asymmetric bromine-lithium exchange: … bromine-lithium exchange: besides the diamine ligands, the...
TRANSCRIPT

Asymmetric Bromine-Lithium Exchange: besides the diamine ligands, the organolithium matters
Jézabel Praz, Julien Graff, Léo Egger, Laure Guénée, Simon Wagschal, E. Peter Kündig and Alexandre Alexakis*
Supporting Information
I) General remarks II) General procedure III) Supplementary results
• Derivatives of binaphtyl diamine • Asymmetric Chlorine-Lithium Exchange • Asymmetric Iodine-Lithium Exchange • Scope of Substrate
IV) Experimental part V) Spectroscopic and Chromatographic data VI) X-ray Crystallography
I) General remarks 1H (500 MHz, 400 MHz or 300 MHz), 13C (126 MHz or 100 MHz) spectra were recorded on either a Bruker 300 MHz, 400 or 500 MHz spectrometer at room temperature and are reported as chemical shift (δ) in ppm relative to solvent peak. Spin multiplicity are reported as singlet (s), doublet (d), triplet (t), brs (broad singlet) and multiplet (m). Coupling constant J is given in Hertz (Hz). The evolution of the reaction was followed by GC-MS Hewlett Packard (EI mode) HP6890-5973 or analytical thin-layer chromatography (TLC, visualisation was performed with UV, anisaldehyde or KMnO4). Optical rotation were measured at 22°C in a 10 cm cell in the stated solvent; [α]D values are given in 10-1 deg.cm2g-1 (concentration c given as g/100 mL). Melting points (mp) were measured in open capillary tubes and are uncorrected. Electrospray mass spectra were obtained by the Sciences Mass Spectrometry (SMS) platform at the Faculty of Sciences (University of Geneva) on a QStar pulsar instrument from AB/MDS Sciex, ESI (positif). Impact electron mass spectra were obtained by the Faculty of Sciences (University of Zürich) on a double-focusing magnetic sector mass spectrometer MAT 95 from Finnigan. Enantiomeric excesses were determined by chiral-GC, HP6890, (capillary column 10 psi H2) or chiral super fluid chromatography (SFC) with an appropriate program using a gradient of methanol or isopropanol. Temperature programs are described as follows: initial temperature (°C)-initial time (min)-temperature gradient (°C/min)-final temperature (°C); retention times (RT) are given in minutes. Flash chromatography was performed using silica gel 32-63 µm, 60 Å. Reactions were generally carried under atmosphere of nitrogen or argon using flame-dried glassware. All solvents were reagent-grade and dried over aluminium oxide columns using following standard procedure and TMSCl (98%, Acros) distilled on calcium hydride. nBuLi (1.6 M in hexanes, Acros), sBuLi (1.3 M in cyclohexane/hexanes 98:2, Acros), tBuLi (1.6 M in pentane, Acros), DMF (99.8%, Extra Dry, over Molecular Sieves, Acros) ,(S)-5,5’,6,6’,7,7’,8,8’-Octahydro-1,1’-bi-2-naphtol (Acros), Trifluoromethanesulfonic anhydride (99%, Aldrich), Pd(OAc)2 (99%,
Electronic Supplementary Material (ESI) for ChemComm.This journal is © The Royal Society of Chemistry 2015

STREM), dppp (Sigma-Aldrich), LiAlH4 (95%, Acros), PCC (98%, Sigma-Aldrich), were purchased and used as received. Following substrates were prepared according to literature procedures 1a [1], 1b-d[2], 9[3]. The following product 2 1 has been described previously and was identified by comparison of its physical and spectroscopic datas (1H, 13C NMR, GC/MS, chiral-GC and SFC). Ligands (L2 [4], L3 1, L4 [5]) were synthesized as described in the literature.
II) General Procedure
Asymmetric Halide-Lithium Exchange
XX
XX
1) 0.5 equiv L* 5 equiv RLi Toluene, -78 °C1.5 h
2) 8 equiv DMF, -78 °CXX
O
O
R R
RR
Sub-Stoichiometric pathway
In a dry Schlenk tube was added the diamine (0.055 mmol, 0.5 equiv) followed by toluene (1.4 mL), then the mixture was cooled to -78 °C for 10 min and RLi (5 equiv) was added dropwise to it. After 15 min the substrate (0.11 mmol, 1 equiv) dissolved in toluene (0.6 mL) was added dropwise and the whole was stirred during 1h30 at -78 °C. Then still at low temperature, DMF was added dropwise (8 equiv) and the whole was allowed to reach room temperature. The reaction was quenched with 1 M HCl and the product extracted by ether. The organic layers were dried over sodium sulfate and the solvents concentrated under reduced pressure to give the crude bisaldehyde.
Enantiomeric excess was determined by chiral GC or SFC.
1 Q. Perron, A. Alexakis, Adv. Synth. Catal. 2010, 352, 2611-2620. 2 J. Graff, T. Debande, J. Praz, L. Guénée, A. Alexakis, Org. Lett. 2013, 15, 4270 – 4273. 3 M. Abboud, V. Mamane, W. Aubert, C. Lecomte, Y. Fort, J. Org. Chem. 2010, 75, 3224. 4 J.C. Kizirian, N. Cabello, L. Pinchard, J. C. Caille, A. Alexakis, Tetrahedron 2005, 61, 8939-8946. 5 J. Praz, L. Guénée, S. Aziz, A. Berkessel, A. Alexakis, Adv. Synth. Catal. 2012, 354, 1780-1790.

III) Supplementary results
Derivatives of binaphtyl diamines
NN
L12
Ph
Ph
Ph
Ph
L9N
N
L10
NN
Ph
Ph
L11
NN
NN
L13
Ph
Ph
NN
Ph
Ph
nBuLi58% Conv.,
9 % ee
sBuLi >99% Conv.,
7%ee
83% 2a, 15% 3, 2% 4. L14
N N
sBuLi >99% Conv.
Racemic
L15
BrBr
BrBr
1) 0.5 equiv L* 5 equiv RLi Toluene, -78°C 1.5 h
2) 8 equiv DMF, -78 °C
BrO
BrO
1a 2a
O
BrO
3
O
O
4
O O
O
nBuLi >99% Conv.,
6%ee
sBuLi >99% Conv.
48%ee
51% 2a, 44% 3, 5% 4.
nBuLi86% Conv.,
8%ee
sBuLi >99% Conv.
60%ee
sBuLi >99% Conv.
Racemic
93% 2a, 7% 3.
sBuLi >99% Conv.
10%ee
60% 2a, 36% 3, 4% 4.
sBuLi 86% Conv.
4% ee
Diamines L9 and L10 gave moderate enantiomeric excesses, 48% and 60% respectively. However, the diamine with only one chiral unit L9 was less efficient, in term of selectivity and amount of desired product 2, than the flexible diamine L10 with a tropos unit. When a steric hindrance was added in 3,3’-position on the binaphytl moieties L12, the amount of desired product 2a increased to 93% but the selectivity decreased drastically to racemic mixture. Same observation in term of selectivity was done for the asymmetric diamine L11. Instead of increasing the bulkiness in the binaphtyle moieties, two stereogenic centers were added on the diamine backbone L13, L14. These two diamines with a trans 1,2-diphenylethane spacer gave poor enantiomeric excesses (4% ee, 7% ee) but higher proportion of bis-exchange products with sBuLi (83% vs. 69% for the Mazaleyrat-Cram diamine L5). The conversion of the reaction with the diamine (aS, S, S, aS)-L13 was not total. The 1,3-

diamines were less explored as chiral ligand in asymmetric synthesis but good results were obtained with these ligands.6 These diamines contain larger spacer, which increases their conformational lability. We supposed that with the 2,2-dimethylpropane a better selectivity could be obtained, due to Thorpe-Ingold effect. Unfortunately, the 1,3-diamine L15 was not an optimal ligand for this exchange. After all these tests, the Mazaleyrat-Cram diamine L5 and the dehydrogenated binaphtyl diamine L6 remain the most efficient ligands of this binaphtyl diamine family for this asymmetric bromine-lithium exchange reaction.
Asymmetric Chlorine-Lithium Exchange
Recently a paper of McNulty et al. described the synthesis of biaryl phosphane ligands by a chemoselective mono-Suzuki-Miyaura cross-coupling reaction on dichlorobenzene substrates, followed by chlorine-lithium exchange and a subsequent trapping with suitable dialkyl or diaryl chlorophosphane.[7]
In the literature just few examples are described as a chlorine-lithium exchange. Normally, aryl chlorides tend to undergo deprotonation leading to benzynes, rather than halogen-lithium exchange.[8 ] In special conditions, when deprotonation is impossible a chlorine-lithium exchange could occurs.[9]
ClCl
ClCl
Way 21) 2 equiv L2.2 equiv sBuLi Ether, -78 °C,1 h and -40 °C, 2.5 h
2) 8 equiv DMF, -78 °C
ClO
ClO
ClClCl
ClCl
ClCl
Cl
O O
O5 6a 6b 6c
or
Way 11) 2 equiv L2.2 equiv sBuLi Toluene, -78 °C, 2 h
2) 8 equiv DMF, -78 °C
TMEDA NN
Ph
N
Ph
N
L2 L3
NN
L4
L=
Table 6
Entry Solvent sBuLi (equiv)
L (equiv) ratio 6b: 6c [a] yield (%) ee (%)[c]
1 Toluene 2.2 TMEDA(1) 0 : 100 38 rac. 2[b] Ether 2.2 TMEDA(2) 100 : 010 21 - 3 Toluene 2.2 L4(1) 100 : 010 21 - 4 Toluene 2.2 L4(2) 100 : 010 26 -
6 J.-C. Kizirian, Chem. Rev. 2008, 108, 140. 7 E. Ullah, J. McNulty, A. Robertson, Eur. J. Org. Chem. 2012, 2127-2131. 8 J. Clayden, in Organolithium: Selectivity for Synthesis; J. E. Baldwin, R. M. Williams, Eds.; Pergamon: Oxford. 2002. 9 a) G. Körbich, Angew. Chem., Int. Ed. Engl. 1967, 6,41-52; b) F. Tellier, R. Sauvêtre, J. F. Normant, Y. Deromsee, Y. Jeannin, J. Organomet. Chem. 1987, 331, 281-298.

