.6 98 2.3 south)
“Bisessar, 1982; reprinted with kind permission from Kluwer Academic Publishers.

ARSENIC IN THE SOIL ENVIRONMENT: A REVIEW 185
of the surface (0-50 mm) of a pasture soil contaminated by runoff by preserving liquor runoff from an adjacent timber-treatment plant. Total concentrations of Cr, Cu, and As ranged from 86 to 1260,70 to 1233, and 79 to 1265 mg kg-', respec- tively. They found no significant relationship between soil As, Cu, and Cr con- centrations and urease activity, whereas a significant relationship was observed be- tween As, Cu, and Cr (? values = As, 0.923; Cu, 0.933; Cr, 0.872) and a decline in sulphatase activity. However, many factors may influence the inhibitory effect of metals on the soil microflora, with soil type being an important factor in deter- mining the bioavailability of the metals (Maliszewska et al., 1985).
A. BIOTRANSFORMATION OF As IN SOILS
The biotransformation of As in soils has been recognized for many years. Two microbial processes, oxidation and reduction, are of current interest because of their possible application for bioremediation of contaminated soil. A number of re- view articles have discussed As and its transformation in various environments (Cullen and Reimer, 1989; Ehrlich, 1996), and these articles provide a more in depth review of the subject than is provided here.
Bacterial oxidation was first identified by Green in 19 18 wh en a bacterium from a cattle-dipping fluid was isolated. Many other heterotrophic As"' oxidizing bac- teria have since been characterized, with many bacterium classified as Bacillus or Pseudomonas spp. Studies by Osborne and Ehrlich (1976) reported that Alcali- genesfaecalis was able to oxidize As"' to As", although it was not clear if their organism derived energy from this process. The oxidation of reduced As species has been less widely studied than the processes of microbial reduction of As.
Various bacteria, fungi, and algae organisms that are able to reduce As com- pounds have been identified. The reduction of AsV to As"' has been reported to be carried out by Pseudomonasjuorescens under anaerobic conditions, wine yeast, rumen bacteria, and cyanobacteria (Cullen and Reimer, 1989). Cheng and Focht ( 1979) have also identified that Pseudornonas and Alcaligenes were able to pro- duce arsine gas directly from As" solutions in glucose and urea-enriched soils un- der anaerobic conditions. The ability of organisms to reduce inorganic As species directly has only been reported so far for bacteria. The bacterial methylation of in- organic As has been extensively studied in methanogenic bacteria. Methanogenic bacteria are a morphologically diverse group that produce methane as their pri- mary metabolic end product under anaerobic conditions (Tamaki and Franken- berger, 1992). McBride and Wolfe (1971) reported that cell extracts of whole cells of the Methanobacterium strain MOH, growing anaerobically, reduced and methy- lated As" to dimethylarsine. Anaerobic biomethylation of As only proceeds to di- methylarsine, which is stable in the absence of 0, but is rapidly oxidized under aerobic conditions (Cullen and Reimer, 1989). The pathway of As" methylation

186 E. SMITH ETAL.
initially involves the reduction of AsV to As"', with the subsequent methylation of As"' to dimethylarsine by coenzyme M (Frankenberger and Losi, 1995).
In addition to bacteria, several fungi species have the ability to reduce As species. It is well known that fungi and algae are able to methylate As. Toxic trimethylarsine gas is volatilized, and the liberalization of this gas by molds grow- ing on wallpaper decorated with As pigments led to a number of poisoning inci- dents in the 1800s in England and Germany (Challenger, 1945). Cox and Alexan- der (1973a) reported that three fungal species, Candida humicola, Gliocladium roseurn, and Penicilliurn sp. were capable of transforming methylarsonic and di- methylarsonic acids to trimethylarsine. Further work by Cox and Alexander (1973b) showed that other anions, notably phosphate, inhibited the formation of trimethylarsine from inorganic As and methylarsonic acid.
The extent to which microbial activity is involved in the transformation and movement of As in the soil is difficult to quantify. Woolson (1977) detected the gen- eration of alkyarsines, notably dimethyarsine and trimethylarsine, in both anaero- bic and aerobic conditions in the laboratory. Furthermore, the rate of formation of volatile compounds from the three As compounds applied to the soil (74As-sodium arsenate, I4C-MSMA, and 14C-cacodylic acid) was fastest under aerobic condi- tions, with the nature of the As compounds influencing the rate of formation. Hass- ler el al. (1984) suggested that, in addition to methylating As compounds, microbi- ological mobilization of As may occur under certain soil conditions. Woolson and Kearney (1973) found that the loss of 14C-cacodylic acid, applied to a range of soils and incubated over several months, was influenced by soil type, concentration of cacodylic acid applied, and soil moisture levels. Losses were attributed to the pro- duction of methylarsines and were greatest at the higher rates of cacodylic applied (100 mg As kg-I) in anaerobic conditions and in sandy soils.
Few studies have investigated the long-term effects of microbial transforma- tions of As species. In many instances, microbial influences on As sorption and desorption processes have been largely ignored in laboratory studies. Under short- term laboratory studies and with previously uncontaminated soils, the microor- ganisms may have little influence on sorption processes in soils. However, there may be important implications for long-term studies, or where the soil microflora and microfauna have been predisposed to As when studying As sorption-desorp- tion processes in laboratory situations.
