chapter-3 an efficient, commercially viable and greener...
TRANSCRIPT

76 Chapter-3
CHAPTER-3
AN EFFICIENT, COMMERCIALLY VIABLE AND
GREENER PROCESS FOR THE PREPARATION
OF RANOLAZINE: AN ANTIANGINAL AGENT

77 Chapter-3
3.1 INTRODUCTION
Angina also known as Angina pectoris is indication for heart
disease caused by lack of blood circulation to the heart. The most
widespread reason for the angina is Atherosclerosis. In coronary heart
disease patients, arteries become narrow and stiff when compared with
the healthy heart arteries. These narrow and stiff arteries cause
difficulties to reach oxygen rich blood for heart. About 17 million
Americans are suffering with coronary heart diseases and about 9
millions are suffering with chronic angina.
Ranolazine1-4 is the one of the medicament used to manage
chronic angina, developed by Roche Bioscience (formerly Syntex) and
marketed by CV Therapeutics. USFDA was approved Ranolazine 2 under
brand name of Ranexa® in January 27, 2006. Subsequently European
medical agency (EMEA) approved in July 09, 2008. Latter on it was
approved in few other developing countries. Ranexa ® is available in
market in the form of 500 mg and 1000 mg film coated tablet and the
maximum daily dosage should be less than 2.0g. Over dosage of Ranexa
® lead to dizziness, nausea, and vomiting. Worldwide sales of Ranexa®
by December 2011 is about 400 millions USD (~2000 crores) with the
consumption of 1, 00, 678 kg. Major contribution is from USA i.e. about
300 millions USD. The above mentioned particulars of the angina drugs
motivated us to develop efficient, cost effective and moderately greener
process for the synthesis of ranolazine.

78 Chapter-3
3.1.1 PRODUCT PROFILE
1. Generic name : Ranolazine
2. Chemical structure :
N
NO
HN O
O
OH
3. Chemical names :
(±)-N- (2, 6-dimethylphenyl)-4-[2-
hydroxy-3-(2-methoxyphenoxy)
propyl]-1-piperazine acetamide
4. Molecular formula : C24H33N3O4
5. Molecular weight : 427.54
6. CAS No : 142387-99-3
7. Therapeutic category : Anti Angina
8. Indication : Chronic angina
3.1.2 PHYSICAL CHARACTERISTICS
1. Description of API : White to off white solid
2. Melting point : 119-120 °C
3. Solubility of API : Dichloromethane and methanol
(FDA Label)
3.1.3 MARKET INFORMATION
1. Applicant : Roche Bioscience (formerly Syntex)
2. Patentee : Roche Bioscience (formerly Syntex)

79 Chapter-3
3. Marketed by : CV Therapeutics
4. Brand name : Ranexa
5. USFDA Approval date : January 27, 2006
3.2 LITERATURE REVIEW
Amongst the various synthetic routes described for the
preparation of Ranolazine, some of the key approaches are discussed
here under. Kluge.F.A et al 5 have reported two synthetic approaches for
preparation of Ranolazine 2 using commercially available 2-Methoxy
phenol 25 and 2, 6-dimethyl aniline 20 as key starting materials. The
first synthetic route commenced with the synthesis of methyl oxirane
derivative 27. Key intermediate methyl oxirane derivative 27 was
synthesized from 25 and epichlorohydrin 26 in presence of NaOH
employing Williamson reaction conditions. Thus obtained 27 treated
with piperazine 23 in ethanol to obtain hydroxyl piperazine derivative
33. Thereafter, reaction of hydroxyl piperazine derivative 33 with phenyl
acetamide derivative 22 in dimethylformamide afforded dihydrochloride
salt of ranolazine 2, which was treated with ammonia to furnish
ranolazine 2(Scheme 3.1).
Second synthetic path way for the preparation of ranolazine
involves the condensation of piperazinyl acetamide intermediate 24 and
methyl oxirane 27 in mixture of methanol and toluene (Scheme 3.2).

80 Chapter-3
O
OHOCl+
O
O
NaOH
1,4-dioxane
O
Ethanol
NH
HN
25 26 27
23
O
O
33
N
OH NH
NH2
ClCl
O
+Triethyl amine
dichloromethane
HN
Cl
O
N
NO
HN O
O
OH
20 21 22
. 2HCl
DMF
Ammonia
N
NO
HN O
O
OH
2. 2HCl
2
Scheme 3.1: Synthesis of ranolazine 2 (product patent route)
O
OHOCl+
O
O
NaOH
1,4-dioxane
O
Ethanol
NH
HN
25 26 27
23NH2
ClCl
O
+TEA
DCM
HN
Cl
O
20 21 22
MethanolToluene
N
NO
HN O
O
OH
2
HN
N
O NH
24
Scheme 3.2: Synthesis of ranolazine 2 (product patent route)

81 Chapter-3
Mingfieng.S et al reported7 similar approach for the synthesis of
Ranolazine 2 utilizing hydroxy propyl halide intermediate 94 instead of
methyl oxirane compound 27. The requisite hydroxy propyl halide
intermediate 94 prepared by reacting 2-methoxy phenol 25 with 1, 3-
dichloropropan-2-ol 93 in presence of NaOH and mixture of ethanol &
water as shown in Scheme 3.3.
O
OH
+
NaOH
Ethanol, Water
Ethanol
NH
HN
25 9394
23NH2
ClCl
O
+TEA
DCM
HN
Cl
O
20 21 22
N
NO
HN O
O
OH
2
HN
N
O NH
24
Cl Cl
OH
O
O X
OHX=Cl, Br
K2CO3
MethanolToluene
Scheme 3.3: Synthesis of ranolazine 2
Eva.C.A et al.6 discovered an alternative synthetic path way for
preparation of Ranolazine. As depicted in Scheme 3.3 reaction of phenyl
acetamide derivative 22 with diethanolamine in presence of
triethylamine and subsequent chlorination using thionyl chloride
furnishes dichloro compound 91. Condensation of dichloro compound
91 with amino isopropanol derivative 92 provided Ranolazine 2. Amino
isopropanol derivative 92 is achieved by reaction of methyl oxirane
compound 27 with ammonia.

82 Chapter-3
HN
Cl
O
22
HN
N
O
90
DiethanolamineTEA, MIBK, NaI OH
OH
Thionyl chlorideChloroform
HN
N
O
91
Cl
Cl
O
O
NaOH
1,4-dioxane
92
O
O
O27NH2
OH
Aq. acetoneTriethyl amine
N
NO
HN O
O
OH
2
Scheme 3.4: Synthesis of ranolazine 2
The above discussed reported synthetic pathways for the
preparation of ranolazine 2 suffer from the intrinsic disadvantages such
as
(i) moderate yield and purities
(ii) longer reaction times
(iii) lengthy operations and tedious workup procedures
(iv) formation of large number of impurities.
(v) usage of carcinogenic and expensive solvents like 1, 4 dioxane and
ethers
(vi) highly energy consumed operations like fractional distillation at
elevated temperature (above 200 ˚C).
Drawbacks associated with the reported procedures motivated us
to develop an efficient, commercially viable, robust, moderately greener
and large scale synthesis for ranolazine 2.

83 Chapter-3
3.3 PRESENT WORK
3.3.1 OBJECTIVE
To develop an efficient, commercially viable, robust, moderately
greener and large scale synthesis for ranolazine 2. In addition,
comprehensive study of Ranolazine 2 and its key intermediates impurity
profile including identification, synthesis and characterization of all
potential impurities.
3.3.2 RESULT AND DISCUSSION
As per the retero synthetic pathway synthesis of ranolazine 2
involves majorly four reactions (Figure 3.1).
N
NO
HN O
O
OH
N
NHO
HN
+
O
O
O
O
OHOCl+
O
HN
+HN
NH
NH2
+ Cl
O
ClCl
1
Figure 3.1: Retro-synthetic pathway for ranolazine.
1. Formation of N-alkyl linkage.
2. Williamson ether synthesis.
3. Piperazine condensation reaction.
4. Chloro acetyl chloride condensation reaction.
All the raw materials (2, 6-dimethyl aniline, chloroacetyl chloride,
piperazine, 2-methoxy phenol and epichlorohydrin) obtained from the
retro synthetic pathway were commercially available in tonnage scale.

