denovo drug design
TRANSCRIPT

By,Somasekhar guptha


A drug can be discovered by following approach
1. modification of known molecule.2. Synergestic or additive drugs in combination.3. Screening of wide variety of drugs obtained from
natural sources, banks of previously discovered chemical entities.
4. Identification or elucidation of entirely new target for druG.
5.Rational drug design.6.Genetic approaches.
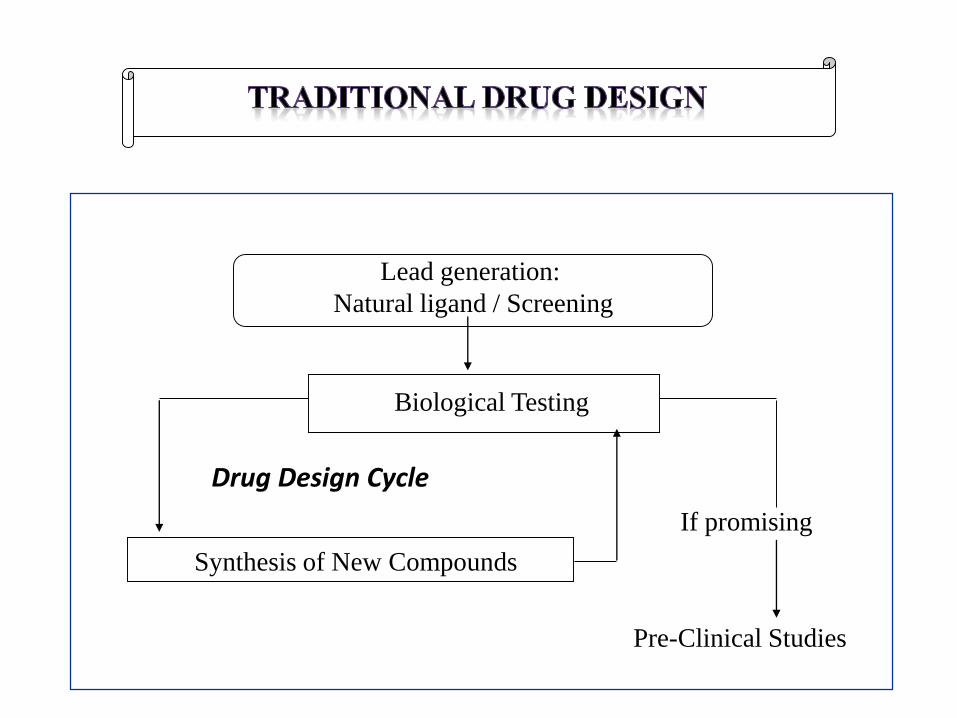
Lead generation:
Natural ligand / Screening
Biological Testing
Synthesis of New Compounds
Drug Design Cycle
If promising
Pre-Clinical Studies

• A lead compound is a small molecule that serves as the starting point for an optimization involving many small molecules that are closely related in structure to the lead compound .
• Many organizations maintain databases of chemical compounds.• Some of these are publically accessible others are proprietary• Databases contain an extremely large number of compounds (ACS
data bases contains 10 million compounds)• 3D databases have information about chemical and geometrical
features» Hydrogen bond donors» Hydrogen bond acceptors» Positive Charge Centers» Aromatic ring centers» Hydrophobic centers

There are two approaches to this problem
• A computer program AutoDock (or similar version Affinity (accelrys)) can be used to search a database by generating “fit” between molecule and the receptor
• Alternatively one can search 3D pharmacophore

• Drug design and development
• Structure based drug design exploits the 3D structure of the target or a pharmacophore
– Find a molecule which would be expected to interact with the receptor. (Searching a data base)
– Design entirely a new molecule from “SCRATCH” (de novo drug/ligand design)
• In this context bioinformatics and chemoinformaticsplay a crucial role

Structure-based Drug Design (SBDD)
Molecular Biology & Protein Chemistry
3D Structure Determination of Target
and Target-Ligand Complex
Modelling
Structure Analysis
and Compound DesignBiological Testing
Synthesis of New Compounds
If promising
Pre-Clinical
Studies
Drug Design Cycle
Natural ligand / Screening

