bicf variant analysis tools - biohpc portal home · bicf variant analysis tools using the biohpc...
TRANSCRIPT

BICFVariantAnalysisTools
Using the BioHPC Workflow Launching Tool Astrocyte
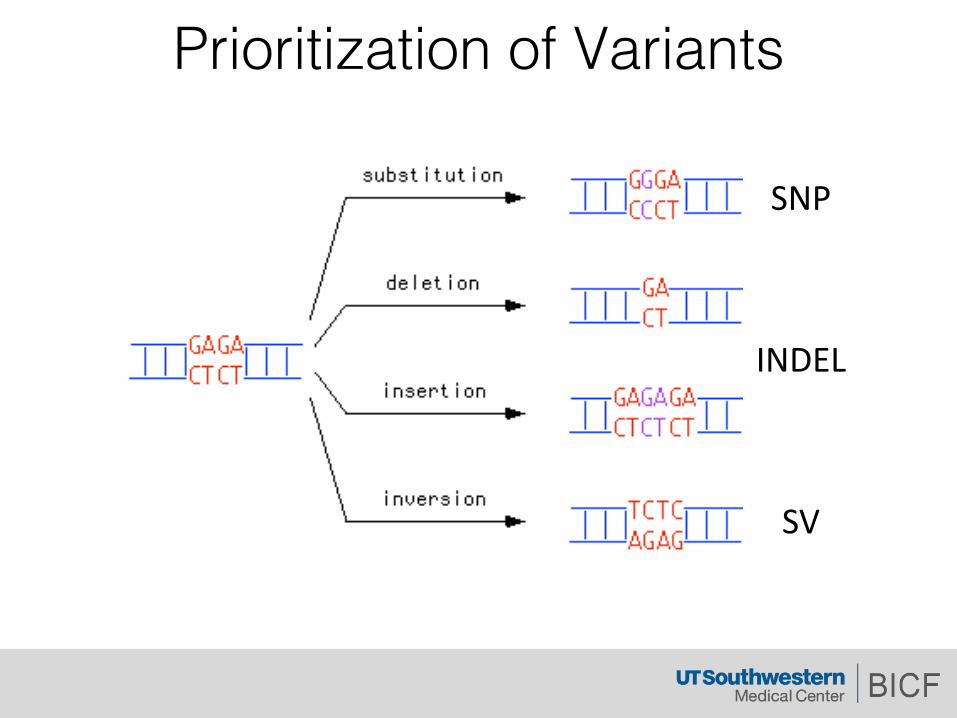
Prioritization of Variants
SNP
INDEL
SV

Allows groups to give easy-access to their analysis pipelines via the web
Astrocyte– BioHPCWorkflowPlatform
StandardizedWorkflows
SimpleWebForms
Onlinedocumentation&resultsvisualization*
WorkflowsrunonHPCclusterwithoutdeveloperoruserneedingclusterknowledge
Slidecontribution:DavidTrudgian@BioHPC
astrocyte.biohpc.swmed.edu

https://astrocyte.biohpc.swmed.edu/brand/bicf/browse/
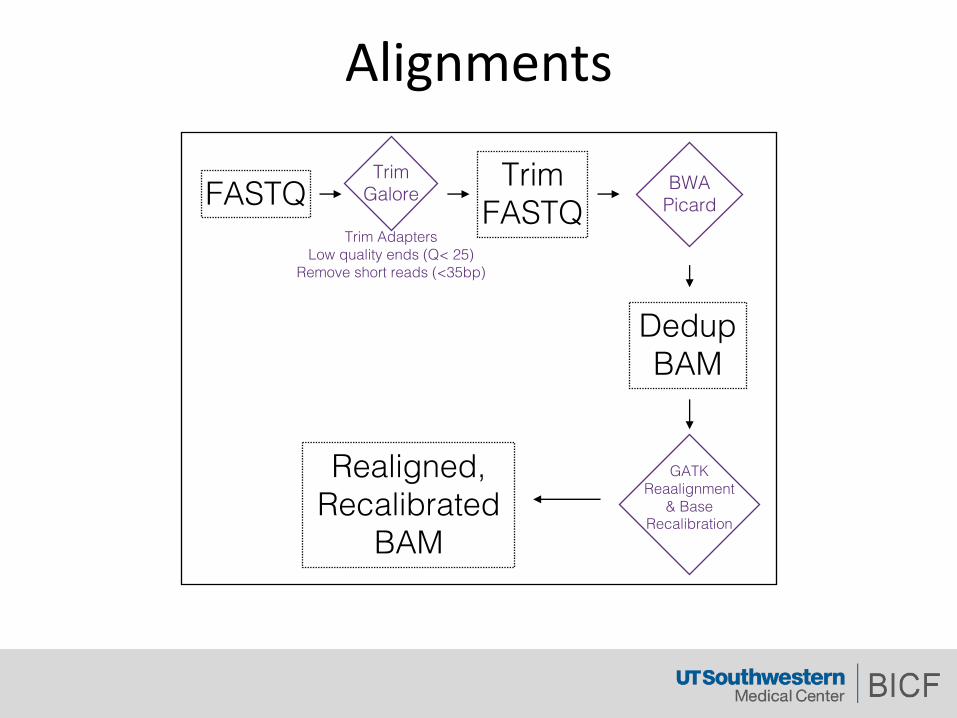
Alignments
FASTQTrim Adapters
Low quality ends (Q< 25)Remove short reads (<35bp)
TrimFASTQ
Trim Galore
DedupBAM
BWAPicard
Realigned, Recalibrated
BAM
GATK Reaalignment
& Base Recalibration

