bipolar disorder - the upswing in research and treatment
TRANSCRIPT


BIPOLAR DISORDER:THE UPSWING IN RESEARCH
AND TREATMENT
Prelims 8/4/05 2:31 pm Page i

Prelims 8/4/05 2:31 pm Page ii

BIPOLAR DISORDER:THE UPSWING IN RESEARCH
AND TREATMENT
EDITED BY
COLM MCDONALDResearch Training Fellow
Division of Psychological MedicineInstitute of Psychiatry
London, UK
KATJA SCHULZEResearch Worker
Division of Psychological MedicineInstitute of Psychiatry
London, UK
ROBIN M MURRAYProfessor of Psychiatry
Division of Psychological MedicineInstitute of Psychiatry
London, UK
MAURICIO TOHENProfessor of Psychiatry
Department of PsychiatryHarvard Medical School
Belmont, Massachusetts, USA
LONDON AND NEW YORK
European Foundation for Psychiatryat The Maudsley
Prelims 8/4/05 2:31 pm Page iii

© 2005 Taylor & Francis, an imprint of the Taylor & Francis Group
First published in the United Kingdom in 2005by Taylor & Francis,an imprint of the Taylor & Francis Group, 2 Park Square, Milton ParkAbingdon, Oxon OX14 4RN, UK
Tel.: +44 (0) 20 7017 6000Fax.: +44 (0) 20 7017 6699Website: www.tandf.co.uk
All rights reserved. No part of this publication may be reproduced, stored in a retrievalsystem, or transmitted, in any form or by any means, electronic, mechanical, photocopy-ing, recording, or otherwise, without the prior permission of the publisher or in accordancewith the provisions of the Copyright, Designs and Patents Act 1988 or under the terms ofany licence permitting limited copying issued by the Copyright Licensing Agency, 90Tottenham Court Road, London W1P 0LP.
Although every effort has been made to ensure that all owners of copyright material havebeen acknowledged in this publication, we would be glad to acknowledge in subsequentreprints or editions any omissions brought to our attention.
British Library Cataloguing in Publication Data
Data available on application
Library of Congress Cataloging-in-Publication Data
Data available on application
ISBN 1-84184-501-9 (Hardback)
Distributed in North and South America by
Taylor & Francis2000 NW Corporate BlvdBoca Raton, FL 33431, USA
Within Continental USATel.: 800 272 7737; Fax.: 800 374 3401Outside Continental USATel.: 561 994 0555; Fax.: 561 361 6018E-mail: [email protected]
Distributed in the rest of the world byThomson Publishing ServicesCheriton HouseNorth WayAndover, Hampshire SP10 5BE, UKTel.: +44 (0) 1264 332424E-mail: [email protected]
Composition by Parthenon PublishingPrinted and bound by T. G. Hostench S.A., Spain
Prelims 8/4/05 2:31 pm Page iv

Contents
Preface ix
Foreword xiii
Contributors xv
Section 1. Do we know the clinical course and epidemiology?
1 The clinical epidemiology of bipolar disorder: a 35-year 1incidence study in south-east LondonNoel Kennedy and Robin M Murray
2 The functional outcome of bipolar disorder 9Mauricio Tohen and Julie M Niswander
Section 2. Is bipolar disorder a brain disease?
3 Brain abnormalities in bipolar disorder: do they exist and 21do they change?E Serap Monkul and Jair C Soares
4 Structural magnetic resonance imaging studies in bipolar 27disorder: a meta-analysisColm McDonald, Jolanta Zanelli, Robin M Murray andNoel Kennedy
5 Are subcortical regions too expansive in bipolar disorder? 37An examination of the nature of prefrontal corticolimbicabnormalities in individuals with bipolar disorderMary L Phillips
6 The Maudsley Bipolar Disorder Project: insights intopathophysiology 51Sophia Frangou
Prelims 8/4/05 2:31 pm Page v

vi Bipolar disorder: the upswing in research & treatment
7 Is any of this real? The word from the grave 59Paul J Harrison
Section 3. Happy genes, blue genes, any genes?
8 How can bipolar disorder be genetically related to both 69schizophrenia and unipolar depression?Peter McGuffin
9 Recent advances in genetics of bipolar disorder 77Daniel J Müller and James L Kennedy
10 Is there a genetic basis to the brain abnormalities of bipolar 93disorder?Colm McDonald
11 Transgenic mouse models for affective disorders based on 103the neurotrophin hypothesisPeter Gass
Section 4. Cortisol: hero or villain?
12 Is the hypothalamic–pituitary–adrenal axis at last paying 115dividends?David A Cousins and Allan H Young
13 Stress on the brain: neuropathology and cortisol 123dysregulation in bipolar disorderDavid Cotter
14 Cortisol in Chicago (from crime of passion to celebrity 129headline)Carmine M Pariante
15 Biological factors sustaining hypothalamic–pituitary– 135adrenal axis overactivation in affective disorder: focuson vasopressinTimothy G Dinan, Sinead O’Brien and Lucinda Scott
Section 5. What is the role of psychology?
16 Cognitive dysfunction: cause or consequence of bipolar 145disorder?Samuel R Chamberlain and Barbara J Sahakian
Prelims 8/4/05 2:31 pm Page vi

Contents vii
17 The neural basis of cognitive function in bipolar disorder 157Vivienne Curtis
18 Psychological treatments: does the evidence stack up? 165Jan Scott
Section 6. Improving the patient’s lot
19 Lithium, the forgotten drug 175Mario Maj
20 Advantages and disadvantages of atypical antipsychotics or 181valproate in bipolar disorderJohn Cookson
21 Is electroconvulsive therapy still given in bipolar disorder 193and does repetitive transcranial magnetic stimulation offermore?Andrew Mogg, Savitha Eranti, Graham Pluck andDeclan M McLoughlin
22 Improving outcome by selecting effective long-term 201treatmentPaul Grof
23 Is what we offer to patients half acceptable? 211Rachel Perkins
Index 219
Prelims 8/4/05 2:31 pm Page vii

Prelims 8/4/05 2:31 pm Page viii

Preface
Bipolar disorder is a recurrent illness which, without appropriate treat-ment, can have a devastating impact on the lives of those affected and theirfamilies. The World Health Organization has estimated that bipolar disor-der is the sixth leading cause of years lived with disability (schizophreniacame ninth) and yet historically it has been relatively under-researched.This situation has greatly changed in recent years as bipolar disorder hasbecome a focus for research, with consequent advances in our understand-ing of the aetiology and management of the disorder. This ‘upswing’ in theresearch and treatment of the illness was discussed at a recent EuropeanFoundation for Psychiatry at the Maudsley (EFPM) meeting held at theInstitute of Psychiatry. The theme of the conference, and of this resultantvolume, is the new research and clinical developments in bipolar disorderresearch across multiple disciplines.
The book comprises six sections. In the first, studies are presented thatemploy the powerful tools of epidemiology to identify how the incidence andage of onset of bipolar disorder are influenced by temporal variation anddemographic variables. The long-term course of the disorder is also con-sidered with a particular emphasis on the relative failure of treatments toaffect important functional outcomes for patients, as distinct from thesymptomatic improvements which clinicians tend to emphasize.
The second section is devoted to investigations attempting to locate thebrain regions that are structurally and functionally impaired in bipolar dis-order at a macroscopic and microscopic level. Although Kraepelin consid-ered the syndrome of ‘manic depressive insanity’ that he described as abrain disease, for much of the past century the neurobiological basis of theillness has been denied or ignored. However, the application of recentneuroimaging techniques has reinvigorated this field, and bipolar disorderhas been associated with subtle deviations of brain structure, and with evi-dence of impaired functioning of critical brain regions implicated in the pro-cessing of cognitive and affective stimuli. In the final chapter of this section,these neuroimaging findings are integrated with the emerging neuropatho-logical literature, most of which emanates from the invaluable resource ofpostmortem tissue provided by the Stanley Medical Research Institute, to
Preface 7/4/05 3:33 pm Page ix

x Bipolar disorder: the upswing in research & treatment
develop an understanding of the structural and functional abnormalities ata cellular level.
It has long been known that bipolar disorder is highly heritable but, aswith many other psychiatric syndromes, progress in identifying the suscep-tibility genes has been slow. Section 3 includes chapters which review thegenetics of the disorder. The topics covered include the genetic overlapbetween bipolar disorder and its related syndromes, schizophrenia andunipolar depression, how abnormalities of brain structure reflect theimpact of susceptibility genes, the depressive-like behaviour of transgenicmice, and the associations of allelic variation in candidate genes withinlinked regions and the clinical syndrome.
A central strand of research into affective disorders has been dysfunc-tion of the hypothalamic–pituitary–adrenal axis. There is clear evidence forimpaired regulation of cortisol secretion from the adrenal glands in bothunipolar and bipolar disorder, and the precise mechanisms whereby thisemerges and its interaction with other components of the axis are excitingareas of current research. Section 4 deals with excessive cortisol secretionin bipolar disorder, addressing its origins, how it is sustained, and itsneurotoxic effects. Whether it causes or compensates for the clinical syn-drome, as well as its potential for manipulation to therapeutic benefit, isalso reviewed.
Section 5 addresses the psychology of bipolar disorder, including themanner in which impaired cognitive processing of emotionally colouredstimuli can contribute to the symptoms of affective disorder. There is abun-dant evidence for cognitive dysfunction during episodes of bipolar illness,but even in remission neurocognitive abnormalities frequently persist. Thisreflects trait-like dysfunction in the neural networks subserving these func-tions and these are explored using functional imaging studies. The finalchapter of this section considers the role of psychological treatments in themanagement of bipolar disorder, and in particular the evidence that cogni-tive behavioural psychotherapy can help to prevent patients relapsing intoepisodes of illness.
The last section is devoted to management of the illness. Bipolar disor-der is clinically heterogeneous and optimal treatment strategies must payheed to the individual patient’s characteristics. Although the number andtype of medications available is expanding, important roles remain for lithi-um and for monotherapy in maintenance treatment. Other issues discussedinclude the use of antipsychotics and valproate as monotherapy or in com-bination for the treatment of mania. Novel strategies such as repetitivetranscranial magnetic stimulation for bipolar depression and the use of
Preface 7/4/05 3:33 pm Page x

Preface: bipolar disorder: the upswing in research and treatment xi
long-term efficacy studies to tailor treatment on the basis of patients’ clini-cal characteristics are also reviewed. The final word from Rachel Perkinscommunicates a unique perspective since she suffers herself from bipolardisorder and is also employed as a senior manager of mental health servic-es within the UK National Health Service. She discusses the practical prob-lems of living with manic depression which are really of importance topatients, most of which are ignored by standard services, with a focus onthe critical issue of protecting employment.
The aim of the EFPM is to provide high-quality postgraduate educationin psychiatry. It is an independent body aiming to provide up-to-date knowl-edge to Europe’s foremost clinical and academic professionals in mentalhealth. Eli Lilly and Company provided a non-restrictive educational grantfor the conference. This book will be of interest to clinicians and academicswithin psychiatry, psychology and neuroscience as well as other mentalhealth professionals interested in bipolar disorder. The editors believe thatthe expert contributions capture the considerable progress that has beenmade in our understanding of this devastating condition in recent years. Weare very grateful to all the contributors and hope that the reader will also bestimulated and informed by the wide range of research approaches that thisbook encompasses.
Colm McDonaldKatja Schulze
Robin M MurrayMauricio Tohen
Preface 7/4/05 3:33 pm Page xi

Preface 7/4/05 3:33 pm Page xii

Foreword
Research on the determinants, treatments and consequences of bipolar dis-order has shown a very welcome increase over the last 15 years. In 1990the seminal Handbook of Bipolar Disorders by Goodwin and Jamieson waspublished which documented all the studies that had been done on bipolardisorder at that time. Although this book became the bible for thoseresearching bipolar disorder and was both of a high intellectual standingand comprehensive in its scope, re-reading it now reveals the uncertainty wehad about the phenotype and the rather poor quality of a lot of studies thatwere done in bipolar disorder. Many studies were on heterogeneous sam-ples and there was a lack of clarity on outcomes. Over the succeeding 15years there have been considerable advances in firming up the classificationand typology of the disorder. The increased number of published studieshas led to much firmer conclusions about the nature of this disorder andits treatment. Another change that has occurred in these 15 years is a muchgreater understanding of the complexity of bipolar disorder. In the past, theKraepelinian view that the patient was either manic or depressed or well,held sway, and the fact that lithium worked gave psychiatrists a false senseof security. The reality is much more complex with considerable subsyn-dromal difficulties in many patients and failure to recover fully in theeuthymic phase evident for many. It is also apparent that while the phar-macological and psychological treatment of bipolar disorder have improvedconsiderably, major problems with partial or complete treatment resistanceand problems with side-effects and/or compliance remain. This book is anexcellent summary of progress in all the important areas of bipolar disor-der sometimes using state of the art technology and its title ‘Upswing’ mir-rors this. The meeting was an exciting event with new science and gooddebate and this volume reflects that excellence. The volume also producesinteresting pointers to where we may be in 15 years time. There is causefor guarded optimism.
Professor Nicol FerrierDepartment of Psychiatry, School of Neurology,
Neurobiology and Psychiatry, Royal Victoria Infirmary, Newcastle upon Tyne, NE1 4LP, UK
Foreword 7/4/05 3:34 pm Page xiii

Foreword 7/4/05 3:34 pm Page xiv

Contributors
Samuel R Chamberlain Department of Psychiatry, University ofCambridge School of Clinical Medicine, Addenbrooke’s Hospital, HillsRoad, Cambridge CB2 2QQ, UK
John Cookson BM DPhil FRCPsych FRCP, The Royal LondonHospital, St Clement’s, 2A Bow Road, London E3 4LL, UK
David Cotter MB BCh MRCPsych PhD, Department of Psychiatry,Royal College of Surgeons in Ireland, Education and ResearchCentre, Beaumont, Dublin 9, Ireland
David A Cousins BMedSci MB BS MRCP MRCPsych, StanleyResearch Centre, School of Neurology, Neurobiology and Psychiatry,University of Newcastle upon Tyne, Leazes Wing, Royal VictoriaInfirmary, Newcastle upon Tyne NE1 4LP, UK
Vivienne Curtis Division of Psychological Medicine, Institute ofPsychiatry, King’s College London, de Crespigny Park, Denmark Hill,London SE5 8AF, UK
Timothy G Dinan MD PhD DSc, Department of Psychiatry, UniversityCollege, Cork, Ireland
Savitha Eranti MRCPsych, Section of Old Age Psychiatry, Institute ofPsychiatry, King’s College London, de Crespigny Park, Denmark Hill,London SE5 8AF, UK
Sophia Frangou Section of Neurobiology of Psychosis, Division ofPsychological Medicine, Institute of Psychiatry, King’s CollegeLondon, de Crespigny Park, Denmark Hill, London SE5 8AF, UK
Peter Gass MD, Central Institute for Mental Health, UniversitätHeidelberg J 5, D-68159 Mannheim, Germany
Contributors 7/4/05 3:34 pm Page xv

xvi Bipolar disorder: the upswing in research & treatment
Paul Grof MD PhD FRCPC, Bipolar Research Unit, University ofOttawa, Royal Ottawa Hospital, 1145 Carling Avenue, Ottawa,Ontario, Canada K17 7K4
Paul J Harrison Department of Psychiatry, University of Oxford,Warneford Hospital, Oxford OX3 7JX, UK
James L Kennedy MD MSc FRCP(C), Neurogenetics Section, Centrefor Addiction and Mental Health, Department of Psychiatry,University of Toronto, 250 College Street, Toronto, Ontario, CanadaM5T 1R8
Noel Kennedy MB MD MSc(Med) MRCPsych, Section of GeneralPsychiatry, Division of Psychological Medicine, Institute ofPsychiatry, King’s College London, de Crespigny Park, Denmark Hill,London SE5 8AF, UK and St. Patrick’s Hospital, Dublin, Ireland
Colm McDonald MRCPsych PhD, Division of Psychological Medicine,Institute of Psychiatry, King’s College London, de Crespigny Park,Denmark Hill, London SE5 8AF, UK
Peter McGuffin MB, PhD, FRCP, FRCPsych, FMedSci, Social, Geneticand Developmental Psychiatry Centre, Institute of Psychiatry, King’sCollege London, de Crespigny Park, Denmark Hill, London SE5 8AF,UK
Declan M McLoughlin PhD MRCPI MRCPsych, Section of Old AgePsychiatry, Institute of Psychiatry, King’s College London, deCrespigny Park, Denmark Hill, London SE5 8AF, UK
Mario Maj MD PhD, Department of Psychiatry, University of Naples,Italy
Andrew Mogg MRCPsych, Section of Old Age Psychiatry, Institute ofPsychiatry, King’s College London, de Crespigny Park, Denmark Hill,London SE5 8AF, UK
E Serap Monkul Division of Mood and Anxiety Disorders, Departmentof Psychiatry, University of Texas Health Science Center at SanAntonio, 7703 Floyd Curl Drive, San Antonio, TX 78229, USA
Contributors 7/4/05 3:34 pm Page xvi

Contributors xvii
Daniel J Müller MD, Neurogenetics Section, Centre for Addiction andMental Health, Department of Psychiatry, University of Toronto, 250College Street, Toronto, Ontario, Canada M5T 1R8 and Departmentof Psychiatry, Charité University Medicine Berlin, Campus CharitéMitte, Berlin, Germany
Robin M Murray MB MD DSc FRCPsych, Section of GeneralPsychiatry, Division of Psychological Medicine, Institute ofPsychiatry, King’s College London, de Crespigny Park, Denmark Hill,London SE5 8AF, UK
Julie M Niswander PhD, Lilly Research Laboratories, Indianapolis,IN, USA
Sinead O’Brien MRCPsych, Department of Psychiatry, UniversityCollege, Cork, Ireland
Carmine M Pariante MD, MRCPsych, PhD, Stress, Psychiatry andImmunology Laboratoy (SPI-LAB), Institute of Psychiatry, King’sCollege London, 1 Windsor Walk, London SE5 8AF, UK
Rachel Perkins BA MPhil PhD, South West London and St George’sMental Health NHS Trust, Springfield University Hospital, Tooting,London SW17 7DJ, UK
Mary L Phillips Section of Neuroscience and Emotion, Division ofPsychological Medicine, Institute of Psychiatry, King’s CollegeLondon, de Crespigny Park, Denmark Hill, London SE5 8AF, UK
Graham Pluck PhD, Section of Old Age Psychiatry, Institute ofPsychiatry, King’s College London, de Crespigny Park, Denmark Hill,London SE5 8AF, UK
Barbara J Sahakian FMedSci, Department of Psychiatry, Universityof Cambridge School of Clinical Medicine, Addenbrooke’s Hospital,Hills Road, Cambridge CB2 2QQ, UK
Katja Schulze Division of Psychological Medicine, Institute ofPsychiatry, King’s College London, de Crespigny Park, Denmark Hill,London SE5 8AF, UK
Contributors 7/4/05 3:34 pm Page xvii

Jan Scott Division of Psychological Medicine, Institute of Psychiatry,King’s College London, de Crespigny Park, Denmark Hill, LondonSE5 8AF, UK
Lucinda Scott PhD MRCPsych, Department of Psychiatry, UniversityCollege, Cork, Ireland
Jair C Soares MD, Division of Mood and Anxiety Disorders,Department of Psychiatry, University of Texas Health Science Centerat San Antonio, 7703 Floyd Curl Drive, San Antonio, TX 78229, USA
Mauricio Tohen MD DrPH, Department of Psychiatry, HarvardMedical School, McClean Hospital, 115 Mill Street, Belmont, MA02478, USA
Allan H Young MB ChB MPhil PhD FRCPsych, Stanley ResearchCentre, School of Neurology, Neurobiology and Psychiatry, Universityof Newcastle upon Tyne, Leazes Wing, Royal Victoria Infirmary,Newcastle upon Tyne NE1 4LP, UK
Jolanta Zanelli MSc, Division of Psychological Medicine, Institute ofPsychiatry, King’s College London, de Crespigny Park, Denmark Hill,London SE5 8AF, UK
Contributors 7/4/05 3:34 pm Page xviii

The clinical epidemiologyof bipolar disorder:a 35-year incidence studyin south-east London Noel Kennedy and Robin M Murray
c h a p t e r 1
Introduction
Numerous studies have established the basic clinical epidemiology ofschizophrenia.1 For example, our research group has carried out a numberof epidemiological studies of schizophrenia in Camberwell, south-eastLondon, and have found that the incidence of narrowly defined schizophre-nia was higher in males than females and that the age at onset of schizo-phrenia was earlier in men than women, echoing findings from other cen-tres.1,2 We have also shown that the incidence of schizophrenia has doubledin Camberwell since 1964, although this increase may be idiosyncratic tosouth London.3 It may result at least in part from the influx of migrants tothis area, as the incidence of schizophrenia is approximately six times high-er among the Black African and African–Caribbean population in southLondon compared with the White population.4 These elevated incidencerates have not been reported in Caribbean countries and may involve com-plex biological, psychological and social factors.5
In contrast to schizophrenia, very little is currently known about thebasic clinical epidemiology of bipolar disorder. The few published incidencestudies, based on small sample sizes, have reported a wide variation in inci-dence rates of first-episode mania from 1.7 to 4.5 per 100000 populationper year.6 Furthermore, studies to date may have underestimated true inci-dence.6 Data from the Epidemiological Catchment Area Survey (ECA) havesuggested that the risk of developing mania may be increasing in recent gen-erations, although these data may be subject to recall bias.7 Similarly,whether there exist gender differences in incidence or age at onset of bipolar
Ch 01 7/4/05 3:34 pm Page 1

2 Bipolar disorder: the upswing in research & treatment
disorder is uncertain; earlier studies showed little difference in age at onsetbetween men and women, but more recent studies, using strict operationalcriteria, have tended to show a later onset in women.8 It also remains uncer-tain whether migrants have a higher incidence of bipolar disorder as well asschizophrenia, although earlier incidence studies have suggested that thismay be so.9,10
Given that our research group have already conducted a number of inci-dence studies of schizophrenia in Camberwell, we similarly undertook todescribe the basic clinical epidemiology of bipolar disorder in this definedcatchment area11,12 (and N. Kennedy et al, unpublished) and specifically toaddress the following questions:
1. What is the overall incidence of bipolar disorder in Camberwell andhas it increased since 1965? (N. Kennedy et al, unpublished)
2. What is the peak incidence of bipolar disorder by age and are there dif-ferences in incidence or age at onset by gender?11
3. Are incidence rates of bipolar disorder higher among African andAfrican–Caribbean migrants than Whites in south London?12
Method
Camberwell is an inner-city area that rates highly on deprivation indices.The total population has declined over the years, from 171000 in 1960 toapproximately 120000 currently. The population has also become increas-ingly ethnically diverse with currently over a fifth of the population being ofAfrican–Caribbean or Black African origin.
To describe the clinical epidemiology of bipolar disorder in Camberwell,all adults living in this defined catchment area who presented to psychiatricservices between 1965 and 1999 with mania, hypomania, bipolar disorderor any possible psychosis were identified from the Cumulative CamberwellPsychiatric Case Register.3 Patients admitted to hospitals outside the areawould normally have been transferred to local hospitals or services for con-tinuing care and these records were also identified in this search. Patientswere excluded if they were not resident in the catchment area, had present-ed previously with a psychotic or manic episode, had a clear organic causefor their symptoms, or had an onset before 16 years of age. All case recordswere examined and the Operational Checklist for Psychotic Disorders, ver-sion 3.4 (OPCRIT)13 completed for the year following presentation. TheOPCRIT checklist, based on the Present State Examination,14 was then usedto generate DSM-IV diagnoses for cases using the accompanying computer
Ch 01 7/4/05 3:34 pm Page 2

The clinical epidemiology of bipolar disorder 3
program. Those who met DSM-IV criteria for bipolar I disorder (BPI) ormania became our cases. Bipolar disorder was defined as fulfilling DSM-IVcriteria for mania with or without a previous treated depressive episode inprimary or secondary care, and mania was defined as fulfilling criteria formania without a previously treated depressive episode. Data concerning thegeneral population of Camberwell were ascertained from the 1961–91 cen-suses with intermediate years interpolated. All population data were strati-fied by age and gender and corrected for under-enumeration.
Results
Over the 35-year period, 246 cases fulfilled criteria for DSM-IV BPI from1443 possible cases of mania, hypomania or psychosis identified. Of these246 cases, 78% had their first psychiatric presentation with mania, where-as 22% had had a previous treated depressive episode before the onset offirst mania. Another 12% described a probable previous depression thatwas not treated; 141 (57%) cases were female and 106 (43%) were male(female :male rate ratio 1.21). Almost a fifth (16%) were not admitted dur-ing index mania and almost three-quarters (72%) were psychotic duringtheir first manic episode. Mean and median ages at onset for mania were 33and 28 years, respectively, with mean and median ages at onset for bipolardisorder, also including prior treated depressive episodes, being 31 and 26years, respectively. Peak age at onset was in the 21–25 and 26–30 agegroups followed by a second much smaller peak in incidence by age in mid-life.
The standardized incidence rates of bipolar disorder and mania were6.5 and 5.2 per 100000 population, respectively, with females having asomewhat higher incidence of bipolar disorder (female :male rate ratio1.21). Table 1.1 shows the number of cases of bipolar disorder and maniaover each of seven 5-year time bands over the course of this study. The num-ber of cases increased modestly though significantly over the course of thestudy, particularly during the earlier time periods. This was reflected by asignificant rate ratio linear trend, which summarized the increase in riskduring each time period for both bipolar disorder (p=0.034) and mania(p=0.013).
We found that women had a later onset of mania and bipolar disorder by5 and 4.5 years, respectively, and that this difference remained significanteven after adjusting for a number of potentially confounding pre-morbidvariables such as ethnicity, family history, developmental abnormalities,
Ch 01 7/4/05 3:34 pm Page 3

4 Bipolar disorder: the upswing in research & treatment
pre-morbid functioning and employment. There also appeared to be signif-icant differences between men and women in age at onset, as for schizo-phrenia, with onset being an average of 4–5 years later in women.1 However,age of onset distributions appeared to be different in bipolar disorder andschizophrenia. In schizophrenia men predominated in those with an earlyonset and women were much more likely than men to have an onset in latelife with little difference being found in mid-life.4 By contrast, age at onsetdifferences in bipolar disorder in this study were largely accounted for bywomen having a higher incidence in mid-life, with men and women havingsimilar incidences in early and late life.
We could not directly estimate incidence of bipolar disorder by ethnicityin this 35-year study, as, prior to the 1981 census, population data by eth-nicity were based on head of household unadjusted for age or gender,whereas subsequent data are much more accurate. Therefore, to investigateincidences of bipolar disorder in different ethnic groups, our group con-ducted a 2-year (1997–99) prospective incidence study of first-episode psy-chosis and mania, the AESOP study, which included the entire Camberwell
Table 1.1 Number of cases of DSM-IV bipolar I disorder and mania during
each of seven 5-year time periods over the course of the study
Time period Bipolar I disorder Mania (no previous depression)
1965–69 23 18
1970–74 33 22
1975–79 37 28
1980–84 43 36
1985–89 36 28
1990–94 38 34
1995–99 36 28
Table 1.2 Incidence ratios of first-episode bipolar disorder by ethnicity in
south London (AESOP Study 1997–99)
Ethnicity South London Adjusted rate ratio irr (95% CI)
White 1
Overall non-White 5.5 (2.8–10.6)
Black Caribbean 7.6 (3.7–15.8)
African 4.4 (1.7–11.1)
Ch 01 7/4/05 3:34 pm Page 4

The clinical epidemiology of bipolar disorder 5
catchment area. As seen from Table 1.2, rates of bipolar disorder among theBlack Caribbean and Black African population in London were 7.6 and 4.4times that of the White population over this 2-year time period, confirmingearlier data that, as for schizophrenia, rates of mania were elevated inmigrant groups in south London.
Discussion
In this 35-year incidence study, overall incidence rates for mania were muchhigher than in previous studies (Table 1.3).15–19 Why were our rates ofmania so high? First, previous studies had a number of methodological lim-itations, which the current study was designed to overcome. For example,some previous studies excluded non-admitted15,16 or non-psychoticcases,18,19 which would have excluded almost a fifth and over a quarter ofour sample, respectively. Similarly, previous studies had an upper age limitof either 60 or 65 years, which would have excluded almost a tenth of oursample.15,16,18 Second, almost 40% of our sample over the course of thestudy and over 20% of the current population of Camberwell are of BlackCaribbean or Black African origin. The influx of ethnic minority groups maypartially have accounted for the high incidence of mania in this study, asincidence of bipolar disorder appears to be higher in these ethnic groups insouth London. Finally, other factors such as the effects of inner-city living orhigher risk of abuse of drugs such as cannabis have been suggested as con-tributing to the high rates of schizophrenia in urban areas1 and potentiallycould also contribute to high incidences of mania. Similarly, our findingfrom the AESOP study, that rates of bipolar disorder are higher among
Table 1.3 Incidence studies of first-episode mania*
Incidence per 105 per year
Incidence studies Number of cases Male Female All
Leff et al (1976)15 63 3.1 2.2 2.6
Daly et al (1995)16 30 4.1 4.9 4.5
Veijola et al (1996)17 2 — — 1.7
Brewin et al (1997)18 22 1.5 4.1 2.8
Scully et al (2002)19 8 3.7 0.6 2.2
Current 35-year incidence study 194 5.1 5.2 5.2
*Data from two incidence studies omitted as some cases may overlap with current study.
Ch 01 7/4/05 3:34 pm Page 5

6 Bipolar disorder: the upswing in research & treatment
those of Black African or Black Caribbean origin in south London, mayreflect social disadvantage, more adverse life events or higher rates of illicitdrug abuse in such groups.5
Age at onset of mania and bipolar disorder in this study, with peak onsetin the late twenties, was similar to that reported in previous incidence stud-ies and later than reported in prevalence studies such as the ECA.20 Thismay reflect delays between onset of this disorder (as reported by prevalencestudies) and initial presentation to psychiatric services (as reported in inci-dence studies).20 A significant increase in the incidence of mania and bipo-lar disorder was seen over the three decades of this study. Results from thefew previous studies describing change in incidence of mania over shortertime periods have been equivocal.21–23 However, cohort data from the ECAhave suggested that the risk of developing mania has been increasing inrecent generations.7 The increase in incidence observed in this study wasmuch more modest than the increased incidence of schizophrenia seen inthe same catchment area over a similar time period. Interestingly, theincrease in incidence was mainly seen over the first 20 years of this study,which was also the time of greatest population change and migration.Therefore, the increase in incidence over time, similar to schizophrenia,may reflect environmental factors such as migration, socioeconomic changeor increasing abuse of alcohol or illicit drugs.
In contrast to the gender differences in age at onset found in the currentstudy, early studies of bipolar disorder have generally found little differencein age of onset between men and women.24 However, early studies did notgenerally use strict operational criteria and were generally based on con-secutive admissions or outpatient attendances rather than on epidemiolog-ical samples, and therefore they may have been less representative of typi-cal patients with bipolar disorder than the current sample. Furthermore, anumber of more recent cross-sectional studies using strict operational cri-teria have found that women have a later onset of both bipolar disorder andmania.8 Similar age at onset differences, with men having a significantly ear-lier onset than women, have been described in schizophrenia. However, inschizophrenia men predominate in those with an onset in early life andwomen in those with an onset in late life. Gender differences in age at onsetof schizophrenia have been explained by (1) abnormal neurodevelopmentmore commonly affecting men; or (2) the protective effects of higher oestro-gen levels in women, with a decline during the menopause leading to a surgein incidence in late life among women.1,2 By contrast, the major gender dif-ferences in age at onset in bipolar disorder observed in this study were inmid-life, so the explanations for gender differences in age at onset of bipolar
Ch 01 7/4/05 3:34 pm Page 6

The clinical epidemiology of bipolar disorder 7
disorder are likely to be different. Women may delay seeking treatment com-pared with men or social factors such as urban living, deprivation or sub-stance misuse may particularly affect young men and therefore influence ageat onset differences between men and women.
Acknowledgements
We wish to acknowledge the contributions of Dr Jane Boydell, ProfessorDavid Castle, Professor Peter Jones, Professor Nori Takei, Professor Jimvan Os and Professor Simon Wessely, for their advice and assistance in con-ducting this study and Professor Peter McGuffin for use of the OPCRIT pro-gram. The study was supported by a grant from the Stanley Institute forMedical Research to Professor Murray, and a grant from the PsychiatryResearch Trust to Dr Kennedy.
References
1. Murray RM, Jones PB, Susser E et al, The Epidemiology of Schizophrenia.Cambridge University Press: Cambridge, 2002.
2. Häfner H, Gender differences in schizophrenia. Psychoneuroendocrinology2003; 28:17–54.
3. Boydell J, van Os J, Lambri M et al, Incidence of schizophrenia in south-eastLondon between 1965 and 1997. Br J Psychiatry 2003; 182:45–49.
4. Castle DJ, Wessely S, van Os J, Murray RM, The effect of gender on age at onsetof psychosis. In: Goldberg D, ed. Psychosis in the Inner City: The CamberwellFirst Episode Study. Maudsley Monographs no. 40. Psychology Press: Hove,1998:27–36.
5. Sharpley M, Hutchinson G, McKenzie K, Murray RM, Understanding the excessof psychosis among the African–Caribbean population in England. Review ofcurrent hypotheses. Br J Psychiatry 2001; 178(Suppl 40):s60–68.
6. Lloyd T, Jones PB, The epidemiology of first-onset mania. In: Tsuang MT, TohenM, eds. Textbook in Psychiatric Epidemiology. 2nd edn. Wiley-Liss: New York,2002:445–458.
7. Lasch K, Weissman M, Wickramaratne P et al, Birth-cohort changes in the ratesof mania. Psychiatr Res 1990; 33:31–37.
8. Arnold LM, Gender differences in bipolar disorder. Psychiatr Clin North Am2003; 26:595–620.
9. Der G, Bebbington PE, Depression in inner London: a register study. SocPsychiatry 1987; 22:73–84.
10. Van Os J, Takei N, Castle DJ et al, The incidence of mania: time trends in rela-tion to gender and ethnicity. Soc Psychiatry Psychiatr Epidemiol 1996;31:129–136.
Ch 01 7/4/05 3:34 pm Page 7

8 Bipolar disorder: the upswing in research & treatment
11. Kennedy N, Boydell J, Kalidindi S et al, Gender differences in incidence and ageat onset of mania and bipolar disorder over a 35-year period in Camberwell,England. Am J Psychiatry 2005; 162:257–262.
12. Lloyd T, Kennedy N, Fearon P et al, Incidence of bipolar affective disorder inthree UK cities: results from the AESOP study. Br J Psychiatry 2005;186:126–131.
13. McGuffin P, Farmer A, Harvey I, A polydiagnostic application of operational cri-teria in studies of psychotic illness. Development and reliability of the OPCRITsystem. Arch Gen Psychiatry 1991; 48:764–770.
14. Wing JK, Cooper JE, Sartorius N, Measurement and Classification ofPsychiatric Symptoms. An Instruction Manual for the PSE and CategoProgram. Cambridge University Press: New York, 1974.
15. Leff JP, Fischer M, Bertelsen A, A cross-national epidemiological study ofmania. Br J Psychiatry 1976; 129:428–437.
16. Daly I, Webb M, Kaliszer M, First admission incidence study of mania,1975–1981. Br J Psychiatry 1995; 167:463–468.
17. Veijola J, Rasanen P, Isohanni M et al, Low incidence of mania in northernFinland. Br J Psychiatry 1996; 168:520–521.
18. Brewin J, Cantwell R, Dalkin T et al, Incidence of schizophrenia in Nottingham.A comparison of two cohorts, 1978–80 and 1992–94. Br J Psychiatry 1997;171:140–144.
19. Scully PJ, Quinn JF, Morgan MG et al, First-episode schizophrenia, bipolar dis-order and other psychosis in a rural Irish catchment area: incidence and gen-der in the Cavan–Monaghan study at 5 years. Br J Psychiatry 2002;43(Suppl):S3–S9.
20. Bebbington P, Ramana R, The epidemiology of bipolar affective disorder. SocPsychiatry Psychiatr Epidemiol 1995; 30:279–292.
21. Mander AJ, Diagnosis change, lithium use and admissions for mania inEdinburgh. Acta Psychiatr Scand 1989; 80:434–436.
22. Eagles JM, Whalley LJ, Ageing and affective disorders: the age at first onset ofaffective disorders in Scotland, 1969–1978. Br J Psychiatry 1985;147:180–187.
23. Parker G, O’Donnell M, Walter S, Changes in the diagnosis of the functional psy-chosis associated with the introduction of lithium. Br J Psychiatry 1985;146:377–382.
24. Goodwin FK, Jamison KR, Course and outcome. In: Goodwin FK, Jamison KR,eds. Manic Depressive Illness. Oxford University Press: New York, 1990:127–156.
Ch 01 7/4/05 3:34 pm Page 8

The functionaloutcome ofbipolar disorderMauricio Tohen and Julie M Niswander
c h a p t e r 2
Introduction
The longitudinal course of bipolar disorder is defined by recurrent manicand depressive mood episodes. Clinicians treating patients with bipolardisorder are observant of the severe impact these mood episodes have onthe lives of patients and their families, including job performance and per-sonal relationships and responsibilities. Despite the magnitude of theimpact of mood episodes on day-to-day function, most bipolar disorderstudies conducted to date have examined measures of symptoms and syn-dromal outcome as opposed to more patient-relevant functional improve-ment. Nonetheless, the study of functional outcome is critical: a recentstudy has shown that, 12 months after hospitalization for bipolar disorder,syndromal recovery measured 61.0%, whereas functional recovery wasreported at only 36.0%.1 Restoration of pre-episode quality of life and levelof functioning is of primary importance to the bipolar patient and shouldtherefore serve as the guiding principle in the study and treatment of bipo-lar disorder. This chapter discusses prospective studies conducted atMcLean Hospital in Belmont, Massachusetts that examined functionalrecovery and sought to determine predictors of functional outcome inpatients with bipolar disorder.
Outcomes and identification of predictors
A 4-year prospective follow-up
Following patients in a naturalistic setting provides insight on the longitudi-nal course of bipolar disorder. Although there are no definitive predictors of
Ch 02 7/4/05 3:35 pm Page 9

10 Bipolar disorder: the upswing in research & treatment
future course in bipolar disorder, the outcome after recovery of an indexmanic episode may identify factors that predict continued remission,interepisode symptoms and functional outcomes. This section presentsfindings from a naturalistic study conducted in the mid-to-late 1980s at theMcLean Hospital (Belmont, Massachusetts, USA), the largest psychiatricteaching facility at Harvard Medical School.2
In this study, a cohort of 75 patients who met the DSM-III criteria forbipolar disorder and had recovered from an index manic episode at time ofdischarge were followed for 4 years with assessments at 6 and 48 monthsafter discharge. The patients in this study were ≥17 years of age, 97%(n=73) of them being Caucasian, and they did not include patients withmixed or rapid-cycling symptoms; 32% (n=24) of patients were experienc-ing their first affective episode. Syndromic recovery was defined as the pres-ence of no more than two DSM-III ‘B’ criteria for an affective episode of mildintensity (<3) and absence of ‘A’ criteria. Relapse was indicated by meetingthe DSM-III criteria for an episode of mania or depression after havingachieved remission (recovery of at least 6 consecutive weeks). A key out-come to this study was that no patients were lost to follow-up.
In this study, the probability of remaining in remission was 51% at theend of the first year and 44% at 24 months, 33% at 36 months, and 28% at48 months (Figure 2.1). Interestingly, most relapses occurred in the firstyear, with only 23% (17 of 75) more patients relapsing by 48 months; theprobability of remaining in remission increased the longer the patient hadmaintained recovery. After an index manic episode, cumulative probabilitiessuggested that the risk of relapsing into depression was highest during theinitial 9 months after recovery, but the risk of relapse into mania remainedrelatively constant during the 4 years of follow-up.
Several risk factors were significantly associated with a shorter time inremission. The presence of psychotic features during the index manicepisode predicted time to relapse. At 6 months, the probability of remain-ing in remission was similar for patients stratified by the absence or pres-ence of psychotic features (67% vs. 63%, respectively; Figure 2.2). However,over time, the probability of remaining in remission differed, based on thepresence of psychosis during the index episode; at 3 years, the probabilityof remaining in remission was 52% for patients without psychotic featuresand 26% for patients with psychotic features. Differentiation of mood con-gruence of psychotic features had additional prognostic relevance as mood-incongruent psychotic features may predict a shorter time in remission3.The median time in remission for patients with mood-incongruent psychot-ic mania was 8 months, contrasting with 33 months for patients having
Ch 02 7/4/05 3:35 pm Page 10

The functional outcome of bipolar disorder 11
experienced mood-congruent psychotic features during the index episode(Figure 2.3).
In addition to the presence of psychotic features, other predictors ofpoor outcome included depressive symptoms during the index episode, ahistory of mood episodes, and a history of alcoholism (Table 2.1). Theserisk factors were significantly associated with a shorter time in remission.Furthermore, depressive symptoms during the index episode predicted timeto relapse into a depressive episode, whereas a history of one or more pre-vious episodes (mania or depression) significantly reduced time to relapseinto a manic episode.
As both occupational and residential status are indicators of the abilityto live productively and independently, these markers serve as measures offunctional outcome. Table 2.2 lists the functional outcomes of patients atboth 6 and 48 months as assessed by the Modified Vocational Status Index(MVSI) and Modified Location Code Index (MLCI). Of 72 patients, 60%(n=43) were able to work or study, and 64% (n=46) had independent
Figure 2.1 The cumulative probability of remaining in remission for the numberof patients with bipolar disorder who have not relapsed after recovery from anindex manic episode up to a given time (dark purple). The probabilities of notrelapsing into a manic (grey) or major depressive (light purple) episode are alsorepresented. (Derived from reference 2.)
75------------ 53------------ 52------------ 51------------ 51 -------------------------- 50-------------------------- 4775------------ 66------------ 58------------ 55------------ 54 -------------------------- 44-------------------------- 4275------------ 48------------ 38------------ 34------------ 33 -------------------------- 25-------------------------- 21
Number of patients at risk
Major depressive episodeManic episodeManic or major depressive episode
Months at risk (t)0 6 12 18 24 30 36 42 48
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
P (t
)
Ch 02 7/4/05 3:35 pm Page 11

12 Bipolar disorder: the upswing in research & treatment
residential status 6 months after discharge. However, at 48 months of fol-low-up, 28% (n=20) were unable to work or study, and 19% (n=14) ofpatients were not able to live independently. Further analyses identified sev-eral variables to be significant predictors of an unfavourable outcome (Table2.2). Risk factors associated with a poor occupational status 6 months afterdischarge included a history of one or more previous episodes and a historyof alcoholism. These factors continued significantly to predict a poor occu-pational status at 48 months. In addition, psychotic features during theindex episode also predicted poor occupational status at 48 months.Predicting an unfavourable residential status at 6 months was a history ofalcoholism. Other factors associated with unfavourable living status at 48months included a history of one or more previous episodes and male sex.
The results of this study suggest that patients with bipolar disorderimprove between 6 months and 4 years after an index manic episode; how-ever, functional outcome remains less than ideal. A key finding of this studywas that predictors of functional outcome varied depending not only on thetype of outcome measured but also on the time at which the outcome was
Figure 2.2 The cumulative probability of not relapsing into any mood episode upto a particular time for patients with bipolar disorder stratified by the presence(purple) and absence (grey) of psychotic features during an index manic episode.(Derived from reference 2.)
54----------- 34------------ 26----------- 22----------- 21 ------------------------- 14--------------------------- 1121----------- 14------------ 12----------- 12----------- 12 ------------------------- 11--------------------------- 10
Number of patients at risk
Without psychotic featuresWith psychotic features
Months at risk (t)0 6 12 18 24 30 36 42 48
1.0
0.9
0.8
0.7
0.6
0.5
0.4
0.3
0.2
0.1
P (t
)
Ch 02 7/4/05 3:35 pm Page 12

The functional outcome of bipolar disorder 13
assessed. However, as in all naturalistic studies, a significant uncontrolledvariable in this study was treatment. Not controlling for treatment has theadvantage of obtaining information on treatments received by patientsunder non-controlled circumstances. In addition, findings of naturalisticstudies may be more readily generalizable. At discharge, 97% (73 of 75) ofpatients were treated with at least one psychotropic drug and 92% (n=69)
Figure 2.3 The cumulative probability of not relapsing into any mood episode upto a particular time for patients with bipolar disorder experiencing mood-congruent (purple) and mood-incongruent (grey) psychotic features during anindex manic episode. (Derived from reference 3.)
24-----------17----------15-----------14----------14 -----------------------11-------------------------830-----------17----------11------------8------------7 -------------------------3-------------------------3
Number of patients at risk
Patients with mood-congruent psychotic maniaPatients with mood-incongruent psychotic mania
Months at risk (t)0 6 12 18 24 30 36 42 48
1.0
0.9
0.8
0.7
0.6
0.5
0.4
0.3
0.2
0.1
0.0
P (t
)
Table 2.1 Risk factors associated with a shorter time in remission after an
index manic episode
Predictors of time to relapse HRa* p Value
Psychotic features during index episode 2.2 0.05
Depressive symptoms during index episode 2.0 0.04
History of alcoholism 3.9 0.02
*Hazard ratio was adjusted (HRa) simultaneously for all variables with Cox regression.
Derived from reference 2.
Ch 02 7/4/05 3:35 pm Page 13

14 Bipolar disorder: the upswing in research & treatment
of patients received lithium. Fifty-five per cent of patients (n=41) receivedan antipsychotic; 15% (n=11), an antidepressant; and 11% (n=8), an anti-convulsant. At 48 months, 79% (57 of 72) of patients were receiving at leastone medication, with 67% (n=48) taking lithium; 46% (n=33), an antipsy-chotic; 19% (n=14), an antidepressant; and 21% (n=15), an anticonvul-sant.
Table 2.2 Functional outcomes and predictors of outcome after an index
manic episode
Functional outcomes n (%) ORa p Value
6 months
Occupational status
Able to work/study (<2 MVSI) 43 (60) — —
Unable to work/study (>3 MVSI) 29 (40) — —
Predictors of poor outcome
One or more previous mood episodes — 5.6 0.001
History of alcoholism — 10.3 0.03
Poor occupational status at baseline — 15.0 0.05
Residential status
Able to live independently (<3 MLCI) 46 (64) — —
Unable to live independently (>4 MLCI) 26 (36) — —
Predictors of poor outcome
History of alcoholism — 14.7 0.02
48 months
Occupational status
Able to work/study (<2 MVSI) 52 (72) — —
Unable to work/study (>3 MVSI) 20 (28) — —
Predictors of poor outcome
One or more previous mood episodes — 5.4 0.05
History of alcoholism — 8.2 0.04
Psychotic features during index episode — 9.0 0.04
Residential status
Able to live independently (<3 MLCI) 58 (81) — —
Unable to live independently (>4 MLCI) 14 (19) — —
Predictors of poor outcome
One or more previous mood episodes — 4.9 0.01
Male sex — 6.0 0.02
ORa, adjusted odds ratio; MVSI, Modified Vocational Status Index; MLCI, Modified
Location Code Index. Derived from reference 2.
Ch 02 7/4/05 3:35 pm Page 14

The functional outcome of bipolar disorder 15
The McLean–Harvard first-episode study
To study the evolution of bipolar disorder comprehensively, the longitudinalfollow-up of first-episode patients is critical. A prospective study of bipolardisorder patients that commences near illness onset provides data less con-founded by prolonged illness and pre-defined poor outcomes. In a natura-listic setting, the quantification of recovery and identification of predictorsof outcome are especially valuable. The McLean–Harvard First-EpisodeMania Study4 analysed 166 hospitalized patients experiencing their firstmanic (75.3%, n=125) or mixed (24.7%, n=41) episode, with 88.6%(n=147) experiencing psychotic features. Patients were recruited for thisstudy between 1989 and 1996.
Syndromic recovery was defined as a severity rating of ≤3 for the DSM-IV ‘A’ criterion for mania, with no ‘B’ criterion rated >3 and no two ‘B’ cri-teria rated at 3; Clinical Global Impressions scores were required to be ≤2.Patients presenting with an initial mixed episode fulfilled recovery criteriafor a manic and a depressive episode. Syndromal remission was achievedby maintaining recovery for at least 8 weeks. Furthermore, symptomaticrecovery, reflecting minimal symptom severity, was defined by a total YoungMania Rating Scale score of ≤5 and the Hamilton Depression Rating scalescore of ≤8. The functional recovery from a first lifetime mood episoderequired that both occupational level and residential status return to orexceed their highest levels during the pre-intake year. As defined, 59.7% (92of 154) of patients who attained syndromal recovery after initial hospital-ization achieved remission and remained in remission by the end of 2 yearsof follow-up. Conversely, 5.8% (n=9) experienced an early relapse, and34.4% experienced a new episode (mania: n=24; mixed: n=5; depression:n=24). Overall, the median latency to 50% risk of any new episode was26.3 weeks. The risks of new manic or depressive episode were equal inincidence (20.1%); however, the time to new depression was shorter at 17.7weeks versus 31.6 weeks to new mania.
In this cohort, survival-computed proportions reflected 85.5% (142 of166) of patients recovered syndromically at 6 months (Table 2.3).Nevertheless, only 39.5% (60 of 152) of patients achieved functional recov-ery at this time point. At the 2-year follow-up, nearly all patients achievedsyndromal recovery (97.6%, n=162), with 71.7% (66 of 92) of patientsachieving symptomatic recovery. However, only 43.1% (59 of 137) ofpatients functionally recovered, as measured by residential and occupa-tional status. Several factors were associated with syndromal and function-al recovery: a shorter initial hospitalization, below-median baseline depres-sion ratings and being female predicted a shorter time to syndromal
Ch 02 7/4/05 3:35 pm Page 15

16 Bipolar disorder: the upswing in research & treatment
Tab
le 2
.3R
ecov
ery
and
pre
dic
tors
of
reco
very
fol
low
ing
a fi
rst
life
tim
e m
anic
/mix
ed m
ood
ep
isod
e
6 m
onth
s2
year
s
Reco
very
n/N
%n/
N%
Pred
icto
rs o
f re
cove
ryRa
tio*
p Va
lue
Synd
rom
al14
2/16
685
.516
2/16
697
.6Sh
orte
r in
itial
hos
pita
lizat
ion
1.99
0.00
1
Belo
w-m
edia
n ba
selin
e de
pre
ssio
n ra
tings
1.65
0.00
8
Fem
ale
sex
1.72
0.00
8
Func
tiona
l60
/152
39.5
59/1
3743
.1>
30 y
ears
3.28
0.00
1
Shor
ter
initi
al h
osp
italiz
atio
n2.
820.
006
*Pre
dic
tors
of
syn
dro
mal
rec
over
y ar
e d
escr
ibed
as
haz
ard
rat
ios
(Cox
reg
ress
ion
).
Pred
icto
rs o
f fu
nct
ion
al r
ecov
ery
are
des
crib
ed a
s od
ds
rati
os (
logi
stic
reg
ress
ion
). D
eriv
ed f
rom
ref
eren
ce 4
.
Ch 02 7/4/05 3:35 pm Page 16
The functional outcome of bipolar disorder 17
recovery (Table 2.3). The likelihood of achieving functional recovery was sig-nificantly associated with being older than 30 years and having a shorter ini-tial hospitalization.
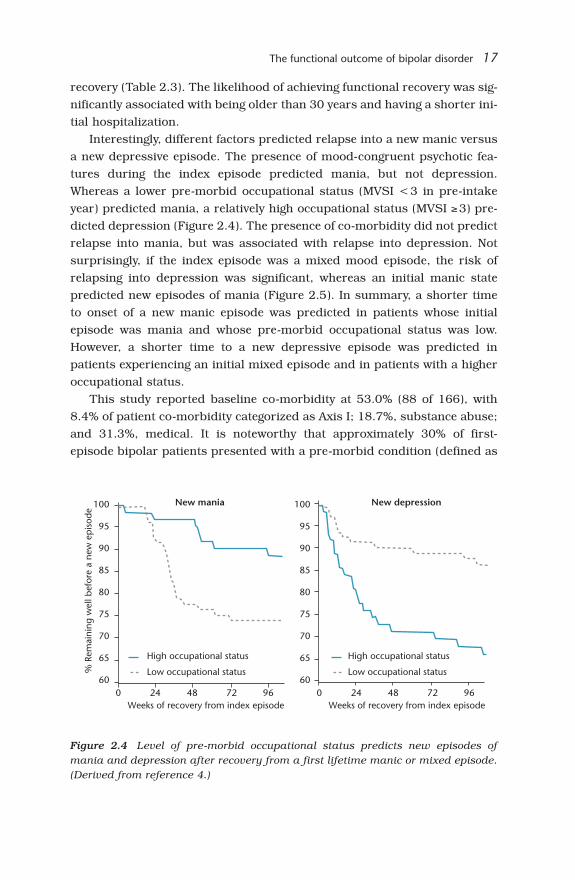
Interestingly, different factors predicted relapse into a new manic versusa new depressive episode. The presence of mood-congruent psychotic fea-tures during the index episode predicted mania, but not depression.Whereas a lower pre-morbid occupational status (MVSI <3 in pre-intakeyear) predicted mania, a relatively high occupational status (MVSI ≥3) pre-dicted depression (Figure 2.4). The presence of co-morbidity did not predictrelapse into mania, but was associated with relapse into depression. Notsurprisingly, if the index episode was a mixed mood episode, the risk ofrelapsing into depression was significant, whereas an initial manic statepredicted new episodes of mania (Figure 2.5). In summary, a shorter timeto onset of a new manic episode was predicted in patients whose initialepisode was mania and whose pre-morbid occupational status was low.However, a shorter time to a new depressive episode was predicted inpatients experiencing an initial mixed episode and in patients with a higheroccupational status.
This study reported baseline co-morbidity at 53.0% (88 of 166), with8.4% of patient co-morbidity categorized as Axis I; 18.7%, substance abuse;and 31.3%, medical. It is noteworthy that approximately 30% of first-episode bipolar patients presented with a pre-morbid condition (defined as
Figure 2.4 Level of pre-morbid occupational status predicts new episodes ofmania and depression after recovery from a first lifetime manic or mixed episode.(Derived from reference 4.)
100
95
90
85
80
75
70
65
600 24 48 72 96
Weeks of recovery from index episode
% R
emai
ning
wel
l bef
ore
a ne
w e
pis
ode
High occupational status
Low occupational status
100
95
90
85
80
75
70
65
600 24 48 72 96
Weeks of recovery from index episode
High occupational status
Low occupational status
New mania New depression
Ch 02 7/4/05 3:35 pm Page 17

18 Bipolar disorder: the upswing in research & treatment
a medical condition that required medical treatment or medical pharmaco-logical treatment). Furthermore, in a separate study, patients with the firstlifetime manic episode aged 65 years and over (n=14) were significantlymore likely to have a pre-morbid neurological condition as compared withbipolar patients of similar age (n=36).5
At discharge, almost all patients were prescribed a psychotropic med-ication (95.2%, n = 158). Although treatments varied widely, 75.3%(n=125) of patients received an antipsychotic, and lithium was prescribedto 68.7% (n=114) of patients. At 2-year follow-up, lithium was the most fre-quent treatment, although 35.6% (48 of 135) of patients were taking nomedication. The use of antidepressants increased during the 2 years offollow-up. No specific treatment was associated with time to syndromalrecovery; similarly, no treatment was significantly associated with a shorterlatency to a new mood episode.
Overall, patients experiencing a first lifetime manic episode improvedduring a 2-year follow-up, with 97.6% achieving syndromal recovery.Nonetheless, only 72% reported symptomatic recovery, and functionalrecovery was attained by fewer than 50% of patients. These findings suggestthat patients with bipolar disorder recover syndromically before they recov-er functionally; symptom severity improves initially followed by a return topre-episode functioning that requires additional time to achieve.Furthermore, in this cohort of patients, substance abuse or dependenceoccurred with a co-morbidity of 18%, relatively low compared with a 60%
Figure 2.5 Time to onset of new episode of mania or depression predicted by theindex episode. (Derived from reference 4.)
100
95
90
85
80
75
70
65
60
55
500 24 48 72 96
Weeks of recovery from index episode
% R
emai
ning
wel
l bef
ore
a ne
w e
pis
ode
Index: manic
Index: mixed
New mania New depression100
95
90
85
80
75
70
65
60
55
500 24 48 72 96
Weeks of recovery from index episode
Index: manic
Index: mixed
Ch 02 7/4/05 3:35 pm Page 18

The functional outcome of bipolar disorder 19
co-morbidity in multiple-episode patients.6 This finding suggests that, insome patients with bipolar disorder, mania may develop first and is fol-lowed by a substance use disorder.7
Conclusions
Prospective, naturalistic study designs produce a non-biased description ofpatient outcomes in a clinical setting whereby treatment is not determinedby the investigator. Results of such studies advance our understanding ofbipolar disorder and help identify illness characteristics and risk factorsthat may predict outcomes and aid in developing optimal treatment inter-vention.
The studies presented here suggest a disparity in recovery of patientswith bipolar disorder. Although achieving syndromic recovery, manypatients continue to experience symptoms; subsyndromal morbidityencroaches on work and personal life, accounting for reduced occupationaland residential status and poor functional outcome. The McLean–HarvardFirst-Episode Mania Study found that, after a first lifetime manic/mixedepisode, only 43% of patients achieved functional recovery at 2 years.4
Previous studies on effective functioning in bipolar patients have reportedsimilar poor outcomes. Accordingly, only 36% of patients with one or noprevious hospitalizations return to pre-morbid function at 12 months;1 asecond study described 27% of patients with good overall functioning at the2-year follow-up.8 Keck et al reported that 24% of patients with a history ofprevious manic or mixed episodes achieved functional recovery at sometime between discharge and 12 months of follow-up.9 Moreover, 26% ofpatients with bipolar disorder have been described as having a good overalloutcome at 2 years, increasing to 47% at 4.5 years.10 Although the majorityof these studies were conducted before the availability of today’s newerdrugs, treatments for bipolar disorder, even in first-episode mania, havebeen less than ideal. Treatments are needed that are both effective atimproving functional outcome and safe in a patient population with signifi-cant medical and substance abuse co-morbidities.
The analysis of functional outcome predictors identifies patients at riskfor poor psychosocial recovery. According to the studies presented here, apoor functional outcome was predicted by one or more previous episodes,a history of alcoholism and the presence of psychotic features during theindex episode. In contrast, a functional recovery was predicted by having ashorter initial hospitalization and being older than 30. Additionally, other
Ch 02 7/4/05 3:35 pm Page 19

