computational modelling of enzyme selectivity
TRANSCRIPT

ACTAUNIVERSITATIS
UPSALIENSISUPPSALA
2017
Digital Comprehensive Summaries of Uppsala Dissertationsfrom the Faculty of Science and Technology 1530
Computational modelling ofenzyme selectivity
PAUL BAUER
ISSN 1651-6214ISBN 978-91-513-0005-4urn:nbn:se:uu:diva-326108

Dissertation presented at Uppsala University to be publicly examined in A1:111 BMC,Husargatan 3, Uppsala, Wednesday, 13 September 2017 at 09:00 for the degree of Doctor ofPhilosophy. The examination will be conducted in English. Faculty examiner: Prof KennethRuud (University of Tromsø, Department of Chemistry CTCC).
AbstractBauer, P. 2017. Computational modelling of enzyme selectivity. Digital ComprehensiveSummaries of Uppsala Dissertations from the Faculty of Science and Technology 1530.104 pp. Uppsala: Acta Universitatis Upsaliensis. ISBN 978-91-513-0005-4.
Enantioselective reactions are one of the ways to produce pure chiral compounds. Understandingthe basis of this selectivity makes it possible to guide enzyme design towards more efficientcatalysts. One approach to study enzymes involved in chiral chemistry is through the use ofcomputational models that are able to simulate the chemical reaction taking place. The potatoepoxide hydrolase is one enzyme that is known to be both highly enantioselective, while stillbeing robust upon mutation of residues to change substrate scope. The enzyme was used toinvestigate the epoxide hydrolysis mechanism for a number of different substrates, using theEVB approach to the reaction both in solution and in several enzyme variants. In addition tothis, work has been performed on new ways of performing simulations of divalent transitionmetals, as well as development of new simulation software.
Keywords: enantiomer, epoxide hydrolase, chiral catalysis, empirical valence bond approach,method development
Paul Bauer, Department of Cell and Molecular Biology, Structure and Molecular Biology,596, Uppsala University, SE-751 24 Uppsala, Sweden.
© Paul Bauer 2017
ISSN 1651-6214ISBN 978-91-513-0005-4urn:nbn:se:uu:diva-326108 (http://urn.kb.se/resolve?urn=urn:nbn:se:uu:diva-326108)

It doesn’t matter how beautiful your theory is, it doesn’t matter howsmart you are. If it doesn’t agree with experiment, it’s wrong.
Richard P. Feynman


List of papers
This thesis is based on the following papers, which are referred to in thetext by their Roman numerals.
I Duarte, F., Bauer, P., Barrozo, A., Amrein, B. A., Purg, M., Åqvist,J., Kamerlin, S. C. L. (2014) Force Field Independent Metal ParametersUsing a Nonbonded Dummy Model. J. Phys. Chem. B, 118:4351–4362
II Amrein, B. A., Bauer, P., Duarte, F., Janfalk Carlsson, Å., Naworyta,A., Mowbray, S. L., Widersten, M., Kamerlin, S. C. L. (2015)Expanding the Catalytic Triad in Epoxide Hydrolases and RelatedEnzymes. ACS Catalysis, 5:5702–5713
III Bauer, P., Janfalk Carlsson, Å., Amrein, B. A., Dobritzsch, D.,Widersten, M., Kamerlin, S. C. L. (2016) Conformational Diversity andEnantioconvergence in Potato Epoxide Hydrolase 1. Org. Biomol.Chem., 14:5639–5651
IV Janfalk Carlsson, Å, Bauer, P., Dobritzsch, D., Nilsson, M., Kamerlin,S. C. L., Widersten, M. (2016) Laboratory-Evolved Enzymes ProvideSnapshots of the Development of Enantioconvergence inEnzyme-Catalyzed Epoxide Hydrolysis. ChemBioChem, 17:1693–1697
V Janfalk Carlsson, Å, Bauer, P., Dobritzsch, D., Kamerlin, S. C. L.,Widersten, M. (2017) Epoxide Hydrolysis as a Model System forUnderstanding Flux Through a Branched Reaction Scheme. Submitted
VI Bauer, P., Barrozo, A., Amrein, B. A., Purg, M., Esguerra, M.,Wilson, P., Major, D. T., Åqvist, J., Kamerlin, S. C. L. (2017) QVersion 6, a Comprehensive Toolkit for Empirical Valence Bond andRelated Free Energy Calculations.Manuscript
Reprints were made with permission from the publishers.

Additional Publications
VII Janfalk Carlsson, Å., Bauer, P., Ma, H., Widersten. M. (2012)Obtaining Optical Purity for Product Diols in Enzyme–CatalyzedEpoxide Hydrolysis: Contributions from Changes in both Enantio– andRegioselectivity. Biochemistry, 51:7627–7637
VIII Satpati, P., Bauer, P., Åqvist, J. (2014) Energetic Tuning by tRNAModifications Ensures Correct Decoding of Isoleucine and Methionineon the Ribosome. Chem. Eur. J., 20:10271–10275
IX Repič, M., Vianello, R., Purg, M., Duarte, F., Bauer, P., Kamerlin, S.C. L., Mavri, J. (2014) Empirical Valence Bond Simulations of theHydride Transfer Step in the Monoamine Oxidase B CatalyzedMetabolism of Dopamine. Proteins, 82:3347–3355
X Barrozo, A., Duarte, F., Bauer, P., Carvalho, A. T. P., Kamerlin, S.C. L. (2015) Cooperative Electrostatic Interactions Drive FunctionalEvolution in the Alkaline Phosphatase Superfamily. J. Am. Chem.Soc., 137:9061–9076
Reprints were made with permission from the publishers.

Contribution report
Contributions to the articles that are part of this thesis are listed here
Paper I Performed part of the simulations and analysed data.Contributed to the writing of the manuscript.
Paper II Performed part of the simulations and the sequence analysis.Analysed data and contributed to the writing of the manuscript.
Paper III Performed all simulations and simulation data analysis.Contributed to writing of the manuscript.
Paper IV Performed all docking simulations and analysedcorresponding data. Contributed to the writing of the manuscript.
Paper V Performed all simulations and simulation data analysis.Contributed to the writing of the manuscript.
Paper VI Contributed to the programming and test simulations.Contributed to the writing of the manuscript.


Contents
1 Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 131.1 Enzyme catalysis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 14
1.1.1 Reaction kinetics . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 151.1.2 Michaelis–Menten kinetics . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 161.1.3 Complex reaction kinetics . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 181.1.4 Determination of individual rate constants . . . . . . . . . . . 19
1.2 Physical chemistry of enzyme reactions . . . . . . . . . . . . . . . . . . . . . . . . . . . . 191.2.1 Linear free energy relationships . . . . . . . . . . . . . . . . . . . . . . . . . . . . 191.2.2 Isotope effects . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 201.2.3 Temperature and pH dependence . . . . . . . . . . . . . . . . . . . . . . . . . 211.2.4 Relating rates to free energy . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 221.2.5 The origins of enzyme catalysis . . . . . . . . . . . . . . . . . . . . . . . . . . . . 24
1.3 Protein structure determination . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 271.4 Stereochemistry . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 28
1.4.1 Compound classification . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 281.4.2 Enantiomers and stereoisomers . . . . . . . . . . . . . . . . . . . . . . . . . . . . 29
2 Hydrolase enzymes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 332.1 α/β hydrolase enzymes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 332.2 Epoxide hydrolases and dehalogenases . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 352.3 Solanum tuberosum epoxide hydrolase I . . . . . . . . . . . . . . . . . . . . . . . . . . . . 36
3 Computational study of biological systems . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 413.1 Quantum mechanics methods . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 41
3.1.1 Wave function methods . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 413.1.2 Density Functional Theory methods . . . . . . . . . . . . . . . . . . . . . 44
3.2 Classical methods . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 453.2.1 Molecular mechanics . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 453.2.2 Estimation of free energies . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 48

3.3 Multiscale models . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 503.3.1 QM/MM approaches . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 513.3.2 The Empirical Valence Bond approach . . . . . . . . . . . . . . . . . 51
4 Present Investigation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 55
5 Catalytic mechanism of Solanum tuberosum epoxide hydrolase 1(StEH1) based epoxide hydrolysis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 565.1 Previous kinetic studies . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 565.2 Hydrolysis of trans–stilbene oxide (TSO) (Paper II) . . . . . . . . . . 575.3 Investigation of styrene oxide (SO) binding modes and
enantioconvergence (Paper III) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 615.4 Study of substrate flexibility for (2,3–epoxypropyl)benzene
(EPB) (Paper IV) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 665.5 Investigation of reduced regioselecivity when hydrolysing
MeSO (Paper V) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 69
6 Method development for chemical modelling . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 736.1 Divalent transition metal models (Paper I) . . . . . . . . . . . . . . . . . . . . . . . 736.2 Updates to the Q simulation program (Paper VI) . . . . . . . . . . . . . 76
7 Concluding Remarks . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 82
8 Populärvetenskaplig Sammanfattning på svenska . . . . . . . . . . . . . . . . . . . . . . . 84
9 Acknowledgements . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 86

Abbreviations
CIP Cahn, Ingold and PrelogDFT Density Functional TheoryEC Enzyme CommissionEVB Empirical Valence BondFEP Free Energy PerturbationKIE Kinetic Isotope EffectMM Molecular MechanicsQCP Quantum Classical PathQM Quantum MechanicsTST Transition State TheoryUS Umbrella Sampling
EnzymesCOMT Catechol O–MethyltransferaseDHA DehalogenaseEH Epoxide HydrolaseLEH Limonene Epoxide HydrolasesEH Soluble Epoxide HydrolaseStEH1 Solanum tuberosum Epoxide Hydrolase 1
SubstratesEPB (2,3–Epoxypropyl)benzeneMeSO trans–Methylstyrene OxideSO Styrene OxideTSO trans–Stilbene Oxide


1. Introduction
Enzymes are Nature's main way of performing catalysis.1 Together withother natural catalysts such as catalytic RNA,2 they accelerate all thechemical reactions that life depends on. The activity of enzymes hasbeen refined over the course of evolution to be both specific for the chem-istry that is being catalysed, as well as selective for the substrates thatare accepted for it.3,4 On the other hand, promiscuous activities havebeen observed for enzymes involved in scavenging of chemicals, or thosetasked with detoxification of xenobiotic compounds.5 One of the strikingselectivities that have been observed in all domains of life is the close–to–absolute preference of L–amino acids.1 Both the L– and D–amino acidsare two different three–dimensional representations (enantiomers) of thesame chemical structure, only differing in the orientation of the atomsin space.6 Still, nature generally only incorporates the L form into pro-teins, with the result that the proteins built from the amino acid subunitsinclude the structural information of the orientation in addition to thechemical properties. Enzymes involved in the task of providing the ma-terial for building new proteins are some of the most selective known,with amino acyl tRNA synthases only accepting correct amino acids andcorresponding messenger RNA molecules.7 In enzyme catalysis, highlyselective enzymes are usually involved in reactions that are essential forthe survival of the organism.8 The host has to rely on the exact reactionto be catalysed with the correct substrate, resulting in high evolution-ary pressure to create a selective enzyme. Promiscuous enzymes providedifferent advantages for their host organisms. They make it possible tolet established enzymes explore new chemistry, while keeping a possiblycritical native reaction present.3,9 Enzymes may be promiscuous in thechoice of substrate,10 catalysed reaction,3,11 reaction pathway12 or finalproduct.13,14 Understanding what causes enzymes to be promiscuous or
13

selective is of general interest for both medical research and for the chem-ical industry. A new specific inhibitor could be built from the knowledgeof the specific interactions involved in catalysis,15 for example. Or, in adifferent case, the evolution of an enzyme could be traced through dif-ferent ancestral versions of it based on the kind and proficiency of itspromiscuous activities.3
In all of those cases, a detailed understanding of the enzymatic mech-anism is needed. This can be made possible through both experimentaland theoretical studies of the chemistry involved. Experimental studieshere are often limited in the resolution of the information they provide,while theoretical studies are limited by the models used to describe thesystem under investigation. This has often been the case in studies ofenzymatic enantioselectivity, where only minimal changes in the interac-tions with a substrate can lead to large changes in selectivity16–18 thatcan be difficult to explain from experiments alone.19 At the same time,finding and understanding new enantioselective reactions is one of the fo-cus topics in chemistry, with numerous articles describing new reactionsbeing published (a short selection of recent publications from the Ameri-can Chemical Society, Guo et al.,20 de la Torre et al.,21 Tanabe et al.22).Still, better understanding of reactions is needed to make it possible torationally design new functions, or to change the current function purelyby design. Only full understanding will make it possible to further refineenzyme design approaches that are used today.23–25
1.1 Enzyme catalysisEnzymes function as natural catalysts by accelerating the reaction rate ofa transformation without being consumed in the process.1 Mankind's useof this kind of catalysis dates back to early examples of fermentation,26,27
but understanding of it was only starting after studies on the cellular or-ganisation of life and the discovery of the fermenting agents.28 Additionaldevelopment on the nature of chemical reactions followed from researchin the fields of organic and physical chemistry. Here, the previous real-
14

isation that biological molecules can be synthesized in the same way asinorganic species29 generated new interest in understanding how natureachieves the reaction outcome. One path towards understanding enzymecatalysis is by studying the rate by which certain reactants are turnedover and finding patterns between those rates and the physicochemicalproperties of the substrates.
1.1.1 Reaction kineticsThe rate law for a general chemical reaction (Scheme 1.1) can be definedas the change of either the concentration of the reactants or the productsover time (Eq. 1.1).
S k P
Scheme 1.1. Basic chemical reaction from reactants S to products P
v = −d[S]dt
= [S] ·k = d[P ]dt
(1.1)
This equation is valid for any uni–molecular reaction as long as there isno reverse reaction from the product P back towards the substrate.6 Theexact kinetics of a reaction can be determined from observed experimentalrates, giving information about the number and kind of the participatingmolecules. In case of an enzymatic reaction, those simplified schemes areoften not sufficient, as reactions can include several steps from the sub-strate towards the product. These steps are not necessarily observable,making it difficult to assign a single, global rate constant to the reac-tion. An example of an enzyme reaction is given in Scheme 1.2. Here,
E+Sk1
k−1ES
k2 E+P
Scheme 1.2. Example enzymatic reaction, leading from the free molecules, over theenzyme substrate complex to the product.
only the product formation might be observable, or the substrate deple-
15

tion. A possible way to analyse this kind of reaction is the application ofMichaelis–Menten kinetics.
1.1.2 Michaelis–Menten kineticsThe rate laws for the kinetics first described by Leonor Michaelis andMaud Menten30 can be applied to the example reaction in Scheme 1.2to obtain the overall rate constant for the reaction kcat and the so–calledMichaelis constant KM, the substrate concentration where the reactionvelocity is half of the maximum velocity. The basic assumptions are thatthe concentration of the catalyst E is much lower than the concentrationof the substrate S, and that the reaction is proceeding essentially irre-versibly towards the formation of the product P . The complete systemcan then be described using a set of differential equations, shown belowas equations 1.2 to 1.5. The concentration of the reactive species ES isgiven by the rate of formation from the free molecules k1, together withthe unproductive dissociation k−1 and the actual chemical step k2.
d[ES]dt
= k1 · [E][S]−k−1 · [ES]−k2 · [ES] (1.2)
The rate of substrate depletion is the difference between the forwardreaction towards the reactive complex ES k1 and the dissociation back tothe free species k−1.
− d[S]dt
= k1 · [E][S]−k−1 · [ES] (1.3)
The rate of product formation then only depends on the concentration ofthe reactive complex ES.
d[P]dt
= k2 · [ES] (1.4)
Finally, the concentration of free enzyme depends again only on the ratesof formation and depletion of the reactive complex ES.
d[E]dt
= −k1 · [E][S]+k−1 · [ES]+k2 · [ES] (1.5)
16

To solve those equations, additional assumptions about the nature of thedifferent steps in the scheme have to be made. In the original analysis ofMichaelis and Menten,30 it was assumed that the rate of formation of thereactive complex from the free molecules is identical to its dissociation,with the species in instantaneous equilibrium.
k1[E][S] = k−1[ES] (1.6)
Together with the conservation law for the catalyst in Equation 1.7, thisresults in the Michaelis–Menten description for the rate of product for-mation in Equation 1.8. Here, the value of KD is defined as the ratiobetween the rates k−1/k1, the binding constant in this case.
[E]0 = [E]+ [ES] (1.7)
v = d[P]dt
= k2[ES] = k2[E]0[S]
KD +[S] = vmax[S]KD +[S] (1.8)
A later derivation of the rate law was performed by Briggs and Haldane.31
In this case, the assumption in Equation 1.6 was changed to the assump-tion that the reactive complex ES is now in dynamic equilibrium with therest of the system.
k1[E][S] = k2[ES]+k−1[ES] (1.9)
This changes Equation 1.9 to include the rate of product formation intothe constant in the denominator, resulting in the Michaelis constant KM
in Equation 1.10 and the final equation for the enzymatic rate in Equation1.11.
KM = k−1 +k2k1
(1.10)
v = vmax[S]KM +[S] (1.11)
From those equations, basic kinetic parameters such as the maximum rateand the Michaelis constant KM can be obtained.
vmax = kcat · [E]0 (1.12)
17

Using those constants, the catalytic proficiency and the substrate affinityof the enzyme under investigation can be classified. It is also possible toobtain the combined value of the specificity constant kcat/KM. This valuecorresponds to the enzyme activity under any substrate concentration,making it also possible to evaluate the activity under conditions presentin the natural environment.8,32
1.1.3 Complex reaction kineticsThe rate laws given in the paragraph above are valid for the simple re-action given in Scheme 1.2, but need to be adapted for more complexreactions involving multiple reacting species and chemical steps. In thosecases, the King–Altman method33 can be applied to derive the definitionfor the constants in the Michaelis–Menten equation from the individualrate constants. By arranging the different species of the reaction in acircular scheme, the rates for the formation of the individual moleculescan be dissected to the basic rates. This can be of importance in thevalidation of a proposed reaction mechanism, as the rate constants fromthe individual steps need to be in agreement with the observed rate con-stant for the full reaction. One example of such a more complex reaction(Scheme 1.3) results in the rate Equation 1.13.
E+Sk1
k−1ES
k2k−2
EIk3 E+P
Scheme 1.3. A more complex enzymatic reaction, involving an intermediate on thepath to the final product
v =[E]0 · [S] · k2k3
k2 +k−2 +k3
k−1 (k−2 +k3)k1 (k2 +k−2 +k3)+ [S]
(1.13)
18

