conversion p-coumarate caffeate streptomyces nigrifaciensrepository.ias.ac.in/4594/1/321.pdf ·...
TRANSCRIPT

Biochem. J. (1972) 130, 425-433Printed in Great Britain
Conversion ofp-Coumarate into Caffeate by Streptomyces nigrifaciensPURIFICATION AND PROPERTIES OF THE HYDROXYLATING ENZYME
By A. M. D. NAMBUDIRI and J. V. BHATMicrobiology and Pharmacology Laboratory, Indian Institute of Science, Bangalore-12, India
and P. V. SUBBA RAODepartment of Biochemistry, Indian Institute of Science, Bangalore-12, India
(Received 8 May 1972)
1. An enzyme responsible for the conversion of p-coumarate into caffeate was purified97-fold from Streptomyces nigrifaciens. The enzyme had a molecular weight of 18000 asdetermined by Sephadex G-100 gel filtration and was homogeneous on polyacrylamide-gel electrophoresis. 2. The preparation exhibited both p-coumarate hydroxylase andcaffeate oxidase activities. 3. Stoicheiometry of the reaction indicated a mono-oxygenase-mediated catalysis consuming 1 mol of 02/mol of substrate hydroxylated. 4. NADH,NADPH, tetrahydropteroylglutamate or ascorbate act as electron donors for thereaction, ascorbate being inhibitory at higher concentrations. 5. The optimum enzymeactivity was at about pH7.7 and 40°C, with an activation energy of 39kJ/mol.6. Monophenols such asp-hydroxyphenylpropionate, p-hydroxyphenylacetate, L-tyrosineand DL-p-hydroxyphenyl-lactate were also hydroxylated by the preparation, in additionto p-coumarate. 7. The enzyme was a copper protein having 0.38% copper in a boundform. 8. Thiol-group inhibitors did not affect the reaction. 9. The relationship of theenzyme to other hydroxylases is discussed.
The metabolism of aromatic compounds is ofteninitiated by hydroxylation, since these compoundsare generally oxygen deficient and need to be oxygen-ated to become biologically active or more soluble inwater (Hayaishi, 1969). Many micro-organisms,particularly saprophytic ones, have been reported toutilize a great variety of naturally occurring aromaticcompounds for their growth, but only a few enzymesconcerned with the hydroxylation of the aromaticring have been characterized. Those characterizedinclude phenylalanine hydroxylase (Guroff & Ito,1963), melilotate hydroxylase (Levy, 1967), salicylatehydroxylase (Yamamoto et al., 1965), m-hydroxy-benzoate hydroxylase (Premakumar et al., 1969) andp-hydroxybenzoatd hydroxylase (Hosokawa &Stanier, 1966). No enzyme isolated from any micro-organism, however, is known to hydroxylate p-
coumarate. In the present paper we report the purific-ation and properties ofan enzyme from Streptomycesnigrifaciens that can hydroxylate p-coumarate andrelated phenolic acids substituted in thepara position,Enzyme preparations from higher plants (Sato,
1966; Vaughan & Butt, 1969a,b) and mushroom(Sato, 1969) catalyse hydroxylation of p-coumarateand contain o-diphenol oxidase activity in additionto their ability to hydroxylate monophenols.Such phenolases have also been found in bacteria
and microfungi, although their purification hasbeen achieved only from the fungal sources (Flinget al., 1963; Herzfeld & Esser, 1969); most of the
Vol. 130
studies carried out on them have been confined to thephysical properties and the o-diphenol oxidaseactivity of the enzyme. In addition, interpretation ofthe results on the hydroxylase activity of the enzymeis made difficult because of the dual functions of theseenzymes. Ascorbate, which is usually employed asa reductant in the hydroxylation, also reduces theo-quinone subsequently produced, thus facilitatingthe accumulation of the dihydroxyphenol in themedium. Vaughan & Butt (1969b) were able tomeasure the hydroxylase activity of their spinachpreparation by providing excess of reductant toprevent the further oxidation of caffeate. The Strepto-myces enzyme was also found to contain diphenoloxidase activity, which is characteristic of a typicalphenolase. It was, however, possible for us to studythe hydroxylase reaction selectively, since caffeateoxidase activity could be completely inhibited byp-coumarate, the substrate for the hydroxylation step.
Materials and Methods
Materials
Chemicals. Alcohol dehydrogenase (from yeast),p-hydroxymercuribenzoate, NADPH, NADH andH4PteGlu* were obtained from Sigma ChemicalCo., St. Louis, Mo., U.S.A. Mercaptoethanol, GSHand ascorbic acid were supplied by British Drug
* Abbreviation: H4PteGlu, tetrahydropteroylglutamate.
425

A. M. D. NAMBUDIRI, J. V. BHAT AND P. V. SUBBA RAO
Houses Ltd., Poole, Dorset, U.K., and dithiothreitolwas a gift from P-L Biochemicals, Milwaukee, Wis.,U.S.A. The aromatic chemicals were purchased fromKoch-Light Laboratories Ltd., Colnbrook, Bucks.,U.K., and recrystallized before use and SephadexG-100 was from Pharmacia, Uppsala, Sweden. Allother chemicals used were analytical grades availablecommercially.
Organism. A number of species of Streptomyceswere isolated from soil by enrichment culture byusing p-coumarate as sole source of carbon. Theorganism used throughout the present work wasidentified as Streptomyces nigrifaciens.
