chapter- 2.pdf - shodhganga
TRANSCRIPT

10
Chapter-2
2.1. Stages during quality control testing
Quality control is of crucial importance to the pharmaceutical industry
and for this reason numerous checks are made at every stage of production
to ensure that the quality is not compromised and that the code of Good
Manufacturing Process is adhered to quality control procedure include,
Sampling of raw materials.
All incoming raw materials are initially quarantined, and samples are
taken and tested to ensure that the material meets strict purity guidelines.
This testing involves both microbiological and chemical testing, as is laid out
in the relevant Pharmacopeia [1].
In process checks:
The manufacturing staff carries out checks on such things as, purity
of intermediates, Uniformity at intermediate levels and the parameters which
will effect on the production. At hourly intervals the quality control staff
takes samples to check for contamination and to ensure that the
composition is as expected.
Final product checking:
Checking the similar parameters to those measured during
production. Validated methods are used for final product checking.
Monitoring cleaning:
When a batch of certain drugs has been made, all equipment that has
been used must be cleaned. When the next pharmaceutical to be made on

11
that line is going to different, this cleaning must be particularly thorough to
prevent contamination. In this instance, after cleaning the quality control
staff take swabs off each piece of equipment and test them to see if they can
detect the presence active previously used.
Only when the equipments so clean that the previous active is
undetectable or below certain level which will not cause any effect on the
next production can the production of the next pharmaceutical commence.
The results of these tests are recorded on the batch records for the
pharmaceutical, as well as the name and batch number of the
pharmaceutical made immediately prior on the same production line.
2.2. Active Pharmaceutical Ingredient (API) (or Drug Substance)
Any substance or mixture of substances intended to be used in the
manufacture of a drug (medicinal) product and that, when used in the
production of a drug, becomes an active ingredient of the drug product.
Such substances are intended to furnish pharmacological activity or other
direct effect in the diagnosis, cure, mitigation, treatment, or prevention of
disease or to affect the structure and function of the body.
Modern medicines for human use are required to meet exacting
standards that relate to their quality, safety and efficacy. The evaluation of
the above factors in practice depends on the existence of adequate methods
for quality control of the drug substance and drug product. Thus great
demands are placed on the analytical methods that are used for the
determination of active drug and its related impurities in bulk drug
substance.

12
Drug substances are generally not administered in their native form.
The medicine is the whole pharmaceutical formulation (also called as dosage
form) in which the drug, that is active substance, combined with other
ingredients (also called as excipients) to form a convenient form of
administration, such as tablet, capsule, injection, ointment etc.
During manufacturing of the drugs by products, unreacted raw
materials, degradation products are the major impurities in drug
substances. These impurities often possess unwanted pharmacological or
toxicological effects like genotoxicity, carcinogenity due to which any benefit
from the administration of the drug is outweighed. In addition to above, the
impurities can be formed during the storage of drug substances due to
degradation. So it is very important to monitor the level of impurities present
in the drug substance during manufacturing, batch release and during its
storage.
It is very important for the analyst to develop a suitable analytical
method for the drug substance, which can monitor the level of impurities
and content during its manufacturing and release. The target of the analyst
is not only to test and release the batch and must ensure that the same
method shall also be used during the stability testing of drug substance
(stability-indicating).
An “expiry date” will be mentioned on the prescription or over-the-
counter (OTC) pharmaceutical products. Before this date, the product should
remain fully effective under normal storage conditions. The efficacy and
safety of pharmaceuticals cannot be ensured unless the quality of the
pharmaceuticals is maintained during their specified shelf lives. The analysis

13
of stability samples must be carried out with the use of stability-indicating
analytical methods (SIAMs).
A SIAM is a quantitative analytical procedure used to detect a
decrease in the amount of active pharmaceutical ingredient (API) present due
to degradation. During the Quality Control testing, batch release and
stability studies of a drug substance the HPLC technique is routinely used to
separate and quantitate the analytes of interest.
2.3. Drugs
Drug is a chemical substance that is used in the diagnosis, cure,
relief, treatment, or prevention of a disease. In medicine, a drug refers to any
substance with a potential to prevent or cure disease or enhance physical or
mental welfare and in pharmacology, to any chemical agent that alters the
biochemical and physiological processes of tissues or organisms [2]. The
medicinal value of plants has been recognized by almost every society on this
planet. In the earlier days, plant material (leaves or bark) extracts,
particularly provided the main source of folk medicines.
The idea that the effect of a drug in human body is mediated by
specific interactions of the drug molecule with biological macromolecules,
(proteins or nucleic acids in most cases), led scientists to consider that
individual chemicals may function well for interacting with biological
macromolecules and results in controlling their malfunctions. This made the
beginning of the modern era in pharmacology that pure chemicals, instead of
crude extracts, can be used as better drugs.
2.3.1. Drug development

