development of ruthenium catalysts for water oxidation1038808/fulltext01.pdf · list of...
TRANSCRIPT

Development of Ruthenium Catalysts for Water Oxidation
Tanja M. Laine

© Tanja M. Laine, Stockholm University 2016 Cover picture: Above the sylvan glade ISBN print 978-91-7649-508-7 ISBN PDF 978-91-7649-509-4 Printed in Sweden by Holmberg, Malmö 2016 Distributor: Department of Organic Chemistry, Stockholm University

“A thinker sees his own ac-tions as experiments and questions – as attempts to find out something. Success and failure are for him an-swers above all.” Friedrich Nietzsche


i
Abstract
An increasing global energy demand requires alternative sources to fossil fuels. A promising alternative for generation of such fuels is artificial photo-synthesis. Although the process is different from the natural photosynthetic process, the basic principles are the same, i.e. to convert solar energy into chemical energy. By achieving this, energy can be stored in bonds, which at a later stage can be released upon combustion. The bottleneck in the artifi-cial photosynthesis is the water oxidation. The aim of this research has been to develop catalysts for water oxidation that are stable, yet efficient. The mo-lecular catalysts are comprised of ligands which have a great influence on the catalyst structure and activity. These ligands are often based on polypyridyl or other nitrogen-containing aromatic compounds. This thesis describes the development of molecular ruthenium catalysts and the evaluation of their ability to mediate chemical and photochemical oxidation of water. Previous work from our group has shown that the introduction of negatively charged groups into the ligand frameworks lowers the redox potentials of the metal complexes. This enables water oxidation with [Ru(bpy)3]3+-type oxidants (bpy = 2,2’-bipyridine), which can be photochemically generated from the corre-sponding [Ru(bpy)3]2+ photosensitizer. Hence, all the designed ligands herein contain negatively charged groups in the ruthenium coordination sphere. The first part of this thesis describes the development of two mononuclear ruthenium complexes and the evaluation of their activity towards water oxi-dation. Both catalysts displayed low redox potentials, allowing for water ox-idation to be driven either chemically or photochemically. The second part of the thesis is a structure–activity relationship study on sev-eral analogues of mononuclear ruthenium complexes. The complexes were active for water oxidation and their redox potentials displayed a linear rela-tionship with the Hammet σmeta parameter. It was also found that the com-plexes form high-valent Ru(VI) states which are responsible for mediating O–O bond formation.

ii
The last part of the thesis describes the development of a dinuclear ruthe-nium complex and its catalytic performance in chemical and photochemical water oxidation. It was found that the O–O bond formation proceeds via a radical coupling, to yield a bridging peroxide intermediate, i.e. an I2M–type of mechanism.

iii
List of Publications
This thesis is based on the following papers, referred to by their Roman nu-merals I–IV. Reprints were made with the kind permission from the publish-ers. The contribution by the author to each publication is clarified in Appen-dix A. I. Water Oxidation by Single-Site Ruthenium Complexes: Using Ligands
as Redox and Proton Transfer Mediators Markus D. Kärkäs, Torbjörn Åkermark, Eric V. Johnston, Shams R. Karim, Tanja M. Laine, Bao-Lin Lee, Tobias Åkermark, Timofei Privalov, Björn Åkermark Angew. Chem. Int. Ed. 2012, 51, 11589–11593
II. Molecular Ruthenium Water Oxidation Catalysts Carrying Non-Inno-
cent Ligands: Mechanistic Insight through Structure-Activity Relation-ships and Quantum Chemical Calculations Markus D. Kärkäs, Rong-Zhen Liao, Tanja M. Laine, Torbjörn Åkermark, Shams Ghanem, Per E. M. Siegbahn, Björn Åkermark Catal. Sci. Technol. 2016, 6, 1306–1319
III. Efficient Photochemical Water Oxidation by a Dinuclear Molecular Ru-
thenium Complex Tanja M. Laine, Markus D. Kärkäs, Rong-Zhen Liao, Torbjörn Åkermark, Bao-Lini Lee, Erik A. Karlsson, Per E. M. Siegbahn, Björn Åkermark Chem. Commun. 2015, 51, 1862–1865
IV. A Dinuclear Ruthenium-Based Water Oxidation Catalyst: Use of Non-
Innocent Ligand Frameworks for Promoting Multi-Electron Reactions Tanja M. Laine, Markus D. Kärkäs, Rong-Zhen Liao, Per E. M. Siegbahn, Björn Åkermark Chem. Eur. J. 2015, 21, 10039–10048

iv
Publications not included in this thesis:
On the Mechanism of Water Oxidation Catalyzed by a Dinuclear Ru-thenium Complex: A Quantum Chemical Study Rong-Zhen Liao, Markus D. Kärkäs, Tanja M. Laine, Björn Åkermark, Per E. M. Siegbahn Catal. Sci. Technol. 2016, 6, 5031–5041
Water Oxidation Catalyzed by Molecular Di- and Nonanuclear Fe Com-plexes: Importance of a Proper Ligand Framework Biswanath Das, Bao-Lin Lee, Erik A. Karlsson, Torbjörn Åkermark, Andrey Shatskiy, Serhiy Demeshklo, Rong-Zhen Liao, Tanja M. Laine, Matti Haukka, Erica Zeglio, Ahmed F. Abdel-Magied, Per Siegbahn, Franc Meyer, Markus D. Kärkäs, Eric V. Johnston, Ebbe Nordlander, Björn Åker-mark Dalton Trans. 2016, 45, 13289–13293
Cobal Selenium Oxohalides: Catalysts for Water Oxidation Faiz Rabbani, Henrik Svengren, Iwan Zimmermann, Shichao Hu, Tanja M. Laine, Wenming Hao, Björn Åkermark, Torbjörn Åkermark, Mats Johnsson Dalton Trans. 2014, 43, 3984–3989
An Oxofluoride Catalyst Comprised of Transition Metals on a Metalloid for Application in Water Oxidation Henrik Svengren, Shichao Hu, Ioannis Athanassidis, Tanja M. Laine, Mats Johnsson Chem. Eur. J. 2015, 21, 12991–12995
Catalyst-Solvent Interactions in a Dinuclear Ru-based Water Oxidation Catalyst Andrey Shatskiy, Reinar Lomoth, Ahmed Abdel-Magied, Wangchuk Rab-ten, Tanja M. Laine, Hong Chen, Junliang Sun, Pher Andersson, Markus D. Kärkäs, Eric V. Johnston, Björn Åkermark Dalton Trans. 2016, Accepted for publication

v
Chemical and Photochemical Water Oxidation Mediated by an Effi-cient Single-Site Ruthenium Catalyst Ahmed F. Abdel-Magied, Andrey Shatskiy, Rong-Zhen Liao, Tanja M. Laine, Wael A. A. Arafa, Per E. M. Siegbahn, Markus D. Kärkäs, Björn Åkermark, Eric V. Johnston ChemSusChem 2016, DOI:10.1002/cssc.201601171
Book chapter:
Visible Light-Driven Water Oxidation Catalyzed by Ruthenium Com-plexes Markus D. Kärkäs, Tanja M. Laine, Eric V. Johnston, Björn Åkermark In “Applied Photosynthesis – New Progress” InTech, 2016, ed. Mohammad Mahdi Najafpour

vi

vii
Abbreviations
bpy 2,2’-bipyridine CAN cerium ammonium nitrate CV cyclic voltammetry bda 2,2’-bipyridine-6,6’-diacarboxylic acid deeb 4,4’-diethylester-2,2’-bipyridine DLS dynamic light scattering dmp 4,4’-dimethyl-2,2’-bipyridine DPV differential pulse voltammetry HOMO highest occupied molecular orbital I2M interaction of two M–O units LFER linear free-energy relationship LUMO lowest unoccupied molecular orbital OEC oxygen evolving complex NADP nicotinamide adenine dinucleotide phosphate NHE normal hydrogen electrode P680 reaction center in photosystem II PCET proton-coupled electron transfer PS photosensitizer PEC photoelectrochemical cell pic 4-methyl-pyridine (4-picoline) PS II photosystem II SCE saturated calomel electrode terpy 2,2’:6’,2’’-terpyridine TOF turnover frequency TON turnover number TyrZ tyrosine WNA water nucleophilic attack WOC water oxidation catalyst

viii

ix
Table of Contents
1. Introduction ...................................................................................... 1 1.1 Natural Photosynthesis ........................................................................ 1 1.2 Artificial Photosynthesis ....................................................................... 4 1.3 Molecular Water Oxidation Complexes – Chemical Oxidation ............ 6
1.3.1 Ruthenium Complexes .................................................................. 6 1.3.2 Earth-Abundant Catalysts for Oxidation of Water ..................... 11
1.4 Light-Driven Water Oxidation ............................................................ 13 1.4.1 Ruthenium Complexes for Light-Driven Water Oxidation .......... 15
1.5 Mechanistic Pathways for the O–O Bond Formation ......................... 19 1.5.1 Water Nucleophilic Attack (WNA) .............................................. 19 1.5.2 Interaction of Two Metal Units (I2M) ......................................... 20
1.6 Aim of Thesis ...................................................................................... 20
2. Mononuclear Ruthenium Complexes for Water Oxidation (Paper I) ............................................................................................................ 21
2.1 Introduction ........................................................................................ 21 2.3 Results and Discussion ....................................................................... 22
2.3.1 Synthesis and Characterization .................................................. 22 2.3.2 Electrochemical Studies .............................................................. 23 2.3.3 Catalytic Activity ......................................................................... 25 2.3.4 Ligand Exchange ......................................................................... 28
2.4 Summary ............................................................................................ 29
3. Mononuclear Ruthenium Water Oxidation Catalysts: Mechanistic Insight through Structure-Activity Relationships (Paper II) ................ 30
3.1 Introduction ........................................................................................ 30 3.2 Results and Discussion ....................................................................... 32
3.2.1 Synthesis and Characterization .................................................. 32 3.2.2 Catalytic Performance ................................................................ 32 3.2.3 Electrochemical Studies .............................................................. 35 3.2.4 Structure-Activity Relationships ................................................. 36
3.3 Summary ............................................................................................ 43
4. A Dinuclear Ruthenium Complex for Chemical and Photochemical Water Oxidation (Paper III and IV) ..................................................... 44
4.1 Introduction ........................................................................................ 44

x
4.2 Results and Discussion ....................................................................... 45 4.2.1 Synthesis and Characterization .................................................. 45 4.2.2 Quantum Chemical Calculations ................................................. 47 4.2.3 Electrochemical Studies .............................................................. 51 4.2.3 Electrochemical Studies .............................................................. 51 4.2.4 Water Oxidation Catalysis .......................................................... 55 4.2.5 Proposed Catalytic Cycle ............................................................. 58
4.3 Summary ............................................................................................ 60
Acknowledgements ............................................................................ 61
Appendix A: Contribution list ............................................................. 63
References .......................................................................................... 64

1
1. Introduction
An increased energy demand depends mainly on the growth of the popula-tion and economy. The global energy consumption is expected to double from 13.5 TW in 2001 to 27 TW by year 2050.1 Although fossil fuels are ex-pected to last for several centuries, their consumption lead to increased CO2 levels. Different models have predicted different scenarios with moderate to severe raises of the sea level. In any case, it is certain that the CO2 levels in the atmosphere are increasing and have reached levels that the planet has not faced during the past 650 000 years. The only way to lower the CO2 emis-sion is to switch to carbon-free or carbon-neutral renewable energy sources. An alternative to fossil fuels is artificial photosynthesis, i.e. conversion of so-lar energy into chemical energy.1
1.1 Natural Photosynthesis Photosynthesis is the conversion of solar energy into chemical energy by re-ducing CO2 to carbohydrates (Equation 1.1) and is a process that occurs in plants, algae and cyanobacteria. In nature, absorption of sunlight occurs in the cofactor-protein complexes photosystem I (PS I) and photosystem II (PS II). PS II is embedded in the thylakoid membrane and is surrounded by pe-ripheral subunits, chlorophylls and light-harvesting proteins. Chlorophyll a and b absorb sunlight and the energy is subsequently transmitted to the re-action center P680 in PS II. The excited P680 (P680*) transfers an electron to the adjacent pheophytin and the electron is, in several steps, transported across the thylakoid membrane to ferredoxin-NADP+ oxidoreductase for the reduc-tion of NADP+ to NADPH. The oxidized P680, P680
+•, now has a redox potential of ca 1.25 V vs. NHE and abstracts an electron from the adjacent amino acid residue, tyrosine (TyrZ). TyrZ abstracts electrons in several consecutive steps from the oxygen evolving complex (OEC) (Figure 1.3). The central feature of

2
the OEC is a Mn4CaO5 cluster (Figure 1.2), which after transferring four elec-trons retrieves the electrons by oxidizing two molecules of water to one mol-ecule of molecular oxygen. The mechanism by which OEC interacts with wa-ter has been thoroughly studied, showing that four electrons are abstracted, one electron at the time, before molecular oxygen is released.2
6 CO2 + 7 H2O + hv C6H12O6 + 6 O2 + H2O (1.1)
Figure 1.1. Simplified cartoon of the photosynthetic system. The electron flow across the thylakoid membrane is represented by the black arrows. The electrons are ulti-mately abstracted from water and transported to NADP+ for carbon fixation. Re-printed with permission from the publisher.3 In the catalytic cycle, each redox state of OEC is referred to as an “S-state” and was first described by Kok and co-workers.4 As four electrons are trans-ferred, four protons are shuffled to the surrounding amino acid residue(s) to avoid high-energy intermediates, thus enabling a low-energy pathway for the reaction (Figure 1.3).2d,4

3
Figure 1.2. Structure of the oxygen evolving complex (OEC) containing the Mn4CaO5 cluster in a distorted chair form. Reprinted from Umena et al. Nature 2011, 473, 55-61, with kind permission from the publisher.2d
S0
S1S4
S3 S2
TyrZ
TyrZ
TyrZ
TyrZ
TyrZTyrZ
TyrZ
TyrZ
P680
P680
P680
P680
P680
P680P680
P680
hv
hv
hv
hv
2 H2O
O2
Figure 1.3. The Kok cycle showing the five oxidation states (denoted as S) of the tetranuclear Mn-cluster.