84 Chapter-3
Based on the retro synthetic pathway way (Figure 3.1) 2-((2-
methoxyphenoxy) methyl)oxirane 27 and N-(2, 6-dimethylphenyl)-2-
(piperazin-1-yl) acetamide 24 are identified as key synthetic fragments
for the preparation of ranolazine 2. In view of this detailed study was
carried out to overcome the disadvantages associated with the key
synthetic fragments 27 & 24.
3.3.2.1 SYNTHESIS OF 2-((2-METHOXYPHENOXY)METHYL)OXIRANE
27
Methyl oxirane compound 27 is also used as a key synthetic
fragment for synthesis of methocarbamol, moprolol and mephenoxalone
8 along with the ranolazine 2 (Figure 3.2).
O
O
O
O
O
O O NH2
O
OH
Methocarbamol
Mephenoxalone
Moprolol
NH
O
OHHN
O
Figure 3.2: API prepared from methyl- oxirane 27 as key intermediate.
Reported processes for the synthesis of 27 involved the reaction of
2-methoxyphenol 25 with epichlorohydrin 26 in presence of NaOH and
1, 4- dioxane by employing Williamson ether reaction conditions. These

85 Chapter-3
processes sfuffer from the intrinsic disadvantages such as (i) moderate
yield (~60%) and less purity (~60%), (ii) use of carcinogenic and highly
expensive organic solvents like 1, 4-dioxane and ethers and (iii)
purification of crude compound using highly energy consumed fractional
distillation.9-10
Formation impurities during the synthesis of methyl oxirane
compound 27 plays a significant role on the yield and purity of the 27.
To minimize the formation of impurities in 27, identification of all
potential impurities observed during the synthesis of 27 is desired.
3.3.2.1.1 Reaction mechanism
Reaction of 2-methoxy phenol 25 with epichlorohydrin 26 usually
proceed via two mechanisms i.e. either (i) direct nucleophilic
substitution (SN2) of chlorine with phenoxide (path-1)or (ii) opening of
epoxide ring with phenoxide (path-2) followed by intramolecular
nucleophilic substitution (SNi) of chlorine with alkoxide ion (Scheme 3.6).
O
OHNaOH
O
O Na O
O
OSN2
Ring opening
O
O Cl
O Na
Intramolecularcyclisation
SNi
path 1
path 225
3
27
ClO
26
ClO
Scheme 3.6: Possible mechanism for synthesis of 27

86 Chapter-3
3.3.2.1.2 Identification of impurities
Crude compound 27 was prepared by reproducing the process
reported in literature and thus obtained crude compound 27 was
analysed in LC-MS. Based on the reaction mechanism (Scheme 3.6),
reaction conditions and molecular weight obtained in LC-MS analysis,
impurities were anticipated as chloro hydroxy compound 28, diphenyl
compound 29 and dihydroxy compound 30 (Figure 3.3). These
impurities were synthesized and confirmed by their spectral data (Mass,
IR and NMR).
O
O Cl
OH O
O OH
OHO
O O
OH O
28 29 30
Figure 3.3: Structures of related compounds 28, 29 and 30.
Based on the chemistry knowledge, reaction mechanism and
process R&D experience the following process parameters are identified
as key parameters for the synthesis of methyl oxirane compound 27 and
these conditions are plays key role for the formation of above mentioned
potential impurities.
(a) solvent,
(b) epichloro hydrin 26 mole ratio
(c) NaOH mole ratio

87 Chapter-3
3.3.2.1.3 Solvent screening for synthesis of 27
Several solvents including non polar solvents (toluene), polar
solvents (water), chlorinated solvents (dichloromethane), polar aprotic
solvents (N, N-dimethylformamide), mixture of toluene & water and
water & 1, 4-dioxane are examined. Better yield and purity was obtained
in water medium (Table 3.1, entry 3) as compared with the other organic
solvents. By considering the experimental results, cost, economic
significance and greener components water was identified as suitable
solvent for the synthesis of 27.
Table 3.1: Synthesis of 27 using different solvents
Entry Solvent*
Yield of
27
(%)
Purity by HPLC (%)
27
(%)
28
(%)
29
(%)
30
(%)
1. Water & 1,4-Dioxane 96 72.6 24.0 --- ---
2. Toluene &water 94 58.4 1.3 23.2 0.1
3. Water 96 84.0 7.8 3.7 1.5
4. Toluene 76 62.0 3.8 26.5 1.2
5. Dimethylformamide 96 81.5 0.3 13.7 0.04
6. Dichloromethane 93 70.4 4.1 12.4 3.7
7. Neat 63 54.8 0.0 19.6 0.3
3.3.2.1.3 Impact of epichlorohydrin 26 mole ratio in synthesis of 27

88 Chapter-3
Various mole ratios of epichlorohydrin 26 were screened to check
the impact of the 26 in preparation of 27. Experimental results revealed
that higher amount of diphenyl impurity 29 observed when lesser
amount of epichlorohydrin 26 was used (Table 3.2, entry 1). This
indicates that usage of less equivalence of 26 facilitate the availability of
unreacted 2-methoxyphenoxide in reaction mixture. Unreacted 2-
methoxyphenoxide remained in reaction mixture would have reacted
with 27 and leading to increase in diphenyl impurity 29. Optimal results
were obtained when 3.0 mole ratio of epichloro hydrin 26 was used
(Table 3.2, entry 3).
Table 3.2: Synthesis of 27 using different mole ratios of epichlorohydrin 3
Entry Compound 26 mole ratio
Yield of 27
(%)
Purity by HPLC (%)
27 (%) 28 (%) 29 (%) 30 (%)
1. 1.0+ 84 45.3 0.1 34.7 0.4
2. 2.0 94 82.5 1.3 10.0 1.8
3. 3.0 99 85.5 2.5 5.3 1.9
4. 5.0 99 85.4 9.6 2.2 1.1
3.3.2.1.4 Impact of NaOH mole ratio in synthesis of 27
Experimental results obtained by using different mole ratios of
NaOH as shown in Table 3.3, indicates that; 1) by decreasing the NaOH
mole ratio, higher level of chloro hydroxy compound 28 is observed, 2)
by increasing the sodium hydroxide mole ratio higher level of diphenyl

89 Chapter-3
impurity 29 were found. These results manifested that reaction was
preceded through two steps, first step is opening of epoxide ring with
phenoxide and second step is the intramolecular cyclization of chlorine
with alkoxide as shown in Path 2, Scheme 3.6. Optimum results were
obtained with 1.5 mole ratio of NaOH (Table 3.3, entry 4) when
compared with the other mole ratios of NaOH.
Table 3.3: Synthesis of 27 using different mole ratios of NaOH
Entry NaOH mole ratio
Yield of
27 (%)
Purity by HPLC (%)
27 (%) 28 (%) 29 (%) 30 (%)
1. 0.5 99 39.5 55.8 1.2 1.8
2. 1.0 93 76.2 18.8 2.3 0.9
3. 1.25 99 84.4 6.0 4.3 1.6
4. 1.50 99 85.5 2.5 5.3 1.9
5. 2.0 94 81.3 0.8 9.1 2.6
6. 3.0 94 67.9 0.3 20.0 2.9
In view of the above experimental results to further improve the
yield and purity of compound 27, NaOH was added in two lots rather
than in single lot since reaction was preceded through in-situ chloro
hydroxyl compound 28. In first lot, 0.5 mole ratio NaOH was used for
the synthesis of chloro hydroxyl compound 28 and second lot 1.0 mole
ratio NaOH was used for intramolecular cyclization of chloro hydroxyl
compound 28 obtained in first step. This modification facilitates us to
90 Chapter-3
improve the yield and purity of 27 (Table 3.4, entry 1) by controlling all
potential impurities. Formation of diphenyl impurity 29 in methyl
oxirane compound 27 is significantly controlled to below 1.0% by
removing the aqueous layer (containing unreacted 2-methoxyphenoxide),
which is obtained after completion of first step. Dihydroxy compound 30
was reduced by employing the aqueous NaOH solution washings to the
organic phase containing mixture of methyl oxirane compound 27 and
epichlorohydrin 26. Excess epichlorohydrin 26 (2.0 mole ratio of 26
remain in organic phase) was collected from the organic phase by
distillation at below 80 °C under reduced pressure and reused for the
preparation of 27.
Table 3.4: Synthesis of 27 by addition of NaOH in single lot and two
lots.
Entry Sodium hydroxide mode of addition
Yield of
27
(%)
Purity by HPLC (%)
27
(%)
28
(%)
29
(%)
30
(%)
1. Two lots (0.5 mole ratio first and 1.0 mole ratio in second lot)
95 95.9 0.5 0.6 0.2
2. Single lot (1.5 mole ratio)
99 85.5 2.5 5.3 1.9
By included all optimum conditions methyl oxirane compound 27
was prepared consistently in laboratoary and same process was
implemented in commercial scale (Table 3.5).