• So the two important features in drug design are
Target-receptor
lead- ligand
so there are different approaches based on structural availibility
receptor ligand approach comments
known known DOCK receptor based
Programmes-AUTO-DOCK
known unknown De novo based GROW, LEGEND
unknown known Ligand based QSAR
unknown unknown Combinationalbased

De novo means start afresh, from the beginning, from the scratch
It is a process in which the 3D structure of receptor is used to design newer molecules
It involves structural determination of the lead target complexes and lead modifications using molecular modeling tools.
Information available about target receptor but no existing leads that can interact.
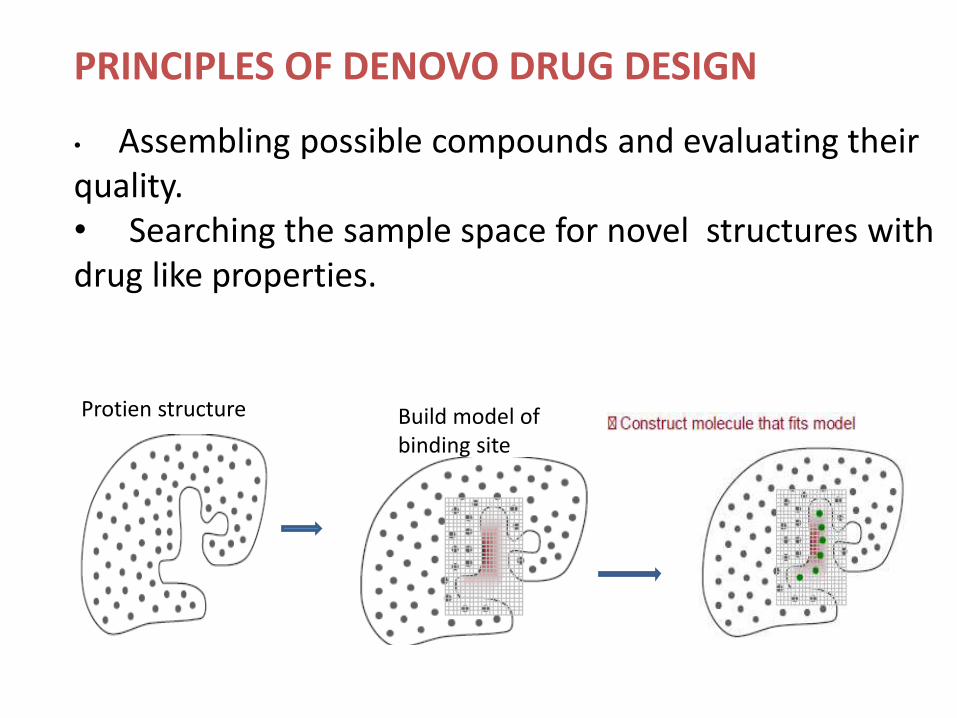
PRINCIPLES OF DENOVO DRUG DESIGN
• Assembling possible compounds and evaluating their quality.• Searching the sample space for novel structures with drug like properties.
Build model of binding site
Protien structure

COMPUTER BASED DRUG DESIGN CONSISTS OF FOLLOWING STEPS :-
1) Generation of potentiol primary constraints2) Derivation of interaction sites3) Building up methods4) Assay (or ) scoring5) Search strategies6) Secondary target constraints

These are the molecules which set up a framework for the desired structure with the required ligand receptor interactions
- These are of 2 types:
1) receptor based:-interactions of the receptor form basis for the
drug design.
2) ligand based:- ligand to the target functions as a key.
Primary target constraints

• In denovo design, the structure of the target shouldbe known to a high resolution, and the binding to sitemust be well defined.
• This should defines not only a shape constraint buthypothetical interaction sites, typically consisting ofhydrogen bonds, electrostatic and other non-covalentinteractions.
• These can greatly reducing the sample space, ashydrogen bonds and other anisotropic interactions candefine specific orientations.

