click to edit master title style irys data analysis january 10 th, 2014
TRANSCRIPT

Click to edit Master title styleClick to edit Master title styleIrys data analysis
January 10th, 2014

Irys Workflow – Data Analysis
Genome Map (.cmap)
Single molecule
maps (.bnx)
Sample Anchoring (.xmap)
irys™ ICS
irysView™
Image processing
Short NGS Contigs
RefSeqReference
Genome Map 2
Structural variation detection
Sequence Assembly Validation
Sequence contig
scaffolding
Integration AnalysisScanning
Sequence scaffolding without de novo assembly
Using a reference (eg hg19)Using a second genome mapUsing NGS contigs
Gross assembly quality (reiterate)Missing sites, extra sites, interval differences structural differences Consed
Alignment in irysviewmanual editingAGP outputConversion to FASTAReimport superscaffolds to reiterate
Mapping based variant calling
Two color applications:
epigenetics, DNA damage
Assembly

workshops
• De novo assembly (Using irysview (Alex); Python/command line – Heng/Ernest)
• SV detection – Warren/Andy

4
Core workflow: Data QC: basic molecule stats

5
Core workflow: Data QC: molecule quality report
Always consider the mapping rate with respect to the stringency setting
Mapping rate helps us estimate the useful coverage depth as well as data quality

6
Stretch normalization
• Evaporation (increasing [salt]) during the scanning prolonged of version 2 chips results in shortening of molecules in nanochannels. This can be corrected for by measuring the average stretch in each scan and correcting with a normalization factor.
• Determining average stretch:– Internal ruler based normalization– Reference mapping based normalization

7
Core workflow: De novo assembly: optArg
From molecule quality report and .err file
p value based on genome size or as stringent as possible
Stringencies vary based on step

8
No reference?
• With no reference, we can run a de novo assembly based on expectations and data QC observations:– Expected genome size– Site density (in silico)– Label density (empirical)– Molecule n50 (empirical)
• Run de novo assembly (relaxed)• Use the result of the de novo assembly to run molecule
quality report• Update error characteristics (stretch normalization) and rerun
de novo assembly
9
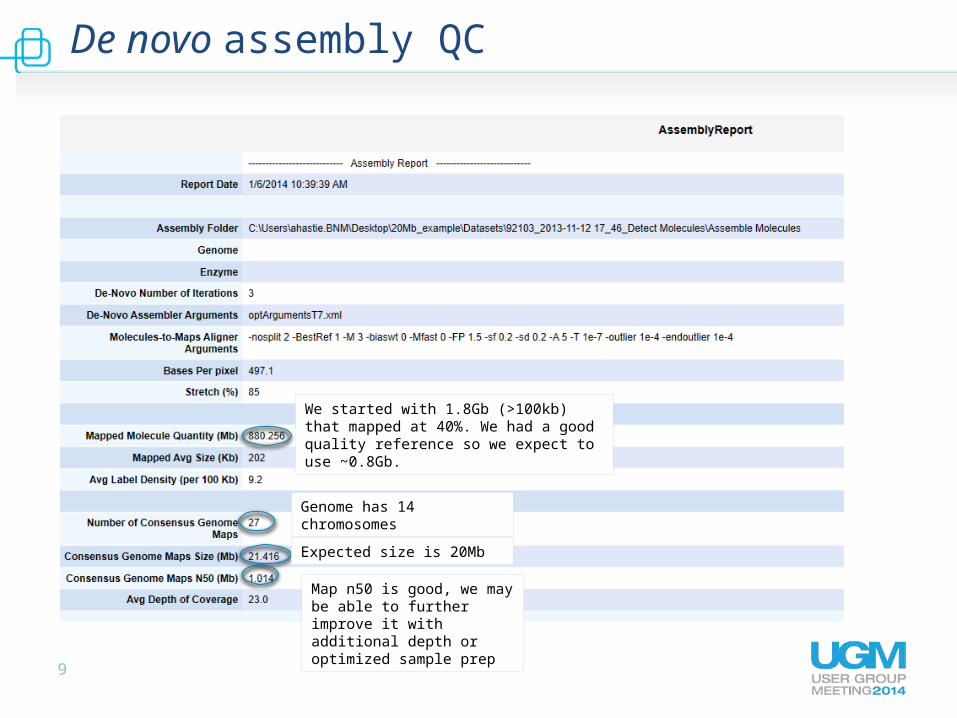
De novo assembly QC
We started with 1.8Gb (>100kb) that mapped at 40%. We had a good quality reference so we expect to use ~0.8Gb.
Genome has 14 chromosomes
Expected size is 20Mb
Map n50 is good, we may be able to further improve it with additional depth or optimized sample prep
10
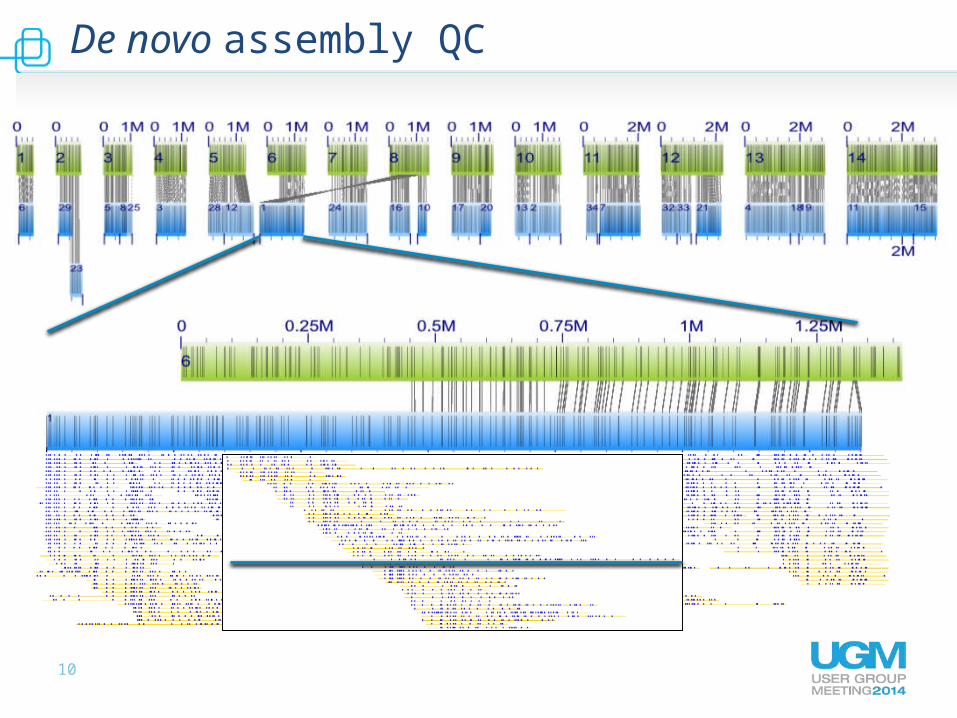
De novo assembly QC