Methods
U.v. and i.r. spectroscopy. The u.v. spectra wererecorded on a Unicam SP. 500 spectrophotometer.A Perkin-Elmer (Infracord) model 137 spectrometerwas used to determine the i.r. spectra of the syntheticand isolated enzymic products (in a Nujol phase).Medium and conditions of growth. S. nigrifaciens
was grown on a nutrient medium containing maltextract (3 %), peptone (0.5 %) andyeast extract (0.1 %).The pH of the medium was adjusted to 7.0 with 1 M-NaOH. The flasks (500ml) containing 100ml ofmedium were autoclaved for 10min at 120°C andinoculated with a heavy suspension of spores. Forpreparation of cell extracts the cells were harvestedby filtering through Terylene cloth and were washedseveral times with water. Fresh cells were alwaysused for enzyme preparation.Enzyme assays. Enzyme assays were done at 30°C.
One unit of enzyme activity is defined as the amountthat catalyses the transformation of 1,utmol of thesubstrate or the formation of 1 ,tmol of product/minunder the conditions of the assay. Specific activity isexpressed as milliunits of the enzyme/mg of protein.Measurement of hydroxylase activity. The
hydroxylase activity was measured by incubating areaction mixture (1 ml) containing tris-HCI buffer,pH7.7 (30,urmol), p-coumaric acid (2,umol), NADH(2,umol) and 0.2ml of enzyme (15-20 milliunits) at30°C for 15min. The reaction was stopped by theaddition of 1 ml of0.5M-HCI, and the caffeate formedwas determined by the method described by Amow(1937) for the measurement of 3,4-dihydroxyphenyl-alanine (dopa).
Assay of o-diphenol oxidase activity. A reactionmixture (1 ml) consisting of tris-HCl buffer, pH7.7(30,umol), caffeic acid (0.25,umol) and the enzymepreparation (15-20 milliunits) was incubated for15min at 30°C. After the reaction had been stoppedwith 1 ml of 0.5M-HCI, the disappearance of caffeatewas determined colorimetrically (Arnow, 1937).02 consumption. The conventional Warburg tech-
nique (Umbreit et al., 1964) was used to measure 02consumption during the hydroxylation reactions. 02
consumption was also measured with the Gilsonmodel K Oxygraph to establish stoicheiometry.
Determination of protein. The method of Lowryet al. (1 95 1) was used for the determination ofprotein.Dry bovine serum albumin was the standard.
Determination of copper. The copper content ofthe purified enzyme was determined by the methodof Stark & Dawson (1958).
Determination of molecular weight. The molecularweight of the enzyme was determined by the methodof Andrews (1964) by filtering the protein througha calibrated column (1.5cmx40cm) of SephadexG-100 with a flow rate of 18ml/h. The column wasequilibrated with 25mM-sodium phosphate buffer(pH7); the purified enzyme (3mg of protein) wasloaded on to the column and 2ml fractions werecollected. The standard proteins used were cyto-chrome c (mol.wt. 12500), pepsin (mol.wt. 35500),hexokinase (mol.wt. 47000) and bovine serumalbumin fraction V (mol.wt. 60000).
Polyacrylamide-gel electrophoresis. Polyacryl-amide-gel electrophoresis was done essentially bythe method of Davis (1964) with glycine-tris buffer,pH 8.6, and L-alanine buffer, pH4.3. Electrophoresiswas for 40min at 5°C with a current of 6mA/tube.The gel, after removal from the tube, was stainedfor 45min with Amido Black and de-stained in7% (w/v) acetic acid.
Purification of the enzyme. All operations weredone at 0-40C.
Step I: crude extract. Washed cells of S. nigrifaciens(20g) were suspended in 60ml of 50mM-sodiumphosphate buffer, pH7.0. Batches (30ml) were treatedfor 10min in a 10kHz Raytheon sonic oscillatorcooled with circulating iced water and then they werecentrifuged for 20min at 8000g. The clear super-natant (80ml), having a specific activity of 3-6 milli-units/mg of protein, was used as crude extract.
Step II: protamine sulphate treatment. The super-natant solution from step I (76.5ml) was treateddropwise with 0.1 vol. of a 2% (w/v) solution of pro-tamine sulphate dissolved in 25mM-sodium phos-phate buffer, pH7.0 (8.5ml). After being stirredgently for 10min, the suspension was centrifuged at12000g for 10min and the precipitate was discarded.This procedure removed a large amount of inactiveprotein and material absorbing at 260nm.
Step III: first ammonium sulphate fractionation.To the above supernatant (75ml) solid (NH4)2SO4(18.4g) was added with stirring to bring the solutionto 40% saturation. The pH was maintained at 7 witha dilute solution ofNH3. After the mixture had stoodfor 20min in an ice bath the precipitate was removedby centrifugation and discarded. The concentrationof the supematant fluid was raised to 70% saturationby a further addition of solid (NH4)2SO4 (15.4g).After the mixture had stood for lh the resultingprecipitate was collected by centrifugation, dissolved
1972
426

CONVERSION OF p-COUMARATE INTO CAFFEATE BY STREPTOMYCES
in the minimumn volume of 25 mM-sodium phosphatebuffer, pH 7.0 (8 ml), and desalted by passage througha column (2cmx26.4cm) of Sephadex G-25 (beadform) that had been previously equilibrated with25mM-sodium phosphate buffer, pH7.0. The activeprotein fraction was collected in 17ml ofeffluent afterthe void volume (36ml).
Step IV: DEAE-cellulose treatment. The enzyme
30 40 50 iElution volume (ml)
Fig. 1. Sephadex G-100 gel elution pattern of the p-coumarate hydroxylating enzyme from S. nigrifaciens
Details are given in the text. ----, Protein;enzyme activity
responsible for the hydroxylation of p-coumaratewas not adsorbed on DEAE-cellulose under theexperimental conditions used. Hence the followingrapid procedure involving DEAE-cellulose was usedfor the removal of some inert protein.