5 Toluene 2.2 L3(1) 0 : 100 33 rac. 6 Toluene 2.2 L3(2) 58 : 42 17 : 13 rac. 7 Toluene 2.2 L2 (1) 72 : 28 18 : 7 rac. 8[d] Ether 2.5 - 100 : 100 4 rac. [a] Determined by 1H NMR spectroscopy and GC-MS; [b] Mixture of alkylate compounds; [c] Determined by chiral SFC; [d] Conditions of McNulty et al.: way 2.[4]
The tetrachlorobiphenyl substrate 5 was used as prochiral target to perform chlorine-lithium exchange by using two ways: the first was the same procedure using to perform Br-Li exchange, then the second was the same used by McNulty et al. Both of them gave orthometallate substrates. Yields were really low and no enantioselectivities were observed for all chiral ligands.
Way 1
A solution of the 2,2',6,6'-tetrachloro-1,1'-biphenyl 5 (32 mg, 0.11 mmol) in toluene (0.6 mL) was added to a solution of sBuLi (1.3 M, 0.18 mL, 0.24 mmol) and diamine (1 or 2 equiv) in 1.4 mL of toluene at -78 °C. After 2h the reaction was quenched at -78 °C with DMF (68 µL, 0.88 mmol). HCl (1 mL, 1 M) was added and the aqueous layer was extracted 3 times with ether. The organic phases were combined, dried over sodium sulphate, filtered and finally concentrated under reduced pressure.
Way 2
The sBuLi (1.3 M, 0.42 mL, 0.55 mmol) was added slowly to a solution of 2,2',6,6'-tetrachloro-1,1'-biphenyl 5 (32 mg, 0.11 mmol) in ether (2 mL) at -78 °C under argon. The reaction mixture was stirred for 1h at -78 °C and subsequently for 2 h at -45 °C. Then the mixture was quenched dropwise at -78 °C with DMF (0.136 mL, 1.76 mmol) and was allowed to warm to room temperature then HCl (1 mL, 1 M) was added and the aqueous layer was extracted 3 times with ether. The organic phases were combined, dried over sodium sulfate, filtered and finally concentered under reduced pressure.
Asymmetric Iodine-Lithium Exchange
II
II
1) 0.5 equiv L 2.2 equiv nBuLi Toluene, -78 °C1.5 h
2) 8 equiv DMF, -78 °C
II
O
O
Ph
N
Ph
N
L3
Ph
O
Ph
O
L7
TMEDAL=
7 8
Table 2
Entry Solvent nBuLi (equiv)
L (equiv) Conv. [a] ee (%)[b]
1 Toluene 2.2 TMEDA(2.2) >99% rac. 2 Toluene 2.2 L3(0.5) 20% rac. 3 Toluene 2.2 L7(0.5) 50% rac.
[a] Determined by 1H-NMR spectroscopy and GC-MS; [b] Determined by chiral SFC.

Only racemic mixtures were obtained with chiral ligands L3, L7 when iodine-lithium exchange was performed on the 2,2',6,6'-tetraiodo-1,1'-biphenyl 7.
Scope of Substrate
BrBr
BrBr
BrBr
1a : R1 = H1b : R1 = Me1c : R1 = OMe1d : R1 = TMS1e : R1 =1f : R1 = CF3
R1
R1 R1
R1
O
O
1) 0.5 equiv L *5 equiv RLi Toluene, -78 °C1.5 h
2) DMF, -78 °C
TMS
2a : R1 = H2b : R1 = Me2c : R1 = OMe2d : R1 = TMS2e : R1 =2f : R1 = CF3
TMS
Entry Sub.
R1 L* RLi Prod Conv [b] (%)
Yield (%)
ee[a]
(%)
1 1a H L3 nBuLi 2a >99 89 72 2[c] 1a H L4 sBuLi 2a >99 25 94 3[c] 1a H L5 sBuLi 2a >99 50 97 4 1a H L7 nBuLi 2a >99 89 80 5 1b Me L3 nBuLi 2b >99 96 80
6[c] 1b Me L4 sBuLi 2b >99 21 74 7[c] 1b Me L5 sBuLi 2b >99 74 73 8 1b Me L7 nBuLi 2b >99 77 52 7 1c OMe L3 nBuLi 2c >99 67 59
8[c] 1c OMe L4 sBuLi 2c >99 31 77 9 1c OMe L5 sBuLi 2c >99 57 47
10 1c OMe L7 nBuLi 2c >99 77 58 11 1d TMS L3 nBuLi 2d >99 64 26
12[c] 1d TMS L4 sBuLi 2d >99 34 58 13[c] 1d TMS L5 sBuLi 2d >99 49 41 14 1d TMS L7 nBuLi 2d >99 82 74
15[d], [c] 1d TMS L5 sBuLi 2d 66 63 76 16[e] 1d TMS L5 sBuLi 2d >99 73 Rac. 17 1e ≡-TMS L3 nBuLi 2e >99 92 59
18 [f] 1e ≡-TMS L4 sBuLi 2e >99 - - 19[d], [g] 1e ≡-TMS L4 sBuLi 2e 57 20 69
20[f] 1e ≡-TMS L5 sBuLi 2e >99 - - 21[d], [g] 1e ≡-TMS L5 sBuLi 2e 46 17 85
22 1e ≡-TMS L7 nBuLi 2e >99 67 10 23 1f CF3 L3 nBuLi 2f >99 54 28
24[h] 1f CF3 L4 sBuLi 2f >99 4 - 25[d] 1f CF3 L4 sBuLi 2f 78 18 14 26 1f CF3 L5 sBuLi 2f >99 10 Rac.
27[d] 1f CF3 L5 sBuLi 2f >99 36 10 28 1f CF3 L7 nBuLi 2f >99 67 14
[a]Determined by chiral GC or SFC using a chiral stationary phase. [b]Determined by 1H NMR spectroscopy. [c] By-products due to tris, tetra exchange. [d] 2.2 equiv of sBuLi instead of 5 equiv. [e] 0.01 equiv of L*. [f] Only tetra exchange product. [g] Partial conversion, mono exchange product also observed. [h] Majority of tris and tetra exchange products.