20 Bipolar disorder: the upswing in research & treatment
related studies have identified higher pre-morbid function and highersocioeconomic class with favourable functional outcomes.1,9
To summarize, in the current treatment of bipolar disorder, sympto-matic improvement is not correlative with functional improvement. Thisfinding necessitates a greater clinical emphasis on functional outcomes.Functional recovery must be the hallmark of drug development, clinicalstudy design and treatment intervention, whereby patients with bipolar dis-order may enjoyably and responsibly return to personal, family and worklife.
References
1. Strakowski SM, Keck PE Jr, McElroy SL et al, Twelve-month outcome after afirst hospitalization for affective psychosis. Arch Gen Psychiatry 1998;55:49–55.
2. Tohen M, Waternaux CM, Tsuang MT, Outcome in mania. A 4-year prospectivefollow-up of 75 patients utilizing survival analysis. Arch Gen Psychiatry 1990;47:1106–1111.
3. Tohen M, Tsuang MT, Goodwin DC, Prediction of outcome in mania by mood-congruent or mood-incongruent psychotic features. Am J Psychiatry 1992;149:1580–1584.
4. Tohen M, Zarate CA Jr, Hennen J et al, The McLean–Harvard First-EpisodeMania Study: prediction of recovery and first recurrence. Am J Psychiatry2003; 160:2099–2107.
5. Tohen M, Shulman KI, Satlin A, First-episode mania in late life. Am JPsychiatry 1994; 151:130–132.
6. Regier DA, Farmer ME, Rae DS et al, Comorbidity of mental disorders withalcohol and other drug abuse. Results from the Epidemiologic Catchment Area(ECA) Study. JAMA 1990; 264:2511–2518.
7. Strakowski SM, DelBello MP, The co-occurrence of bipolar and substance usedisorders. Clin Psychol Rev 2000; 20:191–206.
8. Goldberg JF, Harrow M, Grossman LS, Course and outcome in bipolar affec-tive disorder: a longitudinal follow-up study. Am J Psychiatry 1995;152:379–384.
9. Keck PE Jr, McElroy SL, Strakowski SM et al, 12-month outcome of patientswith bipolar disorder following hospitalization for a manic or mixed episode.Am J Psychiatry 1998; 155:646–652.
10. Goldberg JF, Harrow M, Consistency of remission and outcome in bipolar andunipolar mood disorders: a 10-year prospective follow-up. J Affect Disord2004; 81:123–131.
Ch 02 7/4/05 3:35 pm Page 20

Brain abnormalities inbipolar disorder: do theyexist and do they change?E Serap Monkul and Jair C Soares
c h a p t e r 3
This chapter focuses on recent work that has utilized in vivo brain imaging
to understand the mechanisms involved in bipolar disorder. Structural
magnetic resonance imaging (MRI) and neurochemical studies with mag-
netic resonance spectroscopy (MRS) have identified changes in prefrontal
cortex regions, which are highly interconnected with limbic portions of the
brain, including medial temporal lobe regions and the striatum.1 Some of
these regions or the connections between them may be impaired and possi-
bly result in the mood dysregulation that we see in patients who have mood
disorders.2,3
One of the regions of interest is the anterior cingulate, which is thought
to be involved in the pathophysiology of mood disorders. In some of our
prior work we measured the cingulate gyrus, subdivided into specific
regions, and found a reduction in the grey matter content in the left anteri-
or cingulate in untreated bipolar patients compared with healthy controls.4
Reduction in anterior cingulate grey matter volumes4,5 and density6,7 is a
consistently reported finding in recent studies. Cingulate findings are also
present in children and adolescents with bipolar disorder,5 and there is evi-
dence that lithium may protect or reverse grey matter changes in this par-
ticular brain region.4
Drevets and colleagues found a pronounced reduction in grey matter pri-
marily on the left side in a specific part of the anterior cingulate gyrus, lying
ventral to the genu of the corpus callosum, called the ‘subgenual prefrontal
cortex’, in subjects with familial mood disorder compared with healthy con-
trols.8 We have not been able to replicate this finding in a recent study, with
familial and non-familial bipolar patients, although our sample involved
patients who were generally less severely ill9 than those of Drevets et al.8
Ch 03 7/4/05 3:36 pm Page 21

22 Bipolar disorder: the upswing in research & treatment
Another region that has attracted much attention for research on the
pathophysiology of mood disorders is the amygdala, which is involved in
regulation of emotions such as fear and anxiety. There are intriguing reports
by different groups who have found enlargement of the amygdala in bipolar
disorder.10–12 The neuropathology underlying such enlargement is still
unclear.
The other topic to discuss briefly relates to the hyperintense lesions,
which are non-specific markers, and generally reflect changes in the water
content in the brain. They are present in normal aging, dementia, epilepsy,
schizophrenia, and several other neuropsychiatric disorders.13 In the liter-
ature on mood disorders, there are several studies suggesting that patients
with bipolar disorder have these lesions at higher rates compared with well-
matched healthy controls,2,13 although controversy exists as to whether
individuals with bipolar disorder are more likely than other psychiatric
patients to have these lesions.14,15 These lesions seem to be more directly
involved in late life depression,16 and their importance may perhaps be
related to disrupting pathways that interconnect those brain circuits that
modulate mood.2
The other technique to discuss is MRS, which utilizes the same hard-
ware as MRI. MRS results are in the form of graphs from which one can
quantitate certain chemicals of interest, such as myoinositol, N-acetylaspartate
(NAA), choline-containing compounds and many others.17 NAA is a known
specific marker of neuronal viability and functioning, and any type of brain
insult will result in decreased levels of NAA in the brain. There are also data
showing that, if those insults are removed, levels of NAA go back up. There
have been two published studies in the bipolar literature suggesting a
decrease in NAA levels in the dorsolateral prefrontal cortex.18,19 Most of the
anatomical studies did not find detectable anatomical changes in the dor-
solateral prefrontal cortex, so decreased NAA levels could be an early mark-
er of neuronal impairment in this particular brain region, before anatomi-
cal changes take place. The question we asked in our most recent studies
had to do with whether young bipolar patients (children and adolescents
who have bipolar disorder) had such a change already early on, or whether
it was something that developed over time (J.C. Soares, unpublished work).
This was a cross-sectional study, and bipolar children/adolescents (mean
age 13 years, well matched with healthy controls) had a reduction of NAA
levels in the dorsolateral prefrontal cortex, which is consistent with what
has been reported previously in adults18 and children.19 In the same
Ch 03 7/4/05 3:36 pm Page 22

Brain abnormalities in bipolar disorder 23
sample, there was a pronounced reduction in grey matter not only on the
left side, but also on the right side, so the reduction in grey matter volumes
seems to be even more extensive in this sample with early onset of disease.
It is also of interest that these children primarily had familial bipolar dis-
order, as almost all of them had a first-degree relative with bipolar disorder,
most often their parent. These findings suggest that some changes might be
already present in the prefrontal cortex at the onset of the symptoms or
early in the disease process.
Recently we published a cross-sectional study where we reported that,
over time, patients with bipolar disorder may lose more grey matter com-
pared with healthy controls.20 This study revealed an inverse relationship
between age and total grey matter volume in bipolar disorder patients, sig-
nificantly more pronounced than would be expected in healthy controls,
suggesting that bipolar patients are losing grey matter at faster rates than
healthy individuals.
Both age and length of illness appear to be important factors in some of
the structural imaging changes that we find in bipolar patients. Further
work needs to be done to examine their relationship with regional brain
changes, with the hypothesis that, perhaps in some of those regions
involved in mood regulation, one would see more striking relationships with
age and length of illness.
Another interesting finding has emerged from our recent structural MRI
studies in children and adolescents with bipolar disorder. Although amyg-
dala volumes appear to be reduced in young bipolar patients21–23 in contrast
to adult patients,10–12 we24 found a direct correlation between age and amyg-
dala volumes in a patient group with mean age of 16, suggestive of neu-
rodegenerative and/or compensatory mechanisms as the disease pro-
gressed. This was not a follow-up study, but it is an intriguing finding that
suggests that perhaps during adolescence there are abnormal neurodevel-
opmental processes affecting the medial temporal lobe structures in bipolar
patients.
In conclusion, bipolar disorder, in both the paediatric and the adult age
groups, seems to involve frontolimbic brain abnormalities, both structural
and functional. The relationship of these with specific illness domains,
course and treatment response has not yet been characterized. Further
studies will be needed to characterize the role of such abnormalities in the
pathophysiology of the illness and their origin, which could be neuro-
developmental and/or neurodegenerative.
Ch 03 7/4/05 3:36 pm Page 23

24 Bipolar disorder: the upswing in research & treatment
Acknowledgements
This work was partly supported by NIH grants MH 01736, MH 068766 andM01-RR-01346 (UTHSCSA GCRC), NARSAD, the Veterans Administration(VA Merit Review), and the Krus Endowed Chair in Psychiatry (UTHSCSA).
References
1. Tekin S, Cummings JL, Frontal-subcortical neuronal circuits and clinical neu-ropsychiatry: an update. J Psychosom Res 2002; 53:647–654.
2. Soares JC, Mann JJ, The anatomy of mood disorders – review of structuralneuroimaging studies. Biol Psychiatry 1997; 41:86–106.
3. Strakowski SM, DelBello MP, Adler C et al, Neuroimaging in bipolar disorder.Bipolar Disord 2000; 2:148–164.
4. Soares JC, Sassi RB, Brambilla P et al, Decreased left anterior cingulate vol-umes in untreated bipolar disorder patients. Presented at the Society forNeuroscience Meeting, Orlando, FL: 2–7 November, 2002.
5. Kaur S, Sassi R, Axelson D et al, Anatomical MRI study of cingulate cortex inadolescent bipolar patients. Biol Psychiatry 2003; 53:72S.
6. Doris A, Belton E, Ebmeier KP et al, Reduction of cingulate gray matter densi-ty in poor outcome bipolar illness. Psychiatry Res 2004; 130:153–159.
7. Lyoo IK, Kim MJ, Stoll AL et al, Frontal lobe gray matter density decreases inbipolar I disorder. Biol Psychiatry 2004; 55:648–651.
8. Drevets WC, Price JL, Simpson JR Jr et al, Subgenual prefrontal cortex abnor-malities in mood disorders. Nature 1997; 386:824–827.
9. Brambilla P, Nicoletti MA, Harenski K et al, Anatomical MRI study of subgenu-al prefrontal cortex in bipolar and unipolar subjects. Neuropsychopharma-cology 2002; 27:792–799.
10. Altshuler LL, Bartzokis G, Grieder T et al, An MRI study of temporal lobe struc-tures in men with bipolar disorder or schizophrenia. Biol Psychiatry 2000;48:147–162.
11. Strakowski SM, DelBello MP, Sax KW et al, Brain magnetic resonance imagingof structural abnormalities in bipolar disorder. Arch Gen Psychiatry 1999;56:254–260.
12. Brambilla P, Harenski K, Nicoletti M et al, MRI investigation of temporal lobestructures in bipolar patients. J Psychiatr Res 2003; 37:287–295.
13. Altshuler LL, Curran JG, Hauser P et al, T2 hyperintensities in bipolar disor-der: magnetic resonance imaging comparison and literature meta-analysis. AmJ Psychiatry 1995; 152:1139–1144.
14. Moore PB, Shepherd DJ, Eccleston D et al, Cerebral white matter lesions inbipolar affective disorder: relationship to outcome. Br J Psychiatry 2001;178:172–176.
15. Breeze JL, Hesdorffer DC, Hong X et al, Clinical significance of brain white mat-ter hyperintensities in young adults with psychiatric illness. Harvard RevPsychiatry 2003; 11:269–283.
Ch 03 7/4/05 3:36 pm Page 24

Brain abnormalities in bipolar disorder 25
16. Videbech P, MRI findings in patients with affective disorder: a meta-analysis.Acta Psychiatr Scand 1997; 96:157–168.
17. Stanley JA, In vivo magnetic resonance spectroscopy and its application to neu-ropsychiatric disorders. Can J Psychiatry 2002; 47:315–326.
18. Winsberg ME, Sachs N, Tate DL et al, Decreased dorsolateral prefrontal N-acetyl aspartate in bipolar disorder. Biol Psychiatry 2000; 47:475–481.
19. Chang KD, Adleman N, Dienes K et al, Decreased N-acetyl aspartate in childrenwith familial bipolar disorder. Biol Psychiatry 2003; 53:1059–1065.
20. Brambilla P, Harenski K, Nicoletti M et al, Differential effects of age on braingray matter in bipolar patients and healthy individuals. Neuropsychobiology2001; 43:242–247.
21. Caetano SC, Olvera R, Hunter K et al, Abnormal amygdala volumes in pediatricbipolar disorder. Biol Psychiatry 2004; 55:111S.
22. Blumberg HP, Kaufman J, Martin A et al, Amygdala and hippocampal volumesin adolescents and adults with bipolar disorder. Arch Gen Psychiatry 2003;60:1201–1208.
23. DelBello MP, Zimmerman ME, Mills NP et al, Magnetic resonance imaginganalysis of amygdala and other subcortical brain regions in adolescents withbipolar disorder. Bipolar Disord 2004; 6:43–52.
24. Caetano SC, Nicoletti MA, Hatch JP et al, Associations of age and length of ill-ness with hippocampus and amygdala volumes in mood disorder patients. BiolPsychiatry 2004; 55:109S.
Ch 03 7/4/05 3:36 pm Page 25

Ch 03 7/4/05 3:36 pm Page 26

Structural magnetic resonanceimaging studies in bipolardisorder: a meta-analysis
Colm McDonald, Jolanta Zanelli, Robin M Murray andNoel Kennedy
c h a p t e r 4
Introduction
Bipolar disorder is frequently described in reviews as being associated withsubtle structural brain abnormalities, but it is also acknowledged that theexisting literature is sparse.1,2 The most consistently reported brain abnor-malities are increased rates of white matter hyperintensities and mild later-al ventricular enlargement, with studies often disagreeing on other regionalvolume deviations. This disagreement is contributed to by the small num-bers of subjects studied, sample heterogeneity and a dependence on tests ofsignificance with inconsistent ‘positive’ and ‘negative’ findings. Meta-analysisis a powerful technique for integrating quantitative data from several stud-ies; the resultant increase in sample size provides more statistical power todetect subtle volume deviations and provide a more accurate estimate of theeffect size. Previous meta-analyses have found that bipolar disorder is asso-ciated with preservation of cerebral size3 and increased rates of white mat-ter hyperintensities.4,5 However, these prior meta-analyses were mostlybased on computed tomography (CT) studies or qualitative ratings of mag-netic resonance imaging (MRI) studies. In this chapter we present results ofa meta-analysis of regional brain volume abnormalities of subjects withbipolar disorder based on studies using high-resolution MRI and completecoverage of each brain region.
Methodology
A systematic search of electronic databases supplemented by manualsearches was used to identify relevant studies. Studies were included if they
Ch 04 7/4/05 3:36 pm Page 27

28 Bipolar disorder: the upswing in research & treatment
(1) were published in full in a peer-reviewed journal by December 2003; (2)compared a group of subjects with bipolar disorder and a normal compar-ison group; (3) reported means and standard deviations of regional volu-metric brain measurements; and (4) reported on a brain structure that wasevaluated by at least two other studies, and where data were available for atleast 50 bipolar patients and controls in total. A rigorous standard ofmethodology was applied to ensure high precision, and studies were notincluded if they reported only area measurements, or if volumetric meas-urements were based on non-contiguous slices or incomplete coverage ofthe brain structure.
As a readily interpretable measure of effect size, we used the ratio of themean volume of the bipolar group divided by the mean volume of the con-trol group. The meta-analysis was based on the logarithm of the ratio of thegroup means, which is more likely to be normally distributed. A meta-analysis of the logarithm of the volume ratios of all the measured structureswas carried out using a random effects model. This assumes the ‘popula-tion’ of studies to have variable true effects that are normally distributed,and aims to estimate the overall mean of this distribution of effect sizes. Thedelta method was used to obtain variance of the logarithm of this effectsize.6
For each brain region we also calculated I2 as a test of heterogeneity.7
This measure is an estimate of the percentage of total variation across stud-ies that are due to true heterogeneity rather than chance, where larger val-ues imply increasing heterogeneity. When five or more studies were includ-ed, the possibility of publication bias was investigated using the Egger test8
to test for the presence of a surplus of low-precision studies providing effectsizes of magnitude greater than the average. We adopted a significance levelof p<0.05 for all analyses. Further details on the methodology aredescribed by McDonald et al.9
Results
Twenty-six studies met all the criteria for inclusion in the meta-analysis andincluded 404 patients with bipolar disorder. The majority (77%) of studieshad been published since 1998. Most studies used DSM-III-R or DSM-IV cri-teria for diagnosis, and most were of patients with recurrent episodes of ill-ness rather than first-episode mania. In all of the studies that providedinformation on medication, the majority or all of the patients were med-icated with mood stabilizers and/or other psychotropic medication at thetime of scanning.
Ch 04 7/4/05 3:36 pm Page 28

Structural magnetic resonance imaging studies in bipolar disorder 29
Table 4.1 demonstrates the comparison of regional brain volumes ofbipolar disorder patients and controls for the structures included in themeta-analysis. Patients with bipolar disorder had significantly larger totallateral ventricular volume than controls. Ventricular volume was dividedinto right and left in all but one of these studies, and right lateral ventriclevolume was found to be significantly greater in patients with bipolar disor-der than in comparison subjects, whereas left lateral ventricular volume dif-ference did not reach significance. A forest plot demonstrating the individ-ual study results for right lateral ventricular volume is shown in Figure 4.1.There was no significant difference between bipolar patients and compari-son subjects in third ventricular volume. No significant differences wereseen in total brain volume or in the volume of any regional cortical, sub-cortical or limbic brain structures. No significant publication bias wasdetected in the studies reporting lateral ventricular volume enlargement(Figure 4.2).
High heterogeneity was detected for several of the structures studied,especially the third ventricle, amygdala, left subgenual prefrontal cortex andleft hippocampus–amygdala.
Discussion
This meta-analysis of methodologically robust regional morphometric MRIstudies demonstrates that bipolar disorder is indeed associated with later-al ventricular enlargement, especially on the right, but with no significantdifferences in the other structures examined. Elkis and colleagues10 previ-ously performed a meta-analysis on studies using a broad category of affec-tive disorders and reported an association with increased ventricular sizewhich was less prominent than that found in schizophrenia. Most studiesincluded in that meta-analysis were based upon CT data, and the majorityof patients had unipolar depression. The present study is consistent withthis previous meta-analysis but builds upon the findings by identifyingincreased ventricular volume in a more homogeneous sample of patientswith bipolar disorder. The finding of more prominent right-sided lateralventricular enlargement echoes evidence that bipolar disorder is more like-ly to be associated with right-sided cerebral pathology, for example whenmania occurs in association with cerebral trauma, temporal lobe epilepsyor stroke.11
This meta-analysis also confirms that global cerebral volume ispreserved in bipolar disorder, in contrast to schizophrenia, which is
Ch 04 7/4/05 3:36 pm Page 29

30 Bipolar disorder: the upswing in research & treatment
Table 4.1 Comparison of regional brain volumes of bipolar disorder sub-
jects and controls from 26 studies (adapted from reference 9)
Bipolar
disorder/ % p Heterogeneity Egger test
Structure control Volume* Value I2 (p value) (p value)
Whole brain 239/292 99 0.26 44% (0.05) 0.06 (0.97)(11 studies) (96–101)
Total lateral ventricles 160/168 113 0.03 5% (0.39) –0.37 (0.84)(6 studies) (101–127)
Right lateral ventricle 143/152 114 0.03 0% (0.52) –2.10 (0.26)(5 studies) (101–128)
Left lateral ventricle 143/152 108 0.32 26% (0.24) 0.07 (0.97)
(5 studies) (93–124)
Third ventricle 139/161 113 0.16 67% (0.006) 0.38 (0.87)
(6 studies) (95–135)
Hippocampus 245/273 99 0.24 2% (0.43) 0.12 (0.90)(8 studies) (96–101)
Amygdala 115/175 101 0.90 83% (0.001) –4.56 (0.54)(5 studies) (91–112)
Left hippocampus– 158/247 102 0.76 91% (0.001) –0.37 (0.91)
amygdala (8 studies) (92–112)
Right hippocampus– 158/247 100 0.85 58% (0.01) 1.86 (0.21)
amygdala (8 studies) (96–104)
Left subgenual 95/76 80 0.31 98% (0.001) —prefrontal (4 studies) (52–123)
Right subgenual 95/76 94 0.36 51% (0.10) —prefrontal (4 studies) (82–108)
Left temporal lobe 178/215 101 0.62 52% (0.03) –1.93 (0.51)(6 studies) (97–105)
Right temporal lobe 178/215 99 0.55 46% (0.06) –3.93 (0.20)(6 studies) (96–102)
Thalamus 129/124 103 0.26 58% (0.05) 1.82 (0.60)(5 studies) (98–109)
Caudate 172/187 102 0.42 35% (0.14) 0.41 (0.73)(7 studies) (97–107)
Putamen 165/154 102 0.20 0% (0.95) –1.15 (0.02)(6 studies) (99–106)
Globus pallidus 76/74 105 0.38 61% (0.05) —(3 studies) (95–116)
*Volume for subjects with bipolar disorder in relation to volume for control subjects; the
brain volume of control subjects is assumed to be 100%. Significant p values in bold.
Ch 04 7/4/05 3:36 pm Page 30

Structural magnetic resonance imaging studies in bipolar disorder 31
characterized by a small decrease in brain size.12 This is consistent with aprevious meta-analysis of seven studies of brain volume in bipolar disorderby Hoge et al,3 three of which were included in the present meta-analysis(the other four were CT studies or MRI studies from which complete volu-metric measurements of the cerebrum could not be derived).
Figure 4.1 Forest plot of meta-analysis results for right lateral ventricular volume.
0.4
0.6
0.8
1
1.2
1.4
1.6
1.8
2
Overall Swayze
90
Swayze
90
Strak
99
McInt
01
Bramb
01
Strak
02
Effe
ct s
ize
Study
Figure 4.2 Funnel plot of seven studies reporting total lateral ventricular volumeresults, demonstrating no publication bias.
0
Log
effe
ct
Standard error of log effect size
0.5
0
–0.5
0 0.1 0.2 0.3
Ch 04 7/4/05 3:36 pm Page 31

32 Bipolar disorder: the upswing in research & treatment
One of the most striking findings is the volume preservation in bipolardisorder of most regions assessed. This is in sharp contrast to schizophre-nia, where a recent meta-analysis using similar methodology detected wide-spread volume deficits of up to 9% in some brain regions.12 For example,hippocampal volume was specifically measured in nearly 250 patients withbipolar disorder included in the current meta-analysis but did not differ atall from controls. Recent meta-analyses have demonstrated volume deficitof the hippocampus in both schizophrenia12 and major depressive disor-der,13 and this study therefore provides powerful support for the existenceof morphometric distinctions between the major diagnoses within the spec-tra of psychotic and affective disorders. The pathophysiology of hippocam-pal volume deficit in depression has been linked to hypercortisolaemia,13
and in schizophrenia to the impact of susceptibility genes,14 obstetric com-plications15 and glutamatergic excitotoxic damage.16 Either such factors donot impact upon hippocampal volume in bipolar disorder or other featuresassociated with the illness or its treatment have a protective effect.
Conventional antipsychotic medications are known to be associated withbasal ganglia enlargement but recent evidence suggests that their use inpatients experiencing their first episode of psychosis may be linked to vol-ume deficit of other grey matter regions including the frontal and temporalcortex.17 Since these medications are more likely to be used chronically inschizophrenia but intermittently in bipolar disorder during manic relapse,it is possible that the consistent presence of grey matter deficit in schizo-phrenia and its relative absence in bipolar disorder is contributed to by dif-ferential treatment with conventional antipsychotics. Alternatively, othermedications used to treat bipolar disorder may reverse or ameliorate pri-mary structural changes associated with the illness, as indicated by evi-dence that lithium is neurotrophic and can increase grey matter volume invivo.18,19
Given the apparent volume preservation of subcortical grey matter struc-tures surrounding the ventricles in bipolar disorder, what is the likely cor-responding tissue loss underlying the ventricular enlargement? One possi-bility is that white matter pathology with associated volume deficit isresponsible for this. Increased rates of white matter hyperintensities are aconsistent feature of bipolar disorder5 and emerging evidence using com-putational morphometry analysis of structural MRI scans points to exten-sive white matter volume deficits.20 Furthermore, gene expression profilingstudies of frontal cortical tissue have identified specific down-regulation ofgenes related to myelination and oligodendrocyte function in bipolar disor-der.21 These data suggest that a potentially critical component in the
Ch 04 7/4/05 3:36 pm Page 32

Structural magnetic resonance imaging studies in bipolar disorder 33
pathophysiology of the illness is disturbed structural connectivity of dis-tributed neural networks.
Heterogeneity
Another principal finding is the very high levels of heterogeneity which werefound for several structures, including the amygdala, third ventricle (whichis largely bordered by the thalamus) and left subgenual prefrontal cortex.These structures have been a focus of interest in bipolar disorder research,since they form part of a limbic–thalamic–cortical circuit which, togetherwith a limbic–striatal–pallidal–thalamic circuit, is thought to play a role inthe pathophysiology of mood disorders.1 Functional imaging studies indi-cate that bipolar disorder is characterized by overactivity of limbic and sub-cortical structures and underactivity of the prefrontal cortex in response toemotionally salient stimuli, consistent with a hypothesis whereby impairedprefrontal regulation linked to excessive subcortical activity underlies mooddysregulation in bipolar disorder.22 A pattern of reduced subgenual pre-frontal cortex volume and increased amygdala/thalamic volume wouldtherefore be consistent with the functional imaging findings, if one pre-sumes that functional activity is likely to be correlated with regional volume(which remains largely uninvestigated). Although some studies support thispattern of volume deviation, this meta-analysis demonstrates that the struc-tural imaging literature is wholly inconsistent regarding these critical struc-tures. Indeed, studies of the same structures included in the meta-analysisoften demonstrate effects in opposing directions. Five studies were includ-ed which examined amygdala volume; two studies reported significant vol-ume deficit and three studies reported significant volume enlargement ofthis structure, with no reports of volume equivalence. Quantitatively com-bining these studies as in the present meta-analysis unsurprisingly demon-strates that no conclusive effect has emerged and that very high hetero-geneity exists.
Probable sources of heterogeneity include methodological differencesand clinical sample variation. Although patients fulfilled criteria for opera-tionally defined bipolar disorder, there were considerable clinical variationswithin both the individual studies and the meta-analysis as a whole inchronicity, severity, phase of illness, types of treatment and co-morbid con-ditions such as substance abuse, all of which could potentially impact uponregional volume as measured by MRI. The meta-analysis therefore under-lines the particular need in bipolar disorder research for future structuralmorphometry studies to capture more homogeneous clinical samples.Study designs likely to be more informative include those focusing on
Ch 04 7/4/05 3:36 pm Page 33

34 Bipolar disorder: the upswing in research & treatment
(1) first-episode medication-naive patients; (2) patients during the euthymic
phase of illness; (3) patients who experience psychotic symptoms during ill-
ness exacerbation; (4) subjects at high genetic risk of illness, such as first-
degree relatives and offspring; and (5) longitudinal studies which can exam-
ine the effects of age, duration of illness, differing mood-stabilizing medica-
tions and number of illness episodes on brain morphometry.
Conclusion
Quantitatively combining the results of high-quality structural MRI studies
of regional brain volume demonstrates that bipolar disorder is associated
with lateral ventricular enlargement that is more prominent on the right,
but with preservation of most of the other brain regions commonly chosen
for measurement. Considerable heterogeneity exists in the results of stud-
ies to date and it remains unclear whether volumetric deviations exist in
regions such as the amygdala, thalamus and subgenual prefrontal cortex in
bipolar disorder, either as a whole or in subsamples defined by symptom
profile, medication status, chronicity or severity. Research into the struc-
tural morphometry of bipolar disorder remains at an early stage of devel-
opment. Considerable further research is warranted in this field before we
can conclusively describe the brain structural deviations of this disorder.
Acknowledgement
Colm McDonald is supported by the Wellcome Trust.
References
1. Soares JC, Mann JJ, The anatomy of mood disorders – review of structuralneuroimaging studies. Biol Psychiatry 1997; 41:86–106.
2. Strakowski SM, Adler CM, DelBello MP, Volumetric MRI studies of mood dis-orders: do they distinguish unipolar and bipolar disorder? Bipolar Disord2002; 4:80–88.
3. Hoge EA, Friedman L, Schulz SC, Meta-analysis of brain size in bipolar disor-der. Schizophr Res 1999; 37:177–181.
4. Altshuler LL, Curran JG, Hauser P et al, T2 hyperintensities in bipolar disor-der: magnetic resonance imaging comparison and literature meta-analysis. AmJ Psychiatry 1995; 152:1139–1144.
Ch 04 7/4/05 3:36 pm Page 34

Structural magnetic resonance imaging studies in bipolar disorder 35
5. Videbech P, MRI findings in patients with affective disorder: a meta-analysis.Acta Psychiatr Scand 1997; 96:157–168.
6. Dunn G, Design and Analysis of Reliability Studies: The StatisticalEvaluation of Measurement Errors. Edward Arnold: London, 1989.
7. Higgins JP, Thompson SG, Deeks JJ, Altman DG, Measuring inconsistency inmeta-analyses. BMJ 2003; 327:557–560.
8. Egger M, Davey Smith G, Schneider M, Minder C, Bias in meta-analysis detect-ed by a simple, graphical test. BMJ 1997; 315:629–634.
9. McDonald C, Zanelli J, Rabe-Hesketh S et al, Meta-analysis of magnetic reso-nance imaging brain morphometry studies in bipolar disorder. Biol Psychiatry2004; 56:411–417.
10. Elkis H, Friedman L, Wise A, Meltzer HY, Meta-analyses of studies of ventricu-lar enlargement and cortical sulcal prominence in mood disorders.Comparisons with controls or patients with schizophrenia. Arch GenPsychiatry 1995; 52:735–746.
11. Flor-Henry P, Lateralized temporal–limbic dysfunction and psychopathology.Ann NY Acad Sci 1976; 280:777–797.
12. Wright IC, Rabe-Hesketh S, Woodruff PWR et al, Meta-analysis of regional brainvolumes in schizophrenia. Am J Psychiatry 2000; 157:16–25.
13. Campbell S, Marriott M, Nahmias C, MacQueen GM, Lower hippocampal vol-ume in patients suffering from depression: a meta-analysis. Am J Psychiatry2004; 161:598–607.
14. Seidman LJ, Faraone SV, Goldstein JM et al, Left hippocampal volume as a vul-nerability indicator for schizophrenia: a magnetic resonance imaging morpho-metric study of nonpsychotic first-degree relatives. Arch Gen Psychiatry 2002;59:839–849.
15. Schulze K, McDonald C, Frangou S et al, Hippocampal volume in familial andnonfamilial schizophrenic probands and their unaffected relatives. BiolPsychiatry 2003; 53:562–570.
16. McCarley RW, Wible CG, Frumin M et al, MRI anatomy of schizophrenia. BiolPsychiatry 1999; 45:1099–1119.
17. Dazzan P, Morgan K, Orr KG et al, Different effects of typical and atypicalantipsychotics on grey matter in first episode psychosis: the AESOP study.Neuropsychopharmacology in press.
18. Moore GJ, Bebchuk JM, Wilds IB et al, Lithium-induced increase in humanbrain grey matter. Lancet 2000; 356:1241–1242.
19. Sassi RB, Nicoletti M, Brambilla P et al, Increased gray matter volume inlithium-treated bipolar disorder patients. Neurosci Lett 2002; 329:243–245.
20. McDonald C, Bullmore E, Sham P et al, Regional volume deviations of brainstructure in schizophrenia and psychotic bipolar disorder: a computationalmorphometry study. Br J Psychiatry in press.
21. Tkachev D, Mimmack ML, Ryan MM et al, Oligodendrocyte dysfunction inschizophrenia and bipolar disorder. Lancet 2003; 362:798–805.
22. Phillips ML, Drevets WC, Rauch SL, Lane R, Neurobiology of emotion percep-tion II: Implications for major psychiatric disorders. Biol Psychiatry 2003;54:515–528.
Ch 04 7/4/05 3:36 pm Page 35

Ch 04 7/4/05 3:36 pm Page 36

Are subcortical regionstoo expansive inbipolar disorder? An examination of the nature of prefrontal corticolimbicabnormalities in individuals with bipolar disorder
Mary L Phillips
c h a p t e r 5
Introduction
Bipolar disorder affects up to 1.5% of the population of the USA,1 with ill-ness relapse rates estimated at between 37% and 44% per year,2,3 a totalmortality elevated by 58% (predominantly from suicide and cardiovasculardisease4), and syndromic recovery after 1 year following manic or mixedepisodes only at 48%.5 Whilst it is clear clinically that mood dysregulation,or affective instability, is a key symptom of the disorder, the nature of theneural mechanism underlying this abnormality remains poorly understood.Clarification of this mechanism will be crucial for the future development ofeffective therapeutic interventions for this common but poorly treated dis-order.
In this chapter, the focus is therefore upon a discussion of findings fromstudies that have employed neuroimaging techniques to measure neuralresponses to emotionally salient stimuli in individuals with the disorder. Itwill conclude with a postulated neural mechanism for the affective instabil-ity in the disorder based on these findings.
Neural responses to emotionally salient stimuli inindividuals with bipolar disorder
The accurate recognition of facial expressions is crucial for successful inter-personal function in the social environment.6 Facial expressions represent
Ch 05 7/4/05 3:36 pm Page 37

38 Bipolar disorder: the upswing in research & treatment
ecologically valid, emotionally salient stimuli. In healthy individuals, find-ings from neuroimaging studies have implicated a network of predominant-ly anterior limbic regions in the response to and appraisal of emotionalstimuli. These regions include the amygdala, but also other areas: the ven-tral striatum, hippocampus and anterior insula.7,8–12 Dorsal and ventrome-dial prefrontal cortical regions have been implicated in the regulation ofthese responses, although further studies are required to clarify the neuralmechanisms associated with the regulation of responses to facial expres-sions and other emotionally salient material.13
Previous studies of euthymic and remitted individuals with bipolar dis-order indicate impaired fear14 and enhanced disgust recognition15 in facialexpressions of adolescent individuals with bipolar disorder, a tendency tomisinterpret the faces of peers as being angry,16 and of manic individualswith the disorder, both specific impairments in the recognition of fear anddisgust of unfamiliar others,17 and generalized deficits in the recognition ofall emotional expressions.18 Other studies employing tasks examining indi-rect biases in the identification of material as emotional or neutral, includ-ing the emotional Stroop task19 and the affective go/no-go task,20 have indi-cated negative attentional biases in depressed individuals with bipolar dis-order,19 and both negative and positive attentional biases in manic individ-uals.19,20 These findings suggest deficits in the recognition of emotive stim-uli, including negative facial expressions, in euthymic individuals with bipo-lar disorder and those in a mood episode.
There has been limited examination in individuals with bipolar disorderof the neural mechanism underlying this abnormality in processing emo-tional stimuli. Instead, studies in individuals with the disorder have focusedupon the examination of neural responses during rest and performance ofexecutive and memory tasks. Findings from these studies suggest dysfunc-tional prefrontal cortical–subcortical interactions in euthymic, in additionto symptomatic, individuals with the disorder at rest and during perform-ance of such tasks. Reports include predominant reductions in activity indorsal and ventral prefrontal cortical regions,21–26 but increases in activitywithin the dorsal anterior cingulate gyrus27–29 and subcorticalregions,28,30,31, which have been positively correlated with mania severi-ty.26,28,29 Earlier studies provided conflicting findings regarding sub-cortical–temporal cortical activity in manic individuals at rest.32–34 Of thefew studies directly comparing euthymic with mood episode individuals,some reports have indicated an amelioration of abnormal neural responsesin euthymic individuals during executive task performance,22,24,25,28
although others have suggested greater impairments in prefrontal cortical
Ch 05 7/4/05 3:36 pm Page 38

Are subcortical regions too expansive in bipolar disorder? 39
activity in euthymic compared with depressed individuals with bipolardisorder.26
Regarding neural responses to emotional stimuli, our recent findings inremitted individuals with bipolar disorder, using a facial expression para-digm, indicate increased activity within limbic and subcortical regions, pre-dominantly to expressions of fear and happiness, in the absence of anydeficits in facial expression recognition35 (Figure 5.1). These findings sup-port earlier reports of increased subcortical (amygdala) activity to fearfulexpressions14 in remitted individuals. Our findings also indicate subsyn-dromal depression-related abnormalities, namely a positive correlation
Figure 5.1 The figure depicts brain slices in healthy individuals (CON), individu-als with bipolar disorder (BD) and individuals with major depressive disorder(MDD) in responses to facial expressions of happiness (A) and fear (B) contrastedwith neutral faces. The graphs demonstrate that BD compared with CON and MDDdemonstrated increased activity within the left amygdala (blue), ventral striatum(putamen) and ventromedial prefrontal cortex (PFC; pink) in response to happyfaces, and increased activity within the left amygdala in response to fearful faces(blue).
L amygdala/ putamen
ventromedial PFC
L amygdala/ ventrolateral PFC
CON BD MDD
CON BD MDD
0.04
0
0.04
0
0.04
0
Neu
ral r
esp
onse
Neu
ral r
esp
onse
A B
Ch 05 7/4/05 3:36 pm Page 39

40 Bipolar disorder: the upswing in research & treatment
between depression severity and hippocampal response to sad expressions,in remitted individuals with bipolar disorder. We also demonstratedincreased ventromedial prefrontal cortical responses in these individuals,particularly in response to expressions of mild happiness.
Neural responses during mood induction in individuals with bipolardisorder
Positive and negative mood states can be induced with specific mood-induction paradigms, including the use of facial expressions and/or auto-biographical memory scripts, associated with activity within the ventralstriatum and ventromedial prefrontal cortex in healthy individuals,36,37 andemotive scenes from a standardized series (e.g. the International AffectivePicture Series, IAPS38), associated with subcortical limbic responses inhealthy and anxiety-disordered populations.7,39 Few studies to date,however, have examined neural responses during mood induction para-digms in individuals with bipolar disorder. We have developed a moodinduction paradigm involving autobiographical scripts to induce happy orsad mood, followed by presentation of emotion-congruent facial expres-sions.40 Using this paradigm, we have demonstrated in individuals withmajor depressive disorder during happy mood induction an absence of thenormal increase in autonomic response (as measured by skin conductancerecordings, SCR), and increased activity within the dorsomedial and ven-tromedial prefrontal cortex, regions associated with regulation of emotion-al responses.13 Other studies using similar mood induction paradigms havedemonstrated relative decreases in activity within these regions during sad
mood induction in euthymic and depressed individuals with bipolar disor-der.41 One study, employing emotive scenes, has demonstrated in depressedindividuals with bipolar disorder increased subcortical responses to posi-tive and negative scenes during affect generation.42
Neural responses during performance of other emotion-processingtasks in individuals with bipolar disorder
Interestingly, other studies employing an affective go/no-go paradigm, inwhich individuals respond to emotional target words (either happy or sad)and inhibit responses to emotional distractors (either happy or sad) havedemonstrated in manic individuals with bipolar disorder decreased ventro-medial prefrontal cortical responses during semantic task versus ortho-graphic go/no-go task performance, but increased ventrolateral prefrontalcortical responses to emotional versus neutral targets, and elevated ventral
Ch 05 7/4/05 3:36 pm Page 40

Are subcortical regions too expansive in bipolar disorder? 41
and medial prefrontal cortical responses to emotional distractors.43
Employing this paradigm, a similar pattern of increased response within
the ventral anterior cingulate gyrus to sad targets, and increased ventrolat-
eral prefrontal cortical response to sad distractors, was demonstrated in
individuals with major depressive disorder.44
Summary
Together, findings from studies examining neural responses during mood
induction and to non-facial, emotionally salient stimuli (emotional scenes
and words), indicate decreased activity in a dorsomedial prefrontal cortical
region during sad mood induction, increased subcortical activity to emo-
tional scenes, and increases in predominantly ventral regions of the pre-
frontal cortex to emotional word targets and distractors in individuals with
bipolar disorder. This complex pattern of decreases and increases in pre-
frontal cortical and subcortical responses requires further study, but does
include relative decreases in activity in regions associated wth mood regu-
lation (e.g. ventromedial–dorsomedial prefrontal cortices), but relative
increases in activity in regions assoicated with decision-making about and
affective responses to emotional material (subcortical regions, ventral ante-
rior cingulate gyrus, ventromedial–ventrolateral prefrontal cortices).
Structural neural abnormalities in individuals withbipolar disorder
Findings to date in individuals with bipolar disorder regarding regions
important for emotion processing, including the amygdala and hippocam-
pus, have been variable, with studies reporting volume increases, decreas-
es, or no abnormality within the amygdalae, and volume decreases or no
abnormalities in the hippocampi.45–48 Other studies have reported
increased ventral striatal (caudate nucleus and putamen) volumes,46,49 and
decreased middle, superior and inferior, including subgenual cingulate, pre-
frontal cortical volumes,50–52 although yet others have reported no signifi-
cant differences in prefrontal cortical volumes between individuals with
bipolar disorder and healthy volunteers.53 Interestingly, a recent study has
provided further evidence for increased grey matter volumes in the bilater-
al thalamus, insulae and cortical regions involved in the response to emo-
tional stimuli and mood generation in individuals with bipolar disorder.54
Ch 05 7/4/05 3:36 pm Page 41

42 Bipolar disorder: the upswing in research & treatment
The effect of previous illness history andmedication on persistent abnormalities in thesefunctional neural responses
To date, previous studies have suggested that the magnitude of executivedysfunction within remitted individuals may be associated with longer ill-ness duration and number of illness episode,55,56 particularly manicepisodes,57,58 suggestive of a positive correlation between prefrontal corticaldysfunction and these clincal variables, whilst a history of psychosis hasbeen associated with greater verbal memory impairment.57 Structural neu-roimaging studies suggest that enlarged ventricular volumes and decreasedputamen size59–61 are associated with an increased number of previousepisodes of illness. There are discrepant findings regarding the effect of psy-chotropic medication upon neurocognitive function in individuals with bipo-lar disorder, however. Neuroleptic medication has been associated withattentional impairments in healthy volunteers,62 but also with no impair-ment in attention in individuals with psychiatric disorders.63 There are con-flicting findings regarding the effect of lithium on cognitive function,64–68 butlittle effect of other mood stabilizers,69 or antidepressants,70 on cognitivefunction, whilst citalopram has been associated with a ‘normalization’ of theotherwise increased recognition of fear in euthymic individuals with a pre-vious history of major depressive disorder.71 The effect of psychotropicmedication upon structural and functional neuroanatomy in individualswith bipolar disorder is largely unknown. Long-term use of lithium has beenassociated with an increase in volume of the subgenual cingulate gyru.72,73
Neuroleptic medication levels have been positively correlated with meanregional cerebral blood flow at rest74 and prefrontal cortical activation dur-ing decision-making in manic individuals with bipolar disorder,27 whilst arelative reduction in subcortical activity has been demonstrated in manicand depressed individuals with bipolar disorder taking neuroleptic andmood-stabilizing medications compared with unmedicated individuals.30
Whilst findings in individuals with major depressive disorder, predomi-nantly at rest, have indicated increased prefrontal cortical (dorsolateral andventromedial prefrontal cortex) and decreased limbic, hipppocampal andsubgenual cingulate gyral responses after successful treatment with med-ication,75–78 although a reversed pattern after successful cognitive behav-ioural79 and interpersonal therapy,80,81 it remains to be determined whethersimilar changes in neural response to emotionally salient stimuli occur inindividuals with bipolar disorder after treatment.
Ch 05 7/4/05 3:36 pm Page 42

Are subcortical regions too expansive in bipolar disorder? 43
The predictive value of these functional neuralabnormalities for the clinical course
Few studies have examined the extent to which neurocognitive function pre-dicts clinical outcome in individuals with bipolar disorder. Previous studieshave indicated that greater cognitive dysfunction per se,82,83 a history ofpsychotic symptoms associated with greater cognitive dysfunction84 and anincreased number of white matter hyperintensities,85 may be associatedwith poorer clinical outcome in the disorder.
Conclusion
Recent functional neuroimaging data indicate that a pattern of increasedlimbic (amygdalar and ventral striatal) responses to ecologically valid‘probes’ of activity within neural systems important for emotion processing(facial expressions in others) is indeed present in remitted individuals withbipolar disorder, and therefore may exist as a persistent marker of the dis-order.35 The relationship between this abnormal pattern of neural responseto facial expressions and any deficits in facial expression identification per
se remains unclear, however. Other data suggest increased subcortical activ-ity during mood generation with positively and negatively valenced scenes indepressed individuals,42 and decreased medial prefrontal cortical activityduring sad mood generation with autobiographical retrieval in euthymicand depressed individuals with bipolar disorder.41 The increased subcorti-cal and limbic response to facial expressions and during positive and nega-tive mood generation with emotive scenes, together with the decreasedmedial prefrontal cortical response during sad mood induction with auto-biographical memory retrieval, suggests in individuals with bipolar disordera reduced regulation by prefrontal cortical regions of subcortical responsesto emotive stimuli, and during mood generation procedures. Findings alsoindicate structural volume increases in subcortical, and volume decreasesin prefrontal cortical, regions.
Taken together, these findings suggest a potential neural mechanismunderlying the affective instability in bipolar disorder86 (Figure 5.2). Here,it has been hypothesized that the affective instability in the disorder mayresult from a combination of increased activity within subcortical and lim-bic regions implicated in the initial appraisal of emotive stimuli (amygdalaventral striatum, anterior insula), resulting in increased activity in regions
Ch 05 7/4/05 3:36 pm Page 43

44 Bipolar disorder: the upswing in research & treatment
associated with mood generation and decisions about emotional material(ventromedial and ventrolateral prefrontal cortices, ventral anterior cingu-late gyrus), and reduced activity within regions implicated in the regulationof these responses (dorsal and ventromedial prefrontal cortices). Futureresearch employing emotion processing paradigms and neuroimaging tech-niques will help to clarify further the nature of the dysfunction in neural sys-tems underlying mood regulation in the disorder.
Figure 5.2 (A) Neural structures important for appraisal of emotionally salientinformation, mood generation and the regulation of emotional behaviour. A pre-dominantly ventral system is important for the identification of emotional infor-mation and the generation of affect state (green), whilst a predominantly dorsalsystem is important for selective attention and regulation of behaviour responsesto emotional stimuli (orange). The arrows (red) represent the reciprocal functionalrelationship that exists between these two distinct but parallel neural systems.PFC, prefrontal cortex; ACG, anterior cingulate gyrus. (B) Model for the neuralbasis of the affective instability in individuals with bipolar disorder. Enlargedrather than decreased amygdalar volumes, and increased amygdalar activity dur-ing performance of attentional tasks and in response to emotional stimuli, togeth-er with reduced prefrontal cortical volumes and reduced prefrontal metabolismduring task performance and at rest, would be consistent with increased but dys-functional identification of emotional stimuli and mood generation of emotionalstates, and an impaired regulation of the subsequent emotional behaviour (repre-sented by the reduction in size of the red arrows).
Appraisal AmygdalaAmygdala
InsulaInsulaThalamusThalamus
Ventral striatumVentral striatum
MoodVentrolateral PFCVentrolateral PFCVentromedial PFCVentromedial PFC
Ventral ACGVentral ACG
Stimulus presentation
RegulationDorsomedial PFCDorsomedial PFC
Dorsal ACGDorsal ACGVentromedial PFCVentromedial PFC
Appraisal AmygdalaAmygdala
InsulaInsulaThalamusThalamus
Ventral striatumVentral striatum
MoodVentrolateral PFCVentrolateral PFCVentromedial PFCVentromedial PFC
Ventral ACGVentral ACG
Stimulus presentation
RegulationDorsomedial PFCDorsomedial PFC
Dorsal ACGDorsal ACGVentromedial PFCVentromedial PFC
A B
Ch 05 7/4/05 3:36 pm Page 44

Are subcortical regions too expansive in bipolar disorder? 45
References
1. Kessler RC, McGonagle KA, Zhao S et al, Lifetime and 12-month prevalence ofDSM-III-R psychiatric disorders in the United States. Results from the NationalComorbidity Survey. Arch Gen Psychiatry 1994; 51:8–19.
2. O’Connell RA, Mayo JA, Flatow L, Cuthbertson B, O’Brien BE, Outcome ofbipolar disorder on long-term treatment with lithium. Br J Psychiatry 1991;159:123–129.
3. Gitlin MJ, Swendsen J, Heller TL, Hammen C, Relapse and impairment inbipolar disorder. Am J Psychiatry 1995; 152:1635–1640.
4. Angst F, Stassen HH, Clayton PJ, Angst J, Mortality of patients with mood dis-orders: follow-up over 34–38 years. J Affect Disord 2002; 68:167–181.
5. Keck PE Jr, McElroy SL, Strakowski SM et al, 12-month outcome of patientswith bipolar disorder following hospitalization for a manic or mixed episode.Am J Psychiatry 1998; 155:646–652.
6. Darwin C, The Expression of the Emotions in Man and Animals, 3rd edn.Harper Collins: London, 1872/1998.
7. Mataix-Cols D, Cullen S, Lange K et al, Neural correlates of anxiety associatedwith obsessive–compulsive symptom dimensions in normal volunteers. BiolPsychiatry 2003; 53:482–493.
8. Morris JS, Frith CD, Perrett DI et al, A differential neural response in thehuman amygdala to fearful and happy facial expressions. Nature 1996;383:812–815.
9. Phillips ML, Young AW, Senior C et al, A specific neural substrate for perceivingfacial expressions of disgust. Nature 1997; 389:495–498.
10. Sprengelmeyer R, Rausch M, Eysel UT, Przuntek H, Neural structures associ-ated with recognition of facial expressions of basic emotions. Proc R Soc LondB Biol Sci 1998; 265:1927–1931.
11. Calder AJ, Lawrence AD, Young AW, Neuropsychology of fear and loathing. NatRev Neurosci 2001; 2:352–363.
12. Surguladze SA, Brammer MJ, Young AW et al, A preferential increase in theextrastriate response to signals of danger. Neuroimage 2003; 19:1317–1328.
13. Phillips ML, Drevets WC, Rauch SL, Lane R, Neurobiology of emotion percep-tion I: The neural basis of normal emotion perception. Biol Psychiatry 2003;54:504–514.
14. Yurgelun-Todd DA, Gruber SA, Kanayama G et al, fMRI during affect discrimi-nation in bipolar affective disorder. Bipolar Disord 2000; 2:237–248.
15. Harmer CJ, Grayson L, Goodwin GM, Enhanced recognition of disgust in bipo-lar illness. Biol Psychiatry 2002; 51:298–304.
16. McClure EB, Pope K, Hoberman AJ et al, Facial expression recognition in ado-lescents with mood and anxiety disorders. Am J Psychiatry 2003;160:1172–1174.
17. Lembke A, Ketter TA, Impaired recognition of facial emotion in mania. Am JPsychiatry 2002; 159:302–304.
18. Getz GE, Shear PK, Strakowski SM, Facial affect recognition deficits in bipolardisorder. J Int Neuropsychol Soc 2003; 9:623–632.
Ch 05 7/4/05 3:36 pm Page 45

46 Bipolar disorder: the upswing in research & treatment
19. Lyon HM, Startup M, Bentall RP, Social cognition and the manic defense: attri-butions, selective attention, and self-schema in bipolar affective disorder. JAbnorm Psychol 1999; 108:273–282.
20. Murphy FC, Sahakian BJ, Rubinsztein JS et al, Emotional bias and inhibitorycontrol processes in mania and depression. Psychol Med 1999; 29:1307–1321.
21. Baxter LR Jr, Phelps ME, Mazziotta JC et al, Cerebral metabolic rates for glu-cose in mood disorders. Studies with positron emission tomography andfluorodeoxyglucose F 18. Arch Gen Psychiatry 1985; 42:441–447.
22. Baxter LR Jr, Schwartz JM, Phelps ME et al, Reduction of prefrontal cortex glu-cose metabolism common to three types of depression. Arch Gen Psychiatry1989; 46:243–250.
23. Ketter TA, Kimbrell TA, George MS et al, Effects of mood and subtype on cere-bral glucose metabolism in treatment-resistant bipolar disorder. BiolPsychiatry 2001; 49:97–109.
24. Martinot JL, Hardy P, Feline A et al, Left prefrontal glucose hypometabolism inthe depressed state: a confirmation. Am J Psychiatry 1990; 147:1313–1317.
25. Blumberg HP, Stern E, Ricketts S et al, Rostral and orbital prefrontal cortexdysfunction in the manic state of bipolar disorder. Am J Psychiatry 1999;156:1986–1988.
26. Blumberg HP, Leung HC, Skudlarski P et al, A functional magnetic resonanceimaging study of bipolar disorder: state- and trait-related dysfunction in ventralprefrontal cortices. Arch Gen Psychiatry 2003; 60:601–609.
27. Rubinsztein JS, Fletcher PC, Rogers RD et al, Decision-making in mania: a PETstudy. Brain 2001; 124:2550–2563.
28. Blumberg HP, Stern E, Martinez D et al, Increased anterior cingulate and cau-date activity in bipolar mania. Biol Psychiatry 2000; 48:1045–1052.
29. Goodwin GM, Cavanagh JT, Glabus MF et al, Uptake of 99mTc-exametazimeshown by single photon emission computed tomography before and after lithi-um withdrawal in bipolar patients: associations with mania. Br J Psychiatry1997; 170:426–430.
30. Caligiuri MP, Brown GG, Meloy MJ et al, An fMRI study of affective state andmedication on cortical and subcortical brain regions during motor perform-ance in bipolar disorder. Psychiatry Res 2003; 123:171–182.
31. Berns GS, Martin M, Proper SM, Limbic hyperreactivity in bipolar II disorder.Am J Psychiatry 2002; 159:304–306.
32. al Mousawi AH, Evans N, Ebmeier KP et al, Limbic dysfunction in schizophre-nia and mania. A study using 18F-labelled fluorodeoxyglucose and positronemission tomography. Br J Psychiatry 1996; 169:509–516.
33. O’Connell RA, Van Heertum RL, Luck D et al, Single-photon emission comput-ed tomography of the brain in acute mania and schizophrenia. J Neuroimaging1995; 5:101–104.
34. Gyulai L, Alavi A, Broich K et al, I-123 iofetamine single-photon computedemission tomography in rapid cycling bipolar disorder: a clinical study. BiolPsychiatry 1997; 41:152–161.
35. Lawrence NS, Williams AM, Surguladze S et al, Subcortical and ventral pre-frontal cortical neural responses to facial expressions distinguish patients withbipolar disorder and major depression. Biol Psychiatry 2004; 55:578–587.
Ch 05 7/4/05 3:36 pm Page 46

Are subcortical regions too expansive in bipolar disorder? 47
36. Mayberg HS, Liotti M, Brannan SK et al, Reciprocal limbic–cortical functionand negative mood: converging PET findings in depression and normal sad-ness. Am J Psychiatry 1999; 156:675–682.
37. Breiter HC, Gollub RL, Weisskoff RM et al, Acute effects of cocaine on humanbrain activity and emotion. Neuron 1997; 19:591–611.
38. Lang PJ, Bradley MM, Cuthbert BN, International Affective Picture System(IAPS). NIMH Center for the Study of Emotion and Attention: University ofFlorida, Gainesville, 1997.
39. Phillips ML, Senior C, David AS, Perception of threat in schizophrenics withpersecutory delusions: an investigation using visual scan paths. Psychol Med2000; 30:157–167.
40. Keedwell PA, Andrew C, Williams SCR et al, The neural correlates of depres-sion. Biol Psychiatry 2003; 53:1S–217S.
41. Kruger S, Seminowicz D, Goldapple K et al, State and trait influences on moodregulation in bipolar disorder: blood flow differences with an acute mood chal-lenge. Biol Psychiatry 2003; 54:1274–1283.
42. Mahli GS, Lagopoulos J, Ward PB et al, Cognitive generation of affect in bipo-lar depression: an fMRI study. Eur J Neurosci 2004; 19:741–754.
43. Elliott R, Ogilvie A, Rubinsztein JS et al, Abnormal ventral frontal responseduring performance of an affective go/no go task in patients with mania. BiolPsychiatry 2004; 55:1163–1170.
44. Elliott R, Rubinsztein JS, Sahakian BJ, Dolan RJ, The neural basis of mood-congruent processing biases in depression. Arch Gen Psychiatry 2002;59:597–604.
45. Brambilla P, Harenski K, Nicoletti M et al, MRI investigation of temporal lobestructures in bipolar patients. J Psychiatr Res 2003; 37:287–295.
46. Strakowski SM, DelBello MP, Sax KW et al, Brain magnetic resonance imagingof structural abnormalities in bipolar disorder. Arch Gen Psychiatry 1999;56:254–260.
47. Altshuler LL, Bartzokis G, Grieder T et al, Amygdala enlargement in bipolardisorder and hippocampal reduction in schizophrenia: an MRI study demon-strating neuroanatomic specificity. Arch Gen Psychiatry 1998; 55:663–664.
48. Blumberg HP, Martin A, Kaufman J et al, Frontostriatal abnormalities in ado-lescents with bipolar disorder: preliminary observations from functional MRI.Am J Psychiatry 2003; 160:1345–1347.
49. Aylward EH, Roberts-Twillie JV, Barta PE et al, Basal ganglia volumes and whitematter hyperintensities in patients with bipolar disorder. Am J Psychiatry1994; 151:687–693.
50. Sax KW, Strakowski SM, Zimmerman ME et al, Frontosubcortical neuroanato-my and the continuous performance test in mania. Am J Psychiatry 1999;156:139–141.
51. Lopez-Larson MP, DelBello MP, Zimmerman ME et al, Regional prefrontal grayand white matter abnormalities in bipolar disorder. Biol Psychiatry 2002;52:93–100.
52. Sharma V, Menon R, Carr TJ et al, An MRI study of subgenual prefrontal cor-tex in patients with familial and non-familial bipolar I disorder. J Affect Disord2003; 77:167–171.
Ch 05 7/4/05 3:36 pm Page 47

