contributions of epstein-barr nuclear antigen 1 (ebna1 ... · i investigated whether this is due to...
TRANSCRIPT

Contributions of Epstein-Barr Nuclear Antigen 1 (EBNA1) and the Family of Repeats (FR) Region to oriP-Mediated
Replication and Segregation Functions in Nasopharyngeal Carcinoma
by
Natalia Thawe
A thesis submitted in conformity with the requirements for the degree of Master of Science
Graduate Department of Molecular Genetics University of Toronto
© Copyright by Natalia Thawe 2012

ii
Contributions of Epstein-Barr Nuclear Antigen 1 (EBNA1) and the
Family of Repeats (FR) Region to oriP-Mediated Replication and
Segregation Functions in Nasopharyngeal Carcinoma
Natalia Thawe
Master of Science
Department of Molecular Genetics
University of Toronto
2012
Abstract
The Epstein-Barr virus (EBV) EBNA1 protein mediates the replication and mitotic
segregation of the EBV genomes via interactions with the viral oriP sequences. C666-1 is the
only known nasopharyngeal carcinoma (NPC) cell line that stably maintains EBV in culture and
I investigated whether this is due to differences in oriP-mediated functions in replication and
segregation. I found that both C666-1 and EBV-negative NPC cell lines can replicate and
maintain oriP plasmids for extended periods but that high EBNA1 levels interfered with plasmid
segregation. The segregation element within oriP was recently shown to contain 29 repeated
sequences instead of the 20 repeats in initial oriP isolates. I compared the functions of oriP with
20 or 29 repeats and found that the higher number of repeats decreased plasmid replication but
increased plasmid maintenance, consistent with a segregation effect. Finally, I identified a
potential role for promyelocytic leukemia nuclear bodies in oriP plasmid replication.

iii
ACKNOWLEDGEMENTS
First and foremost, I would like to thank God for giving me strength and leading me on
this journey. I would like to express my sincere gratitude to my supervisor, Dr. Lori Frappier, for
giving me the opportunity to pursue my MSc degree in her lab. Her passion for EBNA1 and
EBV first inspired me to join her lab as a fourth year project student after listening to her lecture
during the third year of my undergraduate degree. I am thankful for her guidance and support
over the years as well as during the process of writing this thesis. I would also like to thank my
committee members, Dr. Brigitte Lavoie and Dr. Laurence Pelletier, for their support, great
ideas, and constructive critiques. They have always given me great suggestions and input, and
helped me be more attentive when I approach scientific problems and experiments.
I am also so grateful to all the members of the Frappier Lab, both past and present, for
their support, help, guidance, and friendship. I want to thank Niro Sivachandran, Shan Wang,
Teresa Sanchez, Kathy Shire, Jennifer Yinuo Cao, and Natasha Malik for their amazing
friendships, honesty, and all our great laughs and conversations all the time. You guys were not
only a pillar of support, but a sisterhood that I will always treasure. The lab would not have been
nearly as enjoyable without you. I especially want to thank Kathy Shire for all her wisdom,
patience, kindness, and of course, all her baked goods! I will definitely need the recipe for that
carrot cake! Thank you for some great memories, Frappier Lab!
I want to dedicate this thesis to my family: my father, my mother, and my brother. This
has all been possible because of your constant love and support all these years. Thank you for
listening to all my complaints, understanding me, celebrating with me when my experiments
work, and for picking me back up when I feel low. You are the best part of my life and you mean
so much to me. I am especially grateful to my parents. Mom and Dad, thank you for everything
that you have done for me and for all the opportunities that you have given me. I thank God
everyday for being born into this family and for having you as parents. I hope that I have made
you proud and that I will continue to do so in the future. Matthew, you are the best brother that I
could ever ask for. Thank you for giving me a shoulder to lean on, keeping me calm, toughening
me up, and making me laugh. Mom, Dad, Matt, I love you and thank you!

iv
TABLE OF CONTENTS
ABSTRACT ii
ACKNOWLEDGEMENTS iii
TABLE OF CONTENTS iv
LIST OF TABLES vi
LIST OF FIGURES vii
LIST OF ABBREVIATIONS viii
CHAPTER I. INTRODUCTION 1
I.1 The Epstein-Barr Virus 1
I.1.1 Brief Overview 1
I.1.2 Primary Infection 2
I.1.3 Latent EBV Infection 2
I.1.4 EBV-associated Diseases 5
I.1.4.1 Nasopharyngeal Carcinoma 5
I.1.4.2 Gastric Carcinoma 6
I.2 EBV Latent Origin of Replication, oriP 7
I.2.1 Dyad Symmetry (DS) Element 7
I.2.2 Family of Repeats (FR) Element 9
I.2.3 Cellular Proteins Recruited to oriP 11
I.3 Epstein-Barr Nuclear Antigen 1 (EBNA1) 14
I.3.1 Overview of Domain Structures 14
I.3.2 EBNA1 Functions 17
I.3.2.1 DNA Replication 17
I.3.2.2 DNA Segregation 20
I.3.2.2.1 EBNA1 Domains Contributing to Plasmid 20
Segregation
I.3.2.2.2 Involvement of EBP2 in EBNA1-Mediated 21
Plasmid Segregation
I.3.2.2.3 The Mitotic Tethering or “Piggybacking” 22
Model
I.3.2.2.4 Equal Partitioning of EBV Episomes to 23
Daughter Cells Following Mitosis
I.3.2.3 Transcriptional Regulation 24
I.3.2.4 Roles in Cell Transformation and Immortalization 26
I.4 Roles of Promyelocytic Leukemia (PML) bodies in Viral Replication 28
I.5 EBV Genome Maintenance in Nasopharyngeal Carcinoma 30
I.6 Thesis Rationale 31

v
CHAPTER II. MATERIALS AND METHODS 33
II.1 Cell Culture 33
II.2 Plasmid Constructs 33
II.3 Immunofluorescence Microscopy 35
II.4 Plasmid Replication and Maintenance Assays 35
II.5 Plasmid Labelling 37
II.6 Southern Blotting 37
II.7 Cell Proliferation Assay 38
II.8 Antibodies and Western Blotting 38
II.9 Monitoring Episomal Maintenance 39
CHAPTER III. RESULTS 40
III.1 pBFRGC6-Based Plasmids Do Not Autonomously Replicate in the 40
NPC Cell Line, CNE2Z
III.2 Higher EBNA1 Expression Can Inhibit Plasmid Maintenance Without 44
Inhibiting Plasmid Replication Efficiency in NPC Cells
III.3 Higher EBNA1 Expression Does Not Reduce EBV Copy Number 49
in C666-1 or AGS/EBV Cell Lines
III.4 Additional 9 Repeats within the FR Element Decrease Plasmid 51
Replication Efficiency but Improve Plasmid Segregation
III.5 Silencing of PML Protein Leads to a Reduction in Replication Efficiency 55
in NPC cells
CHAPTER IV. DISCUSSION 57
IV.1 FR-Bound EBNA1 May Enable Expression of GAL4-Cdc6 Fusion Protein 57
and Facilitate Replication of pBFRGC6-Based Plasmids
IV.2 Higher Levels of EBNA1 do not Inhibit Plasmid Replication and are not 58
Cytotoxic to CNE2Z Cells.
IV.3 Additional 9 Repeats within the FR Element May Confer an Advantage 60
in EBV Maintenance
IV.4 Possible Role of PML in Plasmid DNA Replication? 63
IV.5 Future Directions 64
CHAPTER V. REFERENCES 66

vi
LIST OF TABLES
Table 1 Epstein-Barr virus latency and expression profiles 5

vii
LIST OF FIGURES
Figure 1 The Epstein-Barr virus genome 4
Figure 2 Organization of the EBV latent origin of replication, oriP 8
Figure 3 EBNA1 functional domains 15
Figure 4 Plasmids used in this study 34
Figure 5 Schematic representation of protocol for plasmid replication and 42
maintenance assays
Figure 6 The pBFRGC6 plasmid does not autonomously replicate 43
Figure 7 Plasmid maintenance assays in CNE2Z and C666-1 cells 45
Figure 8 Higher levels of EBNA1 do not affect plasmid replication in 46
CNE2ZE cells
Figure 9 Higher levels of EBNA1 increase plasmid replication in C666-1 47
Figure 10 Higher levels of EBNA1 do not affect the copy number of EBV 50
episomes in C666-1 or AGS/EBV cell lines
Figure 11 The extra 9 repeats reduce plasmid replication efficiency 53
Figure 12 The extra 9 FR repeats improve plasmid segregation 54
Figure 13 PML silencing reduces the plasmid replication efficiency in 56
CNE2Z cells

viii
LIST OF ABBREVIATIONS
2D Two-dimensional
aa Amino acid
AT Adenine-thymine
BARF 1 BamHI rightward frame I
BART BamHI rightward transcript
BL Burkitt’s lymphoma
bp Base pairs
Brd4 Bromodomain protein 4
BrdU Bromodeoxyuridine
BSA Bovine serum albumin
BZLF1 BamHI Z leftward frame 1
BPV Bovine papillomavirus
CAT Chloramphenicol acetyltransferase
CEN Centromeric
ChIP Chromatin immunoprecipitation
CK2 Casein kinase 2
CMV Cytomegalovirus
Co-IP Co-immunoprecipitation
CTD C-terminal domain
DAPI 4’-6-Diamidino-2-phenylindole
DNA Deoxyribonucleic acid
dNTPs Deoxynucleotide triphosphate
dCTP Dyad symmetry
DTT Dithiothreitol
EBER Epstein-Barr expressed ribonucleic acid
EBNA1 Epstein-Barr nuclear antigen 1
EBNA1-LP Epstein-Barr nuclear antigen leader protein
EBP2 EBNA1-binding protein 2
EBV Epstein-Barr virus
ECL Enhanced chemiluminescence

ix
EDTA Ethylenediaminetetraacetic acid
FISH Fluorescence in-situ hybridization
FR Family of repeats
FBS Fetal bovine serum
GAPDH Glyceraldehyde-3-phosphate dehydrogenase
GC Gastric carcinoma
GFP Green fluorescent protein
Gly-Ala Glycine-alanine
Gly-Arg Glycine-arginine
USP7 Ubiquitin specific protease 7
HMG-1 High mobility group 1
IE Immediate-early
IM Infectious mononucleosis
Kb Kilobase
kD Kilodalton
KSHV Kaposi’s sarcoma-associated herpes virus
LANA1 Latency-associated nuclear antigen 1
LCLs Lymphoblastoid cell lines
LMP Latent membrane protein
MCM Minichromosome maintenance
MHC Major histocompatibility complex
miRNA microRNA
NAP1 Nucleosome assembly protein 1
NLS Nuclear localization signal
NPC Nasopharyngeal carcinoma
ORC Origin recognition complex
oriP Origin of plasmid replication
PAR Poly-ADP-ribosylation
PARP Poly-ADP-ribose polymerase
PBS Phosphate buffered saline
PCR Polymerase chain reaction
PML Promyelocytic leukemia

x
PML NBs Promyelocytic leukemia nuclear bodies
RARα Retinoic acid receptor α
RPA Replication protein A
SDS Sodium dodecyl suphate
SDS-PAGE SDS polyacrylamide gel electrophoresis
siRNA Small interfering RNA
shRNA Short hairpin RNA
SSC Saline sodium citrate
SV40 Simian virus 40
TAF1β Template activatin factor 1β
TR Terminal repeat
TRF Telomeric repeat binding factor
WHO World Health Organization

1
CHAPTER I. INTRODUCTION
I.1 Epstein-Barr Virus
I.1.1 Brief Overview
Epstein-Barr virus (EBV) is a well-studied member of the herpesvirus family which
encompasses over one hundred diverse enveloped viruses that are divided into three
classifications termed alpha, beta, and gamma. These classifications are mainly assigned
according to distinguishing characteristics of a herpesvirus such as their genome sequence and
biological cycle, and host specificity. Despite the different classes, all herpesviruses share a
common architecture composed of a linear double-stranded DNA genome core encased within an
icosahedral capsid structure. The capsid is coated by a proteinaceous layer referred to as
tegument, and an outer lipid bilayer membrane or envelope, which together form a virion. The
biological cycle of herpesviruses involves two stages: a latent (or nonproductive) infection, or a
lytic (productive) infection. Herpesviruses are capable of infecting a broad range of vertebrate
host species; however, as yet, only eight herpesviruses specific to humans have been identified
and have been characterized for their pathogenicity and associated diseases.
Anthony Epstein and Yvonne Barr first discovered EBV viral particles from cancer tissue
biopsies in 1964 while analyzing electron micrographs of Burkitt’s lymphoma cells (Epstein et
al. 1964). EBV was the first herpesvirus genome to be entirely sequenced (Lear et al. 1992) and
is very wide-spread, as greater than 95% of the adult population worldwide is infected with this
virus. EBV, also known as human herpesvirus 4, is classified as a member of the gamma
herpesvirus family that also includes Kaposi’s sarcoma associated herpesvirus (KSHV). These
are the only two gamma herpesviruses known to infect humans. An EBV virion consists of a 172
Kb linear genome surrounded by a nucleocapsid composed of 162 capsomeres, which in turn is
enclosed by tegument and a typical herpesvirus envelope containing viral-derived glycoproteins.
Upon internalization by a host cell, the linear EBV genome is taken up into the nucleus of the
cell and circularizes by ligating its two termini. The termini vary in length and encode a series of
500 bp direct repeats; hence, a characteristic number of terminal repeats is associated with each
EBV infection. The size of the terminal repeats remains constant with each clonal infection event
such that when an infected cell divides the resulting daughter cells will continue to maintain
identical number of terminal repeats. The extrachromosomal circular EBV genome associates

2
with core histone proteins and is packaged into nucleosomes, similar to cellular chromatin (Shaw
et al. 1979).
I.1.2 Primary Infection
Primary infection with EBV usually occurs during early childhood and is typically
asymptomatic. The infected individual remains as a carrier of EBV for their lifetime. Known as
the “kissing disease” due to its transmission via saliva, EBV initially enters the body and infects
the mucosal epithelial cells of the oropharynx wherein the virus replicates and infectious virions
are produced. During this transient phase of lytic replication, approximately 80 viral proteins
involved in viral replication and virion production are expressed. The virions subsequently
encounter infiltrating B-lymphocytes at the submucosal layer and establish latent infection (Kieff
1996; Rickinson and Kieff 1996). In order to infect and be taken up by circulating B-cells, the
main viral envelope glycoprotein gp350/220 binds to its receptor CD21 on the host cell surface
(Fingeroth et al. 1984). Studies have shown that the entry of EBV into host cell is facilitated by
the viral envelope glycoprotein gp42, which associates with the major histocompatibility
complex (MHC) class II protein (Li et al. 1997). Once the virion enters the cytoplasm of the cell,
it is transported to a nuclear pore through which the EBV genome is released into the host
nucleus and circularizes, which is necessary for latent infection.
If exposure to EBV is delayed until adolescence or adulthood, primary infection can
result in a self-limiting lymphoproliferative disorder called infectious mononucleosis (IM)
following a one month incubation stage (Gerber et al. 1972). Clinical features of IM include sore
throat, fever, lethargy, pharyngitis, and lymphadenopathy (Gerber et al. 1972; Kutok and Wang
2006). Once this acute lytic phase of infection subsides, EBV persists latently in a pool of
circulating B-cells within the host for their lifetime. Importantly, the lytic phase of the EBV life
cycle can be reinitiated upon expression of either of two immediate-early (IE) viral genes
encoding, BZLF1 and BRLF1 which are two regulators that transactivate genes involved in lytic
replication (Miller et al. 2007).
I.1.3 Latent EBV Infection
EBV is predominantly found in a latent state within resting or proliferative cells, in which
multiple copies of the virus are maintained as circular episomes which express restricted sets of

3
viral genes. B-cells are typically not the favoured location for EBV lytic infection, instead the
virus establishes a latent infection within this reservoir (Young and Murray 2003). Viral latency
is distinguished by infection without the production of virions. EBV latency has been classified
into four programs (latency 0, I, II, and III), each of which has a characteristic gene expression
profile and infects specific cell lineages (Rickinson and Kieff 1996). During latent infection, at
least 12 different viral genes can be expressed including the six nuclear antigen proteins or
EBNAs (EBNA1, EBNA2, EBNA3A, EBNA3B, EBNA3C, and EBNA leader protein (LP)),
three latent membrane proteins (LMP1, LMP2A, LMP2B), two small, non-coding RNAs
(EBER1 and EBER2), and the secreted BARF1 protein (Rickinson and Kieff 1996). In addition,
numerous micro RNAs are expressed from the EBV genome from the BHRF1 and BART
regions of the genome. Subsets of these latency genes are expressed for each latency state of
EBV infection, however, EBERs are expressed throughout all latency programs (see Table 1 and
Figure 1).
In the resting B-lymphocytes of the majority of the EBV-infected population, EBV is
predominantly kept in latency 0, a quiescent state in which only EBER1, EBER 2 and BART
RNAs are expressed. No viral proteins are usually expressed in the latency 0 program, although
LMP2A expression is infrequently observed (Thorley-Lawson et al. 1996; Thorley-Lawson
2001). In healthy adults, the restricted gene expression profile of latency 0 allows EBV to remain
silent and efficiently evade the host immune system (Klein et al. 2007).
Latency I is typically associated with both EBV-positive Burkitt’s lymphoma (BL) and
gastric carcinoma (GC) (Rickinson and Kieff 1996); however, latency I infection of memory B
cells is a common form of EBV infection in healthy individuals (Thorley-Lawson and Allday
2008). In addition to expression of EBV miRNAs, BARTs, and EBERs, the only viral protein
expressed during latency I is EBNA1, which is necessary for latent EBV genome persistence
(Rickinson and Kieff 2001). Latency II is similar to latency I in that EBNA1 is the only nuclear
antigen expressed, and its expression occurs from the Qp promoter (Rickinson and Kieff 1996).
In addition to EBNA1, all the LMPs are expressed as well as BARTs, miRNAs, and EBERs. The
latency II program was first recognized in EBV-positive nasopharyngeal carcinoma and
Hodgkin’s lymphoma tissue specimens and has not been observed in healthy individuals
(Fahraeus et al. 1988; Deacon et al. 1993).

4
Figure 1. The Epstein-Barr virus genome. Diagram showing the position and transcription of
the latent EBV genes on the double-stranded viral DNA episome. The latent origin of replication
(oriP) is depicted in orange. The large green solid arrow heads represent exons encoding the six
nuclear antigens (EBNAs 1, 2, 3A, 3B and 3C, and EBNA-LP) and the three latent membrane
proteins (LMPs 1, 2A and 2B), the direction of gene transcription is also indicated by the arrow.
Transcription of the gene encoding EBNA-LP occurs from an inconsistent number of repetitive
exons. LMP2A and LMP2B genes are composed of multiple exons, which are positioned on
either side of the terminal repeat (TR) region. The small blue arrows at the top represent the
genes encoding the two non-polyadenylated RNAs EBER1 and EBER2, which are consistently
expressed during all EBV latencies The long, thin outer green arrow represents EBV
transcription during latency III, in which all six EBNAs are transcribed from either the Cp or Wp
promoter. Differential splicing of the same long primary RNA transcript generates the individual
mRNAs for the different EBNAs. The inner, shorter red arrow represents the Qp promoter-
derived EBNA1 transcript during Latencies I and II. The BamHIA region at the top encodes
BARF0 and BARF1 and many microRNAs. (Modified from Yang, LS & Rickinson, AB, Nat
Rev Cancer, 2004)
172 kb

5
Finally, latency III, also referred to as the “growth program”, involves the expression of all six
EBNAs, the three LMPs, and the aforementioned viral RNA transcripts. Expression of the
six EBNAs occurs from either the Cp or Wp promoters which generate a precursor mRNA
transcript that is differentially spliced to give rise to individual transcripts for each EBNA
(Rickinson and Kieff 1996). Due to the fact that the five EBNAs other than EBNA1 are highly
immunogenic, latency III is not commonly observed in healthy individuals. Conversely, the
latency III program is associated with lymphoblastoid cell lines (LCLs), acute infectious
mononucleosis, and lymphoproliferative disorders. LCLs are generated in vitro when EBV-
infected resting B-cells are immortalized and transformed (Rickinson and Kieff 2001).
Table 1. Epstein-Barr virus latency and expression profiles
Note: Latency programs (I-III) all express EBERs
Latency Program EBNA proteins LMP proteins Associated Disease(s)
0 --- --- ---
I EBNA1
(Qp promoter) --- Burkitt’s lymphoma
II EBNA1
(Qp promoter) LMP1, LMP2A
NPC, GC, Hodgkin’s
diseases
III All 6 EBNAs
(Cp or Wp promoters)
LMP1, LMP2A,
LMP2B
IM, lymphoproliferative
disorders
I.1.4 EBV-associated Diseases
EBV is causally associated with infectious mononucleosis and the development and
progression of several human malignancies of lymphocyte or epithelial origin. EBV infection has
been linked with various types of cancers including Burkitt’s lymphoma, Hodgkin’s diseases,
gastric carcinoma, and nasopharyngeal carcinoma. Furthermore, EBV infection causes several
lymphoproliferative disorders upon immunosuppression including oral hairy leukoplakia
amongst AIDs and organ transplant patients (Delecluse et al. 2007). In the interest of my thesis I
will discuss two EBV-associated epithelial tumours: nasopharyngeal carcinoma and gastric
carcinoma, with more emphasis on the first disease.
I.1.4.1 Nasopharyngeal Carcinoma
Nasopharyngeal carcinoma or NPC is an epithelial tumour that occurs within the
nasopharynx and is marked by the development of a growth or swelling in the neck, hearing loss,
and/or blockage of the nasal pathway. NPC is endemic in South-East Asia, Indonesia, and some

6
regions of Northern Africa, where it is responsible for nearly 20% of all cancers amongst adults
(Shah and Young 2009). There is high incidence for the development of NPC observed amongst
Chinese descendents, which suggests that there may be genetic factors that contribute to the
onset of this disease. In addition to genetic predisposition, some environment carcinogenic
agents are suspected to contribute to NPC development including some dietary components and
preservatives (e.g. salted fish and nitrosamines) (Huang et al. 1978; Yu et al. 1986).
The World Health Organization (WHO) has previously categorized NPC into two
different histological types: keratinizing (WHO type I) and non-keratinizing (WHO types 2 and
3) squamous cell carcinomas (Shah and Young 2009). Non-keratinizing squamous carcinomas
can be further subdivided based on their cell differentiation state; namely carcinomas composed
of differentiated or undifferentiated cells are referred to as WHO2 or WHO3, respectively
(Shanmugaratnam 1978). Undifferentiated NPC, which account for approximately 80% of all
NPC cases, have been consistently linked with latent EBV infection since the mid-1960s (Young
and Murray 2003; Shah and Young 2009). In fact, most NPC arise from the monoclonal
proliferation of latent EBV-infected epithelial cells, as determined by analysis of EBV terminal
repeat length. Early serological studies and in situ hybridization were the first to confirm an
association between the EBV virus and NPC (zur Hausen et al. 1970; Henle and Henle 1976). It
is believed that a single EBV-infected progenitor cell continuously divides, undergoing clonal
expansion and develops into a tumour (Raab-Traub and Flynn 1986; Raab-Traub 2002). The
EBV latency II program is associated with NPC such that EBNA1, LMP2A, EBERs, miRNA,
and possibly LMP1 and/or the secreted BARF1 protein are expressed (Brooks et al. 1992; Seto et
al. 2005). However, the individual contribution of each of these factors to NPC development and
progression is not fully known.
I.1.4.2 Gastric Carcinoma
EBV latent infection is associated with approximately ten percent of all gastric carcinoma
cases. Gastric cancer is the fourth most common cancer worldwide and it is indiscriminate of
regional and ethnic differences (Uozaki and Fukayama 2008). The remaining ~90% of GC is
commonly linked to the presence of the pathogen Helicobacter pylori. EBV-infected GC cells
are typically located in the upper middle area of the stomach, whereas H.pylori-associated GC is
usually found in the lower section of the stomach. EBV-associated GC share some common