DEAE-cellulose was washed by the method ofPeterson & Sober (1962) and suspended in 25mM-sodium phosphate buffer, pH7.0. A portion (20ml)of the suspension was filtered on a Buchner funnel.The enzyme preparation obtained after passagethrough the Sephadex G-25 column (17ml) wasadded to washed DEAE-cellulose cake (1 g ofDEAE-cellulose/15ml of column effluent) and the mixturewas stirred mechanically for 30min. The suspensionwas filtered on a Buchner funnel and the filtrate wascollected.
Step V: second ammonium sulphate precipitation.To theDEAE-cellulose supernatant (17ml) was addedsolid (NH4)2SO4 (9.9g) with mechanical stirring togive 80% saturation. The mixture was equilibratedfor 1 h. The resulting precipitate was collected bycentrifuging at 10000g for 15min and dissolved in25mM-sodium phosphate buffer, pH7.0 (8ml).
Step VI: Sephadex G-100 filtration. A 5ml portionof the concentrated enzyme from step V was chrom-atographed on a column (2cnx 20cm) of SephadexG-100. The column was eluted with 25mm-sodiumphosphate buffer, pH7.0, at an average flow rate of30ml/h. Fractions (4ml) were collected and assayedfor p-coumarate hydroxylase activity. The elutionpattern of the enzyme from Sephadex G-100 is shownin Fig. 1. The active fractions were colourless.The enzyme preparations at various stages of
Table 1. Purification of the p-coumarate hydroxylating system from S. nigrifaciens
p-Coumarate hydroxylase activity was measured by incubating, for 15min at 300C, a reaction mixture (1 ml)containing tris-HCl buffer, pH7.7 (30,umol), p-coumaric acid (2,umol), NADH (2,umol), ethanol (2.5,umol),alcohol dehydrogenase (100 milliunits) and a suitable amount of enzyme. Other conditions for the assay of p-coumarate hydroxylase activity as well as the conditions for determination of caffeate oxidase activity were asdescribed in the Materials and Methods section.
Purification stepI: crude extract
II: protamine sul-
Tvc
PotaAlun(ml)7675
phateIII: first (NH4)2S04 17
(40-70% satn.)IV: DEAE-cellulose 17V: second 8
(NH4)2S04(0-80% satn.)
VI, Sephadex G-100 4
Vol. 130
d Totalne protein
(mg)425.6217.5
72.4
51.036.8
0.84
Total activity(milliunits)
p-Coumaratehydroxylase
20262700
2380
30262453
360
Caffeateoxidase13681795
1593
18511634
240
Specific activity(milliunits/mg of protein)
p-Coumaratehydroxylase
4.712.4
32.9
59.566.6
428.5
Caffeateoxidase
3.28.2
22.0
36.344.5
285.6
a00.OI4)
Hydroxylase/oxidase
activity ratio1.491.39
1.45
1.641.50
1.50
427
A. M. D. NAMBUDIRI, J. V. BHAT AND P. V. SUBBA RAO
purification were found to have both p-coumaratehydroxylase (monophenol oxidase) and caffeateoxidase (diphenol oxidase) activities. Table 1 sum-marizes the yields and specific activities of hydroxyl-ase and diphenol oxidase activities for each step ofthe purification procedure. An overall yield of 18%through step I to step VI was achieved with 96-and 89-fold increases in the specific activities of p-coumarate hydroxylase and caffeate oxidase activitiesrespectively. The ratio of hydroxylase activity tocaffeate oxidase activity was more-or-less constantthroughout the purification.
Isolation of theproduct ofp-coumaric acid hydroxy-lation. A large-scale incubation mixture (200ml)consisting of sodium phosphate buffer, pH 7.7(7mmol), p-coumaric acid (0.8mmol), ascorbic acid(0.4mmol) and purified enzyme from step VI (40ml)was incubated at 30°C with occasional stirring.At 15min intervals 0.1 M-ascorbic acid (1 ml each time)was added. After 2h the pH of the reaction mixturewas adjusted to 2 with 1 M-HCl and it was extractedtwice with equal volumes of peroxide-free ether.The ethereal layer was evaporated to dryness afterbeing shaken with anhydrous Na2SO4. The procedureadopted for the isolation of caffeic acid from theresidue was essentially similar to that described bySato (1969). The crude product was dissolved in0.2M-sodium acetate (25ml). After the addition of0.2M-EDTA (0.5ml) and alumina (5g) to the solutionthe pH was adjusted to 8.5 by dropwise addition of2M-NH3, then the suspension was stirred for 5min.The slurry was transferred to a column and washedwith distilled water (100ml). This process removedp-coumaric acid, whereas caffeic acid was retainedon the column. Subsequent elution of the columnwith 0.6M-acetic acid (30ml) followed by extractionof the eluate with peroxide-free ether resulted in thetransfer of caffeic acid into the ether phase. Theorganic layer was shaken with anhydrous Na2SO4and evaporated to dryness. The residue was dis-solved in the minimum volume of hot water andrecrystallized twice to yield 20mg of the product.Both authentic caffeic acid and the isolated productmelted at 195°C and there was no change in meltingpoint on admixture. The i.r. and u.v. spectra of theproduct and the authentic compound were alsoidentical.
ResultsProperties of the purified p-coumarate-hydroxylatingenzyme
Homogeneity. The enzyme was found to yield onlyone protein band on disc electrophoresis at bothalkaline pH (8.6) and acid pH (4.3).