1.1.4 Determination of individual rate constantsThe individual rates of a multi–step reaction can be obtained to analysewhich are the steps that actually limit the overall rate, and are thus ofinterest for either understanding the enzymatic function, or for studieson how to modify it.34,35 Determining those rates depends on the abilityto observe any kind of signal during an experiment that can be relatedto the appearance or disappearance of the subspecies involved in the stepunder investigation, for example a spectroscopic or fluorescent signal fromgroups on the enzyme or the molecule being converted.36 One challengein studying those individual steps is that the reactions often reach steadystate in a short amount of time, making it necessary to use methods thatallow for rapid measurements, such as stopped flow36 or quench flow.37
The observed rates can then be related to the fundamental rate constants,according to what kind of observable has been measured and at whichpoint of the reaction the rate was observed.35
1.2 Physical chemistry of enzyme reactionsEnzyme reactions follow the same basic laws that regulate the rates ofthe reactions in organic chemistry. This means they can be subjectedto the same kind of analysis of their reaction properties as any otherreaction.6 Of general interest are the presence and nature of reactive in-termediates, the pathway used to perform a given reaction and the natureof the enzymatic transition states.38 Information from investigations intothe principle chemical behaviour can later be used to modify both theenzyme catalyst and/or the substrate to attempt to improve propertiessuch as the reaction rate or binding constants. The additional informa-tion can also help in understanding complicated reaction schemes andidentify parts of the enzyme that are favouring the reaction.
1.2.1 Linear free energy relationshipsOne possible investigation of the physical chemistry of a reaction is thevariation of either substrate or catalyst by varying chemical groups in-
19

fluencing the reaction.39 Those kinds of investigations are searching forthe linear dependency between observed rate constants and other exper-imental observables, such as equilibrium constants. From these kindsof analysis, information regarding the nature of the transition state ofthe reaction can be obtained.40–42 In the case of enzymatic reaction, thedetermination of these kind of relations is more difficult. Often, severalreaction steps are involved in the full reaction from the initial reactants tothe products. Also, modifications to the enzyme catalyst or reactants canhave additional effects besides the desired effect introduced by changingthem for the study. One example for this has been the case of reactionscatalysed by alkaline phosphatase.40,43 A possible relation that can beanalysed is the slope of the plot of the Hammett parameter σ ∝ K
K0,44
against the logarithm of the rate constant.
logk = A+Bσ (1.14)
Here, the rate k is related to a physical property σ by a constant factor B
and an offset A. Depending on the kind of reaction and the nature of thesubstituents, different slopes B are expected and will point to differentreaction mechanisms.6
1.2.2 Isotope effectsA different possible investigative pathway to determine the nature of (en-zyme) reaction transition states is the analysis of the effects caused byexchanging the atoms involved in the reaction for a different isotope.45–48
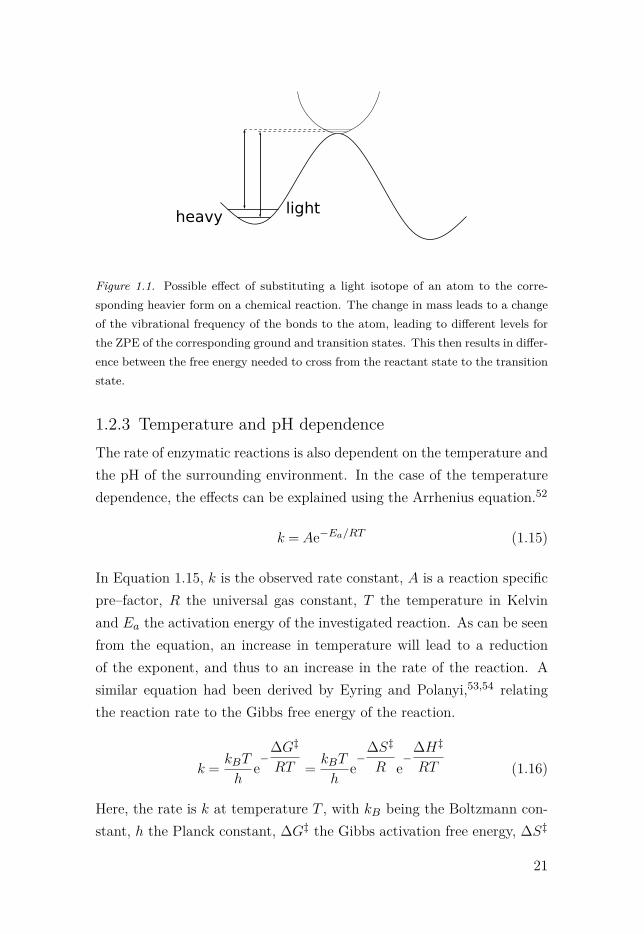
Depending on if kinetic or equilibrium effects are observed, the resultingeffects are either presented as kinetic isotope effect (KIE) or equilibriumisotope effects (EIE). In case of a study involving KIEs, a simple expla-nation for the observed effect can be given using Figure 1.1. Dependingon which atoms are exchanged and how those atoms contribute to thereactions, either normal or inverse isotope effects can be observed.49 Theobserved effect can then indicate a certain reaction mechanism and help todistinguish between several different possibilities. In reactions involvinglight atoms such as hydrogen, high values for kinetic isotope effects canalso indicate contributions from quantum mechanical tunnelling.48,50,51
20
lightheavy
Figure 1.1. Possible effect of substituting a light isotope of an atom to the corre-sponding heavier form on a chemical reaction. The change in mass leads to a changeof the vibrational frequency of the bonds to the atom, leading to different levels forthe ZPE of the corresponding ground and transition states. This then results in differ-ence between the free energy needed to cross from the reactant state to the transitionstate.
1.2.3 Temperature and pH dependenceThe rate of enzymatic reactions is also dependent on the temperature andthe pH of the surrounding environment. In the case of the temperaturedependence, the effects can be explained using the Arrhenius equation.52
k = Ae−Ea/RT (1.15)
In Equation 1.15, k is the observed rate constant, A is a reaction specificpre–factor, R the universal gas constant, T the temperature in Kelvinand Ea the activation energy of the investigated reaction. As can be seenfrom the equation, an increase in temperature will lead to a reductionof the exponent, and thus to an increase in the rate of the reaction. Asimilar equation had been derived by Eyring and Polanyi,53,54 relatingthe reaction rate to the Gibbs free energy of the reaction.
k = kBT
he
−ΔG‡
RT = kBT
he
−ΔS‡
R e−
ΔH‡
RT (1.16)
Here, the rate is k at temperature T , with kB being the Boltzmann con-stant, h the Planck constant, ΔG‡ the Gibbs activation free energy, ΔS‡
21

the activation entropy and ΔH‡ the activation enthalpy. The advan-tage of this relationship is the ability to directly obtain the energeticparameters from the experiments, allowing the comparison to theoreticalmethods evaluating the energetics of an reaction. In enzyme catalysis,the temperature not only acts by modulating the rate of the chemicalreaction, but also the stability of the enzyme. Therefor all enzymes areonly active in a relatively narrow window of accepted temperatures.8
Enzymatic reactions are also influenced by the proton strength (pH) ofthe environment. The concentrations of hydronium ions H3O+ given bythe relationship pH = −log10cH3O+ affects the protonation state of pro-tein amino acids according to their pKa, as defined by the Henderson–Hasselbalch equation.55
pH = pKa +log10
([A−][HA]
)(1.17)
Here, [A−] is the concentration of conjugated base of the acid HA. Thechange in protonation state of residues involved in the mechanism canlead to observable changes in the reaction rate. These changes can thenbe analysed to identify the nature of the participating residues and thekind of catalysis taking place. In the case of reactions involving acids orbases, pH–rate profiles can be helpful in determining how those residuesaffect the reaction, with a number of examples given in Figure 1.2. There,examples for the different observable rates are given for general and spe-cific acid and base catalysis.6 Similar to temperature effects, the pH ofthe environment also affects the protein stability, which will additionallyinfluence the reaction rates.56
1.2.4 Relating rates to free energyA main issue for the study of enzymatic reactions in terms of physicalchemistry is the relation of the experimentally observed rates to the freeenergy of the system that can be obtained from theoretical approaches.The concept of transition state theory (TST) has been developed by
22

pH = -log10 cH+
log 1
0 k
B)
pH = -log10 cH+
log 1
0 k
D)
A)
pH = -log10 cH+
log 1
0 k
C)
pH = -log10 cH+
log 1
0 k
Figure 1.2. Examples for the different pH versus rate profiles observed for the differentkinds of general (A,C) or specific (B,D) acid (A,B) and base (C,D) catalysis. In actualreactions, more complicated observations can arise from the combination of differentgroups that observe different forms of catalysis. The inversion points of the profilescan be an indicator for the nature of the amino acid being involved in the reaction,by comparing the points to the pKa values of possible candidates
Eyring and Polanyi and is the basis of the Eyring equation 1.16 men-tioned above. The basic equation is shown below as Equation 1.18 withthe only difference to Equation 1.16 being the inclusion of the pre–factorκ.
k = κkBT
he
−ΔG‡
RT (1.18)
This reaction specific value includes both the recrossing probability andthe contributions from quantum mechanical tunnelling to the reaction.57
The limitations of TST are the assumption that all chemical species in-volved in the reaction are at chemical equilibrium and that the effectsfrom quantum behaviour are not significant.58,59 In cases where generalTST is no longer valid, different additional treatments are needed to accu-rately describe the relation between reaction rate and energy.60,61 Otherexperimental observables can also be related to the energetics of a re-action, such as the equilibrium constants K to the free energy through
23

the relation ΔG = −RT lnK or the ratio of of the specificity constantskcat/KM.
ΔΔG = −RT ln(kcat/KM)1
(kcat/KM)2 (1.19)
This allows the direct comparison between the energetics of two differentreactions.
1.2.5 The origins of enzyme catalysisA catalyst is any molecule that increases the reaction rate of a chemicalreaction, meaning it reduces the free energy barrier between the reactantand transition state without itself being transformed by the reaction.6 Inthe case of enzyme catalysis, the enzyme functions as this catalytically–active molecule by first forming a complex between the enzyme catalystand the reactants, which is then converted during the chemical step intothe enzyme product complex. An example of this is shown in a free energydiagram in Figure 1.3 A for a simple one step transformation. The way bywhich the enzyme catalysts are able to reduce the free energy differencehas been the focus of biochemical research since the dawn of this field.8
Early models to explain enzyme catalysisAmong the first proposals on enzyme catalysis has been Fischer's modelbased on substrate binding,62 the “Lock and Key” model, and the in-duced fit model by Koshland following after this.63 Those models had incommon that they proposed that optimal interactions between the en-zyme and the substrate are responsible for the catalytic activity. Thoseinteractions were either supposed to be caused by the perfect shape com-plementarity between enzyme and substrate,62 or caused by a change ofenzyme conformation after binding to the substrate.63 Those explana-tions have now fallen out of favour, as enzymes have been shown not tobind tightly to the substrates but to analogues resembling the transitionstate of the reaction,15 as first proposed by Pauling.64
24

Catalysis through ground state destabilisationIn addition to Paulings theory on catalysis through transition state stabil-isation, theories were developed to include destabilizing effects from theenzyme on the reactant state to lower the free energy difference betweenit and the transition state.65,66 Several example reactions have been pro-posed to be catalysed by this effects.67,68 The general idea of the proposalis that the interactions between catalyst and reactants lead to distortionsin the reactants to a more transition state like structure. The basic prin-ciple is shown in Figure 1.3 B, indicating that in the idea of pure desta-bilisation, only the reaction state energies change, while the transitionstate stays the same. This approach to explain catalysis has later beenchallenged in multiple cases, showing that the contributions from groundstate effects were minor compared to effects on the transition state.69–72
Catalysis through transition state stabilisationThe idea that a catalyst improves the reaction rate by tightly binding tothe transition state has been first proposed by Linus Pauling, as men-tioned above.64 Figure 1.3 C gives a basic example, showing that thecatalysed transition state is lower in energy compared to the uncatal-ysed reaction. The way by which the stabilisation is occurring has alsobeen a topic of debate, with the ideas of electrostatic stabilisation73
and preorganisation74 (Figure 1.3 D) currently supported by differentmodels75–79 and experiments.72,80–82 The theory of transition state sta-bilisation through electrostatic interactions proposes that the active sitecharge environment is arranged so as to achieve optimal stabilisation ofthe transition state compared to reactant or product states. The theorywas reinforced by coupling it with the concept of active site preorganisation,73
meaning that the charges involved in the stabilisation do not have to re-arrange during the reaction in the catalyst, and thus reduce the penaltycaused by the reorganisation present for the same reaction in solution.
25

Reaction Coordinate
Free
Ene
rgy
Catalysis
A)
RS
TS
Free
Ene
rgy
RS
TSFr
ee E
nerg
y
TS PS
N+
O-
O
OP
O
OH O
OH -
++
-
-
P
O-
OH -
OH ON+
O-
O
O
+ +
-
P
O
OH
OH ON+
O-
O
O-
++
-
RS
B) C)
D)
Figure 1.3. (A) Visualization of the free energy profile for a simple one step transfor-mation. The catalytic effect is the difference between the free energy of the catalysedreaction in red and the uncatalysed reaction in black. (B) and (C) Simplified exam-ples for ground state destabilisation and transition state stabilisation. In both casesthe catalysed reaction in red reduces the free energy difference between catalysedreaction in red and the uncatalysed reaction in black. (D) Example visualization forelectrostatic preorganisation. The active site charges (red spheres) are organized insuch a way as to achieve optimal stabilisation of the reactant charges (blue spheres)at the transition state.
26

Non–thermodynamic explanations for catalysisIn recent years, cases of enzyme reactions have been found that indicatedeparture from the model of catalysis through transition state stabilisa-tion. These include cases of enzymes showing exceptionally large KIEsfor hydride transfer reactions.51 It has been proposed that those enzymesachieve a fine–tuning of catalysis by modulating the width and shape ofthe free energy barrier that the hydride has to cross, instead of reducingit.48,83 While the approach has been seen as a favourable explanation forcatalysis in some systems, other studies have proposed that the contribu-tions from tunnelling are minor compared to those from transition statestabilisation.84,85
1.3 Protein structure determinationThe structure of any catalyst has the greatest impact on its actual func-tion, with enzymes being no exception. Structural information has helpedin investigating a large number of different enzymatic mechanisms due tothe ability to see which groups are likely to interact with substrate mo-lecules and which are unable to do so.8 Obtaining the coordinates of allatoms in a protein involves, in most cases, the creation of a protein crystal,followed by X–ray diffraction studies to determine the electron density,which are then used to fit the atomic coordinates.86 Other methods arethe study of smaller proteins using nuclear magnetic resonance (NMR),or cryo–electronmicroscopy for larger complexes.87,88 Even though struc-tural information is the starting point for a large number of different pos-sible investigations, it can also be misleading in the study of a mechanism,as the conditions involved in the experiments leading to the structure donot have to correlate with the conditions either during a kinetic experi-ment, or the natural environment of the protein.Another important aspect of enzymatic activities is the stability of thecatalyst, as it is the case for classical organic catalysts.8 Studying stabil-ity can also lead to further insights concerning the functions of differentstructure elements, as they might be involved in providing necessary sta-bility, or can be mutated to achieve it. The study of enzyme temperature
27

dependence has given new insights here concerning those effects, as en-zymes adapted to higher or lower system temperatures have been shownto employ different catalytic strategies.89,90
1.4 StereochemistryThe systematic determination of molecular structures based on crystalli-sation or spectroscopic methods is a central factor to the characterisationof molecules.6 Different to basic composition analysis, the structural de-termination not only gives information about from what the compoundis built, but also how. As organic molecules can be extremely complex,there are numerous structures that can be formed from exactly the samechemicals.91 There are several levels of chemical redundancy, from com-pounds having the same chemical composition, to chemicals having thesame kind of functional groups (so–called stereoisomers),6 and moleculeshaving the same kind of bonding patterns and only differing in the orien-tation of the substituents in space (enantiomers).6
1.4.1 Compound classificationFischer rulesSome of the first studies on different isomeric forms of molecules had beenperformed by Emil Fischer on the nature of different sugar molecules,92
with the classification of all different sugar isomers as the result. Fischeralso introduced the projection scheme named after him93 to simplify thevisualisation of complex organic molecules. This classification organisesthe molecules along the longest axis, as visualised in Figure 1.4 (A) for asimple aldehyde.
Cahn, Ingold and Prelog projection rulesMore advanced classification had been developed later by Cahn, Ingoldand Prelog (CIP)94 for the general classification of organic compoundsand not just carbohydrates. The CIP classification first assigns the dif-ferent stereo centres of a molecule, atoms with substituents that can be
28

exchanged to result in a different isomer of the molecule of interest. Then,the substituents are arranged in space such as to point the atom with thelowest mass away from the viewer, with the remaining ones pointing outof the plane towards the observer. The identifiers are then assigned ac-cording to the relative position of the order from lowest to highest masssubstituent. An example is given in Figure 1.4 (B).
CH2
O
H OH
OH
H1
2
Cl
BrF
H
A) B)
Figure 1.4. (A) D-Glyceraldehyde represented using the Fischer projection. Thecarbon at the highest oxidation state is oriented to the top (1), with thelast chiral carbon (2) indicating the configuration of the molecule. (B) (S)–Bromo(chloro)fluoromethane in CIP representation.
Compound namingIn the Fischer classification, molecules are separated into the L (levo) andD (dextro) forms, indicating the orientation of the heavier substituenton the penultimate carbon either on the left (L) or on the right (D).In case of the CIP classification, compounds are classified as either S(sinister) or R (rectus), depending on whether the sequence of the atomsmentioned above is clockwise (R) or counter clockwise (S). For moleculesinvolving the orientations of substituents on double bonds, the CIP rulescan be used to classify a system as either E or Z type, indicating if thesubstituents are on the same site of the molecule (Z), or on opposite sites(E). This is similar to the cis or trans classification of organic molecules.
1.4.2 Enantiomers and stereoisomersIt is possible to classify different forms of the same basic chemical compo-sition depending on the kind and amount of symmetry operations needed
29

to superimpose them onto each other.95 In terms of different stereoiso-mers, this can be a rotational axis or plane, while enantiomers can not besuperimposed on each other using a mirror plane. A visualisation of thisis provided in Figure 1.5 A for stereoisomerism and B for an enantiomer.Molecules containing different enantiomeric forms are termed chiral, fromthe Greek term chiros for handed. The difference between the two classeslies within the ability to chemically differentiate between different formsof molecules built from the same components. In case of stereoisomers, itis still possible to use chemical classification to identify different forms, aschanges in the arrangement can lead to differences such as melting pointor spectroscopic signals. Those differences can be used to both identifyand purify the separate forms of the molecules. This is in general nolonger possible for different enantiomers, as the exchange between twosubstituents does not change the chemical properties of the molecule.6
The main observed effect for enantiomers is the different rotation of po-larized light, an effect first observed in the investigation of sugars andrelated compounds in the 19th century.
CH3O
H
HO
CH3
trans-(E)-methyl styreneoxide
cis-(Z)-methyl styreneoxide
A)
(L)-Cysteine
OO-
NH3+
SH
H
(D)-Cysteine
O O-
NH3+
SH
H
Mirror plane
B)
Figure 1.5. (A) Different stereoisomers of methyl Styrene oxide (B) Enantiomers ofcysteine separated by a mirror plane.
Enantiomers in biochemistryAs enzymes are build up from chiral amino acid building blocks, theythemselves are chiral molecules, with the spatial organisation in bindingor active sites being determined by the exact positioning and orientationof the amino acid side chains.1 All common amino acids used in living
30

cells are the L enantiomers in the Fischer projection (except glycine, anachiral molecule due to the missing side chain).1 This naturally resultsin differences when binding molecules that are themselves chiral, com-ing from the orientations of the molecules and the side–chains when theycome into contact during an interaction. For a simplified example seeFigure 1.6. This visualisation shows how the different enantiomers willbind in the same active site, with only one of them showing favourableinteractions. In an enzyme (or any other molecule selectively binding to
Di erent interactions of chiral mirror images
O
H
H
H
S O
H H
H
R
Hydrogen bond
Apolar interaction
Apolar interaction
- Stacking
- Stacking
- Stacking
Hydrogen bond
Apolar interaction
Apolar interaction
- Stacking
- Stacking
- Stacking
Figure 1.6. Simplified visualisation of the interactions between two different enan-tiomers and a binding site. Only in the case of the (S)–enantiomer is there a favourableinteraction between the oxygen and the site labelled ”hydrogen bond“. In case of the(R)–enantiomer, this interaction becomes unfavourable.
a ligand), this kind of difference in binding interaction can be the decid-ing point between an effective binder or one that binds only loosely orin a transient manner. Prominent examples for this kind of behaviourcan be found in the effectiveness of pharmaceuticals, where the differ-ence in enantiomers can often be decisive when it comes to the finaleffectiveness.96
31

Industrial and pharmaceutical use of enantiomersThe differences between the orientation of a single functional group mightnot seem to be of too much importance for many, but as has been shownabove it makes all the difference between a functional organism and arandom ensemble of molecules. Chemically the production of enantiopurecompounds is a challenging task, with the discovery of a new pathway thatleads to enantiopure compounds still being frequently the focus of majorpublications.20–22 A large number of current pharmaceutically importantmolecules are chiral and the production of any of the precursors for thosemolecules as enantiopure compounds will make sure that no further pu-rification will be necessary. The pressure on changing current synthesisprotocols towards producing the pure chiral forms of drug molecules hasbeen increased since the decision of the Federal Drug Administration inthe United States declared that new compounds that can be produced inpure form have to be enantiopure.97 While this has been possible througheither chiral organic synthesis (some examples can be found in Breuer etal.,98 Heitbaum et al.,99 Nestl et al.100) or through purifying the reactionproducts,101 those approaches often involve expensive additional steps,while biocatalytic approaches can avoid those expenses.
32