M. CONCLUSIONS
Arsenic is widely distributed in nature, with traces of As in the soil almost uni- versal. The behavior of As in the soil is dominated by As speciation. In soil solu- tions the inorganic As species predominate, either as AsV or As"'. Under moder- ately oxidizing conditions (> + 100 mV) AsV will predominate, whereas under

ARSENIC IN THE SOIL ENVIRONMENT: A REVIEW 187
moderately reducing conditions As111 will predominate. The conversion of As from one oxidation state to another in soils is also affected by other soil parameters, in- cluding pH and microbial activity. The importance of microbial activity in con- trolling As species present in soil environments is well acknowledged, but the pathways and kinetics of the microbial processes are not well understood.
An understanding of the physiochemical behavior of As in the soil is important for quantifying the persistence and bioavailability of As in the environment. The sorption of As onto soil surfaces plays an important role in mediating the avail- ability of As in the environment. Iron, Al, and to a lesser extent Mn oxides are im- portant soil constituents in controlling soil solution concentrations of As. Soil pH has a major influence on the availability of As. Arsenic is apolyprotic acid, and pH has a major influence on the valence charge of the As ion in soil solution and hence on the As adsorbed.
In general, bioaccumulation of As to hazardous levels for human and animal consumption in the edible portions of plants seldom occurs because of the phyto- toxic effects before such hazardous levels are reached. Plants accumulate the high- est concentrations of As in plant roots. Unlike other elements such as P, As is not generally translocated to other parts of the plant. However, some plant species have been found to translocate As to a greater extent than others.
Future research of As in soils is needed to understand factors controlling the na- ture of As species in the soil solution, as well as the role of microbes in control- ling As speciation. Although adsorption processes have been well studied, further work is needed in understanding desorption processes and the factors that influ- ence the kinetics of these processes. Scope also exists for studies on both plant and microbial uptake of As and the possible use of plants as low-cost, long-term means of remediation of As-contaminated sites.
ACKNOWLEDGMENTS
We would like to thank Primary Industries for South Australia and the Co-operative Research Cen- tre for Soil and Land Management for their support.
REFERENCES
Adriano, D. C.. Page, A. L.. E1seewi.A. A., Chang. A. C.. and Straughan, I. (1980). Utilization and dis- posal of fly ash and other residues in terristrial ecosystems: Areview. J. Environ. Quol. 9,333-344.
Ajwa, H. A,, and Tabatabai, M. A. (1995). Metal-induced sulfate adsorption by soils: I. Effect of pH and ionic strength. Soil Sci. 159,3242.
Anastasia, E B., and Kender, W. J. (1973). The influence of soil arsenic on the growth of lowbeny bush. J. Environ. Quol. 2,335-337.
Anderson, M. A,, and Malotky, D. T. (1979). The adsorption of protolizable anions on hydrous oxides at the isoelectric pH. J. Colloid Interface Sci. 72,413427.

188 E. SMITH ETAL.
Anderson, M. A., Ferguson, J. F., and Gavis. J. (1976). Arsenate adsorption on amorphous aluminium hydroxide. J . Colloid Inferface Sci. 54,391-399.
Australian Bureau of Statistics. (1995). “Imports of arsenic by all countries and states.” Adelaide, Aus- tralia.
Australian and New Zealand Environment and Conservation Council (ANZECC). (1 992). “Australian and New Zealand Guidelines for the Assessment and Management of Contaminated Sites.” Aust. Govt. Pub. Service, Canberra.
Bardgett, R. D., Speir, T. W., Ross, D. J., Yeates, G. W., and Kettles, H. A. (1994). Impact of pasture contamination by copper, chromium, and arsenic timber preservative on soil microbial properties and nematodes. Biol. Fen. Soils. 18,7 1-79.
Barrow, N. J. (1974). On the displacement of adsorbed anions from soil: 2. Displacement of phosphate by arsenate. Soil Sci. 117,28-33.
Barrow, N. J. (1983). A mechanistic model for describing the sorption and desorption of phosphate by soil. J . Soil Sci. 34,733-750.
Barrow, N. J. (1984). Modelling the effects of pH on phosphate sorption by soils. J . Soil Sci. 35, 283-297.
Barrow, J., Bowden, J. W., Posner. A. M., and Quirk, J. P. (1980). Describing the effects of electrolyte on adsorption of phosphate by a variable charge surface. Aust. J . Soil Res. 18,395-404.
Barzi, F., Naidu, R., and McLaughlin, M. J. (1996). Contaminants and the Australian soil environment. In “Contaminants and the Australian Soil Environment in the Australasia-Pacific Region” (Naidu et al., eds.), pp. 45 1484. Kluwer Academic Publishers, Dordrecht.
Belzile, N., and Tessier, A. (1990). Interactions between arsenic and iron oxyhydroxides in lacustrine sediments. Geochim. Cosmochim. Acfa 54, 103-109.
Bencko, V., and Symon, K. (1977). Health aspects of burning coal with a high arsenic content. Envi- ron. Res. 13,378-385.
Beretka, J., and Nelson, P. (1994). The current state of utilisation of fly ash in Australia. In “Ash-A Valuable Resource,” Vol. I , pp. 51-63. South African Coal Ash Association.
Bernard, D. W. (1983). Arsenic leachate from an abandoned smelter, Delero. Ontario, Canada. In “Pa- pers of the International Conference on Groundwater and Man,” Vol. 2, pp. 29-41. Aust. Govt. Pub. Service, Canberra.
