cse182-l9 gene finding (dna signals) genome sequencing and assembly
Post on 20-Dec-2015
222 views
TRANSCRIPT

CSE182-L9
Gene Finding (DNA signals)Genome Sequencing and
assembly
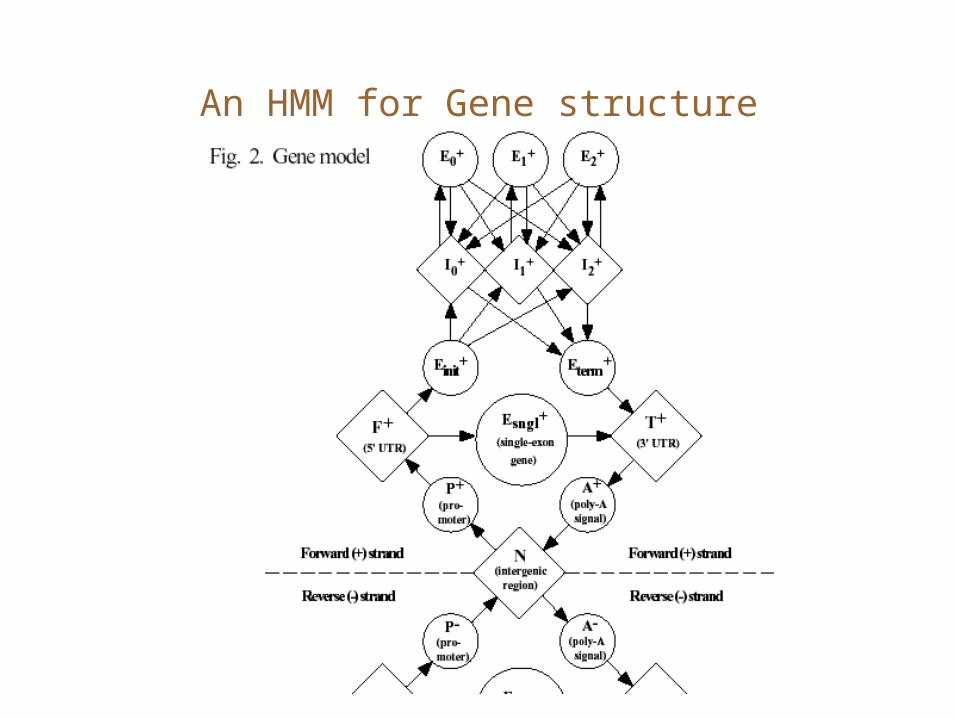
An HMM for Gene structure

Gene Finding via HMMs
• Gene finding can be interpreted as a d.p. approach that threads genomic sequence through the states of a ‘gene’ HMM. – Einit, Efin, Emid,
– I, IG (intergenic)
Einit
I
Efin
Emid
IG
Note: all links are not shown here
i

Generalized HMMs, and other refinements
• A probabilistic model for each of the states (ex: Exon, Splice site) needs to be described
• In standard HMMs, there is an exponential distribution on the duration of time spent in a state.
• This is violated by many states of the gene structure HMM. Solution is to model these using generalized HMMs.
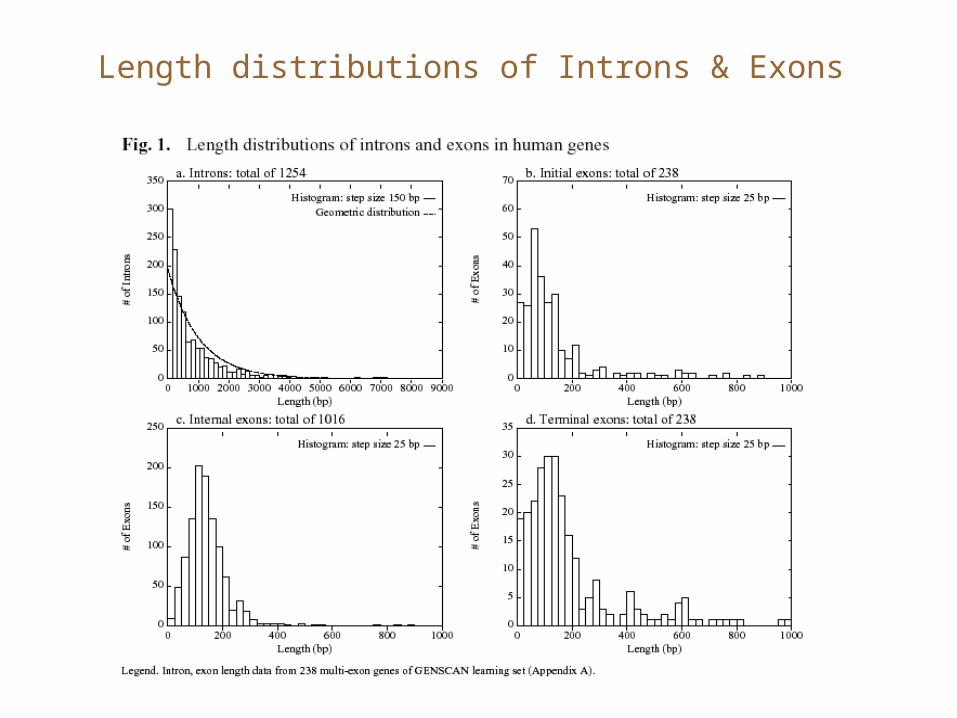
Length distributions of Introns & Exons

