cancer research protein tyrosine phosphatase receptor type ... · tumor and stem cell biology...
TRANSCRIPT

Tum
ProFunDow
MarcoAndreFabrizMaria
Abst
Intro
Chrchroninantcountcasesfrom aresultshas asignalphoryling ac
Author2Depar3ARC-NVeronae Ariosdi DiagFrances
Note: SOnline
AuthorexperiminformacipitatioQPCR
Cance8896
Published OnlineFirst October 19, 2010; DOI: 10.1158/0008-5472.CAN-10-0258
Canceresearch
or and Stem Cell Biology
tein Tyrosine Phosphatase Receptor Type γ Is actional Tumor Suppressor Gene Specifically
R
nregulated in Chronic Myeloid Leukemia
Della Peruta1, Giovanni Martinelli4, Elisabetta Moratti1, Davide Pintani1, Marzia Vezzalini1,a Mafficini1,3, Tiziana Grafone4, Ilaria Iacobucci4, Simona Soverini4, Marco Murineddu5,
io Vinante2, Cristina Tecchio2, Giovanna Piras5, Attilio Gabbas5, Monne5, and Claudio Sorio1,3ractChr
phospCD34+
lines Mexpresand inBCR/Aat theincludwith aK562 c
ation of ktivity of B
s' Affiliatiotment of Clinet Researc, Italy; 4Instto Seragnolnostica Bioco” Hospita
upplementa(http://cance
contributionents, analytion (Bolognn studies. Danalysis,
r Res; 70(
Downloa
onic myelogenous leukemia (CML) is the most common myeloproliferative disease. Protein tyrosinehatase receptor type γ (PTPRG) is a tumor suppressor gene and a myeloid cell marker expressed bycells. Downregulation of PTPRG increases colony formation in the PTPRG-positive megakaryocytic cellEG-01 and LAMA-84 but has no effect in the PTPRG-negative cell lines K562 and KYO-1. Its over-
sion has an oncosuppressive effect in all these cell lines and is associated with myeloid differentiationhibition of BCR/ABL-dependent signaling. The intracellular domain of PTPRG directly interacts withBL and CRKL, but not with signal transducers and activators of transcription 5. PTPRG is downregulatedmRNA and protein levels in leukocytes of CML patients in both peripheral blood and bone marrow,ing CD34+ cells, and is reexpressed following molecular remission of disease. Reexpression was associatedloss of methylation of a CpG island of PTPRG promoter occurring in 55% of the patients analyzed. Inell line, the DNA hypomethylating agent 5-aza-2′-deoxycytidine induced PTPRG expression and causedibition of colony formation, partially reverted by downregulation of PTPRG expression. These findings
an inhestablish, for the first time, PTPRG as a tumor suppressor gene involved in the pathogenesis of CML, suggestingits use as a potential diagnostic and therapeutic target. Cancer Res; 70(21); 8896–906. ©2010 AACR.
Fornaturasignalof enzThe
belongfamilyand 6To daare knforma
duction
onic myelogenous leukemia (CML), also known asc myeloid or chronic myelocytic leukemia, is a malig-cancer of the bone marrow myeloid lineage. It ac-s for ∼15% to 20% of all cases of leukemia (1–1.5per 100,000 population per year; ref. 1) and originatespluripotential stem cell in which a 9:22 translocationin the production of BCR/ABL fusion protein. Thisconstitutive tyrosine kinase activity and deregulatestransduction pathways, leading to leukemia (2). Phos-
ey residues is required for the full transform-CR/ABL (3).
phospPTPs
ns: 1Department of Pathology and Diagnostics andical and Experimental Medicine, University of Verona;h Center, University of Verona, Policlinico G.B. Rossi,itute of Hematology and Medical Oncology, «Lorenzoi», University of Bologna, Bologna, Italy; and 5Centromolecolare e Citogenetica Emato-Oncologica, “Sanl, ASL3, Nuoro, Italy
ry data for this article are available at Cancer Researchrres.aacrjournals.org/).
statement: M. Della Peruta: designed and performedzed data. G. Martinelli: patient selection and clinicala). E. Moratti: performed experiments, coimmunopre-. Pintani: performed clonogenic assays. M. Vezzalini:
clonogenic and proliferation assays. T. Grafone:
performmanagand pa(VeronacytomeM. Monin patie
CorresDiagnoLe Gra802712
doi: 10
©2010
21) November 1, 2010
Research. on March 11, 202cancerres.aacrjournals.org ded from
this reason, much attention has been focused onlly occurring negative regulators of tyrosine kinaseing: the protein tyrosine phosphatase (PTP) familyymes.human PTP family contains 107 members, 38 of whichto the phosphotyrosine-specific (“classic”) PTP sub-(subdivided in receptor- and nonreceptor-like types)
1 belong to the so-called “dual-specific phosphatases.”te, only two tyrosine phosphatases, PTP1B and SHP-1,own to dephosphorylate and partially inhibit the trans-tion potential of BCR/ABL (4, 5). Serine-threonine
hatase PP2A is inhibited in blast crisis CML (6). Thesebelong to the nonreceptor class of enzymes. Recently,ed flow cytometry. I. Iacobucci: BCR/ABL analysis. S. Soverini:ement of clinical data (Bologna). M. Murineddu: flow cytometrytient selection. F. Vinante: patient selection and clinical information). C. Tecchio: performed research. A. Mafficini: performed flowtry. G. Piras: statistical analysis. A. Gabbas: provided clinical data.ne: patient selection and clinical information, methylation studiesnts (Nuoro). C. Sorio: designed research, wrote manuscript.
ponding Author: Claudio Sorio, Department of Pathology andstics, General Pathology Section, University of Verona, Stradazie 8, 37134 Verona, Italy. Phone: 39-45-8027688; Fax: 39-45-7; E-mail: [email protected].
.1158/0008-5472.CAN-10-0258
American Association for Cancer Research.