14
In the latter part of the nineteenth century, biologically-active organic
molecules began to be isolated in relatively pure form for medicinal use. For
example, salicylic acid, the precursor of Aspirin, was isolated in 1874 from
willow bark. Various more potent painkillers, such as Morphine and
Codeine, were isolated from the opium poppy. The anti-malarial agent
Quinine was separated from cinchona (china bark). The leaves of the purple
foxglove plant provided an excellent source of digitalis that was purified for
use against heart disease. The synthesis of the first synthetic
pharmaceutical drug, aspirin, occurred in the latter half of the nineteenth
century,
Alexander Fleming [3] noticed that a fungus growing on an agar plate
had inhibited the growth of bacteria in the year 1928. Ten years later,
Howard Florey and Ernst Chain [3] isolated the chemical penicillin that was
affecting the bacteria. Penicillin is still most widely used antibiotic for the
treatment of bacterial diseases. The discovery of the antibiotics
Streptomycin, Chloramphenicol, and chlortetracycline followed the discovery
of penicillin.
The introduction of Salvarsan for the treatment of syphilis by the
German hematologist Paul Ehrlich in 1911 [4] paved the way for the
introduction modern drugs like sulphonamide in 1930. Recent advances in
X-ray crystallography, NMR spectroscopy, mass spectrometry, developments
in electrophoresis, ultracentrifugation, chromatography etc. helped the
discovery of additional chemical entities with therapeutic activities.
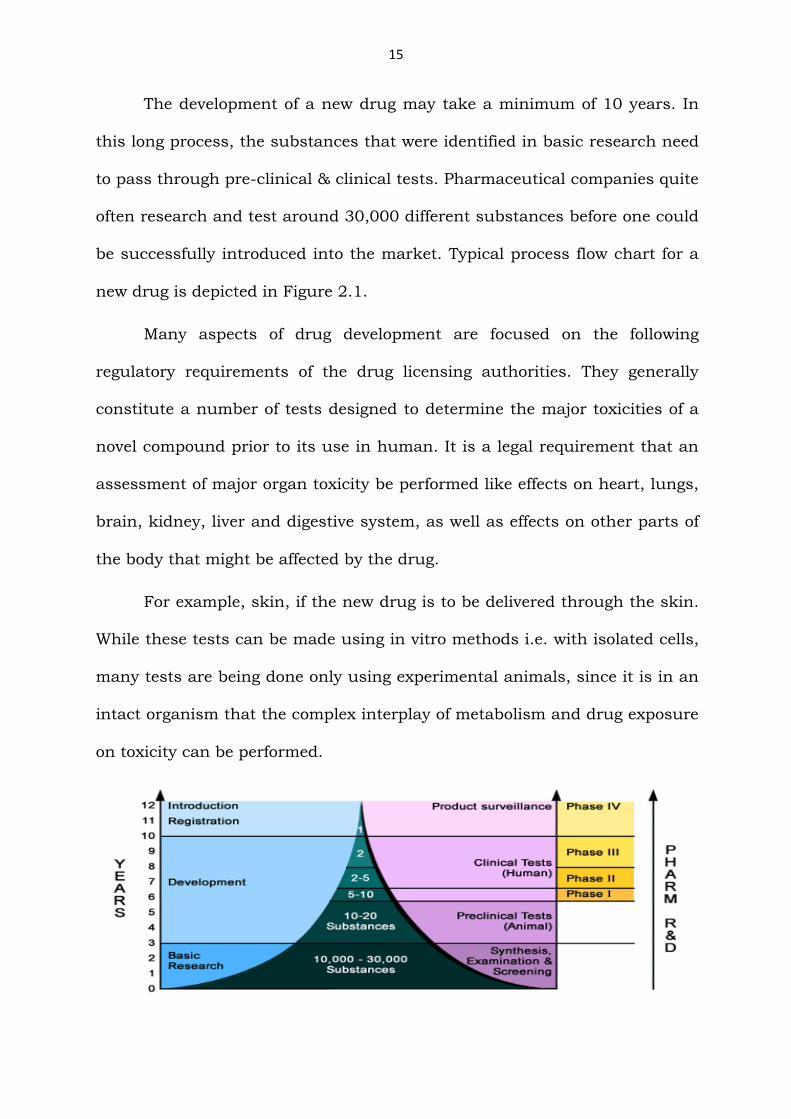
15
The development of a new drug may take a minimum of 10 years. In
this long process, the substances that were identified in basic research need
to pass through pre-clinical & clinical tests. Pharmaceutical companies quite
often research and test around 30,000 different substances before one could
be successfully introduced into the market. Typical process flow chart for a
new drug is depicted in Figure 2.1.
Many aspects of drug development are focused on the following
regulatory requirements of the drug licensing authorities. They generally
constitute a number of tests designed to determine the major toxicities of a
novel compound prior to its use in human. It is a legal requirement that an
assessment of major organ toxicity be performed like effects on heart, lungs,
brain, kidney, liver and digestive system, as well as effects on other parts of
the body that might be affected by the drug.
For example, skin, if the new drug is to be delivered through the skin.
While these tests can be made using in vitro methods i.e. with isolated cells,
many tests are being done only using experimental animals, since it is in an
intact organism that the complex interplay of metabolism and drug exposure
on toxicity can be performed.

16
Figure 2.1: Typical process flow chart for the development of a new
drug
It is reported that average cost may be around $800 million to bring a
new drug in to the market for the beneficial use of people [5]. By law, drugs
are divided into two categories: prescription drugs and nonprescription
drugs [6]. Prescription drugs are those considered safe for use only under
medical supervision and may be dispensed only with a prescription from a
licensed professional Doctor with governmental privileges to prescribe.
Nonprescription drugs, those considered safe for use without medical
supervision and are sold over the counter. In the United States, the Food
and Drug Administration (FDA) is the government agency that decides which
drugs require a prescription and which may be sold over the counter.
2.4. Impurities and their origin
Impurities in drugs are the unwanted chemicals that remain with the
drug or active pharmaceutical ingredients (APIs), or develop during
formulation, or developed upon aging of both API and formulated products
[7]. The presence of these unwanted chemicals even in small amounts may
influence the efficacy and safety of the pharmaceutical products. Impurities
can have a strong adverse effect on human being due to undesirable
pharmacological and toxicological action, which can prevail over the positive
effect of the medicine, and can hinder the positive effect of the main
medicinal substance.
2.4.1. Nature of impurities

17
Medicines are the formulated forms of active pharmaceutical
ingredients. There are two types of impurities in medicines: (a) Impurities
associated with the active pharmaceutical ingredients and (b) Impurities that
are created during formulation and or with aging or that are related to the
formulated forms. Impurities associated with the active pharmaceutical
ingredients are classified into three categories as specified in the
International Conference of Harmonization (ICH) guidelines [8, 9 ].
Organic impurities that are either process and/or drug-related
Inorganic impurities
Residual solvents
2.4.2. Organic impurities
Organic impurities may come up in the manufacturing process and/or
during storage of the drug substance. They may generate during the
synthetic processes of a drug substance and due to degradation reactions of
the drug substances by the environmental parameters like temperature,
humidity, sunlight. The impurities can be originated from the following. (a)
Starting materials, (b) Intermediates, (c) By-products, (d) Enontiomers, (e)
Degradation products, (f) Reagents, ligands and catalysts
Starting materials & Intermediates: These are the most common
impurities found in every API unless a proper care is taken in every step
involved throughout the multi-step synthesis. Although, at the end of each
process step the products are always washed with solvents, there is every
chance that small concentration of these solvents remains with the main
drug. The non reacting impurities in the starting materials may also remain

