cited2 modulates breast cancer metastatic ability...
TRANSCRIPT

Oncogenes and Tumor Suppressors
CITED2 Modulates Breast Cancer MetastaticAbility through Effects on IKKaSwaathi Jayaraman, Michele Doucet,Wen Min Lau, and Scott L. Kominsky
Abstract
Previously, we identified the transcriptional coactivator CIT-ED2 as a potential facilitator of bone metastasis using a murinemammary cancermodel. Extending these studies to humanbreastcancer, it was observed that CITED2 mRNA expression wassignificantly elevated in patient specimens of metastatic breastcancer relative to primary tumors, with highest levels inmetastasisto bone relative to non-bone sites. To further evaluate CITED2functions in breast cancer metastasis, CITED2 expression wasstably reduced in the human breast cancer cell lines MDA-MB-231 and MDA-MB-468, which are metastatic in animal models.While CITED2 knockdown had no effect on cell proliferation,cell migration and invasion were significantly reduced, as wasthe establishment of metastasis following intracardiac admin-istration in athymic nude mice. To explore the mechanismbehind these effects, gene expression following CITED2 knock-down in MDA-MB-231 cells by cDNA microarray was per-formed. As confirmed at the mRNA and protein levels in both
MDA-MB-231 and MDA-MB-468 cells, expression of the NF-kBregulator IKKawas significantly reduced, along with several NF-kB targets with known roles in metastasis (OPN, MMP9, uPA,SPARC, IL11, and IL1b). Furthermore, ChIP assay revealedrecruitment of CITED2 to the promoter of IKKa, indicating adirect role in regulating its expression. Consistent with reducedIKKa expression, CITED2 knockdown inhibited both canonicaland noncanonical NF-kB signaling. Finally, restoration of IKKaexpression following CITED2 knockdown in MDA-MB-231 andMDA-MB-468 cells rescued their invasive ability. Collectively,these data demonstrate that CITED2 modulates metastaticability in human breast cancer cells, at least in part, throughthe regulation of IKKa.
Implications: The current study highlights the role of CITED2in facilitating breast cancer metastasis, partly via regulation ofIKKa. Mol Cancer Res; 14(8); 730–9. �2016 AACR.
IntroductionBreast cancer is themost frequently diagnosed cancer inwomen
worldwide and the second most commonly occurring canceroverall. Although primary tumorsmay be effectively treated whendetected early, metastatic disease is largely incurable and repre-sents the ultimate cause of mortality in breast cancer patients. It isestimated that approximately 6% of patients already have met-astatic disease at the time of diagnosis while approximately 20%to 50% of patients who are initially diagnosed with early-stagebreast cancer will eventually develop metastasis (1). Sadly, themedian survival time for patients with metastatic breast cancer isonly 18 to 30months. Despite recent research efforts, elucidationof the critical drivers of metastasis and their mechanism of actionis lacking. Filling this knowledge gap is essential to the develop-ment of novel therapeutic modalities and improving the clinicalmanagement of this disease.
The Cbp/p300–interacting transactivator with Glu/Asp–richcarboxy-terminal domain-2 (CITED2) is a non-DNA–bindingtranscriptional coactivator that was originally discovered for its
role in development (2–5). As a transcriptional coactivator, CIT-ED2 interactswith several transcription factors, such as p300/CBP,Lhx2, TFAP2, Smad2/Smad3, PPARg, and estrogen receptor,modulating their ability to activate gene transcription (6–11).Beyond its involvement in development, CITED2 has also beenreported to play a role in cancer, including that of the skin, colon,and lung (12–14). Recently, we identified CITED2 as a potentialfacilitator of breast cancer bone metastasis using a murine mam-mary cancer model (15). Although our preliminary analysis ofprimary human breast tumor tissues revealed significantly higherlevels of CITED2mRNA relative to normal mammary epithelium(15), its expression pattern in metastatic lesions and functionalcontribution to human breast cancer metastasis remain unclear.
In this study, we investigated the role of CITED2 in humanbreast cancer metastasis. Here, we show that in breast cancerpatients, CITED2 expression is significantly elevated inmetastaticlesions relative to primary tumors, with highest levels in bonemetastasis. Furthermore, utilizing two highly invasive breastcancer cell lines, we show that stable knockdown of CITED2significantly reduces tumor migration and invasion in vitro andthe establishment of metastasis in vivo. Finally, we provide evi-dence that CITED2 mediates metastatic ability in human breastcancer cells, at least in part, by regulating the expression of IKKa.
Materials and MethodsCell lines, tissues, and treatment
The human breast cancer cell lines MDA-MB-231 and MDA-MB-468 were obtained from ATCC (2014) and were authenticat-ed using DNA profiling and cytogenetic analysis by the cell bank.Cells were utilized for the experiments within 6 months from the
DepartmentofOrthopaedicSurgery, JohnsHopkinsUniversity Schoolof Medicine, Baltimore, Maryland.
Note: Supplementary data for this article are available at Molecular CancerResearch Online (http://mcr.aacrjournals.org/).
Corresponding Author: Scott L. Kominsky, Johns Hopkins University School ofMedicine, 720 Rutland Avenue, Ross 232, Baltimore, MD 21205. Phone: 410-502-6417; Fax: 410-502-6414; E-mail: [email protected]
doi: 10.1158/1541-7786.MCR-16-0081
�2016 American Association for Cancer Research.
MolecularCancerResearch
Mol Cancer Res; 14(8) August 2016730
on July 10, 2018. © 2016 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst May 23, 2016; DOI: 10.1158/1541-7786.MCR-16-0081

time of resuscitation. MDA-MB-231 cells were maintained inRPMI medium (Gibco) supplemented with 10% FBS (AtlantaBiologicals), and MDA-MB-468 cells were maintained in DMEMmedium (Gibco) supplemented with 10% FBS and 1% L-gluta-mine (Gibco). To identify genes that are regulated by the NF-kBpathway, cells were treated with 10 mmol/L of PS1145 (Sigma-Aldrich) for 16 hours at 37�C.
Normal mammary epithelium samples, kindly provided byDr. Saraswati Sukumar (Johns Hopkins University School ofMedicine, Baltimore, MD), were prepared from reduction mam-moplasty specimens of women with no breast abnormalities.Normal and tumor tissues were obtained from the SurgicalPathology Division of the Johns Hopkins Hospital (Baltimore,MD) following the approval of the Institutional Review Board(IRB) of the Johns Hopkins University School of Medicine. For allspecimens, required written informed patient consents wereobtained as approved by the IRB.
TransfectionTo study the effects of CITED2 in human breast cancer metas-
tasis, MDA-MB-231 and MDA-MB-468 cells were infected withthe lentiviral shRNA expression vector pLKO.1-puro (Addgeneplasmid 8453) containing siRNA sequence specific for scrambledor CITED2. The CITED2 siRNA sequence has been describedpreviously (9, 16). Stable cells were selected in the presence of1mg/mLpuromycin (Sigma-Aldrich) for oneweek andutilized forsubsequent experiments.
For experiments involving reexpression of IKKa, shCITED2-expressing cells were transiently transfected with a 3:1 ratio ofX-tremeGENE HP DNA Transfection Reagent (Roche) andpCR3.1-FLAG-IKKa vector [a kind gift from Hiroyasu Nakano,Juntendo University School of Medicine, Tokyo, Japan (Addgeneplasmid 15467; ref. 17)] or empty vector in OPTI-MEM medium(Gibco) for 24, 48, or 72 hours.