4
1.2 Artificial Photosynthesis Converting solar energy into chemical energy is a promising alternative to fossil fuel. One example of a potential renewable fuel that can be produced from this process is molecular hydrogen, where the source for protons for could be derived from water splitting. Molecular hydrogen can be used as clean fuel, since combustion of hydrogen only yields water without emission of greenhouse gases or other air pollutants. The solar to chemical energy conversion can be put into practice with a photo-electrochemical cell (PEC) (Figure 1.4).5 A PEC is constructed of two half-cells, a semiconductor-based photoanode where oxidation of water occurs and a cathode for reduction of protons into molecular hydrogen gas. In the first PEC developed by Honda and Fujishima5, the semiconductor consisted of TiO2 and the cathode of a Pt-electrode. When the photoanode absorbs a photon an electron is excited from the valence band to the conduction band, from where it migrates through the bulk of the semiconductor, via an external circuit. The positive hole formed in the valence band of the photoanode will subsequently be filled with an electron stemming from the oxidation of water. The produced protons migrate through the membrane to the cathode where they are re-duced into molecular hydrogen. As mention above, these systems have been extensively studied, nevertheless, the solar-to-hydrogen conversion efficien-cies for the developed PECs are not sufficient enough to be cost-effective and commercialized.6 The highest solar-to-hydrogen efficiency for n-type oxide-based PEC reported to date reaches 7.1%.7 The yearly average supply of en-ergy from the sun that reaches earth is about 3.0 x 1024 J, which is around 10 000 times higher than the average global energy consumption. Therefore, covering 0.1% of earth’s surface with solar cells with an efficiency of 10% would supply enough energy for the current demand.6
Water splitting is a thermodynamically uphill reaction. The light-driven water splitting requires therefore an efficient and stable water oxidation catalyst (WOC) that supplies the semiconductor with electrons. The WOC has to be capable of donating four electrons in order to oxidize two water molecules into molecular oxygen. The two-half reactions, i.e. the oxidation of water and reduction of protons, with the corresponding redox potentials are summa-rized in Equations 1.2–1.4.8

5
2 H2O O2 + 4 H+ (1.2)
E° = 1.23 – (0.059 pH) V vs. NHE 4 H+ + 4 e- 2 H2
(1.3) E° = 0.0 – (0.059 pH) V vs. NHE 2 H2O O2 + 2 H2 (1.4) E° = 1.23 V vs. NHE A difficulty with semiconductor that are based on inorganic oxides is the modification of their band levels, whereas the tuning of highest occupied mo-lecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) of metal complexes with organic ligand frameworks can be done by modifying the molecular structure. By employing organic metal complexes as WOCs and as photosensitizer (PS)9,15b the energy levels of these can be tuned to match each other in a three–component system for water oxidation that occurs on the anode side in a PEC (Figure 1.4).10 Several transition-metal complexes have been developed that are able to catalyze water oxidation.
PS WOC
2 H2O
O2 + 4 H+
2 H2
hv
e-e-
Figure 1.4. General depiction of a photoelectrochemical cell (PEC). A water oxidizing catalyst (WOC) abstracts electrons from water, which are subsequently transferred to the photosensitizer and to the semiconductor. However, these do not currently meet the requirements neither in terms of efficiency nor stability for light-driven water oxidation.10 In the development

6
of WOCs the evaluation of the catalysts can be eased by testing these for water oxidation in a less complex two-component system. In the two-com-ponent system the photosensitizer is replaced with a sacrificial oxidant, thus light can be omitted from the measurements. The oxidant abstracts electrons from the WOC, after which the WOC reaches a high-valent oxidation state that catalyzes water oxidation. This two-component system is referred to as chemical oxidation. A commonly employed oxidant is cerium(IV) ammonium nitrate (CAN, Ce(IV)), which is a strong one-electron oxidant (E1/2 = 1.6 V vs. NHE).16 An advantage with CAN is that is commercially available and rela-tively stable in acidic aqueous solution (ca pH 1). A drawback with CAN is that the conditions are quite different from the light-driven water oxidation with photosensitizers. Furthermore, upon reduction to Ce(III) cerium oxide is formed, which could, potentially, interfere with the catalysis.11 Another oxi-dant, less commonly used, is the milder one-electron oxidant tris(2,2’-bipyr-idine)ruthenium(III) complex, [Ru(bpy)3]3+ (E1/2 = 1.26 V vs. NHE; bpy = 2,2’-bipyridine),12 which is stable at relatively low pH (pH < 4) and is well charac-terized.13,14,15 Drawbacks with the [Ru(bpy)3]3+-type oxidants is that these are sensitive to decomposition and quite expensive. The benefit however, is that the conditions for the chemically driven water oxidation with [Ru(bpy)3]3+ are similar to the light-driven three-component system with [Ru(bpy)3]2+-type photosensitizers that are frequently used for water oxidation.
1.3 Molecular Water Oxidation Complexes – Chemical Oxidation 1.3.1 Ruthenium Complexes The dinuclear ruthenium complex cis,cis-[(bpy)2(H2O)RuORu(H2O)(bpy)2]4+ (1) “blue dimer”, was the first molecular catalyst reported for oxidation of water (Figure 1.5).17 The complex was developed by Meyer and co-workers in the early 1980s and has been extensively studied ever since. The catalytic reaction for water oxidation were driven electrochemically and chemically, using cerium(IV) ammonium nitrate (Ce(IV)) (E1/2 = 1.6 V vs. NHE),16 as the sacrificial oxidant.17 Chemically-driven water oxidation using Ce(IV) resulted in a turnover number (TON) of 13.218 and a turnover frequency (TOF) of 4.2 x 10–3 s–1.19 Cyclic voltammetry (CV) of ruthenium complex 1 displayed sev-eral redox waves with pH dependence for various oxidation states, which is
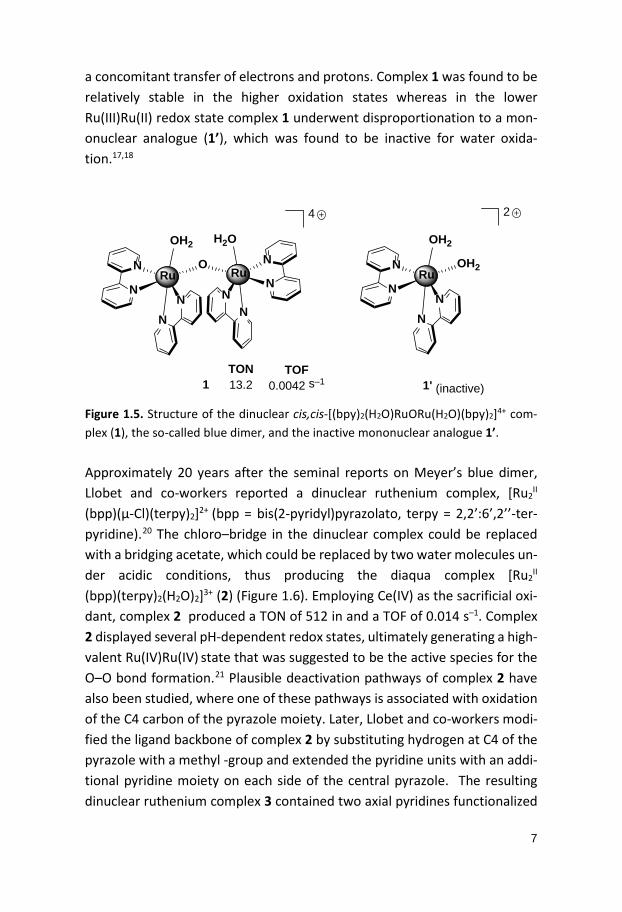
7
a concomitant transfer of electrons and protons. Complex 1 was found to be relatively stable in the higher oxidation states whereas in the lower Ru(III)Ru(II) redox state complex 1 underwent disproportionation to a mon-onuclear analogue (1’), which was found to be inactive for water oxida-tion.17,18
N
N N
N
NN
NN
Ru Ru
OH2 H2O
O
1
4
N
NN
N
Ru
OH2
OH2
1' (inactive)
2
TON13.2
TOF0.0042 s–1
Figure 1.5. Structure of the dinuclear cis,cis-[(bpy)2(H2O)RuORu(H2O)(bpy)2]4+ com-plex (1), the so-called blue dimer, and the inactive mononuclear analogue 1’. Approximately 20 years after the seminal reports on Meyer’s blue dimer, Llobet and co-workers reported a dinuclear ruthenium complex, [Ru2
II (bpp)(µ-Cl)(terpy)2]2+ (bpp = bis(2-pyridyl)pyrazolato, terpy = 2,2’:6’,2’’-ter-pyridine).20 The chloro–bridge in the dinuclear complex could be replaced with a bridging acetate, which could be replaced by two water molecules un-der acidic conditions, thus producing the diaqua complex [Ru2
II (bpp)(terpy)2(H2O)2]3+ (2) (Figure 1.6). Employing Ce(IV) as the sacrificial oxi-dant, complex 2 produced a TON of 512 in and a TOF of 0.014 s–1. Complex 2 displayed several pH-dependent redox states, ultimately generating a high-valent Ru(IV)Ru(IV) state that was suggested to be the active species for the O–O bond formation.21 Plausible deactivation pathways of complex 2 have also been studied, where one of these pathways is associated with oxidation of the C4 carbon of the pyrazole moiety. Later, Llobet and co-workers modi-fied the ligand backbone of complex 2 by substituting hydrogen at C4 of the pyrazole with a methyl -group and extended the pyridine units with an addi-tional pyridine moiety on each side of the central pyrazole. The resulting dinuclear ruthenium complex 3 contained two axial pyridines functionalized
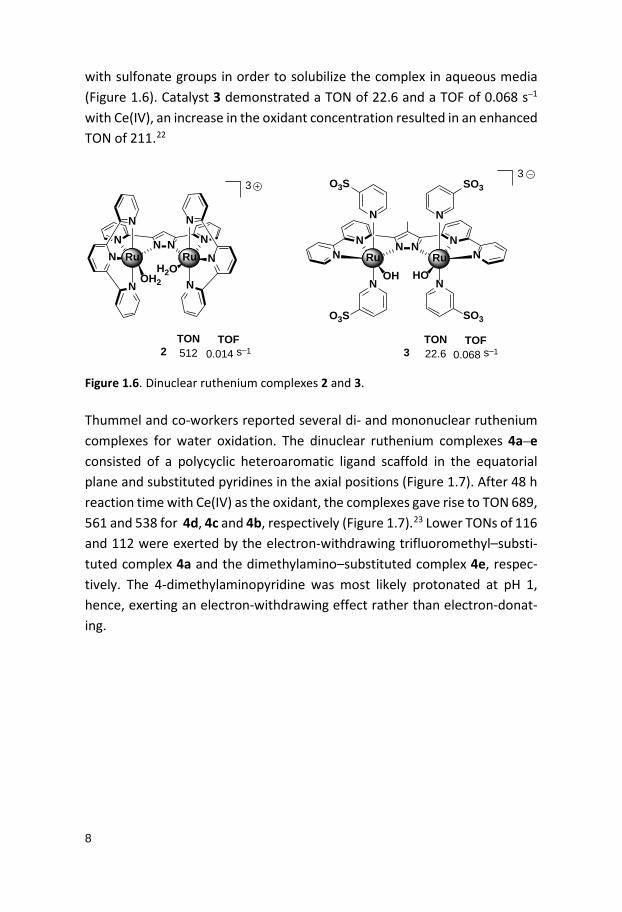
8
with sulfonate groups in order to solubilize the complex in aqueous media (Figure 1.6). Catalyst 3 demonstrated a TON of 22.6 and a TOF of 0.068 s–1 with Ce(IV), an increase in the oxidant concentration resulted in an enhanced TON of 211.22
NN
N
N
N N NRu
N
N
N
Ru
OH2H2O
3
2
N NN NN NRu Ru
OH HO
N N
NN
O3S SO3
SO3O3S
3
3TON512
TOF0.014 s–1
TON22.6
TOF0.068 s–1
Figure 1.6. Dinuclear ruthenium complexes 2 and 3. Thummel and co-workers reported several di- and mononuclear ruthenium complexes for water oxidation. The dinuclear ruthenium complexes 4a–e consisted of a polycyclic heteroaromatic ligand scaffold in the equatorial plane and substituted pyridines in the axial positions (Figure 1.7). After 48 h reaction time with Ce(IV) as the oxidant, the complexes gave rise to TON 689, 561 and 538 for 4d, 4c and 4b, respectively (Figure 1.7).23 Lower TONs of 116 and 112 were exerted by the electron-withdrawing trifluoromethyl–substi-tuted complex 4a and the dimethylamino–substituted complex 4e, respec-tively. The 4-dimethylaminopyridine was most likely protonated at pH 1, hence, exerting an electron-withdrawing effect rather than electron-donat-ing.

9
N N
N
N
NRu
O
N
tBu N
R
R
H
H
5a: R = CF3
5b: R = CH3
5c: R = NMe2
2
N NN NN N
NNRu Ru
N N
N N
Cl
3
R R
RR
4a: R = CF34b: R = CH34c: R = t-Bu4d: R = OMe4e: R = NMe2
TON116538561689112
TON7058050
Figure 1.7. Polypyridyl-based ruthenium complexes 4 and 5a–c and the TON. Three mononuclear ruthenium complexes 5a–c, with -CF3, -CH3 or -N(CH3)2 groups on the axial pyridines were also compared (Figure 1.7). The highest TON of 580 was obtained for complex 5b containing 4-methylpyridine whereas complexes 5a and 5c gave TONs of 70 and 50, respectively. Interest-ingly, complex 5c was shown to have the highest first-order kinetics with a rate constant (k) of 7.8 x 10–4 s–1, which is more than twice as high as for complexes 5a and 5b (k5a = k5b = 3.5 x 10–4 s–1).24 Subsequent work by Åkermark, Sun and co-workers, reported in 2010, in-volved the design of a ligand with a central pyridazine moiety with peripheral pyridines, functionalized with carboxylate groups.26 Previous studies had shown that incorporation of negatively charged ligands, such as carboxylate groups, can lower the redox potential of the metal complexes.24,25 Surpris-ingly a dinuclear ruthenium complex 6 (Figure 1.8) with an “anti” structure, i.e. having the two ruthenium centers residing on opposite sides of the cen-tral pyridazine, was formed (Figure 1.8).26 Despite the “anti” structure, the complex gave rise to a TON of 4700 after 20 h and and a TOF of 0.28 s–1 using Ce(IV) oxidant. By designing a novel ligand with a central phtalazine moiety the cis dinuclear complex 7 could be synthesized (Figure 1.8).27 Complex 7, produced a TON 10400 and TOFs 1.2 s–1, under identical reaction conditions as complex 6.