48 Bipolar disorder: the upswing in research & treatment
53. Brambilla P, Nicoletti MA, Harenski K et al, Anatomical MRI study of subgenu-al prefrontal cortex in bipolar and unipolar subjects. Neuropsychopharma-cology 2002; 27:792–799.
54. Lochhead RA, Parsey RV, Oquendo MA, Mann JJ, Regional brain gray mattervolume differences in patients with bipolar disorder as assessed by optimizedvoxel-based morphometry. Biol Psychiatry 2004; 55:1154–1162.
55. Clark L, Iversen SD, Goodwin GM, Sustained attention deficit in bipolar disor-der. Br J Psychiatry 2002; 180:313–319.
56. Cavanagh JT, Van Beck M, Muir W, Blackwood DH, Case–control study of neu-rocognitive function in euthymic patients with bipolar disorder: an associationwith mania. Br J Psychiatry 2002; 180:320–326.
57. Martinez-Aran A, Vieta E, Reinares M et al, Cognitive function across manic orhypomanic, depressed, and euthymic states in bipolar disorder. Am JPsychiatry 2004; 161:262–270.
58. Zubieta JK, Huguelet P, O’Neil RL, Giordani BJ, Cognitive function in euthymicbipolar I disorder. Psychiatry Res 2001; 102:9–20.
59. Ali SO, Denicoff KD, Altshuler LL et al, Relationship between prior course of ill-ness and neuroanatomic structures in bipolar disorder: a preliminary study.Neuropsychiatry Neuropsychol Behav Neurol 2001; 14:227–232.
60. Brambilla P, Harenski K, Nicoletti M et al, Differential effects of age on braingray matter in bipolar patients and healthy individuals. Neuropsychobiology2001; 43:242–247.
61. Strakowski SM, Adler CM, DelBello MP, Volumetric MRI studies of mood dis-orders: do they distinguish unipolar and bipolar disorder? Bipolar Disord2002; 4:80–88.
62. Mehta MA, Sahakian BJ, McKenna PJ, Robbins TW, Systemic sulpiride inyoung adult volunteers simulates the profile of cognitive deficits in Parkinson’sdisease. Psychopharmacology (Berl) 1999; 146:162–174.
63. King DJ, Psychomotor impairment and cognitive disturbances induced by neu-roleptics. Acta Psychiatr Scand Suppl 1994; 380:53–58.
64. Kocsis JH, Shaw ED, Stokes PE et al, Neuropsychologic effects of lithium dis-continuation. J Clin Psychopharmacol 1993; 13:268–275.
65. Ananth J, Gold J, Ghadirian AM, Long-term effects of lithium carbonate on cog-nitive functions. J Psychiatr Eval Treat 1981; 3:551–555.
66. Engelsmann F, Katz J, Ghadirian AM, Schachter D, Lithium and memory: along-term follow-up study. J Clin Psychopharmacol 1988; 8:207–212.
67. Ferrier IN, Stanton BR, Kelly TP, Scott J, Neuropsychological function ineuthymic patients with bipolar disorder. Br J Psychiatry 1999; 175:246–251.
68. Kessing LV, Cognitive impairment in the euthymic phase of affective disorder.Psychol Med 1998; 28:1027–1038.
69. Devinsky O, Cognitive and behavioral effects of antiepileptic drugs. Epilepsia1995; 36(Suppl 2):S46–S65.
70. Thompson PJ, Annual Review of Human Psychopharmacology 1991; 6:79–9071. Bhagwagar Z, Cowen PJ, Goodwin GM, Harmer CJ, Normalization of enhanced
fear recognition by acute SSRI treatment in subjects with a previous history ofdepression. Am J Psychiatry 2004; 161:166–168.
Ch 05 7/4/05 3:36 pm Page 48

Are subcortical regions too expansive in bipolar disorder? 49
72. Manji HK, Moore GJ, Chen G, Clinical and preclinical evidence for the neu-rotrophic effects of mood stabilizers: implications for the pathophysiology andtreatment of manic-depressive illness. Biol Psychiatry 2000; 48:740–754.
73. Harrison PJ, The neuropathology of primary mood disorder. Brain 2002;125:1428–1449.
74. Silfverskiold P, Risberg J, Regional cerebral blood flow in depression andmania. Arch Gen Psychiatry 1989; 46:253–259.
75. Mayberg HS, Brannan SK, Tekell JL et al, Regional metabolic effects of fluoxe-tine in major depression: serial changes and relationship to clinical response.Biol Psychiatry 2000; 48:830–843.
76. Bench CJ, Friston KJ, Brown RG et al, Regional cerebral blood flow in depres-sion measured by positron emission tomography: the relationship with clinicaldimensions. Psychol Med 1993; 23:579–590.
77. Kennedy SH, Evans KR, Kruger S et al, Changes in regional brain glucosemetabolism measured with positron emission tomography after paroxetinetreatment of major depression. Am J Psychiatry 2001; 158:899–905.
78. Goodwin GM, Austin MP, Dougall N et al, State changes in brain activity shownby the uptake of 99mTc-exametazime with single photon emission tomographyin major depression before and after treatment. J Affect Disord 1993;29:243–253.
79. Goldapple K, Segal Z, Garson C et al, Modulation of cortical–limbic pathwaysin major depression: treatment-specific effects of cognitive behavior therapy.Arch Gen Psychiatry 2004; 61:34–41.
80. Brody AL, Saxena S, Stoessel P et al, Regional brain metabolic changes inpatients with major depression treated with either paroxetine or interpersonaltherapy: preliminary findings. Arch Gen Psychiatry 2001; 58:631–640.
81. Martin SD, Martin E, Rai SS et al, Brain blood flow changes in depressedpatients treated with interpersonal psychotherapy or venlafaxine hydrochlor-ide: preliminary findings. Arch Gen Psychiatry 2001; 58:641–648.
82. Martinez-Aran A, Penades R, Vieta E et al, Executive function in patients withremitted bipolar disorder and schizophrenia and its relationship with func-tional outcome. Psychother Psychosom 2002; 71:39–46.
83. Martinez-Aran A, Vieta E, Colom F et al, Cognitive impairment in euthymicbipolar patients: implications for clinical and functional outcome. BipolarDisord 2004; 6:224–232.
84. Tohen M, Hennen J, Zarate CM Jr et al, Two-year syndromal and functionalrecovery in 219 cases of first-episode major affective disorder with psychoticfeatures. Am J Psychiatry 2000; 157:220–228.
85. Moore PB, Shepherd DJ, Eccleston D et al, Cerebral white matter lesions inbipolar affective disorder: relationship to outcome. Br J Psychiatry 2001;178:172–176.
86. Phillips ML, Drevets WC, Rauch SL, Lane R, Neurobiology of emotion percep-tion II: Implications for major psychiatric disorders. Biol Psychiatry 2003;54:515–528.
Ch 05 7/4/05 3:36 pm Page 49

Ch 05 7/4/05 3:36 pm Page 50

The Maudsley BipolarDisorder Project:insights intopathophysiologySophia Frangou
c h a p t e r 6
The structural neuroanatomy of bipolar disorder
Neuroanatomical changes in bipolar disorder are mostly regional andinvolve the prefrontal cortex (PFC), the anterior cingulate and the amygdala.Reductions in grey matter volume (or density) have been found in the dor-sal PFC and they appear to be more pronounced on the left.1–3 Similarly,reduction has been reported in the ventral PFC particularly in Brodmannareas 44 and 47.1,3 Volume (or density) decreases in the left cingulate gyrushave been reported in some3,4 but not all studies.1,5 In contrast, the volumeof the amygdala has appeared to be enlarged bilaterally or only on the leftin recent studies.1,5–7
The functional neuroanatomy of bipolar disorder
Resting-state functional imaging studies, despite differences in methodolog-ical approaches, have found reduced activity in the dorsal PFC, mostly onthe left8 and increased activity in the amygdala in depressive states.9 Manicstates have been associated with decreased activity in the ventral PFC andincreased activity in the anterior cingulate cortex (ACC).10,11 Trait-relateddecreases in brain activation have also been reported within the left ventralPFC (Brodmann areas 47 and 10).12 However, the relative contributions ofdorsal and ventral prefrontal functioning in bipolar disorder remainunclear.
Ch 06 7/4/05 3:37 pm Page 51

52 Bipolar disorder: the upswing in research & treatment
The Maudsley Bipolar Disorder Project
The Maudsley Bipolar Disorder Project started in 1999 and is ongoing. Ithas a case–control design and comprises interconnected modules focusingon cognition, and structural and functional neuroanatomy.13–15 Diagnosis inpatients and its absence in controls was established using the StructuredClinical Interview for DSM-IV Axis I Disorders; symptomatology wasassessed using the 31-item Hamilton Depression Rating Scale (HAMD), andthe Mania Rating Scale (MRS) from the Schedule for Affective Disorders andSchizophrenia – Change Version. All participants had detailed cognitive test-ing that assessed the domains of general intelligence (IQ), memory and exec-utive function.
Here we present data from the functional imaging component of thestudy. For this component, 14 participants were selected from the pool ofBDI patients and matched controls form the Maudsley Bipolar DisorderProject. Patients were selected on the following criteria: (1) remitted clini-cal status; (2) monotherapy with mood stabilizer; and (3) test performanceless than 0.5 standard deviation below the control mean for all tests. Onlyeight patients fulfilled these requirements. Seven agreed to participate andwere individually matched to an equal number of controls on age, gender,years of education and IQ. The mean age of the seven participating patients(five women and two men) was 37±5.88 years and mean years of educationwere 12.43±2.64. Their mean age of onset was 21.6±6.5 years and they hadexperienced an average of 9±2.6 episodes. On the day of their functionalmagnetic resonance imaging (fMRI) assessment, patients’ mean scores onthe HAMD and MRS were 5.14±0.29 and 0.14±3.37, respectively. Theywere on treatment with lithium (n=4; dose range 600–1000mg/day) andsodium valproate (n=3; dose range 750–1000mg/day). Their mean fullscale IQ was 102.57±16.21. The mean age of controls was 39±5.88; yearsof education were 13.43±3.26, and mean full scale IQ was 104.71±15.88.All participants were right handed.
All participants underwent fMRI whilst performing cognitive paradigmsselected on their relative specificity in engaging dorsal and ventral PFC. TheN-back letter-sequencing task, a verbal working memory task, has beendemonstrated to engage a wide network of brain regions with dorsal PFCactivation being the most consistent finding across different experimentalmanipulations.16 The Iowa Gambling Task, initially developed to testdecision-making in individuals with PFC lesions,17 has been shown toengage the bilateral ventral prefrontal regions.18 Gradient-echo echoplanarmagnetic resonance images were acquired by using a 1.5T GE
Ch 06 7/4/05 3:37 pm Page 52

The Maudsley Bipolar Disorder Project 53
Neurovascular Signa MR system (General Electric, Milwaukee, WI, USA) fit-ted with 40mT/m high-speed gradients and were analysed on a SPARC Ultra10 workstation (Sun Microsystems, Palo Alto, CA, USA) using MATLAB(version 5.3, The Mathworks Inc, Natick, MA, USA) and SPM99 software(Statistical Parametric Mapping, The Wellcome Department of CognitiveNeurology, London; http://www.fil.ion.ucl.ac.uk/spm).
N-back task
During the N-back task, in the ‘control’ or 0-back condition, participantsresponded by button press when a designated target letter appeared (letter‘X’). In the three active conditions, the 1-back, 2-back and 3-back, the tar-get letter was defined as any letter that was identical to the one presented inthe preceding 1, 2, or 3 trials, respectively. All stimuli were visually pre-sented to subjects by means of a prismatic mirror as they lay in the scannerand responses were monitored. A series of 13 letters was presented with aninterstimulus interval of 2.3 seconds during each epoch. There were 18epochs in all, each lasting 30 seconds with the total experiment time of9 minutes. All conditions were matched for number of target letters pre-sented. The order of the tasks was pseudorandomized to avoid any system-atic order effects. Reaction time to target letters and accuracy were record-ed.
Patients and controls did not differ in reaction time or accuracy (Figure6.1). During the active conditions (1-, 2-, 3-back tasks minus the 0-backtask) no significant differences were seen in patients and controls in the pat-tern and degree of brain regions activated. When we examined the effect ofmemory load, in the controls increased activation was seen bilaterally to thesuperior (BA 6) and middle frontal gyrus (BA 9,46) and the anterior cingu-late gyrus (BA 32) as well as the right superior parietal lobule (BA 7). Inbipolar disorder patients the effect of increasing memory load was localizedto the left superior parietal lobule (BA 7) and the right middle frontal gyrus(BA 10).
Therefore, it seems that, in bipolar disorder, dorsal PFC dysfunction issubtle and only becomes apparent with increasing mental load.
Gambling task
During the Decision-Making Task, subjects were instructed to select from96 cards arranged in four piles (decks) in order to win ‘pretend’ money.Unknown to the subjects, deck A had frequent, small magnitude punish-ments, deck B had infrequent, but higher punishments, deck C had
Ch 06 7/4/05 3:37 pm Page 53

54 Bipolar disorder: the upswing in research & treatment
frequent, small rewards, and deck D had infrequent, higher rewards.Subjects were asked to select a card from a deck of their choice, once every5 seconds, and the win or loss associated with their choice appeared visu-ally on the screen. Each ‘active’ condition lasted 1 minute and was alternat-ed eight times with the ‘control’ condition, when subjects were required toselect cards as before, but were informed that no money would be won orlost during this epoch; total experiment time was 16 minutes. Performancemeasures were the net global outcome score (net score) calculated by sub-tracting the total number of cards selected from the disadvantage decks(A+B) from the total number of cards selected from the advantage decks(C+D).
In controls activation associated with incentive decision making was evi-dent in the left superior frontal gyrus (BA 6), the bilateral middle(BA 8,9,46) and inferior (BA 44,45) frontal gyri, and the right superior pari-etal lobule (BA 7). Patients demonstrated activation of the left superior(BA 7) and inferior (BA 40) parietal lobules and the right superior frontal
Figure 6.1 Cerebral activation in controls (A) and patients (B) during the N-backtask.
A B
Ch 06 7/4/05 3:37 pm Page 54

The Maudsley Bipolar Disorder Project 55
gyrus (BA 10). Compared with controls, patients showed significantly lessactivation in the frontal cortices (Figure 6.2).
Human lesion studies suggest that the interface between the dorsal andventral PFC is crucial for incentive decision making.19 Studies in primateshave indicated that reward-related dorsal PFC modulation is probably driv-en by brain areas within the ventral PFC which are primarily involved withprocessing and representing incentive information. This hypothesis is sup-ported by electrophysiological measurements in primates. Wallis andMiller20 recorded neuronal activity from the dorsal and ventral PFC of rhe-sus monkeys while choosing between pictures associated with differentamounts of a juice reward. They found that, whilst both dorsal and ventralPFC were involved in encoding the incentive value (reward) of the differentstimuli (pictures), neural activation peaked earlier in the ventral PFC, sug-gesting that this region generated incentive information that enters the dor-sal PFC where it is used to influence behavioural response. According tothis model, deficits in the ventral PFC should attenuate reward information
Figure 6.2 Cerebral activation in controls (A) and patients (B) during theGambling Task.
A B
Ch 06 7/4/05 3:37 pm Page 55

56 Bipolar disorder: the upswing in research & treatment
in the dorsal PFC. The findings of the Gambling Task in the bipolar disor-
der sample in our study are consistent with this model.
In summary, this study provides evidence of PFC dysfunction as a trait
deficit in bipolar disorder that is not predicted by task performance. It also
suggests that dorsal PFC dysfunction may be more subtle than ventral, and
that the greatest deficit may be seen in tasks crucially dependent on the
interaction between dorsal and ventral PFC.
References
1. Frangou S, Hadjulis M, Chitnis X et al, The Maudsley Bipolar Disorder Project:brain structural changes in bipolar 1 disorder. Bipolar Disord 2002;4:123–124.
2. Lopez-Larson MP, DelBello MP, Zimmerman ME et al, Regional prefrontal grayand white matter abnormalities in bipolar disorder. Biol Psychiatry 2002;52:93–100.
3. Lyoo IK, Kim MJ, Stoll AL et al, Frontal lobe gray matter density decreases inbipolar I disorder. Biol Psychiatry 2004; 55:648–51.
4. Hirayasu Y, Shenton ME, Salisbury DF et al, Subgenual cingulate cortex volumein first-episode psychosis. Am J Psychiatry 1999; 156:1091–1093.
5. Brambilla P, Nicoletti MA, Harenski K et al, Anatomical MRI study of subgenu-al prefrontal cortex in bipolar and unipolar subjects. Neuropsychopharma-cology 2002; 27:792–799.
6. Strakowski SM, DelBello MP, Sax KW et al, Brain magnetic resonance imagingof structural abnormalities in bipolar disorder. Arch Gen Psychiatry 1999;56:254–260.
7. Altshuler LL, Bartzokis G, Grieder T et al, An MRI study of temporal lobe struc-tures in men with bipolar disorder or schizophrenia. Biol Psychiatry 2000;48:147–162.
8. Baxter LR, Schwartz JM, Phelps ME et al, Reduction of prefrontal cortex glu-cose metabolism common to three types of depression. Arch Gen Psychiatry1989; 46:243–250.
9. Drevets WC, Price JL, Bardgett ME et al, Glucose metabolism in the amygdalain depression: relationship to diagnostic subtype and plasma cortisol levels.Pharmacol Biochem Behav 2002; 71:431–47.
10. Blumberg HP, Stern E, Ricketts S et al, Rostral and orbital prefrontal cortexdysfunction in the manic state of bipolar disorder. Am J Psychiatry 1999; 156:1986–1988.
11. Blumberg HP, Stern E, Martinez D et al, Increased anterior cingulate and cau-date activity in bipolar mania. Biol Psychiatry 2000; 48:1045–52.
12. Blumberg HP, Leung HC, Skudlarski P et al, A functional magnetic resonanceimaging study of bipolar disorder: state- and trait-related dysfunction in ventralprefrontal cortices. Arch Gen Psychiatry 2003; 60:601–9.
Ch 06 7/4/05 3:37 pm Page 56

The Maudsley Bipolar Disorder Project 57
13. Frangou S, Raymont V, Bettany D, The Maudsley Bipolar Disorder Project. Apharmaco-epidemiological survey of prescribing patterns in bipolar I disorder.Bipolar Disord 2002; 4:378–385.
14. Donaldson S, Goldstein LH, Landau S et al, The Maudsley Bipolar DisorderProject: The effect of medication, family history, and duration of illness on IQand memory in bipolar 1 disorder. J Clin Psychiatr 2003; 64:86–93.
15. Raymont V, Bettany D, Frangou S, The Maudsley Bipolar Disorder Project.Clinical characteristics of bipolar disorder I in a catchment area derived treat-ment sample. Eur Psychiatry 2003; 18:13–17.
16. Fletcher PC, Henson RN, Frontal lobes and human memory: insights from func-tional neuroimaging. Brain 2001; 124:849–881.
17. Bechara A, Damasio AR, Damasio H, Anderson SW, Insensitivity to future con-sequences following damage to human prefrontal cortex. Cognition 1994;50:7–15.
18. Ernst M, Bolla K, Mouratidis M et al, Decision-making in a risk-taking task: APET study. Neuropsychopharmacology 2002; 26:682–691.
19. Bechara A, Damasio H, Tranel D, Anderson SW, Dissociation of working mem-ory from decision making within the human prefrontal cortex. J Neurosci1998; 18:428–37.
20. Wallis JD, Miller EK, Neuronal activity in primate dorsolateral and orbital pre-frontal cortex during performance of a reward preference task. Eur J Neurosci2003; 18:2069–2081.
Ch 06 7/4/05 3:37 pm Page 57

Ch 06 7/4/05 3:37 pm Page 58

Is any of this real?The word fromthe gravePaul J Harrison
c h a p t e r 7
Introduction
As with all messages from the grave, we have to be sceptical when dis-cussing the neuropathology of bipolar disorder. So far, there have beensome intriguing whispers, but they remain difficult to hear, and we still can-not be sure how best to decipher them. In this chapter, I cover the main find-ings from postmortem studies of bipolar disorder, and try to link them withsome other themes. I have not attempted to provide a systematic overviewof the subject; for this see two recent reviews.1,2
At the start it is important to point out that neuropathological studies ofbipolar disorder only began a few years ago. It is no exaggeration to saythere were no data worth discussing until a study was published in 1998.3
The reason why the field is so recent is largely a practical one: people sim-ply did not collect enough brains from patients who had suffered from bipo-lar disorder, with adequate characterization and appropriate controls,4 tocarry out any kind of quantitative, or molecular, studies. It is thanks to theStanley Foundation (now the Stanley Medical Research Institute) that thesituation changed in the mid-1990s, because of their funding of an autopsyseries of brains from patients (15 in each group) with bipolar disorder aswell as schizophrenia, major depression and control subjects.5 This serieswas made available to investigators across the world. Part of the deal wasthat one had to study all 60 subjects in all experiments (the material wascoded), and so people began to study bipolar disorder. A significant pro-portion of the world literature on the neuropathology of bipolar disordernow comes from this series. The gene expression component of this workhas been meta-analysed on a region-by-region basis.6,7
Ch 07 7/4/05 3:43 pm Page 59

60 Bipolar disorder: the upswing in research & treatment
A few other practical issues are also worth noting. First, the concepts,methods, targets and interpretation of the research have been much influ-enced by the experience with schizophrenia which had emerged over thepreceding 10–15 years, and by the fact that it is the latter disorder that isoften of primary interest. The majority of papers therefore discuss bipolardisorder findings in terms of similarities (of which there are many) and dif-ferences (of which there are relatively few) with those of schizophrenia. Atthe same time, however, comparison with other mood disorders cannot beneglected, and this is emphasized in some publications. Thus, for example,some papers concentrate on bipolar versus unipolar mood disorder, oremphasize the importance of familiality not polarity.3 These different waysof cutting the cake (or comparing two cakes) makes the literature somewhatconfusing. The second point is that neuropathologists have to pick a partic-ular part of the brain to study: the brain is too big (and too structurally com-plex even within a single area) to examine the whole thing. Because one hasto start somewhere, research has naturally focused on where researchersthink the positive findings are most likely to be. (As the man said whenasked why he robbed banks: ‘they’re where the money is’.)
The most relevant clues have been those from brain imaging, and hencemany of the neuropathological data are in the anterior cingulate cortex andorbitofrontal cortex, because of the many data implicating these regions(Figure 7.1). The best example of this kind is the combined study of mag-netic resonance imaging (MRI) and positron emission tomography (PET)showing smaller volumes and reduced metabolic activity in mood disorderin the ‘subgenual’ part of the cingulate cortex,8 which led the same group tocarry out the landmark postmortem study mentioned.3 Whilst this ‘candi-date region’ approach may have been inevitable, it does of course lead to acircularity in the argument – it means we do not know where the pathologyof bipolar disorder might be centred because people have not looked, toanything like the same extent, in many other areas of the brain – comparethe cerebellum in schizophrenia: hardly an area which many people impli-cated, nor were interested in, a decade ago, yet where there is now good evi-dence for structural and functional involvement.
Neuropathology of bipolar disorder: findings andinterpretations
There are three basic kinds of neuropathology reported in bipolar disorder(Table 7.1). The first observation, as reported by Öngür and colleagues,3
Ch 07 7/4/05 3:43 pm Page 60

Is any of this real? The word from the grave 61
was unexpected: there were fewer glial cells in the subgenual cingulate cor-tex. The decrease was also seen in major depression subjects with a familyhistory. The finding has been replicated, albeit equivocally, in subsequentstudies of the cingulate cortex and other prefrontal areas.2,9,10 The secondkind of abnormality affects neurons, in the same areas and in the hip-pocampus, which may be slightly fewer in number, or smaller.2,10,11 Thethird aspect is ‘synaptic pathology’, where one uses molecular markers ofsynaptic terminals as one way of getting at the connectivity between the neu-rons. Three studies have shown a reduction in the expression of synapticproteins, two in the anterior cingulate cortex and one in the hippocam-pus.2,12
So these are the elements of pathology of which we need to consider thesignificance. Before doing so, I should again emphasize that these are verypreliminary studies, often on small numbers of subjects, or the same sub-jects. The studies face the various difficulties of postmortem brain studies,meaning that one has always to be alert to confounders of different kinds,
Figure 7.1 Areas of the cerebral cortex implicated in bipolar disorder by neuro-pathological studies. (A) Lateral surface of the left hemisphere; (B) medial surfaceof the right hemisphere; (C) coronal section through the left hemisphere at the levelof the dashed line in (A) and (B). The numbers refer to Brodmann areas. Thesubgenual region of the anterior cingulate cortex is sg24. For detailed discussionsee reference 2.
24b
24a
24c
sg24
C
32
24′
12
25
33
2432′
B
9
10 46
1147
A
Ch 07 7/4/05 3:43 pm Page 61

62 Bipolar disorder: the upswing in research & treatment
for example those related to the cause of death or autopsy delay.4 Thoughsurmountable, these can jeopardize the robustness of the results, given thesubtlety of the differences being investigated. Nevertheless, for the rest of thechapter I shall assume that the above findings are basically true, and dis-cuss what they may mean.
The first question concerns the clinicopathological correlations. It hasbeen suggested that grey matter volumes may change with the duration ofthe illness. We also know that bipolar disorder runs an episodic course.Therefore, the neuropathology could be the pathology of having had bipolardisorder and its sequelae – most of the subjects died after having manyyears of illness, so we are not looking at first episode or unmedicated sub-jects. We also need to bear in mind that patients die at different phases ofthe illness: some were euthymic when they died, many were depressed, anda few may have been manic. It therefore seems unlikely that we are lookingat the pathology of abnormal mood per se. Instead it is more likely that weare looking at the pathology of one of the more enduring aspects of bipolar
Table 7.1 Main themes in bipolar disorder neuropathology
Strength of evidence
Brain areas involved
Anterior cingulate cortex ++++
Orbitofrontal cortex +++
Dorsolateral prefrontal cortex +++
Hippocampus ++
Amygdala +
Main features
Reduced number or density of glia +++
Smaller neurons ++
Decreased synaptic markers ++
Reduced neuronal density ++
Clinical correlations
Independent of mood state ++
May be related to duration of illness +
May be ameliorated by mood stabilizers +
Overlaps with schizophrenia ++
Overlaps with major depression ++
Ch 07 7/4/05 3:43 pm Page 62

Is any of this real? The word from the grave 63
disorder. That could be the neurocognitive abnormalities, at least some ofwhich persist during euthymia; or, it could be the neuropathology of vul-nerability to mood disturbance and of the circuits that regulate mood.Alternatively, we may be studying the neuropathology of the genetic predis-position to bipolar disorder (see below) – there have never been any post-mortem studies of unaffected relatives or obligate carriers of bipolar disor-der.
The next point concerns anatomical specificity and precision. For exam-ple, even within the anterior cingulate cortex, to which Brodmann assigneda single number (area 24), it is clear that there is anatomical, functional andneurochemical heterogeneity13,14 (see Figure 7.1). Thus, while there is atemptation to simplify things when we talk about this part of the brain as ifit were a single area, it is not. To illustrate, a student working with me dida very careful counting study at different sites within area 24.15 As onemoves from the back of area 24 round to the front, one goes from having acortex which is thick with a deep layer 5, and many large neurons and glia,to a thinner cortex with a shallow layer 5, smaller neurons, and fewer glia.The proportion of different neuron types also changes. This heterogeneityhas a number of implications. One is practical – you must be very carefulwhere you are sampling from, because if sampling is not from exactly thesame area in the different groups, the differences can be larger than the dif-ferences being reported between bipolar disorder and controls. The secondpoint, parenthetically, is that the metabolic activity of an area of the brain isdetermined to a large extent by its cellular and synaptic composition. Thus,some of the differences in activity seen in functional imaging studies mayreflect the local cellular and synaptic content. Therefore, however pretty thepseudocolour blobs are, it is necessary to think exactly where the blob is,and to what extent the local cytoarchitecture affects the interpretation.
Another issue relates to therapeutics. We have heard already that lithi-um may ‘grow your brain’, based on MRI findings, and the animal literaturesuggests that lithium may enhance neurogenesis.16 Obviously, such effectsare of potential interest, not least to the pharmaceutical industry and clini-cians. If it is true that some of the therapeutic effects of mood stabilizers aredue to long-term alterations in synaptic and neuronal plasticity, this hasimplications not only for therapeutics but also for interpreting the extantneuropathology. That is, perhaps the medication received by the patientsreversed or at least reduced some of the neuronal, synaptic and glialdeficits. In other words, there would be more rather than less pathology ifdrug-free patients were studied. One paper of ours produced weak evidencesupportive of this notion.17
Ch 07 7/4/05 3:43 pm Page 63

64 Bipolar disorder: the upswing in research & treatment
Next, I return to the most striking and arguably well-replicated finding,the reduction of glia. Until recently these cells were rather boring; they werenot thought to do very much except fill in the spaces between neurons, sup-port them metabolically, and help the brain to monitor and respond toinjury, infection, etc. In fact, glia are inextricably linked to neuronal andsynaptic structural and functional integrity. Manipulating one affects theother. Glia can even release and uptake ‘neuro’transmitters and expressmany receptors; about the only thing they cannot do which a neuron can isto discharge an action potential. In this light, a loss of glia can have a rangeof causes and consequences, any one or more of which may underlie theirinvolvement in bipolar disorder.9 For example, if a glial deficit were a pri-mary event, it is plausible, even likely, that synapses and neurons willappear to be structurally abnormal in some way (i.e. be visible neuropatho-logically), as well as being functionally abnormal, which is presumably whatis being seen in in vivo studies. This hypothetical scenario is shown inFigure 7.2. Equally, there could be a primary change in neuronal organiza-tion and activity, leading to secondary glial changes. Complicating mattersfurther, glia are not a single entity, but exist as at least three fundamentallydistinct classes of cell (astrocytes, oligodendrocytes and microglia). Eachhas its own origins, roles and pathological implications. The present evi-dence in bipolar disorder particularly implicates the oligodendrocytes,which regulate myelination and may relate to the evidence for myelinabnormalities in bipolar disorder.18
The final and most fundamental question concerns the cause(s) of theneuropathology. Prefacing Chapter 8, I am going to suggest that the struc-tural changes in bipolar disorder are genetic in origin. That is, some of thepathology might be directly related to the genetic predisposition to bipolardisorder, just as has been postulated for schizophrenia.19,20 Indeed, thegenetics of schizophrenia is (partly) the genetics of bipolar disorder, andindividual genes associated with schizophrenia are now being associatedwith bipolar disorder too, such as G72 and COMT. Interestingly, the genesfor schizophrenia all appear (at least with the eye of faith) to have a com-mon effect upon synaptic plasticity and synaptic function, and so by extrap-olation this principle may apply, to a greater or lesser extent, to bipolar dis-order. If this is true, then it is relatively easy to see how they may impactupon the kinds of abnormality outlined here. However, before getting car-ried away with this notion, or indeed aligning bipolar disorder too closelywith schizophrenia, we must also bear in mind that the morphologicalabnormalities seen in bipolar disorder are at least as similar to those ofmajor depression as they are to schizophrenia (e.g. the glial loss, smaller
Ch 07 7/4/05 3:43 pm Page 64

Is any of this real? The word from the grave 65
neurons); this is somewhat harder to explain in terms of shared geneticdiathesis. Indeed, the question of the relationships between bipolar disor-der, major depression and schizophrenia remain just as unclear neu-ropathologically as they are in every other respect. We should also bear inmind that MRI has revealed another structural aspect of bipolar disorder,namely, an excess of signal hyperintensities in the subcortical white matter.These have yet to be studied neuropathologically – in elderly unipolardepression they are focal areas of ischaemia and infarction21 – and suchlesions do not fit as neatly into a genetic, developmental model.
Figure 7.2 A hypothetical glia-based origin of neuropathology in bipolar disorder.
Genetic factors
Environmental factors
Gliogenesis Glial toxicity
Glial deficit
Formation or maintenance of synapses
Neuronal size and density
Dendrites and dendritic spines
Altered circuitry
Altered neurotransmission Impaired
plasticity
MOOD DISORDER
HPA axis dysfunction
Ch 07 7/4/05 3:44 pm Page 65

66 Bipolar disorder: the upswing in research & treatment
Summary
Neuropathological studies are providing evidence that there is a structuralcomponent to the neurobiology of bipolar disorder, and the first clues as towhat its nature might be. The evidence is accumulating, but remains pre-liminary. To modify the metaphor in the title I was given, there is somethingin the graveyard, but much more digging will be needed to identify what itis and how it got there. In addition, a separate dig will be needed to identi-fy the white matter neuropathology, and to establish whether it belongs tothe same corpse.
References
1. Vawter MP, Freed WJ, Kleinman JE, Neuropathology of bipolar disorder. BiolPsychiatry 2000; 48:486–504.
2. Harrison PJ, The neuropathology of primary mood disorder. Brain 2002;125:1428–1449.
3. Öngür D, Drevets WC, Price JL. Glial reduction in the subgenual prefrontal cor-tex in mood disorders. Proc Natl Acad Sci USA 1998; 95:13290–13295.
4. Lewis DA, The human brain revisited: Opportunities and challenges in post-mortem studies of psychiatric disorders. Neuropsychopharmacology 2002;26:143–154.
5. Torrey EF, Webster M, Knable M et al, The stanley foundation brain collectionand neuropathology consortium. Schizophr Res 2000; 44:151–155.
6. Knable MB, Torrey EF, Webster MJ, Bartko JJ, Multivariate analysis of pre-frontal cortical data from the Stanley Foundation Neuropathology Consortium.Brain Res Bull 2001; 55:651–659.
7. Knable MB, Barci BM, Bartko JJ et al, Abnormalities of the cingulate gyrus inbipolar disorder and other severe psychiatric illness: postmortem findingsfrom the Stanley Foundation Neuropathology Consortium and literature review.Clin Neurosci Res 2002; 2:171–181.
8. Drevets WC, Price JL, Simpson JR Jr et al, Subgenual prefrontal cortex abnor-malities in mood disorders. Nature 1997; 386:824–827.
9. Cotter DR, Pariante CM, Everall IP, Glial cell abnormalities in major psychiatricdisorders: The evidence and implications. Brain Res Bull 2001; 55:585–595.
10. Rajkowska G, Halaris A, Selemon LD, Reductions in neuronal and glial densi-ty characterize the dorsolateral prefrontal cortex in bipolar disorder. BiolPsychiatry 2001; 49:741–752.
11. Benes FM, Kwok EK, Vincent SL, Todtenkopf MS, Reduction of nonpyramidalcells in sector CA2 of schizophrenics and manic depressives. Biol Psychiatry1998; 44:88–97.
12. Eastwood SL, Harrison PJ, Hippocampal synaptic pathology in schizophrenia,bipolar disorder and major depression: a study of complexin mRNAs. MolPsychiatry 2000; 5:425–432.
Ch 07 7/4/05 3:44 pm Page 66

Is any of this real? The word from the grave 67
13. Vogt BA, Nimchinsky EA, Vogt LJ, Hof PR, Human cingulate cortex: surface fea-tures, flat maps, and cytoarchitecture. J Comp Neurol 1995; 359:490–506.
14. Paus T, Primate anterior cingulate cortex: where motor control, drive and cog-nition interface. Nat Rev Neurosci 2002; 2:417–424.
15. Gittins RM, Harrison PJ, A quantitative morphometric study of the humananterior cingulate cortex. Brain Res 2004; 1013:212–222.
16. Manji HK, Moore GJ, Chen G. Clinical and preclinical evidence for the neu-rotrophic effects of mood stabilizers: implications for the pathophysiology andtreatment of manic-depressive illness. Biol Psychiatry 2000; 48:740–754.
17. Eastwood SL, Harrison PJ, Synaptic pathology in the anterior cingulate cortexin schizophrenia and mood disorders: a review and a Western blot study ofsynaptophysin, GAP-43 and the complexins. Brain Res Bull 2001; 55:569–578.
18. Tkachev D, Mimmack M, Ryan MM et al, Oligodendrocyte dysfunction in schiz-ophrenia and bipolar disorder. Lancet 2003; 362:798–804.
19. Harrison PJ, Owen MJ, Genes for schizophrenia? Recent findings and theirpathophysiological implications. Lancet 361:417–419.
20. Harrison PJ, Weinberger DR, Schizophrenia genes, gene expression and neu-ropathology: on the matter of their convergence. Mol Psychiatry 2005;10:40–68.
21. Thomas AJ, O’Brien JT, Davis S et al, Ischemic basis for deep white matterhyperintensities in major depression – a neuropathological study. Arch GenPsychiatry 2002; 59:785–792.
Ch 07 7/4/05 3:44 pm Page 67

Ch 07 7/4/05 3:44 pm Page 68

How can bipolar disorderbe genetically relatedto both schizophreniaand unipolar depression?Peter McGuffin
c h a p t e r 8
Familial overlap
This chapter addresses the seemingly complicated issue of how bipolar dis-order (BPD) can be genetically related to both schizophrenia and unipolardepression (UPD). The least controversial aspect of this question concernsthe overlap between UPD and BPD which, until the publication of a familystudy independently in the same year, 1966, by Angst1 and Perris andD’Elia2 were usually lumped together. In Table 8.1 the family study resultspublished over approximately the following 20 years are summarized.These show a fairly consistent pattern which is that, if the starting point isa proband or index case with bipolar disorder, there is an increase in the
Table 8.1 Affective disorder in first-degree relatives of bipolar and unipolar
probands (data from studies reviewed by McGuffin and Katz, 19863)
Relatives
Proband Number Age-corrected*
type of studies n at risk Bipolar Unipolar
Bipolar 12 3710 7.8 (1.5–17.9) —
3648 — 11.4 (0.5–22.4)
Unipolar 7 2319 0.6 (0.3–2.1) 9.1 (5.9–18.4)
*Corrected denominator (Bezugsziffer) to allow for relatives who have not lived through
the period of risk. †Weighted means.
Morbid risk† (range): %
Ch 08 7/4/05 3:44 pm Page 69

70 Bipolar disorder: the upswing in research & treatment
frequency of both UPD and BPD among relatives, whereas in families ascer-tained via a UPD proband there was an excess only of UPD.
This pattern of findings was influential in convincing most researchersand clinicians that UPD and BPD were to some extent aetiologically differ-ent. Subsequently, in the 1980s, some groups began to take a more adven-turous approach to family studies and began looking at the overlap withother disorders, particularly schizophrenia and schizoaffective disorder.For example, Gershon et al4 found that, in the relatives of patients withschizoaffective disorder, but not in the relatives of patients with schizo-phrenia, there was an increase in BPD. They also found that the incidenceof major depressive disorder was increased in the relatives of both types ofproband.
Nevertheless, the received wisdom from twin studies was that pairs ofidentical individuals always show the same phenotype if they are concor-dant for psychosis.5 It was therefore a surprise to my colleagues and me tocome across a set of triplets (the ‘Z.’ triplets) where the proband was listedon the Maudsley twin register with a diagnosis of manic depression or BPDwhereas his two identical triplets had been diagnosed elsewhere as suffer-ing from schizophrenia.6 We therefore set out to test the hypothesis that thiscould be explained by diagnostic error. We did this both by interviewing thetriplets using two separate research interviews, the Present StateExamination7 and the Schedule for Affective Disorders and Schizophrenia,8
and by obtaining ‘blind’ diagnostic assessments from three distinguishedexperts. The blind assessments were made from case abstracts of each ofthe triplets’ histories and description of mental state from which all
Figure 8.1 A diagnostic hierarchy?
Schizophrenia
Organic Dx
Bipolar Dx
Unipolar Dx
Other non-psychotic Dx
Ch 08 7/4/05 3:44 pm Page 70

Genetic relationship to both schizophrenia and unipolar depression 71
identifying information, including current age, had been removed. Theabstracts were presented to the assessors along with case abstracts basedon three completely unrelated ‘decoy’ patients. To our surprise, bothapproaches, the expert panel judgements and the diagnoses from theresearch interviews, led to the triplets’ hospital diagnoses being confirmed.Furthermore, blood testing looking at several genetic polymorphisms madethe probability very low that the triplets were not monozygotic. We conclud-ed that, although genetically identical individuals such as the Z. triplets whohad non-identical psychoses may be rare, we had effectively performed a‘black swan’ observation, refuting the orthodox Kraepelinian idea that BPDand schizophrenia are totally distinct at the genetic level. (The philosopherof science Karl Popper once proposed that the way to test the hypothesis ‘allswans are white’ is to search for a black swan.)
Explanatory hypotheses
If we accept that at a familial and perhaps at a genetic level there is a degreeof overlap between schizophrenia, BPD and UPD, how can we explain theobserved patterns? The simplest hypothesis is that we are dealing not withseveral but with a single disorder – a ‘unitary psychosis’. A variant on thistheme is that there is a continuum of psychosis with schizophrenia at oneend and unipolar depression at the other. Neither of these hypotheses workswell, either in the sense of explaining the observed data in a way that bringsnew insights, or of appealing to practical clinical commonsense. I will there-fore concentrate on two more interesting hypotheses that are amenable totesting and refutation. These are the notion of a diagnostic hierarchy andthe alternative notion of overlapping sets of polygenes. The idea of a diag-nostic hierarchy has been highly influential in modern psychiatric classifi-cation schemes. Although the authors of current systems such as DSMIV9
have been at pains to produce diagnostic categories that are ‘atheoretical’,all of the diagnostic criteria in DSMIV contain exclusion clauses that areimplicitly based on a diagnostic hierarchy, principally that shown in Figure8.1. Here, disorders resulting from a demonstrable brain lesion or meta-bolic imbalance take precedence over any ‘functional’ disorders, and func-tional disorders are then ranked with schizophrenia at the top, followed bybipolar disorder, unipolar disorder and then other forms of illness.
Translating such a model into genetic terms is fairly straightforward ifwe invoke the notion of thresholds on a continuum of liability. The simplesingle threshold model is illustrated in Figure 8.2.
Ch 08 7/4/05 3:44 pm Page 71

72 Bipolar disorder: the upswing in research & treatment
Here it is assumed that liability to develop a disorder is continuouslydistributed in the population and contributed to by multiple factors, bothgenetic and environmental, such that it will tend to have a normal distribu-tion. Only those individuals who at some point exceed the threshold areclassed as affected. When the disorder is familial, relatives of affectedprobands have an increased mean liability such that more of them liebeyond the threshold than occurs in the general population.10 This can beextended to multiple thresholds by considering broader versus narrowerforms of illness with probands who have the narrow and more extremeforms tending to have more relatives affected than probands with broaderforms.11 This is shown diagrammatically in Figure 8.3.
Such a model has been fitted to family data by Gershon et al12 where itwas hypothesized that the disorder beyond the most extreme threshold isschizoaffective disorder, with bipolar I, bipolar II and unipolar disorderlying beyond successively broader thresholds. It was found that the modelexplained the data satisfactorily. However, the main problem with such anapproach is that a large family dataset may be required in order to rejectthe model. Twin data can sometimes provide a more stringent test.McGuffin et al13 applied a two-threshold model, in which bipolar disorderwas the narrow form of illness and unipolar disorder the broad form, to
Figure 8.2 The polygenic liability-threshold model.
Affected
Liability
Population
Relatives
Ch 08 7/4/05 3:45 pm Page 72

Genetic relationship to both schizophrenia and unipolar depression 73
twin data consisting of 67 pairs ascertained through bipolar probands and177 pairs ascertained through unipolar probands. They were able to rejectconclusively a model where the two forms for affective disorder are on thesame continuum of liability. They then went on to fit a rather different kindof model, testing the extent to which liability to the two forms of disorderhave correlated genetic and environmental bases. They concluded that thegenetic component of UPD and BPD is substantially correlated but thatmost of the genetic variation in liability to the manic syndrome is specific tomania.
Fitting such a model requires a departure from conventional nosology, inthat subjects who have had both manic and depressive symptoms are clas-sified as having two syndromes, whereas subjects who have had only manicor only depressive symptoms are classified as having a single syndrome.The approach has been applied also to schizophrenia, schizoaffective dis-order and bipolar disorder in a twin study14 where the usual hierarchicalrules were suspended and subjects were classed as having one, two or allthree disorders. The principal finding was that there is a set of genes thatcontribute to all three syndromes and specific genes that contribute only tobipolar disorder or to schizophrenia. Interestingly, there was no evidence ofa specific set of genes contributing to schizoaffective disorder, but there wasevidence of a specific environmental effect. These findings have been viewedas somewhat controversial and a commentary in the same issue of the jour-nal suggested that the authors’ interpretation that there are overlapping setsof genes contributing to both sides of the Kraepelinian dichotomy was incor-rect.15 However, there is now emerging evidence from molecular studies thatat least some of the same genes do indeed contribute to both schizophreniaand bipolar disorder.
Figure 8.3 A multiple threshold model.
Relatives affected++
Relatives affected+++
Relatives affected +
Ch 08 7/4/05 3:45 pm Page 73

74 Bipolar disorder: the upswing in research & treatment
The molecular findings
Modern gene finding studies are beginning to yield useful results in bothschizophrenia and affective disorders.16 The positional cloning approachhas been described in greater detail elsewhere in this volume (Chapter 9).Essentially positional cloning consists of searching through the genome, the23 pairs of chromosomes, using linkage analysis, narrowing down theregion of interest by linkage disequilibrium mapping and then findingpotential candidates within the refined region with which further associa-tion studies are performed to confirm or reject the hypothesis of a role inthe disorder. The starting point then is linkage, and two major sets of meta-analyses have recently been performed in order to try to make sense of thelarge body of data that now exists. One such meta-analysis17 implicated tworegions on chromosomes 13q and 22q in both schizophrenia and bipolardisorder and a third region on chromosome 8 that was specific to schizo-phrenia.
Subsequently there has been much interest in candidate genes withinthese regions. A search across the 13q region in schizophrenia resulted inthe identification of a novel primate-specific gene known as G72, the geneproduct of which was found to interact in vitro, using a method called yeast2 hybrid analysis, with D-amino acid oxidase (DAAO).18 There was also sta-tistical evidence of epistasis (gene–gene interaction) between G72 and theDAAO gene. There have now been several studies supporting the associationbetween G72 and schizophrenia19–21 but there have also been several stud-ies that have found an association between G72 and bipolar disorder.20,22,23
So far, a number of other positional candidates have been implicated onlyin schizophrenia,16 but the prediction that follows on from the twin analy-sis described earlier is that future molecular studies will uncover sets ofgenes that contribute either to schizophrenia or to bipolar disorder andgene sets contributing to both syndromes.
Conclusions
Modern genetic evidence does not support the idea that schizophrenia andthe affective disorders are completely distinct at an aetiological level.Neither does it support the idea that there is a ‘continuum’ of psychosis withschizophrenia at one end and BPD at the other. Rather, there appear to beoverlapping gene sets. That is, there are genes that contribute both to schiz-ophrenia and to BPD, and genes that contribute only to one or other
Ch 08 7/4/05 3:45 pm Page 74

Genetic relationship to both schizophrenia and unipolar depression 75
syndrome. BPD also shows genetic overlap with UPD but a simple hierar-chical, or two-threshold model, where BPD is a more extreme and severeform on a single continuum of liability does not explain the available twindata. There does appear to be a sizeable overlap between the genes con-tributing to unipolar and bipolar disorders, but most of the genetic liabilityto the manic syndrome is specific to mania. This is a result that, as Beardenet al24 have pointed out in a recent review, has major implications for gene-finding studies.
References
1. Angst J, Zur aetilogie und nosologie endogener depressiver psychosen.Monographen ans der Neurologie und Psychiatrie, N112. Springer: Berlin,1966.
2. Perris C, D’Elia G, A study of bipolar (manic-depressive) and unipolar recur-rent depressive psychoses X: mortality, suicide, and life cycles. Acta PsychiatrScand 1966; 42(194 Suppl):172–183.
3. McGuffin P, Katz R, Nature, nurture and affective disorders. In: Deakin JW (ed),The Biology of Depression. Gaskell Press: London, 1986:26–51.
4. Gershon ES, Delisi LE, Hamovit J et al, A controlled family study of chronicpsychoses. Schizophrenia and schizoaffective disorder. Arch Gen Psychiatry1988; 45:328–336.
5. Gottesman II, Shields J, Schizophrenia – the Epigenetic Puzzle. CambridgeUniversity Press: Cambridge, 1982.
6. McGuffin P, Reveley A, Holland A, Identical triplets: non-identical psychosis? BrJ Psychiatry 1982; 140:1–6.
7. Wing JK, Cooper JE, Sartorius N, The Measurement and Classification ofPsychiatric Symptoms. Cambridge University Press: Cambridge, 1974.
8. Endicott J, Spitzer RL, A diagnostic interview: the schedule for affective disor-ders and schizophrenia. Arch Gen Psychiatry 1978; 35:837–844.
9. American Psychiatric Association. Diagnostic and Statistical Manual ofMental Disorders, Fourth Edition (DSMIV). American Psychiatric AssociationPress; Washington DC, 1994.
10. Falconer DS, The inheritance of liability to certain diseases, estimated from theincidence among relatives. Ann Hum Genet 1965; 29:51–76.
11. Reich T, James JW, Morris CA, The use of multiple thresholds in determiningthe mode of transmission of semi-continuous traits. Ann Hum Genet 1972;36:163–184.
12. Gershon ES, Hamovit J, Guroff JJ et al, A family study of schizoaffective bipo-lar I, bipolar II, unipolar, and normal control probands. Arch Gen Psychiatry1982; 39:1157–1167.
13. McGuffin P, Rijsdijk F, Andrew M et al, The heritability of bipolar affective dis-order and the genetic relationship to unipolar depression. Arch Gen Psychiatry2003; 60:497–502.
Ch 08 7/4/05 3:45 pm Page 75

76 Bipolar disorder: the upswing in research & treatment
14. Cardno AG, Rijsdijk FV, Sham PC et al, A twin study of genetic relationshipsbetween psychotic symptoms. Am J Psychiatry 2002; 159:539–545.
15. Kendler KS, Hierarchy and heritability: the role of diagnosis and modeling inpsychiatric genetics. Am J Psychiatry 2002; 159:515–518.
16. Elkin A, Kalidindi S, McGuffin P, Have schizophrenia genes been found? CurrOpin Psychiatry 2004; 17:107–113.
17. Badner JA, Gershon ES, Meta-analysis of whole-genome linkage scans of bipo-lar disorder and schizophrenia. Mol Psychiatry 2002; 7:405–411.
18. Chumakov I, Blumenfield M, Guerassimenko O et al, Genetic and physiologicaldata implicating the new human gene G72 and the gene for D-amino acid oxi-dase in schizophrenia. Proc Natl Acad Sci USA 2002; 99:13675–13680.
19. Wang X, He G, Gu N et al, Association of G72/G30 with schizophrenia in theChinese population. Biochem Biophys Res Commun 2004; 319:1281–1286.
20. Schumacher J, Jamra RA, Freudenberg J et al, Examination of G72 and D-amino-acid oxidase as genetic risk factors for schizophrenia and bipolar affec-tive disorder. Mol Psychiatry 2004; 9:203–207.
21. Addington AM, Gornick M, Sporn AL et al, Polymorphisms in the 13q33.2 geneG72/G30 are associated with childhood-onset schizophrenia and psychosis nototherwise specified. Biol Psychiatry 2004; 55:976–980.
22. Chen YS, Akula N, Detera-Wadleigh SD et al, Findings in an independent sam-ple support an association between bipolar affective disorder and the G72/G30locus on chromosome 13q33. Mol Psychiatry 2004; 9:87–92.
23. Hattori E, Liu C, Badner JA et al, Polymorphisms at the G72/G30 gene locus,on 13q33, are associated with bipolar disorder in two independent pedigreeseries. Am J Hum Genet 2003; 72:1131–1140.
24. Bearden CE, Reus VI, Freimer NB, Why genetic investigation of psychiatric dis-orders is so difficult. Curr Opin Genet Dev 2004; 14:280–286.
Ch 08 7/4/05 3:45 pm Page 76

Recent advancesin genetics ofbipolar disorderDaniel J Müller and James L Kennedy
c h a p t e r 9
Introduction
It has long been recognized that manic depression or bipolar disorder (BD)runs in families and therefore a familial and/or genetic component in theaetiology of BD has been postulated. Epidemiological research involvingfamily, twin and adoption studies led to the observation that genetic factorsdo confer susceptibility to BD.1 The overall prevalence of BD appears to beabout 1% in the general population. In contrast, first-degree relatives havea 5–10-fold increased risk of developing BD, while it appears that the riskfor second-degree relatives falls between risks for first-degree relatives andthe general population. Concordance for BD has consistently been found tobe significantly higher in monozygotic than in dizygotic twins (about 50%versus 10%, which is similar to the rate for first-degree relatives).1,2 Thesefindings point to some important conclusions: the clustering of BD in fam-ilies is based on the presence of genetic factors; however, since penetranceis incomplete, the presence of specific non-genetic (epigenetic and/or envi-ronmental) factors is likely to influence the occurrence of BD.3 Finally, it isgenerally accepted, based on statistical models, that not one but many (orat least several) genes of moderate or small effect contribute to BD.4
From symptoms to syndromes: the pitfalls ofphenotype definition
The recognition and distinction of manic symptoms as pathognomonic fea-tures of BD (e.g. elevated mood, pressured speech, grandiose delusions) canusually be achieved with high reliability among clinicians. However, as with
Ch 09 7/4/05 3:46 pm Page 77

78 Bipolar disorder: the upswing in research & treatment
many other complex disorders, the clinical presentation of BD itself is high-ly heterogeneous. This heterogeneity is likely to be caused by a variety ofspecific genes that may interact with neuronal circuitries or synapticprocesses. It is likely that only through the combination of different genes isa threshold reached and disorder-related symptoms appear. It remains amatter of debate whether affective and psychotic symptoms may representa continuum that ranges from dysthymia through depression, bipolar II andbipolar I towards schizoaffective disorder and schizophrenia.5 Therefore,and since genes ‘do not read’ DSM-IV or ICD-10, it seems plausible thatgenetic studies of BD must take into account the heterogeneity of BD andshould focus on core symptoms that are known to be ‘familial’ or genetical-ly determined. However, only recently have researchers begun to focus onsymptoms, endophenotypes (such as imaging or electrophysiological tech-niques) or to incorporate covariates in their analyses with some promisingpreliminary findings. Response pattern to mood stabilizers is anotherpromising clinical measure to refine the phenotype of patients with BD.6
How to find genes involved in bipolar disorder:principles of genetic linkage and association studies
It should be emphasized here that genetic mechanisms in the aetiology ofBD are poorly understood. Whether or not the susceptibility is associatedwith DNA sequence variation alone or in combination with epigenetic mech-anisms (i.e. gene expression controlled by potentially reversible changes inDNA methylation and/or chromatin structure3) remains unknown at pres-ent. Nonetheless, the first step is to analyse genomic DNA sequence varia-tion patterns in subjects affected with BD compared with non-affected indi-viduals. In Mendelian disorders, distinct genetic variations have alreadybeen proven to be causative for the occurrence of a given disease. Therefore,it is reasonable to hypothesize that distinct genetic variants will increase thesusceptibility to a complex disorder such as BD. However, genes that areinvolved in the aetiology of BD remain unknown and facts are more com-plicated when one considers that BD risk conferring sequence variation mayoccur anywhere in the genomic DNA (e.g. in promoter regions, in intronicand/or exonic regions or in intragenic regions). Finally, sequence variationsmay be harmless (in terms of increasing disease risk) but may also affectgene expression, protein structure, or mRNA stability.7 The functional rele-vance of any sequence variation needs to be established in further analyses.
Ch 09 7/4/05 3:46 pm Page 78

Recent advances in genetics of bipolar disorder 79
Two complementary approaches are widely applied in medical geneticsto identify genes involved in the aetiology of a given disorder: linkage analy-sis and genetic association studies.
Linkage analyses are based on the principle that two loci are linked ifthey are close to each other and are co-inherited, meaning that they are nottransmitted independently. Thus, in linkage analyses two chromosomal lociare considered: one that may harbour the unknown locus (or putative gene)and one that is a marker trait for which the site is known. Typically, largefamily pedigrees are chosen for linkage studies using trait markers span-ning the whole genome. Subsequently, analyses seek to determine thosemarkers that are typically inherited (and are ‘linked’) to the disorder.Finally, conclusions can be drawn that indicate the broader location of sus-ceptibility genes. Statistical analyses are divided into parametric and non-parametric linkage analyses, depending upon whether parameters (such asmode of inheritance, level of penetrance) are known or included alterna-tively based on a priori hypotheses. Furthermore, analyses may be per-formed considering two markers or multiple markers (i.e. two point or mul-tipoint analyses). Estimates of significance levels are generally given in logof the odds ratio (LOD) scores (for parametric analyses) or NPL scores (fornon-parametric analyses).8,9 The LOD score is defined by the probabilitythat the locus is linked to disease divided by the probability that the locusis not linked to disease given the observed data. Specific guidelines havebeen proposed on how to interpret or classify LOD scores.10 Based on thesecriteria, LOD scores above 2.2 are suggestive of significant findings, where-as a LOD score above 3.6 will establish significance of findings obtained inlinkage analyses. However, interpretation is more complex, as individualresults need to be put into the context of findings obtained from independ-ent studies. Therefore, LOD scores above 1.0 are generally reported in link-age studies, as such scores may receive importance in the presence of sim-ilar (or higher) LOD scores from independent studies.
Genetic association studies typically compare DNA sequence variationsbetween unrelated cases and controls. Controls can either be recruited froma healthy population (i.e. not being affected by the disorder; this is alsocalled case–control design) or from healthy family members (so-calledfamily-based design). The latter approach has the advantage that familymember controls are not prone to population stratification, meaning thatfalse-positive findings are less likely to be due to allelic heterogeneity adher-ent in specific populations. Association studies can either be performedwith candidate genes that have been proved (or have been supposed) to playan aetiological role in a given disorder (and are called functional candidate
Ch 09 7/4/05 3:46 pm Page 79

80 Bipolar disorder: the upswing in research & treatment
genes) or may be chosen on the basis of their specific location that hasemerged from linkage studies (and are called positional candidate genes).11
Problems with linkage and association studies derive from the commonlyaccepted assumption that genetic susceptibility to BD is heterogeneous interms of loci and alleles. Locus heterogeneity means a disorder will developif a combination of genes in locus A and/or B (and/or C, etc.) confers sus-ceptibility to BD. Thus, locus heterogeneity will cause problems (in terms ofdifficulties of replicating initial findings) in linkage studies. Allelic hetero-geneity means that different polymorphisms in one gene will confer suscep-tibility to BD, comparable to the scenario wherein many different variantsof one gene all cause cystic fibrosis. Hence, allelic heterogeneity will hamperreplication findings in allelic association studies.
It should be noted that linkage and association studies are not juxta-posed strategies but are to be seen as complementary approaches. Once achromosomal locus has been identified, this region will ideally be narroweddown with more markers towards the highest signals. Once that ‘hot spot’site with highest linkage signals has been identified, subsequent case–control studies should be carried out to identify those specific genetic vari-ations that confer susceptibility to BD. In the presence of only few com-pelling candidate genes, these positional approaches have been suggested torepresent the most powerful strategies in genetic studies of BD.12
Other genetic mechanisms that could be involved in the aetiology of BDinclude some types of chromosomal aberrations, dynamic mutations(expanded trinucleotide repeat sequences), or mitochondrial mutations.The latter are not likely to be involved in the aetiology of BD, although thiscannot be excluded at this point.13,14 Finally, the phenomenon of epistasis(gene–gene interactions) currently receives much attention. However, thereare no widely accepted statistical models for epistasis, although this field israpidly progressing.
Linkage studies in bipolar disorder
Linkage analyses from numerous genome scans have yielded several regionsthat may harbour susceptibility genes for bipolar disorder. Not surprising-ly, results arising out of these studies are hampered by inconsistent find-ings, as comparisons among study findings remain complicated, owing tomultiple study heterogeneities (e.g. sample sizes, ascertainment strategies,molecular genetic techniques and so on). The most interesting findings,indicated either by highly significant findings in single studies or through
Ch 09 7/4/05 3:46 pm Page 80

