digeorge syndrome: part catch 22digeorge syndrome:partofcatch22 an aberrant right subclavian artery....
TRANSCRIPT

8 Med Genet 1993; 30: 852-856
SYNDROME OF THE MONTH
DiGeorge syndrome: part of CATCH 22
D I Wilson, J Burn, P Scambler, J Goodship
AbstractDiGeorge syndrome (DGS) comprisesthymic hypoplasia, hypocalcaemia, out-flow tract defects of the heart, and dys-morphic facies. It results in almost allcases from a deletion within chromo-some 22qll. We report the clinical find-ings in 44 cases. We propose thatDiGeorge syndrome should be seen as thesevere end of the clinical spectrumembraced by the acronym CATCH 22syndrome; Cardiac defects, Abnormalfacies, Thymic hypoplasia, aeft palate,and Hypocalcaemia resulting from 22qlldeletions.(J7 Med Genet 1993;30:852-6)
In 1965 DiGeorge described a child withhypoparathyroidism and recurrent infectionsalong with three necropsy cases of absent thy-mus and parathyroid glands at an immunologymeeting.' The phenotype of the syndrome thatwas named after him was widened to includecongenital heart disease and facial dysmor-phism. The first clue to the aetiology ofDGS came when de la Chapelle reported afamily with a chromosome translocation,t(20;22)(ql1;q 1), in which four subjects withunbalanced products had DGS.2 It is now wellestablished that the vast majority of casesresult from deletions within chromosome22q 1.5 Here we report the phenotypicfeatures in our series of 44 cases of DGS andset this in the broader clinical context of 22ql 1deletions, for which we now propose the acro-nym CATCH 22 syndrome.
Division of HumanGenetics, University ofNewcastle upon Tyne,19/20 ClaremontPlace, Newcastle uponTyne NE2 4AA, UK.D I WilsonJ BurnJ Goodship
Molecular MedicineUnit, Institute of ChildHealth, London WC1N1EH, UK.P Scambler
Correspondence toDr Goodship.Received 21 May 1993.Revised version accepted12 July 1993.
SubjectsThis clinical study is based on 44 children withDiGeorge syndrome (21 female and 23 male)who were referred by members of the BritishPaediatric Association, the British PaediatricCardiac Association, and the Clinical GeneticsSociety. Children were considered to havefulfilled the diagnostic criteria if they had atleast three out of the following four features:cardiac defect, hypocalcaemia, evidence ofthymus hypoplasia, and dysmorphic features.One of these children has features of bothNoonan syndrome and DiGeorge syndromeand has already been described in detail.6
Cardiac abnormalitiesEach child was investigated in a regional pae-diatric cardiothoracic centre by echocardio-
graphy or cardiac catheterisation or both. Ofthe 44 children, 42 had a cardiac defect (table1). In 35 cases the cardiac anatomy was con-firmed at operation or necropsy. The majorityhad obstructive lesions, predominantly affect-ing the left outflow tract. Eighteen had inter-rupted aortic arch (IAA) type B and one IAAtype C. Two had a hypoplastic left heart andone coarctation of the aorta.The right outflow tract was also affected in a
significant proportion of children, the lesionsagain being primarily obstructive. Five hadpulmonary atresia with a ventricular septaldefect (VSD), three had tetralogy of Fallot,one had double outlet right ventricle, and twohad mild isolated pulmonary valve stenosis.
It was common for the children to havemore than one structural defect, for example,VSD in association with IAA. There werethree children who had isolated septal defects.These were a VSD, an atrioventricular septaldefect, and a secundum atrial septal defect.A significant proportion of children were
found to have an aberrant right subclavianartery or a right aortic arch. For example,GOS4 presented with hypocalcaemia but sub-sequently developed dysphagia secondary to
Table 1 Cardiac abnormalities. The major defects arelisted in bold print and the associated abnormalities inthe indented lists.
