dia china 2017 optimizing clinical trials with advanced tools
TRANSCRIPT

Optimizing Clinical Trials with Advanced Tools
CAPT E. Dennis Bashaw, Pharm.D. Dir. Division of Clinical Pharmacology-3
Office of Clinical PharmacologyOffice of Translational Sciences
US Food and Drug Administration
Session 901:STRATEGY OF CLINICAL DEVELOPMENT

2
• The presentation today should not beconsidered, in whole or in part as beingstatements of policy or recommendationby the US Food and Drug Administration.
• Throughout the talk, representativeexamples of commercial products will bementioned. No commercial endorsementis either implied or intended.

3
Outline
• 21st Century Challenges in Drug Development
• Beyond Classical PK/PD
• Case Studies
–Secukinumab
–Edoxaban
• Lessons Learned
• Conclusions and Closing Thoughts

4
21ST CENTURY CHALLENGES IN DRUG DEVELOPMENT
5
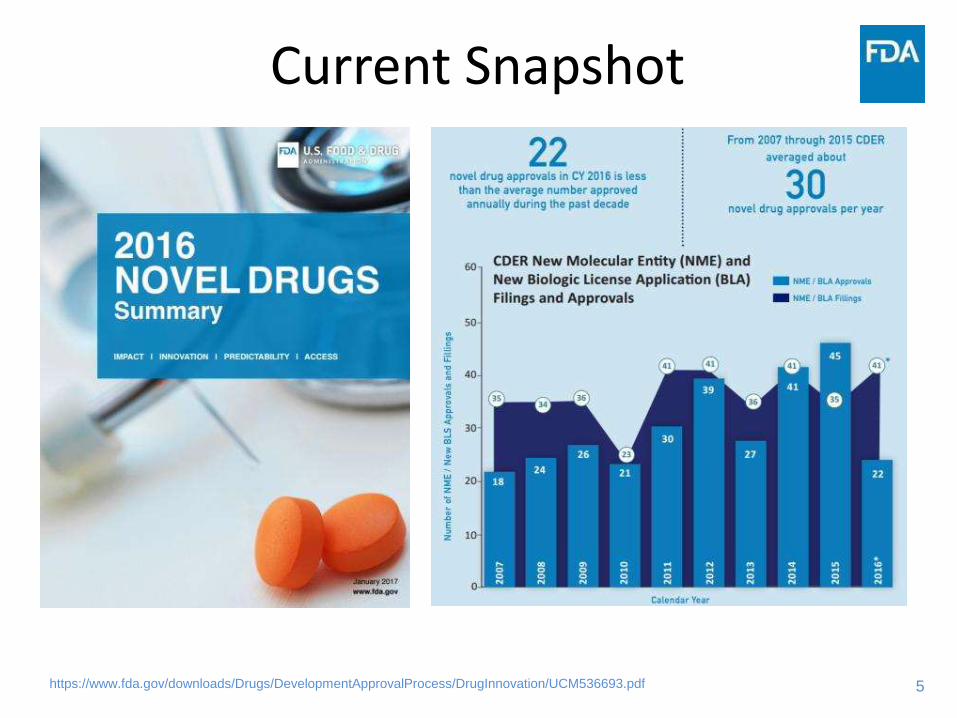
Current Snapshot
https://www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/DrugInnovation/UCM536693.pdf
6
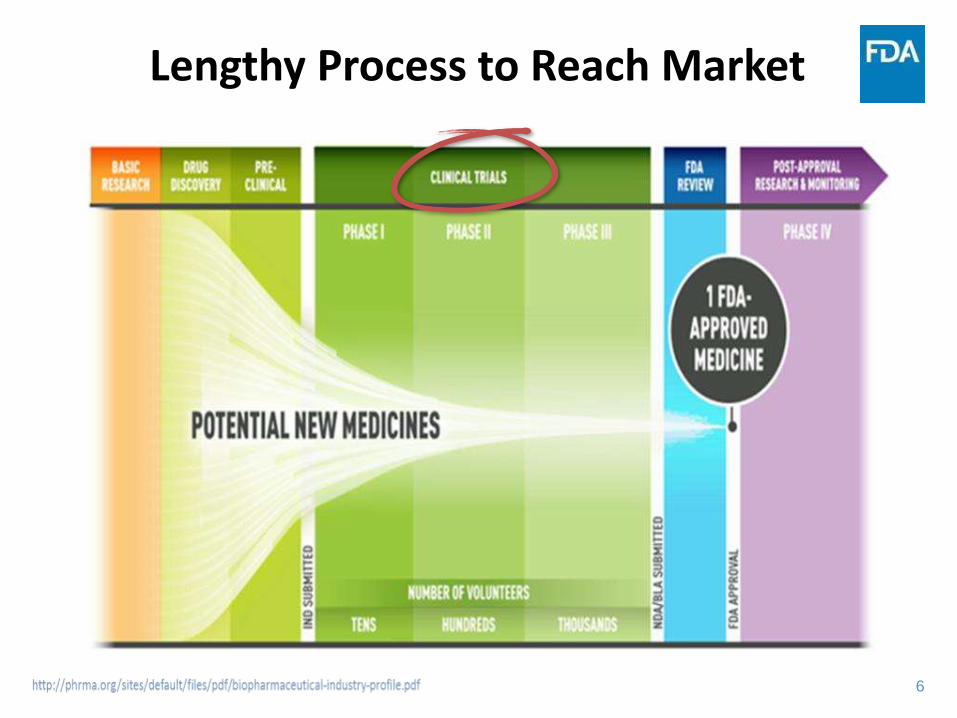
Lengthy Process to Reach Market

