cdk inhibitors upregulate bh3-only proteins to sensitize ... filetherapeutics, targets, and chemical...
TRANSCRIPT

Therapeutics, Targets, and Chemical Biology
CDK Inhibitors Upregulate BH3-Only Proteins to SensitizeHuman Myeloma Cells to BH3 Mimetic Therapies
Shuang Chen1, Yun Dai1, Xin-Yan Pei1, Jennifer Myers1, Li Wang1, Lora B. Kramer1, Mandy Garnett1,Daniella M. Schwartz1, Florence Su1, Gary L. Simmons1, Justin D. Richey1, Dustin G. Larsen1,Paul Dent2, Robert Z. Orlowski5, and Steven Grant1,3,4
AbstractBH3mimetic drugs induce cell death by antagonizing the activity of antiapoptotic Bcl-2 family proteins. Cyclin-
dependent kinase (CDK) inhibitors that function as transcriptional repressors downregulate the Bcl-2 familymember Mcl-1 and increase the activity of selective BH3mimetics that fail to target this protein. In this study, wedetermined whether CDK inhibitors potentiate the activity of pan-BH3 mimetics directly neutralizing Mcl-1.Specifically, we evaluated interactions between the prototypical pan-CDK inhibitor flavopiridol and the pan-BH3mimetic obatoclax in multiple myeloma (MM) cells in which Mcl-1 is critical for survival. Coadministration offlavopiridol and obatoclax synergistically triggered apoptosis in both drug-na€�ve and drug-resistant MM cells.Mechanistic investigations revealed that flavopiridol inhibited Mcl-1 transcription but increased transcriptionof Bim and its binding to Bcl-2/Bcl-xL. Obatoclax prevented Mcl-1 recovery and caused release of Bim fromBcl-2/Bcl-xL and Mcl-1, accompanied by activation of Bax/Bak. Whether administered singly or in combinationwith obatoclax, flavopiridol also induced upregulation of multiple BH3-only proteins, including BimEL, BimL,Noxa, and Bik/NBK. Notably, short hairpin RNA knockdown of Bim or Noxa abrogated lethality triggered by theflavopiridol/obatoclax combination in vitro and in vivo. Together, our findings show that CDK inhibitionpotentiates pan-BH3 mimetic activity through a cooperative mechanism involving upregulation of BH3-onlyproteins with coordinate downregulation of their antiapoptotic counterparts. These findings have immediateimplications for the clinical trial design of BH3 mimetic-based therapies that are presently being studiedintensively for the treatment of diverse hematopoietic malignancies, including lethal multiple myeloma. CancerRes; 72(16); 4225–37. �2012 AACR.
IntroductionMultiple myeloma (MM) is an incurable accumulative dis-
ease of plasma cells characterized by dysregulation of Bclfamily members (1). These apoptosis regulatory proteins aredivided into pro- and antiapoptotic groups. The former con-sists of multidomain proteins (e.g., Bak and Bax) and BH3-onlyproteins (e.g., Bim, Bid, Puma, Noxa, Bad, Bik, Bmf, and Hrk,
etc.). The latter includes multidomain proteins, for example,Bcl-2, Bcl-xL, and Mcl-1 (2). Although Bax and Bak are abso-lutely required for apoptosis, BH3-only proteins, which convertnoxious stimuli into death signals, consist of "activators" (e.g.,Bim) and "sensitizers/derepressors" (e.g., Noxa, Bik; ref. 2).Evidence implicating BH3-only proteins in anticancer agent-induced apoptosis (3, 4) prompted the development ofBH3 mimetics that bind to and inactivate antiapoptotic Bclproteins (5).
One such agent, ABT-737, binds avidly to Bcl-2/Bcl-xL, butnot Mcl-1 (6). Consequently, relative levels of Bcl-2/Bcl-xLversus Mcl-1 determine susceptibility to this agent (7). Mcl-1 is highly expressed in MM (e.g., in 51% patients at diagnosisand 81% at relapse), and high Mcl-1 expression correlates withpoor clinical outcome (8). Mcl-1 also plays an important role inresistance to agents such as bortezomib (9). Recently, the novelpan-BH3 mimetic obatoclax has been developed, which inaddition to other antiapoptotic proteins, antagonizes theactivity of Mcl-1 in various tumors types (10, 11), includinghematologic malignancies such as MM. Preclinical in vitrostudies inMM showed single-agent activity and additivity withother agents, but limited in vivo bioactivity when administeredalone (12).
Cyclin-dependent kinases (CDK) regulate cell-cycle progres-sion and transcription (13). Pan-Cdk inhibitors such as
Authors' Affiliations: 1Division of Hematology/Oncology, Department ofMedicine,Departments of 2Neurosurgery and 3Biochemistry, VirginiaCom-monwealth University and the Massey Cancer Center; 4Institute of Molec-ularMedicine,Richmond, Virginia; and 5Lymphoma/Myeloma,University ofTexas, MD Anderson Cancer Center, Houston, Texas
Note: Supplementary data for this article are available at Cancer ResearchOnline (http://cancerres.aacrjournals.org/).
S. Chan and Y. Dai have made equal contributions to this article.
Corresponding Authors: Steven Grant, Division of Hematology/Oncolo-gy, Virginia Commonwealth University, PO Box 980035, Richmond, VA23298. Phone: 804-828-5211; Fax: 804-828-8079; E-mail:[email protected]; andYunDai, VirginiaCommonwealthUniversity,MasseyCancerCenter, Room234GoodwinResearchBuilding, 401College Street,Richmond, VA 23298. Phone: 804-828-5168; Fax: 804-827-3781; E-mail:[email protected]
doi: 10.1158/0008-5472.CAN-12-1118
�2012 American Association for Cancer Research.
CancerResearch
www.aacrjournals.org 4225
on April 13, 2017. © 2012 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 12, 2012; DOI: 10.1158/0008-5472.CAN-12-1118

flavopiridol (alvocidib) act in part by inhibiting Cdk9, a kinaseinvolved in RNA polymerase II (Pol II)-mediated transcriptionelongation (13). Consequently, Cdk inhibitors block gene tran-scription and downregulate short-lived proteins includingMcl-1, promoting apoptosis (14, 15). Clinical trials have sug-gested activity for flavopiridol singly or in combination withother agents in patients with hematopoietic malignancies,including MM (16–18). Recently, several new-generationpan-Cdk inhibitors (e.g., CYC202, SCH727965), which alsotarget Cdk9, have entered clinical trials (13).
Although pan-Cdk inhibitors have been shown to potentiateABT-737 lethality in transformed cells by downregulating Mcl-1 (7), it is unknown whether synergistic interactions wouldoccur with pan-BH3 mimetics such as obatoclax, which bindto/inactivate Mcl-1 (10). To address this question, we exam-ined interactions between the pan-Cdk inhibitor flavopiridoland obatoclax in human MM cells. Here we report thatflavopiridol synergistically increases obatoclax lethality indiverse MM cells, including those resistant to novel agents,in the presence of stromal cell factors, and in primary CD138þ
MM samples, but not in their normal counterparts. Signifi-cantly, obatoclax/flavopiridol coadministration, in sharp con-trast to obatoclax alone, displays marked in vivo activity andincreases survival in multiple murine systems. From a mech-anistic standpoint, the unexpected upregulation of multipleBH3-only proteins, including BimEL, BimL, Noxa, and Bik/NBK, cooperates with downregulation of antiapoptotic pro-teins (e.g., Mcl-1, Bcl-xL) to play a significant functional role inlethality. Collectively, these findings provide proof-of-principlefor a novel anti-MM strategy in which pan-Cdk inhibitors arecombined with pan-BH3 mimetics and highlight the criticalimportance of interplay between pro- and antiapoptotic pro-teins in synergistic interactions between such agents.
Materials and MethodsCells and reagents
Human MM U266 and RPMI8226 cells were obtained fromAmerican Type Culture Collection (ATCC) and maintained asbefore (19). Both were authenticated (Basic STR ProfilingService, ATCC 135-X) by ATCC immediately after thisstudy was completed. Bortezomib-resistant cells (PS-R) weregenerated by continuously culturing U266 cells in increasingconcentrations of bortezomib (beginning at 0.5 nmol/Land increasing in stepwise increments of 0.2 nmol/L) until20 nmol/L, and maintained in medium containing 15 nmol/Lbortezomib. A revlimid-resistant RPMI8226 (R10R) cell linewas similarly established and maintained in 10 mmol/L revli-mid (20). Dexamethasone-sensitive (MM.1S) and -resistant(MM.1R) cell lines were provided by Dr Steven T. Rosen(Northwestern University, Chicago, IL). U266/Mcl-1 andRPMI8226/Bcl-xL cells were established by stably transfectingfull-length humanMcl-1 andBcl-xL cDNA, respectively (19). Allexperiments used logarithmically growing cells (3—5 � 105
cells/mL). MycoAlert (Lonza) assays were carried out, showingthat all cell lines were free of Mycoplasma contamination.
Bonemarrow sampleswere obtainedwith informed consentaccording to the Declaration of Helsinki and Virginia Com-monwealth University Institutional Review Board approval
from 4 patients with MM undergoing routine diagnosticaspirations. CD138þ cells were separated using a MACS mag-netic separation technique (Miltenyi Biotech). Normal CD34þ
hematopoietic progenitor cells were isolated from 2 cord blood(CB) samples; purity and viability were >90%, by flow cyto-metry and trypan blue exclusion, respectively.
Obatoclax, flavopiridol, and SCH 727965 were generouslyprovided by GeminX Pharmaceuticals, Sanofi-Aventis, andMerck, respectively, and by the National Cancer Institute, NIH.Cycloheximide (CHX) andMG-132were purchased fromSigmaand Calbiochem respectively, dissolved in dimethyl sulfoxide(DMSO), aliquoted, and stored at �20�C. In all experiments,final DMSO concentrations did not exceed 0.1%. Recombinanthuman interleukin (IL)-6, IGF-1, BAFF, and APRIL wereobtained from PeproTech.
Procedures for in vitro studiesFor procedures related to flow cytometry, terminal deox-
ynucleotidyl transferase–mediated dUTP nick end labeling(TUNEL) staining, quantitative real-time PCR (qRT-PCR),immunoblot, coimmunoprecipitation, subcellular fraction-ation, Bak and Bax conformational change, and RNA interfer-ence see Supplementary Materials and Methods (7).
Animal studiesAnimal studieswere approved by theVirginia Commonwealth
University The Institutional Animal Care and Use Committee,and carried out in accordance with the U.S. Department ofAgriculture and Department of Health and Human Services, andthe NIH. Three mouse models were used in this study.
Model #1—subcutaneous (s.c.) flank murine model. AthymicNCr-nu/nu mice (NCI) were s.c. innoculated in the right rearflank with 5 � 106 RPMI8226 cells stably transfected with aconstruct encoding luciferase. Treatment was administeredafter luciferase activity was detected.
Model #2—s.c. dual-side flank murine model. NOD/SCID-gmice (Jackson Laboratories) were s.c. inoculated in 2 oppositeflanks with 1 � 107 U266 cells stably transfected with con-structs encoding short hairpin RNA (shRNA) targeting eitherBim (shBim, left flank) or scrambled sequence negative control(shNC, right flank).
Model #3—intravenous (i.v.) orthotopic murine model. NOD/SCID-g mice (Jackson Laboratories) were i.v. injected with5� 106 U266 cells stably transfected with constructs encodingluciferase.
Obatoclax mesylate (GX15-070MS) was freshly reconstitutedwith 5% dextrose for injection (USP) and administered viaintramuscular (i.m.) or intraperitoneal (i.p.) injection. flavopir-idol inDMSOwas diluted in 0.9% saline and administered via i.p.injection. Control animals were injected with equal volumes ofvehicle. Mice were monitored for tumor growth every other dayvisually or with the use of an IVIS 200 imaging system (XenogenCorporation). Measurement of animal body weight was carriedout every other day throughout the study to monitor toxicity.Tumor volumes were calculated using the formula (L � W2)/2,with L and W representing length and width respectively, andwhen tumor size reached 2,000 mm3, mice were euthanizedin accordance with institutional guidelines.
Chen et al.