7
similarities with NPC. First, the gene expression correlating to the EBV latency II program is
typically exhibited in GC-associated tumours such that EBNA1, LMP2A, BARF1, miRNA and
EBERs are all expressed, but unlike in NPC, LMP1 is not expressed (Fukayama et al. 2008;
Fukayama 2010). Second, GC is believed to develop from the monoclonal proliferation of an
EBV-infected gastric epithelial cell.
I.2 EBV Latent Origin of DNA Replication, oriP
In latent infection, EBV genomes replicate once per cell cycle during cellular S phase
using the cellular DNA replication machinery (Yates and Guan 1991; Chaudhuri et al. 2001).
The viral episomes then evenly segregate to the daughter cells during cell division using an ill-
defined mechanism that involves tethering to mitotic host chromosomes, thereby enabling the
maintenance of constant viral genome copy number. One viral protein, EBNA1 (Epstein-Barr
nuclear antigen 1) and the viral DNA sequence, oriP, are the two factors necessary to maintain
EBV genomes at a stable copy number (Yates et al. 1985).
It will soon be approaching three decades since the discovery of oriP, the EBV latent
origin of DNA replication. OriP, a 1.8kbp viral DNA sequence, was identified from a screen of
multiple EBV genome fragments that were tested for their ability to support autonomous
replication and stable maintenance of plasmids in latent EBV-infected human cells (Yates et al.
1984). Provided that EBNA1 is present, plasmids containing the oriP sequence were shown to be
stably maintained in human cells without selection at a loss rate of ~2-5% per cell division
(Yates et al. 1984; Sugden et al. 1985; Yates et al. 1985). Mutational studies on oriP found that
the sequence is composed of two cis-acting elements; the dyad symmetry (DS) element and
family of repeats (FR), each with distinct functions in bidirectional replication initiation, and
episomal maintenance and transactivation, respectively (Reisman et al. 1985; Reisman and
Sugden 1986; Gahn and Schildkraut 1989).
I.2.1 Dyad Symmetry (DS) Element
The DS element, whose name originates from the 65 bp sequence with a dyad symmetry
within it, is a 120 bp sequence that is located nearly 1Kb from the FR and consists of two sets of
two closely paired EBNA1-binding sites (Rawlins et al. 1985; Reisman et al. 1985). In addition
to the DS and FR regions, oriP contains three copies of a 9 bp sequence, known as nonamers,
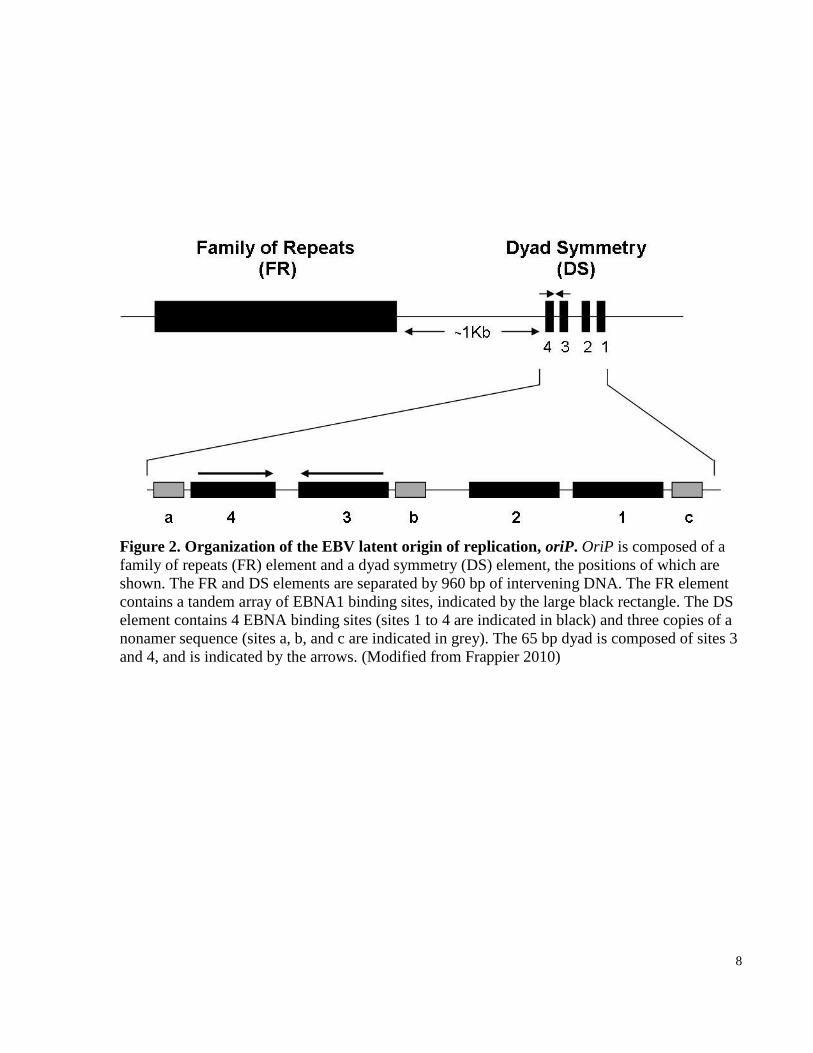
8
Figure 2. Organization of the EBV latent origin of replication, oriP. OriP is composed of a
family of repeats (FR) element and a dyad symmetry (DS) element, the positions of which are
shown. The FR and DS elements are separated by 960 bp of intervening DNA. The FR element
contains a tandem array of EBNA1 binding sites, indicated by the large black rectangle. The DS
element contains 4 EBNA binding sites (sites 1 to 4 are indicated in black) and three copies of a
nonamer sequence (sites a, b, and c are indicated in grey). The 65 bp dyad is composed of sites 3
and 4, and is indicated by the arrows. (Modified from Frappier 2010)

9
two of which flank either side of the DS element and the third is found within it, between
EBNA1 sites 2 and 3 (Niller et al. 1995). The sequences of these nonamers bear a resemblance to
telomeric repeats and were identified as binding sites for telomeric repeat binding factors 1 and 2
(TRF1 and TRF2, respectively), which are recruited in a cell cycle-dependent manner (Deng et
al. 2002; Deng et al. 2003).The four EBNA1-binding sites are each made up of an imperfect18-
bp palindromic sequence that is constitutively bound by EBNA1 dimers throughout the cell cycle
(Hsieh et al. 1993). Although each pair of EBNA1-binding sites (combination of sites 1+2 or
sites 3+4) within the DS can serve as a minimal plasmid replicator, as determined through
mutational studies, efficient replication from the DS is only attained with both pairs of EBNA1-
binding sites and their flanking nonamer repeats (Yates et al. 2000; Koons et al. 2001). The
spatial organization of the two EBNA1-binding sites within each pair is also critical for origin
function such that even altering the 3 bp that separates sites 1 and 2 or sites 3 and 4 abrogates
DS-mediated replication initiation (Harrison et al. 1994; Bashaw and Yates 2001).
Replication initiation occurs at or near the DS region on oriP-containing plasmids, which are
dependent on the DS, not only for their replication but also for their stable maintenance in human
cells in the presence of EBNA1 (Gahn and Schildkraut 1989; Wysokenski and Yates 1989;
Harrison et al. 1994; Little and Schildkraut 1995; Yates et al. 2000). Unlike oriP plasmids,
replication of the EBV genome does not rely solely on the DS element. In fact, several lines of
evidence using 2D gel electrophoresis and small molecule analysis of replicated DNA (SMARD)
have demonstrated the presence of extensive replication initiation zones that exist outside of oriP
which are used to various degrees in different EBV-positive cancer cell lines (Gahn and
Schildkraut 1989; Little and Schildkraut 1995; Norio and Schildkraut 2001; Norio and
Schildkraut 2004). A typical example is EBV replication within Raji cells where only a small
percentage (>10%) of initiation events take place at or close by the DS (Little and Schildkraut
1995; Norio and Schildkraut 2001; Chau and Lieberman 2004; Norio and Schildkraut 2004).
I.2.2 Family of Repeats (FR) Element
The FR element is comprised of an array of imperfect tandem repeats of a 30 bp DNA
sequence, each of which contains a 12 bp AT-rich sequence as well as the 18 bp palindromic
sequence to which EBNA1 dimers bind (Rawlins et al. 1985; Reisman et al. 1985; Frappier and
O'Donnell 1991). The affinity with which EBNA1 binds to its recognition sites within the DS

10
and FR differ due to minor variations in their respective 18 bp palindromic sequences (Jones et
al. 1989; Ambinder et al. 1990). In fact, EBNA1 exhibits a stronger binding affinity to its
recognition sites within the FR compared to those found in the DS element, even when the
sequences between the sites in the FR are mutated (Jones et al. 1989; Ambinder et al. 1990;
Frappier and O'Donnell 1991).
Despite the fact that the DS and FR elements both contain EBNA1 binding sites, they
play separate roles during EBV latency. Upon EBNA1-binding, the FR directs the segregation of
EBV genomes or oriP plasmids during mitosis and serves as a transcriptional enhancer even in
the absence of the DS (Reisman and Sugden 1986; Kanda et al. 2001; Kapoor et al. 2001). In a
recent study, the FR, and not the DS, was shown to be involved in primary EBV infection,
contributing to EBV establishment and human B-cell transformation (Deutsch et al. 2010). Both
EBNA1 as well as EBV genomes have been shown to associate with cellular metaphase
chromosomes (Harris et al. 1985; Petti et al. 1990). The binding of EBNA1 to the FR element
allows for tethering of viral episomes and FR-containing plasmids to condensed human
chromosomes (often referred to as “piggybacking”) as they both segregate during mitosis
(Krysan et al. 1989; Wu et al. 2000; Kanda et al. 2001; Sears et al. 2004). This function is critical
for the faithful segregation and maintenance of the virus in proliferating cells. Furthermore, in
the presence of EBNA1, the FR facilitates nuclear uptake of oriP plasmids and enables their
stable retention, thereby preventing plasmid loss during cell division (Langle-Rouault et al. 1998;
Sears et al. 2003). Although the FR was initially believed to enhance DS-mediated DNA
replication initiation (Wysokenski and Yates 1989), several studies suggest that it is more
probable that the observed increase in replication efficiency in the presence of the FR is as a
result of better retention of oriP plasmids. Finally, additional 2D gel electrophoresis studies have
shown that EBNA1-binding to the FR creates a significant replication fork impediment and can
serve as a replication termination site, which has recently been implicated in EBV genome
maintenance (Gahn and Schildkraut 1989; Ermakova et al. 1996; Dheekollu et al. 2007).
A decade ago, careful analysis of the length of the FR region found in EBV strains
derived from several lymphoblastoid cell lines and B-lymphocytes using PCR amplification
revealed that the number of 30 bp repeats within the FR varies between EBV strains (Fruscalzo
et al. 2001). Importantly, the characteristic number of repeats found in each strain is strictly
maintained during long-term cell passaging, which suggests that maintenance for the FR repeat

11
length may be crucial for the persistence of each strain of EBV (Fruscalzo et al. 2001). Recent
studies by Ali and his colleagues demonstrated that varying the number of EBNA1 binding sites
via FR repeat number affects FR-associated functions (Ali et al. 2009). The extensively studied
B95-8 EBV strain was found to contain 29 repeats within its FR region. This FR region was
previously thought to contain 20 repeats, as this is the number of repeats present in the oriP
DNA sequence which was originally cloned from the B95-8 strain (Baer et al. 1984; Fruscalzo et
al. 2001). The extra 9 repeats, which are believed to have been deleted during the sub-cloning of
EBV genome fragments into plasmids, contribute 252 bp to the 3’ end of the B95-8 FR region
and potentially form a stem-loop DNA structure (Yates et al. 1984; Fruscalzo et al. 2001).
Relative to the FR containing 20 repeats, the full-length FR (29 repeats) was shown to improve
B-cell transformation, but reduced the FR transactivation function and had no effect on plasmid
replication (Ali et al. 2009). Together, these findings are particularly remarkable given that seven
to nine copies of the 30 bp repeat are sufficient to constitute a fully functional FR element
(Wysokenski and Yates 1989).
I.2.3. Cellular Proteins Recruited to oriP
During latent infection, EBNA1 is the only viral protein that functions in EBV genome
replication. However, EBNA1 is an atypical DNA virus origin-binding protein that lacks DNA
helicase activity and is incapable of unwinding and activating viral origin sequences (Frappier
and O'Donnell 1991). Consequently, EBV is dependent on host cellular replication proteins to
replicate its genomes. Indeed, a collection of studies have found subunits from the cellular origin
recognition complex (ORC) and minichromosome maintenance (MCM) complex to be recruited
to the DS element within oriP and function in a cell cycle-dependent manner (Chaudhuri et al.
2001; Dhar et al. 2001; Schepers et al. 2001).
ORC is an evolutionarily well-conserved complex of six subunits that binds to replication
origins in an ATP-dependent manner and initiates the assembly of replication machinery (Bell
and Dutta 2002). Using chromatin immunoprecipitation (ChIP) assays, human ORC subunits
were detected in regions at or near the DS element in vivo (Chaudhuri et al. 2001; Schepers et al.
2001). However, footprinting assays found no direct interaction between ORC and DNA at oriP
(Nieduszynski 2001). Nevertheless, the requirement for ORC in oriP-mediated replication was
demonstrated using oriP plasmids which failed to replicate in cells carrying a hypomorphic

12
mutation in the ORC2 gene (Dhar et al. 2001). Subsequently, EBNA1 and its association with
oriP were found to be factors necessary for efficient ORC recruitment to the DS element (Julien
et al. 2004). There is in vivo evidence that suggests that ORC recruitment to the DS might be
facilitated by the interaction of EBNA1 with the ORC2 subunit (Dhar et al. 2001). However,
previous findings showed that EBNA1 may not directly interact with ORC2, but may interact
indirectly through G-quartet RNA (Norseen et al. 2008).
The first step in the replication initiation cascade is the association of the ORC complex with
replication origins during the cellular G1 phase, followed by the recruitment of Cdt1, Cdc6, and
the MCM complex to form what is known as the pre-replicative complex (pre-RC) (Bell and
Dutta 2002). The MCM complex is the cellular replicative helicase consisting of six subunits that
function together in unwinding the DNA at a replication fork, enabling subsequent DNA
synthesis (Ishimi 1997; Labib et al. 2001). The MCM complex moves along the DNA just ahead
of the replication fork after initiation and to maintain fork progression (Arias and Walter 2007).
Once dissociated, the Cdt1 interacting protein, Geminin, prevents the MCM complex from being
reloaded at replication origins until the end of mitosis (Arias and Walter 2007). Based on the
following observations, it is also highly likely that the MCM complex serves as the helicase for
EBV genome replication during latency. First, both MCM2 and MCM3 subunits are recruited to
oriP in a cell cycle-regulated manner. Similar to cellular replication origins, these subunits
associate with DNA during G1 and dissociate during S phase (Chaudhuri et al. 2001; Zhou et al.
2005). Second, ectopic expression of Geminin was shown to reduce oriP-mediated replication
and oriP replication function was rescued by Cdt1 overexpression (Dhar et al. 2001). Therefore,
in order to replicate its genome during latency, EBV appears to utilize host cellular replication
machinery through recruitment of the pre-RC complex to its replication origin, oriP, via EBNA1.
To date, there has been a series of findings that demonstrate the involvement of several
cellular factors, in addition to the members of the pre-RC, in oriP-based replication and
segregation. First, the nonamers juxtaposed to the DS element in oriP were shown to increase
oriP replication efficiency through EBNA1-dependent binding of TRF2, which was found to
facilitate the recruitment of the ORC complex to oriP (Atanasiu et al. 2006). TRF2 was recently
found to interact with the DNA damage checkpoint protein and member of the ATM (ataxia-
telangiectasia-mutated) pathway, Chk2. Chk2 is capable of phosphorylating TRF2, which in turn
is necessary for the direct interaction between the ORC1 subunit and TRF2 (Zhou et al. 2010).

13
Using oriP-containing plasmids, Chk2 was shown to be involved in regulating oriP replication
as well as plasmid maintenance, and it was proposed that Chk2 phosphorylation of TRF2 was
important for these functions, (Zhou et al. 2010). Although both TRF1 and TRF2 bind the oriP
nonamer sequences, they have opposing functions. Unlike TRF2, which interacts with ORC and
stimulates replication initiation at the DS element, TRF1 does not interact with ORC and appears
to antagonize TRF2 function, thereby disrupting DS-mediated replication (Deng et al. 2003).
Studies on the temporal regulation of EBV genome replication have shown that replication at
oriP initiates at mid-to-late S phase, which is important for stable maintenance of EBV episomes
(Zhou et al. 2009). This group also demonstrated the importance of TRF2 in the timing of
replication initiation at oriP, such that TRF2 depletion led to both earlier oriP replication and the
instability of EBV genomes. Ultimately, these results contribute to a model where EBNA1
stimulates the association of TRF2 with its nonamer binding sites within the DS, to which ORC
is subsequently recruited, thereby promoting replication initiation at oriP.
EBNA1, which constitutively binds its recognition sites within oriP, is able to hinder the
progression of replication forks through the FR region (Dhar and Schildkraut 1991; Hsieh et al.
1993; Aiyar et al. 2009). It has been suggested that impeding replication fork migration through
the FR is functionally linked to EBV episomal maintenance and copy number (Dhar and
Schildkraut 1991; Aiyar et al. 2009). The evolutionarily conserved human proteins, Timeless
(Tim) and Tipin (Timeless-interacting protein) are involved in preserving the stability of
replication forks once they reach barriers such as repetitive DNA sequences or secondary
structure (Gotter et al. 2007). There have also been studies that suggest that yeast homologues of
these two proteins may play a role in sister chromatid cohesion as well as segregation of
metaphase chromosomes (Mayer et al. 2004; Leman et al. 2010). Very recently, Tim and Tipin
have been shown to be recruited to both the FR and DS elements of oriP in S-phase using ChIP
assays in cell lines latently infected with EBV, including Raji. This localization of Tim and Tipin
was dependent on the expression of EBNA1, which was found to weakly bind Tim during late S-
phase (Dheekollu and Lieberman 2011). Dheekollu and Lieberman found that transient or stable
depletion of Tim from cells, using siRNA and shRNA methods, respectively, resulted in reduced
oriP DNA replication as well as loss of EBV episomes. This study suggested that Tim plays a
role in the EBV life cycle, contributing to stability of replication forks at the EBNA1-bound FR
barrier as well as the EBV episomal maintenance.

14
I.3 Epstein-Barr Nuclear Antigen 1 (EBNA1)
The viral protein EBNA1, as derived from the well-studied B95-8 EBV strain, is
composed of 641 amino acids (aa) (Rickinson and Kieff 2001). Since being identified in 1973,
EBNA1 has been shown to be critical for latent EBV infection. Consistent with this is the fact
that EBNA1 is the sole trans-acting viral protein that is essential for oriP replication and
segregation functions, and it is the only viral factor that is expressed in nearly all forms of EBV
latency and its associated diseases (Yates et al. 1985; Kieff 1996). In addition to the requirement
of EBNA1 for EBV episomal maintenance, it is important for the transcriptional activation of
some of the EBV latency genes (Rickinson and Kieff 2001).
I.3.1 Overview of Domain Structures
EBNA1 is composed of several distinct regions that contribute to its various functions
(Figure 3). The N-terminus of EBNA1 contains the first glycine-arginine (Gly-Arg) rich region
from amino acids 33-53, after which a central glycine-glycine-alanine (Gly-Ala) rich repeat
region is found (aa 90-325). The second Gly-Arg rich region is within the core of EBNA1
between aa 325-376. Lastly, the C-terminus of EBNA1 encodes a nuclear localization signal or
NLS (aa 379-386) as well as its DNA-binding and dimerization domain (aa 459-607).
The precise number of amino acids found in EBNA1 is dependent on the size of its N-
terminal Gly-Ala repeat sequence, which varies amongst different strains of EBV (Baer et al.
1984). This large Gly-Ala repeat has been associated with evasion of host immune response.
These repeats inhibit efficient presentation of EBNA1-derived peptides on MHC Class I proteins
and as a result, circulating cytotoxic T cells cannot detect and target EBNA1 (Blake et al. 1997).
Another mechanism involves inhibiting the generation of defective ribosomal products or DRiPs
of EBNA1. DRiPs are protein fragments produced as a result of incomplete translation and they
are now recognized to be the source of EBNA1 peptides presented on MHC Class I molecules as
opposed to proteasomal degradation products (Yin et al. 2003; Fahraeus 2005; Tellam et al.
2007). Importantly, the deletion of this Gly-Ala region has no effects on EBNA1 replication,

15
Figure 3. EBNA1 functional domains. A schematic representation of the domain structure of
wild-type EBNA1 as well as EBNA1ΔG/A (lacking the large Gly-Ala repeat region), which is
used in the studies discussed in this thesis. The functional elements of interest are indicated. The
regions of EBNA1 that are known to be required (black boxes) or contribute but are not essential
(grey boxes) for its replication, segregation, or transactivation functions are shown here.
(Modified from Frappier 2010).

16
segregation, or transcriptional regulatory functions (Yates et al. 1985; Yates and Camiolo 1988).
Consequently, many laboratories, including ours, use an EBNA1 with the Gly-Ala repeat region
deleted, which maintains all of wild-type EBNA1 functions.
EBNA1 stably binds as a homodimer to its recognition sites within the DS and FR via its
C-terminus between amino acids 459 to 607, constituting its DNA-binding and dimerization
domain (Ambinder et al. 1991; Frappier and O'Donnell 1991; Shah et al. 1992). This DNA-
binding and dimerization domain is responsible for recognizing the 18 bp palindromic sequences
found within the DS and FR elements (Summers et al. 1996). Analysis of crystal structures of
this region of EBNA1 bound and unbound to one of its 18 bp palindromic recognition sequences
reveals the presence of two domains: a core eight-stranded antiparallel β-barrel (aa 504-604) and
a flanking domain (aa 461-503) that is comprised of an α-helix oriented perpendicularly to DNA
(Bochkarev et al. 1995; Bochkarev et al. 1996; Bochkarev et al. 1998). The core β-barrel domain
is not only responsible for EBNA1 homodimerization which is necessary for DNA-binding, but
it also contains four α-helices (one pair per EBNA1 monomer) (Bochkarev et al. 1996). Two
helices, one from each monomer pair, together make sequence-specific contacts and serve to
recognize EBNA1 binding sites; hence, they are referred to as recognition helices (Cruickshank
et al. 2000). The core β-barrel and flanking domains of EBNA1 function together to allow stable
interaction with DNA recognition sequences, such that the core domain makes contacts with the
major groove of the DNA, while the flanking domain simultaneously inserts into the minor
groove (Cruickshank et al. 2000). Not surprisingly, this C-terminal region is fundamental for all
EBNA1-associated functions at oriP. For example, studies by Kirchmaier and Sugden showed
that the EBNA1 C-terminal fragment can out-compete full-length EBNA1 for binding to oriP
plasmids, thus functioning as a dominant-negative inhibitor of EBNA1 (Kirchmaier and Sugden
1997). Interestingly, the EBNA1 DNA-binding and dimerization region was found to be
structurally homologous to the DNA-binding domain of the papillomavirus E2 protein,
regardless of the fact that the two viral protein have little or no sequence homology (Hegde et al.
1992; Edwards et al. 1998). The DNA-binding domain of E2 also contains a recognition helix
that, similar to that of EBNA1, facilitates all of the sequence-specific interactions inherently
associated with the domain (Hegde et al. 1992). Therefore, together the core β-barrel and
flanking domain allow EBNA1 to stably bind and recognize its target sites within oriP.