Recent advances in genetics of bipolar disorder 81
replication of initial findings, include chromosomal regions 1q31-q32,
4p15-16, 6pter-p24, 8q24, 10p13, 10p14, 10q21-26, 12q23-24.1,
13q31-32, 16p13, 18p11.2, 18q12, 18q21-23, 20p11.2-q11.2, 21q22,
22q11-q13 and Xq24-28.5,8,14,15 It is interesting to note that linkage analy-
sis on chromosome 18 indicated a parent-of-origin effect, implying complex
mechanisms such as mitochondrial mutations or genomic imprinting to be
involved in the aetiology of BD.16–19
Most of the genome scans that have been performed were included in
two recent large meta-analyses.20,21 Badner and Gershon21 found the
strongest evidence for linkage for chromosomes 13q and 22q. Depending
on their disease models (i.e. including patients with bipolar II and major
depression), Segurado et al20 found most significant results on chromo-
somes 8q, 9p22.3-21.1; 10q11.21-22.1, 14q24.1-32.12 and 18p-q. How-
ever, none of these regions achieved genomewide statistical significance.
Reasons for inconsistent findings among the analyses may be due to
methodological factors that varied considerably in terms of dataset selec-
tion, disease modelling, statistical analyses, etc. Furthermore, as pointed
out by Segurado et al,20 negative findings do not disprove linkage and there-
fore more efforts are needed to identify BD susceptibility loci in the future.
Moreover, it should be noted that these chromosomal regions represent mil-
lions of base-pairs. On average, a new genetic polymorphism occurs once
every thousand base-pairs. Thus, hundreds of DNA markers are typically
needed to narrow down the region of interest to the true risk-conferring
gene variant. Such investigations are usually laborious, costly and time con-
suming. However, these efforts are currently underway and are likely to
unearth new genes associated with BD.
Candidate gene association studies in bipolardisorder
Studies focusing on particular genes using case–control association studies
have yielded some positive findings, but also negative (non-replication) find-
ings. Thus, the significance of specific results needs to be weighed in light
of several important parameters in order to estimate their true relevance. It
remains difficult to depict algorithms (or guidelines) as to how to interpret
findings arising from association studies. Nonetheless, each study should at
least address some important questions:
Ch 09 7/4/05 3:46 pm Page 81

82 Bipolar disorder: the upswing in research & treatment
1. Sample size (of cases and controls): Are samples large enough interms of statistical power? (Discussed in more detail by reference 22).
2. Ascertainment: Were samples selected in ethnically homogeneous pop-ulations? Were samples ascertained randomly or systematically? Werediagnoses, symptoms and course of disorder all assessed by inter-viewers trained to conduct clinical (semi-) structured interviews andchart assessments reliably?
3. Selection of the candidate gene: How has the gene been selected (func-tional or positional candidate gene)? Are there any (reasonable)assumptions or findings emerging from neurobiological research pro-viding at least some evidence that the gene in question is involved inthe pathophysiology of BD? Are there any supportive findings derivingfrom linkage analyses to pinpoint the particular gene as a positionalcandidate gene?
4. Gene structure/selection of genetic polymorphisms: Has the genealready been investigated in terms of its structure (e.g. promoter,intron/exon boundaries, etc.)? Is there any evidence that the polymor-phism itself bears functional relevance (e.g. modulation of gene expres-sion)? Are there polymorphisms with minor allele frequencies highenough for statistical analyses?
5. Genotyping: High-quality assurance needs to be used in terms of tech-nological equipment and data management (i.e. supervision, cross-checking, etc.)
6. Statistical analyses: Depending on the number of gene variants thatwere chosen for the analyses, appropriate strategies and tests have tobe chosen carefully (e.g. haplotype analyses, problems of multiple test-ing, etc.)
7. Initial (positive) finding – or replication? (Rule of thumb: An initialpositive finding is interesting, but only further replications will deter-mine the true ‘significance’.)
The observer of molecular genetic studies in BD will quickly realize thatmost studies suffer from at least some methodological flaws, althoughnumerous candidate gene studies have been performed. Most of our currentknowledge on genetics of BD is hypothesis-based and some importantaspects remain yet to be determined. For example, the pathophysiology ofBD remains largely unknown. Hence, a functional candidate cannot bedetermined with certainty, unless each of the perhaps 30 000 human genesare to be considered as candidate genes. Moreover, the functional relevanceof a genetic polymorphism is often impossible to determine; if a polymor-phism is found to be functionally relevant in vitro, this is not necessarily
Ch 09 7/4/05 3:46 pm Page 82

Recent advances in genetics of bipolar disorder 83
true in vivo. Conversely, if a test indicates that a gene polymorphism is notfunctionally relevant in vitro, this may not be so in vivo – or may be due tolack of sensitivity of the given test.
Neurotransmitter systems
Based on current knowledge in psychopharmacology, most associationstudies focused on a priori functional candidate genes that are related to themechanism of action of monoamine neurotransmitters such as serotonin,noradrenaline (norepinephrine) or dopamine. Polymorphisms of thecatechol-o-methyl transferase (COMT) gene, monoamine oxidase A (MAOA)gene and the serotonin transporter (5-HTT) gene revealed most interestingresults even though these findings could not be replicated in all studies (e.g.references 11, 23–32). These findings are most consistent with the notionthat these genes are likely to play a minor role (i.e. with odds ratios <2) andmay perhaps be associated with yet undefined subgroups of patients affect-ed with BD.
In the dopamine system, the genetic mechanism of imprinting was sug-gested in the aetiology of BD by Muglia et al,33 who found evidence of a riskeffect of functional variable number of tandem repeats (VNTRs) in exon IIIof the DRD4 receptor gene. In that report, the risk was transmitted throughmaternal, but not paternal, transmission.
G72/G30 locus (and DAAO) and brain-derived neurotrophic factor
A large variety of functional and positional candidate genes have beenanalysed for association with BD over the years. Here we focus on two can-didate genes that we believe preliminary studies suggest consistently inter-esting findings.
G72/G30 locus and DAAO
Chumakov et al34 identified the gene G72 on chromosome 13q33.2, a regionthat has previously been found to be linked with schizophrenia and BD insome studies. G72 overlaps with another gene, G30 and therefore this locuswas named the G72/G30 locus. Chumakov et al34 demonstrated that theG72 protein interacts as activator on the D-amino acid oxidase (DAAO)enzyme. In turn, DAAO oxidizes D-serine, an activator of N-methyl-D-aspartate (NMDA)-type glutamate receptors. Modulation of NMDA receptorsmay be involved in the aetiology of schizophrenia.35,36 Thus, the G72/G30locus may be regarded as a positional and functional candidate gene for
Ch 09 7/4/05 3:46 pm Page 83

84 Bipolar disorder: the upswing in research & treatment
schizophrenia and bipolar disorder. A shared genetic susceptibility betweenBD and schizophrenia has been suggested for some time.37 In their initialstudy, Chumakov et al34 identified polymorphisms of the G72 gene associ-ated with schizophrenia without including patients with BD. In contrast,Hattori et al38 included patients with BD and found significant associationswith markers at and around the G72/G30 locus. Importantly, similar sig-nificant findings were found independently at the G72/G30 locus in patientswith BD in subsequent studies by Chen et al39 and Schumacher et al.40 Eventhough findings varied with respect to specific DNA markers and statisticalresults among these studies, the results relating to the G72/G30 locusshould be regarded as an important development in the search for suscep-tibility genes for BD. Nonetheless, further studies are clearly needed.
Brain-derived neurotrophic factor
The brain-derived neurotrophic factor (BDNF) gene is located on chromo-some 11p14.1.41 One study by Detera-Wadleigh et al42 identified moderatepairwise parametric LOD scores of 1.84 at marker D11S915 (11p15-p14)and of 1.62 at marker D11S904 (11p14-p13) under the assumption of adominant model of inheritance with 50% penetrance. The affection statusmodel in this study was restricted to bipolar I, bipolar II (with major depres-sion) and schizoaffective disorder. However, non-parametric multipointlinkage analyses revealed no comparable findings in the same study.Besides that study, one other recent study by Zandi et al43 detected a signalof highest linkage on chromosome 11p15.5. Nonetheless, there is com-pelling evidence that BDNF is involved in a variety of complex mechanismsthat are thought to be relevant in BD. The mechanism of action of BDNF ismultifunctional and complex44 and this paragraph will only provide a shortsummary. BDNF is a neuronal growth factor that was found to be critical forthe normal development of central 5HT neurons in mice. For example,BDNF-deficient mice display an aggressive hyperphagic phenotype;45 symp-toms that are observed in mood disorders and have long been hypothesizedto be associated with serotonin dysfunction. Serotonin dysfunction is likelyto play a major role in depression, anxiety and suicidal behaviour, which arecommon symptoms in BD. An increased BDNF expression has beenobserved in the frontal cortex and hippocampus after treatment with anti-depressants, lithium, or valproic acid in rats.46 In human postmortem stud-ies, increased hippocampal BDNF immunoreactivity was found in patientstreated with antidepressant medications.47 In contrast, decreased levels ofBDNF mRNA and protein in postmortem frontal cortex and hippocampuswere detected in suicide completers versus controls.48 Serum levels of
Ch 09 7/4/05 3:46 pm Page 84

Recent advances in genetics of bipolar disorder 85
BDNF were found to increase significantly after antidepressant medicationwas administered in patients with major depression compared with untreat-ed patients with major depression or controls. Moreover, a significant neg-ative correlation between serum BDNF and Hamilton Rating Scale forDepression (HAM-D) scores was noted in patients.49 In summary, thesefindings clearly suggest that modulation of BDNF is associated with symp-toms pertaining to depression and/or BD. Moreover, symptom-relievingdrugs might act by modulating (i.e. increasing) BDNF expression or BDNFdistribution.
The BDNF gene is relatively large (approximately 67kb) with seven prob-able promoters and fifteen alternatively spliced exons producing 11 differ-ent transcripts creating five variations of the BDNF protein (NCBI Aceview).The BDNF gene structure is thus complex and the gene-map has been mod-ified over the course of time.44,50,51 The map we present here (Figure 9.1)was constructed from the primer sequences used in our ongoing studiesand exon sequence given on NCBI Aceview.
The most prominent polymorphism that has been studied extensively is
a G to A substitution that results in an amino acid substitution (valine to
methionine) in codon 66 (named Val66Met polymorphism). The Val66Met
polymorphism is found in alternatively spliced exons 13 through 15 (previ-
ously called exon 11,44 exon 6,50 or exon 551) one of which is contained in
Figure 9.1 Map of brain-derived neurotrophic factor (BDNF) gene with location ofVal66Met and GT(n) repeat polymorphisms.
BDNF located on (–) strand
(+) strand direction
(GT)n rep
eat
val66met (rs6265) A
(Met)/G
(val)
Exon: 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15
Ch 09 7/4/05 3:46 pm Page 85

86 Bipolar disorder: the upswing in research & treatment
all BDNF transcript variants. This polymorphism has been proven to be of
functional relevance since in cultured hippocampal neurons the Val allele
(vBDNF) has been shown to increase intraneuronal BDNF peptide secretion
and distribution compared to the Met allele (mBDNF).52 Using fluorescent
microscopy techniques, neurons expressing vBDNF were shown to express
BDNF in the cell body and distal processes (dendrites). In contrast, mBDNF
was mainly localized in cell bodies. Furthermore, it appeared that neurons
expressing mBNDF displayed an impaired (i.e. decreased) secretion of
BDNF (see Figure 4 A–C in reference 52).
The Val allele was found to be associated with BD in two independent
studies simultaneously.53,54 In the study of Neves-Perreira et al,53 an intron-
ic polymorphism, a GT(n) repeat upstream of the Val66Met polymorphism
was found to be associated with BD. Although these studies varied in
methodology55 they both included large samples of similar ethnic groups
and used a family-based association approach. In another family-based
association study, by Geller et al,56 a significant association was found with
the Val allele and BD in children with a prepubertal and early adolescent BD
phenotype. Negative findings used case–control designs and samples
derived from other ethnic backgrounds.50,57–60 However, in a recent study
using a case–control design and larger samples of patients with major
depression and BD, significant associations were detected with three mark-
er haplotypes including the Val66Met polymorphism.61 One study, by
Strauss et al,62 reported a negative finding with the Val66Met polymorphism
in adults with childhood-onset mood disorders, but a significant association
with the GT(n) repeat and haplotype containing the Val allele.
Inconsistent findings among these studies indicate that BDNF apparent-
ly plays a role but not in all patients affected with BD; more likely to be in
specific – yet undefined – subgroups of patients. It has long been recognized
that BD represents a heterogeneous disorder in terms of clinical and/or epi-
demiological characteristics (e.g. sex, age at onset, psychotic symptoms,
course of disorder, response pattern to medication, etc.). Preliminary find-
ings in our follow-up studies indicate that our significant findings with the
BDNF gene in BD are mainly driven by those patients who displayed ‘rapid
cycling’ (i.e. the occurrence of four or more mood episodes (major depres-
sive, manic, mixed or hypomanic) within a time frame of 12 months).63
Furthermore, significant findings between the BDNF gene and psychi-
atric disorders other than BD (e.g. schizophrenia,60,64 obsessive–compulsive
disorder,51 adult attention deficit hyperactivity disorder65) indicate that
Ch 09 7/4/05 3:46 pm Page 86

Recent advances in genetics of bipolar disorder 87
BDNF is likely to be associated with a broader range of symptoms that are
encountered in a variety of neuropsychiatric disorders. In conclusion, the
meaningfulness of the BDNF gene in BD needs to be further elucidated by
focusing on specific characteristics adherent to subgroups of BD patients,
and also symptom sharing with other known disorders in neuropsychiatry.
Summary and outlook
There is unequivocal evidence that genetic factors play a major role in the
aetiology of BD. More precisely, we expect that a yet unknown number of
gene variants confer susceptibility to BD that will lead to exacerbation of
manic or depressive episodes in conjunction with environmental factors
such as stress or major life events.
The search for vulnerability genes over the past 20 years has not been as
successful as was initially hoped. This time period has left behind many
inconsistent findings and some false leads. However, rather than looking
back in dismay it is perhaps more important to learn lessons from the past
studies in order to achieve better interpretation of current findings and
address the correct questions in the future. Therefore, we emphasized on
the interpretation of study findings rather than presenting each single locus
or candidate gene that has been studied; understanding the strengths and
weaknesses of genetic studies, as well as integrating genetic results with
neurobiology and clinical sciences is more valuable than simply tabulating
each and every LOD score and p value. The findings in BDNF tell us an
important and demonstrative story. The BDNF gene is most likely not to be
a risk factor for BD per se but a risk factor for BD in certain groups of
patients. Sources of inconsistent findings may be that – aside from the
problem of large sample sizes that are needed to identify genes with small
effects (and most samples were relatively small) – patients ascertained for
family-based studies may differ from patients ascertained for case–control
samples.66 The group of patients may be defined by certain characteristics
(e.g. earlier age at onset) or specific course of disorder (e.g. rapid cycling) or
distinct symptoms (e.g. depressive symptomatology), or a combination of
such features related to BD. Definite answers are likely to be obtained in the
near future, provided that the field is prepared to take into consideration
the lessons learned from the past. We are confident that the detection of
genetic factors underlying BD will become a reality over the coming years.
Ch 09 7/4/05 3:46 pm Page 87

88 Bipolar disorder: the upswing in research & treatment
Acknowledgements
This work was supported by a Canadian Institutes of Health Research
(CIHR) operating grant to JLK, and a CIHR postdoctoral fellowship award
to DJM. Thanks to Matthew Lanktree who assisted in this work.
References
1. Smoller JW, Finn CT, Family, twin, and adoption studies of bipolar disorder.Am J Med Genet C Semin Med Genet 2003; 123:48–58.
2. Belmaker RH, Bipolar disorder. N Engl J Med 2004; 351:476–486.3. Petronis A, Epigenetics and bipolar disorder: new opportunities and chal-
lenges. Am J Med Genet C Semin Med Genet 2003; 123:65–75.4. Kelsoe JR, Arguments for the genetic basis of the bipolar spectrum. J Affect
Disord 2003; 73:183–197.5. Tsuang MT, Taylor L, Faraone SV, An overview of the genetics of psychotic mood
disorders. J Psychiatr Res 2004; 38:3–15.6. Alda M, Pharmacogenetic aspects of bipolar disorder. Pharmacogenomics
2003; 4:35–40.7. Duan J, Wainwright MS, Comeron JM et al, Synonymous mutations in the
human dopamine receptor D2 (DRD2) affect mRNA stability and synthesis ofthe receptor. Hum Mol Genet 2003; 12:205–216.
8. Sklar P, Linkage analysis in psychiatric disorders: the emerging picture. AnnuRev Genomics Hum Genet 2002; 3:371–413.
9. Schulze TG, McMahon FJ, Genetic linkage and association studies in bipolaraffective disorder: a time for optimism. Am J Med Genet C Semin Med Genet2003; 123:36–47.
10. Lander ES, Kruglyak L, Genetic dissection of complex traits: guidelines forinterpreting and reporting linkage results. Nat Genet 1995; 11:241–247.
11. Craddock N, Dave S, Greening J, Association studies of bipolar disorder.Bipolar Disord 2001; 3:284–298.
12. Gershon ES, Kelsoe JR, Kendler KS, Watson JD, A scientific opportunity.Science 2001; 294:957.
13. Fortune MT, Kennedy JL, Vincent JB, Anticipation and CAG*CTG repeat expan-sion in schizophrenia and bipolar affective disorder. Curr Psychiatry Rep2003; 5:145–154.
14. Baron M, Manic-depression genes and the new millennium: poised for discov-ery. Mol Psychiatry 2002; 7:342–358.
15. Cichon S, Schumacher J, Muller DJ et al, A genome screen for genes predis-posing to bipolar affective disorder detects a new susceptibility locus on 8q.Hum Mol Genet 2001; 10:2933–2944.
16. McMahon FJ, Stine OC, Meyers DA et al, Patterns of maternal transmission inbipolar affective disorder. Am J Hum Genet 1995; 56:1277–1286.
Ch 09 7/4/05 3:46 pm Page 88

Recent advances in genetics of bipolar disorder 89
17. Gershon ES, Badner JA, Detera-Wadleigh SD et al, Maternal inheritance andchromosome 18 allele sharing in unilineal bipolar illness pedigrees. Am J MedGenet 1996; 67:202–207.
18. Nothen MM, Cichon S, Rohleder H et al, Evaluation of linkage of bipolar affec-tive disorder to chromosome 18 in a sample of 57 German families. MolPsychiatry 1999; 4:76–84.
19. McInnis MG, Lan TH, Willour VL et al, Genome-wide scan of bipolar disorderin 65 pedigrees: supportive evidence for linkage at 8q24, 18q22, 4q32, 2p12,and 13q12. Mol Psychiatry 2003; 8:288–298.
20. Segurado R, Detera-Wadleigh SD, Levinson DF et al, Genome scan meta-analysis of schizophrenia and bipolar disorder, part III: Bipolar disorder. Am JHum Genet 2003; 73:49–62.
21. Badner JA, Gershon ES, Meta-analysis of whole-genome linkage scans of bipo-lar disorder and schizophrenia. Mol Psychiatry 2002; 7:405–411.
22. Risch N, Merikangas K, The future of genetic studies of complex human dis-eases. Science 1996; 273:1516–1517.
23. Kirov G, Norton N, Jones I et al, A functional polymorphism in the promoter ofmonoamine oxidase A gene and bipolar affective disorder. Int JNeuropsychopharmacol 1999; 2:293–298.
24. Kunugi H, Ishida S, Kato T et al, A functional polymorphism in the promoterregion of monoamine oxidase-A gene and mood disorders. Mol Psychiatry1999; 4:393–395.
25. Preisig M, Bellivier F, Fenton BT et al, Association between bipolar disorder andmonoamine oxidase A gene polymorphisms: results of a multicenter study. AmJ Psychiatry 2000; 157:948–955.
26. Ho LW, Furlong RA, Rubinsztein JS et al, Genetic associations with clinicalcharacteristics in bipolar affective disorder and recurrent unipolar depressivedisorder. Am J Med Genet 2000; 96:36–42.
27. Mundo E, Walker M, Tims H et al, Lack of linkage disequilibrium between sero-tonin transporter protein gene (SLC6A4) and bipolar disorder. Am J MedGenet 2000; 96:379–383.
28. Jones I, Craddock N, Candidate gene studies of bipolar disorder. Ann Med2001; 33:248–256.
29. Rotondo A, Mazzanti C, Dell’Osso L et al, Catechol o-methyltransferase, sero-tonin transporter, and tryptophan hydroxylase gene polymorphisms in bipolardisorder patients with and without comorbid panic disorder. Am J Psychiatry2002; 159:23–29.
30. Gutierrez B, Arias B, Gasto C et al, Association analysis between a functionalpolymorphism in the monoamine oxidase A gene promoter and severe mooddisorders. Psychiatr Genet 2004; 14:203–208.
31. Mendlewicz J, Massat I, Souery D et al, Serotonin transporter 5HTTLPR poly-morphism and affective disorders: no evidence of association in a largeEuropean multicenter study. Eur J Hum Genet 2004; 12:377–382.
32. Lasky-Su JA, Faraone SV, Glatt SJ, Tsuang MT, Meta-analysis of the associationbetween two polymorphisms in the serotonin transporter gene and affective dis-orders. Am J Med Genet B Neuropsyhchiatr Genet 2005; 133:110–115.
Ch 09 7/4/05 3:46 pm Page 89

90 Bipolar disorder: the upswing in research & treatment
33. Muglia P, Petronis A, Mundo E et al, Dopamine D4 receptor and tyrosinehydroxylase genes in bipolar disorder: evidence for a role of DRD4. MolPsychiatry 2002; 7:860–866.
34. Chumakov I, Blumenfeld M, Guerassimenko O et al, Genetic and physiologicaldata implicating the new human gene G72 and the gene for D-amino acid oxi-dase in schizophrenia. Proc Natl Acad Sci USA 2002; 99:13675–13680.
35. Mundo E, Tharmalingham S, Neves-Pereira M et al, Evidence that the N-methyl-D-aspartate subunit 1 receptor gene (GRIN1) confers susceptibility to bipolardisorder. Mol Psychiatry 2003; 8:241–245.
36. Sivagnansundaram S, Müller DJ, Gubanov A et al, Genetics of schizophrenia:Current strategies. Clin Neurosci Res 2003; 3:5–16.
37. Berrettini W, Evidence for shared susceptibility in bipolar disorder and schizo-phrenia. Am J Med Genet C Semin Med Genet 2003; 123:59–64.
38. Hattori E, Liu C, Badner JA et al, Polymorphisms at the G72/G30 gene locus,on 13q33, are associated with bipolar disorder in two independent pedigreeseries. Am J Hum Genet 2003; 72:1131–1140.
39. Chen YS, Akula N, Detera-Wadleigh SD et al, Findings in an independent sam-ple support an association between bipolar affective disorder and the G72/G30locus on chromosome 13q33. Mol Psychiatry 2004; 9:87–92; image 5.
40. Schumacher J, Jamra RA, Freudenberg J et al, Examination of G72 and D-amino-acid oxidase as genetic risk factors for schizophrenia and bipolar affec-tive disorder. Mol Psychiatry 2004; 9:203–207.
41. Hanson IM, Seawright A, van Heyningen V, The human BDNF gene mapsbetween FSHB and HVBS1 at the boundary of 11p13-p14. Genomics 1992;13:1331–1333.
42. Detera-Wadleigh SD, Badner JA, Berrettini W et al, A high-density genome scandetects evidence for a bipolar-disorder susceptibility locus on 13q32 and otherpotential loci on 1q32 and 18p11.2. Proc Natl Acad Sci USA 1999;96:5604–5609.
43. Zandi PP, Willour VL, Huo Y et al, Genome scan of a second wave of NIMHgenetics initiative bipolar pedigrees: chromosomes 2, 11, 13, 14, and X. Am JMed Genet B Neuropsychiatr Genet 2003; 119:69–76.
44. Green E, Craddock N, Brain-derived neurotrophic factor as a potential risklocus for bipolar disorder: evidence, limitations, and implications. CurrPsychiatry Rep 2003; 5:469–476.
45. Lyons WE, Mamounas LA, Ricaurte GA et al, Brain-derived neurotrophicfactor-deficient mice develop aggressiveness and hyperphagia in conjunctionwith brain serotonergic abnormalities. Proc Natl Acad Sci USA 1999;96:15239–15244.
46. Fukumoto T, Morinobu S, Okamoto Y et al, Chronic lithium treatment increas-es the expression of brain-derived neurotrophic factor in the rat brain.Psychopharmacology (Berl) 2001; 158:100–106.
47. Chen B, Dowlatshahi D, MacQueen GM et al, Increased hippocampal BDNFimmunoreactivity in subjects treated with antidepressant medication. BiolPsychiatry 2001; 50:260–265.
48. Dwivedi Y, Rizavi HS, Conley RR et al, Altered gene expression of brain-derivedneurotrophic factor and receptor tyrosine kinase B in postmortem brain of sui-cide subjects. Arch Gen Psychiatry 2003; 60:804–815.
Ch 09 7/4/05 3:46 pm Page 90

Recent advances in genetics of bipolar disorder 91
49. Shimizu E, Hashimoto K, Okamura N et al, Alterations of serum levels of brain-derived neurotrophic factor (BDNF) in depressed patients with or without anti-depressants. Biol Psychiatry 2003; 54:70–75.
50. Nakata K, Ujike H, Sakai A et al, Association study of the brain-derived neu-rotrophic factor (BDNF) gene with bipolar disorder. Neurosci Lett 2003;337:17–20.
51. Hall D, Dhilla A, Charalambous A et al, Sequence variants of the brain-derivedneurotrophic factor (BDNF) gene are strongly associated with obsessive–compulsive disorder. Am J Hum Genet 2003; 73:370–376.
52. Egan MF, Kojima M, Callicott JH et al, The BDNF val66met polymorphismaffects activity-dependent secretion of BDNF and human memory and hip-pocampal function. Cell 2003; 112:257–269.
53. Neves-Pereira M, Mundo E, Muglia P et al, The brain-derived neurotrophic fac-tor gene confers susceptibility to bipolar disorder: evidence from a family-based association study. Am J Hum Genet 2002; 71:651–655.
54. Sklar P, Gabriel SB, McInnis MG et al, Family-based association study of 76candidate genes in bipolar disorder: BDNF is a potential risk locus. Brain-derived neutrophic factor. Mol Psychiatry 2002; 7:579–593.
55. Schulze TG, Hardy J, McMahon FJ, Inconsistent designs of association studies:a missed opportunity. Mol Psychiatry 2003; 8:770–772.
56. Geller B, Badner JA, Tillman R et al, Linkage disequilibrium of the brain-derived neurotrophic factor Val66Met polymorphism in children with a prepu-bertal and early adolescent bipolar disorder phenotype. Am J Psychiatry 2004;161:1698–1700.
57. Hong CJ, Huo SJ, Yen FC et al, Association study of a brain-derivedneurotrophic-factor genetic polymorphism and mood disorders, age of onsetand suicidal behavior. Neuropsychobiology 2003; 48:186–189.
58. Kunugi H, Iijima Y, Tatsumi M et al, No association between the Val66Met poly-morphism of the brain-derived neurotrophic factor gene and bipolar disorderin a Japanese population: a multicenter study. Biol Psychiatry 2004;56:376–378.
59. Skibinska M, Hauser J, Czerski PM et al, Association analysis of brain-derivedneurotrophic factor (BDNF) gene Val66Met polymorphism in schizophrenia andbipolar affective disorder. World J Biol Psychiatry 2004; 5:215–220.
60. Neves-Pereira M, Cheung JK, Pasdar A et al, BDNF gene is a risk factor forschizophrenia in a Scottish population. Mol Psychiatry 2005; 10:208–212.
61. Cichon S, Schumacher J, Abou Jamra R et al, Supportive evidence for a rela-tionship between genetic variations at the brain-derived-neurotrophic factor(BDNF) locus and depressive symptoms in affective disorder and schizophre-nia. Am J Hum Genet (Neuropsych Genet) 2004; 130B:27.
62. Strauss J, Barr CL, George CJ et al, Association study of brain-derived neu-rotrophic factor in adults with a history of childhood onset mood disorder. AmJ Med Genet 2004; 131B:16–19.
63. Müller DJ, De Luca V, Sicard T et al, ‘Rapid cycling’ mainly determines signifi-cant findings between the BDNF gene and bipolar disorder. Am J Hum Genet(Neuropsych Genet) 2004; 130B:45.
64. Muglia P, Vicente AM, Verga M et al, Association between the BDNF gene andschizophrenia. Mol Psychiatry 2003; 8:146–147.
Ch 09 7/4/05 3:46 pm Page 91

92 Bipolar disorder: the upswing in research & treatment
65. Lanktree M, Muglia P, Squassina A et al, A potential role for brain derived neu-rotrophic factor (BDNF) in adult ADHD. Am J Hum Genet (Neuropsych Genet)2004; 130B:96.
66. Schulze TG, Muller DJ, Krauss H et al, Caught in the trio trap? Potential selec-tion bias inherent to association studies using parent–offspring trios. Am J MedGenet 2001; 105:351–353.
Ch 09 7/4/05 3:46 pm Page 92

Is there a genetic basisto the brain abnormalitiesof bipolar disorder?Colm McDonald
c h a p t e r 1 0
‘…I could demonstrate a hereditary taint (for manic depressive
insanity) in about 80 per cent of cases observed in Heidelberg.’
Emil Kraepelin1
Familial liability to bipolar disorder has been recognized since the clinicalsyndrome was introduced a century ago, and twin studies have furtherdemonstrated that such familiality is related to genetic transmission of theillness.2 The heritability of operationally defined bipolar disorder is esti-mated to be over 80%, similar to that of other psychotic disorders such asschizophrenia,2,3 with the remaining variance attributable to non-sharedenvironmental risk factors. Clearly, susceptibility genes must exist for thisillness, but progress in identifying such genes has been exceedingly slow,owing to the complexity of genetic transmission. There are unlikely to beany genes of major effect for bipolar disorder, expect perhaps in some rarepedigrees, and instead inheritance is probably due to many genes of smalleffect. Furthermore, the molecular genetics research of bipolar disorder,like many other psychiatric disorders, is hindered by the inaccuracy of phe-notypic definition; we lack laboratory diagnostic tests, even at postmortem,and the use of the clinical syndrome alone as the phenotype is problematicbecause of the effects of genetic heterogeneity and reduced penetrance.
What do susceptibility genes do?
Susceptibility genes do not directly produce the symptoms of bipolar disor-der. Genes code for proteins and such proteins impact upon brain functionand brain structure at some level. The symptoms by which we diagnose the
Ch 10 7/4/05 3:47 pm Page 93

94 Bipolar disorder: the upswing in research & treatment
illness emerge from the underlying neurobiological abnormalities producedby susceptibility genes. Therefore, an alternative and potentially comple-mentary approach to the use of the qualitative clinical syndrome in molec-ular genetic studies is to identify those neurobiological markers of braindysfunction or abnormal structure which are produced by susceptibilitygenes and to utilize these as alternative phenotypes. This approach is gain-ing momentum within schizophrenia research but has rarely been appliedto bipolar disorder.
Such ‘endophenotypes’, which are presumed more proximal to geneaction than the clinical syndrome, can be identified by employing studydesigns that examine how candidate neurobiological markers vary amongsubjects with increasing genetic risk. The first such design within schizo-phrenia research examined how biological variables differed in subjectswith a strong family history, who were presumably enriched with suscepti-bility genes, compared with those with no family history of illness. Thisdesign is problematic not least since the presence of illness itself, withaccompanying disease progression, chronic medication and other healtheffects, can impact upon the biological variables being measured. A morepowerful design is to use unaffected subjects who are at a higher genetic riskfor the illness, such as co-twins, first-degree relatives or offspring. Such rel-atives are free from any medication or disease effects but share 50% of theaffected patient’s genes (or 100% in the case of unaffected monozygotic co-twins) and are thus more likely to manifest the effects of susceptibility genesupon neurobiological markers than are control subjects with no family his-tory of illness. Further power to detect genetic effects can be attained byintroducing a gradient of genetic liability among the unaffected relativesstudied, for example including relatives from both multiply affected andsingly affected families, or unaffected co-twins from both monozygotic anddizygotic pairs. In support of this approach, there is accumulating evidencewithin the schizophrenia literature that unaffected first-degree relativesfrom multiply affected families have more severe abnormalities of potentialcognitive and morphometric endophenotypic measures than unaffected rel-atives from singly affected families.4,5
Why might brain morphometry represent a usefulendophenotype?
Potential endophenotypic neurobiological markers should be: (1) heritablethemselves; (2) measurable in both affected and unaffected subjects; (3)
Ch 10 7/4/05 3:47 pm Page 94

Is there a genetic basis to the brain abnormalities of bipolar disorder? 95
manifest whether or not the illness is active; (4) associated with the illnessin the general population; and (5) found more frequently in unaffected rela-tives of patients than controls.6 Brain morphometry meets some of thesecriteria. Magnetic resonance imaging (MRI) brain scans are easily measura-ble in unaffected individuals, and several MRI studies of healthy twins havedemonstrated high degrees of genetic control over volumetric measure-ments of the cerebrum, grey and white matter and several subregions of thebrain.7,8 The morphometry of bipolar disorder is relatively under-researched compared to schizophrenia; however, the illness is associatedwith subtle deviation from normal brain anatomy at the group level whenpatients with bipolar disorder are compared with controls (see Chapter 4for a meta-analysis of structural brain deviations in bipolar disorder). Themost consistent evidence supports mild enlargement of lateral ventricularvolume and higher rates of white matter hyperintensities, with some evi-dence for reduced volume of parts of the prefrontal lobe, such as the ante-rior cingulate gyrus.9,10 There has been little attempt to date to identifybrain abnormalities in the unaffected relatives of bipolar disorder patientsin order to elucidate any effect of bipolar disorder susceptibility genes ongross brain structure.
The Maudsley Family Study of Psychosis
This large study was initially developed to identify endophenotypic markersof schizophrenia by examining a range of brain structural and functionalmeasurements in patients with schizophrenia and their unaffected relativesat varying genetic risk. Recently, the approach has also been extended topsychotic bipolar disorder. T1-weighted MRI brain scans were successfullyobtained in 37 patients who fulfilled DSM-IV criteria for bipolar I disorderand 50 of their unaffected first-degree relatives. The patients had all experi-enced psychotic symptoms at some stage during episodes of illness exacer-bation. All the subjects were from multiply affected families, in that thepatients had at least one other relative with psychotic bipolar disorder oranother functional psychotic disorder among their first- and/or second-degree relatives. The patients had a mean age of 41 years and mean dura-tion of illness of 18 years. All were outpatients at the time of assessment and33 were taking mood-stabilizing medication, mostly lithium.
Although all families were multiply affected, the density of illness withinthe families varied considerably. In order to model variation in genetic riskamong subjects, we calculated a continuous measure of likely genetic liabil-
Ch 10 7/4/05 3:47 pm Page 95

96 Bipolar disorder: the upswing in research & treatment
ity using a quantitative scale.11 This measure is based upon a multifactori-al liability threshold model of illness, which assumes that liability within thepopulation is normally distributed, and utilizes information about affectionstatus from all adult members in each pedigree as far as second degree fromthe index patient, taking into account family size and genetic relatedness toaffected individuals. Examples of the genetic liability scores produced forfamilies of differing density are demonstrated in Figure 10.1.
This measure of genetic liability was then used to predict regional vol-ume variation of grey and white matter throughout the entire brain derivedfrom computational morphometry analyses of the MRI scans of all patientsand relatives. Optimized voxel-based morphometry12 was used to segmentMRI images into grey matter, white matter and cerebrospinal fluid and tonormalize grey and white matter maps to templates derived from healthycontrols matched on demographic variables. Non-parametric analyses con-trolling for the confounds of age, gender and subject group and utilizingcluster level statistics to account for multiple testing at the voxel level wereemployed.
Figure 10.1 Genetic liability scores in families with differing density of illness. (A)An unaffected parent (arrowed), who appears to be transmitting genetic suscepti-bility to her son, has a relatively high genetic liability of 0.63. (B) Although stillmultiply affected, the large number of unaffected individuals in this pedigree isreflected by the relatively low genetic liability score of 0.10 for an unaffected sib-ling (arrowed).
A
B
0.03 0.03 1.95 –0.08
0.01 0.01 0.01 0.01 0.21 0.63 0.42 0.42 1.99
1.75 0.19 0.19
0.03 0.03 0.26 0.26
0.01 0.01 0.01 0.01 0.21 0.32 1.68 0.12 0.12 0.12 0.12
0.09 –0.03 –0.03 0.11 –0.02 1.68 0.10 –0.03
0.010.01 0.010.01 0.010.01 0.010.01 0.01 0.01 0.010.01
Ch 10 7/4/05 3:47 pm Page 96

Is there a genetic basis to the brain abnormalities of bipolar disorder? 97
Grey matter
Increasing genetic liability for bipolar disorder was associated with reducedgrey matter volume in a discrete region comprising the right medial frontallobe, including the pregenual and subgenual sections of the anterior cingu-late gyrus, and the ventral striatum (Figure 10.2A). The relationshipbetween increasing genetic liability and reduced volume in these regionswas present in both patients and their unaffected relatives, since no inter-action existed between subject group and genetic liability score upon region-al grey matter volume defined by the cluster in a multiple regression analy-sis employing multilevel modelling (B, –0.27, 95% CI –1.85–1.30, p=0.72;Figure 10.2B). This indicates that the association was not determined sole-ly by abnormalities in the patients. The anterior cingulate gyrus and ventralstriatum are components of brain circuits which are critical in the regula-tion of normal emotion13 and abnormalities of this region have been previ-ously detected in familial bipolar disorder using structural and functionalneuroimaging.14 The present study demonstrates that volume deficit is notsolely a disease-related phenomenon but reflects the impact of susceptibili-ty genes upon this area, since volume deficit is identified also in those unaf-fected relatives at highest genetic liability for bipolar disorder.
White matter
Increasing genetic liability for bipolar disorder was also associated with dis-tributed white matter volume deficits involving the anterior corpus callosumas well as bilateral frontal, left temporoparietal and right parietal regions(Figure 10.3A). The relationship between increasing genetic liability andreduced volume in these regions was present in both patients and theirunaffected relatives, since no interaction existed between subject group andgenetic liability score upon first principal component factor scores, whichsummarized (explaining 80.7% of the variance) the highly intercorrelatedregional white matter volumes defined by the clusters, in a multiple regres-sion analysis employing multilevel modelling (B, 0.11, 95% CI –1.82–2.03,p=0.72; Figure 10.3B). This indicates that the association was not deter-mined solely by abnormalities in the patients. These hemispheric whitematter regions are characteristically occupied by major intrahemispherictracts: the superior longitudinal fasciculus, which connects the frontal lobeto the temporal, parietal and occipital lobes; and the inferior longitudinalfasciculus, which connects the temporal pole to the occipital lobe.
Ch 10 7/4/05 3:47 pm Page 97
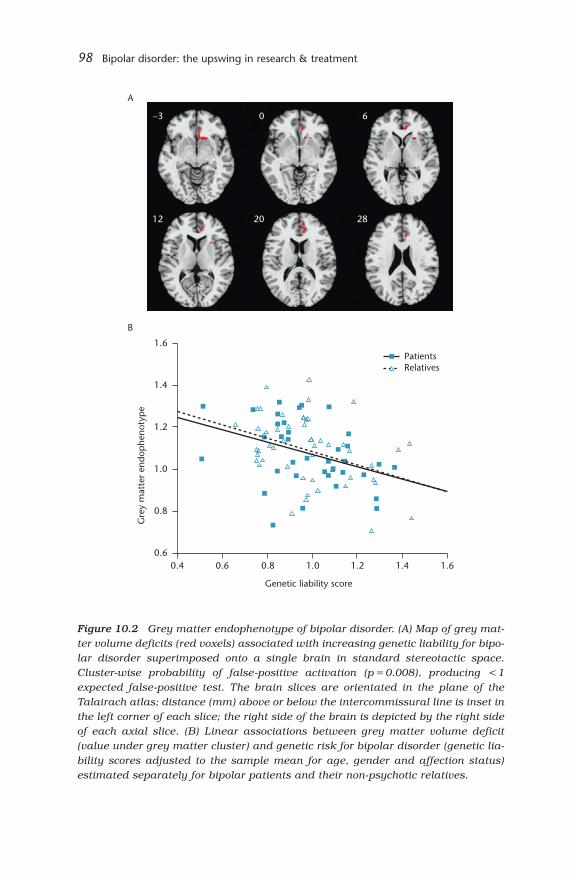
98 Bipolar disorder: the upswing in research & treatment
Figure 10.2 Grey matter endophenotype of bipolar disorder. (A) Map of grey mat-ter volume deficits (red voxels) associated with increasing genetic liability for bipo-lar disorder superimposed onto a single brain in standard stereotactic space.Cluster-wise probability of false-positive activation (p=0.008), producing <1expected false-positive test. The brain slices are orientated in the plane of theTalairach atlas; distance (mm) above or below the intercommissural line is inset inthe left corner of each slice; the right side of the brain is depicted by the right sideof each axial slice. (B) Linear associations between grey matter volume deficit(value under grey matter cluster) and genetic risk for bipolar disorder (genetic lia-bility scores adjusted to the sample mean for age, gender and affection status)estimated separately for bipolar patients and their non-psychotic relatives.
0.4 0.6 0.8 1.0 1.2 1.4 1.6
Genetic liability score
1.6
1.4
1.2
1.0
0.8
0.6
Gre
y m
atte
r en
dop
heno
typ
e
PatientsRelatives
A
B
–3 0 6
12 20 28
Ch 10 7/4/05 3:49 pm Page 98

Is there a genetic basis to the brain abnormalities of bipolar disorder? 99
Figure 10.3 White matter endophenotype of bipolar disorder. (A) Map of whitematter volume deficits (red voxels) associated with genetic liability for bipolar dis-order superimposed onto a single brain in standard stereotactic space. Cluster-wise probability of false-positive activation (p=0.01), producing <1 expectedfalse-positive test. The brain slices are orientated in the plane of the Talairachatlas; distance (mm) above or below the intercommissural line is inset in the leftcorner of each slice; the right side of the brain is depicted by the right side of eachaxial slice. (B) Linear associations between systemic white matter volume deficit(first principal component factor scores, which summarize correlated white matterdeficit) and genetic risk for bipolar disorder (genetic liability scores adjusted to thesample mean for age, gender and affection status) estimated separately for bipo-lar patients and their non-psychotic relatives.
0.4 0.6 0.8 1.0 1.2 1.4 1.6
Genetic liability score
3
2
1
0
–1
–2
–3
–4
Whi
te m
atte
r en
dop
heno
typ
e
A
B
PatientsRelatives
0 6 12 20
28 35 42 50
Ch 10 7/4/05 3:54 pm Page 99

100 Bipolar disorder: the upswing in research & treatment
White matter abnormalities in bipolar disorder are supported by convergentfindings from case–control studies using varying research methodologies.Studies have consistently demonstrated increased rates of hyperintensitiesdetectable on T2-weighted MRI images;9 volume deficit of left hemisphericwhite matter has been reported;15 reduced fractional anisotropy and macro-molecular density in the prefrontal white matter has been detected usingdiffusion tensor imaging16 and magnetization transfer imaging17 respective-ly; abnormalities of myelination and oligodendrocyte function are indicatedby down-regulation of gene expression in postmortem prefrontal tissue.18
The present study indicates that white matter abnormalities are not con-fined to the impact of the illness or its treatment, but that some suscepti-bility genes for bipolar disorder are likely to be associated with reduced vol-ume of inter- and intrahemispheric white matter tracts. Interestingly, anoverlapping endophenotype comprising left temporoparietal white mattervolume reduction was identified for schizophrenia using similar methodol-ogy, consistent with the hypothesis that shared susceptibility genes for psy-chosis in general produce disturbed left frontotemporal anatomical connec-tivity.11
What are the implications of identifyingmorphometric endophenotypes of bipolardisorder?
These results from the Maudsley Family Study of Psychosis demonstratethat susceptibility genes for bipolar disorder do have an impact upon grossbrain structure which is detectable from MRI scans. Volume deficit in corti-cal and subcortical regions involved in mood regulation and in more wide-spread inter- and intra-hemispheric white matter tracts is associated withincreasing genetic liability, not only in patients but also in unaffected rela-tives who have never experienced the illness of bipolar disorder or its treat-ment. The identification of these morphometric endophenotypes is likely torepresent more proximal effects of susceptibility genes for bipolar disorderthan the clinical syndrome, and we will now use them as alternative pheno-types in molecular genetic studies in two ways: (1) allelic variation in geneswhich may confer susceptibility to bipolar disorder identified by studiesusing the clinical syndrome as the phenotype, will now be tested for associ-ation with the morphometric endophenotypes to assess whether such genet-ic variation impacts upon these brain structures; (2) the morphometricendophenotypes identified by this study will themselves be used as
Ch 10 7/4/05 3:54 pm Page 100

Is there a genetic basis to the brain abnormalities of bipolar disorder? 101
phenotypes in molecular genetic studies informed by the neurobiology of the
tissue classes and regions identified – for example, functional polymor-
phisms in genes coding for myelination and oligodendrocyte function are
suitable candidates for association studies using the white matter endophe-
notype identified by the present study.
In conclusion, genetic liability for psychotic bipolar disorder is manifest
in volumetric deficit of grey matter in the right anterior cingulate and ven-
tral striatum, and of white matter in bilateral frontal and temporoparietal
regions. The identification of these morphometric endophenotypes provides
a neuroanatomical substrate for molecular genetic studies of bipolar disor-
der and should facilitate the identification of the elusive genes that confer
susceptibility to this highly heritable illness.
Acknowledgement
Colm McDonald is supported by the Wellcome Trust.
References
1. Kraepelin E, Manic Depressive Insanity and Paranoia. [Translated by R.M.Barclay] E&S Livingstone: Edinburgh, 1921:165.
2. McGuffin P, Rijsdijk F, Andrew M et al, The heritability of bipolar affective dis-order and the genetic relationship to unipolar depression. Arch Gen Psychiatry2003; 60:497–502.
3. Cardno AG, Marshall EJ, Coid B et al, Heritability estimates for psychotic dis-orders: the Maudsley twin psychosis series. Arch Gen Psychiatry 1999;56:162–168.
4. Faraone SV, Seidman LJ, Kremen WS et al, Neuropsychological functioningamong the nonpsychotic relatives of schizophrenic patients: the effect of genet-ic loading. Biol Psychiatry 2000; 48:120–126.
5. McDonald C, Grech A, Toulopoulou T et al, Brain volumes in familial and non-familial schizophrenic probands and their unaffected relatives. Am J MedGenet 2002; 114:616–625.
6. Leboyer M, Bellivier F, Nosten-Bertrand M et al, Psychiatric genetics: search forphenotypes. Trends Neurosci 1998; 21:102–105.
7. White T, Andreasen NC, Nopoulos P, Brain volumes and surface morphology inmonozygotic twins. Cereb Cortex 2002; 12:486–493.
8. Wright IC, Sham P, Murray RM et al, Genetic contributions to regional variabil-ity in human brain structure: methods and preliminary results. Neuroimage2002; 17:256–271.
Ch 10 7/4/05 3:54 pm Page 101

102 Bipolar disorder: the upswing in research & treatment
9. Bearden CE, Hoffman KM, Cannon TD, The neuropsychology and neuroanato-my of bipolar disorder: a critical review. Bipolar Disord 2001; 3:106–150.
10. McDonald C, Zanelli J, Rabe-Hesketh S et al, Meta-analysis of magnetic reso-nance imaging brain morphometry studies in bipolar disorder. Biol Psychiatry2004; 56:411–417.
11. McDonald C, Bullmore ET, Sham PC et al, Association of genetic risks for schiz-ophrenia and bipolar disorder with specific and generic brain structuralendophenotypes. Arch Gen Psychiatry 2004; 61:974–984.
12. Good CD, Johnsrude IS, Ashburner J et al, A voxel-based morphometric studyof ageing in 465 normal adult human brains. Neuroimage 2001; 14:21–36.
13. Rolls ET, The Brain and Emotion. Oxford University Press: Oxford, 1999.14. Drevets WC, Price JL, Simpson JR Jr et al, Subgenual prefrontal cortex
abnormalities in mood disorders. Nature 1997; 386:824–827.15. Kieseppa T, van Erp TG, Haukka J et al, Reduced left hemispheric white mat-
ter volume in twins with bipolar I disorder. Biol Psychiatry 2003; 54:896–905.16. Adler CM, Holland SK, Schmithorst V et al, Abnormal frontal white matter
tracts in bipolar disorder: a diffusion tensor imaging study. Bipolar Disord2004; 6:197–203.
17. Bruno SD, Barker GJ, Cercignani M et al, A study of bipolar disorder usingmagnetization transfer imaging and voxel-based morphometry. Brain 2004;127:2433–2440.
18. Tkachev D, Mimmack ML, Ryan MM et al, Oligodendrocyte dysfunction inschizophrenia and bipolar disorder. Lancet 2003; 362:798–805.
Ch 10 7/4/05 3:54 pm Page 102

Transgenic mouse modelsfor affective disordersbased on theneurotrophin hypothesisPeter Gass
c h a p t e r 1 1
Introduction
According to the monoaminergic hypothesis, depression is related to animpairment of neurotransmission by serotonin, norepinephrine (noradren-aline) and dopamine. These deficiencies can result from several mecha-nisms: (1) decreased synthesis or increased degradation of neurotransmit-ters; (2) altered expression or function of the respective neurotransmitterreceptors; or (3) impairment of signal transduction systems activated bypost-synaptic receptors. Most antidepressant drugs act primarily via thefirst mechanism, aiming to improve monoaminergic transmission byincreasing the presence of neurotransmitters inside the synapse. It is stillunclear how the various antidepressants with their different modes ofaction (e.g. serotonin or norepinephrine re-uptake or degradationinhibitors) finally induce emotional and behavioural improvements.
Common to all classes of antidepressants is also that their onset ofaction takes 2–3 weeks. Therefore, a key biological mechanism must beresponsible for the fact that only chronic administration has mood-elevatingeffects, while enhancement of serotoninergic or noradrenergic neurotrans-mission occurs within minutes after drug intake. Recent concepts for patho-genesis and therapy claim slowly developing plasticity changes induced bychronic alterations in monoaminergic neurotransmission. Two biologicalsystems have attracted special attention: (1) the stress-responsive hypo-thalamic–pituitary–adrenal (HPA) system, which is disinhibited in manypatients with major depressive episodes;1,2 and (2) the neurotrophin brain-derived neurotrophic factor (BDNF), which has been implicated in
Ch 11 7/4/05 3:56 pm Page 103

104 Bipolar disorder: the upswing in research & treatment
hippocampal maladaptative processes related to depressive episodes.3,4
Interestingly, regulatory mechanisms that control stress hormones and hip-
pocampal BDNF expression are linked to each other. This has led to the so-
called ‘neurotrophin hypothesis of depression’.
The neurotrophin hypothesis of depression
The CREB–BDNF–TrkB pathway
A variety of signalling molecules (e.g. neurotransmitters, growth factors)
that act via cAMP- or Ca2+-activated kinases, or by direct activation of tyro-
sine kinases, lead to the phosphorylation of the transcription factor cAMP
response element (CRE)-binding protein (CREB). This activation results in
elevations in CRE-mediated gene transcription of downstream target genes
such as BDNF. BDNF exerts its effects mainly by activation of tyrosine recep-
tor kinase B (TrkB) (Figure 11.1). Neurotrophins and especially BDNF influ-
ence structural plasticity and have trophic effects on neurons.
Clinical and experimental observations have led to the concept that a
deficiency in BDNF contributes to the pathophysiology of depression.3,4
BDNF has a high concentration in brain regions thought to be involved in
the pathogenesis of depression, such as the hippocampus or neocortex.5
Moreover, CREB-mediated BDNF induction is a downstream effect of chron-
ic but not acute antidepressive treatment.3,4
Stress decreases the activity of the CREB–BDNF–TrkB pathway
Chronic stress, and a subsequent rise in plasma corticosteroids, is regard-
ed as a major cause in the pathogenesis of depressive disorders.6
Accordingly, physical and psychosocial stresses have been used to induce a
depression-like syndrome in rodents.7,8 Stress or corticosteroid injections
cause a decrease in BDNF mRNA in the hippocampus and other brain areas
thought to be involved in the pathogenesis of depression, most probably by
activation of glucocorticoid receptors.9–11 This effect is mitigated by electro-
convulsive therapy (ECT) or by chronic administration of antidepressants.10
Similar to the ECT findings in vivo, up-regulation of BDNF triggered by
depolarization in cultured hippocampal neurons is blocked by dexametha-
sone, a synthetic glucocorticoid.12 In a genetic rat model of depression, the
Flinders Sensitive Line, several brain regions have lower BDNF levels as
control lines.13
Ch 11 7/4/05 3:56 pm Page 104

Transgenic mouse models for affective disorders 105
At the clinical level, postmortem investigations show that untreateddepressive patients have lower levels of CREB in the temporal cortex thanhealthy subjects.14,15 Moreover, patients with major depression have lowerBDNF serum concentrations than controls, and these levels are negativelycorrelated with scores in clinical depression rating scales.16 Altogether,human and animal studies suggest that the depressive state is positivelycorrelated with CREB and BDNF expression, because reduced levels arecaused by stress exposure and reversed by antidepressant treatment.
Antidepressants activate the CREB–BDNF–TrkB pathway
Chronic but not acute treatment with antidepressants increases BDNF andCREB mRNA levels in the hippocampus.10,17 Long-term ECT also induces
Figure 11.1 The neurotrophin hypothesis of depression. Chronic stress combinedwith a rise in plasma corticosteroid levels is regarded as a major factor in thepathogenesis of depressive disorders. Corticosteroids can downregulate brain-derived neurotrophic factor (BDNF) expression through an activation of glucocorti-coid receptors (GR). Decreased BDNF expression leads to reduced tyrosine recep-tor kinase B (TrkB) activation, which, directly or via other transmitter systems,decreases the activation of the transcription factor cAMP response element bindingprotein (CREB). By a vicious cycle, the reduced CREB activity further diminishesBDNF expression. Antidepressants block the stress-induced decrease in BDNFexpression and are also able directly to enhance CREB, BDNF and TrkB expressionand signalling. ECT, electroconvulsive therapy.
Depression
Glucocorticoids
Stress
GR
BDNF
TrkB
CREB
Plasticity & trophic effects
Monoaminergic neurotransmitters & growth factors
KinasesAntidepressants & ECT
+
+
+
–
Ch 11 7/4/05 3:56 pm Page 105

106 Bipolar disorder: the upswing in research & treatment
the expression of BDNF and TrkB in limbic brain regions.10,18,19 Moreover,chronic antidepressant treatment induces TrkB phosphorylation, which inturn enhances CREB activation.20 Because the CREB–BDNF–TrkB pathwayis a closed circle, it is difficult to determine which molecular changes arethe cause and which are the effect (Figure 11.1). Results from animal mod-els suggest that antidepressants do not only prevent a stress-induceddecrease in CREB–BDNF–TrkB signalling, but are also able to activate thispathway.
At the clinical level, higher concentrations of CREB and BDNF are foundin patients under antidepressive medication than in untreated patients.14,15
Patients with a major depressive disorder on antidepressant medicationshow increased TrkB expression when compared with healthy subjects.21
Successfully treated patients also show a significant increase of CREB phos-phorylation in T lymphocytes.22 Thus, current clinical data also suggest thatantidepressant effects are mediated by activation of the CREB–BDNF–TrkBpathway.
Neurotrophins have antidepressant-like effects
BDNF, when infused near the raphe nucleus, the main source of serotoner-gic innervation of the hippocampus, has an antidepressant-like effect in thelearned-helplessness model of depression.23 This paradigm uses uncon-trollable and inescapable stress to evoke physiological and behaviouralabnormalities similar to those observed in human depression.24 BilateralBDNF infusion into the hippocampus also produces antidepressant-likeeffects in the learned-helplessness paradigm and in the forced swim test.25
In summary, while antidepressive drugs activate the CREB–BDNF–TrkBpathway, exogenous application of neurotrophins can also induce anantidepressant-like effect.
Transgenic mice
Several strains of mice with altered or disrupted expression of CREB, BDNF
or TrkB have been used to analyse individual steps of the
CREB–BDNF–TrkB signalling cascade, and to test the predictions made by
the neurotrophin hypothesis of depression. Generally, one would expect a
predisposition for depression, if this pathway were compromised. A battery
of behavioural tests has been developed to define syndromically – similarly
to the human classification systems – a depression-like state in mice.26 The
Ch 11 7/4/05 3:56 pm Page 106

Transgenic mouse models for affective disorders 107
following emotional states or behaviours can be investigated by tests: ‘anhe-
donia’ (sucrose preference test), ‘despair’ (forced swim test, tail suspension
test), diminished interest (novel object approach), anxiety (elevated T-maze,
dark–light-box test, openfield test) and general activity (openfield test, activ-
ity boxes).
Transgenic mice with compromised CREB expression
A complete knockout of all isoforms of the CREB gene (α, ∆ and β) leads to
postnatal death due to respiratory failure, preventing analyses of adult ani-
mals.27 Therefore, most behavioural experiments have been performed in
mice lacking the α and ∆ isoforms (so-called CREBα∆ mutant mice), leading
to a more than 90% reduction of CRE-binding activity in the brain.28–30 In
contrast to the predictions made by the CREB–BDNF–TrkB hypothesis,
CREBα∆ mutant mice demonstrated significantly less despair behaviour
than controls.30 Similar results are described in mice with inducible over-
expression of a dominant-negative CREB form, leading to decreased CREB
activity in the forebrain.31 These animals show reduced depression-like
behaviours in the learned-helplessness paradigm. In accordance with these
findings, inducible overexpression of CREB evokes increased depression-
like behaviours.31 Thus, opposed to expectations from clinical and experi-
mental data, reduced CREB expression or function does not induce despair
behaviour but rather has an antidepressant-like effect. Despite the para-
doxical reduction of behavioural despair in CREBα∆ mutant mice, treatment
with antidepressants results in a further decrease of the despair reaction, to
a similar extent as observed in wild-type mice, indicating that the mecha-
nisms that underlie the behavioural effects of antidepressant drugs are less
dependent on CREB activation.30 However, the antidepressant-induced
upregulation of BDNF in the hippocampus and cortex is absent in CREBα∆
mutant animals,30 demostrating that CREB is an upstream regulator of
BDNF and may be a critical downstream mediator of transcriptional effects
of antidepressants. Surprisingly, these data also suggest an uncoupling
between transcriptional BDNF upregulation and direct behavioural effects
of antidepressants.
Transgenic mice with reduced BDNF expression
Conventional BDNF knockout mice do not survive to adulthood, because of
severe developmental defects. Behavioural experiments are therefore
restricted to heterozygous mice (BDNF+/–) or mice with a conditional
Ch 11 7/4/05 3:56 pm Page 107