Cancer Res; 72(16) August 15, 2012 Cancer Research4226
on April 13, 2017. © 2012 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 12, 2012; DOI: 10.1158/0008-5472.CAN-12-1118
Statistical analysisValues represent the means� SD for at least 3 independent
experiments carried out in triplicate. The significance ofdifferences between experimental variables was determinedusing the Student t test or 1-way ANOVA with Tukey–Kramermultiple comparisons test. The significance of P values was<0.05 (�),<0.01 (��), or<0.001 (���) wherever indicated. Analysisof synergism was carried out according to median dose effectanalysis using the software Calcusyn (Biosoft). Kaplan–Meieranalysis of mouse survival or hind-leg paralysis was carried outusing IBM SPSS Statistics software.
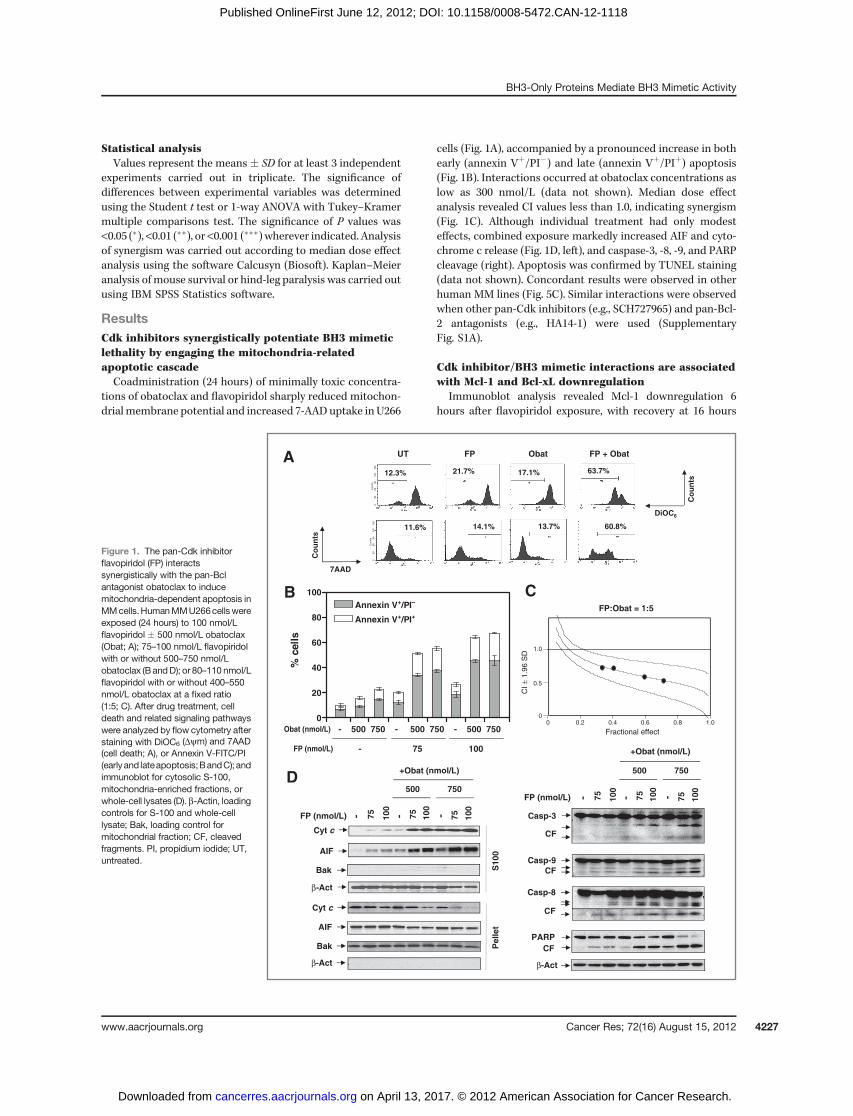
ResultsCdk inhibitors synergistically potentiate BH3 mimeticlethality by engaging the mitochondria-relatedapoptotic cascadeCoadministration (24 hours) of minimally toxic concentra-
tions of obatoclax and flavopiridol sharply reduced mitochon-drialmembrane potential and increased 7-AAD uptake in U266
cells (Fig. 1A), accompanied by a pronounced increase in bothearly (annexin Vþ/PI�) and late (annexin Vþ/PIþ) apoptosis(Fig. 1B). Interactions occurred at obatoclax concentrations aslow as 300 nmol/L (data not shown). Median dose effectanalysis revealed CI values less than 1.0, indicating synergism(Fig. 1C). Although individual treatment had only modesteffects, combined exposure markedly increased AIF and cyto-chrome c release (Fig. 1D, left), and caspase-3, -8, -9, and PARPcleavage (right). Apoptosis was confirmed by TUNEL staining(data not shown). Concordant results were observed in otherhuman MM lines (Fig. 5C). Similar interactions were observedwhen other pan-Cdk inhibitors (e.g., SCH727965) and pan-Bcl-2 antagonists (e.g., HA14-1) were used (SupplementaryFig. S1A).
Cdk inhibitor/BH3 mimetic interactions are associatedwith Mcl-1 and Bcl-xL downregulation
Immunoblot analysis revealed Mcl-1 downregulation 6hours after flavopiridol exposure, with recovery at 16 hours
Figure 1. The pan-Cdk inhibitorflavopiridol (FP) interactssynergistically with the pan-Bclantagonist obatoclax to inducemitochondria-dependent apoptosis inMMcells.HumanMMU266cellswereexposed (24 hours) to 100 nmol/Lflavopiridol � 500 nmol/L obatoclax(Obat; A); 75–100 nmol/L flavopiridolwith or without 500–750 nmol/Lobatoclax (B andD); or 80–110 nmol/Lflavopiridol with or without 400–550nmol/L obatoclax at a fixed ratio(1:5; C). After drug treatment, celldeath and related signaling pathwayswere analyzed by flow cytometry afterstaining with DiOC6 (Dym) and 7AAD(cell death; A), or Annexin V-FITC/PI(earlyand lateapoptosis;BandC); andimmunoblot for cytosolic S-100,mitochondria-enriched fractions, orwhole-cell lysates (D). b-Actin, loadingcontrols for S-100 and whole-celllysate; Bak, loading control formitochondrial fraction; CF, cleavedfragments. PI, propidium iodide; UT,untreated.
A
0
20
40
60
80
100
Annexin V+/PI–
Annexin V+/PI+
- 500 750 - 500 750 - 500 750
- 75 100
Obat (nmol/L)
FP (nmol/L)
% c
ell
s
B
β-Act β-Act
β-Act
Casp-9
CF
Casp-8
CF
PARP
CF
Casp-3
CF
75
10
0
- 75
10
0
- 75
10
0
-FP (nmol/L)
500 750
+Obat (nmol/L)
FP:Obat = 1:5
75
10
0
- 75
10
0
- 75
10
0
-FP (nmol/L)
Cyt c
AIF
Pe
lle
t S
10
0
500 750
+Obat (nmol/L)
C
D
Bak
Cyt c
AIF
Bak
Cou
nts
030
60
9
0
120
1
50
Cou
nts
030
60
9
0
120
1
50
12.3%
11.6%
21.7% 17.1% 63.7%
14.1% 13.7% 60.8%
UT FP Obat FP + Obat
7AAD
Co
un
ts
DiOC6
Co
un
ts
0 0.2 0.4 0.6 0.8 1.00
0.5
1.0
Fractional effect
CI ±
1.9
6 S
D
BH3-Only Proteins Mediate BH3 Mimetic Activity
www.aacrjournals.org Cancer Res; 72(16) August 15, 2012 4227
on April 13, 2017. © 2012 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 12, 2012; DOI: 10.1158/0008-5472.CAN-12-1118
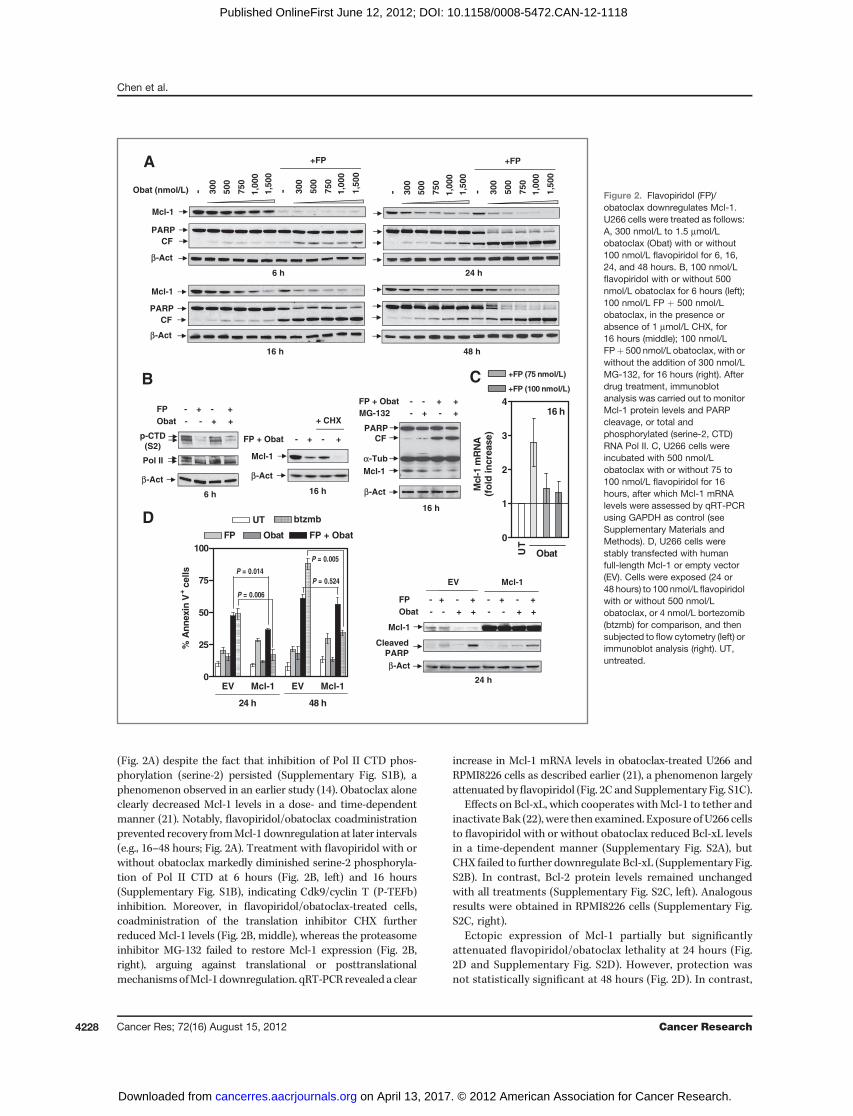
(Fig. 2A) despite the fact that inhibition of Pol II CTD phos-phorylation (serine-2) persisted (Supplementary Fig. S1B), aphenomenon observed in an earlier study (14). Obatoclax aloneclearly decreased Mcl-1 levels in a dose- and time-dependentmanner (21). Notably, flavopiridol/obatoclax coadministrationprevented recovery fromMcl-1 downregulation at later intervals(e.g., 16–48 hours; Fig. 2A). Treatment with flavopiridol with orwithout obatoclax markedly diminished serine-2 phosphoryla-tion of Pol II CTD at 6 hours (Fig. 2B, left) and 16 hours(Supplementary Fig. S1B), indicating Cdk9/cyclin T (P-TEFb)inhibition. Moreover, in flavopiridol/obatoclax-treated cells,coadministration of the translation inhibitor CHX furtherreduced Mcl-1 levels (Fig. 2B, middle), whereas the proteasomeinhibitor MG-132 failed to restore Mcl-1 expression (Fig. 2B,right), arguing against translational or posttranslationalmechanismsofMcl-1 downregulation. qRT-PCR revealed a clear
increase in Mcl-1 mRNA levels in obatoclax-treated U266 andRPMI8226 cells as described earlier (21), a phenomenon largelyattenuated byflavopiridol (Fig. 2C and Supplementary Fig. S1C).
Effects on Bcl-xL, which cooperates withMcl-1 to tether andinactivate Bak (22), were then examined. Exposure ofU266 cellsto flavopiridol with or without obatoclax reduced Bcl-xL levelsin a time-dependent manner (Supplementary Fig. S2A), butCHX failed to further downregulate Bcl-xL (Supplementary Fig.S2B). In contrast, Bcl-2 protein levels remained unchangedwith all treatments (Supplementary Fig. S2C, left). Analogousresults were obtained in RPMI8226 cells (Supplementary Fig.S2C, right).