TypesofVariation
Germline Somatic

GermlineWorkflows
GATK VCF
SAM VCF
SV VCF
Lumpy
SS VCF
Speed Seq
GATK Haplotype
CallerSamtools Mpileup
Platypus
Platypus VCF+ + + = Union
VCFHotspot
VCF
DedupBAM
Realigned, Recalibrated
BAM

KeyFiles• VCF file — SNPs/Indels for each sample
• SampleID.annot.vcf.gz• Coverage Histogram for each sample
• SampleID.coverage_histogram.png• Cumulative Distribution Plot for all samples
• coverage_cdf.png• QC for all samples
• sequence.stats.txt• Structural Variants (unfiltered)
• SampleID.sssv.sv.vcf.gz.annot.txt

RecommendedFilteringforGermlineTesting
• ExAC POPMAX AF (0.01-0.05) -• depends on rarity of the phenotype of the
proband• Depth >10• LOF or Misssense (Coding Changes)• Alt Read Ct > 3• Mutation Allele Frequency (MAF) > 0.15• If novel:
– Called by 2+ callers

AccuracyinGIABSample
Sample Fixed Adapters SNV-SN Indel-SN SNV/Indel PPV
NA12878_1_HFVC2BBXX Fresh 1 99.6% 100% 98.9%
NA12878_2_HFVC2BBXX Fresh 1 99.6% 100 98.7%
NA12878_1_HFYWMBBXX Fresh 1 99.7% 100 98.8%
NA12878_2_HFYWMBBXX Fresh 1 99.6% 100 98.5%
GM12878_Fresh_1adapter Fresh 1 99.6% 100 98.6%
GM12878_Fresh_4adapter Fresh 4 99.6% 100 99.0%
GM12878_FFPE_1adapter FFPE 1 99.5% 100 98.5%
GM12878_FFPE_4adapter FFPE 4 99.6% 100 98.4%

Normal Tumor
Tumors are Heterogeneous

SomaticWorkflows
Mutect VCF
VarScan VCF
SS VCF
Speed SeqMuTect2 VarScan Virmid
Virmid VCF+ + + = Union
VCFShimmer
VCF
Shimmer
+
Check Mate
QC Pairs Same
Subject
DedupBAM
Realigned, Recalibrated
BAM

KeyFiles
• VCF file — SNPs/Indels for each sample• TumorID_NormalID.annot.vcf.gz
• Match Check File• TumorID_NormalID_matched.txt

RecommendedFilteringforSomaticMutations
• ExAC POPMAX AF > 0.01• Depth < 20• LOF or Misssense• MAF (Normal) * 10.< MAF (Tumor)• In COSMIC > 5 Subject
– Tumor: Alt Read Ct < 3– Tumor: MAF < 0.01
• Others– Tumor: Alt Read CT < 8– Tumor: MAF < 0.05– Tumor: Called by 2+ callers

SimulatedDatasetstoEvaluateSensitivityandSpecificityofSomaticMutationCalling
• We generated 3 sets of 18 SNVs and 16 Indels • We inserted each set into 4 normal alignment
files (1 cell line (Depth of Coverage) and 3 Saliva samples (Depth of Coverage) using BamSurgeon
• We calculated the observed mutation allele frequency (MAF) using bamreadct
• We ran our somatic mutation workflow using the original bam (Normal) and the altered bam (Tumor)