Bhumbla, D. K., and Keefer. R. F. (1994). Arsenic mobilization and bioavailability in soils. In “Ar- senic in the Environment, Part I : Cycling and Characterization.” (J. 0. Nriagu, ed.), pp. 51-82. Wiley & Sons, New York.
Bisessar, S. (1982). Effects of heavy metals on microorganisms in soils near a secondary lead smelter. Waf. Air Soil Poll. 17,305-308.
Bishop, R. F., and Chisholm, D. (1961). Arsenic accumulation in Annapolis Valley orchards. Can. J . Soil Sci. 42,77-80.
Bohn, H. L. (1976). Arsenic Eh-pH diagram and comparisons to soil chemistry of phosphorus. Soil Sci.
Bolan, N. S., Syers, J. K., and Tillman, R. W. (1986). Ionic strength effects on surface charge and ad-
Braman, R. S., and Foreback, C. C. (1973). Methylated forms of arsenic in the environment. Science
Brannon, J. M., and Patrick, Jr., W. H. (1987). Fixation, transformation, and mobilization of arsenic in sediments. Environ. Sci. Technol. 21,450459.
Carbonell Barrachiina, A., Burl6 Carbonell, F., and Mataix Beneyto, J. (1996). Kinetics of arsenite des- orption in Spanish soils. Commun. Soil Sci. Plant Anal. 27, 3101-31 17.
Challenger, F. (1945). Biological methylation. Chem. Rev. 36,315-361. Cired in Hassler, R. A,, Klein, D. A., and Meglen, R. R. (1984). Microbial contributions to soluble and volatile arsenic dynam- ics in retorted oil shale. J. Environ. Qual. 13,466470,
121, 125-127.
sorption of phosphate and sulphate by soils. J . Soil Sci. 37,379-388.
182,1247-1 249.

ARSENIC IN THE SOIL ENVIRONMENT: A REVIEW 189
Charter, R. A., Tdbatabai, M. A., and Schafer, J. W. (1995).Arsenic, molybdenum, selenium, and tung- sten contents of fertilizers and phosphate rocks. Commun. Soil Sci. Planr Anal. 26, 3051- 3062.
Chen, Jen Chen, and Lin. Li-Ju. (1994). Human carcinogenicity and atherogenicity induced by chron- ic exposure to inorganic arsenic. In “Arsenic in the Environment: Part 11. Human Health and Ecosystems Effects.” (J. 0. Nriagu, ed.), pp. 109-157. Wiley & Sons, New York.
Cheng, C. N., and Focht. D. D. (1979). Production of arsine and methylarsines in soil and in culture. Appl. Environ. Microbiol. 38,494-498.
Chilvers, D. C.. and Peterson, P. J. (1987). Global cycling of arsenic. In “Lead, Mercury, Cadmium, and Arsenic in the Environment” (T. C. Hutchinson and K. M. Meema. eds.), pp. 279-301. Wiley & Sons, New York.
COX. D. P.. and Alexander, M. ( 1973a). Production of trimethylarsine gas from various arsenic com- pounds by three sewage fungi. Bull. Environ. Contam. Toxicol. 9 , 8 6 8 8 .
COX, D. P., and Alexander, M. (1973b). Effect of phosphate and other anions on trimethylarsine for- mation by Candida humicola. Appl. Microbiol. 25,408-413.
Crecelius, E. A., Johnson, C. J., and Hoffer, G. C. (1974). Contamination of soils near a copper smelter by arsenic, antimony, and lead. Wat. Air Soil Poll. 3,337-342.
Cullen, W. R., and Reimer, K. J. (1989). Arsenic speciation in the environment. Chem. Rev. 89,
Curtin, D., Syers, J. K., and Bolan, N. S. (1992). Phosphate sorption by soil in relation to exchange- able cation composition and pH. Ausr. J. Soil Rex 31, 137-149.
Das, D., Samanta, G., Mandal, B. K.. Chowdhury, T. R., Chanda, C. R., Chowdhury, P. R., Basu, G. K., and Chakraborti, D. (1996). Arsenic in groundwater in six districts of West Bengal, India. Environ. Geochem. and Health. 18,5-15.
Davenport, I. R., and Peryea, F. J. (1991). Phosphate fertilizers influence leaching of lead and arsenic in soil contaminated with lead arsenate. War. Air Soil Poll. 57-58, 101-110.
Department of Mines. Perth, Western Australia. (1980). “Mineral Resources of Western Australia,” p. 21. Dept. of Mines, Perth, Western Australia.
Deuel, L. E., and Swoboda, A. R. (1972). Arsenic solubility in a reduced environment. Soil Sci. Am. Proc. 36,276-278.
DiGiano. F. A.. Baldauf, G., Frick, B., and Sontheimer, H. (1978). A simplified competitive equilibri- um adsorption model. Chem. Eng. Sci. 33,1667-1673.
DIPMAC Report. (1992). “Report on the Management of Contaminated Waste at the Cattle Dip Sites in North Eastern New South Wales.” NSW Gov.. Sydney, Australia.
Doelman, P., and Haanstra, L. (1984). Short-term and long-term effects of cadmium, chromium, cop- per, nickel, lead, and zinc on soil microbial respiration in relation to abiotic soil facetors. Plant and Soil 79,3 17-327.