0. © 2010 American Association for Cancer

PTPROwith BPTP
like PTprecu(9–11tumorbreastgenetiHer
supprehibitindownrcytesinvolvin thePTPRGpeutic
Mate
TissuePati
BolognCMLthe PhmenttakenimatinfromdiseasCD34+
remisavailabdata. Wwith tclassif
be inBIOM
CellsK56
CulturDSMZtantsthe cuysis anFicoll-(flow c
Cell tFul
cDNAPTPRwhere(NP_0To a
withinthat mwith t(28, 2proces
TransHEK
p210B
pLNL-sity ofPTPRGperforcordinDNA (
Tab
AgeFemBcr-HemPlatWBPeriPeriBMPalp
Abb
PTPRG in Chronic Myeloid Leukemia
www.a
Published OnlineFirst October 19, 2010; DOI: 10.1158/0008-5472.CAN-10-0258
t, a receptor-like PTP, has been found to interfereCR/ABL signaling in K562 cells (7).receptor type γ (PTPRG) is a member of the receptor-Ps (8), is expressed in myeloid cells, including CD34+
rsors, and can affect hematopoietic differentiation). It is a candidate tumor suppressor gene in solids (12–14) owing to its reduced expression in ovarian,, and lung tumors (15, 16). Somatic mutations and epi-c silencing were reported (17–21).e, we show that PTPRG acts as a functional tumorssor gene in CML, interacting with BCR/ABL and in-g downstream signaling events. PTPRG is specificallyegulated in peripheral blood and bone marrow leuko-of CML patients, at least in part by a mechanisming hypermethylation of a PTPRG CpG island located5′ untranslated region. These findings imply thatmight represent a potential diagnostic and thera-target in CML.
rials and Methods
samplesents were recruited at the hematology departments ofa, Nuoro, and Verona, Italy among newly diagnosedpatients during the first chronic phase. Presence ofiladelphia chromosome and p210BCR-ABL rearrange-was a prerequisite for enrollment. CML samples wereat diagnosis and after the initiation of therapy withib mesylate (IM). Age- and sex-matched samplesindividuals diagnosed as not affected by malignante were used as a reference group. Analysis of thepopulation for the group of patients in molecular
sion was not possible, as these samples were notle at the time of evaluation. Table 1 reports clinicalritten informed consent was obtained in accordance
he Declaration of Helsinki. Cytogenetic response wasied as complete, and the patients were considered to
transfewere c
le 1. Clinical data of patients
DiagnMed
54.alAb 69.og 10.ele 346.C ( 218.ph 4
reviations: WBC, white blood cells; BM, bone marrow.
acrjournals.org
Research. on March 11, 202cancerres.aacrjournals.org Downloaded from
complete cytogenetic remission (CCR) based on theED 2 standardized protocols.
2 (22) and MEG-01 (23) were from American Typee Collection; KYO-1 (24) and LAMA-84 (25) were from. HEK293 was purchased from Invitrogen. K562 transfec-were selected, adding 0.50 mg/mL G418 (Invitrogen) tolturemedium. All were characterized by cytogenetic anal-d antigen expression. Human samples were derived frompurified cells (mRNA analysis) or whole blood samplesytometry analysis).
ransfection and selection of CML cell linesl-length (FL) human PTPRG (26) and antisense (AS)and cell lines were described (10, 27). The D1028A
G cDNA was obtained by site-directed mutagenesis,in the Asp 1028 codon was replaced with an Ala codon02832.3) and verified by sequencing.void the source of error associated with clonal variationcell lines, we selected a K562 clone (named B4)aintains the capability to differentiate when treatedhe well-known inducers sodium butyrate and hemin9) and performed all the transfection and selectionses starting from this well-characterized clone.
fection of HEK293 cells293 cells were transfected with FL cDNA coding for
CR/ABL cDNA in pLNL 5LX cytomegalovirus, derived fromXHC (30), kindly provided by Dr. Paolo Vigneri (Univer-Catania, Italy). FL human PTPRG cDNA and D1028AcDNA were previously described. Transfection was
med with Lipofectamine 2000 reagent (Invitrogen) ac-g to themanufacturer's instructions using 1.5μg plasmidfor single transfection) or 1 + 1μg plasmidDNA (for double
ction) following the manufacturer's instructions. Culturesultured for 48 hours, washed with cold TBS, and lysed.RemisMed
60.42
0.1011.3
193.74.77
Cancer Res; 70(21) Nov
0. © 2010 American Association for C
osis (n = 22)ian (range)
sion (n = 8)ian (range)
12 (47–63)
(33–84) e (%) 12.5 33.4 l/Abl (%) 13 (30.84–100) (0–1) lobin level (g/dL) 99 (7–13.8) (5.90–15.5) ts (×109/L) 25 (6–589) (88–291) ×109/L) 75 (137–296) (0.13–7.73) eral blood blasts (%) .4 (2–6) 0 eral blood basophils (%) .6 (0–15) (0.002–0.01) ph 7 0.006blasts (%) 3.2 (1–4) 0able splenomegaly (%) 57.14% 0.00
ember 1, 2010 8897
ancer

ImmuCell
descriTris-H5% gly23-gauand el(MilliptyrosiPY99anti-Cand anvatorsSTAT5weredetect30 min100 mwith a
ImmuFast FTot
(Sigmaple) whours(Sigmaincubaand su
ExpredomaPTP
(aminclonedA (Invquencand th2 μg otein (EK562 lwasheand W
ClonoK56
LAMAH4230ferredeach wWh
5-aza-platedproducloneswas h100 μthe in
AldricwereAbsorplate
XenogIn v
ing 18were p
ReverquantTot
ing tosion was dePTPRPTPNin SupTo
levelsformugues (tween(T) ansum oratiopressiequalspatienmediadiffere
MethyGen
tion uaccordwerefor 30(methPCR)7 minand 1fromresolv
FlowCell
anti-PmonodescriDickiperfor
StatisDat
Della Peruta et al.
Cance8898
Published OnlineFirst October 19, 2010; DOI: 10.1158/0008-5472.CAN-10-0258
noblotting and antiseras (20 × 106/1 mL) were solubilized in lysis buffer (LB) asbed (10) or directly lysed in sample buffer [40 mmol/LCl (pH 6.8), 183 mmol/L β-mercaptoethanol, 1% SDS,cerol], heated at 95°C for 5 minutes, passed through age needle to fragment DNA, resolved on SDS-PAGE,ectroblotted onto polyvinylidene difluoride membranesore Corp.). The antibodies used were antiphospho-ne [clone 4G10 (Upstate Biotechnology) and clone(Santa Cruz Biotechnology)]; anti-PTPRG P4 (26),rk-L (H-62), and anti-c-Abl (Santa Cruz Biotechnology);ti-phospho-Crk-L (Tyr207), signal transducers and acti-of transcription 5 (STAT5), and rabbit anti-phospho-(Tyr694; Cell Signaling, Boston). Bound antibodies
visualized using the enhanced chemiluminescenceion system (Amersham). Membranes were treated forutes at 65°C in 0.5 mmol/L Tris (pH 6.7), 2% SDS, andmol/L β-mercaptoethanol and washed before probingdditional antibodies.
noprecipitation with Protein G-Sepharose 4lowal protein content was assessed using Bradford assay). Cell lysates (400 µg of total protein for each sam-ere incubated with 3 µg of specific antibodies for 3at 4°C. Twenty microliters of protein G-Sepharose) for each sample were added, and the mixture wasted for 1 hour at 4°C with gentle rocking, washed,bjected to SDS-PAGE.