Inhibition ofcaffeate oxidase activity of the purifiedenzyme by p-coumarate. Since electrophoretically
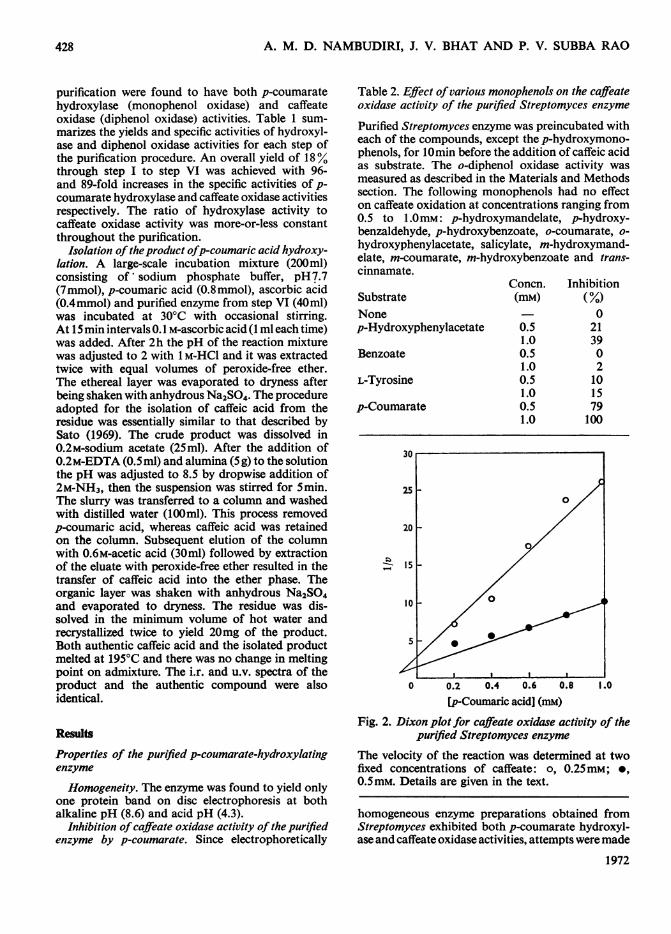
Table 2. Effect ofvarious monophenols on the caffeateoxidase activity of the purified Streptomyces enzyme
Purified Streptomyces enzyme was preincubated witheach of the compounds, except the p-hydroxymono-phenols, for 10min before the addition of caffeic acidas substrate. The o-diphenol oxidase activity wasmeasured as described in the Materials and Methodssection. The following monophenols had no effecton caffeate oxidation at concentrations ranging from0.5 to 1.OmM: p-hydroxymandelate, p-hydroxy-benzaldehyde, p-hydroxybenzoate, o-coumarate, o-hydroxyphenylacetate, salicylate, m-hydroxymand-elate, m-coumarate, m-hydroxybenzoate and trans-cinnamate.
Concn. InhibitionSubstrate (mM) (%)None 0p-Hydroxyphenylacetate 0.5 21
1.0 39Benzoate 0.5 0
1.0 2L-Tyrosine 0.5 10
1.0 15p-Coumarate 0.5 79
1.0 100
0.2 0.4 0.6 0.8[p-Coumaric acid] (mM)
Fig. 2. Dixon plot for caffeate oxidase activity of thepurified Streptomyces enzyme
The velocity of the reaction was determined at twofixed concentrations of caffeate: o, 0.25mM; 0,0.5mM. Details are given in the text.
homogeneous enzyme preparations obtained fromStreptomyces exhibited both p-coumarate hydroxyl-ase and caffeate oxidase activities, attempts were made
1972
428

CONVERSION OF p-COUMARATE INTO CAFFEATE BY STREPTOMYCES
to inhibit the second reaction selectively. The effectof various structural analogues on caffeate dis-appearance in the presence of purified enzyme issummarized in Table 2. p-Coumarate was the mostpotent inhibitor, and it completely suppressed theoxidase activity at a concentration of 1.5mM. Theinhibition, as shown in Fig. 2 was competitive.Because of this interesting property of the enzyme itwas consequently possible to study the p-coumaratehydroxylase reaction without interference from the
Table 3. Specificity of the purified p-coumaratehydroxylating enzyme from S. nigrifaciens for various
monophenols
02 uptake was measured by using the conventionalWarburg technique. Each flask contained, in a totalvolume of 3.2ml, tris-HCl buffer (90,umol), 0.6mlof enzyme (from step VI), 6,umol of NADH and6,umol of substrate in the side arm. The central wellcontained 0.2ml of 20% (w/v) KOH. The substratewas tipped at zero time and incubation was at 30°Cin an atmosphere of air.
Substratep-Coumaratep-Hydroxyphenylpropionatep-HydroxyphenylacetateL-TyrosineDL-p-Hydroxyphenyl-lactate
Relative activity100170685940
o-diphenol oxidase oxidizing the caffeate formed.Complete inhibition of the o-diphenol oxidase re-action was ensured by adding 2mM-p-coumarate tothe standard reaction mixture.
Stability. The purified enzyme from step VI in25mM-sodium phosphate buffer, pH7 (0.2-0.25mgof protein/ml) lost 89% of its activity in 72h onstorage at 4°C. Freezing of the enzyme at -20°Cresulted in total loss of activity in 12h.
Molecular weight. The molecular weight of theStreptomyces enzyme possessing p-coumarate hy-droxylase activity, as determined by gel filtration isapprox. 18000.
U.v. spectrum. The purified enzyme gave a u.v.spectrum characteristic of a simple protein withoutshowing any apparent bound flavin prosthetic group.