BioinformaticsSomaticMutationSensitivity
Somatic Germline FP
SNV Obs MAF > 5%
100% novel and known hotspots
80.5% novel, 88.3% known
hotspotsGermline: 0; Somatic: 0
Indel MAF > 5% and Alt Read CT >
8
86.2% novel, 95.4% known
hotspots
86.3% novel, 87.5% known
hotspotsGermline: 0; Somatic: 0
Indel MAF > 10% and Alt Read CT >
893.2% novel, 100%
known hotspots100% novel and known hotspots
Germline: 0; Somatic: 0
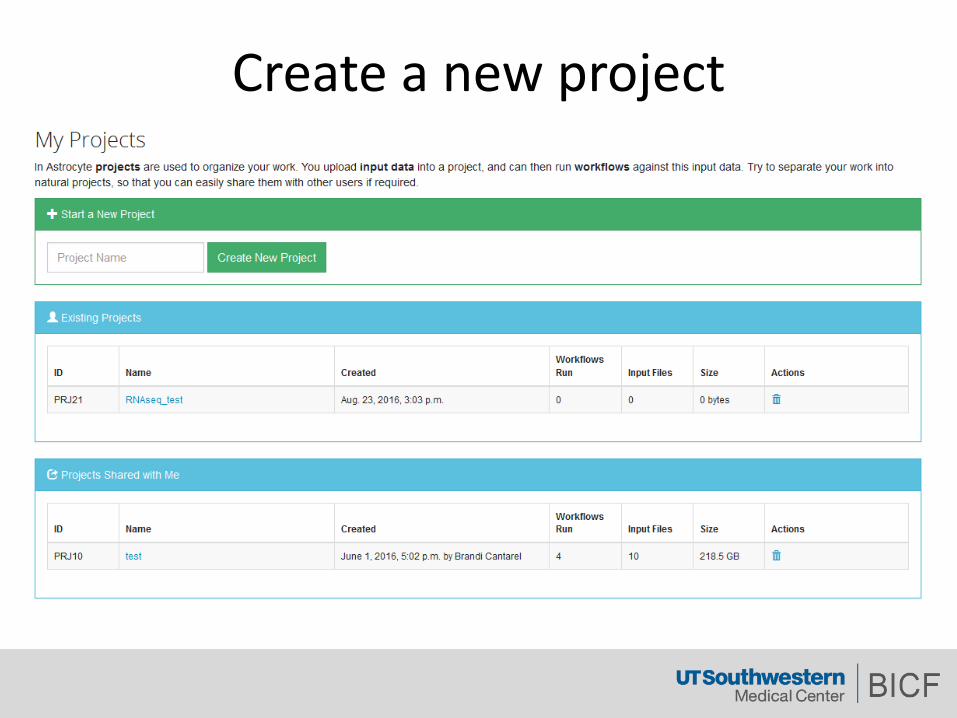
Createanewproject

Adddatatoyourproject

Adddatatoyourproject
ForNGSexperiment,thisisrecommended.

MakeyourdesignfileGermlineWorkflowSampleID
This ID will be used to name all workflow produced files ie S0001 will produce S0001.bam
FullPathToFqR1
Name of the fastq file R1 (not the full path)
FullPathToFqR2
Name of the fastq file R2 (not the full path)
SampleID FullPathToFqR1 FullPathToFqR2
GM12877 GM12877_S124_R1_001.fastq.gz GM12877_S124_R2_001.fastq.gz
GM12878 GM12878_S124_R1_001.fastq.gz GM12878_S124_R2_001.fastq.gz
GM12879 GM12879_S124_R1_001.fastq.gz GM12879_S124_R2_001.fastq.gz