IV) Experimental part
2 ,2’,6,6’-tetrachlorobiphenyl 5 [10 ]: To a solution of diisopropylamine (3.2 mL,
22.8 mmol) in dry THF (30 mL) at 0 °C was added nBuLi (1.6 M, 11.9 mL, 19 mmol) and the mixture was stirred at this temperature during 30 min before cooling to -78 °C. The 1,3-dichlorobenzene (2.3 mL, 19 mmol) was added to the mixture at -78 °C. After 2h at this temperature, a solution of CuCN, LiCl (9.5
mmol), in THF (22 mL) was added. After 1.5 h, benzoquinone (3.1 g, 29 mmol, under solid form) was added to the mixture and the whole was stirred for 15 min at -78 °C, then warmed slowly to room temperature overnight. The solvent was removed and water was added. The mixture was extracted with DCM, dried over sodium sulfate and the solvent evaporated. The crude was purified by chromatography column (pentane/ether 9:1) and recrystallized in acetonitrile. A white solid was obtained (2.8 g, 52% yield). 1H NMR (300 MHz, CDCl3) δ: 7.45 (d, J = 8.5 Hz, 4H), 7.33 (dd, J = 8.9, 7.1 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ: 135.3, 135.2, 130.37, 128.07.
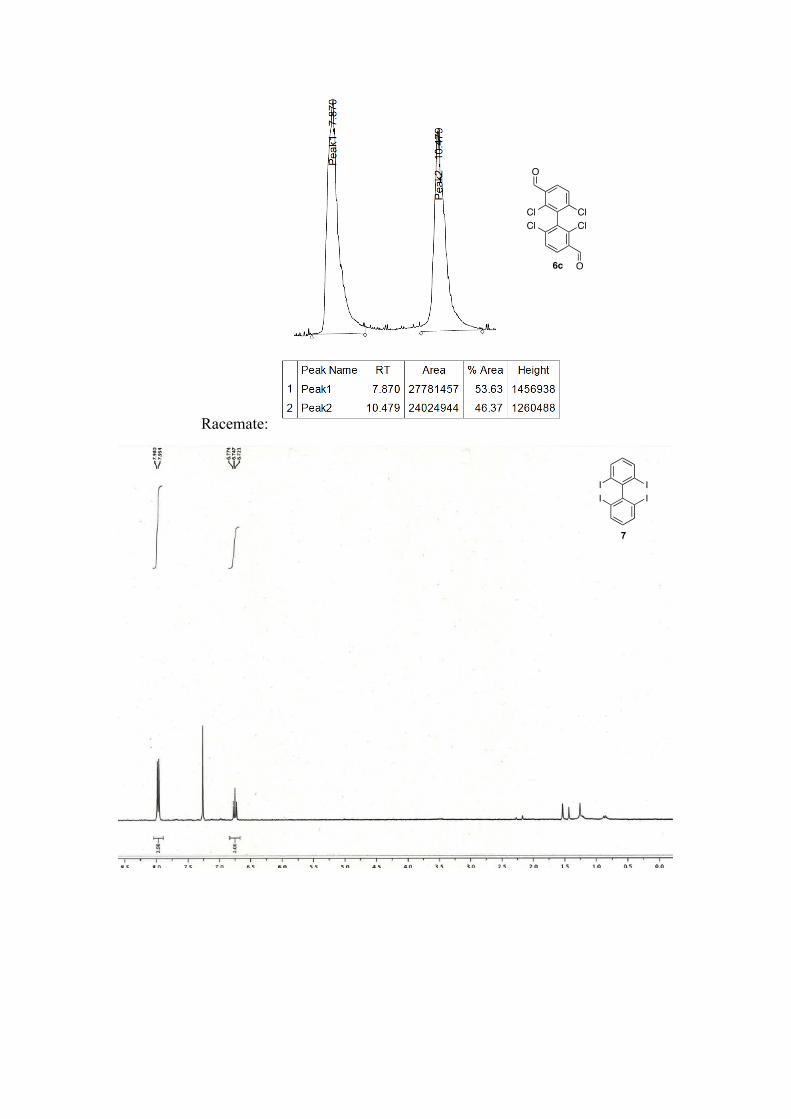
2, 2', 6, 6'-tetrachloro-[1,1'-biphenyl]-3,3'-dicarbaldehyde 6c: Purification by column chromatography (pentane/ether) to afford a white solid in 38% yield, ee = Racemic; 1H NMR (500 MHz, CDCl3) δ: 10.49 (s, 2H), 8.03 (dd, J = 8.4, 1.1 Hz, 2H), 7.62 (d, J = 8.4 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ: 188.66, 141.28, 138.69, 135.75, 131.86, 130.61, 128.95.
Enantiomeric excesses were determined by SFC Chiralpak OD, 200 bar, 2mL.min-1, 10% 1% /min to 25% of isopropanol in CO2, 40 °C, t1 = 7.87 min, t2 = 10.47 min for 6c.
2,2',6,6'-tetraiodo-1,1'-biphenyl 7 11: To a flame-dried Schlenck tube was added 1 g (2.13 mmol, 1 equiv.) of the substrate 2,2',6,6'-tetrabromo-1,1'-biphenyl followed by 25 mL of dry toluene and 1.27 mL (8.52 mmol, 4 equiv) of distilled TMEDA. The resulting mixture was cooled to -78 °C during 15 min. Then 21 mL (21.3 mmol, 10 equiv.) of sBuLi (1.0M in cyclohexane/hexanes) were added
dropwise. The reaction was carried during 20 minutes at -78 °C. Then it was quenched with 4.3 g of I2 (17.04 mmol, 8 equiv) and allowed to reach room temperature. Then it was quenched with Na2S2O3 and followed by an extraction with AcOEt. A column chromatography (Pentane) followed by a recristallization in (trifluoromethyl)benzene was used to get 253 mg of the desired product as white crystals in 18% yield. 1H NMR (400 MHz, CDCl3) δ: 7.97 (d, J=10.4 Hz, 4H), 6.75 (t, J=10.4 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ: 153.7, 139.2, 131.2, 98.7.
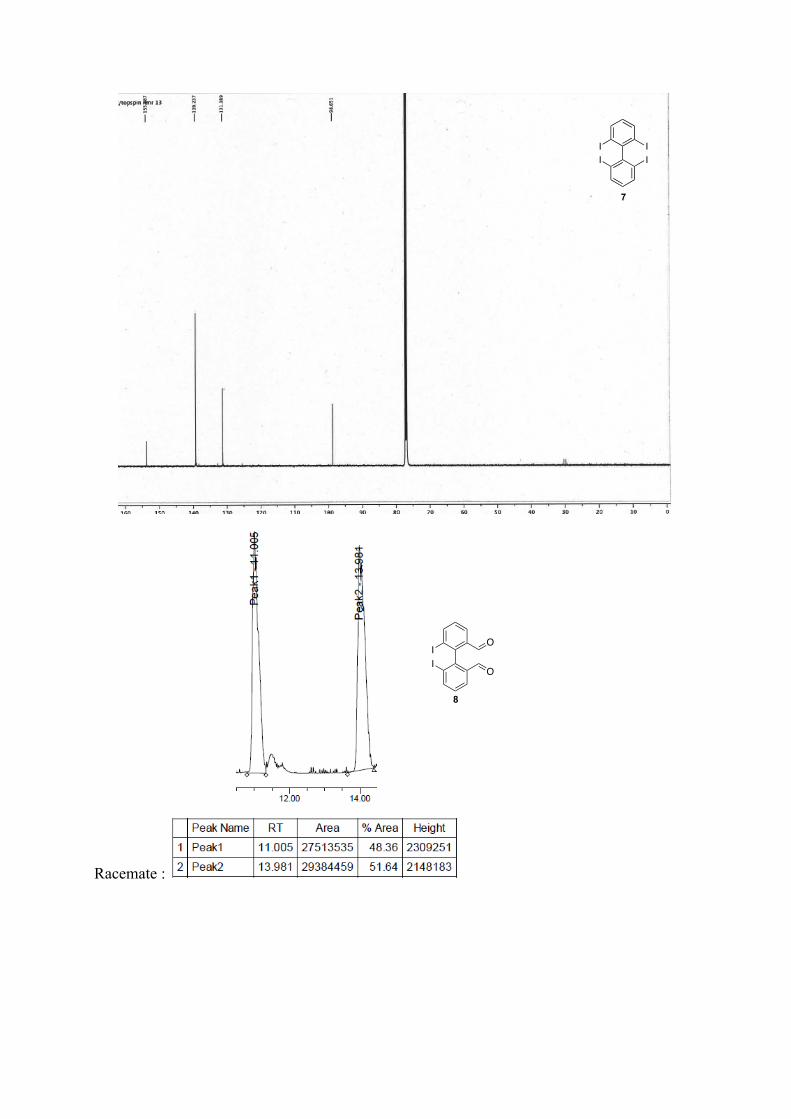
6,6'-diiodo-[1,1'-biphenyl]-2,2'-dicarbaldehyde 8: Following the general procedure for bromine-lithium exchange. Crude mixture is purified by column chromatography (pentane/ether, 9:1) White solid. Racemic. 1H NMR (400 MHz, CDCl3) δ: 9.54 (s, 2H), 8.24 (dd, J=8.0, 1.2 Hz, 2H), 8.06 (dd, J=7.6, 1.2 Hz, 2H), 7.40 (t, J=7.6 Hz, 2H). 13C NMR (400 MHz, CDCl3) δ: 190.4, 147.4, 144.7,
10 S. C. Waller, E. A. Mash, Organic Preparations and Procedure Int., 1997, 29, 679-685. 11 S. Sandin, J. Org. Chem. 1969, 34,456-457.
ClCl
ClCl
5
ClClCl
Cl
O
O6c
III I
7
II
8
O
O