ssion and purification of PTPRG intracellularin and pull-down assayRG intracellular domain (ICD) and D1028A mutanto acid residues 797–1145 of NP_002832 sequence) werein the T7-based HisG-tagged vector expression pRSETitrogen) using the unique BamHI and EcoRI sites, se-ed, and expressed in BL21 (DE3) pLysS Escherichia coli,e recombinant proteins were purified. Approximatelyf PTPRG and 10 μg of enhanced green fluorescent pro-GFP) affinity purified protein were added to 500 μg ofysed in LB for 3 hours at 4°C. The beads were collected,d three times with LB, and then subjected to SDS-PAGEestern blotting with specific antibodies as described.
genic and proliferation assays2 (3 × 103 cells/mL), KYO-1 (2.4 × 103 cells/mL),-84, and MEG-01 (2 × 104 cells/mL) in MethoCult(StemCell Technologies) were transfected and trans-in 24-well plates with 0.5 mg/mL G418. After 8 days,ell was scored for the presence and number of colonies.en indicated, K562 cells were exposed to 2 μmol/L2′-deoxycytidine (DAC) for 24 hours, washed, and thenin a drug-free medium. The same experiment was re-ced in mock-transfected K562, and two independentwere stably transfected with PTPRG-AS cDNA that
arvested and plated in a 96-well plate (5 × 103 cell/
L/well) or in MethoCult as described previously. Atdicated time points, 10 μL of 5 mg/mL MTT (Sigma-samplconsid
r Res; 70(21) November 1, 2010
Research. on March 11, 202cancerres.aacrjournals.org Downloaded from
h) resuspended in sterile PBS were added, and cellslysed according to the manufacturer's instructions.bance at 570 nm was read in an ELX808iu Ultra Micro-Reader (Bio-Tek Instruments, Inc.).
rafting in nude miceivo studies using 4-week-old nu/nu Swiss mice weigh-to 22 g (Charles River) for each experimental conditionerformed exactly as described (31).
se transcription-PCR and real-timeitative PCRal RNA was isolated using TRIZOL (Invitrogen) accord-the manufacturer's indications. PTPRG cDNA expres-as analyzed by PCR and quantitative PCR (QPCR)
scribed (11). The primers used for amplification ofC-CD45, PTPRJ-CD148, PTPRE-PTPε, PTPRU-PTPμ,1-PTP1B, PTPN6-SHP-1, and PPP2R4-PP2A are shownplementary Table S1.evaluate the change of PTPRG and BCR/ABL mRNAin CML patients during follow-up, we applied thela (T − U)/(T + U) adapted from Mauri and collea-32). For each individual, the difference (T − U) be-the mRNA levels of both PTPRG and BCR/ABL ond before treatment (U) with IM was divided by thef the same values (T + U). When T equals U, theis zero, which corresponds to no change in the ex-on level between the two conditions. This ratio−1.0 when no expression is detectable in the treatedt (T = 0). When U = 0, the ratio will be +1.0. Inter-te ratio values between −1.0 and +1.0 correspond tont expression levels.
lation-specific PCRomic DNA (1 μg) was subjected to bisulfite modifica-sing the CpGenome DNA modification kit (Chemicon)ing to the manufacturer's protocol. PCR conditions95°C for 8 minutes followed by 45 cycles at 95°Cseconds and an annealing temperature of 60°C
ylation-specific PCR) or 61°C (unmethylation-specificfor 1 minute followed by a final extension at 72°C forutes. PCR products (168 bp for unmethylated PTPRG66 bp for methylated PTPRG) amplifying the region−576 to −410 relative to the transcription start wereed in a 2.5% agarose gel.
cytometry of purified cells and whole bloodlines and whole blood samples were stained with theTPRGantibody chPTPRG IgY or preimmune IgY andwithclonal anti-CD34 PE (clone AC136, Miltenyi Biotech) asbed (10). Flow cytometry was performed on a Bectonnson FACScan flow cytometer. Data analysis wasmed with FCS Express V3 software (De Novo Software).
tical analysisa analysis was performed using unpaired and one-
e t test (GraphPad Instat software). A P < 0.05 wasered statistically significant.Cancer Research
0. © 2010 American Association for Cancer

Resu
PTPRclonoK56
Cult. Ecolonigrownsion cume olinkedsion in
PTPR[PTPNPTP
positivexpresto 40%sion ban incandMkaryoc
Figureas descvolumean invaCML ceG418. R
PTPRG in Chronic Myeloid Leukemia
www.a
Published OnlineFirst October 19, 2010; DOI: 10.1158/0008-5472.CAN-10-0258
lts
G expression specifically correlates with decreasedgenic capability and growth in CML cell lines2, KYO-1, LAMA-84, and MEG-01 were plated in Metho-ach well was scored for the presence and number ofes after 8 days. Fig. 1A shows the number of colonies(top) and their volumes (bottom). Only PTPRG expres-orrelates with a reduced clonogenic capability and vol-f the colonies (Fig. 1B). The choice of the PTP panel was
either to a receptor-like structure and known expres- humanll lines were transfected with empty vector (mock), PTPRG FL, and AS cDNA andesults are percentages (%) of colonies grown compared with the control group
acrjournals.org
Research. on March 11, 202cancerres.aacrjournals.org Downloaded from
E-PTPε, and PTPRU-PTPμ) or to their role in CML1-PTP1B (4), PTPN6-SHP-1 (5), and PPP2R4-PP2A (6)].RG affects the clonogenic capability of both PTPRG-e and -negative cell lines. As shown in Fig. 1C, PTPRGsion reduced the clonogenicity in all cell lines from 31%relative to controls. Down-modulation of PTPRG expres-y transfection of an AS PTPRG construct (27) resulted inrease of colony number only in PTPRG-positive LAMA-84EG-01 cell lines. Interestingly, these cell lines are ofmega-ytic origin, and it is known that PTPRG is expressed in
megakaryocytes (16). The construct is capable to down-hematopoietic lineages (PTPRC-CD45, PTPRJ-CD148, regulate PTPRG as shown in Supplementary Table S2.
1. PTPRG expression and clonogenic capability in CML cell lines. A, colony formation assay. Cells were plated in 24-well plates in MethoCultribed. Top, clonogenic capability was evaluated as the ratio between the number of cells plated and the number of colonies developed. Bottom,of colonies (n = 3). B, reverse transcription-PCR analysis of PTPs. Expression of the indicated phosphatases. ACTB expression was used asriant control in all experiments (one representative of a minimum of two experiments). C, effect of PTPRG modulation on clonogenic capability.
, the day after, plated in MethoCult in the presence of 0.5 mg/mL(cell transfected with empty plasmid, 100%). n = 3.
Cancer Res; 70(21) November 1, 2010 8899
0. © 2010 American Association for Cancer

PTPRK562Clo
and Dmentaslightland wentiatthe ceD1028formeveryclonesWil
p210B
substrthat rand isdeadindicain K5depen
BCR/As
transfmighttaggedto affiWT aBCR/Aspecifprecipof anyproteithe Wsametainspresencomplteins a
PTPR
Figurewere chanalysismock acells (ripercentxenogrconditiowere sestrippinand Ponceau staining, which showed equal protein loading (n = 3).