11
De novo assembly QC
Higher stringency assembly
The higher stringency assembly misses some of the genome but resolves the chimera

Click to edit Master title styleClick to edit Master title style
12
Applications:
Sequence anchoring

12 Mb Streptomyces Genome Assembly with Various Technologies Total Mb Contigs N50 (kb)
9.08 124 92
11.38 97 154
11.63 20 918
11.87 1 11,870
DNA sequence scaffolding
BioNano Genomics
NGS + Cosmids
Short-Read NGS Only
3rd-Gen Reads

Sequence anchoring
Illumina + cosmids: 11.38Mb, 97 contigs, n50 length: 154kb , 11.38Mb anchored
Illumina: 9.08Mb, 124 contigs, n50 length: 92kb , 8.9Mb anchored
Pac Bio: 11.63Mb, 20 contigs, n50 length: 918kb , 11.63Mb anchored
1 Mb
Validate sequence assemblyFind errorsScaffold/Orient/Size gaps
Output FASTA or AGP (soon)

Click to edit Master title styleClick to edit Master title style
15
Applications:
Structural variation

Structural Variation-Insertion/Deletion Calls (vs hg19)
<10 20 30 40 50 60 70 80 90 100
110
120
130
140
150
160
170
180
190
200
210
220
230
240
>250
0
10
20
30
40
50
60
70
80
Insertion and Deletion size distribution in a normal human genome
Deletion (n=210)
Insertion (n=280)
SV Size (kb)
# of
cal
ls 95 regions in BioNano GenomeMaps correspond to N-based gaps in hg19 (not included in graph). The gaps may contain repeats and polymorphic regions, where SV enriches.

Structural Variant Examples: Insertions and Deletions
Genome Map
hg19M
olec
ules
+4.9kb
Genome Map
hg19
Mol
ecul
es
-176,265 kb#h SmapEntryID QryContigID RefcontigID1 RefcontigID2 QryStartPos QryEndPos RefStartPos RefEndPos Orientation Confidence Type
#f int int int int float float float float string float string net size 1 282 6 6 1,483,278 1,488,217 75,697,428 75,878,632 + -1 delete 176,265
4.9 kb region 181.2 kb region
#h SmapEntryID QryContigID RefcontigID1 RefcontigID2 QryStartPos QryEndPos RefStartPos RefEndPos Orientation Confidence Type#f int int int int float float float float string float string net size
4 577 6 6 1,093,571 1,111,027 13,122,638 13,135,195 + -1 insert 4,899
17.5 kb region 12.6 kb region

workshops
• De novo assembly (Using irysview (Alex); Using Python/command line – Heng/Ernest)– OptArg- iterations, stringencies, merging, ref mapping– Output
• .err file• Alignref• Visualization of genome maps to molecules• Identification of chimeras
• SV detection – Warren/Andy– Explain the SV detection application (consider IP issues)– Discuss stringency parameters– Show resulting table
• ranges• explain types

19