136.3, 131.0, 129.5, 102.9.
Enantiomeric excesses were determined by SFC Chiralpak AZ, 200 bar, 2mL.min-1, 1%/min to 25% of isopropanol in CO2, 40°C, t1= 11.05 min, t2 = 11.98 min for 8.
(S)-6,6'-dibromo-[1,1'-biphenyl]-2,2'-dicarbaldehyde 2a [1]: Following the
general procedure for bromine-lithium exchange. Crude mixture is purified by column chromatography (pentane/ether, 9:1). Spectroscopic data consistent with those reported in the literature. White solid, 50% yield. 97% ee with L5, sBuLi. [α]22
D: -20.2 (c = 0.33, in CHCl3, 97% ee (S)).
Enantiomeric excess was determined by GC: CP-Chirasil-Dex CB, 25m, T: 140-100-1-170- 60 min, t1 = 181.07 min, t2 = 182.7 min for 2a.
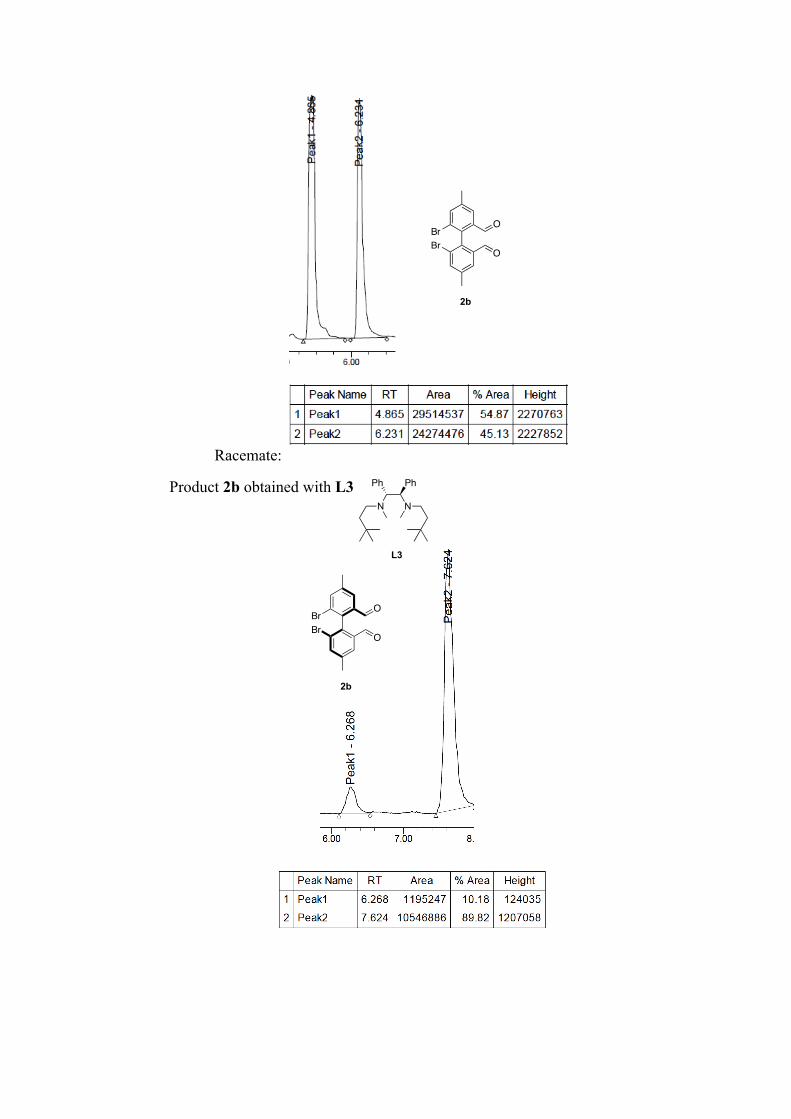
(R)-6,6'-dibromo-4,4'-dimethyl-[1,1'-biphenyl]-2,2'-dicarbaldehyde 2b: Following the general procedure for bromine-lithium exchange. Crude mixture is purified by column chromatography (pentane/ether, 9:1). White solid, 96 % yield. 80% ee with L3, nBuLi. 1H NMR (400 MHz, CDCl3) δ: 9.57 (s, 2H), 7.81 (d, J=7.8 Hz, 4H), 2.50 (s, 6H). 13C NMR (100 MHz, CDCl3) δ: 190.1, 141.1, 138.6, 137.5, 136.1, 128.8, 125.6, 21.0. M.p.: 138.7-142.8 °C HR-MS: m/z =[M]+• Observed 395.91774, calcd. for C16H12Br2O2 395.9177. [α]22
D: + 15.91 (c = 0.4, in CHCl3, 80% ee (R) with L3, nBuLi).
Enantiomeric excesses were determined by SFC Chiralpak AS, 200 bar, 2mL.min-1, 10% 1%/ min to 25% of isopropanol in CO2, 40 °C, t1 = 6.3 min, t2 = 7.6 min for 2c.
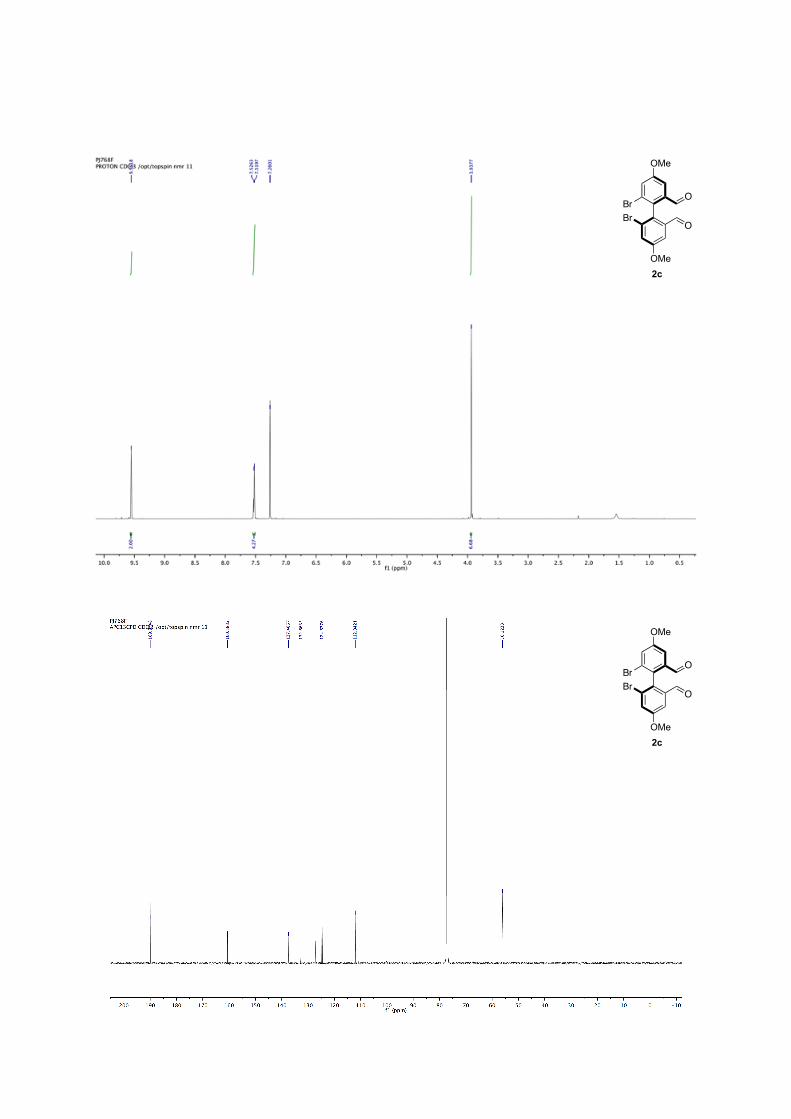
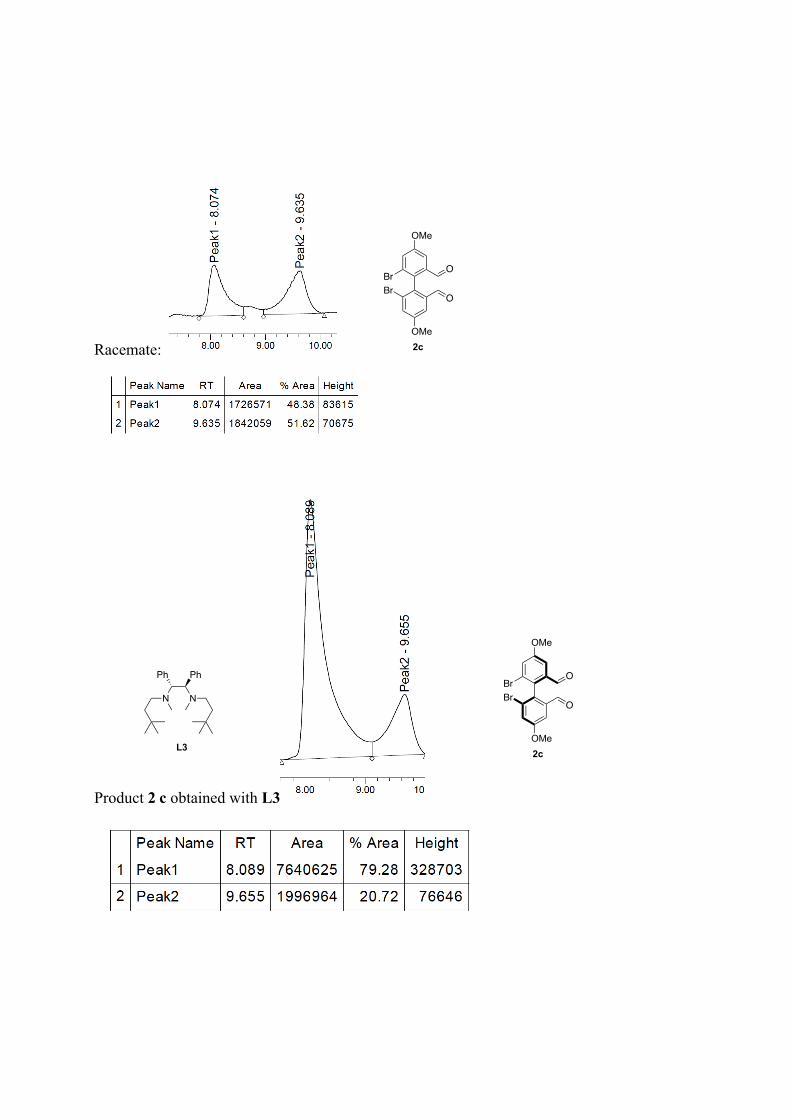
(R)-6,6'-dibromo-4,4'-dimethoxy-[1,1'-biphenyl]-2,2'-dicarbaldehyde 2c: Following the general procedure for bromine-lithium exchange. Crude mixture is purified by column chromatography (pentane/ether, 8:2) yellowish solid, 67% yield. 59% ee with L3, nBuLi. 1H NMR (500 MHz, CDCl3) δ: 9.55 (s, 2H), 7.52 (d, J = 3.3 Hz, 4H), 3.94 (s, 6H). 13C NMR (126 MHz, CDCl3) δ: 189.97, 160.58, 137.49, 132.66, 124.68, 112.04, 56.12. M.p.: 128.5-131.1°C. HR-MS: m/z =[M]+• Observed 427.90772, calcd. for C16H12Br2O4 427.9082. [α]22
D: + 28.74 (c = 0.95, in CHCl3, 59% ee (R) with L3, nBuLi). Enantiomeric excesses were determined by SFC Chiralpak = OZ, 200 bar, 2mL.min-1, 10% -1%/min to 20% of MeOH in CO2, 40 °C, t1 = 6.3 min, t2 = 7.6 min for 2c.
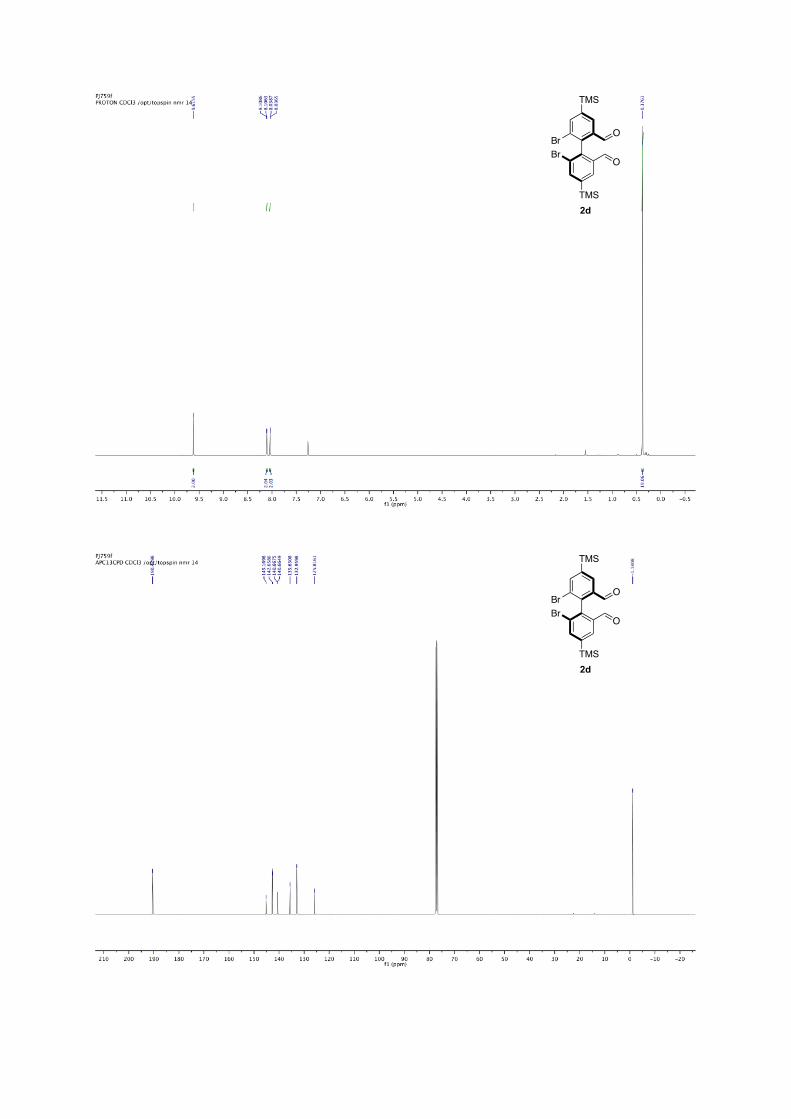
6,6'-dibromo-4,4'-bis(trimethylsilyl)-[1,1'-biphenyl]-2,2'-dicarbaldehyde
2d: Following the general procedure for bromine-lithium exchange. Crude mixture is purified by column chromatography (pentane/ether, 9:1) white solid, 82% yield. 74% ee with L7, nBuLi. 1H NMR (500 MHz, CDCl3) δ: 9.62 (s, 2H), 8.07 (dd, J = 34.9, 1.1 Hz, 4H), 0.38 (s, 18H). 13C NMR (126 MHz, CDCl3) δ: 190.48, 145.17, 142.66, 140.67, 135.63, 132.96, 125.82, -1.14. M.p.: 202.3-207.2°C HR-MS: m/z =[M-Me]+• Observed 496.9424, calcd. for C19H21Br2O2Si2
496.9426. [α]22D: + 28.8 (c = 1.45, in CHCl3, 74% ee (R) with L7, nBuLi).
Enantiomeric excesses were determined by SFC Chiralpak = OZ, 200 bar, 2mL.min-1, 10%, 1%/min to 20% of MeOH in CO2, 40 °C, t1 = 2.5 min, t2 = 4.3 min for 2d.
BrBr
O
O
2a
BrBr
O
O
2b
BrBr
O
O
TMS
TMS2d
BrBr
O
O
OMe
OMe2c