Derivation of Interaction Sites:• A key step to model the binding site as accurately as possible.
• This starts with an atomic resolution structure of the active site.• Programs like UCSF , DOCK define the volume available to aligand by filling the active site with spheres.
• Further constraints follow, using positions of H-bond acceptorsand donors.
• Other docking algorithms, such as FLOG, GOLD, and FlexiDock 16use an all-atom representations to achieve fine detail.
• Ray-tracing algorithms, such as SMART,represent another strategy.

1) Growing2) Linking3) Lattice Based sampling4) Molecular dynamics based
methods

GROWING:-

• A single key building block is the starting point or seed.
• Fragments are added to provide suitable interactions to both key sites and space between key sites
• These include simple hydrocarbon chains, amines, alcohols, and even single rings.
• In the case of multiple seeds, growth is usually simultaneous and continues until all pieces have been integrated into a single molecule.

Linking


The fragments, atoms, or building blocks are either placed at key interaction sites
(or)
pre-docked using another program
They are joined together using pre-defined rules to yield a complete molecule.
Linking groups or linkers may be predefined or generated to satisfy all required conditions .

Lattice based method
• The lattice is placed in the binding site, and atomsaround key interaction sites are joined using the shortestpath.
• Then various iterations, each of which includestranslation, rotation or mutation of atoms, are guided by apotential energy function, eventually leading to a targetmolecule.


Molecular Dynamics Methods:
• The building blocks are initially randomly placed and then byMD simulations allowed to rearrange.
• After each rearrangement certain bonds were broken and the process repeated.
• During this procedure high scoring structures were stored for later evaluation.

SCORING
• Each solution should be tested to decide which is the most promising.this is called as scoring.
• Programs such as LEGEND18, LUDI19, Leap-Frog16, SPROUT20, HOOK21, and PRO-LIGAND22 attempt this using different scoring techniques
• These scoring functions vary from simple stericconstraints and H-bond placement to explicit force fields and empirical or knowledge-based scoring methods.

• Programs like GRID and LigBuilder3 set up a grid in thebinding site and then assess interaction energies by placingprobe atoms or fragments at each grid point.
• Scoring functions guide the growth and optimization ofstructures by assigning fitness values to the sampled space
• Scoring functions attempt to approximate the binding freeenergy by substituting the exact physical model with simplifiedstatistical methods.
• Force fields usually involve more computation than theother types of scoring functions eg:- LEGEND

• Empirical scoring functions are a weighted sum of individualligand–receptor interactions.
• Apart from scoring functions, attempts have been made touse NMR, X-ray analysis and MS to validate the fragments.
• Essential to know which path to follow
Types:-
1) Combinatorial search algorithms
• reducing the effective size of the solution space • explore the space efficiently.

2) Breadth first• keep all possible solutions per step, and solves them all to endstructures.• can only be done in a limited fashion, as an exhaustive searchwould not be feasible.
3)Depth first • select the highest scoring solution per stepand proceeds.• This strategy may spontaneously generate nonsensesolutions.
• Usually combinations of these last two are used;for example, a breadth-first search could be done until thesolution space is relatively limited and in subsequent steps depth-first searcheswould be used.

4) Monte Carlo algorithms • based on random sampling, and are usually tied with the“Metropolis criterion”.• After each modification, the partial solution is eitheraccepted if it is a better solution or rejected, based on thedifference of scoring of the modified versus the unmodifiedstructure.
5)Evolutionary Algorithms • model natural processes, such as selection, recombination,mutation, and migration, and work in a parallel manner.
• The number of partial solutions is not fixed and “evolves”based on the “fitness” of the solutions.

• Binding affinity alone does not suffice to make an effective drugmolecule.• Essential properties include effective Absorption, Distribution,Metabolism, Excretion and Toxicity (ADMET).
•In general, an orally active drug has
• (a) not more than 5 hydrogen bond donors (OH and NH groups),• (b) not more than 10 hydrogen bond acceptors (notably Nand O),• (c) a molecular weight under 500 Da• (d), a LogP (log ratio of the concentrationsof the solute in thesolvent) under 5.•Using these rules as a filter, the resulting compounds are morelikely to have biological activity.