91 Chapter-3
Table 3.5: Synthesis of 27 by included all optimal conditions.
Entry Yield of
27
(%)
Purity by HPLC (%)
27
(%)
25
(%)
26
(%)
28
(%)
29
(%)
30
(%)
1. 95 97.2 0.2 0.2 0.1 0.4 0.3
2. 94 98.3 0.2 0.3 0.1 0.3 0.4
3. 94 98.2 0.2 0.1 0.1 0.3 0.3
The improved process having some advantages over the existing
processes including
(a) Yield was improved from ~60% to ~95%,
(b) Purity was improved from ~60% to ~98%
(c) Usage of carcinogenic and partially recoverable solvents like 1,4-
dioxane and ethers were avoided.
(d) All the potential impurities were identified, synthesized and
controlled in 27 at below 0.5% level.
(e) Highly energy consumed operations like fractional distillation at
more than 200 ˚C is avoided.
(f) Excess epichlorohydrin 26 (2.0 mole ratios) used for the reaction
was recovered and reused for synthesis of 27.
3.3.2.2 SYNTHESIS OF N-(2, 6-DIMETHYLPHENYL)-2-(PIPERAZIN-1-
YL) ACETAMIDE 24

92 Chapter-3
Preparation of N-alkyl piperazine derivative 24 involves the
chloroacylation of 2, 6-dimethyl aniline 20 using chloroacetyl chloride
21 to obtain N-chloroacetyl dialkyl aniline compound 22. Thus obtained
compound 22 was treated with piperazine 23 to afford N-alkyl
piperazine derivative 24.
After screening the various key reaction conditions (solvent, base,
temperature), we have synthesized dialkyl aniline compound 22 with
96% of the yield and 99.8% of purity using 0.5 mole ratio of Na2CO3 as
base in DCM solvent system. This step was earlier reported with the 80%
yield and ~95% purity using triethylamine as base. In addition to the
enhancement of yield and purity, tedious workup process in the reported
process was avoided by adopted simple crystallization technique.
Reaction of N-chloroacetyl dialkyl aniline compound 22 with
piperazine 23 suffers with few disadvantages such as (i) moderate yield ~
70%, (ii) No information available about the purity of N-alkyl piperazine
derivative 24, (iii) formation of large amount of bis acetamide compound
31 and (iv) usage of excess mole ratio of 23 leads to formation of
dihydroxy piperazine analogue 34 in subsequent reaction.
In order to improve the yield and purity of 24, the key parameters
like solvent and piperazine mole ratios are examined. Experimental
results revealed that acetone as solvent (Table 3.6, entry 3) using 3.0
mole ratio of 23 (Table 3.7, entry 3) offered compound 24 with optimum
yield (76%) and purity (99.6%) as shown in Table 3.6 and Table 3.7.

93 Chapter-3
Table 3.6: Synthesis of 24 using different solvents
Entry Solvent
Temp. ( °C)
Time (hrs)
Yield of 24 (%)
Purity of 24 (%)
Purity of 31 (%)
1. Methanol 60-65 2 76 99.5 0.1
2. Toluene 60-65 2 71 98.8 0.2
3. Acetone 50-55 2.5 71 98.7 0.4
4. Aq. HCl 60-65 2.5 61 98.7 0.4
5. Water 60-65 2 66 98.9 0.9
6. IPA 60-65 4 56 96.6 2.9
7. DMF 60-65 1.5 --- --- ---
Table 3.7: Synthesis of 24 using different mole ratios of piperazine
Entry Piperazine 23 Mole ratio
Yield of 24 (%)
Purity of 24 (%)
Purity of 31 (%)
1. 1.0 21 63.8 34.6
2. 2.0 53 98.0 1.8
3. 3.0 76 99.6 0.1
4. 4.0 77 99.5 0.1
By incorporated the all optimal conditions, we could able to
synthesize compound 24 with 76% of yield, 99.6% of purity and 0.3% of
piperazine 23. Excess piperazine 23 present in N-alkyl piperazine
compound 24 reacted with the methyl oxirane compound 27 in
subsequent step and leds to formation of related compounds 33 and 34.

94 Chapter-3
HN
O
N
NNH
O
31
O
O
HON
NH
33
O
O
HON
NOH
O
O34
Figure 3.4: Structures of related compounds 31, 33 and 34.
Attempts were made to remove the related compounds 33 and 34
in ranolazine 2 but we could not succeed since solubility profile of
ranolazine 2 and related compound 34 are similar. In view of this,
piperazine content in compound 24 was restricted to below 0.008% to
avoid the formation of the related compounds 33 and 34 in subsequent
stages by employing acidic (HCl or H3PO4) water washings to the crude
compound 24.
NaOHWater
Methanol
AcetoneMethanol80%
96%
97% 77%
O
OHOCl+
O
O
O
NH
HN
25 26 27
23NH2
ClCl
O
+TEA
DCM
HN
Cl
O
20 21 22
N
NO
HN O
O
OH
1
HN
N
O NH
24
2
Scheme 3.5: Improved synthesis of Ranolazine 2.

95 Chapter-3
After developing the efficient process for the synthesis of key
intermediates of 2 (methyl oxirane compound 27 and N-alkyl piperazine
derivative 24), our focus was moved towards final condensation step.
3.3.2.3 SYNTHESIS OF RANOLAZINE 2
Synthesis of ranolazine 2 involves condensation of methyl oxirane
compound 27 and N-alkyl piperazine derivative 24. Previously reported
processes involves (i) usage of mixture of solvents for reaction, (ii)
laborious workup procedures like distillation, extractions, washings and
recrystallization, (iii) purification of crude compound 2 by isolate
dihydrochloride salt of 2 as intermediate or usage of commercially not
viable techniques such as column chromatography.
In order to improve the process efficiency, various solvents were
screened for the synthesis of compound 2. Experimental results revealed
that acetone and acetonitrile offered compound 2 with better yield and
purity when compared with the other solvents depicted in Table 3.8. By
considering cost and solubility, acetone was selected as suitable solvent
for the synthesis of 2. This modification avoided laborious workup
procedure like distillation, extraction and washings and allowed us to
isolate crude compound 2 by simple crystallization at lower temperature
(0-5 º C). Thus obtained crude compound 2 was subjected to purification
in mixture of methanol and acetone (1:4 volumes) to afford pure
ranolazine 2(99.9%).

96 Chapter-3
Table 3.8: Synthesis of 2 using different solvents
Entry Solvent
Temp. ( °C)
Time (hrs)
Yield of 2 (%)
Purity of 2 (%)
1. Methanol 60-65 4.5 78 92.0
2. Ethyl acetate 75-80 13.5 80 97.0
3. Acetone 50-55 12 88 98.0
4. Acetonitrile 80-85 8 88 98.0
5. Water 95-100 7.5 61 73.0
6. IPA 80-85 8.5 71 94.0
7. DMF 75-80 4.5 89 94.0
3.4 Related substances of Ranolazine
As per the ICH guidelines, proposed limits for any known or
unknown impurity in ranolazine is less than or equal to 0.05% since
Ranolazine 2 was high dosage pharmaceutical compound (maximum
daily dosage is less than or equal to 2000 mg). The stringent regulatory
requirements indicate the importance of the potential impurities during
the synthesis of Ranolazine 2. Till now no literature precedence
available for the identification, synthesis, characterization and
controlling of the possible potential impurities formed during the
synthesis of ranolazine 2 and its key intermediates. In view of this,
comprehensive study on related compounds formed in the synthesis of
crude compound 2, and its key starting materials 27 & 24 is desired.

97 Chapter-3
In this perspective to find out the possible process related
impurities in compound 2 and its key intermediates 27 & 24, crude
compound 2 and its key intermediates 27 & 24 are analysed by Liquid
Chromatographic-Mass Spectrometry (LC-MS). Based on molecular
weight obtained in LC-MS, reaction mechanism and reaction conditions
probable impurities were anticipated and their structures were described
in figure 3.5.
O
O Cl
OH O
O OH
OHO
O O
OH O
28 29 30
HN
N
O NNH
OHN
Cl
O
Cl
31 32
O
HO
O
N
NH
O
HO
O
N
N
O
OH
O
33
34
HN
N
O N N
OH N
HN
O
HN
N
O NOH
OHN
N
O NOH
OO
O
35
36 37
Figure 3.5: Structures of ranolazine related substances

98 Chapter-3
These impurities were synthesized, characterized by spectral data
such as Mass, IR and NMR. Root cause for formation of these impurities
were identified and controlled in below 0.05% in final active
pharmaceutical ingredient and as well as in finished pharmaceuticals.
3.4.1 Synthesis and root cause for formation of related substance 28.
Related compound 28 is process related impurity formed during
synthesis of methyl oxirane compound 27. As explained above O-
alkylation of compound 25 with 26 proceeds via an in situ chloro
hydroxyl compound 28. This compound was restricted to below 1.0% in
methyl oxirane compound 27 and further controlled in ranolazine 2 at
below 0.05% level. Compound 28 was synthesized in two ways. First
synthetic route involves the reaction of 2-methoxy phenol 25 with
epichlorohydrin 26 using 0.5 mole ratios of sodium hydroxide.
Alternatively compound 28 was prepared by treating 27 with HCl.
OCl
26
O
OHNaOH
O
O Na
O
O Cl
OH
H2O25 28
O
O
O27
HCl, water
O
O Cl
OH
28
Path-1
Path-2
Scheme 3.7: synthesis of compound 28