2. Hydrolase enzymes
Hydrolase enzymes are one of the basic families of enzymes, catalysingthe breaking of molecular bonds by the addition of water.1 All enzymesfrom this group are classified under the Enzyme Commission number 3,with a multitude of different functions and structural folds. The focus ofthis chapter will be on one of the subfamilies in this class of enzymes, theα/β hydrolases.
2.1 α/β hydrolase enzymesThe family of enzymes known as α/β hydrolases are one of the largerenzyme families that have been studied extensively for their involvementin a large number of important biological processes.1,102 Members of thefamily include the ubiquitous peptidases and esterases103 involved in re-actions like peptide bond cleavage and ester metabolism.1,102
Enzyme structureMembers of the enzyme family are characterised by their recognisableprotein architecture, composed of a β sheet sandwiched between a num-ber of α helices that connect the different β strands with each other. Anexample structure is shown in Figure 2.1 for the Homo sapiens acetyl-choline esterase enzyme. From the structure, the general arrangement ofthe α helices and β sheets mentioned above can be seen. The general foldused to build the enzyme structure is the Rossmann fold.
Catalytic mechanismAll the hydrolytically active enzymes in the family function through a gen-eral reaction mechanism involving a general base histidine and a chargerelay acid (aspartate or glutamate).1,102 Those two residues in concert
33

Figure 2.1. Three dimensional structure of Homo sapiens acetylcholine esterase(PDB: 4PQE). β strands are colored in dark red, α helices in dark blue. Structurevisualized in Chimera104 and rendered using POVRay.105
are then able to activate an active site nucleophile. The nature of thisnucleophile depends on the enzyme, with generic esterases and peptidasesoften using serine or threonine102 nucleophiles. The primary or secondaryalcohol can be deprotonated by the histidine polarized by the charge relayacid, generating a potent alcoxy moiety that can attack the carbonyl inesters or peptides.106,107 There are several variations of this general mech-anism, but the canonical structure of nucleophile, histidine, and chargerelay acid is common to all members.103 The residues are arranged to formthe catalytic machinery of the enzyme, called catalytic triad, visualisedin Figure 2.2.
NNHO-
O Nnuc
hisacid
Figure 2.2. Canonical structure of the catalytic triade in α/β hydrolase enzymes,build from a nucleophile (nuc), catalytic base histidine (his) and charge relay acid(acid)
34

2.2 Epoxide hydrolases and dehalogenasesOne subfamily of the larger class of α/β hydrolases with a distinct ac-tive site variation is the epoxide hydrolase (EH) and dehalogenase (DHA)tree. Here, the active site nucleophile is a carboxylate anion instead of analcohol, with the most commonly found amino acid being aspartate.108
This change in nucleophile is needed to perform the catalysed reactionof either epoxide ring opening or dehalogenation, to again form a re-active ester intermediate that can be further hydrolysed, as shown inthe epoxide hydrolase reaction mechanism in Figure 2.4. The remainingresidues in the catalytic triad are the same as for the other subfami-lies. This subfamily of the larger family of α/β hydrolases is of majorinterest in the development of biocatalysts, as epoxides, diols and halo-genated carbon compounds are important precursors for a large numberof pharmacologically active molecules,100 that can pose difficulties in or-ganic synthesis due to their high reactivity. The enzymes themselves arehighly selective for the catalysis of their native reaction, reducing the needfor purification of the desired reaction products from chemically similarbyproducts.100,109,110
DehalogenasesThe DHA subfamily of α/β hydrolases contains enzymes that are ableto break the halide – carbon bonds in their substrate molecules, by firstforming a covalent intermediate that is subsequently hydrolysed to thecorresponding alcohol.111,112 The enzymes belong to the EC class 3.8.1.5,with the important distinction that the reaction causes the inversion ofconfiguration at the position of the halide–carbon bond. The active sitesof these enzymes are similar to those of the EH class, with the differencethat the leaving group halide is stabilised by a set of slightly polar residuesin the binding pocket,111 instead of being still part of the substrate inEH enzymes. Enzymes in this class are able to act on a large numberof different halogenated compounds,113 which makes them interesting forindustrial applications.100
35

Epoxide hydrolasesThe other member of the DHA and EH sub–branch of the α/β hydro-lase superfamily gave the name of this class, soluble epoxide hydrolase(sEH). Enzymes from this class share the common fold of other α/β hy-drolases and are distinct from other ether–metabolising enzymes by form-ing a covalent bond to the substrate molecule before releasing the productdiol.114–116 This is one of the main discerning factors to other, non–α/β
epoxide hydrolases, such as limonene epoxide hydrolase (LEH).117 LEHenzymes are built around a different protein fold and catalyse the re-action by directly adding water to one of the epoxide carbons.118,119 Incontrast, sEH, as well as the related, but membrane–associated microso-mal epoxide hydrolase, do not directly hydrolyse the epoxide, but ratherforms a reactive ester intermediate with an active site nucleophile such asaspartate. The resulting ester is later hydrolysed by water and the wateroxygen being incorporated into the amino acid side chain.120 Due to theester intermediate, the enzyme can act on a variety of substrates usinga common hydrolysis pathway. The sEH reaction is further facilitatedby two tyrosine residues being positioned at the ceiling of the active site,with the aim of stabilising the epoxide oxygen after ring opening.121,122
The stabilisation of the tetrahedral intermediate formed during the es-ter hydrolysis is performed similar to all α/β hydrolase enzymes, usingbackbone amides to counteract the charge build up on one of the oxygenatoms.78,102
2.3 Solanum tuberosum epoxide hydrolase IThe Solanum tuberosum epoxide hydrolase 1 (StEH1) is a well–characterizedmember of the epoxide hydrolase family, with the main function thoughtto be in the cutin biosynthesis pathway.123 The enzyme consists of 321residues, with a total weight of 36 kDa. The three–dimensional structureof the enzyme is shown in Figure 2.3. In the active site, residues D105,H300 and D265 form the catalytic triad,124 with residues Y154 and Y235
situated at the roof of the active site.115,122,125 The two conserved ty-rosine residues provide the stabilisation of the epoxide ring oxygen and
36

Y154Y235
D105
H300
D265
Figure 2.3. Rendering of StEH1. The general α/β hydrolase fold is clearly visible.The inset shows the enzyme active site, with the residues involved in the catalytic triadand the tyrosine stabilizing the epoxide oxygen shown as sticks. Figure generated fromPDB 2CJP in Chimera104 and rendered with POVRay105
are thought to be involved in the mechanism to stabilise the intermediateepoxide oxy–anion (see Figure 2.4). The enzyme is interesting for bio-catalytic applications due to its ability to perform the enantioconvergentconversion of a set of smaller epoxide substrates,16,108,126 and its appli-cability in kinetic resolution with different substrate classes.17,126 This,together with the favourable stability and easy handling124,126 has alreadylead to some applications that employ variants of related enzymes,100 aswell as studies that try to link products from the epoxide hydrolase re-action to further catalytic steps,127,128 aiming at providing a pathwayto produce optical pure diols and metabolites that are precursors for anumber of different pharmaceutical compounds.
37

Previous mechanistic workThe aim of previous studies on the StEH1 enzyme have been focusedon understanding the mechanism of the enzyme in reactions with a setof phenyl substituted substrates, as well as long chain fatty acid epox-ides thought to be close to the native substrate (for an overview pleasesee Widersten et al.126). During those studies, the structure of the en-zyme was solved129 and the main residues were identified that are involvedin the reaction mechanism as shown in Figure 2.4.124,130 The hydrolytic
235
HO H
OHR1
OH R2
NHNH
H300
Y154
OH
Y235
OH
D105
OH O
OH2OHR1
OH R2
O
1 2(II)
(III)
Y
OHHO H
D105
OH ONH
NH
H300
Y154
OH
HO H
ProductComplex
Tetrahedral Intermediate
Substrate-free Enzyme-H+
O R2R1
Michaelis ComplexY154
OH
D105
O- ON
NH
H300H O
H
Y235
OH
O
R1
R2
HO H (I)
AlkylenzymeIntermediate
HO H
D105
O OR1
O R2
NNH
H300H O
H
Y235
OH
Y154
OH
-
HO H
D105
O OHR1
O R2
OHNHNH
H300
Y235
OH
Y154
OH
Solvent H+
-
R1
R2
Figure 2.4. Mechanism of StEH1, adapted with permission from Amrein et al..131
Residue numbers are based on the StEH1 enzyme.
step of the reaction was proposed as rate limiting124 for the hydrolysis,with the actual ring opening step being fast and leading to accumulationof the covalent alkyl–enzyme intermediate. The presence of the inter-mediate also made it possible to determine that the active site tyrosineresidues play an important part during the ring opening reaction,122 sta-bilising a negative charge that exists on the same time scale as the covalent
38

intermediate. This observation changed the previously–held perceptionthat the tyrosine residues are acting directly as Brönstedt acids dur-ing the ring opening reaction.116,121,125 The experimental data showeda number of unusual features of the enzyme, such as the existence ofslight hysteresis for the hydrolysis of one substrate that could only be ex-plained through invoking multiple binding modes,19 as well as changes inenantio- and regioselectivity under different temperatures and at differentbuffer pH.132 Early studies of the enzyme–substrate complex with trans–stilbene oxide showed several residues likely being involved in substratebinding,130 that were later used to select sites for future directed evolutionexperiments.17,18 Several characterised variants of the enzyme exhibitedchanges in both enantio– and regioselectivity for the target substrate ofthe experiments, as well as for a set of other test substrates.17,18
Substrate promiscuityThe initial experiments on StEH1 showed that the enzyme has the abil-ity to convert a number of different fatty acid and phenyl substitutedsubstrates,126 with further experiments indicating that the enzyme isable to accept a wide variety of different phenyl substituted epoxides,126
with some examples shown in Figure 2.5. The ability of an enzyme to
OCH3
O
O
O
trans-stilbeneoxide (TSO) trans-methylstyreneoxide (MeSO)
styreneoxide (SO)(2,3)-epoxypropylbenzene (EPB)
Figure 2.5. Size comparison between the different epoxide hydrolase substrates trans–stilbene oxide, trans–methylstyrene oxide, (2,3–epoxypropyl)benzene and styrene ox-ide.
act on several different substrates is grouped under the ambiguous term“promiscuity”, with studies focusing on enzymes catalysing several dis-
39

tinct reactions,3,9 or acting on different substrates but using a commonmechanism.133,134 Studies of enzyme promiscuity can help to identify con-served mechanisms and to identify the evolutionary origin of biochemicalreactions.3,133,135,136 They can also give starting points for further stud-ies on enzyme directed evolution or rational design by identifying enzymesite architectures that are related to a given activity, even if it is a non-natural reaction.137 In terms of enantioselectivity, the observed selectivitypatterns for a promiscuous activity can give insights on how other relatedor unrelated substrates might react or how the preference for a given sub-strate or reaction product can be improved. One important note here isthat the enzyme shows a large preference towards substrates in the transconfiguration, with substrates such as cis–stilbeneoxide showing only min-imal activity.124 The shape of the active site might be the reason for thispreference, due to steric clashes between the substrate phenyl rings in thecis configuration and the active site residues.
Use in biocatalysisEspecially due to it's independence from cofactors and its high stabil-ity, the wild–type StEH1 enzyme already has potential for biocatalyticapplications.126 Nevertheless, the most interesting property is the enanti-oconvergence when hydrolysing small, phenyl substituted epoxides.138,139
This makes it possible to obtain the pure chiral vicinal diol product froma racemic mixture of the substrate that can itself be prepared using in-expensive epoxidation reactions.98 This intrinsic selectivity has alreadybeen employed in the case of a related enzyme from Aspergillus niger,that was combined with a LEH enzyme from Rhodococcus erythropolisto produce enantiopure diols.140 Other proposed applications involve thegeneration of hydroxy–aldehydes and ketones from the diol products, inmulti–step biocatalytic applications.126–128 In general, the ability to ad-just the enzyme selectivity17,18 could allow the adjustment to any numberof different substituted substrates to produce pure fine chemicals.
40

3. Computational study of biological systems
The study of physics and chemistry using computational approaches datesback to the first half of the 20th century. In this time, the first theoreticalcalculations were still based on manual calculation and derivation of thenewly–discovered equations governing quantum mechanics.141 The firstcomputer–based approaches followed shortly after this, with the calcu-lation of basic molecular properties.142 Around the same time, the firstcalculations were performed on the behaviour of large atomic systemsusing classical approaches,143 even though they had to be done withoutthe help of electronic computers in some cases.144 In this chapter, a num-ber of the current methods and principles involved in the calculation ofexperimental observables in chemistry will be explained, together withexamples for their applications. As the field is too vast to cover all of it,the focus is mainly on methods employed during the work presented inthis thesis.
3.1 Quantum mechanics methods3.1.1 Wave function methodsCurrently, the most precise way to calculate how particles interact witheach other is through the use of quantum mechanics (QM). In the wavefunction description of QM, the current state of a system can be obtainedby solving the electronic Schrödinger equation.
HΨ = EΨ (3.1)
Here, H is the Hamiltonian operator for the system in question, actingon the wave function Ψ, with E being the energy of Ψ.145 If Ψ is known,Equation 3.1 can be solved analytically. This is usually not the case
41

except for trivial systems, making it necessary to approximate certainaspects of the system under investigation.146 The most commonly usedone is the Born-Oppenheimer approximation,147 where the movementsof nuclei and electrons are treated separately, due to the difference inmass between them. This means that the Hamiltonian for the electronicinteractions (Equation 3.2) can be solved first, with the movement ofthe nuclei later calculated using the average energy 〈Ve〉 of the electrons(Equation 3.3).
He = −∑
i
12 ·∇2
i −∑
i
∑A
ZA
ri,A+
∑i
∑i�=j
1ri,j
(3.2)
Hn = −∑A
12 ·∇2
A +∑A
∑B �=A
ZAZB
rA,B+ 〈Ve〉 (3.3)
The electronic wave function (Equation 3.4) must then be solved to findlowest possible energy, corresponding to the ground state of the systembeing investigated for a given set of coordinates r for the electrons andR for the nuclei.
HeΨe (r,R) = EeΨe (r,R) (3.4)
This can be achieved using the variational principle, stating that any trialwave function found that satisfies Equation 3.4 will be either higher inenergy or equal to the true value.146 The equation is solved numericallyuntil a given convergence criterium is achieved for the current coordinates.This still requires a trial function to be used as the starting point forEquation 3.4. One approach to create this function is as the product ofall individual electron wave functions.
Ψe (r1, . . . ,rN ) = ψ1 (r1) · · ·ψN (rN ) (3.5)
The result of Equation 3.5 would then be the wave function for the system,but it would not be anti–symmetric. To be an actual representation ofa physical system, it will need to satisfy the Pauli exclusion principle,148
meaning that no two electrons can be in the same state at the same time.This is usually achieved by the use of Slater determinants that include theantisymmetry of the electron wave function by changing the sign for the
42

interaction when exchanging two electrons. Each of the one electron wavefunctions ψi can be approximated as a linear combination of functions.
ψi (r) =∑
k
ck,iφk (r) (3.6)
A complete set of functions φk in Equation 3.6 compose a so called “basisset”, that describes the electronic structure, while the coefficients ck,i areoptimised to generate the lowest energy wave function.
Methods to solve the Schrödinger equationAs there is no analytical solution to the Schrödinger equation for multi–electron systems, methods have been employed that take the approxima-tion from Equations 3.2 to 3.6 into account to numerically approximatethe correct results. One of the early methods has been the Hartree–Fockapproach, where the Hamiltonian for the multi–electron wave functionfrom Equation 3.2 is split into individual, one electron Hamiltonians.149
Those so–called Fock operators are composed out of the the one electroninteraction terms from Equation 3.2 and a repulsion term between thecurrent electron and all other electrons of the system as a mean field.
hi = −12 ·∇2
i −∑
i
∑A
ZA
ri,A+
∑j �=i
(Jj (i)+ Kj (i)
)(3.7)
In Equation 3.7, Jj (i) is the repulsive interaction between two elec-trons, while Kj (i) is the exchange operator caused by the exclusionprinciple.146,150 Solving the equations like this will result in the Hartree–Fock structure and energy of the system. The energies obtained from theapproach will still be not the correct ones, as the method only approx-imates the correct electron–electron interaction energies from Equation3.2. Obtaining the more precise values is only possible by either ex-plicitly calculating the interactions using methods such as configurationinteraction151 or coupled cluster,152 or by adding correction terms to theequation using perturbation theory.153,154 All those approaches have incommon that they are computationally expensive when exact, meaningthat they can usually only be applied to calculate the properties of smallmolecules or atoms.
43

3.1.2 Density Functional Theory methodsA completely different approach to calculate the energetics of moleculeshas been the advance of methods based on the calculation of the electrondensity, instead of the electron wave function. This kind of approach,named DFT for Density Functional Theory, has been first proposed atthe beginning of the 20th century,155 but was not further explored untilthe work of Hohenberg and Kohn156 and a bit later Kohn and Sham157
that first made its application for the study of chemistry possible. InDFT the ground state energy E0 of a system is given by the followingterm.150
E0 = Ev [ρ0] =∫
ρ0 (r)v (r)dr+T [ρ0]+V ee [ρ0] (3.8)
Here, Ev indicates the dependence of the energy on the so called “exter-nal potential” generated from the positions of the nuclei acting on theelectrons in the system. The first part of the equation is the averageattraction of the nuclei towards the electrons, depending on the electrondensity, with the second and third terms being the electron kinetic en-ergy and electron–electron interaction energy. The last two terms areindependent of the first term, and also depend solely on the electron den-sity of the system. The first Hohenberg–Kohn theorem156 was used toshow that it is possible to use the variational approach to find the correctelectron density from a trial density, but not how to calculate the density.The Kohn–Sham approach developed later157 showed that it is possibleto calculate the correct density, starting from a system of independent,non–interacting electrons, giving the electron density ρs. They redefinedthe total electron kinetic energy as.
T [ρ] = ΔT [ρ]+T s [ρ] (3.9)
In Equation 3.9, ΔT is the difference between the kinetic energy of thestarting system and the real system. A similar approach was used toredefine the electron–electron interaction energy V ee as follows.
V ee [ρ] = 12
∫∫ρ(r1)ρ(r2)
r12dr1dr2 +ΔV ee [ρ] (3.10)
44

Here, ΔV is again a correction for the electron–electron interaction energyfrom the non–interacting to the real system. The two terms ΔT and ΔV
are combined to form the so–called exchange correlation function of theelectron density.
EXC [ρ] = ΔT [ρ]+ΔV [ρ] (3.11)
In DFT, the main problem is finding the correct term for Equation 3.11,as the other terms in Equation 3.8, substituted by using Equations 3.9 and3.10 are mathematically exact.146 Several different ways have been used toobtain this functional, resulting in the plethora of different DFT function-als. A possible way to obtain estimates for this functional is parametri-sation to reproduce the properties of known compounds, with differentnumber of parameters used in different methods.158,159 The search forthe “correct” EXC functional is still an ongoing process, as pure fitting ofparameters can lead to notable errors in the predicted properties.160,161
3.2 Classical methods3.2.1 Molecular mechanicsThe calculation of molecular properties using any of the methods men-tioned above is usually too computationally expensive for systems such asenzymes in solvent, even though simulations of around a thousand atomsare now possible.162 In molecular mechanics (MM), the interactions be-tween atoms in a molecule are approximated using classical descriptionsto allow for more efficient calculations.141 The way to describe the in-teractions is through a set of analytical functions called a force field.Force fields are build from individual functions and parameters to defineinteraction strength and equilibrium values for the different possible in-teractions. One of the first description of such a system has been providedby Levitt and Lifson,163 giving rise to the canonical form of the force field
45

equation.
V =∑
bonds
12kb · (r − r0)2 +
∑angles
12kφ · (φ−φ0)2
+∑
dihedrals
kψ · (1+cos(n ·ψ − δ))
+∑
impropers
12kθ · (θ − θ0)2
+∑
atompairs
qiqj
4πε0rij+ Aij
r12ij
+ Bij
r6ij
(3.12)
In Equation 3.12, the constants kb, kφ, kψ and kθ stand for the force con-stants of the bond and angle harmonic springs, the proper torsion forceand the improper torsion harmonic spring, respectively. The values of qi
and qj are the partial atomic charges for two atoms, ε0 is the gas per-mittivity, while Aij and Bij are the repulsive and attractive term of theLennard–Jones potential,164 usually used to model van der Waals interac-tions. To calculate the energy of a molecule for a given set of coordinates,all terms in Equation 3.12 are solved, resulting in the potential energy ofthe system.
The ensemble conceptTo relate energies calculated as, for example, using Equation 3.12 with ex-perimental observables, the field of statistical mechanics defines so–calledpartition functions that define the number of possible configurations thata system can assume.165 Those functions can then be combined with ex-ternal conditions that produce the ensemble. The ensemble defines a set ofconfigurations that can be obtained under the constraints of the externalconditions that are used, called the available phase space. Several differ-ent ensembles can be chosen to describe a system, such as one confinedby a constant number of particles, constant volume and constant energy.This so called microcanonical ensemble166 then applies a weighting factorof β = 1
kT to each of the different configurations, with the probability ofbeing in a state A being related to the energy of the state.
P (A) ∝ e−βV (A) (3.13)
46