10
N NN NOO O ORu Ru
N N
N N
Cl
7
N NNN
OO
OO
Ru
Ru
N
NN
N
N
N
6TON4700
TOF0.28 s–1
TON10400
TOF1.2 s–1
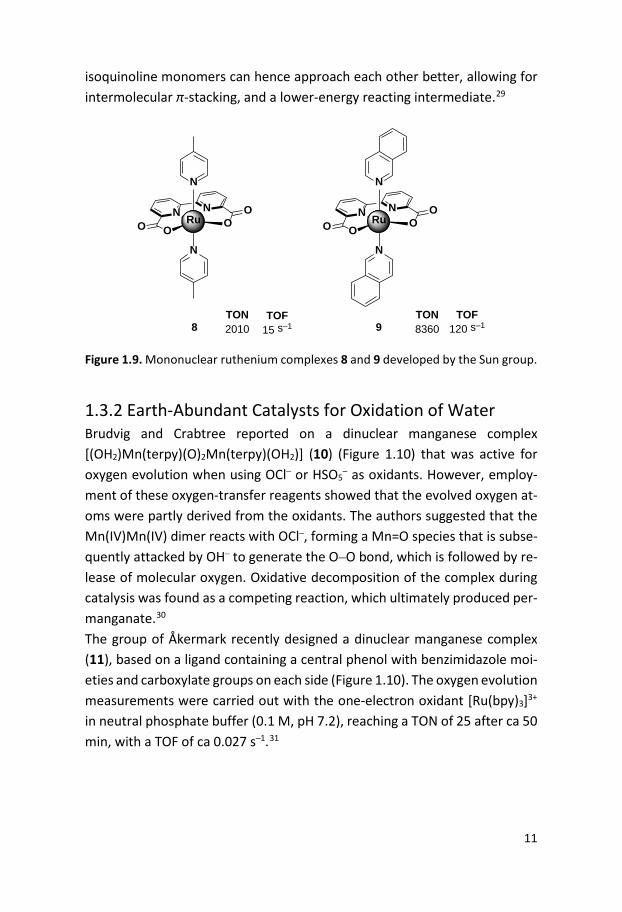
Figure 1.8. Ruthenium complexes 6 and 7 containing carboxylate groups. In 2009, a mononuclear ruthenium complex, [Ru(bda)(pic)2] (bda = 2,2’–bi-pyridine-6,6’–dicarboxylic acid) (8, Figure 1.9), was reported by the Sun group.28 Complex 8 has a distorted octahedral structure with a relatively large O-Ru-O angle of ca 123°. This large angle served as an open coordina-tion site, allowing coordination of water to the complex at the Ru(IV) state without a ligand exchange process. A rare seven-coordinate dimeric complex µ–(HOHOH) (Ru(IV)(bda)(pic)2)2(PF6)3 could also be isolated, which sup-ported that water coordination did not require ligand exchange reactions. The catalytic performance of the complex for water oxidation was tested chemically, using Ce(IV). Complex 8 gave rise to a TON of 2010 with a TOF of 15 s–1 and displayed a second order rate dependence on catalyst concentra-tion.28 A few years later the Sun group reported a mononuclear ruthenium complex (9, Figure 1.9) based on the same equatorial bda ligand but with axial isoquinolines instead of picolines.29 This complex catalyzed water oxi-dation under the same conditions as the previously reported complex 8, giv-ing rise to an impressive TOF of 120 s–1 and a TON of 8360. The kinetic meas-urements revealed a second order dependence on catalyst concentration and electrochemical studies suggested that O–O bond formation occurs at the Ru(V) state. Mechanistic studies revealed a binuclear Ru(IV)O–ORu(IV) peroxo intermediate responsible for the oxygen evolution. Computational studies suggested that the bridging O–O distance in the picoline complex are longer than for the isoquionline complex 9, due to sterical clash of the methyl groups in picoline, which results in a higher-energy reacting complex. The
11
isoquinoline monomers can hence approach each other better, allowing for intermolecular π-stacking, and a lower-energy reacting intermediate.29
N N
OO OO
Ru
N
N
8
N N
OO OO
Ru
N
N
9TON2010
TOF15 s–1
TON8360
TOF120 s–1
Figure 1.9. Mononuclear ruthenium complexes 8 and 9 developed by the Sun group.
1.3.2 Earth-Abundant Catalysts for Oxidation of Water Brudvig and Crabtree reported on a dinuclear manganese complex [(OH2)Mn(terpy)(O)2Mn(terpy)(OH2)] (10) (Figure 1.10) that was active for oxygen evolution when using OCl– or HSO5
– as oxidants. However, employ-ment of these oxygen-transfer reagents showed that the evolved oxygen at-oms were partly derived from the oxidants. The authors suggested that the Mn(IV)Mn(IV) dimer reacts with OCl–, forming a Mn=O species that is subse-quently attacked by OH– to generate the O–O bond, which is followed by re-lease of molecular oxygen. Oxidative decomposition of the complex during catalysis was found as a competing reaction, which ultimately produced per-manganate.30 The group of Åkermark recently designed a dinuclear manganese complex (11), based on a ligand containing a central phenol with benzimidazole moi-eties and carboxylate groups on each side (Figure 1.10). The oxygen evolution measurements were carried out with the one-electron oxidant [Ru(bpy)3]3+ in neutral phosphate buffer (0.1 M, pH 7.2), reaching a TON of 25 after ca 50 min, with a TOF of ca 0.027 s–1.31

12
ONNH
NH
N
O OO OMn Mn
OOO
11
N
N
N
N
N
N
Mn MnO
O
H2O
OH2
3
10TON25
TOF0.027 s–1
TON4
TOF–
Figure 1.10. The dinuclear manganese complexes 10 and 11. Iron complexes have also been applied for water oxidation. However, these have often been reported to be unstable in acidic or basic aqueous solutions, and prone to ligand dissociation. When employing Ce(IV) as oxidant the lig-ands have shown to undergo oxidative decomposition, whereas in basic so-lution the formation of iron hydroxide nanoparticles have been reported which are potential catalysts for water oxidation.32 Several mononuclear iron complexes have been reported to catalyze water oxidation. In 2011 the groups of Costas and Fillol developed a series of active iron based complexes for oxidation of water.33 Two of the most active com-plexes, [Fe(mep)(OTf)2] (12; mep = N,N’-dimethyl-N,N’-bis-(2-pyridylmethyl)-ethane-1,2-diamine) and [Fe(mcp)(OTf)2] (13; mcp = N,N’-dimethyl-N,N’-bis-(2-pyridyl-methyl)-cyclohexane-1,2-diamine), contain neutral tetradentate ligands and have coordinating triflates in a cis fashion, which allows for easy access to open coordination sites for water (Figure 1.11). These complexes exhibited the highest activity among the reported iron-based WOCs with TONs of 360 and 145 within three hours and initial TOFs of 0.23 and 0.14 s–1 for 13 and 12, respectively, when driven by Ce(IV).33
NN Fe
N
OTfOTf
12
N
NN Fe
N
OTfOTf
13
N
TON145
TOF0.14 s–1
TON360
TOF0.23 s–1
Figure 1.11. Molecular structures of iron complexes 12 and 13.

13
In 2014, the cobalt salen complex 14 was reported by Ma and co-workers (Figure 1.12). The complex exhibited a TON of 194 and a TOF of 2.0 s–1 after 3 min when applying [Ru(bpy)3]3+ as an oxidant in borate buffer at pH 9.0. The stability of the complex was also examined and suggested that nanopar-ticles were formed within the detection limit of dynamic light scattering (DLS), supporting that the complex decomposes under the investigated reac-tion conditions. Further analysis of the reaction solution revealed formation of CO2 and NOx gases and 1H NMR studies of the reaction mixture disclosed that the complex decomposed completely.34
N N
O OCo
14TON194
TOF2.0 s–1
Figure 1.12. Cobalt salen complex 14.
1.4 Light-Driven Water Oxidation Several WOCs have been evaluated for light-driven water oxidation employ-ing a three–component system consisting of catalyst, photosensitizer and a sacrificial electron acceptor, such as persulfate (S2O8
2–). [Ru(bpy)3]2+-type complexes (15a–d, Figure 1.13) are commonly used as photosensitizers since these are relatively stable photochemically, water soluble and have been well-studied for their photophysical properties. The excited state lifetimes for these [Ru(bpy)3]2+ photosensitizers are long enough to enable bimolecu-lar reactions, such as electron transfer to an acceptor via an oxidative quenching mechanism.35

14
NN
N
NNN
Ru
2R1
R1
R1
R1
R2
R2
15a: R1, R2 = H15b: R1 = H, R2 = CO2Et15c: R1 = CO2Et, R2 = H15d: R1, R2 = Me
E (RuIII/RuII) = 1.26 V
E (RuIII/RuII) = 1.40 V
E (RuIII/RuII) = 1.54 V
E (RuIII/RuII) = 1.10 V
15a-d
Figure 1.13. Structure of [Ru(bpy)3]2+-type photosensitizers (15a–d) typically em-ployed in light-driven water oxidation.40,41 The absorption of visible light at 452 nm by these photosensitizers results in the generation of a high-energy spin-allowed singlet [Ru(bpy)3]2+* excited state, which is assigned as a metal-to-ligand charge transfer (MLCT).37 The initially produced singlet excited state can undergo intersystem crossing (ISC) to give a lower energy long-lived exited triplet state (τ = 600 ns), which can undergo oxidation to [Ru(bpy)3]3+ (Equation 1.5) in presence of an electron acceptor (Figure 1.14), such as persulfate. The persulfate is a sacrificial elec-tron acceptor that is decomposed to sulfate and a sulfate radical (Equation 1.6), which has a relatively high potential (E = 2.4 V vs. NHE36) and can directly oxidize [Ru(bpy)3]2+ to produce a second equivalent of [Ru(bpy)3]3+ (Equation 1.7). The [Ru(bpy)3]2+/S2O8
2– system has been extensively studied and em-ployed in light-driven water oxidation as the electron transfer from [Ru(bpy)3]2+* to S2O8
2– has shown to be irreversible.37

15
PSII
PS* PSIII
hv
(MLCT)
S2O82- SO4
2- + SO4
WOCOx
WOCRed
E1/2 RuIII/RuII* = -0.81 V E1/2
RuIII/RuII = 1.26 V
Figure 1.14. Three-component light-driven water oxidation consisting of [Ru(bpy)3]2+ as photosensitizer, S2O82– as sacrificial electron acceptor and a WOC.37b [Ru(bpy)3]2+ + hv [Ru(bpy)3]2+* (1.5) [Ru(bpy)3]2+* + S2O8
2– [Ru(bpy)3]3+ + SO42– + SO4
•– (1.6) [Ru(bpy)3]2+ + SO4
•– [Ru(bpy)3]3+ + SO42– (1.7)
1.4.1 Ruthenium Complexes for Light-Driven Water Oxidation Shortly after that the “blue dimer”, [(bpy)2(H2O)RuORu(OH2)(bpy)2]4+ (1), was reported as an active catalyst for chemical water oxidation by Meyer and co-workers, the two dicarboxylic acid analogues 16a,b (Figure 1.15) were ap-plied in light-driven water oxidation, employing [Ru(bpy)3]2+-type photosen-sitizers.38,39 The activity of complex 16a was tested in a system with the strongly oxidizing [Ru(deeb)3]2+ (deeb = 4,4’-diethylester-2,2’-bipyridine) (15c) photosensitizer and S2O8
2– as the acceptor in neutral phosphate buffer. In the presence of complex 16a, oxygen evolution could be observed with a TOF of approximately 0.0027 s–1 within 15 min. According to spectrophoto-

16
metric results complex 16a stayed intact during the reaction, indicating sta-bility under the abovementioned conditions.38 Regarding the analogue 16b, a total amount of 10 µmol of oxygen was produced after 2 h with a TON of 50 in sodium bicarbonate buffer at pH 9.15, using the [Ru(bpy)3]2+ photosen-sitizer (15a). Oxygen evolution was immediately triggered upon illumination, resulting in a TOF of 0.014 s–1.39
N
N N
N
NN
NN
Ru Ru
OH2 H2O
O
16a: R = COOH
4
R
R
R R
R
R
R R
N
N N
N
NN
NN
Ru Ru
OH2 H2O
O
4
R
R
R
R
R
RR
R
16b: R = COOHTON2.43
TOF0.0027 s–1
TON50
TOF0.014 s–1
Figure 1.15. The “blue dimer” derivatives 16a and 16b. The previously mentioned dinuclear ruthenium complex 6 (Figure 1.8), from Sun and Åkermark groups, has also been evaluated in light-driven water oxi-dation using Na2S2O8 as the electron acceptor.40 Relatively high TONs and TOFs were obtained with the more strongly oxidizing photosensitizers 15b (TON = 245, TOF = 0.22 s–1) and 15c (TON = 370, TOF = 0.26 s–1) than with the mild photosensitizer 15a (TON = 185, TOF = 0.11 s–1). A decrease in pH was noted after the catalytic runs. Adjusting the pH to 7.2 and addition of new photosensitizer resulted in continued oxygen evolution, indicating that the studied WOC was still active.40 The dinuclear ruthenium complex 7 with a cis-conformation (Figure 1.8) was shown to mediate light-driven water oxidation with a TON of 580 and a TOF of 0.83 s–1 with the strongly oxidizing photosen-sitizer [Ru(deeb)3]2+ (15c).40 Sun and co-workers reported that the mononuclear ruthenium complex 8 (Figure 1.9) was an active catalyst for light-driven water oxidation when em-ploying either [Co(NH3)5Cl]2+ or S2O8
2– as sacrificial acceptors with photosen-sitizer 15a in phosphate buffer.41 In the case of S2O8
2–, a TOF of >0.35 s–1 was obtained, and was higher than for [Co(NH3)5Cl]2+ for which a TOF of 0.15 s–1 was measured. A plausible cause for this difference in activity could be that

17
the electron transfer to S2O82– is faster.41 Substituting 15a with the stronger
photosensitizer 15b, and phosphate buffer to borate buffer, while employing S2O8
2- as an acceptor resulted in a TON of 251 and 232, and a TOF of 0.33 and 0.32 s–1 for 8 and 9, respectively.42 Llobet and co-workers reported that pyrazolate-based dinuclear ruthenium complexes 17 (Figure 1.16) and 3 (Figure 1.6) were also capable of mediating light-driven water oxidation.43 Their ligand design furnished water soluble and oxidatively stable complexes, where the catalysts reach high TON and TOF for light-driven water oxidation. They have implemented carboxylated and sulfonated pyridine ligands as spectator ligands for complex 17 and 3, respectively, making the complexes fully soluble in aqueous media. Carbox-ylate functionalized complexes can further be heterogenized and applied onto photoanodes, which is important for further development of devices for overall water splitting.43
N NN NN NRu Ru
OH2 H2O
N N
NN
17
COO COO
COOCOO
TON2362
TOF9.2 s–1
Figure 1.16. Dinuclear ruthenium complex 17.
The catalytic activity for light-driven water oxidation was measured in phos-phate buffer at pH 7, using the [Ru(bpy)(deeb)2]2+ (15b) (E = 1.40 V vs. NHE) photosensitizer together with sodium persulfate as the sacrificial electron ac-ceptor. Complex 3 gave rise to a TON of 2373 and a TOF of 11.1 s–1 in the first run. The experiments were repeated two times, resulting in a total TON of 5300. Complex 17 exhibited similar activity, of the same magnitude, a TON of 2362 and a TOF of 9.2 s–1. The activity of 3 and 17 were also compared with the previously reported complexes 2 and 8 (Figure 1.6 and 1.9) under the same conditions. Catalysts 2 and 8 yielded TONs of 67 and 405 and TOFs

18
of 0.13 and 0.36 s–1, respectively. An explanation for the lower activity of 2 is presumably the higher onset potential for water oxidation, occurring at 1.60 V vs. NHE, which is close to the redox potential of the photosensitizer. A rea-son for the lower activity of complex 8 could be the different mechanisms of action; the O–O bond formation occurs via a water nucleophilic attack for 3, whereas an intermolecular coupling of two Ru–O species takes place for cat-alyst 8. The lower concentrations thus decrease the reaction rate when the rate-determining step is bimolecular. Another important factor that can af-fect the oxygen evolution is the kinetics of the electron transfer from the WOC to the photosensitizer.43,44 The mononuclear polypyridyl ruthenium complex 5b (Figure 1.7) with a naphtyridyl pyridine ligand framework, developed by Thummel, has also been shown to catalyze light-driven water oxidation.45 Using [Ru(bpy)3]2+ (15a) as photosensitizer and S2O8
2– as sacrificial electron acceptor produced a TON of 83 and a TOF of 0.12 s–1 in phosphate buffer at pH 7. Increasing the pH of the buffer to 7.8 resulted in a slightly higher TON of 103. The catalytic activity was tested at different catalyst concentrations and showed a linear relationship, indicating first-order behavior for catalyst 5b.45
Table 1.1. TON and TOF for complexes for light-driven water oxidation.
Complex TON TOF (s–1) Photosensitizer 2 67 0.16 15 b 3 2373 11.1 15b
5b 83 0.12 15a 6 185 0.11 15a 6 245 0.22 15b 6 370 0.26 15c 7 580 0.83 15c 8 251 0.33 15b 9 332 0.32 15b
16a 2.43 0.0027 15c 16b 50 0.014 15a 17 2362 9.2 15b