Ectopic expression of Mcl-1 partially but significantlyattenuated flavopiridol/obatoclax lethality at 24 hours (Fig.2D and Supplementary Fig. S2D). However, protection wasnot statistically significant at 48 hours (Fig. 2D). In contrast,
Mcl-1
PARP
CF
ββ-Act
Obat (nmol/L) - 30
0
50
0
75
0
1,0
00
1,5
00
- 30
0
50
0
75
0
1,0
00
1,5
00
+FP
Mcl-1
PARP
CF
β-Act
A
6 h
16 h
24 h
48 h
B
FP + Obat - - + +
MG-132 - + - +
Mcl-1
α-Tub
PARP
CFp-CTD
(S2)
FP - + - +
Obat - - + +
Pol II
6 h
β-Act
FP + Obat - + - +
+ CHX
Mcl-1
β-Act
- 30
0
50
0
75
0
1,0
00
1,5
00
- 30
0
50
0
75
0
1,0
00
1,5
00
+FP
16 h
16 h
0
1
2
3
416 h
ObatUT
+FP (75 nmol/L)
+FP (100 nmol/L)
Mcl-
1 m
RN
A(f
old
in
cre
ase)
C
β-Act
FP - + - + - + - +
Obat - - + + - - + +
Mcl-1
β-Act
Cleaved
PARP
EV Mcl-1
D
EV Mcl-1 EV Mcl-10
25
50
75
100
UT
FP Obat FP + Obat
24 h 48 h
btzmb
P = 0.014
P = 0.006
P = 0.524
P = 0.005
% A
nn
exin
V+ c
ells
24 h
Figure 2. Flavopiridol (FP)/obatoclax downregulates Mcl-1.U266 cells were treated as follows:A, 300 nmol/L to 1.5 mmol/Lobatoclax (Obat) with or without100 nmol/L flavopiridol for 6, 16,24, and 48 hours. B, 100 nmol/Lflavopiridol with or without 500nmol/L obatoclax for 6 hours (left);100 nmol/L FP þ 500 nmol/Lobatoclax, in the presence orabsence of 1 mmol/L CHX, for16 hours (middle); 100 nmol/LFPþ 500 nmol/L obatoclax, with orwithout the addition of 300 nmol/LMG-132, for 16 hours (right). Afterdrug treatment, immunoblotanalysis was carried out to monitorMcl-1 protein levels and PARPcleavage, or total andphosphorylated (serine-2, CTD)RNA Pol II. C, U266 cells wereincubated with 500 nmol/Lobatoclax with or without 75 to100 nmol/L flavopiridol for 16hours, after which Mcl-1 mRNAlevels were assessed by qRT-PCRusing GAPDH as control (seeSupplementary Materials andMethods). D, U266 cells werestably transfected with humanfull-length Mcl-1 or empty vector(EV). Cells were exposed (24 or48 hours) to 100 nmol/L flavopiridolwith or without 500 nmol/Lobatoclax, or 4 nmol/L bortezomib(btzmb) for comparison, and thensubjected to flow cytometry (left) orimmunoblot analysis (right). UT,untreated.
Chen et al.
Cancer Res; 72(16) August 15, 2012 Cancer Research4228
on April 13, 2017. © 2012 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 12, 2012; DOI: 10.1158/0008-5472.CAN-12-1118
cells overexpressing Mcl-1 were substantially resistant tobortezomib at both 24 and 48 hours (Fig. 2D), consistentwith previous reports (9). Bcl-xL overexpression partially butsignificantly protected cells from flavopiridol/obatoclaxlethality at both 24 and 48 hours (Supplementary Fig.S2E). Together, these findings suggest that Mcl-1 and Bcl-xL downregulation plays a significant but limited functionalrole in flavopiridol/obatoclax lethality.
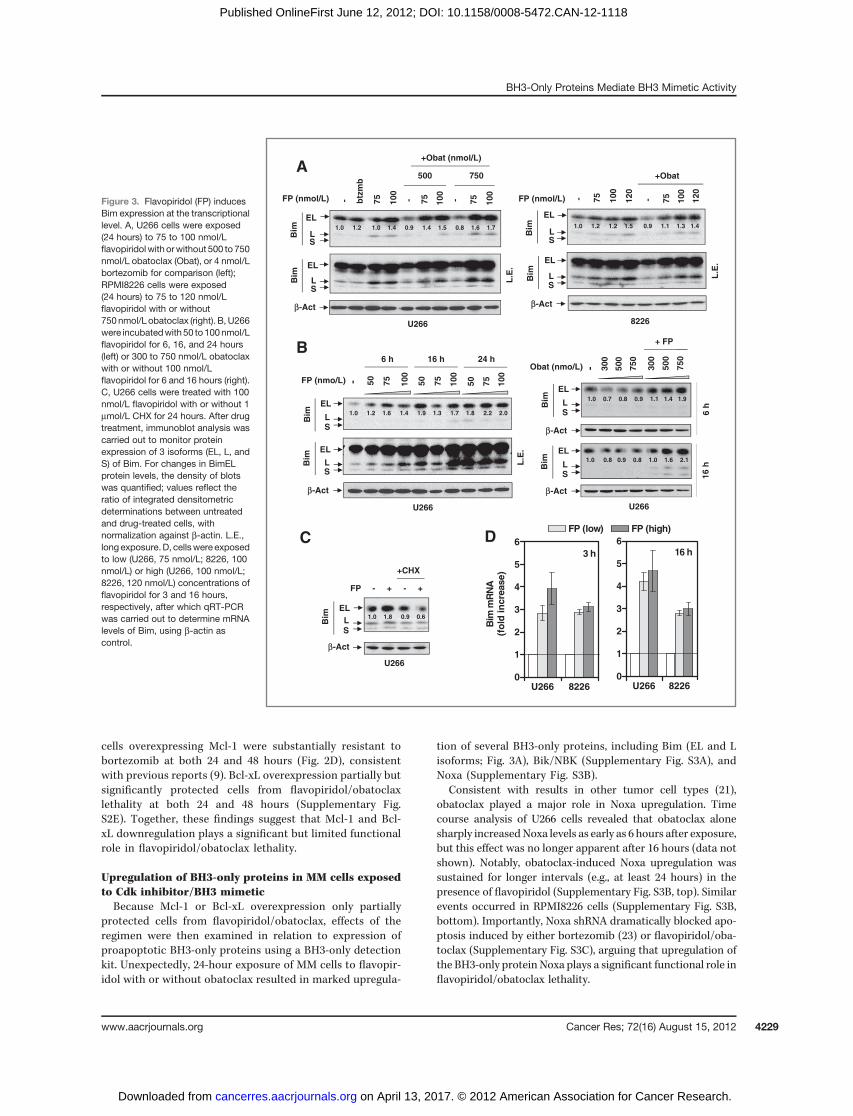
Upregulation of BH3-only proteins in MM cells exposedto Cdk inhibitor/BH3 mimeticBecause Mcl-1 or Bcl-xL overexpression only partially
protected cells from flavopiridol/obatoclax, effects of theregimen were then examined in relation to expression ofproapoptotic BH3-only proteins using a BH3-only detectionkit. Unexpectedly, 24-hour exposure of MM cells to flavopir-idol with or without obatoclax resulted in marked upregula-
tion of several BH3-only proteins, including Bim (EL and Lisoforms; Fig. 3A), Bik/NBK (Supplementary Fig. S3A), andNoxa (Supplementary Fig. S3B).
Consistent with results in other tumor cell types (21),obatoclax played a major role in Noxa upregulation. Timecourse analysis of U266 cells revealed that obatoclax alonesharply increasedNoxa levels as early as 6 hours after exposure,but this effect was no longer apparent after 16 hours (data notshown). Notably, obatoclax-induced Noxa upregulation wassustained for longer intervals (e.g., at least 24 hours) in thepresence of flavopiridol (Supplementary Fig. S3B, top). Similarevents occurred in RPMI8226 cells (Supplementary Fig. S3B,bottom). Importantly, Noxa shRNA dramatically blocked apo-ptosis induced by either bortezomib (23) or flavopiridol/oba-toclax (Supplementary Fig. S3C), arguing that upregulation ofthe BH3-only protein Noxa plays a significant functional role inflavopiridol/obatoclax lethality.
Figure 3. Flavopiridol (FP) inducesBim expression at the transcriptionallevel. A, U266 cells were exposed(24 hours) to 75 to 100 nmol/Lflavopiridol with orwithout 500 to750nmol/L obatoclax (Obat), or 4 nmol/Lbortezomib for comparison (left);RPMI8226 cells were exposed(24 hours) to 75 to 120 nmol/Lflavopiridol with or without750 nmol/L obatoclax (right). B, U266were incubatedwith50 to100nmol/Lflavopiridol for 6, 16, and 24 hours(left) or 300 to 750 nmol/L obatoclaxwith or without 100 nmol/Lflavopiridol for 6 and 16 hours (right).C, U266 cells were treated with 100nmol/L flavopiridol with or without 1mmol/L CHX for 24 hours. After drugtreatment, immunoblot analysis wascarried out to monitor proteinexpression of 3 isoforms (EL, L, andS) of Bim. For changes in BimELprotein levels, the density of blotswas quantified; values reflect theratio of integrated densitometricdeterminations between untreatedand drug-treated cells, withnormalization against b-actin. L.E.,longexposure.D, cellswere exposedto low (U266, 75 nmol/L; 8226, 100nmol/L) or high (U266, 100 nmol/L;8226, 120 nmol/L) concentrations offlavopiridol for 3 and 16 hours,respectively, after which qRT-PCRwas carried out to determine mRNAlevels of Bim, using b-actin ascontrol.
L.E
.
U266
EL
Bim
SL
A
EL
Bim
SL
+Obat
FP (nmol/L) 12
0
12
0
EL
SL
EL
SL L
.E.
ββ-Act
8226
C
B- btz
mb
500 750
75
10
0
- 75
10
0
- 75
10
0
FP (nmol/L)
+Obat (nmol/L)
- 75
10
0
- 75
10
0
Bim
Bim
EL
Bim
S
L
FP - + - +
+CHX
β-Act
U266 82260
1
2
3
4
5
6
FP (low)
3 h
FP (high)
Bim
mR
NA
(fo
ld in
cre
ase)
D
U266
U266 82260
1
2
3
4
5
616 h
FP (nmo/L)
6 h 16 h 24 h- 50
75
10
0
50
75
10
0
50
75
10
0
L.E
.