IAA(type B)/VSDIAA(type C)/VSDRAAARSABicuspid AoVASDMuscular VSDTV/AoV hypoplasiaTruncus, AVSD
TruncusRAAARSASmall LPA
Pulmonary atresia/VSDRAAALSAMAPCAVascular ring
Fallot's tetralogyARSARAADORVAbsent PV
L hypoplastic heartMild PVSASD (secundum)AVSD/CoarctationVSDARSA (isolated)Coarctation
18
2552
521
134
221
l
l
DORV = double outlet right ventricle; ARSA = aberrant rightsubclavian artery; ALSA = aberrant left subclavian artery;PVS = pulmonary valve stenosis; AoV = aortic valve; TV = tri-cuspid valve; RAA = right aortic arch; LPA = left pulmonaryartery; MAPCA = multiple anomalous pulmonary collateralarteries.
852
on March 5, 2020 by guest. P
rotected by copyright.http://jm
g.bmj.com
/J M
ed Genet: first published as 10.1136/jm
g.30.10.852 on 1 October 1993. D
ownloaded from

DiGeorge syndrome: part of CATCH 22
an aberrant right subclavian artery. Althoughthese abnormalities are clinically less severethan major obstructive lesions, they still rep-resent abnormal development of the pharyn-geal arch arteries. IAA type B may result frommaldevelopment of the fourth left branchialarch artery and an aberrant right subclavianartery may arise from maldevelopment of thefourth right branchial arch artery. These twolesions are likely to have a similar embryologi-cal origin but have very different clinical mani-festations.
HypocalcaemiaChildren were considered to be hypocalcaemicif the total ionised serum calcium was less thanthe normal range for the laboratory perform-ing the analysis. Forty children had docu-mented hypocalcaemia out of the 44 cases ofDGS. The serum calcium was not measured inone child as it was clear that she would dierapidly from her cardiac lesion. In three chil-dren hypocalcaemia was not documented de-spite two having received intensive monitor-ing.
In four children the presenting feature was aseizure secondary to hypocalcaemia. A furthernine had a seizure after the diagnosis of acardiac defect.
Information about the onset of hypocalcae-mia was available in 24 cases. In 20 of these,onset was within the first 14 days of life but inone child it was first documented at 5 years ofage when a child presented with hypocalcae-mic convulsions six weeks after operative re-pair of Fallot's tetralogy.The hypocalcaemia resolved in 26 of the 40
cases. Four of the hypocalcaemic childrendied. In the remaining 10 cases treatment hascontinued. Parathormone was measured innine children when they were hypocalcaemicand found to be inappropriately low in eight ofthese.
Figure 1 This figure shows the difficulty in assessing the dysmorphic features in thesick neonate, though the appearance of the ears is helpful in this ventilated neonate.
Thymic abnormalitiesThirty-three children had either low T cells orthymus hypoplasia. Five children were con-sidered to have normal thymus/immunefunction and in six children the data wereinadequate to assess immune function. Thethymus gland could not be identified in 18children undergoing sternotomy for repair of acardiac defect or at necropsy and in six casesthe thymus was considered inappropriatelysmall even accounting for the shrinkage thatoccurs in severely ill neonates. Interestinglythere were several children in whom no thy-mus was identified at operation whose T cellnumbers were only just below the lower limitof normal. The assessment of the thymusgland by chest x ray was often found to beincorrect at surgery or necropsy.Two children had virtually no CD3 positive
cells and both died. With the exception ofthese two cases none of the children had severeinfections related to immunodeficiency,though frequent common infections occurredup to 2 or 3 years of age followed by spon-taneous improvement.
Absolute numbers of T lymphocytes (CD3antigen positive) were between 15 and 50% ofnormal in the majority of cases. While theabsolute counts of both CD4 antigen positiveand CD8 antigen positive were reduced, thelow lymphocyte count was largely explainedby the CD4 count. Formal phytohaemaggluti-nin studies were carried out in only a minorityof children. However, a poor response wasoften noted to the phytohaemagglutinin stimu-lation carried out for karyotyping.
Facial featuresWhile all of the children had some dysmorphicfeatures these had not been noted by the refer-ring clinician in a number of cases. Sometimesthis was because a neonate required assistedventilation and facial features were obscured,as shown in fig 1. In other cases it was becauseof the subtlety of the dysmorphic features.