19
1.5 Mechanistic Pathways for the O–O Bond Formation Oxidizing water to molecular oxygen, as mentioned earlier, requires the re-moval of four electrons, i.e. the catalyst has to take part of four-electron transfer processes for each molecule of oxygen that is formed. Insight into O–O bond formation scenarios is vital in the development of efficient and stable catalysts. Various reaction pathways for different transition-metal complexes have been postulated throughout the years. Mechanistic studies on ruthenium complexes disclose two main classes of reaction pathways, one that involves a water nucleophilic attack (WNA) on a metal-oxo (M–O) unit and another pathway that involves the interaction of two M–O units (I2M) (Scheme 1.1).46 As mentioned earlier, water oxidation involves removal of electrons and protons, either consecutively or concerted. Through a con-certed proton-coupled electron transfer (PCET) pathway, high-energy inter-mediates can be bypassed. The ability of a ligand to participate in hydrogen bonding with water can affect the energies and stabilize the intermediates along the reaction pathway. The ligand coordination sphere around the metal, its geometry, its flexibility/rigidity, the electron-donating ability and the electron transfers within the complex can dramatically affect the reaction pathway. The reaction steps for the WNA and I2M can have variations, de-pending on the properties of the complex, yet there are some common fea-tures for each mechanistic route, that are described below in Sections 1.5.1 and 1.5.2.46
1.5.1 Water Nucleophilic Attack (WNA) In the WNA mechanism (Scheme 1.1), a high oxidation state of the metal, such as Ru(IV)-oxo or Ru(V)-oxo species are obtained through several con-secutive oxidation events, thus making the metal-oxo unit more electrophilic. Upon nucleophilic attack by water on the high-valent metal-oxo unit a metal hydroperoxide (Mn–OOH) species is formed. An additional oxidation and deprotonation step subsequently generates a Mn+1–OO– intermediate, which is accompanied with release of O2.46

20
1.5.2 Interaction of Two Metal Units (I2M) Two metal-oxo units can undergo both intra- and intermolecular coupling, where intramolecular coupling is only possible for multinuclear complexes (Scheme 1.1). The I2M mechanism involves the coupling of two metal-oxo units, i.e. coupling of two M–O oxyl radicals or coupling of one M–O oxyl rad-ical with another M–O unit of non-radical character to afford a peroxo M–O–O–M intermediate with subsequent release of O2.46
The rate-determining step has often been ascribed to either the oxygen atom transfer to generate M–O–O or M–O–O–M, or the release of O2.46
M O
MO
OHH
M OMO
H+
M OHOWNA mechanism
I2M mechanism OM
Scheme 1.1. General mechanistic pathways for O–O bond formation.
1.6 Aim of Thesis The aim of this thesis has been to develop mono- and dinuclear ruthenium catalysts and study their ability to oxidize water. An aspiration has been to design ligands and establish a possible correlation between structure, activ-ity and mechanistic pathways. The main ambition has been to study the ca-talysis under relatively mild conditions, i.e. performing oxygen evolution re-actions in aqueous phosphate buffer solutions at neutral pH and with fairly mild oxidants ([Ru(bpy)3]3+-type oxidants), which have relatively low redox potentials and can be generated photochemically.

21
2. Mononuclear Ruthenium Complexes for Water Oxidation (Paper I)
2.1 Introduction Imidazole, carboxylate, and in particular phenol groups have been reported to be efficient proton and electron transfer mediators.47 These functional groups have important functions in natural systems and have been reported to facilitate proton-coupled electron transfers (PCET) reactions, i.e. simulta-neous transfer of both a proton and electron from a substrate to an acceptor. PCETs are prevalent processes in nature as well as in artificial systems, for which several compounds have been reported to undergo PCET pathways. By transferring both electrons and protons, charge accumulation can be avoided and thereby surpassing high-energy intermediates. PCET is a collo-quial term that does not disclose the mechanism of the transfers, whether the transfers of electrons and protons are concerted or stepwise. Overall, PCET is of fundamental importance in water oxidation reactions, to avoid charge build-up.47 Therefore a ligand design with units that could be involved in these processes can play an important role for the catalytic performance. We therefore envisioned that complexes 18 and 19 (Figure 2.1), which are based on ligands 20 and 21 (Scheme 2.1) containing carboxylate, imidazole and phenols would be interesting as WOCs. Furthermore, negatively charged groups in the coordination sphere of a ruthenium center have been reported to lower the redox potentials in comparison to ruthenium complexes based on neutral ligands.48 The sacrificial oxidant must have a sufficient redox po-tential to oxidize the WOC. Hence, lowering the redox potential of a catalyst enables the application of an oxidant with a lower redox potential than the commonly used Ce(IV) (E1/2 = 1.6 V vs. NHE)16 such as [Ru(bpy)3]3+ (E1/2 = 1.26 V vs. NHE),41 to allow for light-driven water oxidation. An advantage regard-ing the synthesis is that the ligands can be easily accessed from commercially available starting materials in one step.

22
L
NN
O
ORu
L
LO
18
NNO
ORuLL
L
19
L =N
Figure 2.1. Structure of ruthenium complexes 18 and 19.
2.3 Results and Discussion 2.3.1 Synthesis and Characterization A facile one-step procedure, starting from commercially available 2-amino-3-nitrobenzoic acid (22) and 2-amino-3-nitrophenol (23) and salicylaldehyde (24), provided the desired ligands 20 and 21 in good yields (Scheme 2.1). An aqueous ethanolic solution with sodium dithionite as an electron donor per-mits a reductive cyclization of the nitro-group and subsequent formation of the benzimidazole units.49 The corresponding ruthenium complexes were synthesized by refluxing a MeOH solution containing ligand 20 or 21 and Ru(DMSO)4Cl2 with subsequent addition of 4-picoline, and afforded com-plexes 18 and 19 in 48% and 72% yield, respectively. Analysis of the com-plexes by 1H NMR exhibited spectra that are typical for paramagnetic species. Addition of ascorbic acid resulted in relatively well-resolved signals, which support the structures of complexes 18 and 19. The Ru(III) oxidation state of the complexes was further supported by solid-state electron paramagnetic resonance (EPR) at 298 K with g-values of 2.18 and 2.14 for complexes 18 and 19, respectively. These values agree well with a low spin d5 ruthenium complex.50

23
L
NN
O
ORu
L
LO
18
NNO
ORuLL
L
19
COOHNH2
NO2
OH
H
OHN
N
HO
HO
O
HNNHO
HO
21
20
OH
H
OOHNH2
NO2
Na2S2O4
EtOH/H2O 1:1reflux, 24 h80%
Na2S2O4
EtOH/H2O 1:1reflux, 24 h64%
22 24
23 24
1. Ru(DMSO)4Cl2Et3N, MeOHreflux, 24 h
2. 4-picoline (L)reflux, 48 h48%
1. Ru(DMSO)4Cl2Et3N, MeOHreflux, 24 h
2. 4-picoline (L)reflux, 48 h72%
+
+
Scheme 2.1. Synthetic routes to ligands 20 and 21 and the corresponding ruthenium complexes 18 and 19.
2.3.2 Electrochemical Studies Electrochemical studies were performed in neutral phosphate buffer (0.1 M, pH 7.2). Differential pulse voltammetry (DPV) displayed four redox peaks around 0.57, 0.75, 1.17 and 1.40 V vs. NHE for complex 18 whereas complex 19 exhibited only two peaks at 0.60 and 0.80 V vs. NHE (Figure 2.2). Both complexes exhibited an onset potential for water oxidation in the interval of 1.2–1.4 V vs. NHE. A plausible reason for the different number of peaks ob-served for the complexes could be that the structure of the complex 18 changes as it is oxidized within the time frame of the experiment; the struc-

24
tural change could for example involve a picoline-water ligand exchange. An-other reason could be that complex 18 reaches a higher oxidation state than 19 under the applied conditions. Nevertheless, the active catalysts promote electrocatalytic oxidation of water at similar redox potentials as seen from the cyclic voltammograms (Figure 2.3).
Figure 2.2. Differential pulse voltammograms of complexes 18 (––) and 19 (– –) (26 µM) in an aqueous phosphate buffer solution (0.1 M, pH 7.2). The measurements were using a glassy carbon disk as the working electrode and Ag/AgCl as the refer-ence electrode.
Figure 2.3. Cyclic voltammograms of complexes 18 (––) and 19 (– –) (26 µM) in an aqueous phosphate buffer solution (0.1 M, pH 7.2). The background is the aqueous phosphate buffer solution (––). The measurements were recorded at a scan rate of 0.1 V s-1 using a glassy carbon disk as working electrode and Ag/AgCl as reference electrode.
6
8
10
12
14
16
18
0.5 0.7 0.9 1.1 1.3
E (V vs. NHE)
I ( µ
A)
7
8
9
10
11
12
13
0.45 0.65 0.85 1.05
E (V vs. NHE)
I ( µ
A)
7
17
27
37
1 1.2 1.4

25
2.3.3 Catalytic Activity The catalytic activity of complexes 18 and 19 was investigated in aqueous phosphate buffer solutions (0.1 M, pH 7.2). At a catalyst concentration of 3.3 µM the complexes gave TONs of 210 and 50 and initial TOFs of 0.77 and 0.30 s–1, respectively, when applying the one-electron oxidant [Ru(bpy)3]3+ (Figure 2.4). Decreasing the catalyst concentration of 18 to 0.33 µM resulted in a TON of 1100 and an initial TOF of 3.4 s–1, whereas complex 19 exhibited a lower TON of 150 and an initial TOF of 0.28 s–1 at the same catalyst concen-tration. The highest amount of evolved oxygen was obtained for complex 18 at a catalyst concentration of 0.033 µM and afforded a TON of ca 4000 within 17 min and an initial TOF of ca 7 s–1. An important aspect was the suitability of the developed complexes to drive water oxidation using visible light. Eval-uation of complexes 18 and 19 to mediate photochemical water oxidation was therefore initially carried out with the milder photosensitizer [Ru(bpy)3]2+(E1/2 (Ru(III)/Ru(II)) = 1.26 V vs. NHE)41 and subsequently with the [Ru(bpy)2(deeb)]2+ (15b) analogue that has a higher oxidation potential (E1/2(Ru(III)/Ru(II)) = 1.40 V vs. NHE).40 These photochemical systems require a sacrificial electron acceptor and sodium persulfate (Na2S2O8) was selected since the [Ru(bpy)3]2+/Na2S2O8 systems have been well studied.37 A TON of 20 was reached after ca 17 min with the mild [Ru(bpy)3]2+ photosensitizer for both complexes 18 and 19 (10 µM) (Figure 2.5) whereas employing the stronger [Ru(bpy)2(deeb)]2+ photosensitizer resulted in a TON of approxi-mately 200 under otherwise identical conditions. To verify that the source of oxygen atoms in the evolved molecular oxygen is derived from water, isotop-ically labelled water (H2
18O) was used for the catalytic reactions. Indeed, the detected amounts of 18,18O2, 18,16O2, and 16,16O2 correlated well with the cal-culated values for both complexes 18 and 19 and support that both oxygen atoms in the evolved oxygen are derived from water (Figure 2.6 and 2.7).

26
Figure 2.4. Chemical water oxidation catalyzed by ruthenium complexes 18 (upper) and 19 (lower). Conditions: Reactions were performed in an aqueous phosphate buffer solution (0.1 M, pH 7.2, 0.50 mL) containing the oxidant [Ru(bpy)3](PF6)3 (5.1 mg, 5.1 µmol) and the catalysts 18 or 19 at the following concentrations: ● = 33 µM, ▲ = 3.3 µM, ♦ = 0.33 µM, ■ = 0.033 µM. Oxygen evolution was measured by mass spectrometry.

27
Figure 2.5. Light-driven water oxidation catalyzed by ruthenium complexes 18 (●) and 19 (▲). Conditions: Catalyst 18 and 19 (10 µM) in aqueous phosphate buffer solutions, [Ru(bpy)2(deeb)](PF6)2 (0.38 mM) and Na2S2O8 (20 mM). Oxygen evolution was measured by mass spectrometry.
Figure 2.6. Light-driven water oxidation catalyzed by ruthenium complex 18 in iso-topically labeled water (H218O). The reaction was carried out in an aqueous phos-phate buffer solution (0.1 M, pH 7.2) together with complex 18 (0.05 mM), [Ru(bpy)2(deeb)](PF6)2 (0.27 mM) and Na2S2O8 (10 mM). ● = 16,16 O2, ▲ = 16,18 O2, ■ = 18,18 O2 and the solid lines are theoretical values. Oxygen evolution was measured by mass spectrometry.

28
Figure 2.7. Light-driven water oxidation catalyzed by ruthenium complex 19 in iso-topically labeled water (H218O). The reaction was carried out in an aqueous phos-phate buffer solution (0.1 M, pH 7.2) together with 19 (0.05 mM), [Ru(bpy)2(deeb)](PF6)2 (0.27 mM) and Na2S2O8 (10 mM). ● = 16,16 O2, ▲ = 16,18 O2, ■ = 18,18 O2 and the solid lines are theoretical values. Oxygen evolution was measured by mass spectrometry.
2.3.4 Ligand Exchange High-resolution mass spectrometry (HRMS) revealed peaks correlating to the protonated species of the complexes [18 + H]+, [19 + H]+ and to the loss of picolines [18 – L + H]+ and [19 – L + H]+. These results suggest that loss of picoline could occur and support a possible ligand change. A computational study on the picoline-water exchange was therefore carried out. The equa-torial position has in previous studies been reported as an accessible position for the exchange.51 According to the computational study, the ligand ex-change reaction of an equatorial picoline for water for complexes 18 and 19 was slightly endothermic by +5 kcal/mol. The ligand exchange of complex 19 was further analyzed by HRMS. Complex 19 was mixed with an excess of the one-electron oxidant [Ru(bpy)3]3+. The resulting reaction mixture revealed a signal in positive mode that corre-sponded to [(HL)(pic)2Ru(V)=O]+ species of 19 (Figure 2.8) providing further support to the picoline-water ligand exchange.

29
Figure 2.8. High-resolution mass spectrum of [(HL)(pic)2Ru(V)=O]+ of complex 19 (left) and proposed structure (right).
2.4 Summary Two novel mononuclear ruthenium complexes 18 and 19 based on anionic ligands have been developed and their performance for catalytic water oxi-dation has been studied. Both complexes displayed onset potentials around 1.2–1.4 V vs. NHE and were able to drive water oxidation chemically or pho-tochemically under relatively mild conditions, using either the oxidant [Ru(bpy)3]3+ or the photosensitizers [Ru(bpy)3]2+ (15a) [Ru(bpy)2(deeb)]2+ (15b) at neutral pH.
NNHO
ORu
L
L
O

30
3. Mononuclear Ruthenium Water Oxidation Catalysts: Mechanistic Insight through Structure-Activity Relationships (Paper II)
3.1 Introduction A fundamental aspiration in any mechanistic study is to examine whether there exists a relationship between the structural change of a molecule and its reactivity. Studying analogues and the changes in reactivity provide valu-able information of how sensitive the reaction is to changes in the catalyst structure. Establishing a linear free-energy relationship (LFER), by finding a correlation between properties of complexes and an equilibrium constant or rate constant of a reaction can give insight to the reaction mechanism. An early example of LFER is the Hammett relationship.52 In the 1930s Hammett studied the acidity of various benzoic acid derivatives and found a correlation between the different substituents in the aromatic ring (R in Scheme 3.1) and their acid–base equilibria. Hammett subsequently defined a scale of the acid-ity and was able to express a relationship between the substituents and the acidity of benzoic acid derivatives where the unsubstituted benzoic acid served as a reference (Scheme 3.1). The scale laid the foundation to a library of various benzoic acid derivatives, where the logarithm of the ratio of an equilibrium constant is a function of the reaction constant (ρ) and substitu-ent constant (σ) (Equation 3.1 and 3.2). The reaction constant (ρ) is a measure of how susceptible an equilibrium is to alterations in electronic effects. The greater the value of ρ the more sus-ceptible the reaction is to electronic perturbation. A positive ρ value suggests that negative charge is built up during the reaction and will therefore be sta-bilized by electron withdrawing groups whereas negative ρ values means that positive charge is built up during the reaction and is therefore favored by electron-donating groups.

