chemical composition and gibbs standard free energy of...
TRANSCRIPT

Clay Minerals (1999) 34, 499-510
Chemical composition and Gibbs standard free energy of formation of Fe(II)-Fe(III)
hydroxysulphate green rust and Fe(II) hydroxide
PH. R E F A I T , C. B O N , L. S I M O N , G. B O U R R I I ~ * ' * * , F. T R O L A R D * J. B E S S I I ~ R E AND J . - M . R. G ] ~ N I N 1
Laboratoire de Chimie Physique pour l'Environnement, UMR 7564 CNRS-Universitk H. Poincard, Equipe sur la Rdactivit~ des Espbces du Fer and Ddpartement de Science des Mat&iaux, ESSTIN, 405, rue de Vandoeuvre, F 54600 Villers-les-Nancy, *INRA - UR de Science du Sol et de Bioclimatologie, 65 rue de Saint Brieuc, F 35042 Rennes Cedex, and **UPR 4661 CNRS-Univ. Rennes 1, Gkoscienees Rennes, Campus de Beaulieu, F 35042 Rennes Cedex, France
(Received 20 January 1997, revised 15 October 1998)
A B S T RA C T: The redox potential and pH of aerated suspensions of iron(li) hydroxide in sulphate- containing aqueous solutions are measured during the oxidation process. Plateaux corresponding to the equilibrium conditions between Fe(OH)2(s) and Fe(II)-Fe(III) hydroxysulphate GR2(SO2-)(s) on the one hand, and between GR2(SO]-)(s~ and FeOOH(s) on the other hand, are displayed. Potentiometry, voltammetry, pH-metry and M6ssbauer spectroscopy are applied to follow all reactions. The thermodynamic meaning of the measured potential of the first plateau which corresponds to the GR2(SO42 )(s)/Fe(OH)2(~) equilibrium is demonstrated. The chemical composition of GR2(SO42 )(s) is found to be Fe~Fe~II(oH)12SOg.nH20 all along the oxidation process implying that this compound must be considered as a pure phase with a well-defined composition. The Gibbs standard free energy of formation or ehemical potential g~ 2 )(s)] in 'anhydrous form' (n = 0) is determined at -3790_+10 kJ tool 1. A consistent value of g~ at -490___ 1 kJ mol I is obtained.
The Fe(II)-Fe(III) hydroxysulphate, with formula [FeIIFem(OH)lz]z+.[SO4.nH20] 2 (Hansen et al.,
1994; G6nin et al., 1996) is more commonly known as green rust two and des igna ted GR2(SO ] )(s). As with other green rust (GR) compounds, it is a layered double hydroxysalt, consisting of hydroxide sheets, [Fe4Fezn III(oH)12 ]2+, charged positively due to the presence of ferric cations, which alternate regularly with negatively charged interlayers, [SO4.nH20] 2-, composed of
1 Corresponding author
sulphate anions and water molecules. The GRs have been observed as a corrosion product of steel (G6nin et al., 1991) and proposed in ochre sludge (Bender Koch & Morup, 1991). Their occurrence in anaerobic soils and sediments was suggested by several workers (e.g. Ponnamperuma et al., 1967), but it was only very recently that the definite presence of a GR compound as a mineral in hydromorphic soils was demonstrated (Trolard et
al., 1996, 1997), even though its precise nature still remains uncertain, i.e. in relation to the type of anions involved (G6nin et al., 1998b). Of special interest is the reduction of nitrite and nitrate by green rust compounds, i.e. the possibility that GRs
�9 1999 The Mineralogical Society

500 Ph. Refait et al.
could influence the denitrification of soils and sediments. Hansen et al. (1994, 1996) focused on the role that GR2(SOeZ-)(s) could play; they showed that it should be able to reduce nitrite and nitrate since their estimation of the Gibbs standard free energy (free enthalpy) of formation or chemical potential translated in terms of redox potential infers the fact that the reduction of nitrite is thermodynamically possible. Some other works were also devoted to the chemical and electro- chemical properties of GR2(SO2-)(~) and various computations of the Gibbs standard free energy of formation have been proposed (Detournay et al.,
1975; Olowe & G~nin, 1989; Refait & G~nin, 1994) but the chemical formula considered for GR2(SOZ-)(s) was erroneously referred to as [Fe~lFe~n(OH)1412+.[SO4-nH2O12 ; thus, these results cannot be used. In contrast, the composition [Fe~IFem(OH)12]2+.[SO4.nH20] 2- was recently ascertained and the standard free energy of formation re-evaluated by G~nin et al. (1996) at -3770_+4 kJmo1-1 while Hansen et al. (1994) obtained -3669_+4 kJ tool 1, taking the number n of water molecules incorporated in the interlayers to be equal to zero for easier comparison (add -237.18 kJ mol 1 per water molecule).
The method we used, originally devised by Detournay et al. (1975) and also applied to other GRs (Refait & G6nin, 1993; Drissi et al., 1995), consists of an electrochemical survey of the oxidation of an aerated Fe(OH)2(s) suspension. If iron(II) hydroxide is initially precipitated from iron(II) sulphate and sodium hydroxide, GR2( SO] )(s) is obtained at the end of a first oxidation stage, provided that a sufficient excess of dissolved iron(II) sulphate is left in solution after precipitation of Fe(OH)2(s) (Olowe & G~nin, 1989, 1991). The pH and redox potential of the solution do not vary during the process and their values were assumed to verify equilibrium conditions between Fe(OH)2(~) and GR2(SO42 )(s~, allowing the computation of the standard equilibrium potential of the reaction and of the standard chemical potential I.t~ The measured redox potential was assumed to be close to that of the equilibrium, but the difference between them, which can govern the accuracy of the g~ 2 )(~)] value, was not established.
Since some discrepancy exists between the value we found and that given by Hansen et al. (1994), it appears necessary to validate the procedure used in our own method. This determination implies a
thorough study of the electrochemical behaviour of GR2(SO ] )(s~, realized here by voltammetry. It allows us to propose a reliable and definite estimation of the standard chemical potential of the Fe(II)-Fe(III) hydroxysulphate. Moreover, the characterization by M6ssbauer spectroscopy of the solid phases which form during the oxidation process will allow us to follow the chemical composition of GR2(SOZ-)(s) with time, a key point in discovering whether true equilibrium is reached.