Quantitative qRT-PCRTotal RNA from tissue samples and cell lines was extracted
using TRIzol (Invitrogen), and cDNA was generated using areverse transcription system (Promega). The qRT-PCR parametershave been described previously (11). Amplification of 36B4 wasused as an internal control. Relative expression between sampleswas calculated by the comparative Ct method. The primersequences used were: CITED2 (sense) 50-ACCATCACCCTGCC-CACC-30, (antisense) CGTAGTGTATGTGCTCGCCCA; IKKa(sense) 50-GGCTTCGGGAACGTCTGTC-30, (antisense) 50-TTTG-GTACTTAGCTCTAGGCGA-30; OPN (sense) 50-GAAGTTTCGCA-GACCTGACAT-30, (antisense) 50-GTATGCACCATTCAACTCCT-CG-30;MMP9 (sense) 50-GGGACGCAGACATCGTCATC-30, (anti-sense) 50-TCGTCATCGTCGAAATGGGC-30; uPA (sense) 50-GGGAATGGTCACTTTTACCGAG-30, (antisense) 50-GGGCATG-GTACGTTTGCTG-30; SPARC (sense) 50-AGCACCCCATTGACG-GGTA-30, (antisense) 50-GGTCACAGGTCTCGAAAAAGC-30; IL11(sense) 50-CGAGCGGACCTACTGTCCTA-30, (antisense) 50-GCCCAGTCAAGTGTCAGGTG-30; IL1b (sense) 50-ATGATGGCT-TATTACAGTGGCAA-30, (antisense) 50-GTCGGAGATTCGTAGC-TGGA-30; 36B4 (sense) 50-GAAGGCTGTGGTGCTGATGG-30,(antisense) 50-CCCCTGGAGATTTTAGTGGT-30.
IHCFormalin-fixed and paraffin-embedded tissue sections were
deparaffinized in xylene (Fisher Scientific) and rehydrated through
a graded series of ethanol (Pharmco-AAPER). Sections wereimmersed in antigen retrieval solution (Dako) and heated in asteamer for 20 minutes. Cooled sections were washed with PBS(Gibco), and endogenous peroxidase activity was quenched byimmersing sections in 3% hydrogen peroxide (Fischer Scientific)for 12 minutes and washed with PBS. Sections were blocked byincubation with protein block solution (Dako) for 30 minutes atroom temperature and incubated at 4�C for 18 hours with goatanti-CITED2 (1:500; Everest Biotech). Sections were then sequen-tially incubated for 15 minutes at room temperature with strepta-vidin–biotin complex, tyramide amplification reagent, and strep-tavidin–horseradish peroxidase (HRP) from the DACO CSAKit (Vector Laboratories). To visualize proteins, the chromogen3, 3-diaminobenzamindine (DAB; Open Biosystems) was addedfor two minutes at room temperature. Sections were subsequentlywashed in water and counterstained with hematoxylin Gill No.3 (Sigma-Aldrich).
Western blot analysisTotal protein extracts from cell lines were obtained as
described previously (15). Cytoplasmic and nuclear extractswas processed using NE-PER cytoplasmic and nuclear extrac-tion reagents (Thermo Scientific) according to the manufac-turer's instructions. Conditioned medium was collected bymaintaining the cells in serum-free medium. Samples wereresolved using SDS-PAGE, transferred to nitrocellulose mem-brane (Bio-Rad), and probed with sheep anti-CITED2 (1:250;R&D Systems), rabbit anti-IKKa, anti-IkBa, anti-p65, anti-RelB,anti-HDAC1 (1:1,000; Cell Signaling Technology), mouse anti-GAPDH (1:10,000; kindly provided by Dr. Shanmugasun-daram Ganapathy Kanniappan, Johns Hopkins UniversitySchool of Medicine, Baltimore, MD), or actin (1:1,000; Sig-ma-Aldrich) antibodies. Membranes were incubated with HRP-conjugated antibody against sheep (1:2,000; R&D Systems),rabbit, or mouse (1:2,000; GE Healthcare) IgG, and bindingwas revealed by chemiluminescence detection (Millipore).
Proliferation assayThe in vitro proliferation of cells was determined by MTS assay
using 0.2 mg/mLMTS reagent (Promega) as described previously(15). Data for each time point were obtained in triplicate perexperimental condition.
Migration and invasion assayA 24-well plate containing either 8.0-mmpore cell culture insert
(BD Falcon) or 8.0-mm pore transwell inserts precoated with100 mL Matrigel (BD Falcon) was utilized for the migration andinvasion assays, respectively. Tumor cells (2.5 � 104 cells) wereseeded in the upper chamber in medium containing 0% FBS andthe bottom chamber filled with medium containing either 0% or20% FBS as the chemoattractant. After 16 hours (in case ofmigration) or 48 hours (in case of invasion), cells in the upperchamber were removed with cotton swabs. Cells that migrated orinvaded to the lower surface of the insert were fixed in 100% coldmethanol (Fischer Scientific),washed inPBS, and stainedwith 2%crystal violet (Harleco). Three representative images from eachwell were captured at 100� magnification by light microscopy,and the total number of migrated or invaded cells per image wascounted using ImageJ imaging software (NIH, Bethesda, MD).Data are representative of at least two independent experimentsperformed in triplicates per experimental condition.
CITED2 Modulates Human Breast Cancer Metastasis
www.aacrjournals.org Mol Cancer Res; 14(8) August 2016 731
on July 10, 2018. © 2016 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst May 23, 2016; DOI: 10.1158/1541-7786.MCR-16-0081

In vivo assessment of breast cancer metastasisTumor cells (1� 105 cells) from each group were injected into
the left cardiac ventricle of five-week-old athymic nude mice[Taconic; for MDA-MB-231 cells, n ¼ 9 (scramble) and 10(shCITED2); for MDA-MB-468 cells, n ¼ 10 (scramble) andn ¼ 7 (shCITED2)]. Two weeks later, the establishment oftumor-induced osteolysis in the bone was analyzed by obtainingdigital radiographic images of the femur and tibia twice a weekusing a Faxitron MX-20 X-ray unit (Faxitron X-ray Corp.) untiltermination of the experiment. The experiment was terminatedwhen animals became moribund. The osteolytic area in theradiographic images was measured using MetaMorph imageanalysis software (Meta Imaging Series version 6.1, UniversalImaging Corp.). Tumor lesions within the bone were analyzedby H&E staining of bone sections decalcified in 10% EDTA(Sigma-Aldrich). Brain, liver, and lungs were harvested andmain-tained in Bouin's fixative (RICCA Chemical) for 24 hours andcounted for the total number of macrometastatic lesions.
All animal experiments were carried out in accordance with theNational Research Council's "Guide to the Care and Use ofLaboratory Animals." Animal use was approved by the JohnsHopkins Animal Care and Use Committee, animal welfare assur-ance #A3272-01, protocol #MO10M450.
Microarray analysiscDNA expression between scramble and shCITED2 MDA-MB-
231 cells was compared using Agilent Human GE 4 � 44 K v2microarray (G4845A). Log2-transformed signal intensities, with-out background subtraction were imported into GeneSpring GX10 software (Agilent Technologies) and (quantile) normalizedwithin the sample type. Differentially expressed genes inshCITED2-expressing cells relative to scramble cells were identi-fied based on �2-fold change in gene expression. Quality assess-ment of samples and microarray analysis were conducted at theSidney Kimmel Cancer Center Microarray Core Facility at JohnsHopkins University School of Medicine (Baltimore, MD; sup-ported by NIH grant P30 CA006973 entitled Regional OncologyResearch Center). Microarray data are deposited in the ArrayExpress database (www.ebi.ac.uk/arrayexpress) under accessionnumber E-MTAB-4267.
ELISA analysisOPN and IL11 ELISA immunoassay (R&D Systems) were
performed on serum-free tumor-conditioned media obtainedfrom cell lines according to the manufacturer's instructions.
Electrophoretic mobility shift assayElectrophoretic mobility shift assay (EMSA) was performed on
nuclear cell lysates using the LightShift EMSA and Chemilumines-centDetectionKit (ThermoScientific) based on themanufacturer'sinstructions using NF-kB (50-Biotin-AAGTTGAGGGGACTTTCC-CAGGCT-30 and 50-Biotin-AGCCTGGGAAAGTCCCCTCAACTT-30) oligonucleotides.Oct1 (50-Biotin-TGTCGAATGCAAATCACTA-GAA-30 and 50-Biotin-TTCTAGTGATTTGCATTCGACA-30) wasused as the loading control.
Chromatin immunoprecipitationChromatin immunoprecipitation (ChIP) was performed on
nuclear cell lysates using the SimpleChIPEnyzmaticChromatin IPKit (Cell Signaling Technology) based on the manufacturer'sinstructions. The promoter primer sequence used for IKKa was
(sense) 50-GTGGTTCCGTTCAGCCCT-30, (antisense) 50-TGCTC-GCGCGTCTTTG-30.