Dowdy, R. H., and Volk, V. V. (1984). Movement of heavy metals in soils. In Chemical Mobility and Reactivity in Soil Systems (0. W. Nelson et al., eds.), pp. 229-240. SSSA Special Publication No. 1 1. ASA and SSSA, Madison, WI.
Dudas, M. J. (1987). Accumulation of native arsenic in acid sulphate soils in Alberta. Can. J. Soil Sci. 67,317-331.
Dudas, M. J., and Pawluk, S. (1980). Natural abundances and mineralogical partitioning of trace ele- ments in selected Alberta soils. Can. J. Soil Sci. 60,763-771.
Ehrlich, H. L. (1996). Geomicrobiology, 3rd ed., pp. 276-286. Marcel Dekker, New York. Elkhatib, E. A,, Bennett, 0. L.. and Wright, R. J. (1984). Arsenite sorption and desorption in soils. Soil
Fendorf, S., Eick, M. J., Grossl, P., and Sparks, D. L. (1997). Arsenate and chromate retention mecha-
Fergus, I. F. (1955). A note on toxicity in some Queensland soils. Queensland J . Ag. Sci. 12,95-100.
7 13-764.
Sci. Soc. Am. J . 48, 1025-1 030.
nisms on goethite. I. Surface structure. Environ. Sci. Technol. 31,315-320.

190 E. SMITH ETAL.
Fordham, A. W., and Norrish, K. ( 1979). Arsenate-73 uptake by components of several acidic soils and its implications for phosphate retention. Aust. J. Soil Res. 17,307-316.
Fordham, A. W., and Norrish, K. (1983). The nature of soil particles particularly those reacting with arsenate in a series of chemically treated samples. Ausr. J. Soil Rex 21,455477.
Fowle 111, J. R., Abemathy, C. 0.. Mass, M. J., McKinney, J. D., North, D. W., Ohanian, E. V., and Uthus, E. (1991). Arsenic research needs. Trace Subst. Environ. Healrh 25,257-271.
Fowler, B. A. (1977). Toxicology of Environmental Arsenic. In “Toxicology of Trace Elements.” (R. A. Coyer and M. A. Mehlman, eds.), pp. 79-122. Wiley & Sons, New York.
Frank, R., Braun, H. E., Ishida, K., and Suda, P. (1976). Persistent organic and inorganic pesti- cide residues in orchard soils and vineyards of Southern Ontario. Can. J. Soil Sci. 56, 463- 484.
Frankenberger, W. T., and Losi, M. E. (1995). Applications of bioremediation in the cleanup of heavy metals and metalloids. In “Bioremediation,” (H. D. Skipper and R. F. Turco, eds.), pp. 173-210. SSSA Special Publication 43 Soil Sci. SOC. Am ., Madison, WI.
Freeman, J. S., and Rowell, D. L. (1981). The adsorption and precipitation of phosphate onto calcite. J. Soil Sci. 32,7544.
Frost, R. R., and Griffin, R. A. (1977). Effect of pH on adsorption of arsenic and selenium from land- fill leachate by clay minerals. Soil Sci. SOC. Am. J . 41,53-57.
Garcia-Rodeja, I. , and Gil-Sortes, F. (1995). Laboratory study of phosphate desorption kinetics in soils of Galicia (N.W. Spain). Commun. Soil Sci. Plant Anal. 26,2023-2040.
Gilmor, J. T., and Wells, B. R. (1980). Residual effects of MSMA on sterility in rice cultivars. Agron. J. 72, 10661067.
Goldberg, S. (1986). Chemical modelling of arsenate adsorption on aluminium and iron oxide miner- als. Soil Sci. SOC. Am. J . 50, 1154-1 157.
Goldberg, S. (1992). Use of surface complexation models in soil chemical systems. Adv. Agron. 47,
Goldberg, S., and Glaubig, R. A. (1988). Anion sorption on a calcareous montmorillonitic soil-Ar- senic. Soil Sci. Am. J . 52, 1297-1300.
Goodroad, L. L., and Caldwell, A. C. (1979). Effects of phosphorus fertilizer and lime on the As. Cr, Pb, and V content of soils and plants. J. Environ. Quul. 8,493496.
Green, H. H. (1918). Rep. Ver. Res. S. Afr 516 (1918), 595 Cired in Cullen, W. R., and Reimer, K. J. (1989). Arsenic speciation in the environment. Chem. Rev. 89,713-764.
Grossl, P. R., Eick, M. Sparks, D. L., Goldberg, S., and Ainsworth, C. C. (1997). Arsenate and chro- mate retention mechanisms on goethite. 2. Kinetic evaluation using a pressure-jump relaxation technique. Environ. Sci. Technol. 31,32 1-326.
Gustafsson, J. P., and Jacks. G. (1995). Arsenic geochemistry in forest soil profiles as revealed by sol- id-phase studies. Appl. Geochem. 10,307-3 15.
Harrison, J. B., and Berkheiser, V. E. (1982). Anion interactions with freshly prepared hydrous iron ox- ides. Cluys Clay Mine,: 30,97-102.
Harter, R. D., and Naidu, R. (1995). Role of metal-organic complexation in metal sorption by soils. Adv. Agmn. 55,219-264.