Tipsonmakingyourdesignfile• Use tab as delimiter
– Excel save as “Text (tab delimited)”• If no SubjectID, use same number/character for
all rows• SampleID and SampleName • If no FqR2, leave them empty• For all contents, no “-”• For all contents, no spaces• Columns names MUST be exactly the same as
documented
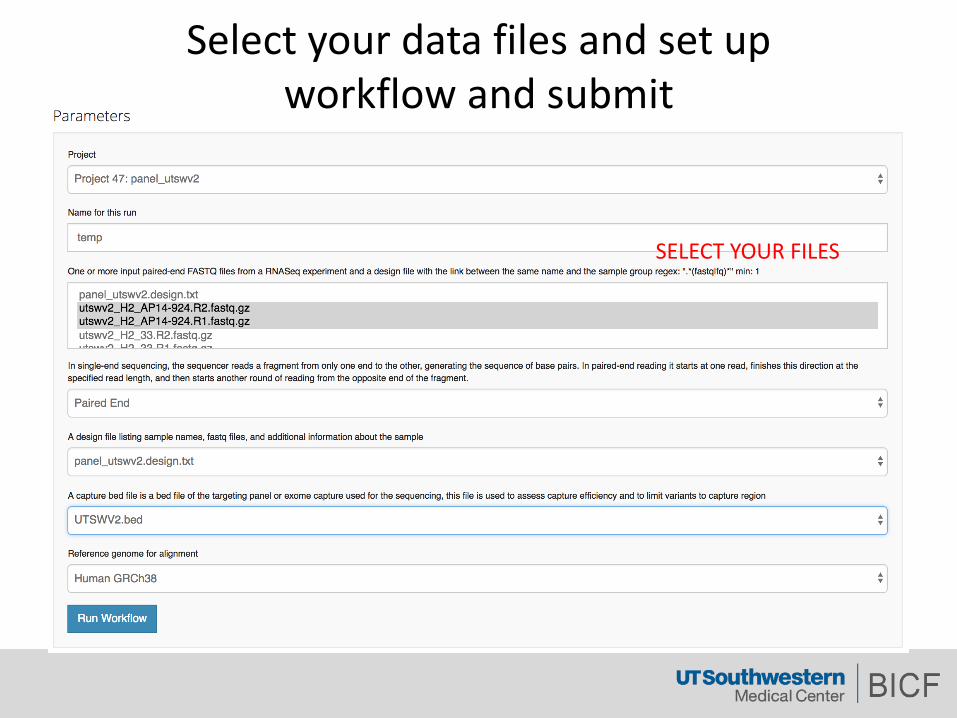
Selectyourdatafilesandsetupworkflowandsubmit
SELECTYOURFILES

Projectisrunning

Timelineofthewholerun

Workingwiththeoutput
ExportalloutputtoAstrocyteOutgoingDirectory

HowtoTransferdatatoRunSomaticWorkflow
• Mount BioHPC on your computer (see BioHPC Introduction slides)
• Login into Cluster

MakeyourdesignfileSomaticWorkflowTumorID
NormalID
The TumorID and NormalID are used for naming the files TumorID_NormalID.annot.vcf.gz
TumorBam
Name of the bam file for the Tumor sample
NormalBam
Name of the bam file for Normal sample
TumorID NormalID TumorBAM NormalBAM
Patient1_tumor Patient1_normal p1_tumor.bam p1_normal.bam
Patient2_tumor Patient2_normal p2_tumor.bam p2_normal.bam

Commonerrorsandsolutions
• Make sure the delimiter is tab• Make sure the column name are the same
as mentioned in documentation• Make sure the file names match

Common errors and solutions
• Not all files are uploaded
• It’s about the proxy setting
• Use auto-detect proxy

Downstreamvisualizationofvariantsfromauserperspective
LingCaiQBRC/CRI-GMDP

Visualizationtools
• IGV– Somaticmutationexamplefromacancersample
• gene.iobio– Germlinemutationexamplesfromgeneticdiseasepatientsamples
IGVuserguide:http://software.broadinstitute.org/software/igv/book/export/html/6
gene.iobio tutorials:http://gene.iobio.io/help_resources.html

UsingIGVonBioHPC – gettingstarted1. LaunchaWebGUI sessionfrom“WebVisualization”under“Cloud
Services”fromBioHPC portal2. Openterminalandtypeincommand
moduleloadIGV/2.3.90;igv.sh
3. Specifygenome(shouldmatchtothereferencegenomefromwhichthevariantswerecalled)

UsingIGVonBioHPC – loadingfilesandsearch1. File->LoadfromFile->Select
2. Search• Alocus(forexample,chr5:90,339,000-90,349,000)• Agenesymbolorotherfeatureidentifier(e.g.,DPYDorNM_10000000)• Amutation(EGFR:T790MorEGFR:2369C>T)
Havetheindexfilesinthesamefolder!