99 Chapter-3
The ESI-Mass spectrum of chloro hydroxyl compound 28 (Fig.3.6)
displayed a protonated molecular ion at 217 m/z with positive segment
polarity and ammonium adduct molecular ion at 234.2 m/z. The IR
spectrum (Fig.3.7), absorption at 3458 cm-1 for alcoholic hydroxyl group,
2937 cm-1 for aliphatic alkyl group, 1594 & 1508 cm-1 for alkene group
and 1250 & 1225 for alkyl ether group were observed. The 1H NMR
spectrum (figure 3.8) shows the methyloxy protons at δ 3.8 (s, 3H, CH3),
methylene protons attached to chlorine at 3.68-3.72 (dd, 2H, CH2),
methylene protons attached to phenoxy group at δ 4.05-4.09 (dd, 2H,
CH2), CH proton attached to alcohol group at δ 4.2 (m, 1H, CH), hydroxyl
proton at δ 5.5 (s, 1H, OH) and aromatic protons at δ 6.8-7.0 (m, 4H, Ar-
H).
Figure 3.6: ESI mass spectrum of compound 28
O
O Cl
OH

100 Chapter-3
Figure 3.7: IR spectrum of compound 28
Figure 3.8: 1H NMR spectrum of compound 28
O
O Cl
OH
O
O Cl
OH

101 Chapter-3
3.4.2 Synthesis and root cause for formation of related substance 29.
Related compound 29 is formed as impurity during synthesis of
27, a key intermediate for the preparation of 2. Compound 29 was
derived from reaction of residual amount of 2-methoxy phenoxide
sodium ion with the 27 (scheme 3.8).
O
OHNaOH
O
O Naring
opening
O
O Cl
O Na
25
26
O
O O
OH O
29
SNi
ring opening
O
O
O27
Scheme 3.8: Root cause for formation of 29
Formation of related compound 29 was high when excess amount
of (more than 1.5 mole ratio) NaOH was used for synthesis of 27. This
impurity was controlled in below 0.05% in ranolazine 2 and it was
prepared by reaction of 2-methoxy phenol 25 with epichlorohydrin 26 in
presence of NaOH and 1, 4-dioxane at 100 °C.
O
O O
OH O
29
NaOH, 1,4-Dioxane
100 0 C
O
OH
25
26
Scheme 3.9: synthesis of compound 29

102 Chapter-3
The ESI-Mass spectrum of diphenyl compound 29 (Fig.3.9)
displayed a protonated molecular ion at 305 m/z. The IR spectrum
(Fig.3.10), absorption at 2922 cm-1 for alkyl group, 1593 & 1508 cm-1 for
alkene group and 1252 & 1223 for arylalkyl ether group were observed.
The 1H NMR spectrum (figure 3.11) showed the methyloxy protons peak
at δ 3.7 (s, 6H, CH3), methylene protons attached to phenoxy group at δ
4.05-4.2 (dd, 4H, CH2), CH proton attached to alcohol group at δ 4.2 (m,
1H, CH), hydroxyl proton at δ 5.3 (s, 1H, OH) and aromatic protons at δ
6.8-7.0 (m, 8H, Ar-H).
Figure 3.9: ESI mass spectrum of compound 29
O
O O
OH O

103 Chapter-3
Figure 3.10: IR spectrum of compound 29
Figure 3.11: 1H NMR spectrum of compound 29
O
O O
OH O
O
O O
OH O

104 Chapter-3
3.4.3 Synthesis and root cause for formation of related substance 30.
Dihydroxy compound 30 is process related impurity observed in
synthesis of methyl oxirane compound 27. Reaction of chloro hydroxy
compound 28 (in situ intermediate) with NaOH in presence of water
leads to the formation of compound 30. This impurity was restricted to
below 0.5% in methyl oxirane compound 27 by employing basic water
washings and further controlled to below 0.05% in final ranolazine 2.
Compound 30 was prepared by reaction of 2-methoxy phenol 25 with
glycidol 95 in presence of NaOH (Scheme 3.10).
NaOHO
OH25
O
O OH
OH
30
OHO+
95
Scheme 3.10: synthesis of compound 30
In ESI-Mass spectrum, sodium adduct of dihydroxy compound 30
(Fig.3.12) displayed at 220.9 m/z. In the IR spectrum (Fig.3.13),
absorption at 3274 cm-1 for alcoholic group, 2928 cm-1 for aliphatic alkyl
group, 1595 & 1508 cm-1 for aromatic alkene group and 1257 & 1227
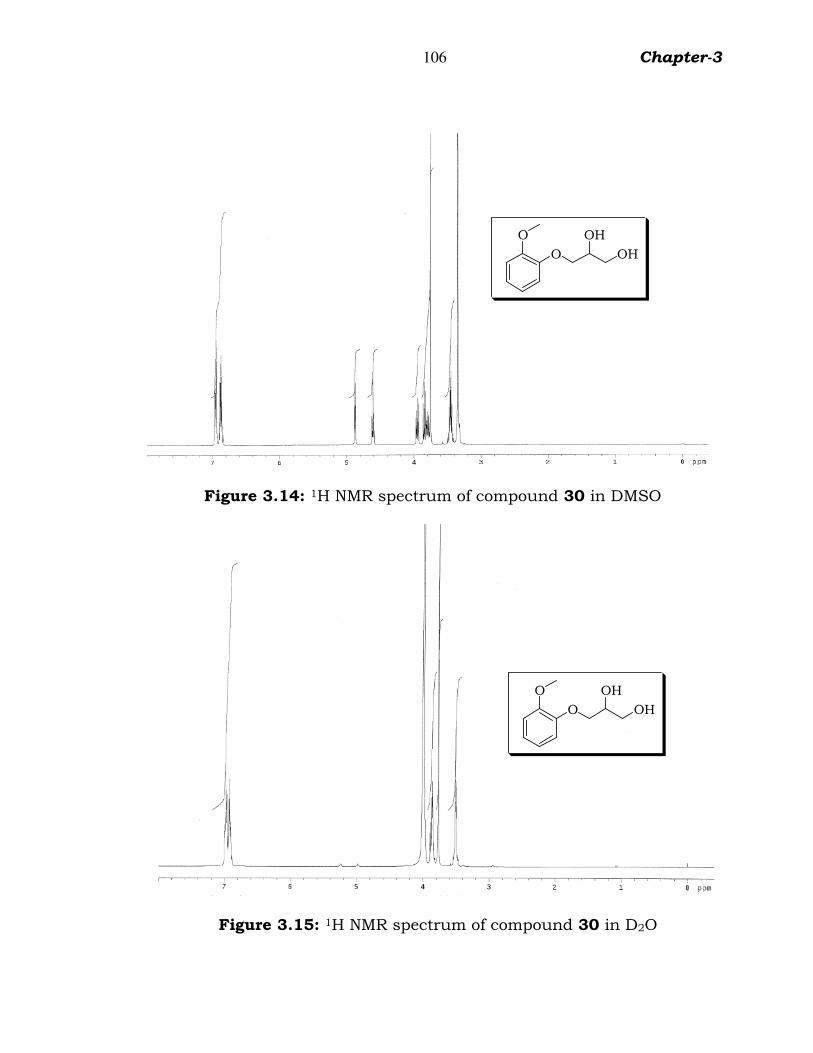
for alkyl ether group were observed. The 1H NMR spectrum (figure 3.14)
in DMSO showed methyloxy protons at δ 3.7 (s, 3H, CH3), hydroxyl
protons at δ 4.6 (s, 1H, OH) & 4.9 (s, 1H, OH) and aromatic protons at δ
6.8-7.0 (m, 4H, Ar-H). Hydroxy protons δ 4.6 (s, 1H, OH) & 4.9 (s, 1H,
OH) were absent when the compound was analysed 1H NMR spectrum
(figure 3.15) by using duterated water (D2O).