Hassler, R. A., Klein, D. A., and Meglen, R. R. (1984). Microbial contributions to soluble and volatile arsenic dynamics in retorted oil shale. J. Environ. Qual. 13,466-470.
Hess, R. E., and Blanchar. R. W. (1976). Arsenic stability in contaminated soils. Soil Sci. SOC. Am. J .
Heylar, K. R., Munns, D. N., and Burau, R. G. (1976). Adsorption of phosphate by gibbsite. II. For-
Hillier, G. (1980). Arsenic. In “Australian Mineral Industry Annual Review for 1978,” p. 50. Aust.
Hiltbold, A. E., Hajek, B. F.. and Buchanan, G. A. (1974). Distribution of arsenic in soil profiles after
233-330.
40,847-852.
mation of a surface complex involving divalent cations. J. Soil Sci. 27,35 1-323.
Govt. Publishing Service, Canberra.
repeated applications of MSMA. Weed Sci. 22,272-275.

ARSENIC IN THE SOIL ENVIRONMENT: A REVIEW 191
Hingston, F. J., Atkinson, R. J., Posner. A. M., and Quick, J. P. (1967). Specific adsorption of anions. Narure 215, 1459-1461.
Hingston, F. J., Posner. A. M., and Quirk, J. P. (1971). Competitive adsorption of negatively charged ligands on oxide surfaces. Discuss. Faraday Soc. 52,334-342.
Hingston. F. J., Posner, A. M., and Quirk, J. P. (1972). Anion adsorption by goethite and gibbsite. I: The role of the proton in determining adsorption envelopes. J. Soil Sci. 23, 177-191.
Jacobs, L. W., Syere, J. K., and Keeney. D. R. (1970). Arsenic sorption by soils. Soil Sci. SOC. Am. Proc. 34,750-754.
Jiang, Q. Q.. and Singh, B. R. (1994). Effect of different forms and sources of arsenic on crop yield and arsenic concentration. War. Air Soil Poll. 74,32 1-343.
Johnson, L. R., and Hiltbold, A. E. (1969). Arsenic content of soil and crops following use of methanearsonate herbicides. Soil Sci. Soc. Am. Proc. 33,279-282.
Kabata-Pendias, A., and Pendias, H. (1992). “Trace Elements in Soils and Plants,” 2nd ed. CRC Press, Boca Raton, FL.
Kiss, A. M., Oncsik, M., Dombovari, J. Veres. S., and Acs, G. (1992). Dangers of arsenic drinking and irrigation water to plants and humans. Antagonism of arsenic and magnesium. Acra Agron. Hung. 41,3-9.
Leblanc, M., Achard, B., Ben Othman. D., and Luck, J. M. ( I 996). Accumuulation of arsenic fron acidic mine waters by ferruginous bacterial accretions (stromatolites). Appl. Geochem. 11,541-554.
Leonard, A. (1991 ).Arsenic. In “Metals and Their Compounds in the Environment. Occurrence, Analy- sis, and Biological Relevance,” 2nd ed. (E. Merian, in cooperation with Clarkson et al., eds.), pp. 751-773. Weinheim, VCH.
Li, X., and Thornton, I. (1993). Arsenic, antimony, and bismuth in soil and pasture herbage in some old metalliferous mining areas in England. Environ. Geochem. Health 15, 135-144.
Livesey, N. T., and Huang, P. M. (1981). Adsorption of arsenate by soils and its relation to selected chemical properties and anions. Soil Sci. 131,88-94.
Loebenstein, J. R. (1993). Arsenic. In “Minerals Yearbook 1990,” pp. 167-170. US. Govt. Printing Oftice, Washington, D.C.
Lookman, R., Freese, D.. Merckx, R., Vlassak, K., and Van Riemsdijk, W. H. (1995). Long-term ki- netics of phosphate release from soil. Environ. Sci. Technol. 29, 1569-1575.
Lu, F, J. (1990). Blackfoot disease: Arsenic or humic acid? Lancer 336, 115-1 16. Lumsdon, D. G., Fraser, A. R., Russell, J. D., and Livesey, N. T. (1984). New infrared band assign-
ments for the arsenate ion adsorbed on synthetic goethite (a-FeOOH). J. Soil Sci. 35, 381-386. Lund, U., and Fobian, A. (1991). Pollution of two soils by arsenic, chromium, and copper, Denmark.
Geodema 49,83-103. MacLean, K. S., and Langille, W. M. (1981). Arsenic in orchard and potato soils and plant tissue. Plant
and Soil 61,4 1 3 4 1 8. Maliszewska, W., Dec, S.. Wierzbicka, H., and Wozniakowska, A. (1985). The influence of various
heavy metal compounds on the development and activity of soil micro-organisms. Environ. Pol- hi. 9,: A. 37, 195-2 15.
Malone, C. R . (1971). Responses of soil microorganisms to non-selective vegetation control in a fes- cue meadow. Soil Bid . Biochem. 3, 127-1 3 I .
Manful, G. A,, Verloo, M., and De Spiegeleer, F. (1989). Arsenate sorption by soils in relation to pH and selected anions. Pedologie 39,5548.
Manning, B. A,, and Goldberg, S. (1996). Modeling competitive adsorption of arsenate with phosphate and molybdate on oxide minerals. Soil Sci, Soc. Am. J. 60, 121-131.