Statistical analysisDifferences in themigratory and invasive ability, average tumor
area and osteolytic area, and protein expression between exper-imental conditions were compared by unpaired Student t test.Differences in the mRNA expression of prometastatic genes in theshCITED2-expressing cells relative to scramble cells normalized to1.0 were compared by one sample t test. CITED2 mRNA expres-sion in tissues and the results of the invasion assay upon IKKareexpression were analyzed by ANOVA and Tukey multiple com-parison test. P < 0.05 was considered statistically significant. Forall figures, � denotes P < 0.05, �� denotes P < 0.01, and ��� denotesP < 0.001.
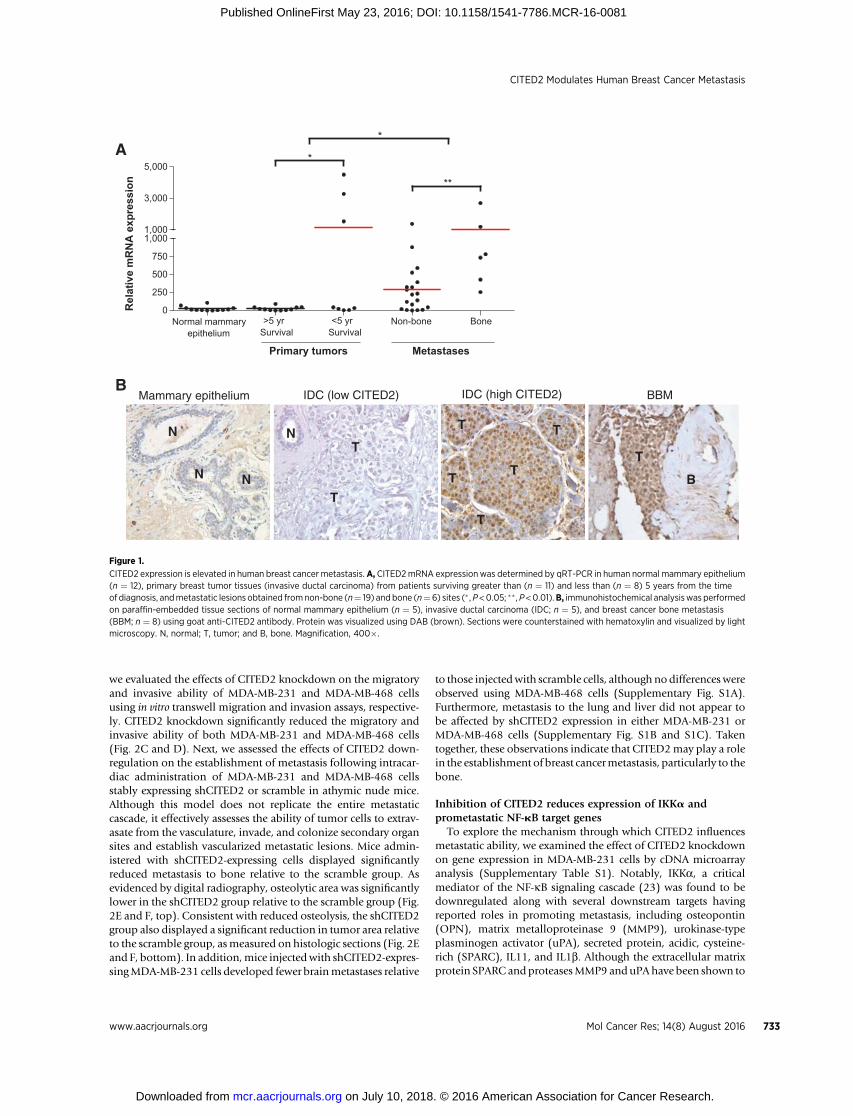
ResultsCITED2 expression is elevated in breast cancer metastasis
Previously, we presented evidence that CITED2 expression issignificantly elevated in primary human breast tumors relative tonormal mammary epithelium and is negatively correlated withsurvival (11, 15). Extending our analysis to metastatic lesions,CITED2 mRNA expression was significantly higher in humanbreast cancer metastases relative to primary breast tumors byqRT-PCR analysis (Fig. 1A). This difference appeared to be dueto the fact that CITED2 levels in metastases were more frequentlyelevated beyond those observed in normal mammary epitheliumas compared with primary tumors, many of which displayedCITED2 levels equivalent to that in normal. Consistent withmRNA results, this expression pattern was also appreciated at theprotein level in a limited subset of samples by immunohisto-chemical analysis (Fig. 1B). Finally, among metastases, higherexpression of CITED2 mRNA was observed in metastasis to bonerelative to non-bone sites. Taken together, these data demonstratethat CITED2 expression is frequently elevated in metastaticlesions of breast cancer patients, with highest levels in bonemetastasis.
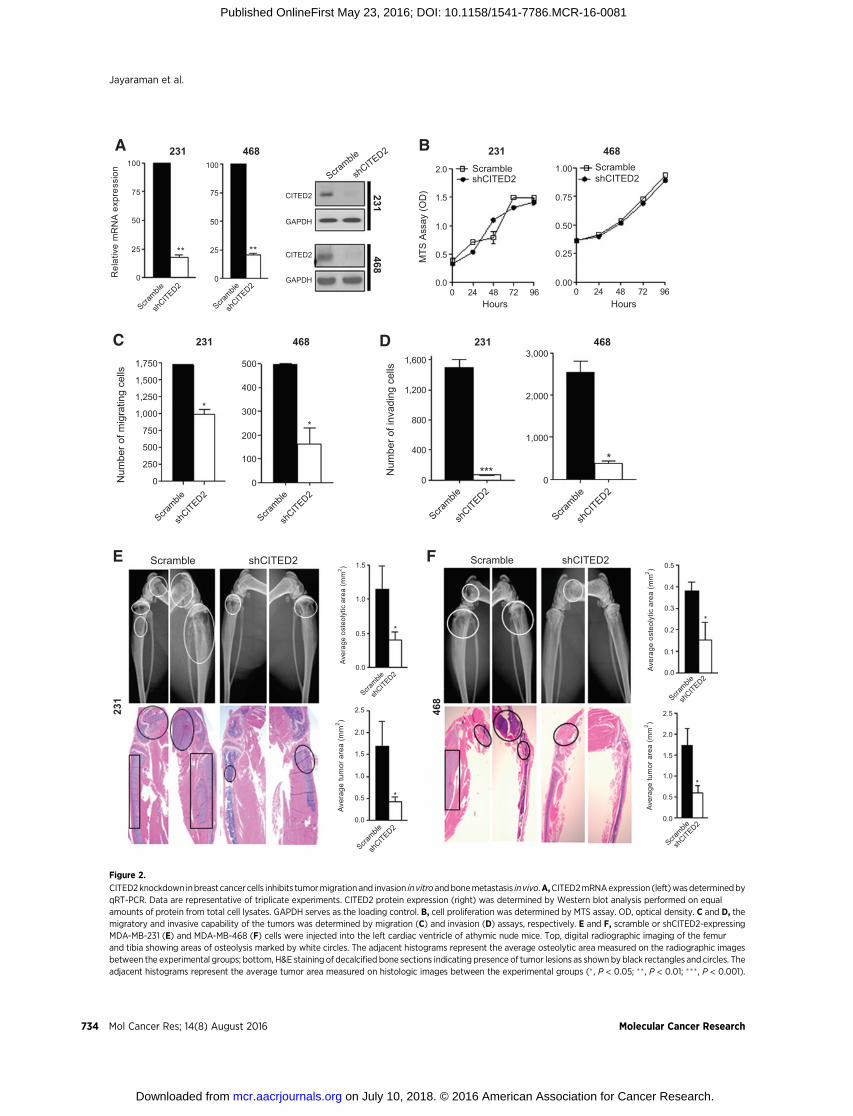
Downregulation of CITED2 inhibits breast cancer metastasisTo explore the role of CITED2 in breast cancer metastasis, we
utilized the human breast cancer cell lines MDA-MB-231 andMDA-MB-468. These cell lines are highly invasive in vitro, readilyestablish metastases following systemic administration in animalmodels (18–22), and as we have shown previously, expresselevated levels of CITED2 relative to human mammary epithelialcells and breast cancer cell lines that are nonmetastatic in animalmodels (15). MDA-MB-231 and MDA-MB-468 cells were stablyinfected with a lentiviral expression vector containing eithershRNA specific for CITED2 (shCITED2) or scrambled shRNA(scramble), and levels of CITED2were assessed at both themRNAand protein levels (Fig. 2A). Stable expression of shCITED2resulted in a greater than 75% reduction in CITED2 expressionin both MDA-MB-231 and MDA-MB-468 cells by qRT-PCR andWestern blot analysis. Prior to examining the effect of CITED2on metastatic progression, we first examined whether reducingCITED2 expression altered the rate of cell proliferation in vitro. Asdetermined by MTS assay, shCITED2 cells exhibited a similargrowth rate to that of scramble cells, indicating that knockdownofCITED2 did not affect cell growth in either cell line (Fig. 2B). Tobegin exploring CITED2 involvement in metastatic progression,
Jayaraman et al.