Generalized HMM for gene finding
• Each state also emits a ‘duration’ for which it will cycle in the same state. The time is generated according to a random process that depends on the state.

Forward algorithm for gene finding
j i
qk
€
Fk (i) = P qkj<i
∑ (X j ,i) fqk ( j − i +1) alkl∈Q
∑ Fl ( j)
Emission Prob.: Probability that you emitted Xi..Xj in state qk (given by the 5th order markov model)
Forward Prob: Probability that you emitted i symbols and ended up in state qk
Duration Prob.: Probability that you stayedin state qk for j-i+1 steps

De novo Gene prediction: Summary
• Various signals distinguish coding regions from non-coding
• HMMs are a reasonable model for Gene structures, and provide a uniform method for combining various signals.
• Further improvement may come from improved signal detection
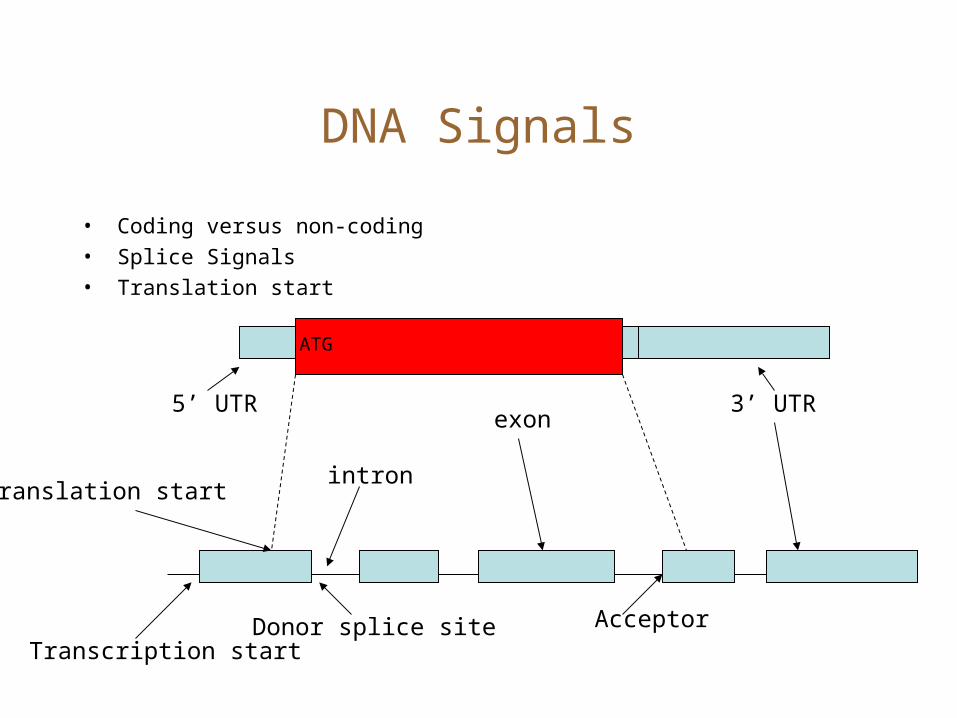
DNA Signals
• Coding versus non-coding• Splice Signals• Translation start
ATG
5’ UTR
intron
exon3’ UTR
AcceptorDonor splice siteTranscription start
Translation start