Marin, A. R., Masscheleyn, P. H., and Patrick Jr., W. H. (1992). The influence of chemical form and concentration of arsenic on rice growth and tissue arsenic concentration. Plant and Soil 139,
Marin, A. R., Masscheleyn, P. H., and Patrick Jr., W. H. (1993). Soil redox-pH stability of arsenic specis 175-183.
and its influence on arsenic uptake by rice. Plant and Soil 152,245-253.

192 E. SMITH ETAL.
Masscheleyn, P. H., Delaune, R. D., and Patrick Jr., W. H. (1991). Effect of redox potential and pH on arsenic speciation and solubuility in a contaminated soil. Environ. Sci. Technol. 25, 1414-1418.
McBride, B. C., and Wolfe, R. S. (1971). Biosynthesis of dimethylarsine by Methnnobacrerium. Biochem. 10,43124317.
McLaren, R. G.. Carey, P. L., Cameron, K. C., Adams, J. A,, and Sedcole. J. R. (1994). Effect of soil properties and contact period on the leaching of copper, chromium, and arsenic through undis- turbed soils. In “15th World Cong. Soil Sci.,” Vol. 3a, pp. 156-169. Intl. Soc. Soil Sci. and The Mexican Soc. of Soil Sci., Acapulco, Mexico.
McMillan, M. G. (1988). Weed control in cotton: Agdex 151/640. NSW Agriculture and Fisheries. Melamed, R., Jurinak, J. J., and Dudley, L. M. (1995). Effect of adsorbed phosphate on transport of ar-
senate through an oxisol. Soil Sci. Am. J. 59, 1289-1294. Merry, R. H., Tiller, K. G., and Alston, A. M. (1983). Accumulation of copper, lead, and arsenic in some
Australian orchard soils. Aust. J . Soil Res. 21,549-561. Merry, R. H., Tiller, K. G., and Alston, A. M. (1986). The effects of contamination of soil with copper,
lead, and arsenic on the growth and composition of plants. I. Effects of season, genotype, soil tem- perature, and fertilizers. Plant and Soil 91, 115-128.
Naidu. R., and Rengasamy, P. (1993). Ion interactions and constraints to plant nutrition in Australian sodic soils. Aust. J. Soil Res., 31,801-819.
Naidu, R., Syers, J. K., Tillman, R. W., and Kirkman, J. H. (1990). Effect of liming and added phos- phate on charge characteristics of acid soils. J . Soil Sci. 41, 157-164.
Naidu, R., Merry, R. H., Churchman, G. J., Wright, M. J., Murray, R. S., Fitzpatrick, R. W., and Zarci- nas, B. A. (1993). Sodicity in South Australia-A review. Aust. J . Soil Res. 31,911-929.
Naidu. R., Bolan, N. S., Kookana, R. S., and Tiller, K. G. (1994). Ionic-strength and pH effects on the sorption of cadmium and the surface charge of soils. Eur J. Soil Sci. 45,419429.
Naidu, R., Smith, J., Smith, L. H., Tiller, K. G., and McDougall, K. W. (1995). ArsenicDDT residues at cattle tick contaminated sites: Preliminary investigation. CSIRO Division of Soils Technical Report No. 60. Adelaide, Australia.
National Academy of Sciences. ( I 977). “Medical and Biologic Effects of Environmental Pollutants: Arsenic.” National Research Council, Washington, D.C. Cited in Leonard, A. (1991). Arsenic. In “Metals and Their Compounds in the Environment. Occurrence, Analysis. and Biological Rele- vance” 2nd ed. (E. Merian, in cooperation with Clarkson er al., eds.), pp. 751-773. Weinheim, VCH.
National Food Authority. (1993). “Australian Food Standards Code: March, 1993.” Australian Govt. Pub. Service, Canberra.
National Research Council of Canada. (1978). “Effects of Arsenic in the Canadian Environment.” NRCC no. I539 1. Ottawa, Canada.
Natush, D. F. S., Bauer, C. F.. Matusiewicz, H., Evans, C. A., Baker, J., Loh, A., Linton, R. W., and Hopke, P. K. (1975). Characterization of trace metals in fly ash. In “International Conference on Heavy Metals in the Environment.” Vol. 11. pp. 553-575. Toronto.
Nriagu, J. 0. (1994). Arsenic: Historical perspectives. I n “Arsenic in the Environment: Part 1. Cycling and Characterization” (J. 0. Nriagu, ed.), pp. 3-15. Wiley & Sons, New York.
Nriagu, J. 0.. and Pacyna, J. M. (1988). Quantitive assessment of worldwide contamination of air, wa- ter, and soils by trace metals. Nature 333, 134-139.
O’Neill, P. (1990). Arsenic. In “Heavy Metals in Soils” (B. J. Alloway, ed.), pp. 83-99. Blackie. Lon- don.
O’Neill, P. (1995). Arsenic. In “Heavy Metals in Soils,’’ 2nd ed. (B. J. Alloway, ed.), pp. 105-121. Blackie, London.
Onken, B. M., and Hossner, L. R. (1995). Plant uptake and determination of arsenic species in soil so- lution under flooded conditions. J. Environ. Quaf. 24,373-38 l .
Osborne, F. H., and Ehrlich, H. L. (1976). Oxidation of arsenite by a soil isolate of alcaligenes. J . Appl. Bacreriol. 41.295-305.