7
Updated Drug Development Cost Figures
J Health Econ. 2016 May;47:20-33. doi: 10.1016/j.jhealeco.2016.01.012
8
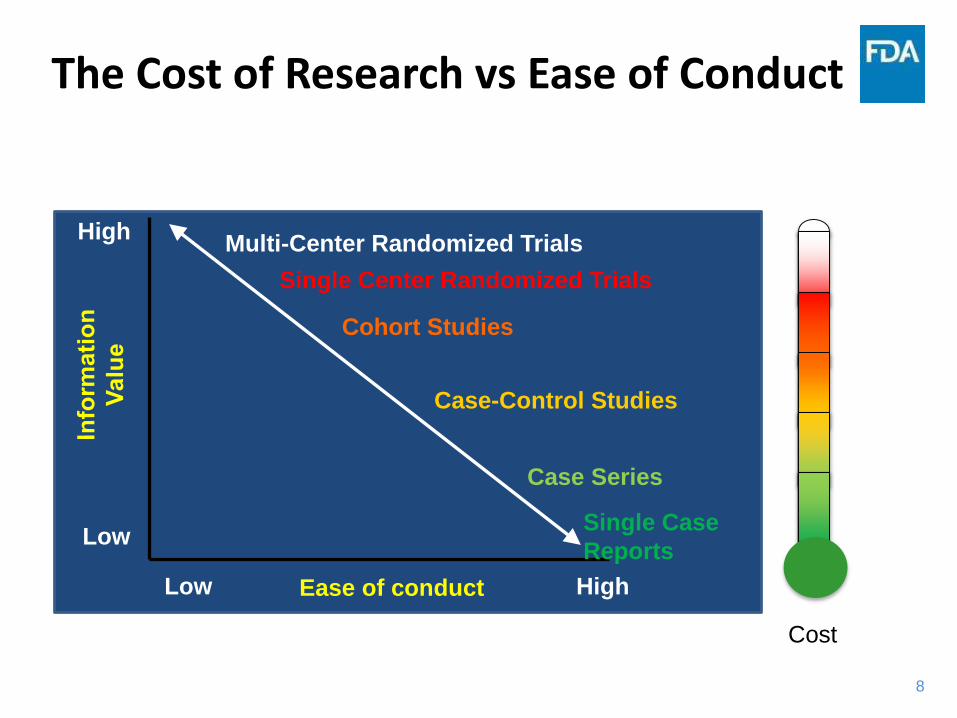
The Cost of Research vs Ease of Conduct
High
Low
Single Center Randomized Trials
Low
Single Case
Reports
Cohort Studies
Case-Control Studies
Case Series
HighEase of conduct
Multi-Center Randomized Trials
Cost

9
Why Replicate Trials?
• Guidance for Industry-Providing Clinical Evidence of
Effectiveness for Human Drugs and Biological Products
– Congress adopted the 1962 Drug Amendments, Section 505(d) of the Act uses the plural form in defining “substantial evidence” as “adequate and well-controlled investigations, including clinical investigations.” See also use of “investigations” in section 505(b) of the Act, which lists the contents of a new drug application. which included a provision requiring manufacturers of drug products to establish a drug’s effectiveness by "substantial evidence." Substantial evidence was defined in section505(d) of the Act as “evidence consisting of adequate and well-controlled investigations, including clinical investigations, by experts qualified by scientific training and experience to evaluate the effectiveness of the drug involved, on the basis of which it could fairly and responsibly be concluded by such experts that the drug will have the effect it purports or is represented to have under the conditions of use prescribed, recommended, or suggested in the labeling or proposed labeling thereof.”

10
BEYOND CLASSICAL PK/PD
Patrick Muller and Mark Milton, “The Determination and Interpretation of the Therapeutic Index in Drug Development”
Nature Reviews Drug Discovery, 2012, vol. 11, pg 751-751

11
The Triad of Drug Innovation
• Knowledge Management
– Leveraging information• Biomarker Qualification
• Disease History
• Innovative Trial Designs
– Moving away from the “2 replicate trials” standard
• Innovative Analysis
– Pharmacometrics• Modeling and Simulation
– Pharmcogenomics• Patient Factors
– Pharmacovigilance

12
Tools vs Utility• In the last 20yrs, the term “precision medicine” has been
growing in the literature as a goal to real patient individualization
• Ideally precision medicine should include not only patient care but also encompass drug development
– Identify and include actionable information on patient factors
– Empower the physician to use state of the art diagnostics and biomarkers
Huang S-M, Temple R, Clin Pharmacol Ther. 2008

13
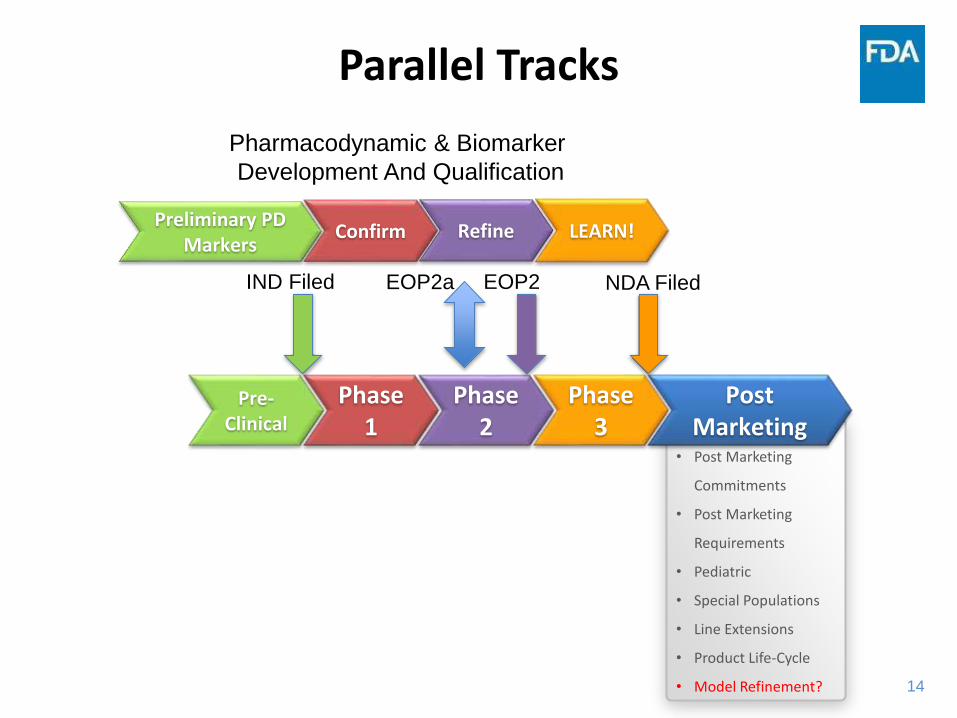
Linkage of Patient Factors to PK and PD
14
• Post Marketing
Commitments
• Post Marketing
Requirements
• Pediatric
• Special Populations
• Line Extensions
• Product Life-Cycle
• Model Refinement?
Parallel Tracks
Post Marketing
Phase 3
Phase 2
Phase 1
Pre-Clinical
EOP2IND Filed EOP2a
Preliminary PD Markers
LEARN!Confirm Refine
Pharmacodynamic & Biomarker
Development And Qualification
NDA Filed
15
SECUKINUMABDOSE RANGING-PHASE II TO PHASE III
16
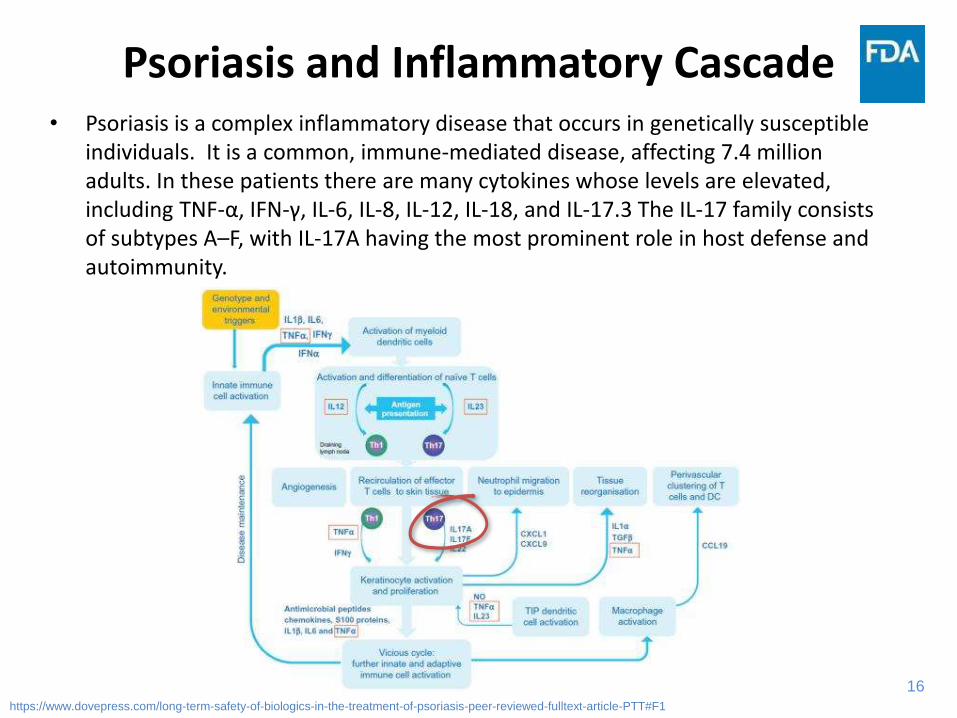
Psoriasis and Inflammatory Cascade• Psoriasis is a complex inflammatory disease that occurs in genetically susceptible
individuals. It is a common, immune-mediated disease, affecting 7.4 million adults. In these patients there are many cytokines whose levels are elevated, including TNF-α, IFN-γ, IL-6, IL-8, IL-12, IL-18, and IL-17.3 The IL-17 family consists of subtypes A–F, with IL-17A having the most prominent role in host defense and autoimmunity.
https://www.dovepress.com/long-term-safety-of-biologics-in-the-treatment-of-psoriasis-peer-reviewed-fulltext-article-PTT#F1