Several other external conditions can be applied, such as the more com-mon use of constant temperature instead of constant energy, resulting inthe canonical ensemble, or the change of the constant number of particlesto a constant chemical potential, with the grand canonical ensemble asthe result.165 It is then possible to calculate a given property for a largenumber of different structures within an ensemble to obtain the ensem-ble average 〈A〉 of a physical observable A. If sufficient structures havebeen sampled, this value will represent the true physical observable of agiven property under the constraints of the force field used to describethe system.
Molecular dynamicsOne possibility to obtain different configurations of a system is the inte-gration of Newtons equations of motion to propagate the system in time,and let it explore the available phase space. This involves both the cal-culation of the individual forces on all atoms, as the derivatives of theenergies over the positions.
Fi = δU
δri(3.14)
From those forces, new velocities can be calculated for each atom veloci-ties, and are then used obtain new coordinates for a defined time interval.Several algorithms exist to perform this stepwise procedure, with one ofthem being the leap–frog method.167 Here, the positions and velocities ofthe particles are not adjusted at the same time, but offset by a half stepof the simulation.
r(t+Δt)=r(t)+v(
t+ Δt
2
)·Δt
v(
t+ Δt
2
)=v
(t− Δt
2
)+ F(t)
mΔt
(3.15)
Other methods include the verlet and velocity–verlet algorithm, and theBeeman algorithm.165
Monte Carlo methodsA completely different approach to obtain different configurations in anensemble is the generation of non–deterministic configurations that are
47

accepted according to energy compared to a previous state.165,168 Thosemethods are not dependent on the propagation of the system throughtime, but instead explore the available phase space directly. The advan-tage of this kind of approach is that the previous configuration does notimpose any limits on the nature of the next one, as only the energies needto be similar. The disadvantage is that the number of sampling pointsneeded for systems containing large number of coupled degrees of free-dom is usually too high to fully explore phase space and obtain sufficientsampling for the calculation of ensemble averages.169
Molecular dockingOne approach to evaluate possible interaction sites between a receptorand ligand has been the concept of molecular docking.170–172 In this ap-proach, possible interactions between the ligand and receptor are anal-ysed according to a scoring function, after the generation of random ori-entations of the ligand within a defined search volume of the receptor.Scoring functions can be based on empirical estimates for the interactionstrengths,173 on molecular force fields,174 or on previous knowledge aboutpossible interactions.175 The approach allows the rapid evaluation of dif-ferent binding modes for ligands, both to identify new possible interactionpartners and to analyse differences between potential binding positions.
3.2.2 Estimation of free energiesFree energy perturbationAs mentioned in Section 1.2.4, the rates of chemical reactions can bedirectly related to the free energy differences between different states ofthe system in question. This means that the computational evaluationof this difference is the main goal when applying theoretical methodsto study a chemical reaction mechanism. One of the earliest proposedmethods on how to obtain those energies from a sampled distributionwas proposed by Zwanzig,176 using the following approach.
ΔG = −RT ln⟨
e−
(U2 −U1
RT
)⟩1
(3.16)
48

In Equation 3.16, the free energy difference between two states is ob-tained by calculating the ensemble average of the potential energy dif-ference U2 − U1, sampled on state 1. The requirement to obtain the freeenergy is then only that both states are sufficiently sampled in the phasespace available to state 1, meaning that state 2 has to be similar to it.This limitation can be avoided if the difference between the two states isreduced through the introduction of intermediate states. If those inter-mediate states represent the actual path between states 1 and 2, then thefree energy of the complete change can be evaluated as the sum over allintermediate steps 1 ≤ i ≤ n, with the energy difference Vi,i+1 betweentwo points i and i+1 being defined in Equation 3.17.
Vi,i+1 = Ui+1 −Ui (3.17)
The total free energy is then given by Equation 3.18.
ΔG = −RTn−1∑i=1
ln⟨
e−
(Vi,i+1RT
)⟩i
(3.18)
The term for this general approach is free energy perturbation (FEP).
Thermodynamic integrationA very similar approach to the FEP method is to use a scaled Hamilto-nian to interpolate between two different states. This approach, calledthermodynamic integration, scales the total energy of one state with anadditional factor λ ranging from zero to one that indicates the position onthe reaction coordinate.165 Here, a value of λ = 1 would indicate that thesystem is only sampling configuration 1, while a value of λ = 0 indicatesthe system being in state 2.
U (λ) = λU1 +(1−λ)U2 (3.19)
The total free energy for moving from state 1 to 2 is then the integralover all individual values of λ.
ΔG =1∫
0
⟨δU
δλ
⟩λ
dλ (3.20)
This approach makes it possible to create unphysical intermediate statesbetween the different end points, to drive the system from one state tothe other.
49

Umbrella samplingA general issue for calculations involving FEP is the choice of the inter-mediate states to allow sufficient sampling. This is especially the case forthe sampling for rare events like binding of substrates or the crossing ofa transition state.165 The method of umbrella sampling (US) is anotherapproach to sufficiently sample the system in or near such a state, byapplying an external biasing potential along a reaction coordinate thatdefines the path between two states.177 Here, the actual potential energyof a state U1 is modified by an extra term W shown in Equation 3.21.
U (r)total = U (r)1 +W (r) (3.21)
This addition then results in a total energy as a function of the normalposition dependent potential energy, and the extra term W that alsodepends on the current configuration. The energy associated with thisbiasing potential has to be later removed from the total energy whenevaluating the free energy difference, using methods such as the weightedhistogram analysis.178
3.3 Multiscale modelsOne major problem for the description of reactive chemistry in solutionis that the computational cost is too high for models that are able toactually describe the chemical events of bond breaking and forming (seeSection 3.1). At the same time, classical methods are able to approxi-mately calculate the interactions between large numbers of particles, butare limited to the force field description employed. As this description isusually static it can not account for changes in the bonding pattern (ex-cluding reactive force fields such as ReaxFF179). The idea of combiningthose two approaches by embedding a small system treated using moreaccurate theory into a larger system treating the atoms in a more approx-imate way had been first employed by Warshel and Karplus180 and latergeneralised by Warshel and Levitt.181 Several possible ways of combiningtwo system can be chosen, with two of them being detailed further here.
50

3.3.1 QM/MM approachesThe most common use of multiscale models is by directly treating a part ofthe system using a QM method embedded into the larger region treatedusing classical physics.182,183 Both the QM and classical region can betreated in different ways by using different levels of electronic structuretheory for the QM part and by being able to employ different force fieldmethods for the classical region. The interactions between the two regionsare treated using a given embedding scheme. It is possible to eithercalculate the interactions in the QM region using only the chosen QMmethod (additive QM/MM),184 with the MM treatment only being usedfor the outside region.
UQM/MM = UQM (QM)+UMM (MM)+Ucoupl (QM to MM) (3.22)
The term Ucoupl in Equation 3.22 refers to the interaction between theregions treated using the respective QM and MM approaches and caninvolve different levels of approximations to bridge the regions. The ad-vantage of this approach is that the MM force field does not need anyinformation about the reactive region, but it can lead to problems withwhen crossing the boundary between them. In the subtractive scheme,the MM energies are calculated for for the whole region, including theQM part, and are subtracted from the total energy later. This treatmentmakes the difference between the QM and MM energy of the reactiveregion a correction to the total energy of the system.185 One example forthe application of this would be the ONIOM method.186
UQM/MM = UMM (QM and MM)+UQM (QM)−UMM (MM) (3.23)
This means that both the QM and MM calculations can be fully inde-pendent from each other, but it introduces the issue of having a suitableforce field description for the reactive part in the MM region, somethingthat might not be available.
3.3.2 The Empirical Valence Bond approachA different approach to the methods mentioned above has been pre-sented by Warshel and co–workers in the Empirical Valence Bond (EVB)
51

model.187 This approach, originally based on Marcus theory for electrontransfer,188 assumes that the energy of a system can be treated by defin-ing the different states as independent valence bond states, connected byreaction specific coupling terms.189 The Hamiltonian shown in 3.24 thendescribes the ground state of the system.
H =
⎡⎣ ε1 Hij
Hji ε2
⎤⎦ (3.24)
In this case, ε1 and ε2 are the energies of the valence bond states for a twostate system, and Hij = Hji are defined as the coupling elements. TheHamiltonian can then be used to solve the ground state energy eigenvalueproblem, as shown in Equation 3.25.∣∣∣∣∣∣
ε1 −Eg Hij
Hij ε2 −Eg
∣∣∣∣∣∣ = 0 (3.25)
Here, Eg is the ground state energy. Solving this leads to the secularEquation 3.26, with solutions for the ground state and first excited stateenergies.189
Eg = 12 · (ε1 + ε2)− 1
2 ·√
(ε1 − ε2)2 +4 ·H2ij (3.26)
In EVB, the Hamiltonian elements of ε1 and ε2 can be approximatedby using molecular force field functions to represent the individual va-lence bond states,187,190 while the coupling elements can either be simplefunctions of the distance between reacting atoms, or more complicatedfunctions191–193 to describe the interaction between different states. Inthis formulation, the EVB would still face similar limitations like the othermethods mentioned above, for example that the sampling of rare eventslike the crossing of a transition state would not be sufficient to analysethe free energy of such a process.187 The solution of this problem was thecoupling of the description in Equation 3.24 with a reaction coordinatebuilt from the linear interpolation of the states of the system.194 Similarto Equation 3.19, the system is changed from one state to the other bymixing the force field terms for the individual states along the coordinateof λ from zero to one.
Uλ = λUε1 +(1−λ)Uε2 (3.27)
52

At every value of λ, the free energy difference between those windows iscalculated similar to Equation 3.18 in the following way.
ΔG(λi) = −RT lni−1∑n=0
⟨e
−Un+1 −Un
RT
⟩n
(3.28)
In addition to Equation 3.28, the free energy difference between the statesfor a given set of coordinates r, called free energy gap, can be calculated.
Ugap (r) = Uε2 (r)−Uε1 (r) (3.29)
The energy gap can then be used as a generalised reaction coordinate,73,195
by splitting the calculated energy values into several bins Xbin that spanthe complete range of it. For each of the individual bins, the differencebetween the EVB ground state energy (Equation 3.26) and the free en-ergy from Equation 3.28 can be calculated as average aver all λ windowsthat populate this part of the energy gap.
ΔGEV B (Xbin) = −RT ln⟨
e−
Eg (Xbin)−Ui (bin)RT
⟩λi
(3.30)
Together with Equation 3.28, the potential of mean force can then be cal-culated for all points on the energy gap reaction coordinate by combiningit with Equation 3.30.194
ΔG(Xbin) = ΔG(λi)+ΔGEV B (Xbin) (3.31)
For a different number of EVB states, the Equation 3.24 and 3.26 canbe generalized for the matrix diagonalization to give the correct groundstate energies for the individual states.
Analysis of energy contributionsThe relevant points corresponding to the actual reactant, transition andproduct states can be extracted from the full EVB potential of meanforce to be analysed concerning the effect of the protein environment onthe reacting system at those points. One possible investigation concernsthe energy contributions of individual amino acid side chains. The linear
53

response approximation is used to obtain the interaction free energy dif-ferences between the selected states.196 Equation 3.32 gives the generalmethod to obtain those energies, with U1 and U2 indicating the potentialenergies of the interaction in a given state, while the average 〈〉i indicatesthe ensemble average sampled in a given state.
ΔGLRA = 12 · (〈U2 −U1〉2 + 〈U2 −U1〉1) (3.32)
This kind of relationship allows a quick evaluation of the free energydifferences between different states. In a similar fashion, the so–called“reorganisation energy” λ (Equation 3.33), based again on Marcus theoryfor electron transport,188 can be used to estimate the amount of changein interactions have been needed to achieve the transition between thedifferent states.197,198
λ = 12 · (〈U2 −U1〉2 −〈U2 −U1〉1) (3.33)
Both of those approaches can be used as a quick estimation for the freeenergy of the processes being studied.
EVB and quantum classical path approachOne possibility to extend the reach of the EVB approach has been theinclusion of quantum effects using path integral methods.165,199,200 Onesuch method has been the quantum classical path (QCP) approach, inwhich the classical coordinates are used as the central coordinates forthe generation of ring polymers, that can then be used to evaluate theenergies in a path integral formulation.201–203 The advantage of this kindof formulation is that classical simulations can be used as the basis forthe evaluation of QM effects such as quantum tunnelling corrections tothe kinetic isotope effect obtained from isotopic exchange. One problemof this approach has been the correct generation of the free particle dis-tribution to describe the ring polymer, an issue that has bee successfullyapproached by the development of new ways to generate it directly.204
54

4. Present Investigation
The focus of this thesis is both the study of the enantioconvergent reactionmechanism of the Solanum tuberosum epoxide hydrolase 1 enzyme, andthe development of new methods for performing chemical simulations.First, the work on the epoxide hydrolase mechanism from Paper II willbe presented, starting with the investigation of the hydrolysis of trans–stilbene oxide and the mapping of the active site residues that have majoraffect on the reaction pathways chosen in the enzyme. This is followed bythe investigation of the smallest substrate styrene oxide in Paper III andthe origins of the enantioconvergence in the native enzyme and possiblereasons for differences observed in active site mutants. Substrate bindingof the only not phenyl substituted epoxide (2,3–epoxypropyl)benzene isstudied in Paper IV, together with the presentation of several new struc-tures of StEH1 variants that had been created during the previous workon directed evolution of the enzyme.17,18 The final part of the epoxidehydrolase project is the work on the substrate trans–methylstyrene ox-ide in Paper V, investigating the branched reaction scheme previouslyproposed for the hydrolysis of this substrate.132 The remaining parts arefocused on the work involving method development. First, the resultsfrom the parametrization of a new model for the classical simulations ofdivalent transition metals in Paper I. Following this, a new version of theQ simulation software is presented in Paper VI. Here, the new algorithmsthat have been implemented are showcased, together with results of thoseapproaches applied to a number of model systems.
55

5. Catalytic mechanism of StEH1 basedepoxide hydrolysis
Epoxide hydrolase enzymes have been one of the focal points of researchinto enantioselective epoxide hydrolysis, with a particular focus on the en-zymes from Solanum tuberosum,205 Rattus norvegius206 and Aspergillusniger.16 All those enzymes share the common epoxide hydrolase mech-anism, using carboxylic ester side chains for the nucleophilic ring open-ing and the charge relay system based on a catalytic histidine to facil-itate the breakdown of the formed intermediate.114 The special inter-est in StEH1 stems mostly from its surprising ability to enantioconver-gently hydrolyse several substrates,126 while the native reaction has seenonly limited study.205 Even though the mechanism of the enzyme hasbeen explored in depth,114,124,207 several question still remained concern-ing the nature of the enantio– and regioselectivity,116,130,132,207 with alarger focus on finding new enzyme variants to perform desired chemicaltransformations.16,17,110,208 The aim of Papers II to V was to extend theknowledge about the EH mechanism and to explain several of the kineticobservations that had been found for the wild type enzyme and its mutantforms.18
5.1 Previous kinetic studiesAs mentioned above, the previous studies of sEH enzymes have mostlybeen focused on exploiting to malleability of the system and its robust-ness for mutation.16,17,208 While there have been mechanistic studies inthe past employing both mixed and pure QM methods,116,207 as wellas experimental approaches, some of the observed effects in the kineticstudies still lacked complete explanation.130,132 Especially, the hyperbolic
56

kinetic behaviour observed in the hydrolysis of some substrates could onlybe partially explained19,132 and the inverse pH profile seen for a mutantenzyme has been a mystery.130 Computational models have been wellsuited to study those kinds of problems, as they can give a microscopicunderstanding of the process and identify all favourable and unfavourableinteractions that are involved.
5.2 Hydrolysis of trans–stilbene oxide (TSO) (Paper II)The aim of Paper II was to cover all relevant parts of the epoxide hydrol-ysis mechanism in StEH1 shown in Figure 2.4 and to provide the basicunderstanding needed to perform future studies on this system. It wasalso intended as a benchmark for the EVB approach to verify the ac-curacy when studying epoxide hydrolysis. The substrate trans–stilbeneoxide was chosen due to its relatively large molecular volume filling outthe active site, leading to only one productive binding mode that neededto be analysed.
Computational study of intermediate breakdownThe simulation of the reaction of the hydrolytic breakdown of epoxidesin StEH1 was initiated from the common point of the alkyl–enzyme in-termediate (subspecies II in Figure 2.4). This starting point was chosento ensure that all sampled reaction pathways lead to the common exper-imentally observed structure and to minimize issues from the samplingof unproductive binding modes in the reactant state. As previous workshowed that the active site charge balance needs to be attained by pro-tonating one of the residues around the nucleophile,207 we investigatedpossible candidates while keeping the active site base in the neutral formneeded for the hydrolytic step of the mechanism.106,116,207,209 Initial cal-culations showed that the charge balance was indeed needed for the EVBmodel to reproduce the experimentally observed values.
Active site protonation strongly affects reactionBased on sequence alignments in the sEH and DHA superfamilies (Figure5.1), we decided to protonate the conserved histidine proximal to the ac-
57

tive site nucleophile and evaluated the effect on the calculated activationenergies (Table 5.2).
Figure 5.1. Sequence alignment based on a curated data set of EH superfamilyenzymes from the ESTHER database.210 The residues next to the nucleophile werechosen for the center of the figure, showing that the adjacent histidine is highlyconserved. Reproduced with permission from Amrein et al.131
It was found that the protonation of H104 made it possible to fullyreproduce the observed catalytic effects for the native enzyme and a setof mutants previously characterised, shown in Figure 5.2. The effectfrom the histidine residue was also found using empirical estimates ofthe pKa of the active site residues shown in Table 5.1. Interestingly,the mutation of the glutamate proximal to the conserved histidine didchange the pKa estimates (even though the empirical estimates are likelyoverestimating the shift), clearly indicating a different interaction patternand a likely change in protonation state. Additionally, we observed thatthe mutated residue is sampling different conformations depending on thesubstrate enantiomer, providing a possible explanation for the inversionof the observed pH profile of the hydrolysis reaction.
Intrinsic regioselectivityStudying the regioselectivity of the nucleophilic attack on the epoxidering was one of the main aims for the project to establish a baseline forinvestigating the other substrates that the enzyme is able to hydrolyse.As TSO is fully symmetric and as its hydrolysis leads to fully symmetricproducts, the regioselectivity cannot be studied experimentally. Still, our
58

ResidueSystem state
Apo RS Int TDWild type
E35 3.6 2.3±0.3 2.4±0.4 2.6±0.4H104 7.3 8.2±0.7 6.7±1.1 8.0±0.5D105 1.8 6.1±1.0 n.d. n.d.D265 2.7 2.6±0.3 2.5±0.4 2.6±0.4H300 11.0 7.1±1.5 7.3±0.7 6.5±0.5
E35QH104 –– 2.1±0.7 2.5±0.5 2.8±0.5D105 –– 5.8±1.0 n.d. n.d.D265 –– 3.0±0.7 2.7±0.6 3.0±0.6H300 –– 8.4±1.1 7.2±0.1 7.3±0.1
Table 5.1. Estimated values for the pKa of the active site residues in the wild–type and E35Q variants of StEH1. Apo – Ligand free enzyme; RS – Reactantstate; Int – Covalent acyl–intermediate state; TD – Tetrahedral intermediatestate. Adapted with permission from Amrein et al.131
calculations showed that the enzyme has a strong preference to open theepoxide ring at the position distal to the active site base histidine (carbonC2 in Figure 2.4). What could also be seen was that the rate limitinghydrolysis can effectively block some of the reaction pathways by beingprohibitively expensive to cross, leading to the preferred reaction havingto pass through a different pathway. While being just of casual interestfor this study, it opened a possible explanation for the kinetics with othersubstrates.132
Mutant effectsThe last test of the EVB model was the correct reproduction of the effectof several characterised active site mutants other than the E35Q andH300N mutants mentioned above.124,211 The correct modelling of thetyrosine mutants of the enzyme, as well as the effect from residues involvedin a water channel leading to the active tyrosine, would further cement
59