108 Bipolar disorder: the upswing in research & treatment
knockout. BDNF+/– mice with about 50% reduced BDNF levels are behav-iourally indistinguishable from control littermates in a test battery for loco-motor, exploratory, anxiety- and depression-related behaviours.32,33
According to the theory, one would expect that these mice would be moresusceptible to developing depression-like symptoms. However, they do notdiffer in their hedonic capacity (sucrose consumption) nor in behaviouralmeasures for despair.32,33 The HPA-system as a key marker for majordepressive episodes is also not affected in these animals.33 A coping deficitof BDNF+/– mice in the learned-helplessness paradigm has been attributedto an impaired pain sensitivity.32 The fact that all other measures of thestress response are unchanged in BDNF+/– mice is perplexing, consideringthe proposed role of BDNF in the pathophysiology of depression. A possibleexplanation may be that a 50% reduction of BDNF is not sufficient to inducestrong behavioural effects, and a compensatory mechanism may addition-ally occur during development. Indeed, mice with a complete forebrain-spe-cific conditional BDNF knockout display increased anxiety-related behav-iours.34 Other behavioural tests more related to anhedonia, despair andcoping should be performed with this strain.
Interestingly, there is an uncoupling between baseline behaviours andantidepressant-induced behavioural alterations in BDNF mutant mice.Acute treatment with imipramine fails to reduce the despair reaction ofBDNF+/– mice in the forced swim test that is seen in wild-type mice, sug-gesting that BDNF directly mediates the behavioural effects of antidepres-sant drugs.20
Transgenic mice with reduced TrkB expression
Mice with a forebrain-specific disruption of TrkB receptors also lackdepression-like behaviours and HPA-system alterations.35 Similar resultsare obtained with mice constitutively overexpressing a dominant negativeform of TrkB, leading to a reduced TrkB activation in the brain.20 However,similar to BDNF+/– mice, antidepressants do not reduce the despair reac-tion in TrkB-deficient mice.20 These findings confirm that antidepressantsdirectly influence despair behaviour in mice via BDNF–TrkB signalling.20
The temporal profile of these experiments – with behavioural alterationsobserved as early as 30 minutes after drug treatment – strongly indicatesthat these effects occur at a post-transcriptional level. This interpretation isin line with findings in CREB-mutant mice (see above), which exhibit a nor-mal reduction of despair behaviour in response to antidepressants, buthave regular BDNF levels under baseline conditions.
Ch 11 7/4/05 3:56 pm Page 108

Transgenic mouse models for affective disorders 109
Discussion
Based on clinical and experimental observations, the neurotrophin hypoth-esis of depression as conceptualized originally made the following predic-tions: (1) reduced activity of the CREB–BDNF–TrkB pathway is implicatedin the pathogenesis of depression; and (2) activation of theCREB–BDNF–TrkB pathway is part of the molecular mechanisms of anti-depressive therapy.3,4 This concept has been challenged by studies withtransgenic mice. According to the neurotrophin hypothesis, mice with genet-ic disruptions of any part of this pathway are expected to displaydepression-like behaviours. However, none of several mouse strains investi-gated has exhibited such behaviours, some mice being even less ‘depressive’than the controls.
This surprising result could be due to factors that may mask or preventa depressive phenotype. First, a complete knockout of genes coding for anypart of the pathway is not compatible with survival to adulthood. Therefore,most studies have been performed with heterozygous mice or strains withincomplete downregulation of the targeted genes. This reduction in geneexpression may not have been sufficient to result in behavioural alterations.However, this interpretation seems unlikely regarding the results obtainedwith many other mouse strains where a 50% under- or overexpression ofgenes has caused strong behavioural effects. Second, mice carrying a trans-gene or mutation since early embryogenesis may develop compensatorymechanisms to overcome the effects of the targeted genes. Third, mice withgenetic modifications of the CREB–BDNF–TrkB pathway have been mostlyinvestigated under basal conditions. Subjecting them to stress-induceddepression models may unmask a predisposition to develop depression-likebehaviours. Fourth, depression is a multi-genetic disease and inactivation ofone gene may not suffice to induce a depressive state.
The fact that mice with mutations impairing the CREB–BDNF–TrkBpathway do not show any features of depression challenges the hypothesisthat this signalling pathway plays a major role in the pathogenesis ofdepression. This critical view is supported by further experimental evi-dence. Contradictory to the concept that stress causes a decreased activityof the CREB–BDNF–TrkB cascade, the forced swim test induces a long-lasting increase in CREB phosphorylation in several rat brain areasincluding the hippocampus.36 Immobilization stress produces a rapidincrease in BDNF mRNA and protein in the hypothalamus.37 Chronicunpredictable and uncontrollable stress also evokes an increase in TrkBexpression in hippocampal neurons. These results have been interpreted as
Ch 11 7/4/05 3:56 pm Page 109

110 Bipolar disorder: the upswing in research & treatment
compensatory adaptation to the stress-induced BDNF decrease that could
serve to protect the neurons from damage.38 Furthermore, two genetic rat
models of depression, the congenital learned-helplessness rats as well as
the depressive Flinders strain, exhibit unaltered levels of BDNF in the hip-
pocampus.13,39
Despite conflicting results on the role of the CREB–BDNF–TrkB pathway
in the pathogenesis of depression, this signalling cascade seems to be clear-
ly involved in the mechanisms of antidepressive therapy. Thus, chronic anti-
depressive treatment evokes an increase in BDNF expression.10 This
increase is absent in mice with impaired CREB function,30 indicating that
CREB is important in mediating the transcriptional effects of antidepres-
sants. On the other hand, mice with reduced BDNF expression (BDNF+/–) as
well as mice with impaired TrkB function do not show the normal reduc-
tion of behavioural despair seen in wild-type mice following antidepressant
treatment, indicating that BDNF-mediated TrkB activation is necessary for
at least some of the behavioural effects of antidepressants.20 These findings
in transgenic mice together with the direct antidepressive effects of BDNF in
mouse models of depression23,25 and the increased BDNF levels in patients
on antidepressive medication15 provide cumulative evidence for a central
role of this neurotrophin and its receptor in the molecular mechanisms of
antidepressive therapy.
There are several possible biological mechanisms that could mediate the
antidepressant effects of BDNF. On a molecular level, the mitogen-activated
protein (MAP) kinase cascade is activated by BDNF–TrkB signalling and
induces the plasticity-related transcription factor c-fos. On a cellular level,
BDNF is involved in synaptic remodelling and functioning.40,41 Such cellu-
lar adaptations may also improve monoaminergic signalling in the fore-
brain, which is a major participant in the pathogenesis of depression
according to the monoaminergic hypothesis. Mice that are compromised in
BDNF–TrkB signalling, represent a valuable tool to study molecular and cel-
lular downstream effects of this signalling pathway. The utility of BDNF
itself as an antidepressant in humans is limited, owing to the restrictions by
the blood–brain barrier. Therefore, pharmacological strategies have to be
developed to generate small-molecule agents that increase the expression
and promote the release of BDNF more specifically and more efficiently than
currently available antidepressants.
Ch 11 7/4/05 3:56 pm Page 110

Transgenic mouse models for affective disorders 111
References
1. Holsboer F, Barden N, Antidepressants and hypothalamic–pituitary–adrenocortical regulation. Endocr Rev 1996; 17:187–205.
2. Nemeroff CB, The corticotropin-releasing factor (CRF) hypothesis of depres-sion: new findings and new directions. Mol Psychiatry 1996; 1:336–342.
3. Duman RS, Heninger GR, Nestler EJ, A molecular and cellular theory ofdepression. Arch Gen Psychiatry 1997; 54:597–606.
4. Altar CA, Neurotrophins and depression. Trends Pharmacol Sci 1999;20:59–61.
5. Lewin GR, Barde YA, Physiology of the neurotrophins. Annu Rev Neurosci1996; 19:289–317.
6. Holsboer F, The corticosteroid receptor hypothesis of depression.Neuropsychopharmacology 2000; 23:477–501.
7. Porsolt RD, Animal models of depression: utility for transgenic research. RevNeurosci 2000; 11:53–58.
8. Willner P, Mitchell PJ, The validity of animal models of predisposition todepression. Behav Pharmacol 2002; 13:169–188.
9. Barbany G, Persson H, Regulation of neurotrophin mRNA expression in the ratbrain by glucocorticoids. Eur J Neurosci 1992; 4:396–403.
10. Nibuya M, Morinobu S, Duman RS, Regulation of BDNF and trkB mRNA in ratbrain by chronic electroconvulsive seizure and antidepressant drug treatments.J Neurosci 1995; 15:7539–7547.
11. Schaaf MJ, Hoetelmans RW, de Kloet ER, Vreugdenhil E, Corticosterone regu-lates expression of BDNF and trkB but not NT-3 and trkC mRNA in the rat hip-pocampus. J Neurosci Res 1997; 48:334–341.
12. Cosi C, Spoerri PE, Comelli MC et al, Glucocorticoids depress activity-dependent expression of BDNF mRNA in hippocampal neurones. Neuroreport1993; 4:527–530.
13. Angelucci F, Aloe L, Vasquez PJ, Mathe AA, Mapping the differences in the brainconcentration of brain-derived neurotrophic factor (BDNF) and nerve growthfactor (NGF) in an animal model of depression. Neuroreport 2000;11:1369–1373.
14. Dowlatshahi D, MacQueen GM, Wang JF, Young LT, Increased temporal cortexCREB concentrations and antidepressant treatment in major depression.Lancet 1998; 352:1754–1755.
15. Chen B, Dowlatshahi D, MacQueen GM et al, Increased hippocampal BDNFimmunoreactivity in subjects treated with antidepressant medication. BiolPsychiatry 2001; 50:260–265.
16. Karege F, Perret G, Bondolfi G et al, Decreased serum brain-derived neu-rotrophic factor levels in major depressed patients. Psychiatry Res 2002;109:143–148.
17. Nibuya M, Nestler EJ, Duman RS, Chronic antidepressant administrationincreases the expression of cAMP response element binding protein (CREB) inrat hippocampus. J Neurosci 1996; 16:2365–2372.
18. Duman RS, Vaidya VA, Molecular and cellular actions of chronic electroconvul-sive seizures. J ECT 1998; 14:181–193.
Ch 11 7/4/05 3:56 pm Page 111

112 Bipolar disorder: the upswing in research & treatment
19. Zetterstrom TS, Pei Q, Grahame-Smith DG, Repeated electroconvulsive shockextends the duration of enhanced gene expression for BDNF in rat brain com-pared with a single administration. Brain Res Mol Brain Res 1998;57:106–110.
20. Saarelainen T, Hendolin P, Lucas G et al, Activation of the TrkB neurotrophinreceptor is induced by antidepressant drugs and is required for antidepressant-induced behavioural effects. J Neurosci 2003; 23:349–357.
21. Bayer TA, Schramm M, Feldmann N et al, Antidepressant drug exposure isassociated with mRNA levels of tyrosine receptor kinase B in major depressivedisorder. Prog Neuropsychopharmacol Biol Psychiatry 2000; 24:881–888.
22. Koch JM, Kell S, Hinze-Selch D, Aldenhoff JB, Changes in CREB-phosphorylation during recovery from major depression. J Psychiatr Res2002; 36:369–375.
23. Siuciak JA, Lewis DR, Wiegand SJ, Lindsay RM, Antidepressant-like effect ofbrain-derived neurotrophic factor (BDNF). Pharmacol Biochem Behav 1997;56:131–137.
24. Shanks N, Anisman H, Strain-specific effects of antidepressants on escapedeficits induced by inescapable shock. Psychopharmacology 1989;99:122–128.
25. Shirayama Y, Chen AC, Nakagawa S et al, Brain-derived neurotrophic factorproduces antidepressant effects in behavioural models of depression. JNeurosci 2002; 22:3251–3261.
26. Gass P, Reichardt HM, Strekalova T et al, Mice with targeted mutations of glu-cocorticoid and mineralocorticoid receptors: models for depression and anxi-ety? Physiol Behav 2001; 73:811–825.
27. Rudolph D, Tafuri A, Gass P et al, Impaired fetal T cell development and peri-natal lethality in mice lacking the cAMP response element binding protein. ProcNatl Acad Sci USA 1998; 95:4481–4486.
28. Blendy JA, Kaestner KH, Schmid W et al, Targeting of the CREB gene leads toup-regulation of a novel CREB mRNA isoform. EMBO J 1996; 15:1098–1106.
29. Graves L, Dalvi A, Lucki I et al, Behavioural analysis of CREB alphadelta muta-tion on a B6/129 F1 hybrid background. Hippocampus 2002; 12:18–26.
30. Conti AC, Cryan JF, Dalvi A et al, cAMP response element-binding protein isessential for the upregulation of brain-derived neurotrophic factor transcrip-tion, but not the behavioral or endocrine responses to antidepressant drugs. JNeurosci 2002; 22:3262–3268.
31. Newton SS, Thome J, Wallace TL et al, Inhibition of cAMP response element-binding protein or dynorphin in the nucleus accumbens produces anantidepressant-like effect. J Neurosci 2002; 22:10883–10890.
32. MacQueen GM, Ramakrishnan K, Croll SD et al, Performance of heterozygousbrain-derived neurotrophic factor knockout mice on behavioral analogues ofanxiety, nociception, and depression. Behav Neurosci 2001; 115:1145–1153.
33. Chourbaji S, Hellweg R, Brandis D et al, Mice with reduced brain-derivedneurotrophic factor expression show decreased choline acetyltransferase activ-ity, but regular brain monoamine levels and unaltered emotional behavior. MolBrain Res 2004; 121:28–36.
Ch 11 7/4/05 3:56 pm Page 112

Transgenic mouse models for affective disorders 113
34. Rios M, Fan G, Fekete C et al, Conditional deletion of brain-derived neu-rotrophic factor in the postnatal brain leads to obesity and hyperactivity. MolEndocrinol 2001; 15:1748–1757.
35. Zörner B, Wolfer DP, Brandis D et al, Forebrain-specific trkB-receptor knock-out mice: behaviorally more hyperactive than ‘depressive’. Biol Psychiatry2003; 54:972–982.
36. Bilang-Bleuel A, Rech J, De Carli S et al, Forced swimming evokes a biphasicresponse in CREB phosphorylation in extrahypothalamic limbic and neocorti-cal brain structures in the rat. Eur J Neurosci 2002; 15:1048–1060.
37. Rage F, Givalois L, Marmigere F et al, Immobilization stress rapidly modulatesBDNF mRNA expression in the hypothalamus of adult male rats. Neuroscience2002; 112:309–318.
38. Nibuya M, Takahashi M, Russell DS, Duman RS, Repeated stress increases cat-alytic TrkB mRNA in rat hippocampus. Neurosci Lett 1999; 267:81–84.
39. Vollmayr B, Henn FA, Learned helplessness in the rat: improvements in validi-ty and reliability. Brain Res Brain Res Protoc 2001; 8:1–7.
40. Poo MM, Neurotrophins as synaptic modulators. Nat Rev Neurosci 2001;2:24–32.
41. Manji HK, Quiroz JA, Sporn J et al, Enhancing neuronal plasticity and cellularresilience to develop novel, improved therapeutics for difficult-to-treat depres-sion. Biol Psychiatry 2003; 53:707–742.
Ch 11 7/4/05 3:56 pm Page 113

Ch 11 7/4/05 3:56 pm Page 114

Is the hypothalamic–pituitary–adrenalaxis at last payingdividends?David A Cousins and Allan H Young
c h a p t e r 1 2
Psychiatry is the medical specialty concerned with pathological human psy-chology. Throughout its history, attempts have been made to explain psy-chological experiences in terms of neurophysiology and neuropathology.Such practice is probably overly reductionistic, as it is not legitimate toexplain all psychological problems in terms of biological processes.1 Even ifthe mind–brain debate concludes in favour of materialism, the subjectiveexperience of consciousness will remain. What then is the role of biologicalpsychiatry? Biological psychiatry has two main purposes: to understand thepathophysiology of psychiatric illness, and to use that understanding todevelop therapeutic strategies. Some of the progress made in these regardswill be the subject of this chapter, with specific reference to thehypothalamic–pituitary–adrenal (HPA) axis in bipolar affective disorder.
Pathophysiology of bipolar affective disorder
There is an overwhelming amount of information in this field that has yet tofit into a cohesive paradigm. A simple overarching model would incorporatethe influence of genes, environment and stress, and the ongoing effects ofthe illness itself. Stress, in particular its main substrate system the HPAaxis, has the potential to integrate many of our findings. We will first reviewthe structure and function of the HPA axis, the evidence for its disturbancein bipolar affective disorder and finally the consequences of that distur-bance.
Ch 12 7/4/05 3:57 pm Page 115

116 Bipolar disorder: the upswing in research & treatment
The HPA axis
The structural and functional organization of the HPA axis is well estab-lished and is represented in Figure 12.1. In response to stress, neurosecre-tory cells in the paraventricular nucleus of the hypothalamus secrete corti-cotrophin releasing hormone (CRH) into the microportal circulatory system
Figure 12.1 The hypothalamic–pituitary–adrenal axis. Purple triangle, glucocorti-coid receptors; CRH, corticotrophin releasing hormone; ACTH, adrenocorticotrophichormone.
Midbrain
AmygdalaHippocampus
Hypothalamus
Pituitary
Adrenals
Cortisol
ACTH +ve
CRH +ve
+ve
–ve
–ve
–ve
±ve±ve
Ch 12 7/4/05 3:57 pm Page 116

Is the hypothalamic–pituitary–adrenal axis at last paying dividends? 117
of the pituitary stalk. CRH acts on the anterior pituitary, which in turn
releases adrenocorticotrophic hormone (ACTH) into the systemic circula-
tion. ACTH governs the release of cortisol from the adrenal cortex. Cortisol
is the end product of the HPA axis and has numerous central and periph-
eral effects, including completion of a feedback loop. The HPA axis is high-
ly regulated by this feedback, and by neuronal inputs to the hypothalamus
from a number of brain regions (amygdala, hippocampus and certain mid-
brain nuclei). It is through these neuronal inputs that the system responds
to both physical and psychological stressors.
At a cellular level, the effects of cortisol are mediated through intracel-
lular glucocorticoid receptors, of which there are two subtypes: mineralo-
corticoid receptor (MR) and glucocorticoid receptor (GR). Activated recep-
tors move from the cytosol to the nucleus and interact with transcription
factors or bind to specific DNA, thus promoting the expression of various
genes. GRs are ubiquitously distributed throughout the body and brain,
MRs less so, being found in the kidney and the limbic system. The relative
contribution of the receptor subtypes in the regulation of the HPA axis
remains unclear. MRs have a high affinity for cortisol and aldosterone; GRs
have low affinity for cortisol but avidly bind synthetic steroids. It is pro-
posed that MRs regulate basal cortisol secretion when hormonal levels are
low, and that GRs become increasingly important as levels rise and the MRs
become saturated. GRs may therefore be pivotal in the response to circadi-
an rhythms and to stress.
The HPA axis is vulnerable in a way that mirrors our understanding of
patients’ vulnerabilities. There are genetic effects; genes control the system
at various different levels including hormone production and receptor
expression. The potential interaction from multiple genes of small effect is
large and may predispose to, and perpetuate, abnormal stress reactions.
There are early environmental effects; it is well established that the HPA axis
is open to influence by early life events in utero and the postnatal period.
There are precipitating factors; both physical and psychological stress
results in increased production of cortisol. This is useful in times of acute
stress but is maladaptive in the long term at cellular and system levels.
There are maintaining factors; serotonergic and dopaminergic neurotrans-
mitter systems are thought to be modulated by cortisol, and ongoing hyper-
cortisolaemia may have a detrimental effect upon neuronal function.
Increased frequency of episodes of illness with ongoing time and stress
(eventually entering rapid cycling) could potentially be mediated through the
HPA axis.2
Ch 12 7/4/05 3:57 pm Page 117

118 Bipolar disorder: the upswing in research & treatment
HPA dysfunction in bipolar affective disorder
Abnormalities have been demonstrated and replicated at all levels of theHPA axis in patients suffering from affective disorders. Structurally there isenlargement of the pituitary and adrenal glands. Functionally there areincreased levels of cortisol (present in plasma, urine and cerebrospinalfluid), an exaggerated cortisol response to ACTH, and hypersecretion ofCRH. This pattern of abnormalities is suggestive of an impaired feedbackloop, possibly resulting from GR abnormalities such as decreased receptornumber and/or function. Such abnormalities have been confirmed in post-mortem studies.3 The functional integrity of the receptors can be assessedusing the dexamethasone suppression test (DST), with reports of cortisolnon-suppression in affective disorders supporting a primary GR abnormal-ity.4 The ability of the DST reliably to distinguish between controls andpatients with bipolar disorder may be dramatically increased by being com-bined with a CRH infusion. This dexamethasone (DEX)/CRH test is provingpromising, with significant abnormalities in the HPA axis being demon-strated in patients with bipolar affective disorder.2 Importantly, these abnor-malities are present during both illness and recovery.
Cognitive impairment and hypercortisolaemia
Patients with affective disorder commonly report impairment of cognitivefunction, typically in terms of poor attention, concentration and memory.Baddeley proposed a now popular model of working memory in which acentral executive (the ‘attentional controller’) co-ordinates two slave sys-tems, the ‘phonological loop’ and the ‘visuospatial scratch pad’. This systemis probably served by a wide variety of brain structures including the frontallobes and hippocampal formations. It has been suggested that dysfunctionof the central executive would manifest as impaired set shifting, planning,verbal fluency and response inhibition. Recently, systematic evaluations ofcognitive function in patients with affective disorders have identified specif-ic deficits in executive function, learning and memory tasks.5 These findingswere thought to be independent of drugs effects. Although these deficits canimprove on remission of affective symptoms, the impairment in executivefunction has been shown to persist in a cohort of patients with bipolar affec-tive disorder, prospectively verified as being euthymic.5,6 This challenges theview that patients with severe mood disorders make a full interepisoderecovery.
Animal studies have demonstrated that chronic administration of gluco-corticoids results in abnormalities of learning and memory, with associated
Ch 12 7/4/05 3:57 pm Page 118

Is the hypothalamic–pituitary–adrenal axis at last paying dividends? 119
atrophy of neurons in the hippocampal formation.7 Observation of patientswith pathologically high cortisol levels, such as in Cushing’s disease, pro-vides a natural experiment. It is now established that such patients have sig-nificant cognitive impairments.8 More objective evidence can be drawn fromexperimental models using normal volunteers. Chronic administration ofhydrocortisone to normal male volunteers results in cognitive impairmentsthat appear to be mediated in part via the effects on the frontal lobe. Further,although such subjects have faster reaction times, they make more errorsin keeping with observations of behaviour at times of stress. Recently, it hasbeen demonstrated that the frontal lobes are adversely affected by cortisoland suffer a pattern of degeneration similar to that occurring in the hip-pocampus. It is therefore feasible that the neurodegenerative effects of cor-tisol may underlie some of the cognitive deficits observed in patients withsevere affective disorders.9
Developing therapeutic strategies
There is substantial evidence that HPA axis dysfunction is important in thepathophysiology of mood disorders and the associated cognitive deficits.Modulation of this dysfunction and counteraction of the effects of hypercor-tisolaemia may provide potential treatments for mood disorders.Antidepressant effects have been demonstrated with agents possessingantiglucocorticoid effects,10 steroid synthesis inhibitors11 and pituitary glu-cocorticoid receptor agonists.12
Glucocorticoid receptor antagonists
As previously discussed, one possible explanation for the HPA axis dys-function seen in bipolar affective disorder is a primary abnormality of GRnumber and/or function. A reduction in GR number and/or function isthought to underlie the hypercortisolaemia through impaired negative feed-back. Naturally, the question arises of how the hypercortisolaemia exerts itsdetrimental effect when its mediator, the glucocorticoid receptor, is not func-tioning. There are a number of possible explanations. First, the hypercortis-olaemia may be sufficient to override the impaired GR function with theHPA system re-establishing equilibrium at higher cortisol levels. Second,the receptors regulating HPA feedback (hippocampus and hypothalamus)may be dysfunctional, but other receptors (frontal lobes, etc.) may be intact.Thus, the normal GRs in some brain regions would be exposed to the
Ch 12 7/4/05 3:57 pm Page 119

120 Bipolar disorder: the upswing in research & treatment
detrimental effects of high cortisol levels whilst the receptors governingfeedback would be unable to respond. Finally, it is possible that the adverseeffects of cortisol are mediated through secondary mechanisms or non-receptor-mediated processes.
A novel therapeutic strategy that is attracting increasing attention is theuse of GR antagonists such as mifepristone (Mifeprex, RU-486).13
Administration of GR antagonists is thought to result in an immediateantiglucocorticoid effect followed by a compensatory upregulation of GRnumbers. This upregulation could lead to improved negative feedback,effectively ‘resetting’ the HPA axis. Animal studies have confirmed suchincreases in receptor number following administration of mifepristone.7 GRantagonists have been shown to be beneficial in severe depression, and thatonly a brief period of treatment may be adequate to restore normal HPAfunction.13
We have recently conducted an exploratory trial investigating the effectsof mifepristone as a treatment of bipolar affective disorder. We hypothesizedthat the GR antagonist would improve neurocognitive functioning andreduce depressive symptoms in our patients. To test this hypothesis, weundertook a randomized, double-blind, placebo-controlled trial with across-over design in which mifepristone was administered as an adjunctiveagent. Twenty patients, aged 18–65 years, with a diagnosis of bipolar affec-tive disorder (confirmed using the Structured Clinical Interview for DSM-IV14) were recruited. All patients had residual depressive symptoms.Patients’ medication had been unchanged for 6 weeks prior to entry, andremained so throughout the study. Patients were randomly assigned toreceive either mifepristone 600mg/day for 7 days or placebo, administeredin a double-blind manner. At day 21, the groups crossed over and receivedthe alternative treatment. Neurocognitive function was assessed at baselineand 21 days after each treatment. Mood ratings were performed at baselineand at weekly intervals thereafter.
Fourteen days after the mifepristone administration phase, assessmentof neurocognitive function revealed a significant improvement compared tobaseline. Patients showed a reduced error rate on the spatial working mem-ory task of the magnitude of 20%. The degree of improvement correlatedpositively with baseline cortisol output prior to mifepristone administra-tion. Patients also improved in terms of verbal fluency and spatial recogni-tion memory. No changes were observed at any time points following theplacebo phase. Similarly, mifepristone was associated with improvementsin depression rating scales and the Brief Psychiatric Rating Scale (BPRS) at14 days. No change was seen in the placebo phase. These results support
Ch 12 7/4/05 3:57 pm Page 120

Is the hypothalamic–pituitary–adrenal axis at last paying dividends? 121
the hypothesis that mifepristone, a GR antagonist, improves neurocognitivefunction and has a putative antidepressant action in patients with bipolaraffective disorder.
Conclusion
Abnormalities in the function of the HPA axis, with resultant hypercortisol-aemia, have been consistently demonstrated and replicated in bipolar affec-tive disorder. These abnormalities can persist despite recovery from depres-sion. Neurocognitive impairments, in particular abnormalities on tests offrontal lobe function, are present in patients with bipolar affective disorder.Similarly, these can persist into euthymia.
Hypercortisolaemia is associated with cognitive impairment and neu-rodegeneration, as demonstrated in animal studies and normal human vol-unteers. It is reasonable to suggest that the disruption in mood and cogni-tion seen in bipolar patients could arise, in part, from HPA axis dysfunction.The HPA axis can be successfully manipulated with good therapeutic effect,such that GR antagonists may become a viable treatment option in thefuture. Large-scale trials will be required to confirm this.
References
1. Fish F, Clinical Psychopathology: Signs and Symptoms in Psychiatry. JohnWright and Sons: Bristol, 1967.
2. Watson S, Gallagher P, Ritchie JC et al, Hypothalamic–pituitary–adrenal axisfunction in patients with bipolar disorder. Br J Psychiatry 2004; 18:496–503.
3. Webster MJ, O’Grady J, Orthmann J, Weickert CS, Decreased glucocorticoidsreceptor mRNA levels in individuals with depression, bipolar disorder andschizophrenia. Schizophr Res 2000; 41:111–112.
4. Zhou DF, Shen YC, Shu LN, Lo HC, Dexamethasone suppression test and uri-nary MHPG X SO4 determination in depressive disorders. Biol Psychiatry1987; 22:883–891.
5. Thompson JM, Gray JM, Hughes JH et al, A component process analysis ofworking memory dysfunction in bipolar affective disorder. Bipolar Disord2001; 3:60 (abstr).
6. Ferrier IN, Thompson JM, Cognitive impairment in bipolar affective disorder:implications for the bipolar diathesis. Br J Psychiatry 2002; 180:293–295.
7. Lupien SJ, McEwan BS, The acute effects of corticosteriods on cognition: inte-gration of animal and human model studies. Brain Res Bain Res Rev 1997;24:1–27.
Ch 12 7/4/05 3:57 pm Page 121

122 Bipolar disorder: the upswing in research & treatment
8. Wolkowitz OM, Reus VI, Weingartner H et al, Cognitive effects of corticosteroids.Am J Psychiatry 1990; 147:1297–1303.
9. Sapolsky RM, Krey LC, McEwan BS, The neuroendocrinology of stress andaging: the glucocorticoid cascade hypothesis. Endocr Rev 1986; 7:284–301.
10. Wolkowitz OM, Reus VI, Keebler R et al, Double-blind treatment of majordepression with dehydroepiandrosterone. Am J Psychiatry 1999; 156:646–649.
11. Ravaris CL, Nelpa I, Huang M et al, Effect of ketoconazole on a hypophysec-tomized, hypercortisolemic, psychotically depressed woman. Arch GenPsychiatry 1988; 45:966–967.
12. Arana GW, Santos AB, Laraia MT et al, Dexamethasone for the treatment ofdepression: a randomized, placebo-controlled, double-blind trial. Am JPsychiatry 1995; 152:265–267.
13. Murphy BE, Filipini D, Ghadirian JL, Possible use of glucocorticoid receptorantagonists in the treatment of major depression: preliminary results usingRU486. J Psychiatry Neurosci 1993; 18:209–213.
14. First MB, Spitzer RL, Gibbon M et al, Structured Clinical Interview for DSM-IV Axis I Disorders (SCID). New York State Psychiatric Institute, BiometricsResearch: New York, 1999.
Ch 12 7/4/05 3:57 pm Page 122

Stress on the brain:neuropathology andcortisol dysregulationin bipolar disorderDavid Cotter
c h a p t e r 1 3
Introduction
Considering the prevalence and importance of major depressive disorder(MDD) and bipolar disorder (BPD), it is surprising that we have only veryrecently started to look carefully at the cerebral cellular architecture ofthese disorders. Structural imaging investigations of living subjects withmood disorders can point us in the direction of pathology. These imaginginvestigations have shown a variety of important changes. In the case ofMDD, the subgenual anterior cingulate cortex, the hippocampus and thefrontal cortex have been shown to be reduced in volume. In BPD, the hip-pocampal volume has also been shown to be reduced, and the amygdalamay be increased in size. Functional imaging investigations have also pro-vided clues to the neuronal circuits implicated in mood disorders and pointto involvement of the corticolimbic and the corticostriatal networks.
Neuropathology of major depression and bipolardisorder
Neuronal size correlates with the extent of a neuron’s efferent and afferentconnections, and consequently reduced neuronal size points to functionaland/or structural dysconnectivity. Neuronal number is important, as adeficit would have functional implications and may reflect abnormal cell
Ch 13 7/4/05 3:58 pm Page 123

124 Bipolar disorder: the upswing in research & treatment
death or apoptosis, such as occurs in some neurodegenerative disorders, ormay reflect altered neurogenesis. In MDD and BPD, there is evidence forreduced neuronal size in the anterior cingulate cortex, the dorsolateral pre-frontal cortex and the orbitofrontal cortex. There may also be a reduceddensity of larger neurons in MDD and BPD, which probably equates with areduced median or mean neuronal size. Reduced density of smallerinterneurons in BPD has also been described.
Deficits in glial cell numbers and density are observed in MDD, and to alesser extent in BPD. This is exciting, because the finding is relatively new(the main finding and the replications have occurred within the past 4 years)and potentially important in terms of providing new therapeutic strategies.Glial cells have traditionally been viewed as neuronal supporting cells in thecentral nervous system – so-called ‘mind glue’ with their primary roles inglutamatergic neurotransmission, glucose metabolism and neurotrophicsupport largely ignored. However, over the past few years several independ-ent studies have shown a cortical glial cell deficit in affective disorders, andthese observations have come at a time when the important role of glial cellsin normal cortical function has been fundamentally re-evaluated. Theorbitofrontal cortex, anterior cingulate cortex and dorsolateral prefrontalcortex are the regions in which these changes have been observed. This isin keeping with the functional and structural imaging studies, and indeedthe neuropsychological investigations that have already implicated thesebrain regions. Glial cells are composed of three different cell types(microglia, oligodendroglia and astrocytes) and it is not yet clear which ofthese is responsible for the observed deficit in MDD. Support for the possi-bility that astrocyte deficits are responsible for the glial cell changes havebeen provided by proteomics investigations based on the StanleyFoundation Brain series, although others have also found evidence forreductions in markers of oligodendroglia.
Synaptic changes have also been observed in BPD and MDD. However,because postmortem tissue is not always suitable for quantitation of den-drites and synapses, they have been evaluated through a number of meth-ods including direct assessment of silver-stained dendrites and dendriticspines, immunocytochemical staining of dendrites and quantitation of geneproducts localized to synaptic and presynaptic compartments. Three synap-tic proteins (synaptophysin, complexin I and growth-associated protein-43(GAP-43)) have been shown to be reduced in the anterior cingulate cortex inBPD, with only complexin II reduced in MDD. In the prefrontal cortex inMDD, GAP-43 and synaptophysin are unaltered and there are no studies yetof BPD. Studies of the hippocampus have shown reduced levels of synaptic
Ch 13 7/4/05 3:58 pm Page 124

Stress on the brain: neuropathology and cortisol dysregulation 125
associated protein-25 and complexin I and II in BPD, with no changes inMDD. The consensus of these studies is that synaptic pathology is presentin mood disorders in the cortical limbic regions and that the changes aremore marked in BPD than MDD.
Magnetic resonance imaging (MRI) investigations have shown a strongassociation between subcortical white matter hyperintensities (WMH) onT2-weighted images and both MDD and BPD. These lesions are observedchiefly in the deep white matter, around the basal ganglia and in the periven-tricular region. In MDD these lesions are more common in the elderly withvascular disease. In both elderly MDD and BPD subjects, WMHs confer apoorer prognosis. Diffusion tensor imaging has shown that WMH reflectdamage to white matter tracts, leading to the suggestion that the mood dis-order in these subjects is due to interruption of the frontal cortical–subcor-tical connections. While the neuropathological basis of these lesions has notyet been characterized, it is likely that mood disorder of this type is neu-ropathologically distinct.
Evidence for a common neuropathology in majordepression, bipolar disorder and schizophrenia
There are some important similarities in the neuropathology of schizophre-nia, MDD and BPD, which suggest that a common process of change isinvolved in each disorder. Macroscopic neuroanatomical investigations ofbrain pathology in schizophrenia, BPD and MDD show differences that aregenerally quantitative rather than qualitative. For example, ventriculardilatation and reduced hippocampal and frontal brain volumes are seen inschizophrenia, but they are also present to a lesser degree in MDD and BPD.The single main departure from this pattern is that the volume of the amyg-dala may be specifically enlarged in BPD, possibly because of drug treat-ment. Microscopically, reductions in dendritic spine density, neuronal size,and synaptic proteins have been described in mood disorders as well as inschizophrenia. More recently it has become apparent that glial cell loss maybe a feature of MDD, BPD and schizophrenia, depending, possibly, on thepresence of co-existing affective symptoms and on which region of the brainis investigated.
This similar pattern of changes in cortical cellular architecture in schiz-ophrenia and mood disorders suggests that a common pathophysiologymay underlie aspects of these psychiatric diseases. What aspects of illness,common to MDD, BPD and schizophrenia, could cause changes in keeping
Ch 13 7/4/05 3:58 pm Page 125

126 Bipolar disorder: the upswing in research & treatment
with the known cellular changes described above? Glucocorticoid-relatedneurotoxicity is a candidate that needs to be considered.
A role for glucocorticoids in the neuronal changesof mood disorders and schizophrenia?
There is substantial evidence that hyperactivity of the hypothalamic–pituitary-adrenal (HPA) axis is involved in the pathogenesis of mood disor-der. Impaired function of the glucocorticoid receptor (GR) and subsequentaltered feedback inhibition by endogenous glucocorticoids probably repre-sents the mechanism by which the HPA axis is activated in depression. Incontrast, to date there has been no firm evidence that HPA hyperactivity ispart of the pathogenesis of schizophrenia. This may be because few studieshave assessed subjects during the acute phase of the schizophrenic illnesswhich is the period of the illness most likely to be associated with a stress-related elevation of glucocorticoids. Recently, however, reduced GR geneexpression has been described in the frontal cortex in schizophrenia andmajor depression, providing the first firm evidence that HPA axis abnor-malities are a feature of these disorders.
There are several other lines of investigation that support the view thatglucocorticoid-related neurotoxicity may be implicated in depression andschizophrenia. First, in vitro investigations have shown that high levels ofglucocorticoid hormones result in reduced neuronal volume and dendriticarborization, and these changes have been observed in both disorders.Second, elevated plasma glucocorticoid levels are associated with hip-pocampal volume reductions in MDD, post-traumatic stress disorder,Cushing’s disease and normal aging, and such reductions have beenobserved in the phase of a first psychotic episode. Third, the functionaleffect of glucocorticoids on reducing hippocampal glial cell activation andproliferation mirrors the glial deficit observed in MDD, BPD and possiblyschizophrenia. Consequently, the glial deficit found in these disorders mayalso relate to glucocorticoid effects.
Conclusion
Evidence is accumulating that there are brain changes occurring during andpossibly after the period of the first presentations of BPD and schizophre-nia, i.e. changes that are not developmental in the traditional sense.
Ch 13 7/4/05 3:58 pm Page 126

Stress on the brain: neuropathology and cortisol dysregulation 127
Furthermore, these changes are not specific to schizophrenia, in terms of
either macroscopic or microscopic brain structure, for they are also pres-
ent to a generally milder degree in subjects with BPD and MDD, and they
are in keeping with glucocorticoid-related brain changes. These brain
changes may be epiphenomena secondary to stress-related changes in
glucocorticoid hormones and not primary pathogenetic pathways.
Nevertheless, they could have crucial clinical effects through diminishing
neuronal and cortical function and so complicate recovery from the primary
illness. These changes may possibly be reversed by therapies that protect
from glucocorticoid-related neurotoxicity or which act to promote neuro-
protective cell-signalling pathways. It will now be important to understand
these illnesses in terms of both early and late events that involve both devel-
opmental and atrophic processes.
Bibliography
Altshuler LL, Bartzokis G, Grieder T et al, Amygdala enlargement in bipolar disorderand hippocampal reduction in schizophrenia: an MRI study demonstrating neu-roanatomic specificity. Arch Gen Psychiatry 1998; 55:663–664.
Cotter D, Mackay D, Landau S et al, Glial cell loss and reduced neuronal size in theanterior cingulate cortex in major depressive disorder. Arch Gen Psychiatry 2001;58:545–553.
Cotter DR, Pariante CM, Everall IP, Glial cell abnormalities in major psychiatric dis-orders: the evidence and implications. Brain Res Bull 2001; 55:585–595.
Drevets WC, Neuroimaging and neuropathological studies of depression: implicationsfor the cognitive–emotional features of mood disorders. Curr Opin Neurobiol 2001;11:240–249.
Eastwood SL, Harrison PJ, Synaptic pathology in the anterior cingulate cortex inschizophrenia and mood disorders. A review and a Western blot study of synapto-physin, GAP-43, and the complexins. Brain Res Bull 2001; 55:569–578.
Harrison PJ, The neuropathology of schizophrenia. A critical review of the data andtheir interpretation. Brain 1999; 122:593–624.
Harrison PJ, The neuropathology of primary mood disorder. Brain 2002;125:1428–1449.
Ismail K, Murray RM, Wheeler MJ, O’Keane V, The dexamethasone suppression testin schizophrenia. Psychol Med 1998; 28:311–317.
Lawrie SM, Whalley H, Byrne M et al, Brain structure change and psychopathology insubjects at high risk of schizophrenia. Schizophr Res 2000; 41:11.
Ch 13 7/4/05 3:58 pm Page 127

128 Bipolar disorder: the upswing in research & treatment
McCarley RW, Wible CG, Frumin M et al, MRI anatomy of schizophrenia. BiolPsychiatry 1999; 45:1099–1119.
Pantellis C, Velakoulis D, Suckling J et al, Left medial temporal lobe volume reduc-tion occurs during the transition from high risk to first episode psychosis. SchizophrRes 2000; 41:35.
Pariante CM, Miller AH, Glucocorticoid receptors in major depression: relevance topathophysiology and treatment. Biol Psychiatry 2001; 49:391–404.
Pearlson GD, Structural and functional brain changes in bipolar disorder: a selectivereview. Schizophr Res 1999; 39:133–140; discussion 162.
Rajkowska G, Miguel-Hidalgo JJ, Wei J, Morphometric evidence for neuronal andglial prefrontal cell pathology in major depression. Biol Psychiatry 1999;45:1085–1098.
Rappaport JL, Giedd JN, Blumenthal J et al, Progressive cortical change during ado-lescence in childhood-onset schizophrenia. A longitudinal magnetic resonance imag-ing study. Arch Gen Psychiatry 1999; 56:649–654.
Rosoklija G, Toomayan G, Ellis SP et al, Structural abnormalities of subicular den-drites in subjects with schizophrenia and mood disorders. Arch Gen Psychiatry2000; 57:349–356.
Sapolsky R, The possibility of neurotoxicity in the hippocampus in major depression:a primer on neuron death. Biol Psychiatry 2000; 48:755–765.
Webster MJ, O’Grady J, Orthmann C, Weickert C, Decreased glucocorticoid receptormRNA levels in individuals with depression, bipolar disorder and schizophrenia.Schizophr Res 2000; 41:111.
Weinberger DR, Implications of normal brain development for the pathogenesis ofschizophrenia. Arch Gen Psychiatry 1987; 44:660–669.
Ch 13 7/4/05 3:58 pm Page 128

Cortisol in Chicago(from crime of passionto celebrity headline)Carmine M Pariante
c h a p t e r 1 4
Cortisol, the stress hormone that is life-saving in a variety of circumstances,is considered a ‘criminal’ by part of the scientific community. It has beenaccused of killing the brain, inducing hippocampal atrophy, blocking neu-rogenesis, causing cognitive abnormality, even of making one depressed.This has been a real press campaign against the poor hormone! In the 2003award movie, Chicago, it takes a very good lawyer to get two beautiful mur-derers out of prison and ‘rehabilitated’ in the newspapers. Clearly, cortisolalso needs a good lawyer.
So, who is the defendant? The hypothalamic–pituitary–adrenal (HPA)axis, producing cortisol, is the main stress hormonal response system. Asshown in Figure 14.1, HPA axis activity is governed by the secretion ofcorticotrophin releasing hormone (CRH) from the hypothalamus. CRH acti-vates the secretion of adrenocorticotrophic hormone (ACTH) from the pitu-itary. ACTH, in turn, stimulates the secretion of glucocorticoids (cortisol inhumans) from the adrenal glands, two small organs situated on top of thekidneys. Glucocorticoids interact with their corticosteroid receptors − theglucocorticoid receptor (GR) and the mineralocorticoid receptor (MR) − inalmost every tissue in the body, and the best-known physiological effect isthe regulation of energy metabolism. By binding to corticosteroid receptorsin the brain, glucocorticoids also inhibit the secretion of CRH from thehypothalamus and ACTH from the pituitary (negative feedback).
There is a variety of data produced over the past 30 years showing thatthe most severe phases of mental disorders such depression, mania andschizophrenia are associated with hyperactivity of the HPA axis and
Ch 14 7/4/05 3:58 pm Page 129

130 Bipolar disorder: the upswing in research & treatment
increased concentrations of cortisol in the bloodstream. This hyperactivityhas been demonstrated by findings such as the lack of suppression of cor-tisol secretion by dexamethasone, the increased level of CRH in the brain,the increased volume of the anterior pituitary gland, the increased volumeof the adrenal gland and, of course, the increased level of the circulating cor-tisol.1–3 Indeed, we have recently demonstrated increased volume of thepituitary gland in two independent samples of patients at their first episodeof psychosis, including psychotic depression, psychotic mania and schizo-phrenia.4,5 In fact, during a situation of stress, the pituitary cells producingACTH (and other hormones participating in the stress response) proliferate,and the pituitary becomes macroscopically larger. In our first study,4 in anAustralian sample, the first-episode subjects had pituitary volumes thatwere 10% larger than those of controls. However, because they were receiv-ing antipsychotics, we could not exclude the hypothesis that the pituitaryhyperplasia could be related, at least in part, to increased function ofprolactin-producing cells activated by neuroleptic treatment. However, inthe second study,5 conducted in London as part of the AESOP First-OnsetPsychosis Study, this effect was present even in neuroleptic-free first-
Figure 14.1 Schematic diagram of the hypothalamic–pituitary–adrenal axis,describing regulation and negative feedback (–) of cortisol via glucocorticoid recep-tors (GRs) and mineralocorticoid receptors (MRs). CRF, corticotrophin releasing fac-tor; AVP, arginine vasopressin; ACTH, adrenocorticotrophic hormone.
Amygdala Midbrain Hippocampus
Hypothalamus
CRFAVP
Pituitary
ACTH
GRs
GRs/MRs
GRs
Circulating cortisol
Adrenal cortex
–/+
–
–
–
Ch 14 7/4/05 3:58 pm Page 130

Cortisol in Chicago 131
episode patients (>15%), although it was more evident in patients receivingtypical antipsychotics (>30%).
Having demonstrated that the severe phases of affective and psychoticdisorders are characterized by HPA axis hyperactivity, we argue that there isno conclusive evidence that these elevated levels of cortisol can cause thepsychiatric symptoms or induce the brain functional and anatomical abnor-malities that are described in these patients. Indeed, we claim that the oppo-site is true: that patients have a hyperactive HPA axis as a compensatorymechanism, because their brain is resistant to the effects of circulating cor-tisol. The accepted explanation for HPA axis hyperactivity in depression andduring stress (and possibly in psychosis) is reduced function of the GR andthe MR that mediate the inhibition of CRH and ACTH secretion.3,6
According to this model, because of the reduced function of these receptors,the circulating cortisol is unable to inhibit HPA axis activity successfully(‘glucocorticoid resistance’). Consistent with this model, antidepressants(including lithium) directly increase the function of corticosteroid receptorsin the brain, thus increasing the effects of cortisol on the brain, enhancingthe negative feedback and reducing HPA axis activity.3,7,8 The reduction ofGR levels in the brains of patients with schizophrenia and bipolar disor-der,9,10 and the reduction of GR function in patients with depression and insubjects experiencing chronic stress,3,6 all suggest that elevated plasma cor-tisol levels represent a compensatory strategy. Moreover, recent studies haveindicated that levels of cortisol in the brain of animals and humans are reg-ulated by efflux systems at the blood–brain barrier, that are influenced bypsychotropic drugs.7,11 For example, we have shown that only 20% of cir-culating cortisol can access the brain in the guinea pig, an animal that, likehumans, has cortisol as the main stress hormone.7 This indicates thatperipheral cortisol levels, as often assumed in studies, may not necessarilydictate cerebral levels. Furthermore, our work suggests that a mechanismby which antidepressants increase cortisol effects on the brain is by regu-lating these transporters and increasing the access of cortisol to the brain.7
Finally, further evidence ‘defending’ cortisol comes from studies using HPAaxis manipulation to induce antidepressant effects. In fact, treatment withGR and MR agonists, including cortisol, has antidepressant effects inhumans,12–14 again suggesting that these patients do not experience ‘toomuch cortisol’ in their brain. Surprisingly, even the therapeutic effects ofRU-486, a GR antagonist, in the treatment of psychotic depression15 andbipolar disorder16 could be explained by the fact that this drug completelyblocks the negative feedback, and thus causes a persistent elevation of cor-tisol levels and therefore an increase in cortisol signal in the brain.
Ch 14 7/4/05 3:58 pm Page 131

132 Bipolar disorder: the upswing in research & treatment
Clearly, whether patients with major depression, bipolar disorder or
schizophrenia have elevated or lowered activation of the corticosteroid
receptors in the brain is yet to be conclusively elucidated. Nevertheless, we
believe that the putative ‘neurotoxic’ effects of cortisol have been
undeservedly emphasized. Defending cortisol is not only about getting an
innocent out of Chicago jails (see Figure 14.2) but also about following all
the available leads – in all directions – to understand the pathophysiology of
mental disorders.17,18
Acknowledgements
Our research is funded by the UK Medical Research Council (MRC), the
National Alliance for Research on Schizophrenia and Depression
(NARSAD), the Rosetrees Trust and the Psychiatry Research Trust.
References
1. Pariante CM, Depression, stress and the adrenal axis. J Neuroendocrinol 2003;15:811–812.
2. Cotter D, Pariante CM, Stress and the progression of the developmental hypoth-esis of schizophrenia. Br J Psychiatry 2002; 181:363–365.
Figure 14.2 Who is innocent?
Cortisol
Ch 14 7/4/05 3:58 pm Page 132

Cortisol in Chicago 133
3. Pariante CM, Miller AH, Glucocorticoid receptors in major depression: rele-vance to pathophysiology and treatment. Biol Psychiatry 2001; 49:391–404.
4. Pariante CM, Vassilopoulou K, Velakoulis D et al, Pituitary volume in psychosis.Br J Psychiatry 2004; 185:5–10.
5. Pariante C, Brudaglio F, Danese A et al, Increased pituitary volume in patientsof the AESOP First-Onset Psychosis Study. Schizophr Res 2004; 67(Suppl1):99–100.
6. Raison CL, Miller AH, When not enough is too much: the role of insufficient glu-cocorticoid signaling in the pathophysiology of stress-related disorders. Am JPsychiatry 2003; 160:1554–1565.
7. Pariante CM, Thomas SA, Lovestone S et al, Do antidepressants regulate howcortisol affects the brain? 2003 Curt Richter Award Paper. Psychoneuro-endocrinology 2004; 29:423–447.
8. Pariante CM, Papadopoulos AS, Poon L et al, Four days of citalopram increasesuppression of cortisol secretion by prednisolone in healthy volunteers.Psychopharmacology (Berl) 2004; 177:200–206.
9. Xing GQ, Russell S, Webster MJ, Post RM, Decreased expression of mineralo-corticoid receptor mRNA in the prefrontal cortex in schizophrenia and bipolardisorder. Int J Neuropsychopharmacol 2004; 7:143–153.
10. Webster MJ, Knable MB, O’Grady J et al, Regional specificity of brain gluco-corticoid receptor mRNA alterations in subjects with schizophrenia and mooddisorders. Mol Psychiatry 2002; 7:985–94, 924.
11. de Kloet ER, Vreugdenhil E, Oitzl MS, Joels M, Brain corticosteroid receptorbalance in health and disease. Endocr Rev 1998; 19:269–301.
12. Dinan TG, Lavelle E, Cooney J et al, Dexamethasone augmentation intreatment-resistant depression. Acta Psychiatr Scand 1997; 95:58–61.
13. Bouwer C, Claassen J, Dinan TG, Nemeroff CB, Prednisone augmentation intreatment-resistant depression with fatigue and hypocortisolaemia: a caseseries. Depress Anxiety 2000; 12:44–50.
14. DeBattista C, Posener JA, Kalehzan BM, Schatzberg AF, Acute antidepressanteffects of intravenous hydrocortisone and CRH in depressed patients: a double-blind, placebo-controlled study. Am J Psychiatry 2000; 157:1334–1337.
15. Belanoff JK, Flores BH, Kalezhan M et al, Rapid reversal of psychotic depres-sion using mifepristone. J Clin Psychopharmacol 2001; 21:516–521.
16. Young AH, Gallagher P, Watson S et al, Improvements in neurocognitive functionand mood following adjunctive treatment with mifepristone (RU-486) in bipo-lar disorder. Neuropsychopharmacology 2004; 29:1538–1545.
17. Juruena MF, Cleare AJ, Pariante CM, Hypothalamic pituitary adrenal axis,glucocorticoid receptor function and relevance to depression. Rev BrasPsiquiatr 2004; 26:189–201.
18. Juruena MF, Cleare AJ, Bauer ME, Pariante CM, Molecular mechanisms of GRsensitivity and relevance for affective disorders. Acta Neuropsychiatrica 2003;15:354–367.
Ch 14 7/4/05 3:58 pm Page 133

Ch 14 7/4/05 3:58 pm Page 134

Biological factors sustaininghypothalamic–pituitary–adrenalaxis overactivation in affectivedisorder: focus on vasopressin
Timothy G Dinan, Sinead O’Brien and Lucinda Scott
c h a p t e r 1 5
Overactivity of the hypothalamic–pituitary–adrenal (HPA) axis characterizedby hypercortisolism, adrenal hyperplasia and abnormalities in negativefeedback is the most consistently described biological abnormality inmelancholic depression.1 Corticotrophin releasing hormone (CRH) andarginine vasopressin (AVP) are the main secretagogues of the HPA/stresssystem. CRH is a peptide of 41 amino acid residues, originally discoveredand sequenced by Vale et al,2 which is produced in the medial parvicellularneurones of the paraventricular nucleus of the hypothalamus. These neu-rones project to the external zone of the median eminence, where CRH isreleased into the portal vasculature to act on CRH1 receptors at the anteri-or pituitary. CRH and AVP act synergistically in bringing about adrenocorti-cotrophin (ACTH) release from the corticotrophs of the anterior pituitarywhich in turn stimulates cortisol output from the adrenal cortex. Followingits identification in 1954, vasopressin, a nonapeptide, was considered theprincipal factor in the regulation of ACTH release but, with the subsequentelucidation of the structure of CRH and the domination of the oneneurone/one transmitter principle, the role of CRH came to overshadow thatof AVP. This dominance has been reflected in neuroendocrine studies con-ducted in depression.
Arginine vasopressin neuroanatomy
AVP is released following a variety of stimuli, including increasing plasmaosmolality, hypovolaemia, hypotension and hypoglycaemia. It has powerful
Ch 15 7/4/05 3:59 pm Page 135

136 Bipolar disorder: the upswing in research & treatment
antidiuretic and vasoconstrictor effects. AVP has also been implicated inlearning and memory processes.3 Our knowledge of the functional activityand pharmacology of AVP and its receptors in the regulation of HPA activi-ty rests largely on studies conducted in rodents. AVP is released from themagnocellular system and from the parvicellular neurones of the paraven-tricular nucleus (PVN). AVP produced by the parvicellular neurones of thePVN is secreted into the pituitary portal circulation from axon terminalsprojecting to the external zone of the median eminence.4 AVP-containing cellbodies in the PVN are co-localized with CRH-containing neurones. In con-trol non-stressed rats, within the pool of CRH neurosecretory cells, 50% co-express AVP.5 CRH+/AVP– neurones are mostly found medially and ventral-ly, whereas CRH+/AVP+ cells are located more dorsally and laterally in thePVN. The neurosecretory granules in CRH-neurones undergo remarkablechanges in size and peptide composition under experimental manipulationsof the HPA axis.
The PVN serves as an important relay site. It receives projections fromascending catecholaminergic pathways including noradrenergic projectionsfrom the nucleus of the solitary tract and from the locus coeruleus. The PVNalso receives input from areas of the limbic system, notably the bed nucle-us of the stria terminalis, the hippocampus and amygdala. In primates,including humans, high levels of immunoreactive AVP neurones have beendemonstrated in these areas and also in the pituitary intermediate lobe,granular layers of the cerebellum and dentate gyrus. Lower levels are foundin the medial habenula, adenohypophysis, area postrema, pineal, subforni-cal and subcommisural organs.
Whilst it is evident that almost all of the CRH in the hypophyseal portalblood originates from the hypothalamic PVN, the precise origin of AVP ismore controversial. It is thought that the bulk of AVP derives from the PVN.However, morphological and neurochemical studies suggest that AVP fromsupraoptic magnocellular AVP-secreting cells also access the hypophysealportal blood,4 although this has not been definitively shown in humans.
Vasopressin receptors
As with other peptide hormones, AVP exerts its effects through interactionwith specific plasma membrane receptors, of which three major subtypeshave been identified. V1a receptors are widely distributed on blood vessels,and have also been found in the central nervous system (CNS), including thePVN. V2 receptors are predominantly located in the principal cells of the
Ch 15 7/4/05 3:59 pm Page 136

HPA axis overactivation in affective disorder 137
renal collecting system, although there is some evidence for central V2receptors also. The ACTH-releasing properties occur via the V3 (V1b) recep-tor subtype. In situ hybridization studies reveal that V3 receptor mRNA isexpressed in the majority of pituitary corticotrophs, in multiple brainregions and a number of peripheral tissues including kidney, heart, lung,breast and adrenal medulla.6
Synergism of corticotrophin releasing hormoneand arginine vasopressin
Vasopressin has ACTH-releasing properties when administered alone inhumans, a response which may be dependant on the ambient endogenousCRH level. Following the combination of AVP and CRH, a much greaterACTH response is seen and both peptides are required for maximalpituitary–adrenal stimulation. The precise nature of this synergism isincompletely understood, with most information deriving from animal stud-ies. It has been demonstrated that CRH, through cAMP, increases POMC
gene transcription and peptide synthesis and storage. There may also bedistinct corticotroph populations in the anterior pituitary, some of whichrequire both AVP and CRH for ACTH release. Antoni4 suggests that AVPplays a role in stimulating the primary nuclear transcripts induced by CRHat the pituitary corticotroph.
Corticotrophin releasing hormone and vasopressinregulation by glucocorticoids
Glucocorticoids play a pivotal feedback role in the regulation of the HPA.Two types of cortisol-binding sites have been described in the brain. Thetype 1 receptor, which is indistinguishable from the peripheral mineralo-corticoid receptor, is distributed principally in the septohippocampalregion. The type 2 or glucocorticoid receptor has a wider distribution.These receptor systems provide negative feedback loops at a limbic, hypo-thalamic and pituitary level. Overall the type 1 receptor is thought to medi-ate tonic influences of cortisol or corticosterone, whilst the type 2 receptormediates stress-related changes in cortisol levels. Under normal conditionsthe responsiveness of parvicellular neurones to stress is under marked inhi-bition by the low resting levels of glucocorticoids.
Ch 15 7/4/05 3:59 pm Page 137