DNA signal example:
• The donor site marks the junction where an exon ends, and an intron begins.
• For gene finding, we are interested in computing a probability – D[i] = Prob[Donor site at position i]
• Approach: Collect a large number of donor sites, align, and look for a signal.

PWMs
• Fixed length for the splice signal.• Each position is generated independently
according to a distribution• Figure shows data from > 1200 donor
sites
321123456321123456AAGAAGGTGTGAGTGAGTCCGCCGGTGTAAGTAAGTGAGGAGGTGTGAGGGAGGTAGTAGGTGTAAGGAAGG

Improvements to signal detection
• Pr[GGGTGTAA] is a donor site? – 0.5*0.5
• Pr[CCGTGTAA] is a donor site?– 0.5*0.5
• Is something wrong with this explanation?
GGGTGTAAGGGTGTAAGGGTGTAAGGGTGTAACCGTGTGGCCGTGTGGCCGTGTGGCCGTGTGG
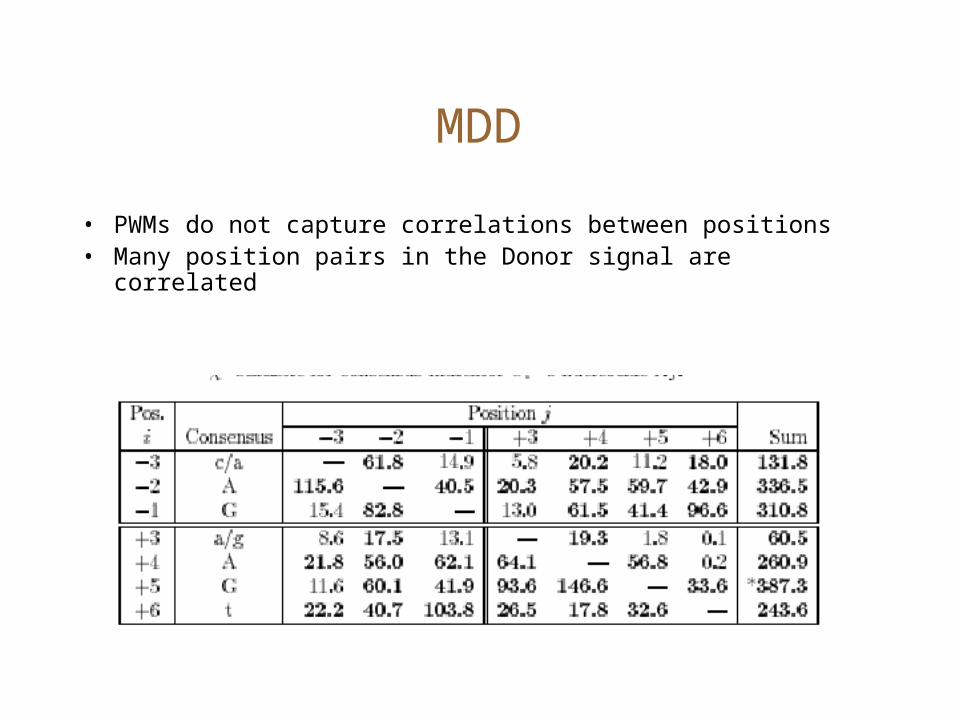
MDD
• PWMs do not capture correlations between positions• Many position pairs in the Donor signal are correlated