Della Peruta et al.
Cance8900
Published OnlineFirst October 19, 2010; DOI: 10.1158/0008-5472.CAN-10-0258
G expression has an oncosuppressive effect in thecell line both in vitro and in vivones expressing comparable protein levels of PTPRG1028A mutant cDNA were selected (Fig. 2A; Supple-ry Table S2). Protein expression was comparable buty lower than those recorded in human monocytesas associated with the increase of the myeloid differ-ion marker CD13 (Fig. 2B). Clonogenic capability oflls was decreased in FL compared with mock- andA-transfected cells. In in vivo proliferation assay per-d in xenografted nude mice, K562 FL clone formedsmall tumors compared with mock and D1028A(Fig. 2C).
d-type (WT) PTPRG induces a reduction of total andCR-ABL-specific tyrosine phosphorylation of its directate CRKL (33–35) and of STAT5, a transcription factorepresents a downstream target of BCR/ABL (36, 37),not occurring in mock-transfected and phosphatase-(D1028A)–transfected clones (Fig. 2D). These resultste that the tumor suppressor effect of PTPRG
62 cells is mediated by interference with BCR/ABL-dent signaling.
PTPtrans
r Res; 70(21) November 1, 2010
Research. on March 11, 202cancerres.aacrjournals.org Downloaded from
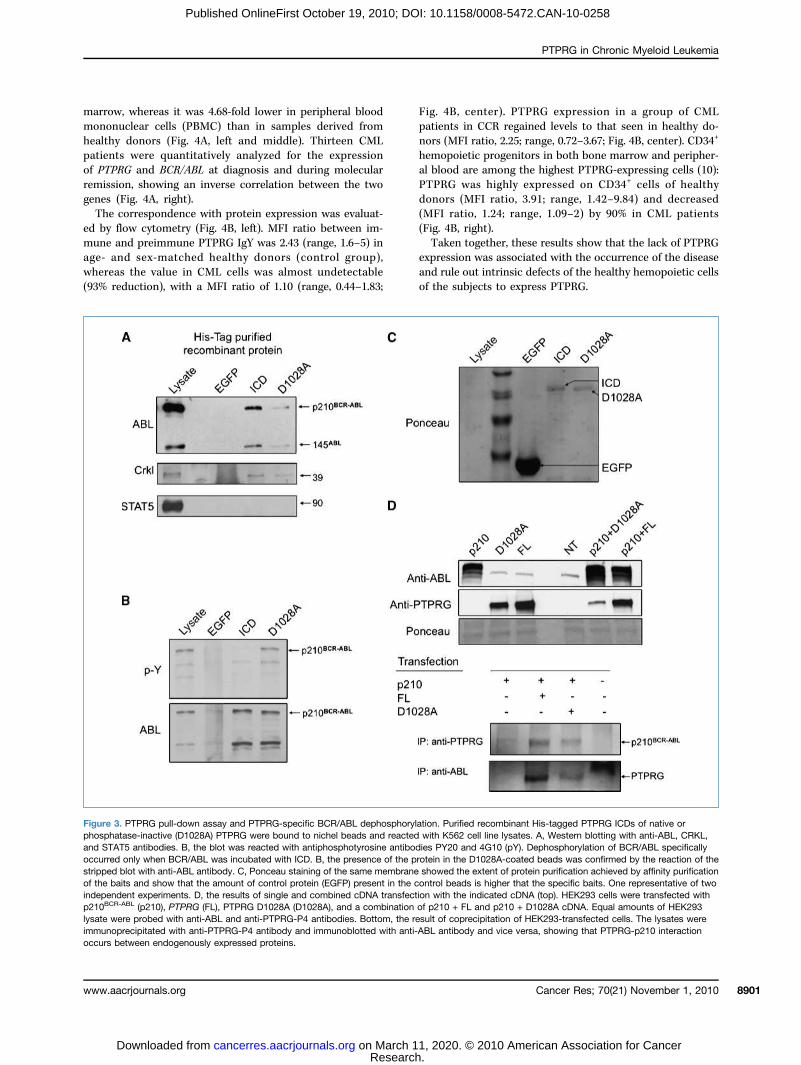
ABL is a substrate of PTPRGBCR/ABL dephosphorylation occurred in PTPRG-ected K562 (Fig. 2D), we hypothesized that BCR/ABLrepresent a direct substrate for PTPRG. Histidine-EGFP protein (mock), WT, and D1028A ICD bound
nity resin were then incubated with K562 cell lysates.nd D1028A ICD precipitated a complex containingBL, ABL, and its direct substrate CRKL (Fig. 3A). Theicity of the interaction was supported by the lack of co-itation of STAT5 with the complex as well as by the lacksignal in the lane containing larger amounts of EGFPn (mock; Fig. 3A and C). BCR/ABL precipitated fromT PTPRG ICD is specifically dephosphorylated as theprotein bound to the catalytic inactive enzyme main-its tyrosine phosphorylated status, thus ruling out thece of other tyrosine phosphatase activities within theex (Fig. 3B). The interaction also occurs when both pro-re coexpressed within HEK293 (Fig. 3D).
G expression in CML patients
2. PTPRG expression in K562 cells inhibits clonogenic capability and p210BCR-ABL signaling. A, stable transfectants (mock, FL, and D1028A cells)aracterized by immunoblotting analysis with anti-P4 antibody (arrow indicates the band corresponding to FL PTPRG) and by flow cytometricwith chPTPRG antibody (n = 4). B, level of PTPRG expression detected in blood monocytes compared with the level of expression in K562
nd FL cell lines (left). Glycophorin (top) and CD13 (bottom) expression in mock, PTPRG cDNA (FL), and PTP inactive (D1028A) transfected K562ght). C, top, clonogenic assay in MethoCult. Cells were plated at 2 × 103/mL, and after 8 d, each well was scored for the number, expressed as aage relative to the mock-transfected cells. K562 cells mock (left), PTPRG FL (middle), and PTPRG D1028A (right) are shown (n = 7). Bottom,aft assay in nu/nu mice: 10 million cells per mouse (mock-, FL-, and D1028A-transfected cells, five mice each) were inoculated for each experimentaln. Tumor volumes were recorded at the indicated time points. D, immunoblotting analysis of K562 cell lines. Same amounts of whole-cell lysatesparated by SDS-PAGE, transferred to nitrocellulose, and probed with a mixture of antiphosphotyrosine antibodies (PY99 and 4G10) and afterg with anti-ABL, anti-CRKL, anti-phospho-CRKL (p-CRKL), anti-STAT5, and phospho-STAT5 (pSTAT5) antibodies as described. Bottom, actin
RG downregulation also occurred in CML patients:cript level was 12.2-fold in Ficoll-purified bone
Cancer Research
0. © 2010 American Association for Cancer
marromonohealthpatienof PTPremissgenesThe
ed bymuneage- awhere(93% r
Fig. 4patiennors (hemopal blooPTPRdonor(MFI(Fig. 4Tak
expres
Figurephosphand SToccurrestrippedof the bindepenp210BC
lysate wimmunooccurs
PTPRG in Chronic Myeloid Leukemia
www.a
Published OnlineFirst October 19, 2010; DOI: 10.1158/0008-5472.CAN-10-0258
w, whereas it was 4.68-fold lower in peripheral bloodnuclear cells (PBMC) than in samples derived fromy donors (Fig. 4A, left and middle). Thirteen CMLts were quantitatively analyzed for the expressionRG and BCR/ABL at diagnosis and during molecularion, showing an inverse correlation between the two(Fig. 4A, right).correspondence with protein expression was evaluat-flow cytometry (Fig. 4B, left). MFI ratio between im-and preimmune PTPRG IgY was 2.43 (range, 1.6–5) innd sex-matched healthy donors (control group),
as the value in CML cells was almost undetectable and ruprecipitated with anti-PTPRG-P4 antibody and immunoblotted with anti-ABL anbetween endogenously expressed proteins.