17
Case 1: Secukinumab
• Human interleukin-17A antagonist indicated for the treatment of moderate to severe plaque psoriasis in adult patients who are candidates for systemic therapy or phototherapy
– adults with psoriatic arthritis
– adults with active ankylosing spondylitis
• Dose-ranging in Phase II can help identify subgroups who may benefit from altered dosing
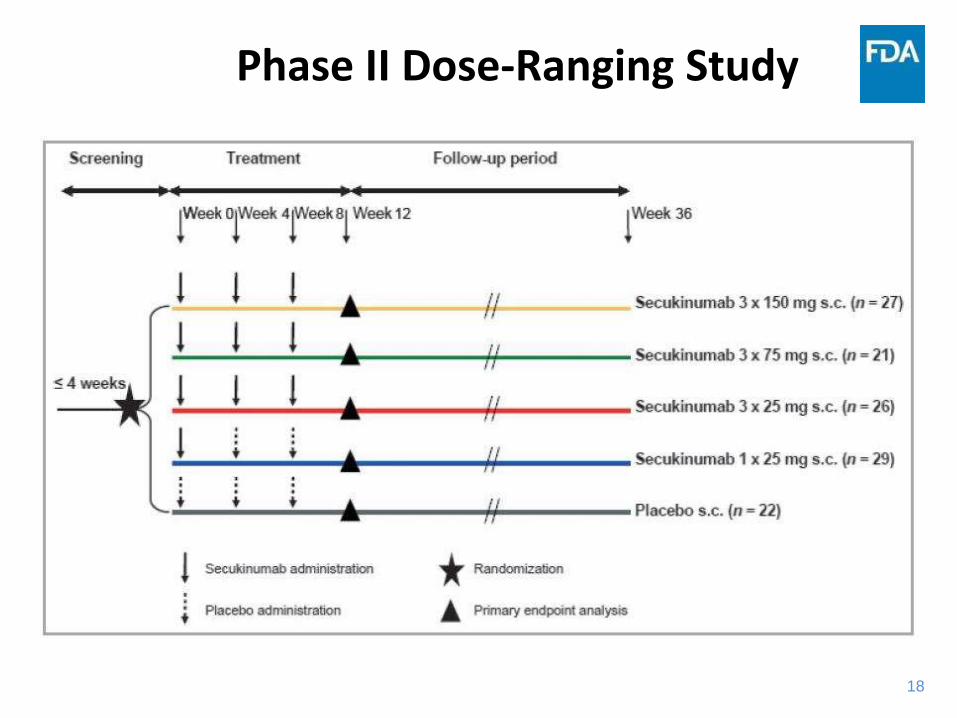
18
Phase II Dose-Ranging Study