Maximal Dependence Decomposition
• Choose the position i which has the highest correlation score.
• Split sequences into two: those which have the consensus at position i, and the remaining.
• Recurse until <Terminating conditions>– Stop if #sequences is ‘small enough’
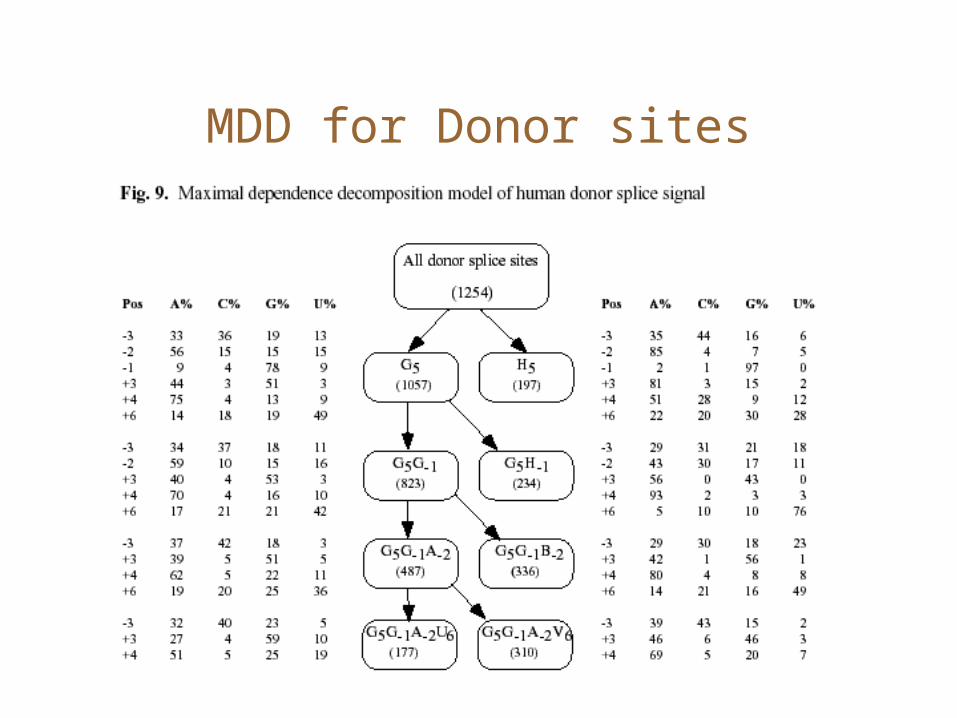
MDD for Donor sites

Gene prediction: Summary
• Various signals distinguish coding regions from non-coding
• HMMs are a reasonable model for Gene structures, and provide a uniform method for combining various signals.
• Further improvement may come from improved signal detection

How many genes do we have?
Nature
Science
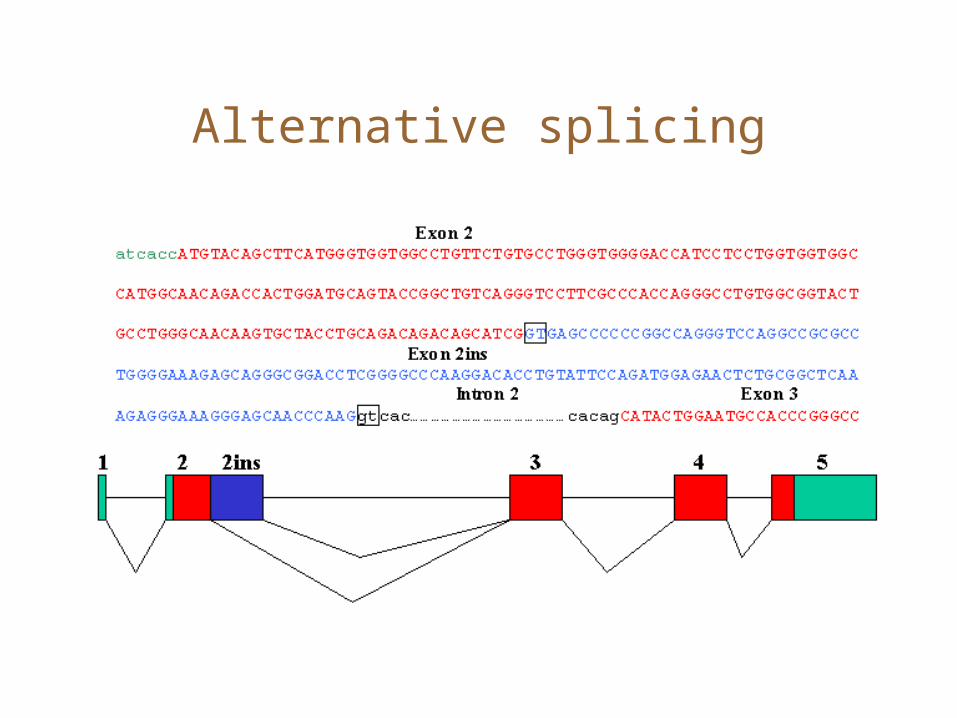
Alternative splicing