EL
β-Act
SL
EL
SL
Bim
Bim
β-Act
Obat (nmo/L) - 30
0
50
0
75
0
30
0
50
0
75
0
+ FP
EL
SLB
im
EL
SLB
im
β-Act
β-Act
6 h
16
h
U266 U266
1.0 1.2 1.0 1.4 0.9 1.4 1.5 0.8 1.6 1.7 1.0 1.2 1.2 1.5 0.9 1.1 1.3 1.4
1.0 1.2 1.6 1.4 1.9 1.3 1.7 1.8 2.2 2.0
1.0 0.7 0.8 0.9 1.1 1.4 1.9
1.0 0.8 0.9 0.8 1.0 1.6 2.1
1.0 1.8 0.9 0.6
BH3-Only Proteins Mediate BH3 Mimetic Activity
www.aacrjournals.org Cancer Res; 72(16) August 15, 2012 4229
on April 13, 2017. © 2012 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 12, 2012; DOI: 10.1158/0008-5472.CAN-12-1118
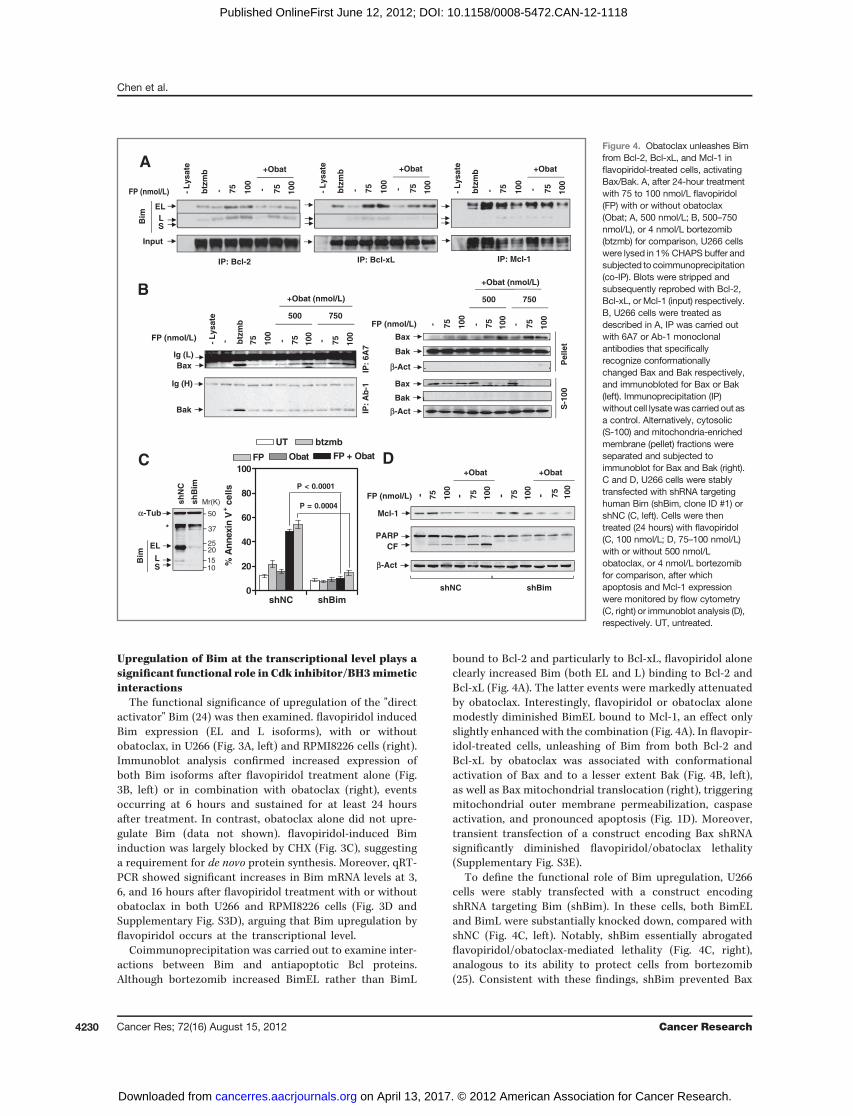
Upregulation of Bim at the transcriptional level plays asignificant functional role in Cdk inhibitor/BH3mimeticinteractions
The functional significance of upregulation of the "directactivator" Bim (24) was then examined. flavopiridol inducedBim expression (EL and L isoforms), with or withoutobatoclax, in U266 (Fig. 3A, left) and RPMI8226 cells (right).Immunoblot analysis confirmed increased expression ofboth Bim isoforms after flavopiridol treatment alone (Fig.3B, left) or in combination with obatoclax (right), eventsoccurring at 6 hours and sustained for at least 24 hoursafter treatment. In contrast, obatoclax alone did not upre-gulate Bim (data not shown). flavopiridol-induced Biminduction was largely blocked by CHX (Fig. 3C), suggestinga requirement for de novo protein synthesis. Moreover, qRT-PCR showed significant increases in Bim mRNA levels at 3,6, and 16 hours after flavopiridol treatment with or withoutobatoclax in both U266 and RPMI8226 cells (Fig. 3D andSupplementary Fig. S3D), arguing that Bim upregulation byflavopiridol occurs at the transcriptional level.
Coimmunoprecipitation was carried out to examine inter-actions between Bim and antiapoptotic Bcl proteins.Although bortezomib increased BimEL rather than BimL
bound to Bcl-2 and particularly to Bcl-xL, flavopiridol aloneclearly increased Bim (both EL and L) binding to Bcl-2 andBcl-xL (Fig. 4A). The latter events were markedly attenuatedby obatoclax. Interestingly, flavopiridol or obatoclax alonemodestly diminished BimEL bound to Mcl-1, an effect onlyslightly enhanced with the combination (Fig. 4A). In flavopir-idol-treated cells, unleashing of Bim from both Bcl-2 andBcl-xL by obatoclax was associated with conformationalactivation of Bax and to a lesser extent Bak (Fig. 4B, left),as well as Bax mitochondrial translocation (right), triggeringmitochondrial outer membrane permeabilization, caspaseactivation, and pronounced apoptosis (Fig. 1D). Moreover,transient transfection of a construct encoding Bax shRNAsignificantly diminished flavopiridol/obatoclax lethality(Supplementary Fig. S3E).
To define the functional role of Bim upregulation, U266cells were stably transfected with a construct encodingshRNA targeting Bim (shBim). In these cells, both BimELand BimL were substantially knocked down, compared withshNC (Fig. 4C, left). Notably, shBim essentially abrogatedflavopiridol/obatoclax-mediated lethality (Fig. 4C, right),analogous to its ability to protect cells from bortezomib(25). Consistent with these findings, shBim prevented Bax
50
2025
37
1510
D+Obat +Obat
FP (nmol/L) - 75
10
0
- 75
10
0
- 75
10
0
- 75
10
0ββ-Act
shNC shBim
PARP
CF
Mcl-1
EL
Bim
SL
A
-btz
mb
-L
ys
ate
75
10
0
- 75
10
0
FP (nmol/L)
+Obat
Input
B
Bax
Bax
Bak
S-1
00
P
ell
et
Bak
β-Act
β-ActIP:
6A
7
- btz
mb
-L
ys
ate
Ig (H)
IP:
Ab
-1
Bak
500 750
75
10
0
- 75
10
0
- 75
10
0FP (nmol/L)
Ig (L)
Bax
+Obat (nmol/L) 500 750
75
10
0
- 75
10
0
- 75
10
0
FP (nmol/L)
+Obat (nmol/L)
C
-
EL
α-Tub
sh
Bim
sh
NC
LS
*
shNC shBim0
20
40
60
80
100
UT
FP Obat FP + Obat
btzmb
P < 0.0001
P = 0.0004
% A
nn
exin
V+ c
ells
Bim
Mr(K)
-btz
mb
-L
ys
ate
75
10
0
- 75
10
0
+Obat
-btz
mb
-L
ys
ate
75
10
0
- 75
10
0
+Obat
IP: Bcl-2 IP: Bcl-xL IP: Mcl-1
Figure 4. Obatoclax unleashes Bimfrom Bcl-2, Bcl-xL, and Mcl-1 inflavopiridol-treated cells, activatingBax/Bak. A, after 24-hour treatmentwith 75 to 100 nmol/L flavopiridol(FP) with or without obatoclax(Obat; A, 500 nmol/L; B, 500–750nmol/L), or 4 nmol/L bortezomib(btzmb) for comparison, U266 cellswere lysed in 1%CHAPS buffer andsubjected to coimmunoprecipitation(co-IP). Blots were stripped andsubsequently reprobed with Bcl-2,Bcl-xL, or Mcl-1 (input) respectively.B, U266 cells were treated asdescribed in A, IP was carried outwith 6A7 or Ab-1 monoclonalantibodies that specificallyrecognize conformationallychanged Bax and Bak respectively,and immunobloted for Bax or Bak(left). Immunoprecipitation (IP)without cell lysatewas carried out asa control. Alternatively, cytosolic(S-100) and mitochondria-enrichedmembrane (pellet) fractions wereseparated and subjected toimmunoblot for Bax and Bak (right).C and D, U266 cells were stablytransfected with shRNA targetinghuman Bim (shBim, clone ID #1) orshNC (C, left). Cells were thentreated (24 hours) with flavopiridol(C, 100 nmol/L; D, 75–100 nmol/L)with or without 500 nmol/Lobatoclax, or 4 nmol/L bortezomibfor comparison, after whichapoptosis and Mcl-1 expressionwere monitored by flow cytometry(C, right) or immunoblot analysis (D),respectively. UT, untreated.
Chen et al.
Cancer Res; 72(16) August 15, 2012 Cancer Research4230
on April 13, 2017. © 2012 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 12, 2012; DOI: 10.1158/0008-5472.CAN-12-1118

conformational change and translocation (SupplementaryFig. S3F), caspase activation, and PARP cleavage (Fig. 4D)induced by flavopiridol/obatoclax. However, coexposure toflavopiridol/obatoclax induced nearly equivalent Mcl-1downregulation in both shNC and shBim cells (Fig. 4D).Analogous results were obtained when another Bim shRNAwas used (Supplementary Fig. S3C). Together, these findingsraise the possibility that unleashing of upregulated Bim fromantiapoptotic proteins (e.g., Bcl-2, Bcl-xL, and Mcl-1) byobatoclax contributes to synergistic interactions. They alsoargue that in the setting of downregulation of antiapoptoticproteins, upregulation of BH3-only proteins such as Bim playa critical functional role in lethality.
TheCdk inhibitor/BH3mimetic regimen is active againstMM cells displaying conventional or novel forms of drugresistance, as well as primary MM cellsIn addition to drug resistance because of Bcl family dysre-
gulation, microenvironmental factors also confer resistance inMM (26). To address these issues, U266 cells were cultured inthe presence of HS-5 cells (a human bone marrow stromal cellline; ref. 27), HS-5-conditionalmedium (CM), or both. AlthoughCM slightly reduced Bim levels, coculture with HS-5 markedlydownregulatedBim (Fig. 5A). Notably, HS-5 plus CMessentiallyabolished Bim expression, raising the possibility that Bimdownregulation represents a mechanism underlying stromalcell–mediated drug resistance (28). Importantly, neither HS-5with or without CM prevented flavopiridol/obatoclax lethality(P > 0.05, Fig. 5B, left). Furthermore, addition of IL-6, BAFF,APRIL, or IGF-1 also failed to attenuate flavopiridol/obatoclaxlethality (P > 0.05, Fig. 5B, right), suggesting that the flavopir-idol/obatoclax regimen overcomes drug resistance related tomicroenvironmental factors (e.g., stromal cells, cytokines, andgrowth factors).Dexamethasone-resistant (MM.1R) and revlimid-resistant
(R10R) cells exhibited roughly equivalent sensitivity to flavo-piridol/obatoclax, compared with their drug-na€�ve counter-parts (MM.1S and RPMI8226) respectively (Fig. 5C). Interest-ingly, bortezomib-resistant U266 cells (PS-R), which wereresistant to 20 nmol/L bortezomib (Fig. 5D, bottom), displayeda clear increase in Mcl-1 levels, accompanied by a dramaticreduction in Bim expression (24), particularly the EL isoform(top). Significantly, PS-R cells exhibited no cross-resistance toflavopiridol/obatoclax, compared with parental cells (Fig. 5D,bottom), suggesting that MM cells exhibiting either conven-tional or novel forms of drug resistance remain fully suscep-tible to this regimen.Although sensitivity to individual agents varied between
primary CD138þ MM specimens isolated from differentpatients, cotreatment with flavopiridol/obatoclax sharplyincreased cell death (Fig. 5E). However, toxicity towardCD138� bone marrow cells was minimal in all samples. Inter-estingly, despite differences between samples, CD138þ cellsdisplayed relatively higher levels of Bim compared with theirCD138� counterparts (data not shown; ref. 29). Furthermore,flavopiridol or obatoclax alone displayed only modest toxicitytoward normal CB CD34þ cells, whereas combined treatmentdid not increase lethality (Fig. 5E), suggesting that this regimen
is active against and relatively selective toward primary MMcells.