19
Phase II Study Results
• Subgroup analysis showed an increased benefit with regardless of body weight, but highest response was in patients with body weight <90 kg
• Results from this trial and other sources support benefit of exploring a higher 300 mg dose in Phase III
Endpoint PlaceboSecukinumab
25 (x1) mg 25 (x3) mg 75 (x3) mg 150 (x3) mgPASI 75 9% 3% 19% 57% 82%IGA 0/1 9% 0% 12% 33% 48%
Papp et al, 2012
20
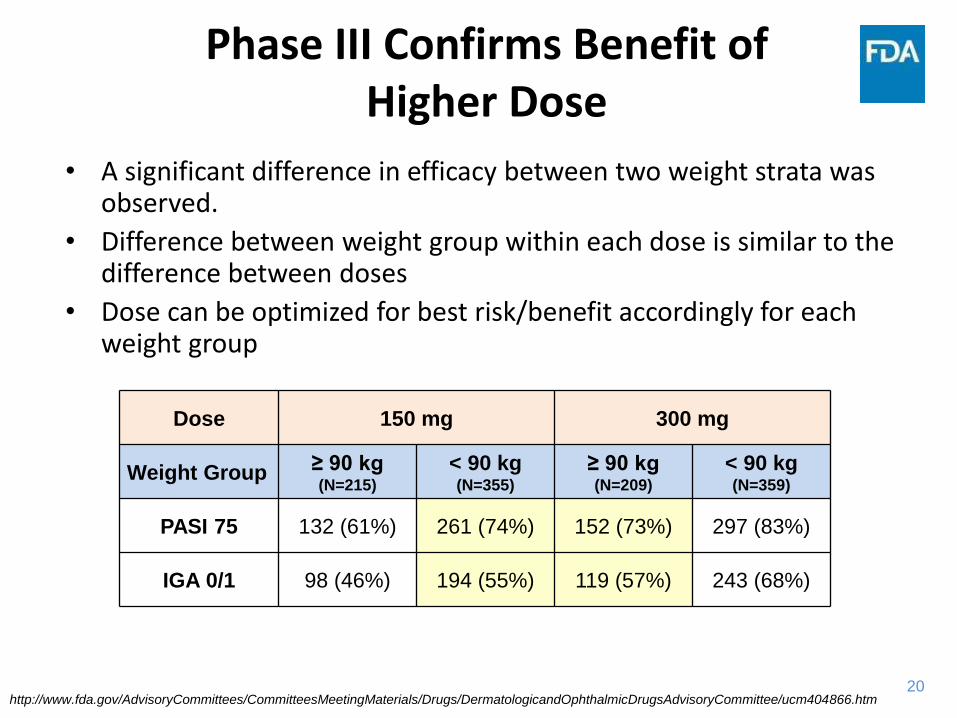
Phase III Confirms Benefit of Higher Dose
• A significant difference in efficacy between two weight strata was observed.
• Difference between weight group within each dose is similar to the difference between doses
• Dose can be optimized for best risk/benefit accordingly for each weight group
Dose 150 mg 300 mg
Weight Group ≥ 90 kg (N=215)
< 90 kg (N=355)
≥ 90 kg (N=209)
< 90 kg (N=359)
PASI 75 132 (61%) 261 (74%) 152 (73%) 297 (83%)
IGA 0/1 98 (46%) 194 (55%) 119 (57%) 243 (68%)
http://www.fda.gov/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/DermatologicandOphthalmicDrugsAdvisoryCommittee/ucm404866.htm

21
Case 1: Outcome
• Phase III trial results support safety and efficacy of both doses
• Patients with higher body weight or those that are non-responsive at the 300 mg dose may benefit from a further increase in dosing
• Sponsor agreed to conduct a post-marketing study to evaluate a higher dose
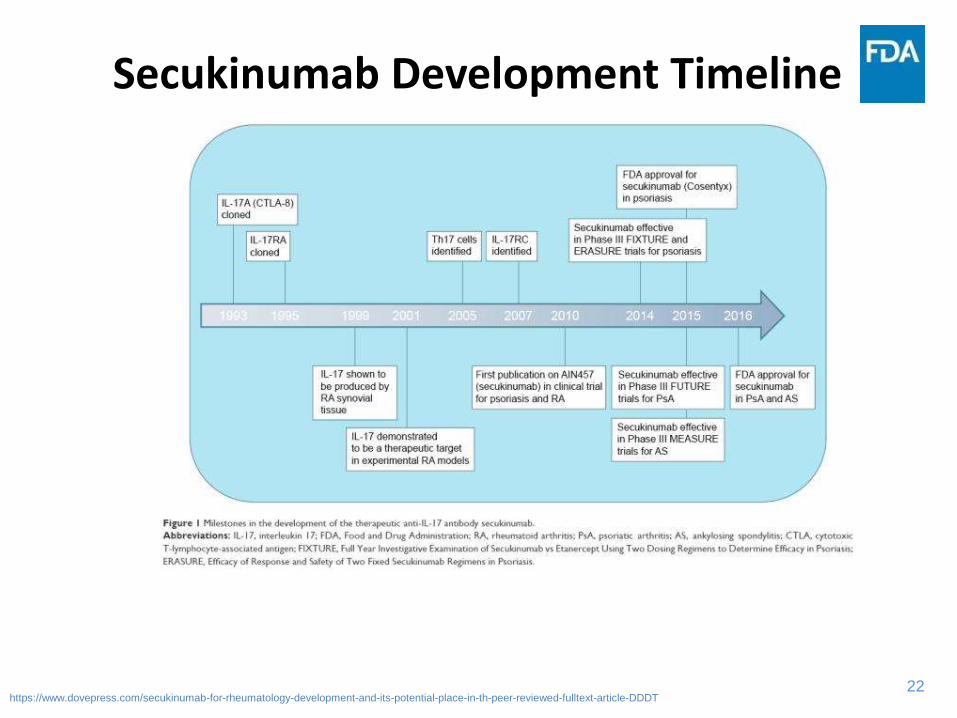
22
Secukinumab Development Timeline
https://www.dovepress.com/secukinumab-for-rheumatology-development-and-its-potential-place-in-th-peer-reviewed-fulltext-article-DDDT
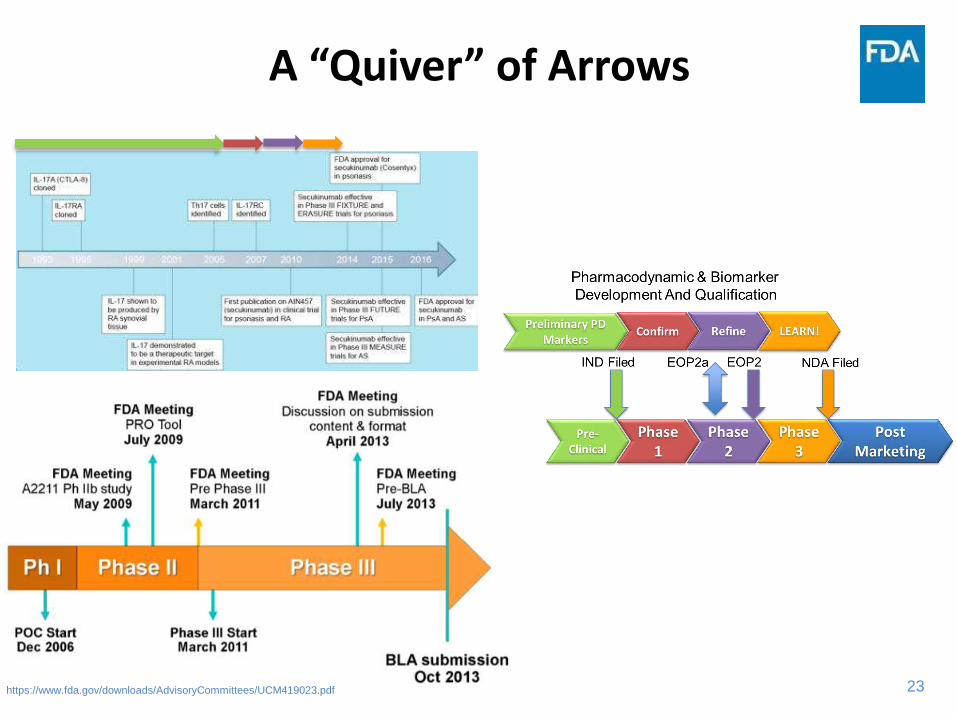
23
A “Quiver” of Arrows
https://www.fda.gov/downloads/AdvisoryCommittees/UCM419023.pdf
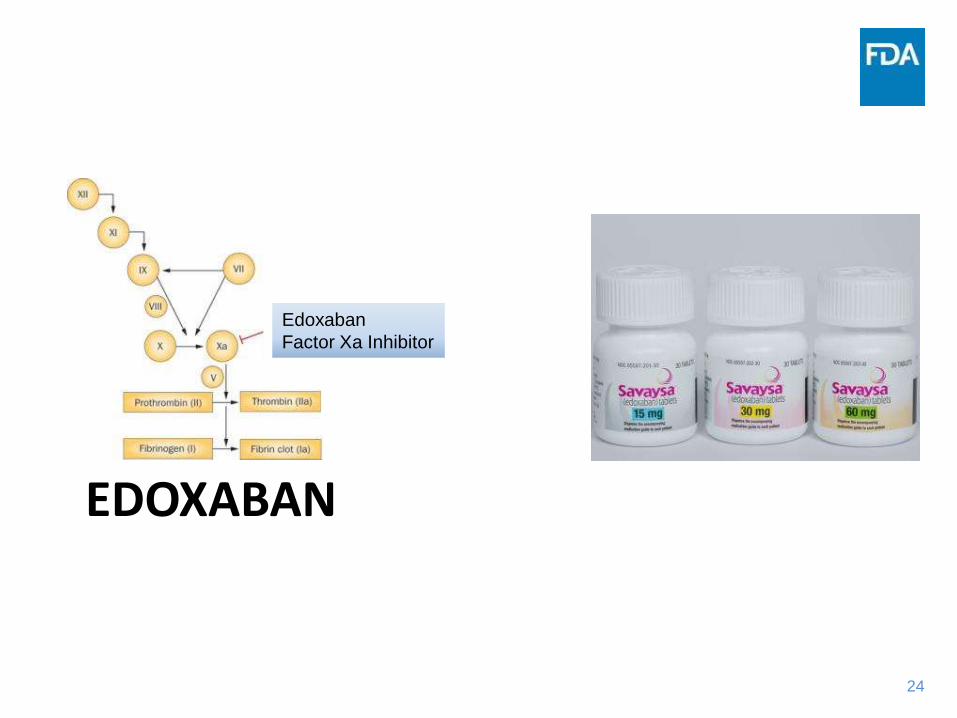
24
EDOXABAN
Edoxaban
Factor Xa Inhibitor