M E T H O D S
The Fe(OH)2(s) suspensions were obtained by mixing 100 ml of an aqueous solution of melanterite FeSO4.7H20 (Normapur| provided by PROLABO | and containing a maximum impurity content of 1%, in particular <0.02% of Fe(III), with 100 ml of a solution of NaOH (Rectapur | with a minimum NaOH content of 97% and a maximum NazCO3 content of 1%. The FeSO4.7H20 and NaOH concentrations were fixed at 0.12 tool 1-1 and 0.20 tool 1-1, respectively. No specific precautions were taken to de-aerate these solutions, implying that some Fe(III) was present as soon as the precipitation of Fe(OH)2(s) was complete. The ratio [FeSO4.7H20]/[NaOH] of 0.60 provided an excess of dissolved Fe(II) equal to 0.02 tool 1 1. The suspension was aerated at the interface between solution and atmosphere and magnetic stirring ensured a progressive oxidation of the precipitate at a rotation speed of 550 rpm. Homogeneous oxidation was apparent. A thermostat controlled the temperature, kept at 25+0.5~ Reactions were monitored by recording the pH and the redox potential of the solution, measured by use of a Pt wire (1 mm in diameter and 10 mm in length) or a silver rod (5 mm diameter) using a saturated calomel electrode as reference.
Voltammetry was carried out with a Radiometer- Tacussel | potentiostat monitored by a PIL 101-T pilot and connected to an XY-t recorder, Using a conventional three-electrode electrochemical cell. Though a saturated calomel electrode was used as reference, all data, i.e. Eh, were given with respect to the standard hydrogen electrode. A Pt wire electrode identical to that used for the zero-current potential vs. time measurements was used as a working electrode and its potential varied at a slow sweep rate of 1 mV s 1. Different sets of experiments were performed, always starting from
i i

Fe(II)-Fe(III) hydroxysulphate
the zero-current potential either with reduction or oxidation. Experiments were made with aerated or de-aerated suspensions by keeping a flow of nitrogen (Airgaz | 99.999% min., O2 <2 vpm) through the cell. The inductively coupled plasma- atomic emission spectrometry (ICP-AES) method was used to measure the Fe and S concentrations. Two ml of the suspension were sampled and filtered through a membrane of cellulose acetate (pore size 0.45 lain) and a glass fritt (Millipore| Nitric acid was then added to the solutions in order to avoid the precipitation of Fe(III) oxyhydroxides, which could result from oxidation of Fe(II) in neutral or basic solutions. The Perkin-Elmer | Plasma 2000 apparatus used here was mounted with two monochromators, one going from 160 to 800 nm and the other from 160 to 400 nm. The detection limits were 3 btg 1-1 and 50 ttg 1-1 for Fe and S, respectively. The analyses were carried out at 238.204 nm for Fe species and 180.731 nm for S species, and four measurements were performed for each species in each sample. The calibration was made with four standards.
Transmission M6ssbauer spectroscopy (TMS), using a constant acceleration drive unit and a 57Co 7-source of 50 mCi embedded in a Rh matrix, was carried out to analyse the compounds during the oxidation process. The samples to be analysed were filtered and prepared under N2 atmosphere and set in a cryostat at 15 K under He atmosphere preventing any oxidation. To achieve analyses at such temperatures, a closed M6ssbauer cryogenic workstation with vibrations isolation stand manu- factured by Cryo Industries of America | was used.
R E S U L T S
Redox potential, pH, [Fe] and IS] vs time c u r v e s
Typical zero-current potential Eh and pH vs. time curves (Fig. 1) are composed of three plateaux A, B and C separated by two sharp jumps at Tg and Tf: These sharp variations indicate the end of a reaction stage. During the first one ending at Tg, Fe(II) hydroxide oxidizes into GR2(SO]-)(s) and during the second one, from Tg to Tf, GR2(SO4Z-)(s) oxidizes into 7-FeOOH(s) and ~-FeOOH(s ). More details concerning these oxidation processes can be found elsewhere (Olowe & G~nin, 1991). The potential Eh and pH remain quite constant during the advancement of the reactions testifying that the
green rust and Fe(II) hydroxide 501
oxidation processes Fe(OH)2(s) ~ GR2(SO 2 )(s) (plateau A) and GR2(SO42-)(s) --+ FeOOH(s) (plateau B) are independent of the pH according to:
5 Fe(OH)z(s) + Fe 2+ + SO 2 + 1/2 02 + H20 "+ FelIFem(OH)IzSO4(s) (1)
FeltFeln(OH)laSO4(s) + 314 O 2 ---->
5 FeOOH(s) + Fe 2+ + SO 2- + 7/2 H20 (2)
The influence of oxygen was tested by de- aerating the suspensions once the middle of plateau A is reached. The potential and pH were recorded for five hours under a N atmosphere but almost no changes were observed (cf Detournay et aL, 1975), staying constant within 5 mV and 0.1 unit, respectively. This demonstrates that the measured parameters are independent of the presence of oxygen.
Values of Eh (standard hydrogen electrode) measured in the middle of plateau A for 12 experiments are reproducible to within 7 mV and equal to Eh(A) = -0.497 V with pH(A) = 8.0 + 0.1. It is important to stress that the value of Eh(A) is independent of the nature of the electrode, e.g. Pt or Ag, of its area and of the stirring conditions of the solution. These results favour the thermodynamic meaning of the measured potential (Chariot, 1959). Measurements of Eh(B) in plateau B are realized in the domain limited between times 100 and 170 mn, since the pH decreases thereafter, whereas the electrode potential stays constant up to 250 mn. They are less reproducible than previously and depend upon the electrode, meaning that the
0.5
0.3
0.1
-0.1
-0.3
-0.5
Eh (V)
(e) T f ~ pH
-A- . . . . . ~C-"-- ~' Eh - - / ( g i 8 Tgl % 7
\\ 6 (a)(b) (c) Tf '
~.C 5 I I I I
0 60 120 180 240 300
time (min)
FIG. 1. Zero-current potential Eh (S.H.E.) and pH vs. time curves recorded during the oxidation of an aerated suspension of Fe(II) hydroxide. A, B and C are the various plateaux when Eh and pH stay constant; Tg and Tf correspond to the ends of reaction stages and (a)-(g) indicate the times at which the precipitates,
analysed by TMS, were sampled.

502 Ph. Refait et al.
e l e c t r o n i c t r a n s f e r is s lower . V a l u e s o f E h ( B ) - - 0 . 0 7 5 _ 0 . 0 1 V and pH(B) = 6.7+0.1 are measured. The same is done on the last plateau, C, after Tt-where ferric oxyhydroxides are the only solid phases which do not evolve. Values of Eh(C) = 0.40_+0.05 V and pH(C) = 3.9_+0.1 are measured.