((2,2',6,6'-tetrabromo-[1,1'-biphenyl]-4,4'-diyl)bis(ethyne-2,1-diyl))bis(trimethylsilane) 1e: At room temperature, a flame-dried Schlenk tube was charged with 557 mg (0.773 mmol, 1 equiv) of 2,2',6,6'-tetrabromo-4,4'-diiodobiphenyl, 27.3 mg (0.039 mmol, 0.05 equiv) of PdCl₂(PPh₃)₂ and 14.5 mg (0.077 mmol, 0.1 equiv) of CuI in 11 mL of dry NEt₃. After a few minutes, 334 µL (2.32 mmol, 3 equiv) of ethynyltrimethylsilane, previously dissolved in 2.8 mL of dry NEt₃, were added to the mixture. After 24 hours at room temperature, the reaction was quenched with H2O and extracted with AcOEt. Then a column chromatography (100% Pentane) was used to get 297 mg of the desired product as a white powder in 58% yield. 1H NMR (400 MHz, CDCl3) δ: 7.74 (s, 4H),
0.25 (s, 18H). 13C NMR (100 MHz, CDCl3) δ: 141.9, 135.0, 126.5, 124.0, 101.7, 98.5, 0.0. M.p.: 197.2-198.4 °C. HR-MS: m/z =[M]+• Observed 661.7951, calcd. for C22H22Br4Si2 661.7953.
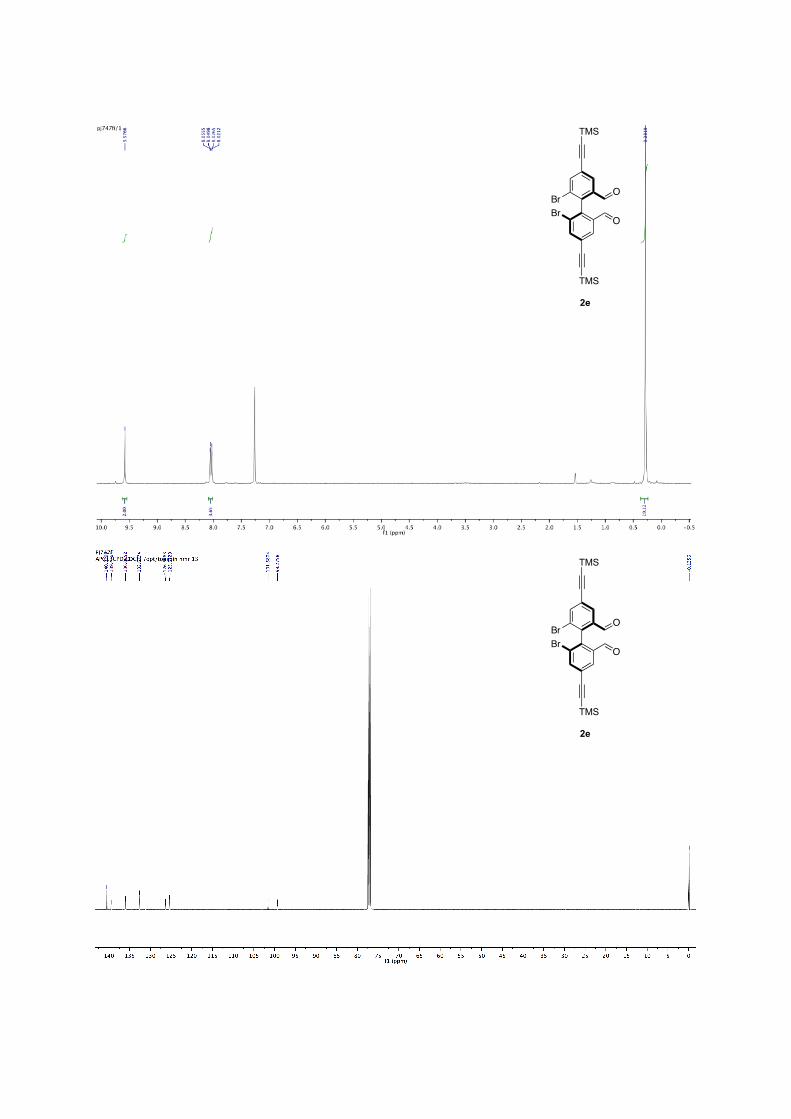
(R)-6,6'-dibromo-4,4'-bis((trimethylsilyl)ethynyl)-[1,1'-biphenyl]-2,2'-dicarbaldehyde 2e: Following the general procedure for bromine-lithium exchange. Crude mixture is purified by column chromatography (pentane/ether, 9:1) white solid, 17% yield. 85% ee with L5, sBuLi. 1H NMR (300 MHz, CDCl3) δ: 9.58 (s, 2H), 8.04 (d, J = 7.0 Hz, 4H), 0.28 (s, 18H). 13C NMR (100 MHz, CDCl3) δ: 189.02, 140.53, 139.48, 136.01, 132.68, 126.37, 125.33, 101.58, 99.28, -0.14. M.p.: 164-166°C HR-MS: m/z =[M]+• Observed 559.9661, calcd. for C24H24Br2O2Si2 559.9661. [α]22
D: + 1.97 (c = 0.39, in CHCl3, 85% ee (R) with L5, sBuLi) Enantiomeric excesses were determined by SFC Chiralpak = OD, 200 bar,
2mL.min-1, 10% 1%/min to 25% of isopropanol in CO2, 40 °C, t1 = 11.4 min, t2 = 12.4 min for 2e.
1,3-dibromo-5-(trifluoromethyl)benzene: A solution of 5.76 g of 5-(trifluoromethyl)benzene-1,3-diamine (32.7 mmol, 1 equiv.) in aq. 48% HBr (150 mL) was stirred at room temperature for 18 h. Then a solution of 18 g of sodium
nitrite (262 mmol, 8 equiv) in 200 mL of water was added dropwise to the reaction medium over 60 min at 0 °C. After the reaction mixture was stirred for another 30 min at 0 °C. Then a suspension of 15 g of CuBr (105 mmol, 3.2 equiv) in aq. 48% HBr solution (300 mL) was added dropwise to the reaction medium over 60 min at 0 °C. After, the all mixture was stirred at 70 °C for one hour. Then it was poured in ice water and extracted with DCM. Then a column chromatography (Pentane) was used to get 8.96 g of the desired product as a colorless oil in 90 % yield. 1H NMR (400 MHz, CDCl3) δ: 7.82 (s, 1H), 7.70 (s, 2H). 13C NMR (100 MHz, CDCl3) δ: 120.4, 123.3, 127.3 (q, J=3.8 Hz), 133.6 (q, J=34.1 Hz), 137.5. 19F NMR (172 MHz, CDCl3) δ: -63.1.
2,2',6,6'-tetrabromo-4,4'-bis(trifluoromethyl)-1,1'-biphenyl 1f: At -78 °C, 6.73 g (22.1 mmol, 1 equiv) of 1,3-dibromo-5-(trifluoromethyl)benzene was added in a 44 mL solution of dry THF of LDA (13.8 mL, 22.1 mmol, 1 equiv of nBuLi (1.6 M in hexanes) and (3.1 mL, 22.1 mmol,) 1 equiv of distilled diisopropylamine mixed during 45 minutes at 0 °C) and stirred for 2 hours. Then, still at -78 °C, a solution of 1g (11.05 mmol, 0.5 equiv) of CuCN and 555 mg (13.26 mmol, 0.6 equiv) of dry LiCl in 26.5 mL of dry THF was added to the
mixture and stirred for another 2 hours. Finally 3.56 g (33.15 mmol, 1.5 equiv) of
BrBr
BrBr
1e
TMS
TMS
BrBr
O
O
2e
TMS
TMS
BrBr
BrBr
CF3
CF31f
BrBr
CF3