ARSENIC IN THE SOIL ENVIRONMENT: A REVIEW 193
Oscarson. D. W., Huang, P. M., Defosse, C., and Herbillon. (1981). Oxidative power of Mn(IV) and Fe(II1) oxides with respect to As(1II) in terristrial and aquatic environments. Nature 291, 50-51.
Oscatson, D. W.. Huang, P. M., Liaw, W. K., and Hammer, U. T. (1983a). Kinetics of oxidation of ar- senite by various manganese dioxides. Soil Sci. SOC. Am. J. 46,644-648.
Oscarson, D. W., Huang. P. M., and Hammer, U. T. (1983b). Oxidation and sorption of arsenite by man- ganese dioxide as influenced by surface coatings of iron and aluminium oxides and calcium car- bonate. War. Air Soil Poll. 20,233-244.
Peryea. F. J. (1991). Phosphate induced release of arsenic from soils contaminated with lead arsenate. Soil Sci. SOC. Am. J . 55, 1301-1306.
Peryea, F. J., and Creger, T. L. (1994). Vertical distribution of lead and arsenic in soils contaminated with lead arsenate pesticide residues. War. Air Soil Poll. 78, 297-306.
Peterson, P. J., Benson, L. M., and Porter, E. K. (1979). Biogeochemistry of arsenic on polluted sites in SW England. In “Heavy Metals in the Environment, London, September 1979,” pp. 198-201, CEP Consultants Ltd., Edinburgh.
Petito, C. T., and Beck, B. D. (1990). Evaluation of evidence of nonlinearities in the dose-response curve for arsenic carcinogenesis. Trace Subsr. Environ. Health 24, 143-176.
Pierce, M. L., and Moore, C. B. (1980). Adsorption of arsenite and arsenate on amorphous iron hy- droxide from dilute aqueous solutions. Environ. Sci. Technol. 14,214-216.
Pierzynski, G. M., Schnoor, J. L., Banks, M. K., Tracy, J. C., Licht, L. A., and Erickson, L. E. (1994). Vegetative remediation at superfund sites. In “Mining and its Environmental Impact” (R. E. Hes- ter and R. M. Harrison, eds.), pp. 49-69. Royal SOC. of Chem., Cambridge.
Pignatello, J. I., and Xing, B. (1996). Mechanisms of slow sorption of organic chemicals to natural par- ticles. Environ. Sci. Tech. 30, 1-11.
Ragaini, R. C.. Ralston, H. R., and Robert, N. (1977). Environmental trace metal contamination in Kel- log, Idaho, near a lead smelting complex. Environ. Sci. Tech. 11,773-781.
Rimstidt, J. D., Chermak, J. A,, and Gagen, P. M. (1994). Rates of reaction of galena, sphalerite, chal- copyrite, and arsenopyrite with Fe(1n) in acidic solutions. In “Environmental Geochemistry of Sulfide Oxidation” (C. N. Alpers and D. W. Blowes, eds.), pp. 2-13. Am. Chem. Soc., Washing- ton, D.C.
Rittle, K., Dreiver, J., and Colberg, P. (1995). Precipitation of arsenic during bacterial sulfate reduc- tion. Geomicrobiol. J. 13, 1-1 1 .
Roy, W. R., Hassett, J. J., and Griffin, R. A. (1986). Competitive interactions of phosphate and molyb- date on arsenate adsorption. Soil Sci. 142,203-210.
Sachs, R. M., and Michael, J. L. (1971). Comparative phytotoxicity among four arsenical herbicides. Weed Sci. 19,558-564.
Sadiq, M. (1986). Solubility relationships of arsenic in calcareous soils and its uptake by corn. Plant and Soil 91,241-248.
Sadiq, M., Zaidi, T. H.. and Mian, A. A. (1983). Environmental behaviour of arsenic in soils: Theoret- ical. Wat. Air Soil Poll. 20,369-377.
Sadler. R. Olszowy, H.. Shaw, G.. Biltoft, R., and Connell, D. (1994). Soil and water contamination by arsenic from tannery waste. War. Air Soil Poll. 78, 189-198.
Schuthess, C. P., and Huang, C. P. (1990). Adsorption of heavy metals by silicon and aluminium sur- faces on clay minerals. Soil Sci. SOC. Am. J . 54,679-688.
Scott, M. J. , and Morgan, J. J. (1995). Reactions of oxide surfaces. 1. Oxidation of As(II1) by synthet- ic birnessite. Environ. Sci. Technol. 29, 1898-1905.
Sheppard, S. C. (1992). Summary of phytotoxic levels of soil arsenic. War. AirSoil Poll. 64,539-550. Shiendorf, C., Rebhun, M., and Sheintuch, M. (1981). A Freundlich-type multicomponent isotherm. J .
Shuman, L. M. (1976). Zinc adsorption isotherms for soil clays with and without iron oxides removed.
Speir. T. W., August, J . A., and Feltham, C. W. (1992b). Assessment of the feasibility of using CCA
Colloid Interface Sci. 79, 136-142.
Soil Sci. SOC. Am. J. 40,349-352.

194 E. SMITH ETAL.
(copper, chromium, and arsenic)-treated and boric acid-treated sawdust as soil admendments. 11. Soil biochemical and biological properties. Plant and Soil 142,249-258.
Steevens, D. R., Walsh. L. M., and Keeney, D. R. (1972). Arsenic phytotoxicity on a Plainfield sand as affected by ferric sulfate or aluminium sulfate. J . Environ. Qual. 1,301-303.