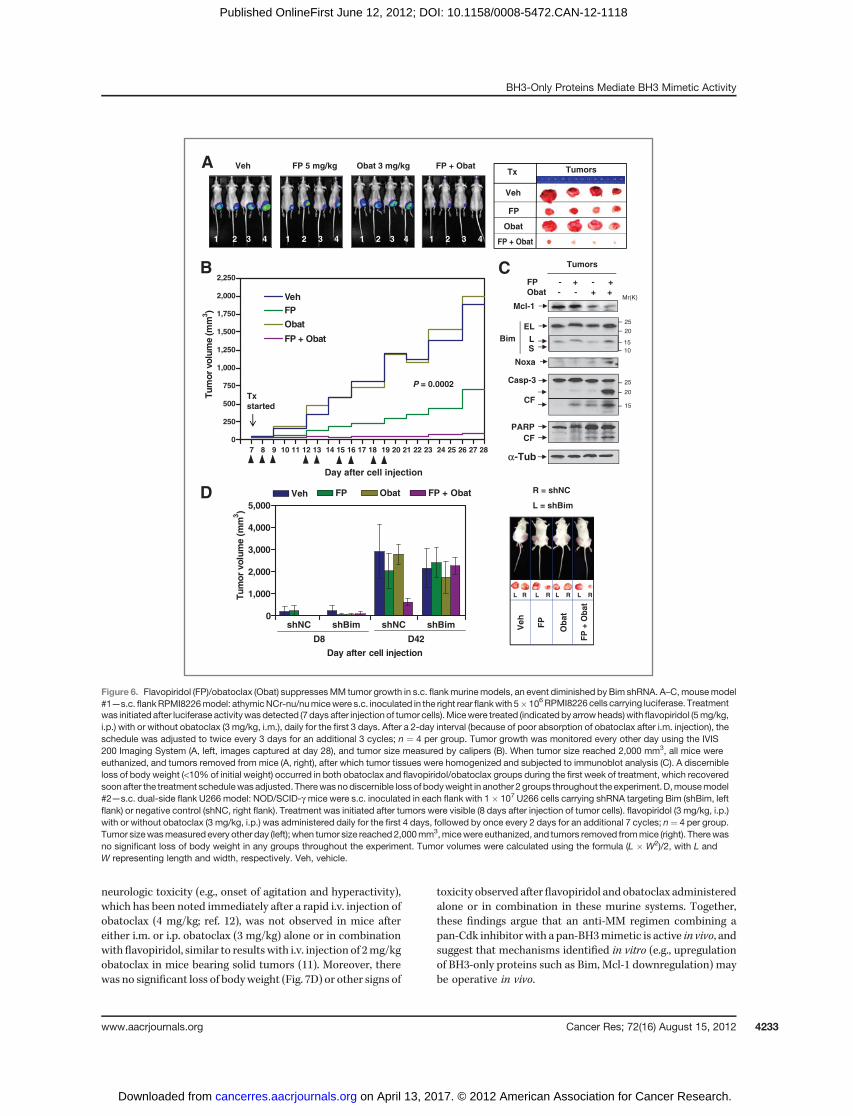
The Cdk inhibitor/BH3 mimetic regimen displaysmarked in vivo antitumor activity through a Bim-dependent mechanism
Obatoclax reportedly lacked in vivo single-agent bioactivityin mice bearing s.c. KMS12PE human MM tumors (12). Todetermine whether the flavopiridol/obatoclax regimen exhi-bits in vivo activity, athymic nude mice were inoculated in theflank with RPMI8226 cells carrying a luciferase gene. Whentumors became visible, mice were treated with flavopiridol (5mg/kg, i.p.) with or without obatoclax (3 mg/kg, i.m.). Consis-tent with previous reports (12), obatoclax (3 mg/kg) alone hadno effect on tumor growth, manifested by luciferase activity(Fig. 6A, left). However, whereas flavopiridol (5 mg/kg) aloneexerted modest but discernible effects, combined treatmentsubstantially suppressed tumor growth. The size of tumorsexcised from mice confirmed pronounced tumor growth sup-pression with combined treatment (Fig. 6A, right). Moreover,tumor size measurements yielded concordant results (P ¼0.0002; Fig. 6B). Interestingly, immunoblot analysis of tumortissues revealed that flavopiridol/obatoclax coadministrationdownregulated Mcl-1 and upregulated Bim (particularly the Lisoform) and Noxa, accompanied by caspase-3 activation andPARP cleavage (Fig. 6C), consistent with in vitro observations.
To determine whether BH3-only protein upregulation (e.g.,Bim) plays a significant functional role in flavopiridol/obato-clax lethality in vivo, NOD/SCID-g mice were inoculated s.c.with U266 cells stably transfected with shBim or shNC respec-tively (Fig. 4C) in each flank, after which flavopiridol (3 mg/kg,i.p.) with orwithout obatoclax (3mg/kg, i.p.) was administered.flavopiridol/obatoclax coadministration markedly suppressedgrowth of shNC tumors (Fig. 6D, right flank), analogous toresults observed in the previously described flank model (Fig.6A–C). Notably, whereas a slight reduction in tumor size wasobserved in the obatoclax group, no obvious growth suppres-sion was observed by flavopiridol alone or with obatoclax inshBim tumors (Fig. 6D, left flank), showing an importantfunctional role for Bim in flavopiridol/obatoclax lethalityin vivo.
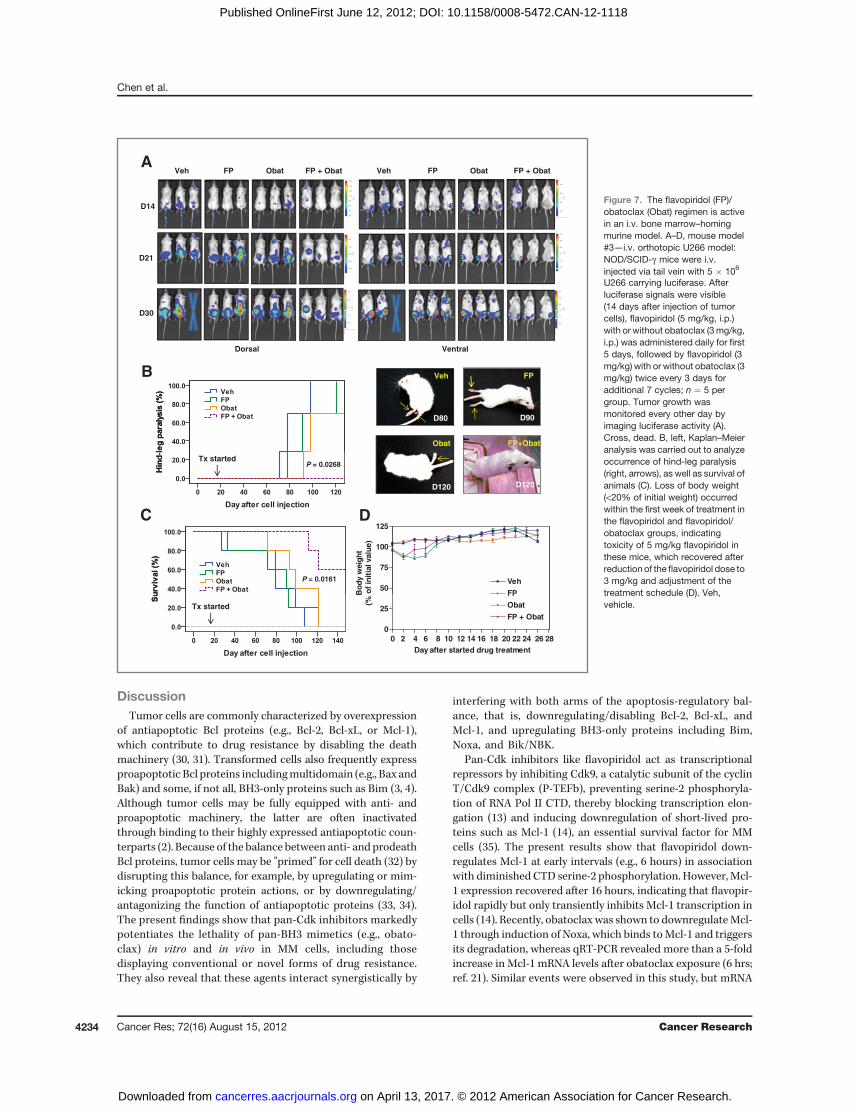
The bone marrow microenvironment plays a critical role insurvival, growth, and drug resistance of MM cells (26). Theactivity of the flavopiridol/obatoclax regimen was thereforeassessed in an animal model in which human MM cells formbone marrow lesions, leading to bone disease at late intervals.In this orthotopic murine model, NOD/SCID-g mice were i.v.injected with U266 cells stably transfected with a luciferasegene, after which homing and growth of tumor cells weredynamically monitored by imaging luciferase activity. Notably,U266 cells homed to bone marrow and then formed lesions atskeletal sites (Fig. 7A), without detectable lesions in otherorgans, findings confirmed by IHC staining for human CD138(data not shown). At later intervals (9–13 weeks after cellinjection), inoculated mice displayed hind-leg paralysis (Fig.7B, right), a classic indicator of bone disease. After luciferasesignals were visible, flavopiridol (5 mg/kg, i.p.) with or withoutobatoclax (3 mg/kg, i.p.) was administered daily for 5 days,
BH3-Only Proteins Mediate BH3 Mimetic Activity
www.aacrjournals.org Cancer Res; 72(16) August 15, 2012 4231
on April 13, 2017. © 2012 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 12, 2012; DOI: 10.1158/0008-5472.CAN-12-1118
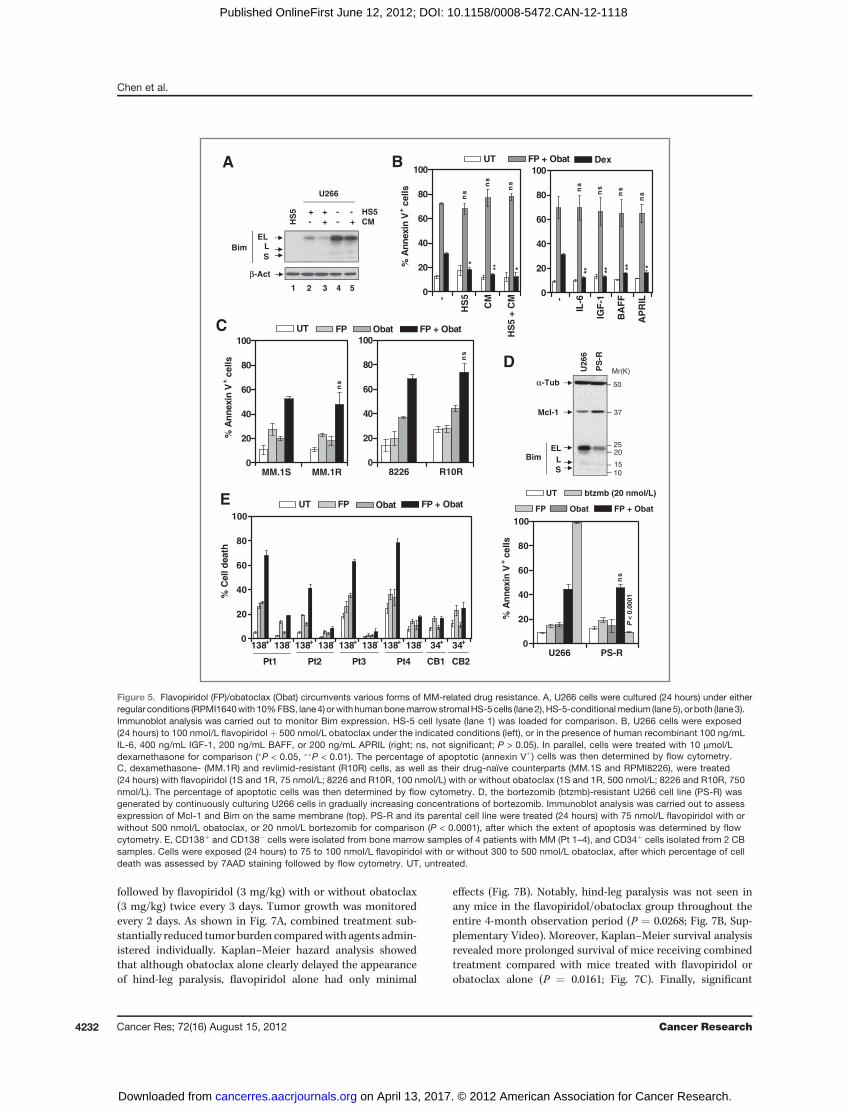
followed by flavopiridol (3 mg/kg) with or without obatoclax(3 mg/kg) twice every 3 days. Tumor growth was monitoredevery 2 days. As shown in Fig. 7A, combined treatment sub-stantially reduced tumor burden comparedwith agents admin-istered individually. Kaplan–Meier hazard analysis showedthat although obatoclax alone clearly delayed the appearanceof hind-leg paralysis, flavopiridol alone had only minimal
effects (Fig. 7B). Notably, hind-leg paralysis was not seen inany mice in the flavopiridol/obatoclax group throughout theentire 4-month observation period (P ¼ 0.0268; Fig. 7B, Sup-plementary Video). Moreover, Kaplan–Meier survival analysisrevealed more prolonged survival of mice receiving combinedtreatment compared with mice treated with flavopiridol orobatoclax alone (P ¼ 0.0161; Fig. 7C). Finally, significant
A
-
IL-6
IGF
-1
BA
FF
AP
RIL
0
20
40
60
80
100
UT FP + Obat Dex
ns
**
ns
**
ns
**
ns
*
-
HS
5
CM
HS
5 +
CM
0
20
40
60
80
100
% A
nn
exin
V+ c
ells
ns
*
ns
ns
** *
EL
Bim L
S
HS
5 + + - - HS5
- + - + CM
U266
β-Act
2025
15
37
50
10
Mcl-1
α-Tub
U2
66
PS
-R
ELBim
S
L
U266 PS-R0
20
40
60
80
100
UT
FP Obat FP + Obat
btzmb (20 nmol/L)
% A
nn
exin
V+ c
ells
P <
0.0
001
ns
DMr(K)
B
138+
138-
138+
138-
138+
138-
138+
138-
34+
34+0
20
40
60
80
100
UT FP Obat FP + Obat
Pt1 Pt2 Pt3 Pt4 CB1 CB2
% C
ell d
eath
C
E
MM.1S MM.1R0
20
40
60
80
100
UT FP Obat FP + Obat
% A
nn
exin
V+ c
ells
ns
8226 R10R0
20
40
60
80
100
ns
1 2 3 4 5
Figure 5. Flavopiridol (FP)/obatoclax (Obat) circumvents various forms of MM-related drug resistance. A, U266 cells were cultured (24 hours) under eitherregular conditions (RPMI1640with 10%FBS, lane4) orwith humanbonemarrowstromalHS-5cells (lane2),HS-5-conditionalmedium (lane5), or both (lane3).Immunoblot analysis was carried out to monitor Bim expression. HS-5 cell lysate (lane 1) was loaded for comparison. B, U266 cells were exposed(24 hours) to 100 nmol/L flavopiridol þ 500 nmol/L obatoclax under the indicated conditions (left), or in the presence of human recombinant 100 ng/mLIL-6, 400 ng/mL IGF-1, 200 ng/mL BAFF, or 200 ng/mL APRIL (right; ns, not significant; P > 0.05). In parallel, cells were treated with 10 mmol/Ldexamethasone for comparison (�P < 0.05, ��P < 0.01). The percentage of apoptotic (annexin Vþ) cells was then determined by flow cytometry.C, dexamethasone- (MM.1R) and revlimid-resistant (R10R) cells, as well as their drug-naïve counterparts (MM.1S and RPMI8226), were treated(24 hours) with flavopiridol (1S and 1R, 75 nmol/L; 8226 and R10R, 100 nmol/L) with or without obatoclax (1S and 1R, 500 nmol/L; 8226 and R10R, 750nmol/L). The percentage of apoptotic cells was then determined by flow cytometry. D, the bortezomib (btzmb)-resistant U266 cell line (PS-R) wasgenerated by continuously culturing U266 cells in gradually increasing concentrations of bortezomib. Immunoblot analysis was carried out to assessexpression of Mcl-1 and Bim on the same membrane (top). PS-R and its parental cell line were treated (24 hours) with 75 nmol/L flavopiridol with orwithout 500 nmol/L obatoclax, or 20 nmol/L bortezomib for comparison (P < 0.0001), after which the extent of apoptosis was determined by flowcytometry. E, CD138þ and CD138� cells were isolated from bone marrow samples of 4 patients with MM (Pt 1–4), and CD34þ cells isolated from 2 CBsamples. Cells were exposed (24 hours) to 75 to 100 nmol/L flavopiridol with or without 300 to 500 nmol/L obatoclax, after which percentage of celldeath was assessed by 7AAD staining followed by flow cytometry. UT, untreated.