Comparative methods
• Gene prediction is harder with alternative splicing.• One approach might be to use comparative methods to
detect genes• Given a similar mRNA/protein (from another species,
perhaps?), can you find the best parse of a genomic sequence that matches that target sequence• Yes, with a variant on alignment algorithms that penalize
separately for introns, versus other gaps.• There is a genome sequencing project for a different Hirudo
species. You could compare the Hirudo ESTs against the genome to do gene finding.

Comparative gene finding tools
• Procrustes/Sim4: mRNA vs. genomic• Genewise: proteins versus genomic• CEM: genomic versus genomic• Twinscan: Combines comparative and de novo
approach.• Mass Spec related?
– Later in the class we will consider mass spectrometry data.
– Can we use this data to identify genes in eukaryotic genomes? (Research project)

Databases
• RefSeq and other databases maintain sequences of full-length transcripts/genes.
• We can query using sequence.
Course• Sequence Comparison
(BLAST & other tools)• Protein Motifs:
– Profiles/Regular Expression/HMMs
• Discovering protein coding genes– Gene finding HMMs– DNA signals (splice
signals)
• How is the genomic sequence itself obtained?
Protein sequence analysis
ESTs
Gene finding

Silly Quiz
• Who are these people, and what is the occasion?
QuickTime™ and aTIFF (Uncompressed) decompressorare needed to see this picture.

Genome Sequencing and Assembly

DNA Sequencing
QuickTime™ and aTIFF (Uncompressed) decompressorare needed to see this picture.
• DNA is double-stranded
• The strands are separated, and a polymerase is used to copy the second strand.
• Special bases terminate this process early.

Sequencing
• A break at T is shown here.
• Measuring the lengths using electrophoresis allows us to get the position of each T
• The same can be done with every nucleotide. Fluorescent labeling can help separate different nucleotides
QuickTime™ and aTIFF (Uncompressed) decompressorare needed to see this picture.

• Automated detectors ‘read’ the terminating bases.
• The signal decays after 1000 bases.
QuickTime™ and aTIFF (Uncompressed) decompressorare needed to see this picture.