18
with the drug unless the manufacturers are very careful about the
impurities.
In Paracetamol bulk drug substance; there is a limit test for p-
aminophenol, which is the starting material. This is clear from the synthesis
of Paracetamol shown in Figure 2.2. Impurities present in the staring
materials could also follow the same reaction pathways similar to the
starting material, and the reaction products could carry over to the final
product as process impurities. Knowledge of the impurities in the starting
materials helps to identify related impurities in the final product, and to
understand the formation mechanisms of these related process impurities.
By-products: In synthetic organic chemistry, getting a single end
product with 100% pure is very rare; there is always a chance of having
some by-products. In the case of Paracetamol bulk, shown in Figure 2.2
diacetylated paracetamol is the by-product which will remain as process
related impurity in the drug.
Figure 2.2: Synthesis route of Paracetamol showing the origin of
impurities.
Enantiomeric impurities: The single enantiomeric form of a chiral
drug is now considered as an improved chemical entity that may offer a
better pharmacological profile and an increased therapeutic index. For the

19
manufacturers of single enantiomeric drug, the undesirable stereo isomers
in the drug are considered in the same manner as the other organic
impurities. The prominent single isomer drugs, which are being marketed,
includes levocetrizine , esomeprazole, Eszopiclone etc. [10,11,12].
Degradation products: Impurities can also be formed due to
degradation of the drug during manufacturing of bulk drugs. However,
degradation of drug resulting from storage or formulation to different dosage
forms or aging are common impurities in the medicines. Also degradation
occurs by few pathways but the most often seen mechanism is hydrolysis.
Many drugs will show tendency of decomposition by hydrolysis.
In case of aspirin [13] the degradation of aspirin in various buffer
solutions was studied and the degradants are identified as salicylic acid and
acetic acid Figure 2.3. Oxidation and photolysis are the next two
mechanisms that degrade the main drug. From Figure 2.4, it can be seen
that exposure of Ciprofloxacin to light will lead to photolysis and formation
of Ethylene diamine analog of Ciprofloxacin [14].
Figure 2.3: Degradation of Aspirin by hydrolysis

20
N
O
OH
O
F
N
NH
N
O
OH
O
F
NH
NH2
Sun Ligjt
Figure 2.4: Degradation of Ciprofloxacin by photolysis
Reagents, Ligands and Catalysts: These chemicals are less commonly found
in pharmaceutical compounds; however, in some cases they may pose a
problem as impurities .Chemical reagents, ligands, and catalysts used in the
synthesis of a drug substance can be carried over to the final products as
trace level impurities. For example, carbonic acid chloromethyl tetrahydro-
pyran-4-yl ester (CCMTHP), which is used as an alkylation agent in the
synthesis of ß lactam drug substance, is observed as an impurity in the final
product.
Metal-based catalysts promote many chemical reactions. For example,
use of Ziegler-Natta catalyst may result in Ti or TiO2 as impurity in the
main drug, Grubb‟s catalyst ruthenium, and Adam‟s catalyst platinum. In
some cases, reagents or catalysts may react with intermediates or final
products to form by-products. Pyridine, a catalyst used in the course of
synthesis of mazipredone, reacts with an intermediate to form a pyridinium
impurity [15].
2.4.3. Impurities that are formed during formulation
Apart from bulk drug-related impurities, the formulated form of API
may contain impurities that originated from various ways. Impurities arising

21
from excipients present in the drug product or extracted or leached from the
container system, due to environmental factor such as temperature, light
particularly UV light, humidity and storage conditions of the drug called
aging. A known impurity, 1-(2,6-diclorophenyl)indolin-2-one is formed in the
production of a parenteral dosage form of diclofenac sodium, if it is
terminally sterilized by autoclave. It is the condition of the autoclave method
(i.e., 123 + 2°C) that enforced the intramolecular cyclic reaction of diclofenac
sodium forming the indolinone derivative and sodium hydroxide.
In general, liquid dosage forms are very much susceptible to both
degradation and microbiological contamination. In this regard, water
content, pH of the solution/suspension, compatibility of anions and cations,
mutual interactions of ingredients, and the primary container are critical
factors. Ciprofloxacin (0.3% preparation) is available on the market for
topical ophthalmic use. If the ophthalmic solutions are exposed to sun light,
they may lead to formation of a degradation product, i.e. ethylenediamine
derivative of ciprofloxacin.
2.4.4. Inorganic impurities
Inorganic impurities arise from the manufacturing process, from
reagent, ligands, and catalysts. Inorganic salts, heavy metals, filter aids and
charcoal falls under this category. Metal residues also fall under this
category. Metal residues in pharmaceutical substances or the drug products
may originate from several sources like metal catalysts and metal reagents
used during the synthesis of the active pharmaceutical substance and the
excipients, manufacturing equipment and piping, bulk packaging and the

22
environment. As per European medicine agency recommendation [16], limits
for metal residues in the pharmaceutical substance are presented in Table
2.1.
Table 2.1: Specification limits for metal residues in pharmaceutical
products
Classification Oral Exposure Parenteral Exposure
Permitted
Daily
Exposure
(μg/day)
Concentration
(ppm)
Permitted
Daily
Exposure
(μg/day)
Concentratio
n
(ppm)
Class 1A: Pt, Pd
Class 1B: Ir, Rh, Ru,
Os
Class 1C: Mo, Ni, Cr, V
Metals of significant
safety concern
100
100
250
10
10
25
10
10
25
1
1
2.5
Class 2: Cu, Mn
Metals with low safety
concern
2500 250 250
25
Class 3: Fe, Zn
Metals with minimal
safety concern
13000 1300 1300 130
2.4.5. Residual solvents
Residual solvents may be present in pharmaceutical substance as they
are used or produced in the manufacture of the drug substances or
excipients, or in the preparation of drug products. Appropriate selection of
the solvent for the synthesis of the drug substance may enhance the yield, or
determine characteristics such as crystal form, purity, and solubility.