Mol Cancer Res; 14(8) August 2016 Molecular Cancer Research732
on July 10, 2018. © 2016 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst May 23, 2016; DOI: 10.1158/1541-7786.MCR-16-0081
we evaluated the effects of CITED2 knockdown on the migratoryand invasive ability of MDA-MB-231 and MDA-MB-468 cellsusing in vitro transwell migration and invasion assays, respective-ly. CITED2 knockdown significantly reduced the migratory andinvasive ability of both MDA-MB-231 and MDA-MB-468 cells(Fig. 2C and D). Next, we assessed the effects of CITED2 down-regulation on the establishment of metastasis following intracar-diac administration of MDA-MB-231 and MDA-MB-468 cellsstably expressing shCITED2 or scramble in athymic nude mice.Although this model does not replicate the entire metastaticcascade, it effectively assesses the ability of tumor cells to extrav-asate from the vasculature, invade, and colonize secondary organsites and establish vascularized metastatic lesions. Mice admin-istered with shCITED2-expressing cells displayed significantlyreduced metastasis to bone relative to the scramble group. Asevidenced by digital radiography, osteolytic area was significantlylower in the shCITED2 group relative to the scramble group (Fig.2E and F, top). Consistent with reduced osteolysis, the shCITED2group also displayed a significant reduction in tumor area relativeto the scramble group, asmeasured on histologic sections (Fig. 2Eand F, bottom). In addition,mice injectedwith shCITED2-expres-singMDA-MB-231 cells developed fewer brainmetastases relative
to those injectedwith scramble cells, although nodifferenceswereobserved using MDA-MB-468 cells (Supplementary Fig. S1A).Furthermore, metastasis to the lung and liver did not appear tobe affected by shCITED2 expression in either MDA-MB-231 orMDA-MB-468 cells (Supplementary Fig. S1B and S1C). Takentogether, these observations indicate that CITED2may play a rolein the establishment of breast cancermetastasis, particularly to thebone.
Inhibition of CITED2 reduces expression of IKKa andprometastatic NF-kB target genes
To explore the mechanism through which CITED2 influencesmetastatic ability, we examined the effect of CITED2 knockdownon gene expression in MDA-MB-231 cells by cDNA microarrayanalysis (Supplementary Table S1). Notably, IKKa, a criticalmediator of the NF-kB signaling cascade (23) was found to bedownregulated along with several downstream targets havingreported roles in promoting metastasis, including osteopontin(OPN), matrix metalloproteinase 9 (MMP9), urokinase-typeplasminogen activator (uPA), secreted protein, acidic, cysteine-rich (SPARC), IL11, and IL1b. Although the extracellular matrixprotein SPARC andproteasesMMP9 anduPAhave been shown to
A
B
0
250
500
750
1,000
Primary tumors
*
1,000
3,000
5,000
Metastases
**
Normal mammaryepithelium
>5 yrSurvival
<5 yrSurvival
BoneNon-bone
*R
elat
ive
mR
NA
exp
ress
ion
Mammary epithelium IDC (high CITED2)IDC (low CITED2) BBM
N
T
TT
T
T
N B
T
N
N
T
T
Figure 1.
CITED2 expression is elevated in human breast cancer metastasis. A, CITED2mRNA expressionwas determined by qRT-PCR in human normal mammary epithelium(n ¼ 12), primary breast tumor tissues (invasive ductal carcinoma) from patients surviving greater than (n ¼ 11) and less than (n ¼ 8) 5 years from the timeof diagnosis, andmetastatic lesions obtained fromnon-bone (n¼ 19) andbone (n¼6) sites (� ,P<0.05; �� ,P<0.01).B, immunohistochemical analysiswas performedon paraffin-embedded tissue sections of normal mammary epithelium (n ¼ 5), invasive ductal carcinoma (IDC; n ¼ 5), and breast cancer bone metastasis(BBM; n ¼ 8) using goat anti-CITED2 antibody. Protein was visualized using DAB (brown). Sections were counterstained with hematoxylin and visualized by lightmicroscopy. N, normal; T, tumor; and B, bone. Magnification, 400�.
CITED2 Modulates Human Breast Cancer Metastasis
www.aacrjournals.org Mol Cancer Res; 14(8) August 2016 733
on July 10, 2018. © 2016 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst May 23, 2016; DOI: 10.1158/1541-7786.MCR-16-0081
A
Scramble
shCITED2
0
25
50
75
100
Rel
ativ
e m
RN
A e
xpre
ssio
n
231 468
Scramble
shCITED2
0
25
50
75
100Scra
mble
CITED2
GAPDH
CITED2
GAPDH
231 468
shCITED2
0 24 48 72 960.0
0.5
1.0
1.5
2.0
Hours
MTS
Ass
ay (O
D)
B
0 24 48 72 960.00
0.25
0.50
0.75
1.00
Hours
ScrambleshCITED2
231 468
shCITED2Scramble
Scramble
shCITED2
0
250
500
750
1,000
1,250
1,500
1,750
Num
ber o
f mig
ratin
g ce
lls
Scramble
shCITED2
0
100
200
300
400
500
C 231 468
Scramble
shCITED2
0
400
800
1,200
1,600
Num
ber o
f inv
adin
g ce
lls
Scramble
shCITED2
0
1,000
2,000
3,000D 231 468
E FScramble shCITED2 Scramble shCITED2
Scramble
shCITED2
0.0
0.5
1.0
1.5
Aver
age
oste
olyt
ic a
rea
(mm
2 )
Scramble
shCITED2
0.0
0.1
0.2
0.3
0.4
0.5
Aver
age
oste
olyt
ic a
rea
(mm
2 )
231
468
Scramble
shCITED2
0.0
0.5
1.0
1.5
2.0
2.5
Aver
age
tum
or a
rea
(mm
2 )
Scramble
shCITED2
0.0
0.5
1.0
1.5
2.0
2.5
Aver
age
tum
or a
rea
(mm
2 )
** **
*
*
****
*
**
*
Figure 2.
CITED2knockdown inbreast cancer cells inhibits tumormigrationand invasion invitroandbonemetastasis invivo.A,CITED2mRNAexpression (left)wasdeterminedbyqRT-PCR. Data are representative of triplicate experiments. CITED2 protein expression (right) was determined by Western blot analysis performed on equalamounts of protein from total cell lysates. GAPDH serves as the loading control. B, cell proliferation was determined by MTS assay. OD, optical density. C and D, themigratory and invasive capability of the tumors was determined by migration (C) and invasion (D) assays, respectively. E and F, scramble or shCITED2-expressingMDA-MB-231 (E) and MDA-MB-468 (F) cells were injected into the left cardiac ventricle of athymic nude mice. Top, digital radiographic imaging of the femurand tibia showing areas of osteolysis marked by white circles. The adjacent histograms represent the average osteolytic area measured on the radiographic imagesbetween the experimental groups; bottom, H&E staining of decalcified bone sections indicating presence of tumor lesions as shown by black rectangles and circles. Theadjacent histograms represent the average tumor area measured on histologic images between the experimental groups (� , P < 0.05; �� , P < 0.01; ���, P < 0.001).
Jayaraman et al.