105 Chapter-3
Figure 3.12: ESI mass spectrum of compound 30
Figure 3.13: IR spectrum of compound 30
O
O OH
OH
O
O OH
OH
106 Chapter-3
Figure 3.14: 1H NMR spectrum of compound 30 in DMSO
Figure 3.15: 1H NMR spectrum of compound 30 in D2O
O
O OH
OH
O
O OH
OH

107 Chapter-3
3.4.4 Synthesis and root cause for formation of related substance 31.
Bis acetamide compound 31 is process related compound formed
during the reaction of N-acetyl alkyl aniline 22 with piperazine 23. High
amount of compound 31 was observed when 1.0 and 2.0 mole ratios of
piperazine 23 were used for the synthesis of N-alkyl piperazine derivative
24. Formation of compound 31 was controlled to below 7% during the
reaction of 22 and 23 by using 3.0 mole ratios of 23. Thus observed
compound 31 (below 7%) eliminated from the reaction mixture by simple
filtration and this impurity was restricted to below 0.05% in ranolazine
2. Compound 31 was prepared by treating N-acetyl alkyl amine 22 with
piperazine 23 in methanol.
Methanol
NH
HN
23
HN
Cl
O
22
HN
N
O N
31
NH
O
Scheme 3.11: synthesis of compound 31
The ESI-Mass spectrum, displayed bis acetamide compound 31
(Fig.3.16) protonated molecular ion peak of at 409.2 m/z. The IR
spectrum (Fig.3.17), absorption at 3303 cm-1 for amine group, 2943 cm-1
for aliphatic alkyl group and 1682 for amide group were observed. The
1H NMR spectrum (figure 3.18) in CDCl3 showed the methyl protons at δ
2.2 (s, 12H, CH3), methylene protons in piperazine δ 2.8 (t, 8H, CH2),

108 Chapter-3
methylene protons attached to piperazine at δ 3.2 (s, 4H, CH2) and
aromatic protons at δ 7.1-7.2 (m, 6H, Ar-H).
Figure 3.16: ESI mass spectrum of compound 31
Figure 3.17: IR spectrum of compound 31
HN
N
O NNH
O
HN
N
O NNH
O

109 Chapter-3
Figure 3.18: 1H NMR spectrum of compound 31
3.4.5 Synthesis and root cause for formation of related substance 32.
Related compound 32 is process related impurity, formed during
the synthesis of dialkyl aniline compound 32 due to the contamination
of chloroacetyl chloride with dichloroacetyl chloride. This impurity was
controlled in ranolazine 2 at below 0.05% and it was prepared by
reaction of compound 20 with dichloroacetyl chloride 96 in presence of
sodium carbonate (Scheme 3.12).
NH2
+ ClCl
O
Cl
Dichloromethane
Na2CO3
20 96
HN
Cl
Cl
O
32
Scheme 3.12: synthesis of compound 32
HN
N
O NNH
O

110 Chapter-3
The ESI-Mass spectrum, exhibited deprotonated (negative
segment) molecular ion peak of compound 32 (Fig.3.19) at 232 m/z. In
the IR spectrum (Fig.3.20), peaks at 3247 cm-1 for amine group and
1675 cm-1 for amide keto group were observed. The 1H NMR spectrum
(figure 3.21) in CDCl3 shows the methyl protons at δ 2.3 (s, 6H, CH3),
CH proton attached to dichloro compound δ 6.1 (s, 1H, CH), aromatic
protons at δ 7.1-7.2 (m, 4H, Ar-H) and amine proton at δ 7.7 (s, 1H, NH).
Figure 3.19: ESI mass spectrum of compound 32
Figure 3.20: IR spectrum of compound 32
HN
Cl
Cl
O
HN
Cl
Cl
O

111 Chapter-3
Figure 3.21: 1H NMR spectrum of compound 32
3.4.6 Synthesis and root cause for formation of related substance 33.
Hydroxy piperazine compound 33 is observed as impurity during
the reaction of N-alkyl piperazine derivative 24 with the methyl oxirane
compound 27. This impurity was formed by reaction of residual quantity
of piperazine 23 present in N-alkyl piperazine derivative 24 with the
methyl oxirane compound 27 during the synthesis of ranolazine 2.
Formation of compound 33 in ranolazine 2 was restricted to below
0.05% by controlling piperazine 23 in compound 24 to below 0.02% and
it was prepared by treating methyl oxirane compound 27 with piperazine
23 in methanol (Scheme 3.13).
HN
Cl
Cl
O

112 Chapter-3
Methanol
NH
HN
23
O
O
O27
O
O
HON
NH
33
Scheme 3.13: synthesis of compound 33
The ESI-Mass spectrum, displayed protonated molecular ion peak
of compound 33 (Fig.3.22) at 267 m/z. The IR spectrum (Fig.3.23)
absorption at 3436 cm-1 for amine group, 2976 cm-1 for aliphatic alkyl
group and 1221 for C-N group were observed. The 1H NMR spectrum
(figure 3.24) in CDCl3 shows the methyloxy protons at δ 3.1 (s, 3H, CH3),
methylene protons in piperazine δ 2.6-2.8 (t, 8H, CH2) and aromatic
protons at δ 6.8-7.0 (m, 4H, Ar-H).
Figure 3.22: ESI mass spectrum of compound 33
O
O
HON
NH

113 Chapter-3
Figure 3.23: IR spectrum of compound 33
Figure 3.24: 1H NMR spectrum of compound 33
O
O
HON
NH
O
O
HON
NH

114 Chapter-3
3.4.7 Synthesis and root cause for formation of related substance 34.
Compound 34 is process related compound, derived by reaction of
compound 33 with methyl oxirane compound 27. As discussed above,
compound 33 is process related impurity formed by reaction of residual
quantity of piperazine 23 present in N-alkyl piperazine derivative 24
with the methyl oxirane compound 27. Related compound 33 and
compound 34 in ranolazine 2 was controlled to below 0.05% by using
compound 24 having less than 0.02% of piperazine 23. Residual
quantity of 23 present in compound 24 was eliminated by employing
acidic water (phosphoric acid) washings. Compound 34 was prepared by
reaction of methyl oxirane compound 27 with piperazine 23 in methanol
(Scheme 3.14).
Methanol
NH
HN
23
O
O
O27
O
O
HON
NOH
O
O34
Scheme 3.14: synthesis of compound 34
The ESI-Mass spectrum, exhibited protonated molecular ion peak
of compound 34 (Fig.3.25) at 447.2 m/z. In the IR spectrum (Fig.3.26),
peaks at 3179 cm-1 for alcoholic group, 2933 cm-1 for aliphatic alkyl
group, 1250 for ether group and 1222 for C-N group were observed. The
1H NMR spectrum (figure 3.27) in CDCl3 shows the piperazine methylene
protons at δ 2.5-2.6 (m, 8H, CH2), methylene protons attached to
piperazine at δ 2.5 (d, 4H, CH2), methyloxy protons at δ 3.8 (s, 3H, CH3),
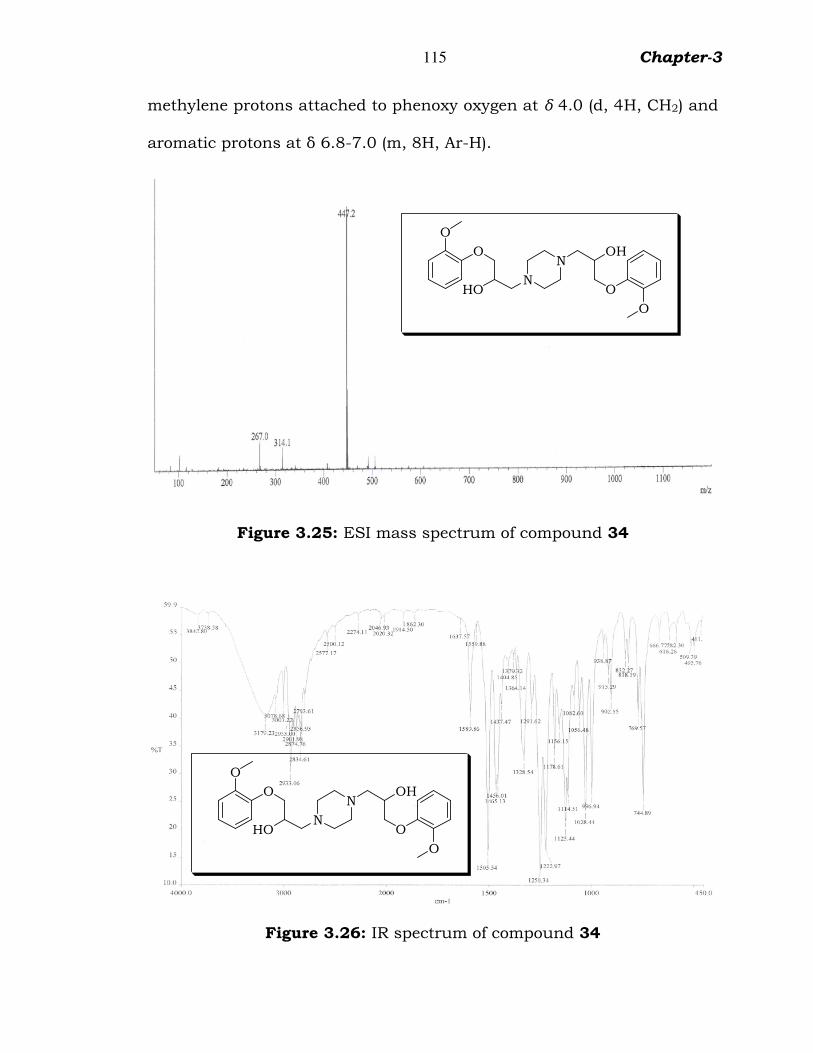
115 Chapter-3
methylene protons attached to phenoxy oxygen at δ 4.0 (d, 4H, CH2) and
aromatic protons at δ 6.8-7.0 (m, 8H, Ar-H).
Figure 3.25: ESI mass spectrum of compound 34
Figure 3.26: IR spectrum of compound 34
O
O
HON
NOH
O
O
O
O
HON
NOH
O
O