25
Case 2: Edoxaban
• Indicated to reduce the risk of stroke and systemic embolism in patients with non-valvular atrial fibrillation
– Also indicated for treatment of deep vein thrombosis and pulmonary embolism
• Edoxaban inhibits free factor Xa and prothrombinase activity and inhibits thrombin-induced platelet aggregation. Inhibition of factor Xa in the coagulation cascade reduces thrombin generation and thrombus formation.
• Anti-coagulant class associated with steep bleeding and stroke dose and exposure-response curves
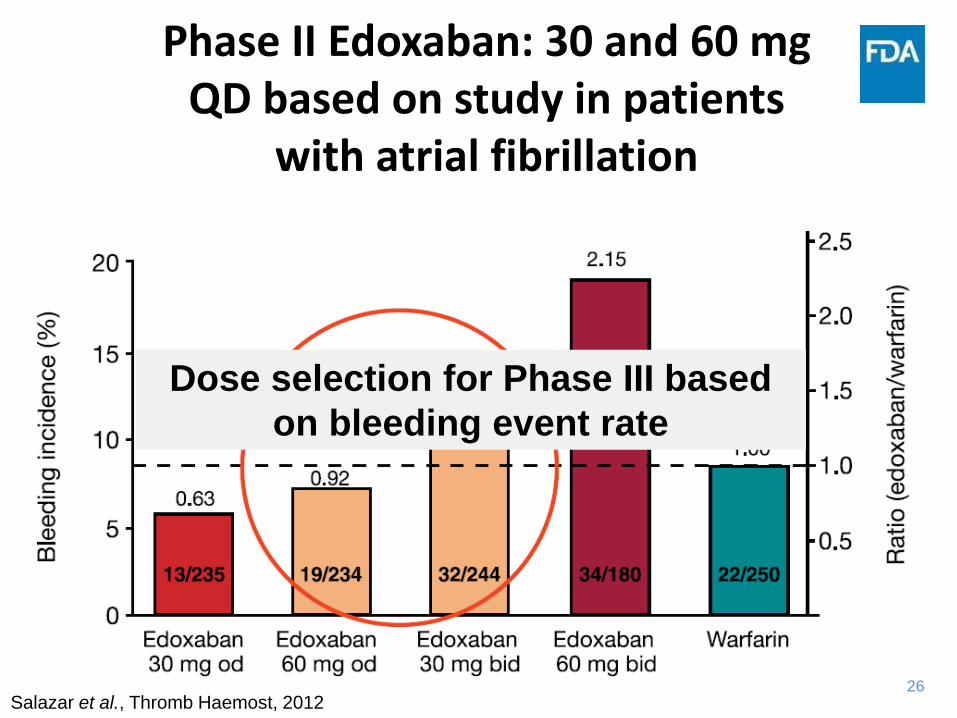
26
Phase II Edoxaban: 30 and 60 mg QD based on study in patients
with atrial fibrillation
Salazar et al., Thromb Haemost, 2012
Dose selection for Phase III based
on bleeding event rate
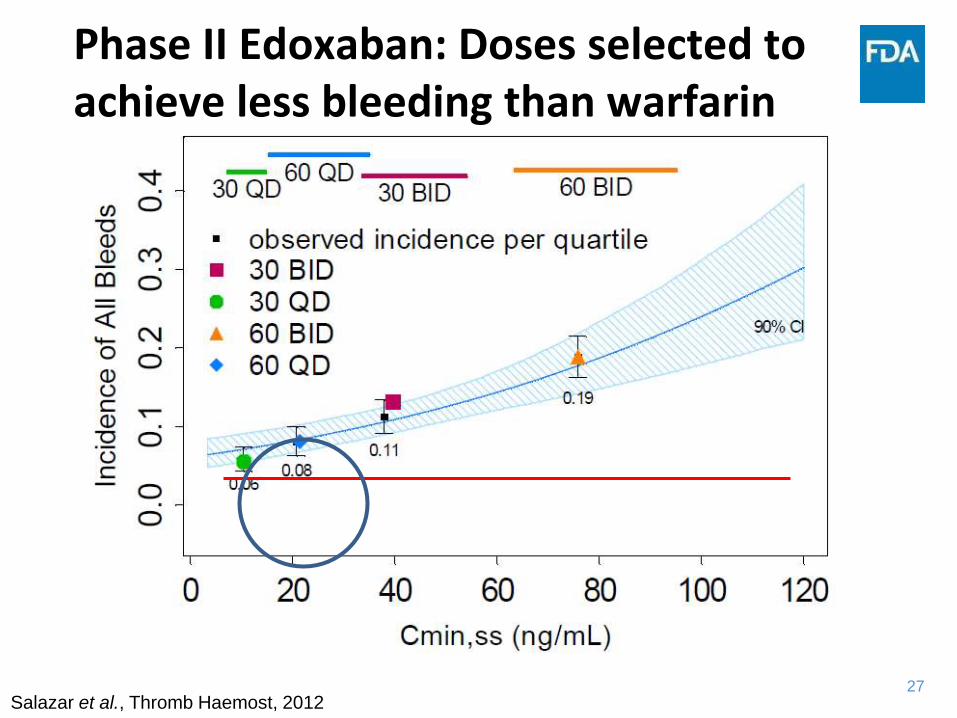
27
Phase II Edoxaban: Doses selected to achieve less bleeding than warfarin
Salazar et al., Thromb Haemost, 2012
28
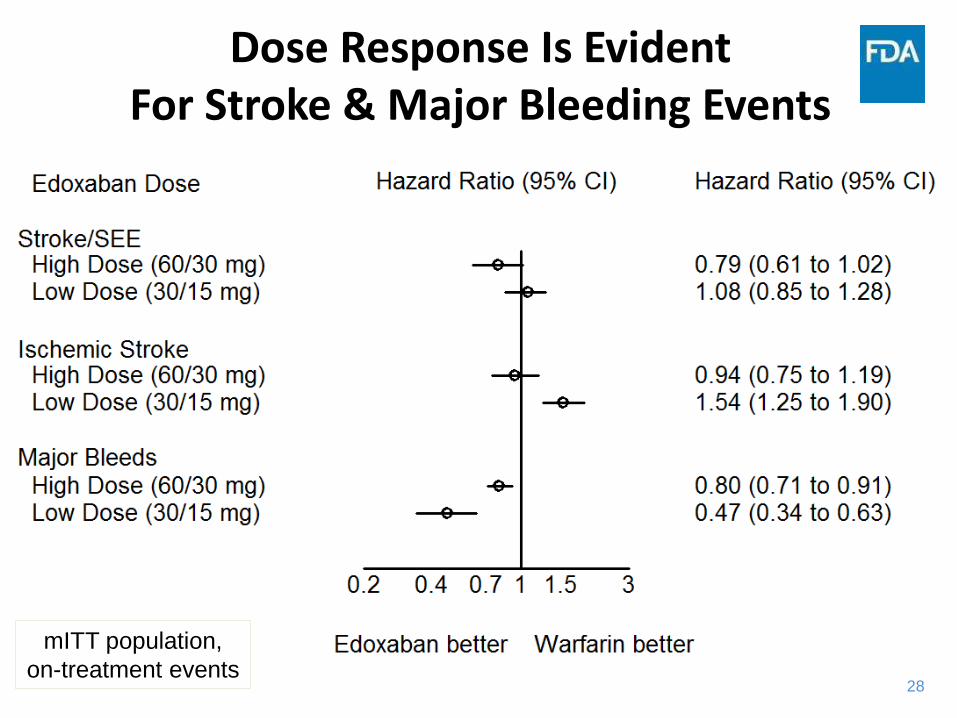