METHOD PROGRAMS AVAILABLE
Site point connection method
LUDI
Fragment connection method
SPLICE, NEW LEAD, PRO-LIGAND
Sequential build up methods
LEGEND, GROW, SPORUT
Random connection and disconnection methods
CONCEPTS, CONCERTS, MCDNLG

Applications:-
• Design of HIV I protease inhibitors• Design of bradykinin receptor antagonist• Catechol ortho methyl transferase inhibitor
Ex:- entacapone and nitecapone• Estrogen receptor antagonist• Purine nucleoside phosphorylase inhibitors

HIV 1 protease inhibitor
• HIV 1 protease is an enzyme crucial for replication of HIV virus
• Inhibitors include saquinavir, ritonavir, indinavar
• These drugs have been developed using this appraoch
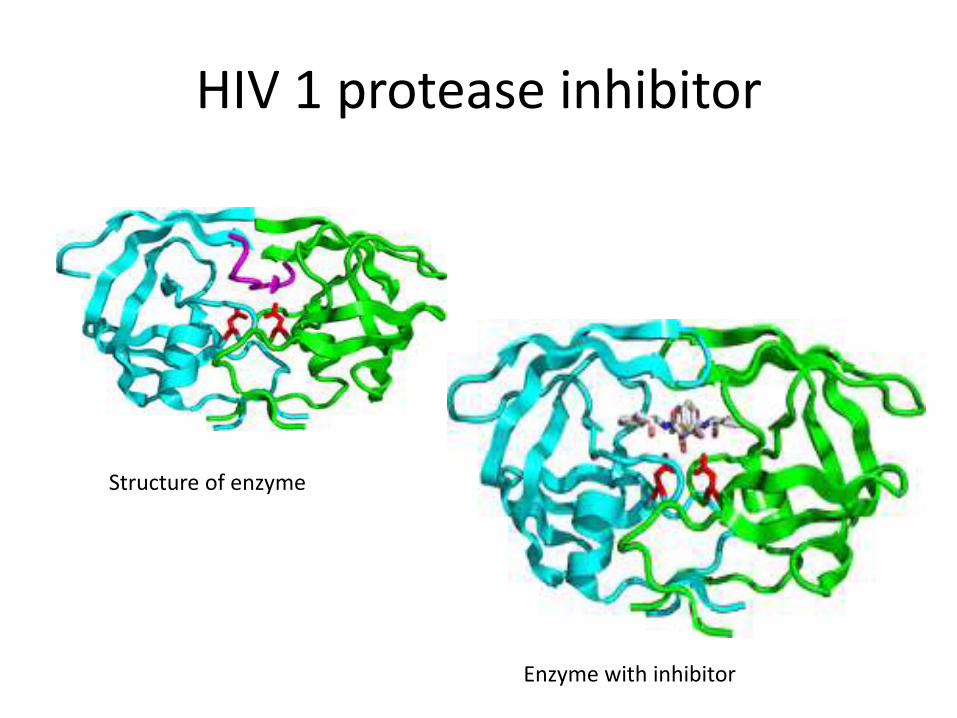
HIV 1 protease inhibitor
Structure of enzyme
Enzyme with inhibitor

limitations
• Rather slow and inefficient
• Ignores synthetic feasibility while constructing structures
• Cannot be a sole basis for drug design

• Although a relatively new design method, de novo
design will play an ever-increasing role in modern drugdesign.
• Though yet not able to automatically generate viabledrugs by itself, it is able to give rise to novel and oftenunexpected drugs
• when coupled with HTS, is proving to reduce drugdesign turn aroundtime.

References • Foye’s principles of medicinal chemistry-sixth edition,
published by lippincott williams and wilkins, page num-69-70
• Sanmati K jain and avantika agarwal- “ De novo drug design:anoverview”. Indian journal of pharmaceutical sciences.,2004,66[6]:721-728
• http://www.bama.ua.edu/~chem/seminars/student_seminars/spring06/papers-s06/luthra-sem.pdf
• http://journal.chemistrycentral.com/content/2/S1/
• http://en.wikipedia.org/wiki/HIV-1_protease
• Schneider G.; Fechner U.; “Computer-based de novo design of drug-like molecules.” Nat Rev Drug Discov 2005 Aug; 4(8), 649-63. Review