23
Therefore, the solvent may sometimes be a critical parameter in the
synthetic process. Since there is no therapeutic benefit from residual
solvents, all residual solvents should be removed to the extent possible to
meet product specifications. The solvents are not completely removed by
practical manufacturing techniques.
International conference on Harmonization (ICH) classified the solvents
based on the toxicity of solvents in to three classes, Class-1, Class-2, and
Class-3 [17]. Solvents which are known human carcinogens, strongly
suspected human carcinogens, and environmental hazards are classified
into Class 1, solvents that are to be avoided. The details of solvents,
concentration limits in the main drug and the effects they cause if in excess
are presented in the Table 2.2.
Table 2.2: Class 1 solvents and their specification limits in
pharmaceutical products
Solvent Concentration limit (ppm) Concern
Benzene 2 Carcinogen
Carbon tetrachloride 4 Toxic and environmental
hazard
1,2-Dichloroethane 5 Toxic
1,1-Dichloroethene 8 Toxic
1,1,1-
Trichloroethane 1500 Environmental hazard
Class 2 solvents: Solvents to be limited, Non-genotoxic animal carcinogens or
possible causative agents of other irreversible toxicity such as neurotoxicity

24
or teratogenicity. These are the solvents suspected of other significant but
reversible toxicities. The details of these solvents are presented in Table 2.3.
Class 3 solvents: Solvents with low toxic potential to human; no health-based
exposure limit are classified as Class 3. An exposure of 50 mg or more per
day and concentration limit is 5000 ppm is permitted for these solvents. The
list of such solvents is presented in Table 2.4.
Table 2.3: Class 2 solvents and their specification limits in
pharmaceutical products
Solvent Permitted Daily
Exposure (mg/day)
Concentration
limit (ppm)
Acetonitrile 4.1 410
Chlorobenzene 3.6 360
Chloroform 0.6 60
Cyclohexane 38.8 3880
1,2-Dichloroethene 18.7 1870
Dichloromethane 6.0 600
1,2-Dimethoxyethane 1.0 100
N,N-Dimethylacetamide 10.9 1090
N,N-Dimethylformamide 8.8 880
1,4-Dioxane 3.8 380
2-Ethoxyethanol 1.6 160
Ethylene glycol 6.2 620
Formamide 2.2 220
Hexane 2.9 290
Methanol 30.0 3000
2-Methoxyethanol 0.5 50
Methylbutyl ketone 0.5 50
Methylcyclohexane 11.8 1180
N-Methylpyrrolidone 5.3 530
Nitromethane 0.5 50
Pyridine 2.0 200
Sulfolane 1.6 160
Tetrahydrofuran 7.2 720
Tetralin 1.0 100

25
Toluene 8.9 890
1,1,2-Trichloroethene 0.8 80
Xylene 21.7 2170
Table 2.4: List of Class 3 solvents
Solvents
Acetic acid Ethanol 3-Methyl-1-butanol
Acetone Ethyl acetate Methyl ethyl ketone
Anisole Ethyl ether Methyl isobutyl ketone
1-Butanol Ethyl formate 2-Methyl-1-propanol
2-Butanol Formic acid Pentane
Butyl acetate Heptane 1-Pentanol
tert-Butylmethyl ether Isobutyl acetate 1-Propanol
Cumene Isopropyl acetate 2-Propanol
Dimethyl sulfoxide Methyl acetate Propyl acetate
2.5. Quality Control of drugs
Quality is the most important factor for the products and
manufacturing firms. Quality control is a process by which entities review
the quality of all factors involved in production. Quality control is a
mechanism for ensuring that the product conforms to predetermined
specifications. Quality is important in every product but it is vital in
medicine as it involves life of human beings. Unlike ordinary consumer‟s
goods, there is no second quality in drugs.
Synthetic bulk drugs are produced in the production plants in large
quantities using different manufacturing technologies. The purity of a drug
and the nature and quantity of any impurities present in that drug are very

26
important for the modern pharmaceutical analyst. Synthetic precursors, by-
products, un-reacted raw materials, intermediates and products of
degradation are the potential components of manufacturing processes likely
to be present as impurities in the bulk drugs and their formulations.
Impurities in reagents or starting materials may be carried out intact
through the synthesis or react to produce new impurities. These impurities
often pose unwanted pharmacological or toxicological effects due to which
any benefit from the administration of the drug is outweighed. Impurities in
reagents or starting materials may be carried out intact through the
synthesis or react to produce new impurities.
Threshold for impurities specified in ICH impurities guideline [7],
which is mentioned in the following Table 2.5. In most of the cases, the daily
dosage of drugs may be less than 2 grams per day; hence general limit for
any unknown impurities should be less than 0.10% and for any identified
impurity limit is not more than 0.15%. As per ICH recommendation, the limit
of quantification (LOQ) requirement should be at least 50% target
concentration, in case of unknown impurities, the LOQ requirement is less
than 0.05%. Hence, the sensitivity of preferred method for determination of
impurities is very critical.
Table 2.5: ICH guidelines for impurities present in a drug
Maximum
Daily Dose
Reporting
Threshold
Identification
Threshold
Qualification
Threshold
2g/day 0.05% 0.10% or 1.0 mg per day
intake (whichever is
lower)
0.15% or 1.0 mg per
dayintake (whichever is
lower)

27
> 2g/day 0.03% 0.05% 0.05%
2.5.1. Different measurement techniques for impurities determination
a. Organic impurities
Majorly organic impurities like related substances, raw materials used
in process intermediates, by products and degradants are being determined
frequently by using thin layer chromatography (TLC), high performance
liquid chromatography (HPLC), and in case of volatile, thermally stable
materials, Gas chromatography (GC) is used. Other chromatographic and
electrophoretic techniques such as supercritical fluid chromatography,
capillary electrophoresis and capillary electro chromatography are also being
used. Mass spectroscopy (MS) and Nuclear magnetic Resonance (NMR) can
also contribute in the identification and determination of the impurities [14].
b. Inorganic impurities
For determination or inorganic impurities like chlorides, sulfate,
sulfites etc conventional gravimetric and titremetric techniques may be
adopted. Ion chromatographic techniques are being employed as they are
very specific and sensitive. The heavy metal concentration is reported to be
estimated by the conventional sulfide precipitation techniques [18]. As the
above techniques lack the sensitivity, specificity, and poor recovery to
monitor properly low levels of these metals, for the determination of toxic