Chen et al.
Cancer Res; 72(16) August 15, 2012 Cancer Research4232
on April 13, 2017. © 2012 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 12, 2012; DOI: 10.1158/0008-5472.CAN-12-1118
neurologic toxicity (e.g., onset of agitation and hyperactivity),which has been noted immediately after a rapid i.v. injection ofobatoclax (4 mg/kg; ref. 12), was not observed in mice aftereither i.m. or i.p. obatoclax (3 mg/kg) alone or in combinationwithflavopiridol, similar to results with i.v. injection of 2mg/kgobatoclax in mice bearing solid tumors (11). Moreover, therewas no significant loss of bodyweight (Fig. 7D) or other signs of
toxicity observed afterflavopiridol and obatoclax administeredalone or in combination in these murine systems. Together,these findings argue that an anti-MM regimen combining apan-Cdk inhibitor with a pan-BH3mimetic is active in vivo, andsuggest that mechanisms identified in vitro (e.g., upregulationof BH3-only proteins such as Bim, Mcl-1 downregulation) maybe operative in vivo.
10
25
20
15
20
25
15
A Veh FP 5 mg/kg Obat 3 mg/kg FP + Obat
B
0
250
500
750
1,000
1,250
1,500
1,750
2,000
2,250
Day after cell injection
Veh
FP
Obat
FP + Obat
7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28
Tu
mo
r vo
lum
e (
mm
3)
α-Tub
Mcl-1
EL
BimSL
Noxa
Casp-3
CF
PARP
CF
C
Mr(K)
Veh
FP
Obat
FP + Obat
Tx
Veh
FP
Ob
at
FP
+ O
ba
t
R = shNC
L = shBim
1 2 3 4 1 2 3 4 1 2 3 4 1 2 3 4
Tumors
shNC shBim shNC shBim0
1,000
2,000
3,000
4,000
5,000
Veh FP Obat FP + Obat
D8 D42
Day after cell injection
Tu
mo
r vo
lum
e (
mm
3)
D
Tx
started
Tumors
P = 0.0002
FP - + - +
Obat - - + +
L R L R L R L R
Figure 6. Flavopiridol (FP)/obatoclax (Obat) suppressesMM tumor growth in s.c. flankmurinemodels, an event diminished by Bim shRNA. A–C,mousemodel#1—s.c. flankRPMI8226model: athymicNCr-nu/numicewere s.c. inoculated in the right rear flankwith 5�106RPMI8226 cells carrying luciferase. Treatmentwas initiated after luciferase activitywas detected (7days after injection of tumor cells).Micewere treated (indicated by arrowheads)with flavopiridol (5mg/kg,i.p.) with or without obatoclax (3 mg/kg, i.m.), daily for the first 3 days. After a 2-day interval (because of poor absorption of obatoclax after i.m. injection), theschedule was adjusted to twice every 3 days for an additional 3 cycles; n ¼ 4 per group. Tumor growth was monitored every other day using the IVIS200 Imaging System (A, left, images captured at day 28), and tumor size measured by calipers (B). When tumor size reached 2,000 mm3, all mice wereeuthanized, and tumors removed from mice (A, right), after which tumor tissues were homogenized and subjected to immunoblot analysis (C). A discernibleloss of body weight (<10% of initial weight) occurred in both obatoclax and flavopiridol/obatoclax groups during the first week of treatment, which recoveredsoonafter the treatment schedulewas adjusted. Therewasnodiscernible loss of bodyweight in another 2 groups throughout the experiment. D,mousemodel#2—s.c. dual-side flank U266 model: NOD/SCID-g mice were s.c. inoculated in each flank with 1� 107 U266 cells carrying shRNA targeting Bim (shBim, leftflank) or negative control (shNC, right flank). Treatment was initiated after tumors were visible (8 days after injection of tumor cells). flavopiridol (3 mg/kg, i.p.)with or without obatoclax (3 mg/kg, i.p.) was administered daily for the first 4 days, followed by once every 2 days for an additional 7 cycles; n¼ 4 per group.Tumor sizewasmeasured every other day (left); when tumor size reached 2,000mm3,micewere euthanized, and tumors removed frommice (right). Therewasno significant loss of body weight in any groups throughout the experiment. Tumor volumes were calculated using the formula (L � W2)/2, with L andW representing length and width, respectively. Veh, vehicle.
BH3-Only Proteins Mediate BH3 Mimetic Activity
www.aacrjournals.org Cancer Res; 72(16) August 15, 2012 4233
on April 13, 2017. © 2012 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 12, 2012; DOI: 10.1158/0008-5472.CAN-12-1118
DiscussionTumor cells are commonly characterized by overexpression
of antiapoptotic Bcl proteins (e.g., Bcl-2, Bcl-xL, or Mcl-1),which contribute to drug resistance by disabling the deathmachinery (30, 31). Transformed cells also frequently expressproapoptotic Bcl proteins includingmultidomain (e.g., Bax andBak) and some, if not all, BH3-only proteins such as Bim (3, 4).Although tumor cells may be fully equipped with anti- andproapoptotic machinery, the latter are often inactivatedthrough binding to their highly expressed antiapoptotic coun-terparts (2). Because of the balance between anti- andprodeathBcl proteins, tumor cells may be "primed" for cell death (32) bydisrupting this balance, for example, by upregulating or mim-icking proapoptotic protein actions, or by downregulating/antagonizing the function of antiapoptotic proteins (33, 34).The present findings show that pan-Cdk inhibitors markedlypotentiates the lethality of pan-BH3 mimetics (e.g., obato-clax) in vitro and in vivo in MM cells, including thosedisplaying conventional or novel forms of drug resistance.They also reveal that these agents interact synergistically by
interfering with both arms of the apoptosis-regulatory bal-ance, that is, downregulating/disabling Bcl-2, Bcl-xL, andMcl-1, and upregulating BH3-only proteins including Bim,Noxa, and Bik/NBK.
Pan-Cdk inhibitors like flavopiridol act as transcriptionalrepressors by inhibiting Cdk9, a catalytic subunit of the cyclinT/Cdk9 complex (P-TEFb), preventing serine-2 phosphoryla-tion of RNA Pol II CTD, thereby blocking transcription elon-gation (13) and inducing downregulation of short-lived pro-teins such as Mcl-1 (14), an essential survival factor for MMcells (35). The present results show that flavopiridol down-regulates Mcl-1 at early intervals (e.g., 6 hours) in associationwith diminished CTD serine-2 phosphorylation. However, Mcl-1 expression recovered after 16 hours, indicating that flavopir-idol rapidly but only transiently inhibits Mcl-1 transcription incells (14). Recently, obatoclax was shown to downregulateMcl-1 through induction of Noxa, which binds toMcl-1 and triggersits degradation, whereas qRT-PCR revealed more than a 5-foldincrease in Mcl-1 mRNA levels after obatoclax exposure (6 hrs;ref. 21). Similar events were observed in this study, but mRNA
160
140
120
100
80
60
40
20
x103
500
400
300
200
100
x103
3.0
500
400
300
200
100
x103
2.5
2.0
1.5 x106
2.5
2.0
1.5
1.0
0.5
x106
3.0
2.5
2.0
1 5
x106
Veh FPB
1.0
0.5
1.5
1.0
0.5
D80 D90
Obat FP+Obat
D120 D120
C
Tx started
-
P = 0.0268
D
P = 0.0161
50
75
100
125
Veh
FPBo
dy w
eig
ht
of
init
ial valu
e)
Tx started
0
25
FP
Obat
FP + Obat
0 2 4 6 8 10 12 14 16 18 20 22 24 26 28
Day after started drug treatment
B
(%
Veh FP Obat FP + Obat Veh FP Obat FP + Obat
Dorsal Ventral
D14
D21
D30
A
Figure 7. The flavopiridol (FP)/obatoclax (Obat) regimen is activein an i.v. bone marrow–homingmurine model. A–D, mouse model#3—i.v. orthotopic U266 model:NOD/SCID-g mice were i.v.injected via tail vein with 5 � 106
U266 carrying luciferase. Afterluciferase signals were visible(14 days after injection of tumorcells), flavopiridol (5 mg/kg, i.p.)with or without obatoclax (3mg/kg,i.p.) was administered daily for first5 days, followed by flavopiridol (3mg/kg) with or without obatoclax (3mg/kg) twice every 3 days foradditional 7 cycles; n ¼ 5 pergroup. Tumor growth wasmonitored every other day byimaging luciferase activity (A).Cross, dead. B, left, Kaplan–Meieranalysis was carried out to analyzeoccurrence of hind-leg paralysis(right, arrows), as well as survival ofanimals (C). Loss of body weight(<20% of initial weight) occurredwithin the first week of treatment inthe flavopiridol and flavopiridol/obatoclax groups, indicatingtoxicity of 5 mg/kg flavopiridol inthese mice, which recovered afterreduction of the flavopiridol dose to3 mg/kg and adjustment of thetreatment schedule (D). Veh,vehicle.
Chen et al.