Sequencing Genomes: Clone by Clone
• Clones are constructed to span the entire length of the genome.
• These clones are ordered and oriented correctly (Mapping)
• Each clone is sequenced individually
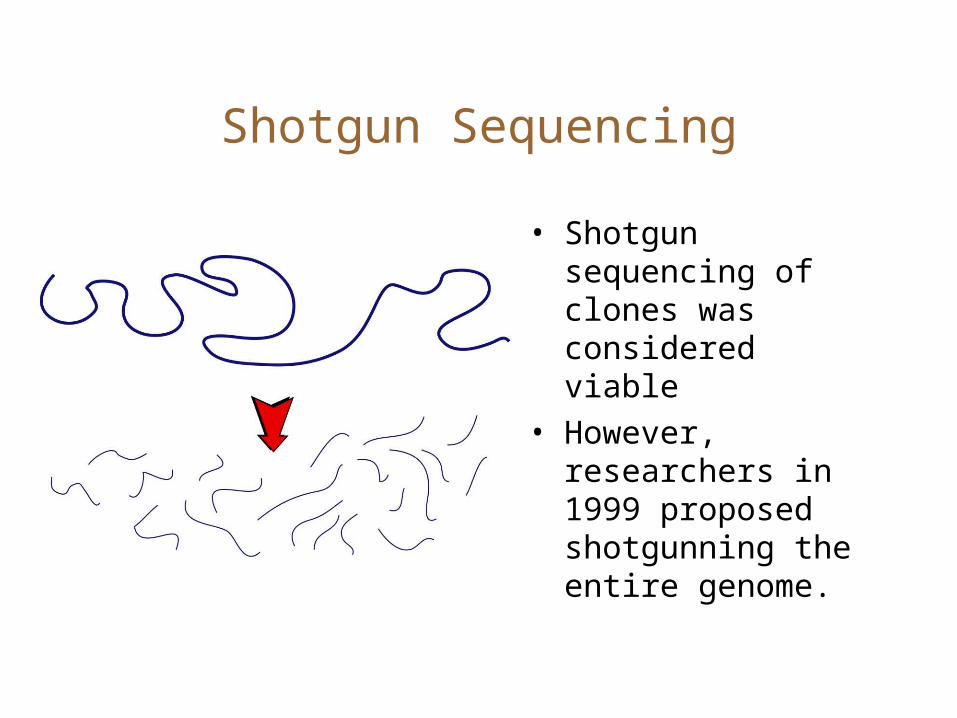
Shotgun Sequencing
• Shotgun sequencing of clones was considered viable
• However, researchers in 1999 proposed shotgunning the entire genome.
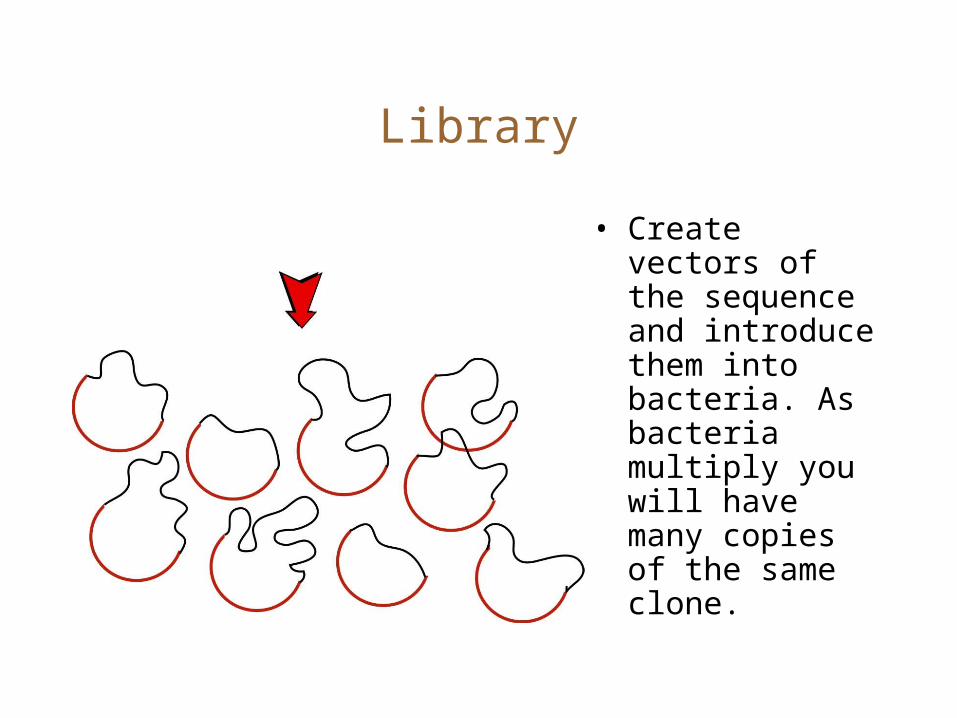
Library
• Create vectors of the sequence and introduce them into bacteria. As bacteria multiply you will have many copies of the same clone.
Sequencing