138 Bipolar disorder: the upswing in research & treatment
The sensitivity of CRH and AVP transcription to glucocorticoid feedbackis markedly different.7 CRH mRNA and CRH1 receptor mRNA levels arereduced by elevated glucocorticoids, whereas V3 receptor mRNA levels andcoupling of the receptor to phospholipase C are stimulated by glucocorti-coids, effects which may contribute to the refractoriness of AVP-stimulatedACTH secretion to glucocorticoid feedback.
Effects of chronic stress on corticotrophin releasinghormone/arginine vasopressin
Immobilization stress and the use of hierarchically structured colonies bothfunction as ‘psychological stress’ paradigms. Studies employing these para-digms in rats have revealed a shift of the hypothalamic CRH/AVP signal infavour of AVP; there is enhanced AVP synthesis in CRH-producing cells ofthe parvicellular neurones of the PVN and AVP storage in the CRH-containing nerve terminals in the external zone of the median eminence. Anincreased proportion of hypothalamic neurones co-expressing AVP is alsoobserved. In contrast, CRH production remains unaltered or decreased. Asimilar pattern of response is seen following prolonged immobilizationstress where a shift towards an increased AVP/CRH ratio is observed.
Direct manipulations of the HPA underscore this thesis. Tizabi andAguilera,8 using a minipump infusion of CRH, revealed a reduction in CRHreceptor number which was enhanced by the simultaneous infusion of AVP.Repeated immobilization stress was found to bring about sustained eleva-tions in V3 receptor mRNA in the pituitary, suggesting an upregulation ofAVP receptors in situations of chronic stress. This may explain the pre-served enhanced response to novel superimposed stress in these animalmodels.
Vasopressin in major depression
Studies of HPA function in depression have revealed numerous abnormali-ties. These abnormalities are most pronounced in depressives with melan-cholic features.9 A potential role for AVP in affective illness was put forwardin 1978 by Gold and Goodwin.10 The hypothesis was based on the obser-vations that (1) AVP deficiency produces deficits of behaviour which arereversed when the peptide is replaced, and (2) well-developed systems existfor distribution of AVP throughout the CNS, rendering AVP a suitable
Ch 15 7/4/05 3:59 pm Page 138

HPA axis overactivation in affective disorder 139
candidate for involvement in complex behavioural systems. They also
described the symptom complexes in affective illness that AVP is known to
influence, notably memory processes, pain sensitivity, synchronization of
biological rhythms and the timing and quality of rapid eye movement (REM)
sleep.
A role for AVP was supported not only by the above spectrum of symp-
toms but also by dynamic tests of HPA activity, and, in particular, the
‘DEX/CRH’ test. Dexamethasone (DEX), a potent synthetic glucocorticoid,
binds primarily to the glucocorticoid receptor on anterior pituitary corti-
cotrophs and, by feedback inhibition, suppresses ACTH and cortisol secre-
tion.11 In the DEX/CRH test, when healthy subjects are treated with dexa-
methasone prior to CRH infusion, the release of ACTH is blunted and the
extent of blunting is proportional to the dose of DEX. Paradoxically, when
depressives are pretreated with DEX they show an enhanced ACTH
response to CRH. It was postulated that vasopressin (VP)-mediated ACTH
release was responsible for this finding. The combined DEX/CRH test is
estimated to have a sensitivity of 80% in differentiating healthy subjects
from depressives.12
Basal measures and symptom profile
There are relatively few data on plasma AVP levels in depression. An early
report found no change in plasma AVP levels in depression.13 In contrast,
van Londen et al14 reported basal plasma levels of AVP to be elevated. In
that study, 52 major depressives and 37 healthy controls were compared;
AVP concentrations were found to be higher in the depressed cohort, with
greater elevation in in-patient compared with out-patient depressives and in
those with melancholic features. A number of studies have shown a signifi-
cant positive correlation between peripheral plasma levels of AVP and
hypercortisolaemia in patients with unipolar depression.14
Postmortem studies
A postmortem study of depressed subjects reported an increased number
of vasopressin-expressing neurones in paraventricular hypothalamic neu-
rones.15 Raadsheer et al16 have also shown an increase in number of CRH-
neurones in depressives.
Ch 15 7/4/05 3:59 pm Page 139

140 Bipolar disorder: the upswing in research & treatment
Dynamic tests of hypothalamic–pituitary–adrenalfunction and the vasopressin-ergic system
Dinan et al17 examined a cohort of depressed subjects on two separate occa-sions, with CRH alone, and with the combination of CRH and desmopressin(DDAVP). A significant blunting of ACTH output to CRH alone was noted.Following the combination of CRH and DDAVP, the release of ACTH indepressives and healthy volunteers was indistinguishable. It was concludedthat, whilst the CRH1 receptor is downregulated in depression, a concomi-tant upregulation of the V3 receptor takes place. This is consistent with theanimal models of chronic stress, described above, in which a switchingfrom CRH to AVP regulation is observed. It is interesting that, in CRH1receptor-deficient mice, basal plasma AVP levels are significantly elevated,AVP mRNA is increased in the PVN and there is increased AVP-likeimmunoreactivity in the median eminence.18
In a recent study we have provided further evidence for the upregulationof the anterior pituitary V3 receptor.19 Fourteen patients with major depres-sion and 14 age- and sex-matched healthy comparison subjects wererecruited. Desmopressin 10µg was given intravenously and ACTH and cor-tisol release was monitored for 120 minutes. The mean±SEM ACTHresponse in the depressives was 28.4±4.3ng/l and in the healthy subjectswas 18.8±4.9ng/l (p=0.04). The mean±SEM cortisol response in thedepressives was 261.8±46.5nmol/l and in the healthy subjects it was107.3±26.1nmol/l (p<0.01). This suggests that patients with majordepression have augmented ACTH and cortisol responses to desmopressinindicating enhanced V3 responsivity.
Treatment with fluoxetine decreases cerebrospinal fluid (CSF) levels ofCRH and AVP.20 Chronic fluoxetine administration has also been shown toreduce hypothalamic AVP secretion in vitro. In contrast, Heuser and co-workers21 examined CSF concentrations of CRH, AVP and somatostatin indepressed patients and in healthy controls prior to, and following,amitriptyline treatment. In treatment responders, CSF CRH was reducedwith no effect of amitriptyline on AVP or somatostatin levels. Larger studiesare required before any conclusions can be drawn about an antidepressanteffect on central AVP activity.
The question arises as to what type of involvement AVP may have in HPAdysregulation in depressive illness. Is a change in AVP regulation a biologi-cal correlate, a component of aetiology or a response to another hormonaldisequilibrium? An understanding of major depression as a disorder of thestress system is fundamental in attempting to elucidate these possibilities.
Ch 15 7/4/05 3:59 pm Page 140

HPA axis overactivation in affective disorder 141
Activation of the HPA in situations of stress is a normal homeostatic mech-
anism. In healthy subjects this response ceases when it is no longer biolog-
ically relevant. It is on this basis that we suggest an important role for AVP
in the pathophysiology of major depression. Extrapolating from animal
models of chronic stress, in which there is a switch from CRH to AVP regu-
lation of ACTH release, a similar occurrence in humans is readily concep-
tualized. Supportive evidence includes the demonstration in depressed sub-
jects of increased numbers of CRH neurones in the PVN co-expressing AVP
and the ability of DDAVP to normalize the blunting of CRH-induced ACTH
release, suggesting pituitary V3 receptor upregulation.17 Of importance also
is the relative refractoriness of AVP-stimulated ACTH release to circulating
glucocorticoids. This would suggest that, in major depression, the constant
forward drive of the HPA axis may depend on AVP activity unrestrained by
elevated ambient cortisol levels. The switch in emphasis from CRH to AVP-
ergic pituitary–adrenal regulation in situations of chronic stress results in a
continued activation of the HPA axis after the initial CRH-mediated response
to stress is biologically appropriate. This recruitment of AVP as the primary
regulator of the HPA axis in chronic stress conditions may explain the
hypercortisolaemia that is demonstrated in depressed subjects, even when
CRH/ACTH release is reduced, probably secondary to pituitary CRH recep-
tor downregulation. The V3 receptor would seem an appropriate target site
for the development of future antidepressants. Preliminary studies of a V3
antagonist in animal models of depression provide support for this view.22
Acknowledgement
Timothy Dinan is in receipt of a project grant from the Health Research
Board to study vasopressin in depression.
References
1. Rubin R, Dinan TG, Scott LV, The neuroendocrinology of affective disorders. In:Pfaff D, Arnold AP, Etgen AM, Fahrbach SE, Moss RL, Rubin RT (eds),Hormones, Brain and Behaviour. Academic Press: New York, 2001.
2. Vale W, Spiess J, Rivier C, Rivier J, Characterisation of a 41 residue ovine hypo-thalamic peptide that stimulates secretion of the corticotropin and beta-endorphin. Science 1981; 213:1394–1399.
Ch 15 7/4/05 3:59 pm Page 141

142 Bipolar disorder: the upswing in research & treatment
3. De Wied D, Diamant M, Fodor M, Central nervous system effects of the neuro-hypophyseal hormones and related peptides. Front Neuroendocrinol 1993;14:251–302.
4. Antoni FA, Vasopressinergic control of pituitary–adrenal secretion comes ofage. Front Neuroendocrinol 1993; 14:76–122.
5. Whitnall M, Vasopressin co-exists in half of the corticotropin-releasing factorneurones in the external zone of the median eminence in normal rats.Neuroendocrinology 1987; 45:420–424.
6. Grazzini E, Lodboerer AM, Perez-Martin A et al, Molecular and functional char-acterization of the V1b receptor in rat adrenal medulla. Endocrinology 1996;137:3906–3914.
7. Ma XM, Lightman S, Aguilera G, Vasopressin and corticotropin-releasing hor-mone gene responses to novel stress in rats adapted to repeated restraint.Endocrinology 1999; 140:3623–3632.
8. Tizabi Y, Aguilera G, Desensitization of the hypothalamic–pituitary–adrenal axisfollowing prolonged administration of corticotropin-releasing hormone andvasopressin. Neuroendocrinology 1992; 56:611–618.
9. Dinan TG, Glucocorticoids and the genesis of depressive illness: a psychobio-logical model. Br J Psychiatry 1994; 164:365–371.
10. Gold PW, Goodwin FK, Vasopressin in affective illness. Lancet 1978;10:1233–1236.
11. Cole MA, Kim PJ, Kalman BA, Spencer RL, Dexamethasone suppression ofcorticosteroid secretion: evaluation of the site of action by receptor measuresand functional studies. Psychoneuroendocrinology 2000; 25:151–167.
12. Heuser I, Yassouridis A, Holsboer F, The combined dexamethasone/CRH test: arefined laboratory test for psychiatric disorders. J Affect Dis 1994; 4:93–101.
13. Gjerris A, Hammer M, Vendsborg P et al. Cerebrospinal fluid vasopressin-changes in depression. Br J Psychiatry 1995; 147:696–701.
14. van Londen L, Goekoop J, van Kempen G et al, Plasma levels of argininevasopressin elevated in patients with major depression. Neuropsycho-pharmacology 1997; 17:284–292.
15. Purba J, Hoogendijk W, Hoffman M, Swaab D, Increased numbers ofvasopressin- and oxytocin-containing neurons in the paraventricular nucleus ofthe hypothalamus in depression. Arch Gen Psychiatry 1996; 53:137–143.
16. Raadsheer F, Hoogendijk W, Hofman M, Swaab B, Increased number ofcorticotropin-releasing hormone expressing neurons in the paraventricularnucleus of the hypothalamic paraventricular nucleus of depressed patients.Neuroendocrinology 1996; 18:436–444.
17. Dinan TG, Lavelle E, Scott L et al, Desmopressin normalises the blunted ACTHresponse to corticotropin-releasing hormone in melancholic depression: evi-dence of enhanced vasopressinergic responsivity. J Clin Endocrinol Metab1999; 84:2238–2246.
18. Muller MB, Landgraf R, Preil J et al, Selective activation of the hypothalamicvasopressinergic system in mice deficient for the corticotropin-releasing hor-mone receptor 1 is dependent on glucocorticoids. Endocrinology 2000;141:4262–4269.
Ch 15 7/4/05 3:59 pm Page 142

HPA axis overactivation in affective disorder 143
19. Dinan TG, O’Brien S, Lavelle E, Scott LV, Further evidence of enhanced vaso-pressin V3 receptor responses in melancholic depression. Psychol Med 2004;34:169–172.
20. De Bellis M, Gold PW, Geriacoti T et al, An association of fluoxetine treatmentwith reductions in CSF corticotropin-releasing hormone and arginine vaso-pressin in patients with depression. Am J Psychiatry 1996; 150:656–657.
21. Heuser I, Bissette G, Dettling M et al, Cerebrospinal fluid concentrations ofcorticotropin-releasing hormone, vasopressin, and somatostatin on depressedpatients and healthy controls: response to amitryptiline treatment. DepressAnxiety 1998; 8:71–79.
22. Griebel G, Simiand J, Serrandeil-Le Gal C et al, Anxiolytic- and antidepressant-like effects of the non-peptide vasopressin V1b receptor antagonist,SSR149415, suggest an innovative approach for the treatment of stress-relateddisorders. Proc Natl Acad Sci USA 2002; 30:6370–6375.
Ch 15 7/4/05 3:59 pm Page 143

Ch 15 7/4/05 3:59 pm Page 144

Cognitive dysfunction:cause or consequenceof bipolar disorder?Samuel R Chamberlain and Barbara J Sahakian
c h a p t e r 1 6
Bipolar disorder is characterized by cycling between depression, euthymiaand mania. The argument that alterations or dysfunctions in cognition area core phenomenon comes from evidence on several levels. Cognitive dys-function is central to the diagnosis of depressive and manic episodes usingthe Diagnostic and Statistical Manual IV (DSM-IV). Distractibility and poordecision-making are included in the diagnostic criteria for manic episodes,and diminished ability to concentrate and indecisiveness are included in thecriteria for depression. Another tier of evidence comes from psychologicalmodels, in which abnormalities in cognition are often held to be important.In Beck’s Cognitive Model,1 aberrant cognitive schema develop during child-hood and are activated in later years by stressful and unpleasant life events(Figure 16.1). The activation of these aberrant schema leads to systematicerrors in logic, and the well-known triad of negative belief directed at self,world and future. Cognitive therapies developed from these psychologicalmodels2 aim to correct dysfunctional attitudes and negative automaticthoughts. From a top-level perspective, the emotional states of people withmania and depression form two extremes of an affective spectrum (Figure16.2). The emotional status of a given individual can vary on this spectrumin response to life events: towards dysphoria in response to relationshipbreak-up, or towards euphoria when celebrating achievements. However,the extreme emotional states of people with mania or depression differ inthat the moods are disproportionate and cause gross impairments in socialfunctioning. Indeed, the deficits in social functioning, and behaviour of bipo-lar patients more broadly, are in themselves suggestive of cognitive dys-function. Collectively, whether one examines affective disorder from theperspective of overt syndromic behaviour, DSM-IV diagnosis, or psycho-logical models, cognitive dysfunction is central to our understanding. The
Ch 16 7/4/05 3:59 pm Page 145

146 Bipolar disorder: the upswing in research & treatment
development of advanced neurocognitive testing coupled with functionalneuroimaging has in recent years facilitated the reliable investigation of spe-cific cognitive profiles between patient groups. It has been possible to iden-tify different cognitive dysfunctions that are (1) common to depression andmania; (2) capable of differentiating depression from mania; and (3) shownto persist into full clinical remission.
Early measures of cognitive functioning, including the Wisconsin CardSorting Test (WCST),3 revealed cognitive inflexibility in manic anddepressed patients. However, these findings are difficult to interpret as theyconfound multiple areas of cognition, and there is an ongoing need for moresensitive and specific neurocognitive tests. With the advent of automatedneurocognitive testing, such as the Cambridge Neuropsychological TestAutomated Battery (CANTAB),4 broad and substantial cognitive deficits havebeen identified that are common to manic and depressed patients. Frequentfindings include deficits on measures of attention, executive functioning,memory, and psychomotor speed (Figure 16.3) (see reference 10 for review).Importantly, the deficits appear to worsen with increasing age, and it maybe necessary to augment the usual psychopharmacological treatment of eld-erly depressed patients with agents such as methylphenidate11 or modafinil,
Figure 16.1 Beck’s cognitive model of depression: an overview.
Cognitive distortions
Activation of schema by life events in adolescence or
adulthood
Negative cognitive schema: SELF, WORLD, FUTURE
Aversive events during childhood
Overgeneralization (e.g. 'this always happens to me')
All-or-nothing thinking (e.g. 'whole week's work is
useless because of one error')
Arbitrary inference (e.g. 'my relative didn't
answer my call. She must be ignoring me')
Minimization (e.g. 'he complimented me because he was having a
good day')
Selective abstraction (disregarding positive comments from people and placing great
emphasis on criticisms)
Personalization (attributing events/comments
to self)
Environmental factors (e.g. family background)
Genetic susceptibility
Ch 16 7/4/05 3:59 pm Page 146

Cognitive dysfunction 147
the latter of which has been shown to reverse some of the cognitive dys-functions found in schizophrenia.12 The DSM-IV diagnosis of mania anddepression allows for considerable flexibility within the criteria, and there-fore it should not be surprising that the precise pattern of cognitive dys-function is somewhat variable. As neurocognitive tasks continue to be devel-oped and modified in the light of clinical and research findings, it becomesincreasingly feasible to identify trait markers for specific disorders. Much ofour research work is geared towards the development of neurocognitivetasks capable of identifying trait and state markers, in order to detect dis-ease onset, monitor treatment response, and assess psychopharmacologicaltreatment efficacy. Advanced neurocognitive batteries are now commonlyused in psychiatric research worldwide. In future, we foresee a central rolefor these approaches in the everyday assessment of psychiatric patients,particularly as many psychiatric disorders, including schizophrenia, arebeing reconstrued as neurocognitive disorders in the light of recent researchfindings.
Figure 16.2 The affective spectrum, and bipolar cycling. An individual’s positionon the affective continuum is determined by genetic, social and other factors; it canfluctuate in day-to-day life. Mania and major depressive disorder (MDD) representtwo extremes of this spectrum. In bipolar disorder, there is cycling between MDDand manic states, with ‘euthymia’ as an intermediate mood state.
Depression Mania
Euthymia
Euthymia
(1) Grandiosity(2) Overactivity(3) Distractibility(4) Socially
inappropriate behaviour
(5) Increased appetite(6) Impaired insight
(1) Persistent low mood
(2) Feelings of: - helplessness - hopelessness - worthlessness
(3) Anhedonia
Dysphoria
Euphoria
Ch 16 7/4/05 3:59 pm Page 147

148 Bipolar disorder: the upswing in research & treatment
In order to assess whether cognitive dysfunctions in bipolar disorder arecause or consequence of the overt pathology, we need to ask whether thebroad and substantial cognitive deficits common to both these conditionsare ‘primary’ deficits, or can be explained by more fundamental impair-ments in cognition. For example, can cognitive deficits be explained by anarrowing of the attentional focus to specific depression-relevant environ-mental stimuli or thoughts, or can they be related to an abnormal responseto negative feedback? Additionally, we need to examine whether a subset ofthe cognitive dysfunctions found in mania differ in character from thosefound in depression. Multiple studies have failed to identify a different pat-tern of deficits on measures of attention, memory, visuospatial ability andexecutive functioning. Where might we expect differences to manifest? On abehavioural level, both disorders share great overlap in terms of impair-ment in everyday functioning, but mania and depression are considered tobe at two extremes of the affective spectrum. Whereas depression is char-acterized by persistent low mood, anhedonia and negative interpersonal
Figure 16.3 Neurocognitive dysfunctions in unipolar and bipolar patients. Thecognitive deficits in unipolar disorder are broad and variable, and appear to beworse with increasing age. Bipolar patients demonstrate a degree of cognitive dys-function equivalent to older unipolar patients.
Study a b c d e
Age 32 37.5 50 72 43
HAM-D 22 23 22 30 26
Pattern recognition memory
Spatial recognition memory
Motor latency
Delayed matching to sample (DMTS)
Tower of London (% correct)
New Tower of London (% correct)
New Tower of London (latency)
Set-shifting (ID/ED) ED deficits
a Sweeney et al, 20005
b Purcell et al, 19976
c Elliott et al, 19967
d Beats et al, 19968
e Rubinsztein et al, 20009
No deficits
Deficits
Not performed
Increasing age (unipolar patients) Bipolar patients
Ch 16 7/4/05 3:59 pm Page 148

Cognitive dysfunction 149
bias, mania is characterized by grandiose ideas, distractibility, socially inap-propriate behaviour and an excessive positive interpersonal bias. On thisbasis, it would seem logical that differential cognitive characteristics mightbe found on neurocognitive tasks where affective processes are involved –i.e. on ‘hot’ cognitive tasks13 involving the processing of emotionallycoloured stimuli, or where subjects are given negative feedback about theirongoing performance. Indeed, key differences have been identified betweenmanic and depressed patients on tasks examining attentional processingbias, response to negative feedback, and decision-making.
In go/no-go tasks, subjects give a rapid motor response to stimuli that fitinto a target category, and withhold motor responses to stimuli in distrac-tor categories. The affective version of this task uses positively valenced(happy) words, negatively valenced (sad) words, and emotionally neutral(‘cold’) words. This allows attentional processes to be investigated, and isparticularly important, given that the mood states of patients with bipolardisorder are indicative of dysfunctions within attentional processes. Healthycontrols undertaking the affective go/no-go task show a differential neuralresponse in the subgenual cingulate when responding to emotionally ‘hot’words compared with neutral ‘cold’ words.14 This neural region is known tofunction abnormally in bipolar disorder.15 Depressed patients respondmore rapidly to sad versus happy words, and manic patients respond morerapidly to happy versus sad words,16 consistent with attentional biastowards negative environmental stimuli in depression, and towards positiveenvironmental stimuli in mania. Acute tryptophan depletion in healthy con-trols has been found to cause a slowing of response to happy words in thesame go/no-go task, demonstrating a central role for serotonin in the mod-ulation of these aspects of attentional processing.17 These attentional bias-es are linked with the ruminations and triad of negativity central to Beck’scognitive approach. Excitingly, it has been possible to identify putative neu-ral substrates underlying these attentional abnormalities in depressedpatients, in whom elevated neural responses to sad target words are foundin the rostral anterior cingulate extending to the anterior medial prefrontalcortex. Additionally, depressed patients show a differential neural responsein the right lateral orbitofrontal cortex when responding to sad distractorwords (Figure 16.4).18 The findings agree with previous work demonstrat-ing the importance of the medial prefrontal and orbitofrontal cortex in emo-tional processing and behavioural inhibition. These structures are compo-nents of relatively segregated basal ganglia–thalamocortical loops, withstrong connections to limbic regions known to be important in emotionalregulation (for example, see reference 19).
Ch 16 7/4/05 3:59 pm Page 149

150 Bipolar disorder: the upswing in research & treatment
When depressed patients make errors in their daily life, such errors areviewed in a pessimistic light and serve as focal points for rumination andnegative automatic thoughts. The attributional psychological model pro-vides a formalization for these findings, in which depressed patients inap-propriately attribute the cause of failure to global, stable and internal fac-tors rather than specific, unstable, external factors.20 For example, after fail-ing an exam, rather than thinking ‘I failed one exam, I will be able to passre-takes if I work harder, and it was a difficult exam’, typically a depressedpatient might think ‘I failed one exam because I’m a failure’. These negativeautomatic thoughts contribute to the hopelessness and pervasive, persistentnegative mood. Novel cognitive tasks have been developed that can renderobjective measures of these abnormal responses in patients, by examiningthe effect of negative feedback on task performance. In the New Tower ofLondon task (NTOL) (Figure 16.5), two sets of ‘snooker’ balls are present-ed on the screen, and the subject has to decide on the minimum number ofmoves necessary to make the bottom set of balls match the top set within aspecified rule-set. After thinking through the problem, the subject selectsthe corresponding number on the screen. When the subject responds incor-rectly, the computer gives negative feedback, and allows the subject to thinkthrough the problem again and select a new response. Although patients
Figure 16.4 Affective go/no-go task – functional magnetic resonance imaging acti-vations comparing depressed patients with healthy controls. From reference 18,with permission from American Medical Association.
Response to sad versus happy targets: medial prefrontal cortex extending from rostral anterior
cingulate to the medial prefrontal cortex
Responses to sad versus happy distractors: right lateral
orbitofrontal cortex
Ch 16 7/4/05 3:59 pm Page 150

Cognitive dysfunction 151
with depression, schizophrenia and Parkinson’s disease all show impair-ments on this and similar tasks, in terms of percentage problems solvedcorrectly, only depressed patients show an abnormal response to negativefeedback;21,22 they are far more likely to fail a given problem if they have just
Figure 16.5 Neurocognitive tasks of use in differentiating mania from depressionin bipolar disorder. CANTAB, Cambridge Neuropsychological Test AutomatedBattery.
• Per cent correct impaired in bipolar disorder, schizophrenia and Parkinson's disease
• Abnormal response to negative feedback in depression
• Task performance dependent on relatively frontal cortical structures
• Abnormal response to false negative feedback in depression
• Depressed and manic patients: impairment on latency and strategy
• Only manic patients demonstrate impaired quality of decision-making
• Per cent correct impaired in bipolar disorder, schizophrenia and Parkinson's disease
• Abnormal response to negative feedback in depression
• Task performance dependent on relatively posterior cortical structures
Delayed Matching-to-Sample Text (DMTS) (CANTAB)
New Tower of London (NTOL) (CANTAB)
Probabilistic reversal learning (CANTAB) Decision-making task
Ch 16 7/4/05 3:59 pm Page 151

152 Bipolar disorder: the upswing in research & treatment
been given negative feedback. These findings are replicated on the DelayedMatching-To-Sample Test of Recognition Memory (DMTS),4 in which sub-jects memorize an abstract complex pattern and select the correct patternfrom other patterns at a later time (Figure 16.5). Similarly, Steffens et alhave found an abnormal response to negative feedback in a different task inelderly depressed patients.23 False negative feedback, in which subjects aretold that they made an incorrect response when their response was in factcorrect, selectively impairs depressed patients’ performance on a proba-bilistic reversal learning task (Figure 16.5). On this task, subjects have toselect an arbitrarily assigned correct pattern from a choice of two. Aftereach choice, feedback is given as to whether the choice was correct or incor-rect. However, in a minority of cases (20% of trials) subjects are given falsefeedback. It is made clear in the task instructions before administering thistask that the computer will give false feedback some of the time, yetdepressed patients’ performance is still found to be persistently impaired byfalse negative feedback. Even on the last 10 trials, false negative feedback isstill disrupting depressed patients, whereas controls are just ignoring it.The abnormal response to negative feedback in depression found on thesevarious tasks is indicative of dysfunctional reward systems, and this is like-ly to contribute to the anhedonia and loss of motivation reported at a syn-dromic level.
Bipolar patients in the manic phase show impaired decision-making,with excessive involvement in pleasurable activities with high potential fornegative consequences. For example, they might opt to go on spendingsprees with the family savings or make unwise business decisions. In manyways, these behaviours are akin to those found in patients withorbitofrontal/ventromedial prefrontal cortical damage consequential tobrain injury.24,25 These patients often demonstrate normal performance on‘cold’ cognitive tasks of memory, learning and executive function – such asthe Wisconsin Card Sorting Task3 – but abnormal performance on ‘hot’gambling tasks. In one version of a computerized gambling task, subjectsmake decisions based on variably weighted probabilities (Figure 16.5). A‘token’ is hidden behind a red or blue box, and the subject bets a variablenumber of points on which coloured box the token is hidden behind. Bothmanic and depressed patients’ performance on this task is worse than con-trols’ in terms of response latency, and total number of points accumulated.However, only manic patients made suboptimal decisions – they make poordecisions that run contrary to logic whereas depressed patients do not.26
The extent of this deficit is correlated with the severity of the mania record-ed by Young’s Mania Score, and functional imaging points to abnormalities
Ch 16 7/4/05 3:59 pm Page 152

Cognitive dysfunction 153
in the dorsal anterior cingulate, frontal polar and right inferior frontal cor-tex as neural substrates of these suboptimal decisions in manic patients.27
Manic patients also behave in a risky way, in that they often lose all accu-mulated points on this task.26
In contrast to the broad and variable cognitive dysfunctions found dur-ing manic and depressive episodes, recent studies have identified specificcognitive dysfunctions that persist even when patients are considered to befully recovered clinically. These residual cognitive deficits include: psy-chomotor slowing, impaired visual recognition memory and impaired sus-tained attention.8,9,28–31 One study found psychomotor slowing on a spatialworking memory task that persisted into recovery in patients with seasonalaffective disorder (SAD), despite other measures of cognitive dysfunctionreturning to baseline.28 Residual deficits have also been identified in bipo-lar patients who had been in remission for at least 4 months and met care-ful screening criteria, on a test of visuospatial recognition memory.9 TheRapid Visual Information Processing (RVIP) task4 is a measure of sustainedattention, and requires subjects to detect odd or even sequences of digits byobserving a white box in the centre of the screen. The task has been shownto activate a right frontoparietal neural network in healthy controls.32 Digitsappear within the white box one at a time (Figure 16.6). Euthymic patientsshow impaired sustained attention, as measured by RVIP, even when theextent of remission is at peak.31 It has been argued that this may representa trait marker for the development of bipolar disorder. These residualdeficits may prove to be the greatest barrier to patient rehabilitation, as hasbeen found to be the case in schizophrenia,10,33,34 and serve to limit the useof psychological treatments in certain contexts.
Cognitive dysfunction is central to our understanding of bipolar disor-der, as evidenced by the syndromic behaviour, DSM-IV diagnostic criteria,psychological models and neurocognitive findings. In particular, thecoupling of advanced neurocognitive testing with functional imaging is lead-ing an upsurge in research towards the synthesis of endophenotypes –intermediate constructs capable of being both downstream of overt syn-dromic behaviour and upstream of biochemical abnormalities and genetics.In many respects then, cognitive dysfunctions and their neural substratescan be viewed as being cause rather than consequence of the overt mani-festation of bipolar disorder. The finding of residual cognitive deficits per-sisting into full clinical remission has huge ramifications for treatment, asdo the differential performances of manic and depressed patients on ‘hot’processing tasks. Advanced neurocognitive testing batteries are likely toplay a key role in the generation of more objective means of detecting
Ch 16 7/4/05 3:59 pm Page 153

154 Bipolar disorder: the upswing in research & treatment
disease onset, monitoring recovery and relapse, and assessing the efficacyof novel and established pharmacological and psychological interventions,towards the synthesis of more efficient treatment algorithms for these high-ly prevalent and debilitating disorders.
References
1. Beck AT, The past and future of cognitive therapy. J Psychother Pract Res1997; 6:276–284.
2. Parker G, Roy K, Eyers K, Cognitive behavior therapy for depression? Choosehorses for courses. Am J Psychiatry 2003; 160:825–834.
3. Berg E, A simple objective technique for measuring flexibility in thinking. J GenPsychol 1948; 39:15–22.
4. Cambridge Cognition, Cambridge Neuropsychological Test Automated Battery(CANTAB). www.camcog.com.
5. Sweeney JA, Kmiec JA, Kupfer DJ, Neuropsychologic impairments in bipolarand unipolar mood disorders on the CANTAB neurocognitive battery. BiolPsychiatry 2000; 48:674–684.
6. Purcell R, Maruff P, Kyrios M, Pantelis C, Neuropsychological function in youngpatients with unipolar major depression. Psychol Med 1997; 27:1277–1285.
7. Elliott R, Sahakian BJ, McKay AP et al, Neuropsychological impairments inunipolar depression: the influence of perceived failure on subsequent perform-ance. Psychol Med 1996; 26:975–989.
8. Beats BC, Sahakian BJ, Levy R, Cognitive performance in tests sensitive tofrontal lobe dysfunction in the elderly depressed. Psychol Med 1996; 26:591–603.
Figure 16.6 Rapid visual information processing tasks (RIVP) (CambridgeNeuropsychological Test Automated Battery (CANTAB)).
• Impaired performance in bipolar patients even at peak of remission
• Impaired sustained attention, as measured by RVIP, may represent a trait marker for bipolar disorder
Ch 16 7/4/05 3:59 pm Page 154

Cognitive dysfunction 155
9. Rubinsztein JS, Michael A, Paykel ES, Sahakian BJ, Cognitive impairment inremission in bipolar affective disorder. Psychol Med 2000; 30:1025–1036.
10. Tavares JV, Drevets WC, Sahakian BJ, Cognition in mania and depression.Psychol Med 2003; 33:959–967.
11. Lavretsky H, Kumar A, Methylphenidate augmentation of citalopram in elderlydepressed patients. Am J Geriatr Psychiatry 2001; 9:298–303.
12. Turner DC, Clark L, Pomarol-Clotet E et al, Modafinil improves cognition andattentional set shifting in patients with chronic schizophrenia. Neuropsycho-pharmacology 2004; 29:1363–1373.
13. Roiser J, Rubinsztein JS, Sahakian B, Cognition in depression. Psychiatry2003; 2:43–47.
14. Elliott R, Rubinsztein JS, Sahakian BJ, Dolan RJ, Selective attention to emo-tional stimuli in a verbal go/no-go task: an fMRI study. Neuroreport 2000;11:1739–1744.
15. Drevets WC, Ongur D, Price JL, Neuroimaging abnormalities in the subgenualprefrontal cortex: implications for the pathophysiology of familial mood disor-ders. Mol Psychiatry 1998; 3:220–226, 190–191.
16. Murphy FC, Sahakian BJ, Rubinsztein JS et al, Emotional bias and inhibitorycontrol processes in mania and depression. Psychol Med 1999; 29:1307–1321.
17. Murphy FC, Smith KA, Cowen PJ et al, The effects of tryptophan depletion oncognitive and affective processing in healthy volunteers. Psychopharmacology(Berl) 2002; 163:42–53.
18. Elliott R, Rubinsztein JS, Sahakian BJ, Dolan RJ, The neural basis of mood-congruent processing biases in depression. Arch Gen Psychiatry 2002;59:597–604.
19. Alexander GE, DeLong MR, Strick PL, Parallel organization of functionally seg-regated circuits linking basal ganglia and cortex. Annu Rev Neurosci 1986;9:357–381.
20. Abramson LY, Metalsky GI, Alloy LB, Hopelessness depression: A theory-basedsubtype of depression. Psychol Rev 1989; 96:358–372.
21. Elliott R, Baker SC, Rogers RD et al, Prefrontal dysfunction in depressedpatients performing a complex planning task: a study using positron emissiontomography. Psychol Med 1997; 27:931–942.
22. Elliott R, Sahakian BJ, Herrod JJ et al, Abnormal response to negative feed-back in unipolar depression: evidence for a diagnosis specific impairment. JNeurol Neurosurg Psychiatry 1997; 63:74–82.
23. Steffens DC, Wagner HR, Levy RM et al, Performance feedback deficit in geri-atric depression. Biol Psychiatry 2001; 50:358–363.
24. Starkstein SE, Mayberg HS, Berthier ML et al, Mania after brain injury: neuro-radiological and metabolic findings. Ann Neurol 1990; 27:652–659.
25. Bechara A, Tranel D, Damasio H, Characterization of the decision-makingdeficit of patients with ventromedial prefrontal cortex lesions. Brain 2000;123:2189–2202.
26. Murphy FC, Rubinsztein JS, Michael A et al, Decision-making cognition inmania and depression. Psychol Med 2001; 31:679–693.
27. Rubinsztein JS, Fletcher PC, Rogers RD et al, Decision-making in mania: a PETstudy. Brain 2001; 124:2550–2563.
Ch 16 7/4/05 3:59 pm Page 155

156 Bipolar disorder: the upswing in research & treatment
28. O’Brien JT, Sahakian BJ, Checkley SA, Cognitive impairments in patients withseasonal affective disorder. Br J Psychiatry 1993; 163:338–343.
29. Silverstein ML, Harrow M, Bryson GJ, Neuropsychological prognosis and clin-ical recovery. Psychiatry Res 1994; 52:265–272.
30. Ferrier IN, Stanton BR, Kelly TP, Scott J, Neuropsychological function ineuthymic patients with bipolar disorder. Br J Psychiatry 1999; 175: 246–251.
31. Clark L, Iversen SD, Goodwin GM, Sustained attention deficit in bipolar disor-der. Br J Psychiatry 2002; 180:313–319.
32. Coull JT, Frith CD, Frackowiak RS, Grasby PM, A fronto-parietal network forrapid visual information processing: a PET study of sustained attention andworking memory. Neuropsychologia 1996; 34:1085–1095.
33. Goldberg TE, Gold JM, Greenberg R et al, Contrasts between patients withaffective disorders and patients with schizophrenia on a neuropsychologicaltest battery. Am J Psychiatry 1993; 150:1355–1362.
34. Zarate CA Jr, Tohen M, Land M, Cavanagh S, Functional impairment and cog-nition in bipolar disorder. Psychiatr Q 2000; 71:309–329.
Ch 16 7/4/05 3:59 pm Page 156

The neural basisof cognitive functionin bipolar disorderVivienne Curtis
c h a p t e r 1 7
Despite the notion of apparent ‘recovery’ between episodes of bipolar disor-der,1 impairments in cognitive task performance have been reported duringremission.2,3 These deficits have been observed across a range of cognitivedomains including declarative memory,4,5 attention6 and executive func-tion.7–9 However, when residual symptomatology is controlled for a specificdeficit in attention, executive function predominates.2,3,6,8 This implies thata dysfunction of the neuronal substrates underlying cognitive operationsmay exist alongside alterations in limbic function. The use of cognitive par-adigms to elicit information about neural networks has been establishedwithin the literature relating to schizophrenia and is now beginning to beapplied to studies of bipolar disorder.
To date, only a handful of studies have examined working memory func-tion in remitted bipolar disorder patients. Kusumo and Vaughan10 reportedthat ‘well’ bipolar disorder patients showed delay-dependent deficits on theBrown–Peterson paradigm, a test of retention in short-term (working) mem-ory. Asarnow and MacCrimmon11 reported that bipolar disorder patients‘free from major symptoms’, were impaired on an attentional target detec-tion task only when the targets were embedded in longer stimulus arrays.In tasks specifically testing the integrity of the phonological loop, euthymicbipolar subjects have been shown to have no impairments of the Sternbergparadigm and the digits forward sub-test of the WAIS-R, but subjects haveshown impairment on the digits backwards sub-test which is an index of theintegrity of the central executive.8,12 Harmer et al13 observed impaired per-formance in remitted bipolar subjects undertaking a sustained attentiontask that was independent of working memory load, and have proposed thatdeficits in sustained attention rather than working memory may representa core feature of bipolar disorder. Taken together, these studies converge on
Ch 17 7/4/05 4:00 pm Page 157

158 Bipolar disorder: the upswing in research & treatment
a general concept of dysfunction within the attentional and executive controlsystems in euthymic bipolar disorder.
The combination of a neuropsychological and neuroimaging approachcan enable us to understand these dysfunctions further and investigate theeffect of disease trait on the relationships between neuroanatomy and neu-ropsychology.
We have recently undertaken a functional magnetic resonance imaging(fMRI) study using two tasks: the two-back task (dependent on the integrityof the central executive) and the Sternberg paradigm (dependent on theintegrity of the phonological loop) in matched groups of euthymic bipolarpatients.14 Twelve right-handed euthymic bipolar I males receiving lithiumcarbonate monotherapy were recruited and matched with 12 controls. Thetwo-back task comprised a single working memory load contrasted with abaseline vigilance condition (Figure 17.1). The Sternberg paradigm used aparametric design incorporating variable working memory load with fixeddelay between presentation of an array of items to be remembered and a tar-get item (Figure 17.2). Functional activation data were acquired during per-formance of the tasks and were analysed to produce brain activation mapsrepresenting significant group differences in activation (ANOVA). Load-response curves were derived from the Sternberg task data set. There wereno significant differences between the groups in demographics, clinical
Figure 17.1 Two-back task.
Task parameters:• Random series of letters displayed on the screen at the rate of 1 every 2000 ms• Inter-stimulus interval of 1000 ms• Subject to press response button when the letter on the screen is the same as
that occurring two previously
1000 ms1000 ms
1000 ms1000 ms
1000 msa
b
a
'YES'
Ch 17 7/4/05 4:00 pm Page 158

The neural basis of cognitive function in bipolar disorder 159
symptomatology or on-line performance of the tasks. In the two-back task,compared with controls, bipolar disorder patients showed reductions inbilateral frontal, temporal and parietal activation, and increased activationswith the left precentral, right medial frontal and left supramarginal gyri. Nobetween-group differences were observed in the Sternberg task at any work-ing memory load.
Common criticisms of clinical fMRI studies are that the results are con-founded by differences in clinical state, medication and task performancebetween patient and control groups. Within our study we tried to limit theseconfounds at source wherever possible. Thus, we used standardized diag-nosis to reduce variability within the subject groups and we activelyscreened subjects to detect the presence of significant misuse of alcohol orillicit substances. No statistically significant differences in training require-ments for either group were observed outside the scanner or during on-lineperformance. In addition, our bipolar subjects had been euthymic for atleast 4 weeks prior to scanning, and did not differ from controls on stan-dardized clinical ratings prior to testing, facilitating an investigation of thebipolar ‘trait’ without ‘state’ confounds.
One potential confounding factor is medication. Although the bipolarsubjects had previously been exposed to different medications, they were all
Figure 17.2 The Sternberg task.
Task parameters:• On each trial 1–5 digits are presented to the participant• Interval of 500 ms in which a plus sign is presented• Single probe letter presented for 1500 ms• Subject to state whether probe digit was a member of the original memory set
(via dual response box)• 1500 ms inter-trial interval
1500 ms
500 ms1500 ms
1500 ms
-384-+
4
'YES'
Ch 17 7/4/05 4:00 pm Page 159

160 Bipolar disorder: the upswing in research & treatment
established on lithium monotherapy for at least 6 months prior to the scan.While no studies of the effect of lithium on the blood oxygen level depend-ent (BOLD) response in humans have been undertaken, chronic lithiumadministration has been demonstrated in the rat aorta to increase acetyl-choline endothelium-dependent relaxation, and also to increase vascularcontraction.15,16 Therefore, it is possible that there is an effect of lithium onvascular responsiveness and, consequently, on neurovascular coupling.Future specific investigations of the influence of medication and lithium inparticular on cognitive activation effects will be required to quantify such atheoretical confound.
There have been relatively few functional imaging studies using cognitiveactivation tasks in euthymic patients with bipolar disorder. The findingswith respect to frontal lobe function have varied depending on experimentaldesign and clinical state, but the anterior cingulate, supplementary motorarea and dorsal and ventrolateral prefrontal cortices appear to be regularlyimplicated in both increases and decreases of network activation in bipolarsubjects.2,17–19
During performance of the two-back task, neural networks were activat-ed which were in keeping with those reported by other neuroanatomic20,21
and neuroimaging studies of working memory in healthy controls.22 Theprefrontal regions which showed an attenuated response in the bipolar sub-jects are in keeping with those previously implicated in both the mainte-nance and the manipulation of information held in the working memorystore in studies of healthy subjects23–26 and in other psychiatric groups27
and mood states.19 Furthermore, both structural neuroimaging and post-mortem studies have suggested morphological alterations in these regionsin bipolar subjects.2
The bipolar subjects showed regions of attenuated response beyond theleft frontal lobes in the right middle temporal gyrus, cuneus/precuneus andthe cerebellum. These regions have been inconsistently reported in otherfunctional imaging working memory studies and may represent a variabili-ty of recruitment of other nodes within the working memory network. Therewere few regions of increased activation in our bipolar group during thetwo-back task. These included the left supramarginal gyrus, which has beenimplicated as part of the phonological loop.28–30 This altered pattern ofresponse suggests that changes are not simply due to increased effort, butthis might reflect bipolar subjects’ attempt to draw upon intact slave systemresources to support executive task performance, and intact working mem-ory capacity may scaffold euthymic bipolar disorder patients’ perform-ance.25 Our subjects also had some small regions of increased activation
Ch 17 7/4/05 4:00 pm Page 160

The neural basis of cognitive function in bipolar disorder 161
within the frontal lobes, a finding reported previously during a verbal flu-ency task.18 This hyperactivation of the frontal cortices has also beenreported in schizophrenic subjects during working memory tasks and maybe a reflection of inefficient use of the prefrontal networks.31 Callicott et al32
have suggested that in schizophrenic subjects the pattern of load responserelationships may vary across different nodes of the working memory net-work and that regions may move through hyperfrontality to a hypofrontalresponse. In this way our findings of both hyper- and hypofrontality duringtask performance may be a reflection of compromised neural strategies ina subject group performing at their ceiling.
While the Sternberg task has been studied less extensively than the two-back task (and with a greater heterogeneity of experimental designs) it hasbeen shown to generate load-dependent changes in activation in the dorso-lateral prefrontal cortex which are felt to reflect encoding of items.33,34
These findings have not been widely replicated, either within the literatureor in our study, although the neural networks subserving task performancein our subjects were in keeping with those seen in neuroimaging studies ofworking memory tasks.35 Furthermore, we have found no regions of signif-icant difference between bipolar and control groups while performing theSternberg task. Despite the lack of a load-dependent increase in the BOLDsignal evident in this study there was a main effect of memory load on theresponse time variables, suggesting that this paradigm served its paramet-ric function. However, as neural networks were engaged to a similar degreein both groups, the current results indicate that the range of task difficultyfell within the range of achievable performance. It is therefore possible thatBOLD signal changes may be no more sensitive as a measure of neural net-work function than performance in the behavioural task and so significantdecrements in activation for the Sternberg task may accompany only signif-icant decrements in performance.
This study demonstrates how combining functional imaging can teach usabout the neural basis of cognitive function in bipolar disorder and identi-fies a task-dependent alteration of prefrontal lobe function which we suggestis related to central executive rather than phonological loop function. Thisfinding reinforces the importance of using more than one cognitive activa-tion paradigm in a patient group to increase our understanding of the rela-tionship between task demand and neural activity. While we have controlledfor the confounding effects of mood symptoms, future studies of bipolarsubjects are required to exclude additional confounds such as medication.However, our findings support the notion that, in bipolar disorder, failure toengage frontoexecutive function represents a core deficit.
Ch 17 7/4/05 4:00 pm Page 161

162 Bipolar disorder: the upswing in research & treatment
References
1. Goodwin FK, Jamison KR, Manic Depressive Illness. Oxford University Press:New York, 1990.
2. Bearden CE, Hoffman KM, Cannon D, The neuropsychology and neuroanatomyof bipolar affective disorder: a critical review. Bipolar Disord 2001; 3:106–150.
3. Ferrier IN, Thompson JM, Cognitive impairment in bipolar affective disorder:implications for the bipolar diathesis. Br J Psychiatry 2002; 180:293–295.
4. Van Gorp WG, Altshuler L, Theberge DC et al, Cognitive impairment ineuthymic bipolar patients with and without prior alcohol dependence Arch GenPsychiatry 1998; 55:41–46.
5. Rubinsztein JS, Michael A, Paykel ES, Sahakian BJ, Cognitive impairment inremission in bipolar affective disorder. Psychol Med 2000; 30:1025–1036.
6. Clark L, Iverson SD, Goodwin GM, Sustained attention deficit in bipolar disor-der. Br J Psychiatry 2002; 180:313–319.
7. Hawkins KA, Hoffmann RE, Quinlan DM et al, Cognition, negative symptoms,and diagnosis: a comparison of schizophrenic, bipolar, and control samples. JNeuropsychiatry Clin Neurosci 1997; 9:81–89.
8. Ferrier IN, Stanton BR, Kelly TP, Scott J, Neuropsychological function ineuthymic patients with bipolar disorder. Br J Psychiatry 1999; 175:246–251.
9. Zubieta JK, Huguelet P, O’Neil RL, Giordani BJ, Cognitive function in euthymicbipolar I disorder. Psychiatry Res 2001; 102:9–20.
10. Kusumo KS, Vaughan M, Effects of lithium salts on memory. Br J Psychiatry1977; 131:453–457.
11. Asarnow RF, MacCrimmon DJ, Span of apprehension deficits during thepostpsychotic stages of schizophrenia. A replication and extension. Arch GenPsychiatry 1981; 38:1006–1011.
12. Thompson JM, Gray JM et al, A component process analysis of working mem-ory dysfunction in bipolar affective disorder. J Psychopharmacol 2000;14(Suppl):A24.
13. Harmer CJ, Clark L, Grayson L, Goodwin GM, Sustained attention deficit inbipolar disorder is not a working memory impairment in disguise.Neuropsychologia 2002; 40:1586–1590.
14. Monks PJ, Thompson JM, Bullmore ET et al, A functional MRI study of work-ing memory task in euthymic bipolar disorder: evidence for task-specific dys-function. Bipolar Disord 2004; 6:550–564.
15. Dehpour AR, Ghafourifar P, Samenian J et al, The effect of lithium onendothelial-dependent relaxation in rat isolated aorta. Gen Pharmacol 1995;26: 1003–1007.
16. Ullian ME, Walsh LG, Wong KC, Allan CJ, Potentiation of angiotensin II-stimulated vascular contraction by lithium. Am J Physiol 1995;268:H2009–2016.
17. Frangou S, Raymont V, Kingston J, Shergill S, Investigating the functional inter-face between the dorsal and central prefrontal circuitry in bipolar disorder.Bipolar Disord 2003; 5(Suppl):47–48.
Ch 17 7/4/05 4:00 pm Page 162

The neural basis of cognitive function in bipolar disorder 163
18. Curtis VA, Dixon TA, Morris RG et al, Differential frontal activation in schizo-phrenia and bipolar illness during verbal fluency. J Affect Disord 2001;66:111–122.
19. Blumberg HP, Leung HC, Skullarski P et al, A functional magnetic resonanceimaging study of bipolar disorder: state- and trait-related dysfunction in ventralprefrontal cortices. Arch Gen Psychiatry 2003; 60:601–609.
20. Goldman-Rakic PS, Circuitry of primate prefrontal cortex and regulation ofbehaviour by representational memory. In: Plum F, Mountcastle V (eds),Handbook of Physiology – The Nervous System. American PhysiologicalSociety: Bethesda, 1987:373–417.
21. Alexander GE, Delong MR, Strick PL, Parallel organization of functionally seg-regated circuits linking basal ganglia and cortex. Annu Rev Neurosci 1986;9:357–381.
22. Duncan J, Owen AM, Common regions of the human frontal lobe recruited bydiverse cognitive demands. Trends Neurosci 2000; 23:475–483.
23. Cohen JD, Forman SD, Braver TS et al, Activation of prefrontal cortex in a non-spatial working memory task with functional MRI. Hum Brain Mapp 1994;1:293–304.
24. Awh E, Jonides J, Smith EE et al, Dissociation of storage and rehearsal in ver-bal working memory: Evidence from PET. Psychol Sci 1996; 7:25–31.
25. D’Esposito M, Ballard D, Aguirre GK, Zarahn E, Human prefrontal cortex is notspecific for working memory: a functional MRI study. Neuroimage 1998;8:274–282.
26. Collette F, Salmon E, Van der Linden M et al, Regional brain activity duringtasks devoted to the central executive of working memory. Cogn Brain Res1999; 7:411–417.
27. Goldberg TE, Weinberger DR, Effects of neuroleptic medication on the cogni-tion of patients with schizophrenia: a review of recent studies. J ClinPsychiatry 1996; 57(Suppl 9):62–65.
28. D’Esposito M, Postle BR, Rypma B, Prefrontal cortical contributions to workingmemory: evidence from event-related fMRI studies. Exp Brain Res 2000;133:3–11.
29. Becker JT, Mintun MA, Diehl DJ et al, Functional neuroanatomy of verbal freerecall: a replication study. Hum Brain Mapp 1994; 1:284–292.
30. Palescu E, Frith CD, Frackowiak RS, The neural correlates of the verbal com-ponent of working memory. Nature 1993; 362:342–345.
31. Callicott JH, Bertolino A, Mattay VS et al, Physiological dysfunction of the pre-frontal cortex in schizophrenia revisited. Cereb Cortex 2000; 10:1078–1092.
32. Callicott JH, Mattay VS, Verchinski BA et al, Complexity of cortical dysfunctionin schizophrenia: more than up or down. Am J Psychiatry 2003;160:2209–2215.
33. Braver TS, Cohen JD, Nystrom LE et al, A parametric study of prefrontal cor-tex involvement in human working memory. Neuroimage 1997; 5:49–62.
34. Rypma B, D’Esposito M, The roles of prefrontal brain regions in components ofworking memory: effects of memory load and individual differences. Proc NatlAcad Sci USA 1999; 96:6558–6563.
35. Veltman DJ, Rombouts SARB, Dolan RJ, Maintenance versus manipulation inworking memory revisited: an fMRI study. Neuroimage 2003; 18:247–256.
Ch 17 7/4/05 4:00 pm Page 163

Ch 17 7/4/05 4:00 pm Page 164

Psychological treatments:does the evidencestack up?Jan Scott
c h a p t e r 1 8
Introduction
To put some of the issues of this chapter into context, I will briefly reviewsome of the morbidity and cost issues in bipolar disorders. I will then sum-marize outcomes of randomized controlled trials (RCTs) for bipolar disor-der where psychological therapies have been added to medication and usu-ally, but not exclusively, are compared with treatments as usual. I will thenpresent some preliminary data from the recently completed MedicalResearch Council (MRC) study looking at a similar paradigm using cogni-tive therapy added to treatment as usual, compared with routine psychiatrictreatment. Finally, I will draw some conclusions particularly highlightinghow these data fit in with the recommendations given by treatment guide-lines on bipolar disorders, such as those published by the AmericanPsychiatric Association, the British Association of Psychopharmacology, andthe Royal College of Psychiatrists of Australia and New Zealand Psychiatry(for example, see references 1 and 2).
Burden of disease
Of the top ten leading causes of disability worldwide amongst people aged19–45, six are psychiatric disorders, and unipolar and bipolar disordersoccupy the first and second positions, respectively, both coming aboveschizophrenia in terms of burden.3 Standardized mortality ratios suggestbipolar disorders again exceed schizophrenia if all causes of death are con-sidered.4 Morbidity statistics suggest that about 2% of the population will
Ch 18 7/4/05 4:00 pm Page 165

166 Bipolar disorder: the upswing in research & treatment
experience bipolar I, bipolar II or a bipolar spectrum disorder.4,5 Veryimportantly, patients with bipolar I and II disorders show a great deal ofaxis I and axis II co-morbidity, with about 30–50% demonstrating an anxi-ety, substance misuse or personality disorder.6–8 Suicide attempts are alsovery common, in about a third to a half of these individuals.4 Studies ofsocial adjustment suggest that at least 30% of individuals with bipolar dis-order do not return to their previous employment within a year of anepisode, and in another 30%, if they do return to employment, they do notreturn to their previous level of functioning.9,10 In Britain, people with bipo-lar disorders rather than other severe mental disorders occupy most acutepsychiatric bed days, and this is despite the fact that, in bipolar depression,people are often not admitted to hospital for treatment.8,11 Betweenepisodes, individuals are highly symptomatic; Judd et al12 reported thatpersons with bipolar disorders had syndromal or subsyndromal depressivesymptoms for about 50% of the time during 12 years of weekly follow-upratings. This illustrates the enormous level of morbidity and mortality ofbipolar disorders, yet the National Service Framework for UK Mental HealthServices did not set one target specifically aimed at the care and treatmentof bipolar disorders.
Our MRC study13 highlighted the annual costs of care for individualswith bipolar disorders: 92% received one or more psychiatric out-patientcontacts, the median number being five per year and 98% were receivingmedication, with the median number of prescribed medications being aboutfive (similar to reports from the USA). However, individuals rarely reportedday care, additional support or therapy and few were living in supportedaccommodation. These cost figures contrast sharply with the distribution ofresources in schizophrenia, where about 10–15% of the costs of care and
Figure 18.1 Odds ratios of relapse rates in early trials.
Citation N Total Effect p Value
Cochran 198416 28 0.12 0.02Lam et al 200018 23 0.02 0.00Perry et al 199917 68 0.59 0.34Scott et al 200119 42 0.38 0.17
Fixed Combined (4) 161 0.28 0.00Random Combined (4) 161 0.22 0.01
0.01 0.1 1 10 100
Favours psychotherapy
Ch 18 7/4/05 4:00 pm Page 166

Psychological treatments 167
resources are allocated to psychological treatments or other community-based supports. In bipolar disorders, the majority of the health-care spend(about 53%) was actually accounted for by hospital admissions. This isquite important in the context of psychological treatments because the argu-ment against adjunctive therapy is usually that it is too expensive, but theopportunity cost of a course of about 20 hours of therapy is probably onlyequivalent to that of 2 days of in-patient care and, as evidenced from recentresearch, most psychological treatments can significantly reduce hospital-izations compared with usual psychiatric treatment.
Psychological treatment studies
A review of psychological treatment studies in bipolar disorders over thepast 30 or 40 years shows that, up until very recently, the research was notof a high standard. Between 1960 and 1995, there were about 20 studiesand the average sample size was about 20–25 subjects.14,15 Few of themwere randomized trials, but the studies provided some indication that, withfamily therapy and group therapy, adherence to medication was improvedand individuals with bipolar disorders did better subjectively if theyreceived these additional inputs. However, there were no data thataddressed the issue of relapse or measured social or functional outcomesin specific terms. The situation has changed quite dramatically in the past4–5 years and there are now 17 or 18 RCTs ongoing across the world andfour or five of those trials have over 100 participants, which may be a smallnumber in terms of drug trials, but is a large number in terms of many psy-chological treatment studies. About eight of the completed studies have pro-duced data that are accessible to be put into a pooled analysis of relapserates for psychological treatments compared with usual psychiatric treat-ment (either routine or standardized).
The first set of studies is put into rank order of the size of the studiesand includes four or five relatively small-scale studies using a variety ofapproaches, but predominantly either cognitive behaviour therapy (CBT) orcognitive and behavioural techniques, or interpersonal social rhythms ther-apy (IPSRT).16 These small-scale studies demonstrate that psychologicaltreatments appear to have some benefit in preventing relapse, althoughthere is a hint that the briefer interventions17,18 are more effective in pre-venting mania rather than depression. As shown in Figure 18.1, a meta-analysis of relapse rates using a fixed effects model shows that, when datafrom all the studies are combined,17–20 the odds ratio for relapse in the
Ch 18 7/4/05 4:00 pm Page 167

168 Bipolar disorder: the upswing in research & treatment
intervention as compared with the control group is 0.31 (95% confidenceinterval (CI) 0.15–0.64; p<0.002). A similar statistically significant result isobtained for the random effects model.
The three largest studies published in the literature used either CBT,21
family therapy22 or group psycho-education.23 A separate meta-analysis ofoutcome data from these RCTs using fixed and random effects modelsdemonstrates that the odds ratio (OR) for relapse in the active as comparedwith the control treatment groups (OR 0.37; 95% CI 0.23–0.60; p<0.001)is similar to that reported for the earlier studies (Figure 18.2). There appearto be some differences in odds ratios between studies, but these may relateto sample characteristics (e.g. proportion of participants who met criteriafor bipolar I or bipolar II disorders) as well as similarities or differences inthe style and content of the treatments. Importantly, these interventions allhave a significant effect on rates of depressive relapses. Perhaps for thetreatment of syndromal and subsyndromal symptoms of bipolar depressionit is beneficial to use more complex and more extended interventions.
The problem with these studies, and the same is true of the majority ofmedication trials, is that they report treatment efficacy in less representa-tive groups of patients with bipolar disorders, rather than effectiveness witha clinical sample more typical of those being treated in general adult psy-chiatry services in the UK. Many of the RCTs were single centre or special-ist centre studies, which often recruited selected sub-populations of indi-viduals with bipolar disorders. In some studies, participants had to beeuthymic for more than 1 year to be included and individuals with co-morbid disorders were largely excluded. Some RCTs utilized self-report,postal returns of self-ratings or unblinded researcher assessments of out-comes, and the trial results often reported cross-sectional analyses ofrelapse rates rather than longitudinal data such as weekly symptom fluctu-ations or survival curve analyses. The outcome data reported in some of the
Figure 18.2 Odds ratios of relapse rates in recent larger-scale trials.
Citation N Total Effect p Value
Colom et al 200323 120 0.41 0.02Lam et al 200321 96 0.26 0.00Miklowitz et al 200022 101 0.46 0.08
Fixed Combined (3) 317 0.37 0.00Random Combined (3) 317 0.37 0.00
0.01 0.1 1 10 100
Favours psychotherapy
Ch 18 7/4/05 4:00 pm Page 168