28
metal impurities United States Pharmacopeia [19] proposed inductively
coupled plasma–atomic emission spectrometer ICP-AES and inductively
coupled plasma mass spectrometer ICP-MS methods.
c. Residual solvents
For Class 3 solvents, non-specific method such as loss on drying may
be used. Class 1 and Class 2 solvents are to be determined using
chromatographic techniques such as gas chromatography. For making
injection, “Head space” sampling technique is being employed to avoid
interferences of the product and to achieve desired sensitivity [20].
2.6. ICH activity and quality guidelines
The ICH was established in 1990 as a joint regulatory and industry
project to improve, through harmonization, the efficiency of the process for
developing and registering new medicinal products in Europe, Japan and the
United States of America, to make these products available to patients with a
minimum of delay. The six parties involved in ICH process represent the
regulatory bodies and pharmaceutical industry in the three regions, Europe,
Japan and the USA.
The parties involved from these three regions are – European
Commission (EC), European Federation of Pharmaceutical Industries and
Associations (EFPIA); Ministry of Health, Labor and Welfare (MHLW), Japan
Pharmaceutical Manufacturers Association (JPMA); and US Food and Drug
Administration (FDA), and the Pharmaceutical Research and Manufacturers
of America (PhRMA).
The fourth International Conference on Harmonization in 1997

29
marked the completion of the first phase of activities. In the first phase the
exercise was directed towards elimination of redundant and duplicates
technical requirements for registration in individual countries, and lay down
of minimum standards applicable uniformly irrespective of where the
product is manufactured and/or marketed in the three regions. This was
done with development of over 40 guidelines covering Efficacy, Quality and
Safety aspects of drug development. The list of „Quality‟ guidelines is given in
Table 2.6 [21].
Table 2.6: List of topics, codes and corresponding quality guidelines
developed by ICH
Topics /
Code Quality guidelines
Q1A(R2) Stability Testing of New Drug Substances and Products
Q1B Stability Testing: Photo stability Testing of New Drug Substances and Products
Q1C Stability Testing for New Dosage Forms
Q1D
Bracketing and Matrixing Designs for Stability Testing of Drug Substances and
Drug Products
Q1E Evaluation of Stability Data
Q1F Stability Data Package for Registration in Climatic Zones III and IV
Q2(R1) Validation of Analytical Procedures: Text and Methodology
Q3A(R2) Impurities in New Drug Substances
Q3B(R2) Impurities in New Drug Products
Q3C(R4) Impurities: Guideline for Residual Solvents
Q4 Pharmacopeias
Q5A(R1)
Viral Safety Evaluation of Biotechnology Products Derived From Cell Lines of
Human or Animal Origin
Q5B
Quality of Biotechnological Products: Analysis of the Expression Construct in
Cells Used for Production of R-DNA Derived Protein Products
Q5C
Quality of Biotechnological Products: Stability Testing of Biotechnological/
Biological Products
Q5D
Derivation and Characterization of Cell Substrates Used for Production of
Biotechnological/Biological Products
Q5E Comparability of Biotechnological/Biological Products Subject to Changes in Their Manufacturing Process
Q6A
Specifications: Test Procedures and Acceptance Criteria for New Drug
Substances and New Drug Products: Chemical Substances

30
Q6B Specifications: Test Procedures and Acceptance Criteria for Biotechnological/Biological Products
Q7 Good Manufacturing Practice Guide for Active Pharmaceutical Ingredients
Q8(R2) Pharmaceutical Development
Q9 Quality Risk Management
Q10 Pharmaceutical Quality system
2.7. Techniques applied during quality control testing
In the field of drug analysis, the analytical investigation of bulk drug
materials, the intermediates in their synthesis, products of drug research,
drug formulations, impurities and degradation products, and biological
samples containing the drugs and their metabolites is a very important area
of research. From the point of view of public health, the safety, efficacy, and
economy of drug therapy are extremely important issues.
The importance of drug impurity and stability-related issues has also
been characterized by a number of books and articles devoted to this subject
[22].The determination of drugs and metabolites in biological samples [23],
with particular attention to toxicological and forensic analysis, requires
special techniques and a special manner of thinking [24, 25], as reflected by
many books and articles on these issues.
Drug discovery requires a solid analytical background, with a great
variety of methods to be used. Innumerable drug-related chapters have also
appeared in general analytical books and special issues of scientific journals.
During synthesis of drugs and during their stability studies lot of impurities
may form which maybe organic, inorganic and residual solvents.
2.7.1. Application of Chromatographic Techniques

31
Rapid development of analytical methodology in pharmaceutical and
biomedical analyses has led to various forms of high-performance liquid
chromatography (HPLC) becoming undoubtedly the most important
methods. The theoretical and practical foundations for this method were laid
down at the end of 1960s and the beginning of 1970s [26]. The latter decade
was the period that saw a rapid spread of this technique.
In pharmaceutical analysis, the HPLC method shares its importance
with various techniques. HPLC has been used to solve no less than 50% of
the problems, leaving the other 50% to about 15 other chromatographic,
spectroscopic, and other methods, about 10% to gas chromatography (GC),
5% to thin-layer chromatography (TLC), 10% to ultraviolet (UV)
spectrophotometry, and the rest to electro analytical methods.
The contribution of HPLC in drug analysis has further increased in
this long period. For example, in 1983, a UV detector was applied almost
exclusively, leaving a little share to refractive index, fluorimetric, and
electrochemical detection, but in 2005, a mass spectrometer was applied as
a detector in about one-third of the analysis.
HPLC coupled with mass spectrometry, HPLC/MS (MS) or LC/MS (MS)
due to high sensitivity and selectivity have become the predominant method
in bioassays and pharmacokinetic and metabolic studies, as well as in the
structure elucidation of drug impurities and degradation products. A new
development in the field of HPLC/MS has been the introduction of column
packing with ultrafine particles (< 2 um)[27,28,29], enabling short columns
(5 cm or less) to be used, and rapid analyses (e.g., 5 minutes or even less

32
than 1 minute) to be carried out by ultra performance liquid
chromatography(UPLC).
In the compendial analysis of small organic molecules, the breakthrough
of HPLC was also extremely rapid. In the twenty-ninth United States
Pharmacopoeia, HPLC was applied to the assay of bulk drug materials of this
type in about 45% of the monographs [30]. This share was somewhat higher
than that of the non-selective ones, but with titration methods that were less
time-consuming, leaving only about 10% to other methods, mainly the
similar, non-selective UV-Vis spectrophotometry. HPLC and TLC were used
almost exclusively, with almost equal shares for the purity control of bulk
drug materials and the related compounds test.
Even more spectacular was the propagation of HPLC in the assay of
pharmaceutical formulations, which needed specific methods indicating
stability. No other method had spread so rapidly in pharmaceutical analysis.
Among other chromatographic methods the important application field of
modern TLC is the separation of the components of complex mixtures, for
example, impurities and degradation products of drug materials and extracts
of medicinal plants.
The speed and the resolution could be greatly improved by the
introduction of special techniques, such as high-performance thin-layer
chromatography (HPTLC), using ultra thin layers and coatings with ultrafine
particles or over pressured-layer chromatography (OPLC). The development
of densitometers enables classical TLC and the latter techniques could be
successfully used as tools for the quantitative analysis of complex mixtures.