Dose Response Is EvidentFor Stroke & Major Bleeding Events
mITT population,
on-treatment events
29
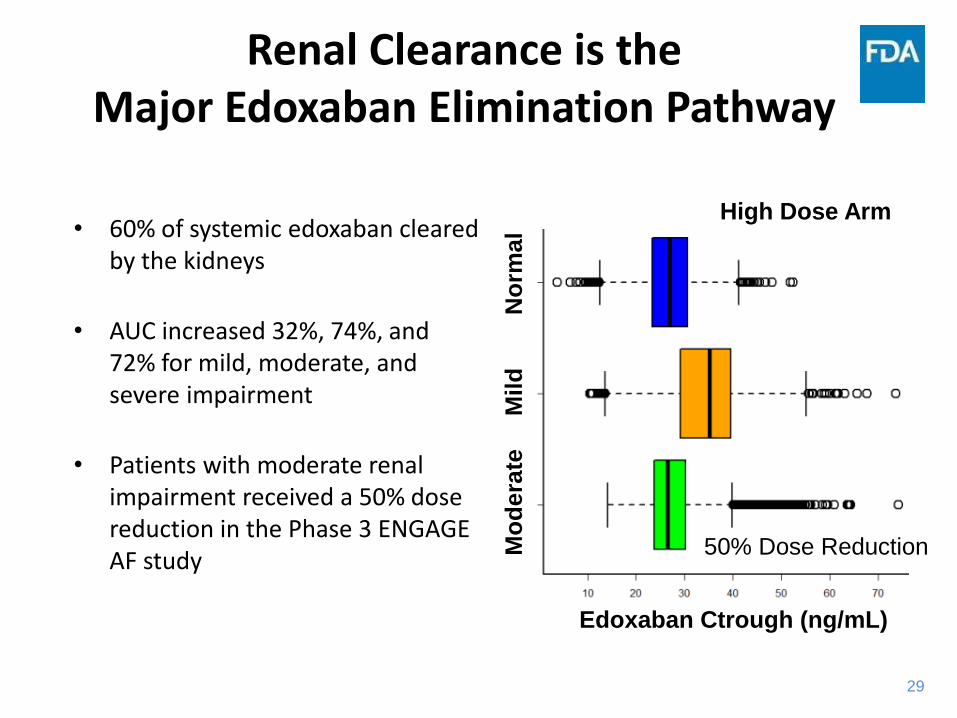
Renal Clearance is the Major Edoxaban Elimination Pathway
• 60% of systemic edoxaban cleared by the kidneys
• AUC increased 32%, 74%, and 72% for mild, moderate, and severe impairment
• Patients with moderate renal impairment received a 50% dose reduction in the Phase 3 ENGAGE AF study
Applicant: CSR U-120, Table 11.2, Edoxaban 60 mg; CSR U-301, Tables 14.2.5.2, 14.5.3.24, Edoxaban 60 mg
Mo
dera
te
M
ild
N
orm
al
Edoxaban Ctrough (ng/mL)
50% Dose Reduction
High Dose Arm
30
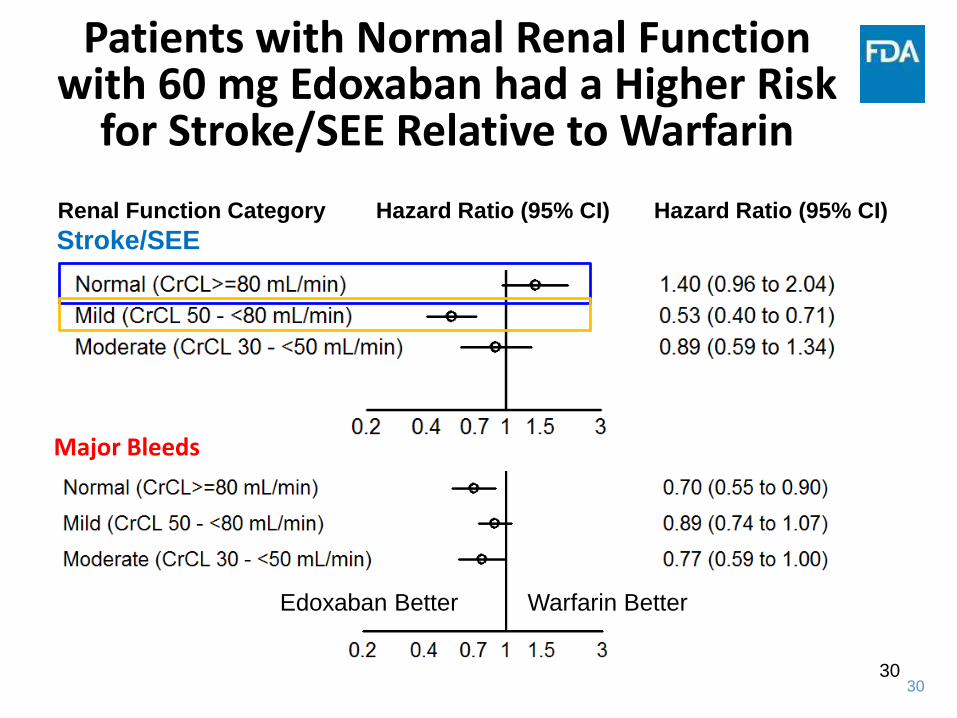
Patients with Normal Renal Function with 60 mg Edoxaban had a Higher Risk
for Stroke/SEE Relative to Warfarin
Applicant: CSR U-120, Table 11.2, Edoxaban 60 mg; CSR U-301, Tables 14.2.5.2, 14.5.3.24, Edoxaban 60 mg
Stroke/SEERenal Function Category Hazard Ratio (95% CI) Hazard Ratio (95% CI)
Edoxaban Better Warfarin Better
Major Bleeds
30