Mol Cancer Res; 14(8) August 2016 Molecular Cancer Research734
on July 10, 2018. © 2016 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst May 23, 2016; DOI: 10.1158/1541-7786.MCR-16-0081
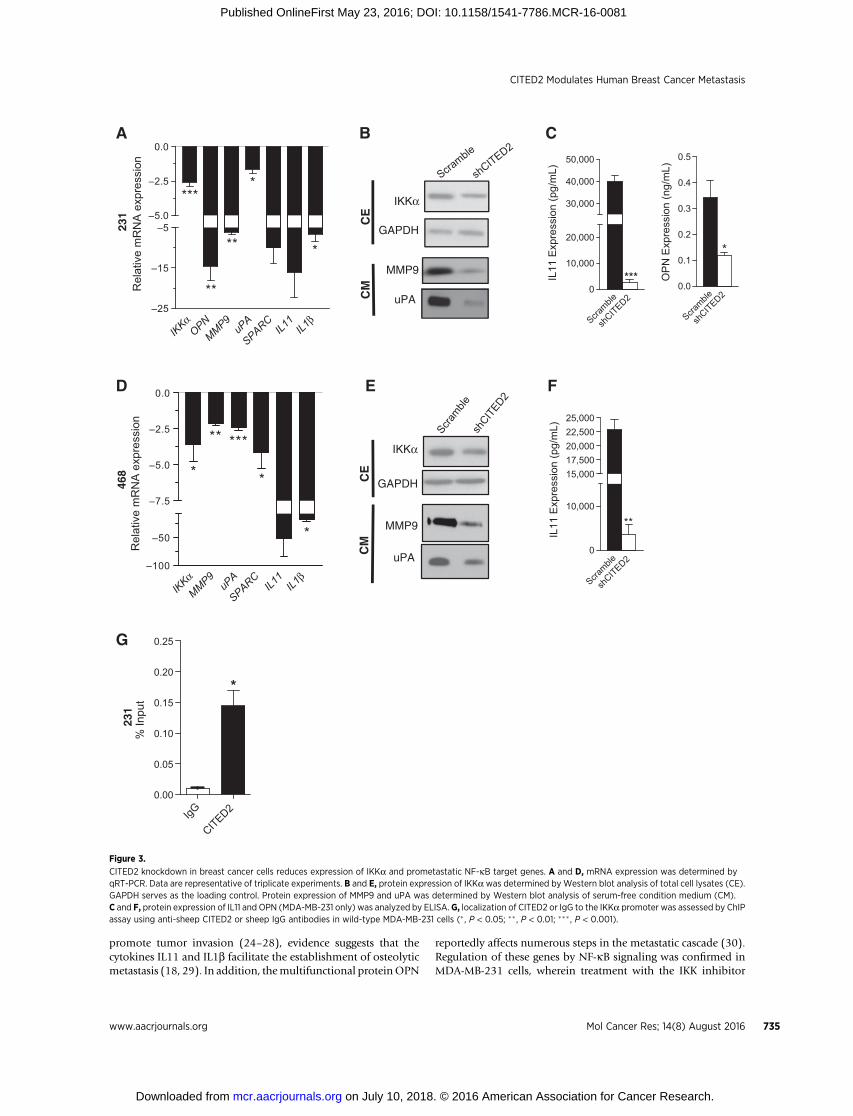
promote tumor invasion (24–28), evidence suggests that thecytokines IL11 and IL1b facilitate the establishment of osteolyticmetastasis (18, 29). In addition, themultifunctional proteinOPN
reportedly affects numerous steps in the metastatic cascade (30).Regulation of these genes by NF-kB signaling was confirmed inMDA-MB-231 cells, wherein treatment with the IKK inhibitor
IKKαMMP9 uP
A
SPARCIL11IL1
β–100
–50
–7.5
–5.0
–2.5
0.0
*
** ***
*
*
Rel
ativ
e m
RN
A e
xpre
ssio
n23
146
8A
D
G
E F
B C
MMP9
uPA
CE
CM
IKKα
GAPDH
MMP9
CM
uPA
IKKα
CE
GAPDH
Scramble
shCITED2
0.0
0.1
0.2
0.3
0.4
0.5
*
OP
N E
xpre
ssio
n (n
g/m
L)
Scramble
shCITED2
Scramble
shCITED2
Scramble
shCIT
ED2
Scramble
shCITED2
0
10,000
20,000
***
30,000
40,000
50,000
IL11
Exp
ress
ion
(pg/
mL)
0
10,000
15,00017,50020,00022,50025,000
**
IL11
Exp
ress
ion
(pg/
mL)
231
IgG
CITED20.00
0.05
0.10
0.15
0.20
0.25
*
% In
put
IKKα OP
NMMP9 uP
A
SPARCIL11IL1
β–25
–15
–5–5.0
–2.5
0.0
***
**
**
*
*
Rel
ativ
e m
RN
A e
xpre
ssio
n
Figure 3.
CITED2 knockdown in breast cancer cells reduces expression of IKKa and prometastatic NF-kB target genes. A and D, mRNA expression was determined byqRT-PCR. Data are representative of triplicate experiments. B and E, protein expression of IKKawas determined byWestern blot analysis of total cell lysates (CE).GAPDH serves as the loading control. Protein expression of MMP9 and uPA was determined by Western blot analysis of serum-free condition medium (CM).C and F, protein expression of IL11 and OPN (MDA-MB-231 only) was analyzed by ELISA. G, localization of CITED2 or IgG to the IKKa promoter was assessed by ChIPassay using anti-sheep CITED2 or sheep IgG antibodies in wild-type MDA-MB-231 cells (� , P < 0.05; �� , P < 0.01; ��� , P < 0.001).
CITED2 Modulates Human Breast Cancer Metastasis
www.aacrjournals.org Mol Cancer Res; 14(8) August 2016 735
on July 10, 2018. © 2016 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst May 23, 2016; DOI: 10.1158/1541-7786.MCR-16-0081
PS1145, which prevents IkBa degradation (31), reduced bothbasal NF-kB signaling by Western blot analysis and expression ofOPN, MMP9, uPA, SPARC, IL11, and IL1b by qRT-PCR (Supple-mentary Fig. S2A and S2B). Moreover, using qRT-PCR andWestern blot/ELISA analyses, we confirmed downregulation ofIKKa along with the aforementioned NF-kB target genes inshCITED2-expressing MDA-MB-231 and MDA-MB-468 cellscompared with scramble cells at both the mRNA and proteinlevels (Fig. 3A–F). Furthermore, by ChIP assay in MDA-MB-231
cells, CITED2 was found to localize to the promoter of IKKa,indicating a potentially direct role for CITED2 in the regulationof its expression. (Fig. 3G). Collectively, these data indicate thatCITED2 knockdown reduces the expression of IKKa and severaldownstream prometastatic genes in breast cancer cells.