acrjournals.org
Research. on March 11, 202cancerres.aacrjournals.org Downloaded from
B, center). PTPRG expression in a group of CMLts in CCR regained levels to that seen in healthy do-MFI ratio, 2.25; range, 0.72–3.67; Fig. 4B, center). CD34+
oietic progenitors in both bone marrow and peripher-d are among the highest PTPRG-expressing cells (10):G was highly expressed on CD34+ cells of healthys (MFI ratio, 3.91; range, 1.42–9.84) and decreasedratio, 1.24; range, 1.09–2) by 90% in CML patientsB, right).en together, these results show that the lack of PTPRGsion was associated with the occurrence of the disease
le out intrinsic defects of the healthy hemopoietic cellseduction), with a MFI ratio of 1.10 (range, 0.44–1.83; of the subjects to express PTPRG.
3. PTPRG pull-down assay and PTPRG-specific BCR/ABL dephosphorylation. Purified recombinant His-tagged PTPRG ICDs of native oratase-inactive (D1028A) PTPRG were bound to nichel beads and reacted with K562 cell line lysates. A, Western blotting with anti-ABL, CRKL,AT5 antibodies. B, the blot was reacted with antiphosphotyrosine antibodies PY20 and 4G10 (pY). Dephosphorylation of BCR/ABL specificallyd only when BCR/ABL was incubated with ICD. B, the presence of the protein in the D1028A-coated beads was confirmed by the reaction of theblot with anti-ABL antibody. C, Ponceau staining of the same membrane showed the extent of protein purification achieved by affinity purificationaits and show that the amount of control protein (EGFP) present in the control beads is higher that the specific baits. One representative of twodent experiments. D, the results of single and combined cDNA transfection with the indicated cDNA (top). HEK293 cells were transfected withR-ABL (p210), PTPRG (FL), PTPRG D1028A (D1028A), and a combination of p210 + FL and p210 + D1028A cDNA. Equal amounts of HEK293ere probed with anti-ABL and anti-PTPRG-P4 antibodies. Bottom, the result of coprecipitation of HEK293-transfected cells. The lysates were
tibody and vice versa, showing that PTPRG-p210 interaction
Cancer Res; 70(21) November 1, 2010 8901
0. © 2010 American Association for Cancer

DemePTP
beenmethy(Fig. 5and cepartiapressimid itransfence oPTPRGremovefficaccells aclone
6.5% ament
ReducexpreCpG
and alationPTPRGof a rpatien
Discu
Figureby QPCwith IMnormaliwas evmedianratio values of PTPRG antigen in CD34 cells from peripheral blood of healthy donors (control) and CML patients. Data are average geometric MFI ratiosbetween the chPTPRG-stained sample and the preimmune chicken IgY. In the absence of PTPRG antigen expression, the MFI ratio equals one.
Della Peruta et al.
Cance8902
Published OnlineFirst October 19, 2010; DOI: 10.1158/0008-5472.CAN-10-0258
thylating agents induce PTPRG expressionRG downregulation by epigenetic modification hasreported in various cancer types (18–21). The hypo-lating agent (DAC) induced reexpression of PTPRGA) followed by a marked inhibition of colony formationll proliferation/survival in semisolid and liquid medially reversed by the concomitant inhibition of PTPRG ex-on through transfection with a PTPRG AS carrying plas-n both transiently (not shown) as well as stablyected K562 cell line clones (Fig. 5B and C). The pres-f AS construct was effective in inhibiting DAC-inducedexpression as evaluated after 3 and 6 days from the
al of the drug (Fig. 5C). Flow cytometry confirmed they of the construct: the percentage of PTPRG-positive
t 3 and 6 days after DAC treatment in control K562was 13.2% and 16.2%, respectively, which dropped toIn tween
r Res; 70(21) November 1, 2010
Research. on March 11, 202cancerres.aacrjournals.org Downloaded from
nd 1.1% at 3 and 6 days, respectively, on DAC treat-of AS-transfected clones.
ed promoter methylation and recovery of PTPRGssion in a subset of patientsmethylation analysis in the same patients at diagnosis
fter successful treatment (all characterized by upregu-of PTPRG expression) indicated that the recovery ofexpression is associated with reduced methylation
egion of its promoter in a substantial fraction ofts (Fig. 5D).
ssion
4. PTPRG mRNA and protein expression in patients. A, expression of PTPRG mRNA in samples from healthy donors or CML patients was evaluatedR: bone marrow (BM; left) and PBMC (center). Right, opposite variation of PTPRG and BCR/ABL mRNA levels in 13 CML patients on treatment. The difference of mRNA levels during (T) and before (U) treatment for each patient has been divided by the sum of the same values, yieldingzed fold change values within the range (−1, +1). B, left, expression of PTPRG antigen in samples from healthy donors (control) and CML patientsaluated by flow cytometry of whole blood (gate on the left) with chPTPRG antibody; an example of the analysis is shown. Center, geometricfluorescent intensity (MFI) ratio values of the samples analyzed, including controls and CML at diagnosis and at CCR. Right, geometric MFI
+
his study, we first observed a specific association bet-loss of PTPRG expression and increased clonogenic
Cancer Research
0. © 2010 American Association for Cancer

capabothertwo Cof gr
reducof PT
FigureA, exprmediumOne repThe celafter 8 d(magnifC, samK562 ccells foD, PTPamplificwhereaunder t
PTPRG in Chronic Myeloid Leukemia
www.a
Published OnlineFirst October 19, 2010; DOI: 10.1158/0008-5472.CAN-10-0258
ility in CML cell lines that did not occur with sevenphosphatases analyzed. The observation that the
ML cell lines that express PTPRG were still capableowing in methylcellulose, although at a muchgrowthypot
s patients 6 to 11 were unmethylated. No alterations in the methylation pattern of Phe same experimental conditions.