Pathwaydeprotonated H104 protonated H104
ExperimentΔG‡ ΔG0 ΔG‡ ΔG0
(R,R)–TSOC1 13.0±1.7 −8.7±1.9 18.3±1.1 3.4±1.6
14.4C2 11.6±1.3 −8.1±1.3 14.7±1.0 0.2±1.4
(S,S)–TSOC1 12.1±2.1 −6.2±2.6 15.2±0.6 2.2±1.0
16.0C2 13.6±2.1 −8.7±2.2 16.9±0.8 0.4±1.1
Table 5.2. Calculated free energy values observed for the StEH1 wild–typeenzyme with different H104 protonation states. All energies are for the firstreaction step in the mechanism from Figure 2.4, given in kcal/mol and averagedover 10 independent calculations. Adapted with permission from Amrein etal.131
the accuracy of the method. Our results reproduce the observed mutanttrends in all cases (Figure 5.2), indicating the our way of modelling thereaction is sufficiently accurate to capture those effects.
ConclusionsThe work on the TSO hydrolysis in StEH1 showed that the mechanismproposed earlier126 can be validated using the EVB model employed inour work. The calculations show that the selectivity for a given reactionpathway is not always defined at the alkylation step of the reaction, butthat the hydrolysis step can act as a kinetic barrier and lead the reactionthrough a different path. What was also possible to see is that the pro-posed function of the active side tyrosine residues as a second oxyanionhole can be reproduced using the computational approach, in agreementwith earlier spectroscopic experiments.122 The most striking observationis that the two residues in close proximity to the active site, but notinvolved in the actual mechanism, have a striking influence on the selec-tivity of the enzyme. This observation may open up new strategies forenzyme engineering in the future.
60

0
5
10
15
20
25
WT Y149F Y154F Y235F E35Q H300N
0
5
10
15
20
25 (R,R) TSO
(S,S) TSO
Figure 5.2. Calculated activation energies for the hydrolysis of TSO in several StEH1variants. All energies are averages over 10 independent simulations, given in kcal/mol.Calculated value are in lighter color, compared to the experimental values using thedarker color. Reproduced with permission from Amrein et al.131
5.3 Investigation of SO binding modes andenantioconvergence (Paper III)
Following the first study to establish the EVB method for the study ofthe StEH1 enzyme, it was decided to tackle a more challenging problem,the study of the enantioconvergence with the smaller substrate styreneoxide (see Figure 2.5 for a comparison between the substrates). Thisstudy was deemed to be more difficult as compared to TSO case due tothe anticipated possibility of multiple binding modes (Figure 5.3). It wasdecided to modify the previously used EVB model to account for theremoval of one of the phenyl rings of the substrate and its replacementby a hydrogen atom, leaving the parameters largely unchanged for theremaining system. The initial point of the calculations was also changedto start from the Michaelis complex to allow the substrate to explore morepossible orientations available to the general binding modes.
61

O
OO-
D105N
NH
H300
NH
W106
O
OO-
D105N
NH
H300
NH
W106
Figure 5.3. Different binding modes for the smaller substrate styrene oxide. Thephenyl ring of the substrate can either interact with the imidazol of histidine 300(Mode 1), or the indol of tryptophan 106 (Mode 2). Adapted from Bauer et al.212
Enantioconvergence in the native enzymeThe kinetic studies on StEH1 showed that the enzyme behaves fully enan-tioconvergent when hydrolysing racemic SO.18,138 The exact cause behindthis behaviour has not been fully understood, but is hypothesized to becaused by the different binding modes available to the substrate.132,138
Finding the reason for the enantioconvergence on the molecular levelwould make more rational design strategies feasible for the further de-velopment of the biocatalytic ability of the enzyme.
Available substrate binding modesWhen studying the two possible binding modes for the substrate, wefound that both systems showed similar stabilities in the boundaries ofour simulation approach for binding in the Michaelis complex. Interest-ingly, analysis of the calculated activation energies showed that thoughone binding mode might be preferred for the alkylation step and allowthe formation of the covalent enzyme intermediate, the reaction becomestrapped by a high barrier for the second step of the reaction and effec-tively traps any substrate bound in this orientation, shown in Table 5.3for the native enzyme. The second binding mode does not show this kindof selectivity barrier and allows the reaction to proceed to the product.
62

Rea
ctio
nSt
ep(R
)–SO
,Mod
e1
(R)–
SO,M
ode
2(S
)–SO
,Mod
e1
(S)–
SO,M
ode
2C
–1C
–2C
–1C
–2C
–1C
–2C
–1C
–2W
ild–t
ype
StEH
1
Alk
ylat
ion
ΔG
‡24
.3±1
.319
.1±0
.719
.5±0
.417
.5±0
.511
.7±0
.215
.8±0
.415
.0±0
.617
.2±1
.2Δ
G0
9.0±
0.3
−7.1
±0.7
7.1±
0.4
−8.2
±0.5
−12.
3±0.
4−1
0.4±
0.5
−4.8
±1.7
−1.4
±3.1
Hyd
roly
sisΔ
G‡
14.9
±1.7
12.8
±1.3
12.6
±0.6
11.9
±1.7
19.3
±0.7
13.6
±1.2
15.1
±0.9
12.2
±1.7
ΔG
08.
7±1.
96.
0±1.
34.
9±0.
94.
5±1.
513
.3±0
.88.
0±1.
58.
1±1.
26.
5±2.
0R
-C1B
1m
utan
t
Alk
ylat
ion
ΔG
‡20
.3±0
.516
.3±0
.427
.3±0
.723
.0±1
.115
.6±1
.019
.8±0
.517
.6±1
.519
.7±0
.9Δ
G0
8.1±
1.1
−8.0
±1.5
18.4
±1.0
0.7±
1.1
−9.2
±0.9
−6.3
±0.6
−3.8
±1.5
−7.1
±1.2
Hyd
roly
sisΔ
G‡
9.0±
1.1
9.0±
1.1
8.1±
0.5
10.8
±1.0
15.0
±0.7
9.8±
1.2
12.8
±1.4
10.9
±1.5
ΔG
0−0
.1±0
.5−0
.7±1
.7−1
.9±1
.02.
3±1.
58.
1±1.
23.
5±1.
34.
8±1.
75.
2±1.
7
Tab
le5.
3.O
verv
iew
over
the
calc
ulat
eden
ergi
esfo
rth
etw
ore
actio
nst
eps
inve
stig
ated
.A
llen
ergi
esar
epr
esen
ted
inkc
al/m
olan
dar
eca
lcul
ated
asth
eav
erag
eov
er10
inde
pend
entf
ree
ener
gyca
lcul
atio
ns,w
ithth
eer
ror
bein
gth
est
anda
rdde
viat
ion.
Ada
pted
from
Bau
eret
al.21
2
63

Again, as before for the larger substrate, the hydrolysis step thus be-comes the deciding point for the reaction outcome, not the actual forma-tion of the covalent enzyme intermediate. We note that an independentstudy by Lind and Himo213 employed DFT to study the mechanism ofthe same StEH1 system after an earlier study on LEH.214 The resultsare in general agreement with our study, even though the authors used adifferent description of the active site together with partial protonation ofthe epoxide during the ring opening reaction. There are, however, issueswith such a representation and such calculations need to be carefully in-vestigated due to known issues with the DFT method employed to modelthe reaction.215,216
Probing of mutant effectsWe were also interested in studying the mutant effects observed for avariant found during older directed evolution studies of StEH1.18 To-gether with a new crystal structure, the EVB approach has been used tostudy the quadruple mutant, showing a shift in regioselectivity when hy-drolysing SO. Our calculations show that the mutant shifts the preferredbinding mode for the hydrolysis of both substrate enantiomers (Table5.3). In case of the (S)–SO enantiomer, this is achieved by unblockingthe hydrolysis pathway in the first binding mode, while the (R)–SO sub-strate can be effectively hydrolysed through a reduction of the free energybarrier of the first reaction step. It has to be noted that the activationenergy for the inversion reaction with the (R)–enantiomer is still higherthan expected from the experimental product ratio, likely due to issueswith the difficult parametrization of the reaction. Still, the pronouncedshift from one binding mode to another, together with the experimentalobservation of no intermediate accumulation, is a good indication thatthe actual reaction proceeds in this fashion.
Interaction differences during catalysisAn investigation of the interactions contributing to catalysis for the pre-ferred pathways showed that the stabilising interactions from the twoactive site tyrosine residues were extremely different between the two
64

substrate enantiomers, as well as between the two tyrosine residues them-selves (Figure5.4). This could also be observed in the investigation of theinteraction distances between the tyrosine oxygen atoms and the epoxideoxygen, with Y154 showing stable interactions during molecular dynamicsequilibration, while Y235 only interacts later over the course of the firstreaction step, and thus showing stabilising interactions.
0 50 100 150 200 250 300 350–10–8–6–4–2
02468
Residue Number
ΔΔG
‡ elec
(RS
>TS)
(R) SO, C 2(S) SO, C 1 Y235
W106
E35
F33
H300
D265Y219
Y154
Figure 5.4. Plot of the electrostatic group contributions of the wild–type enzymetowards the reacting region, calculated for the preferred pathways first reaction stepof SO hydrolysis, in kcal/mol. Adapted from Bauer et al.212
Intermediate stability relates to fluorescence signal strengthOne challenging issue in performing the calculations was the problem ofthe large stabilisation of the formed covalent intermediate relative to theMichaelis complex. We could show from the experimental observationsthat the affinity for the covalent alkyl–intermediate is much stronger forthe (S)–enantiomer compared to the (R)–enantiomeric form, in line witha greater stabilisation of this state being predicted in our calculations.While our calculations are still clearly overestimating this stabilisation,this is an indication that the issue lies within the energetics of the ref-erence reaction as estimated by the DFT calculations and are not anintrinsic problem of the EVB model.
65

ConclusionsThe study of the smaller substrate SO showed that although the enzymecan successfully bind the substrate in different orientations, only the bind-ing mode showing interactions with the conserved tryptophan can leadall the way to products in the wild–type enzyme. This was surprising, asit was expected from structural considerations that the enzymatic enan-tioconvergence stems from binding the substrate enantiomers in differentorientations. It could also be seen that this preference is lost upon re-moval of the tryptophan in an active site quadruple mutant. The muta-tions main effect is from effectively unblocking the second reaction stepfor the binding mode involving the active site base, making it possible toform the products by this pathway.
5.4 Study of substrate flexibility for(2,3–epoxypropyl)benzene (EPB) (Paper IV)
The previous studies of StEH1 directed evolution generated a large num-ber of interesting variants with new kinetic profiles.17,18 Crystallographicstudies have been performed to find structural explanations for the ob-served changes in substrate and regioselectivity. These new structureswhere then used to analyse the binding of the substrate used as the evo-lutionary selection pressure, (2,3–epoxypropyl)benzene.
Challenges with binding studies involving EPBAs the Michaelis constants for EPB have been estimated to be high com-pared to the other substrates (see Janfalk Carlsson et al.18), with a fastfirst reaction step as indicated by an inability to capture the burst phaseof the signal for the formation of the covalent enzyme intermediate, itwas expected to be difficult to study binding effects and chemistry usingmolecular dynamics. When it was attempted to perform full atomisticsimulations of the substrate in the active site, it was not possible to ob-tain stable conformations in the active site. As similar difficulties wereexpected when performing studies involving EVB, we investigated if moresimple methods might be used to study different binding conformations,
66

without the problems involved with sampling all available binding con-formations. The final approach that we decided on has been the use ofmolecular docking, with the added constraint that the active site tyrosineresidues need to be in hydrogen bond distance to the epoxide oxygen.The method of choice has been Glide docking,217 allowing full flexibil-ity of the side chains in the active site. Even though docking methodscan be limited in accuracy concerning the interaction strength,218 theycarry sufficient information to investigate the possible conformations ofsubstrates in the active site.172 Due to this, it was decided to use dockingto investigate the enzyme variant dependent substrate positioning.
New structures for StEH1 variantsSeveral new enzyme variants had been characterised during the courseof the previous work on generating new StEH1 variants using directedevolution.18 The crystal structures of several of those variants have beensolved to investigate the changes in the active site upon mutation, to-gether with the singular new structure published before in combinationwith the work on styrene oxide.212 Analysis of those shows that the ter-tiary structure of the enzyme does not change to a noticeable degree,with RMSD values of around 0.2 to 0.5 for the pair–wise comparison ofthe C–α of the variants. The active sites also only vary to small degreesbetween the variants, with a slight increase in active site volume beingobserved when comparing the wild–type enzyme to the mutants (Table5.4), and only limited changes in shape (Figure 5.5).
VariantResidue position Active site volume
106 109 141 155 189 266 Å3
wild–type W L V I F L 2065R–C1 W L K V F L 2225
R–C1B1 L Y K V F L 2222R–C1B1D33 L Y K V L L 2258
R–C1B1D33E6 L Y K V L G 2506
Table 5.4. Active site volumes for the different variant crystal structures.Reproduced from Janfalk Carlsson et al.219
67

This slight increase caused a reshaping of the active site cavity, asshown in Figure 5.5 in columns A and B.
Figure 5.5. Active site cavity and primary docking position found for the EPB sub-strate in the different enzyme variants. Reproduced from Janfalk Carlsson et al.219
68

Docking results point to different binding modesThe docking results for all the new crystallised variants show that EPBexplores slightly different binding modes compared to the previous studyof the substrate styrene oxide. This is very likely caused by the increasedflexibility of the methylene group between the phenyl ring and the epoxidegroup. Comparing the structures, we can see that there is satisfactorycorrelation between the distance of the nucleophile towards the preferredepoxide carbon for the ring–opening reaction (Figure 5.5 C).
5.5 Investigation of reduced regioselecivity whenhydrolysing MeSO (Paper V)
One major remaining puzzle in understanding the kinetics of StEH1 hasbeen the observation that the hydrolysis of trans–methylstyrene oxideleads to the observation of two distinct rates in the spectroscopic measure-ments for the formation of the covalent intermediate when hydrolysing(S,S)–MeSO, while only one hydrolysis product is being observed.19,132,220
At the same time, the hydrolysis of (R,R)–MeSO shows only one signalfor the formation of the intermediate, but two products are observedwhen analysing the reaction outcome. The general explanation for thisobservations has been a branched reaction scheme (Figure 5.6), but theexact causes have been unknown. It was decided to test the hypothe-
E'
E
E'S
ES
E'-alkyl
E-alkyl
E' + diol
E + diolKS, [S]
KS', [S]
k2
k-2
k6
k-6
k3
k7
k0k-0 k-5 k5
Figure 5.6. Proposed kinetic mechanism for the hydrolysis of MeSO in StEH1. Thecrossing between the states ES and E´S has been invoked to explain the observedhysteresis when hydrolysing the substrate.19
sis of the branched reaction pathway by employing the EVB method to
69

study MeSO hydrolysis in StEH1, with emphasis on the different bindingmodes studied for the smaller substrate SO, following a similar approachto perform the calculations.
Sampling of binding conformationsAs the study of SO showed that the convergence of the calculations islargely dependent on the stability of the initial substrate binding posi-tions, the computational approach was modified to allow more extensivesampling of the Michaelis complex. To this end, all individual energy cal-culations were performed after fully relaxing a system with independentinitial conditions before conducting EVB calculations for the two studiedsteps of the mechanism. This allowed the sampling of more individualstates while still giving convergent results for the free energy calculations(Table 5.5). The same calculations were also performed for one of thepreviously studied mutants of the enzyme showing a shift in the hydrol-ysis behaviour,18 again being able to identify the main pathways leadingfrom the substrate to the products (Table 5.5).
VariantPathway and Reaction Step
ΔG‡ C1 Step 1 ΔG‡ C1 Step 2 ΔG‡ C2 Step 1 ΔG‡ C2 Step 2Calc Exp Calc Exp Calc Exp Calc Exp
WT (R,R)–MeSO 15.1±0.5 n.d. 17.8±0.3 16.8 13.5±0.7 n.d. 18.1±0.3 16.8WT (S,S)–MeSO 12.2±0.4 14.2 16.5±0.2 14.9 13.3±0.3 14.2 20.9±0.2 16.5
R–C1 (R,R)–MeSO 10.9±0.3 n.d. 16.1±0.3 15.8 9.8±0.4 n.d. 14.1±0.2 15.8R–C1 (S,S)–MeSO 9.9±0.2 13.1 14.8±0.2 15.1 10.4±0.4 13.1 19.6±0.3 15.8
Table 5.5. Calculated energies in kcal/mol for the favoured pathways leadingto product formation for both the native and mutant StEH1 enzyme. The errorsare given as the standard error of the mean over 30 independent calculations.
Branched reaction pathways blocked at intermediate hydrolysisThe results of the calculations showed a striking explanation for the obser-vation of the differences between observed rates and products. Dependingon the chosen substrate, the enzyme again can initially follow a number ofreaction pathways towards the covalent intermediate (Figure 5.7). Oncethe alkyl–intermediate is formed, only one, in case of (S,S)–MeSO, ofthose covalent intermediates can become hydrolysed to product, while theother species are kinetically trapped. In case of (R,R)–MeSO, the differ-
70

Wild-Type
R-C1
(R,R) - MeSO
(S,S) - MeSO
E'
E
E'S
ES
E'-alkyl
E-alkyl
E' + diol
E + diol
KS', [S]
k2
k-2
k6
k-6
k3
k7
k0k-0
[S]
E'
E
E'S
ES
E'-alkyl
E-alkyl
E' + diol
E + diol
KS', [S]
k2
k-2
k6
k-6
k3
k7
k0k-0
[S]
E'
E
E'S
ES
E'-alkyl
E-alkyl
E' + diol
E + diol
KS', [S] k6
k-6
k3
k7
k0k-0
[S]
E'
E
E'S
ES
E'-alkyl
E-alkyl
E' + diol
E + diol
KS', [S] k6
k-6
k3
k7
k0k-0
[S]
Wild-Type
R-C1
Figure 5.7. Schematic depiction of available and blocked reaction pathways for thehydrolysis of MeSO enantiomers in the StEH1 variants.
ences in observed energies for the first and second reaction steps indicatethat formation of the alkylenzyme intermediate might be rate limiting forone of the pathways, meaning that no signal would be expected to be ob-served for this step, while the hydrolysis pathways towards both productsshow strikingly similar energetics, leading to the observed, almost equalproduct ratios.18
ConclusionsOur calculations show that the experimental observations for the hydrol-ysis of MeSO fits into a similar model as has been deduced for the smaller
71

substrate SO212 that had already been hinted at in the first EVB studyof the large substrate TSO.131 The surprising finding has been that theenergy barriers needed to cross the non–favoured pathways are more pro-nounced for this substrate as compared to SO, giving further indicationthat the initial proposal for the branched reaction scheme for MeSO hy-drolysis proposed by Lindberg et al.132 is indeed a valid explanation.This opens further avenues for the engineering and modification of thoseindividual pathways, aimed at blocking or unblocking selected pathways.
72