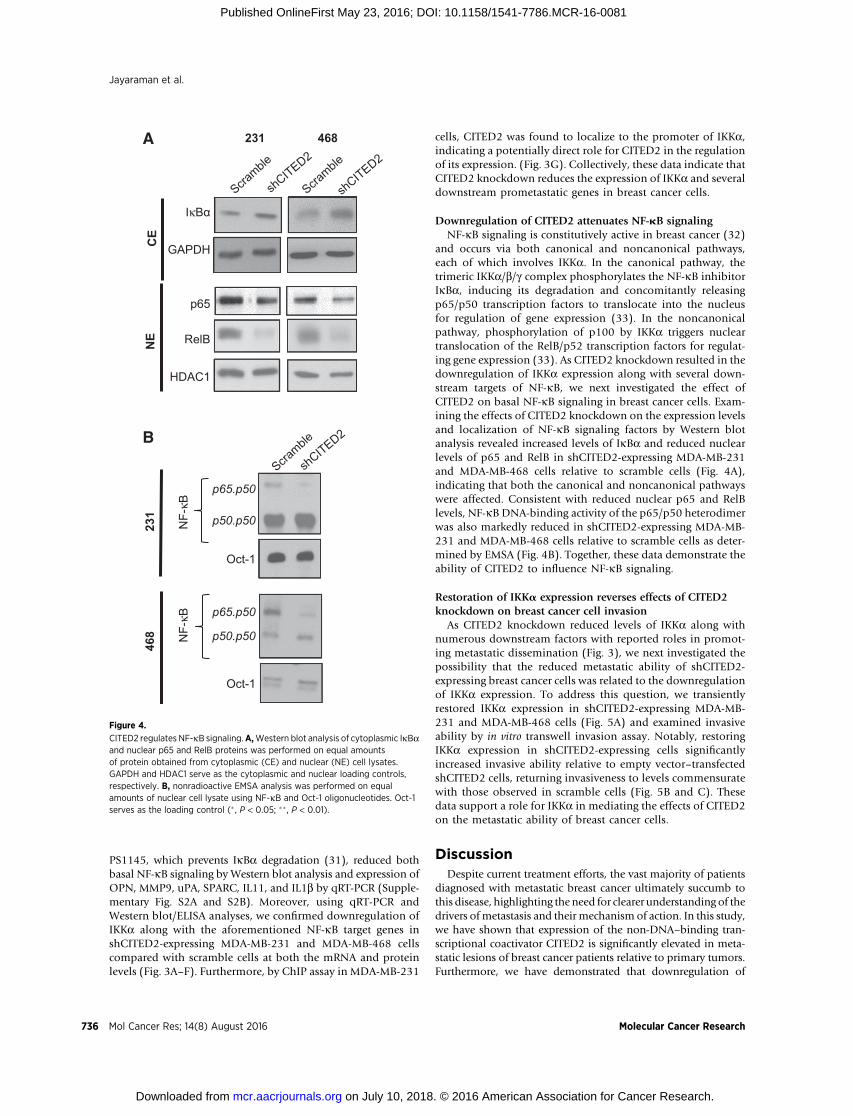
Downregulation of CITED2 attenuates NF-kB signalingNF-kB signaling is constitutively active in breast cancer (32)
and occurs via both canonical and noncanonical pathways,each of which involves IKKa. In the canonical pathway, thetrimeric IKKa/b/g complex phosphorylates the NF-kB inhibitorIkBa, inducing its degradation and concomitantly releasingp65/p50 transcription factors to translocate into the nucleusfor regulation of gene expression (33). In the noncanonicalpathway, phosphorylation of p100 by IKKa triggers nucleartranslocation of the RelB/p52 transcription factors for regulat-ing gene expression (33). As CITED2 knockdown resulted in thedownregulation of IKKa expression along with several down-stream targets of NF-kB, we next investigated the effect ofCITED2 on basal NF-kB signaling in breast cancer cells. Exam-ining the effects of CITED2 knockdown on the expression levelsand localization of NF-kB signaling factors by Western blotanalysis revealed increased levels of IkBa and reduced nuclearlevels of p65 and RelB in shCITED2-expressing MDA-MB-231and MDA-MB-468 cells relative to scramble cells (Fig. 4A),indicating that both the canonical and noncanonical pathwayswere affected. Consistent with reduced nuclear p65 and RelBlevels, NF-kB DNA-binding activity of the p65/p50 heterodimerwas also markedly reduced in shCITED2-expressing MDA-MB-231 and MDA-MB-468 cells relative to scramble cells as deter-mined by EMSA (Fig. 4B). Together, these data demonstrate theability of CITED2 to influence NF-kB signaling.
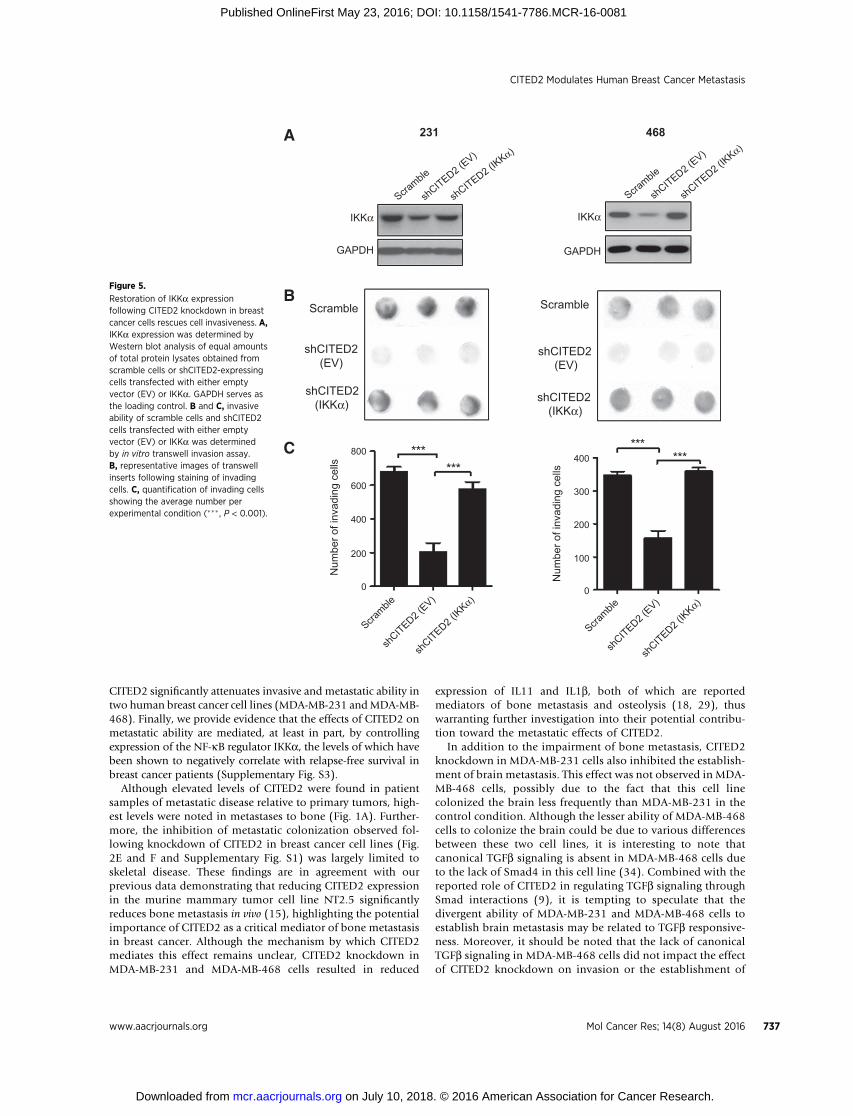
Restoration of IKKa expression reverses effects of CITED2knockdown on breast cancer cell invasion
As CITED2 knockdown reduced levels of IKKa along withnumerous downstream factors with reported roles in promot-ing metastatic dissemination (Fig. 3), we next investigated thepossibility that the reduced metastatic ability of shCITED2-expressing breast cancer cells was related to the downregulationof IKKa expression. To address this question, we transientlyrestored IKKa expression in shCITED2-expressing MDA-MB-231 and MDA-MB-468 cells (Fig. 5A) and examined invasiveability by in vitro transwell invasion assay. Notably, restoringIKKa expression in shCITED2-expressing cells significantlyincreased invasive ability relative to empty vector–transfectedshCITED2 cells, returning invasiveness to levels commensuratewith those observed in scramble cells (Fig. 5B and C). Thesedata support a role for IKKa in mediating the effects of CITED2on the metastatic ability of breast cancer cells.
DiscussionDespite current treatment efforts, the vast majority of patients
diagnosed with metastatic breast cancer ultimately succumb tothis disease, highlighting the need for clearer understanding of thedrivers of metastasis and their mechanism of action. In this study,we have shown that expression of the non-DNA–binding tran-scriptional coactivator CITED2 is significantly elevated in meta-static lesions of breast cancer patients relative to primary tumors.Furthermore, we have demonstrated that downregulation of
IκBα
GAPDHCE
p65
RelB
HDAC1
NE
231 468A
B
468
2
31 NF-
κBN
F-κB
Oct-1
p65.p50
p50.p50
p65.p50
p50.p50
Oct-1
Scramble
Scramble
shCITED2
Scramble
shCITED2
shCITED2
Figure 4.