33
The introduction and rapid spread of HPLC and HPLC/MS decreased
the importance of GC and GC/MS in pharmaceutical analysis. Nevertheless,
these are still important techniques in many fields of drug analysis [31],
where the analytes are volatile and thermally stable. In the past 15 to 20
years a new field application is being used for the determination of residual
solvents in drugs. Almost always the headspace technique is used to fulfill
the demanding requirements, that is, determination of solvents at the 10-
ppm level; and down to the ppm level in the case of carcinogenic or genotoxic
solvents.
2.7.2. Application of Capillary Electrophoresis
Since the introduction of the commercially available instruments
capillary electrophoresis (CE), related methods such as micellar electro
kinetic chromatography (MEKC), micro emulsion electro kinetic
chromatography (MEEKC), and capillary electrochromatography (CEC), have
attracted great interest in pharmaceutical analysis as possible alternatives
or amendments to HPLC [32].
This share is an underestimation, as there are researchers specializing
in CE analysis. It is an overestimation because, despite CE having several
advantages such as a flat flow profile that results in an extremely high
column efficiency, due to its limitations with regard to its general
applicability, it does not yet seem to be a real rival to HPLC in the practice of
compendial-industrial pharmaceutical analysis.
The separation and quantification of enantiomeric mixtures are among
the greatest challenges of the past years in pharmaceutical and biomedical

34
analysis. The main problems to be solved are, to determine the enantiomeric
purity of drugs being used in therapy as pure enantiomers and the
simultaneous determination of the components of race-mates in the
biological samples [33]. The latter type of enantiomeric separation has been
successfully adapted to CE. The present situation can be characterized by
the spread of this technique and the continuous development and
commercialization of new types of chiral HPLC columns [34].
2.7.3. Application of Spectroscopy
In pharmaceutical and biomedical analysis, the development of
nuclear magnetic resonance (NMR) and mass spectrometry (MS), along a
road paved with Nobel Prizes, has also been successfully exploited. The
dramatic decrease in the demanding requirements for sample size and the
solution of the difficult problems of interfacing these techniques with
chromatographic (and electrophoretic) separation methods have greatly
expanded their field of application.
In addition to the offline applications that are still widely used, online
HPLC/MS, HPLC/ NMR, HPLC/NMR/MS, and other hyphenated methods,
are becoming leading methods, for example, the structure elucidation of
drug impurities, degradation products, metabolites, and bioactive
components in natural products. Due to its high sensitivity and selectivity,
HPLC/MS/MS has become the predominant method, even in the
quantitation of these minor components (e.g., in pharmacokinetic and
bioequivalence studies) [35].

35
UV spectroscopy [36] is observable due to the availability of diode-
array detectors attached to HPLC and TLC densitometers, both suitable for
obtaining good-quality spectra, which are often useful; in identifying
impurities for example. As for the quantitative analytical application of this
technique, approximately 10% of the share in pharmacopoeias for the assay
of bulk drug materials and pharmaceutical formulations is very slowly
decreasing.
The most important field of application of infrared (IR) [37] and near-
infrared (NIR) spectroscopy [38] is the identification of drugs. IR has greatly
decreased (almost completely eliminated) the importance of the classical
color tests, while NIR is a method of increasing importance in the in-process
control of manufacturing pharmaceutical formulations. IR and Raman
spectroscopy, together with solid-phase NMR, X-ray diffraction, and thermal
methods are the up-to-date methods in solid-phase characterization, which
is of great importance in developing pharmaceutical formulations, with
optimal bioavailability [39].
In recent times FTIR spectroscopy has been coupled with ATR
(attenuated Total Reflection).FTIR spectroscopic imaging in ATR mode is a
powerful tool for studying biomedical samples and dissolution of
pharmaceutical formulations and drug release. One of the key advantages of
ATR-FTIR imaging is that is requires minimal or no sample preparation prior
to spectral measurements. Consequently, this approach is particularly
suitable to measure substances with strong infrared absorption such as
water.

36
The application of ATR-FTIR imaging also allows for the
characterization of biomedical materials in tissue engineering. Also the
quantitative information about the spatial distribution of chemical
components on pharmaceutical tablets in contact with water, as a function
of time, provides an important basis for building new mathematical models
for the optimization of controlled drug delivery. ATR-FTIR imaging is suitable
for imaging of realistic tablets in contact with aqueous solutions because of
the shallow penetration of the evanescent wave into the sample.
In pharmacopoeias, for the study of toxic metal impurities, the
classical sulfide and other limit tests are still widely used. At present, the
rapidly increasing importance of the much more selective and sensitive
atomic spectroscopic methods can be observed, such as, graphite furnace
atomic absorption spectrometry (GF-AAS), inductively coupled plasma
atomic emission spectrometry (ICP/AES), and mass spectrometry (1CP/ MS).
2.7.4. Other Analytical Techniques
The classical titrations non-selective method is still widely used in
compendial analysis for the assay of bulk drug materials. Even in the USP,
where the breakthrough of HPLC has been much faster, more than 40% of
the low molecular weight organic compounds are determined by aqueous or
non-aqueous titration. Other electro analytical methods have always been
only modestly important in pharmaceutical analysis. Classical polar graphic
methods using toxic mercury electrodes are being driven out from practice
and replaced by new electrodes, for example, glassy carbon electrodes
modified with carbon nanotubes, which provide highly sensitive analyses.