acrjournals.org
Research. on March 11, 202cancerres.aacrjournals.org Downloaded from
ed extent, prompted us to speculate that the levelPRG expression/activity could determine the
h capability and differentiation of these cells. Thishesis was confirmed by the results of the PTPRG5. PTPRG promoter hypermethylation in K562 cell lines and primary cells. Involvement of PTPRG in the oncosuppressive effect of DAC.ession of PTPRG in K562 after 3 and 6 days of treatment with DAC; cells were exposed to 2 μmol/L DAC for 24 h and then replated in drug-free. RNA was extracted on the third and sixth days after treatment to verify PTPRG gene expression by PCR (ATCB, 22 cycles; PTPRG, 40 cycles).resentative experiment of three experiments. B, cells were transfected with mock and PTPRG AS cDNA vector and selected for G418 resistance.ls were then exposed to 2 μmol/L DAC. At 24 h after drug addition, cells were washed with drug-free medium and then replated at 3 × 103/mL;, each well was scored for colony number and expressed as a percentage relative to untreated transfected cells. Top, a detail of the colonies grownication, 10×); bottom, MTT-stained wells (n = 3). The same experiment was reproduced in transiently transfected K562 clones (mock and AS).e cells of B cultured in liquid medium. The percentage of live cells after DAC treatment was calculated, setting the value of the mock and ASell lines treated with vehicle (DMSO) to 100%. Top, MTT staining was performed at 3 and 6 d. Bottom, at the same time points, an aliquot ofr each experimental condition was analyzed for PTPRG expression by flow cytometry (reported in the text) and mRNA levels by QPCR. n = 2.RG methylation-specific PCR in CML patients. All patient (1–11) samples at diagnosis were hypermethylated for PTPRG. Examples of PCRation data are shown: M, methylated amplicon; UM, unmethylated amplicon. Patients 1 to 5 were hypermethylated at the follow-up after treatment,
TPRG were observed in non-CML disease (n = 5, data not shown)
Cancer Res; 70(21) November 1, 2010 8903
0. © 2010 American Association for Cancer

modumineddownrnogenlinesK56
the min cloing PTtotal,phospwas fothe aphospBCR/Aproteoactivadephowith PexpresCRK
hibitiorect inBCR/Aacts asphoryenzymportioof theboth tof sig(EGFPtify totion isbe deABL,(36, 3A n
the owhereof healowingexpresthe dABL mthat trecoveby PTsimilaidentitranslscriptwithcourseies. Mprincision mdition
of thiducibMe
volvedtherapposedstimuexpresvelopmpropoDow
methydescriwithtreatmCMLdepention mic druobservstill nationhyperthis ped byislandbe invanalyapproThe
this isis stillexpresleadinOve
denceindicapleteveloplevelsmal meffectThe
new player in the pathogenesis of CML and suggest it as apoten
Disclosure of Potential Conflicts of Interest
No p
Ackn
We tof the K
Della Peruta et al.
Cance8904
Published OnlineFirst October 19, 2010; DOI: 10.1158/0008-5472.CAN-10-0258
lation experiments, wherein overexpression deter-a reduction of clonogenic capability whereas
egulation was associated with an increase in clo-ic capability only in the two PTPRG-positive cellsMEG-01 and LAMA-84.2 cell line showed activity-dependent increase ofyeloid differentiation marker CD13 and decreasenogenic capability both in vitro and in vivo follow-PRG cDNA transfection, associated with a reducedBCR/ABL-, CRKL-, and STAT5-specific tyrosinehorylation. No detectable PTPRG degradationund at variance with other studies reporting that
ssociation of BCR/ABL with the serine-threoninehatase PP2A leads to SHP-1 activation followed byBL dephosphorylation and proteasome-dependentlysis (6). PTPRG likely acts through a different pathwayting myeloid differentiation and a different pattern ofsphorylation and/or protein association in comparisonP2A/SHP-1 whose mRNA, at variance with PTPRG, issed in K562 (Fig. 1B).L dephosphorylation directly links PTPRG to the in-n of BCR/ABL (35). Pull-down assay confirms a di-teraction between the ICD domain of PTPRG andBL along with its direct substrate CRKL. BCR/ABLs a bona fide substrate for PTPRG, as it is depho-lated only by precipitation with the catalytic activee. This interaction occurs most likely in the ABLn of the aberrant kinase, as suggested by the abilitybait to coprecipitate ABL, and also occurs when
argets are coexpressed in HEK293 cells. The absencenal using higher amounts of an unrelated protein) and the absence of STAT5 within the complex tes-the specificity of the interaction. This last observa-in agreement with the fact that, although known to
pendent on phosphorylation and activation by BCR/STAT5 was never reported to coprecipitate with it8, 39).ear-complete lack of expression was associated withccurrence of CML in primary patient samples,as we observed a recovery to values close to thoselthy donors for both mRNA and protein levels fol-molecular remission. This shows that the lack of
sion was not due to other factors independent ofisease status. The inverse correlation with BCR/RNA levels before and after treatment suggests
he loss of the oncogenic clone is followed by thery of a nonneoplastic hemopoiesis characterizedPRG expression. Along these lines, we show that,r to the well-established molecular analysis forfication of the t(9;22)(q34.1;q11.21) chromosomalocation (40), the measurement of the PTPRG tran-might offer a new diagnostic tool in associationthe former. This potential application needs, of, a more extensive validation by multicenter stud-oreover, we provide for the first time the proof ofple that flow cytometric analysis of PTPRG expres-
ight represent a potentially useful and unique ad-al biomarker for CML, with the advantages typical
technicby Drsacknow
r Res; 70(21) November 1, 2010
Research. on March 11, 202cancerres.aacrjournals.org Downloaded from
s application, such as ease of use, enhanced repro-ility, and cost-effectiveness.mbers of the tyrosine phosphatase family are in-in the pathogenesis of CML (4–6, 41, 42), and a
eutic strategy based on PTP activation has been pro-(43). Having identified a molecular target, ligand-lated activation of the residual PTPRG moleculessed on the surface of Ph+ myeloid blasts and the de-ent of specific chemical activators can be readily
sed.nregulation of PTPRG expression associated withlation of specific promoter regions has been recentlybed in various cancers (18–21). This result, alongthe protective effect of the AS construct on DACent, is in accordance with these findings and addsto the list of neoplastic diseases where methylation-dent PTPRG downregulation occurs. This observa-ight explain the molecular mechanism of epigenet-gs found to be active in CML (44). Even if weed this association in 55% of the patients analyzed,ot all of them show the effect. However, an associ-between PTPRG downregulation and promoter
methylation within this region was reported, despitehenomenon occurring in 27% of the patients affect-cutaneus T-cell lymphoma (20). It is likely that CpGs within other PTPRG promoter regions mightolved, and this possibility needs to be thoroughlyzed in future studies using high-throughputaches.loss of PTPRG expression in CD34+ cells indicates thatan early event in the pathogenesis of CML, although itunclear if the maintenance of critical levels of PTPRGsion can act as a “gatekeeper” in the molecular eventsg to the clinical manifestation of CML.rall, our findings provide the first compelling evi-of the tumor-suppressive effect of PTPRG in CML,ting that downregulation, but not necessarily a com-loss of PTPRG expression, is associated with the de-ment of CML and that restoration of expressionsimilar to, but even lower than those present in nor-yeloid cells seems to exert a strong oncosuppressivein Ph+ cells.se results point for the first time to PTPRG as a relevant
tial target for diagnostic and therapeutic applications.