benzoquinone were added and stirred for 1 hour then warmed slowly to reach room temperature. The reaction was quenched with distilled water and followed by an extraction with AcOEt. Then a column chromatography (100% Pentane) was used to get 1.62 g of the desired product as a white solid in 24% yield. 1H NMR (400 MHz, CDCl3) δ: 7.94 (s, 4H). 13C NMR (100 MHz, CDCl3) δ: 144.6, 133.4 (q, J=33.8 Hz), 128.9 (t, J=3.6 Hz), 124.4, 123.5, 120.7. 19F NMR (172 MHz, CDCl3) δ: -62.9. M.p.: 97.2-99.3. HR-MS: m/z =[M]+• Observed 605.69058 calcd. for C14H4Br4F6 605.69062.
(R)-6,6'-dibromo-4,4'-bis(trifluoromethyl)-[1,1'-biphenyl]-2,2'-
dicarbaldehyde 2f: Following the general procedure for bromine-lithium exchange. Crude mixture is purified by column chromatography (pentane/ether, 9:1) white solid, 66% yield. 34% ee with L8, PhLi. 1H NMR (400 MHz, CDCl3) δ: 9.69 (s, 2H), 8.24 (ddt, J = 11.4, 1.8, 0.8 Hz, 4H). 13C NMR (101 MHz, CDCl3) δ: 188.15, 142.56, 136.35, 134.76 (q, J=3.7 Hz), 133.32, 129.19, 127.19 (q, J = 3.6 Hz), 125.89. 19F NMR (282 MHz, CDCl3) δ: -63.04.
M.p. 97.2-99.3. HR-MS: m/z =[M]+• Observed 502.8550 calcd.for C16H6Br2F6 O2 502.8546. [α]22
D: -7.36 (c =0.6, in CHCl3, 34% ee (R) with L8, PhLi).
The catalysis dialdehyde product was then exposed to NaBH4 (3 equiv.) in dry MeOH to get the corresponding dialcohol, which was separated by chiral SFC using a stationary phase. 1H NMR (400 MHz, CDCl3) δ: 7.93 (s, 2H), 7.84 (s, 2H), 4.43 (d, J=12.8 Hz, 2H), 4.23(d, J=12.8 Hz, 2H). Enantiomeric excesses were determined by SFC Chiralpak = OJ, 200 bar, 2mL.min-1, 10 1%/min to 20% of MeOH in CO2, 40°C, t1= 7.6 min, t2 = 8.5min for 2fbis.
Ligand 6
We started the synthesis with the enantiopure dehydrogenated BINOL 25 by using a procedure developed by Lacour et al. for the synthesis of azepines. [9]
OTfOTf
COPd(OAc)2, dppp
DIPEA, MeOH, DMSO
72 h, 85 °C, 2.5 bar
CO2MeCO2Me
OH
OH
LiAlH4
Et2O
PCCDCM
H2NNH2
NaBH3CN
MeOH, AcOH cat.rt, 24 h
80%
NN
8 9 10 11
L6
OHOH
Tf2O, pyridine
DCM0°C to rt98% 62% 99%
83%
O
O
12
Synthesis of 1-((S)-8,9,10,11,12,13,14,15-octahydro-3H-dinaphtho[2,1-c:1',2'-e]azepin-4(5H)-yl)-2-(8,9,10,11,12,13,14,15-octahydro-3H-dinaphtho[2,1-c:1',2'-e]azepin-4(5H)-yl)ethane L6.
BrBr
O
O
CF3
CF32f

4a, 5,5’, 6,6’, 7, 7’, 8, 8a, 8’-Decahydro-[1,1’-binaphtalene]-2,2’-diylbis(trifluoromethanesulfonate) 9 [12
]: Under argon, (S)-H8-BINOL 19[13
] (2 g, 6.75 mmol) was dissolved in 40 mL of dry DCM in a Schlenk flask. Analytical-grade pyridine (1.54 mL, 17.55 mmol) was added and the solution was cooled to 0 °C. Then triflic anhydride (2.5 mL, 14.85 mmol) was added
slowly via syringe over 5 min. The reaction mixture was allowed to warm up to 25 °C and kept stirred at 25 °C for 12 h. At the conclusion of the reaction, the mixture was passed through a pad of silica gel and washed with 1:30 ethyl acetate/hexanes until no more product was eluted out (monitored by TLC). Concentration of the filtrate gave the pure product (3.7 g, 6.6 mmol, 98%) as white solid. 1H NMR (300 MHz, CDCl3) δ: 7.22 (d, J=8.5 Hz, 2H), 7.15 (d, J=8.5 Hz, 2H), 2.85 (t, J=6.3 Hz, 4H), 2.53-2.19 (m, 4H), 1.91-1.62 (m, 8H). 19F (172MHz, CDCl3) δ: -74.78.
Dimethyl 4a,5,5',6,6',7,7',8,8a,8'-decahydro-[1,1'-binaphthalene]-2,2'-dicarboxylate 10 [14
]: Bistriflate (3.7 g, 6.6 mmmol), methanol (13.4 mL, 330 mmol, 50 equiv) and diisopropylethylamine (5.07 mL, 29 mmol, 4.4 equiv) were dissolved in 33 mL of dimethyl sulfoxide. This homogeneous solution was transferred via cannula to a Fisher-Porter flask charged with Pd(OAc)2 (222 mg, 0.99 mmol, 0.15 equiv), dppp (0.412 g, 0.99 mmol, 0.15 equiv) and
a stir bar under an argon atmosphere. The vessel was then sealed and quickly cycled three times between a vacuum and an atmosphere of CO. The CO pressure in the flask was increased to 1.5 atm and partially immersed in an 80 °C oil bath. TLC followed the reaction at roughly 24 h intervals following the release of pressure and removal from heat. After complete consumption of all starting material (48 – 72 h) the reaction was purged with nitrogen and cooled to room temperature. The contents were transferred to a flask for removal of a majority of excess solvents in vacuo. The reduced mixture was diluted with ethyl acetate (80 mL), washed with water (3 x 20 mL), saturated NaCl solution (20 mL), and dried over anhydrous MgSO4. The filtered solution was concentrated in vacuo and purified by flash column chromatography (5:1 hexanes:EtOAc) to afford a white solid (1.55 g, 62% yield). 1H NMR (400 MHz, CDCl3) δ: 7.76 (d, J=8.0 Hz, 2H), 7.12 (d, J=8.0 Hz, 2H), 3.57 (s, 6H), 2.86 (q, J=6.3 Hz, 4H), 2.24-2.08 (m, 2H), 1.99 (dt, J=16.9, 6.1 Hz, 2H), 1.80-1.49 (m, 8H). 13C NMR (100 MHz, CDCl3) δ: 167.27, 141.65, 141.46, 135.09, 127.79, 126.86, 126.26, 51.41, 30.16, 27.15, 23.00, 22.34.
(4a,5,5',6,6',7,7',8,8a,8'-decahydro-[1,1'-binaphthalene]-2,2'-diyl)dimethanol 118: To a suspension of LiAlH4 (7.66 mmol, 0.29 g, 2.0 equiv) in dry diethyl ether (30 mL) (S)-2,2’-bis(carbomethyl)-5,5’,6,6’,7,7’,8,8’-octahydro-1,1’-binaphthyl (3.83 mmol) was added as a solution (30 mL) at 0 °C. After the addition, the resulting mixture was stirred at room temperature overnight, refluxed for 0.5 h and recooled to 0 °C. Water
(20 mL) was added carefully via an addition funnel and concentrated HCl was added slowly until the mixture became homogeneous. Diethyl ether (20 mL) was added and the resulting two phases were separated. The aqueous layer was extracted with ether (2x20 mL). The combined organic layers were washed with 10% aqueous solution of NaHCO3 (30 mL), brine 12 R. Novikov, G. Bernardinelli, J. Lacour, Adv. Synth. Catal. 2008, 350 ,1113-1124 13 A. Korostylev, V. I. Tararov, C. Fischer, A. Monsees, A. Börner, J. Org. Chem. 2004, 69,3220-3221. 14 A. K. Unni, N. Takenaka, H. Yamamoto, V. H. Rawal, J. Am. Chem. Soc. 2005, 127 ,1336-1337.
OTfOTf
9
CO2MeCO2Me
10
OH
OH
11