Substrate specificity. p-Coumarate was not the onlymonophenolic acid hydroxylated by the Strepto-myces enzyme: p-hydroxyphenyl-lactate, p-hydroxy-phenylpropionate, DL-p-hydroxyphenylacetate and L-tyrosine were also substrates (Table 3). Several othermonophenols, including DL-p-hydroxymandelate, p-hydroxybenzoate, p-hydroxybenzaldehyde and p-hydroxybenzyl alcohol, were not hydroxylated.Monophenols having the hydroxyl group in theortho or the meta position were not substrates forthe purified hydroxylating enzyme.
Substrate specificity for reductant. There was nodetectable hydroxylase activity when the purifiedenzyme was incubated with the substrate in theabsence of any cofactors under standard assay con-ditions. Addition of various reductants, however,
Table 4. Effect of various reductants on the hydroxylation of p-coumarate and p-hydroxyphenylacetate by thepurified hydroxylating enzyme from S. nigrifaciens
Assay conditions were as given in the Materials and Methods section except that different reductants were usedat the stated concentrations. Homoprotocatechuate was the product formed from p-hydroxyphenylacetate. Itsformation was studied in the same way as was that of caffeate.
Concn. Caffeate formed(mM) (nmol)0.5 1851.0 2201.5 2202.0 2200.5 2001.0 2201.5 2202.0 2150.5 1351.0 1751.5 1602.0 1200.5 1801.0 1801.5 1802.0 215
Homoprotocatechuate formed(nmol)
911241481481717262618019020020075867575
ReductantNADH
NADPH
Ascorbate
H4PteGlu
Vol. 130
429
A. M. D. NAMBUDIRI, J. V. BHAT AND P. V. SUBBA RAO
0~~~~~V 0.15
0.10
U 0.05
0 0.4 0.8 1.2 1.6 2.0
Concentration of reductant (mM)
Fig. 3. Effect of concentration of reductants on thehydroxylation of p-coumarate by the Streptomyces
enzyme
Conditions of assay were as given in the Materialsand Methods section except that different concentra-tions of reductants were used. *, NADH; o,ascorbate.
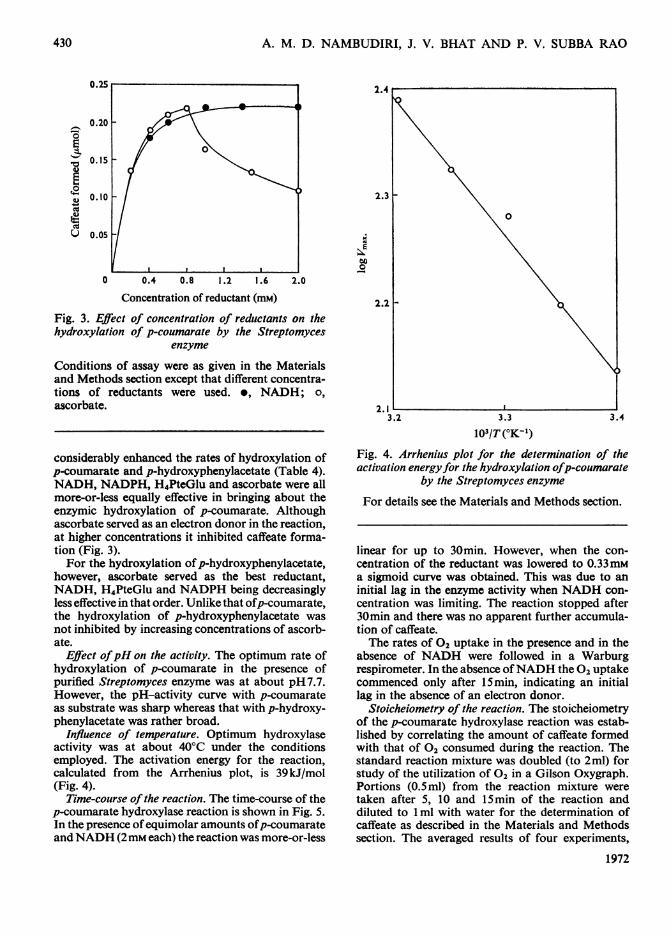
considerably enhanced the rates of hydroxylation ofp-coumarate and p-hydroxyphenylacetate (Table 4).NADH, NADPH, H4PteGlu and ascorbate were allmore-or-less equally effective in bringing about theenzymic hydroxylation of p-coumarate. Althoughascorbate served as an electron donor in the reaction,at higher concentrations it inhibited caffeate forma-tion (Fig. 3).For the hydroxylation ofp-hydroxyphenylacetate,
however, ascorbate served as the best reductant,NADH, H4PteGlu and NADPH being decreasinglyless effective in that order. Unlike that ofp-coumarate,the hydroxylation of p-hydroxyphenylacetate wasnot inhibited by increasing concentrations of ascorb-ate.
Effect ofpH on the activity. The optimum rate ofhydroxylation of p-coumarate in the presence ofpurified Streptomyces enzyme was at about pH7.7.However, the pH-activity curve with p-coumarateas substrate was sharp whereas that with p-hydroxy-phenylacetate was rather broad.
Influence of temperature. Optimum hydroxylaseactivity was at about 40°C under the conditionsemployed. The activation energy for the reaction,calculated from the Arrhenius plot, is 39kJ/mol(Fig. 4).