17
Although the DNA-binding and dimerization domain of EBNA1 is necessary for its
replication function, additional regions of EBNA1 contribute significantly to this activity.
EBNA1 contains two stretches of Gly-Arg repeats (amino acids 33-53 and amino acids 325-376),
found at the N-terminus and in the middle of EBNA1, respectively. To date, the role of the N-
terminal Gly-Arg repeat (aa33-53), which is found between the 8-to-67 region, remains elusive.
However, using an EBNA1 deletion mutant, the 8-to-67 region was found to contribute to
EBNA1-mediated transactivation, segregation, and mitotic attachment functions (Avolio-Hunter
and Frappier 1998; Wu et al. 2002). Amino acids 33-89, which encompass the N-terminal Gly-
Arg repeat, were shown to function cooperatively with the central Gly-Arg repeat (aa325-376) to
bring the DS and FR elements together upon binding by EBNA1, a phenomenon referred to as
DNA looping or linking (Middleton and Sugden 1992; Mackey et al. 1995; Avolio-Hunter and
Frappier 1998). Moreover, both regions 33-89 and 325-376 are necessary for facilitating ORC
recruitment to the DS element (Norseen et al. 2008). Previous mutational studies have shown
that the Gly-Arg-rich region between amino acids 325-376 of EBNA1 is also required for mitotic
chromosome attachment and its segregation function, but this region is dispensable for plasmid
replication (Shire et al. 1999; Wu et al. 2002).
I.3.2 EBNA1 Functions
I.3.2.1 EBNA1 DNA Replication Function
Despite its apparent lack of intrinsic enzymatic activity, EBNA1 facilitates the initiation
of DNA replication at the DS element of oriP. Similar to host cellular chromatin, EBV circular
genomes associate with histones and are assembled into nucleosomal structures (Shaw et al.
1979). Studies from ten years ago elucidated the mechanism by which EBNA1 gains access to its
DNA recognition sequence at oriP by destabilizing the nucleosome structure and effectively
displacing the core histones from the DNA (Avolio-Hunter et al. 2001).
EBNA1 homodimers assemble in a cooperative fashion on neighbouring recognition sites
at the DS such that the nearby dimers interact via their DNA binding and dimerization domains
(Summers et al. 1996). It is thought that cooperative binding of EBNA1 dimers to their
recognition sites in the DS causes unwinding and alterations to the DNA structure (Bochkarev et
al. 1996). Notably, the interaction between an individual recognition site with the DNA binding
and dimerization domain of EBNA1 not only causes local distortion and DNA unwinding, but

18
induces a smooth bend in the DNA, which are also characteristic of an origin-binding protein
(Bochkarev et al. 1996). Consistent with these observations, previous studies have confirmed the
presence of local distortions in DNA upon interaction between EBNA1 with its recognition sites
at the DS using potassium permanganate (KMnO4) sensitivity (Frappier and O'Donnell 1992).
Potassium permanganate oxidizes pyrimidine bases in both single-stranded DNA or distorted
double-stranded DNA (Bui et al. 2003). Essentially, the EBNA1-DS interaction leads to changes
in the DNA structure such that two thymine residues, each from a single EBNA1 binding site,
become exposed and susceptible to permanganate oxidation (Frappier and O'Donnell 1992;
Hsieh et al. 1993; Summers et al. 1997). These studies also predict that similar twisting of the
DNA occurs when EBNA1 binds to its other recognition sites that do not possess a thymine
residue and will therefore, not display sensitivity to potassium permanganate.
Studies by Frappier and O’Donnell (Frappier and O'Donnell 1991) made use of electron
microscopy to visualize EBNA1-oriP interactions and demonstrated the formation of looped
DNA structures upon binding of EBNA1 to the oriP sequence. Importantly, the FR and DS
elements were specifically brought into close proximity through interactions between EBNA1
complexes at both sites. If more than one oriP molecule is present, the respective EBNA1
complexes can interact and create a bridge between the DNA molecules, whereas interactions
between EBNA1 complexes at the DS and FR elements on one oriP molecule can effectively
loop out the intervening DNA (Frappier and O'Donnell 1991; Su et al. 1991; Middleton and
Sugden 1992). Both modes of interactions have been shown to be mediated by residues 327-377
and 40-89 of EBNA1 and together they stabilize binding of EBNA1 at oriP, which allows for
assembly of host replication machinery at the DS element (Su et al. 1991; Frappier et al. 1994;
Avolio-Hunter and Frappier 1998). Due to the fact that these two regions are associated with
EBNA1 DNA replication, segregation, and transcriptional activation functions, it is possible that
the looping out and cross-linking of DNA by EBNA1 may be functionally important (Mackey
and Sugden 1999; Shire et al. 1999).
It is believed that a combination of both nucleosome disruption and distortion of DNA at
oriP by EBNA1 enables it to facilitate replication initiation by the cellular machinery. Consistent
with this hypothesis, replication protein A (RPA), which binds, coats, and stabilizes single-
stranded DNA, has been shown to interact with EBNA1 and be recruited by EBNA1 to oriP,
which suggests an early role for RPA in replication initiation at the DS (Zhang et al. 1998).

19
Moreover, modifications of EBNA1 appear to play a role in modulating its DNA replication
function. Poly(ADP-ribose) polymerase 1(PARP1) is a highly abundant, NAD-dependent
enzyme that is involved with a variety of cellular processes including DNA repair and apoptosis
(Krishnakumar and Kraus 2010). PARP1 is a chromatin-associated enzyme that catalyzes the
covalent attachment of ADP-ribose polymers (PAR) to protein substrates which can modulate
their respective functions and activities (Krishnakumar and Kraus 2010). Other member of the
PARP family is the telomere-associated poly(ADP-ribose) polymerase 1 and 2 (TNKS1 and
TNKS2), which have not only been shown to associate with telomere repeat sequences, but also
EBNA1 and both the DS and FR elements of oriP (Deng et al. 2002; Deng et al. 2005).
Recruitment of PARP1 and TNKS1 to the oriP region was shown to be dependent on EBNA1
(Deng et al. 2002; Deng et al. 2005; Tempera et al. 2010). EBNA1 has been shown to be a
substrate of PARP1 and TNKS1 and is subject to PARylation by both proteins, which negatively
regulates EBNA1 replication function (Deng et al. 2005; Tempera et al. 2010). An enhancement
of oriP-mediated replication activity was observed upon small hairpin RNA (shRNA) targeted
depletion of either TNKS1 or PARP1, which suggests that both proteins serve to inhibit oriP
replication in a PARylation (of EBNA1) -dependent manner (Deng et al. 2005; Tempera et al.
2010). Moreover, an increase in association of ORC2, EBNA1, MCM3 at oriP was observed
when PARP1 was silenced by targeted shRNA or functionally inhibited using pharmacological
agents (Tempera et al. 2010). Finally, these studies suggest that there may be DNA structural
changes at the DS element which are caused by PARylation of EBNA1 and decrease its binding
affinity for its recognition sites and reduce ORC recruitment to oriP (Deng et al. 2005; Tempera
et al. 2010).
TAFIβ, part of the NAP family of nucleosome assembly proteins, was found to interact
with EBNA1 through affinity chromatography and to localize to both the DS and FR elements of
oriP in an EBNA1-dependent manner by ChIP analysis (Holowaty et al. 2003; Wang and
Frappier 2009). Wang and Frappier not only showed that TAFIβ was important for EBNA1
transactivation function, but demonstrated a role for TAFIβ in suppressing EBNA1-mediated
DNA replication from the DS element of oriP (Wang and Frappier 2009). Therefore, these
studies indicate that the interaction of TAFIβ with EBNA1 on DS negatively regulates
replication from the DS.

20
I.3.2.2 EBNA1 DNA Segregation Function
Maintaining latent EBV infection in proliferating cells is due to EBNA1-facilitated
replication of the viral genome and its ability to efficiently segregate the viral episome to each
daughter cell during cell division in order to maintain a stable copy number of EBV episomes.
The EBV genome does not contain a centromeric sequence; therefore, segregation of the viral
episome is unlike the mechanism used by host chromosomes. Segregation of EBV genomes
requires only two viral factors: the trans-acting EBNA1 protein and the cis-acting FR element of
oriP (Lupton and Levine 1985; Krysan et al. 1989). Consistent with this, the introduction of the
FR element into plasmids that contain heterologous replication origin sequences ensures their
stability, although not indefinitely, in human or yeast cells expressing EBNA1 (Krysan et al.
1989; Simpson et al. 1996; Kapoor et al. 2001).
I.3.2.2.1 EBNA1 Domains Contributing to Plasmid Segregation
The most widely-accepted model to explain EBV faithful transmittance in host cells is
the mitotic tethering model. This model proposes that FR-bound EBNA1 tethers EBV episomes
to condensed cellular chromosomes to allow their equal partitioning when sister chromatids
separate to daughter cells at the end of mitosis. Whereas, binding of EBNA1 dimers to the FR
element within the EBV episome is mediated through its C-terminal DNA-binding and
dimerization domain, attachment of EBNA1 to mitotic chromosomes involves sequences in the
central and N-terminal regions. By fusing N-terminal EBNA1 peptides to green fluorescent
protein (GFP), three regions of EBNA1 (aa 8-54, 72-84, 328-368) were found that can
independently interact with metaphase chromosomes (Marechal et al. 1999). Similarly, using
GFP-tagged EBNA1 fragments, another group found that amino acids 1-89 and 323-386 of
EBNA1 each demonstrated a weak association with mitotic chromosomes, but, collectively, a
more robust interaction was observed with an EBNA1 peptide encompassing both regions (Hung
et al. 2001). Subsequent studies using deletions of the aforementioned regions in the context of
full-length EBNA1 showed that amino acids 325-376 have a key role in EBNA1’s segregation
function by contributing significantly to its chromosomal attachment (Wu et al. 2000; Nayyar et
al. 2009). This was demonstrated by analyzing the oriP plasmid maintenance ability in human
and yeast cells and mitotic chromosomal attachment of the EBNA1Δ325-376 mutant which was
defect in both capacities but still retained its DNA replication activity (Shire et al. 1999; Wu et

21
al. 2000; Wu et al. 2002). These studies provided clear evidence that the replication and
segregation functions of EBNA1 are distinctive and separable.
Comparing the oriP plasmid maintenance ability of wild-type EBNA1 and its deletion
mutant EBNA1Δ8-67 showed that this region (aa 8-67) may contribute to the binding of EBNA1
to cellular mitotic chromosomes. The EBNA1Δ8-67 mutant was shown to interact with
metaphase chromosomes less well than wild-type as shown by immunofluorescence staining, and
a four-fold decrease in the maintenance of the oriP plasmid was observed with the mutant (Wu et
al. 2002). The specific residues between amino acids 8-67 that are involved in EBNA1’s
chromosomal attachment and segregation function have not yet been defined, although deletion
of the Gly-Arg repeat found between amino acids 33-53 did not affect plasmid maintenance,
therefore, this sequence is not important for chromosome attachment or segregation (Wu et al.
2002).
I.3.2.2.2 Involvement of EBP2 in EBNA1-Mediated Plasmid Segregation
A yeast two-hybrid screen previously identified the EBNA1-interactor, human EBNA1-
binding protein 2 (hEBP2), which was shown through a reconstituted Saccharomyces cervisiae
system to support EBNA1-mediated plasmid partitioning and attachment of EBNA1 to mitotic
chromosomes (Shire et al. 1999; Kapoor et al. 2001; Kapoor and Frappier 2003). Importantly,
the hEBP2-EBNA1 interaction involves the region between amino acids 325-376 and 8-67 of
EBNA1 and abrogation of this interaction in the yeast system led to decreases in EBNA1 mitotic
attachment and partitioning of oriP plasmids (Shire et al. 1999; Nayyar et al. 2009). Our
laboratory has also demonstrated that EBNA1 is subject to serine phosphorylation in vivo
between amino acids 325-376. Using serine-to-alanine substitutions and aspartic acid residues
that mimic phosphorylation, phosphorylation of serine residues within this region was shown to
influence EBNA1-hEBP2 interactions in yeast to affect the segregation function of EBNA1 in
both yeast and mammalian cells (Shire et al. 2006). In addition, the importance of EBP2 for
EBNA1 attachments to mitotic chromosomes was confirmed in human cells. Specifically, EBP2
silencing in human cell lines led to release of both EBNA1 and oriP plasmids from metaphase
chromosomes into the soluble cell fraction (Kapoor et al. 2005)
By immunofluorescence microscopy, EBNA1 associates with metaphase chromosomes
ahead of EBP2 and their subsequent interaction occurs within the second half of mitosis (Nayyar

22
et al. 2009). This may tie in with the observations of Sears et al. which suggest that EBNA1 can
associate with metaphase chromosomes independent of EBP2 (Sears et al. 2004). This group
focused on the Gly-Arg repeats between 33-53 and 325-376 and drew a parallel between these
repeats and the AT-hooks found in cellular proteins and associate with AT-rich DNA sequences.
They demonstrated that the EBNA1 Gly-Arg repeats are capable of binding AT-rich DNA in
vitro (Sears et al. 2004). Interestingly, when the N-terminus of EBNA1 is substituted with the
cellular protein HMG1a, this fusion protein still maintains plasmid replication and segregation
abilities with efficiencies that are comparable to that of wild-type EBNA1 (Hung et al. 2001;
Sears et al. 2003). Moreover, the HMG1a protein utilizes its Gly-Arg repeats, which are also
referred to as AT-hooks, to associate with cellular chromosomes (Aravind and Landsman 1998).
Therefore, although EBP2 is required for stability of the attachment of EBNA1 at mitotic
chromosomes, EBNA1 may also possess its own chromosome-binding activity which contributes
to the strength of the interaction.
I.3.2.2.3 The Mitotic Tethering or “Piggybacking” Model
Grogan et al. were the first group whose findings alluded to the ability of EBV episomes
to piggyback onto host cellular chromosomes during mitosis, ensuring their stable transmission
to both daughter cells during cell division (Grogan et al. 1983). Their immunofluorescence
microscopy studies in EBV-positive Raji cells illustrated the localization of EBNA1 to
condensed mitotic chromosomes (Grogan et al. 1983). Additional fluorescence in situ
hybridization (FISH) experiments demonstrated the association of EBV episomes with cellular
metaphase chromosomes in Burkitt’s lymphoma cell lines (Harris et al. 1985). Moreover, this
association of EBV episomes was shown to be dependent on EBNA1, as it is the only latent viral
protein with this mitotic chromosome-binding activity (Petti et al. 1990).
Soon after, EBV-based plasmids such as the oriP plasmid were used to help elucidate the
mechanism by which the EBV episome-metaphase chromosome interaction occurs. Similar to
EBV episomes, oriP plasmids were shown to localize to host chromosomes and this association
was dependent on EBNA1 expression as well as interactions between EBNA1 and oriP
(Simpson et al. 1996; Kanda et al. 2001). Specifically, the FR element of oriP (on a plasmid) is
sufficient to mediate its chromosomal attachment and stable segregation during cell division
(Krysan et al. 1989; Kanda et al. 2001). Moreover, the interaction between EBNA1 and

23
condensed mitotic chromosomes is indispensable for oriP plasmid segregation, as the
EBNA1Δ325-376 mutant, which is nuclear but unable to bind mitotic chromosomes, could not
partition oriP plasmids (Shire et al. 1999; Wu et al. 2000; Hung et al. 2001). Finally, by
substituting the this region of EBNA1 with the chromosome-binding sequences of high-mobility-
group-I (HMG-1) or histone H1, Hung et al. demonstrated that region 325-376 of EBNA1 was
sufficient and essential for EBNA1 mitotic chromosomal attachment and EBNA1-mediated oriP
plasmid maintenance (Hung et al. 2001). The stable maintenance of oriP-containing plasmids
requires a combination of both their replication and segregation during cell division.
The mitotic tethering or “piggybacking” model may be a common mechanism utilized by
other double-stranded DNA viruses that exist in low copy numbers in infected cells. For
example, only other identified human gamma herpesvirus, Kaposis sarcoma associated
herpesvirus (KSHV) as well as bovine papillomavirus (BPV) also appear to use the same
partitioning mechanism whereby their extrachromosomal, circular genomes attach to cellular
chromosomes and are evenly segregated to daughter cells. LANA1 and E2, which are trans-
acting viral origin binding proteins structurally homologous to EBNA1 for KSHV and BPV,
respectively, have also been shown to tether viral genomes to host mitotic chromosomes
(Lehman and Botchan 1998; Ballestas et al. 1999; Cotter and Robertson 1999; Ilves et al. 1999;
Barbera et al. 2004; Silla et al. 2005). Similar to EBNA1, both LANA and E2 tethering activities
have been shown to be facilitated by interactions with cellular proteins (Cotter and Robertson
1999; You et al. 2004; Barbera et al. 2006; You et al. 2006).
I.3.2.2.4 Equal Partitioning of EBV Episomes to Daughter Cells Following Mitosis
EBV episomes, ranging from 5 to 200 in number, persist stably at a constant copy
number in several latent EBV-infected cell lines (Sternas et al. 1990; Kieff 1996). Maintaining a
constant copy number of EBV episomes and oriP plasmids throughout several cell divisions
requires a system that ensures their equal distribution between daughter cells during cell division.
Utilizing a random mechanism for EBV genome partitioning in infected cells would likely be
inefficient for the virus, which is stably maintained throughout the lifetime of its human host.
Past observations by Harris et al. using FISH demonstrated a stochastic association of oriP
plasmids with cellular mitotic chromosomes, such that the plasmids did not localize to specific
areas on the chromosomes (e.g. centromeric or telomeric regions) (Harris et al. 1985). Although

24
oriP plasmids and EBV episomes do not appear to be targeted to any identifiable chromosomal
regions, FISH studies in Burkitt’s lymphoma cells have shown that signals corresponding to
EBV episomes were symmetrically localized on sister chromatids (Delecluse et al. 1993).
Consistent with this finding, by combining FISH and immunofluorescence techniques Kanda et
al. showed that EBNA1 localized with oriP plasmids and both, together, were found
symmetrically distributed on the surfaces of sister chromatids during mitosis (Kanda et al. 2007).
Close examination revealed paired EBNA1 dots in G2 phase that appear as a result of
concatenated oriP plasmids, which are formed upon replication of circular DNA molecules and
are eventually disconnected by topoisomerase II, as confirmed using a targeted inhibitor (Kanda
et al. 2007). Accordingly, this group proposed an association between the utilization of
catenation as a means to pair replicated plasmids and their equal partitioning during cell division.
I.3.2.3 EBNA1 Transcriptional Regulation Function
In addition to its functions in DNA replication and segregation, EBNA1 can activate the
transcription of some EBV latency genes and autoregulate its own expression. Early studies
demonstrated that in the presence of EBNA1, the FR element increased the expression of
reporter genes when placed upstream or downstream of the promoter, which suggested that the
FR element can serve as an enhancer element (Lupton and Levine 1985; Reisman and Sugden
1986). Studies performed by Wysokenski and Yates subsequently showed that only 6-7 EBNA1
recognition sites within the FR region were necessary for the transcriptional enhancer function of
the FR element (Wysokenski and Yates 1989).
When bound to the FR element of oriP, EBNA1 can activate transcription from the EBV
Cp and Wp promoters, from which all viral nuclear antigen (EBNA) genes, including the
EBNA1 gene, are transcribed (Sugden and Warren 1989; Gahn and Sugden 1995). EBNA1 was
also found to activate transcription from latent promoters that regulate expression of the viral
LMP1 protein, which is essential for EBV-mediated B cell transformation (Gahn and Sugden
1995; Kieff 1996). Besides its C-terminal DNA-binding and dimerization domain, two other
regions of EBNA1, amino acids 61-83 and 325-376, have been shown to be responsible for its
transcriptional activation function (Mackey and Sugden 1999; Ceccarelli and Frappier 2000; Wu
et al. 2002; Kennedy and Sugden 2003). While the EBNA1Δ61-83 mutant was found to be
defective in transcription regulation, it still maintained the DNA replication and segregation

25
functions, showing that EBNA1-mediated transcription is distinct from its other functions (Wu et
al. 2002). Further analysis for the 325-376 transcriptional activation region showed that four
serine-to-aspartate substitutions abolished the transcriptional activation function of EBNA1. This
mutational analysis not only provided evidence that sequences other than the Gly-Arg stretches
in this region were important for EBNA1-mediated transcription, but also showed the potential
for regulation of this function by phosphorylation of the four serine residues (Shire et al. 2006).
Not only does EBNA1 regulate expression of the other five EBNA proteins, it also
regulates its own expression. During latency III phase of EBV infection, expression of all six
EBNAs is dependent on regulation at the Cp promoter (Rogers et al. 1990; Woisetschlaeger et al.
1990; Altmann et al. 2006), however, the Qp promoter is solely responsible for EBNA1
expression throughout latencies I and II, during which the other five EBNAs are not expressed
(Schaefer et al. 1995). This repression is mediated by binding of EBNA1 to its two recognition
sites downstream of the Qp promoter for which EBNA1 has a lower affinity compared to the
sites found in the DS and FR elements of oriP (Ambinder et al. 1990; Sample et al. 1992). It is
possible that this may serve as an autoregulatory mechanism such that when EBNA1 levels
exceed a threshold and the EBNA1-binding sites within DS and FR elements are saturated,
EBNA1 will then bind its recognition sites near the Qp promoter to inhibit its own expression in
latencies I and II. Recent findings by Yoshioka et al. (Yoshioka et al. 2008) have suggested that
EBNA1-mediated repression at the Qp promoter does not occur by direct inhibition as formerly
understood, but rather by post- or co-transcriptionally inhibiting processing of pre-mRNA
transcripts.
Several cellular proteins might contribute to the transcriptional activation function of
EBNA1. Many cellular proteins including the mitochondrial protein P32/TAP, NAP1, and
TAF1β, have been shown to interact with the 325-376 transcriptional activation region of
EBNA1 (Holowaty et al. 2003). P32/TAP possesses a C-terminal transcriptional activation
domain which not only associates with the transcription factor TFIIB, but also mediates its
interactions with EBNA1 (Yu et al. 1995; Wang et al. 1997). P32/TAP is known to interact with
residues 40-60 and 325-376 of EBNA1 (Wang et al. 1997), and also has been shown to localize
to the EBV oriP region in vivo (Van Scoy et al. 2000). Therefore, P32/TAP has been implicated
in playing a role in EBNA1-mediated transcriptional activation and latent replication of the EBV
genome. NAP1 is a highly conserved nucleosome assembly protein that is well-known for its

26
involvement in viral transcription, specifically for the bovine papillomavirus and more recently,
for EBV. Previous studies have shown that binding of NAP1 to the bovine papillomavirus
protein E2, which is a functional homologue of EBNA1, increases its transcriptional activity
(Rehtanz et al. 2004). More recently, NAP1 has been shown using reporter assays and RNA
interference, as well as ChIP to be important for EBNA1-mediated transactivation and to localize
to the FR element of oriP, respectively (Wang and Frappier 2009). Bromodomain protein 4
(Brd4) has been previously implicated in E2-mediated transcriptional activation of bovine
papillomavirus (BPV) (You et al. 2004; McPhillips et al. 2006). Our laboratory has previously
found that Brd4 can interact with region 61-83 of EBNA1, which is responsible for the
transcriptional activation function of EBNA1 (Lin et al. 2008). Brd4 was also shown to be
preferentially localized to the FR transcriptional enhancer element in EBV genomes (Lin et al.
2008). In addition, both depletion and overexpression of Brd4 was shown to inhibit the
transcriptional activation activity of EBNA1 via the FR enhancer element (Lin et al. 2008). This
is thought to be a result of disrupting ternary complex formation, and it also demonstrates a role
for Brd4 in EBNA1-mediated transcriptional activation (Lin et al. 2008).
I.3.2.4 EBNA1: Roles in Cell Transformation and Immortalization
During latent infection, the controlled expression of a limited set of latent viral genes
enables EBV to immortalize its host cell, which in turn can initiate the development of lymphoid
and epithelial tumours (Rickinson and Kieff 1996). Multiple lines of evidence have led to the
hypothesis that EBNA1 is an important factor involved in the transformation and
immortalization EBV-infected cell. However, uncovering whether EBNA1 contributes directly
to these events has proven to be challenging due to its required expression for EBV genome
maintenance in proliferating cells.
EBNA1 is the sole viral protein that is expressed in all EBV-associated tumours and all
EBV latency states in proliferating cells (Kieff and Rickinson 2001; Frappier 2010), raising the
possibility that it is involved in the proliferation of EBV-infected cells. Wilson et al. provided
supporting evidence by showing that expression of EBNA1 in the B-cells of transgenic mice
lines stimulates cell proliferation and increases the incidence of developing B-cell lymphomas
(Wilson et al. 1996). However, this finding was questioned by Kang et al. in three studies where
they showed that EBNA1 expression in B-cells of transgenic mice did not increased tumour

27
incidence and cell survival (Kang et al. 2001; Kang et al. 2005; Kang et al. 2008). Years later,
another study showed that expression of a dominant negative version of EBNA1 in a Burkitt’s
lymphoma cell line increased apoptosis (Kennedy et al. 2003). Consistent with a potential
oncogenic role for EBNA1, a reduction in cell proliferation was observed in Raji Burkitt’s
lymphoma and nasopharyngeal carcinoma cells when EBNA1 expression was diminished using
RNA interference (Hong et al. 2006; Yin and Flemington 2006). Moreover, determining the
precise role of EBV infection and the contribution to the expressed latent genes including
EBNA1 to the pathogenesis of gastric carcinoma (GC) and nasopharyngeal carcinoma (NPC) has
yet to be fully resolved. However, studies in our laboratory have recently provided some insight
into this by examining the role of EBNA1 in the development of GC by comparing the EBV- and
EBNA1-negative parental GC cell line with its two derivatives that are either EBV-positive or
EBNA1-positive. In the both the EBNA1- and EBV-positive GC cell lines, EBNA1 was found to
disrupt promyelocytic leukemia (PML) nuclear bodies, which are known to have functions in
p53 activation, tumour suppression, and apoptosis (Sivachandran et al. 2011). As a result, a
decrease in p53 activation and DNA damage-induced apoptosis were observed in the presence of
EBNA1, which provided evidence for its role in promoting cell survival and contributing to GC
development (Sivachandran et al. 2011). Importantly, the contribution of EBNA1 to PML loss
was confirmed in GC, where EBV-positive GC biopsies were shown to have less PML staining
than EBV-negative biopsies (Sivachandran et al. 2011).
Other EBNA1-mediated changes to the cellular environment which may be conducive for
increased cell proliferation and survival have also been reported. Studies in our laboratory have
demonstrated specific interactions between EBNA1 and ubiquitin specific protease 7 (USP7), as
well as casein kinase 2 (CK2) (Holowaty et al. 2003; Holowaty et al. 2003), mediated by amino
acids 395-450 and 387-394 of EBNA1, respectively (Holowaty et al. 2003; Sivachandran et al.
2010). USP7 is well-known for its key role in regulating apoptosis upon cellular stress through
binding, deubiquitinating, and as a result, stabilizing the tumour suppressor protein p53, whose
levels accumulate and induce cellular apoptosis and cell cycle arrest (Li et al. 2002; Cummins et
al. 2004; Cummins and Vogelstein 2004; Li et al. 2004). Saridakis et al. found that EBNA1
encourages cell survival through its ability to tightly associate with USP7 with an affinity ten-
fold greater than that of p53, thereby effectively out-competing p53 for binding to USP7, which
in turn results in destabilization and proteasomal degradation of p53 (Saridakis et al. 2005).