6. Method development for chemical modelling
6.1 Divalent transition metal models (Paper I)A large number of proteins are dependent on metal ions bound to theprotein structure, acting either as anchors to stabilise the protein,221 oras active partner in the chemical mechanism in the active site.221 Tosuccessfully study metal ions, both the coordination chemistry and theelectrostatic interactions need to be physically accurate, as an inaccuratedescription will lead to simulation artefacts.222
Bonded metal modelsA possible approach to model transition metal ions in classical simu-lations is the use of explicit bonds within the actual metal centre. Inthose representations, the ligands surrounding the ion are connected toit through explicit harmonic bonds and angles to keep the coordinationgeometry constant.223 In addition to this, charges and van der Waalsinteractions can also be readjusted to fit QM calculations of the metalcentre in question.224 The advantage of this kind of model is that the ex-act coordination geometry can be reproduced, as well as the exact chargesfrom the QM model. The main issue here is that the model will be limitedto only the one metal centre that has been parametrized in QM, meaningthat each new ligand sphere will need a new parametrisation.
Soft sphere modelsA more simple approach is the representation of the metal as a van derWaals sphere with the appropriate charge, with parameters such as to re-produce both the correct solvent coordination sphere and electrostatics.225
An improvement to this approach has been the development of the socalled 12 − 6 − 4 models226 that added an extra term to the usual rep-resentation of the London interactions as the 12 − 6 potential.164 This
73

allows more fine tuning of the interactions. The advantage of both ap-proaches are that they are relatively easy to parametrise and transferablebetween different systems having different ligand spheres. A problem ofthe original soft sphere representation for transition metals is that thesize of the ligand sphere and the electrostatic properties do not followa linear trend for them due to crystal field effects.227 Then, the simplemodels will fail to represent either the geometry of the metal centre orthe electrostatics of it.228 The problem for the 12 − 6 − 4 model is thatit requires modifications to existing software to include the calculationof the extra term, making it impossible to use this in approach in anymolecular dynamics software that has not been adapted to it.
Dummy models for transition metal ionsThe dummy model originally proposed by Åqvist and Warshel,229 andlater refined by the Warshel group,230 simulated transition metals bysurrounding the central metal particle by a set of particles only interactingthrough their charge. A schematic representation is shown in Figure6.1. Those “dummy” atoms are linked to the central particle carryingthe remaining charge and providing the van der Waals interactions tothe ligands. A main feature of the metal model is that it can re–orientfreely to interact with the metal ligands, just like the case of the softsphere model or metals described using modified van der Waals potentials,while still being able to reproduce the correct geometries and interactionenergies229,230 and not needing extensive work on the simulation softwareto incorporate the new potential form.226
Improved dummy model parametersBased on the original dummy model, we decided to re–evaluate the pub-lished parameters for the manganese (Mn2+), magnesium (Mg2+) andzinc (Zn2+) ions, as well as extend the model to nickel (Ni2+), iron (Fe2+)and cobalt (Co2+) and provide a dummy model for calcium (Ca2+).Based on the shared geometric parameters and charge distributions shownin Table 6.1, we fitted the van der Waals parameters of the central par-ticle to reproduce both the radial distribution function in water, and thecalculated solvation free energy. One challenge here had been the se-lection of an appropriate reference set for the solvation free energies, as
74

Figure 6.1. Schematic depiction of the octahedral dummy model. The central par-ticle M is surrounded by a shell of 6 dummy particles that only interact throughelectrostatic interactions. The dummy particles are connected to the metal throughharmonic springs and angles, with the complete model allowed to rotate freely.
different references provide largely different values even for simple casessuch as the Mg2+ ion.231–233 We decided to continue to use the extensivetabulated values of Noyes, as they were used in the original parametri-sation of the dummy models and are consistent with the values obtainedby Rosseinsky.233 Our parameters provided excellent fits both for the ge-ometry of the metal and its ligands, as well as for the hydration usingdifferent water models.
Atom Mass Charge AvdW BvdW
Calculated ExperimentalΔGsolv M2+ – O ΔGsolv M2+ – O
Dummy 3.00 +0.5 0.05 0.00 – – – –Ni 40.69 −1.0 113.00 84.00 −451.9±0.2 2.13±0.04 −451.8 2.12Co 40.93 −1.0 61.00 31.00 −492.7±0.1 2.06±0.03 −492.8 2.06Zn 47.39 −1.0 68.00 38.00 −480.5±0.1 2.08±0.03 −481.0 2.08Mn 36.94 −1.0 171.00 35.00 −483.4±0.1 2.08±0.03 −483.3 2.08Fe 37.85 −1.0 70.00 10.00 −436.9±0.2 2.19±0.03 −436.4 2.20Mg 6.30 −1.0 63.00 9.00 −454.4±0.1 2.12±0.04 −454.2 2.10Ca 22.08 −1.0 350.00 15.00 −379.9±0.2 2.38±0.02 −379.5 2.39−2.46
Table 6.1. Parameters, resulting calculated and experimental observablesfor the different metal models resulting from this study. All charges arein values of elemental charge, the van der Waals parameters are given inkcal1/2 ·mol−1/2·Å6 for AvdW and kcal1/2 ·mol−1/2·Å3 for BvdW . Energies inkcal/mol and distances in Å are calculated over five independent simulations.Reproduced with permission from Duarte et al.234
75

Modelling protein–dummy model interactionsAfter the reproduction of the behaviour in water, the real test of themodel was to introduce it in different protein systems. For this firststudy, human and Escherichia coli Glyoxylase I had been chosen as thetest systems, as previous studies had shown selective interactions withalmost all of the newly parametrised metals.235 Our classical moleculardynamics simulations were able to reproduce the key metal–ligand dis-tances observed in the crystal structures for both the studied proteinsystems, giving further evidence for the robustness of the models.
ConclusionsThe dummy model could be shown to be an extremely valuable tool forthe simulation of metal proteins. In addition to the work done usingthose models in EVB studies,134,136,236,237 several other groups have alsocontinued work on them238–240 to use those simple models to study metalproteins. Still, the used models are only an approximation of the ac-tual interactions between a metal and the surrounding ligands, as anyreal description would need to take into account the polarization andcharge transfer effects that take place. The models are a starting pointfor further improvement of the way classical models are able to treatthose issues, where further improvements in force field development,241
especially towards fully polarisable force fields, might lead to even betterdescriptions.
6.2 Updates to the Q simulation program (Paper VI)While there are several standard molecular simulation packages availabletoday,242–244 there is still the need for specialised software to performcertain kinds of simulations.182,183,245 One such program is the Q packagedeveloped originally by Åqvist and co–workers246 to perform EVB andFEP simulations. The aim of this study was to improve the current stateof the program, and implement new methods to perform and analysethose simulations.
76

New methods to study protein chemistry using EVBOne challenging aspect of studying chemical reactions is the exact quan-tification of nuclear quantum effects. The observation of the substitutioneffects from exchanging isotopes of a certain elements in chemical com-pounds has been used for decades in the study of transition states andreaction pathways.47,48 The calculation of those effects has been challeng-ing due to the computational limitations of the different QM methods thatcan be employed to evaluate them. One possibility to quantify the effectshas been the use of path integral methods.199,200 Those approaches havebeen proven to be accurate in determining the quantum mechanical ener-gies of systems, but come at the price of increased computational costs as-sociated with sampling the free particle distributions of the particles beingstudied. One approach to reduce the computational cost has been the useof classical particle distributions or positions followed by the calculationof the path integral energy from the free particle distribution centred onthe classical position.201,202 The centroid bisection method for the quan-tum classical path approach first presented by Hwang and Warshel201 hasbeen implemented as both a post processing method and as an additionalenergy calculation routine during dynamics in the Q software. The freeparticle sampling by the bisection method as presented by203,204 has beenchosen to allow efficient exploration of free particle space, with the energyanalysis being performed using the EVB approach.
Implementation detailsThe QCP approach has been implemented as an additional module avail-able when performing energy calculations in the Q program.246 If theuser requires the calculation of path integral corrections, additional en-ergy calculations are performed at the time when the classical energiesare saved during the simulations, or during the post–processing of coor-dinate files. Here, the classical coordinates are first transformed to thebead representation as in Major and Gao204 and Major and Gao,203 usingthe de Broglie wavelength Λ to calculate the particle density depending
77

on the number of beads P .
Λ =(
βh2
2mP
)1/2
(6.1)
This is done for each bead i in an iterative fashion.
ρ(xi,xm,β/P ) =( 1
4πΛ2
)1/2e(xi−xm)2/(2Λ2) (6.2)
The position xm is then based of the positions of the surrounding beads.
xm = xi−1 +xi+12 (6.3)
The QM energy in the QCP method is taken as the average over boththe classical coordinate ensemble and the free particle ensemble obtainedfor the ring polymer.201 In the program, the current coordinates are firsttransformed into the ring polymer beads according to the atom mass m
and the free particle distribution equilibrated for a number of steps de-fined by the user. Afterwards, energies are calculated for each bead posi-tion, also scaled according to a user defined isotopic mass if desired.
Elimination and methyl–transfer reactions as test caseAs test cases for the new function, the E2 elimination of halo-alcoholsin ethanol,247 and the transmethylation of of S–adenosyl by a catecholsubstrate in catechol O–methyltransferase were studied.248,249 The calcu-lated and experimental kinetic isotope effect are presented in Table 6.2.The method proves to be accurate in determining KIE values for thosetwo different cases and is hoped to be useful in all kind of studies involvingisotope effects.
Calculation of virtual group deletionOne approach to quantify the effects of the active site residues towardscatalysis has been the concept of calculating the individual residue “groupcontributions” at the different stationary points and using methods suchas the linear response approximation to calculate the actual free energies.196
A problem of this approach has been that it only involves a small num-ber of points sampled to calculate the energies from. This could lead toconvergence problems and might not carry sufficient information aboutthe magnitude of the individual residue contribution towards catalysis.
78

System ΔG‡light ΔG‡
heavy KIEcalc KIEexp
COMT 16.0±0.2 15.9±0.2 0.80 0.79Dehalogenation 20.0±0.1 21.3±0.1 8.7 7.1
Table 6.2. Test of the QCP calculation implemented in the new Q softwareversion. Energies are presented in kcal/mol. The calculation for the COMTsystem involved 50 replicates, with 20 data points at each classical configura-tion and 64 quantum beads for each classical particle, spread over 51 samplingwindows. Each classical configuration was sampled again over 50 free particleconfigurations. In case of the E2 elimination, the approach was changed, using10 replicates with 500 classical data points and 8 quantum beads, again spreadover 51 sampling windows. The free particle distribution was sampled 10 timesfor every classical configuration.
Implementation detailsThe new routine has been added in such a way that an arbitrary numberof residues or atoms can be specified that should be excluded from anadditional energy calculation. When the classical energy is saved, thoseatoms are passed to an additional energy calculation, but for the subtrac-tion of their contribution.
Etotal =Eqq,el +Eqq,vdw +Eqp,el +Eqp,vdw
+Epp,el +Epp,vdw +Ebond +Eangle +Etorsion
Eexclude =ET otal −Eq−excqq,el −Eq−exc
qq,vdw −Eq−excqp,el −Eq−exc
qp,vdw
(6.4)
Only the non–bonded interactions are removed in this case, leading toan idealised system where the residue does not interact with the reactionregion, as would be the case for a glycine mutant.
Epoxide hydrolase test systemAs an example for the use of this routine, the residue contribution ofthe two active site tyrosine residues in StEH1122 are calculated for thefavoured pathway at the epoxide ring–opening step (see Figure 2.4). Thefinal values for the estimated and explicitly calculated differences to theactivation free energy are shown in Table 6.3, indicating that the methodcan be useful to quantify the catalytic contributions in cases like this.
79

System Y154F Y235F Y154F/Y235FΔΔG‡
est 0.22 1.86 2.09ΔΔG‡
calc 1.5 3.7 NA
Table 6.3. Group contribution estimated (est) and explicitly calculated (calc)activation free energy differences for the epoxide ring opening reaction of(R,R)–TSO. Explicitly calculated values are those reported in Amrein et al.,131
with energies given in kcal/mol. NA – not explicitly calculated.
Algorithmic improvementsIn addition to the new methods mentioned above, we also updated a num-ber of the implemented algorithms in the software, adding support for theLINCS250 constraint algorithm, support for arbitrary temperature coup-ling groups and new thermostats. The inclusion of LINCS will make itpossible to improve the performance of the code when using constraints onsolute atoms, while the addition of arbitrary temperature scaling makes itpossible to scale ions together with the solute, improving the behaviour ofsimulations under periodic boundary conditions with counter ions. Theaddition of the Langevin165 and Nose–Hoover165 thermostats can alsoimprove the behaviour of simulations needing more refined coupling to atemperature bath.
Support for additional solventsThe solvent handling has been improved to add support for solvent mo-lecules of any size and number of atoms (under the limitation that theymay not be larger than the cut–offs employed). The parameters of sev-eral solvents available for the OPLS–AA force field in Gromacs244,251,252
have been converted to the format used in Q, with the results for ethanolshown in Figure 6.2. The drop in density at the edge of the calculationsphere is related to the polarisation correction246 that is not available forthe more complex solvents.
ConclusionsA new version of the Q simulation software is being presented with anumber of newly–implemented methods and updates to the existing al-
80

0 5 10Distance [Å]
0
0.5
1
1.5
2
OH-OHOH-HOHO-HO
Ethanol, 30Å sphereRadial distribution functions
10 20 30Radius [Å]
0
0.005
0.01
0.015
Num
ber d
ensit
y
g(r)
Sphere density2Å shell densityTarget density
Ethanol SCAAS density2 ns sampling
Figure 6.2. Plotted radial distribution functions (A) and solvent densities at differentradii in the solvent sphere (B).
gorithms. The hope is that this will lead to a better performance ofthe software, as well as the ability to study a large number of problemsefficiently using the FEP and EVB approaches.
81

7. Concluding Remarks
The study of biological systems using computational models is now justone part of the full investigation of new enzymes, if the mechanism ofaction has to be understood. Explaining the reason for the selectivityand efficiency is needed for the successful engineering of new enzymevariants and to see the system in respect to its natural function. Whileexperimental methods are able to determine the reaction rates and helpto distinguish between different mechanisms, they are often not able toquantify the how exactly the rate enhancements are achieved. Compu-tational methods have become more and more established in the recentyears to bridge this gap in our knowledge about enzyme function andmade it possible to both quantify and visualise the effects that the en-zyme surrounding has on the catalysed reaction. One challenge that stillremains is addressing the problem that observed differences in enzyme ac-tivity often relate to only small changes in the energetics of the reactions,making it necessary to use methods that are able to distinguish thosedifferences in energy. The studies presented here show that the EVBmethod is both able to describe the difficult chemistry of the investigatedsystems, while still successfully differentiating between the different sub-strate forms. The investigation of the StEH1 enzyme has shown thatthe observed regio– and enantioselectivity is an intrinsic property of thesystem. Even the largest of the studied substrates exhibits the selectivityobserved for the smaller substrates, an observation that had not beenpreviously possible with the experimental methods. A combination ofsequence and computational chemistry has shown that two residues lo-cated next to the general base residue have major impact on the catalysisby modulating the charge balance in the active site. In addition to this,the viability of the proposed reaction mechanism involving the negativelycharged epoxide oxygen atom next to the active site tyrosine residues has
82

been validated with the used models, showing the correct change in ac-tivity upon in–silico mutation. It is hoped that the new insight into thesystem will make it possible to further extend the previous attempts toengineer the enzyme towards improved enantioselectivity, with the com-putational models as basis for experimental studies.
The kind of calculations needed to perform this kind of study are stillcomputationally expensive due to the extensive sampling needed to obtainconvergent free energy profiles. The work on method development wasaimed at both providing precise models for the challenging calculationsinvolving transition metals, as well as improving the software used toperform the calculations. The inclusion of new methods to study chemicalreactions and the improvements in the algorithms used for the simulationof molecular dynamics is hoped to extend the reach of the computationalapproach and make the simulations more accessible for a wider audience.
83

8. Populärvetenskaplig Sammanfattning påsvenska
Enzymbiokatalys är ett av de aktuella forskningsämnen som fångar intres-set av såväl de forskargrupper som söker förstå hur enzymer fungerar, somav industrin på jakt för att hitta billigare alternativ till de katalysatorersom vanligen används i organisk syntes. Det gemensamma behovet avbåda fälten är att förstå hur katalysatorerna fungerar för att kunna för-bättra dem och utöka omfattningen av deras användning.
Att förstå hur enzymer fungerar har varit målet för biokemister sedanforskningsområdets födelse, med experimentella och teoretiska tillväg–agångssätt som förbättrar och förstärker vår kunskap om deras natur.Experimentellt arbete har alltid varit begränsat till vad som kan ob-serveras i laborationsmiljö, medan teoretisk forskning på katalysens naturhar begränsats av noggrannheten i metoderna och deras relation till deexperiment som de försöker beskriva. I idealfallet leder detta till ett direktförhållande mellan de två tillvägagångssätten och fullständig förståelse förkatalysatorn som undersöks. Men då naturen är mer komplicerad kan imånga fall bara delar av pusslet samlas in som då måste kombineras sågott det går för att bygga den slutgiltiga bilden, även om vissa bitar kansaknas.
En familj av enzymer som har varit av intresse har varit de som kanbryta ner föreningar genom tillsats av vattenmolekyler, så kallade hydro-lasenzymer. Det finns en mängd olika former av dessa enzymer som kangrupperas i ett antal olika familjer utifrån hur de är uppbyggda, och där-för antas använda samma mekanismer för att utföra sina reaktioner. Enav dessa familjer heter α/β hydrolasfamiljen, där alla enzymer är byggdaenligt samma generella struktur.
84

En mindre antal av detta större enzymkonglomerat har varit av intresseför att de kan producera vissa kemikalier, kallade vicinala dioler, som kananvändas som utgångsmaterial för att producera läkemedelsföreningar.Dessa enzym (epoxid hydrolas, EH) enzymer har också varit av intressepå grund av deras specifika verkningsmekanism, vilket involverar flera dis-tinkta kemiska species innan den slutliga produkten har bildats. Exper-imentella studier har spridit ljus över det generella sättet dessa enzymerfungerar, men har utelämnat några saknade ”pusselbitar“ som varit svåraatt förklara från endast de experimentella observationerna.
Huvuddelen av arbetet i denna avhandling handlar om beräkningsstudienav dessa enzymer, vilken syftar till att fylla i fler delar av pusslet genomatt utföra beräkningar på möjliga reaktionsmekanismer som leder till pro-dukterna. Studierna förklarar hur enzymet kan välja att följa vissa vägaroch undvika andra, och hur dessa mönster förändras vid förändringari enzymet självt. Den huvudsakliga modellen som har använts för attundersöka detta beteende tillät oss att både observera de kemiska reak-tionerna som skedde och hur enzymerna reagerar på förändringar genomatt simulera så många olika strukturer som möjligt.
Vårt arbete har visat att detta tillvägagångssätt leder till en bättre för–ståelse för hur reaktioner händer i detta system. En annan del av studiernahar varit att förbättra programvaran som används för att utföra beräkning-arna. Även om det generella tillvägagångssättet inte har behövt förän-dras sedan det utformades så har några av de ytterligare metoderna ochkoncepten betydligt förbättrats, speciellt när det kommer till beräkning,prestanda och metodomfattning. I förlängningen av detta har både nyamodeller att utföra storskaliga beräkningar, liksom nya simuleringsme-toder introducerats.
85