Time-course of the reaction. The time-course of thep-coumarate hydroxylase reaction is shown in Fig. 5.In the presence ofequimolar amounts ofp-coumarateand NADH (2mM each) the reaction was more-or-less
Fig. 4. Arrhenius plot for the determination of theactivation energyfor the hydroxylation ofp-coumarate
by the Streptomyces enzyme
For details see the Materials and Methods section.
linear for up to 30min. However, when the con-centration of the reductant was lowered to 0.33mMa sigmoid curve was obtained. This was due to aninitial lag in the enzyme activity when NADH con-centration was limiting. The reaction stopped after30min and there was no apparent further accumula-tion of caffeate.The rates of 02 uptake in the presence and in the
absence of NADH were followed in a Warburgrespirometer. In the absence ofNADH the 02 uptakecommenced only after 15min, indicating an initiallag in the absence of an electron donor.
Stoicheiometry of the reaction. The stoicheiometryof the p-coumarate hydroxylase reaction was estab-lished by correlating the amount of caffeate formedwith that of 02 consumed during the reaction. Thestandard reaction mixture was doubled (to 2ml) forstudy of the utilization of 02 in a Gilson Oxygraph.Portions (0.5ml) from the reaction mixture weretaken after 5, 10 and 15min of the reaction anddiluted to 1 ml with water for the determination ofcaffeate as described in the Materials and Methodssection. The averaged results of four experiments,
1972
4
-
3.3
103/T(0K-1)3.4
430

CONVERSION OF p-COUMARATE INTO CAFFEATE BY STREPTOMYCES
'0 0.25
0.20
0.15I
'U 0.10 _
0.05 }
0 15 30 45 60 75 90 105 120Time (min)
Fig. 5. Time-course of caffeate formation in thepresence ofpurifiedp-coumarate-hydroxylating enzyme
from S. nigrifaciensConditions of assay were as given in the Materialsand Methods section except that two concentrationsof NADH were used: o, 2mm; *, 0.33mM.
Table 6. Effect ofvarious inhibitors on the hydroxyla-tion of p-coumarate by the purified Streptomyces
enzyme
For details see the text.
Concn. InhibitionInhibitor (mM) (o%)None 0oca'-Bipyridyl 0.1 0
1.0 21EDTA 0.1 5
1.0 24o-Phenanthroline 0.1 0
1.0 44Salicylaldoxime 0.1 0
1.0 41Diethyldithiocarbamate 0.1 70
1.0 1008-Hydroxyquinoline 0.1 11
1.0 77N-Ethylmaleimide 0.1 0
1.0 0p-Hydroxymercuribenzo- 0.1 0
ate 1.0 0
Table 5. Stoicheiometry of the p-coumarate hydroxyl-ase reaction
02 consumption was measured in a Gilson Oxygraphmodel B. Samples were taken at different times andassayed for the formation ofcaffeate.Other conditionsare given in the Materials and Methods section.
Time Caffeate formed(mnin) (jumol)
5
1015
0.100.220.44
02 consumed
(,umol)0.1240.2250.450
presented in Table 5, show that 1 mol of 02 was con-sumed per mol ofcaffeate formed; this is in agreementwith the stoicheiometry normally encountered inhydroxylation reactions catalysed by mono-oxy-genases.
Reaction kinetics. A double-reciprocal plot ofinitialvelocity against p-coumarate concentration waslinear, indicating that Michaelis-Menten saturationkinetics are characteristic with the substrate. Thecalculated apparent Km value was 0.37mM for p-
coumarate.Copper content. The Streptomyces enzyme possess-
ing p-coumarate hydroxylase activity is a copperprotein. Although colourless, the purified enzymepreparation contained 0.38% of copper, expressedas a percentage of the concentration of protein.
Vol. 130
Effect ofmetal ions. A study of the effect of variousmetal ions showed that none of the bivalent cationstested, including Fe2+, Cu2+, Mn2+ and Hg2+ hadeither stimulatory or inhibitory effects on theformation of caffeic acid.
Inhibition studies. The effect of various inhibitorson the hydroxylation ofp-coumarate is presented inTable 6. Even though the enzyme activity was notenhanced by any metal ions, the potent inhibition bymetal-chelating agents such as o-phenanthroline,salicylaldoxime, diethyldithiocarbamate and 8-hy-droxyquinoline suggests that a tightly bound metalion, probably copper, is required for enzyme activity.The inhibition, however, was found to be irreversible,as neither Cu2+ nor any other metal ion could restorethe activity.
Thiol-group inhibitors such as N-ethylmaleimideand p-hydroxymercuribenzoate had no effect on thereaction even at concentrations as high as 1 mM.
Effect of thiol-reducing agents. Thiol-reducingagents such as GSH, 2-mercaptoethanol and di-thiothreitol at concentrations of 0.01-0.1 mm totallyinhibited caffeate formation. Preincubation withCu2+ or NADH did not prevent the inhibition, nordid Cu2+ reverse the inhibition.