28
Consequently, with the p53-dependent apoptosis inhibited through destabilization of the protein,
increased cell survival and decreased sensitivity to DNA damage were observed in cells
expressing EBNA1 (Saridakis et al. 2005; Sivachandran et al. 2008). EBNA1 has been shown to
localize to promyelocytic leukemia (PML) nuclear bodies (NBs) during native EBV latent
infection in both NPC and GC cell lines and disrupt the foci by facilitating the proteasomal
degradation of PML protein (Sivachandran et al. 2010; Sivachandran et al. 2011). This activity is
dependent on both USP7 and CK2, which have been shown to negatively regulate PML nuclear
bodies. Previous studies have demonstrated localization of CK2 to PML nuclear bodies, where it
phosphorylates PML protein, targeting it for degradation, an activity that is taken advantage of
by EBNA1 (Scaglioni et al. 2006; Sivachandran et al. 2010). As discussed above, EBNA1-
mediated disruption of PML nuclear bodies negatively affects PML-associated functions
including p53 activation and apoptosis, which promotes cell survival following treatment with a
DNA damaging agent (Sivachandran et al. 2011).
Another mechanism employed by EBNA1 to alter the cellular environment involves
increasing the expression of STAT1 and decreasing TGFβ-mediated transcription (Wood et al.
2007). STAT1 responds to interferon-γ (IFN-γ) stimulation and activates transcription of sets of
genes involved in several cellular processes including immune regulation (Najjar and Fagard
2010). Associations have been made between constitutive activation of STAT pathway family
members and hematopoietic diseases (Najjar and Fagard 2010). Furthermore, activation of the
TGFβ signalling cascade is important for determining the fate of a cell, by playing a role in the
control of cell growth and differentiation (Inman 2011). Therefore, EBNA1 may affect the
function of key players in major cellular processes to promote an environment that is favourable
for persistent latent EBV infection.
I.4 Roles of Promyelocytic Leukemia (PML) Bodies in Viral Replication
Promyelocytic leukemia (PML) protein is recognized as a cellular tumour suppressor that
forms PML nuclear bodies (NBs). Consequently, the depletion of PML protein leads to
disruption and dispersal of the nuclear bodies as well as other cellular factors that associate with
these structures or foci. PML nuclear bodies are involved in diverse and important cellular
processes including apoptosis, antiviral response, transcription, and DNA damage repair (Everett
2001; Dellaire and Bazett-Jones 2004; Everett et al. 2006; Bernardi and Pandolfi 2007; Everett

29
and Chelbi-Alix 2007). In fact, the loss of PML or PML function is associated with several
cancers including acute promyelocytic leukemia (from which its name originates) where a
chromosomal translocation event generates a fusion protein that combines PML with the retinoic
acid receptor α and does not retain any PML-associated functions (Di Croce 2005). In the
interest of my thesis, I will briefly discuss some previous reports of the involvement of PML in
the replication of viruses.
Upon detection of a viral infection, the host cell elicits an interferon (IFN) response,
whereby IFN-regulated cellular effector proteins, such as PML, are upregulated and mediate
antiviral activities. Studies have provided evidence for a PML role in restricting lytic viral
replication by using PML-knockout mice and PML-depletion using siRNA in human cells
(Djavani et al. 2001; Blondel et al. 2002; Crowder et al. 2005). PML-knockout mouse embryonic
fibroblasts (MEFs) were found to be not only sensitive to infection with rabies virus (Blondel et
al. 2002), but could better support replication of the lymphocytic choriomeningitis virus (LCMV)
as compared to PML-positive fibroblasts (Djavani et al. 2001). In addition, PML suppresses
herpes simplex virus 1 (HSV-1) lytic infection but is overcome by the HSV ICP0 protein,
allowing replication to proceed (Everett et al. 2006). Moreover, using live cell imaging of HSV-1
infection, Everett et al. demonstrated the relocalization of PML NBs toward HSV-1 viral
genomes as they enter the host nucleus (Everett and Murray 2005). In addition, PML NBs have
been shown to associate with and sequester the nucleocapsids of the varicella zoster virus (VZV)
in different cell types (Reichelt et al. 2011).
While PML is usually suppressive for viral replication, there is some evidence that it can
also play a positive role in the replication of some viral DNA. For example, the SV40 virus is
well-known for initiating replication of its genome at sub-nuclear sites within close proximity to
fully functional PML NBs (Ishov and Maul 1996; Tang et al. 2000). Recent studies by Boichuk
et al. have demonstrated that depletion of PML protein by siRNA reduces the in vivo replication
efficiency of SV40 viral genomes in COS-1 monkey kidney cells and human U2OS cells
(Boichuk et al. 2011). Therefore PML can play diverse roles in viral replication.
As mentioned above, our laboratory has shown that EBV EBNA1 protein interacts with
and disrupts PML NBs in latent infection, thereby increasing cell survival and possibly
contributing to tumourigenesis. Consistent with results for other herpesviruses, our laboratory
has recently found that PML has a repressive function in EBV reactivation and lytic DNA

30
replication (Sivachandran et al. 2011 submitted). However, whether or not PML plays a role in
the latent replication of EBV genomes has not yet been explored.
I.5 EBV Genome Maintenance in Nasopharyngeal Carcinoma
The basis for the differential maintenance of the EBV genome within B-lymphocytes and
NPC cell lines has remained elusive for years. Unlike EBV-infected lymphoblastoid cell lines
(LCLs) which experience minimal loss of the virus, undifferentiated NPC cannot stably maintain
the EBV genome in long-term culture (Knox et al 1995, refs). For example, well-used NPC cell
lines such as CNE-1 and CNE-2 (also known as CNE2Z) were initially EBV-positive biopsies
obtained from Chinese patients, however, each of these three cell lines eventually lost the virus
in culture (Sun et al. 1992). Moreover, in the absence of drug selection, the EBV genome is
rapidly lost from epithelial cells infected with the virus in vitro. Until recently, the mechanism by
which the EBV episomes are lost in NPC cell lines has been poorly understood. Dittmer et al.
have shed some light on this topic using the EBV-positive NPC cell line HONE-1 and shown
that loss of the EBV episome occurs in two successive and discrete phases (Dittmer et al. 2008).
The primary phase involves rapid loss of the complete EBV genome from a large subset of cells:
up to 25% of the infected cell population (Dittmer et al. 2008). The reason and mechanism by
which the EBV genome is lost in this initial phase has yet to be deciphered. Subsequently, in the
second phase the EBV genome undergoes various recombination and deletion events that
eventually impair the crucial functions of oriP, EBNA1 and the Qp promoter, thereby disrupting
the maintenance of the EBV genome (Dittmer et al. 2008).
Finally, recent studies have shown that providing a cellular environment that is
permissive to EBV genome amplification may be important for long-term EBV maintenance in
culture (Shannon-Lowe et al. 2009). By comparing B-lymphocytes to NPC and AGS cell lines,
this group demonstrated that the EBV genome could amplify in both B-cells and AGS cells after
infection and EBV-positive cells from both cell lines expressed EBNA1 and stably maintained
the viral genome; however, this was not observed in NPC cells (Shannon-Lowe et al. 2009). The
authors proposed these observations may be attributed to differences in cell lineages as well as
the environmental conditions found within each of the three cell lines (Shannon-Lowe et al.
2009). C666-1 is the only NPC cell line that consistently maintains the EBV virus in tissue
culture without selection (Cheung et al. 1999). It is likely that these cells remain EBV-positive

31
due to inherent differences between all other NPC cell lines with respect to their
microenvironment, expression of cellular factors, and any accumulated chromosomal mutations.
I.6 Thesis Rationale
All undifferentiated NPC biopsies test positive for the presence of the EBV virus, however
with the exception of the C666-1 cell line, these cells cannot maintain the EBV genomes when grown
in tissue culture. Mitotic chromosome attachment and segregation of the EBV episome and oriP-
containing plasmids requires binding of EBNA1 to recognition sites within the FR region of the oriP
sequence. Our laboratory has previously identified which regions of EBNA1 are involved in its
segregation function, but interpreting their roles is complicated by the fact that EBNA1 is also
required for the replication of the oriP plasmids being monitored. Using the autonomously
replicating pBFRGC6 plasmid, I sought to more directly examine the contribution of
EBNA1sequences to segregation. In addition, studies in our laboratory have shown that higher levels
of EBNA1 cause the viral-based plasmid, oriPE, which contains the oriP sequence, to be lost more
rapidly from both CNE2Z (EBV-negative) and C666-1 cells, the latter of which endogenously
expresses low levels of EBNA1. Since plasmid and episomal maintenance requires a combination of
replication and mitotic segregation both of which are mediated by EBNA1, one of the objectives of
my research was to determine whether this reduction in oriP plasmid maintenance was due to effects
on replication, segregation, or cell proliferation/viability. Using plasmid replication assays as well as
cell proliferation assays, I found that higher levels of EBNA1 are not inhibitory to oriP plasmid
replication and do not affect the growth rate of treated cells, suggesting that the higher EBNA1 levels
were negatively impacting segregation. In addition, I investigated whether or not higher EBNA1
levels might also reduce EBV episomal maintenance (as seen with oriP plasmids). I found no
significant changes to the EBV episomal copy number within the EBV-positive epithelial cell lines,
C666-1 and AGS/EBV.
Another objective was to determine whether increasing the number of repeats in the FR
from 20 to 29 affected EBNA1-mediated replication and segregation. Ali et al. (Ali et al. 2009)
has recently cloned the full-length FR region from the EBV-positive cell line, B95-8, which contains
29 repeats in contrast to the 20 repeats encoded in the oriP plasmids commonly used. I found the
extra 9 repeats reduced plasmid replication efficiency, but improved long-term plasmid maintenance.
Finally, our laboratory has found that PML bodies have a repressive function in EBV lytic
replication; however, whether or not PML has a function in latent replication is not known. By

32
comparing PML-positive and –negative cell lines, I have examined whether the presence of PML
has any effect on plasmid replication and found that PML positively contributes to plasmid
replication in the NPC cell line, CNE2Z. Together my findings have helped to improve our
understanding of EBNA1 replication and segregation functions in NPC cells.

33
CHAPTER II: MATERIALS AND METHODS
II.1 Cell Culture
CNE2Z is an EBV-negative nasopharyngeal carcinoma cell line, derived from an NPC
that lost the EBV episome in culture (Sun et al. 1992). CNE2ZE is a stable cell line derived from
CNE2Z cells and contain an integrated EBNA1 expression cassette fused to a hygromycin B-
resistance gene. CNE2ZE cells stably express low levels of EBNAΔG/A (Sivachandran et al.
2008). CNE2Z-shPML is a stable cell line that was depleted of PML protein using a shRNA-
lentivirus (Sarkari et al. 2011). C666-1 is an EBV-positive nasopharyngeal carcinoma cell line
described elsewhere (Cheung et al. 1999). CNE2Z, CNE2ZE, and CNE2Z-shPML cell lines were
all maintained in alpha-minimal essential medium (α-MEM), whereas C666-1 cells were grown
in RPMI 1640 (Sigma). In all cases, the media were supplemented with 10% fetal bovine serum
(FBS) (GIBCO) and 1% L-Glutamine (L-Q) (GIBCO) and cells were incubated at 37°C. For
CNE2E cells, 0.5 mg of hygromycin B was included per mL of medium.
II.2 Plasmid Constructs
The pBFRGC6 plasmid was provided by Anindya Dutta’s laboratory (University of
Virginia). This plasmid is derived from the pBudCE4.1 plasmid (Invitrogen) and contains a
GAL4 DNA-binding region (5 copies of GAL4 recognition sequence), a GAL4-Cdc6 fusion
protein expression cassette (under control of the high expression cytomegalovirus (CMV)
promoter), and the FR element from oriP (see Figure 4A). EBNA1 and the EBNA1 mutants
Δ61-83, Δ325-376, and Δ395-450 were generated by PCR amplification of these EBNA1
sequences from pc3OriPE, pc3OriPEΔ61-83, pc3OriPEΔ325-376, or pc3OriPEΔ395-450 and
insertion between the Not I and Kpn I sites of pBFRGC6 (performed by Niro Sivachandran) and
are under the control of the high expression EF-1α promoter (Figure 4B). I subcloned the
EBNA1Δ8-67 sequence from pc3OripEΔ8-67 into pBFRGC6 as described above. The sequences
of all the pBFRGC6-based plasmids were verified by DNA sequencing.
EBNA1 and EBNA1Δ387-394 were expressed from the CMV promoter in the pc3DNA3
plasmid (Invitrogen) that also contained the EBV oriP viral DNA sequence (see Figure 4C).
These plasmids are referred to as oriPE, oriPEΔ387-394, oriP (does not express EBNA1) (see

34
Figure 4. Plasmids used in this study. Schematic representation of pBFRGC6, pBFRGC6-
EBNA1, pc3OriPE, pc3OriP, FR20, and FR29 plasmids used in this study. The DS and FR
elements of oriP as well as the 5X GAL4 DNA binding sites are indicated by the black boxes.
The pBFRGC6-derived plasmids encoding EBNA1 mutants are the same as the pBFRGC6-
EBNA1 plasmid represented above except that the EBNA1 cDNA is replaced with that of one of
its mutants.

35
Figure 4D), respectively. The construction of these plasmids has been previously described
(Shire et al. 1999; Wu et al. 2002; Sivachandran et al. 2010).
B95-8-derived FR elements containing 20 and 29 copies of the EBNA1 recognition
sequence were subcloned into the low-copy number pMBL19 plasmid between Mlu I and Xcm I
sites (performed by Teru Kanda’s group), generating FR20 and FR29 plasmids, respectively.
Both plasmids contain an EGFP expression cassette between Bam HI and Not I sites as well as
the DS element of oriP between Spe I and Eco RV sites (see Figure 4E and 4F).
II.3 Immunofluorescence Microscopy
Cells adhered to cover slips were fixed with 3.7% formaldehyde (SIGMA) in phosphate-
buffered saline (PBS) solution for 20 minutes followed by two 5 minute washes using PBS. For
cells transfected with FR20 or FR29 plasmids, GFP fluorescence emission from transfected cells
was then visualized (excitation, 480/440 nm, emission 527/30 nm). For cells transfected with any
of the other plasmids, cell membranes were then permeabilized by a 5 minute incubation with
1% Triton X-100 in PBS. Cover slips were washed twice (each 2-10 min) and blocked with 50
μL of 4% bovine serum albumin (BSA) in PBS for 30 minutes to 1 hour. The samples were then
stained with rabbit polyclonal anti-EBNA1 R4 serum (Holowaty et al. 2003) at 1:200 dilution in
4% BSA-PBS for 1 hour, followed by three 10 minute washes in PBS. Fluorescein
isothiocyanate-conjugated goat anti-rabbit secondary antibody (Alexafluor 488; Invitrogen)
(1:500 dilution in 4% BSA-PBS) was applied to the cover slips and incubated in the dark for 45
minutes with three subsequent 10 minute washes. Cells were then stained overnight with
ProLong Gold anti-fade DAPI (Invitrogen) for visualization of cellular nuclei and analysis the
following day. The ratio of transfected to untransfected cells was determined for several fields of
view, allowing for calculation of transfection efficiency. Cells were analysed using the 63x oil
immersion objective lens (NA 1.4) on a Leica DM IRE2 inverted fluorescent microscope.
II.4 Plasmid Replication and Maintenance Assays
For experiments involving the CNE2Z cell line, using Lipofectamine 2000 (Invitrogen) in
a 1:2 ratio (DNA:Lipofectamine), 1 x 106 cells in one 10-cm dish were transfected with 6 μg of
pc3OriP, pc3OriPE, pc3OriPEΔ387-394, pBFRGC6, or EBNA1-expressing derivatives of

36
pBFRGC6. Using the same approximate cell density and the same amount of plasmid DNA, both
CNE2Z-E and C666-1 cells were transfected with PCAN, pc3OriP or pc3OriPE, while CNE2Z-E
cells were also transfected with FR20 or FR29. CNE2ZshPML cells were transfected with
pc3OriP or pc3OriPE. For all replication assays, 24 hours post-transfection, the treated cells were
transferred to a 15-cm dish and subsequently harvested 48 hours later. For all maintenance
assays, 24 hours post-transfection, the treated cells were transferred to 15-cm dish and
subsequently re-plated every 3 cell doublings and samples were harvested weekly for up to 6
weeks or 42 cell doublings. Plasmids were isolated from transfected cells by Hirt’s method (Hirt
1967). Treated cells were harvested by centrifugation at 1100 rpm for 10 min and the cell pellet
(~5 x 106 cells) was resuspended with 350 µL of 1X phosphate buffered saline (PBS) solution.
The cells were then washed with 1X PBS, the supernatant was removed, and resuspended again
with 350 µL of 1X PBS. Subsequently, 350 µL of 2X Hirt’s solution (final concentration 0.6%
SDS, 10 mM EDTA, 10 mM Tris-HCl, pH 7.5) were added and the samples were gently inverted
10 times and incubated at 25ºC for 10 min. Following the incubation, 140 µL of 5 M NaCl were
added to each lysate sample, which were then gently inverted 10 times and incubated overnight
at 4ºC. The following day, the samples were centrifuged for 1 hour at 15,000 rpm at 4ºC. The
supernatants were transferred to fresh 1.5 mL microfuge tubes containing 6 µL of RNase A (10
mg/mL, Fermentas) and incubated at 37ºC for 1 hour. Subsequently, 8 µL of Proteinase K (20
mg/mL, Fermentas) were then added and samples were incubated at 50ºC for at least 2 hours. To
extract DNA from samples, ~700 µL of phenol:chloroform (1:1) were added, after which
samples were vortexed for 30 seconds and then centrifuged at 13,200 rpm for 3 min. The
supernatants were then transferred to a fresh 1.5 mL microfuge tube to which 700 µL of
chloroform was added. The samples were then vortexed for 30 seconds and centrifuged at 13,200
rpm for 3 min. The supernatants were then transferred to a new 1.5 mL microfuge tube and 700
µL of isopropanol were added. The samples were mixed well by inversion and incubated at -
20ºC overnight. The precipitated DNA was then collected by centrifugation at 15,000 rpm for 45
min at 4ºC, washed twice with 70% ethanol, and air dried. Finally, the DNA was resuspended in
a master mix solution (30 µL) containing 1 μL of Kpn I (or Xho I or Mlu I) (New England
Biolabs), 3 μL of 10X NEB Buffer, and 26 μL of a mixture of Tris pH 8.5 and dH2O (in a 14:3
ratio).