31
Case 2: Outcome
• Concerns identified with stroke event rate in patients with normal renal function
• Such subjects would have lower exposure given edoxaban’sprimary route of elimination
• Boxed warning included in labeling based on renal function >90 mL/min

32
Labeling Outcome
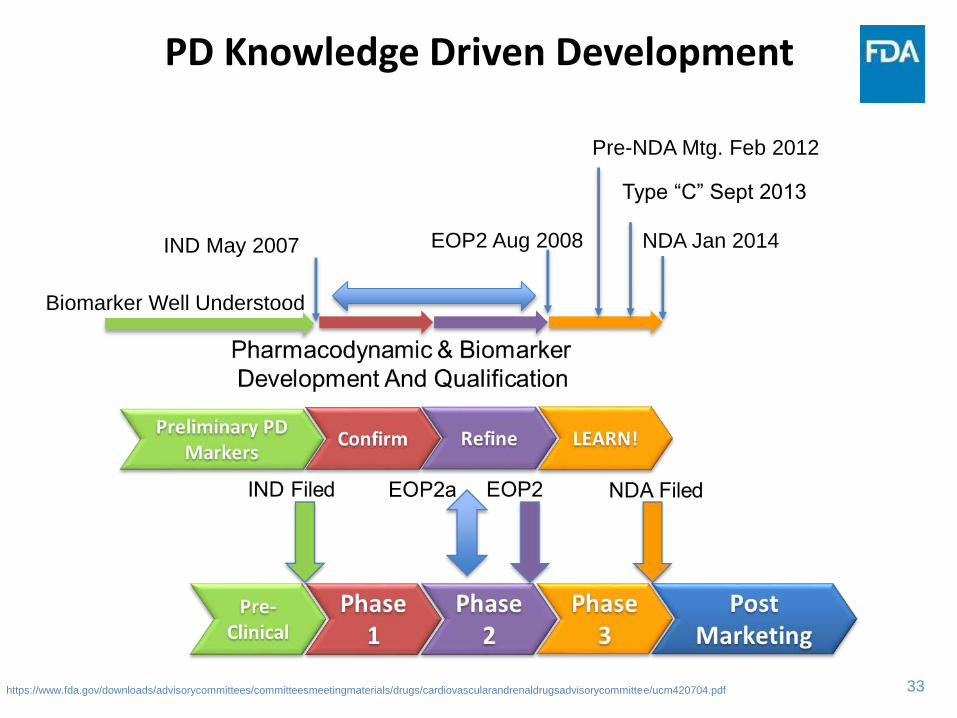
33
PD Knowledge Driven Development
https://www.fda.gov/downloads/advisorycommittees/committeesmeetingmaterials/drugs/cardiovascularandrenaldrugsadvisorycommittee/ucm420704.pdf
Biomarker Well Understood
IND May 2007 NDA Jan 2014
Type “C” Sept 2013
Pre-NDA Mtg. Feb 2012
EOP2 Aug 2008