9. Acknowledgements
The work in this thesis would not have been possible without the helpfrom numerous people, both at Uppsala University and beyond. I wantto try to thank all of them here, but please do not be upset if I forgotsomeone!
The first person to thank is my supervisor,Prof. Lynn Kamerlin (aka The boss). Thank you for taking the riskin having me as your student, even though I was a starting experimen-tal biochemist that did not even had his master studies finished! Thankyou for allowing me to try out work on different projects (even though anumber of them never did work out in the end) and trusting me enoughto work on the program code everyone depends on. I also want to thankyou for allowing me to visit different groups outside of Uppsala during mytime as your student, giving me the opportunity to broaden my horizons!
Following this, I also want to thank my co–supervisor,Prof. Mikael Widersten (aka The other boss). Thank you for allow-ing me to still be around the experimental lab and to be able to ask youat every time I needed help with connecting my research to experimentaldata.
Fernanda, thank you for guiding me during the first years of my PhD,you have been the best Postdoc anyone could have dreamed of, alwayshelping me with my questions concerning computational chemistry andteaching me as best as I could possibly understand about it.
All past and present members of the Kamerlin lab, thank you fordealing with me! Alex, thank you so much for helping me both with
86

my understanding of physics and chemistry, for the time at the musicfestival and others! Beat, thanks for your insights in programming andhow to handle the PhD situation, even though we do still have to talkabout who actually owns the epoxide project\s! Miha, thank you forbeing the nagging voice of reason and your dedication to scientific work,as well as music festivals and showing me around in Slovenia! Klaudiaand Irek, thank you for the board game evenings at your place! David,thank you for the discussions about life and work and the great time at allthe parties! Ania, thank you for your insights into QM calculations andsorry for all the bad jokes! Yashraj, thank you for giving me the excuseto visit the USA and to continue the work on the new program version!Also, I hope I did not scare you too much when you started in the lab.Fabian, thank you for the help on our projects, even though I could notfully commit all my time to them! Dennis, thanks for your insights intohow computational chemistry should be done and having me sitting nextto the GPU server! Qinghua, sorry that we did not interact that muchin the end. Thanks for showing us how to approach simulations in otherprograms and that it is possible to actually use the metal models! Xana,thanks for working with me on the PMH project, allowing me to test thecalculations in Cambridge! Chris, thank you for the early work on theprojects we both worked on. Hope you are doing fine in Canada!Now, I also want to thank the members of the Widersten lab, where Istarted my stay here as a student in Uppsala. First and foremost, thanksto Åsa for helping me in the early days as a visiting bachelor student,for working with me on the epoxide projects and always being there toteach me about how to do things in the lab. I can not thank you enough.Next, thanks to Emil for supervising me during my work on the masterthesis and working together on the ADH–A project. One day this shouldalso work out in the end. Huan, thank you for your good mood duringall the days working in the lab! Cissi and Nisse, thank you for beingthe voices of calm an reason during my early days working in the lab!Thilak, thank you for the discussions we had during the time I was inthe lab!To the people at the Department of Cell and Molecular Biology: Prof.
87

Johan Åqvist, thank you for allowing me to work on Q and for yourinsights on computational chemistry! Prof. David van der Spoel,thank you for pointing out answers to questions on molecular simulations,helping me to see issues from a different point of view. Prof. DanThomas Major, thank you for helping me with the work on the QCPimplementation in Q. Mauricio, thank you for your help with the Q code!Masoud, thanks for your opinions and insights on EVB simulations andwhen to avoid them!To my friends in the department and Uppsala in general: Mirthe, thankyou for being a good friend all the time I have been here in Sweden andyes, I did finish before you! Petar, I can only be amazed by how youmanage your work here while still being relaxed about life in general.Thanks for showing that this is possible, too! Zaza, thank you for beinga good friend, especially during my early days here! Felipe, thank you forthe geeky discussions and the soccer sessions! Alexandra, thank you foralways cheering me up with your good mood! Thank you for dealing withall my complaints about science and life in general. Showgy, thanks forthe fun discussions at the different beer clubs, keep on rocking! Danna,great to have met you just before I was done, hope Uppsala will treat youwell during your PhD!Special thanks to Madde, for helping me with the swedish summary ofthe thesis (read: rewriting what I initially wrote). Good luck to you andDavid here in Uppsala!I also want to thank all the master students that partially had to sufferunder my lack of supervision, Nils, Malin and Arina. I hope you willdo great in your future career in science.Some people asked to remain anonymous, so I will respect their wishes.You know who you are, thank you!This work has been supported by the Sven och Lilly Lawski Foundation,the European Research Council and the Swedish National Infrastructurefor Supercomputing.
88

Bibliography
(1) Berg, J. M.; Tymoczko, J. L.; Stryer, L., Biochemistry; W.H. Freeman:Basingstoke, 2012.
(2) Fedor, M. J.; Williamson, J. R. Nature Reviews Molecular Cell Biology2005, 6, 399–412.
(3) Tawfik, D. S.; Khersonsky, O. Annual Review of Biochemistry 2010,79, 471–505.
(4) Khersonsky, O.; Roodveldt, C.; Tawfik, D. Current Opinion in ChemicalBiology 2006, 10, 498–508.
(5) Harayama, S.; Kok, M.; Neidle, E. L. Annual Review of Microbiology1992, 46, 565–601.
(6) Anslyn, E. V.; Dougherty, D. A., Modern Physical Organic Chemistry;University Science Books: 2006, 157 to 162.
(7) Ibba, M.; Söll, D. Annual Review of Biochemistry 2000, 69, 617–650.
(8) Fersht, A., Structure and Mechanism in Protein Science: A Guide toEnzyme Catalysis and Protein Folding; W. H. Freeman: 1999.
(9) Babtie, A.; Tokuriki, N.; Hollfelder, F. Current Opinion in ChemicalBiology 2010, 14, 200–207.
(10) Copley, S. D. Trends in Biochemical Sciences 2015, 40, 72–78.
(11) Pabis, A.; Kamerlin, S. C. L. Current Opinion in Structural Biology2016, 37, 14–21.
(12) Bornscheuer, U. T.; Kazlauskas, R. J. Angewandte Chemie Interna-tional Edition 2004, 43, 6032–6040.
(13) Major, D. T.; Freud, Y.; Weitman, M. Current Opinion in ChemicalBiology 2014, 21, 25–33.
(14) Jia, M.; Potter, K. C.; Peters, R. J. Metabolic Engineering 2016, 37,24–34.
(15) Wolfenden, R. Nature 1969, 223, 704–705.
89

(16) Reetz, M. T.; Bocola, M.; Wang, L.-W.; Sanchis, J.; Cronin, A.; Arand,M.; Zou, J.; Archelas, A.; Bottalla, A.-L.; Naworyta, A.; Mowbray, S. L.Journal of the American Chemical Society 2009, 131, 7334–43.
(17) Gurell, A.; Widersten, M. Chembiochem : A European Journal of Chem-ical Biology 2010, 11, 1422–9.
(18) Janfalk Carlsson, Å.; Bauer, P.; Ma, H.; Widersten, M. Biochemistry2012, 51, 7627–7637.
(19) Lindberg, D.; Gogoll, A.; Widersten, M. FEBS Journal 2008, 275,6309–6320.
(20) Guo, Y.-A.; Liang, T.; Kim, S. W.; Xiao, H.; Krische, M. J. Journal ofthe American Chemical Society 2017, 139, 6847–6850.
(21) De la Torre, A.; Kaiser, D.; Maulide, N. Journal of the American Chem-ical Society 2017, 139, 6578–6581.
(22) Tanabe, J.; Taura, D.; Ousaka, N.; Yashima, E. Journal of the AmericanChemical Society 2017, 139, 7388–7398.
(23) Rohl, C. A.; Strauss, C. E.; Misura, K. M.; Baker, D. In, 2004, pp 66–93.
(24) Richter, F.; Leaver-Fay, A.; Khare, S. D.; Bjelic, S.; Baker, D. PLoSONE 2011, 6, ed. by Uversky, V. N., e19230.
(25) Amrein, B. A.; Steffen-Munsberg, F.; Szeler, I.; Purg, M.; Kulkarni, Y.;Kamerlin, S. C. L. IUCrJ 2017, 4, 50–64.
(26) Salque, M.; Bogucki, P. I.; Pyzel, J.; Sobkowiak-Tabaka, I.; Grygiel, R.;Szmyt, M.; Evershed, R. P. Nature 2012, 493, 522–525.
(27) Legras, J.-.-.-L.; Merdinoglu, D.; Cornuet, J.-.-.-M.; Karst, F. MolecularEcology 2007, 16, 2091–2102.
(28) Buchner, E.; Rapp, R. Berichte der Deutschen Chemischen Gesellschaft1899, 32, 2086–2094.
(29) Wöhler, F. Annalen der Physik und Chemie 1828, 88, 253–256.
(30) Michaelis, L.; Menten, M. L. Biochem. Z 1913, 49, 352.
(31) Briggs, G. E.; Haldane, J. B. S. Biochemical Journal 1925, 19, 338–339.
(32) Albe, K. R.; Butler, M. H.; Wright, B. E. Journal of Theoretical Biology1990, 143, 163–195.
90

(33) King, E. L.; Altman, C. The Journal of Physical Chemistry 1956, 60,1375–1378.
(34) Segel, I. H., Enzyme Kinetics: Behavior and Analysis of Rapid Equilib-rium and Steady-State Enzyme Systems; Wiley Classics Library; Wiley:1993.
(35) Kyte, J., Mechanism in Protein Chemistry; Garland Pub.: 1995.
(36) Schechter, A. N. Science 1970, 170, 273–80.
(37) Barman, T. E.; Bellamy, S. R. W.; Gutfreund, H.; Halford, S. E.; Lionne,C. Cellular and Molecular Life Sciences 2006, 63, 2571–2583.
(38) Williams, I.; Williams, N., Advances in Physical Organic Chemistry;Academic Press: 2014; Vol. 48.
(39) Chapman, N., Advances in Linear Free Energy Relationships; SpringerScience & Business Media: 2012.
(40) Hollfelder, F.; Herschlag, D. Biochemistry 1995, 34, 12255–12264.
(41) Rosta, E.; Kamerlin, S. C. L.; Warshel, A. Biochemistry 2008, 47, 3725–3735.
(42) Duarte, F.; Barrozo, A.; Åqvist, J.; Williams, N. H.; Kamerlin, S. C. L.Journal of the American Chemical Society 2016, 138, 10664–10673.
(43) Duarte, F.; Geng, T.; Marloie, G.; Al Hussain, A. O.; Williams, N. H.;Kamerlin, S. C. L. The Journal of Organic Chemistry 2014, 79, 2816–2828.
(44) Hammett, L. P. Journal of the American Chemical Society 1937, 59,96–103.
(45) Northrop, D. B. Biochemistry 1975, 14, 2644–2651.
(46) Sen, A.; Kohen, A. Journal of Physical Organic Chemistry 2009, 23,613–619.
(47) Vardi-Kilshtain, A.; Nitoker, N.; Major, D. T. Archives of Biochemistryand Biophysics 2015, 582, 18–27.
(48) Meisner, J.; Kästner, J. Angewandte Chemie International Edition 2016,55, 5400–5413.
(49) Westaway, K. C. In Advances in Physical Organic Chemistry, 2006;Vol. 41, pp 217–273.
91

(50) Nagel, Z. D.; Klinman, J. P. Chemical Reviews 2006, 106, 3095–3118.
(51) Klinman, J. P. Journal of Physical Organic Chemistry 2010, 23, 606–612.
(52) Arrhenius, S. Zeitschrift für Physikalische Chemie 1889, 4, DOI: 10.1515/zpch-1889-0108.
(53) Eyring, H. The Journal of Chemical Physics 1935, 3, 107–115.
(54) Evans, M. G.; Polanyi, M. Transactions of the Faraday Society 1935,31, 875.
(55) Po, H. N.; Senozan, N. M. Journal of Chemical Education 2001, 78,1499.
(56) Talley, K.; Alexov, E. Proteins: Structure, Function, and Bioinformatics2010, n/a–n/a.
(57) Pu, J.; Gao, J.; Truhlar, D. G. Chemical Reviews 2006, 106, 3140–3169.
(58) Miller, W. H. The Journal of Chemical Physics 1975, 62, 1899–1906.
(59) Pineda, J.; Schwartz, S. Philosophical Transactions of the Royal SocietyB: Biological Sciences 2006, 361, 1433–1438.
(60) Truhlar, D. G.; Garrett, B. C. Annual Review of Physical Chemistry1984, 35, 159–189.
(61) Gao, J.; Truhlar, D. G. Annual Review of Physical Chemistry 2002, 53,467–505.
(62) Fischer, E. Berichte der Deutschen Chemischen Gesellschaft 1894, 27,2985–2993.
(63) Koshland, D. E. Proceedings of the National Academy of Sciences 1958,44, 98–104.
(64) Pauling, L. Chemical & Engineering News 1946, 24, 1375–1377.
(65) Jencks, W. P., Catalysis in Chemistry and Enzymology; Dover Bookson Physics and Chemistry; Dover: 1987.
(66) Menger, F. M. Biochemistry 1992, 31, 5368–5373.
(67) Xiang, S.; Short, S. A.; Wolfenden, R.; Carter, C. W. Biochemistry1997, 36, 4768–4774.
(68) Wu, N.; Mo, Y.; Gao, J.; Pai, E. F. Proceedings of the National Academyof Sciences 2000, 97, 2017–2022.
92

(69) Warshel, A.; Štrajbl, M.; Villà, J.; Florián, J. Biochemistry 2000, 39,14728–14738.
(70) Shurki, A.; Štrajbl, M.; Villà, J.; Warshel, A. Journal of the AmericanChemical Society 2002, 124, 4097–4107.
(71) Callahan, B. P.; Wolfenden, R. Journal of the American Chemical So-ciety 2004, 126, 14698–14699.
(72) Burschowsky, D.; van Eerde, A.; Ökvist, M.; Kienhöfer, A.; Kast, P.;Hilvert, D.; Krengel, U. Proceedings of the National Academy of Sci-ences 2014, 111, 17516–17521.
(73) Warshel, A.; Sharma, P. K.; Kato, M.; Xiang, Y.; Liu, H.; Olsson,M. H. M. Chemical Reviews 2006, 106, 3210–3235.
(74) Warshel, A. Journal of Biological Chemistry 1998, 273, 27035–27038.
(75) Roca, M.; Martí, S.; Andrés, J.; Moliner, V.; Tuñón, I.; Bertrán, J.;Williams, I. H. Journal of the American Chemical Society 2003, 125,7726–7737.
(76) Štrajbl, M.; Shurki, A.; Kato, M.; Warshel, A. Journal of the AmericanChemical Society 2003, 125, 10228–10237.
(77) Kamerlin, S. C. L.; Haranczyk, M.; Warshel, A. The Journal of PhysicalChemistry B 2009, 113, 1253–1272.
(78) Kamerlin, S. C. L.; Sharma, P. K.; Chu, Z. T.; Warshel, A. Proceedingsof the National Academy of Sciences 2010, 107, 4075–4080.
(79) Szefczyk, B.; Mulholland, A. J.; Ranaghan, K. E.; Sokalski, W. A. Jour-nal of the American Chemical Society 2004, 126, 16148–16159.
(80) Preiswerk, N.; Beck, T.; Schulz, J. D.; Milovnik, P.; Mayer, C.; Siegel,J. B.; Baker, D.; Hilvert, D. Proceedings of the National Academy ofSciences 2014, 111, 8013–8018.
(81) Fried, S. D.; Bagchi, S.; Boxer, S. G. Science 2014, 346, 1510–1514.
(82) Wu, Y.; Boxer, S. G. Journal of the American Chemical Society 2016,138, 11890–11895.
(83) Hu, S.; Sharma, S. C.; Scouras, A. D.; Soudackov, A. V.; Carr, C. A. M.;Hammes-Schiffer, S.; Alber, T.; Klinman, J. P. Journal of the AmericanChemical Society 2014, 136, 8157–8160.
93

(84) Kamerlin, S. C. L.; Warshel, A. Journal of Physical Organic Chemistry2010, 23, 677–684.
(85) Kamerlin, S. C. L.; Mavri, J.; Warshel, A. FEBS Letters 2010, 584,2759–2766.
(86) McPherson, A. Methods 2004, 34, 254–265.
(87) Wuthrich, K. Science 1989, 243, 45–50.
(88) Wang, R. Y.-R.; Kudryashev, M.; Li, X.; Egelman, E. H.; Basler, M.;Cheng, Y.; Baker, D.; DiMaio, F. Nature Methods 2015, 12, 335–338.
(89) Isaksen, G. V.; Åqvist, J.; Brandsdal, B. O. Proceedings of the NationalAcademy of Sciences 2016, 113, 7822–7827.
(90) Isaksen, G. V.; Åqvist, J.; Brandsdal, B. O. PLoS Computational Biol-ogy 2014, 10, ed. by MacKerell, A. D., e1003813.
(91) Polishchuk, P. G.; Madzhidov, T. I.; Varnek, A. Journal of Computer-Aided Molecular Design 2013, 27, 675–679.
(92) Fischer, E. Berichte der Deutschen Chemischen Gesellschaft 1891, 24,2683–2687.
(93) Fischer, E. Berichte der Deutschen Chemischen Gesellschaft 1891, 24,1836–1845.
(94) Cahn, R. S.; Ingold, C.; Prelog, V. Angewandte Chemie InternationalEdition in English 1966, 5, 385–415.
(95) Mislow, K.; Siegel, J. Journal of the American Chemical Society 1984,106, 3319–3328.
(96) Silverman, R. B., The Organic Chemistry of Drug Design and DrugAction; Elsevier Science: 2004.
(97) Chirality 1992, 4, 338–340.
(98) Breuer, M.; Ditrich, K.; Habicher, T.; Hauer, B.; Kesseler, M.; Stürmer,R.; Zelinski, T. Angewandte Chemie International Edition in English2004, 43, 788–824.
(99) Heitbaum, M.; Glorius, F.; Escher, I. Angewandte Chemie InternationalEdition 2006, 45, 4732–4762.
(100) Nestl, B. M.; Hammer, S. C.; Nebel, B. A.; Hauer, B. AngewandteChemie International Edition 2014, 53, 3070–3095.
94

(101) Shen, J.; Okamoto, Y. Chemical Reviews 2016, 116, 1094–1138.
(102) Frey, P. A.; Hegeman, A. D., Enzymatic Reaction Mechanisms; OxfordUniversity Press: 2007.
(103) Ollis, D. L.; Cheah, E.; Cygler, M.; Dijkstra, B.; Frolow, F.; Franken,S. M.; Harel, M.; Remington, S. J.; Silman, I.; Schrag, J.; Sussman,J. L.; Verschueren, K. H.; Goldman, A. Protein Engineering, Designand Selection 1992, 5, 197–211.
(104) Pettersen, E. F.; Goddard, T. D.; Huang, C. C.; Couch, G. S.; Green-blatt, D. M.; Meng, E. C.; Ferrin, T. E. Journal of Computational Chem-istry 2004, 25, 1605–1612.
(105) Buck, D. K.; Collins, A. A. POV-Ray - The Persistence of Vision Ray-tracer.
(106) Bender, M. L.; Turnquest, B. W. Journal of the American ChemicalSociety 1957, 79, 1656–1662.
(107) Jencks, W. P.; Carriuolo, J. Journal of the American Chemical Society1961, 83, 1743–1750.
(108) Arand, M.; Cronin, A.; Oesch, F.; Mowbray, S. L.; Alwyn Jones, T.Drug Metabolism Reviews 2003, 35, 365–383.
(109) Archelas, A.; Furstoss, R. Current Opinion in Chemical Biology 2001,5, 112–119.
(110) De Vries, E. J.; Janssen, D. B. Current Opinion in Biotechnology 2003,14, 414–420.
(111) Copley, S. D. Current Opinion in Chemical Biology 1998, 2, 613–617.
(112) Damborský, J.; Koca, J. Protein Engineering 1999, 12, 989–98.
(113) Koudelakova, T.; Bidmanova, S.; Dvorak, P.; Pavelka, A.; Chaloupkova,R.; Prokop, Z.; Damborsky, J. Biotechnology Journal 2013, 8, 32–45.
(114) Jones, A. D. Journal of Biological Chemistry 1995, 270, 26923–26930.
(115) Rink, R.; Janssen, D. B. Biochemistry 1998, 37, 18119–18127.
(116) Hopmann, K. H.; Himo, F. Chemistry - A European Journal 2006, 12,6898–6909.
(117) Arand, M.; Hallberg, B. M.; Zou, J.; Bergfors, T.; Oesch, F.; van derWerf, M. J.; de Bont, J. A. M.; Jones, T. A.; Mowbray, S. L. The EMBOJournal 2003, 22, 2583–2592.
95