CITED2 regulates NF-kB signaling.A,Western blot analysis of cytoplasmic IkBaand nuclear p65 and RelB proteins was performed on equal amountsof protein obtained from cytoplasmic (CE) and nuclear (NE) cell lysates.GAPDH and HDAC1 serve as the cytoplasmic and nuclear loading controls,respectively. B, nonradioactive EMSA analysis was performed on equalamounts of nuclear cell lysate using NF-kB and Oct-1 oligonucleotides. Oct-1serves as the loading control (� , P < 0.05; ��, P < 0.01).
Jayaraman et al.
Mol Cancer Res; 14(8) August 2016 Molecular Cancer Research736
on July 10, 2018. © 2016 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst May 23, 2016; DOI: 10.1158/1541-7786.MCR-16-0081
CITED2 significantly attenuates invasive and metastatic ability intwo human breast cancer cell lines (MDA-MB-231 andMDA-MB-468). Finally, we provide evidence that the effects of CITED2 onmetastatic ability are mediated, at least in part, by controllingexpression of the NF-kB regulator IKKa, the levels of which havebeen shown to negatively correlate with relapse-free survival inbreast cancer patients (Supplementary Fig. S3).
Although elevated levels of CITED2 were found in patientsamples of metastatic disease relative to primary tumors, high-est levels were noted in metastases to bone (Fig. 1A). Further-more, the inhibition of metastatic colonization observed fol-lowing knockdown of CITED2 in breast cancer cell lines (Fig.2E and F and Supplementary Fig. S1) was largely limited toskeletal disease. These findings are in agreement with ourprevious data demonstrating that reducing CITED2 expressionin the murine mammary tumor cell line NT2.5 significantlyreduces bone metastasis in vivo (15), highlighting the potentialimportance of CITED2 as a critical mediator of bone metastasisin breast cancer. Although the mechanism by which CITED2mediates this effect remains unclear, CITED2 knockdown inMDA-MB-231 and MDA-MB-468 cells resulted in reduced
expression of IL11 and IL1b, both of which are reportedmediators of bone metastasis and osteolysis (18, 29), thuswarranting further investigation into their potential contribu-tion toward the metastatic effects of CITED2.
In addition to the impairment of bone metastasis, CITED2knockdown in MDA-MB-231 cells also inhibited the establish-ment of brain metastasis. This effect was not observed in MDA-MB-468 cells, possibly due to the fact that this cell linecolonized the brain less frequently than MDA-MB-231 in thecontrol condition. Although the lesser ability of MDA-MB-468cells to colonize the brain could be due to various differencesbetween these two cell lines, it is interesting to note thatcanonical TGFb signaling is absent in MDA-MB-468 cells dueto the lack of Smad4 in this cell line (34). Combined with thereported role of CITED2 in regulating TGFb signaling throughSmad interactions (9), it is tempting to speculate that thedivergent ability of MDA-MB-231 and MDA-MB-468 cells toestablish brain metastasis may be related to TGFb responsive-ness. Moreover, it should be noted that the lack of canonicalTGFb signaling in MDA-MB-468 cells did not impact the effectof CITED2 knockdown on invasion or the establishment of
A 231 468
IKKα
GAPDH
IKKα
GAPDH
Scramble
Scramble
shCITED2 (E
V)
shCITED2 (E
V)
shCITED2 (IKKα)
shCITED2 (IK
Kα)
BScramble
shCITED2 (EV)
shCITED2 (IKKα)
Scramble
shCITED2 (EV)
shCITED2 (IKKα)
Scramble
shCITED2 (
EV)
shCITED2 (
IKKα)0
200
400
600
800
Num
ber o
f inv
adin
g ce
lls
Scramble
shCITED2 (
EV)
shCITED2 (
IKKα)0
100
200
300
400
Num
ber o
f inv
adin
g ce
lls
C ******
******
Figure 5.
Restoration of IKKa expressionfollowing CITED2 knockdown in breastcancer cells rescues cell invasiveness. A,IKKa expression was determined byWestern blot analysis of equal amountsof total protein lysates obtained fromscramble cells or shCITED2-expressingcells transfected with either emptyvector (EV) or IKKa. GAPDH serves asthe loading control. B and C, invasiveability of scramble cells and shCITED2cells transfected with either emptyvector (EV) or IKKa was determinedby in vitro transwell invasion assay.B, representative images of transwellinserts following staining of invadingcells. C, quantification of invading cellsshowing the average number perexperimental condition (��� , P < 0.001).
CITED2 Modulates Human Breast Cancer Metastasis
www.aacrjournals.org Mol Cancer Res; 14(8) August 2016 737
on July 10, 2018. © 2016 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst May 23, 2016; DOI: 10.1158/1541-7786.MCR-16-0081

bone metastasis. This indicates that these effects were medi-ated in a TGFb-independent manner, further supporting a rolefor NF-kB signaling, which was attenuated in both the MDA-MB-231 and MDA-MB-468 cell lines.
Despite the ability of CITED2 to directly regulate expressionof the NF-kB pathway regulator IKKa, it is not yet clear howCITED2 modulates NF-kB signaling and transcriptional activ-ity in breast cancer cells. Although we did not observe changesin the mRNA expression of NF-kB signaling intermediatesdownstream of IKKa, upstream mediators also exist whoseexpression could be impacted by CITED2. In addition, CITED2is known to interact with CREB-binding protein (CBP) andp300, reported coactivators of p65-mediated gene transcrip-tion (35), suggesting that CITED2 could also regulate theactivity of NF-kB as part of the transcriptional complex.Although the ability of IKKa to restore NF-kB signaling (datanot shown) and rescue tumor invasion in MDA-MB-231 andMDA-MB-468 cells following CITED2 knockdown (Fig. 5B andC) implicates involvement of the NF-kB pathway, it should benoted that IKKa can also exert NF-kB–independent effects(36). Thus, further investigation is required not only to assessthe mechanism whereby CITED2 modulates NF-kB activity,but also to determine the extent of its contribution to theprometastatic effects of CITED2, as well as the ultimate effec-tors of its action. Addressing these questions will not onlyfurther our understanding of CITED2 action in breast cancer
but may also provide new avenues for the prevention andtreatment of metastatic spread.
Disclosure of Potential Conflicts of InterestNo potential conflicts of interest were disclosed.
DisclaimerThe content is solely the responsibility of the authors and does not neces-
sarily represent the official views of the NIH.
Authors' ContributionsConception and design: S. Jayaraman, S.L. KominskyAcquisition of data (provided animals, acquired and managed patients,provided facilities, etc.): S. Jayaraman, M. Doucet, W.M. Lau, S.L. KominskyAnalysis and interpretation of data (e.g., statistical analysis, biostatistics,computational analysis): S. Jayaraman, S.L. KominskyWriting, review, and/or revision of the manuscript: S. Jayaraman,S.L. KominskyStudy supervision: S.L. Kominsky
Grant SupportThe research reported in this article was supported by the NCI of the NIH
under Award number R01CA157687.The costs of publication of this articlewere defrayed inpart by the payment of
page charges. This article must therefore be hereby marked advertisement inaccordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Received March 9, 2016; revised April 26, 2016; accepted May 14, 2016;published OnlineFirst May 23, 2016.
References1. Lu J, Steeg PS, Price JE, Krishnamurthy S, Mani SA, Reuben J, et al. Breast
cancer metastasis: challenges and opportunities. Cancer Res 2009;69:4951–3.
2. Chen Y, Doughman YQ, Gu S, Jarrell A, Aota S, Cvekl A, et al. Cited2 isrequired for the proper formation of the hyaloid vasculature and for lensmorphogenesis. Development 2008;135:2939–48.
3. Qu X, Lam E, Doughman YQ, Chen Y, Chou YT, Lam M, et al. Cited2, acoactivator of HNF4alpha, is essential for liver development. EMBO J2007;26:4445–56.
4. Xu B, Qu X, Gu S, Doughman YQ, Watanabe M, Dunwoodie SL, et al.Cited2 is required for fetal lung maturation. Dev Biol 2008;317:95–105.
5. Yin Z, Haynie J, Yang X, Han S, Kiatchoosakun S, Restivo J, et al. Theessential role of Cited2, a negative regulator for HIF-1alpha, in heartdevelopment and neurulation. Proc Natl Acad Sci U S A 2002;99:10488–93.