31
OH
O
R H2O+ O-
O
R H3O++k1
k2
KR = k1
/ k2 Scheme 3.1. The ionization of benzoic acid with different substituents (R) in either meta or para to the carboxylic acid. The substituent constant, σ, can be expressed as a function of the equilib-rium constants of the substituted benzoic acid (KR) and the unsubstituted benzoic acid (KH).
log �𝐾𝐾𝑅𝑅𝐾𝐾𝐻𝐻� = 𝜎𝜎𝑅𝑅 (3.1)
log �𝐾𝐾𝑅𝑅𝐾𝐾𝐻𝐻� = 𝜌𝜌𝜎𝜎𝑅𝑅 (3.2)
The substituent constant (σ) correlates to the electronic effect exerted by a substituent, reflecting the influence on charge at the reaction center. The position of the substituent in relation to a reaction center can be either para (σpara) or meta (σmeta). The para substituents mainly influence a reaction cen-ter via resonance whereas the predominant electronic effect exerted by meta substituents is essentially inductive. Our group has reported a mononuclear ruthenium complex 18 (as discussed in Paper I) that was active for water oxidation. The subsequent work focused on investigating the substituent effects of several analogues in order to ob-tain a more detailed understanding of the correlation between the structure and catalyst activity. On that account, four analogues 18a–d (Scheme 3.2) with different substituents on the phenol moiety of the ligand were synthe-sized. The catalytic performance of the corresponding ruthenium complexes were examined along with electrochemical studies to assess the redox states of the different species involved in the catalytic cycle. The electrochemical data apprehended were implemented to the investigations of a structure-activity relationship.

32
3.2 Results and Discussion 3.2.1 Synthesis and Characterization Ligands 20a–d were synthesized in one step from commercially available al-dehydes and 2-amino-3-nitrobenzoic acid in fairly good yields (54–70%). The desired complexes were prepared by heating a MeOH solution of the ligands in the presence of [Ru(DMSO)4Cl2] and Et3N to afford the desired ruthenium complexes in 45–60% yield (Scheme 3.2). The complexes were characterized by 1H NMR, HRMS, elementary analysis, UV-vis and IR.
L
NN
O
ORu
L
LO
18a: R = Me (45%)18b: R = H (48%)18c: R = F (49%)18d: R = Br (60%)
HNN
HO
HO
O
20a: R = Me (54%)20b: R = H (80%)20c: R = F (70%20d: R = Br (62%)
1. Ru(DMSO)4Cl2Et3N, MeOHreflux, 24 h
2. 4-picoline (L)reflux, 48 h
R
R
COOHNH2
OH
RNO2
+ H
ONa2S2O4
EtOH/H2O 1:1reflux, 24 h
24 23a: R = Me23b: R = H23c: R = F23d: R = Br
Scheme 3.2. Synthesis of ligands 20a–d and the corresponding mononuclear ruthe-nium complexes 18a–d.
3.2.2 Catalytic Performance The catalytic performance for complexes 18a–d at different concentrations were carried out in aqueous phosphate buffer solutions (0.1 M, pH 7.2) em-ploying [Ru(bpy)3]3+ as a sacrificial one-electron oxidant. Evolution of molec-ular oxygen was instantly triggered upon addition of the oxidant to the cata-lyst solution, resulting in TONs and initial TOFs from 460 to 1700 and from

33
0.86 to 3.1 s–1 for 18a–d (Figure 3.1 and Table 3.2). The non-substituted com-plex 18b exhibited the highest activity with an initial TOF of 3.1 s–1 and high-est TON reaching 1700. The correlation between the initial oxygen evolution rate and catalyst concentration for complexes 18a–d revealed a pseudo first-order dependence, indicating that water oxidation occurs at a single ruthe-nium center.
Figure 3.1. Oxygen evolution during catalytic water oxidation by 18a (upper, left), 18b (upper, right), 18c (lower, left) and 18d (lower, right). The reactions were per-formed in aqueous phosphate buffer (0.1 M, pH 7.2, 0.5 mL) with the oxidant [Ru(bpy)3](PF6)3 (3.0 mg, 3.0 µmol) and the complexes at the following concentra-tions: 4.0 µM (––), 3.3 µM (––) 2.0 µM (––), 0.73 µM (––), 0.33 µM (––) and 0.066 µM (––).

34
Table 3.2 Summary of the catalytic performances of complexes 18a–d. Complex TON (mol O2/molcat)a TOF (mol O2/molcat s–1)a
18a 460 0.86 18b 1700 3.1 18c 1520 2.2 18d 850 1.0
a Reaction conditions: An aqueous phosphate buffer (0.1 M, pH 7.2, 0.5 mL) contain-ing ruthenium complexes 18a–d (0.33 µM) and the oxidant [Ru(bpy)3](PF6)3 (3.0 mg, 3.0 µmol). Aliquots of the reaction mixtures after ca 25 turnovers were analyzed by HRMS, for which neither free ligand nor oxidized ligand species could be de-tected. The corresponding aqua complexes 25a–d were also observed by HRMS, supporting picoline-aqua exchange (Figure 3.2 and 3.3). Furthermore, 1H NMR spectrum of the complexes 18a–d in aqueous media, in the presence of sodium dithionite, supported the ligand exchange and displayed signals corresponding to free picoline. Figure 3.2. High-resolution mass spectroscopy of ruthenium complex 18b in positive mode (top) with the theoretical peak (middle) and suggested structure of aqua com-plex 25b (below).
H2O
NN
O
ORu
L
LO
25b
H

35
Figure 3.3. High-resolution mass spectroscopy of ruthenium complexes 18a,c,d in positive mode (top) with the theoretical peak (middle) and suggested structures of the aqua complexes 25a,c,d (below).
3.2.3 Electrochemical Studies CV and DPV were employed for the electrochemical studies of complexes 18a–d. The measurements were carried out in aqueous phosphate buffer so-lutions (0.1 M, pH 7.2), similar to the catalytic conditions. The first redox events observed by DPV appears around 0.54–0.60 V vs. NHE and are as-signed to the Ru(IV)/Ru(III) couple, with an increasing redox potential as fol-lows: 18d (-Br) < 18c (-F) < 18b (-H) < 18a (-CH3). The same order can be seen for the Ru(V)/Ru(IV) redox species whereas the order of the onset potentials for the catalytic current and the redox potentials of the Ru(VI)/Ru(V) couple were found to be 18a (-CH3) < 18c (-F) < 18b (-H) < 18d (-Br) (Table 3.3).
H2O
NN
O
ORu
L
LO
25a
H2O
NN
O
ORu
L
LO
25c
H2O
NN
O
ORu
L
LO
25d
Me F Br
36
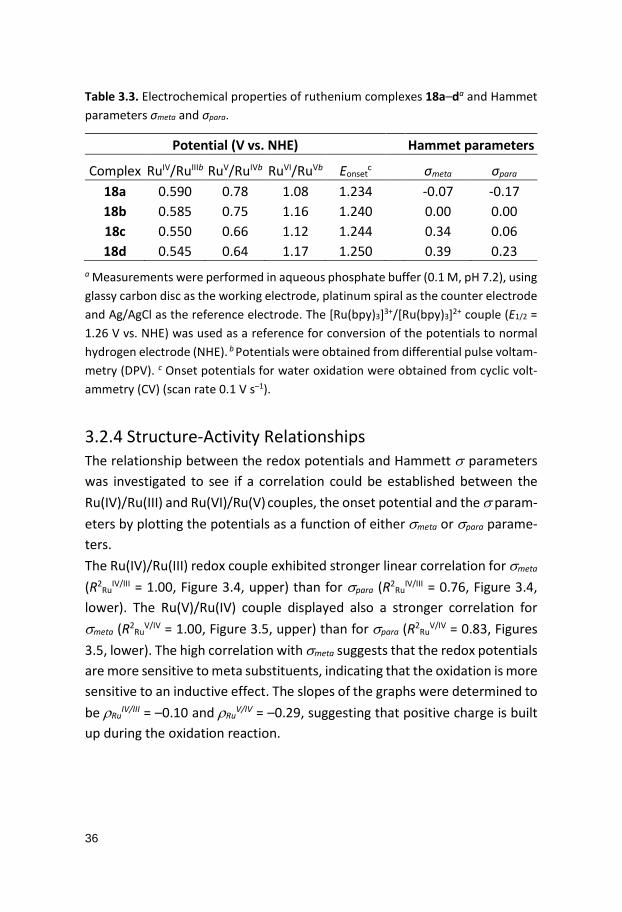
Table 3.3. Electrochemical properties of ruthenium complexes 18a–da and Hammet parameters σmeta and σpara.
Potential (V vs. NHE) Hammet parameters
Complex RuIV/RuIIIb RuV/RuIVb RuVI/RuVb Eonsetc σmeta σpara
18a 0.590 0.78 1.08 1.234 -0.07 -0.17 18b 0.585 0.75 1.16 1.240 0.00 0.00 18c 0.550 0.66 1.12 1.244 0.34 0.06 18d 0.545 0.64 1.17 1.250 0.39 0.23
a Measurements were performed in aqueous phosphate buffer (0.1 M, pH 7.2), using glassy carbon disc as the working electrode, platinum spiral as the counter electrode and Ag/AgCl as the reference electrode. The [Ru(bpy)3]3+/[Ru(bpy)3]2+ couple (E1/2 = 1.26 V vs. NHE) was used as a reference for conversion of the potentials to normal hydrogen electrode (NHE). b Potentials were obtained from differential pulse voltam-metry (DPV). c Onset potentials for water oxidation were obtained from cyclic volt-ammetry (CV) (scan rate 0.1 V s–1).
3.2.4 Structure-Activity Relationships The relationship between the redox potentials and Hammett σ parameters was investigated to see if a correlation could be established between the Ru(IV)/Ru(III) and Ru(VI)/Ru(V) couples, the onset potential and the σ param-eters by plotting the potentials as a function of either σmeta or σpara parame-ters. The Ru(IV)/Ru(III) redox couple exhibited stronger linear correlation for σmeta (R2
RuIV/III = 1.00, Figure 3.4, upper) than for σpara (R2
RuIV/III = 0.76, Figure 3.4,
lower). The Ru(V)/Ru(IV) couple displayed also a stronger correlation for σmeta (R2
RuV/IV = 1.00, Figure 3.5, upper) than for σpara (R2
RuV/IV = 0.83, Figures
3.5, lower). The high correlation with σmeta suggests that the redox potentials are more sensitive to meta substituents, indicating that the oxidation is more sensitive to an inductive effect. The slopes of the graphs were determined to be ρRu
IV/III = –0.10 and ρRuV/IV = –0.29, suggesting that positive charge is built
up during the oxidation reaction.

37
Figure 3.4. The Hammet plots for Ru(IV)/Ru(III) couples as function of σmeta (upper)
and σpara (lower).

38
Figure 3.5. The Hammet plots for Ru(V)/Ru(IV) couples as function of σmeta (upper)
and σpara (lower).
The meta position can affect either one of the nitrogen atoms of the benzim-idazole moiety, hence the substituent can influence either the nitrogen co-ordinated to the metal center or via protonation of the uncoordinated nitro-gen. To investigate the second possibility, i.e. the effect of the substituent on the protonated nitrogen the relationship between pKa values and σmeta was studied. The pKa values for complexes 18a–d were derived from spectropho-tometric titration. The strong correlation between pKa and σmeta (R2 = 0.98) suggests that the protonated nitrogen is sensitive to substituent effects (Fig-ure 3.6). In addition to the stabilization by electron-withdrawing groups of a

39
positive charge build up, an inductive effect facilitates deprotonation, which alone lowers the redox potentials.
Figure 3.6. Hammet plot for the pKa values of complexes 18a–d as a function of σmeta.
The onset potential for the catalytic current, on the other hand, displays a higher linear relationship with σpara (R2 = 0.99) than with σmeta and has a slight positive slope ρonset = 0.04, suggesting that a small negative charge is built up during the reaction (Figure 3.7). However, the initial rate of the oxygen evo-lution and σ-parameters did not display a linear relationship. The scattered values give rise to a positive ρ for complexes 18a and 18b and a change to a negative ρ for complexes 18c and 18d, suggesting that there could be either a change in the rate-determining step (RDS) or a change in mechanism (Fig-ure 3.8).

40
Figure 3.7. Hammet plot for the onset potentials for ruthenium complexes 18a–d as a function of σpara.
Figure 3.8. The initial rate of the oxygen evolution as a function of σmeta (•) and σpara (•). Quantum chemical studies on complex 18b revealed that the displacement of the equatorial picoline to generate 25b is more favorable (by ca 2.0 kcal/mol) than water displacement at the axial position (which would pro-duce 25b’) (Figures 3.9 and 3.10). Deprotonation of the coordinated water in aqua complex 25b was slightly more favored than the deprotonation of the benzimidazole unit. The studies revealed that the oxidation of Ru(III) to Ru(IV) occurs through PCET, and a further investigation of which site is more accessible for deprotonatation — whether it is the aqua or benzimidazole proton — showed that the aqua protons are preferably removed.
0
0,5
1
1,5
2
2,5
3
3,5
-0,3 -0,1 0,1 0,3 0,5
18a
18c18c
18d18a 18d
18b 18b
Initi
al ra
te o
f O2 e
volu
tion
(nm
ol s-1
)
σ

41
H2O
NN
O
ORu
L
LO
25b: R = H
R
L =N
L
NN
O
ORu
OH2
LO
R
25b': R = H
Figure 3.9. Structure of aqua-complexes 25b (equatorial, left) and 25b’ (axial, right).
0.0
10.0
20.0
30.0
0.0
11.714.2
17.019.0Dissociative
pathway
24.027.6Associative
pathway
rela
tive
ener
gy (k
cal m
ol-1
)
Axial picoline exchangeEqutorial picoline exchange
reaction coordinate
Figure 3.10. Potential energy profiles for the picoline–aqua ligand exchange for com-plex 18b, either following an associative or a dissociative pathway at the axial (––) or the equatorial position (––). The Pourbaix diagram of complex 18b was subsequently constructed in the range 1.5 < pH < 7.5. The change, if any, in redox potential in relation to pH can be expressed with the Nernst-equation (Equation 3.3) where m is the number of protons and n the number of electrons, transferred in the redox process. The diagram (Figure 3.11) revealed three one-electron-one-proton coupled events in the interval 2.9 < pH < 6.7, which were assigned to one-electron oxidations of Ru(III) to Ru(VI). Above pH 6.7, the oxidation of Ru(III) to Ru(IV) is associated with the loss of two protons. The following oxidation to Ru(V) is a one-electron-one-proton process, enabling a high-valent Ru(V)

42
complex at relatively low redox potentials (around 0.80 V vs. NHE at neutral pH) and the subsequent oxidation to reach a rare Ru(VI) species occurs at
~1.1 V vs. NHE and is pH independent (Figure 3.11). The involvement of Ru(VI) for complex 18b was further supported by HRMS, which revealed a peak corresponding to a formal Ru(VI)-oxo species (Figure 3.12). The redox potentials as a function of pH were also calculated, revealing that the changes in potential relative to pH for the aqua complex 25b agree fairly well with the experimental values, with slightly higher values for the calculated potentials than the experimentally determined redox potentials.
𝐸𝐸 = 𝐸𝐸° − 𝑚𝑚𝑛𝑛
0.059𝑝𝑝𝑝𝑝 (3.3)
Figure 3.11. Pourbaix diagram of ruthenium complex 18b in the range 1.5 < pH < 7.5. The measurements were performed in Britton-Robinson buffer solution, for which the pH was adjusted with aq. 0.2 M NaOH. The potentials were obtained from dif-ferential pulse voltammetry, using glassy carbondisc as working electrode, platinum spiral as counter electrode and Ag/AgCl as reference electrode. The reference com-pound was [Ru(bpy)3]3+/[Ru(bpy)3]2+ couple (E1/2 = 1.26 V vs. NHE) for conversion of the potentials to vs. NHE.