Discussion
Hydroxylation of p-coumarate is a rare pheno-menon in micro-organisms. One such reaction, ob-served by Power et al. (1965) with Leutimus lepidus,
431

A. M. D. NAMBUDIRI, J. V. BHAT AND P. V. SUBBA RAO
had more bearing on the synthesis of lignin pre-cursors. More recently the hydroxylation of p-coumarate to caffeate and subsequent ring cleavageby Pseudomonas was demonstrated by Seidman etal. (1969). Experiments with whole cells of S. nigri-faciens revealed that this organism convertedp-coumarate into protocatechuate with the inter-mediate formation of caffeate, followed by degrada-tion of the aliphatic side chain (Nambudiri et al.,1969). A purified enzyme from S. nigrifaciens readilyhydroxylated p-coumarate to caffeate. o-Hydroxyla-tion of p-coumarate has also been shown to occurin higher plants (Sato, 1966; Vaughan & Butt,1969a,b; Stafford & Baldy, 1970). A phenolasepreparation from mushroom also catalysed thehydroxylation ofp-coumarate to caffeate (Sato, 1969).A spinach enzyme isolated by Vaughan & Butt
(1967) that catalyses the hydroxylation ofp-coumar-ate was found to contain both monophenol oxidaseand o-diphenol oxidase activities, typical of a phenol-ase type of enzyme (Vaughan & Butt, 1969a,b). Thepurified Streptomyces enzyme also contained both p-coumarate hydroxylase and caffeate oxidase activities.The two activities appear to be the function of thesame enzyme, as the ratio of the hydroxylase activityto the caffeate oxidase activity remained virtually thesame throughout the purification procedure. Themolecular weight of the Streptomyces enzyme, in thepresent studies, was estimated to be about 18000 bySephadex gel filtration. This is the smallest tyrosinasemolecule yet found in Nature and is a little over halfthe molecular weight of the Neurospora enzyme re-ported by Fling et al. (1963) as 33000±2000.The Streptomyces preparation hydroxylated, in
addition to p-coumarate, p-hydroxyphenylpropion-ate, p-hydroxyphenylacetate, L-tyrosine and p-hydroxyphenyl-lactate in decreasing order of activity.In some respects this resembled in substrate specificitya monophenol oxidase isolated from sorghum(Stafford & Baldy, 1970). Although not a classicalpolyphenol oxidase, the sorghum enzyme catalysedthe hydroxylation ofp-coumarate. Compounds suchas p-hydroxybenzoate, p-hydroxymandelate, p-hy-droxybenzyl alcohol and p-hydroxybenzaldehydewere not substrates. However, caffeate and tyrosine,which are substrates for the Streptomyces enzyme,were inhibitory with the sorghum preparation.
Ascorbate serves as a reductant for mono-oxygen-ase reactions catalysed by dopamine ,B-hydroxylase(Kaufman, 1966) and phenolase (Mason et al., 1955).Reduced nicotinamide nucleotides, however, areobligatory participants in many other mono-oxygenase-catalysed hydroxylations (Hayaishi, 1969).2-Amino-4-hydroxy-6,7-dimethyl-5,6,7,8-tetrahydro-pteridine was the most effective hydrogen donorfor the p-coumarate hydroxylating enzyme fromspinach, although NADH, NADPH and ascorbatealso were effective (Vaughan & Butt, 1969a). The
p-coumarate-hydroxylating enzyme isolated fromS. nigrifaciens also showed a requirement for a re-duced cofactor as an electron donor. In the absenceof any reductant the hydroxylation proceeded onlyafter a considerable lag period. As an electrondonor, NADH, NADPH, H4PteGlu and ascorbatewere more-or-less equally effective, but at higherconcentrations ascorbate was inhibitory. Such aninhibition was not observed with spinach (Vaughan& Butt, 1969a) and mushroom (Sato, 1969) phenolasepreparations, and, in fact ascorbate was a suitablereductant for the hydroxylation ofp-coumarate. Theeffectiveness of the electron donor for hydroxylationseems to depend on the nature of the substrate aswell. In the present investigation, with p-hydroxy-phenylacetate as substrate, ascorbate was the mosteffective hydrogen donor without exerting, at the sametime, any inhibitory effect on the reaction. Thereason for the differential effect of higher concentra-tions of ascorbate remains to be explained.The Streptomyces enzyme under study contained
0.38% copper, which is similar to the copper contentsreported for other phenolases (Bauchillaux et al.,1963; Fling et al., 1963; Kertesz & Zito, 1962;Smith & Krueger, 1962). Barron & Singer (1945)showed that phenolase is not a thiol enzyme. Puremushroom phenolase remains fully active even inthe presence of a great excess of p-hydroxymercuri-benzoate (Kertesz & Zito, 1958). Similarly, inhibitionstudies on Neurospora crassa enzyme by Fling et al.(1963) also revealed that p-hydroxymercuribenzoatedid not significantly inhibit the enzyme activity evenat high concentrations. The fact that neither p-hydroxymercuribenzoate nor heavy-metal ions suchas Hg2+ inhibited the Streptomyces enzyme suggeststhat free thiol groups are not involved in the reaction.In agreement with this finding thiol-reducing agentssuch as GSH, P-mercaptoethanol and dithiothreitolcompletely inhibited the enzyme activity even at aconcentration of 0.1 mm. Attempts to prevent theinhibition by preincubation with Cu2+ were noteffective. Inhibition of tyrosinase activity by GSHand cysteine was observed also by Miamoto et al.(1967) with a purified preparation from potato.However, unlike the Streptomyces enzyme, inhibitionof potato phenolase was reversed by the addition ofCU2+. In contrast with the Streptomyces enzyme, themonophenol oxidase from sorghum (Stafford &Baldy, 1970) showed an obligatory requirement for athiol-reducing agent such as ,B-mercaptoethanol,GSH or dithiothreitol.The caffeate oxidase activity of the Streptomyces
enzyme was inhibited by some monophenols such asp-coumarate, p-hydroxyphenylacetate and tyrosine.The o- or m-hydroxy compounds had no effect.Inhibition of catecholase activity of the mammaliantyrosinase by monophenols was reported byMiamoto & Fitzpatrick (1957), and also by
1972
432

CONVERSION OF p-COUMARATE INTO CAFFEATE BY STREPTOMYCES 433
Pomerantz (1963), Osaki (1963) and Kean (1964)in other phenolase systems. p-Coumarate is a potentinhibitor of apple phenolase (Walker, 1969). Inhibi-tion of catecholase activity by benzoate and cinnam-ate has been reported (Kuttner & Wagreich, 1953).Similarly, inhibition of cresolase activity of potatotyrosinase by cinnamate, benzoate and ferulate isknown (Zucker, 1966).The Streptomyces enzyme seems to differ from
previously described systems in that compoundssuch as trans-cinnamate and benzoate did not inhibitthe monophenol oxidase or the diphenol oxidaseactivity. Also the m- and p-hydroxyphenols did nothave any effect on the oxidation of caffeate.Pomerantz (1964) reported that the hydroxylationof tyrosine catalysed by mammalian tyrosinase ex-hibited an apparent substrate (tyrosine) inhibition atconcentrations above 0.8mM. The rate of hydroxyla-tion of p-coumarate by the Streptomyces enzyme,however, was not affected by high concentrations ofp-coumarate.