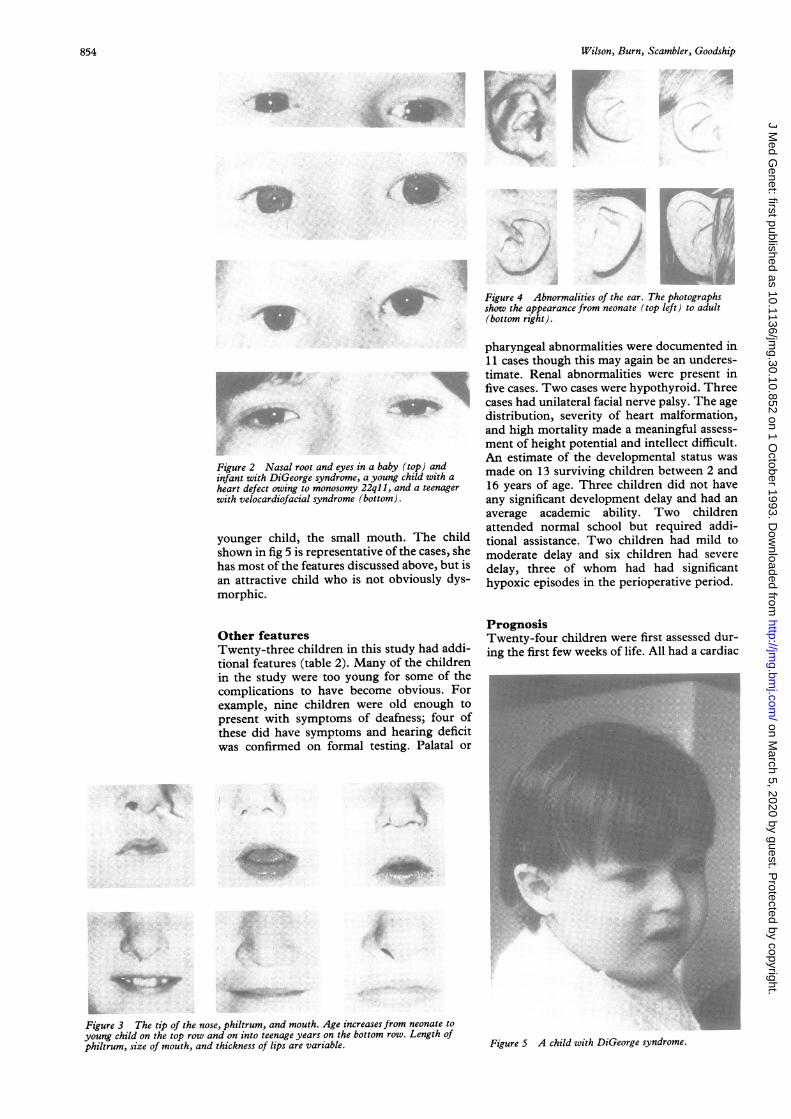
Lateral displacement of the inner canthi wasa frequent finding. The palpebral fissures wereoften short and sometimes narrow. However,the angle of the palpebral fissures was not auseful feature as horizontal, upward, anddownward slanting palpebral fissures were allseen (fig 2).The root and bridge of the nose were wide
and prominent. There was a marked inden-tation on either side of the nasal tip above themidpoint of each nostril. The philtrum wasoften short and poorly modelled but this wasnot a consistent finding, as can be seen fromfig 3.
In the younger children the mouth was oftensmall but this was not so noticeable in the oldercases. The lips were thin in a third of cases.The ears were low set and posteriorly
rotated with deficient upper helices along withan increase in anteroposterior diameter givinga relatively circular ear (fig 4).Most of the features were quite variable.
The features that we found most helpful werethe ear shape, prominent nasal root, and, in the
853
f
-x
.'A
I
on March 5, 2020 by guest. P
rotected by copyright.http://jm
g.bmj.com
/J M
ed Genet: first published as 10.1136/jm
g.30.10.852 on 1 October 1993. D
ownloaded from
Wilson, Burn, Scambler, Goodship~~~~~~~~~~~~~~~~~~~~~~~~~~.