43
Figure 3.12. High-resolution mass spectrum of [(L)Ru(V)=O]+ (left) of complex 18b with proposed structure (right). The computational calculations on the redox states of complex 18b, the elec-trochemical studies with the constructed Pourbaix diagram and the observa-tion of a formal Ru(VI)-oxo species support the formation of a high-valent Ru(VI) complex, which is proposed to be the key intermediate in O–O bond formation.
3.3 Summary A strong correlation was found between the redox potentials and Hammett parameters for the investigated ruthenium complexes 18a–d. The studies gave a notion of how electronic perturbation may alter the reactivity of WOCs for the Ru(IV)/Ru(III), Ru(V)/Ru(IV) redox potentials, the onset poten-tial for water oxidation and the initial rate of the oxygen evolution. These results give a notion that the electronic effects may influence each step dif-ferently; the separate redox processes of the complexes and the multi-elec-tron reaction during water oxidation and overall imparts how sensitive the catalytic reaction can be to substituent effects.
NN
O
ORu
L
LO
R = H
RO

44
4. A Dinuclear Ruthenium Complex for Chemical and Photochemical Water Oxidation (Paper III and IV)
4.1 Introduction The dinuclear ruthenium complex 26 was reported by our group in 2010.53 The complex was based on a heptadentate ligand, containing phenol, pyri-dine and amine moieties (Figure 4.1). Despite the low redox potentials of complex 26, the complex failed to oxidize water. A hypothesis was that the benzylic amines underwent degradation under oxidative conditions. Succes-sively, ligand 27 was developed by replacing the benzylic amines with imid-azoles (Figure 4.2). However, the anticipated dinuclear complexes was not obtained, instead a dimeric complex 28 with a bridging iodide was observed. It is likely that the coordination geometry of ligand 27 was incompatible with accommodation of two ruthenium ions for a dinuclear complex.54
ON N
Ru
O
Ru
OO
N
O
N
OO
26
Previous work Figure 4.1 Dinuclear ruthenium complex 26 developed by Åkermark and co-workers.

45
With these results in mind ligand 29 with a more open coordination geome-try was designed, consisting of a central pyrazole core with adjacent benzim-idazole moieties (Figure 4.2). As anticipated, a dinuclear ruthenium complex 30 could be obtained and the results are described herein.
N
NN
N
ORu
OH
OH
ON
N NH
N
O
O
Ru
H
H
I
L
LL
L
27
OH
HN
N N
HN
OH HO
Previous work
28
N N N
HNNH
N
OOOO
Ru Ru
N N
N N30
picpic
HN N N
HNNH
N
HOOOHO
29
This work
2
Figure 4.2. Previously synthesized ligand 27 that resulted in the dimeric ruthenium complex 28 (upper) and the development of ligand 29 and the corresponding dinu-clear ruthenium complex 30.
4.2 Results and Discussion 4.2.1 Synthesis and Characterization The synthetic pathway started with the oxidation of the commercially avail-able 3,5-dimethylpyrazole (31) to 3,5-pyrazoledicarboxylic acid (32). The es-terification of the acid to the diethyl 3,5-pyrazoledicarboxylate (33) was fol-lowed by a reduction to the corresponding diol (34). Oxidation of the alcohol units to the bisaldehyde 3555 and a subsequent reductive cyclization with 2-
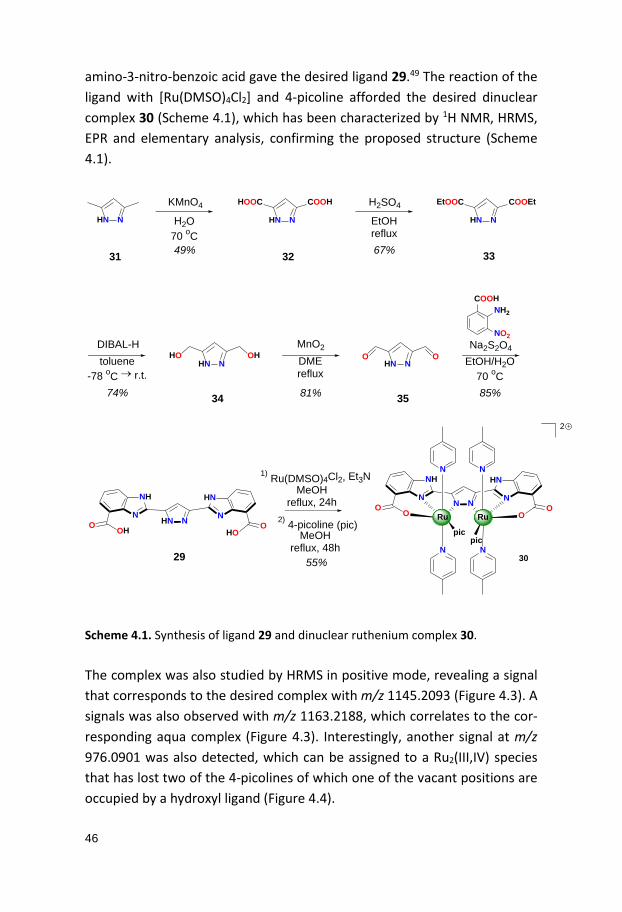
46
amino-3-nitro-benzoic acid gave the desired ligand 29.49 The reaction of the ligand with [Ru(DMSO)4Cl2] and 4-picoline afforded the desired dinuclear complex 30 (Scheme 4.1), which has been characterized by 1H NMR, HRMS, EPR and elementary analysis, confirming the proposed structure (Scheme 4.1).
HN N HN N
HOOC COOH
HN N
EtOOC COOEt
HN NHO OH
HN NO O
KMnO4
H2O70 oC
EtOHreflux
H2SO4
49% 67%31 32 33
DIBAL-Htoluene
-78 oC → r.t.
74%
DMEreflux
81%
MnO2
COOHNH2
NO2Na2S2O4
EtOH/H2O70 oC85%34 35
HN N N
HNNH
N
HOOOHO
1) Ru(DMSO)4Cl2, Et3N
2) 4-picoline (pic)MeOH
reflux, 48h
MeOHreflux, 24h
55%29
N N N
HNNH
N
OOOO
Ru Ru
N N
N N30
picpic
2
Scheme 4.1. Synthesis of ligand 29 and dinuclear ruthenium complex 30. The complex was also studied by HRMS in positive mode, revealing a signal that corresponds to the desired complex with m/z 1145.2093 (Figure 4.3). A signals was also observed with m/z 1163.2188, which correlates to the cor-responding aqua complex (Figure 4.3). Interestingly, another signal at m/z 976.0901 was also detected, which can be assigned to a Ru2(III,IV) species that has lost two of the 4-picolines of which one of the vacant positions are occupied by a hydroxyl ligand (Figure 4.4).

47
Figure 4.3. High-resolution mass spectrum (HRMS) of species of the dinuclear ruthe-nium complex 30 (left) and the proposed structures (right). Measurement was car-ried out in methanol solution.
4.2.2 Quantum Chemical Calculations Computational studies employing density functional theory (DFT) on various species derived from the dinuclear ruthenium complex 30 containing differ-ent numbers of 4-picoline, aqua, hydroxo and oxo ligands at various redox
N N N
NN
N
OOOO
Ru Ru
N N
N N
picpic
Ru2III,III
N N N
NN
N
OOOO
Ru Ru
N N
N N
picpic
Ru2III,III
OH2
N N N
NN
N
OOOO
Ru Ru
N N
N N
Ru2III,IV
OH

48
and spin states were investigated. These studies revealed that the Ru2(III,III) state is accessible at relatively low potentials at pH 7.2 (Table 4.1). The most stable species was found to be the six coordinated picoline complex, [(L)Ru2
III,III(pic)6] (Figures 4.4 and 4.6), hence the energy of this complex was set to zero.
Figure 4.4. Calculated structure of [(L)Ru2III,III (pic)6]+.
Figure 4.5. Calculated structures of dinuclear ruthenium isomers [(L)Ru2III,III(pic)6(OH2)]+ A (left) and B (right). Previous mechanistic study on the mononuclear ruthenium complexes 8 and 9, reported by Sun and co-workers, showed that these complexes operate

49
through seven-coordinated metal centers.56 However, for [(L)Ru2III,III(pic)6]+
seven-coordinating species were not favored. Instead, the pyrazole moiety dissociates upon coordination of water, thus maintaining a six-coordinated configuration. Two isomers of the aqua complex [(L)Ru2
III,III(pic)6(OH2)]+ can co-exist due to similar energies (Figure 4.5). One of the isomers, [(HL)Ru2
III,III(pic)6(OH)]+ (isomer A, Figure 4.5), is protonated at the benzimid-azole nitrogen of the ligand as a hydroxyl-group is coordinating to one of the metal centers. In the other isomer, [(L)Ru2
III,III(pic)6(OH2)]+ (structure B, Figure 4.5), a water molecule is coordinated to the metal, forming a hydrogen bond with the nitrogen of the pyrazole. Isomer A is more stable than isomer B by 0.5 kcal mol–1. The relative energy of isomer A was calculated to be +2.5 kcal mol–1 higher than the reference complex [(L)Ru2
III,III(pic)6]+ (Figure 4.6). A sub-sequent pyrazole coordination to the metal and loss of one picoline results in [(L)Ru2
III,III(pic)5(OH2)]+ and is close to isogonic. A second water can coordi-nate to [(L)Ru2
III,III(pic)6(OH2)]+ in a similar fashion, forming the diaqua inter-mediate [(L)Ru2
III,III(pic)6(OH2)2]+ with a relative energy of +5.4 kcal mol–1. The sequential loss of a picoline and a proton gives complex [(L)Ru2
III,III(pic)4(OH2)(OH)]+ with a relative energy of 1.8 kcal mol–1.
0
2
4
6
[Ru2III,III(pic)6(OH2)]
0.0
+2.5 +2.6
[Ru2III,III(pic)5(OH2)]
H2O pic
+5.4
[Ru2III,III(pic)5(OH2)2]
H2O
pic + H+
+1.8
[Ru2III,III(pic)4(OH)(OH2)]
[Ru2III,III(pic)6]
kcal mol-1
reaction coordinate
Figure 4.6. Energy diagram for the ligand exchange at the Ru2(III,III) state for [(L)Ru2III,IIIpic)6]+ at pH 7.2. The structure [(L)Ru2III,IIIpic)6]+ has the lowest energy, hence is set to zero.

50
The DFT calculations showed that picoline–aqua ligand exchange could occur at the Ru2(II,III) oxidation state as well. The coordination of aqua to the [(L)Ru2
II,III(pic)6] complex, forming [(L)Ru2II,III(pic)6(OH)] complex, requires +1.5
kcal mol-1 of energy. Coordination of a second aqua species and loss of two picolines, forming the corresponding [(L)Ru2
II,III(pic)6(OH)(OH2)] is energeti-cally isogonic, in relation to the reference complex [(L)Ru2
II,III(pic)6] (Figure 4.7). The computational studies on redox potentials of complexes [(H2L)Ru2(pic)6], [(H2L)Ru2(pic)6(OH2)] and [(H2L)Ru2(pic)4(OH2)2] agreed fairly well with the ex-perimental values (Table 4.1 and Table 4.2). The results indicate that a 4-pic-oline-aqua exchange is necessary in order to reach the higher oxidation states Ru2(IV,IV) and Ru2(V,IV).
0
2 [Ru2II,III(pic)6(OH)]
[Ru2II,III(pic)4(OH)(OH2)][Ru2
II,III(pic)6]
4
kcal mol-1
0.0
+1.5H2O
2 pic + H+
H2O
0.0
reaction coordinate
Figure 4.7. Energy diagram for the ligand exchange at the Ru2(II,III) state for [(L)Ru2II,III(pic)6]+ at pH 7.2. The structure [(L)Ru2II,III(pic)6]+ has the lowest energy, hence is set to zero.

51
Table 4.1. Density functional theory calculations on redox potentials (V vs. NHE) for different ruthenium species at pH 7.2.
4.2.3 Electrochemical Studies Five broad oxidation processes were detected by DPV in phosphate buffer (0.1 M, pH 7.2). These peaks were assigned to one-electron oxidation from Ru2(II,II) to Ru2(IV,V) in the range 0.05–1.20 V vs. NHE (Figure 4.8). A catalytic wave for water oxidation was also detected with an onset potential of 1.20 V vs. NHE (Figure 4.9).
4.2.3 Electrochemical Studies Five broad oxidation processes were detected by DPV in phosphate buffer (0.1 M, pH 7.2). These peaks were assigned to one-electron oxidation from Ru2(II,II) to Ru2(IV,V) in the range 0.05–1.20 V vs. NHE (Figure 4.8). A catalytic wave for water oxidation was also detected with an onset potential of 1.20 V vs. NHE (Figure 4.9).
[(H2L)Ru2(pic)6] [(H2L)Ru2(pic)6(OH2)] [(H2L)Ru2(pic)4(OH2)2] Redox couple E Redox couple E Redox couple E
Ru2III,II/Ru2II,II 0.19 Ru2III,II/Ru2II,II 0.05 Ru2III,II/Ru2II,II -
0.15 Ru2III,III/Ru2II,III 0.39 Ru2III,III/Ru2II,III 0.43 Ru2III,III/Ru2II,III 0.14 Ru2III,IV/Ru2III,III 1.07 Ru2III,IV/Ru2III,III 0.49
Ru2III,IV/Ru2III,III 0.66 Ru2IV,IV/Ru2III,IV 0.85 Ru2IV,V/Ru2IV,IV 1.14
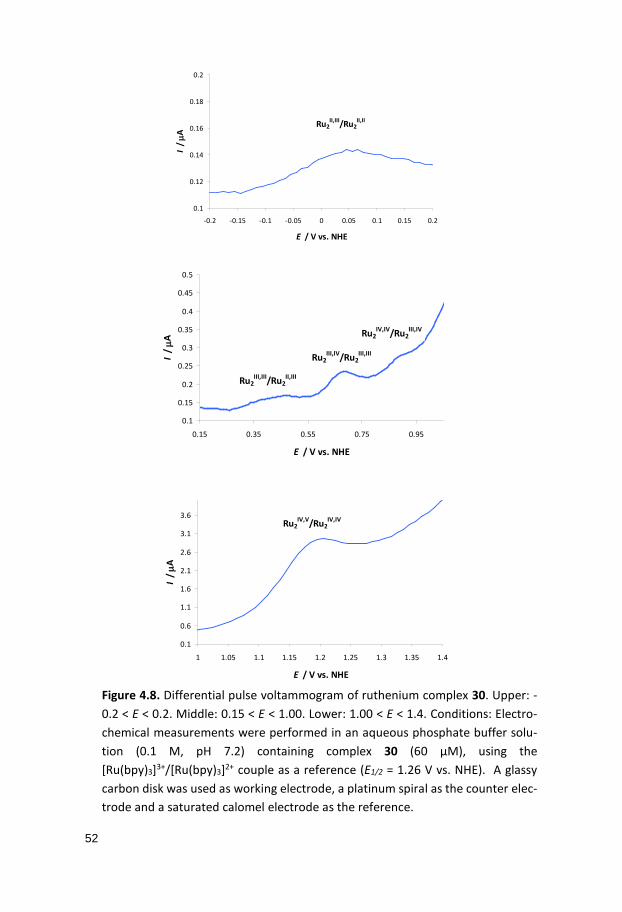
52
Figure 4.8. Differential pulse voltammogram of ruthenium complex 30. Upper: -0.2 < E < 0.2. Middle: 0.15 < E < 1.00. Lower: 1.00 < E < 1.4. Conditions: Electro-chemical measurements were performed in an aqueous phosphate buffer solu-tion (0.1 M, pH 7.2) containing complex 30 (60 μM), using the [Ru(bpy)3]3+/[Ru(bpy)3]2+ couple as a reference (E1/2 = 1.26 V vs. NHE). A glassy carbon disk was used as working electrode, a platinum spiral as the counter elec-trode and a saturated calomel electrode as the reference.
0.1
0.12
0.14
0.16
0.18
0.2
-0.2 -0.15 -0.1 -0.05 0 0.05 0.1 0.15 0.2
E / V vs. NHE
I / µA
Ru2II,III/Ru2
II,II
0.1
0.15
0.2
0.25
0.3
0.35
0.4
0.45
0.5
0.15 0.35 0.55 0.75 0.95
E / V vs. NHE
I / µA
Ru2III,III/Ru2
II,III
Ru2III,IV/Ru2
III,III
Ru2IV,IV/Ru2
III,IV
0.1
0.6
1.1
1.6
2.1
2.6
3.1
3.6
1 1.05 1.1 1.15 1.2 1.25 1.3 1.35 1.4
E / V vs. NHE
I / µA
Ru2IV,V/Ru2
IV,IV