otential conflicts of interest were disclosed.
owledgments
hank Dr. Emanuela Parmagnani for the subcloning and characterization562-B4 cell line and Dr. Maria Antonietta Fancello for her skillful
al assistance. KYO-1 and MEG-01 cell lines were kindly provided. Livia Manzella and Paolo Vigneri (University of Catania). Weledge the role of “Consorzio per gli Studi Universitari di Verona” forCancer Research
0. © 2010 American Association for Cancer

its criticfollowin
Grant
Suppin VeroIG 4667
Thepaymenadvertisthis fac
Refe1. Va
tion229
2. FadTh164
3. Meylaact
4. LaMNKon199
5. Bruph142
6. Neis fact355
7. Moonof
8. Toto
9. Soph19
10. Maγ (pre
11. Listyrcel
12. BacadeBio
13. Laphch50
14. Lubtioren
15. vanph61–
16. Vetraan
17. Watyr11
18. FuT.islaCa
19. Wa
PTPRG in Chronic Myeloid Leukemia
www.a
Published OnlineFirst October 19, 2010; DOI: 10.1158/0008-5472.CAN-10-0258
al support during the earliest phase of the study and throughout theg developments.
Support
ort for this study was provided by the Consorzio Studi Universitarina and by the Associazione Italiana per la Ricerca sul Cancro (grant Rece
OnlineF
nds in putative promoter regions in human malignant melanomas.ncer Res 2006;66:6080–6.ng JF, Dai DQ. [Difference in methylation of genomic DNA
bega
20. vancutum20
21. ChcycphRe
22. KleceJ C
23. OghuPh
24. Ohnowit9:9
25. Semyexh19
26. SoChprosid
27. Liuofin12
28. Wioftio
29. Bethemeest
30. HodruNa
31. GocaprocuCa
32. Marelidema
33. NicIdetyrBlo
34. tenTy19
35. Ha
acrjournals.org
Research. on March 11, 202cancerres.aacrjournals.org Downloaded from
costs of publication of this article were defrayed in part by thet of page charges. This article must therefore be hereby markedement in accordance with 18 U.S.C. Section 1734 solely to indicatet.
ived 01/21/2010; revised 07/28/2010; accepted 07/29/2010; publishedirst 10/19/2010.
).rencesrdiman JW, Harris NL, Brunning RD. The World Health Organiza-(WHO) classification of the myeloid neoplasms. Blood 2002;100:2–302.erl S, Talpaz M, Estrov Z, O'Brien S, Kurzrock R, Kantarjian HM.
e biology of chronic myeloid leukemia. N Engl J Med 1999;341:–72.yn MA III, Wilson MB, Abdi FA, et al. Src family kinases phosphor-te the Bcr-Abl SH3-2 region and modulate Bcr-Abl transformingivity. J Biol Chem 2006;281:30907–16.ontagne KR, Jr., Flint AJ, Franza BR, Jr., Pandergast AM, Tonks
. Protein tyrosine phosphatase 1B antagonizes signalling bycoprotein tyrosine kinase p210 bcr-abl in vivo. Mol Cell Biol8;18:2965–75.echer-Encke B, Griffin JD, Neel BG, Lorenz U. Role of the tyrosineosphatase SHP-1 in K562 cell differentiation. Leukemia 2001;15:4–32.viani P, Santhanam R, Trotta R, et al. The tumor suppressor PP2Aunctionally inactivated in blast crisis CML through the inhibitoryivity of the BCR/ABL-regulated SET protein. Cancer Cell 2005;8:–68.tiwala T, Majumder S, Ghoshal K, et al. PTPROt inactivates thecogenic fusion protein BCR/ABL and suppresses transformationK562 cells. J Biol Chem 2009;284:455–64.nks NK. Protein tyrosine phosphatases: from genes, to function,disease. Nat Rev Mol Cell Biol 2006;7:833–46.rio C, Melotti P, D'Arcangelo D, et al. Receptor protein tyrosineosphatase γ, Ptpγ, regulates hematopoietic differentiation. Blood97;90:49–57.fficini A, Vezzalini M, Zamai L, et al. Protein tyrosine phosphatasePTPγ) is a novel leukocyte marker highly expressed by CD34+cursors. Biomarker Insights 2007;2:217–24.sandrini D, Vermi W, Vezzalini M, et al. Receptor-type proteinosine phosphatase γ (PTP{γ}), a new identifier for myeloid dendriticls and specialized macrophages. Blood 2006;108:4223–31.rnea G, Silvennoinen O, Shaanan B, et al. Identification of arbonic anhydrase-like domain in the extracellular region of RPTPγfines a new subfamily of receptor tyrosine phosphatases. Mol Celll 1993;13:1497–506.Forgia S, Morse B, Levy J, et al. Receptor protein-tyrosineosphatase γ is a candidate tumor suppressor gene at humanromosome region 3p21. Proc Natl Acad Sci U S A 1991;88:36–40.inski J, Hadaczek P, Podolski J, et al. Common regions of dele-
n in chromosome regions 3p12 and 3p14.2 in primary clear cellal carcinomas. Cancer Res 1994;54:3710–3.Niekerk CC, Poels LG. Reduced expression of protein tyrosine
osphatase γ in lung and ovarian tumors. Cancer Lett 1999;137:73.zzalini M, Mombello A, Menestrina F, et al. Expression ofnsmembrane protein tyrosine phosphatase γ (PTPγ) in normald neoplastic human tissues. Histopathology 2007;50:615–28.ng Z, Shen D, Parsons DW, et al. Mutational analysis of theosine phosphatome in colorectal cancers. Science 2004;304:64–6.ruta J, Nobeyama Y, Umebayashi Y, Otsuka F, Kikuchi K, UshijimaSilencing of peroxiredoxin 2 and aberrant methylation of 33 CpG
tween gastric primary cancer and lymph nodes with metastaticstric cancer]. Zhonghua Yi Xue Za Zhi 2006;86:536–9.Doorn R, Zoutman WH, Dijkman R, et al. Epigenetic profiling of
taneous T-cell lymphoma: promoter hypermethylation of multipleor suppressor genes including BCL7a, PTPRG, p73. J Clin Oncol