116 Chapter-3
Figure 3.27: 1H NMR spectrum of compound 34
3.4.8 Synthesis and root cause for formation of related substance 35.
Synthesis of ranolazine involves the reaction of N-alkyl piperazine
derivative 24 and Methyl oxirane compound 27 in acetone. Compound 25
was formed as impurity in the synthesis of ranolazine 2. This compound
was derived from the reaction of two equivalents of N-alkyl piperazine
derivative 24 with the epichlorohydrin 26 present in methyl oxirane
compound 27. Compound 25 was prepared by reaction of N-alkyl
piperazine derivative 24 with epichlorohydrin 26 and it was controlled to
below 0.05% in final ranolazine 2.
HN
N
O N N
OH N
HN
O
35
HN
N
O NH
Cl
O
24
26
Acetone
Scheme 3.15: synthesis of compound 35
O
O
HON
NOH
O
O

117 Chapter-3
The Mass spectrum, displayed protonated molecular ion peak of
compound 35 (Fig.3.28) at 551.4 m/z. In the IR spectrum (Fig.3.29),
peaks at 3394 cm-1 for alcoholic group, 3262 cm-1 for amine group, 1662
cm-1 for amide keto group and 1162 cm-1 for alcohol group were
observed. The 1H NMR spectrum (figure 3.30) in CDCl3 showed the
methyl protons at δ 2.1 (s, 12H, CH3), methylene group protons between
two piperazines at δ 2.2-2.4 (d, 4H, CH2), piperazine methylene protons
at δ 2.5 (m, 16H, CH2), acetanilide methylene protons at δ 3.1 (s, 4H,
CH2), aromatic protons at δ 7.0-7.1 (m, 6H, Ar-H) and amine proton at δ
9.2 (s, 2H, NH).
Figure 3.28: ESI mass spectrum of compound 35
HN
N
O N N
OH N
HN
O

118 Chapter-3
Figure 3.29: IR spectrum of compound 35
Figure 3.30: 1H NMR spectrum of compound 35
3-Methoxy isomer of ranolazine 36 and 4-methoxy isomer of
ranolazine 37 were prepared by following the procedure used for the
synthesis of ranolazine 2 using 3-methoxy phenol and 4-methoxy phenol
HN
N
O N N
OH N
HN
O
HN
N
O N N
OH N
HN
O

119 Chapter-3
as in put material instead of 2-methoxy phenol 25. These isomers were
characterized by spectral techniques such as IR, Mass and NMR.
3.5 CONCLUSION
In conclusion, efficient, commercially viable, robust, moderately
greener and large scale process was developed for the preparation of
ranolazine 2. The possible potential impurities formed during the
synthesis of ranolazine 2 and its key intermediate N-alkyl piperazine
derivative 24 & methyl oxirane compound 27 were identified and root
cause for thier formation also presented. All impurities were synthesized
and characterized by using spectroscopic techniques (Mass, IR and
NMR).
3.6 EXPERIMENTAL SECTION
3.6.1 Process description
3.6.1.1 2-((2-methoxyphenoxy)methyl)oxirane (27)
O
O
O
NaOH solution (prepared by dissolving 16.1 g (0,402 mol) in 100
mL) was added to the reaction mixture containing 2-methoxy phenol
(25) (100 g, 0.805 mol) and water (400 mL) at ambient temperature.
Epichlorohydrin (26) (223.5 g, 2.416 mol) was added to reaction mass at
25-35 °C and stirred at same temperature for 11 hours. Organic phase
was separated, water (400 mL) and NaOH solution containing 32.2 g

120 Chapter-3
(0.805 mol) of NaOH in 100 mL of water was added to the organic phase.
After stirring the reaction mixture at 28 °C for 6 hours, layers were
separated. Organic phase was washed with 5% NaOH solution (200 mL).
Organic phase was distilled at below 85 °C under reduced pressure to
afford 95% of title compound with 98.4% purity. Excess quantity of
compound 26 was collected from distillate and reused.
IR (KBr, cm–1): 2935 (Ali, CH), 1594 and 1509 (C═C, aromatic), 1258
and 1231 (C-O-C, Aryl ethers) & 1125 (C-O epoxide).
1H NMR (400 MHz, DMSO–d6): δH 6.8-7.0 (m, 4H, Ar-H), 3.8-4.3 (dd,
2H, J ═ 5.6 Hz, 5.4 Hz, CH2), 3.7 (s, 3H, CH3), 3.2-3.4 (m, 1H, CH), 2.7-
2.8 (dd, 2H, J ═ 5.6 Hz, 5.3 Hz, CH2).
M/S (m/z): 181 (M+ + H).
3.6.1.2 2-chloro-N-(2,6-dimethylphenyl)acetamide (22)
HN
Cl
O
To the stirring suspension of Na2CO3 (43.8, 0.413 mol), 2,6-
dimethyl aniline 20 (100 g, 0.826 mol) and DCM (500 mL), chloroacetyl
chloride 21 (100 g, 0.99 mol) was added slowly at 12 °C and strirred at
same temperature for 1-2 hours. Water (1.0 L) was added to the reaction
mixture, DCM was distlled completely at below 40 °C under vacuum.
The resultan reaction mixture was cooled to ambient temperature and
stirred for 1 hour. The resulted solid was collected by filtration, washed

121 Chapter-3
with water (200 mL) and dried at 70 °C afforded 97% of title compound
with 99% of purity.
IR (KBr, cm–1): 3214 (Amine, NH), 3037 (Aromatic, =CH), 2973 (Ali, CH),
1645 (Amide, C=O), 1594 and 1537 (Aromatic, C═C).
1H NMR (400 MHz, CDCl3–d6): δH 7.8 (s, 1H, N-H), 7.0-7.3 (m, 3H, Ar-
H), 4.3 (s, 2H, CH2), 2.2 (s, 6H, CH3).
M/S (m/z): 197.9 (M+ + H).
3.6.1.3 N-(2,6-dimethylphenyl)-2-(piperazin-1-yl)acetamide (24)
HN
N
O NH
N-chloroacetyl dialkyl aniline compound (22) (100 g, 0.505 mol),
methanol (300 mL) and piperazine (23) (130.6 g, 1.518 mol) were heated
to 60 °C and stirred at same temperature for 4 hours. Methanol was
completely distilled at below 65 under vacuum. To the resultant reaction
mixture, water (800 mL) was added at 29 °C and stirred for 1 hour.
Separated undesired solid was filtered and washed with water (300 mL).
Filtrate pH was adjusted to 5.0–5.5 by using H3PO4 solution (44%, 140
mL) at 28 °C and stirred for 0.5 hours. Separated piperazine salt was
filtered. Filtrate was washed with water (100 mL) and pH was adjusted
to 10.5–10.8 by using aqueous NaOH solution (20%, 160 mL). DCM (200
mL) was added to the reaction mixture at 28 °C and stirred for 5
minutes. Organic and aqueous phases were separated, aqueous phase

122 Chapter-3
was extracted with DCM (2 x 500 mL) and the combined organic was
washed with water (300 mL). Upto 80% of organic phase was distilled at
below 40 °C, cyclohexane (500 mL) was added to the reaction mass at 40
°C and heated to 70 °C. The obtained reaction mixture was cooled to 28
°C and stirred for 1 hour. The resulted solid was collected by filtration,
washed with cyclohexane (100 mL) and dried at 55 °C to afford 89 g of
title compound with 99.8 % purity.
IR (KBr, cm–1): 3337 (Amine, NH), 3299 (Amine, NH), 2949 (Ali, CH),
1678 (Amide, C=O), 1596 and 1501 (Aromatic, C═C).
1H NMR (400 MHz, CDCl3–d6): δH 8.7 (s, 1H, N-H), 7.0-7.1 (m, 3H, Ar-
H), 3.2 (s, 2H, CH2), 3.0 (t, 4H, CH2), 2.7 (t, 4H, CH2), 2.2 (s, 6H, CH3).
M/S (m/z): 248.1(M+ + H).
3.6.1.4 2-(4-(3-(2-methoxyphenoxy)-2-hydroxypropyl)piperazin-
1-yl)-N-(2,6-dimethylphenyl)acetamide (Ranolazine 2).
N
NO
HN O
O
OH
The suspension of methyl oxirane compound (27) (47.5 g, 0.264
mol), N-alkyl piperazine derivative (24) (50 g, 0.202 mol) and acetone
(250 mL) were stirred at 56 °C for 17 hours. The resultant reaction
mixture was cooled to 3 °C and stirred for 3.5 hours. The resulted solid
was filtered and washed with pre cooled acetone (50 mL). Methanol (7
mL) and wet compound was heated to 60 °C to get the clear solution.