Figure 4 Abnormalities of the ear. The photographs
show the appearance from neonate (top left) to adult
(bottom right).
Figure 2 Nasal root and eyes in a baby (top) andinfant with DiGeorge syndrome, a young child with a
heart defect owing to monosomy 22ql l, and a teenagerwith velocardiofacial syndrome (bottom).
younger child, the small mouth. The childshown in fig 5 is representative of the cases, shehas most of the features discussed above, but isan attractive child who is not obviously dys-morphic.
Other featuresTwenty-three children in this study had addi-tional features (table 2). Many of the childrenin the study were too young for some of thecomplications to have become obvious. Forexample, nine children were old enough to
present with symptoms of deafness; four ofthese did have symptoms and hearing deficitwas confirmed on formal testing. Palatal or
pharyngeal abnormalities were documented in11 cases though this may again be an underes-timate. Renal abnormalities were present infive cases. Two cases were hypothyroid. Threecases had unilateral facial nerve palsy. The age
distribution, severity of heart malformation,and high mortality made a meaningful assess-
ment of height potential and intellect difficult.An estimate of the developmental status was
made on 13 surviving children between 2 and16 years of age. Three children did not haveany significant development delay and had an
average academic ability. Two childrenattended normal school but required addi-tional assistance. Two children had mild to
moderate delay and six children had severe
delay, three of whom had had significanthypoxic episodes in the perioperative period.
PrognosisTwenty-four children were first assessed dur-ing the first few weeks of life. All had a cardiac
..
i.
A*
....7.....
::.: .,
Figure 3 The tip of the nose, philtrum, and mouth. Age increases from neonate to
young child on the top row and on into teenage years on the bottom row. Length ofphiltrum, size of mouth, and thickness of lips are variable. Figure 5 A child with DiGeorge syndrome.
854
on March 5, 2020 by guest. P
rotected by copyright.http://jm
g.bmj.com
/J M
ed Genet: first published as 10.1136/jm
g.30.10.852 on 1 October 1993. D
ownloaded from

DiGeorge syndrome: part of CATCH 22
Table 2 Associated abnormalities.
Neural abnormalitiesSensorineural deafness
Conductive deafnessCerebral hypoplasiaMicrocephalyUnilateral facial nerve palsySuspected hypopituitarismRenal/urogenitalUnilateral renal agenesisUnilateral dilated ureterDilated renal calycesUndescended testesMusculoskeletalScoliosisTalipesPectus excavatumJoint laxityRight lobster claw deformityPalatalBilateral cleft lip and hard palateCleft soft palateSubmucous cleftHigh arched palateVCFS speech without cleftMiscellaneousHypothyroidismColobomaAnteriorly placed anusUmbilical herniaSingle umbilical arteryBilateral single palmar creasesCoronal synostosis
Bilateral 1
Unilateral 2
l
l3
23
2
2
23
23l
defect. Thirteen of these 24 children havedied, 11 as a consequence of their cardiacdefect and two secondary to immuno-deficiency.
In the survivors where development was
assessed, about half had moderate to severe
delay. In half of those with severe develop-mental delay there had been significanthypoxic episodes at the time of operation thatcould have contributed to this.
Recurrence riskParental DNA was analysed in 15 of thedeleted cases in this study. Deletions withinchromosome 22ql 1 were identified in fourmothers and no fathers. None of the fourmothers had a cardiac defect or a history ofconvulsions or frequent infections in child-hood. However, each of these mothers didhave dysmorphic features associated withmonosomy 22ql 1. One of them had two pre-vious children with heart defects.7We have not observed any sibs with a dele-
tion in the absence of a parental deletionthough the numbers of later born sibs remainsmall.
DiscussionThis series of patients was selected on the basisof the DiGeorge phenotype. Criteria for inclu-sion were three of the following: congenitalheart defect, hypoparathyroidism, T lympho-cyte abnormalities, and dysmorphic facies.The cardiac abnormalities were predomi-
nantly obstructive lesions of the outflow tract,particularly IAA. However, the spectrum ofdefects ranged from anatomical abnormalitiesof the subclavian artery with no clinical signi-ficance to left hypoplastic heart. Of note is theabsence of abnormal venous connections inthis group of children.