37
The isolated plasmids were linearized with 1 µL of Xho I (for pc3DNA3-based plasmids),
Mlu I (for pMBL19-based plasmids), or Kpn I (for pBFRGC6-based plasmids) (New England
Biolabs) for at least 3 hours. 9/10ths of the linearized DNA were then further digested with 1 µL
of Dpn I for at least 3 hours, while the remaining 1/10th
of the linearized DNA was used as an
input control for the efficiency of plasmid recovery. Finally, the plasmid samples were separated
on a 0.8% agarose gel by electrophoresis, transferred to Hybond-XL membranes (Amersham) as
described below, and probed with 32
P-labelled pc3OriP. Bands were visualized by
autoradiography and signals were quantified by PhosphorImager analysis using the ImageQuant
software.
II.5 Plasmid Labelling
1 µL of linearized pc3OriP (300 ng/µL), 4 µL of random hexamer primer (0.5 µg/µL)
(Roche), and 8.3 µL of dH2O were incubated at 95ºC for 5min and then placed on ice for 5 min.
Subsequently, 1.25 µL of 1 mM dNTPs (without dCTP), 2.5 µL of NEB Buffer 2 (10 mM Tris-
HCl, 50 mM NaCl, 10 mM MgCl2, 1 mM DTT pH 7.9), 2.4 µL of dH2O, 0.6 µL of Klenow
DNA polymerase, and 5 µL of 3000 Ci/mmol αP32
-dCTP were added and the sample was
incubated at 37ºC for 1 hour 30 min. The labelled plasmid was then added to an
IllustraTM
MicroSpinTM
G-25 column and centrifuged for 1 min at 3000 rpm. The sample was
then incubated at 95ºC for 5 min and then placed on ice for 5 min.
II.6 Southern Blotting
Linearized plasmid DNA fragments from the digestion described above were separated
on a 0.8% agarose gel at 25V for 18 hours. The agarose gel was soaked in 0.25 M HCl for 10min
to allow for depurination of the DNA. The gel was then rinsed twice with dH2O and the duplex
DNA fragments were denatured by soaking the gel in an alkaline solution (0.4 M NaOH and 0.6
M NaCl) for 1 hour. The DNA fragments were then transferred from the agarose gel to a
Hybond-XL membrane in the aforementioned alkaline solution for 1 hour using a vacuum pump.
Subsequently, a 20X SSC solution (3 M NaCl, 0.3 M sodium citrate, pH 7.0) was added for
another 2 hours, after which the membranes were rinsed in a 2X SSC solution. The membranes
were then incubated with blocking buffer (50% formamide, 5X Denhardts, 0.5% sodium dodecyl
sulphate, 6X SSC, and 0.2 mg/mL sheared salmon sperm DNA) in a hybridization oven for 2

38
hours at 42°C, followed by an overnight incubation at 42°C with 32
P-labelled pc3OriP
(approximately 5 x 106 cpm in 15 mL of blocking buffer). The following day, the membranes
were washed twice with a solution containing 1% SDS and 2% SSC at 25°C for 10 min. The
membranes were washed twice for 30 min at 65°C using the same solution, followed by another
wash in a solution containing 0.1% SDS and 0.2% SSC for 10 min at 25°C. The membranes
were then dried and subjected to autoradiography for 1 hour to 3 days, followed by a
quantification of the radiolabelled bands by PhosphorImager analysis using ImageQuant
software.
II.7 Cell Proliferation Assay
The proliferation rates of CNE2Z, CNE2ZE, and C666-1 cells following plasmid
transfection were monitored. In all cases, one day prior to transfection, approximately 1 x 106
cells were seeded in 10 mL of growth medium without antibiotics on a 10-cm dish. CNE2ZE
cells were transfected with 6 μg of pc3OriP, pc3OriPE, FR20, or FR29 plasmids, whereas both
CNE2Z and C666-1 cells were transfected with 6 μg of pc3OriP or pc3OriPE. Plasmids were
transfected into cells using Lipofectamine 2000 (Invitrogen) in a 1:2 ratio (DNA:Lipofectamine).
Approximately 1 x 105 cells were plated in four 6-cm dishes 24 hours later, at which point a
coverslip was obtained and cells were fixed for IF imaging (as described above) and transfection
efficiency was determined. The 6-cm plates were subsequently harvested and counted 1, 3, 5, or
7 days post-transfection using a haemocytometer. The average cell numbers from at least two
independent experiments were plotted with standard deviations.
II.8 Antibodies and Western Blotting
Cells were lysed in high salt buffer (500 mM NaCl, 10% glycerol, 1% Triton X-100, 50
mM HEPES pH 8.0, 1 mM dithiothreitol (DTT), 1X P8340 protease inhibitor cocktail (Sigma)).
Whole cell lysates (50 μg) were separated on a 12% SDS-PAGE gel and transferred to
nitrocellulose. Membranes were blocked for 20 min using 5% non-fat dry milk in PBS with 0.1%
Tween-20 and subsequently incubated with primary antibodies against EBNA1 (mix of OT1X
monoclonal antibody at 1:5000 and R4 rabbit serum at 1:5000 (Holowaty et al. 2003)), actin
(CALBIOCHEM CP01, 1:10,000), or cdc6 (Santa Cruz, sc-13136, 1:2000). Membranes were
washed three times (15 min each), probed with goat anti-mouse (1:5000) or goat anti-rabbit

39
(1:5000) conjugated to horseradish peroxidase secondary antibodies for 1 hour, and then
developed using the enhanced chemiluminescence (ECL) reagents (Perkin Elmer). Where
necessary, membranes were then stripped to remove the initial antibody using 0.1 M glycine pH
2.9 for 15 min, washed three times (10 min each) with PBS-Tween and incubated with the
subsequent antibody.
II.9 Monitoring Episomal Maintenance
C666-1 and AGS/EBV cells on 10-cm plates were transfected with 6 μg of pc3OriP or
pc3OriPE using Lipofectamine 2000 in a 1:2 ratio (DNA:Lipofectamine). One day post-
transfection, the cells were transferred to a 15-cm dish and samples of treated cells (1 x 106 cells
per sample) were harvested weekly for 5 to 6 weeks. Cells were resuspended in 150 μL of RIPA
buffer (50 mM Tris.Cl pH 8.0, 150 mM NaCl, 1% NP40, 0.1% sodium deoxycholate, 1 mM
PMSF and protease inhibitors (P8340, Sigma)) and incubated on ice for 20 min. The samples
were then sonicated and the clarified lysates were diluted to final volume of 500 μL, to which 3
μL of RNase A (10 mg/mL from Fermentas) was added. The samples were then incubated at 37
ºC for 1 hour and then 8 μL of proteinase K (20 mg/mL from Fermantas) was added. The
samples were then incubated at 50°C for at least 2 hours, after which the DNA was extracted by
phenol:chloroform (1:1) and chloroform, and then precipitated with ethanol. The precipitated
DNA was washed twice with 70% ethanol, dried, and resuspended in Tris (pH 8.5). The DNA
was quantified by real-time PCR using primers for the BZLF1 region of EBV and normalized to
the cellular DNA signal from the GAPDH locus (Yuan et al. 2006) (see below for sequences).
Quantitative real-time PCR was performed using 1/4th (for BZLF1) or 1/20th (for GAPDH) of the
template DNA and SsoFastTM
EvaGreenTM
Mix (Bio-Rad) on a Rotorgene qPCR system (Corbett
Research) for 50 cycles of 10 sec at 95ºC, 15 sec at 60ºC,and 25 sec at 72ºC.
Primers:
BZLF1-Forward: CAGCTGAGGTGCTGCATAAGCTTG
BZLF1- Reverse: ACCTTGCCGGCACCTTTGCTATC
GAPDH-Forward: CTC CAA ACA GCC TTG CTT GCT TCG AGA ACC ATT TGC TTC
CCG CTC AGA CGT CTT GAG TGC TAC AGG AAG CTG GCA
GAPDH-Reverse: GAC CCG ACC CCA AAG GCC AGG CTG TAA ATG TCA CCG GGA
GGA TTG GGT GTC TGG GCG CCT CGG GGA ACC TGC CCT

40
CHAPTER III. RESULTS
III.1 pBFRGC6-Based Plasmids do not Autonomously Replicate in the Nasopharyngeal
Carcinoma Cell Line, CNE2Z
EBNA1 is required for the replication and segregation of oriP-containing plasmids and
therefore these plasmids are often used to study the replication and segregation functions of
EBNA1. However, the ability to examine the segregation function of EBNA1 using oriP
plasmids is complicated by the fact that persistence of oriP plasmids (followed as a measure of
segregation) also is dependent on EBNA1-mediated replication. Therefore we sought an
alternative plasmid system that could be used to measure EBNA1-mediated segregation
independent of replication function.
Previously our laboratory identified regions within EBNA1 that contribute to the
maintenance of oriP plasmids. Some of these regions affect the replication function of EBNA1
therefore leading to decreased maintenance of oriP plasmids. In these cases the effect of the
mutation on segregation could not be assessed since plasmid maintenance is the assay for
segregation. To investigate the contribution of these EBNA1 regions to its plasmid segregation
function, I used the pBFRGC6 plasmid system which we obtained from Anindya Dutta’s
laboratory (University of Virginia). This plasmid contains an artificial origin of replication as in
plasmid pFR_Luc, as described in Takeda et al 2005. More specifically, the pBFRGC6 plasmid
contains five GAL4 DNA binding sites and also encodes a fusion protein consisting of the C-
terminal DNA-binding domain of GAL4 fused to the N-terminal domain of Cdc6, a pre-
replicative complex component. Autonomous replication of pBFRGC6 should involve the
binding of the GAL4-Cdc6 fusion protein to the GAL4 recognition sites within the plasmid and
subsequent recruitment of the origin recognition complex (ORC) by Cdc6 as seen for plasmid
pFR_Luc. For purposes of studying the EBNA1 segregation function, the pBFRGC6 plasmid had
been engineered to also contain the family of repeats (FR) element of oriP, which must be bound
by EBNA1 to mediate mitotic segregation of the plasmid. Furthermore, a series of derivatives of
the pBFRGC6 plasmid were developed: an expression cassette was engineered into pBFRGC6
expressing EBNA1 or four deletion mutants of EBNA1 (Δ8-67, Δ61-83, Δ325-376, and Δ395-
450). Therefore, with its ability to autonomously replicate, it was believed that this pBFRGC6
system would allow me to determine the specific regions of EBNA1 that contribute to its
segregation function independently from potential roles in replication.

41
In EBV-negative and EBNA1-negative cell lines (e.g. CNE2Z), all of the pBFRGC6-
based plasmids should autonomously replicate, as this function requires the GAL4-Cdc6 fusion
protein and is EBNA1-independent. Without EBNA1 expression, the parental pBFRGC6
plasmid should not segregate efficiently or be stably maintained in cells as segregation requires
EBNA1 binding to the FR sequence. To confirm that each of the pBFRGC6-based plasmids were
capable of replication prior to examining their segregation ability, I first performed several
replication assays in the CNE2Z cell line (see Figure 5 for a schematic of the experimental
protocol). CNE2Z cells were transfected with oriP, oriPE, or the pBFRGC6-based plasmids,
which were harvested and lysed 3 cell doublings (equivalent to 3 days) post-transfection. The
oriP and oriPE plasmids were used as negative and positive controls, respectively. The oriP
plasmid cannot replicate in CNE2Z cells in the absence of EBNA1, whereas the oriPE plasmid
can replicate in this cell line, as it encodes an EBNA1 expression cassette and overexpresses
EBNA1. In all cases, the plasmids were isolated by the method of Hirt (Hirt 1967) and
linearized. 9/10ths
of the sample was treated with the Dpn I restriction enzyme to test for
replication, while the remainder of the sample (input) was used as an indication of plasmid yield.
Plasmids that have replicated at least once in transfected cells will be resistant to digestion by
Dpn I, whereas plasmids that did not replicate will be digested by Dpn I. This restriction enzyme
cleaves at its GATC recognition sequence when the DNA is methylated on both strands at this
site, which occurs during the propagation of plasmids in bacteria that express Dam methylase
such as E.coli. Mammalian cells do not express Dam methylase and therefore, after one round of
replication (or one cell doubling) the plasmid will be hemi-methylated and will not be cleaved
efficiently by Dpn I. Southern blot analysis showed that plasmids encoding wild-type EBNA1
and the five deletion mutants were able to replicate; however, the parental pBFRGC6 plasmid
(positive control) did not autonomously replicate (Figure 6A). I monitored transfection efficiency
of the EBNA1-expressing plasmids by western blotting using a polyclonal antibody against
EBNA1, whereas uptake of the parental pBFRGC6 plasmid was confirmed by a signal from the
input samples on the Southern blot (lanes 10-12). In order for the pBFRGC6-based plasmids to
autonomously replicate, the GAL4-Cdc6 fusion protein must be expressed. Therefore, I
subsequently examined these samples for expression of the fusion protein by Western blotting
using an antibody against cdc6. I found that GAL4-Cdc6 was only expressed in samples co-

42
Figure 5. Schematic representation of protocol for plasmid replication and maintenance
assays. A. Cells are transfected with test plasmids and harvested 3 cell doublings or weekly post-
transfection to collect samples for replication and plasmid loss assays, respectively. Cells are
lysed (Hirt method (Hirt 1967)), plasmids are isolated, linearized, and, where indicated, were
also incubated with Dpn I to digest unreplicated plasmids. For samples without Dpn I digestion,
1/10th
of the amount of sample was loaded relative to Dpn I-digested samples and used as an
indication of plasmid yield following the isolation protocol. DNA was separated by agarose gel
electrophoresis, Southern blotted, and probed with a 32
P-labeled linearized plasmid to detect
samples. B. Dpn I recognition site and sequences susceptible to digestion. Dpn I recognizes and
cleaves at the DNA sequence, GATC, containing an N6-methyladenine on both DNA strands,
which occurs in bacteria such as E.coli but not mammalian cells. If a plasmid does not replicate
after transfection in mammalian cells, both DNA strands will maintain their methylation and can
be cleaved by Dpn I. However, if the plasmid replicates at least once it will be resistant to Dpn I-
digestion.
A
B

43
Figure 6. The pBFRGC6 plasmid does not autonomously replicate. CNE2Z cells were
transfected with oriP, oriPE and pBFRGC6 plasmids encoding wild-type and EBNA1 deletion
mutants (ΔG/A, Δ8-67, Δ61-83, Δ325-376, Δ395-450) and cells were harvested after 3 cell
doublings (3 days). A. Southern blot comparing the replication efficiencies of pBFRGC6 and
pBFRGC6-based plasmids encoding wild-type EBNA1 and EBNA1 deletion mutants. 9/10ths of
the treated cells were lysed by the Hirt method and low-molecular-weight DNA (including
plasmids) was isolated as previously described. The remaining tenth of the harvested cells were
kept for analysis of EBNA1 and GAL4-Cdc6 expression. Recovered plasmids were linearized
using Xho I (for oriP and oriPE) and Kpn I (for pBFRGC6-based plasmids) and, where
indicated, were also incubated with Dpn I to digest unreplicated plasmids. For samples without
Dpn I digestion, 1/10th
of the amount of sample was loaded relative to Dpn I-digested samples.
DNA was separated by agarose gel electrophoresis, Southern blotted, and probed with 32
P-
labeled oriPE. The position of the Dpn1-resistant plasmid is indicated by the bracket. 300 pg of
oriPE plasmid was used as a size marker (Lane 1). B. Western blot analysis of GAL4-Cdc6 and
EBNA1 expression from transfected CNE2Z cells. Total cells lysates (30 μg) were separated
using a 12% SDS-PAGE gel and probed with mouse anti-EBNA1 and anti-Cdc6 antibodies (top
panel) and actin (bottom panel). Values at the left are molecular size markers in kilodaltons. The
signals were detected by enhanced chemiluminescence (ECL).
B
A B

44
expressing EBNA1 or EBNA1 deletion mutants Δ8-67, Δ61-83, or Δ395-450 and not in the
pBFRGC6 control samples (Figure 6B), suggesting that EBNA1 was enhancing GAL4-Cdc6
expression possibly through binding to the FR element. Due to the fact that the parental
pBFRGC6 plasmid did not autonomously replicate, this system was no longer useful for my
segregation studies and was not further pursued.
III.2 Higher EBNA1 Expression can Inhibit Plasmid Maintenance without Inhibiting
Plasmid Replication Efficiency in Nasopharyngeal Carcinoma Cells
We were initially interested in directly comparing the abilities of EBV-negative and
EBV-positive NPC cell lines, CNE2Z and C666-1, respectively, to maintain oriP plasmids. Since
the CNE2Z cell line was derived from EBV-positive NPC cells that failed to maintain EBV in
culture, we wondered whether these cells were less able to support EBNA1-mediated replication
and segregation from oriP as compared to C666-1 cells that stably maintain EBV. Replication
and segregation of oriP plasmids requires the expression of EBNA1 protein, hence, an
expression cassette for EBNA1 was integrated into CNE2Z cells to generate the stable cell line,
CNE2ZE, which expresses EBNA1 at similar levels to C666-1 cells. Niro Sivachandran in our
laboratory first compared the maintenance of the same oriP plasmid in CNE2ZE and C666-1
cells and found that both cells lines had similar abilities to maintain this plasmid. Unexpectedly,
in both cell lines, she found that the oriP plasmid was maintained for longer periods of time than
the oriPE plasmid (Figure 7A and 7B), which overexpresses EBNA1 from a cytomegalovirus
(CMV) promoter. Hence, the major difference in the experiments comparing the maintenance of
oriP plasmids and oriPE plasmids is that the EBNA1 levels are considerably higher (at least a
thousand fold) when expressed from oriPE than the levels expressed in CNE2ZE or C666-1
cells, which was confirmed by Western blot analysis (Figures 8B and 9B). Therefore, higher
levels of EBNA1 appear to disrupt the maintenance of the oriP plasmids in these cell lines.
OriP plasmid maintenance requires both replication and segregation of the plasmids.
Therefore, I then performed further studies to determine whether or not the decreased
maintenance of oriPE plasmids was due to differences in DNA replication in the presence of
excess EBNA1. To this end, I performed replication assays using Dpn I-resistance as a measure
to compare replication efficiencies of oriP and oriPE plasmids in C666-1 and CNE2ZE cells.

45
Figure 7. Plasmid maintenance assays in CNE2ZE and C666-1 cells. Cells were transfected
with pCAN (negative control), oriP, or oriPE plasmid as indicated and harvested after 3 to 56 or
42 cell doublings (3 to 56 and 6 to 84 days) for CNE2ZE (A) and C666-1 (B) cells, respectively.
Cells were lysed by the Hirt method and plasmids were isolated, linearized using Xho I and,
where indicated, were also incubated with Dpn I to digest unreplicated plasmids. For samples
without Dpn I digestion, 1/10th
of the amount of sample was loaded relative to Dpn I-digested
samples. DNA was separated by agarose gel electrophoresis, Southern blotted, and probed with 32
P-labeled oriP or pCAN for CNE2ZE and C666-1 samples, respectively. The position of the
Dpn I-resistant plasmid is indicated by the asterisk. The amount of Dpn I-resistant plasmid was
quantified using phosphorimager analysis and plotted relative to the 3-cell doubling amount,
which was set to one (histogram). Average values with standard deviation are shown for
CNE2ZE samples.
A
B

46
Figure 8. Higher levels of EBNA1 do not affect plasmid replication in CNE2ZE cells. A.
Transient-replication assays comparing the bands from oriP and oriPE plasmids after 3 cell
doublings (3 days) before (Input) and after (+Dpn I) Dpn I digestion. The positions of Dpn I-
resistant bands are indicated by the asterisk. The histogram shows quantification of Dpn I-
resistant bands relative to input bands from three experiments. Results are plotted relative to the
signal from the oriP plasmid (set to one). Error bars indicate standard deviations. B. Western blot
confirming the overexpression of EBNA1 in oriPE samples compared to endogenous EBNA1 in
CNE2E. Total cell lysates (30 μg) are shown from CNE2E 3 days post-transfection with pCAN,
oriP, or oriPE plasmid. Blots were probed with antibodies against EBNA1 and actin. C.
CNE2ZE cells were transfected with oriP or oriPE plasmid as for replication assays. Cells
(1x105) were then plated in four 6-cm dishes and harvested and counted 1, 3, 5, or 7 days later.
Average cell numbers from three independent experiments are plotted with standard deviations.
A
B
(kDa)
C

47
Figure 9. Higher levels of EBNA1 increase plasmid replication in C666-1. A. Transient-
replication assays comparing the bands from oriP and oriPE plasmids after 3 cell doublings (6
days) before (Input) and after (+Dpn I) Dpn I digestion. The positions of Dpn I-resistant bands
are indicated by the asterisk. The double bands seen in lanes 5 and 8 are due to incomplete
linearization of oriPE, resulting in both nicked (upper band) and linearized (lower band) forms
of the plasmid. The histogram shows quantification of Dpn I-resistant bands relative to input
bands from three experiments. Results are plotted relative to the signal from the oriP plasmid
(set to one). Error bars indicate standard deviations. B. Western blot confirming the
overexpression of EBNA1 in oriPE samples (EBNA1) compared to endogenous EBNA1 in
C666-1 cells (GA), which is larger due to having a long Gly-Ala repeat region. Total cell lysates
C
A
(kDa)
B

48
(30 μg) are shown from C666-1 cells 6 days post-transfection with pCAN, oriP, or oriPE
plasmid. Blots were probed with antibodies against EBNA1 and actin. C. C666-1 cells were
transfected with oriP or oriPE plasmid as for replication assays. Cells (3x105) were then plated
in four 6-cm dishes and harvested and counted 4, 8, 12, and 14 days later. Average cell numbers
from three independent experiments are plotted, with standard deviations.

49
These two cell lines were transfected with the oriP and oriPE plasmids and subsequently
harvested 3 cell doublings post-transfection (equivalent to 3 days and 6 days, respectively). The
plasmids were extracted from the cells and their replication was assessed following
quantification of signals from Southern blots. Comparison of replication signals from oriP and
oriPE showed that the higher EBNA1 levels did not decrease replication
efficiency in either the
CNE2ZE or C666-1 cell lines (Figure 8A and 9A). In fact, relative to the total plasmid taken up
by the cell (input), the oriPE plasmid replicated approximately two-fold more efficiently than the
oriP plasmid in C666-1 cells (Figure 9A).
I also performed cell proliferation assays to assess whether the high levels of EBNA1
impacted the viability and/or the proliferation rate of transfected cells. CNE2ZE and C666-1
cells were transfected with oriP and oriPE plasmids then seeded at equal cell density one-day
post-transfection. Cells were subsequently harvested and counted every 2 cell doublings for a
total of 8 cell doublings. I found that higher levels of EBNA1 were not cytotoxic to CNE2ZE or
C666-1 cells and the proliferation rates of these two cell lines were not significantly affected by
the presence of oriPE compared to oriP (Figure 8C and 9C). The majority of the cells contained
the plasmids with average transfection efficiencies of 60% and 80% for C666-1 and CNE2ZE,
respectively, as determined by immunofluorescence (IF) microscopy for EBNA1 expressed from
the oriPE plasmid. The considerably higher levels of EBNA1 from the oriPE plasmid compared
to the endogenous EBNA1 expression in C666-1 and CNE2ZE cells allows for easy detection of
transfected cells by IF microscopy. Taken together, the results indicate that higher EBNA1 levels
do not negatively affect plasmid replication or cell viability/growth and are therefore most likely
detrimental to plasmid segregation, resulting in decreased oriP plasmid maintenance.
III.3 Higher EBNA1 Levels do not Reduce EBV Copy Number in C666-1 or AGS/EBV Cell
Lines
Since we found that higher levels of EBNA1 expression reduce plasmid maintenance, I
was interested in investigating the possibility that EBNA1 overexpression might lead to the loss
of the endogenous EBV genome in EBV-positive epithelial cell lines, C666-1 and AGS-EBV,
the latter of which is a gastric carcinoma cell line infected in vitro with recombinant EBV. In the
case of C666-1 and AGS-EBV cells, the EBV genome is not integrated but maintained
extrachromosomally as a circular episome, which allowed me to detect any changes in the virus

50
Figure 10. Higher levels of EBNA1 do not affect the copy number of EBV episomes in
C666-1 or AGS/EBV cell lines. C666-1 (A) and AGS/EBV (B) cells were transfected with oriP
or oriPE plasmid, and cells were propagated for the indicated numbers of cell doublings before
they were lysed in radioimmunoprecipitation assay (RIPA) buffer and total DNA was isolated.
Quantitative RT-PCR on the EBV genomes was performed using a primer set for the BZLF1
gene, and results were normalized to the host GAPDH (glyceraldehyde-3-phosphate
dehydrogenase) gene. The histograms show the average levels of the EBV genomes and standard
deviations from two independent experiments for each cell line, where the signal from the 3-
doubling time point is set to one.
A
B

51
copy number. For instance, if the EBV copy number decreased over time in cells transfected
with the oriPE plasmid, this would suggest that maintenance of the virus is perturbed by higher
levels of EBNA1, which may have an expression threshold for stable EBV persistence. To
address this possibility, I transfected these cell lines with oriP and oriPE plasmids and harvested
weekly samples for 6 weeks. Total DNA, including the EBV episomes, was extracted from these
cells and the EBV genomes were quantified by quantitative real-time PCR (RT-PCR) using
primers directed for the viral BZLF1 region. After normalization to the host GAPDH gene, the
results obtained from both AGS-EBV and C666-1 cell lines showed no obvious decrease in the
EBV copy number at any time up to 20 and 24 cell doublings, respectively (Figure 10A and
10B). Also, although higher levels of EBNA1 increased plasmid replication in the C666-1 cells, I
found no significant difference in EBV copy number between the oriP and oriPE transfected
samples after 3 cell doublings, the same time point where replication of the oriPE plasmid had
approximately doubled. Therefore, the level of EBNA1 alone does not itself determine the
efficiency of EBV episome maintenance in C666-1 and AGS/EBV cells.
III.4 Additional 9 Repeats within the FR Region Decrease Plasmid Replication Efficiency
but Improve Plasmid Segregation
Mitotic chromosome attachment and segregation of the EBV episome and oriP-
containing plasmids requires binding of EBNA1 to recognition sites within the FR region of the
oriP sequence. While the FR region was initially believed to contain only 20 repeats of the
EBNA1 binding site, previous studies have demonstrated that the FR region within the EBV
virus actually contains 29 repeats. It is believed that the extra 9 repeats were deleted while the
oriP sequence was cloned into plasmids which were propagated in E.coli.
Not only is the FR region required for maintaining the virus and oriP-containing
plasmids, it is known to function as a transcriptional enhancer and in B-cell transformation.
Recently, Ali et al. characterized some of the effects of the additional 9 repeats using B-
lymphocytes and found that the extra 9 repeats in the FR region had no effect on plasmid
replication, but reduced the transcriptional enhancer activity inherent to the FR region and
improved the efficiency of EBV transformation of B-cells. Although no effects on plasmid
replication were observed in B-cells, due to the inherent differences in ability to maintain EBV
episomes between B-cells and epithelial cells, I was interested in whether the additional 9 FR

52
repeats had any effect on the plasmid replication and segregation ability in NPC cells. To this
end, I first performed a replication assay to determine if the 9 repeats affected plasmid
replication efficiency. CNE2ZE cells were transfected with oriP plasmids encoding an FR region
that contains 20 repeats or 29 repeats, which will be referred to as FR20 and FR29, respectively.
These cells stably express low levels of EBNA1 which allows for replication and segregation of
the two oriP-containing plasmids. The presence of the extra 9 FR repeats was verified by
plasmid digestion with the restriction enzymes, Xcm I and Mlu I producing the expected band
patterns following agarose gel electrophoresis (data not shown). Three days post-transfection, I
harvested the CNE2ZE cells, isolated plasmids from the cells and assessed the replication
efficiencies of the FR20 and FR29 by Southern blotting. The results showed that the replication
efficiency of the FR29 plasmid was reduced by approximately 40% compared to the FR20
plasmid (Figure 11A), as determined by normalizing the replication signal (Dpn I-resistant band)
to the input signal. To confirm that there were no differences in the viability and/or proliferation
of the cells transfected with either plasmid, I performed a cell proliferation assay. As expected, I
found no difference in cell proliferation rates between cells transfected with either plasmid
(Figure 11B). Together, the results from the replication and proliferation assays suggest that the
extra 9 repeats negatively affect the replication of oriP plasmids.
Since the EBV genome contains 29 repeats within the FR element, there is a possibility
that the additional 9 repeats may play a role in maintenance of the virus during its latency. The
endogenous EBV episome within C666-1 cells includes 29 repeats within the FR region. Thus,
investigating whether the oriP plasmid containing 29 FR repeats (FR29) is better maintained will
further our understanding of persistence of the virus in C666-1 cells. Therefore, I compared the
maintenance efficiency of the FR20 and FR29 plasmids using CNE2ZE cells. To this end,
CNE2ZE cells were transfected with the FR20 and FR29 plasmids. Samples were harvested 3
days post-transfection to assess their replication while the remaining cells were continuously
passaged for weekly sample collection for a total of six weeks to analyze plasmid maintenance.
Southern blot results show that the replication signal of the FR20 plasmid decreases more rapidly
than that of the FR29 plasmid (Figure 12). Thus, although the additional 9 repeats within the FR
region reduce plasmid replication efficiency, they appear to confer an advantage to plasmid
segregation and long-term maintenance.