(30 mL) and dried under MgSO4. Evaporation of the solvent under reduced pressure provided 1.28 g, 99% yield of pure product. 1H NMR (400 MHz, CDCl3) δ: 7.26 (d, J=7.8 Hz, 2H), 7.13 (d, J=7.8 Hz, 2H), 4.25 (d, J=11.2 Hz, 2H), 4.08 (d, J=11.2 Hz, 2H), 2.83 (t, J=6.3 Hz, 4H), 2.29-1.88 (m, 4H), 1.81-1.55 (m, 8H).
4a,5,5',6,6',7,7',8,8a,8'-decahydro-[1,1'-binaphthalene]-2,2'-dicarbaldehyde
128: A round-bottomed flask (100 mL), equipped with a magnetic stirring bar, containing PCC (11.3 mmol, 2.45 g, 3.0 equiv) was charged with dry CH2Cl2 (15 mL). A solution of substrate (3.78 mmol, 1.23 g, 1.0 equiv) in CH2Cl2 (15 mL) was added to the resulting suspension in one portion. The resulting dark mixture was vigorously stirred for 3 h at ambient temperature, and then diethyl
ether (30 mL) was added. The mixture was stirred for 10 min, filtered through silica gel plug topped with a layer of Celite, which was then washed with ether. The filtrate was concentrated under reduced pressure to provide a green solid. Subsequent purification by column chromatography on silica gel petroleum ether/ethylacetate 5:1 gave pure product as a colorless solid 1.01 g; yield: 83%. 1H NMR (400 MHz, CDCl3) δ: 9.50 (s, 2H), 7.81 (d, J=8.0 Hz, 2H), 7.28 (d, J=8.1 Hz, 2H), 2.91 (t, J=6.3 Hz, 4H), 2.30-1.97 (m, 4H), 1.93-1.60 (m, 8H). 13C NMR (101 MHz, CDCl3) δ: 191.56, 145.31, 140.80, 136.25, 132.42, 129.88, 125.22, 30.69, 27.63, 22.98, 22.45.
1-((S)-8,9,10,11,12,13,14,15-octahydro-3H-dinaphtho[2,1-c:1',2'-e]azepin-4(5H)-yl)-2-(8,9,10,11,12,13,14,15-octahydro-
3H-dinaphtho[2,1-c:1',2'-e]azepin-4(5H)-yl)ethane L6: To a suspension of bisaldehyde 11 (250 mg, 0.78 mmol, 1.0 equiv) in MeOH (8 mL) ethylenediamine (0.39 mmol 0.5 equiv) was added. After few minutes of stirring, NaBH3CN (98 mg, 3.0
equiv.) and glacial acetic acid (2 drops) were added to the reaction mixture and the resulting colorless solution was stirred for 1 day at ambient temperature. The reaction mixture was quenched by addition of an aqueous solution of NaOH (1 M, 50 mL). DCM (50 mL) was added and the resulting two phases mixture separated. The aqueous phase was extracted with DCM (2x15 mL). The combined organic layers were washed with brine (50 mL), dried (MgSO4), filtered, concentrated under vacuum and purified by column chromatography on silica gel, (DCM:MeOH, 95:5). Dissolution of the product in ether with 50 mL of KOH (1 M) solution, stirring during 2h, following by an extraction, dried to potassium carbonate. 200 mg of a white solid (85% yield, 0.33 mmol) was obtained. 1H NMR (500 MHz, CDCl3) δ: 7.18 - 7.03 (m, 8H), 3.41 (d, J=12.2 Hz, 4H), 2.94 (d, J=12.4 Hz, 4H), 2.85 (q, J=6.8 Hz, 8H), 2.69 (q, J=9.0, 8.4 Hz, 6H), 2.45 (s, 2H), 2.31-2.16 (m, 4H), 1.79 (s, 12H), 1.56 (q, J=7.8 Hz, 4H). 13C NMR (101 MHz, CDCl3) δ: 138.27, 136.96, 135.66, 131.75, 128.09, 126.36, 55.47, 53.49, 29.67, 27.86, 23.04, 22.92. M.p : 106.7-109°C. HR-MS: m/z =[M+H]+ Observed 633.4207, calcd. for C46H52N2 633.4203. [α]25
D: +33.81, (c=1.01, CHCl3).
NN
L6
O
O
12
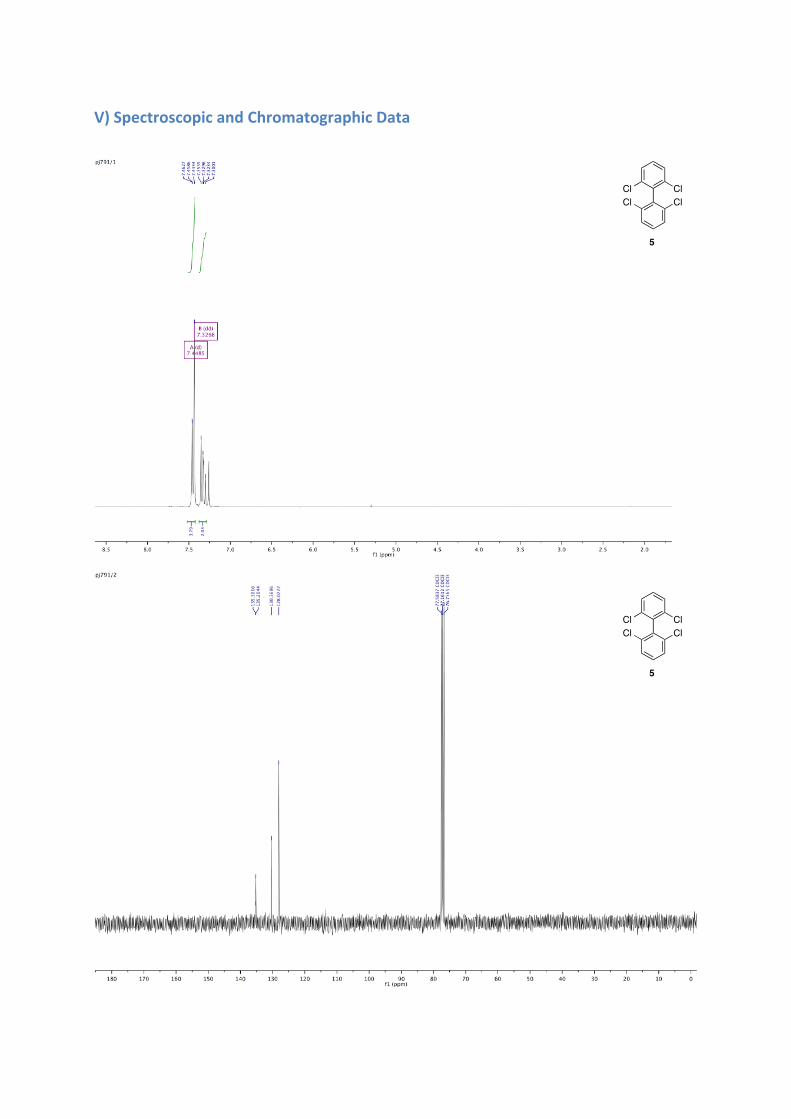
V) Spectroscopic and Chromatographic Data
ClCl
ClCl
5
ClCl
ClCl
5

ClClCl
Cl
O
O6c
ClClCl
Cl
O
O6c
Racemate:
III I
7
ClClCl
Cl
O
O6c
Racemate :
III I
7
II
O
O
8

Racemate :
BrBr
O
O
2a
BrBr
O
O
2a

BrBr
O
O
2b
BrBr
O
O
2b
Racemate:
Product 2b obtained with L3
BrBr
O
O
2b
BrBr
O
O
2b
Ph
N
Ph
N
L3
BrBr
O
O
OMe
OMe2c
BrBr
O
O
OMe
OMe2c
Racemate:
Product 2 c obtained with L3
BrBr
O
O
OMe
OMe2c
BrBr
O
O
OMe
OMe2c
Ph
N
Ph
N
L3
BrBr
O
O
TMS
TMS2d
BrBr
O
O
TMS
TMS2d

Racemate:
Product 2d obtained with L7
BrBr
O
O
TMS
TMS2d
Ph
O
Ph
O
L7
BrBr
O
O
TMS
TMS2d

BrBr
BrBr
1e
TMS
TMS
BrBr
BrBr
1e
TMS
TMS
BrBr
O
O
2e
TMS
TMS
BrBr
O
O
2e
TMS
TMS
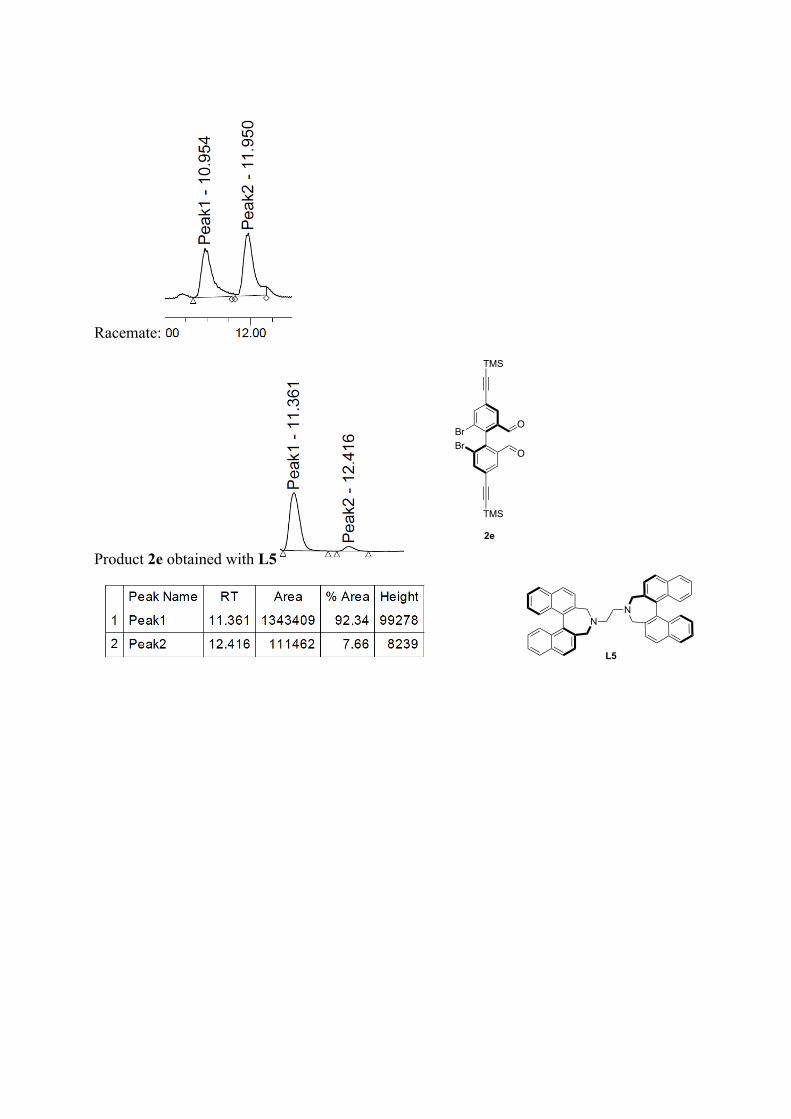
Racemate:
Product 2e obtained with L5
BrBr
O
O
2e
TMS
TMS
NN
L5