Analyses of the concentrations of dissolved Fe and S species were performed by ICP-AES at various moments of the reaction. The precision of the measurements was estimated to be equal to the fluctuations observed after the end of the reaction, i.e. after point Tf, where c[Fe(II)] and c[S] are constant. Similar ly , sys temat ic errors were corrected, the average final values of c[Fe(II)] and c[S] having to be equal to 0.02 and 0.12 mol 1-1, respectively. The corresponding curves of the evolution with time (Fig. 2) are identical. Both species are consumed before Tg, while the sulphate- containing GR2(SO~ )(s) is forming; then, they are released in solution when GR2(SO42 )(s) transforms into ferric oxyhydroxides. From the minima observed at Tg, that is c[Fe] = (3.5_+1)x 10 -3
mol 1 1 and c[S] = (95_+3) x 10 _3 mol 1 -~, the total dissolved Fe(II) and S amounts which are involved in the reactions are A[Fe(II)] = (16.5_+1)x 10 3 mol 1 1 and A[S] - (25_+3) x 10 3 tool 1 1. These values are close to the value 20x 10 3 mol 1-1 implied by the overall balance, if c[Fe(OH)2(s)] is equal to 10 -1 mol 1 1 in eqns. (1) and (2).
Voltammetry
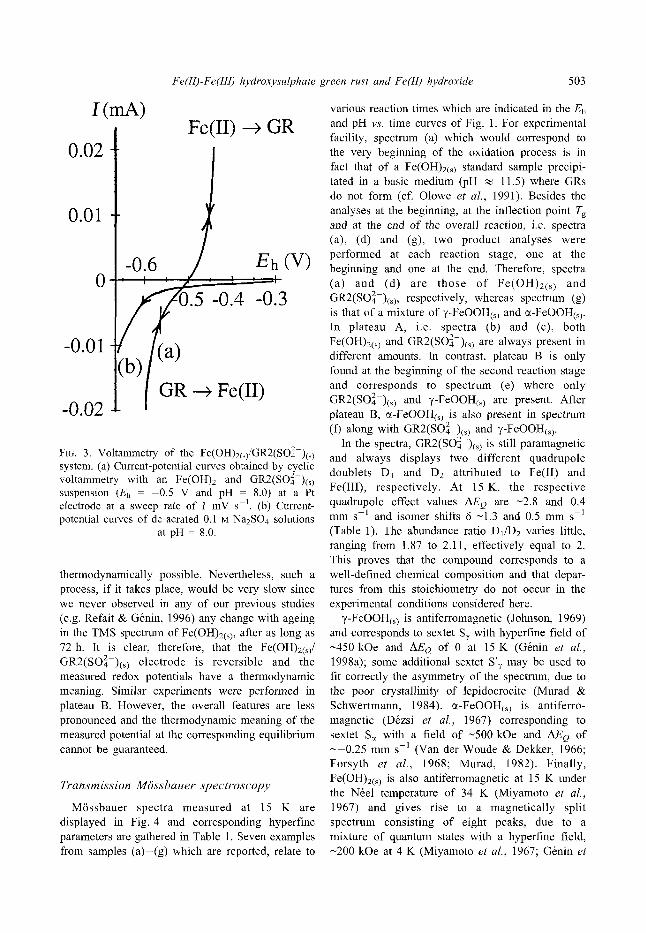
The experiments were first carried out at the middle of plateau A, where the equilibrium conditions between Fe(OH)2(s) and GR2(SO~ )(s) are assumed to be met, the stirring of the suspension being maintained. The potential was restricted, to vary only from -0.655 to -0.425 V, in order to avoid the reduction of Fe(II) hydroxide and the oxidation of GR2(SOZ-)(s). The voltam- metry curves taken at plateau A clearly show the electrochemical transformation of Fe(II) species in GR2(SOZ-)(s) on the one hand, and the reduction of GR2(SO]-)(s) on the other hand (Fig. 3, curve a). Anodic and cathodic curves are drawn by starting from the equilibrium potential at zero intensity at Eh(A) ~ -0 .5 V. Several cycles were performed, but no significant modifications resulted. The behaviour of the i = fiE) curves is the same in the presence or in the absence of O. A preliminary experiment carried out in the absence of Fe species and of O at pH - 8.0 regulated by NaOH in the presence of 10 1M Na2SO4 as an electrolyte, showed that the reduction of H + only takes place below 0.6 V (Fig. 3, curve b). Moreover, if H+/H2 were responsible for the voltammogram of (a), the strong anodic current observed would imply that H2 was present. Since the Fe(OH)2(s)/GR2(SO]-)(s) border is located below the stability field of water, the formation of H from Fe(OH)2(s) is
~,~ 0.13-
g 012- +
, - 0.11
o r o 0.10 O
0.010J 0.005 J 0 . 0 0 0 /
(1) A +
C
+ +§
§247 �9 s +~, B �9 F e
Tg 4' 4' 4' ~ 4'
(;;4',4' 4'4' § 4'4' § 4'4' T i m e ( m i n )
4' 4,
6 5'0 160 150 260 250 360
FIG. 2. Evolution with time of dissolved Fe and S species in solutions analysed by ICP-AES.
Fe(ll)-Fe(Ill) hydroxysulphate green rust and Fe(ll) hydroxide 503
I i
0.02
0.01
0
-0.01
-0.02
mA) Fe(II) > GR
- 0 . 6 _ _ Eh (V) ' ~ J _ _ . _ _ I , - I
-0.4 -0.3
�9 (a)
GR --+ Fe(II)
F~G. 3. Voltammetry of the Fe(OH)2(~)/GR2(SO42 )(s~ system�9 (a) Current-potential curves obtained by cyclic voltammetry with an Fe(OH)2 and GR2(SO]-)(~) suspension (Eh - --0.5 V and pH - 8.0) at a Pt electrode at a sweep rate of 1 mV s 1. (b) Current- potential curves of de-aerated 0.1 M Na2SO 4 solutions
at pH - 8.0.
thermodynamically possible. Nevertheless, such a process, if it takes place, would be very slow since we never observed in any of our previous studies (e.g. Refait & G6nin, 1996) any change with ageing in the TMS spectrum of Fe(OH)2(s> after as long as 72 h. It is clear, therefore, that the Fe(OH)2(s)/ GR2(SO ] )(s~ electrode is reversible and the measured redox potentials have a thermodynamic meaning�9 Similar experiments were performed in plateau B. However, the overall features are less pronounced and the thermodynamic meaning of the measured potential at the corresponding equilibrium cannot be guaranteed�9
Transmission Mdssbauer spectroscopy
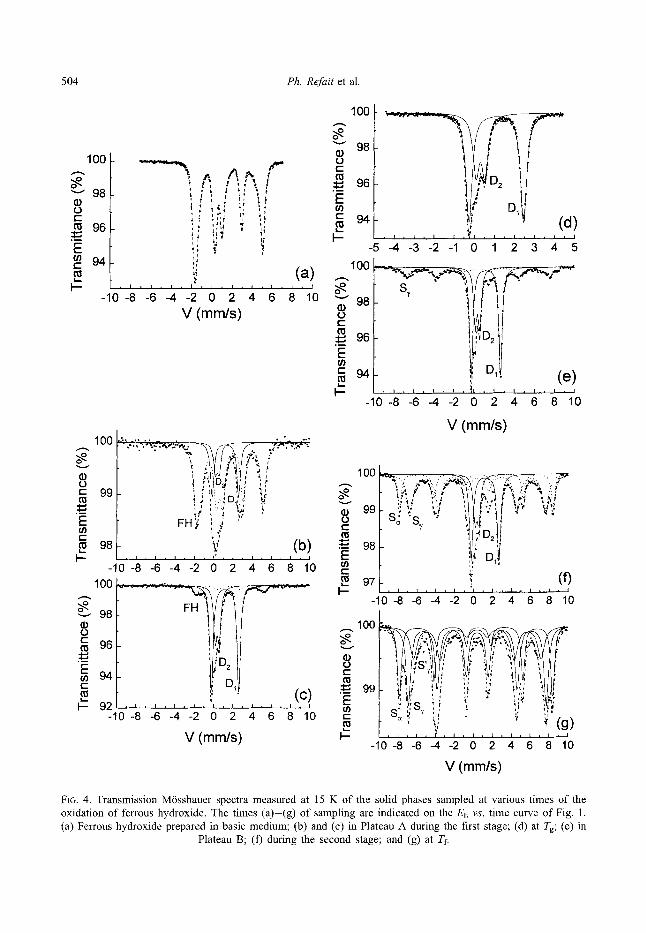
M6ssbauer spectra measured at 15 K are displayed in Fig. 4 and corresponding hyperfine parameters are gathered in Table 1. Seven examples from samples (a)-(g) which are reported, relate to
various reaction times which are indicated in the Eh and pH vs. time curves of Fig. 1. For experimental facility, spectrum (a) which would correspond to the very beginning of the oxidation process is in fact that of a Fe(OH)2(s) standard sample precipi- tated in a basic medium (pH ~ 11.5) where GRs do not form (cf. Olowe et al., 1991). Besides the analyses at the beginning, at the inflection point Tg and at the end of the overall reaction, i.e. spectra (a), (d) and (g), two product analyses were performed at each reaction stage, one at the beginning and one at the end. Therefore, spectra (a) and (d) are those of Fe(OH)2(s) and GR2(SO42-)(~), respectively, whereas spectrum (g) is that of a mixture of 7-FeOOH(s) and ~-FeOOH(s). In plateau A, i.e. spectra (b) and (c), both Fe(OH)2(s) and GR2(SOZ-)(s) are always present in different amounts. In contrast, plateau B is only found at the beginning of the second reaction stage and corresponds to spectrum (e) where only GR2(SO24-)(~) and 7-FeOOH(o are present. After plateau B, ~-FeOOH(s) is also present in spectrum (f) along with GR2(SO] )(s) and 7-FeOOH(s).
in the spectra, GR2(SO42-)(s) is still paramagnetic and always displays two different quadrupole doub le t s D 1 and D2 attributed to Fe(II) and Fe(III), respectively. At 15 K, the respective quadrupole effect values AEQ are -2.8 and 0.4 mm s 1 and isomer shifts 6 ~1.3 and 0.5 m m s 1 (Table 1). The abundance ratio D1/D2 varies little, ranging from 1.87 to 2.11, effectively equal to 2. This proves that the compound corresponds to a well-defined chemical composition and that depar- tures from this stoichiometry do not occur in the experimental conditions considered here.
7-FeOOH(s) is antiferromagnetic (Johnson, 1969) and corresponds to sextet S v with hyperfine field of - 450kOe and AEQ of 0 at 1 5 K (G6nin et al., 1998a); some additional sextet S' r may be used to fit correctly the asymmetry of the spectrum, due to the poor crystallinity of lepidocrocite (Murad & Schwertmann, 1984)�9 ~-FeOOH(s) is antiferro- magnetic (D6zsi et al., 1967) corresponding to sextet S~ with a field o f - 5 0 0 kOe and AEQ of ~-0 .25 mm s -1 (Van der Woude & Dekker, 1966; Forsyth et al., 1968; Murad, 1982). Finally, Fe(OH)2(s) is also antifen-omagnetic at 15 K under the N6el temperature of 34 K (Miyamoto et al., 1967) and gives rise to a magnetically split spectrum consisting of eight peaks, due to a mixture of quantum states with a hyperfine field, -200 kOe at 4 K (Miyamoto et al., 1967; G6nin et
504 Ph. Refait et al.
100
98
t- t~ 96
,=_ E t~ 94 t-
F--- -1(
1
�9 i , i , i , i , i , i , i , i , [ ,
- 8 - 6 - 4 - 2 0 2 4 6 8 10 V (mm/s)
100
(11 o e- 99
E t- O 98
F--
'~ i!:...~D:"
FH~; ;
Z
(b) i I , I , I �9 I , I ~ , I , I , I , I , I
- 1 0 - 8 - 6 - 4 - 2 0 2 4 6 8 10 oo_ ! o~._4 9 8 . FH
o 96
E 94 c
,, (c) I-- 92 ~
- 1 0 - 8 - 6 - 4 - 2 0 2 4 6 8 10
V (mm/s)
lOO
" " 98 o c t~ :~ 96 . m
E t- O
I--
p
94 d) �9 ~ . I , i , E , i , i , i , i , i , i
-5 -4 -3 -2 -1 0 1 2 3 4 5
Sy 98
o c
96 E !:
94 �9 (e)
- 1 0 - 8 - 6 - 4 - 2 0 2 4 6 8 10
V (mm/s)
100
99 o c t~
98 E t/) c E 97 )
I-- - 1 0 - 8 - 6 - 4 - 2 0 2 4 6 8 10
100
o c
99 E taq c )
I--- -10 -8 -6 -4 -2 0 2 4 6 8 10
V (mm/s)
FIG. 4. Transmission M6ssbauer spectra measured at 15 K of the solid phases sampled at various times of the oxidation of ferrous hydroxide. The times (a)-(g) of sampling are indicated on the Eh vs. time curve of Fig. 1. (a) Ferrous hydroxide prepared in basic medium; (b) and (c) in Plateau A during the first stage; (d) at Tg; (e) in
Plateau B; (f) during the second stage; and (g) at Tf.

Fe(II)-Fe(iiI) hydroxysulphate green rust and Fe(II) hydroxide 505
TABLE 1. M6ssbauer hyperfine parameters at 15 K of the precipitates (a)-(g) sampled during the reaction of oxidation of Fe(OH)2(~) in sulphate containing aqueous medium.