53
Figure 4.9. Cyclic voltammogram of ruthenium complex 30. Conditions: Electro-chemical measurements were performed in an aqueous phosphate buffer solution (0.1 M, pH 7.2) containing complex 30 (60 μM), using the [Ru(bpy)3]3+/[Ru(bpy)3]2+ couple as a reference (E1/2 = 1.26 V vs. NHE) at a scan rate of 0.1 V s–1. A glassy carbon disk was used as working electrode, a platinum spiral as the counter electrode and saturated calomel electrode as the reference. The pH dependence in the range 1.5 < pH < 8.5 of the redox processes were also studied using DPV in order to construct the Pourbaix diagram (Figure 4.10). With the help from the quantum chemical calculations on the various species, suggestions on redox-active species and their processes could be given. The oxidation of [(H2L)Ru2
II,II(pic)6] to [(H2L)Ru2II,III(pic)6] under acidic
conditions occurs around 0.4 V vs. NHE and is pH independent in the range 1.5 < pH < 6 and with increasing pH the oxidation of [(H2L)Ru2
II,II(pic)6]2+ to [(HL)Ru2
II,III(pic)6]3+ is proton-coupled. This process seems to coincide with an-other redox event at around 0.38 V vs. NHE at pH 6, which presumably cor-responds to the [(H2L)Ru2
II,III(pic)6(OH2)]3+/[(H2L)Ru2II,II(pic)6(OH2)]2+ couple.
The overlap of the two processes appears as a two-electron-one-proton event with a slope of 29 mV per pH unit. It seems plausible that the [(H2L)Ru2
II,II(pic)6]+ complex together with [(H2L)Ru2II,III(pic)6(OH2)]2+ and
[(H3L)Ru2II,II(pic)6(OH2)]2+ co-exist at potentials E < 0.4 V vs. NHE. A reasonable
assumption is that the aqua species is formed to a greater extent at higher oxidation states and with increasing pH. Several one-electron-one-proton coupled events (with slopes of 59 mV per pH unit) as shown in the Pourbaix diagram occur with increasing potential and pH, highlighting the importance
-20
0
20
40
60
80
100
120
0 0.2 0.4 0.6 0.8 1 1.2 1.4 1.6 1.8
E / V vs. NHE
I / µA

54
of PCET processes for lowering the potentials of the various redox species (Figure 4.10).
Figure 4.10. Pourbaix diagram for the dinuclear ruthenium complex 30 in the range 1.5 < pH < 8.5 with the formal species at different redox states shown. The [(H2L)Ru2II,II(pic)6]+, [(H2L)Ru2II,III(pic)6(OH2)]2+ and [(H3L)Ru2II,II(pic)6(OH2)]2+ complexes co-exist as a mixture, see text for further discussion. Conditions: Potentials were de-termined by DPV. The measurements were carried out in 0.1 M Britton-Robinson buffer solution in the range 1.5 < pH < 8.5. The pH of the solution was changed by using an aqueous 0.2 M NaOH solution.
Table 4.2. Redox potentials of complex 30, obtained by DPVa.
Redox couple E (V vs. NHE) 1 0.02 2 0.35
3 0.47 4 0.64 5 0.86 6 1.12
a Electrochemical potentials were obtained with DPV in an aqueous Britton-Robinson buffer solution (0.1 M). The measurements were carried out using glassy carbon disk as working electrode, a platinum spiral as counter electrode and saturated calomel electrode (SCE) as reference. Potentials were converted to NHE by using the [Ru(bpy)3]3+/[Ru(bpy)3]2+ couple as a standard (E1/2 = 1.26 V vs. NHE)

55
4.2.4 Water Oxidation Catalysis
4.2.4.1 Chemical Water Oxidation The catalytic experiments were initially performed in neutral phosphate buffer (0.1 M, pH 7.2) with [Ru(bpy)3]3+ as a sacrificial one-electron oxidant (E1/2(Ru(III)/Ru(II)) = 1.26 V vs. NHE). The evolved oxygen was detected by real-time mass spectrometry (MS). When the oxidant was added to a solu-tion of the complex, immediate oxygen evolution was triggered, giving rise to a TON of 470 and initial TOF of 1.3 s–1 after 25 min (Figure 4.11). A larger amount of molecular oxygen was produced at the same catalyst concentra-tion when lowering the pH to 6.2, thus resulting in a higher TON of 800 and initial TOF of 0.5 s–1 within the same time frame (Figure 4.11). To identify the source of the oxygen atoms in the evolved oxygen, isotopically labeled water H2
18O was employed in the catalytic experiment. The amounts of 18,16O2 and 16,16O2 were measured (Figure 4.12), and the ratios of the isotopologues were in agreement with the theoretical values, supporting that water is the true source for the produced oxygen.
Figure 4.11. Chemical water oxidation catalyzed by dinuclear ruthenium complex 30 at pH 6.2 and 7.2, using [Ru(bpy)3](PF6)3 as the chemical oxidant. Reactions were performed in aqueous phosphate buffer solutions (0.1 M, 0.50 mL) containing ruthe-nium complex 30 (0.60 μM) and [Ru(bpy3)](PF6)3 (3.0 mg, 3.0 µmol) as the chemical oxidant.

56
Figure 4.12. Chemical water oxidation catalyzed by ruthenium complex 30 in isotop-ically labeled water (8.7% H218O). Reaction was performed in an aqueous phosphate buffer solution (0.1 M, pH 7.2, 0.50 mL, 8.7% H218O) containing catalyst 30 (3.0 μM) and [Ru(bpy)3](PF6)3 (3.0 mg, 3.0 μmol) as the chemical oxidant. 16,16O2 measured (▲), 16,18O2 measured (●).
4.2.4.2 Light-Driven Water Oxidation The ability of the catalyst to mediate light-driven water oxidation in the pres-ence of a [Ru(bpy)3]2+–type photosensitizer was subsequently studied. The stronger [Ru(bpy)2(deeb)]2+ photosensitizer (E1/2(Ru(III)/Ru(II)) = 1.40 V vs. NHE) was initially employed together with Na2S2O8 as the sacrificial electron acceptor, in phosphate buffer at pH 7.2. The experiments were conducted with three different catalyst concentrations and the highest TON of 830 was achieved for the lowest catalyst concentration of 0.3 µM after a 25 min reac-tion time (Figure 4.13). Increasing the catalyst concentration tenfold to 3.0 µM yielded a lower TON of 500. On the other hand, decreasing the pH of the buffer to pH 6.2 resulted in a higher amount of molecular oxygen, giving a TON of 890 (Figure 4.14, Table 4.3). Decreasing the pH further to 5.2 resulted in reduced oxygen production, and similarly, an increase of the pH to 8.2 re-sulted in lower yield of molecular oxygen. The higher TON at pH 6.2 could be due to the increased stability of the photosensitizer at slightly lower pH, whereas a further decrease in pH lowers the driving force to a significant ex-tent and thereby inhibits water oxidation (Figure 4.14). The reduced oxygen

57
production at pH 8.2 could be due to more rapid decomposition of the pho-tosensitizer.
To rule out the possibility of the generated persulfate radical (SO4
–•) to di-rectly oxidize complex 30, the methyl substituted photosensitizer [Ru(dmb)3]2+ ( dmb = 4,4’-dimethyl-2,2’-bipyridine) was employed as photo-sensitizer. This photosensitizer has an oxidation potential of E1/2(Ru(III)/Ru(II)) = 1.10 V vs. NHE, which is not sufficient to oxidize the stud-ied WOC to its active redox state. Indeed, employing [Ru(dmb)3]2+ as photo-sensitizer did not result in any significant amount of evolved oxygen, indicat-ing that SO4
–• is not oxidizing the WOC.
Figure 4.13. Light-driven water oxidation catalyzed by dinuclear ruthenium complex 30 at different catalyst concentrations. Reactions were performed in an aqueous phosphate buffer (0.1 M, pH 7.2, 0.50 mL) containing ruthenium complex 30, [Ru(bpy)2(deeb)](PF6)2 (15b) or [Ru(dmb)3](PF6)2 as photosensitizer (0.60 mM) and Na2S2O8 as sacrificial electron acceptor (23.5 mM).

58
Figure 4.14. Light-driven water oxidation catalyzed by dinuclear ruthenium complex 30 (3.0 µM) at different pH. Reactions were performed in an aqueous phosphate buffer (0.1 M, pH 7.2, 0.50 mL) containing ruthenium complex 30, [Ru(bpy)2(deeb)](PF6)2 (15b) as photosensitizer (0.60 mM) and Na2S2O8 as sacrificial electron acceptor (23.5 mM). Table 4.3. Photochemical water oxidation by dinuclear ruthenium complex 30.
Concentration [µM] TON at pH 7.2 a TON at pH 6.2 b 30 100 - 3 500 890
0.3 830 -
a Conditions: aqueous phosphate buffer solutions (0.1 M, pH 7.2, 0.50 mL) with [Ru(bpy)2(deeb)](PF6)2 (15b) as photosensitizer (0.60 mM) and Na2S2O8 as sacrificial electron acceptor (23.5 mM). b Reaction was carried out in phosphate buffer (0.1 M, pH 6.2, 0.50 mL) with [Ru(bpy)2(deeb)](PF6)2 (15b) as photosensitizer (0.60 mM) and Na2S2O8 as sacrificial electron acceptor (23.5 mM).
4.2.5 Proposed Catalytic Cycle The proposed catalytic cycle for water oxidation by complex 30 is shown in Scheme 4.2 and is based on the experimentally determined electrochemical data along with the computational studies of various intermediates and their relative energies. The complex initially undergoes a ligand exchange to afford an aqua-coordinated complex, upon which three consecutive one-electron one-proton coupled oxidations takes place. The generated

59
[(L)Ru2IV,V(pic)4(O)(OH)]+ species subsequently releases molecular oxygen,
thus returning to the reduced [(L)Ru2III,III(pic)4(OH2)]+ state.
[(L)Ru2III,III(pic)4(OH2)2]+
[(L)Ru2III,IV(pic)4(OH)(OH2)]
+
[(L)Ru2IV,IV(pic)4(OH)2]+
[(L)Ru2IV,V(pic)4(O)(OH)]
+
O2
2 H2O
2 H2O
H+ + e-
H+ + e-
H+ + e-
H+ + e-
2 pic 2 H+ + e-
N N N
HNNH
N
OOOO
Ru Ru
pic pic
pic pic
30
picpic N
pic =
[(H2L)Ru2II,III(pic)6]2+
N N N
NN
N
OOOO
Ru Ru
pic pic
pic pic
OHO
HH
H
N N N
NN
N
OOOO
Ru Ru
pic pic
pic pic
OO
HH
H
N N N
NN
N
OOOO
Ru Ru
pic pic
pic pic
OO
H
H
N N N
NN
N
OOOO
Ru Ru
pic pic
pic pic
H
O O
2
Scheme 4.2. Proposed catalytic cycle for ruthenium complex 30.
A more detailed mechanistic study of 30 was reported in 2016 by Siegbahn and co-workers.57 Calculations on various intermediates derived from the di-nuclear ruthenium complex 30 suggest that high-valent redox states can be reached due to the protonation stages of the complex, to a significant extent by the ligand, allowing lower energy pathways to be accessed. The computa-tional studies on water oxidation investigated two mechanistic pathways for O–O bond formation: WNA and I2M. The data renders an I2M mechanism as the most favorable pathway. The studies entail a detailed mechanistic route, starting with [(L)Ru2
III,III(H2O)(OH)(pic)4] that has a strong hydrogen bond be-tween the equatorial water and hydroxy ligands coordinating to each metal ion. Complex [(L)Ru2
III,III(H2O)(OH)(pic)4] undergoes two PCET events and an one-electron oxidation in order to reach [(HL)Ru2
IV,V(O)2(pic)4], which could be in equilibrium with [(L)Ru2
IV,V(O)(OH)(pic)4] due to an energy difference of

60
merely 0.3 kcal/mol between these isomers, with the latter isomer displaying higher energy. The more favorable isomer, [(HL)Ru2
IV,V(O)2(pic)4], with the proton located on the benzimidzole nitrogen is deprotonated and oxidized to [(L)Ru2
V,V(O)2(pic)4] that subsequently undergoes coupling of the two oxo units to form a bridged peroxido intermediate, [(L)Ru2
III,IV(O)2(pic)4]. This Ru2(III,V) species undergoes a facile one-electron oxidation to [(L)Ru2
IV,IV(O)2(pic)4], which subsequently releases molecular oxygen and is the rate determining step for the catalytic cycle.57
4.3 Summary The dinuclear ruthenium complex 30 was synthesized and evaluated for wa-ter oxidation at neutral pH using [(Ru(bpy)3]3+-type oxidants. Noteworthy is the catalytic performance of 30 in a three-component system for light-driven water oxidation, resulting in high TONs. According to computational studies the aqua-picoline exchange is relatively facile at the Ru2(II,III) and Ru2(III,III) states. The computational results, together with the experimental Pourbaix diagram indicate that several redox-active species may co-exist. Further-more, these results suggest that the ligand participation enables PCET pro-cesses along the reaction pathway. The computational studies further sug-gest that O–O bond formation occurs via a bridging peroxide intermediate, i.e. an I2M-type of mechanism.