6. Bhattacharya S,Michels CL, LeungMK, Arany ZP, Kung AL, LivingstonDM.Functional role of p35srj, a novel p300/CBP binding protein, duringtransactivation by HIF-1. Genes Dev 1999;13:64–75.
7. Glenn DJ, Maurer RA. MRG1 binds to the LIM domain of Lhx2 and mayfunction as a coactivator to stimulate glycoprotein hormone alpha-subunitgene expression. J Biol Chem 1999;274:36159–67.
8. Braganca J, Eloranta JJ, Bamforth SD, Ibbitt JC, Hurst HC, BhattacharyaS. Physical and functional interactions among AP-2 transcription fac-tors, p300/CREB-binding protein, and CITED2. J Biol Chem 2003;278:16021–9.
9. Chou YT, Want H, Chen Y, Danielpour D, Yang YC. Cited2 modulatesTGFbeta-mediated upregulation of MMP9. Oncogene 2006;25:5547–60.
10. Tien ES, Davis JW, Vanden Heuvel JP. Identification of the CREB-bindingprotein/p300-interacting protein CITED2 as a peroxisome proliferatoractivated receptor alpha coregulator. J Biol Chem 2004;279:24053–63.
11. LauWM,DoucetM,HuangD,Weber KL, Kominsky SL. CITED2modulatesestrogen receptor transcriptional activity in breast cancer cells. BiochemBiophys Res Commun 2013;437:261–6.
12. Sun HB, Zhu YX, Yin T, Sledge G, Yang YC. MRG1, the product of amelanocyte-specific gene related gene, is a cytokine-inducible transcriptionfactor with transformation activity. Proc Natl Acad Sci U S A 1998;95:13555–60.
13. Bai L, Merchant JL. A role for CITED2, a CBP/p300 interacting protein, incolon cancer cell invasion. FEBS Lett 2007;581:5904–10.
14. Chou YT, Hsieh CH, Chiou SH, Hsu CF, Kao YR, Lee CC, et al. CITED2functions as a molecular switch of cytokine-induced proliferation andquiescence. Cell Death Differ 2012;19:2015–28.
15. Lau WM, Weber KL, Doucet M, Chou YT, Brady K, Kowalski J, et al.Identification of prospective factors promoting osteotropism in breastcancer: a potential role for CITED2. Int J Cancer 2010;126:876–84.
16. Chou YT, Yang YC. Post-transcriptional control of CITED2by transforminggrowth factor beta. Regulation via Smads and CITED2 coding region. J BiolChem 2006;281:18451–62.
17. Nakano H, Shindo M, Sakon S, Nishinaka S, Mihara M, Yagita H, et al.Differential regulation of IkappaB kinase alpha and beta by twoupstream kinases, NF-kappaB-inducing kinase and mitogen-activatedprotein kinase/ERK kinase kinase-1. Proc Natl Acad Sci U S A 1998;95:3537–42.
18. Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cord�on-Cardo C,et al. A multigenic program mediating breast cancer metastasis to bone.Cancer Cell 2003;3:537–49.
19. Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, et al. Genes thatmediate breast cancer metastasis to lung. Nature 2005;436:518–24.
20. Bos PD, Zhang XH, Nadal C, Shu W, Gomis RR, Nguyen DX, et al. Genesthat mediate breast cancer metastasis to the brain. Nature 2009;459:1005–9.
21. Lau WM, Doucet M, Stadel R, Huang D, Weber KL, Kominsky SL. Enpp1: apotential facilitator of breast cancer bone metastasis. PLoS One 2013;8:e66752.
22. Vantyghem SA, Allan AL, Postenka CO, Al-Katib W, Keeney M, Tuck AB,et al. A new model for lymphatic metastasis: development of a variant ofthe MDA-MB-468 human breast cancer cell line that aggressively metas-tasize to lymph nodes. Clin Exp Metastasis 2005;22:351–61.
23. Adli M, Merkhofer E, Cogswell P, Baldwin AS. IKKalpha and IKKbeta eachfunction to regulate NF-kappaB activation in the TNF-induced/canonicalpathway. PLoS One 2010;5:e9428.
24. Seno T, Harada H, Kohno S, Teraoka M, Inoue A, Ohnishi T. Down-regulation of SPARC expression inhibits cell migration and invasion inmalignant gliomas. Int J Dev Neurosci 1999;17:463–72.
Mol Cancer Res; 14(8) August 2016 Molecular Cancer Research738
Jayaraman et al.
on July 10, 2018. © 2016 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst May 23, 2016; DOI: 10.1158/1541-7786.MCR-16-0081

25. Golembieski WA, Ge S, Nelson K, Mikkelsen T, Rempel SA. IncreasedSPARCexpressionpromotesU87glioblastoma invasion in vitro. Int JOncol2009;34:707–15.
26. Chen J,WangM, Xi B, Xue J, HeD, Zhang J, et al. SPARC is a key regulator ofproliferation, apoptosis and invasion in human ovarian cancer. PLoS One2012;7:e42413.
27. Mehner C, Hockla A, Miller E, Ran S, Radisky DC, Radisky ES. Tumor-cellproduced matrix metalloproteinase 9 (MMP-9) drives malignant progres-sion and metastasis of basal-like triple negative breast cancer. Oncotarget2014;5:2736–49.
28. Tang L,HanX. Theurokinase plasminogen activator system inbreast cancerinvasion and metastasis. Biomed Pharmacother 2013;67:179–82.
29. Liu Q, Russell MR, Shahriari K, Jernigan DL, Lioni MI, Garcia FU, et al.Interleukin-1b promotes skeletal colonization and progression of meta-static prostate cancer cells with neuroendocrine features. Cancer Res2013;73:1–9.
30. Shevde LA, Samant RS. Role of osteopontin in the pathophysiology ofcancer. Matrix Biol 2014;37:131–41.
31. Yemelyanov A, Gasparian A, Lindholm P, Dang L, Pierce JW, Kisselijov F,et al. Effects of IKK inhibitor PS1145 on NF-kB function, proliferation,apoptosis and invasion activity in prostate carcinoma cells. Oncogene2006;25:387–98.
32. Yamaguchi N, Ito T, Azuma S, Ito E, Honma R, Yanagisawa Y, et al.Constitutive activation of nuclear factor-kappaB is preferentially involvedin the proliferation of basal-like subtype breast cancer cell lines. Cancer Sci2009;100:1668–74.
33. Hoesel B, Schmid JA. The complexity of NF-kB signaling in inflammationand cancer. Mol Cancer 2013;12:86.
34. Schutte M, Firuban RH, Hedrick L, Cho KR, Nadasdy GM, WeinsteinCL, et al. DPC4 gene in various tumor types. Cancer Res 1996;56:2527–30.
35. Gerritsen ME, William AJ, Neish AS, Moore S, Shi Y, Collins T. CREB-binding protein/p300 are transcriptional coactivators of p65. Proc NatlAcad Sci U S A 1997;94:2927–32.
36. Huang WC, Hung MC. Beyond NF-kB activation: nuclear functions of IkBkinase a. J Biomed Sci 2013;20:3.
www.aacrjournals.org Mol Cancer Res; 14(8) August 2016 739
CITED2 Modulates Human Breast Cancer Metastasis
on July 10, 2018. © 2016 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst May 23, 2016; DOI: 10.1158/1541-7786.MCR-16-0081

2016;14:730-739. Published OnlineFirst May 23, 2016.Mol Cancer Res Swaathi Jayaraman, Michele Doucet, Wen Min Lau, et al.
αon IKKCITED2 Modulates Breast Cancer Metastatic Ability through Effects
Updated version
10.1158/1541-7786.MCR-16-0081doi:
Access the most recent version of this article at:
Material
Supplementary
http://mcr.aacrjournals.org/content/suppl/2016/06/25/1541-7786.MCR-16-0081.DC1
Access the most recent supplemental material at:
Cited articles
http://mcr.aacrjournals.org/content/14/8/730.full#ref-list-1
This article cites 36 articles, 14 of which you can access for free at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://mcr.aacrjournals.org/content/14/8/730To request permission to re-use all or part of this article, use this link
on July 10, 2018. © 2016 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst May 23, 2016; DOI: 10.1158/1541-7786.MCR-16-0081