Sample (a) Sample (b) Sample (c) AEQ H RA 6 AEQ H RA ~i AEQ H RA
FH 1.4 3.1 175 100 1.4 3.1 175 74.6 1.4 -3.1 175 11.1
. . . . 1.33 2.78 - 16.9 1.34 2.82 60.3 - - - 0.50 0.39 - 8.5 0.53 0.41 - 28.6
1.99 2.11
Ol D2 D~/D2
Sample (d) GR2(SOZ-)(s) obtained at Tg AEQ RA
D1 1.30 2.85 65.2 1)2 0.47 0.45 34.8 D1/D2 1.87
Sample (e) Sample (f) Sample (g) AEQ H RA 6 AEQ H RA ~ AEQ H RA
S~ 0.51 0 440 28.2 0.49 0 445 43.5 0.50 0 454 47 S' v . . . . . . . 0.48 0 420 26
S~ . . . . 0.49 -0.25 505 19.6 0.49 -0.28 504 27
D~ 1.34 2.87 - 47.7 1.34 2.89 - 24.7 . . . . D 2 0.51 0.43 -- 24.1 0.49 0.40 -- 12.2 -- -- Da/O2 1.98 2.02
= isomer shift with respect to metallic et-iron at room temperature in mm s--l; AEQ = quadmpole splitting in mm s-l ; H = hyperfine field in kOe; RA = relative area in %. FH = Fe(OH)2(s~; D1 and D2 = GR2(SO] )(s); S~ and S'~ = 7-FeOOH(s); S~ = et-FeOOH(s).
al., 1986) and 166 kOe at 20 K (Refait et al., 1998), which lie perpendicular to the axis of symmetry of the electric field gradient, and the quadrupole effect AEQ, which is - - 3 mm s 1 (Miyamoto et al., 1967; G6nin et al., 1986), whereas the isomer shift
is 1.4 m m s -1.
D I S C U S S I O N
Chemical formula o f GR2(S02-)C~)
Various authors concluded previously that the Fe(II)/Fe(III) ratio of GR2(SO]-)(s) was close or exactly equal to 2 (Detournay et al., 1975; Cuttler et al., 1990; Schwertmann & Fechter, 1994; G6nin et al., 1996). The TMS analyses confirm that this is the case but also demonstrate that this ratio is kept
constant all along the formation and oxidation process of the GR, from the beginning of the oxidation of Fe(OH)2(s~ to the end of the formation of ferric oxyhydroxides. Therefore, only pure phases are present, and no solid-solution between Fe(OH)2(s~ and GR2(SO42-)(s~ or b e t w e e n GR2(SO]-)(s) and FeOOH(s) is observed. Similar conclusions were drawn recently for Fe(II)-Fe(III) hydroxyoxalate GRl(C2042-)(s) (Refait et al., 1998).
GR2(SO]-)(~) is generally assumed to be a pyroaur i te -s j6greni te - l ike hydroxide (Al lmann, 1970; Taylor, 1973; Hansen et al., 1994) and consists of [Fe~IFe~n(OH)12] 2+ positively charged hydroxide sheets due to the Fe(III) cations, al ternating with negat ively charged interlayers composed of sulphate ions and water molecules. To main ta in the whole e lectroneutral i ty , the

506 Ph. Refait et al.
TABLE 2. Gibbs standard free energy of formation AG~ or go in kJ mo1-1 and thermo-dynamic constants used for calculations and results of this study.
Species AG~'f or g~ References
Fe(OH)2(s) -490 FeI~FelII(OH)~2S04(s~ -3790
7-FeOOH(s) -475.5 Fe 2+ -91.5
FeOH + -277.4
FeSO4aq -823.49 H20 -237.18 SO 2- -744.56
This work This work Computed from Lindsay (1979) Yang, 1982 Bard et al., 1985; Kelsall & Williams, 1991 Wagman et al., 1982; Bard et aL, 1985; Kelsall & Williams, 1991 Bard et al., 1985; Kelsall & Williams, 1991 Bard et al., 1985; Kelsall & Williams, 1991 Bard et at., 1985; Kelsall & Williams, 1991
Complexes log K References
Fe 2+ + H20 = FeOH + + H + -8.98 Computed from the AG~ values Fe z+ + SO 2 = FeSO4aq 2.2 Id Na + + SO,] = NaSO4- 0.7 Jenkins & Monk (1950)
c o m p o s i t i o n o f the in te r l aye r s should be [SO4.rIH20] 2-, the presence of two Fe(III) ions being counterbalanced by one sulphate ion, leading to the overall formula Fe~tFe~H(OH)12SO4-nH20 already proposed (Hansen et al., 1994, 1996; G~nin et al., 1996). The results given by the measurement of Fe and S concentrations can be used to verify this hypothesis. With an initial concentration of Fe(OH)z(s) equal to 0.1 mol 1-1, the amount of Fe in the GR is thus equal to 0.1 + A[Fe], that is 0.1165 mol 1 1. The amount of sulphate in the GR is A[S], that is 0.025 mol 1-1. This gives an Fe/SO4 ratio of 0.1165/0.025 4.66. Since the Fe(II)/Fe(III) ratio is found to be 2 by TMS, the chemical formula given by c[Fe] and c[S] m e a s u r e m e n t s w o u l d b e : Fe~Fe~u(OH)11.48(SO4)l.26.nH20. The departure from the proposed formula is more likely to be due to slight experimental errors. Actually, such a composition would imply a release of OH ions during the formation of GR2(SO42-)(s~ from Fe(OH)2(s~, since the chemical balance would be:
5 Fe(OH)z(s)+Fe 2++ 1.26SO~ + 1 / 2 0 2 + H20 ~ 11 llI Fe4Fe2 (OH)11.48(804)1.26 nt- 0.52 OH- (3)
Since c[Fe(OH)2(~)] is equal to 0.1 tool 1 1, the release of 0.52 OH would correspond to ~0.01 mol 1 -a of OH ions, which would have increased the
pH to -12. This is not observed: formula FelIFe~n(OH)12SO4.nH20 must be maintained (cf. Eqns. 1 & 2)
S t a n d a r d Gibbs energy o f f o r m a t i o n o f GR2(S02-)( .~) and Fe(OH)2(~)
The standard chemical potential of GR2(SO] )(s) can be computed using the equilibrium conditions given by Nernst's law. Before undertaking this determination, it is necessary to dispose of a consistent and reliable set of thermodynamic constants for all species involved in the computa- tions. In particular, the value admitted for ~t~ 2+) is of the utmost importance. On the one hand, M. Pourbaix (1966) and subsequently followed by us recently (Refait & G6nin, 1993; Drissi et al., 1995; G6nin et al., 1996) suggested a value o f -84 .9 kJ mol 1, initially determined by Randall & Frandsen (1932). On the other hand, the value suggested by NBS (Wagman et al., 1982), and used by Hansen et al. (1994) for their detelmination of ~t~ )(~], is larger, - 7 8 . 9 kJ tool - I . Finally, recent studies lead to a smaller value of - - 9 1 . 5 kJ mol a (Yang, 1982; Bard et al., 1985; Kelsall & Williams, 1991), which will be used in this work. Accordingly, the ~t ~ values retained for the other Fe species, listed in Table 2, were also

Fe(II)-Fe(llI) hydroxysulphate green rust and Fe(II) hydroxide 507
taken from the work of Kelsall & Williams (1991); this allows us to have a complete set of consistent values and equations. The activities of dissolved species were computed by means of the MINTEQ A2 computer program (CLAM, 1991) considering the various complexes likely to form in the considered experimental conditions, that is FeOH +, FeSOaaq and NaSO4. The corresponding log K values given in Table 2 were computed using the retained I.t ~ values for the sake of consistency.