123 Chapter-3
Acetone (28 mL) was added to the reaction mixture, cooled to 3 °C and
stirred for 4 hours. The resulted solid was collected by filtration, washed
with pre cooled acetone (5 mL) and dried at 73 °C under vacuum to
afford 59 g of title compound 2 with 99.9% purity.
IR (KBr, cm–1): 3331 (Amine, NH), 3002 (Aromatic, =CH), 2955, 2936
and 2834 (Ali, CH), 1686 (Amide, C=O), 1592 and 1495 (Aromatic, C═C),
1254 and 1022 (Ether, C-O-C) & 1125 (C-N).
1H NMR (500 MHz, DMSO–d6): δH 9.1 (s, 1H, N-H), 6.8-7.1 (m, 6H, Ar-
H), 4.8 (s, 1H, OH), 3.9 (s, 1H, CH), 3.8-3.9 (dd, 2H, J=6.5 Hz, 10.7 Hz,
CH2), 3.8 (s, 3H, CH3), 3.1 (s, 2H, CH2), 2.4-2.6 (m, 10H, CH2) 2.1 (s, 6H,
CH3).
13C NMR (500 MHz, DMSO–d6): 18.23, 39.16, 39.83, 39.50, 39.76,
39.87, 53.18, 53.31, 55.50, 61.13, 61.44, 66.63, 71.96, 112.37, 113.64,
120.74, 120.03, 126.32, 127.62, 134.97, 135.06, 148.36, 149.17,
167.97.
M/S (m/z): 428.4(M+ + H).
CHN analysis: Anal. Calcd for C24H33N3O4 (427.54): C 67.42, H 7.78, N
9.83.; Found: C 67.62 H 7.47, N 9.68.
3.6.1.5 1-(2-methoxyphenoxy)-3-chloropropan-2-ol (28).
O
O Cl
OH

124 Chapter-3
Methyl oxirane compound (27) (10 g, 0.055 mol) and concentrated
HCl (40 mL, 0.383 mol) mixture was stirred at 28 °C for 9 hours.
Toluene (200 mL) followed by water (200 mL) were added to the reaction
mass at 28 °C and stirred for 15 minutes. Organic phase and aqueous
phase were separated, aqueous phase was extracted with toluene (10
mL) and combined organic phase was distilled completely at below 70 °C
under vacuum. The resultant crude was subjected to fractional
distillation under reduced pressure, at vapor temperature 145–150 °C
(bath temperature 215–235 °C) afforded 9.8 g of title compound.
IR (KBr, cm–1): 3458 (Alcohol, OH), 2937 (Ali, CH), 1594 and 1508
(Aromatic, C═C), 1250 and 1225 (C-O-C, Aryl ethers) & 1125 and 1027
(C-O Alcohol).
1H NMR (400 MHz, DMSO–d6): δH 6.8-7.0 (m, 4H, Ar-H), 5.5 (s, 1H,
OH), 4.2 (m, 1H, CH), 4.05-4.09 (dd, 2H, dd, J ═ 4.8, 4.8 Hz, CH2), 3.68-
3.72 (dd, 2H, dd, J ═ 4.8 , 4.8 Hz, CH2), 3.8 (s, 3H, CH3).
M/S (m/z): 217 (M+ + H).
3.6.1.6 1,3-bis(2-methoxyphenoxy)propan-2-ol (29).
O
O O
OH O
NaOH (24.1 g, 0.604 mol) and water (40 mL) followed by
epichlorohydrin (26) (18.6 g, 0.50 mol) were added to the reaction
mixture containing 2-methoxy phenol (25) (50 g, 0.402 mol) and 1, 4-

125 Chapter-3
dioxane (130 mL) at 28 °C. Resultant mixture was heated to 100 °C and
stirred at same temperature for 6 hours. Toluene (100 mL) and water
(100 mL) was added to reaction mass at 28 °C. Organic and aqueous
phases were separated, aqueous phase was extracted with toluene (100
mL) and combined organic phase solvent was evaporated at below 75 °C
under reduced pressure. The obtained crude compound was subjected
to fractional distillation, at vapor temperature 210 °C under reduced
pressure to afford titled compound (60 g).
IR (KBr, cm–1): 3547 (Alcohol, OH), 2922 (Ali, CH), 1593 and 1508
(Aromatic, C═C), 1252 and 1223 (C-O-C, Aryl ethers).
1H NMR (400 MHz, DMSO–d6): δH 6.8-7.0 (m, 8H, Ar-H), 5.3 (s, 1H,
OH), 4.2 (m, 1H, CH), 4.05-4.2 (dd, 4H, J ═ 5.6 Hz, 4.8 Hz, CH2), 3.7 (s,
6H, CH3).
M/S (m/z): 305 (M+ + H).
3.6.1.7 3-(2-methoxyphenoxy)propane-1,2-diol (30).
O
O OH
OH
Mixture of 2-methoxy phenol (25) (20 g, 0.161 mol), NaOH (0.03 g,
0.001mol) and glycidol (95) (12.1 g, 0.163 mol) was heated to 95 °C and
stirred at same temperature for 11 hours. Acetic acid (1.0 mL), DCM (20
mL) followed by water (10 mL) was added to the reaction mixture at 3 °C
and stirred for 30 min. Organic phase and aqueous phase were

126 Chapter-3
separated, aqueous phase was extracted with DCM (20 mL) and
combined organic phase was distilled at below 40 °C under reduced
pressure. Hexane (50 mL) was added to the resultant crude compound
at 27 °C and stirred for 1 hour. Separated solid was filtered, washed
with hexane (20 mL). The obtained wet compound was purified in IPA to
afford titled compound (13.4 g).
IR (KBr, cm–1): 3274 (Alcohol, OH), 2928 (Ali, CH), 1595 and 1508
(Aromatic, C═C), 1257 and 1227 (C-O-C, Aryl ethers) & 1125 and 1039
(C-O Alcohol).
1H NMR (400 MHz, DMSO–d6): δH 6.8-7.0 (m, 4H, Ar-H), 4.9 (s, 1H,
OH), 4.6 (s, 1H, OH), 3.9 (m, 1H, CH), 3.7 (s, 3H, CH3), 3.4-3.5 (dd, 2H, J
═ 5.2 Hz, CH2).
M/S (m/z): 220.9 (M+ + H).
3.6.1.8 1,4-Bis-[(2,6-dimethylphenyl)-aminocarbonylmethyl]-
piperazine (31).
HN
N
O NNH
O
Mixture of N-chloroacetyl dialkyl aniline compound (22) (50 g,
0.253 mol), methanol (150 mL) and piperazine (23) (22 g, 0.255) was
heated to 65 °C and stirred at same temprature for 3 hours. Resulted
reaction mass was cooled to ambient temperature and stirred for 1 hour.

127 Chapter-3
Separated solid was filtered, washed with methanol (50 mL) and dried at
50 °C under vacuum to afford titled compound (50 g).
IR (KBr, cm–1): 3303 (Amine, NH), 2943 (Ali, CH), 1682 (Amide, C=O),
1500 and 1465 (Aromatic, C═C).
1H NMR (400 MHz, CDCl3–d6): δH 7.1-7.2 (m, 6H, Ar-H), 3.2 (s, 4H,
CH2), 2.8 (t, 8H, CH2), 2.2 (s, 12H, CH3).
M/S (m/z): 409.2 (M+ + H).
3.6.1.9 1-(2-methoxyphenoxy)-3-(piperazin-1-yl)propan-2-ol (33).
O
HO
O
N
NH
Methyl oxirane compound (27) (50 g, 0.277) was slowly added to
the reaction mixture containing methanol (250 mL) and piperazine (23)
(96 g, 1.114 mol) at 3 °C. After stirring reaction mixture at 3 °C for 3
hours quenched with water (200 mL). Product was extracted from
aqueous phase with dichloromethane (5 x 50 mL). Acetic acid (32.5 mL)
and water (200 mL) was added to the organic phase and stirred for 15
minutes. Organic and aqueous phases were separated, aqueous
ammonia (55 mL) was added to the aqueous phase and product was
extracted from aqueous phase with DCM (5 x 50 mL). Organic phase
solvent was evaporated completely under vacuum at below 45 °C to
afford titled compound (44.2 g).