Tabatabdi, M. A. (1977). Effects of trace elements on urease activity in soils. Soil Biol. Biochem. 9,
Takamatsu. T., Aoki, H., and Yoshida, T. (1982). Determination of arsenate, arsenite, monomethylar- sonate, and dimethylarsinate in soil polluted with arsenic. Soil Sci. 133,239-246.
Tamaki, S., and Frankenberger Jr., W. T. (1992). Environmental Biochemistry of Arsenic. In “Reviews of Environmental Contamination and Toxicology,” Vol. 124, pp. 79-1 10. Springer-Verlag. New York.
Tammes, P. M., and de Lint, M. M. (1969). Leaching of arsenic from soil. Neth. J. Agric. Sci. 17, 128-132.
Thanabalasingam, P., and Pickering, W. F. (1986). Arsenic sorption by humic acids. Environ. Pollur. Ser: B 12,233-246.
Thomas. R. (1977). Arsenic pollution arising from mining activities in south-west England. In “Inor- ganic Pollution and Agriculture, April 1977,” pp. 126-141. Ministry of Agriculture, Fisheries and Food, London.
9-13.
Tiller, K. G. (1992). Urban soil contamination in Australia. A m . J . Soil Res. 30,937-957. Turner, R. R. (1981). Oxidation state of arsenic in coal ash leachate. Environ. Sci. Technol. 15,
Vaughan, G . T. (1993). Investigation Report CETLHIR148: The environmental chemistry and fate of arsenical pesticides in cattle tick dip sites and banana plantations. CSIRO, Division of Coal and Energy Technology, Centre for Advanced Analytical Chemistry, Sydney, Australia.
Walsh, L. M., and Keeney, D. R. (1975). Behaviour and Phytotoxicity of Inorganic Arsenicals in Soils. In “Arsenical Pesticides” (E. A. Woolson, ed.), pp. 35-53. Am. Chem. SOC., Washington, D.C.
Walsh, L. M., Sumner, M. E., and Keeney, D. R. (1977). Occurrence and distribution of arsenic in soils and plants. Environ. Healrh Perspect. 19,67-71.
Wauchope, R. D. (1975). Fixation of arsenical herbicides, phosphate, and arsenate in alluvial soils. J. Environ. Qual. 4, 355-358.
Waychunas, G . A., Rea, B. A., Fuller, C. C., and Davis, J. A. (1993). Surface chemistry of ferrihydrite: Part 1. EXAFS studies of the geometry of coprecipitated and adsorbed arsenate. Geochim. et Cos- mochim. Acta 51,225 1-2269.
Waychunas, G . A., Fuller, C. C., Rea, B. A., and Davis, J. A. (1996). Wide angle X-ray scattering (WAXS) study of “two-line” ferrihydrite structure: Effect of arsenate sorption and counterion vari- ation and comparison with EXAFS results. Geochim. er Cosmochim. Acta 60,1765-1781.
Wells, B. R., and Gilmor, J. T. (1977). Sterility in rice cultivars as influenced by MSMA rate and wa- ter management. Agron. J . 69,45 1-454.
Wild, A. (1993). Soils and the Environment: An Introduction. Cambridge University Press, Cambridge. Wild, H. ( I 973-74). Arsenic tolerant plants species established on arsenical mine dumps in Rhodesia.
Woolson, E. A. (1973). Arsenic phytotoxicity and uptake in six vegetable crops. WeedSci. 21,524-527. Woolson, E. A. (1977). Generation of alkylarsines from soil. Weed Sci. 25,412416. Woolson, E. A., and Kearney, P. C. (1973). Persistence and reactions of 14C-cacodylic acid in soils. En-
viron. Sci. Technol. 1,47-50. Woolson, E. A., Axely, J. H., and Kearney, P. C. (1971a). Correlation methods between available soil
arsenic, estimated by six methods, and response to corn (Zm mays L.). Soil Sci. Am . Proc. 35,
Woolson, E. A., Axely, J. H., and Kearney, P. C. (1971b). The chemistry and phytotoxicity of arsenic
1062- 1066.
Kirkia 9,265-277.
10 1-1 05.
in soils. 1. Contaminated field soils. Soil Sci. Am. Proc. 35,938-943.

ARSENIC IN THE SOIL ENVIRONMENT: A REVIEW 195
Woolson, E. A., Axely, J. H., and Kearney, P. C. (1973). The chemistry and phytotoxicity of arsenic in
World Health Organization. (198 I). “Environmental Health Criteria 18; Arsenic.” World Health Orga-
Xu. H., Allard, B., and Grimvall, A. (1988). Influence of pH and organic substance on the adsorption
Xu, H., Allard, B.. and Grimvall, A. (1991). Effects of acidification and natural organic materials on
Ydn-Chu, H. (1994). Arsenic distribution in soils. In “Arsenic in the Environment, Part 1: Cycling and
soils: Effects of time and phosphorus. Soil Sci. Am. Proc. 37,254-259.
nization, Geneva.
of As(V) on geologic materials. Waf. Air Soil Poll. 40,293-305.
the mobility of arsenic in the environment. Waf. Air Soil Poll. 57-58,269-278.
Characterization” (J. 0. Nriagu, ed.), pp. 17-49. Wiley & Sons, New York.