Since Fe(OH)2(s) and GR2(SO 2 ){s) are the only solid phases present during the first reaction stage in plateau A (Table 1, Fig. 4b-c) , the standard Gibbs energy of formation of GR2(SO]-)(~) can be determined from the corresponding equilibrium conditions. One can estimate first the standard chemical potential of Fe(OH)2(s) by using the e q u i l i b r i u m cond i t i ons b e t ween Fe 2+ and Fe(OH)2(s) and given by:
Fe 2+ + 2 H20 ~- Fe(OH)2(s) + 2 H + (4) pK = 2 pH(A) + log a[Fe 2+] with (4')
pK = {g~ - I-t~ 2+] 2 la~
where (In 10) x (RT ~ = 5.708 kJ mol 1. As the formation of GR2(SOe-)(s) proceeds,
dissolved Fe species are consumed and the solution becomes undersaturated with respect to Fe(OH)2(~). The equilibrium conditions between Fe 2+ and Fe(OH)2(s) are encountered at the beginning of the reaction, corresponding to pH = 8.0• (Fig. 1), c[Fe] = ( 1 5 • -3 mol 1 1 and c[S] = (113• x10 -3 mol 1 1 (Fig. 2, point 1). From these concentrations, an activity a[Fe 2+] of 2.09x 10 3 is computed. This leads to pK = 1 3 . 3 2 • a n d I . t ~ = - 4 9 0 • kJmol-1 ; this value matches that of -492.0 kJ mo1-1 given by Bard et al., (1985) and by Kelsall & Williams (1991). Thus, even though Fe(OH)2(s) is slightly oxidized as soon as it precipitates, the resulting variations of its chemical potential are kept within the range of the experimental error.
Therefore, the standard Gibbs energy of forma- tion of GR2(SO 2 )(s) can be evaluated from the equilibrium conditions between Fe(OH)2(s) and GR2(SO 2 )(~) met in plateau A. These conditions are given by:
6 Fe(OH)2(s) + SO 2- ,~ FelIFe~1(OH)12SO4(s) + 2e (5)
Eh(A) = E~ 0.029 log a[SO 2 ] (5')
with E~ -{Ia~ )(s)] - It~ SO2-] - 6g~ }/(2 x 96.485) if chemical potentials are expressed in kJ mol - I and Eh in V. Taking Eh(A ) 0.497 + 0.007 V, pH(A) - 8.0 • 0.1, c[Fe] = ( 1 3 + l ) x 10 -3 m o l 1 1 a n d c [S] = (110•215 10 -3 mol 1 - I , the activity of sulphate is obtained at 2.25 x 10 2, which leads to E~ = -0.545_+0.008 V and, taking into account the value of I.t~ determined above with its corresponding error, I.t~ )(s)] becomes - 3 7 9 0 • 10 kJ mo1-1.
This value confirms that GR2(SO 2-) is able to reduce species like NO2 or NO3 into N2, N20 or NH~ spontaneously with formation of 7-FeOOH, as shown by the conditional redox potentials given in Table 3.
Plateau B: verification o f the 7-FeOOH/ GR2(SO24-)(sj equilibrium conditions
Knowing the value of g~ 2 )(s)], it is possible to verify the meaning of parameters measured at plateau B, which would correspond to the equilibrium conditions between GR2(SO 2 )(s) and T-FeOOH(s> But neither NBS (Wagman et al., 1982) nor Kelsall & Williams (1991) gave any value for the chemical potential of lepidocrocite. However, it can be calculated from the log K value given for the reaction:
7-FeOOH(s) + 3 H ~ ~,~ Fe 3+ + 2 HeO (6) since log K =
{g~ bt~ 3+) - 2 ~t~
TABLE 3. Conditional redox potentials for N species and GR2(SO2-). The values given are computed at
pH = 7.0 and all activities equal to 1.
Couples E(V)
NO2/N2 0.96 NO2/N20 0.76 NO3/N2 0.75 NO3/N20 0.60 NOz/NO 3 0.44 NO2/NHI 0.34 NO3/NH~ 0.33 7-FeOOH/GR2(SO ] ) 0.11 GR2(SOZ-)/Fe(OH)2 -0.55

508 Ph. Refait et al.
According to Lindsay (1979), log K = 1.39. With the g~ and g~ ) considered here ( T a b l e 2), it g i v e s l a ~ at - 4 7 5 . 5 kJ mol 1.
The e q u i l i b r i u m c o n d i t i o n s b e t w e e n GR2(SO ] )(s) and 7-FeOOH(s) are given by:
FeI41FenI(OH)12SO4 ~_ 6 y-FeOOH(~) + SOl- + 6 H + + 4 e (7)
Eh(B) = E~ + 0.015 log a[SO]-] - 0.089 pH(B) (7')
with E~ = {g~ + 6 g~ - g~ 2 )(s)]}/(4 x 96.485).
Using the previous procedure and taking pH(B) - 6 . 7 + 0 . 1 , c[Fe] = ( 1 2 + 3 ) x10 -3 m o l l i ,e[S] = (110 + 5)x 10 3 mol 1-1, the activity of sulphate is obtained at 2.26 x 10 -2. Taking into account g~ as d e t e r m i n e d above and g~ = 3790+ 10 kJ mol 1, EOh(B) - 0.50_+0.03 V is found which would lead to Eh(B) - 0.13_+0.03 V according to (7'), a value close to the exper imenta l Eh(B) found at
0.075_+0.01 V.