Psychological treatments 169
earlier studies made it difficult to disentangle relapses from dropouts andit was not always clear in the RCTs whether the results were from intent totreat or per protocol analyses.11
Overall, it seems that there is promising but not conclusive data aboutthe benefits of psychological treatments in bipolar disorders. However, thereis a need for a pragmatic large-scale trial looking at effectiveness, under-taken in a number of centres, with intensive follow-up assessments, includ-ing blind raters and with therapists trained in a way that is comparable towhat usually happens in the NHS, i.e. with therapists competent in deliver-ing psychological therapies then trained to deliver a therapy specificallydesigned for individuals with bipolar disorders. This is what we attemptedin our MRC study.13
Preliminary report on the MRC study
Just over 250 persons with bipolar disorder were recruited to the RCT fromfive centres across Britain. Potential participants were identified via caseregisters, hospital data or Care Programme Approach records and then therelevant community teams were approached to ask whether they wouldallow us to assess the individual for the trial. The main inclusion criterionwas that the individual had had at least one manic episode in the previous12 months, giving them at least a 50% risk of further relapse in the next12–18 months. The sample was fairly typical of bipolar disorder popula-tions (about 60% female), their mean age of onset was about 25 years, mosthad been diagnosed with bipolar disorder for about 17 or 18 years and theyhad had many previous episodes (median 12). Importantly, because we keptour inclusion criteria very broad, the only two factors that led to exclusionwere inability to give informed consent or high risk of suicide. Thus, thepopulation is much more heterogeneous than in any previous RCTs – about30% of our population were currently unwell, about a third had a co-mor-bid axis 1 disorder, about a fifth had co-morbid substance misuse, 7–10%had antisocial personality disorder or borderline personality disorder, halfhad a lifetime history of substance misuse, 21% had a history of violencetowards others, 22% had been in trouble with the police, including a smallnumber who had been in prison, and a number of people (at least one infive) had made significant and severe suicide attempts with intent to killthemselves.
The acute treatment phase lasted 6 months and individuals were ran-domly allocated to usual psychiatric care and treatment or usual treatment
Ch 18 7/4/05 4:00 pm Page 169

170 Bipolar disorder: the upswing in research & treatment
plus 22 sessions of CBT. All subjects were then followed up every 8 weeksfor a further 12 months. The primary outcome measure was time to firstrelapse. The main analysis showed no between-group differences in time tomanic, depressive or any type of relapse. However, if the patient populationwas separated into those with and those without the adverse clinical fea-tures highlighted in this chapter (co-morbidity, severe suicide attempts, per-sistent syndromal or subsyndromal symptoms, multiple episodes, etc.), wediscovered that those subjects with bipolar disorder and additional adverseclinical characteristics had a highly significant reduction in survival timecompared with those with bipolar disorder but no evidence of additionaladversity. The latter sub-group are typical of those who are usually includ-ed in all types of treatment trials, and had comparable reductions in relapserates to those receiving CBT added to treatment as usual, having a relapserate of less than half that of subjects receiving usual treatment alone. Thosepatients with complex bipolar disorders who are difficult to treat have apoorer outcome with and without adjunctive CBT.
Current treatment guidelines
The main treatment guidelines on bipolar disorders, like those on othermood disorders (e.g. American Psychiatric Association24), acknowledge therole and importance of psychological therapies as well as medication.However, the guidelines all suggest that specialist psychological therapiesare a precious resource that should therefore be targeted at the cases thatare most difficult to treat. Whilst the notion that those with the most com-plex problems should be provided with the greatest input seems to makeclinical sense, the data from the RCTs reviewed appear to undermine thisidea. Persons who appear consistently to benefit from adjunctive psycho-logical therapy are those at high risk of recurrence but do not have othercomplications or adverse clinical features that commonly accompany bipo-lar disorders; i.e. the best candidates for psychological therapies are thosewith relatively fewer previous episodes, who are at above average risk of afurther relapse, but who do not appear to have done well with medicationand out-patient support, either because of some ineffectiveness of the pre-scribed medication or because they are not taking medication. The problemis that this group represents only about one in five of the individuals withbipolar disorders in contact with general adult psychiatry services and, veryimportantly, the use of psychological therapies in this group would appearto go against current guideline recommendations on the use of adjunctive
Ch 18 7/4/05 4:00 pm Page 170

Psychological treatments 171
psychological treatments. Further research is now needed to establishwhether those individuals with more complex presentations of bipolar dis-orders require a longer course of therapy, e.g. CBT plus maintenance ses-sions, or whether a different model of psychological therapy needs to beintroduced to help deal with the multiple psychological and social problemsthey confront over and above managing the consequences of bipolar disor-der.
Conclusions
The review of published trials on bipolar disorders offers evidence for theefficacy of adjunctive psychological treatments. Indeed, the evidence is thatadding simple interventions, such as 6–10 sessions targeted at medicationadherence or relapse prevention, is relatively cost-effective, particularly asmanic relapses (the most frequent cause of hospitalizations) are significant-ly reduced. Self-help and psycho-education are also very useful and willprobably avert some of the relapses in some individuals. The extended indi-vidual therapies such as CBT or family therapy appear to be useful for extin-guishing subsyndromal or syndromal symptoms of bipolar depression.However, the main problem at the moment is that the treatment guidelinesfor bipolar disorders are based on extrapolations from unipolar disordersand there is simply not sufficient evidence on effectiveness to support this.The MRC study is the largest study that has ever been undertaken of psy-chological treatments in bipolar disorders and addresses this question ofthe benefits of such therapies in general clinical settings. The findings sug-gest it may be necessary to adapt the current psychological treatment mod-els to take into account the heterogeneity of cases seen in day-to-day prac-tice. At this time we have evidence that suggests that psychological therapieswork very well for the patients with the middle group of outcomes, and that30% do not respond to mood stabilizers alone, but we need more studies toestablish what the best approach will be for the cases of bipolar disordersthat are the most difficult to treat.
References
1. American Psychiatric Association, Practice guideline for the treatment ofpatients with bipolar disorder (revision). Am J Psychiatry 2002; 159(Suppl4):1–50.
Ch 18 7/4/05 4:00 pm Page 171

172 Bipolar disorder: the upswing in research & treatment
2. Goodwin GM; Consensus Group of the British Association for Psycho-pharmacology, Evidence-based guidelines for treating bipolar disorder: recom-mendations from the British Association for Psychopharmacology. JPsychopharmacol 2003; 17:149–173.
3. López AD, Murray CJ, The global burden of disease. Nat Med 1998;4:1241–1243.
4. Angst F, Stassen HH, Clayton PJ, Angst J, Mortality of patients with mood dis-orders: follow-up over 34–38 years. J Affect Disord 2002; 68:167–181.
5. American Psychiatric Association, Diagnostic and Statistical Manual ofMental Disorders, 4th edn. American Psychiatric Association: Washington, DC,1994.
6. Bieling PJ, MacQueen GM, Marriot MJ et al, Longitudinal outcome in patientswith bipolar disorder assessed by life-charting is influenced by DSM-IV per-sonality disorder symptoms. Bipolar Disord 2003; 5:14–21.
7. Black DW, Winokur G, Hulbert J, Nasrallah A, Predictors of immediateresponse in the treatment of mania: the importance of comorbidity. BiolPsychiatry 1988; 24:191–198.
8. Scott J, Psychotherapy for bipolar disorder – An unmet need? Br J Psychiatry1995; 167:581–588.
9. Gitlin MJ, Swendsen J, Heller TL et al, Relapse and impairment in bipolar dis-order. Am J Psychiatry 1995; 152:1635–1640.
10. Harrow M, Goldberg JF, Grossman LS et al, Outcome in manic disorders. Anaturalistic follow-up study. Arch Gen Psychiatry 1990; 47:665–671.
11. Scott J, Cognitive therapy for bipolar disorders. Expert Rev Neurother 2002;2:573–581.
12. Judd LL, Akiskal HS, Schlettler PJ et al, The long-term natural history of theweekly symptomatic status of bipolar 1 disorder. Arch Gen Psychiatry 2002;59: 530–537.
13. Scott J, Cognitive Therapy in Bipolar Disorders. ENCP: Stockholm, Sweden,2004.
14. Gutierrez MJ, Scott J, Psychological treatments for bipolar disorders: A reviewof randomised controlled trials. Eur J Psychiatry 2004; 254:92–98.
15. Scott J, Gutierrez MJ, The current status of psychological treatments in bipo-lar disorders: a systematic review of relapse prevention. Bipolar Disord 2004;6:498–503.
16. Frank E, Interpersonal and social rhythm therapy prevents depressive sympto-matology in bipolar I patients. Bipolar Disord 1999; 1(Suppl):13.
17. Cochran SD, Preventing medical noncompliance in outpatient treatment ofbipolar disorders. J Consult Clin Psychol 1984; 52:873-878.
18. Perry A, Tarrier N, Morriss R et al, Randomised controlled trial of efficacy ofteaching patients with bipolar disorder to identify early symptoms of relapseand obtain treatment. BMJ 1999; 318:149–153.
19. Lam DH, Bright J, Jones S et al, Cognitive therapy for illness: a pilot study ofrelapse prevention. Cogn Ther Res 2000; 24:503–520.
20. Scott J, Garland A, Moorhead S, A pilot study of cognitive therapy in bipolardisorders. Psychol Med 2001; 31:459–467.
Ch 18 7/4/05 4:00 pm Page 172

Psychological treatments 173
21. Lam DH, Watkins ER, Hayward P et al, A randomized controlled study of cog-nitive therapy for relapse prevention for bipolar affective disorder: outcome ofthe first year. Arch Gen Psychiatry 2003; 60:145–152.
22. Miklowitz DJ, Simoneau TL, George EL et al, Family-focused treatment of bipo-lar disorder: 1-year effects of a psychoeducational program in conjunction withpharmacotherapy. Biol Psychiatry 2000; 48:582–592.
23. Colom F, Vieta E, Martinez-Aran A et al, A randomized trial on the efficacy ofgroup psychoeducation in the prophylaxis of recurrences in bipolar patientswhose disease is in remission. Arch Gen Psychiatry 2003; 60:402–407.
24. American Psychiatric Association, Practice guideline for major depressive dis-order in adults. Am J Psychiatry 1993; 150:1–26.
Ch 18 7/4/05 4:00 pm Page 173

Ch 18 7/4/05 4:00 pm Page 174

Lithium,the forgotten drugMario Maj
c h a p t e r 1 9
In this chapter I will address the following questions: (1) Is lithium to someextent a forgotten drug? (i.e. has the use of lithium in bipolar disorderdeclined drastically in recent years?); (2) If so, why has lithium been for-gotten? (i.e. why has its use declined?).
The answer to the first question is yes. Right now, in the USA, lithium isless frequently prescribed than valproate for the treatment of bipolar disor-der. For instance, Scott–Levin’s Physician Drug and Diagnosis Audit showedthat, in May 2002, 1858 prescriptions for bipolar disorder were for lithiumand 2116 were for valproate.1 The decline in the use of lithium is alsoreflected in a similar way in the sale figures in European countries. Forinstance in Italy, in the year 2002, the maintenance treatment for bipolardisorder included in 59% of cases at least one antipsychotic (with olanzap-ine and risperidone surpassing haloperidol as the most frequently pre-scribed drugs of this group); in 37% of cases at least one anticonvulsant(with valproate surpassing carbamazepine as the most frequently pre-scribed drug of this group); in 32% of cases lithium and in 25% of cases anyantidepressant. Several European clinicians probably now share the expe-rience described a few years ago in the USA by Ronald Fieve: ‘For years, inan average week I have seen four to six bipolar patients who have been givensmatterings of all the above drugs (anticonvulsants and new antipsychotics)for a few weeks to 2 years, by one to five psychiatrists. The patient and fam-ily report that lithium has not been used, or was used for a short time andhas not worked’.2
Why has the use of lithium declined so drastically? Five factors have inmy opinion been more or less important, and I will briefly discuss four ofthem. I will not discuss in detail the remaining one, i.e. the doubts recentlyexpressed about the impact of lithium on the course of bipolar disorder inordinary clinical conditions, because this issue has been the subject of sev-eral reviews (for example, reference 3). I do not believe that this factor con-tributed significantly to the recent decline in the use of the drug.
Ch 19 7/4/05 4:01 pm Page 175

176 Bipolar disorder: the upswing in research & treatment
A much more powerful factor has been, in my opinion, the fact that lithi-um treatment is a very demanding one for both the psychiatrist and thepatient. On the psychiatrist’s side, lithium treatment requires training, time,attention and general medical skills; on the patient’s side, it requires col-laboration, tolerance of side-effects, diligence and persistence. The patient’sside, and in particular the burden of side-effects, has been covered exten-sively in the literature, whereas the psychiatrist’s side has not frequentlybeen a focus of attention. I will refer now briefly to the situation of psychi-atric practice in my country (Italy), arguing that some recent developmentsin this practice have worked against the use of a drug with the characteris-tics of lithium. Although the case of Italy may be extreme, some of the devel-opments I will describe are occurring now or are going to occur in severalother European countries. The first and the most significant developmenthas been the move from hospital-based to community-based psychiatricpractice; this means that the vast majority of Italian psychiatrists do notwork now in a hospital environment, but in centres that are sometimes sev-eral hundreds of kilometres away from the nearest hospital. No laboratoryis available in these centres and, if the consultation of another specialist (forinstance, an endocrinologist or a nephrologist) is required, the patient hasto be referred to the nearest hospital. Psychiatric training increasinglyreflects this new situation, with an increasing focus on social psychiatry andcommunity mental health care, and a decreasing emphasis on proper med-ical aspects (for instance, most young psychiatrists in Italy have never seenan electroconvulsive therapy session and several of them are not trainedadequately in the use of lithium). On the other hand, the workload for psy-chiatrists operating in the public sector has increased significantly, whichmeans that much less time can be devoted to the individual patient. Finally,the press and the public have been recently sensitized to incidents in med-ical practice, including those involving the use of drugs, and psychiatrists’alertness to the risk of these incidents has increased significantly. In this sit-uation, it is not surprising that psychiatrists show a preference for drugsthat are easy to use, not problematic, not potentially toxic, unlikely to causeincidents, possibly ‘transnosographic’ (thus not requiring a sophisticateddiagnostic assessment), which do not require frequent physical examina-tions or laboratory tests and which are unlikely to require consultations ofother specialists. Lithium certainly does not correspond to this identikit,nor does clozapine, arguably another forgotten drug, which is received nowby just 8% of Italian patients with a diagnosis of schizophrenia. The exam-ple of Italy may be extreme, but similar problems with training and a pref-erence for drugs that are non-problematic and easy to use have been noted
Ch 19 7/4/05 4:01 pm Page 176

Lithium, the forgotten drug 177
also in the USA. In that country, according to Fieve, ‘most residents are notadequately trained to the subtleties of lithium treatment… Therefore, ongraduation they are poorly equipped and tend to underuse lithium as thefirst line of treatment. Instead, they begin the new bipolar patient on anantiepileptic, since it is easier to use and requires less knowledge’.3
Another powerful factor obviously contributing to the recent decline inthe use of lithium has been the introduction of several new drugs whichhave been found effective in bipolar disorder and which, contrary to lithi-um, are backed by significant commercial interests. The availability of thesenew drugs is, of course, very welcome. For many years, there has been nosignificant alternative for bipolar patients who did not respond or respond-ed partially to lithium or did not tolerate lithium or in whom lithium wascontraindicated. Now several alternatives are available. Of course, drugcompanies producing these drugs pursue profit as their main objective but,if the drugs are effective, it can be said that to a large extent there is a con-vergence of interests between psychiatrists, companies, patients andpatients’ families. It is also understandable that drug companies have theirown marketing strategies and that psychiatrists are a target for these strate-gies, although a code of conduct for both psychiatrists and drug companieswould be very useful in this respect.
I believe, however, that we should make a much more significant effortto preserve the integrity and independence of our scientific publications andof our research in the area of treatment of bipolar disorder. I have witnessedin this area in recent years several unpleasant developments. I have seenseveral biased clinical guidelines, and many biased and tendentious reviewsand editorials. I am aware of some cases of ghostwriting and of someattempts by sponsors to obtain changes in chapters of volumes in favour oftheir drugs. I have seen some cases of double or triple publication of thesame study with different first authors. I have seen several journal supple-ments with papers that had not been peer-reviewed, and I am aware of somecases of selective publication. I have seen several clinical trials with evidentbiases in favour of new drugs and against the comparison drug, which waslithium: for instance, ‘enriched’ designs favouring the drug under testing;trials in which serum lithium levels up to 2.7mEq/l were reached, so thatnot surprisingly there was a very high dropout rate in the lithium group dueto intolerance; reports on clinical trials focusing on secondary outcomemeasures favouring the new drug, so that the trial was presented as posi-tive, even if on the primary outcome measure the drug was not superior toplacebo. These problems and biases affect only a minority of the availablepublications and trials, but I believe that even these relatively few cases are
Ch 19 7/4/05 4:01 pm Page 177

178 Bipolar disorder: the upswing in research & treatment
of concern, because they may impair the credibility of the entire system. A
recent paper published in the British Medical Journal,4 entitled ‘Evidence
b(i)ased medicine – selective reporting from studies sponsored by pharma-
ceutical industry’, focusing on drugs for depression, represents a clear
warning in this respect.
A fourth, less powerful factor contributing to the decline in the use of
lithium has been the recent change in the phenomenology and/or conceptu-
alization of bipolar disorder and the fact that lithium is not equally effective
throughout the ‘bipolar spectrum’. That lithium is less efficacious in atypi-
cal and complicated varieties of bipolar disorder than in the classical
manic–depressive forms is now sufficiently documented. What is not docu-
mented, however, is that lithium is not efficacious in these atypical or com-
plicated varieties, or that other drugs are superior to lithium in these forms,
as is frequently stated in reviews and clinical guidelines. For instance, lithi-
um does exert an impact on bipolar disorder with mood-incongruent psy-
chotic features5 and in rapidly cycling bipolar disorder,6 and a recent meta-
analysis showed no superiority for anticonvulsants over lithium in rapid
cyclers.7
The fifth factor, which is more significant than commonly believed, is
related to the treatment of acute mania. Lithium is regarded as the gold
standard for the treatment of mania, and new drugs proposed for use in
bipolar disorder are first of all tested for their antimanic activity, and usu-
ally compared to placebo or to lithium. The fact is, however, that lithium has
never been the preferred drug for the treatment of acute mania in clinical
practice. Most psychiatrists prefer to use antipsychotics and several of them
show an inclination to continue to prescribe for maintenance treatment the
same drug they used for the manic episode. This is a significant advantage
that new generation antipsychotics may have over lithium, if clinicians will
be convinced that these new drugs are as quick and powerful as standard
antipsychotics in the acute management of mania.
It is clear from the above discussion that lithium has been to some extent
forgotten in the treatment of bipolar disorder. However, did lithium perform
better or worse than other old and powerful drugs challenged by new devel-
opments, such as haloperidol as an antipsychotic or tricyclics as antide-
pressants? It seems that lithium has been less forgotten than these other
drugs. For instance, in Italy, only 26% of patients with a diagnosis of schiz-
ophrenia received haloperidol for maintenance treatment in the year 2002,
and only 7% of those with a diagnosis of depression received tricyclic anti-
depressants. On the research side, a Medline search for the year 2002 iden-
Ch 19 7/4/05 4:01 pm Page 178

Lithium, the forgotten drug 179
tified 943 papers dealing with lithium, versus 446 for haloperidol and 171for amitriptyline.
Finally, I have seen no recent study showing a superiority of haloperidolover second-generation antipsychotics on a significant dimension of schizo-phrenia or a superiority of tricyclics over the new antidepressants on a sig-nificant dimension of depression, whereas I have seen, for instance, a recentstudy showing the significant superiority of lithium over valproate in reduc-ing the risk for suicide in bipolar patients.8 All these may be regarded assignals that lithium has not been, and will probably not be, at least in thenear future, completely forgotten.
References
1. Goodwin FK, Rationale for long-term treatment of bipolar disorder and evi-dence for long-term lithium treatment. J Clin Psychiatry 2002; 63(Suppl10):5–12.
2. Fieve RR, Lithium therapy at the millennium: a revolutionary drug used for 50years faces competing options and possible demise. Bipolar Disord 1999;1:67–70.
3. Maj M, The impact of lithium prophylaxis on the course of bipolar disorder: areview of the research evidence. Bipolar Disord 2000; 2:93–101.
4. Melander H, Ahlqvist-Rastad J, Meijer G, Beermann B, Evidence b(i)asedmedicine – selective reporting from studies sponsored by pharmaceuticalindustry: review of studies in new drug applications. BMJ 2003;326:1171–1173.
5. Maj M, Pirozzi R, Bartoli L, Magliano L, Long-term outcome of lithium prophy-laxis in bipolar disorder with mood-incongruent psychotic features: a prospec-tive study. J Affect Disord 2002; 71:195–198.
6. Baldessarini RJ, Tondo L, Hennen J, Effects of rapid cycling on response tolithium maintenance treatment in 360 bipolar I and II disorder patients. JAffect Disord 2000; 61:13–22.
7. Tondo L, Hennen J, Baldessarini RJ, Rapid-cycling bipolar disorder: effects oflong-term treatments. Acta Psychiatr Scand 2003; 108:4–14.
8. Goodwin FK, Fireman B, Simon GE et al, Suicide risk in bipolar disorder dur-ing treatment with lithium and divalproex. JAMA 2003; 290:1467–1473.
Ch 19 7/4/05 4:01 pm Page 179

Ch 19 7/4/05 4:01 pm Page 180

Advantages anddisadvantages of atypicalantipsychotics or valproatein bipolar disorderJohn Cookson
c h a p t e r 2 0
This chapter concerns the comparison between atypical antipsychotics andvalproate used for acute mania and for the prophylaxis of bipolar disorder.The comparison with classical antipsychotics is also considered. Discussionof antipsychotics and valproate in acute mania is topical in relation to cur-rent guidelines for the management of bipolar disorder. The revisedAmerican Psychiatric Association Guidelines1 propose a choice betweenlithium, valproate or an atypical antipsychotic for less severe mania. Forsevere mania, the Guidelines advise a combination of an antipsychotic andvalproate or lithium. However, it is arguable whether as a first-line treat-ment a combination is better than using one or other of these drugs alone,as is recommended by the Guidelines of the British Association forPsychopharmacology.2
The comparison of antipsychotics and valproate for mania shouldinclude consideration of speed of action, mechanism of action, the spectrumof symptoms that are controlled, what pattern of mania may be affected,whether the presence of psychotic features makes a difference, and whethermixed mania responds differently.
The study of psychotic mania by Keck et al3 indicated that, if a suffi-ciently large dose of valproate (as semisodium valproate 20mg/kg per day)is used from the start, there is a similar improvement with valproate orhaloperidol (0.2mg/kg per day). However, the generalizability of this findingmay be limited, since haloperidol did not show its usual rapid onset ofeffect. There was improvement by only 12% in mania ratings after 24 hours,as opposed to a 30% improvement in 20 minutes after intravenoushaloperidol administration in other studies, which was not due to sedation,
Ch 20 7/4/05 4:02 pm Page 181

182 Bipolar disorder: the upswing in research & treatment
as was confirmed in our own study.4 A second study of valproate loading byHirschfeld et al5 showed a delay in onset of antimanic effect with valproateof about 48 hours. For rapid tranquillization, which is often needed in acutemania, it is necessary to use drugs that provide initial control of the manicstate within minutes or hours, rather than days. A commonly usedapproach is to use combined treatment with 1 or 2mg of lorazepam and 5or 10mg of haloperidol, given intramuscularly. Haloperidol can work quiterapidly but often has unpleasant side-effects, including akathisia, and lessoften dystonia. To avoid the side-effects we require an intramuscular atypi-cal antipsychotic, such as olanzapine or ziprasidone.
In a placebo-controlled trial, Meehan et al studied intramuscular olan-zapine 10mg, with lorazepam 2mg as the active comparator.6 Using a scalemeasuring excitement with items from the Positive and Negative SyndromeScale (PANSS), there was rapid improvement after intramuscular olanzap-ine administration, evident within 30minutes, and greater than with 2mg oflorazepam. Thus, antipsychotics are capable of having an antimanic effectstarting within minutes and developing over 2 hours.
Belmaker and colleagues have reported that valproate controls statusepilepticus quickly when given intravenously, but does not have an immedi-ate antimanic effect.7 This suggests that valproate works indirectly. Thecommon mechanism of action of antipsychotics is blockade of dopaminereceptors. Some, such as haloperidol, also block α1 receptors and olanzap-ine also blocks histamine receptors. Valproate, on the other hand, works onsecond-messenger systems and may reduce dopamine release.8 Lithiummay also reduce dopamine release.9 Thus, a combination of an antipsy-chotic drug and valproate or lithium might be expected to have synergisticeffects, improving mania.
Efficacy in mania
The number needed to treat (NNT) represents the number of patients whomust be treated in order for one patient to achieve the defined response –usually a 50% reduction in score on a scale such as the 11-item YoungMania Rating Scale (YMRS) – as a result of the pharmacological effect of thedrug. The NNT is calculated by dividing the difference in response ratebetween active drug and placebo into 100 and correcting to the next highestinteger. The NNT thus provides a measure of the size of effect that can beexpected of the drug in a clinical situation. For a drug to be a usefulmonotherapy as a first-line treatment in a common and severe disorder
Ch 20 7/4/05 4:02 pm Page 182

Advantages and disadvantages of atypical antipsychotics or valproate 183
such as mania, we have argued that the NNT for 50% improvement in sever-ity should be in the order of 2–4.10
Two studies have compared valproate with placebo in mania. Bowden etal11 randomized patients to placebo, valproate or lithium, using a 50%reduction in scores on a mania rating scale as the criterion for improvement(Table 20.1). The NNT for 50% improvement was 5. Even though valproateis recognized as a useful drug, it was not usually sufficient as monotherapy.The placebo-controlled study by Pope et al12 showed a more dramatic effect.The placebo response rate was low, this being the only modern randomizedtrial in mania, showing such a low placebo response rate, probably becausethe patients were lithium-resistant manic in-patients. The difference inresponse rate was large, giving a NNT of 3, with close confidence intervals.
The first modern controlled trials of antipsychotics in mania were thoseon olanzapine designed under the guidance of Mauricio Tohen and financedby Eli Lilly & Co. These and other large-scale studies of atypical antipsy-chotics answer important questions. In most of the placebo-controlled stud-ies, the criterion for response was a 50% improvement on the YMRS. In thefirst study by Tohen et al,13 olanzapine (up to 15mg a day) had a NNT of 4,with a confidence interval of 3–10.
The antipsychotics that have been shown to improve mania in parallel-group placebo-controlled trials are: haloperidol (in two published studiesas active comparator), olanzapine (in two), risperidone (in three),quetiapine (in two), ziprasidone (in two) and aripiprazole (in two). The mostimpressive result was in a study of risperidone conducted in India byKhanna et al.14 The dropout rate for patients on risperidone (average5.8mg/day) was low, allowing time for a high response rate, and NNT of 3(2–4) (Table 20.2). This is a true reflection of the potential of antipsychoticsin mania: a large and reliable antimanic effect.
Treatment Response rate Difference Number needed 3 weeks Criterion (%) from placebo (%) to treat (95% CI)
Lithium (n = 35) 49
Placebo (n = 73) 25
Divalproex (n = 68) 48
Divalproex (n = 23) 45
Placebo (n = 20) 9
SADS-M, Schedule for Affective Disorders and Schizophrenia; MRS, mania rating scale.
50% less SADS-M
Bowden et al, 199411
50% less MRS
Pope et al, 199112
24 5 (3–22)
23 5 (3–14)
36 3 (2–9)
Table 20.1 Number needed to treat in placebo-controlled trials of valproate inmania
Ch 20 7/4/05 4:02 pm Page 183

184 Bipolar disorder: the upswing in research & treatment
Tab
le 2
0.2
Pla
ceb
o-c
ontr
olle
d p
aral
lel-
grou
p r
and
omiz
ed t
rial
of
mon
oth
erap
y w
ith
ris
per
idon
e in
man
ia
Trea
tmen
tD
urat
ion
Incl
usio
n D
ropo
uts
Dro
pout
s
Oth
er
Resp
onse
Diff
eren
ceN
umbe
r
(mea
n do
se/d
ay)
Aut
hors
crit
eria
for
fo
r ad
vers
e dr
opou
ts(%
)fr
om
need
ed
Num
bers
for
ITT
Site
sC
rite
rion
for
inef
ficac
yev
ents
(%)
plac
ebo
to t
reat
Extr
a dr
ugs
resp
onse
(%)
(%)
Risp
erid
one
3 w
eeks
D
SM-I
V M
anic
4.
83.
42.
873
373
(n=
146)
Khan
na,
Viet
a et
al,
2003
14or
mix
ed(2
–4)
Mea
n m
odal
In
dia
YMRS
≥20
(5.6
mg/
day)
LZP
for
10 d
ays
50%
red
uctio
n
YMRS
Plac
ebo
14.6
2.1
12.5
36
(n=
144)
ITT,
in
ten
tion
to
trea
t; L
ZP,
lor
azep
am;
YM
RS
, Yo
un
g M
ania
Rat
ing
Sca
le.
Ch 20 7/4/05 4:02 pm Page 184

Advantages and disadvantages of atypical antipsychotics or valproate 185
There have been two direct comparisons of olanzapine and valproate inmania, one conducted by Tohen et al for the manufacturers of olanzapine,15
the other for the manufacturers of valproate by Zajecka et al.16 Both stud-ies appear to show that olanzapine produces a slightly greater and fasterimprovement on the YMRS. In the larger of the two studies, which also useda slightly higher dose of olanzapine, there was a significant difference. Thewhole range of manic symptoms improved with both drugs within 3 weeks,the smallest improvement being in the item for insight.
Subdividing the sample into patients with pure or mixed manicepisodes, or into those with or without a rapid-cycling course, or those withor without psychotic features, the degree of improvement was not found todepend on the type of mania, with one exception. Olanzapine was superiorto valproate in the non-psychotic manic patients, whereas for the psychoticmanic patients valproate and olanzapine were equally effective.15 However,the study was not statistically powered to detect differences between sub-groups of manic patients.
Treatment side-effects
In the study by Tohen et al, the side-effects more common with olanzapinethan valproate were drowsiness, dry mouth and increased appetite; theside-effects more common with valproate were gastrointestinal disturbanceand nausea.15 In the study by Zajecka et al, further side-effects, commonerwith olanzapine, were drowsiness, weight gain, rhinitis (from noradrenalineα1 receptor blockade), oedema and slurred speech, probably related todrowsiness.16 Broader evidence would indicate that the side-effects of val-proate include polycystic ovary syndrome (particularly in adolescentwomen), hair loss (usually transient and perhaps counteracted by zinc sup-plementation), fetal valproate syndrome (if used in pregnancy), spontaneousbruising or bleeding, pancreatitis and a risk (especially in children) of liverdamage.
Depression in mania
Most manic patients have additional depressive symptoms and somepatients switch from mania to depression during treatment. This switch hasbeen reported by Zarate et al17 to occur more often when patients are treat-ed with a typical antipsychotic, and anticholinergic medication is withheld,than with placebo.
Ch 20 7/4/05 4:02 pm Page 185

186 Bipolar disorder: the upswing in research & treatment
In the direct comparison between valproate and olanzapine, both drugsreduced depressive symptoms.15 Thus, as mania gets better, the accompan-ying depressive symptoms also usually improve, proportionately with theimprovement in mania.
Some drugs may be better at preventing a switch from mania intodepression. In a comparison by Tohen et al18 of olanzapine with haloperi-dol in mania, patients on haloperidol had a 12% switch rate into depressionwithin 6 weeks, while those on olanzapine had a 5% switch rate. The onlyother comparative study suggesting a lower switch rate in comparison withhaloperidol is that by Brecher et al with quetiapine.19 There was a trend forolanzapine to be better than valproate in preventing a switch into depres-sion, but this was not significant.15
Combination treatment
Several studies have demonstrated an advantage of combining an antipsy-chotic with lithium or valproate, over lithium or valproate alone. Conversely,the addition of valproate to classical antipsychotics (mainly haloperidol) hasbeen shown by Muller-Oerlinghausen et al20 to produce greater improve-ment than addition of placebo.
The majority of these studies involved a design in which patients whohad not responded to one drug administered for at least 3 weeks had addi-tional treatment with the combination of drugs using a placebo control forthe added drug. In some studies a proportion of patients commenced on thecombination without previous treatment, and the control group receivedonly the lithium, valproate or carbamazepine with placebo instead ofantipsychotic. There have been no studies of combination treatment inwhich a control group receive only placebo. It is therefore not possible todetermine directly the size of effect of giving combination treatment to drug-free patients with mania over giving placebo alone, or to determine the sizeof advantage of combination therapy over monotherapy initiated in drug-free patients with mania.
The studies of combination treatment have proved that several antipsy-chotics provided additional efficacy when added to lithium or valproatecompared with lithium or valproate alone. These are: haloperidol, risperi-done, olanzapine and quetiapine. A similar study with ziprasidone was neg-ative.
In combination studies with olanzapine, this drug was shown by Tohenet al21 to provide additional efficacy when added in patients who hadalready received lithium or valproate for at least 3 weeks. The effect was
Ch 20 7/4/05 4:02 pm Page 186

Advantages and disadvantages of atypical antipsychotics or valproate 187
statistically significant in the subgroup on valproate but only at trend levelin the smaller subgroup on lithium.
Risperidone in combination with lithium or valproate was shown bySachs et al22 and Yatham et al23 to be more effective than either lithium orvalproate alone, and as efficacious as haloperidol in combination with eitherlithium or valproate,22 in patients with manic or mixed episodes. Theadvantage of adding risperidone was far more evident in those who hadalready been on a mood stabilizer for at least 2 weeks before randomiza-tion, than in those who started a mood stabilizer shortly before startingrisperidone or placebo.23
In total, these combination studies demonstrate that addition of the sec-ond component of combined treatment, achieves an added response with aNNT of 5 or 6. This would suggest that not every patient needs the combi-nation, but that some patients do better with a combination, and others failto respond even with combined antipsychotic and lithium or valproate.
Maintenance treatment
The only large-scale placebo-controlled maintenance trial of valproate, byBowden et al,24 was very disappointing. The benefit of valproate overplacebo was very small and applied mainly to depressive symptoms in asecondary analysis. In the direct comparison by Tohen et al15 of olanzapineand valproate for acute mania, there was a small difference in favour ofolanzapine, in terms of days spent with symptoms over the subsequent yearof the study. By contrast, there is evidence from Tohen et al25 that contin-ued treatment with olanzapine plus lithium or valproate, after remission ofmania on a combination of the two, led to a lower risk of symptomaticrelapse of bipolar disorder over the subsequent year.
Weight gain
The study of valproate against olanzapine16 demonstrated that olanzapineproduced more weight gain than valproate over the course of 12 weeks. Inthe 1-year follow-up of the study comparing olanzapine and valproate,15 bythe end of the year patients showed similar weight gain with valproate orolanzapine. However, the extent of weight gain on olanzapine in this study(mean 6kg) was considerably less than in studies of olanzapine in schizo-phrenia. Weight gain on olanzapine reached a plateau at about 19 weeks,
Ch 20 7/4/05 4:02 pm Page 187

188 Bipolar disorder: the upswing in research & treatment
which is earlier than in schizophrenia, where the weight gain with olanzap-
ine continued for up to 1 year.
Comparison of classical and atypical antipsychotics
Placebo-controlled monotherapy trials of haloperidol in mania
Two monotherapy studies have included haloperidol as an active compara-
tor: the risperidone study of Eerdekens et al26 and the quetiapine study of
Brecher et al.19
In the study of Eerdekens et al,26 the haloperidol dose started at
4mg/day and was adjusted to 2–12mg/day by day 5. The timecourse of
improvement was similar to that with risperidone, and by day 21 the NNT
for 50% improvement was 8 (95% confidence interval (CI) 4–36). This is far
larger than one would expect with the most commonly used antimanic drug
of the previous decade; this might be either because the mean modal dose
of haloperidol was only 8mg/day, or because the patients in the trial were in
some ways not typical of routine clinic patients and were more resistant to
treatment.
A comparator group on haloperidol (up to 8mg/day) was also included
in the study by Brecher et al19 of quetiapine (up to 800mg/day) and placebo,
analysed at 3 and 12 weeks. At 3 weeks the response rate (50% reduction
in YMRS score) on haloperidol, on a mean dose of only 5.2mg/day, was 55%
compared with 35% on placebo, giving a NNT of 5 (95% CI 3–16). There
were more dropouts on placebo than on haloperidol or quetiapine, so that
the analysis using last observations carried forward was biased in favour of
the active drugs, and especially so after 3 weeks when more patients on
placebo or haloperidol than on quetiapine dropped out. By 12 weeks the
response rate on haloperidol was 70% and on placebo 39%, giving a NNT of
4 (95% CI 3–6). Side-effects in the form of extrapyramidal symptoms were
much more common on haloperidol (59.6%) than on placebo (15.8%), as
was akathisia (33.3% on haloperidol and 5.9% on placebo). Somnolence
occurred more often with haloperidol (9.1%) than placebo (5%).
Comparative randomized controlled trials of antipsychotics in maniawithout placebo
In a comparative trial in mania, in which additional lorazepam was permit-
ted, risperidone showed similar efficacy to haloperidol or lithium.27
Ch 20 7/4/05 4:02 pm Page 188

Advantages and disadvantages of atypical antipsychotics or valproate 189
In the largest randomized comparative study of haloperidol,18 it was
compared with olanzapine over 6 and 12 weeks. Among patients on
haloperidol (up to 15mg/day, mean modal dose at the 6th week 7mg/day),
the proportion responding (50% reduction in YMRS score) at 6 weeks was
74%; the proportion showing syndromal remission (according to DSM-IV)
was 44%. Improvement in mania scores (YMRS) was greater for haloperidol
than for olanzapine at 6 weeks, but not different at 12 weeks.
Extrapyramidal symptoms occur to a much less extent with olanzapine
or quetiapine than with haloperidol. In the largest comparative trial
(n=234) treatment-emergent akathisia was observed in 40% on haloperidol
and 10% on olanzapine, dystonia in 6.8% and 1.3%, and Parkinsonism in
54% and 13%, respectively.
Conclusions
Atypical antipsychotics and valproate are effective in reducing the symp-
toms of mania, although neither is often sufficient as monotherapy to bring
about marked improvement in severe mania. Valproate probably acts a lit-
tle less quickly. Classical antipsychotics remain useful for the rapid control
of a severely agitated manic person, and several are available for use intra-
muscularly, as are olanzapine and ziprasidone. Classical antipsychotics
have the profound disadvantage of inducing extrapyramidal side-effects,
including dystonia and akathisia, which is particularly unwelcome.
Olanzapine and quetiapine may produce a lower switch rate into depres-
sion than haloperidol, and interestingly these two drugs may also have some
antidepressant properties. However, both drugs (in the doses used in clini-
cal trials) seem also to produce slower improvement in mania than
haloperidol. The complexity of diagnosing and treating bipolar mixed states
is discussed by Cookson and Ghalib.28
Antipsychotics are useful in bipolar disorder, both acutely in mania and
in long-term prophylactic treatment. Valproate is useful in severe mania, but
its value in prophylaxis is unproven and requires further investigation.
Intriguingly, there is preliminary evidence that, whereas antipsychotics
and (high doses of) valproate may be equally effective in severe or psychot-
ic mania, antipsychotics including olanzapine may be more effective than
valproate for controlling non-psychotic or milder mania. This might indi-
cate that when patients are educated about what steps to take to avert early
manifestations of recurrence of mania, as described by Perry et al,29 access
Ch 20 7/4/05 4:02 pm Page 189

190 Bipolar disorder: the upswing in research & treatment
to an antipsychotic may be more important than valproate, and certainlymore effective than increasing the dose of lithium they are taking.
References
1. Hirschfeld RMA, Bowden CL, Gitlin MJ et al, Practice guideline for the treat-ment of patients with bipolar disorder (Revision). Am J Psychiatry 2002; 159(4Suppl):1–50.
2. Goodwin GM, Evidence-based guidelines for treating bipolar disorder: recom-mendations from the British Association for Psychopharmacology. JPsychopharmacol 2003; 17:149–173; discussion 147.
3. Keck PE, McElroy SL, Tugrul KC, Bennett JA, Valproate oral loading in thetreatment of acute mania. J Clin Psychiatry 1993; 54:305–308.
4. Cookson JC, Moult PJA, Wiles D, Besser GM, The relationship between pro-lactin levels and clinical ratings in manic patients treated with oral and intra-venous test doses of haloperidol. Psychol Med 1983; 13:279–285.
5. Hirschfeld RM, Allen MH, McEvoy JP et al, Safety and efficacy of oral loadingdivalproex sodium in acutely manic bipolar patients. J Clin Psychiatry 1999;60: 815–818.
6. Meehan K, Zhang F, David S et al, A double-blind, randomized comparison ofthe efficacy and safety of intramuscular injections of olanzapine, lorazepam, orplacebo in treating acutely agitated patients diagnosed with bipolar mania. JClin Psychopharmacol 2001; 21:389–397.
7. Applebaum J, Levine J, Belmaker R, Intravenous fosphenytoin in acute mania.J Clin Psychiatry 2004; 64:408–409.
8. Yatham LN, Liddle PF, Shiah IS et al, PET study of [(18)F]6-fluoro-L-dopauptake in neuroleptic- and mood-stabilizer-naive first-episode nonpsychoticmania: effects of treatment with divalproex sodium. Am J Psychiatry 2002;159:768–774.
9. Ferrie L, Ferrier N, Young AH, 2003. Poster presentation at the SummerMeeting, British Association for Psychopharmacology.
10. Cookson JC, Taylor D, Katona C, Placebo effects, evaluating evidence, and com-bining psychotherapy. In Use of Drugs in Psychiatry: The Evidence FromPsychopharmacology. Gaskell Press: London, 2002:117–131.
11. Bowden C, Brugger AM, Swann AC et al, Efficacy of divalproex vs. lithium andplacebo in the treatment of mania. JAMA 1994; 271:918–924.
12. Pope HG, McElroy SL, Keck PE, Hudson JI, Valproate in the treatment of acutemania: A placebo-controlled study. Arch Gen Psychiatry 1991; 48:62–68.
13. Tohen M, Sanger TM, McElroy SL et al, Olanzapine versus placebo in the treat-ment of acute mania. Olanzapine HGEH Study Group. Am J Psychiatry 1999;156:702–709.
14. Khanna S, Vieta E, Lyons B et al, Risperidone in the treatment of acute bipolarmania: a double-blind, placebo-controlled study of 290 patients. Poster pre-sented at the 16th Congress of the European College of Neuropsycho-pharmacology (ECNP), Prague, September 2003.
Ch 20 7/4/05 4:02 pm Page 190

Advantages and disadvantages of atypical antipsychotics or valproate 191
15. Tohen M, Baker RW, Altshuler LL et al, Olanzapine versus divalproex in thetreatment of acute mania. Am J Psychiatry 2002; 159:1011–1017.
16. Zajecka JM, Weisler R, Sachs G et al, A comparison of the efficacy, safety, andtolerability of divalproex sodium and olanzapine in the treatment of bipolar dis-order. J Clin Psychiatry 2002; 63:1148–1155.
17. Zarate CA, Tohen M, Double-blind comparison of the continued use of antipsy-chotic treatment versus its discontinuation in remitted manic patients. Am JPsychiatry 2004; 161:169–171.
18. Tohen M, Goldberg JF, Gonzalez-Pinto AM et al, A 12-week double-blind com-parison of olanzapine versus haloperidol in the treatment of acute mania. ArchGen Psychiatry 2003; 60:1218–1226.
19. Brecher M, Huizar K, ElBaghdady A, Quetiapine monotherapy for acute maniaassociated with bipolar disorder. Poster presented at the Fifth AnnualInternational Conference on Bipolar Disorder, Pittsburgh, PA, 12–14 June2003.
20. Muller-Oerlinghausen B, Retzow A, Henn FA et al, Valproate as an adjunct toneuroleptic medication for the treatment of acute episodes of mania: a prospec-tive, randomized, double-blind, placebo-controlled, multicenter study. J ClinPsychopharmacol 2000; 20:195–203.
21. Tohen M, Chengappa KN, Suppes T et al, Efficacy of olanzapine in combinationwith valproate or lithium in the treatment of mania in patients partially non-responsive to valproate or lithium monotherapy. Arch Gen Psychiatry 2002;59:62–69.
22. Sachs GS, Grossman F, Ghaemi SN et al, Combination of a mood stabilizerwith risperidone or haloperidol for treatment of acute mania: a double-blind,placebo-controlled comparison of efficacy and safety. Am J Psychiatry 2002;159:1146–1154.
23. Yatham LN, Grossman F, Augustyns I et al, Mood stabilisers plus risperidoneor placebo in the treatment of acute mania: International, double-blind, ran-domised controlled trial. Br J Psychiatry 2003; 182:141–147.
24. Bowden CL, Calabrese JR, McElroy SL et al, A randomized, placebo-controlled12-month trial of divalproex and lithium in treatment of outpatients with bipo-lar I disorder. Divalproex Maintenance Study Group. Arch Gen Psychiatry2000; 57:481–489.
25. Tohen M, Chengappa KN, Suppes T et al, Relapse prevention in bipolar disor-der I disorder: 18-month comparison of olanzapine plus mood stabilizer ver-sus mood stabilizer alone. Br J Psychiatry 2004; 184:337–345.
26. Eerdekens M, Karcher K, Grossman F, Kramer M, Risperidone monotherapy inacute bipolar mania. Poster presented at the Meeting of the World PsychiatricAssociation, 2003.
27. Segal J, Berk M, Brook S, Risperidone compared with both lithium andhaloperidol in mania: a double blind randomized controlled trial. ClinNeuropharmacol 1998; 21:176–180.
28. Cookson JC, Ghalib S, The treatment of bipolar mixed states. In: Mixed States.2004 Marneros A, Goodwin FK, eds.
29. Perry A, Tarrier N, Morriss R et al, Randomised controlled trial of efficacy ofteaching patients with bipolar disorder to identify early symptoms of relapseand obtain treatment. BMJ 1999; 318:149–153.
Ch 20 7/4/05 4:02 pm Page 191

Ch 20 7/4/05 4:02 pm Page 192

Is electroconvulsive therapystill given in bipolar disorderand does repetitivetranscranial magneticstimulation offer more?Andrew Mogg, Savitha Eranti, Graham Pluck andDeclan M McLoughlin
c h a p t e r 2 1
The evidence for the efficacy of electroconvulsive therapy (ECT) in depres-sion is substantial. A recent systematic review and meta-analysis concludedthat ECT is an effective treatment for depression and probably more effec-tive than drug therapy.1 However, there are fewer research data available forthe use of ECT in mania and in bipolar depression. These issues areaddressed below before considering the role of repetitive transcranial mag-netic stimulation (rTMS) as a potential alternative to ECT.
Electroconvulsive therapy and mania
ECT is rarely used to treat mania. In the late 1970s, about 2% of referralsfor ECT in Great Britain were for mania compared with 83% for depres-sion.2 As will be seen, these figures have not changed much in subsequentyears.
Not surprisingly, there have been relatively few studies of ECT for mania.For example, the most recent comprehensive review on the topic was pub-lished nearly a decade ago.3 This review examined the use of ECT in maniasince its introduction about 50 years previously. The authors identified just16 primary studies, most of which were naturalistic case series or compar-isons. Of these studies, seven were before 1950 and lacked certainty aboutboth the diagnosis and the criteria for response to treatment. Data
Ch 21 7/4/05 4:02 pm Page 193

194 Bipolar disorder: the upswing in research & treatment
combined from six retrospective studies published since the 1970s indicat-
ed that ECT was associated with either remission or marked clinical
improvement in 85% of manic patients.
There have been only three prospective randomized controlled studies
examining the efficacy of ECT in mania. Small and colleagues4 randomly
allocated 34 hospitalized patients to receive either lithium carbonate or
bilateral ECT, followed by maintenance lithium carbonate. Blinded clinical
ratings showed greater improvement for patients treated with ECT com-
pared with lithium carbonate during the first 8 weeks but no significant dif-
ference between the groups after 8 weeks. A subsequent trial randomly
assigned patients who had not responded to either lithium or a neuroleptic
to four groups: one pharmacotherapy group (lithium and haloperidol) and
three different ECT groups (bilateral, right unilateral and left unilateral); the
authors reported that 59% of the ECT patients achieved complete remission
compared with none in the pharmacotherapy group.5 Sikdar and colleagues
randomly assigned 30 manic patients to receive eight ECT sessions, either
real or simulated, together with chlorpromazine pharmacotherapy.6 The
group receiving real ECT improved significantly more than the group receiv-
ing chlorpromazine alone. These three prospective trials have all been
small, involving only 91 patients in total. This probably reflects both the dif-
ficulty of recruiting to ECT trials in general and specifically the difficulty of
obtaining informed consent from acutely manic patients.
Combining the data from all the available literature, i.e. both retrospec-
tive and prospective studies, Mukherjee and colleagues found that 80% of
all the patients from this very mixed group either achieved remission or had
a marked clinical improvement.3
Current American Psychiatric Association (APA) guidelines suggest that,
owing to the availability of lithium and anticonvulsant and antipsychotic
agents, ECT in mania is mainly reserved for medication-resistant patients.7
However, it should be noted that to date there have been no comparison
studies of ECT with the newer anti-manic agents. The APA guidelines also
include some specific indications, such as recommending ECT for manic
delirium, and also suggest that ECT may be effective for rapid cycling in
bipolar disorder. However, the evidence base for both these indications is
extremely limited.
Recent guidance from the National Institute of Clinical Excellence (NICE)
in the UK concluded that, although the evidence for ECT in mania is less
robust than in depression, the available data are enough to recommend its
use in mania where other treatments have proved ineffective.8
Ch 21 7/4/05 4:02 pm Page 194

Electroconvulsive therapy and repetitive transcranial magnetic stimulation 195
Electroconvulsive therapy and bipolar depression
As mentioned previously, there is firm evidence that ECT is an effective anti-depressant treatment. However, is there any difference in outcome betweenbipolar depressed patients and unipolar depressed patients? The majorityof studies carried out have been retrospective analyses of case series andthe results have not been consistent across studies. For example, Black etal9 reviewed patient records over a 12-year period in a university hospitaland found that unipolar and bipolar depressed patients received the samenumber of treatments (mean of nine) and that ECT had equivalent efficacyin both groups. In contrast, an earlier study suggested that ECT was moreefficacious in the unipolar group of depressed patients compared with thebipolar group.10
A recent study from New York compiled data from three randomizedcontrolled trials of ECT carried out during the 1990s.11 It included 162patients who had unipolar depression and 66 with bipolar depression, allof whom had a high degree of treatment resistance. The patients wereadmitted to hospital, taken off medications and remained drug-free for atleast a week. They all underwent a stimulus dosing protocol and were ran-domized to bilateral or right unilateral ECT, with a variety of seizure thresh-olds that were the primary focus of the original studies. Taking into accountthe different forms of ECT used, the authors found no difference in outcomebetween the unipolar depressed and bipolar depressed groups; approxi-mately 60% from each group were initial responders to ECT. The one sig-nificant difference between the groups was that the bipolar depressedpatients received fewer ECT treatments than the unipolar depressedpatients, a mean difference of around 1.5 in the number of treatmentsreceived.
Using data collected prospectively on 133 patients over a 7-year period,Grunhaus et al in Israel have recently reported a similar overall responserate (57%) to ECT with no difference between bipolar and unipolardepressed patients.12 Another important observation in this study, relatingto a concern clinicians may have, was that emergent hypomania in bipolardepressed patients treated with ECT was a rare event, experienced by onlyabout 3% of patients.
Findings from London
Most of the published data about ECT and bipolar disorder originates fromthe USA. Some recent data from the South London and Maudsley NHS
Ch 21 7/4/05 4:02 pm Page 195

196 Bipolar disorder: the upswing in research & treatment
Trust in South London provides a British perspective. ECT use at this men-tal health trust, as in the rest of the UK, reached its peak in the mid 1950swhen nearly 35% of all patients admitted had ECT. Following the introduc-tion of neuroleptics and tricyclic antidepressants, this proportion rapidlydeclined and over the past 10 years the use of ECT has reached a plateauwith less than 1% of all patients admitted to hospital now receiving ECT.
We have examined the case notes and ECT records of patients havingECT over a 3-year period from 1999 to 2001. Operationalized diagnoseswere applied on the basis of symptoms recorded in the notes. Over this peri-od, 150 courses of ECT were administered to 130 patients. Figure 21.1summarizes the diagnoses of these patients.
The vast majority of patients (77%) had a diagnosis of unipolar depres-sion; 11% of patients were bipolar depressed while only two patients (1.5%)had mania during this 3-year period. We compared the unipolar and bipo-lar depressed patients on a number of factors (Table 21.1)
There were no differences between the groups in terms of age and sex.They both had similar levels of treatment resistance and the percentage ofpatients per group having treatment without consent under the MentalHealth Act 1983 was equal.
However, there were some interesting differences. There was a trendtowards the mean number of treatments (seven versus nine) being less inthe bipolar depressed group. The mean length of hospital stay of the bipo-lar group was only about 75% that of the unipolar group and there was asignificant difference in terms of the actual response to ECT at the end oftreatment course; 85% of the bipolar group and 57% of the unipolar grouphad either a complete recovery or major improvement following ECT. These
Figure 21.1 Electroconvulsive therapy according to diagnosis, 1999–2001(n=130).
Unipolar depression
Bipolar – depressed
Bipolar – manic
Other
Ch 21 7/4/05 4:02 pm Page 196

Electroconvulsive therapy and repetitive transcranial magnetic stimulation 197
data are broadly in accordance with previous reports and provide furthersupport for considering ECT for patients with a bipolar depressive presen-tation.
Transcranial magnetic stimulation – a potentialalternative to electroconvulsive therapy?
Transcranial magnetic stimulation (TMS) is a potential alternative to ECTfor the treatment of depression.13 Like ECT it electrically stimulates thebrain, but it does not induce a seizure or require anaesthesia. TMS usesFaraday’s principle of electromagnetic induction to induce secondary elec-trical activity within the brain. A brief powerful electrical current is passedthrough a copper wire coil placed in a hand-held device on the scalp. Thiselectric current generates a magnetic field that passes unimpeded throughthe skull and induces secondary electrical currents in the immediatelyunderlying cerebral cortex tissue. TMS can thus be used focally to stimulateselected cortical regions.
Repetitive TMS (rTMS) involves the repeated administration of TMS toinduce sustained effects such as change in mood. ‘Fast’ rTMS (>1Hz) hasan activating effect, while ‘slow’ rTMS (<1Hz) has an inhibitory effect on theunderlying cortex. Much of the therapeutic work using TMS for depressionhas been attempting to target and modify specific neuronal circuitry thoughtto be relevant to mood disorder. Thus, fast rTMS of the left dorsolateral
Table 21.1 Comparison of unipolar and bipolar depressed patients having
electroconvulsive therapy
Unipolar Bipolar t Test
Number of patients 88 13
Mean age (SD) 68 (13) 73 (9) NS
Mean no. of antidepressants (SD) 2.2 (1.3) 2.7 (1.3) NS
Mean no. of treatments (SD) 9 (6.1) 7 (3.6) NS
Mean length of hospital stay (SD) 200 days 145 days NS
(178) (165)
% consenting to treatment 62% 69% NS
% recovery/major improvement 57% 85% p =0.05
NS, not significant.
Ch 21 7/4/05 4:02 pm Page 197

198 Bipolar disorder: the upswing in research & treatment
prefrontal cortex has been widely used in studies of depression. It hasbecome increasingly clear that this can have an antidepressant effect but theextent, duration and how best to harness it remain to be resolved.
Repetitive transcranial magnetic stimulation in bipolar depression
There is rapidly expanding literature about rTMS and unipolar depression,but relatively little is known about rTMS in bipolar depression.13 Thelargest study to date randomized 23 patients with bipolar depression toreceive either real or placebo rTMS over the left prefrontal cortex on a dailybasis for 2 weeks.14 The investigators did not find a significant benefit ofreal rTMS compared with placebo. However, the placebo group used a coilangled away from the scalp rather than a specially designed placebo coil.There is now good evidence from functional neuroimaging studies that theangling away of a real coil still induces stimulation in the brain area beneaththe scalp and hence in this study the placebo coil may not have been a trueplacebo. The study found that rTMS was well tolerated and there were nosignificant adverse effects.
Repetitive transcranial magnetic stimulation in mania
There are very few data available on the use of rTMS in mania. It is inter-esting to note that for rTMS, as for any other antidepressant treatment,there are some case reports of induced manic symptoms, both after indi-vidual treatment sessions and following longer courses of treatment.15,16 Todate there are only two small randomized studies of rTMS in acute mania.The first, from Grisaru and colleagues in Israel, randomized 16 patients toleft or right prefrontal fast rTMS for 2 weeks.17 They reported significantlymore improvement in patients treated with right rather than with left pre-frontal rTMS, suggesting laterality opposite to that seen in depression. Thesame group have performed a larger study in which 25 patients were ran-domized to either real or sham fast rTMS; this time all patients receivedstimulation over the right dorsolateral prefrontal cortex.18 There was no dif-ference between real and placebo treatment groups.
Therapeutic courses of rTMS involve the patient coming to the clinicevery weekday for 2–4 weeks. For patients with mania who are likely to beunsettled, agitated and irritable, it seems likely that practical problemswould arise in trying to deliver the treatment, as a high degree of co-operation is required during treatment sessions lasting about half an hour.
In conclusion, rTMS may have an increasing role to play in the treatmentof bipolar depression, but its use in mania is likely to remain limited.
Ch 21 7/4/05 4:02 pm Page 198