In four of the children the presentingfeature was hypocalcaemic seizures. Almost a
third of the total cases had a hypocalcaemicconvulsion, underlining the importance ofmonitoring serum calcium in this group.While hypocalcaemia developed in the firsttwo weeks of life and was transient in themajority, the fact that one child first becamehypocalcaemic at the age of 5 years shows thatthe clinician must always be alert to this possi-bility. Our clinical impression is that the hypo-calcaemia is transient but longitudinal studiesare needed to clarify this. It would also beinteresting to monitor calcium changes afteracute illnesses in children in whom the calciumlevels had returned to normal. Management ofhypocalcaemia and hypoxia are obviously im-portant to minimise later developmental prob-lems.
Interpretation of the T lymphocyte data isnot easy as normal ranges have not been estab-lished for these age groups and it is known thatthese parameters do change with age. How-ever, 33 children had either low T cellnumbers or evidence of thymus hypoplasia.Two of the group had virtually no T cells andboth died. It is widespread practice to giveirradiated blood products perioperatively tochildren who have cardiac lesions typical ofDGS unless adequate T cell numbers havebeen documented because of the small risk ofgraft versus host disease developing aftertransfusion of whole blood, plasma reducedblood, and platelet transfusions containingimmunocompetent cells. The vaccination ofchildren with DGS with live vaccines is animportant consideration. These vaccines arepotentially fatal in children with significantimmunodeficiency. Equally important is thedrawback of withholding a vaccine from chil-dren with congenital heart disease who shouldreceive them. The relative risks and merits ofvaccination should be considered in each case.As with the case of parathyroid function, lon-gitudinal studies are required.Hearing deficits were identified in five cases
in this study of largely very young children,and so we would recommend a formal assess-ment of hearing. Five cases also had significantrenal abnormalities and again we wouldrecommend screening for renal anomalies andincreased surveillance for urinary tract infec-tions.
Counselling a couple who have a child withDGS depends on whether either parent has adeletion. So far there have been no docu-mented recurrences in the absence of a paren-tal deletion. We counsel a negligible recur-rence risk to such couples. Our limited datasuggest that in approximately 25% of couples adeletion is present. For these couples the riskof a further pregnancy with monosomy 22ql 1is obviously 50%. However, children withlethal DGS represent one end of a broadclinical spectrum. It may be argued that thepossibility of there being few adverse conse-quences make it inappropriate to use deletionanalysis as the sole basis for intervention inpregnancy. Conversely, even in the absence ofthe severe DiGeorge phenotype, significantclinical problems may ensue. Further data onthe effects of maternal transmission, parental
855
on March 5, 2020 by guest. P
rotected by copyright.http://jm
g.bmj.com
/J M
ed Genet: first published as 10.1136/jm
g.30.10.852 on 1 October 1993. D
ownloaded from

Wilson, Burn, Scambler, Goodship
origin of deletions, and the possibility of gen-etic anticipation are needed to clarify theseissues.
In his original report DiGeorge focused onthymic hypoplasia and hypocalcaemia. Theclinical spectrum was widened to include heartdefects and dysmorphic facial appearance. In1976 Kinouchi and Takao reported a group ofchildren with hypertelorism, narrow palpebralfissures, small mouth, nasal speech, and out-flow defects of the heart in Japanese cardiologypublications and suggested the term conotrun-cal anomaly face.8 They also suggested that10% of Japanese children with tetralogy ofFallot have similar facial features. The Austra-lian cardiologist Radford made the observationthat children with truncus arteriosus and aor-tic arch defects but no thymic or parathyroidinvolvement also often have similar facialfeatures.910 In 1978 Shprintzen et al reported agroup of children with overt or submucousclefting of the palate, cardiac abnormalities,and developmental delay in cleft palate publi-cations. 112 Approximately 10% of this groupdevelop psychiatric illness in adulthood. Thedysmorphic features of all of these conditionsshow considerable overlap and deletionswithin chromosome 22ql 1 have now beenidentified in DGS, velocardiofacial syndrome,conotrucal anomaly face, and cases of isolatedcongenital heart disease"5 '>15 (see also the paperby Bum et al on p 822). We think that theseconditions are all part of one clinical spectrumand that the diagnostic label depends upon theage of presentation and the predominant clini-cal manifestation. We propose that each phe-notype retains its established name but that anencompassing term for referring to the groupas a whole should be CATCH 22.16
We thank all the families who have helped inthis work and the clinicians who have referred
them. The work has been supported by TheBritish Heart Foundation, MRC, and TheBorwick Trust. We also wish to thank Dr COley for her helpful comments on the dysmor-phic features and Oonagh Claber for her helpin preparing the manuscript.