34
LESSONS LEARNED

35
Time for Development
• Although both Edoxaban and Secukinumab were developed by different companies and were reviewed by different divisions at the FDA they share at least two things in common:
– They both were approved in 7yrs from the time of initial IND filing.
– They both leveraged information from biomarkers
• The clotting cascade for Edoxaban
• The discovery of IL-17’s role for Secukinumab
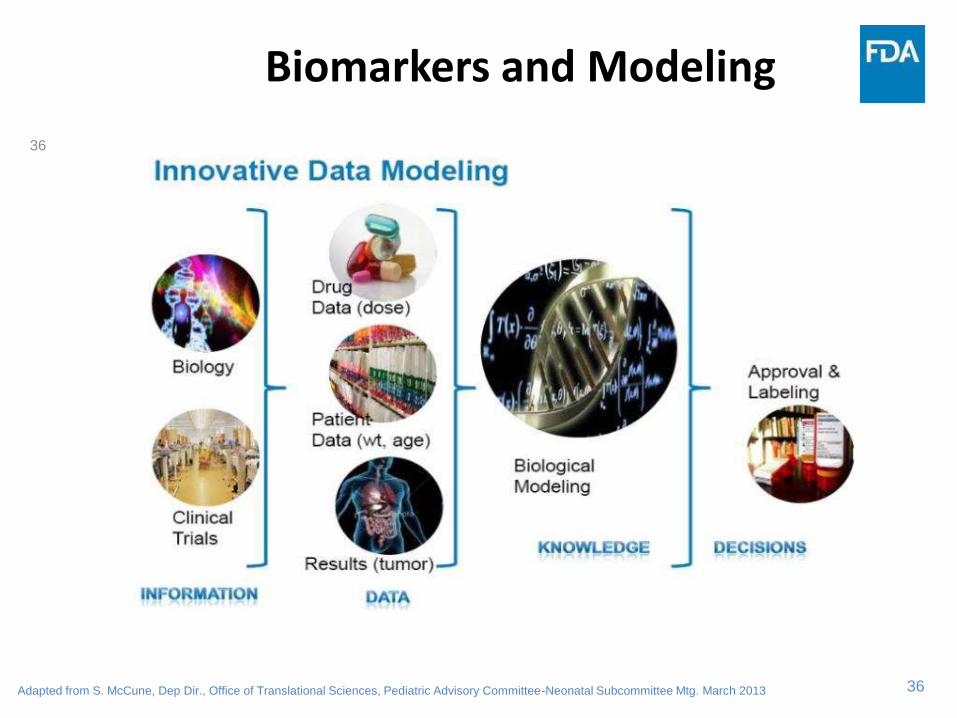
36
36
Biomarkers and Modeling
Adapted from S. McCune, Dep Dir., Office of Translational Sciences, Pediatric Advisory Committee-Neonatal Subcommittee Mtg. March 2013

37
Biomarkers Are the Present and the Future
• Biomarkers– Expand understanding of the disease and its progression
– Provide insight into FUTURE drug development
– Provide the potential for clinician based individualization (i.e., bleeding time vs IL-17A)
– Provide a path forward to a more optimized drug development program
– Provide opportunities for collaboration with regulatory bodies

38
FDA Partnership in Qualification
https://www.fda.gov/drugs/developmentapprovalprocess/drugdevelopmenttoolsqualificationprogram/

39
FDA Qualified Biomarkers
https://www.fda.gov/Drugs/DevelopmentApprovalProcess/DrugDevelopmentToolsQualificationProgram/BiomarkerQualificationProgram/ucm535383.htm

40
CONCLUSIONS AND CLOSING THOUGHTS

41
The Future
• It is easy to say that we are on the edge of a revolution in drug development
– We have ALWAYS been on the EDGE!
– Only the tools and our perspective of them have changed
• Patient factors are being recognized more and more as the key to individualizing not only drug therapy but expectations of therapy.

42
Communicating the Future
• As new advances are made, they must be reflected both in regulatory policy and in patient care
• While we rightly focus on the population, we must not lose sight of the individual patient and the individual physician, nurse, and pharmacist as well– Biomarkers can allow us to link the patient, their genetics, the
ontology of the disease, and the healthcare system into a “holistic” treatment approach
• Clinical Pharmacology can help identify populations and broaden patient utility and safety
• Only by selecting the right biomarkers and identifying the proper dose for the patient population can we make “every patient count” as every patient is a teaching opportunity for us
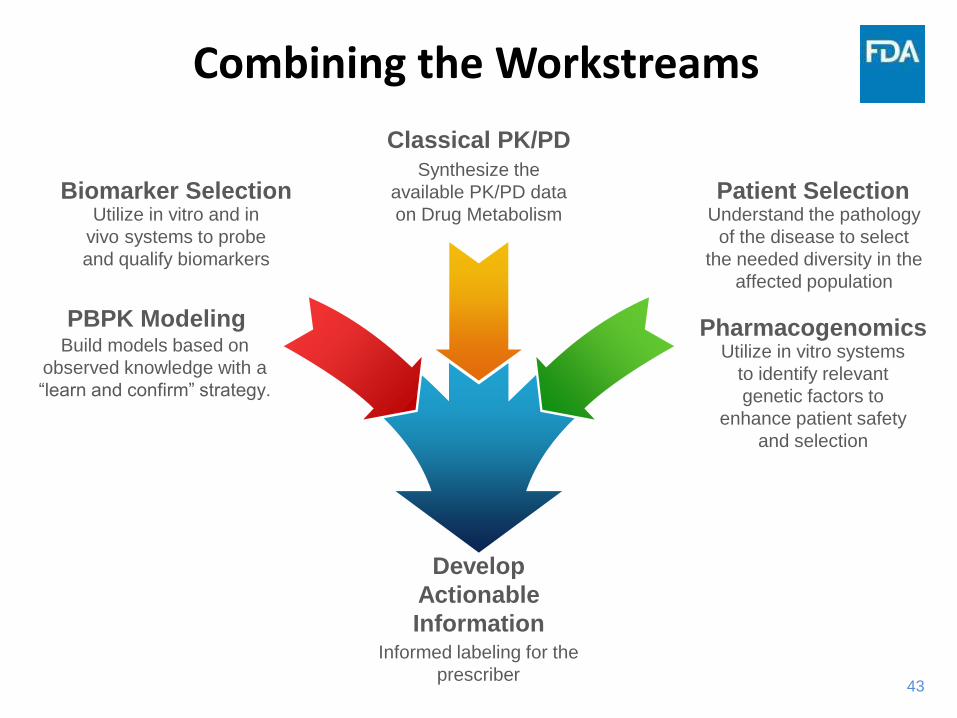
43
Combining the Workstreams
Biomarker SelectionUtilize in vitro and in
vivo systems to probe
and qualify biomarkers
PBPK ModelingBuild models based on
observed knowledge with a
“learn and confirm” strategy.
Classical PK/PDSynthesize the
available PK/PD data
on Drug Metabolism
Develop
Actionable
InformationInformed labeling for the
prescriber
PharmacogenomicsUtilize in vitro systems
to identify relevant
genetic factors to
enhance patient safety
and selection
Patient SelectionUnderstand the pathology
of the disease to select
the needed diversity in the
affected population
44
Development of Safe and Effective Drugs For ALL Requires a Team Effort
Academia
IndustryInternational
Collaboration
Patient
Advocacy
Regulatory
Science
Benefits
To All
Good Science is Everybody’s Business!

45
Contact Information
CAPT Edward D. Bashaw, PharmD.Director, Div. of Clinical Pharmacology-3US FDA10903 New Hampshire AveBuilding 51, Rm [email protected]

46
Acknowledgements
• The Staff of the Division of Clinical Pharmacology-3
• The Office of Clinical Pharmacology
• The Office of Translational Sciences
• The Drug Information Association-China