(118) Van der Werf, M. J.; de Bont, J. A.; Swarts, H. J. Tetrahedron: Asym-metry 1999, 10, 4225–4230.
(119) Hopmann, K. H.; Hallberg, B. M.; Himo, F. Journal of the AmericanChemical Society 2005, 127, 14339–14347.
(120) Borhan, B.; Jones, A. D.; Pinot, F.; Grant, D. F.; Kurth, M. J.; Ham-mock, B. D. Journal of Biological Chemistry 1995, 270, 26923–26930.
(121) Yamada, T.; Morisseau, C.; Maxwell, J. E.; Argiriadi, M. A.; Chris-tianson, D. W.; Hammock, B. D. The Journal of Biological Chemistry2000, 275, 23082–8.
(122) Elfström, L. T.; Widersten, M. Biochemistry 2006, 45, 205–212.
(123) Morisseau, C.; Beetham, J. K.; Pinot, F.; Debernard, S.; Newman,J. W.; Hammock, B. D. Archives of Biochemistry and Biophysics 2000,378, 321–332.
(124) Elfström, L. T.; Widersten, M. Biochemical Journal 2005, 390, 633–640.
(125) Rink, R.; Kingma, J.; Lutje Spelberg, J. H.; Janssen, D. B. Biochemistry2000, 39, 5600–5613.
(126) Widersten, M.; Gurell, A.; Lindberg, D. Biochimica et Biophysica Acta2010, 1800, 316–26.
(127) Blikstad, C.; Dahlström, K. M.; Salminen, T. A.; Widersten, M. ACSCatalysis 2013, 3, 3016–3025.
(128) Hamnevik, E.; Blikstad, C.; Norrehed, S.; Widersten, M. Journal ofMolecular Catalysis B: Enzymatic 2014, 99, 68–78.
(129) Mowbray, S. L.; Elfström, L. T.; Ahlgren, K. M.; Andersson, C. E.;Widersten, M. Protein Science 2006, 15, 1628–1637.
(130) Thomaeus, A.; Carlsson, J.; Aqvist, J.; Widersten, M. Biochemistry2007, 46, 2466–79.
(131) Amrein, B. A.; Bauer, P.; Duarte, F.; Janfalk Carlsson, Å.; Naworyta,A.; Mowbray, S. L.; Widersten, M.; Kamerlin, S. C. L.; Carlsson, Å. J.;Naworyta, A.; Mowbray, S. L.; Widersten, M.; Kamerlin, S. C. L. ACSCatalysis 2015, 5, 5702–5713.
(132) Lindberg, D.; de la Fuente Revenga, M.; Widersten, M. Biochemistry2010, 49, 2297–2304.
96

(133) Mohamed, M. F.; Hollfelder, F. Biochimica et biophysica acta 2013,1834, 417–24.
(134) Barrozo, A.; Duarte, F.; Bauer, P.; Carvalho, A. T. P.; Kamerlin, S. C. L.Journal of the American Chemical Society 2015, 137, 9061–9076.
(135) Khersonsky, O.; Malitsky, S.; Rogachev, I.; Tawfik, D. S. Biochemistry2011, 50, 2683–2690.
(136) Purg, M.; Pabis, A.; Baier, F.; Tokuriki, N.; Jackson, C.; Kamerlin,S. C. L. Philosophical Transactions of the Royal Society A: Mathemat-ical, Physical and Engineering Sciences 2016, 374, 20160150.
(137) Hochberg, G. K. A.; Thornton, J. W. Annual Review of Biophysics2017, 46, 247–269.
(138) Monterde, M. I.; Lombard, M.; Archelas, A.; Cronin, A.; Arand, M.;Furstoss, R. Tetrahedron: Asymmetry 2004, 15, 2801–2805.
(139) Schober, M.; Faber, K. Trends in Biotechnology 2013, 31, 468–478.
(140) Bottalla, A.-L.; Ibrahim-Ouali, M.; Santelli, M.; Furstoss, R.; Archelas,A. Advanced Synthesis & Catalysis 2007, 349, 1102–1110.
(141) Jensen, F., Introduction to Computational Chemistry; Wiley: 2007.
(142) Ransil, B. J. Reviews of Modern Physics 1960, 32, 239–244.
(143) Alder, B. J.; Wainwright, T. E. The Journal of Chemical Physics 1959,31, 459–466.
(144) Bernal, J. D. Proceedings of the Royal Society A: Mathematical, Physicaland Engineering Sciences 1964, 280, 299–322.
(145) Schrödinger, E. Annalen der Physik 1926, 384, 361–376.
(146) Szabo, A.; Ostlund, N. S., Modern Quantum Chemistry: Introductionto Advanced Electronic Structure Theory; Dover Books on Chemistry;Dover Publications: 1989.
(147) Born, M.; Oppenheimer, R. Annalen der Physik 1927, 389, 457–484.
(148) Pauli, W. Zeitschrift für Physik 1925, 31, 765–783.
(149) Hartree, D. R. Reports on Progress in Physics 1947, 11, 305.
(150) Levine, I. N., Quantum Chemistry; Pearson Prentice Hall: 2009.
(151) David Sherrill, C.; Schaefer, H. F. In Advances in Quantum Chemistry;Academic Press: 1999, pp 143–269.
97

(152) Bartlett, R. J.; Musiał, M. Reviews of Modern Physics 2007, 79, 291–352.
(153) Møller, C.; Plesset, M. S. Physical Review 1934, 46, 618–622.
(154) Cremer, D. Wiley Interdisciplinary Reviews: Computational MolecularScience 2011, 1, 509–530.
(155) Thomas, L. H. Mathematical Proceedings of the Cambridge Philosophi-cal Society 1927, 23, 542.
(156) Hohenberg, P.; Kohn, W. Physical Review 1964, 136, B864–B871.
(157) Kohn, W.; Sham, L. J. Physical Review 1965, 140, A1133–A1138.
(158) Becke, A. D. The Journal of Chemical Physics 1993, 98, 5648–5652.
(159) Zhao, Y.; Truhlar, D. G. Theoretical Chemistry Accounts 2008, 120,215–241.
(160) Cohen, A. J.; Mori-Sánchez, P.; Yang, W. Chemical Reviews 2012, 112,289–320.
(161) Medvedev, M. G.; Bushmarinov, I. S.; Sun, J.; Perdew, J. P.; Lyssenko,K. A. Science 2017, 355, 49–52.
(162) Blomberg, M. R. A.; Borowski, T.; Himo, F.; Liao, R.-Z.; Siegbahn,P. E. M. Chemical Reviews 2014, 114, 3601–3658.
(163) Levitt, M.; Lifson, S. Journal of Molecular Biology 1969, 46, 269–279.
(164) Jones, J. E. Proceedings of the Royal Society A: Mathematical, Physicaland Engineering Sciences 1924, 106, 463–477.
(165) Tuckerman, M. E., Statistical Mechanics: Theory and Molecular Simu-lation; Oxford University Press: 2010.
(166) Pathria, R. K.; Beale, P. D., Statistical Mechanics; Elsevier Science:1996.
(167) Van Gunsteren, W. F.; Berendsen, H. J. C. Molecular Simulation 1988,1, 173–185.
(168) Metropolis, N.; Rosenbluth, A. W.; Rosenbluth, M. N.; Teller, A. H.;Teller, E. The Journal of Chemical Physics 1953, 21, 1087–1092.
(169) Jorgensen, W. L.; Tirado-Rives, J. The Journal of Physical Chemistry1996, 100, 14508–14513.
(170) Lengauer, T.; Rarey, M. Current Opinion in Structural Biology 1996,6, 402–406.
98

(171) Kitchen, D. B.; Decornez, H.; Furr, J. R.; Bajorath, J. Nature ReviewsDrug Discovery 2004, 3, 935–949.
(172) Pagadala, N. S.; Syed, K.; Tuszynski, J. Biophysical Reviews 2017, 9,91–102.
(173) Korb, O.; Stützle, T.; Exner, T. E. Journal of Chemical Informationand Modeling 2009, 49, 84–96.
(174) Genheden, S.; Ryde, U. Expert Opinion on Drug Discovery 2015, 10,449–461.
(175) Zheng, M.; Xiong, B.; Luo, C.; Li, S.; Liu, X.; Shen, Q.; Li, J.; Zhu,W.; Luo, X.; Jiang, H. Journal of Chemical Information and Modeling2011, 51, 2994–3004.
(176) Zwanzig, R. W. The Journal of Chemical Physics 1954, 22, 1420–1426.
(177) Torrie, G.; Valleau, J. Journal of Computational Physics 1977, 23, 187–199.
(178) Kumar, S.; Rosenberg, J. M.; Bouzida, D.; Swendsen, R. H.; Kollman,P. A. Journal of Computational Chemistry 1992, 13, 1011–1021.
(179) Van Duin, A. C. T.; Dasgupta, S.; Lorant, F.; Goddard, W. A. TheJournal of Physical Chemistry A 2001, 105, 9396–9409.
(180) Warshel, A.; Karplus, M. Journal of the American Chemical Society1972, 94, 5612–5625.
(181) Warshel, A.; Levitt, M. Journal of Molecular Biology 1976, 103, 227–249.
(182) Van der Kamp, M. W.; Mulholland, A. J. Biochemistry 2013, 52, 2708–2728.
(183) Brunk, E.; Rothlisberger, U. Chemical Reviews 2015, 115, 6217–6263.
(184) Field, M. J.; Bash, P. A.; Karplus, M. Journal of Computational Chem-istry 1990, 11, 700–733.
(185) Chung, L. W.; Sameera, W. M. C.; Ramozzi, R.; Page, A. J.; Hatanaka,M.; Petrova, G. P.; Harris, T. V.; Li, X.; Ke, Z.; Liu, F.; Li, H.-B.; Ding,L.; Morokuma, K. Chemical Reviews 2015, 115, 5678–5796.
(186) Svensson, M.; Humbel, S.; Froese, R. D. J.; Matsubara, T.; Sieber, S.;Morokuma, K. The Journal of Physical Chemistry 1996, 100, 19357–19363.
99

(187) Warshel, A.; Weiss, R. M. Journal of the American Chemical Society1980, 102, 6218–6226.
(188) Marcus, R. A. The Journal of Chemical Physics 1956, 24, 966–978.
(189) Warshel, A., Computer Modeling of Chemical Reactions in Enzymesand Solutions, Wiley Prof; Wiley Interscience: 1997.
(190) Warshel, A.; Weiss, R. M. Annals of the New York Academy of Sciences1981, 367, 370–382.
(191) Chang, Y. T.; Miller, W. H. The Journal of Physical Chemistry 1990,94, 5884–5888.
(192) Carpenter, B. K.; Harvey, J. N.; Glowacki, D. R. Phys. Chem. Chem.Phys. 2015, 17, 8372–8381.
(193) Glowacki, D. R.; Orr-Ewing, A. J.; Harvey, J. N. The Journal of Chem-ical Physics 2015, 143, 044120.
(194) Hwang, J. K.; King, G.; Creighton, S.; Warshel, A. Journal of the Amer-ican Chemical Society 1988, 110, 5297–5311.
(195) Mones, L.; Kulhànek, P.; Simon, I.; Laio, A.; Fuxreiter, M. The Journalof Physical Chemistry B 2009, 113, 7867–7873.
(196) Muegge, I.; Tao, H.; Warshel, A. Protein Engineering 1997, 10, 1363–72.
(197) Warshel, A. Proceedings of the National Academy of Sciences of theUnited States of America 1978, 75, 5250–5254.
(198) Fuxreiter, M.; Mones, L. Current Opinion in Chemical Biology 2014,21, 34–41.
(199) Feynman, R. P.; Hibbs, A. R., Quantum Mechanics and Path Integrals;International series in pure and applied physics; McGraw-Hill: 1965.
(200) Ceperley, D. M. Reviews of Modern Physics 1995, 67, 279–355.
(201) Hwang, J. K.; Warshel, A. The Journal of Physical Chemistry 1993,97, 10053–10058.
(202) Mavri, J.; Liu, H.; Olsson, M. H. M.; Warshel, A. The Journal of Phys-ical Chemistry B 2008, 112, 5950–5954.
(203) Major, D. T.; Gao, J. Journal of Chemical Theory and Computation2007, 3, 949–960.
100

(204) Major, D. T.; Gao, J. Journal of Molecular Graphics and Modelling2005, 24, 121–127.
(205) Morisseau, C.; Hammock, B. D. Annual Review of Pharmacology andToxicology 2005, 45, 311–333.
(206) Arand, M.; Wagner, H.; Oesch, F. The Journal of Biological Chemistry1996, 271, 4223–9.
(207) Lonsdale, R.; Hoyle, S.; Grey, D. T.; Ridder, L.; Mulholland, A. J.Biochemistry 2012, 51, 1774–1786.
(208) Reetz, M. T.; Kahakeaw, D.; Lohmer, R. Chembiochem : A EuropeanJournal of Chemical Biology 2008, 9, 1797–804.
(209) Hopmann, K. H.; Himo, F. The Journal of Physical Chemistry. B 2006,110, 21299–310.
(210) Lenfant, N.; Hotelier, T.; Velluet, E.; Bourne, Y.; Marchot, P.; Chaton-net, A. Nucleic Acids Research 2013, 41, D423–D429.
(211) Thomaeus, A.; Naworyta, A.; Mowbray, S. L.; Widersten, M. ProteinScience : A Publication of the Protein Society 2008, 17, 1275–84.
(212) Bauer, P.; Carlsson, Å. J.; Amrein, B. A.; Dobritzsch, D.; Widersten,M.; Kamerlin, S. C. L. Org. Biomol. Chem. 2016, 14, 5639–5651.
(213) Lind, M. E. S.; Himo, F. ACS Catalysis 2016, 6, 8145–8155.
(214) Lind, M. E. S.; Himo, F. Angewandte Chemie 2013, 125, 4661–4665.
(215) Carlier, P. R.; Deora, N.; Crawford, T. D. The Journal of OrganicChemistry 2006, 71, 1592–1597.
(216) Zhao, Y.; Truhlar, D. G. The Journal of Organic Chemistry 2007, 72,295–298.
(217) Friesner, R. A.; Banks, J. L.; Murphy, R. B.; Halgren, T. A.; Klicic,J. J.; Mainz, D. T.; Repasky, M. P.; Knoll, E. H.; Shelley, M.; Perry,J. K.; Shaw, D. E.; Francis, P.; Shenkin, P. S. Journal of MedicinalChemistry 2004, 47, 1739–1749.
(218) Warren, G. L.; Andrews, C. W.; Capelli, A.-M.; Clarke, B.; LaLonde,J.; Lambert, M. H.; Lindvall, M.; Nevins, N.; Semus, S. F.; Senger, S.;Tedesco, G.; Wall, I. D.; Woolven, J. M.; Peishoff, C. E.; Head, M. S.Journal of Medicinal Chemistry 2006, 49, 5912–5931.
101

(219) Janfalk Carlsson, Å.; Bauer, P.; Dobritzsch, D.; Nilsson, M.; Kamerlin,S. C. L.; Widersten, M. Chembiochem: A European Journal of ChemicalBiology 2016, 17, 1693–1697.
(220) Lindberg, D.; Ahmad, S.; Widersten, M. Archives of Biochemistry andBiophysics 2010, 495, 165–73.
(221) Andreini, C.; Bertini, I.; Cavallaro, G.; Holliday, G. L.; Thornton, J. M.JBIC Journal of Biological Inorganic Chemistry 2008, 13, 1205–1218.
(222) Sousa, S. F.; Fernandes, P. A.; Ramos, M. J. In Kinetics and Dynam-ics: From Nano- to Bio-Scale, Paneth, P., Dybala-Defratyka, A., Eds.;Springer Netherlands: Dordrecht, 2010, pp 299–330.
(223) Vedani, A.; Huhta, D. W. Journal of the American Chemical Society1990, 112, 4759–4767.
(224) Neves, R. P. P.; Sousa, S. F.; Fernandes, P. A.; Ramos, M. J. Journalof Chemical Theory and Computation 2013, 9, 2718–2732.
(225) Åqvist, J. Journal of Molecular Structure: THEOCHEM 1992, 256,135–152.
(226) Li, P.; Merz, K. M. Journal of Chemical Theory and Computation 2014,10, 289–297.
(227) Burgess, J., Metal Ions in Solution; Ellis Horwood series in chemicalscience; Ellis Horwood: 1978.
(228) Joung, I. S.; Cheatham, T. E. The Journal of Physical Chemistry B2008, 112, 9020–9041.
(229) Åqvist, J.; Warshel, A. Journal of Molecular Biology 1992, 224, 7–14.
(230) Oelschlaeger, P.; Klahn, M.; Beard, W. A.; Wilson, S. H.; Warshel, A.Journal of Molecular Biology 2007, 366, 687–701.
(231) Noyes, R. M. Journal of the American Chemical Society 1962, 84, 513–522.
(232) Marcus, Y. Biophysical Chemistry 1994, 51, 111–127.
(233) Rosseinsky, D. R. Chemical Reviews 1965, 65, 467–490.
(234) Duarte, F.; Bauer, P.; Barrozo, A.; Amrein, B. A.; Purg, M.; Åqvist,J.; Kamerlin, S. C. L. The Journal of Physical Chemistry B 2014, 118,4351–4362.
102

(235) He, M. M.; Clugston, S. L.; Honek, J. F.; Matthews, B. W. Biochemistry2000, 39, 8719–8727.
(236) Ben-David, M.; Sussman, J. L.; Maxwell, C. I.; Szeler, K.; Kamerlin,S. C.; Tawfik, D. S. Journal of Molecular Biology 2015, DOI: 10.1016/j.jmb.2015.01.013.
(237) Blaha-Nelson, D.; Krüger, D. M.; Szeler, K.; Ben-David, M.; Kamerlin,S. C. L. Journal of the American Chemical Society 2017, 139, 1155–1167.
(238) Liao, Q.; Kamerlin, S. C. L.; Strodel, B. The Journal of Physical Chem-istry Letters 2015, 6, 2657–2662.
(239) Jiang, Y.; Zhang, H.; Tan, T. Journal of Chemical Theory and Compu-tation 2016, 12, 3250–3260.
(240) Saxena, A.; Sept, D. Journal of Chemical Theory and Computation2013, 9, 3538–3542.
(241) Lemkul, J. A.; Huang, J.; Roux, B.; MacKerell, A. D. Chemical Reviews2016, 116, 4983–5013.
(242) Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M. C.; Xiong, G.; Zhang, W.;Yang, R.; Cieplak, P.; Luo, R.; Lee, T.; Caldwell, J.; Wang, J.; Kollman,P. Journal of Computational Chemistry 2003, 24, 1999–2012.
(243) Brooks, B. R. et al. Journal of Computational Chemistry 2009, 30,1545–1614.
(244) Abraham, M. J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J. C.; Hess,B.; Lindahl, E. SoftwareX 2015, 1-2, 19–25.
(245) Kamerlin, S. C. L.; Warshel, A. Wiley Interdisciplinary Reviews: Com-putational Molecular Science 2011, 1, 30–45.
(246) Marelius, J.; Kolmodin, K.; Feierberg, I.; Åqvist, J. Journal of MolecularGraphics and Modelling 1998, 16, 213–225.
(247) Saunders, W. H.; Edison, D. H. Journal of the American Chemical So-ciety 1960, 82, 138–142.
(248) Zhang, J.; Klinman, J. P. Journal of the American Chemical Society2011, 133, 17134–17137.
(249) Zhang, J.; Kulik, H. J.; Martinez, T. J.; Klinman, J. P. Proceedings ofthe National Academy of Sciences 2015, 112, 7954–7959.
103

(250) Hess, B.; Bekker, H.; Berendsen, H. J. C.; Fraaije, J. G. E. M. Journalof Computational Chemistry 1997, 18, 1463–1472.
(251) Van der Spoel, D.; van Maaren, P. J.; Caleman, C. Bioinformatics 2012,28, 752–753.
(252) Caleman, C.; van Maaren, P. J.; Hong, M.; Hub, J. S.; Costa, L. T.;van der Spoel, D. Journal of Chemical Theory and Computation 2012,8, 61–74.
104


Acta Universitatis UpsaliensisDigital Comprehensive Summaries of Uppsala Dissertationsfrom the Faculty of Science and Technology 1530
Editor: The Dean of the Faculty of Science and Technology
A doctoral dissertation from the Faculty of Science andTechnology, Uppsala University, is usually a summary of anumber of papers. A few copies of the complete dissertationare kept at major Swedish research libraries, while thesummary alone is distributed internationally throughthe series Digital Comprehensive Summaries of UppsalaDissertations from the Faculty of Science and Technology.(Prior to January, 2005, the series was published under thetitle “Comprehensive Summaries of Uppsala Dissertationsfrom the Faculty of Science and Technology”.)
Distribution: publications.uu.seurn:nbn:se:uu:diva-326108
ACTAUNIVERSITATIS
UPSALIENSISUPPSALA
2017