biological inferences from barcoding data timothy g. barraclough establishing a standard dna barcode...
TRANSCRIPT

Biological inferences from barcoding data
Timothy G. Barraclough
Establishing a standardDNA barcode for land plants

Describing and explaining biological diversity
Traditional taxonomy: slow and subjective

Describing and explaining biological diversity
Traditional taxonomy: slow and subjective
Evolutionary methods: model systems

Describing and explaining biological diversity
Traditional taxonomy: slow and subjective
Evolutionary methods: model systems
Barcoding data:
Large samples within and between species

Describing and explaining biological diversity
Traditional taxonomy: slow and subjective
Evolutionary methods: model systems
Barcoding data:
Large samples within and between species
Single marker; lacking conceptual basis;
X biological relevance?

Analysing barcoding data
Empirical approaches: Thresholds; pairwise distances; accuracies
OK for species I.D. but limited for evolutionary inference. Assumes prior knowledge of species.

Analysing barcoding data
Empirical approaches: Thresholds; pairwise distances; accuracies
OK for species I.D. but limited for evolutionary inference. Assumes prior knowledge of species.
Population genetics approaches:Statistical tests of predicted signatures of no gene flow between populations

Population genetics approaches
Pros: biological inference, large body of theory

Population genetics approaches
Pros: biological inference, large body of theory
Cons: - assume neutral coalescence- prior informal species limits- single marker: developed for multi-
locus- computationally intensive

E.g. Rivacindela tiger beetles on salt lakes in Australia
sequence 5 individuals per morphotype per salt lake for mtDNA
Pons, J. et al. In press. Systematic Biology

Genetic signatures of species/speciation
Establishment Time Data needed
1. Allele frequencies<0.5N but* prior groups
2. Fixed differences prior groups
3. Monophyly prior groups
4. Genealogical 2 or more conconcordance unlinked markers
5. Clusters > 1N 1 marker

Likelihood method testing for significant clusters
Among-species branching Within species branching

Among-species branching =
speciation rate, extinction rate, how they vary over timesampling, reconstruction biases

Within species branching =
Coalescence: population size, demographic and selective history, sampling/artefacts?

Birth-death branching models
Relative time since root node
Log (Number of lineages)
x1 x2 x3
Barraclough, T.G. and Nee, S. 2001. Trends Ecol Evol. 16:391-399

Among-species branching, Yule model
Lik(t) = ne-nx
x is waiting interval, n number of lineages during interval is per lineage branching rate

E.g. Human demographic and selective history
Kingman, Hudson, etc. etc.
Coalescent theory
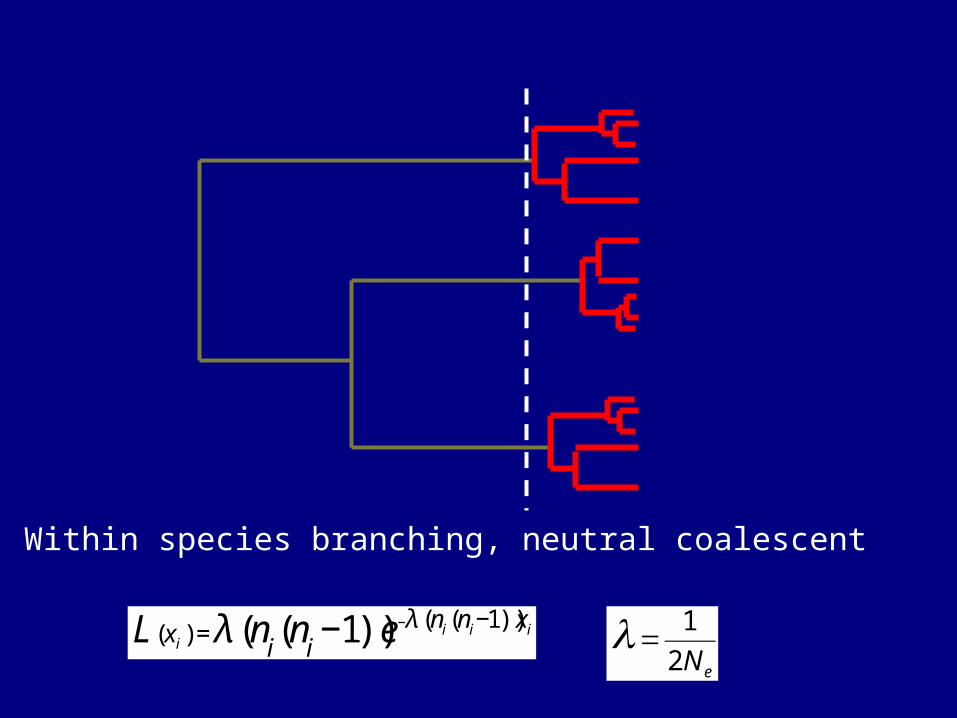
Within species branching, neutral coalescent
€
L(xi ) =λ (ni(ni−1))e−λ (ni(ni−1))xi
€
=1
2N e