C o m m e n t s
The actual fo rmula of GR2(SO]-) (s ) is Fe~IFe~n(OH)12SO4.nH20 but the value of n cannot be ascertained. The actual value of g~ )~yd.] is thus, g~ 2 )hya.] = g~ + n g~ However, by using as a formula, FeIIFenI(OH)12SO4, and its corresponding value for g~ there is no influence on the equilibrium conditions of the reactions assuming a[H20] to be close to 1. Let us write, for instance, eqn. (5) with the actual formula of GR2(SO]-)(s):
6 Fe(OH)2(s ) + SO42- + nH20 FenFem(OH)12SO4(s2.nH20 + 2 e - (8)
Eh = E~ -- 0.029 log a[SO~-] with (8) E%(8) -
{g~ 2 )hyd . ] - - g ~ - -
6 g~ - ng~ x 96.485)
A s g ~ 2 )hya.] = g ~ +
ng~ O) we have E~ = E~ = E~ and eqn. (8) = eqn. (5); the equilibrium conditions are exactly the same.
This is of course true since this so called 'anhydrous form' is totally fictitious and designates GR2(SO]-)(s) without considering the intercalated water molecules.
D i s c u s s i o n o f o t h e r d a t a
The work by Hansen et al. (1994) led to the value of -3669 kJ mo1-1 for GR2(SOZ-)(s) in the ' a n h y d r o u s f o r m ' , i . e . w i t h f o r m u l a Fe~n)Fe~UI)(OH)12SO4. This value appears to be 121 kJ mol 1 larger than that found here. Such a difference corresponds to a difference in the s t a n d a r d e q u i l i b r i u m p o t e n t i a l o f the GR2(SOZ-)(s)/Fe(OH)z(s) couple of 630 mV, which is completely out of the range of the experimental errors. It is possible to illustrate how important this difference is. Using the value given by Hansen et al. (1994), we compute the redox potentials at pH = 7 and [SOl-] = 1 of GR2(SOZ-)(s)/Fe(OH)2(s) with g~ -486.5 kJ mol -a as given by NBS (Wagman et al., 1982) and 7-FeOOH(s)/ G R 2 ( SO2 )(s) w i t h g ~ = -475.5 kJ mo1-1 as computed here from Lindsay (1979). Eh = --0.03 V and -0 .44 V is found, respectively. This means that the formation of 7-FeOOH(s) would take place directly from Fe(OH)2(s) and that GR2(SO 2 )(s) would not form.
The discrepancies between our results and those given by Hansen et al. can be explained. First, the influence of the value considered for the standard free energy of formation of Fe z+ comprises about half of it; the NBS value (Wagman et al., 1982), agreed by Hansen et al., is -78 .9 kJ mol 1 whereas a smaller value of 91.5 kJ mol - l is suggested here. Secondly, Hansen et al. (1994) used a completely different synthesis route to obtain the GR2(SO 2 )(s) product, involving an assumption that could be a source of error: the activity of Fe~q would be controlled by the solubility of ferrihydrite, implying the use of the solubility product of this compound (Fox, 1988), the composition of which is not ascertained. Moreover, it is not demonstrated that the GR compound prepared by Hansen et al. (1994) has the same composition, in particular the same Fe/S ratio, as here. In contrast, the results presented here, which confirm and refine the previous results by G6nin et al. (1996), are experimentally grounded measurements.
C O N C L U S I O N S
The discovery of the mineral 'foug&ite' as being a GR compound in hydromorphic soils (Trolard et al., 1996, 1997) highlights the influence of such Fe(II)- Fe(III) compounds on soil reactivity and the interest of firmly established thermodynamic data (G~nin et

Fe(II)-Fe(III) hydroxysulphate green rust and Fe(II) hydroxide 509
al., 1998b). The methodology for determining the Gibbs standard free energy of formation of G R 2 ( S O 2 )(s) , A G ~ or g~ = --3790+ 10 kJ m o l i in the ' a n h y d r o u s f o r m ' , i .e . c o r r e s p o n d i n g to [Fe~IFeI~II(OH)12]2+.[SO4.nH20] 2- where n is assumed to be 0, yields a reliable value. It also demonstrated that GR2(SO 2 )(s~ is characterized by a well-defined composition, which does not vary all along the oxidation process. The Fe(II)/Fe(III) ratio stays at 1.99-1-0.12 and departures from the stoichiometry of 2 are not observed.
REFERENCES
Allmann R. (1970) Doppelschichtstrukturen mit b r u c i t h i i n l i c h e n S c h i c h t i o n e n [Me(II)l xMe(III)x(OH)2] x+. Chimia, 24, 99 108.
Bard A.J., Parsons R. & Jordan J. (1985) Standard Potentials" in Aqueous Solution. Marcel Dekker Inc., New York.
Bender Koch C. & Morup S. (1991) Identification of green rust in an ochre sludge. Clay Miner. 26, 577-582.
Centre for Exposure Assessment and Modelling (CEAM) (1991) MINTEQ A2. USEPA Office of Research and Development, College Station Rd., Athens, GA, USA.
Chariot G. (1959) Les Rdactions l~lectrochimiques. Masson, Paris.
Cuttler A.H., Man V., Cranshaw T.E. & Longworth G. (1990) A MSssbauer study of green rust precipitates: I. Preparations from sulphate solutions. Clay Miner. 25, 289-301.
Detournay J., De Miranda L., D6rie R. & Ghodsi M. (1975) The region of stability of GRII in the electrochemical potential-pH diagram of iron in sulphate medium. Corros. Sci. 15, 295 306.
DNzsi I., Keszthelyi L., Kulgawczuk D., Molnar & Eissa N.A. (1967) M6ssbauer study of 13- and ~z-FeOOH and their disintegration products. Phys. Stat. Sol. 22, 617-629.
Drissi S.H., Refait Ph., Abdelmoula M. & GNnin J.-M.R. (1995) Preparation and thermodynamic properties of Fe(iI)-Fe(III) hydroxide-carbonate (green rust one), Pourbaix diagram of iron in carbonate-containing aqueous media. Corros. Sci. 37, 2025 2041.
Forsytb J.B., Hedley I.G. & Johnson C.E. (1968) The magnetic structure and hyperfine field of goethite (~-FeOOH). J. Phys. C, 1, 179-188.
Fox L.E. (1988) The solubility of colloidal ferric hydroxide and its relevance to iron concentrations in river water. Geoehim. Cosmochim. Acta, 52, 771-777.
G~nin J.-M.R., Bauer Ph., Olowe A.A. & R~zel D.
(1986) M6ssbauer study of the kinetics of simulated corrosion process of iron in chlorinated aqueous solution around room temperature: the hyperfine structure of ferrous hydroxides and green rust 1. Hyperf Inter. 29, 1355-1358.