05;23:3886–96.eung AK, Lung HL, Hung SC, et al. Functional analysis of a cellle-associated, tumor-suppressive gene, protein tyrosine phos-atase receptor type G, in nasopharyngeal carcinoma. Cancers 2008;68:8137–45.in E, Ben-Bassat H, Neumann H, et al. Properties of the K562ll line, derived from a patient with chronic myeloid leukemia. Intancer 1976;18:421–31.ura M, Morishima Y, Ohno R, et al. Establishment of a novelman megakaryoblastic leukemia cell line, MEG-01, with positiveiladelphia chromosome. Blood 1985;66:1384–92.kubo T, Kamamoto T, Kita K, Hiraoka A, Yoshida Y, Uchino H. Avel Ph1 chromosome positive cell line established from a patienth chronic myelogenous leukemia in blastic crisis. Leuk Res 1985;21–6.igneurin D, Champelovier P, Mouchiroud G, et al. Human chroniceloid leukemic cell line with positive Philadelphia chromosomeibits megakaryocytic and erythroid characteristics. Exp Hematol87;15:822–32.rio C, Mendrola J, Lou Z, LaForgia S, Croce CM, Huebner K.aracterization of the receptor protein tyrosine phosphatase geneduct PTPγ: binding and activation by triphosphorylated nucleo-es. Cancer Res 1995;55:4855–64.S, Sugimoto Y, Sorio C, Tecchio C, Lin YC. Function analysisestrogenically regulated protein tyrosine phosphatase γ (PTPγ)human breast cancer cell line MCF-7. Oncogene 2004;23:56–62.tt O, Sand K, Pekrun A. Butyrate-induced erythroid differentiationhuman K562 leukemia cells involves inhibition of ERK and activa-n of p38 MAP kinase pathways. Blood 2000;95:2391–6.lhacene N, Maulon L, Guerin S, et al. Differential expression ofKell blood group and CD10 antigens: two related membranetallopeptidases during differentiation of K562 cells by phorboler and hemin. FASEB J 1998;12:531–9.ck RA, Miller AD. Retrovirus-mediated transfer and expression ofg resistance genes in human haematopoietic progenitor cells.ture 1986;320:275–7.las JM, Arndt K, Etienne C, et al. SKI-606, a 4-anilino-3-quinoline-rbonitrile dual inhibitor of Src and Abl kinases, is a potent anti-liferative agent against chronic myelogenous leukemia cells inlture and causes regression of K562 xenografts in nude mice.ncer Res 2003;63:375–81.uri P, Scarpa A, Nascimbeni AC, et al. Identification of proteinseased by pancreatic cancer cells by multidimensional proteinntification technology: a strategy for identification of novel cancerrkers. FASEB J 2005;19:1125–7.hols GL, Raines MA, Vera JC, Lacomis L, Tempst P, Golde DW.ntification of CRKL as the constitutively phosphorylated 39-kDosine phosphoprotein in chronic myelogenous leukemia cells.od 1994;84:2912–8.Hoeve J, Arlinghaus RB, Guo JQ, Heisterkamp N, Groffen J.
rosine phosphorylation of CRKL in Philadelphia+ leukemia. Blood94;84:1731–6.milton A, Elrick L, Myssina S, et al. BCR-ABL activity and its
Cancer Res; 70(21) November 1, 2010 8905
0. © 2010 American Association for Cancer

resCrk20
36. Hokinmitra20
37. Quchr
38. FraofSta
39. Ilartyric S
40. Co
tre11
41. Chmytra
42. ShimTCfus
43. Neforph20
44. Ok
Della Peruta et al.
Cance8906
Published OnlineFirst October 19, 2010; DOI: 10.1158/0008-5472.CAN-10-0258
ponse to drugs can be determined in CD34+ CML stem cells byL phosphorylation status using flow cytometry. Leukemia 2006;:1035–9.rita M, Andreu EJ, Benito A, et al. Blockade of the Bcr-Ablase activity induces apoptosis of chronic myelogenous leuke-a cells by suppressing signal transducer and activator ofnscription 5-dependent expression of Bcl-xL. J Exp Med00;191:977–84.intas-Cardama A, Cortes J. Molecular biology of bcr-abl1-positiveonic myeloid leukemia. Blood 2009;113:1619–30.nk DA, Varticovski L. BCR/abl leads to the constitutive activationStat proteins, and shares an epitope with tyrosine phosphorylatedts. Leukemia 1996;10:1724–30.ia RL, Jr., Van Etten RA. P210 and P190(BCR/ABL) induce the
osine phosphorylation and DNA binding activity of multiple specif-TAT family members. J Biol Chem 1996;271:31704–10.tta CV, Bueso-Ramos CE. New insights into the pathobiology anddeaccCa
r Res; 70(21) November 1, 2010
Research. on March 11, 202cancerres.aacrjournals.org Downloaded from
atment of chronic myelogenous leukemia. Ann Diagn Pathol 2007;:68–78.ien W, Tidow N, Williamson EA, et al. Characterization of aeloid tyrosine phosphatase, Lyp, and its role in the Bcr-Abl signalnsduction pathway. J Biol Chem 2003;278:27413–20.imizu T, Miyakawa Y, Iwata S, et al. A novel mechanism foratinib mesylate (STI571) resistance in CML cell line KT-1: role of-PTP in modulating signals downstream from the BCR-ABLion protein. Exp Hematol 2004;32:1057–63.viani P, Santhanam R, Oaks JJ, et al. FTY720, a new alternativetreating blast crisis chronic myelogenous leukemia and Philadel-ia chromosome-positive acute lymphocytic leukemia. J Clin Invest07;117:2408–21.i Y, Kantarjian HM, Gharibyan V, et al. Phase II study of low-dosecitabine in combination with imatinib mesylate in patients with
elerated or myeloid blastic phase of chronic myelogenous leukemia.ncer 2007;109:899–906.Cancer Research
0. © 2010 American Association for Cancer

2010;70:8896-8906. Published OnlineFirst October 19, 2010.Cancer Res Marco Della Peruta, Giovanni Martinelli, Elisabetta Moratti, et al. Chronic Myeloid LeukemiaTumor Suppressor Gene Specifically Downregulated in
Is a FunctionalγProtein Tyrosine Phosphatase Receptor Type
Updated version
10.1158/0008-5472.CAN-10-0258doi:
Access the most recent version of this article at:
Material
Supplementary
http://cancerres.aacrjournals.org/content/suppl/2010/10/18/0008-5472.CAN-10-0258.DC1
Access the most recent supplemental material at:
Cited articles
http://cancerres.aacrjournals.org/content/70/21/8896.full#ref-list-1
This article cites 44 articles, 23 of which you can access for free at:
Citing articles
http://cancerres.aacrjournals.org/content/70/21/8896.full#related-urls
This article has been cited by 3 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://cancerres.aacrjournals.org/content/70/21/8896To request permission to re-use all or part of this article, use this link
Research. on March 11, 2020. © 2010 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 19, 2010; DOI: 10.1158/0008-5472.CAN-10-0258