53
Figure 11. The extra 9 repeats reduce plasmid replication efficiency. A. Transient-replication
assays in CNE2ZE cells comparing the bands from FR20 and FR29 plasmids after 3 cell
doublings (3 days) before (Input) and after (+Dpn I) Dpn I digestion. The positions of Dpn I-
resistant bands are indicated by the asterisk. The histogram shows quantification of Dpn I-
resistant bands relative to input bands from 3 experiments. Results were plotted relative to the
signal from the FR20 plasmid (set to one). Error bars indicate standard deviations. B. CNE2ZE
cells were transfected with FR20 or FR29 plasmid as for replication assays. Cells (1x105) were
then plated in four 6-cm dishes and harvested and counted 1, 3, 5, or 7 days later. Average cell
numbers from two independent experiments are plotted with standard deviations.
C
*
A
C
B
*

54
Figure 12. The extra 9 FR repeats improve plasmid segregation. Cells were transfected with
pCAN (negative control), FR20, or FR29 plasmid as indicated and harvested after 3 to 42 cell
doublings. Cells were then lysed by the Hirt method and plasmids were isolated, linearized using
Xho I (for pCAN) or Mlu I and, where indicated, were also incubated with Dpn I to digest
unreplicated plasmids. For samples without Dpn I digestion, 1/10th
of the amount of the sample
was loaded relative to Dpn I-digested samples. DNA was separated by agarose gel
electrophoresis, Southern blotted, and probed with 32
P-labeled FR20. The position of the Dpn I-
resistant plasmid is indicated by the asterisk. The histogram shows quantification of Dpn I-
resistant bands relative to the 3-cell doubling amount, which was set to one (histogram) from
three experiments. Average values with standard deviation are shown.
*

55
III.5 Silencing of PML Protein Leads to a Reduction in Replication Efficiency in NPC cells.
Recently, our laboratory has found that promyelocytic leukemia (PML) bodies have a
repressive function in EBV lytic replication and that EBNA1 promotes the disruption and
degradation of PML bodies, thereby contributing to EBV reactivation. Whether or not PML
nuclear bodies affect latent EBV replication has not yet been investigated. I wished to determine
if PML affected the replication of oriP-containing plasmids in NPC cells. To this end, I
transfected CNE2Z cells and the same cells in which PML expression had been silenced with
shRNA (CNE2Z-shPML) with oriP (negative control), oriPE, and oriPEΔ387-394 plasmids.
Three days post-transfection, I isolated the plasmids from the two cell lines and analyzed their
replication efficiency using the previously described DpnI-resistance assay. oriPEΔ387-394
encodes for an EBNA1 deletion mutant that does not trigger PML disruption and was used to
limit the loss of PML that occurs when wild-type EBNA1 is expressed. Southern blot analysis
showed that in the absence of PML, replication of oriPE and oriPEΔ387-394 plasmids was
decreased by nearly 10-fold compared to the PML-expressing parental cell line, CNE2Z (Figure
13). Therefore, the results suggest that PML positively contributes to EBNA1-mediated
replication from oriP.

56
Figure 13. PML silencing reduces the plasmid replication efficiency in CNE2Z cells.
Transient-replication assays in CNE2Z and CNE2Z-shPML cells comparing the bands from oriP
(negative control), oriPE, and oriPEΔ387-394 plasmids after 3 cell doublings (3 days) before
(Input) and after (+Dpn I) Dpn I digestion. The positions of DpnI-resistant bands are indicated by
the asterisk. The histogram shows quantification of Dpn I-resistant bands relative to input bands
from 3 and 2 independent experiments for CNE2Z and CNE2Z-shPML cell lines, respectively.
Results were plotted relative to the signal from the oriP plasmid for each cell line. The results for
the CNE2ZshPML cell line were then plotted in relation to the results of the CNE2Z cell. Error
bars indicate standard deviations.
*

57
CHAPTER IV: DISCUSSION AND FUTURE DIRECTIONS
IV.1 FR-bound EBNA1 May Enable Expression of GAL4-Cdc6 Fusion Protein and
Facilitate Replication of pBFRGC6-based Plasmids
Six years ago Takeda et al. demonstrated that the recruitment of cellular factors involved
in pre-RC complex formation alone to DNA is sufficient to initiate replication (Takeda et al.
2005). This group created fusion proteins that combine cellular replication initiation factors such
as Cdc6 and Orc2, with the DNA-binding domain of GAL4 and developed an artificial origin in
vivo on pFRLuc plasmids by the recruitment of the fusion proteins to an element containing
GAL4 DNA-binding sites (Takeda et al. 2005). Originally, assaying the replication activity of
this system involved co-transfection of a plasmid encoding one of the fusion proteins and another
plasmid that contains the GAL4 DNA-binding sites (Takeda et al. 2005). We had initially wanted
to use this system to separate EBNA1 replication and segregation functions, which would allow
me to examine the specific regions of EBNA1 that contribute to its segregation function while
the plasmid autonomously replicates. This group provided us with a single plasmid, pFRGC6,
which contained the GAL4-Cdc6 fusion protein, an element containing 5 copies of the GAL4
DNA-binding site, and the FR element of oriP. The recruitment of GAL4-Cdc6 to the GAL4
DNA-binding sites on the pFRGC6 plasmid should create an artificial origin and allow the
plasmid to autonomously replicate, whereas the FR element should allow the plasmid to
segregate in the presence of EBNA1 during cell division. However, unexpectedly, the plasmid
did not replicate in the absence of EBNA1 and did not express GAL4-Cdc6. On the other hand,
the plasmids that encoded wild-type EBNA1 or its deletion mutants Δ8-67, Δ61-83, Δ395-450
expressed the GAL4-Cdc6 fusion protein and replicated. Past studies using chloramphenicol
acetyltransferase (CAT) gene demonstrated that in the presence of EBNA1 the FR element could
enhance transcription of the CAT gene when positioned upstream or downstream of it (Reisman
and Sugden 1986). Moreover, Gahn et al. previously demonstrated that binding of EBNA1 to the
FR element alone could increase the expression of the EBV LMP gene when separated by 10 Kb
(Gahn and Sugden 1995). Therefore, it is likely that EBNA1 and certain deletion mutants
enhanced the expression of the GAL4-Cdc6 fusion protein through binding to the FR, which also
functions as a transcriptional enhancer element. Finally, the fact that the pFRLuc plasmids,
which lack the FR element, do express the fusion protein and can autonomously replicate raises

58
the possibility that the insertion of the FR element into the pBFRGC6 plasmid may inhibit or
disrupt expression of the GAL4-Cdc6 fusion protein in the absence of EBNA1.
IV.2 Higher Levels of EBNA1 do not Inhibit Plasmid Replication and are not Cytotoxic to
CNE2Z Cells.
The EBV episome can stably persist in B-lymphocytes in tissue culture, however, this is
usually not the case in epithelial cell lines such as NPC. With the exception of the C666-1 cell
line, virtually all undifferentiated NPC cell lines cannot consistently maintain the EBV episome
in culture and as a result, they are all EBV-negative (Cheung et al. 1999). As part of my project,
I was interested in understanding the basis for the difference in EBV maintenance between
C666-1 and other NPC cell lines and specifically, whether it is related to oriP-mediated
replication and segregation functions. A previous student in our laboratory found no apparent
difference in oriP-mediated plasmid replication and maintenance between C666-1 and EBV-
negative NPC cell lines under conditions with low EBNA1 expression; however, higher EBNA1
levels interfered with plasmid segregation during mitosis (Sivachandran et al. 2011). Consistent
with a role in selectively inhibiting plasmid maintenance, using the oriPE plasmid which
overexpresses EBNA1, I showed that higher levels of EBNA1 did not decrease plasmid
replication efficiency in either the C666-1 or CNE2ZE cell line and did not exhibit any cytotoxic
effects. In fact, I found that the oriPE plasmid replicated twice as efficiently as the oriP plasmid
in C666-1 cells. Therefore, with no negative effects on plasmid replication or cell growth rate,
higher EBNA1 expression must be detrimental to another aspect of oriP plasmid maintenance,
most likely segregation. EBV-infected NPC cells exhibit latency program II where EBNA1
expression is controlled by the Qp promoter, which is subject to auto-regulation and maintains
low EBNA1 levels (Sample et al. 1992; Rickinson and Kieff 1996). Therefore, a possible
threshold in EBNA1 expression may exist and contribute to how well oriP plasmids, and
potentially the EBV episome, are stably maintained in NPC cells in long-term culture. Moreover,
FR-bound EBNA1 and its interactions with cellular proteins such as EBP2 are necessary for
efficient oriP plasmid segregation; hence, an excess of EBNA1 may perturb ternary complex
formation and disrupt plasmid partitioning. Also, it has been proposed that EBNA1 may interact
directly with DNA through Gly-Arg repeats which resemble AT-hooks (Sears et al. 2004). If
EBNA1 itself binds to regions on mitotic chromosomes, it is possible that when EBNA1 is in

59
excess non-FR-bound EBNA1 molecules may compete with FR-bound EBNA1 and saturate
sites on chromosomal regions that would normally facilitate the tethering of oriP plasmids and
EBV episomes. As a consequence, this could lead to inefficient plasmid and episomal
segregation and would be reflected in decreased long-term maintenance.
Past studies by Yates et al. demonstrated that oriP plasmids do not replicate more than
once per cell cycle even in the presence of excess EBNA1 (Yates and Guan 1991). Therefore, the
increase in oriPE plasmid replication efficiency observed in C666-1 cells should not be
attributed to plasmid re-replication during cell division and indeed, higher levels of EBNA1
alone may improve plasmid replication efficiency in C666-1 cells. Niro Sivachandran in our
laboratory has also shown that replication efficiency of the oriPE plasmid is comparable between
CNE2Z and C666-1 cells (Sivachandran et al. 2011). C666-1 cells express full-length EBNA1
(including the Gly-Ala repeat region) at low levels from the endogenous EBV episome, which is
capable of replicating and maintaining the oriP plasmid (Figure 7B and 9A). However, from my
findings, I suggest that full-length EBNA1 alone may not mediate replication the oriP plasmid as
efficiently as when EBNA1ΔG/A is co-expressed at much higher levels (Figure 9A histogram) in
C666-1 cells. In accordance with this idea, both oriP and oriPE plasmids were replicated with a
similar efficiency in CNE2ZE cells (Figure 8A histogram). This cell line stably expresses
EBNA1ΔG/A at low levels, which enabled replication of the oriP plasmid (Figure 8A, lane 7).
Interestingly, overexpression of EBNA1ΔG/A in CNE2ZE cells did not improve plasmid
replication as it did in C666-1. Together, these findings imply a possible regulatory role for the
Gly-Ala repeat region in EBNA1-mediated plasmid replication. Alternatively, due to the fact that
C666-1 is the only NPC cell that naturally maintains the EBV episome, it is possible that there
are factors unique to this cell line that may enable increased plasmid replication efficiency in the
presence of excess EBNA1 (Cheung et al. 1999). EBNA1 expression is tightly regulated by the
Qp promoter and is normally expressed at low levels in this cell line, therefore, it may be a
limiting factor for some processes.
Despite its ability to disrupt plasmid segregation, high levels of EBNA1 did not interfere
in EBV episomal segregation or maintenance in C666-1 or the EBV-positive gastric carcinoma
epithelial AGS/EBV cell line. These results suggest that additional factors and even sequences
within the complete EBV episome may contribute to the maintenance of the viral genomes. A
nuclear matrix attachment region (MAR) which traverses the oriP sequence and the EBER

60
genes, has been previously identified within the EBV genome (Jankelevich et al. 1992). It is
possible that the MAR may confer additional long-term stability of the viral episome and
contribute to EBV persistence due to the fact that this region has been shown to increase
maintenance of EBV plasmids up to 2-fold greater than plasmids lacking the MAR region
(Jankelevich et al. 1992; White et al. 2001). Moreover, in addition to EBP2, other cellular factors
may affect the maintenance of the EBV genome, which quite possibly could contain recognition
sites for cellular proteins that function with EBNA1 to tether the large EBV episome to mitotic
chromosomes. Also, there is thought to be a selective pressure on C666-1 to maintain the EBV
genomes due to expression of viral proteins (e.g. EBNA1), miRNAs, EBERs etc. on which these
tumour cells may have become dependent for growth and survival (Shannon-Lowe et al. 2009).
Burkitt’s lymphoma cell lines are dependent on EBV for growth and survival in vivo, and do not
lose the episome in long-term culture (Kennedy et al. 2003; Hammerschmidt and Sugden 2004).
It is thought that upon isolation from tumour biopsies, NPC cell lines (except C666-1) lose their
dependency on EBV-induced growth and survival genes and eventually lose the virus in culture
(Shannon-Lowe et al. 2009).
IV.3 Additional 9 Repeats within the FR Element may Confer an Advantage in EBV
Maintenance
The FR element of oriP is a cis-acting sequence that functions in episomal and plasmid
maintenance through its role in segregation (Yates et al. 1984; Reisman and Sugden 1986). It is
comprised of a tandem array of 30 bp high-affinity recognition sites for EBNA1, which
constitutively binds to each of these sites as a dimer and assembles onto the sequence in a
cooperative manner (Hsieh et al. 1993; Summers et al. 1996). The binding of EBNA1 to the FR
sequence enables the EBV episome and EBV-based plasmids to be piggy-backed or tethered to
host condensed mitotic chromosomes and their subsequent segregation to daughter cells during
cell division (Krysan et al. 1989; Ceccarelli and Frappier 2000; Kanda et al. 2001).
For years it has been believed that the FR element of oriP, as cloned from the genome of
the B95-8 EBV strain, is comprised of 20 copies of the 30 bp repeat (Baer et al. 1984). Indeed,
all available oriP-containing plasmids contain 20 repeats within the FR element, which have
been proven to be sufficient for FR-associated functions. However, ten years ago Fruscalzo et al.
discovered that 252bp of the B95-8 derived-oriP sequence had been deleted, most likely due to

61
its hairpin secondary structure and repeat instability in E.coli during cloning procedures
(Fruscalzo et al. 2001). The 252 bp sequence was found to correspond to 9 additional repeats
within the FR element. Hence, the full-length FR of the B95-8 EBV strain actually contains 29
repeats as opposed to 20 repeats (Fruscalzo et al. 2001). Ali et al. were able to subclone the full-
length FR element (29 repeats) into a low-copy plasmid and their studies in B-lymphocytes
revealed some functional differences between 20 and 29 FR repeats (Ali et al. 2009). In B-cells
the 29 repeats were found to reduce the FR transcriptional enhancer activity, but it improved B-
cell transformation, and did not affect plasmid replication efficiency (Ali et al. 2009).
Conversely, in NPC cells, which are of epithelial origin, I have shown that the presence of the
additional 9 repeats within the FR element reduces plasmid replication efficiency by
approximately 40% as compared to an oriP plasmid with the original 20 repeats. As each 30 bp
FR repeat contains an EBNA1 recognition site, the additional 9 repeats potentially allows for a
45% increase in EBNA1 binding by comparing 20 versus 29 repeats within the FR.
One reason why a larger FR might decrease replication is the ability of the FR, when
bound by EBNA1, to stall the progression of replication forks (Gahn and Schildkraut 1989;
Ermakova et al. 1996). The association of EBNA1 with the FR repeat sequences not only
functions in genome segregation, but is also known to impede the progression of replication
forks traversing through FR repeats within oriP as the EBV genome replicates during S phase
(Gahn and Schildkraut 1989; Ermakova et al. 1996; Aiyar et al. 2009). Studies have shown that
oriP-mediated DNA replication initiation occurs at or in close proximity to the DS element and
replication subsequently stalls at the FR element, which presents a barrier to approaching
replication forks (Gahn and Schildkraut 1989; Dhar and Schildkraut 1991). Although the FR
repeats alone have the inherent ability to hinder the progression of replication forks, it is the
binding of EBNA1 to the FR repeats which forms a protein-DNA complex that is responsible for
creating the highly efficient barrier for replication forks (Dhar and Schildkraut 1991). Moreover,
there is evidence that the number of repeats (and EBNA1-binding sites) within the FR element
determines the efficiency with which the FR functions as a replication fork barrier (Platt et al.
1993; Aiyar et al. 2009). Therefore, the presence of the extra 9 repeats may increase the rate of
replication fork stalling at the FR, thereby decreasing replication efficiency. It is conceivable that
impeding replication fork migration through the FR might serve to limit and maintain EBV copy
number in infected cells (Aiyar et al. 2009). Thus, the reduction in replication efficiency due to

62
the full-length FR may be important during EBV latent infection and maintaining a constant
copy number of several cell generations. Moreover, the EBV EBERs genes, which are heavily
transcribed throughout latent infection, are found immediately 5’ of the FR element and are
transcribed toward the FR and DS regions (Baer et al. 1984). Replication of these genes also
proceeds in the same direction; hence, it is thought that the FR replication barrier may also
function to prevent collisions of replication and transcription forks and resultant DNA damage
(Mirkin and Mirkin 2007). This replication fork barrier function of the EBNA1-FR association
may be analogous to the non-transcribed spacer of the rRNA genes in Saccharomyces cerevisiae,
which contains a replication barrier that forms upon the interaction of the DNA-binding protein
Fob1 with its recognition sites and together they avert head-on replication-transcription fork
collisions (Mirkin and Mirkin 2007).
Although the additional 9 repeats within the FR region reduce plasmid replication
efficiency, I have shown that these extra repeats positively affect plasmid maintenance. By
comparing the long-term maintenance efficiencies of FR20 and FR29 plasmids, which contain
20 and 29 FR repeats, respectively, I have shown that these extra 9 repeats may confer an
advantage in plasmid segregation ability, as the FR29 plasmid was maintained better over 6
weeks than the FR20 plasmid. Together, EBNA1 and cellular proteins (e.g. EBP2) tether EBV
genomes and oriP-containing plasmids to mitotic chromosomes via EBNA1-binding to
recognition sites within the FR element, thereby facilitating plasmid and episomal partitioning
during cell division (Kapoor et al. 2001; Kapoor and Frappier 2003; Kapoor et al. 2005).
Therefore, the full-length FR may improve plasmid segregation by increasing the avidity of the
attachment of plasmids to condensed mitotic chromosomes. Each FR repeat contains a high-
affinity EBNA1 recognition site, hence, it is possible that by increasing the number of EBNA1
binding sites, this will in turn strengthen the adhesion between the oriP plasmids to cellular
chromosomes, which could enhance plasmid segregation efficiency during mitosis. Skalsky et al.
studies of KSHV episomal maintenance have also provided evidence that multiple copies of
LANA-binding sites 1 and 2 (LBS1 and LBS2), which lie in a region consisting of 35 to 45
copies of terminal repeats (referred to as the TR region), may have an episomal maintenance
function similar to that of the FR element of oriP (Skalsky et al. 2007). In fact, this group
demonstrated that increasing the number of LANA-binding sites positively affects long-term
KSHV episomal maintenance efficiency, which is similar to the improved oriP plasmid

63
maintenance that I have found with the additional 9 FR repeats. Moreover, as mentioned
previously, the FR repeats inherently function as natural impediments of the replication fork for
the EBV genome. Dheekollu et al. have recently shown that the replication pausing factor
Timeless (or Tim) is necessary for maintenance of the circular EBV episomes in latent EBV-
infected B-lymphocytes (Dheekollu and Lieberman 2011). Tim is an evolutionarily conserved
proteins that helps stabilize replication forks upon encountering repetitive sequences or DNA
secondary structures and may play a role in sister chromatid cohesion and chromosome
segregation during mitosis (Gotter et al. 2007; Leman et al. 2010; Smith-Roe et al. 2011). Tim
was shown to localize to oriP during cellular S phase and silencing of Tim resulted in an
increase in DNA double-stranded breaks, loss of EBV episomal stability, as well as inhibition of
oriP-mediated replication initiation (Dheekollu and Lieberman 2011). Dheekollu et al. proposed
that there is a linkage between replication fork impediments and maintaining DNA stability and
EBV genome persistence. Therefore, it is conceivable that additional replication fork blockage
conferred by the extra 9 FR repeats as well as increased Tim occupation at oriP may contribute
to the improved segregation and maintenance for the FR29 plasmid.
IV.4 Possible Role of PML in Plasmid DNA Replication?
One of the barriers the cell has developed to defend itself against viral infection is PML
nuclear bodies. Over the years, evidence consistent with both positive and negative roles for
PML in viral replication has been accumulating. Our laboratory has demonstrated that EBNA1-
mediated disruption of PML NBs contributes to EBV reactivation and lytic DNA replication,
which implies a repressive function for PML NBs in these processes (Sivachandran et al.
submitted). EBNA1 disrupts PML NBs and lowers PML protein levels in EBV-negative cells as
well as the latently EBV-infected C666-1 cell line (Sivachandran et al. 2008). I have shown that
depletion of PML by targeted short hairpin RNA drastically reduces the oriP plasmid replication
efficiency in NPC cells, which supports a positive contribution of PML to oriP-mediated
replication. However, I also found that upon treatment with the transfection reagent, CNE2Z
cells depleted of PML exhibited a decrease in proliferation rate as well as a noticeable increase in
cell death as compared to PML-positive CNE2Z cells. In fact, unlike the transfected PML-
positive CNE2Z cells, I did not transfer the CNE2ZshPML cells from a small tissue culture dish
to a larger dish following transfection due to the aforementioned growth differences. It is

64
possible that when cells are depleted of PML and are deficient in PML-associated functions they
are considerably more sensitive to cellular stress, which may eventually lead to an increase in
cell death and explain the growth phenotype that I observed with the CNE2ZshPML cells. In
order to measure plasmid replication efficiency, cells are harvested 3 cell doublings post-
transfection. As oriP plasmids are subject to cellular replication licensing and only replicate once
per cell cycle, a decrease in growth rate will lead to a decrease in the amount of times the
plasmids will undergo replication by the normal 3 cell doubling time point. Therefore, the
decrease in oriP plasmid replication signal observed in CNE2ZshPML cells may be attributed to
a combination of (i) reduction in plasmid replication efficiency due to PML depletion and (ii)
decreased cell proliferation after transfection.
IV.5 Future Directions
Recently, Erle Roberston’s group has found that the kinetochore complex component,
Bub1, can interact with LANA, the KSHV structural homologue of EBNA1 (Xiao et al. 2010).
Using FISH, this group also found that Bub1 co-localizes with circular KSHV episomes as well
as LANA on mitotic chromosomes and particularly at the kinetochore. Moreover, depletion of
Bub1 caused a major reduction in KSHV genome maintenance in KSHV-positive cells (Xiao et
al. 2010). The authors proposed that Bub1 may play an important role in KSHV episomal
segregation and maintenance. It is possible that the cellular Bub1 protein may contribute to EBV
maintenance and possibly via interactions with EBNA1. It would be interesting to investigate
whether or not Bub1 can interact with EBNA1 in vitro and/or in vivo and if Bub1 contributes to
the oriP-mediated segregation function.
In 2004, Kanda et al. cloned the Akata-derived EBV genome into the bacterial artificial
chromosome (BAC) vector and developed a system for producing infectious EBV recombinants
(Kanda et al. 2004). I have found that the extra 9 repeats within the FR element of oriP
negatively affect plasmid replication efficiency but have a positive contribution to plasmid
maintenance. Therefore, it would be valuable to use a similar recombinant EBV system and
determine if the additional 9 repeats are advantageous in long-term maintenance of the entire
EBV episome. This could help our understanding of the reason for which the EBV virus has
evolved to maintain this number of repeats and is capable of persisting for the lifetime of its host.