128 Chapter-3
IR (KBr, cm–1): 3436 (Amine, NH), 2976 (Ali, CH), 1505 and 1456
(Aromatic, C═C), 1221 (C-N).
1H NMR (400 MHz, CDCl3–d6): δH 6.8-7.0 (m, 3H, Ar-H), 4.1 (m, 1H,
CH), 3.1 (s, 3H, CH3), 4.1 (d, 2H, J ═ 4.0 Hz, CH2), 2.4-2.8 (m, 8H, CH2).
M/S (m/z): 267.0 (M+ + H).
3.6.1.10 1-{4-[2-Hydroxy-3-(2-methoxyphenoxy)-propyl]-piperazin-1-
yl}-3-(2-methoxyphenoxy)-propan-2-ol (34).
O
HO
O
N
N
O
OH
O
Mixture of methyl oxirane compound (27) (50 g, 0.277 mol),
piperazine (23) (24 g, 0.278 mol) and methanol (150 mL) were heated to
65 °C and stirred at same temperature for 3 hours. The resultant
reaction mass was cooled to ambient temperature and stirred for 1 hour.
Separated solid was filtered and washed with methanol (25 mL). The
obtained wet compound was purified in methanol (125 mL) and dried at
65 °C under reduced pressure to afford titled compound (14 g).
IR (KBr, cm–1): 3179 (Alcohol, OH), 2933 (Ali, CH), 1589 and 1505
(Aromatic, C═C), 1250 (Ether, C-O-C), 1222 (C-N).
1H NMR (400 MHz, CDCl3–d6): δH 6.8-7.0 (m, 8H, Ar-H), 4.0 (d, 4H, J ═
4.8 Hz, CH2), 3.8 (s, 6H, CH3), 2.5 (d, 4H, J ═ 5.2 Hz, CH2), 2.5-2.6 (m,
8H, CH2).

129 Chapter-3
M/S (m/z): 447.2 (M+ + H).
3.6.1.11 2,2-dichloro-N-(2,6-dimethylphenyl)acetamide (22).
HN
Cl
O
Cl
To the stirring suspension of 2, 6-dimethylaniline (20) (25 g, 0.206
mol), Na2CO3 (21.9 g, 0.206 mol) and DCM (500 mL) were added
dichloroacetyl chloride (96) (36.5 g, 0.248 mol) at 13 °C and stirred at
same temperature for 2 hours. Water (250 mL) was added to the reaction
mixture at 28 °C and DCM was distilled completely at below 45 °C. The
resultant reaction mixture was cooled to 28 °C and stirred for 1 hour.
Separated solid was filtered, washed with water (50 mL) and dried at 70
°C under reduced pressure to get the 38 g of titled compound with the
99 % purity by HPLC.
IR (KBr, cm–1): 3247 (Amine, NH), 3037 (Aromatic, =CH), 2925 (Ali, CH),
1676 (Amide, C=O), 1541 and 1470 (Aromatic, C═C).
1H NMR (400 MHz, CDCl3–d6): δH 7.7 (s, 1H, NH), 7.1-7.2 (m, 3H, Ar-
H), 6.1 (s, 1H, CH), 2.3 (s, 6H, CH3).
M/S (m/z): 232 (M- - H).
3.6.1.12 1,3-Bis-{4-[(2,6-dimethylphenyl)-aminocarbonylmethyl]
piperazin-1-yl}-propan-2-ol (35).

130 Chapter-3
HN
N
O N N
OH N
HN
O
Mixture of N-alkyl piperazine compound (24) (50 g, 0.202 mol),
acetone (500 mL) and epichlorohydrin (26) (29.2 g, 0.316) were heated to
55 °C and stirred at 55 °C for 13 hours. Reaction mass cooled to
ambient temperature and stirred at same temperature for 1 hour.
Separated solid was filtered, the resulted wet compound was
recrystalized from isopropyl alcohol (90 mL) and dried at 55 °C under
vacuum to afford 50 g of the title compound with about 99% of purity.
IR (KBr, cm–1): 3394 (Alcohol, NH), 3262 (Amine, NH), 2937 (Ali, CH),
1662 (Amide, C=O), 1524 and 1475 (Aromatic, C═C) 1162 (Alcohol, C-O).
1H NMR (400 MHz, CDCl3–d6): δH 9.2 (s, 2H, NH), 7.0-7.1 (m, 6H, Ar-H),
3.8-3.7 (m, 1H, CH), 3.1 (s, 4H, CH2), 2.6-2.5 (m, 16H, CH2), 2.2-2.4 (d,
4H, J ═ 12.8 Hz, CH2), 2.1 (s, 12H, CH3).
M/S (m/z): 551.4 (M+ + H).
3.7 REFERENCES
(1) Allely, M. C.; Alps, B. J. Br. J. Pharmacol., 1988, 93, 375-382.
(2) Hale, S. L.; Kloner, R. A. J. Cardiovasc. Pharmacol. Ther., 2006,
11, 249-255.
(3) Fraser, H.; Belardinelli, L.; Wang, L.; Light, P. E.; McVeigh, J. J.;
Clanachan, A. S. J. Mol. Cell. Cardiol., 2006, 41, 1031-1038.
(4) Jerling, M. Clin. Pharmacokinet., 2006, 45, 469-491.

131 Chapter-3
(5) (a) Kluge, A. F.; Clark, R. D.; Strosberg, A. M.; Pascal, J. G.;
Whiting, R. United states patent, US 4,567,264, 1986. (b) Kluge,
A. F.; Clark, R. D.; Strosberg, A. M.; Pascal, J. C.; Whiting, R. L.
European patent, EP 0,126,449, 1987. (c) Kluge, A. F.; Clark, R.
D.; Strosberg, A. M.; Pascal, J. C.; Whiting, R. Canadian patent,
CA 1256874, 1987.
(6) Agai-Csongor, E.; Gizur, T.; Haranyl, K.; Trischler, F.; Demeter-
Szabo, A.; Csehi, A.; Vajda, E.; Szab-Koml si, G. European patent,
EP 483932 A1, 1992.
(7) Lisheng, W.; Xiaoyu, F.; Hong-yuan, Z. Journal of Guangxi
University (Natural Science Edition), 2003, 28, 301-303.
(8) Shu-chun, L.; He-qing, H.; Zhong-jun, L. Chinese Journal of
medicinal chemistry, 2003, 13, 283-285.
(9) ICH harmonized tripartite guideline, Impurities in New Drug
Substances Q3A (R2), current step 4 version dated 25 October
2006.
(10) (a) Reddy, M. S.; Eswaraiah, S.; Satyanarayana, R. Indian patent
application 2942/CHE/2007. (b) Rahul, S.; Venkateswaran, S. C.;
Lalit, W. WO patent 2008/047,388, A2, 2008.
(11) Michel G.; Jef C.; Ivan V.; Dirk D. S.; Stef L. Org. Proc. Reas. &
Dev. 2003, 7, 939-941.
(12) Pchelka B. K.; Loupy A.; Petit A. Tetrahedron, 2006, 62, 10968-
10979.

132 Chapter-3
(13) HPLC method: Symmetry shield RP-18, 250 x 4.6 mm, 5µm; flow:
1 mL/min; eluent A: Water, pH adjusted to 5.0 with dil H3PO4; B:
Acetonitrile and water in the ratio of 800:200 (v/v); Gradient: 0
min: 70% A, 30% B; 5 min: 70% A, 30% B; 35 min: 30% A, 70% B;
55 min: 30% A, 70% B; 57 min: 70% A, 30% B; 65 min: 70% A,
30% B, UV detection at 223 nm.
(14) Wenchao, L.; Yingqi, L.; Xianglin, Z.; Chun, G.; Kai, Z. Chinese
Journal of Pharmaceuticals, 2004, 35, 641-642.
(15) HPLC method: Intertsil ODS-3V, 250 cm x 4.6 mm, 5 µm; flow: 0.8
mL/min; eluent A: buffer (weigh 1.36 g of potassium dihydrogen
ortho phosphate and 1.5 g of n-Hexane sulphonic acid sodium salt
into a 1000 mL of MQ Water, adjust the pH to 3.0 with dil.H3PO4)
and acetonitrile in the ratio of 90:10 (v/v); B: acetonitrile,
methanol and water in the ratio of 600:200:200 (v/v/v); Gradient:
0 min: 75% A, 25% B; 8 min: 75% A, 25% B; 20 min: 60% A, 40%
B; 40 min: 25% A, 75% B; 55 min: 25% A, 75% B; 57 min: 75% A,
25% B; 65 min: 75% A, 25% B, UV detection at 210 nm.
GC method: AT–1701, 30 m x 0.53 mm, 1.2 µm; split ratio: 1:5;
Carrier gas: Helium, 2.5psi (Make up gas: 30.0mL/min); Column
oven temperature: Initially held at 150 °C for 0 min, then
increased to 270 °C at a rate of 25 °C per minute and held at 270
°C for 20 min.

133 Chapter-3
(16) (a) Foye, W. O.; Levine, H. B.; Mc Kenzie, W. L. J. Med. Chem.
1966, 9, 61.
(17) (a) Xiao-lin, C.; Yong-zhou, H. West China Journal of
Pharmaceutical Sciences, 2004, 19, 191-193. (b) Lau, H. P. WO
patent 96/40664, 1996.