BrBr
BrBr
CF3
CF31f
BrBr
BrBr
CF3
CF31f

BrBr
BrBr
CF3
CF31f
BrBr
O
O
CF3
CF32f
BrBr
O
O
CF3
CF32f
BrBr
O
O
CF3
CF32f

Racemate:
Product 2fbis obtained with L8
NN
L8
BrBr
OH
OH
CF3
CF32fbis
BrBr
OH
OH
CF3
CF32fbis

NN
L6
NN
L6

VI) X-Ray Crystallography
The 2,2’,6,6’-tetrabromobiphenyl:
The monocrystal was obtained from hexane-DCM by layer diffusion technique. The (Sa)-2 enantiomer was acquired.
The crystal was mounted on quartz fibers with protection oil. Cell dimensions and intensities were measured at 180K on a STOE IPDS diffractometer with graphite-monochromated Mo[Kα] radiation (λ = 0.71073 Å). Data were corrected for Lorentz and polarization effects and for absorption. The structure was solved by direct methods (SIR97)15 , all other calculation were performed with ShelX97 systems16 and ORTEP 3 programs17. CCDC 859054 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
Crystal data and structure refinement for (Sa)-2,2’,6,6’-tetrabromobiphenyl
Table 1. Crystal data and structure refinement for (Sa)-2 _abs.
Identification code shelxl
Empirical formula C14 H8 Br2 O2
Formula weight 368.02
Temperature 180(2) K
Wavelength 1.54184 Å
Crystal system Orthorhombic
Space group P 21 21 21
Unit cell dimensions a = 8.23880(8) Å α= 90°.
b = 11.64677(12) Å β= 90°.
c = 13.78822(14) Å γ = 90°.
Volume 1323.05(2) Å3
Z 4
Density (calculated) 1.848 Mg/m3
Absorption coefficient 7.701 mm-1
F(000) 712
Crystal size 0.3563 x 0.2831 x 0.2369 mm3
Theta range for data collection 4.97 to 72.57°.
Index ranges -10<=h<=10, -12<=k<=14, -16<=l<=16
Reflections collected 11635 15 A. Altomare, M. C. Burla, M. Camalli, G. L. Cascarano, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, G. Polidori, R. Spagna, J. Appl. Cryst. 1999, 32, 115. 16 G. M. Sheldrick, Acta Cryst. A 2008, A64, 112. 17 L. J. Farrugia, J. Appl. Crystallogr. 1997, 30, 565.

Independent reflections 2604 [R(int) = 0.0438]
Completeness to theta = 66.49° 99.9 %
Absorption correction Analytical
Max. and min. transmission 0.345 and 0.196
Refinement method Full-matrix least-squares on F2
Data / restraints / parameters 2604 / 0 / 165
Goodness-of-fit on F2 1.104
Final R indices [I>2sigma(I)] R1 = 0.0250, wR2 = 0.0671
R indices (all data) R1 = 0.0251, wR2 = 0.0671
Absolute structure parameter 0.00(2)
Extinction coefficient 0.0049(2)
Largest diff. peak and hole 0.638 and -0.460 e.Å-3
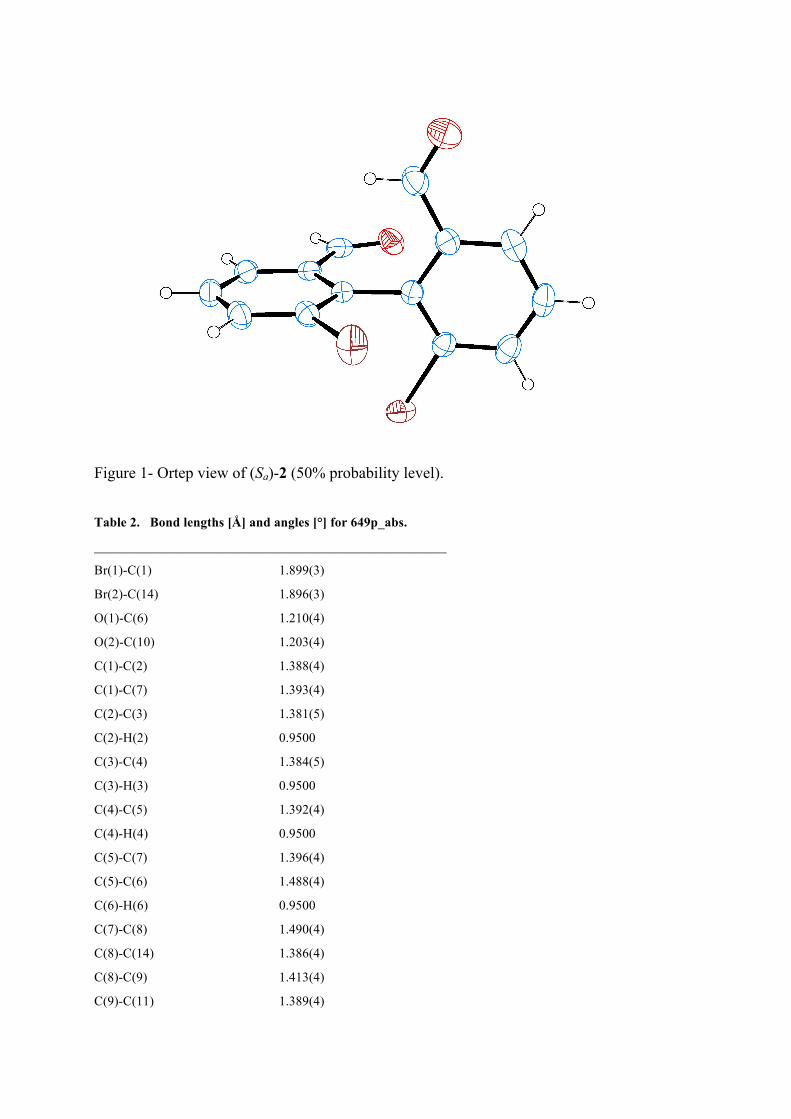
Figure 1- Ortep view of (Sa)-2 (50% probability level). Table 2. Bond lengths [Å] and angles [°] for 649p_abs.
_____________________________________________________
Br(1)-C(1) 1.899(3)
Br(2)-C(14) 1.896(3)
O(1)-C(6) 1.210(4)
O(2)-C(10) 1.203(4)
C(1)-C(2) 1.388(4)
C(1)-C(7) 1.393(4)
C(2)-C(3) 1.381(5)
C(2)-H(2) 0.9500
C(3)-C(4) 1.384(5)
C(3)-H(3) 0.9500
C(4)-C(5) 1.392(4)
C(4)-H(4) 0.9500
C(5)-C(7) 1.396(4)
C(5)-C(6) 1.488(4)
C(6)-H(6) 0.9500
C(7)-C(8) 1.490(4)
C(8)-C(14) 1.386(4)
C(8)-C(9) 1.413(4)
C(9)-C(11) 1.389(4)

C(9)-C(10) 1.476(4)
C(10)-H(10) 0.9500
C(11)-C(12) 1.389(5)
C(11)-H(11) 0.9500
C(12)-C(13) 1.380(5)
C(12)-H(12) 0.9500
C(13)-C(14) 1.399(4)
C(13)-H(13) 0.9500
C(2)-C(1)-C(7) 122.1(3)
C(2)-C(1)-Br(1) 117.5(2)
C(7)-C(1)-Br(1) 120.4(2)
C(3)-C(2)-C(1) 119.3(3)
C(3)-C(2)-H(2) 120.4
C(1)-C(2)-H(2) 120.4
C(2)-C(3)-C(4) 119.9(3)
C(2)-C(3)-H(3) 120.0
C(4)-C(3)-H(3) 120.0
C(3)-C(4)-C(5) 120.5(3)
C(3)-C(4)-H(4) 119.8
C(5)-C(4)-H(4) 119.8
C(4)-C(5)-C(7) 120.5(3)
C(4)-C(5)-C(6) 119.3(3)
C(7)-C(5)-C(6) 120.2(3)
O(1)-C(6)-C(5) 122.8(3)
O(1)-C(6)-H(6) 118.6
C(5)-C(6)-H(6) 118.6
C(1)-C(7)-C(5) 117.7(3)
C(1)-C(7)-C(8) 120.4(2)
C(5)-C(7)-C(8) 121.8(2)
C(14)-C(8)-C(9) 117.2(2)
C(14)-C(8)-C(7) 119.8(2)
C(9)-C(8)-C(7) 123.0(2)
C(11)-C(9)-C(8) 120.4(3)
C(11)-C(9)-C(10) 117.5(3)
C(8)-C(9)-C(10) 122.1(3)
O(2)-C(10)-C(9) 126.3(3)
O(2)-C(10)-H(10) 116.8

C(9)-C(10)-H(10) 116.8
C(12)-C(11)-C(9) 120.8(3)
C(12)-C(11)-H(11) 119.6
C(9)-C(11)-H(11) 119.6
C(13)-C(12)-C(11) 120.0(3)
C(13)-C(12)-H(12) 120.0
C(11)-C(12)-H(12) 120.0
C(12)-C(13)-C(14) 118.9(3)
C(12)-C(13)-H(13) 120.6
C(14)-C(13)-H(13) 120.6
C(8)-C(14)-C(13) 122.7(3)
C(8)-C(14)-Br(2) 119.3(2)
C(13)-C(14)-Br(2) 118.0(2)
_____________________________________________________________
Symmetry transformations used to generate equivalent atoms: