development and approval of drugs and devices epi260 lecture 9 may 23, 2012 richard chin, m.d
TRANSCRIPT

Development and Approval ofDrugs and Devices
EPI260
Lecture 9May 23, 2012
Richard Chin, M.D.

Overview OutlineOverview Outline
• Overview• Classification• Case study• Reporting requirements for FDA, EMEA, and others• Signal detection• REMS and postmarketing surveillance

Overview Patient SafetyOverview Patient Safety
• Safety is paramount• Clinical research is risky• IND is a specific exemption granted by the FDA• Specific adverse event reporting requirements during
clinical trials– Now harmonized
• Reporting requirements after marketing

Defining Adverse EventsDefining Adverse Events
• Adverse events, except targeted adverse events, are not predefined
• Safety is not opposite of efficacy, but rather the complement
• Statistical significance is not required for safety signal– Hard to predefine– Most studies not powered for safety events

Mapping TermsMapping Terms
• MI vs. angina vs. chest pain vs. anterior MI vs. sudden death
• Terms must be mapped to consistent terms• MedDRA - the Medical Dictionary for Regulatory
Activities– Hierarchical and cross referenced mapping system

Defining Adverse EventsDefining Adverse Events
• When does event begin and end?– Exacerbation of asthma– Reinfarction
• What about events that recur?– How many events?
• What events are emergent events?– Exacerbation of condition that existed at baseline
• Who determines causality?– Investigator– Sponsor– Adjudication committee

Adjudication CommitteesAdjudication Committees
• Standardizes adverse events• Similar to DSMB• Necessary and possible if there is a targeted adverse
event• Can be combined with outcome adjudication
committee

Drug EffectsDrug Effects
Treatment
Time
Acute Effect
Initial Treatment
Administration
Treatment Discontinuation
Rebound Effect
Latent Effect
Chronic Effects

Additional Drug EffectsAdditional Drug Effects
• Cumulative• Interactions• Harbinger events

Case Study: Zooming In and OutCase Study: Zooming In and Out
• A promising drug for refractory seizures is being developed
• Unfortunately, it appears to have two drawbacks– Potential to cause arrhythmias– Potential to cause duodenal ulcers
• Therefore, the Phase III safety data is carefully examined to assess potential signal in those two categories

Initial readingInitial reading
• Fortunately, neither concern seems to have been justified, as no signal is apparent
3% 4% 7% 5%0%
20%
40%
60%
80%
100%
Dia
rrhe
al S
ympt
om
Seve
rity
Arrhythmia Duodenal Ulcers
Placebo Active

Illusion: LumpingIllusion: Lumping
• Unfortunately, if the net for arrhythmias is cast more widely to include not just the term, “arrhythmia” but also “sudden death,” “palpitations,” “syncope,” (which can be other presentations of arrhythmia) then a signal becomes apparent
12%
23%
0%
10%
20%
30%
40%
50%
Rat
e
Syncope 2% 7%
Palpitations 6% 9%
Sudden Death 1% 3%
Arrhythmia 3% 4%
Placebo Active
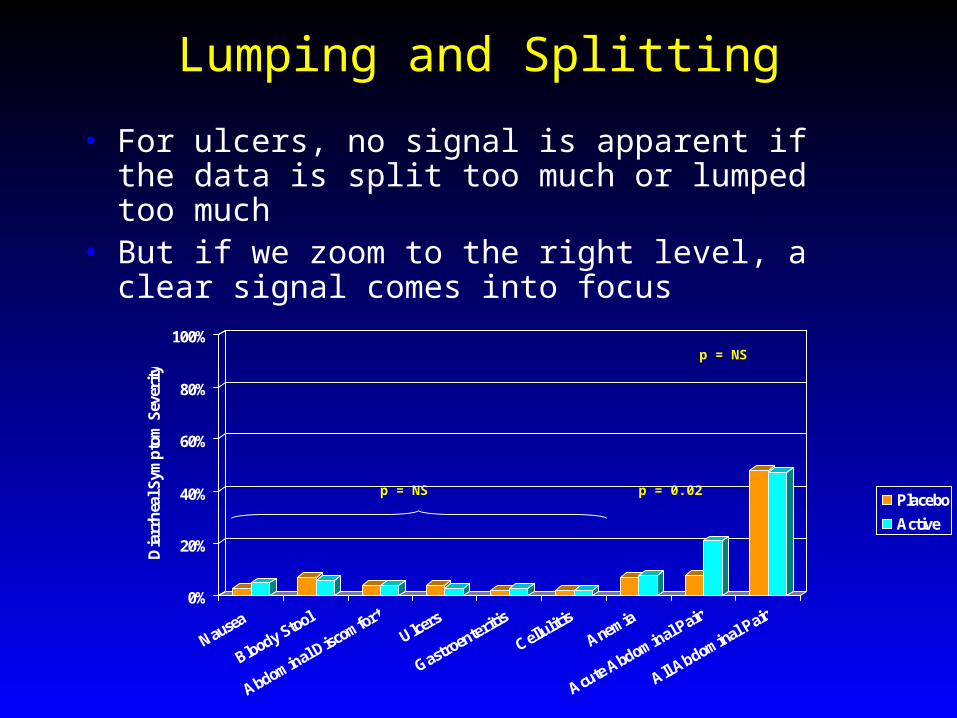
Lumping and Splitting
• For ulcers, no signal is apparent if the data is split too much or lumped too much
• But if we zoom to the right level, a clear signal comes into focus
p = 0.02p = NS
p = NS
0%
20%
40%
60%
80%
100%
Dia
rrh
eal S
ymp
tom
Sev
erit
y
Nausea
Bloody Stool
Abdominal Discomfort
Ulcers
Gastroenteritis
Cellulitis
Anemia
Acute Abdominal Pain
All Abdominal P
ain
Placebo
Active

Types of AEsTypes of AEs
• Premarketing (clinical trial)– Individual– Trend analysis
• Postmarketing– Spontaneous– Trend analysis

Reporting Requirements During TrialsReporting Requirements During Trials
• Serious, Unexpected Adverse Events must be reported AS FAST AS POSSIBLE but no later than– 7 calendar days if life threatening or fatal– 15 calendar days if not life threatening or fatal
• New guidelines specify this applies to events “for which there is a reasonable possibility that the drug caused the adverse event “
• Increased incidence of AE must also be reported

Adverse Event/Drug Reaction Definition Adverse Event/Drug Reaction Definition
• Adverse Event: Any untoward medical occurrence in a subject to whom a medicinal product has been administered, including occurrences which are not necessarily caused by or related to that product
• Suspected adverse reaction : Suspected adverse reaction means any adverse event for which there is a reasonable possibility that the drug caused the adverse event. For the purposes of IND safety reporting, ‘reasonable possibility’ means there is evidence to suggest a causal relationship between the drug and the adverse event.

Reasonable Likelihood Reasonable Likelihood
• A single occurrence of an event that is uncommon and known to be strongly associated with drug exposure (e.g., angioedema, hepatic injury, Stevens-Johnson Syndrome)
• One or more occurrences of an event that is not commonly associated with drug exposure, but is otherwise uncommon in the population exposed to the drug (e.g., tendon rupture)
• An aggregate analysis of specific events observed in a clinical trial (such as known consequences of the underlying disease or condition under investigation or other events that commonly occur in the study population independent of drug therapy) that indicates those events occur more frequently in the drug treatment group than in a concurrent or historical control group

Serious vs. SevereSerious vs. Severe
• Not the same thing• Serious
(a) results in death
(b) is life-threatening
(c) requires hospitalization or prolongation of existing hospitalization
(d) results in persistent or significant disability or incapacity, or
(e) consists of a congenital anomaly or birth defect

Common Terminology Criteria for Adverse Common Terminology Criteria for Adverse Events v3.0 (CTCAE)Events v3.0 (CTCAE)
• Grade 1: Mild AE• Grade 2: Moderate AE • Grade 3: Severe AE• Grade 4: Life-threatening or disabling AE • Grade 5: Death related to AE


Unexpected DefinitionUnexpected Definition
• Not in the Investigator’s Brochure• Not in the Package Insert or SPC• Unclear: Addendum to the IB• May be unexpected if changes occur in
– Rate– Severity– Duration– Other

Emergent Adverse Event DefinitionEmergent Adverse Event Definition
• Sometimes it is not clear when an event began• Examples
– Worsening of event– Event stops and restarts– Repeat of event
• Consistency is critical

Causality and RelatednessCausality and Relatedness
• Categories– Unrelated– Possibly related– Related
• Assessed by investigator and by the sponsor• When is patient considered to be off the drug?• Often, sponsors have Safety Review Boards an
DSMB but CMO has last word• FDA guidelines – sponsor definition prevails• ICH guidelines – either sponsor or investigator

UnblindingUnblinding
• Per ICH guidelines, placebos may qualify as drug• Most sponsors unblind and report only patients who
were on drug

When Does Clock Start?When Does Clock Start?
• Day 1 is the day of the sponsor has knowledge• Sponsor includes anyone at the company, and
anyone at the CRO or other agents of the company• Knowledge occurs when
– a suspected investigational medicinal product– an identifiable subject (e.g. study subject code number)– an adverse event assessed as serious and unexpected, and
for which there is a reasonable suspected causal relationship
– an identifiable reporting source
• Clock stops on the day that the report is received by the regulatory agency

Follow-upFollow-up
• Many sponsors follow SAEs to resolution• At a minimum, the patients should be followed until
end of study

Trend AnalysisTrend Analysis
• Frequency analysis is important• Analysis by subgroups and risk factors is important• Need to monitor event rates and patterns

How to ReportHow to Report
• Medwatch forms• CIOMS forms• EUDRA


Reporting Reporting
• Regulatory Authorities• Investigators• IRB/ECs• Patients
– Reconsent

LogisticsLogistics
• Typical flow of information– P.I. reports to sponsor via EDC– Assigned to case worker in pharmacovigilance group– Data mapped and entered– Follow up information obtained– Medwatch or other form prepared– QC check– Regulatory files with authorities

SOPs and DocumentsSOPs and Documents
• Safety Reporting Agreements: Document outlining how the AEs will be processed between cosponsors or sponsor and CRO
• Annual Safety Reports• Pharmacovigilance SOPs• MedDRA updates

ReconcilingReconciling
• Reconciling with Clinical Database – AE database often separate from clinical database– Reconciliation can be difficult
• Reconciling different versions of MedDRA

Approval and PostmarketingApproval and Postmarketing
• Risk minimization action plans (RiskMAPs)– strategic safety program100designed to meet specific goals
and objectives in minimizing known risks of a product while 101preserving its benefits
• Risk Evaluation and Mitigation Strategies (REMS)– Timetable for Submission of Assessments – Additional Potential Elements
• A Medication Guide
• A patient package insert
• A communication plan to health care providers
– Elements to Ensure Safe Use (ETASU)

Postmarketing ReportingPostmarketing Reporting
• SUSAR: Similar requirements for postmarketing period
• Literature Survey: Sponsors are required to monitor literature for AEs and report them
• Annual reports