37
Another field where remarkable results have been obtained is the
development of ion-specific and molecule-specific sensors. Flow-injection
analysis with various detectors such as spectroscopic, electro analytical
chemiluminescence is often used in the analysis of drug formulations. All
antibiotics, 30 years ago, were determined using microbiological methods.
In modern pharmacopoeias [40], in the majority of cases, these are
replaced by much more selective and informative methods [41], mainly
HPLC. Although the importance of immunoassays has decreased in the
recent years, they are still often used in the determination of some bioactive
compounds in the biological samples. Radioimmunoassay has been greatly
superseded by various enzyme-immunoassay methods.
2.7.5. Application of Microscopy Techniques
A useful instrument for the structural and morphological study of
novel films, nanoparticles, hydrogels, matrices, and porous scaffolds, is
offered by modern microscopy tools, such as, atomic force microscopy (AFM),
scanning electron microscopy (SEM), and confocal Raman microscopy
(CRM).The imaging tools are important for analyzing the surface of soft
organic materials, as understanding these surfaces may help to shed light
on the interactions that occur between crystals and their surrounding
environment[42].
Drug release is the result of a complex interplay between the drug, its
carrier, and the release environment. Study of the surface structure and
morphology of pharmaceutical substances contributes to an understanding
of surface activity and is of critical importance to the pharmaceutical

38
industry. Modern microscopes are tools that are applicable for the collection
of topographic data and morphological investigation of different applicable
pharmaceutical materials.
2.7.6. Future Trends
Globalization of the drug market and the sharpening concurrence
among the drug companies has caused pharmaceutical analysis to become
one of the battlefields in the struggle. The importance of issues related to
drug safety has greatly increased and this has led to the continual increase
of demands with regard to securing the quality of drugs, and often over
securing the safety of drug therapy. It became necessary to harmonize the
demands and analytical strategies [43].
The first step was the establishment in the European Pharmacopoeia,
of which the Sixth Edition is now official. This became the basis of the
national pharmacopoeias of the member states of the European Union. The
next step was the formation of ICH, which was done with the aim of
harmonizing the efforts of registration agencies, principal pharmacopoeias
(Ph. Eur., USP, and Japanese Pharmacopoeia), and pharmaceutical
manufacturers' organizations, to improve the quality of drugs and the safety
and efficacy of drug therapy.
The guidelines issued by ICH are authoritative worldwide with respect
to drug quality issues. It has to be noted that requirements with regard to
the quality of drugs and drug formulations in the drug market are, in
practice, much greater than those prescribed in the pharmacopoeias and
ICH guidelines.

39
The greater change in the pharmacopoeias in the past years has been the
increasing importance of purity tests. At the beginning only a very limited
number of monographs contained tests related to impurities.
Thanks to the development of TLC and HPLC, at present, an
overwhelming majority of the monographs on bulk drugs, and in a fairly high
proportion of those on formulations, contain these tests. The impurity profile
has become the most informative indicator of the quality of bulk drug
materials. At the same time, the importance of assaying bulk drugs has
decreased considerably; moreover, there are opinions that even this
importance is questionable.
The tendencies toward globalization and harmonization mentioned earlier
and the necessity of increasing the safety of drug therapy, have prompted
the validation of analytical methods to the forefront; moreover, it has become
one of the most important issues in contemporary drug analysis. However,
some negative tendencies are also apparent:
A fully validated method meeting the requirements of various guidelines
needs much more analytical data than would be strictly necessary
Many drug analysts, especially among the young generation, feel that the
essence of pharmaceutical analysis is the mass production and handling
of data, rather than problem solving
The way of thinking is changing, with many people, mainly outside the
circles of drug analysts, believing that possession of up-to-date,
automated computerized instruments and validated methods
automatically give good

40
and reliable results. Pharmaceutical analysis is an important field of
activity in the interest of suffering mankind, through increasing the safety
of drug therapy.
2.8. Importance of HPLC in the quality control of Drugs
Thin layer Liquid Chromatography (TLC), Gas Chromatography (GC)
and High Performance Liquid chromatography or Liquid Chromatography
(HPLC or LC) are the important chromatographic techniques, widely used in
the quality control of drugs in the pharmaceutical industry. HPLC is the
dominant method in pharmaceutical analysis in quality control, because it is
most versatile technique, having high resolving power, sensitivity, advanced
column, detector technologies, and commercial availability, flexible
separation modes and capability of computerized optimization of separation.
In the past two decades, LC has replaced GC in most of the QC tests
due to its simplicity and applicability to polar and thermally labile species.
Most synthetic drugs are in this category. HPLC is a powerful separation
method, suited to resolving a large number of similar substances in a short
time. HPLC is especially significant for the pharmaceutical industries
because it allows for both qualitative and quantitative analysis. HPLC has
been used to solve no less than 50% of the problems, leaving the other 50%
to 15 other chromatographic, spectroscopic, and other methods.
A new development in the field of HPLC has been the introduction of
column packing with ultrafine particles (< 2 um), enabling short columns (5
cm or less) to be used, and rapid analyses (e.g., 5 minutes or even less than

41
1 minute) to be carried out by Ultra Fast Liquid Chromatography (UFLC) or
Ultra Performance Liquid Chromatography (UPLC) .
2.9. Importance of Stability- indicating methods
a. Introduction:
Stability studies need to be performed to provide evidence of how the
quality of a drug substance or drug product varies with time under the
influence of variety of environmental factors such as temperature, humidity
and light [44]. As a result of stability testing, a re-test period for the drug
substance or a shelf-life for the drug product can be established and storage
conditions can be fixed for that drug. The analytical test method used for
testing samples which are under stability evolution shall be capable enough
to detect the changes happening in the product [45].
As per US FDA guideline, a stability-indicating method can be defined
as “validated quantitative analytical methods that can detect the changes
with time in the chemical, physical, properties of the drug substance and
drug product, that are specific so that the contents of the active ingredient,
degradation products and other components of interest can be accurately
measured without interference [46].
The purpose of establishing the stability-indicating power of the
method is to ensure that while analyzing the test samples all the related
substances, process impurities and degradation products etc. are separated
from each other and from the peak of the main drug of interest. The
requirement of stability-indicating methods is specified in various regulatory
guidelines. World Health Organization (WHO) stability guidelines specify that