UsingIGVonBioHPC – customizingvisualization• Collapsetracks,displayalternativereadingframes

• Coloralignmentsbyreadstrand• Sortalignmentsbybase
UsingIGVonBioHPC– customizingvisualization

UsingIGVonBioHPC – gettingdetailedinformation• Variant,bamcoverage,read,nucleotidepositionondifferent
transcripts

Usinggene.iobio tovisualizevariantsforgeneticdiseases
Geneticdiseases• Inherited
• Autosomal• Sex-linked
• Denovo
http://www.nature.com/nrg/journal/v18/n10/full/nrg.2017.52.html
http://gene.iobio.io/
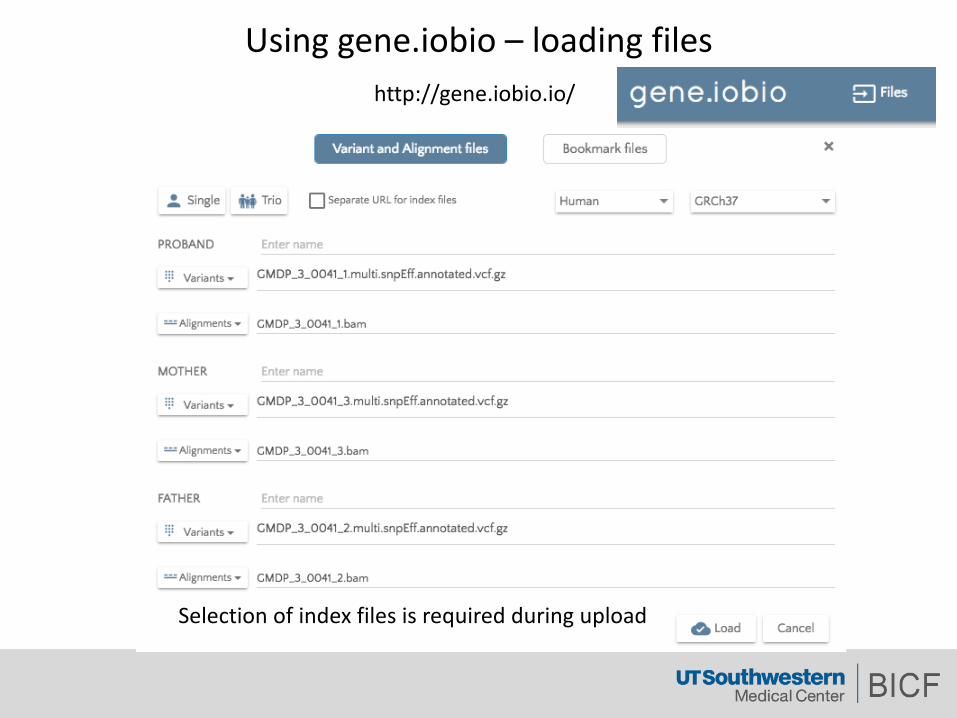
Usinggene.iobio – loadingfiles
Selectionofindexfilesisrequiredduringupload
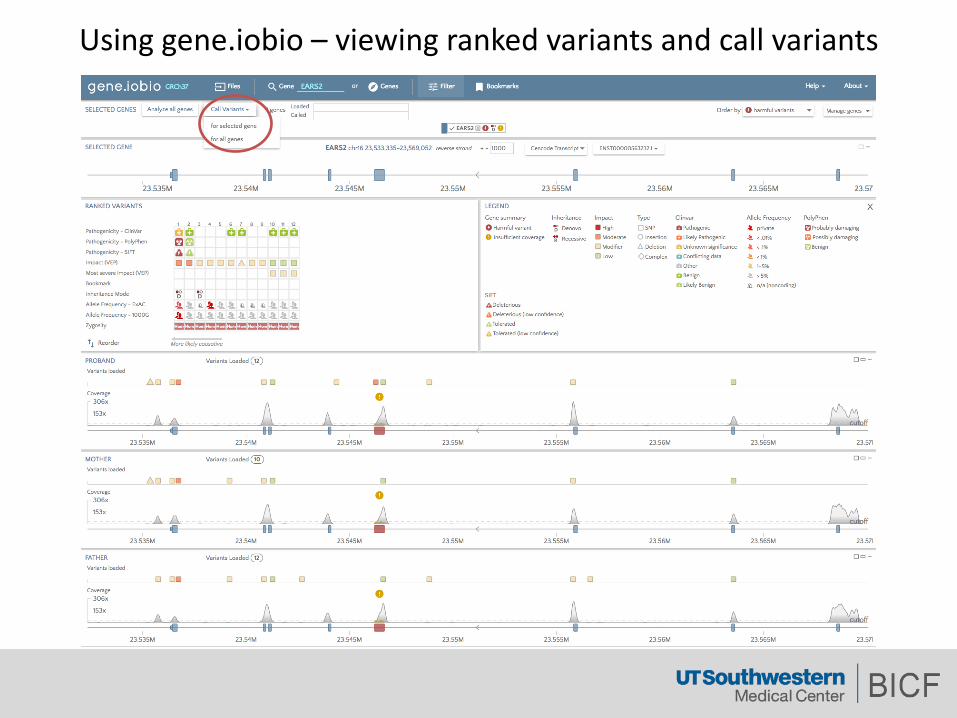
Usinggene.iobio – viewingrankedvariantsandcallvariants