Questions
• Algorithmic: How do you put the genome back together from the pieces? Will be discussed in the next lecture.
• Statistical? • EX: Let G be the length of the genome, and L
be the length of a fragment. How many fragments do you need to sequence?– The answer to the statistical questions had already
been given in the context of mapping, by Lander and Waterman.
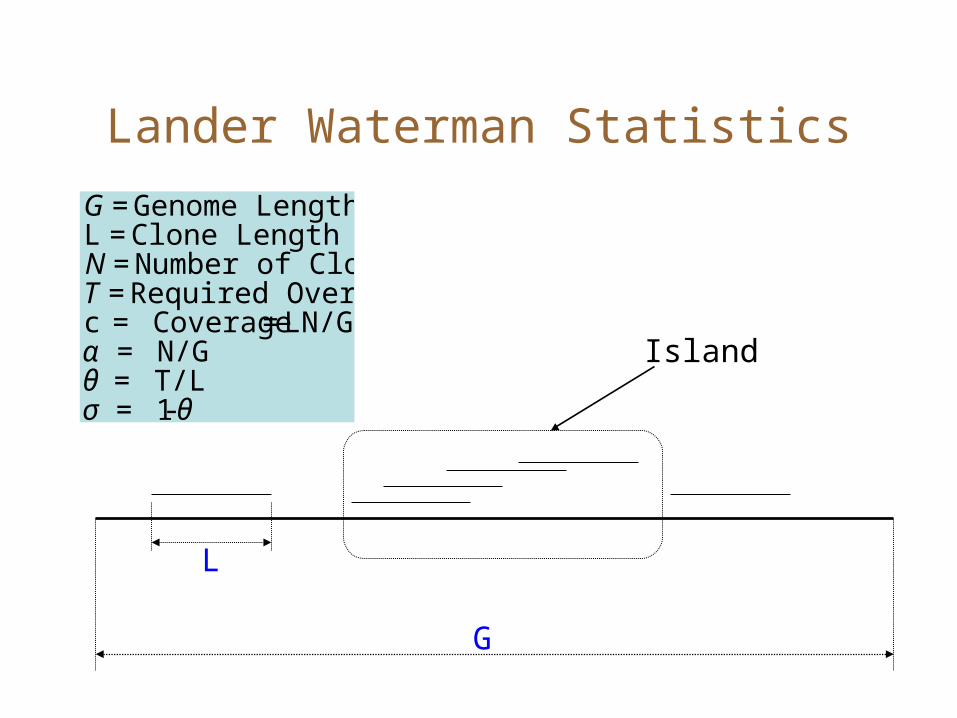
Lander Waterman Statistics
G
L€
G = Genome LengthL = Clone LengthN = Number of ClonesT = Required Overlapc = Coverage = LN/Gα = N/Gθ = T/Lσ = 1-θ
Island
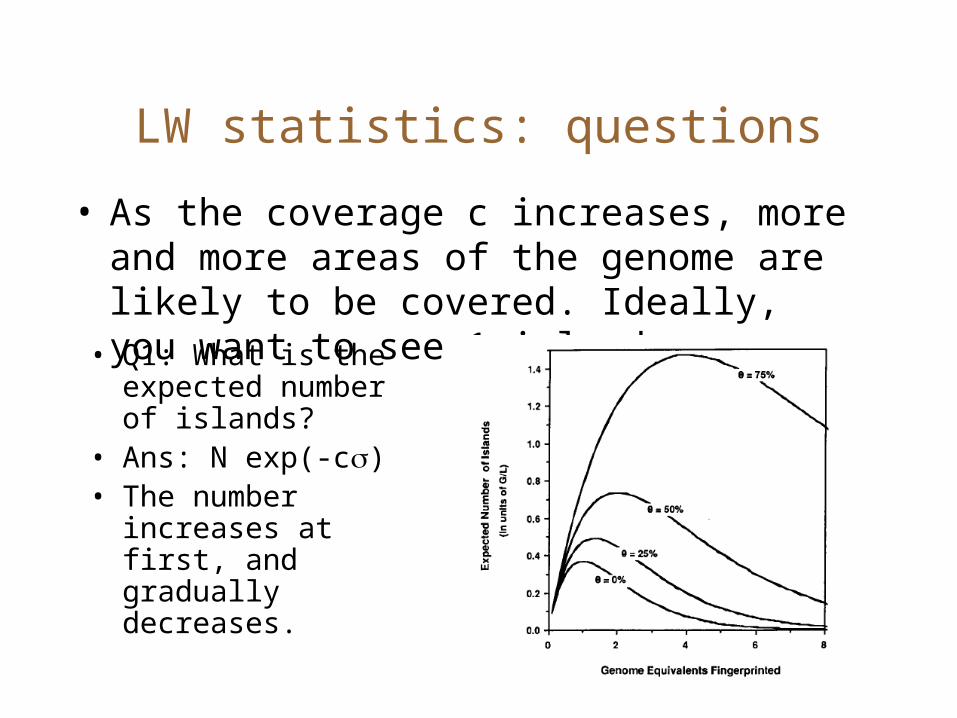
LW statistics: questions
• As the coverage c increases, more and more areas of the genome are likely to be covered. Ideally, you want to see 1 island.• Q1: What is the expected number of islands?
• Ans: N exp(-c)• The number
increases at first, and gradually decreases.