Likelihood method testing for significant clusters
Among-species branching
1
Within species branching
2
=> Compare with no-threshold, single entity model

Complication 1
How to account for infinite range of possible models without fitting and testing all of them?
Solution
Add two scaling parameters optimized to accommodate a large range of specific models

Generalized Yule model
Lik(t) = npe-npt
Among species:
p = 1, constant speciation rate no extinction
p > 1, constant background extinction or recent burst of speciation
p < 1, slowdown model or incomplete sample of species
Within species:
p = 2, neutral coalescent
p > 2, declining populations, recent selective sweep
p < 2, growing populations or balancing selection

Complication 2
Allow for mixture of processes at different times: most recent speciation event could post-date oldest within-species branch
Solution
Likelihoods under mixedmodel
€
L(xi)=b*e−b*xi
€
b* = λk+1
(ni,k+1
)pk+1 + (λ j (n i, j (n i, j−1))
pj )
j=1, k
∑

Model: conclusions
General likelihood model for set of within-species branching processes linked by between-species branching.(written in R statistical programing language)
• Define or optimise species nodes
• Estimate key parameters, e.g. changes through time
• Hypothesis testing
• Confidence intervals
Examples of use
1. Australian tiger beetles
2. Ancient asexual rotifers, bdelloids
3. Barcoding, e.g. plants

Rivacindela tiger beetles on salt lakes
Sampled 5 individuals per morphotype per salt lake

mtDNA tree, 468 individuals, 47 ‘species’
Joan Pons, Jesus Gomez-Zurita, Anabela Cardoso, Daniel Duran, William Sumlin, Alfried Vogler

Method Numb. species
1. Allele frequenciesFst 51
2. Fixed differences PAA 46
3. Monophyly Wiens-Penkrot 47

Likelihood method
48 species(+ 3 /- 1)
Missed embedded species
Recovered singleindividuals

Assumes same population parameters for each species,
Repeated allowing them to vary across species and three categories of values: significantly better fit
Parameter values suggest:
Deficit of recent coalescent events across species Growing populations, past bottleneck
Surprisingly constant levels of variation across speciesBottleneck again? Aridification
Speeding up of apparent speciation rate towards the present

Current work:
Optimisation of species nodes without assuming a threshold
Model does not assume threshold, but easiest way to optimise
Computationally intensive…

Rotifers
Significant fit to transition model
282 clusters(C.I. 273 - 294)
P<<0.0001

Barcoding:
Could use approach to delimit species, e.g.marine bacteria, viruses, ericoid mycorrhiza
Probability of sequence belonging to “species” X, or probability of not belonging to any existing species (repeat across bootstrap/Bayes trees)
Global success of barcoding? incomplete samples, low speciation v. N

How many ambiguous species?
Clade of 100 species of annual plantsAverage effective population sizes of NSpeciation rate of lambda per species per myr
Tmrca = 1N => more recent speciation events ambiguous w.r.t plastid DNA
To have fewer than 5 ambiguous sister pairs
Lambda < 0.05 Myr-1 [N = 1 million]Lambda <5 Myr-1 [N = 10000]

Conclusions
Can use barcode type data to delimit species [limitations]
Can use framework to assess, predict, quantify errors for barcode approaches
Multiple unlinked markers, RI, morphology

Acknowledgements
Mark Chase, Robyn CowanAlfried Vogler, Sean NeeElisabeth Herniou
NERC, Royal Society, Sloan and Moore Foundations, CBOL