Cancer Res; 72(16) August 15, 2012 Cancer Research4234
on April 13, 2017. © 2012 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 12, 2012; DOI: 10.1158/0008-5472.CAN-12-1118

upregulation was largely blocked by flavopiridol. Although themechanism underlying these phenomena is uncertain,increases in Mcl-1 mRNA may represent a compensatoryresponse to downregulation of the protein. Importantly, oba-toclax strikingly blocked recovery from Mcl-1 protein down-regulation after prolonged flavopiridol exposure (e.g., 16–48hours), suggesting that persistent Mcl-1 downregulationfollowing flavopiridol/obatoclax coexposure may reflect 2separate but cooperative mechanisms: (i) Mcl-1 mRNA tran-scription inhibition by flavopiridol; and (ii) promotion ofMcl-1protein degradation through Noxa induction by obatoclax.However, Mcl-1 overexpression only partially attenuated fla-vopiridol/obatoclax-mediated apoptosis at 24 hours, but not atintervals �48 hours, raising the possibility that the flavopir-idol/obatoclax regimen may circumvent Mcl-1-related drugresistance.MM cells exposed to flavopiridol with or without obatoclax
exhibited sharp increases in expression of BH3-only proteinsincluding Bim (EL and L isoforms), Noxa, and Bik/NBK. UnlikeBax/Bak, protein levels of which are relatively stable (2), andwhich must be activated to trigger apoptosis, expression ofBH3-only proteins is tightly regulated, and thus represents acandidate target (3, 36). Many novel agents induce expression(e.g., HDAC inhibitors; ref. 19) or prevent degradation (e.g.,proteasome inhibitors; ref. 37) of BH3-only proteins such asNoxa, Puma, and Bim, thereby directly or indirectly activatingBax/Bak (2). Obatoclax antagonizes Mcl-1 antiapoptotic func-tions by unleashing Bim or Bak from Mcl-1, and also down-regulates Mcl-1 via induction of Noxa (21). This findingshows that obatoclax induced Noxa upregulation at earlyintervals (e.g., 6 hours), and that this event was enhanced andrendered more sustained by flavopiridol. Functionally, bothNoxa and Bim shRNA profoundly diminished flavopiridol/obatoclax lethality. Thus, although downregulation of antia-poptotic proteins (e.g., Mcl-1 and Bcl-xL)may contribute to thelethality of this regimen to a limited extent, upregulation ofBH3-only proteins (e.g., Bim and Noxa) clearly plays a criticalrole in Cdk inhibitor/pan-BH3 mimetic interactions.The marked increase in Bim (both EL and L isoforms) by
flavopiridol with or without obatoclax has, to the best of ourknowledge, not been described previously. Transcriptionalregulation of Bim represents a major mechanism of apoptosisregulation (38). Several findings argue that flavopiridol upre-gulates Bim at the transcriptional level. First, translationinhibition by CHX largely blocked Bim upregulation inducedby flavopiridol. Second, qRT-PCR indicated that flavopiridolinduced a marked increase (e.g., 3- to -5-fold) in Bim mRNAlevels. In this context, transcriptional regulation of Bim iscomplex and multifactorial. For example, growth factor with-drawal-induced Bim upregulation requires JNK activation inneurons, although it depends on the forkhead transcriptionfactor FKHR-L1 in hematopoietic cells (3). Moreover, the bimpromoter is regulated by stress-related transcriptional factorssuch as FOXO and AP-1 family members (38). The mechanismby which a pan-Cdk inhibitor transcriptionally that upregu-lates BH3-only proteins such as Bim is not intuitively obvious.One possibility is that flavopiridol may activate JNK thatupregulates BH3-only proteins to sensitize human myeloma
cells to Bcl-2 antagonists (e.g., HA14-1; ref. 39). Another pos-sibility is that Cdk2 inhibition by pan-Cdk inhibitors mayactivate FOXO1 (40), which induces Bim transcription (41).In addition to transcriptional induction, Bim also exhibitsmultifactorial regulation at the levels of mRNA stability, post-translational modifications, proteasomal degradation, andcellular localization (38, 42). Consequently, the possibility thatadditional mechanisms contribute to Cdk inhibitor-mediatedBim upregulation cannot be excluded.
The observation that Bim shRNA knockdown essentiallyabrogated apoptosis indicates a critical functional role for Bimupregulation in Cdk inhibitor/pan-BH3 mimetic lethality.Mechanisms of Bim-mediated cell death may reflect directactions following displacement/derepression of antiapoptoticproteins by BH3-only "sensitizers," leading to Bax/Bak activa-tion. Alternatively, in the neutralization model, Bim binds toand neutralizes/inactivates all antiapoptotic Bcl proteins thatrepress constitutively active Bax/Bak. Obatoclax releases Bimfrom Bcl-2, Bcl-xL, and Mcl-1 binding, thereby promotingapoptosis (10, 12). Interestingly, flavopiridol upregulatedexpression of Bim but increased its binding to Bcl-2 andBcl-xL, suggesting that upregulated Bim may "prime" cells fordeath (43, 44). Indeed, obatoclax unleashed Bim from Bcl-2,Bcl-xL, andMcl-1, leading to Bax/Bak activation and cell deathin Cdk inhibitor-treated cells. Significantly, the latter event (i.e.,Bax/Bak activation) did not occur in cells in which Bim wasknocked down by shRNA. Notably, in a dual-sided flankmurinemodel that circumvents problems in interpretation related tomouse-to-mouse variability (45), flavopiridol/obatoclax failedto suppress the growth of tumors carrying Bim shRNA, show-ing a functional in vivo role for Bim in lethality. Finally, theobservation that obatoclax released Bim from multiple anti-apoptotic proteins (e.g., Bcl-2, Bcl-xL, and Mcl-1) suggests thatas observed in the case of BH3 mimetics (e.g., ABT-737)administered alone (43), interactions between Bcl familymembers, rather than simply their expression profiles, maybe critical predictors of sensitivity to the Cdk inhibitor/pan-BH3 mimetic regimen in MM.
MM cells interact with the bone marrow microenviron-ment to promote cell proliferation, survival, migration, anddrug resistance. Genetic profiling studies revealed that Bclfamily members represent microenvironment-specific che-motherapeutic response determinants in malignant hemato-poietic cells, and that BH3-only "activators" are necessary forcell death (45). IL-6 and IGF-1, which are critical for MMcell/microenvironment interactions, promote MM cell sur-vival and confer drug resistance through (i) Mcl-1 induction(46, 47) and (ii) Bim downregulation (34, 48). The latterinvolves at least 2 mechanisms, for example, posttransla-tional phosphorylations and/or transcription (48). Notably,flavopiridol/obatoclax was fully active in the presence ofthese cytokines or other autocrine MM survival/growthfactors (e.g., BAFF and APRIL; ref. 49). Direct MM cell–stroma contact also contributes to MM drug-resistance (28).Interestingly, HS-5 coculture and conditioned medium large-ly abrogated Bim expression in MM cells, implicating thisphenomenon as a mechanism of stromal cell–mediated drugresistance. Significantly, flavopiridol/obatoclax lethality
BH3-Only Proteins Mediate BH3 Mimetic Activity
www.aacrjournals.org Cancer Res; 72(16) August 15, 2012 4235
on April 13, 2017. © 2012 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 12, 2012; DOI: 10.1158/0008-5472.CAN-12-1118

persisted under these conditions. flavopiridol/obatoclax alsosuppressed in vivo growth of MM cells that homed to bonemarrow, arguing further that the Cdk inhibitor/pan-BH3mimetic strategy may circumvent bone marrow microenvi-ronment-mediated drug-resistance. Finally, flavopiridol/obatoclax was active against multiple drug-resistant celllines. Interestingly, bortezomib-resistant cells (PS-R) exhib-ited marked Bim downregulation accompanied by modestlyincreased Mcl-1, supporting a recent concept that Bcl-2family member dysregulation determines MM cell suscep-tibility to therapeutic interventions (34). Importantly, thesecells were also fully susceptible to flavopiridol/obatoclax.Collectively, these findings argue that those resistancemechanisms fail to protect MM cells from Cdk inhibitor/pan-BH3 mimetic regimens, which coordinately target bothpro- and antiapoptotic Bcl family members.
The marked lethality of flavopiridol/obatoclax in primaryCD138þ MM specimens, with minimal toxicity toward theirnormal CD138� counterparts or CD34þ hematopoietic cells,raises the possibility that MM cells may be particularlysusceptible to this strategy. Significantly, in marked contrastto obatoclax administered alone, combined treatment pro-duced marked suppression of tumor growth in vivo in both s.c. flank and i.v. orthotopic models. Particularly in the latter,in which MM cells selectively homed to the bone marrowand produced MM-specific bone disease, coadministrationof flavopiridol/obatoclax markedly improved outcomes forthese animals, for example, significantly preventing hind-legparalysis and prolonging survival. Such findings argue thatalthough pan-BH3 mimetics such as obatoclax may notbe sufficient by themselves to induce cell death in vitroor in vivo, additional perturbations (e.g., prolongation ofantiapoptotic protein downregulation in conjunction withupregulation of proapoptotic BH3-only proteins) induced bypan-Cdk inhibitors may trigger a constellation of events thatexceed the apoptotic threshold. In summary, the present
findings provide a preclinical justification for combiningpan-Cdk inhibitors, possibly including new-generation Cdkinhibitors that have recently entered the clinic (13), withpan-BH3 mimetics (e.g., obatoclax; ref. 50) in MM and otherhematologic malignancies. They also highlight the criticalimportance of coordinate disruption of pro- and antiapop-totic arms of the apoptotic regulatory machinery in pro-moting transformed cell death.
Disclosure of Potential Conflicts of InterestNo potential conflicts of interest were disclosed.
Authors' ContributionsConception and design: S. Chen, Y. Dai, P. Dent, R.Z. Orlowski, S. GrantDevelopment of methodology: S. Chen, Y. Dai, X.-Y. Pei, L. WangAcquisition of data (provided animals, acquired and managed patients,provided facilities, etc.): S. Chen, Y. Dai, X.-Y. Pei, J. Myers, L. Wang, L.B.Kramer, M. Garnett, D.M. Schwartz, F. Su, G.L. Simmons, J.D. Richey, D.G. Larsen,R.Z. OrlowskiAnalysis and interpretation of data (e.g., statistical analysis, biostatistics,computational analysis): S. Chen, Y. Dai, X.-Y. Pei, L.Wang, D.M. Schwartz, J.D.Richey, S. GrantWriting, review, and/or revision of themanuscript: S. Chen, Y. Dai, P. Dent,R.Z. Orlowski, S. GrantAdministrative, technical, or material support (i.e., reporting or orga-nizing data, constructing databases): Y. Dai, L.B. Kramer, J.D. RicheyStudy supervision: Y. Dai, S. Grant
Grant SupportThis work was supported by awards CA100866, CA 93738, 1 P50
CA142509-01, and 1 P50 CA130805-01 from the NIH, award 6181-10 fromthe Leukemia and Lymphoma Society of America, and awards from the VFoundation and the Multiple Myeloma Research Foundation. Plasmidpreparation was carried out at the VCU Macromolecule Core Facility,supported, in part, with the funding from NIH-NCI Cancer Center CoreGrant 5P30CA016059-29.
The costs of publication of this article were defrayed in part by thepayment of page charges. This article must therefore be hereby markedadvertisement in accordance with 18 U.S.C. Section 1734 solely to indicatethis fact.
Received March 26, 2012; revised May 25, 2012; accepted May 31, 2012;published OnlineFirst June 12, 2012.
References1. Chauhan D, Velankar M, Brahmandam M, Hideshima T, Podar K,
Richardson P, et al. A novel Bcl-2/Bcl-X(L)/Bcl-w inhibitor ABT-737as therapy in multiple myeloma. Oncogene 2007;26:2374–80.
2. Chipuk JE, Moldoveanu T, Llambi F, ParsonsMJ, Green DR. The BCL-2 family reunion. Mol Cell 2010;37:299–310.
3. Elkholi R, Floros KV, Chipuk JE. The role of BH3-only proteins in tumorcell development, signaling, and treatment. Genes Cancer 2011;2:523–37.
4. Kuroda J, Taniwaki M. Involvement of BH3-only proteins inhematologic malignancies. Crit Rev Oncol Hematol 2009;71:89–101.
5. Azmi AS, Wang Z, Philip PA, Mohammad RM, Sarkar FH. EmergingBcl-2 inhibitors for the treatment of cancer. Expert Opin Emerg Drugs2011;16:59–70.
6. Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ,Belli BA, et al. An inhibitor ofBcl-2 family proteins induces regressionofsolid tumours. Nature 2005;435:677–81.
7. Chen S, Dai Y, Harada H, Dent P, Grant S. Mcl-1 down-regulationpotentiates ABT-737 lethality by cooperatively inducing Bak activationand Bax translocation. Cancer Res 2007;67:782–91.
8. Wuilleme-Toumi S, Robillard N, Gomez P,Moreau P, LeGouill S, Avet-Loiseau H, et al. Mcl-1 is overexpressed in multiple myeloma and
associated with relapse and shorter survival. Leukemia 2005;19:1248–52.
9. Gomez-Bougie P, Wuilleme-Toumi S, Menoret E, Trichet V, RobillardN, Philippe M, et al. Noxa upregulation and Mcl-1 cleavage areassociated to apoptosis induction by bortezomib inmultiplemyeloma.Cancer Res 2007;67:5418–24.
10. Konopleva M, Watt J, Contractor R, Tsao T, Harris D, Estrov Z, et al.Mechanisms of antileukemic activity of the novel Bcl-2 homologydomain-3 mimetic GX15-070 (obatoclax). Cancer Res 2008;68:3413–20.
11. Nguyen M, Marcellus RC, Roulston A, Watson M, Serfass L, MurthyMadiraju SR, et al. Small molecule obatoclax (GX15-070) antagonizesMCL-1 and overcomes MCL-1-mediated resistance to apoptosis.Proc Natl Acad Sci U S A 2007;104:19512–17.
12. Trudel S, Li ZH, Rauw J, Tiedemann RE, Wen XY, Stewart AK.Preclinical studies of the pan-Bcl inhibitor obatoclax (GX015-070) inmultiple myeloma. Blood 2007;109:5430–38.
13. Stellrecht CM,Chen LS. Transcription inhibition as a therapeutic targetfor cancer. Cancers 2011;3:4170–90.
14. Gojo I, Zhang B, Fenton RG. The cyclin-dependent kinase inhibitorflavopiridol induces apoptosis in multiple myeloma cells through
Chen et al.
Cancer Res; 72(16) August 15, 2012 Cancer Research4236
on April 13, 2017. © 2012 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 12, 2012; DOI: 10.1158/0008-5472.CAN-12-1118

transcriptional repression and downregulation of Mcl-1. Clin CancerRes 2002;8:3527–38.
15. MacCallum DE, Melville J, Frame S, Watt K, Anderson S, Gianella-Borradori A, et al. Seliciclib (CYC202, R-Roscovitine) induces celldeath in multiple myeloma cells by inhibition of RNA polymerase II-dependent transcription and downregulation of Mcl-1. Cancer Res2005;65:5399–407.
16. Lin TS, Ruppert AS, Johnson AJ, Fischer B, Heerema NA, AndritsosLA, et al. Phase II study of flavopiridol in relapsed chronic lymphocyticleukemia demonstrating high response rates in genetically high-riskdisease. J Clin Oncol 2009;27:6012–18.
17. Woyach JA, Lozanski G, Ruppert AS, Lozanski A, Blum KA, Jones JA,et al. Outcome of patients with relapsed or refractory chronic lympho-cytic leukemia treated with flavopiridol: impact of genetic features.Leukemia 2012;26:1442–4.
18. Holkova B, Perkins EB, Ramakrishnan V, Tombes MB, Shrader E,Talreja N, et al. Phase I trial of bortezomib (PS-341; NSC 681239) andalvocidib (flavopiridol; NSC 649890) in patients with recurrent orrefractory B-cell neoplasms. Clin Cancer Res 2011;17:3388–97.
19. Chen S, Dai Y, Pei XY, Grant S. Bim upregulation by histone deace-tylase inhibitors mediates interactions with the Bcl-2 antagonist ABT-737: evidence for distinct roles for Bcl-2, Bcl-xL, and Mcl-1. Mol CellBiol 2009;29:6149–69.
20. BjorklundCC,MaW,WangZQ,Davis RE, KuhnDJ, KornblauSM, et al.Evidence of a role for activation of Wnt/beta-catenin signaling in theresistance of plasma cells to lenalidomide. J Biol Chem 2011;286:11009–20.
21. Albershardt TC, Salerni BL, Soderquist RS, Bates DJ, Pletnev AA,Kisselev AF, et al. Multiple BH3 mimetics antagonize antiapoptoticMCL1 protein by inducing the endoplasmic reticulum stress responseand upregulating BH3-only protein NOXA. J Biol Chem 2011;286:24882–95.
22. Willis SN, Chen L, Dewson G, Wei A, Naik E, Fletcher JI, et al. Proa-poptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, untildisplaced by BH3-only proteins. Genes Dev 2005;19:1294–305.
23. Qin JZ, Ziffra J, Stennett L, Bodner B, Bonish BK, Chaturvedi V, et al.Proteasome inhibitors trigger NOXA-mediated apoptosis inmelanomaand myeloma cells. Cancer Res 2005;65:6282–93.
24. Akiyama T, Dass CR, Choong PF. Bim-targeted cancer therapy: a linkbetween drug action and underlying molecular changes. Mol CancerTher 2009;8:3173–80.
25. Li C, Li R, Grandis JR, Johnson DE. Bortezomib induces apoptosis viaBim and Bik upregulation and synergizes with cisplatin in the killing ofhead and neck squamous cell carcinoma cells. Mol Cancer Ther2008;7:1647–55.
26. Anderson KC. Oncogenomics to target myeloma in the bone marrowmicroenvironment. Clin Cancer Res 2011;17:1225–33.
27. Perez LE, Parquet N, Shain K, Nimmanapalli R, Alsina M, Anasetti C,et al. Bone marrow stroma confers resistance to Apo2 ligand/TRAIL inmultiple myeloma in part by regulating c-FLIP. J Immunol 2008;180:1545–55.
28. Nefedova Y, Landowski TH, DaltonWS. Bonemarrow stromal-derivedsoluble factors and direct cell contact contribute to de novo drugresistance of myeloma cells by distinct mechanisms. Leukemia 2003;17:1175–82.
29. Faber A, Corcoran RB, Ebi H, Sequist LV, Waltman BA, Chung E, et al.BIM expression in treatment naive cancers predicts responsiveness tokinase inhibitors. Cancer Discov 2011;1:352–65.
30. Kelly PN, Strasser A. The role of Bcl-2 and its prosurvival relatives intumourigenesis and cancer therapy. Cell Death Differ 2011;18:1414–24.
31. Yip KW, Reed JC. Bcl-2 family proteins and cancer. Oncogene2008;27:6398–406.
32. Letai AG. Diagnosing and exploiting cancer's addiction to blocks inapoptosis. Nat Rev Cancer 2008;8:121–32.
33. Romagnoli M, Seveno C, Wuilleme-Toumi S, Amiot M, Bataille R,Minvielle S, et al. The imbalance between Survivin and Bim mediatestumour growth and correlates with poor survival in patients withmultiple myeloma. Br J Haematol 2009;145:180–9.
34. Gomez-Bougie P, Bataille R, Amiot M. The imbalance between Bimand Mcl-1 expression controls the survival of human myeloma cells.Eur J Immunol 2004;34:3156–64.
35. Derenne S, Monia B, Dean NM, Taylor JK, Rapp MJ, Harousseau JL,et al. Antisense strategy shows that Mcl-1 rather than Bcl-2 or Bcl-x(L)is an essential survival protein of human myeloma cells. Blood 2002;100:194–9.
36. Morales AA, Gutman D, Lee KP, Boise LH. BH3-only proteins Noxa,Bmf, and Bim are necessary for arsenic trioxide-induced cell death inmyeloma. Blood 2008;111:5152–62.
37. Fennell DA, Chacko A, Mutti L. BCL-2 family regulation by the 20Sproteasome inhibitor bortezomib. Oncogene 2008;27:1189–97.
38. Hubner A, Barrett T, Flavell RA, Davis RJ. Multisite phosphorylationregulates Bim stability and apoptotic activity. Mol Cell 2008;30:415–25.
39. Pei XY, Dai Y, Grant S. The small-molecule Bcl-2 inhibitor HA14-1interacts synergistically with flavopiridol to inducemitochondrial injuryand apoptosis in human myeloma cells through a free radical-depen-dent and Jun NH2-terminal kinase-dependent mechanism. Mol Can-cer Ther 2004;3:1513–24.
40. Huang H, Regan KM, Lou Z, Chen J, Tindall DJ. CDK2-dependentphosphorylation of FOXO1 as an apoptotic response to DNA damage.Science 2006;314:294–7.
41. Yang Y, Zhao Y, Liao W, Yang J, Wu L, Zheng Z, et al. Acetylation ofFoxO1 activates Bim expression to induce apoptosis in response tohistone deacetylase inhibitor depsipeptide treatment. Neoplasia2009;11:313–24.
42. Gordon PM, Fisher DE. Role for the proapoptotic factor BIM inmediating imatinib-induced apoptosis in a c-KIT-dependentgastrointestinal stromal tumor cell line. J Biol Chem 2010;285:14109–14.
43. MoralesAA,KurtogluM,MatulisSM, Liu J, SiefkerD,GutmanDM, et al.Distribution of Bim determines Mcl-1 dependence or codependencewith Bcl-xL/Bcl-2 in Mcl-1-expressing myeloma cells. Blood 2011;118:1329–39.
44. Gillings AS, Balmanno K, Wiggins CM, Johnson M, Cook SJ. Apo-ptosis and autophagy: BIM as a mediator of tumour cell death inresponse to oncogene-targeted therapeutics. FEBS J 2009;276:6050–62.
45. Pritchard JR, Gilbert LA, Meacham CE, Ricks JL, Jiang H, Lauffen-burger DA, et al. Bcl-2 family genetic profiling reveals microenviron-ment-specific determinants of chemotherapeutic response. CancerRes 2011;71:5850–8.
46. Jourdan M, De VJ, Mechti N, Klein B. Regulation of Bcl-2-familyproteins in myeloma cells by three myeloma survival factors: interleu-kin-6, interferon-alpha and insulin-like growth factor 1. Cell DeathDiffer 2000;7:1244–52.
47. Zhang B, Potyagaylo V, Fenton RG. IL-6-independent expressionof Mcl-1 in human multiple myeloma. Oncogene 2003;22:1848–59.
48. De BE, Bos TJ, Schuit F, Van Valckenborgh E, Menu E, Thorrez L,et al. IGF-1 suppresses Bim expression in multiple myeloma viaepigenetic and posttranslational mechanisms. Blood 2010;115:2430–40.
49. Moreaux J, Legouffe E, Jourdan E, Quittet P, R�eme T, Lugagne C, et al.BAFF and APRIL protect myeloma cells from apoptosis induced byinterleukin6 deprivation anddexamethasone.Blood2004;103:3148–57.
50. O'BrienSM,ClaxtonDF, CrumpM, Faderl S, Kipps T, KeatingMJ, et al.Phase I study of obatoclax mesylate (GX15-070), a small moleculepan-Bcl-2 family antagonist, in patients with advanced chronic lym-phocytic leukemia. Blood 2009;113:299–305.
BH3-Only Proteins Mediate BH3 Mimetic Activity
www.aacrjournals.org Cancer Res; 72(16) August 15, 2012 4237
on April 13, 2017. © 2012 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 12, 2012; DOI: 10.1158/0008-5472.CAN-12-1118

2012;72:4225-4237. Published OnlineFirst June 12, 2012.Cancer Res Shuang Chen, Yun Dai, Xin-Yan Pei, et al. Myeloma Cells to BH3 Mimetic TherapiesCDK Inhibitors Upregulate BH3-Only Proteins to Sensitize Human
Updated version
10.1158/0008-5472.CAN-12-1118doi:
Access the most recent version of this article at:
Material
Supplementary
http://cancerres.aacrjournals.org/content/suppl/2012/06/12/0008-5472.CAN-12-1118.DC1
Access the most recent supplemental material at:
Cited articles
http://cancerres.aacrjournals.org/content/72/16/4225.full.html#ref-list-1
This article cites 50 articles, 29 of which you can access for free at:
Citing articles
/content/72/16/4225.full.html#related-urls
This article has been cited by 3 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
To request permission to re-use all or part of this article, contact the AACR Publications Department at
on April 13, 2017. © 2012 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 12, 2012; DOI: 10.1158/0008-5472.CAN-12-1118