Analysis: Expected Number Islands
• Computing Expected # islands.• Let Xi=1 if an island ends at position i,
Xi=0 otherwise.• Number of islands = ∑i Xi
• Expected # islands = E(∑i Xi) = ∑i E(Xi)

Prob. of an island ending at i
• E(Xi) = Prob (Island ends at pos. i)
• =Prob(clone began at position i-L+1
AND no clone began in the next L-T positions)
iL
T
€
E(X i) =α 1−α( )L−T
=αe−cσ
€
Expected # islands = E(X i) =i
∑ Gαe−cσ = Ne−cσ

LW statistics
• Pr[Island contains exactly j clones]?• Consider an island that has already begun. With
probability e-c, it will never be continued. Therefore• Pr[Island contains exactly j clones]=
€
(1− e−cσ ) j−1e−cσ
• Expected # j-clone islands
€
=Ne−cσ (1− e−cσ ) j−1e−cσ

Expected # of clones in an island
€
ecσ
Why?
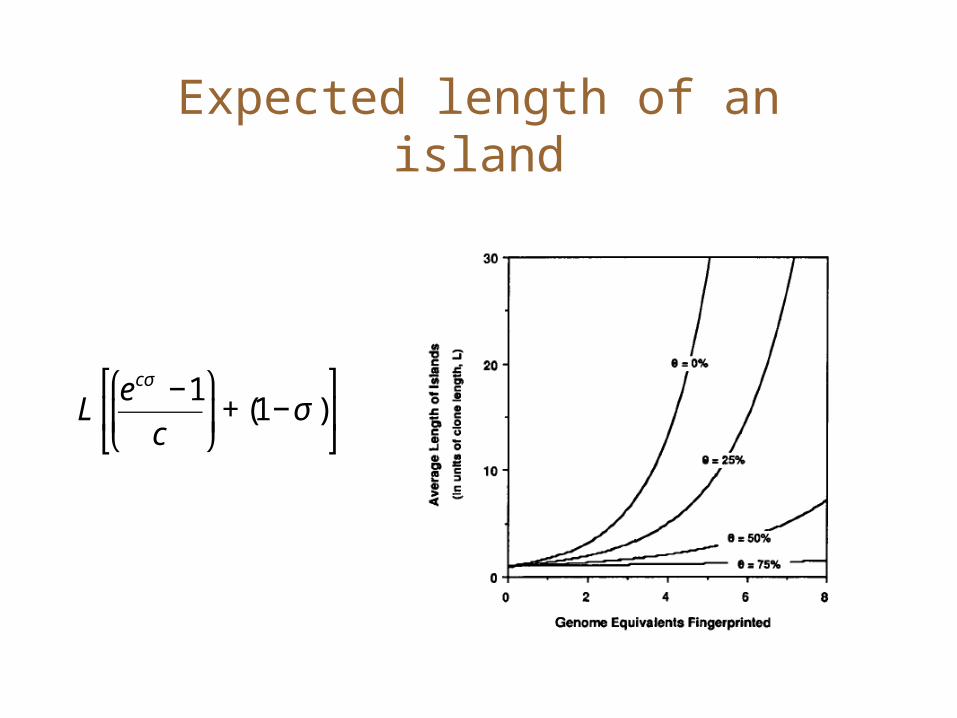
Expected length of an island
€
Lecσ −1
c
⎛
⎝ ⎜
⎞
⎠ ⎟+ (1−σ )
⎡
⎣ ⎢
⎤
⎦ ⎥