42
analytical procedures used for stability monitoring should be fully validated
and stability-indicating.
The ICH guidelines on Good Manufacturing Practices state that the
test procedures used in stability testing should be validated and stability-
indicating [47]. European guidelines state that the testing should cover all
the appropriate, physical, chemical, biological, and microbiological
attributes. Validated stability-indicating analytical procedures should be
applied [48].
United States Pharmacopoeia (USP) has a requirement for adopting a
method to USP, which says that whenever possible, a stability-indicating
procedure should be used for the Assay [49]. Generally, chromatographic
procedures are stability-indicating and titration procedures are not. During
registration of any product with US or Europe agencies, when a non-stability
indicating assay is proposed, a separate stability-indicating impurity
procedure should be provided.
b. Evaluation of stability-indicating power
Stress testing of the active substance can help identify the likely
degradation products, which can in turn help establish the degradation
pathways and the intrinsic stability of the molecule and validate the
stability-indicating power of the analytical procedure used. Stress testing is
likely to be carried out on a single batch of the drug substance. The ICH
guidelines Q1A suggest the following conditions to be employed [50]:
(i) 10°C increments above the accelerated temperatures (e.g.50°C, 60
°C,etc.),

43
(ii) Humidity where appropriate (e.g. 75% or greater),
(iii) Hydrolysis across a wide range of pH values, Oxidation and Hydrolytic
degradation studies need to be performed using 0.1M HCl /NaOH,
oxidation using 3% H2O2. If reasonable degradation is seen study will be
stopped otherwise extended to further harsh condition along with heat
or reflux for longer duration. Alternatively, if total degradation is seen
after subjecting the drug to those conditions, acid/alkali strength can
be decreased along with decrease in the reaction temperature.
(iv) Photolysis, exposing the sample to light providing an overall
illumination of not less than 1.2 million lux hours and an integrated
near ultraviolet radiation with an energy of not less than 200 watt
hours/square meter [51].
The target decomposition of the product shall be about 5 to 10%. The
stressed samples are subjected to analysis under proposed method
conditions to study the separation of degradation products. Well separated
peaks with high resolution are expected for a stability-indicating method.
Also homogeneity of peaks need to be established that to ensure the purity of
peaks and simultaneously to prove no co elution of peaks. For peak
homogeneity, the most popular technique is the Photo Diode Array (PDA)
analysis, the principle of which is the comparison of the spectra of the
analyte peak, taken upslope, at the apex and on the down slope.
If these spectra do not match then the peak is non-homogeneous.
Homogeneity of the peak also can be established by liquid chromatography
mass spectrometry (LC-MS) technique by scanning entire peak for its mass

44
value. Therefore PDA or LC-MS results suggest that if the products are
different but are co-eluting, then suitable modification should be done in the
chromatographic method to achieve a satisfactory resolution.
2.10. Role of Mass Balance during HPLC development
Mass balance correlates the measured loss of a parent drug to the
measured increase in the amount of degradation products [52]. It is a good
quality control check on analytical methods to show that all degradation
products are adequately detected and do not interfere with quantitation of
the parent drug (i.e., stability-indicating methods). Regulatory agencies use
mass balance to assess the appropriateness of the analytical method as a
stability-indicating method and determine whether all degradants have been
accounted for [53].
In mass balance calculations, the loss of parent drug or the amount of
drug remaining is determined from a sample assay, and the measured
increase in degradation products is determined by a related substances
method. The fundamental approach for determining mass balance is to
quantitate the decomposition peaks using degradation methods and then
reconciles the measured loss in the parent drug with the amount of
degradation products. If the loss in potency can be reasonably accounted for
by the amount of degradants measured, then mass balance is achieved.
The assessment of degradation in pharmaceutical products involves
two aspects of analytical measurement. Firstly, a specific or selective
analytical method must be available for accurate assay of parent drug
compound, in order to measure any loss. Second, methodology should be in

45
place for quantification of the degradation products formed. Ideally, when
degradation occurs, the measured amount of parent drug lost should
correlate well with the measured increase in degradation products. This
correlation is referred to as “mass balance”. More recently, the ICH has
provided definition of “mass balance; material balance” as follows [54]
The process of adding together the assay value and levels of
degradation products to see how closely these add up to 100% of initial
value, with due consideration of the margin of analytical precision. The
concept is useful scientific guide for evaluating data, but it is not achievable
in all circumstances.
The focus may instead be on assuring the specificity of the assay, the
completeness of the investigation of route of degradation, and the use, if
necessary, of identified degradants as indicators of the extent of degradation
via particular mechanism [55]. The analyst must balance time and resource
demands to provide the information necessary to understand degradation
without going to extreme measures of quantify components of little interest.
Mass balance in pharmaceutical analysis is very important for several
reasons. By demonstrating the degradative losses of parent drug correlate
well with the measured increase in degradation products unaccounted for.
Conversely, if one observes, for example, a 20% loss of parent drug but only
measures a 5% increase in degradation products, it is likely that additional
degradation products formed are not accurately determined by the given
method(s). Because unknown degradation products could potentially be
toxic or otherwise compromise the safety of drug, it is important to have

46
methods that detect all major degradation products. Thus, safety is the
major reason for the study of mass balance.
Mass balance is also useful in method validation [56]. In order to
demonstrate that analytical methods are stability-indicating, unstressed and
stressed materials are often compared. Any increase in degradation a
product that correlates well with loss of parent drug, aids in demonstrating
that the methods can accurately assess degradation.
Mass balance is also important in understanding alternative
degradation pathways [57]. For example, consider a situation where both
acid-catalyzed and oxidative degradation produces a substantial loss of
parent compound in stress-testing studies. If good mass balance is achieved
for the acid-catalyzed degradation, but not for the oxidative degradation,
further work to better understand the oxidative degradation pathway(s) is
warranted.
It may be that the poor mass balance in the latter case results from
important oxidative products that are unaccounted for or from structures,
which need to be more fully elucidated to understand response factor
differences. Mass balance is an important consideration in assessing
degradation pathways of pharmaceutical products. Often, response factor
differences between degradation products and the parent compound are
responsible for mass balance problems. Relative Response Factor (RRFs)
should be incorporated, when possible, in the quantification of degraded
samples.

47
Mass balance is also an approach to establish the validity of stability-
indicating method. It is the process of adding together the assay value and
levels of degradation products to see how closely these add up to 100% of
the initial value, with due consideration of the margin of analytical error.
Mass balance may not also be established due to formation of volatile
impurities during degradation, non availability of reference standards, some
of the products are strongly bound to stationary phase and do not elute at
all or elute after very long periods, variability of response or reduced
response in UV detector, drug content variation in drug products etc. [58].