G~nin J.-M.R., Olowe A.A., Benbouzid-Rollet N.D., Prieur D., Confente M. & RNsiak B. (1991) The simultaneous presence of green rust 2 and sulphate reducing bacteria in the corrosion of steel sheet piles in a harbour area. Hyperf Inter. 69, 875-878.
GNnin J.-M.R., Olowe A.A., Refait Ph. & Simon L. (1996) On the stoichiometry and Pourbaix diagram of Fe(II)-Fe(III) hydroxy-sulphate or sulphate-con- taining green rust 2; an electrochemical and M6ssbauer spectroscopy study. Corros. Sci. 38, 1751-1762.
GNnin J.-M.R., Abdelmoula M., Refait Ph. & Simon L. (1998a) Comparison of the Green Rust Two lamellar double hydroxide class with the Green Rust One pyroaurite class: Fe(II)-Fe(III) sulphate and selenate hydroxides. Hyperf Inter. (C), 3, 313-316.
GNnin J.-M.R., Bourri6 G., Trolard F., Abdelmoula M., Jaffrezic A., Refait Ph., Maitre V., Humbert B. & Herbillon A.J. (1998b) Thermodynamic equilibria in aqueous suspensions of synthetic and natural Fe(II)- Fe(III) Green Rusts; occurrences of the mineral in hydromorphic soils. Environ. Sei. Technol. 32, 1058-1068.
Hansen H.C.B., Borggaard O.K. & Sorensen J. (1994) Evaluation of the free energy of formation of Fe(II)- Fe(III) hydroxide-sulphate (green rust) and its reduction of nitrite. Geochim. Cosmochim. Acta, 58, 2599-2608.
Hansen H.C.B., Bender Koch C., Nancke-Krogh H., Borggaard O.K. & Sorensen J. (1996) Abiotic nitrate reduction to ammonium: key role of green rust. Environ. Sci. Technol. 30, 2053-2056.
Jenkins I.L. & Monk C.B. (1950) The conductances of sodium, potassium and lanthanum sulphates at 25~ J. Am. Chem. Soc. 72, 2695-2698.
Johnson C.E. (1969) Antiferromagnetism of 7-FeOOH: a MNssbauer effect study. J. Phys. C (Solid St. Phys.) 2, 1996-2002.
Kelsall G.H. & Williams R.A. (1991) Electrochemical behaviour of ferrosilicides (FexSi) in neutral and alkaline aqueous electrolytes, a~ Electrochem. Soc. 138, 931-940.
Lindsay W.L. (1979) Chemical Equilibria in Soils. Wiley Interscience, New York.
Miyamoto H., Shinjo T., Bando Y. & Takada T. (1967) MSssbauer effect of Fe 57 in Fe(OH)2. Bull. Inst. Chem. Res. Kyoto Univ. 45, 333-341.
Murad E. (1982) The characterisation of goethite by MNssbauer spectroscopy. Am. Miner. 14, 273-283.
Mmad E. & Schwertmann U. (1984) The influence of crystallinity on the M6ssbauer spectrum of lepido- crocite. Mineral. Mag. 48, 507-511.

510 Ph. Refait et al.
Olowe A.A. & G~nin J.-M.R. (1989) Potential-pH equilibrium diagrams for the iron-water system in the presence of sulphate ions: Domain of existence of green rust 2. Proc. int. Syrup. Corros. Sci. Engng., CEBELCOR, RT297, 363 390.
Olowe A.A. & G+nin J.-M.R. (1991) The mechanism of oxidation of Fe(II) hydroxide in sulphated aqueous media: importance of the initial ratio of the reactants. Corros. Sci. 32, 965-984.
Olowe A.A., Refait Ph. & G~nin J.-M.R. (1991) Influence of concentration on the oxidation of ferrous hydroxide in basic sulphated medium: particle size analysis of goethite and delta FeOOH. Corros. Sci. 32, 1003-1020.
Ponnamperuma F.N., Tianco E.M. & Loy T. (1967) Redox equilibria in flooded soils: I. The iron hydroxide system. Soil Sci. 103, 374-382.
Pourbaix M. (1966) Atlas of Electrochemical Equilibria in Aqueous Solutions, Pergamon Press Ltd., Oxford.
Randall M. & Frandsen M. (1932) The standard electrode potential of iron and the activity coefficient of Fe(ll) chloride. J. Am. Chem. Soc. 54, 47 54.
Refait Ph. & Gbnin J.-M.R. (1993) The oxidation of Fe(II) hydroxide in chloride-containing aqueous media and Pourbaix diagrams of green rust one. Corros. Sci. 34, 797 819.
Refait Ph. & G~nin J.-M.R. (1994) The transformation of chloride-containing green rust one into sulphated green rust two by oxidation in mixed CI- and SO42 aqueous media. Corros. Sci. 36, 55-64.
Refait Ph. & Gbnin J.-M.R. (1996) Chloride-containing ferrous hydroxides. SIF Conf. Proc. 50, 55 58.
Refait Ph., Charton A. & G6nin J.-M.R. (1998) Identification, composition, thermodynamic and structural properties of a pyroaurite-like Fe(II)- Fe(IIl) hydroxyoxalate green rust. Eur. ~L Solid St. lnorg. 'Chem., 35, 655 666.
Schwertmann U. & Fechter H. (1994) The formation of green rust and its transformation to lepidocrocite. Clay Miner. 29, 87-92.
Taylor H.F.W. (1973) Crystal structures of some double hydroxide minerals. Mineral. Mag. 39, 377-389.
Trolard F., Abdelmoula M., Bourri~ G., Humbert B. & G6nin, J.-M.R. (1996) Evidence of the occurrence of a "Green Rust" component in hydromorphic soils Proposition of the existence of a new mineral: "foug6rite". Comp. Rend. Acad. Sci. Paris, 323, IIA, 1015 1022.
Trolard F., G6nin J.-M.R., Abdelmoula M., Bourri6 G., Humbert B. & Herbillon A.J. (1997) Identification of a green rust mineral in a reductomorphic soil by M6ssbauer and Raman spectroscopies. Geochim. Cosmochim. Acta, 61, 1107-1111.
Van der Woude F. & Dekker A.J. (1966) M6ssbauer effect in ~-FeOOH. Phys. Stat. Sol. 13, 181-193.
Wagman D.D., Evans W.H., Parker V.B., Schumm R.H., Halow l., Bailey S.M., Chumey K.L. & Nutall R.L. (1982) The NBS tables of chemical thermodynamic properties. Selected values for inorganic and C1 and C2 organic substances in SI units. J. Phys. Chem. Ref Data, 11 (suppl. 2).
Yang X. (1982) Problems encountered in the setting-up of equilibrium diagrams of the oxygen-hydrogen- iron system. CEBELCOR, RT263, 177-190.