1 Cooper MD, Peterson RDA, Good RA. A new concept ofthe cellular basis of immunity. J7 Pediatr 1965;67:907-8.
2 De la Chapelle A, Herva R, Koivisto M, Aula P. A deletionin chromosome 22 can cause DiGeorge syndrome. HumGenet 1981;57:253-6.
3 Scambler PJ, Carey AH, Wyse RKH, et al. Microdeletionswithin 22ql 1 associated with sporadic and familialDiGeorge syndrome. Genomics 1991,10:201-6.
4 Driscoll DA, Budarf ML, Emanuel BS. A genetic etiologyfor DiGeorge svndrome: consistent deletions and micro-deletions of 22ql 1. Am J Hum Genet 1992;50:924-33.
5 Carey AH, Kelly D, Halford S, et al. Molecular geneticstudy of the frequency of monosomy 22ql 1 in DiGeorgesyndrome. Am J Hum Genet 1992;51:964-70.
6 Wilson DI, Bennett Britton S, McKeown C, et al. Noonan'sand DiGeorge syndromes with monosomy 22qll. ArchDis Child 1993;68:187-9.
7 Wilson DI, Cross IE, Goodship JA, et al. DiGeorge syn-drome with isolated aortic coarctation and isolated ventri-cular septal defect in three sibs with a 22ql1 deletion ofmaternal origin. Br Heart j 1991;66:308-12.
8 Takao A, Ando M, Cho K, Kinouchi A, Murakami Y.Etiological categorization of common congenital heartdisease. In: Van Praagh R, Takao A, eds. Etiology andmorphogenesis of congenital heart disease. New York:Futura, 1980:253-69.
9 Radford DJ. Spectrum of DiGeorge syndrome in patientswith truncus arteriosis: expanded DiGeorge syndrome.Pediatr Cardiol 1988;9:95-101.
10 Radford DJ. Truncus arteriosis and facial dysmorphism.Aust Paediatr 71985;21:131-3.
11 Shprintzen RJ, Goldberg RB, Lewin ML, et al. A newsyndrome involving cleft palate, cardiac anomalies, typi-cal facies, and learning disabilities: velo-cardio-facial syn-drome. Cleft Palate 7 1978;15:56-62.
12 Shprintzen RJ. Goldberg RB, Young D, Wolford L. Thevelo-cardio-facial syndrome: a clinical and genetic analy-sis. Pediatrics 1981;67:167-71.
13 Scambler PJ, Kelly D, Lindsay E, et al. Velo-cardio-facialsyndrome associated with chromosome 22 deletionsencompassing the DiGeorge locus. Lancet1992;339:1 138-9.
14 Wilson DI, Goodship JA, Burn J, Cross IE, Scambler PJ.Deletions within chromosome 22ql1 in familial congen-ital heart disease. Lancet 1992;340:573-5.
15 Driscoll DA, Spinner NB, Budarf ML, et al. Deletions andmicrodeletions of 22q 11.2 in velo-cardio-facial syndrome.Am J Med Genet 1992;44:261-8.
16 Heller J. Catch 22. London: Jonathan Cape, 1962.
856
on March 5, 2020 by guest. P
rotected by copyright.http://jm
g.bmj.com
/J M
ed Genet: first published as 10.1136/jm
g.30.10.852 on 1 October 1993. D
ownloaded from