Walker (1969) observed that the type of inhibitionof o-diphenol oxidase activity of the apple tyrosinaseby p-coumarate depended on the substrate. Catecholoxidation was competitively inhibited, whereas theinhibition of cholorogenic acid oxidation was non-competitive. Similar differences were also observedby Macrae & Duggleby (1968) in inhibition studieswith apple phenolase. With the Streptomyces enzymethe inhibition was purely competitive when caffeatewas the substrate. No attempts were made to studythe pattern of inhibition with other substrates.
References
Andrews, P. (1964) Biochem. J. 91, 222Arnow, L. E. (1937) J. Biol. Chem. 118, 531Barron, E. S. G. & Singer, T. P. (1945) J. Biol. Chem.
157, 221Bauchillaux, S., McMahil, P. & Mason, H. S. (1963)
J. Biol. Chem. 238, 1699Davis, B. J. (1964) Ann. N.Y. Acad. Sci. 121, 404Fling, M., Horowitz, N. H. & Heinemann, S. F. (1963)
J. Biol. Chem. 238, 2045Guroff, G. & Ito, T. (1963) Biochim. Biophys. Acta 77,
159Hayaishi, 0. (1969) Annu. Rev. Biochem. 38, 688Herzfeld, F. & Esser, K. (1969) Arch. Mikrobiol. 65, 146Hosokawa, K. & Stanier, R. Y. (1966) J. Biol. Chem. 241,
2453
Kaufman, S. (1966) in Biological and Chemical AspectsofOxygenases (Bloch, K. & Hayaishi, O., eds.), p. 261,Maruzen Co., Tokyo
Kean, E. A. (1964) Biochim. Biophys. Acta 92, 602Kertesz, D. & Zito, R. (1958) Proc. Int. Congr. Biochem.
4th. 17, 65Kertesz, D. & Zito, R. (1962) in Oxygenases (Hayaishi,
O., ed.), p. 307, Academic Press, New YorkKuttner, R. & Wagreich, H. (1953) Arch. Biochem.
Biophys. 43, 80Levy, C. L. (1967) J. Biol. Chem. 242, 747Lowry, 0. H., Rosebrough, N. J., Farr, A. L. &
Randall, R. J. (1951) J. Biol. Chem. 193, 265Macrae, A. R. & Duggleby, R. C. (1968) Phytochemistry
7, 855Mason, H. S., Fowlks, W. L. & Peterson, E. W. (1955)
J. Amer. Chem. Soc. 77, 2914Miamoto, M. & Fitzpatrick, T. B. (1957) Nature (London)
179, 199Miamoto, S., Miura, M., Mishima, K. & Immamura, T.
(1967) Kyosilsu Yakke Diagaku Nempo 41Nambudiri, A. M. D., Subba Rao, P. V. & Bhat, J. V.
(1969) Curr. Sci. 38, 402Osaki, S. (1963) Arch. Biochem. Biophys. 100, 378Peterson, E. A. & Sober, H. A. (1962) Methods Enzymol.
5, 3Pomerantz, S. H. (1963) J. Biol. Chem. 238, 2351Pomerantz, S. H. (1964) Biochem. Biophys. Res. Commun.
16, 188Power, D. M., Towers, G. H. N. & Neish, A. C. (1965)
Can. J. Biochem. 43, 1397Premakumar, R., Subba Rao, P. V., Sreeleela, N. S. &
Vaidyanathan, C. S. (1969) Can. J. Biochem. 47, 825Sato, M. (1966) Phytochemistry 5, 385Sato, M. (1969) Phytochemistry 8, 353Seidman, M. M., Toms, A. & Wood, J. M. (1969)
J. Bacteriol. 97, 1192Smith, J. L. & Krueger, R. C. (1962) J. Biol. Chem. 237,
1121Stafford, H. A. & Baldy, R. (1970) Plant Physiol. 45, 215Stark, G. K. & Dawson, G. R. (1958) Anal. Chem. 30, 191Umbreit, W. W., Burris, R. H. & Stauffer, J. F. (1964)Manometric Techniques, 4th. edn., pp. 1-17, BurgessPublishing Co., Minneapolis
Vaughan, P. F. T. & Butt, V. S. (1967) Biochem. J. 104,65P
Vaughan, P. F. T. & Butt, V. S. (1969a) Biochem. J. 111,32P
Vaughan, P. F. T. & Butt, V. S. (1969b) Biochem. J. 113,109
Walker, J. R. L. (1969) Phytochemistry 8, 561Yamamoto, S., Katagiri, M., Maeno, H. & Hayaishi, 0.
(1965) J. Biol. Chem. 240, 3408Zucker, M. (1966) Fed. Proc. Fed. Amer. Soc. Exp. Biol.
25, 758
Vol. 130