65
Alternative splicing of the PML gene generates seven isoforms of PML protein of
varying sizes that share a common N-terminus but differ in their C-terminal cytoplasmic tails
(Jensen et al. 2001). Xueqi Wang in our laboratory has generated derivatives of the
CNE2ZshPML cell line that stably express each PML isoform (I to VI) at levels that are
comparable to that of endogenous PML. My results have suggested that PML may have a
positive role in DNA replication and it is possible that this function could be attributed to a
single or combination of PML isoforms. It would be interesting to analyze plasmid replication
efficiency in each of the derived CNE2ZshPML cell lines to determine if one of the PML
isoforms can fully or partially restore the reduced plasmid replication efficiency to a level that is
comparable to that of the parental PML-positive CNE2Z cell line. First, it is important to
determine the extent that the decreased proliferation rate may have contributed to the
considerable reduction in plasmid replication efficiency that I had observed in transfected
CNE2ZshPML cells. Essentially, the growth rate of a PML-depleted cell line must be
comparable to that of a PML-positive cell line. Transfection reagents can be toxic and cause
stress to treated cells, hence, finding a less toxic/stressful transfection reagent as well as
optimizing the reagent to DNA ratio and cell density conditions may be helpful and reduce the
amount of cell death that I previously observed. Therefore, once a clear association between
PML-depletion and reduced plasmid replication efficiency can be made, this will facilitate the
subsequent determination of which PML isoform(s) may be involved in DNA replication.

66
CHAPTER V: REFERENCES
Aiyar, A., S. Aras, et al. (2009). "Epstein-Barr Nuclear Antigen 1 modulates replication of oriP-
plasmids by impeding replication and transcription fork migration through the family of
repeats." Virol J 6: 29.
Ali, A. K., S. Saito, et al. (2009). "Distinctive effects of the Epstein-Barr virus family of repeats
on viral latent gene promoter activity and B-lymphocyte transformation." J Virol 83(18):
9163-9174.
Altmann, M., D. Pich, et al. (2006). "Transcriptional activation by EBV nuclear antigen 1 is
essential for the expression of EBV's transforming genes." Proc Natl Acad Sci U S A
103(38): 14188-14193.
Ambinder, R. F., M. Mullen, et al. (1991). "Functional domains of Epstein-Barr nuclear antigen
EBNA-1." J. Virol. 65: 1466-1478.
Ambinder, R. F., W. A. Shah, et al. (1990). "Definition of the sequence requirements for binding
of the EBNA-1 protein to its palindromic target sites in Epstein-Barr virus DNA." J.
Virol. 64: 2369-2379.
Aravind, L. and D. Landsman (1998). "AT-hook motifs identified in a wide variety of DNA-
binding proteins." Nucleic Acids Res 26(19): 4413-4421.
Arias, E. E. and J. C. Walter (2007). "Strength in numbers: preventing rereplication via multiple
mechanisms in eukaryotic cells." Genes Dev 21(5): 497-518.
Atanasiu, C., Z. Deng, et al. (2006). "ORC binding to TRF2 stimulates OriP replication." EMBO
Rep 7(7): 716-721.
Avolio-Hunter, T. M. and L. Frappier (1998). "Mechanistic studies on the DNA linking activity
of the Epstein-Barr nuclear antigen 1." Nucl. Acids Res. 26: 4462-4470.
Avolio-Hunter, T. M., P. N. Lewis, et al. (2001). "Epstein-Barr nuclear antigen 1 binds and
destbilizes nucleosomes at the viral origin of latent DNA replication." Nucl. Acids Res.
29: 3520-3528.
Baer, R., A. T. Bankier, et al. (1984). "DNA sequence and expression of the B95-8 Epstein-Barr
virus genome." Nature 310: 207-211.
Ballestas, M. E., P. A. Chatis, et al. (1999). "Efficient persistence of extrachromosomal KSHV
DNA mediated by latency-associated nuclear antigen." Science 284: 641-644.
Barbera, A. J., M. E. Ballestas, et al. (2004). "The Kaposi's sarcoma-associated herpesvirus
latency-associated nuclear antigen 1 N terminus is essential for chromosome association,
DNA replication, and episome persistence." J Virol 78(1): 294-301.

67
Barbera, A. J., J. V. Chodaparambil, et al. (2006). "The nucleosomal surface as a docking station
for Kaposi's sarcoma herpesvirus LANA." Science 311(5762): 856-861.
Bashaw, J. M. and J. L. Yates (2001). "Replication from oriP of Epstein-Barr virus requires exact
spacing of two bound dimers of EBNA1 which bend DNA." J. Virol. 75: 10603-10611.
Bell, S. P. and A. Dutta (2002). "DNA replication in eukaryotic cells." Annu. Rev. Biochem. 71:
333-374.
Bernardi, R. and P. P. Pandolfi (2007). "Structure, dynamics and functions of promyelocytic
leukaemia nuclear bodies." Nat Rev Mol Cell Biol 8: 1006-1016.
Blake, N., S. Lee, et al. (1997). "Human CD8+ T cell responses to EBV EBNA1: HLA class I
presentation of the (GLY-ALA) containing protein requires exogenous processing."
Immunity 7: 791-802.
Blondel, D., T. Regad, et al. (2002). "Rabies virus P and small P products interact directly with
PML and reorganize PML nuclear bodies." Oncogene 21(52): 7957-7970.
Bochkarev, A., J. Barwell, et al. (1996). "Crystal structure of the DNA-binding domain of the
Epstein-Barr virus origin binding protein, EBNA1, bound to DNA." Cell 84: 791-800.
Bochkarev, A., J. Barwell, et al. (1995). "Crystal structure of the DNA binding domain of the
Epstein-Barr virus origin binding protein EBNA1." Cell 83: 39-46.
Bochkarev, A., E. Bochkareva, et al. (1998). "2.2A structure of a permanganate-sensitive DNA
site bound by the Epstein-Barr virus origin binding protein, EBNA1." J. Mol. Biol. 284:
1273-1278.
Boichuk, S., L. Hu, et al. (2011). "Functional Connection between Rad51 and PML in
Homology-Directed Repair." PLoS One 6(10): e25814.
Brooks, L., Q. Y. Yao, et al. (1992). "Epstein-Barr virus latent gene transcription in
nasopharyngeal carcinoma cells: coexpression of EBNA1, LMP1, and LMP2 transcripts."
J Virol 66(5): 2689-2697.
Bui, C. T., K. Rees, et al. (2003). "Permanganate oxidation reactions of DNA: perspective in
biological studies." Nucleosides Nucleotides Nucleic Acids 22(9): 1835-1855.
Ceccarelli, D. F. J. and L. Frappier (2000). "Functional Analyses of the EBNA1 origin DNA
binding protein of Epstein-Barr virus." J. Virol. 74: 4939-4948.
Chau, C. M. and P. M. Lieberman (2004). "Dynamic chromatin boundaries delineate a latency
control region of Epstein-Barr virus." J Virol 78(22): 12308-12319.

68
Chaudhuri, B., H. Xu, et al. (2001). "Human DNA replication initiation factors, ORC and MCM,
associate with oriP of Epstein-Barr virus." Proc. Natl. Acad. Sci. USA 98: 10085-10089.
Cheung, S. T., D. P. Huang, et al. (1999). "Nasopharyngeal carcinoma cell line (C666-1)
consistently harbouring Epstein-Barr virus." Int J Cancer 83: 121-126.
Cotter, M. A. and E. S. Robertson (1999). "The latency-associated nuclear antigen tethers the
Kaposi's sarcoma-associated herpesvirus genome to host genomes to host chromosomes
in body cavity-based lymphoma cells." Virol. 264: 254-264.
Crowder, C., O. Dahle, et al. (2005). "PML mediates IFN-alpha-induced apoptosis in myeloma
by regulating TRAIL induction." Blood 105(3): 1280-1287.
Cruickshank, J., A. Davidson, et al. (2000). "Two domains of the Epstein-Barr virus origin DNA
binding protein, EBNA1, orchestrate sequence-specific DNA binding." J. Biol. Chem.
275: 22273-22277.
Cummins, J. M., C. Rago, et al. (2004). "Tumour suppression: Disruption of HAUSP gene
stabilizes p53." Nature 428: 486-487.
Cummins, J. M. and B. Vogelstein (2004). "HAUSP is required for p53 destabilization." Cell
Cycle 3(6): 689-692.
Deacon, E. M., G. Pallesen, et al. (1993). "Epstein-Barr virus and Hodgkin's disease:
transcriptional analysis of virus latency in the malignant cells." J Exp Med 177(2): 339-
349.
Delecluse, H.-J., S. Bartnizke, et al. (1993). "Episomal and integrated copies of Epstein-Barr
virus coexist in Burkitt's lymphoma cell lines." J. Virol. 67: 1292-1299.
Delecluse, H. J., R. Feederle, et al. (2007). "Epstein Barr virus-associated tumours: an update for
the attention of the working pathologist." J Clin Pathol 60(12): 1358-1364.
Dellaire, G. and D. P. Bazett-Jones (2004). "PML nuclear bodies: dynamic sensors of DNA
damage and cellular stress." Bioessays 26(9): 963-977.
Deng, Z., C. Atanasiu, et al. (2003). "Telomere repeat binding factors TRF1, TRF2, and hRAP1
modulate replication of Epstein-Barr virus OriP." J Virol 77(22): 11992-12001.
Deng, Z., C. Atanasiu, et al. (2005). "Inhibition of Epstein-Barr virus OriP function by tankyrase,
a telomere-associated poly-ADP ribose polymerase that binds and modifies EBNA1." J
Virol 79(8): 4640-4650.
Deng, Z., L. Lezina, et al. (2002). "Telomeric proteins regulate episomal maintenance of
Epstein-Barr virus origin of plasmid replication." Mol. Cell 9: 493-503.

69
Deutsch, M. J., E. Ott, et al. (2010). "The latent origin of replication of Epstein-Barr virus directs
viral genomes to active regions of the nucleus." J Virol 84(5): 2533-2546.
Dhar, S. K., K. Yoshida, et al. (2001). "Replication from oriP of Epstein-Barr virus requires
human ORC and is inhibited by geminin." Cell 106: 287-296.
Dhar, V. and C. L. Schildkraut (1991). "Role of EBNA-1 in arresting replication forks at the
Epstein-Barr virus oriP family of tandem repeats." Mol. Cell. Biol. 11: 6268-6278.
Dheekollu, J., Z. Deng, et al. (2007). "A role for MRE11, NBS1, and recombination junctions in
replication and stable maintenance of EBV episomes." PLoS ONE 2(12): e1257.
Dheekollu, J. and P. M. Lieberman (2011). "The replisome pausing factor Timeless is required
for episomal maintenance of latent Epstein-Barr virus." J Virol 85(12): 5853-5863.
Di Croce, L. (2005). "Chromatin modifying activity of leukaemia associated fusion proteins."
Hum Mol Genet 14 Spec No 1: R77-84.
Dittmer, D. P., C. J. Hilscher, et al. (2008). "Multiple pathways for Epstein-Barr virus episome
loss from nasopharyngeal carcinoma." Int J Cancer 123(9): 2105-2112.
Djavani, M., J. Rodas, et al. (2001). "Role of the promyelocytic leukemia protein PML in the
interferon sensitivity of lymphocytic choriomeningitis virus." J Virol 75(13): 6204-6208.
Edwards, A. M., A. Bochkarev, et al. (1998). "Origin DNA -binding proteins." Curr. Opin.
Struct. Biol. 8: 49-53.
Epstein, M. A., B. G. Achong, et al. (1964). "Virus Particles in Cultured Lymphoblasts from
Burkitt's Lymphoma." Lancet 1(7335): 702-703.
Ermakova, O., L. Frappier, et al. (1996). "Role ot the EBNA-1 protein in pausing of replication
forks in the Epstein-Barr virus genome." J. Biol. Chem. 271: 33009-33017.
Everett, R. D. (2001). "DNA viruses and viral proteins that interact with PML nuclear bodies."
Oncogene 20(49): 7266-7273.
Everett, R. D. and M. K. Chelbi-Alix (2007). "PML and PML nuclear bodies: implications in
antiviral defence." Biochimie 89(6-7): 819-830.
Everett, R. D. and J. Murray (2005). "ND10 components relocate to sites associated with herpes
simplex virus type 1 nucleoprotein complexes during virus infection." J Virol 79(8):
5078-5089.
Everett, R. D., S. Rechter, et al. (2006). "PML contributes to a cellular mechanism of repression
of herpes simplex virus type 1 infection that is inactivated by ICP0." J Virol 80(16):
7995-8005.

70
Fahraeus, R. (2005). "Do peptides control their own birth and death?" Nat Rev Mol Cell Biol
6(3): 263-267.
Fahraeus, R., H. L. Fu, et al. (1988). "Expression of Epstein-Barr virus-encoded proteins in
nasopharyngeal carcinoma." Int J Cancer 42(3): 329-338.
Fingeroth, J. D., J. J. Weis, et al. (1984). "Epstein-Barr virus receptor of human B lymphocytes is
the C3d receptor CR2." Proc Natl Acad Sci U S A 81(14): 4510-4514.
Frappier, L. (2010). EBNA1 in Viral DNA Replication and Persistence. Epstein-Barr Virus:
Latency and Transformation. E. S. Robertson. Norwich, Caister Academic Press: 37-59.
Frappier, L., K. Goldsmith, et al. (1994). "Stabilization of the EBNA1 protein on the Epstein-
Barr virus latent origin of DNA replication by a DNA looping mechanism." J. Biol.
Chem. 269: 1057-1062.
Frappier, L. and M. O'Donnell (1991). "Epstein-Barr nuclear antigen 1 mediates a DNA loop
within the latent replication origin of Epstein-Barr virus." Proc. Natl. Acad. Sci. USA 88:
10875-10879.
Frappier, L. and M. O'Donnell (1991). "Overproduction, purification and characterization of
EBNA1, the origin binding protein of Epstein-Barr virus." J. Biol. Chem. 266: 7819-
7826.
Frappier, L. and M. O'Donnell (1992). "EBNA1 distorts oriP, the Epstein-Barr virus latent
replication origin." J. Virol. 66: 1786-1790.
Fruscalzo, A., G. Marsili, et al. (2001). "DNA sequence heterogeneity within the Epstein-Barr
virus family of repeats in the latent origin of replication." Gene 265(1-2): 165-173.
Fukayama, M. (2010). "Epstein-Barr virus and gastric carcinoma." Pathol Int 60(5): 337-350.
Fukayama, M., R. Hino, et al. (2008). "Epstein-Barr virus and gastric carcinoma: virus-host
interactions leading to carcinoma." Cancer Sci 99(9): 1726-1733.
Gahn, T. and B. Sugden (1995). "An EBNA1 Dependent enhancer acts from a distance of 10
kilobase pairs to increase expression of the Epstien-Barr virus LMP gene." J. Virol. 69:
2633-2636.
Gahn, T. A. and C. L. Schildkraut (1989). "The Epstein-Barr virus origin of plasmid replication,
oriP, contains both the initiation and termination sites of DNA replication." Cell 58: 527-
535.
Gerber, P., S. Lucas, et al. (1972). "Oral excretion of Epstein-Barr virus by healthy subjects and
patients with infectious mononucleosis." Lancet 2(7785): 988-989.

71
Gotter, A. L., C. Suppa, et al. (2007). "Mammalian TIMELESS and Tipin are evolutionarily
conserved replication fork-associated factors." J Mol Biol 366(1): 36-52.
Grogan, E. A., W. P. Summers, et al. (1983). "Two Epstein-Barr viral nuclear neoantigens
distinguished by gene transfer, serology and chromosome binding." Proc. Natl. Acad. Sci.
USA 80: 7650-7653.
Hammerschmidt, W. and B. Sugden (2004). "Epstein-Barr virus sustains Burkitt's lymphomas
and Hodgkin's disease." Trends Mol Med 10(7): 331-336.
Harris, A., B. D. Young, et al. (1985). "Random association of Epstein-Barr virus genomes with
host cell metaphase chromosomes in Burkitt's lymphoma-derived cell lines." J. Virol. 56:
328-332.
Harrison, S., K. Fisenne, et al. (1994). "Sequence requirements of the Epstein-Barr virus latent
origin of DNA replication." J Virol 68(3): 1913-1925.
Hegde, R. S., S. R. Grossman, et al. (1992). "Crystal structure at 1.7Å of the bovine
papillomavirus-1 E2 DNA-binding protein bound to its DNA target." Nature 359: 505-
512.
Henle, G. and W. Henle (1976). "Epstein-Barr virus-specific IgA serum antibodies as an
outstanding feature of nasopharyngeal carcinoma." Int J Cancer 17(1): 1-7.
Hieter, P., C. Mann, et al. (1985). "Mitotic stability of yeast chromosomes: A colony color assay
that measures nondisjunction and chromosome loss." Cell 40: 381-392.
Hirt, B. (1967). "Selective extraction of polyoma DNA from infected mouse cell culture." J. Mol.
Biol. 26: 365-369.
Holowaty, M. N., Y. Sheng, et al. (2003). "Protein interaction domains of the ubiqutin specific
protease, USP7/HAUSP." J. Biol. Chem. 278: 47753-47761.
Holowaty, M. N., M. Zeghouf, et al. (2003). "Protein profiling with Epstein-Barr nuclear antigen
1 reveals an interaction with the herpesvirus-associated ubiquitin-specific protease
HAUSP/USP7." J. Biol. Chem. 278: 29987-29994.
Hong, M., Y. Murai, et al. (2006). "Suppression of Epstein-Barr nuclear antigen 1 (EBNA1) by
RNA interference inhibits proliferation of EBV-positive Burkitt's lymphoma cells." J
Cancer Res Clin Oncol 132(1): 1-8.
Hsieh, D.-J., S. M. Camiolo, et al. (1993). "Constitutive binding of EBNA1 protein to the
Epstein-Barr virus replication origin, oriP, with distortion of DNA structure during latent
infection." EMBO J. 12: 4933-4944.

72
Huang, D. P., H. C. Ho, et al. (1978). "Presence of EBNA in nasopharyngeal carcinoma and
control patient tissues related to EBV serology." Int J Cancer 22(3): 266-274.
Hung, S. C., M.-S. Kang, et al. (2001). "Maintenance of Epstein-Barr virus (EBV) oriP-based
episomes requires EBV-encoded nuclear antigen-1 chromosome-binding domains, which
can be replaced by high-mobility group-I or histone H1." Proc. Natl. Acad. Sci. USA 98:
1865-1870.
Ilves, I., S. Kivi, et al. (1999). "Long-term episomal maintenance of bovine papillomavirus type
1 plasmids is determined by attachment to host chromosomes, which is mediated by the
viral E2 protein and its binding sites." J. Virol. 73: 4404-4412.
Inman, G. J. (2011). "Switching TGFbeta from a tumor suppressor to a tumor promoter." Curr
Opin Genet Dev 21(1): 93-99.
Ishimi, Y. (1997). "A DNA helicase activity is associated with an MCM4,-6,-7 protein
complex." J. Biol. Chem. 272: 24508-24513.
Ishov, A. M. and G. G. Maul (1996). "The periphery of nuclear domain 10 (ND10) as site of
DNA virus deposition." J Cell Biol 134(4): 815-826.
Jankelevich, S., J. L. Kolman, et al. (1992). "A nuclear matrix attachment region organizes the
Epstein-Barr viral plasmid in Raji cells into a single DNA domain." EMBO J 11: 1165-
1176.
Jensen, K., C. Shiels, et al. (2001). "PML protein isoforms and the RBCC/TRIM motif."
Oncogene 20(49): 7223-7233.
Jones, C. H., S. D. Hayward, et al. (1989). "Interaction of the lymphocyte-derived Epstein-Barr
virus nuclear antigen EBNA-1 with its DNA-binding sites." J. Virol. 63: 101-110.
Julien, M. D., Z. Polonskaya, et al. (2004). "Protein and sequence requirements for the
recruitment of the human origin recognition complex to the latent cycle origin of DNA
replication of Epstein-Barr virus oriP." Virology 326(2): 317-328.
Kanda, T., M. Kamiya, et al. (2007). "Symmetrical localization of extrachromosomally
replicating viral genomes on sister chromatids." J Cell Sci 120(Pt 9): 1529-1539.
Kanda, T., M. Otter, et al. (2001). "Coupling of mitotic chromosome tethering and replication
competence in Epstein-Barr virus-based plasmids." Mol. Cell. Biol. 21: 3576-3588.
Kanda, T., M. Yajima, et al. (2004). "Production of high-titer Epstein-Barr virus recombinants
derived from Akata cells by using a bacterial artificial chromosome system." J Virol
78(13): 7004-7015.

73
Kang, M. S., S. C. Hung, et al. (2001). "Epstein-Barr virus nuclear antigen 1 activates
transcription from episomal but not integrated DNA and does not alter lymphocyte
growth." Proc Natl Acad Sci U S A 98(26): 15233-15238.
Kang, M. S., H. Lu, et al. (2005). "Epstein-Barr virus nuclear antigen 1 does not induce
lymphoma in transgenic FVB mice." Proc Natl Acad Sci U S A 102(3): 820-825.
Kang, M. S., V. Soni, et al. (2008). "Epstein-Barr Virus Nuclear Antigen 1 does not cause
lymphoma in C57BL/6J mice." J Virol 82: 4180-4183.
Kapoor, P. and L. Frappier (2003). "EBNA1 partitions Epstein-Barr virus plasmids in yeast by
attaching to human EBNA1-binding protein 2 on mitotic chromosomes." J. Virol. 77:
6946-6956.
Kapoor, P., B. D. Lavoie, et al. (2005). "EBP2 plays a key role in Epstein-Barr virus mitotic
segregation and is regulated by aurora family kinases." Mol Cell Biol 25(12): 4934-4945.
Kapoor, P., K. Shire, et al. (2001). "Reconstitution of Epstein-Barr virus-based plasmid
partitioning in budding yeast." EMBO J. 20: 222-230.
Kennedy, G., J. Komano, et al. (2003). "Epstein-Barr virus provide a survival factor to Burkitt's
lymphomas." Proc. Natl. Acad. Sci. 100: 14269-14274.
Kennedy, G. and B. Sugden (2003). "EBNA-1, a bifunctional transcriptional activator." Mol Cell
Biol 23(19): 6901-6908.
Kieff, E. (1996). Epstein-Barr virus and its replication. Fields Virology. D. M. K. B.N. Fields,
P.M. Howley. Philadelphia, Lippincott-Raven Publishers: 2343-2396.
Kieff, E. and A. B. Rickinson (2001). Epstein-Barr virus and its replication. Fields Virology. D.
M. Knipe and P. M. Howley. Philadelphia, Lippincott Williams and Wilkins. 2: 2511-
2573.
Kirchmaier, A. L. and B. Sugden (1997). "Dominant-negative inhibitors of EBNA1 of Epstein-
Barr virus." J. Virol. 71: 1766-1775.
Klein, E., L. L. Kis, et al. (2007). "Epstein-Barr virus infection in humans: from harmless to life
endangering virus-lymphocyte interactions." Oncogene 26(9): 1297-1305.
Koons, M. D., S. Van Scoy, et al. (2001). "The replicator of the Epstein-Barr virus latent cycle
origin of DNA replication, oriP, is composed of multiple functional elements." J. Virol.
75: 10582-10592.
Krishnakumar, R. and W. L. Kraus (2010). "The PARP side of the nucleus: molecular actions,
physiological outcomes, and clinical targets." Mol Cell 39(1): 8-24.