61
Acknowledgements
I would like to express my sincerest gratitude to the following people: My supervisor, Professor Björn Åkermark. Thank you for accepting me as a graduate student, for seeing potential in me. Your enthusiasm for chemistry and many other topics is truly inspiring. Financial support from the Swedish Research Council, the Knut and Alice Wal-lenberg Foundation, the Carl Trygger Foundation, the Swedish Energy Agency and the Royal Swedish Academy of Agriculture and Forestry. Prof. Pher G. Andersson for proofreading this thesis. My wonderful colleagues: Dr. Markus Kärkäs, for your unlimited guidance, ideas and patience over the years. I am profoundly thankful for your support. Past members of the group; Dr. Bao-Lin Lee, for all the help my first year. I think you have exceptional understanding for others. Dr. Erik Karlsson, for all the advice! Andrey Shatskiy, Dr. Ahmed Fawzy, Dr. Peter Olsén, I am tremendously happy that I have met you and that we shared the work and the lab this last mile. Thank you for your support! Prof. Rong-Zhen Liao, for the numerous calculations and for explaining theo-retical chemistry to me.

62
Dr. Markus Kärkäs, Andrey Shatskiy, Dr. Peter Olsén, Dr. Fredrik Tinnis, Dr. Ahmed Abdel-Magied and Dr. Abraham Mendoza for valuable comments and for helping me to improve this thesis. Dr. Hong Chen, together we chased for crystals. Thank you for your expertise! Co-authors: Markus D. Kärkäs, Torbjörn Åkermark, Bao-Lin Lee, Erik Karlsson, Rong-Zhen Liao, Per E. M. Siegbahn, Eric V. Johnston, Shams R. Karim, Tobias Åkermark, Timofei Privalov. The Girls – Sara Moa, Johanna Löfgren, Tove Kivijärvi, Tove Slagbrand, Elin Stridfeldt, Anuja Nagendiran, Ana Vazquez and Elisa Martínez de Castro –Thi-bault Angles d’Ortoli and Antonio Bermejo-Gómez, for being wonderful and supportive! Let us continue our meetings. My beloved friends for their solid, unconditional support and love: Lotta, Fatma, Valentino, Dario, Maria and Charlie. You are like family to me! Tatiana Skoglund, my mentor and second mother, for always helping me to put things in perspective. Muhammed Ago, Agocuk, aşkim benim, for being an oasis where I can reside at all times. Seni seviyorum! My Mother (with capital M), Hanna Laine, for setting a solid foundation to build on and for always, unconditionally, providing shelter from the storm. Rakas Äiti, eivät sanat riitä kertomaan kuinka kiitollinen olen. Minä rakastan sinua.

63
Appendix A: Contribution list
The author’s contribution to publications I–IV: I. Performed a minor part of the experimental work. II. Performed a significant part of the experimental work. III. Performed a major part of the experimental work and took part in writ-
ing the article. IV. Performed a major part of the experimental work and took part in writ-
ing the article.

64
References
(1) a) U. Siegenthaler, T. F. Stocker, E. Monnin, D. Lüthi, J. Schwander, B. Stauffer, D. Raynaud, J.-M. Barnola, H. Fischer, V. Masson-Delmotte, J. Jouzel, Science 2005, 310, 1313–1317. b) N. S. Lewis, D. G. Nocera, Proc. Natl. Acad. Sci. 2006, 103, 15729–15735. c) British Petroleum, BP Energy Outlook, 2016 Edition, Outlook to 2035.
(2) a) J. P. McEvoy, G. W. Brudvig, Chem. Rev. 2006, 106, 4455–4483. b) W. Lubitz, E. J. Reijerse, J. Messinger, Energy Environ. Sci. 2008, 1, 15–31. c) J. Yano, J. Kern, K. Sauer, M. J. Latimer, Y. Pushkar, J. Biesiadka, B. Loll, W. Saenger, J. Messinger, A. Zouni, V. K. Yachandra, Science 2006, 314, 821–825. d) Y. Umena, K. Kawakami, J.-R. Shen, N. Kamiya, Nature 2011, 473, 55–61.
(3) L. Orr Govindjee, Photosynth. Res. 2010, 105, 167–200. (4) B. Kok, B. Forbush, M. McGloin, Photochem. Photobiol. 1970, 11, 457–475. (5) A. Fujishima, K. Honda, Nature 1972, 238, 37–38. (6) M. Grätzel, Nature 2001, 414, 338–344. (7) X. Shi, H. Jeong, S. J. Oh, M. Ma, K. Zhang, J. Kwon, I. T. Choi, I. Y. Choi, H. K.
Kim, J. K. Kim, J. H. Park, Nat. Commun. 2016, 7, 11943–11948. (8) S. J. A. Moniz, S. A. Shevlin, D. J. Martin, Z.-X. Guob, J. Tang, Energy Environ.
Sci. 2015, 8, 731–759. (9) P. A. Mabrouk, M. S. Wrighton, Inorg. Chem. 1986, 25, 526–531. (10) Z. Yu, F. Lia, L. Sun, Energy Environ. Sci. 2015, 8, 760–775. (11) a) A. R. Parent, R. H. Crabtree, G. W. Brudvig, Chem. Soc. Rev. 2013, 42, 2247–
2252. b) Z. Codolà, L. Gómez, S. T. Kleespies, L. Que Jr, M. Costas, J. Lloret-Fillol, Nat. Commun. 2015, 6, 5865–5873.
(12) W. J. Dressick, Tm J. Meyer, B. Durham, D. P. Rillema, Inorg. Chem. 1982, 21, 3451–3458.
(13) M. Biner, H.-B. Bürgi, A. Ludi, C. Röhr, J. Am. Chem. Soc. 1992, 114, 5197–5203.
(14) A. Lewandowska-Andralojc, D. E. Polyansky, J. Phys. Chem. A, 2013, 117, 10311–10319.
(15) a) K. Kalyanasundaram, Coord. Chem. Rev. 1982, 46, 159–244. b) A. Juris, V. Balzani, Coord. Chem. Rev. 1988, 84, 85–277.
(16) K. Binnemans, Application of tetravalent cerium compounds; Handbook on the Physic and Chemistry of Rare Earths, 2006, Vol 36.

65
(17) a) S. W. Gersten, G. J. Samuels, T. J. Meyer, J. Am. Chem. Soc. 1982, 104, 4029–4030. b) J. A. Gilbert, D.S Eggleston, W. R. Murphy Jr. D. A. Geselowitz, S. W. Gersten, D. J. Hodgson, T. J. Meyer, J. Am. Chem. Soc. 1985, 107, 3855–3864.
(18) J. P. Collin, J. P. Sauvage, Inorg. Chem. 1986, 25, 135–141. (19) K. Nagoshi, S. Yamashita, M. Yagi, M. Kaneko, J. Mol. Catal. A: Chem. 1999,
144, 71–74. (20) C. Sens, I. Romero, M. Rodríguez, A. Llobet, T. Parella, J. Benet–Buchholz, J.
Am. Chem. Soc. 2004, 126, 7798–7799. (21) F. Bozoglian, S. Romain, M. Z. Ertem, T. Todorova, C. Sens, J. Mola, M.
Rodríguez, I. Romero, J. Benet-Buchholz, X. Fontrodona, C. J. Cramer, L. Gagliardi, A. Llobet, J. Am. Chem. Soc. 2009, 131, 15176–15187.
(22) S. Neudeck, S. Maji, I. López, S. Meyer, F. Meyer, A. Llobet, J. Am. Chem. Soc. 2014, 136, 24–27.
(23) Z. Deng, H.-W. Tseng, R. Zong, D. Wang, R. Thummel, Inorg. Chem. 2008, 47, 1835–1848.
(24) R. Zong, R. Thummel, J. Am. Chem. Soc. 2005, 127, 12802–12803. (25) R. Lomoth, P. Huang, J. Zheng, L. Sun, L. Hammarström, B. Åkermark, S.
Styring, Eur. J. Inorg. Chem. 2002, 2965–2974. (26) Y. Xu, T. Åkermark, V. Gyollai, D. Zou, L. Eriksson, L. Duan, R. Zhang, B.
Åkermark, L. Sun, Inorg. Chem. 2009, 48, 2717–2719. (27) Y. Xu, A. Fischer, L. Duan, L. Tong, E. Gabrielsson, B. Åkermark, L. Sun, Angew.
Chem. Int. Ed. 2010, 49, 8934–8937. (28) L. Duan, A. Fischer, Y. Xu, L. Sun, J. Am. Chem. Soc. 2009, 131, 10397–10399. (29) L. Duan, F. Bozoglian, S. Mandal, B. Stewart, T. Privalov, A. Llobet, L. Sun, Nat.
Chem. 2012, 4, 418–423. (30) a) J. Limburg, J. S. Vrettos, H. Chen, J. C. de Paula, R. H. Crabtree, G. W. Bru-
dvig, J. Am. Chem. Soc. 2001, 123, 423–430. b) C. W. Cady, K. E. Shinopoulos, R. H. Crabtree, G. W. Brudvig, Dalton Trans. 2010, 39, 3985–3989.
(31) E. A. Karlsson, B.-L. Lee, T. Åkermark, E. V. Johnston, M. D. Kärkäs, J. Sun, Ö. Hansson, J.-E. Bäckvall, B. Åkermark, Angew. Chem. Int. Ed. 2011, 50, 11715–11718.
(32) a) D. Hong, M. Murakami, Y. Yamada, S. Fukuzumi, Energy Environ. Sci. 2012, 5, 5708–5716. b) D. Hong, S. Mandal, Y. Yamada, Y.-M. Lee, W. Nam, A. Llobet, S. Fukuzumi, Inorg. Chem. 2013, 52, 9522–9531. c) G. Chen, L. Chen, S.-M. Ng, W.-L. Man, T.-C. Lau, Angew. Chem. Int. Ed. 2013, 52, 1789–1791.
(33) J. Lloret Fillol, Z. Codolà, I. Garcia-Bosch, L. Gómez, J. J. Pla, M. Costas, Nat. Chem. 2011, 3, 807–813.
(34) S. Fu, Y. Liu, Y. Ding, X. Du, F. Song, R. Xiang, B. Ma, Chem. Commun. 2014, 50, 2167–2169.
(35) a) J. N. Demas, A. W. Adamson, J. Am. Chem. Soc. 1971, 93, 1800–1801. b) H. D. Gafney, A. W. Adamson, J. Am. Chem. Soc. 1972, 94, 8238–8239. c) N. Sutín, C. Creutz, Pure Appl. Chem. 1980, 52, 2717–2738.

66
(36) A. L. Kaledin, Z. Huang, Y. V. Geletii, T. Lian, C. L. Hill, D.G. Musaev, J. Phys. Chem. A, 2010, 114, 73–80.
(37) a) N. D. Morris, M. Suzuki, T. E. Mallouk, J. Phys. Chem. A, 2004, 108, 9115–9119. b) J. M. R. Narayanam, C. R. J. Stephenson, Chem. Soc. Rev. 2011, 40, 102–114. c) C. K. Prier, D. A. Rankic, D, W. C. MacMillan, Chem. Rev. 2013, 113, 5322–5363. d) D. N. Thompson, A. Ito, T. J. Meyer, Pure Appl. Chem. 2013, 85, 1257–1305.
(38) F. P. Rotzinger, S. Munavalli, P. Comte, J. K. Hurst, M. Grätzel, F.-J. Pern, A. J. Frank, J. Am. Chem. Soc. 1987, 109, 6619–6626.
(39) P. Comte, M. K. Nazeeruddin, F. P. Rotzinger, A. J. Frank, M. Grätzel, J. Mol. Catal. 1989, 52, 63–84.
(40) Y. Xu, L. Duan, L. Tong, B. Åkermark, L. Sun, Chem. Commun. 2010, 46, 6506–6508.
(41) L. Duan, Y. Xu, P. Zhang, M. Wang, L. Sun, Inorg. Chem. 2010, 49, 209–215. (42) L. Wang, L. Duan, L. Tong, L. Sun, J. Catal. 2013, 306, 129–132. (43) S. Berardi, L. Francàs, S. Neudeck, S. Maji, J. Benet-Buchholz, F. Meyer, A.
Llobet, ChemSusChem 2015, 8, 3688–3696. (44) S. Berardi, L. Francàs, S. Neudeck, S. Maji, J. Benet-Buchholz, F. Meyer, A.
Llobet, ChemSusChem. 2015, 8, 3688–3696. (45) A. Lewandowska-Andralojc, D. E. Polyansky, R. Zong, R. P. Thummel, E. Fujita,
Phys. Chem. Chem. Phys. 2013, 15, 14058–14068. (46) a) S. Romain, L. Vigara, A. Llobet, Acc. Chem. Res. 2009, 42, 1944–1953. b) J.
Nyhlén, L. Duan, B. Åkermark, L. Sun, T. Privalov, Angew. Chem. Int. Ed. 2010, 49, 1773–1777. c) L. Tong, L. Duan, Y. Xu, T. Privalov, L. Sun, Angew. Chem. Int. Ed. 2011, 50, 445–449.
(47) a) J. M. Mayer, Annu. Rev. Phys. Chem. 2004, 55, 363–390. b) C. Constentin, M. Robert, J.-M. Savéant, Acc. Chem. Res. 2010, 43, 1019–1029. c) O. S. Wenger, Chem. Eur. J. 2011, 17, 11692–11702. d) D. R. Weinberg, C. J. Gagliardi, J. F. Hull, C. Fecenko Murphy, C. A. Kent, B. C. Westlake, A. Paul, D. H. Ess, D. Granville McCafferty, T. J. Meyer, Chem. Rev. 2012, 112, 4016–4093. e) M. Bourrez, R. Steinmetz, S. Ott, F. Gloaguen, L. Hammarström, Nat. Chem. 2015, 7, 140–145.
(48) M. D. Kärkäs, B. Åkermark, Chem. Rec. 2016, 16, 940–963. (49) D. Yang, D. Fokas, J. Li, L. Yu, C. M. Baldino, Synthesis 2005, 1, 47–56. (50) a) R. E. DeSimone, J. Am. Chem. Soc. 1973, 95, 6238–6244. b) S. Bhattacharya,
A. Chakravorty, Proc. Indian Acad. Sci. 1985, 95, 159–167. (51) L. Duan, Y. Xu, M. Gorlov, L. Tong, S. Andersson, L. Sun, Chem. Eur. J. 2010,
16, 4659–4668. (52) a) L. P. Hammet, J. Am. Chem. Soc. 1937, 59, 96–103. C. b) Hansch, A. Leo, W.
Taft, Chem. Rev. 1991, 91, 165–195. (53) B.-L. Lee, M. D. Kärkäs, E. V. Johnston, A. K. Inge, L.-H. Tran, Y. Xu, Ö. Hansson,
X. Zou, B. Åkermark, Eur. J. Inorg. Chem. 2010, 5462–5470.

67
(54) M. D. Kärkäs, E. V. Johnston, E. A. Karlsson, B.-L. Lee, T. Åkermark, M. Shariatgorji, L. Ilag, Ö. Hansson, J.-E. Bäckvall, B. Åkermark, Chem. Eur. J. 2011, 17, 7953–7959.
(55) V. J. Arán, M. Kumar, J. Molina, L. Lamarque, P. Navarro, E. García-Espana, J. A. Ramírez, S. V. Luis, B. Escuder, J. Org. Chem. 1999, 64, 6135–6146.
(56) L. Duan, F. Bozoglian, S. Mandal, B. Stewart, T. Privalov, A. Llobet, L. Sun, Nat. Chem. 2012, 4, 418–423.
(57) R.-Z. Liao, M. D. Kärkäs, T. M. Laine, B. Åkermark, P. E. M. Siegbahn, Catal. Sci. Technol. 2016, 6, 5031–5041.