Usinggene.iobio – examiningvariants

Usinggene.iobio – transcriptspecificannotation
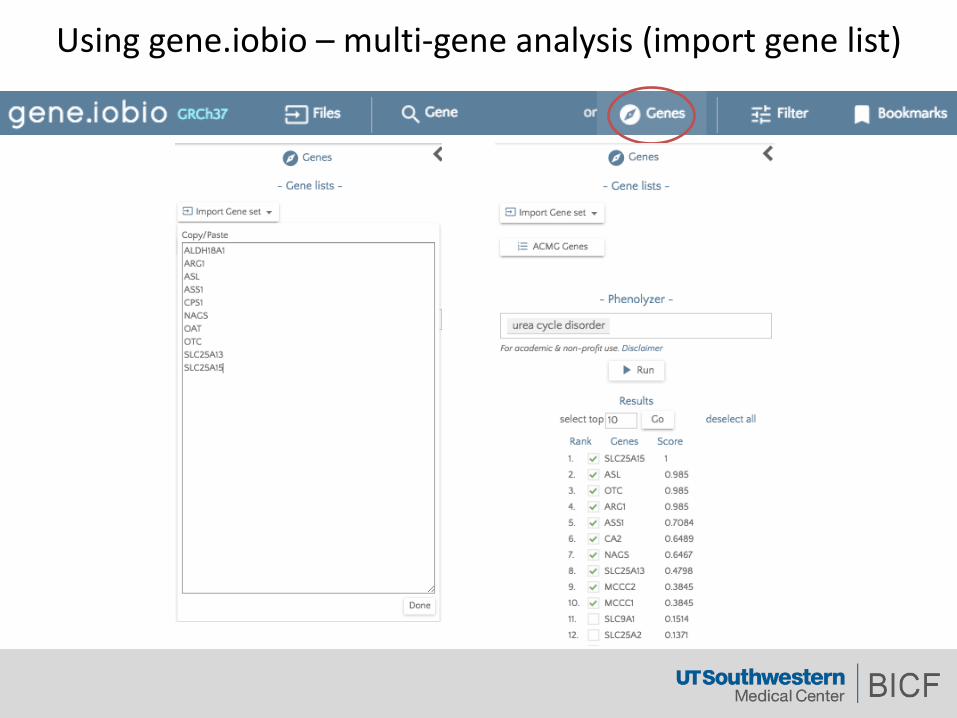
Usinggene.iobio – multi-geneanalysis(importgenelist)

Usinggene.iobio – multi-geneanalysis(Viewingresult)

Usinggene.iobio – multi-geneanalysis(Filteringresult)Loss/Gainoffunctionmutations• Splice• Stopgain/loss• Startgain/loss• CodingframeshiftsNon-synonymousMutations• AminoAcidChangesVariantsLikelytoChangeExpression• TranscriptionFactorBindingSites• miRNATargets

Usinggene.iobio – multi-geneanalysis(bookmarks)

DeterminingGeneticCausesofDiseaseinExomesisNotTrivial• ThecausalvariantisidentifiedinMendelianDisease(Inherited)fromexomesisabout
30%ofcases.
• Ageneticmutationcanexpressarangeofphenotypes(Penetrance)
• Notallfunctionalmutationsareincodingregions(ncRNAsorregulatoryregions)
• Sporaticgeneticdiseasesoftenhaveapolygeniccauses,sometimeswithacombinationofinheritedandsomatic(denovo)mutation
• Mutationscanbelocalizedtoaparticulartissuetypeorregionofthebody(Mosaicism)

https://omictools.com/interpretation5-category
GUItoolsforvariantfiltering(notfree)

Commandlinetoolsforvariantfiltering(free!)
https://gemini.readthedocs.io/en/latest/#
vcftools,bcftools:manipulateVCFpeddy:ped correspondencecheck,ancestrycheck,sexcheck.directly,quicklyonVCF