Electroconvulsive therapy and repetitive transcranial magnetic stimulation 199
References
1. The UK ECT Review Group, Efficacy and safety of electroconvulsive therapy indepressive disorders: a systematic review and meta-analysis. Lancet 2003;361:799–808.
2. Pippard J, Ellam L, Electroconvulsive treatment in Great Britain. Br JPsychiatry 1981; 139:563–568.
3. Mukherjee S, Sackeim HA, Schnur DB, Electroconvulsive therapy of acutemanic episodes: a review of 50 years’ experience. Am J Psychiatry 1994;151:169–176.
4. Small JG, Klapper MH, Kellams JJ et al, Electroconvulsive treatment com-pared with lithium in the management of manic states. Arch Gen Psychiatry1988; 45:727–732.
5. Mukherjee S, Sackeim HA, Lee C, Unilateral ECT in the treatment of manicepisodes. Convuls Ther 1988; 4:74–80.
6. Sikdar S, Kulhara P, Avasthi A, Singh H, Combined chlorpromazine and elec-troconvulsive therapy in mania. Br J Psychiatry 1994; 164:806–810.
7. Rasmussen K, The practice of electroconvulsive therapy: recommendations fortreatment, training, and privileging (second edition). J ECT 2002; 18:58–59.
8. Sanders RD, Deshpande AS, Mania complicating ECT. Br J Psychiatry 1990;157:153–154.
9. Black DW, Winokur G, Nasrallah A, ECT in unipolar and bipolar disorders: anaturalistic evaluation of 460 patients. Convuls Ther 1986; 2:231–237.
10. Homan S, Lachenbruch PA, WinokurG, Clayton P, An efficacy study of electro-convulsive therapy and antidepressants in the treatment of primary depression.Psychol Med 1982; 12:615–624.
11. Daly JJ, Prudic J, Devanand DP et al, ECT in bipolar and unipolar depression:differences in speed of response. Bipolar Disord 2001; 3:95–104.
12. Grunhaus L, Schreiber S, Dolberg OT et al, Response to ECT in major depres-sion: are there differences between unipolar and bipolar depression? BipolarDisord 2002; 4(Suppl 1): 91–93.
13. Lisanby SH, Kinnunen LH, Crupain MJ, Applications of TMS to therapy in psy-chiatry. J Clin Neurophysiol 2002; 19:344–360.
14. Nahas Z, Kozel FA, Li X et al, Left prefrontal transcranial magnetic stimulation(TMS) treatment of depression in bipolar affective disorder: a pilot study ofacute safety and efficacy. Bipolar Disord 2003; 5:40–47.
15. Nedjat S, Folkerts HW, Induction of a reversible state of hypomania by rapid-rate transcranial magnetic stimulation over the left prefrontal lobe. J ECT1999; 15:166–168.
16. Sakkas P, Mihalopoulou P, Mourtzouhou P et al, Induction of mania by rTMS:report of two cases. Eur Psychiatry 2003; 18:196–198.
17. Grisaru N, Chudakov B, Yaroslavsky Y, Belmaker RH, Transcranial magneticstimulation in mania: a controlled study. Am J Psychiatry 1998;155:1608–1610.
18. Kaptsan A, Yaroslavsky Y, Applebaum J et al, Right prefrontal TMS versussham treatment of mania: a controlled study. Bipolar Disord 2003; 5:36–39.
Ch 21 7/4/05 4:02 pm Page 199

Ch 21 7/4/05 4:02 pm Page 200

Improving outcomeby selecting effectivelong-term treatmentPaul Grof
c h a p t e r 2 2
Introduction
If we took the literature on bipolar disorders at face value, we would haveto conclude that over time the outcome of long-term treatment has becomemuch worse. While the earlier reports showed very promising results with75% of bipolar patients benefiting from long-term lithium treatment,1 recentfindings have been very discouraging, suggesting response rates between 20and 35%. However, these numbers are not directly comparable, primarilybecause of major shifts in the patients under study. Bipolar disorders arenow diagnosed much more broadly and more frequently, as a bipolar spec-trum, with negative implications for outcomes. In this brief chapter I willreview the observations indicating that the current unsatisfactory outcomecan be markedly improved by recognizing the striking heterogeneity of bipo-lar disorders and by selecting the treatment for each individual according toa characteristic clinical profile.
Heterogeneity of bipolar disorders
The recognition that the bipolar disorder diagnosis includes a heteroge-neous collection of illnesses has been emerging for some time, both from theclassical studies (e.g. reference 2) and more recent investigations (e.g. ref-erences 3–5). So far this concept has been neglected by mainstream psy-chiatry.
Angst analysed the data from his long-term studies of bipolar illness andfound three subtypes (Dm, MD and Md) that differed markedly not only in
Ch 22 7/4/05 4:02 pm Page 201

202 Bipolar disorder: the upswing in research & treatment
the clinical course, but also in a number of other important clinical charac-
teristics. Patients of the Dm type experienced major depressive episodes
accompanied only by occasional hypomanias; these patients were predomi-
nantly women (over 80%) and their age of onset was in the late thirties on
average, significantly later than the other two groups. The Md group experi-
enced primarily or exclusively hospitalized manic episodes and in compar-
ison with the other two groups had significantly less chronicity, mortality
and suicidal behaviour.
In large multicentre studies Bellivier et al4 identified and replicated three
subtypes of bipolar I patients, based on the age of onset of bipolar illness.
Benazzi6 then reported three similar subgroups from a large series of bipo-
lar II patients. In a comprehensive review Alda5 also found support for the
existence of three main types of bipolar disorder that differed with respect
to clinical presentation, course of illness, family history and possibly long-
term treatment response.
Together these examples of heterogeneity raise a question: can we
improve the outcome of stabilizing treatments by respecting the types of
bipolar disorder? This important question could best be resolved in a
specifically designed, long-term, cross-over clinical trial, with proven stabil-
izing treatments, and focusing on the characteristics of bipolar patients.
However, given the ethical and feasibility dilemmas involved, such a trial
may never happen. In the meantime we need to treat bipolar patients effec-
tively. In addition, evidence-based medicine should not be limited to drug
trials; it has to utilize all valid information.7 Much practically useful infor-
mation about treatment responsiveness of bipolar patients can be gained
from patient-oriented studies, from some drug studies and from extensive
clinical observations.
Heterogeneity of responders
Here I briefly review data extracted from a series of unequivocal responders
to three main types of long-term treatment for bipolar disorders – lithium,
lamotrigine and olanzapine (as a representative of atypical neuroleptics). All
three have been shown in controlled, double-blind trials to be effective in
groups of patients with bipolar disorders.8–10 The body of emerging data
appears to show that unequivocal responders to long-term monotherapies,
such as lithium, lamotrigine and atypical neuroleptics have distinct clinical
profiles. The differences include clinical presentation and course of illness,
Ch 22 7/4/05 4:02 pm Page 202

Improving outcome by selecting effective long-term treatment 203
co-morbidity and in particular family history, thus suggesting that these areclinically relevant subtypes of bipolar disorders.
While the characteristics of unequivocal lithium responders have beenknown for some time,11 the likely features of patients who benefit from lam-otrigine and olanzapine have emerged mainly from two studies, one per-formed in Ottawa, and the other in Halifax. In Ottawa a consecutive seriesof patients was studied who were diagnosed as suffering from bipolar dis-order according to DSM-IV criteria, required long-term prophylaxis withmedication, and were treated in our programme for 3 or more years. Toassess the characteristics of unequivocal responders to prototypic long-term treatment, the diagnosis and co-morbidity of each patient wasassessed with the Schedule for Affective Disorders and Schizophrenia –Lifetime Version (SADS-L) interview and family history with Schedule forAffective Disorders and Schizophrenia – Family History (SADS-FH) com-pleted with the patient. In addition, available and consenting first-degree rel-atives were blindly interviewed using the SADS-L. To be classified as a goodresponder, each patient had to receive a score on the Alda scale12 of 7 orbetter; 112 patients met all criteria and had 756 evaluated relatives.
With regard to family history, loading was strikingly different for eachgroup of responders. When the responders to long-term treatment withthree different medications were compared, only the relatives of lithiumresponders had a significant excess of bipolar disorders. While the first-degree relatives of bipolar patients responding to lamotrigine had an excessof anxiety, panic, substance abuse and alcohol addictions, the relatives ofthose benefiting from olanzapine had no excessive bipolar or anxiety disor-ders but a high rate of chronic psychotic illnesses.
In parallel with the differences in family history between respondergroups, there were corresponding dissimilarities in co-morbid disorders.Like their relatives, the lamotrigine responders also had more problemswith substance and alcohol addictions as well as anxiety disorders, while ahistory of mood-incongruent psychotic symptoms was more prevalentamong the olanzapine responders.
There were also differences in the pre-treatment clinical course in thatlithium responders presented with an episodic, fully remitting course andoften had a predominance of depressive over manic episodes. On the otherhand, lamotrigine and olanzapine responders tended to have mostly a non-episodic course with significant residual symptoms and exacerbations, withpredominance of manic episodes.
The observations on lithium and lamotrigine responders in Ottawa werein good agreement with the findings from Halifax.13
Ch 22 7/4/05 4:02 pm Page 203

204 Bipolar disorder: the upswing in research & treatment
The approaches in the two centres had several aspects in common: forexample, both teams evaluated the outcome of long-term treatment with thesame Alda scale that takes into consideration the risk of recurrences andvarious aspects of treatment such as length and compliance; also, bothteams blindly evaluated the first-degree relatives of the responders, for thepurpose of genetic studies.
Clinical characteristics of responders
From these and earlier studies it is possible to derive the main clinical char-acteristics for bipolar patients responding to each of the major stabilizingtreatments: lithium, olanzapine and lamotrigine. These characteristics canbe useful in clinical practice in order to identify the treatment of choice foran individual bipolar patient.
Lithium
Responders to lithium stabilization present with depressive and manicepisodes of the classical type, without mood-incongruent symptoms, clear-ly sad depressions and often euphoric manias. In their family history, theytend to have bipolar disorders with an episodic course. They, themselves,have an episodic full-remitting course and, if the course has been extensive,one can usually see a predominance of depressions. Finally, these patientshave relatively rare co-morbid conditions (Table 22.1).
Lamotrigine
The characteristics of responders to lamotrigine prophylaxis are different.In the presentation they often have atypical features: the depression isdescribed as emotional emptiness and apathy, indifference, slow motivation
Table 22.1 Characteristics of responder to long-term lithium treatment
Clinical course: episodic, fully remitting, predominance of depressions
Family history: bipolar disorders, with episodic course
Co-morbidity: relatively rare, as in the general population
Presentation: classical, as described in earlier textbooks (e.g. depressions with sad-
ness, manias with euphoria, absence of mood-incongruent psychotic symptoms)
Ch 22 7/4/05 4:02 pm Page 204

Improving outcome by selecting effective long-term treatment 205
and hypomanias as activations without euphoria. These patients often haveanxiety and panic disorders or substance use disorders in their family his-tory; the course of illness is non-episodic and often entails residual symp-toms. Similarly, these patients have substantial co-morbidity similar totheir family history (Table 22.2).
Olanzapine
Olanzapine responders again have atypical features characterizing boththeir depressions and their manias, and one can often identify mood-incongruent psychotic symptoms in their history or in their acute presenta-tion. Family history, if positive, tends to show psychotic disorders or chron-ic psychiatric disorders. The clinical course has residual symptomsbetween the episodes of depressions and manias, and the history, if fullydeveloped, shows more manias than depressions. Co-morbidity is frequent,particularly with alcoholism and substance abuse. We have preliminary evi-dence to suggest that the clinical features of responders to olanzapine maygeneralize to indicate response to other atypical neuroleptics (Table 22.3).
Table 22.2 Characteristics of responder to long-term lamotrigine treatment
Clinical course: non-episodic, with residual symptoms, mostly depressions (often
‘bipolar II’ type)
Family history: anxiety and panic disorders, substance and alcohol addictions
Co-morbidity: high, anxiety and panic disorders, substance and alcohol addic-
tions
Presentation: atypical, non-textbook features (e.g. depressions characterized by
anergia or emotional emptiness, hypomanias by general speeding without
euphoria)
Table 22.3 Characteristics of responder to long-term olanzapine treatment
Clinical course: non-episodic, with residual symptoms, overactive episodes often
more frequent than depression
Family history: non-remitting or psychotic disorders
Co-morbidity: alcohol abuse or addictions
Presentation: atypical, non-textbook features, often history or presence of mood-
incongruent psychotic symptoms
Ch 22 7/4/05 4:02 pm Page 205

206 Bipolar disorder: the upswing in research & treatment
Divalproex
The data on divalproex are missing from these investigations, mainlybecause the evidence from controlled clinical trials for long-term efficacy ofdivalproex is not available.14
Selectivity of responses
There has been a line of investigation documenting that the response tosome stabilizers has been associated with specific clinical correlates, forexample for lithium,11,15 carbamazepine,16 valproate17 and lamotrigine.13 Inaddition, however, there is a growing body of literature indicating that goodresponses to each of these substances are selective and often mutuallyexclusive.
For example, excellent lithium responders failed on long-term carba-mazepine and vice versa.18 Post et al19 made a similar observation: mostpatients with a good acute response to carbamazepine had a clear history ofnon-response to lithium. Bowden et al20 found that previous lithium respon-ders did well on lithium but not on divalproex. Similarly, Swann et al21
noted that responders to valproate had evidence of prior non-response tolithium. Tohen et al22 observed that olanzapine succeeded in patients whohad failed previously on lithium and divalproex. Despite some methodolog-ical limitations of these observations, together they provide a credible pic-ture of a degree of selectivity among these medications. These data do notsupport the often reported clinical impression that many bipolar patientsrequire combination treatment in order to get well.
The issue of selectivity is also indirectly supported by the clustering ofprophylactic responses in families. For lithium there is evidence thatresponse to long-term treatment markedly clusters in families.12 Evidenceis also slowly mounting that affected children of bipolar parents tend to ben-efit markedly from the long-term treatment to which the parent responded(A. Duffy et al, submitted for publication).
Discussion
The critical task in the long-term management of bipolar disorders ismatching the patient and the effective treatment. Much of the current liter-ature stresses that bipolar patients should be treated by a combination of
Ch 22 7/4/05 4:02 pm Page 206

Improving outcome by selecting effective long-term treatment 207
medications and that the right combination can be established by following
an algorithmic sequence, adding one drug at a time. This recommendation
is based on practical experience that many bipolar patients fail on the ini-
tial medication, and respond only after several more drugs are added.
However, such anecdotal observations have more than one possible expla-
nation.
At present, there is much misunderstanding about long-term treatment
of bipolar disorders. In particular, the natural course of atypical forms has
not been well described and there are major misinterpretations about indi-
cations for effective lithium treatment. As a result, the interpretation of the
outcome of treatment is often incorrect.
Clinicians use combinations because they experience treatment failures
during the initial stage of treatment of a bipolar patient. Very frequent ini-
tial failures should be expected. In DSM-IV-diagnosed bipolar disorders,
long-term monotherapy chosen by the current practice of trial-and-error is
effective in one-third of patients at best,23 but can be markedly improved by
treating according to clinical profile of the patient and family as described
above. Observations supporting this are particularly convincing for lithium
and clozapine.
The majority of bipolar patients who have been correctly selected for
lithium treatment and adequately monitored can be completely stabilized
with lithium monotherapy. On the other hand, some patients benefiting sub-
stantially from an atypical neuroleptic or lamotrigine will intermittently or
sometimes chronically require an addition of an antidepressant or another
psychotropic drug.
A combination of medications appears to be indicated, particularly in
bipolar patients who are treatment-resistant to monotherapy, do not toler-
ate it well, or do not have clinical characteristics helpful for a clear treat-
ment choice. Evidence is lacking that combinations of several medications
are necessary in the majority of bipolar patients, despite the current prac-
tice. It is difficult to justify exposing patients to the side-effects of several
drugs if mood stabilization can be achieved without a multiple combination.
Heterogeneity of bipolar disorders poses a major problem for the inter-
pretation of the results of clinical trials. The results of any long-term drug
trial may depend as much on the composition of the patient sample and the
proportion of the subtypes as on the efficacy of the tested drug.
Currently we are developing a computer program that forecasts the
treatment outcome based on the individual bipolar patient’s clinical char-
acteristics. This program should be ready for predictive testing soon.
Ch 22 7/4/05 4:02 pm Page 207

208 Bipolar disorder: the upswing in research & treatment
Conclusions
There are at least three distinct types of bipolar disorder that markedly dif-fer both in clinical characteristics and in treatment outcome.Comprehensive clinical assessment is needed in order to identify the type;the patient can then be matched with a more effective long-term treatment.
Despite prevailing practice, evidence is lacking so far that polypharmacywith multiple stabilizers is necessary for the majority of bipolar patients.However, for patients who can achieve stabilization by one primary medica-tion, whether it is lithium, lamotrigine or atypical neuroleptics, it is difficultto justify exposing them to the side-effects of complex polypharmacy.
References
1. Schou M, Thomsen K, Lithium prophylaxis of recurrent endogenous affectivedisorders. In: Johnson FN (ed), Lithium Research and Therapy. AcademicPress: New York, 1975:63–84.
2. Angst J. The course of affective disorders. II. Typology of bipolar manic–depressive illness. Arch Psychiatrie Nervenkrankheiten 1978; 226:65–73.
3. Angst J, Gamma A, Benazzi F et al, Diagnostic issues in bipolar disorder. EurNeuropsychopharmacol 2004; 13(Suppl 2):S43–S50.
4. Bellivier F, Golmard J, Rietschel M et al, Age at onset in bipolar I affective dis-order: further evidence for three subgroups. Am J Psychiatry 2003;160:999–1001.
5. Alda M, The phenotypic spectra of bipolar disorder. Eur Neuropsycho-pharmacol 2004; 14:S94–S99.
6. Benazzi F, Toward better probing for hypomania of bipolar II disorder. Int JMethods Psychiatr Res 2004; 13:1–9.
7. Jenicek M, Clinical Case Reporting in Evidence-based Medicine, 2nd edn.Oxford UniversityPress: New York, 2001.
8. Schou M, Perspectives on lithium treatment of bipolar disorder: Action, effica-cy, effect on suicidal behaviour. Bipolar Disord 1999; 1:5–10.
9. Calabrese JR, Shelton MD, Rapport DJ et al, Long-term treatment of bipolardisorder with lamotrigine. J Clin Psychiatry 2002; 63(Suppl 10):18–22.
10. Tohen M, Chengappa KNR, Suppes T et al, Olanzapine cotherapy in preventionof recurrence in bipolar disorder. Eur Psychiatry 2002; 17(Suppl 1):109.
11. Grof P, Alda M, Grof E et al, The challenge of predicting response to stabilizinglithium treatment: The importance of patient selection. Br J Psychiatry 1993;163(Suppl 21):16–19.
12. Grof P, Duffy A, Cavazzoni P et al, Is response to prophylactic lithium a famil-ial trait? J Clin Psychiatry 2002; 63:942–947.
13. Passmore M, Garnham J, Duffy A et al, Phenotypic spectra of biopolar disorderin responders to lithium versus lamotrigine. Bipolar Disord 2003; 5:110–114.
Ch 22 7/4/05 4:02 pm Page 208

Improving outcome by selecting effective long-term treatment 209
14. Bowden CL, Calabrese JR, McElroy SL et al, A randomized, placebo-controlled, 12-month trial of divalproex and lithium in treatment of out-patients with bipolar I disorder. Arch Gen Psychiatry 2000; 57:481–489.
15. Grof P, Hux M, Grof E, Arato M, Prediction of response to stabilizing lithiumtreatment. Pharmacopsychiatria 1983; 16:195–200.
16. Greil W, Kleindienst N, Erazo N, Muller-Oerlinghausen B, Differential responseto lithium and carbamazepine in the prophylaxis of bipolar disorder. J ClinPsychopharmacol 1998; 18:455–460.
17. Calabrese JR, Shelton MD, Bipolar diorders and the effectiveness of novel anti-convulsants. J Clin Psychiatry 2002; 63(Suppl 3):5–9.
18. Grof P, Lithium update: selected issues. In: Ayd F, Taylor JT, Taylor BT (eds),Affective Disorders Reassessed. Ayd Medical Publications: Baltimore, 1983.
19. Post RM, Denicoff KD, Frye MA, Everich GS, Re-evaluating carbamazepine pro-phylaxis in bipolar disorder. Br J Psychiatry 1997; 170:202–204.
20. Bowden CL, Brugger AM, Swann AC et al, Efficacy of divalproex versus lithiumand placebo in the treatment of mania. JAMA 1994; 271:918–924.
21. Swann AC, Bowden CL, Calabrese JR et al, Mania: differential effects of previ-ous depressive and manic episodes on response to treatment. Acta PsychiatrScand 2000; 101:444–451.
22. Tohen M, Chengappa KNR, Suppes T et al, Efficacy of olanzapine in combina-tion with valproate or lithium in the treatment of mania in patients partiallynonresponsive to valproate or lithium monotherapy. Arch Gen Psychiatry2002; 59:62–69.
23. Garnham J, Munro A, Teehan A et al, Bipolar disorder: assessing treatmentresponse in a naturalistic setting. J Bipolar Disord 2001; 3(Suppl 1):37.
Ch 22 7/4/05 4:02 pm Page 209

Ch 22 7/4/05 4:02 pm Page 210

Is what we offerto patientshalf acceptable?Rachel Perkins
c h a p t e r 2 3
The catalogue of deficits and dysfunction described in this volume mightsuggest that someone with a diagnosis of bipolar disorder – like myself – haslittle constructive to contribute. It would be easy to dismiss my analysis asnothing other than a manifestation of my psychopathology. However, beforedoing this, it is perhaps worth remembering that I am also a senior providerof mental health services – a consultant clinical psychologist and clinicaldirector – who has written over 150 papers and four books. One might askwhether this is possible in the presence of the cognitive deficits and dys-functions that have been described.
The primary concern of people with bipolar disorder is to be able to livethe lives they wish to live and do the things they want to do.1 While peoplemay desire freedom from debilitating symptoms this is only part of the story– retaining or rebuilding a decent life is at least (if not more) important.People want:
‘…safe, pleasant and affordable housing, well paying and ful-filling jobs … to be treated with dignity and respect, to have con-trol over their lives and to have genuine choices. They want to feelgood about themselves and to have the opportunity to achievethings that all of us do.2
Having a job is central to the physical, psychological and social well-beingof most people;3 therefore, the analysis presented here will focus on employ-ment. However, it must be emphasized that many of the arguments areequally applicable to other important life domains.
Typically, the guiding philosophy – organising principle – of mentalhealth services remains one of ‘symptom’ and ‘cure’: the identification of dif-ficulties and dysfunctions and the reduction of these by pharmacological,
Ch 23 7/4/05 4:03 pm Page 211

212 Bipolar disorder: the upswing in research & treatment
psychological or other means. Within this framework, four implicit assump-tions can be discerned. First, it is assumed that the primary task of theassessment process is to catalogue a person’s symptoms and problems.Second, it is assumed that a person’s symptoms must be controlled beforethey can be helped to resume their life: people must be ‘well’ before they cango back to work and other social roles. Third, it is assumed that until a per-son’s symptoms can be reduced they must be looked after and protectedfrom harm. Fourth, it is assumed that if a person’s symptoms can be elim-inated then they will automatically be able to resume their ‘normal’ lives.
In this chapter I will argue that such assumptions are unfounded andthat a framework based on symptoms and cures provides an inadequatebasis for the development of services that can enable people with bipolardisorder to pursue their ambitions and rebuild decent, satisfying and val-ued lives.
Lives cannot be built on a foundation of deficits and dysfunctions – theyare constructed on a base of talents and possibilities. As Chadwick hasargued:
‘Deficit-obsessed research can only produce theories and atti-
tudes which are disrespectful of clients and are also likely to
induce behaviour in clinicians such that service users are not prop-
erly listened to, not believed, not fairly assessed, are likely treated
as inadequate and are also not expected to become independent
and competent individuals in managing life’s tasks.’4
It is equally, if not more, important that an assessment process identifiesthe strengths that are the building blocks for recovery. A focus on symptomsand problems saps confidence, causes people to lose sight of their potentialand possibilities and can readily result in the hopelessness and despair thatare associated with increased suicide risk.5,6 Mental health services arereplete with people who have given up on themselves and their futures: atragic waste of human potential.
A focus on deficits and dysfunctions too readily results in clinicians hav-ing low expectations about what people with bipolar disorder can achieve.Such low expectations generate a vicious cycle – a self-fulfilling prophecy –that both erodes hope and diminishes opportunity (Figure 23.1). If theexpert professionals believe that people with bipolar disorder are, for exam-ple, unlikely to be able to hold down a job (or at best can only manage low-level, low-stress positions) then this has two effects. First, people with thedisorder believe them and give up applying for jobs – ‘If the experts say Icannot work then what hope is there?’ Second, employers believe them and
Ch 23 7/4/05 4:03 pm Page 212

Is what we offer to patients half acceptable? 213
are reluctant to hire people with the disorder – ‘If the experts say they can-not work then what is the point in employing them?’ If people with bipolardisorder see little point in applying for jobs, and employers are reluctant tohire them, then this guarantees that there will be few people with the diag-nosis in employment. This, in turn, confirms the low expectations ofemployers, clients and mental health professionals in a vicious cycle ofdespondency. All can say ‘I told you so’: interpret low employment rates asconfirmation of the low probability of people with the disorder being able towork.
At a general level, the quest for a ‘bipolar credit’ – akin to ‘schizophreniccredit’4,7 – is as important as the search for deficits. The relationshipbetween creativity and bipolar disorder might furnish a promising basis forsuch endeavours.8,9 At a more specific level, assessments must reveal anindividual’s strengths, possibilities and ambitions if they are to provide auseful foundation for helping them to rebuild their lives.
The assumption that a person’s symptoms must be controlled – theirproblems minimized – is equally problematic. The reduction of symptomsand problems is neither a necessary nor a sufficient condition for enablingpeople to work or resume other social roles.
On the one hand, a person can gain and sustain employment even in theface of continuing, or recurring, symptoms. On the other hand, even if a per-son’s symptoms can be eliminated completely, this does not mean that theywill be able to gain work. Symptom reduction does little to control theprejudice and discrimination that pervade society and operate to exclude
Figure 23.1 The vicious cycle of low professional expectations: a self-fulfillingprophecy.
Expert professionals say that people with bipolar disorder
are unlikely to be able to work
Very few people with bipolar disorder are in employment
Employers believe that people with bipolar disorder cannot work
– so don't employ them
People with bipolar disorder believe that they cannot work and give up trying to get jobs
Ch 23 7/4/05 4:03 pm Page 213

214 Bipolar disorder: the upswing in research & treatment
people from employment (and other social roles) on the basis of a history ofmental disorder as well as its continued presence. However, randomizedcontrolled trials have demonstrated that as many as 70% of those with seri-ous mental health problems can successfully gain and sustain employmentif offered the right kind of support: ‘Individual Placement with Support’; evi-dence-based supported employment.10,11 It is the type of support ratherthan diagnosis or symptoms that are the primary determinant of employ-ment outcomes. Research has failed to find a relationship between any spe-cific client factors – including diagnosis, symptomatology, disability statusor prior hospitalization – and the outcomes of supported employment.11
Furthermore, in the time that it takes for a person’s symptoms to be fullycontrolled, it is likely that people will have lost the things that they value inlife: friends, partners, leisure activities and, most especially, work. Thelonger a person is off work for illness reasons, the less likelihood there isof them returning to work.12 Time is of the essence: after 6 months of sick-ness absence, the probability that a person will return to work falls to 50%;after 1 year to 25% and after 2 years to 10%.13
Although there may be brief periods of acute crisis when it may beappropriate to relieve people of their social roles, these should be kept to anabsolute minimum. The assumption that people cannot work (or engage inother roles) unless or until their symptoms have been controlled is likely toreduce their chances of retaining valued social roles and/or rebuilding theirlives. It is important that services actively assist people to do the things theywant to do at the earliest opportunity, whether or not their symptoms per-sist or recur.
A desire on the part of clinicians to protect those who have continu-ing/recurring symptoms and deficits may, in some instances, be appropri-ate. It is perhaps understandable that some people with bipolar disordermay be reluctant to take risks for fear of precipitating a relapse. However,building a life – pursuing ambitions – necessarily involves taking risks: no-one can ever gain qualifications, get a job, or form a relationship withoutexperiencing anxiety and risking failure. A life in which stress is minimizedin order to reduce the risk of relapse is not necessarily a satisfying one: eachindividual needs to weigh the relative costs and benefits for themselves.Clinicians must be prepared actively to encourage people to explore theirpossibilities and pursue valued ambitions, and to support them in takingthe associated risks, rather than always counselling caution.
If the services are to enable people with bipolar disorder to rebuildmeaningful, satisfying and valued lives then it is necessary to move awayfrom an emphasis on the identification and amelioration of symptoms and
Ch 23 7/4/05 4:03 pm Page 214

Is what we offer to patients half acceptable? 215
deficits. While the importance of treating distressing and disabling symp-toms should not be minimized, such endeavours do not offer an adequateorganizing principle for mental health services or the development of theirunderpinning research base. In place of symptom identification and cure, amore appropriate guiding principle for services might better be framed interms of recovering a meaningful, satisfying and valued life: enabling peopleto do the things they want to do, live the lives they wish to lead and accessthose opportunities that non-disabled citizens take for granted. The treat-ment of symptoms, the reduction of deficits, may form part, but only part,of such an enterprise. Perhaps, after Shepherd,14 it would be preferable toview the treatment of symptoms as part of the assessment process: helpingto define those ongoing impairments that need to be accommodated if a per-son is to do the things they wish to do in life.
Such an approach requires a move away from a focus on deficit and dys-function towards an emphasis on skills and possibilities; a move away froma focus on care towards an emphasis on opportunity; a move away fromprescribing what is good for people (what they should do, what help andsupport they need) to enabling people to take control of their own lives andthe assistance they receive to live them. It also requires a change in the bal-ance of research endeavours.
The identification of deficits and dysfunctions and optimal ways of alle-viating these can no longer take pride of place on the research agenda.Instead, there needs to be an increased focus on the best ways of enhancingsocial role functioning and optimal ways of minimizing the impact of thedisorder on the person’s life: this may be achieved by decreasing symptoms,it may not – that is an empirical question that has not, to date, been ade-quately addressed. Attention is required to issues such as the ways in whichthe hope and optimism necessary for rebuilding a decent life can be fosteredand the ways in which the social (and financial) chaos wrought by episodesof mania or depression can be minimized or mitigated. Under both US andUK disability rights legislation, employers, educators and the providers ofgoods and services are required to make ‘reasonable adjustments/accom-modations’ to facilitate access for people with mental health problems suchas bipolar disorder. Yet very little research has been conducted into thesorts of adjustments that might be most beneficial to those with the disor-der. While there is a mounting body of literature concerning the types ofservices that can best assist people with more serious mental health toretain, regain and sustain employment, none of this has been specific tobipolar disorder and for other areas of people’s lives the research evidenceis virtually non-existent.
Ch 23 7/4/05 4:03 pm Page 215

216 Bipolar disorder: the upswing in research & treatment
When one strays outside the domain of symptoms a vast expanse ofuncharted territory comes into view. Dangerous chasms in knowledge andresearch become evident and these take on a particular significance in rela-tion to the development and refinement of treatment standards and practiceguidelines (such as those being developed by the UK National Institute forClinical Excellence). These can only be based on the best research evidenceavailable. Those things that have attracted the attention of clinical academ-ics and researchers will therefore be included; those areas where researchinterest has been lacking will be excluded. If outcome research has prima-rily addressed the efficacy of different psychological and pharmacologicalinterventions then practice guidelines can focus only on these areas. Thosethings that are equally, if not more, important for people with bipolar dis-order – work, friendship, intimate relationships, parenting, social andleisure activities – will be excluded simply because they have not appearedon research agendas.
While attention remains focused on deficits and dysfunctions, views ofthose with bipolar disorder will remain negative and pessimistic. Whileintervention is considered solely in terms of symptom reduction, then are-nas critical to well-being and quality of life will remain ignored. If what weoffer to people with bipolar disorder is to be half acceptable – if the lives ofpeople with the disorder are really to be improved – then it is imperativethat we broaden the focus of mental health services and the research basethat underpins them.
References
1. Repper J, Perkins R, Social Inclusion and Recovery. A Model for MentalHealth Practice. Ballière Tindall: London, 2003.
2. Bond GR, Psychiatric rehabilitation outcome. In: The Publication Committee ofInternational Association of Psychosocial Rehabilitation Services (IAPSRS),(eds), An Introduction to Psychiatric Rehabilitation. International Associationof Psychosocial Rehabilitation Services: Columbia, MD, 1994: 490–494.
3. Royal College of Psychiatrists, Employment Opportunities and PsychiatricDisability. Council Report CR111. Royal College of Psychiatrists: London, 2002.
4. Chadwick PK, Schizophrenia: The Positive Perspective. In Search of Dignityfor Schizophrenic People. Routledge: London, 1997.
5. Drake RE, Cotton PG, Depression, hopelessness and suicide in chronic schiz-ophrenia. Br J Psychiatry 1986; 148:554–559.
6. Beck AT, Brown G, Berchick RJ et al, Relationship between hopelessness andultimate suicide: a replication with psychiatric outpatients. Am J Psychiatry1990; 147:190–195.
7. Claridge GS, Origins of Mental Illness. Blackwell: Oxford, 1985.
Ch 23 7/4/05 4:03 pm Page 216

Is what we offer to patients half acceptable? 217
8. Jamison KR, Touched with Fire: Manic–depressive Illness and the ArtisticTemperament. The Free Press: New York, 1993.
9. Jamison KR, Manic depressive illness and creativity. Sci Am 1995; 272:62–67.10. Crowther RE, Marshall M, Bond GR, Huxley P, Helping people with severe men-
tal illness to obtain work: systematic review. BMJ 2001; 322:204–208.11. Bond GR, Supported employment: evidence for an evidence-based practice.
Psychiatr Rehabil J 2004; 27:345–359.12. Niemeyer L, Jacobs K, Reynolds-Lynch K et al, Work hardening, past, present
and future – The work programs special interest section national work hard-ening outcome study. Am J Occup Ther 1994; 48:327–339.
13. Clinical Standards Advisory Group, Back Pain. HMSO: London, 1994.14. Shepherd G, Institutional Care and Rehabilitation. Longman: London, 1984.
Ch 23 7/4/05 4:03 pm Page 217

Ch 23 7/4/05 4:03 pm Page 218

Index
adolescents, changes in amygdala of 23impaired recognition of facial expression in 38MRS studies of bipolar disorder in 22
adrenocorticotrophic hormone (ACTH), role in hypothalamic–pituitary–adrenal axis 116, 129, 130, 135
affective instability, neural activity and 43neural basis of 44
affective spectrum 147age,
as predictor of recovery 16at onset 6differences in incidence with 1ECT use and 196gender and age at onset 1, 2, 4grey matter volume and 23hippocampal volume reduction with increasing 126hyperintense lesions and aging 22
alcoholism, as predictor of functional outcome 19occupational status after discharge and 12remission length and 13risk of relapse and 11
amygdala, abnormalities in 22changes in adolescents and children 23heterogeneity within 33increased activity in 51neural responses to facial expression in 38neuroanatomical changes in 51structural changes in 41
anger recognition 38animal studies,
compromised BDNF expression in transgenic mice 107compromised CREB expression in transgenic mice 107compromised TrkB expression in transgenic mice 108cortisol in guinea pigs 131
Index 7/4/05 4:03 pm Page 219

220 Bipolar disorder: the upswing in research & treatment
transgenic mouse models 103–10anterior cingulate,
abnormalities in 21activity and facial expression recognition 38attentional processing in 149increased activity in 51neuroanatomical changes in 51neuron size in 124neuropathology in 60, 61synaptic pathology in 61
anticonvulsants, use and functional outcome 14antidepressants,
cognitive function and 42effect on CREB–BDNF–TrkB pathway 104, 105effect on neurotransmitters 103use and functional outcome 14
antipsychotics, advantages and disadvantages of 181–90basal ganglia enlargement and 32cf lithium in mania 178cf placebo 183cf valproate 181–90classical cf atypical 188combination treatment
with lithium 186with valproate 186
comparative trials 188extrapyramidal symptoms 189grey matter volume changes and 32prescription at discharge 18rapid response to 182use and functional outcome 14
anxiety, co-morbidity with bipolar disorder 166arginine vasopressin,
effects of 135–6in major depressive disorder 138, 139levels in depression 139neuroanatomy 135response to chronic stress 138role in hypothalamic–pituitary–adrenal axis 135role in hypothalamic–pituitary–adrenal axis dysregulation 140synergism with corticotrophin releasing hormone 137
aripiprazole cf placebo 183attentional processes,
dysfunction in 149in bipolar disorder 157
autonomic response, lack in major depressive disorder 40autopsy results 59–66
Index 7/4/05 4:03 pm Page 220

Index 221
basal ganglia, enlargement and antipsychotic use 32BDNF gene 84, 85
see also brain-derived neurotrophic factor; neurotransmittersBeck’s cognitive model of depression 145, 146biased medicine 178bipolar cycling 147bipolar disorder,
advantages and disadvantages of antipsychotics or valproate in 181–90age at onset 6brain abnormalities in 21–4brain volume changes in 27, 30, 31care costs 166cf schizophrenia 4changes in synaptic protein levels in 124, 125clinical epidemiology 1–7clinical epidemiology study in Camberwell 1–7co-morbidities 166cognitive dysfunction and cause or consequence 145–54current treatment guidelines 170ECT use in 193–8functional outcome 9–20gender differences in age at onset 6gender differences in presentation 3genetic basis to brain abnormalities in 93–101genetics of 64, 69–75, 77–88glia-based origin of 65glial cell changes in 124heterogeneity of 201hypothalamic–pituitary–adrenal axis and 115–21
dysfunction in 118imaging studies of 27–34, 37–44incidence rate 3living with 211longitudinal course 9mifepristone use in 120morbidity 165mortality 165neural basis for cognitive function in 157–61neuropathology 59–66
and cortisol dysregulation in 123pathophysiology of 51–6, 115peak age at onset 3postmortem findings 59–66psychological treatments 165–71repetitive transcranial magnetic stimulation use in 193–8services for patients with 215subcortical regions in 37–44transgenic mouse models 103–10
Index 7/4/05 4:03 pm Page 221

222 Bipolar disorder: the upswing in research & treatment
use of lithium in 175–9vicious cycle of low expectations 213
brain abnormalities, activity 37–41amygdala 22anatomical specificity and precision in studies of 63anterior cingulate grey matter reduction 21as endophenotypes 94attentional processing and 149changes in volume 29
by brain region 30cingulate gyrus 21expansion of subcortical region 37–44frontolimbic changes 23functional imaging studies of 158–61genetic basis to 93–101genetic liability and grey matter volume 97, 98genetic liability and white matter volume 97, 99grey matter reduction in children and adolescents with bipolar disorder 22, 23heterogeneity within brain structures 33hyperintense lesions 22in bipolar disorder 21–4meta-analysis of 27–34neural function as predictor 43neuropathology 59–66previous illness history and 42response to medications 42stress and 123–7structural 41
brain morphometry 94brain-derived neurotrophic factor (BDNF) 83, 84
activity 84antidepressant effects of 106, 110map of gene 85response to antidepressants 105response to immobilization stress 109role in depression 103, 104role in other disorders 86transgenic mouse models with compromised expression of 107
Brown–Peterson paradigm 157
Camberwell, clinical epidemiology study in 1–7Cambridge Neuropsychological Test Automated Battery 146, 151cAMP response element-binding protein (CREB)
response to antidepressants 105see also CREB–BDNF–TrkB pathwaytransgenic mouse models with compromised expression of 107
carbamazepine, in lithium responders 206
Index 7/4/05 4:03 pm Page 222

Index 223
children, changes in amygdala of 23MRS studies of bipolar disorder in 22
chlorpromazine, cf ECT 194chromosomal regions involved in bipolar disorder 81chromosome 13q 74, 81chromosome 22q 74, 81cingulate gyrus, abnormalities in 21citalopram and cognitive function 42clinical epidemiology 1–7cognitive behaviour therapy 167, 168
MRC study 170cognitive dysfunction, see under cognitive functioncognitive function,
age and increased deficit 146, 147assessment of deficits in bipolar disorder 148cause or consequence of bipolar disorder 145–54development in childhood 145effect of cortisol on 119effect of glucocorticoids on 118effect of medication on 42effect of mifepristone on 120focus on deficit and dysfunction is negative 212hypercortisolaemia and 121impaired decision-making in mania 152impairment in affective disorders 118in Cushing’s disease 119in recovered bipolar disorder patients 153linked with functional imaging 153measures of 151measures of deficit 146neural basis for 157–61neurocognitive tasks as measure of dysfunction 147residual deficits 153
combination therapies 186use for improved outcome 207
comorbidity, at first episode presentation 17with bipolar disorder 166
complexin-I level in anterior cingulate cortex 124COMT gene 64, 83corticotrophin releasing hormone,
description 135regulation by glucocorticoids 137response to chronic stress 138role in hypothalamic–pituitary–adrenal axis 116, 129, 130synergism with arginine vasopressin 137
Index 7/4/05 4:03 pm Page 223

224 Bipolar disorder: the upswing in research & treatment
cortisol, bad press for 129–32effect on brain 129effect on frontal lobes 119effects at cellular level 117regulation of levels 131
costs, annual care costs for bipolar disorder 166CREB, see under cAMP response element-binding protein CREB–BDNF–TrkB pathway,
description 104response to antidepressants 105response to stress 104role in antidepressive therapy 110role in depression 109, 110
computed tomography (CT) of bipolar disorder 27Cushing’s disease,
cognitive function in 119hippocampal volume reduction in 126
DAAO gene 74decision-making tasks 53, 151declarative memory in bipolar disorder 157Delayed-Matching-to-Sample test 151dementia, hyperintense lesions in 22depression,
arginine vasopressin levels in 139automatic negative thoughts in 150characteristics of 148cognitive dysfunction in cf that in mania 148glucocorticoid receptors for 120hippocampal volume change in 32in mania 185level as predictor of recovery 16neurotrophin hypothesis of 104predictors of cf those for mania 17remission length and presence in index episode 13risk of relapse after 11risk of relapse to 15role of CREB–BDNF–TrkB pathway in 109, 110role of neurotransmitter deficiency in 103syndromal and subsyndromal 166transgenic mouse models for 109
desmopressin, dynamic tests of hypothalamic–pituitary–adrenal axis with 140despair in mice 108diagnosis, hierarchy of 71disgust, recognition of 38dopamine, role in depression 103DRD4 receptor gene 83
Index 7/4/05 4:03 pm Page 224

Index 225
drug companies, power of to change prescribing patterns 177electroconvulsive therapy (ECT)
as an antidepressant 195cf chlorpromazine 194cf lithium 194cf neuroleptics 194clinical trials of 193, 194effect on CREB–BDNF–TrkB pathway 104guidelines for use 194use by diagnosis 196use in bipolar cf unipolar depression 197use in bipolar disorder 193–8use in mania 193
emotion, neural responses to 37neural structures required in processing of 44
emotion-processing tasks, neural responses to 40–1response to emotional target words 40role of amygdala in 22
employment 211focus on dysfunction cf function 212vicious cycle of low expectations 213
endophenotypic markers, genetic liability and grey matter volume 97, 98genetic liability and white matter volume 97, 99implications for identification 100
epidemiology, functional outcome of bipolar disorder 9–20incidence study in south-east London 1–7of schizophrenia 1
epilepsy, hyperintense lesions in 22ethnicity,
differences in schizophrenia incidence 1incidence of bipolar disorder and 4
executive function, impairment in affective disorders 118in bipolar disorder 157previous illness history and 42
facial expression, enhanced recognition of disgust in 38impaired recognition of fear in 38neural responses to in bipolar disorder 37neural responses to recognition 37–40normal brain responses cf bipolar disorder and major depressive disorder 39
familial factors, genetic liability scores 96family studies, familial links of schizoaffective disorder to bipolar disorder 70
Index 7/4/05 4:03 pm Page 225

226 Bipolar disorder: the upswing in research & treatment
family therapy 168fear,
brain responses to recognition of expression 39recognition of 38
fluoxetine 140functional imaging, 43
confounding factors in 159during Iowa Gambling Task 55during N-back task 54linked with cognitive function 153Maudsley Bipolar Disorder Project 52–6of Sternberg paradigm 158, 159of two-back task 158
functional neuroimaging, emotional processing 43functional outcome, definitions of 15
G72 gene 64, 74gambling tasks,
Iowa Gambling Task 52, 53neural function during 55
results in mania 152Wisconsin Card Sorting Task 146, 152
gender, age at onset and 1, 2, 4as predictor of recovery 16differences in incidence with 1differences in schizophrenia incidence 1ECT use and 196
genetic association studies, description 78, 79in bipolar disorder 81
genetics, association studies,
description 78in bipolar disorder 81
basis to brain abnormalities 93–101definition of phenotype 77effects on hypothalamic–pituitary–adrenal axis 117explanation for overlap between disorders 71familial incidence of bipolar and unipolar disorders 69future developments 87genetic liability scores 96glucocorticoid receptor gene expression in mood disorders 126linkage studies,
description 78in bipolar disorder 80
molecular findings in bipolar disorder 74molecular genetics in bipolar disorder 82
Index 7/4/05 4:03 pm Page 226

Index 227
of bipolar disorder 64, 77–88of neurotransmitters in bipolar disorder 83of schizophrenia 64polygenic liability-threshold model 72polymorphism Val66Met 85, 86relation between bipolar disorder schizophrenia and unipolar depression 69–75susceptibility genes 93
glial cells, as basis of neuropathology in bipolar disorder 64, 65changes in bipolar disorder 124changes in major depressive disorder 124decrease in subgenual cingulate cortex 61types and role 124
glucocorticoid receptors 117activity of 129agonists and cortisol 131antagonists for therapy 119gene expression 126impaired function in depression 126
glucocorticoids, effect on cognitive function 118regulation of corticotrophin releasing hormone by 137regulation of vasopressin by 137role in hypothalamic–pituitary–adrenal axis 129, 130role in neuronal changes 126
go/no-go tasks 149imaging of response to 150
grey matter, antipsychotics and volume deficit 32genetic liability and volume 97, 98imaging of 98lithium
and volume changes 32protects against changes in 21
loss with age 23reduction in bipolar disorder 21reduction in children and adolescents with bipolar disorder 22, 23volume changes 41, 51
group psycho-education 168growth-associated protein-43, level in anterior cingulate cortex 124guidelines,
biased 177British cf American 181current treatment guidelines 170for acute mania 181for use of ECT 194
guinea pig, cortisol in 131
Index 7/4/05 4:03 pm Page 227

228 Bipolar disorder: the upswing in research & treatment
haloperidol, cf olanzapine 189cf placebo 183cf valproate in psychotic mania 181combination treatment with 186effect on depression after mania 186placebo-controlled monotherapy 188use with lorazepam 182
happiness, brain responses to recognition of expression 39heterogeneity,
causes of 33in brain 33of bipolar disorder 201of responders 202
hippocampus, BDNF and CREB mRNA levels in 105neuronal changes in 61structural changes in 41synaptic pathology in 61volume changes in 31, 32volume reduction and glucocorticoid receptors 126
hospitalization, length of as a predictor of recovery 16hypercortisolaemia 118
effect on cognitive function 121role of vasopressin in 135–41
hyperintense lesions 22hypothalamic–pituitary–adrenal axis,
biological factors in hyperactivity 135–41control of activity 129, 130cortisol and its receptors 117description of 116development of therapies for dysfunction of 119dynamic tests of 140dysfunction in bipolar disorder 118genetic effects on 117hyperactivity of and mood disorder 126reasons for hyperactivity of 131response to stress 116, 130role in bipolar disorder, 115–21
hypothalamic–pituitary–adrenal system, role in depression 103
imaging, see under computed tomography; functional neuroimaging; magneticresonance imaging; magnetic resonance spectroscopy
imipramine, effect in mice 108incidence 37
clinical epidemiology study in London 1–7increase over time 6variation in 1
Index 7/4/05 4:03 pm Page 228

Index 229
interpersonal social rhythms therapy 167Iowa Gambling Task 52, 53
neural function during 55
Kraepelinian dichotomy 73
lamotrigine, as long-term treatment 202cf lithium 203clinical characteristics of responders 204, 205
lateral ventricles, enlargement of 27, 29right, volume changes in 29, 30, 31total volume changes 31
linkage analysis, description 78, 79in bipolar disorder 80
lithium 175–9as long-term treatment 202cf ECT 194cf lamotrigine 203cf olanzapine 203cf valproate 179clinical characteristics of responders 204cognitive function and 42combination treatment with antipsychotic 186comparison with other drugs 177decline in use 175demands of treatment,
on patient 176on psychiatrist 176
effects on functional imaging studies 160efficacy throughout bipolar spectrum 178grey matter volume changes and 32in carbamazepine responders 206in combination with antipsychotics 187monotherapy cf combination therapies 207neurogenesis and 63prescription at discharge 18reasons for decline in use 176selective publication as reason for decline in use 177subgenual cingulate gyrus changes and 42use and functional outcome 14use in acute mania 178use in Italy 176
lorazepam use with haloperidol 182
McLean–Harvard first-episode study 15–19
Index 7/4/05 4:03 pm Page 229

230 Bipolar disorder: the upswing in research & treatment
magnetic resonance imaging (MRI), brain volume changes on 29for brain abnormalities in bipolar disorder 21–4go/no-go task activation 150meta-analysis 27–34neuropathology studies with 60white matter hyperintensities in bipolar disorder 125white matter hyperintensities in major depressive disorder 125
magnetic resonance spectroscopydescription of technique 22for brain abnormalities in bipolar disorder 21–4
maintenance treatment 187major depressive disorder,
autonomic responses in 40changes in synaptic protein levels in 124, 125genetic links to bipolar disorder and schizophrenia 65glial cell changes in 124hippocampal volume reduction in 126neuropathology of 123position on affective spectrum 147response to facial expression in 39vasopressin in 138
mania, age at onset 6characteristics of 149cognitive dysfunction in cf that in depression 148decision-making impairment in 152depression in 185ECT use for 193efficacy of valproate in 182, 183first-episode,
four-year follow-up after 9–14incidence and gender 5predictors of functional outcome after 14psychosis during 3recovery after 15
incidence rate 3position on affective spectrum 147predictors of cf those for depression 17relapse and recovery from 10repetitive transcranial magnetic stimulation in 198risk of relapse after 10, 11risk of relapse to 15severity and brain activity 38use of lithium as gold standard 178valproate cf antipsychotics for 181
MAOA gene 83Maudsley Bipolar Disorder Project 52–6
Index 7/4/05 4:03 pm Page 230

Index 231
Maudsley Family Study of Psychosis 95medication,
as confounding factors in functional imaging studies 159combination treatments 186effect on neuronal changes on 63fall in lithium usage 175monotherapy cf combination therapies 207valproate cf antipsychotics 181–90
mifepristone, effect on cognitive function 120effect on cortisol levels 131use in bipolar affective disorder 120use in depression 120
migrants, incidence in 2incidence increase and 6
mineralocorticoid receptors 117activity of 129agonists and cortisol 131
mitogen-activated protein (MAP) kinase, role of BDNF in cascade 110Modified Location Code Index 11Modified Vocational Status Index 11molecular genetics in bipolar disorder 82mood congruence,
as predictor of mania but nor depression 17risk of relapse and 10, 13
mood induction, neural responses to 40morbidity 165mortality 37, 165MRC study 169–71MRI, see under magnetic resonance imagingMRS, see under magnetic resonance spectroscopy
N-acetylaspartate (NAA) levels in bipolar disorder 22N-back task 53
neural function during 54neural function,
during Iowa Gambling Task 55during N-back task 54
neural responses to facial expression 37neuroanatomy,
functional 51of arginine vasopressin 135structural 51
neurocognitive dysfunction 146, 148neurogenesis and lithium 63neuroleptics,
attention deficits and 42
Index 7/4/05 4:03 pm Page 231

232 Bipolar disorder: the upswing in research & treatment
cf ECT 194neuropathology 59–66
commonalities between disorders 125of bipolar disorder 123of major depressive disorder 123
neurotransmitters, BDNF 83, 84CREB–BDNF–TrkB pathway 104deficiencies 103G72/G30 locus and DAAO 83genetics of in bipolar disorder 83neurotrophin hypothesis 103
neurotrophin, role in depression 104, 105transgenic mouse models of 103–10
neurotrophins, antidepressant effects of 106New Tower of London task 150, 151norepinephrine, role in depression 103
obsessive–compulsive disorder, BDNF gene in 86occupational status,
alcoholism and 12as predictor of new episodes 17factors affecting 19functional outcome and 14risk of relapse and 11
olanzapine, 182as long-term treatment 202cf haloperidol 189cf lithium 203cf placebo 183cf valproate 185clinical characteristics of responders 205combination treatment with 186controlled trials with 183effect on depression after mania 186in combination with lithium 187side-effects of 185
orbitofrontal cortex, neural responses in 149neuron size in 124neuropathology in 60, 61
outcome, clinical characteristics of responders 204four-year follow-up after first-episode of mania 10functional 9–20
definitions of 15factors affecting 19
Index 7/4/05 4:03 pm Page 232

Index 233
importance of 20predictors of 14risk factors for poor 10, 11, 12
heterogeneity of responders 202, 203improvement of by long-term treatment selection 201neural function as a predictor of 43selectivity of responses 206
paraventricular nucleus 136pathophysiology 51–6
of bipolar disorder 115personality disorder, co-morbidity with bipolar disorder 166phenotype,
definition of 77endophenotypes 93, 94
placebo trials 183polygenes 71polymorphism Val66Met 85, 86positron emission tomography (PET) neuropathology studies 60post-traumatic stress disorder, hippocampal volume reduction in 126postmortem findings 59–66
corticotrophin releasing hormone-expressing neurones 139CREB levels 105phase of illness at time of death and 62vasopressin-expressing neurones 139
predictors, endophenotypic markers 94, 95genetic liability scores 96implications for identification 100neural abnormalities as 43of functional outcome after mania 14of outcome, identification of 9of recovery 16of relapse 13, 17
prefrontal cortex, activity and facial expression recognition 38activity during semantic cf orthographic tasks 40changes in 23decreased activity in 51functional imaging studies 52–6neuroanatomical changes in 51neuron size in 124response to emotion in 38structural changes in 41
presentation, variations in 78probabilistic reversal learning 151psychological treatments,
current treatment guidelines 170
Index 7/4/05 4:03 pm Page 233

234 Bipolar disorder: the upswing in research & treatment
MRC study 169–71studies of 167use in bipolar disorder 165–71
psychosis, as predictor of functional outcome 19during first-episode mania 3hypothalamic–pituitary–adrenal axis hyperactivity in 130, 131remission length and presence in index episode 13risk of relapse and incidence 10, 12verbal impairment in 42
psychotropics, neuroanatomy and 42prescription at discharge 18use and functional outcome 13
quetiapine, cf placebo 183combination treatment with 186
Rapid Visual Information Processing task 153, 154recovery,
after first-episode mania 15definitions of 15predictors of 16syndromal cf functional 15, 19
relapse, after mixed episode 17occupational status as predictor of 17odds ratios for 166, 168predictors of 13risk factors for 10, 11time to onset 18
remission, probability of remaining in 10risk factors for shortened 13risk for relapse from 10
repetitive transcranial magnetic stimulation bipolar disorder use in 193–8description 197in bipolar depression 198in mania 198
residential status, factors affecting 19functional outcome and 14risk of relapse and 11
risperidone, cf placebo 183, 184combination treatment with 186in combination with lithium 187
Index 7/4/05 4:03 pm Page 234

Index 235
in combination with valproate 187RU-486, see under mifepristone
schizoaffective disorder, familial links to bipolar disorder 70schizophrenia
BDNF gene in 86brain volume changes in 29, 31cf bipolar disorder 4clinical epidemiology studies of 1gender differences in age at onset 6genetic relation to bipolar disorder 69–75genetics of 64hyperintense lesions in 22knowledge of cf bipolar disorder 1
seasonal affective disorder 153serotonin,
role in attentional processing 149role in depression 103
serotonin transporter (5-HTT) gene 83side-effects,
avoidance of with antipsychotics 182of lithium 176of olanzapine cf valproate 185weight gain 187
social functioning in bipolar disorder 145status epilepticus, valproate for 182Sternberg paradigm 157
brain regions active during 161functional imaging studies of 158, 159
stress, effect on brain 123–7effect on CREB–BDNF–TrkB pathway activity 104effect on CREB–BDNF–TrkB pathway in mice 109effects on corticotrophin releasing hormone and arginine vasopressin 138hypothalamic–pituitary–adrenal axis response to 116, 130
subcortical regions, activity and facial expression recognition 38activity in response to emotive scenes 40expansion of 37–44
subgenual cingulate cortex, glial cells in 61lithium and 42neuronal changes in 61neuropathology studies with 60structural changes in 41
subgenual prefrontal cortex, grey matter reduction in 21heterogeneity within 33
Index 7/4/05 4:03 pm Page 235

236 Bipolar disorder: the upswing in research & treatment
substance abuse, comorbid with bipolar disorder 17comorbidity with bipolar disorder 166development after mania 19
suicide incidence in bipolar disorder 166susceptibility genes 93synapses,
changes in bipolar disorder 124changes in major depressive disorder 124function of, genetics and 64pathology of, 61synaptic protein levels in anterior cingulate cortex 124
synaptophysin level in anterior cingulate cortex 124
transcranial magnetic stimulation 193–8cf ECT 197
transgenic mouse models 103–10compromised BDNF expression 107compromised CREB expression 107compromised TrkB expression 108CREB–BDNF–TrkB pathway studies in 106
treatment, importance of speed in 214triplets, bipolar disorder and schizophrenia in 70tryptophan, effect on attentional processing 149twin studies 70
bipolar disorder and schizophrenia in triplets 70two-back task,
brain regions active during 160functional imaging studies of 158
tyrosine receptor kinase B (TrkB), response to antidepressants 106role in CREB–BDNF–TrkB pathway 104
unipolar depression, genetic relation to bipolar disorder 69–75
Val66Met polymorphism 85, 86valproate,
advantages and disadvantages of 181–90cf antipsychotics 181–90cf haloperidol in psychotic mania 181cf lithium 175, 179cf olanzapine 185cf placebo 183combination treatment with antipsychotic 186delayed onset 182efficacy in mania 182in combination with antipsychotics 186, 187maintenance treatment 187
Index 7/4/05 4:03 pm Page 236

Index 237
mechanism of action cf antipsychotics 182side-effects of 185weight gain on 187
vasopressin, dynamic tests of vasopressin-ergic system 140in major depressive disorder 138regulation by glucocorticoids 137role in hypothalamic–pituitary–adrenal axis hyperactivity 135–41
vasopressin receptors 136verbal impairment, in psychosis 42
weight gain 187white matter,
genetic liability and volume 97, 99hyperintensities in 27, 65
and outcome 43on MRI 125volume changes and 32
imaging of 99Wisconsin Card Sorting Task 146, 152working memory, in bipolar disorder 157
ziprasidone 182cf placebo 183combination treatment with, 186
Index 7/4/05 4:03 pm Page 237

Index 7/4/05 4:03 pm Page 238