74
Krysan, P. J., S. B. Haase, et al. (1989). "Isolation of human sequences that replicate
autonomously in human cells." Mol. Cell. Biol. 9: 1026-1033.
Kutok, J. L. and F. Wang (2006). "Spectrum of Epstein-Barr virus-associated diseases." Annu
Rev Pathol 1: 375-404.
Labib, K., S. E. Kearsey, et al. (2001). "MCM2-7 proteins are essential components of
prereplicative complexes that accumulate cooperatively in the nucleus during G1-phase
and are required to establish, but not maintain, the S-phase checkpoint." Mol Biol Cell
12(11): 3658-3667.
Lear, A. L, Rowe M, et al. (1992). " The Epstein-Barr virus (EBV) nuclear antigen 1 BamHI F
promoter is activated on entry of EBV-transformed B cells into the lytic cycle." J. Virol.
66 : 7461-7468
Langle-Rouault, F., V. Patzel, et al. (1998). "Up to 100-fold increase of apparent gene expression
in the presence of Epstein-Barr virus oriP sequences and EBNA1: Implications of the
nuclear import of plasmids." J. Virol. 72: 6181-6185.
Lehman, C. W. and M. R. Botchan (1998). "Segregation of viral plasmids depends on tethering
to chromosomes and is regulated by phosphorylation." Proc. Natl. Acad. Sci. USA 95:
4338-4343.
Leman, A. R., C. Noguchi, et al. (2010). "Human Timeless and Tipin stabilize replication forks
and facilitate sister-chromatid cohesion." J Cell Sci 123(Pt 5): 660-670.
Li, M., C. L. Brooks, et al. (2004). "A dynamic role of HAUSP in the p53-Mdm2 pathway."
Mol. Cell 13: 879-886.
Li, M., D. Chen, et al. (2002). "Deubiquitination of p53 by HAUSP is an important pathway for
p53 stabilization." Nature 416(6881): 648-653.
Li, Q., M. K. Spriggs, et al. (1997). "Epstein-Barr virus uses HLA class II as a cofactor for
infection of B lymphocytes." J Virol 71(6): 4657-4662.
Lin, A., S. Wang, et al. (2008). "The EBNA1 protein of Epstein-Barr virus functionally interacts
with Brd4." J Virol 82(24): 12009-12019.
Little, R. D. and C. L. Schildkraut (1995). "Initiation of latent DNA replicatoon in the Epstein-
Barr virus genome can occur at sites other than the genetically defined origin." Mol. Cell.
Biol. 15: 2893-2903.
Lupton, S. and A. J. Levine (1985). "Mapping of genetic elements of Epstein-Barr virus that
facilitate extrachromosomal persistence of Epstein-Barr virus-derived plasmids in human
cells." Mol. Cell. Biol. 5: 2533-2542.

75
Mackey, D., T. Middleton, et al. (1995). "Multiple regions within EBNA1 can link DNAs." J.
Virol. 69: 6199-6208.
Mackey, D. and B. Sugden (1999). "The linking regions of EBNA1 are essential for its support
of replication and transcription." Mol. Cell. Biol. 19: 3349-3359.
Marechal, V., A. Dehee, et al. (1999). "Mapping EBNA1 domains involved in binding to
metaphse chromosomes." J. Virol. 73: 4385-4392.
Mayer, M. L., I. Pot, et al. (2004). "Identification of protein complexes required for efficient
sister chromatid cohesion." Mol Biol Cell 15(4): 1736-1745.
McPhillips, M. G., J. G. Oliveira, et al. (2006). "Brd4 is required for E2-mediated transcriptional
activation but not genome partitioning of all papillomaviruses." J Virol 80(19): 9530-
9543.
Middleton, T. and B. Sugden (1992). "EBNA1 can link the enhancer element to the initiator
element of the Epstein-Barr virus plasmid origin of DNA replication." J. Virol. 66: 489-
495.
Miller, G., A. El-Guindy, et al. (2007). "Lytic cycle switches of oncogenic human
gammaherpesviruses(1)." Adv Cancer Res 97: 81-109.
Mirkin, E. V. and S. M. Mirkin (2007). "Replication fork stalling at natural impediments."
Microbiol Mol Biol Rev 71(1): 13-35.
Najjar, I. and R. Fagard (2010). "STAT1 and pathogens, not a friendly relationship." Biochimie
92(5): 425-444.
Nayyar, V. K., K. Shire, et al. (2009). "Mitotic chromosome interactions of Epstein-Barr nuclear
antigen 1 (EBNA1) and human EBNA1-binding protein 2 (EBP2)." J Cell Sci 122(Pt 23):
4341-4350.
Nieduszynski, C. A., Donaldson, A. D. and Blow, J. J. (2001). "Chromosome replication: from
ORC to fork." Genome biology 2: 4031-4031.4032.
Niller, H. H., G. Glaser, et al. (1995). "Nucleoprotein complexes and DNA 5'-ends at oriP of
Epstein-Barr virus." J. Biol. Chem. 270: 12864-12868.
Norio, P. and C. L. Schildkraut (2001). "Visualization of DNA replication on individual Epstein-
Barr virus episomes." Science 294: 2361-2364.
Norio, P. and C. L. Schildkraut (2004). "Plasticity of DNA replication initiation in Epstein-Barr
virus episomes." PLoS Biol 2(6): e152.

76
Norseen, J., A. Thomae, et al. (2008). "RNA-dependent recruitment of the origin recognition
complex." Embo J 27(22): 3024-3035.
Petti, L., C. Sample, et al. (1990). "Subnuclear localization and phosphorylation or Epstein-Barr
virus latent infection nuclear proteins." Virology 176: 563-574.
Platt, T. H., I. Y. Tcherepanova, et al. (1993). "Effect of number and position of EBNA-1
binding sites in Epstein-Barr virus oriP on the sites of initiation, barrier formation, and
termination of replication." J Virol 67(3): 1739-1745.
Raab-Traub, N. (2002). "Epstein-Barr virus in the pathogenesis of NPC." Semin Cancer Biol
12(6): 431-441.
Raab-Traub, N. and K. Flynn (1986). "The structure of the termini of the Epstein-Barr virus as a
marker of clonal cellular proliferation." Cell 47(6): 883-889.
Rawlins, D. R., G. Milman, et al. (1985). "Sequence-specific DNA binding of the Epstein-Barr
virus nuclear antigen (EBNA1) to clustered sites in the plasmid maintenance region."
Cell 42: 859-868.
Rehtanz, M., H. M. Schmidt, et al. (2004). "Direct interaction between nucleosome assembly
protein 1 and the papillomavirus E2 proteins involved in activation of transcription." Mol
Cell Biol 24(5): 2153-2168.
Reichelt, M., L. Wang, et al. (2011). "Entrapment of viral capsids in nuclear PML cages is an
intrinsic antiviral host defense against varicella-zoster virus." PLoS Pathog 7(2):
e1001266.
Reisman, D. and B. Sugden (1986). "trans Activation of an Epstein-Barr viral transcripitonal
enhancer by the Epstein-Barr viral nuclear antigen 1." Mol. Cell. Biol. 6: 3838-3846.
Reisman, D., J. Yates, et al. (1985). "A putative origin of replication of plasmids derived from
Epstein-Barr virus is composed of two cis-acting components." Mol Cell Biol 5(8): 1822-
1832.
Reisman, D., J. Yates, et al. (1985). "A putative origin of replication of plasmids derived from
Epstein-Barr virus is composed of two cis-acting components." Mol. Cell. Biol 5: 1822-
1832.
Rickinson, A. B. and E. Kieff (1996). Epstein-Barr virus. Fields Virology. D. M. K. B.N. Fields,
P.M. Knipe, P.M. Howley. Philadelphia, Lippincott-Raven Publishers: 2397-2446.
Rickinson, A. B. and E. Kieff (2001). Epstein-Barr virus. Fields Virology. D. M. Knipe and P.
M. Howley. Philadelphia, Lippincott Williams and Wilkins: 2575-2627.

77
Rogers, R. P., M. Woisetschlaeger, et al. (1990). "Alternative splicing dictates translational start
in Epstein-Barr virus transcripts." EMBO J 9(7): 2273-2277.
Sample, J., E. B. D. Henson, et al. (1992). "The Epstein-Barr virus nuclear protein 1 promoter
active in type I latency is autoregulated." J. Virol. 66: 4654-4661.
Saridakis, V., Y. Sheng, et al. (2005). "Structure of the p53 binding domain of HAUSP/USP7
bound to Epstein-Barr nuclear antigen 1 implications for EBV-mediated
immortalization." Mol Cell 18(1): 25-36.
Sarkari, F., X. Wang, et al. (2011). "The herpesvirus associated ubiquitin specific protease,
USP7, is a negative regulator of PML proteins and PML nuclear bodies." PLoS One 6(1):
e16598.
Scaglioni, P. P., T. M. Yung, et al. (2006). "A CK2-dependent mechanism for degradation of the
PML tumor suppressor." Cell 126(2): 269-283.
Schaefer, B. C., J. L. Strominger, et al. (1995). "Redefining the Epstein-Barr virus-encoded
nuclear antigen EBNA-1 gene promoter and transcription initiation site in group I Burkitt
lymphoma cell lines." Proc Natl Acad Sci U S A 92(23): 10565-10569.
Schepers, A., M. Ritzi, et al. (2001). "Human origin recognition complex binds to the region of
the latent origin of DNA replication of Epstein-Barr virus." EMBO J. 20: 4588-4602.
Sears, J., J. Kolman, et al. (2003). "Metaphase chromosome tethering is necessary for the DNA
synthesis and maintenance of oriP plasmids but is insufficient for transcription activation
by Epstein-Barr nuclear antigen 1." J Virol 77(21): 11767-11780.
Sears, J., M. Ujihara, et al. (2004). "The amino terminus of Epstein-Barr Virus (EBV) nuclear
antigen 1 contains AT hooks that facilitate the replication and partitioning of latent EBV
genomes by tethering them to cellular chromosomes." J Virol 78(21): 11487-11505.
Seto, E., L. Yang, et al. (2005). "Epstein-Barr virus (EBV)-encoded BARF1 gene is expressed in
nasopharyngeal carcinoma and EBV-associated gastric carcinoma tissues in the absence
of lytic gene expression." J Med Virol 76(1): 82-88.
Shah, K. M. and L. S. Young (2009). "Epstein-Barr virus and carcinogenesis: beyond Burkitt's
lymphoma." Clin Microbiol Infect 15(11): 982-988.
Shah, W. A., R. F. Ambinder, et al. (1992). "Binding of EBNA-1 to DNA creates a protease-
resistant domain that encompasses the DNA recognition and dimerization functions." J.
Virology 66: 3355-3362.
Shanmugaratnam, K. (1978). "Histological typing of nasopharyngeal carcinoma." IARC Sci
Publ(20): 3-12.

78
Shannon-Lowe, C., E. Adland, et al. (2009). "Features distinguishing Epstein-Barr virus
infections of epithelial cells and B cells: viral genome expression, genome maintenance,
and genome amplification." J Virol 83(15): 7749-7760.
Shaw, J., L. Levinger, et al. (1979). "Nucleosomal structure of Epstein-Barr virus DNA in
transformed cell lines." J. Virol. 29: 657-665.
Shire, K., D. F. J. Ceccarelli, et al. (1999). "EBP2, a human protein that interacts with sequences
of the Epstein-Barr nuclear antigen 1 important for plasmid maintenance." J. Virol. 73:
2587-2595.
Shire, K., P. Kapoor, et al. (2006). "Regulation of the EBNA1 Epstein-Barr virus protein by
serine phosphorylation and arginine methylation." J Virol 80(11): 5261-5272.
Silla, T., I. Haal, et al. (2005). "Episomal maintenance of plasmids with hybrid origins in mouse
cells." J Virol 79(24): 15277-15288.
Simpson, K., A. McGuigan, et al. (1996). "Stable episomal maintenance of yeast artificial
chromosomes in human cells." Mol. Cell. Biol. 16: 5117-5126.
Sivachandran, N., J. Y. Cao, et al. (2010). "Epstein-Barr virus nuclear antigen 1 Hijacks the host
kinase CK2 to disrupt PML nuclear bodies." J Virol 84(21): 11113-11123.
Sivachandran, N., C. W. Dawson, et al. (2011). "Contributions of the Epstein-Barr Virus EBNA1
Protein to Gastric Carcinoma." J Virol.
Sivachandran, N., F. Sarkari, et al. (2008). "Epstein-Barr nuclear antigen 1 contributes to
nasopharyngeal carcinoma through disruption of PML nuclear bodies." PLoS Pathog
4(10): e1000170.
Sivachandran, N., F. Sarkari, et al. (2008). "Epstein-Barr Nuclear Antigen 1 Contributes to
Nasopharyngeal Carcinoma through the Disruption of PML Nuclear Bodies." PLoS
Pathogens 4(10): e1000170.
Sivachandran, N., N. N. Thawe, et al. (2011). "Epstein-Barr virus nuclear antigen 1 replication
and segregation functions in nasopharyngeal carcinoma cell lines." J Virol 85(19):
10425-10430.
Skalsky, R. L., J. Hu, et al. (2007). "Analysis of viral cis elements conferring Kaposi's sarcoma-
associated herpesvirus episome partitioning and maintenance." J Virol 81(18): 9825-
9837.
Smith-Roe, S. L., S. S. Patel, et al. (2011). "Timeless functions independently of the Tim-Tipin
complex to promote sister chromatid cohesion in normal human fibroblasts." Cell Cycle
10(10): 1618-1624.

79
Sternas, L., T. Middleton, et al. (1990). "The average number of molecules of Epstein-Barr
nuclear antigen 1 per cell does not correlate with the average number of Epstein-Barr
virus (EBV) DNA molecules per cell among different clones of EBV-immortalized
cells." J.Virol. 64: 2407-2410.
Su, W., T. Middleton, et al. (1991). "DNA looping between the origin of replication of Epstein-
Barr virus and its enhancer site: stabilization of an origin complex with Epstein-Barr
nuclear antigen 1." Proc. Natl. Acad. Sci. USA 88: 10870-10874.
Sugden, B., K. Marsh, et al. (1985). "A vector that replicates as a plasmid and can be efficiently
selected in B-lymphoblasts transformed by Epstein-Barr virus." Mol Cell Biol 5(2): 410-
413.
Sugden, B. and N. Warren (1989). "A promoter of Epstein-Barr virus that can function during
latent infection can be transactivated by EBNA-1, a viral protein required for viral DNA
replication during latent infection." J Virol 63(6): 2644-2649.
Summers, H., J. A. Barwell, et al. (1996). "Cooperative assembly of EBNA1 on the Epstein-Barr
virus latent origin of replication." J. Virol. 70: 1228-1231.
Summers, H., A. Fleming, et al. (1997). "Requirements for EBNA1-induced permanganate
sensitivity of the Epstein-Barr virus latent origin of DNA replication." J. Biol. Chem.
272: 26434-26440.
Sun, Y., G. Hegamyer, et al. (1992). "An infrequent point mutation of the p53 gene in human
nasopharyngeal carcinoma." Proc Natl Acad Sci U S A 89(14): 6516-6520.
Takeda, D. Y., Y. Shibata, et al. (2005). "Recruitment of ORC or CDC6 to DNA is sufficient to
create an artificial origin of replication in mammalian cells." Genes Dev 19(23): 2827-
2836.
Tang, Q., P. Bell, et al. (2000). "Replication but not transcription of simian virus 40 DNA is
dependent on nuclear domain 10." J Virol 74(20): 9694-9700.
Tellam, J., M. H. Fogg, et al. (2007). "Influence of translation efficiency of homologous viral
proteins on the endogenous presentation of CD8+ T cell epitopes." J Exp Med 204(3):
525-532.
Tempera, I., Z. Deng, et al. (2010). "Regulation of Epstein-Barr virus OriP replication by
poly(ADP-ribose) polymerase 1." J Virol 84(10): 4988-4997.
Thorley-Lawson, D. A. (2001). "Epstein-Barr virus: exploiting the immune system." Nat Rev
Immunol 1(1): 75-82.
Thorley-Lawson, D. A. and M. J. Allday (2008). "The curious case of the tumour virus: 50 years
of Burkitt's lymphoma." Nat Rev Microbiol 6(12): 913-924.

80
Thorley-Lawson, D. A., E. M. Miyashita, et al. (1996). "Epstein-Barr virus and the B cell: that's
all it takes." Trends in Microbiology 4: 204-208.
Uozaki, H. and M. Fukayama (2008). "Epstein-Barr virus and gastric carcinoma--viral
carcinogenesis through epigenetic mechanisms." Int J Clin Exp Pathol 1(3): 198-216.
Van Scoy, S., I. Watakabe, et al. (2000). "Human p32: A coactivator for Epstein-Barr virus
nuclear antigen-1-mediated transcriptional activation and possible role in viral latent
cycle DNA replication." Virol. 275: 145-157.
Wang, S. and L. Frappier (2009). "Nucleosome Assembly Proteins Bind to Epstein-Barr Nuclear
Antigen 1 (EBNA1) and Affect its Functions in DNA Replication and Transcriptional
Activation." J. Virol. 83: 11704-11714.
Wang, Y., J. E. Finan, et al. (1997). "P32/TAP, a cellular protein that interacts with EBNA-1 of
Epstein-Barr virus." Virology 236: 18-29.
White, R. E., R. Wade-Martin, et al. (2001). "Sequences adjacent to oriP improve the persistence
of Epstein-Barr virus-based episomes in B cells." J. Virol. 75: 11249-11252.
Wilson, J. B., J. L. Bell, et al. (1996). "Expression of Epstein-Barr virus nuclar antigen-1 induces
B cell neoplasia in transgenic mice." EMBO 15: 3117-3126.
Woisetschlaeger, M., C. N. Yandava, et al. (1990). "Promoter switching in Epstein-Barr virus
during the initial stages of infection of B lymphocytes." Proc Natl Acad Sci U S A 87(5):
1725-1729.
Wood, V. H., J. D. O'Neil, et al. (2007). "Epstein-Barr virus-encoded EBNA1 regulates cellular
gene transcription and modulates the STAT1 and TGFbeta signaling pathways."
Oncogene 26(28): 4135-4147.
Wu, H., D. F. J. Ceccarelli, et al. (2000). "The DNA segregation mechanism of the Epstein-Barr
virus EBNA1 protein." EMBO Rep. 1: 140-144.
Wu, H., P. Kapoor, et al. (2002). "Separation of the DNA replication, segregation, and
transcriptional activation functions of Epstein-Barr nuclear antigen 1." J Virol 76(5):
2480-2490.
Wysokenski, D. A. and J. L. Yates (1989). "Multiple EBNA1-binding sites are required to form
an EBNA1-dependent enhancer and to activate a minimal replicative origin within oriP
of Epstein-Barr virus." J. Virol. 63: 2657-2666.
Xiao, B., S. C. Verma, et al. (2010). "Bub1 and CENP-F can contribute to Kaposi's sarcoma-
associated herpesvirus genome persistence by targeting LANA to kinetochores." J Virol
84(19): 9718-9732.

81
Yates, J. L. and S. M. Camiolo (1988). "Dissection of DNA replication and enhancer activation
functions of Epstein-Barr virus nuclear antigen 1." Cancer Cells 6: 197-205.
Yates, J. L., S. M. Camiolo, et al. (2000). "The minimal replicator of Epstein-Barr virus oriP." J.
Virol 74: 4512-4522.
Yates, J. L. and N. Guan (1991). "Epstein-Barr virus-derived plasmids replicate only once per
cell cycle and are not amplified after entry into cells." J. Virol. 65: 483-488.
Yates, J. L., N. Warren, et al. (1984). "A cis-acting element from the Epstein-Barr viral genome
that permits stable replication of recombinant plasmids in latently infected cells." Proc.
Natl. Acad. Sci. USA 81: 3806-3810.
Yates, J. L., N. Warren, et al. (1985). "Stable replication of plasmids derived from Epstein-Barr
virus in various mammalian cells." Nature 313(6005): 812-815.
Yates, J. L., N. Warren, et al. (1985). "Stable replication of plasmids derived from Epstein-Barr
virus in various mammalian cells." Nature 313: 812-815.
Yin, Q. and E. K. Flemington (2006). "siRNAs against the Epstein Barr virus latency replication
factor, EBNA1, inhibit its function and growth of EBV-dependent tumor cells." Virology
346(2): 385-393.
Yin, Y., B. Manoury, et al. (2003). "Self-inhibition of synthesis and antigen presentation by
Epstein-Barr virus-encoded EBNA1." Science 301(5638): 1371-1374.
Yoshioka, M., M. M. Crum, et al. (2008). "Autorepression of Epstein-Barr virus nuclear antigen
1 expression by inhibition of pre-mRNA processing." J Virol 82(4): 1679-1687.
You, J., J. L. Croyle, et al. (2004). "Interaction of the bovine papillomavirus E2 protein with
Brd4 tethers the viral DNA to host mitotic chromosomes." Cell 117: 349-360.
You, J., V. Srinivasan, et al. (2006). "Kaposi's Sarcoma-Associated Herpesvirus Latency-
Associated Nuclear Antigen Interacts with Bromodomain Protein Brd4 on Host Mitotic
Chromosomes." J Virol 80(18): 8909-8919.
Young, L. S. and P. G. Murray (2003). "Epstein-Barr virus and oncogenesis: from latent genes to
tumours." Oncogene 22(33): 5108-5121.
Yu, L., P. M. Loewenstein, et al. (1995). "In vitro interaction of the human immunodeficiency
virus type 1 Tat transactivator and the general transcription factor TFIIB with the cellular
protein TAP." J. Virol. 69: 3017-3023.
Yu, M. C., J. H. Ho, et al. (1986). "Cantonese-style salted fish as a cause of nasopharyngeal
carcinoma: report of a case-control study in Hong Kong." Cancer Res 46(2): 956-961.

82
Yuan, J., E. Cahir-McFarland, et al. (2006). "Virus and cell RNAs expressed during Epstein-Barr
virus replication." J Virol 80(5): 2548-2565.
Zhang, D., L. Frappier, et al. (1998). "Human RPA (hSSB) interacts with EBNA1, the latent
origin binding protein of Epstein-Barr virus." Nucl. Acid Res. 26: 631-637.
Zhou, J., C. M. Chau, et al. (2005). "Cell cycle regulation of chromatin at an origin of DNA
replication." Embo J 24(7): 1406-1417.
Zhou, J., Z. Deng, et al. (2010). "Regulation of Epstein-Barr virus origin of plasmid replication
(OriP) by the S-phase checkpoint kinase Chk2." J Virol 84(10): 4979-4987.
Zhou, J., A. R. Snyder, et al. (2009). "Epstein-Barr virus episome stability is coupled to a delay
in replication timing." J Virol 83(5): 2154-2162.
zur Hausen, H., H. Schulte-Holthausen, et al. (1970). "EBV DNA in biopsies of Burkitt tumours
and anaplastic carcinomas of the nasopharynx." Nature 228(5276): 1056-1058.