direct electron transfer from graphite and functionalized gold electrodes to t1 and t2/t3 copper...
TRANSCRIPT

Biochimica et Biophysica Acta 1777 (2008) 1364–1369
Contents lists available at ScienceDirect
Biochimica et Biophysica Acta
j ourna l homepage: www.e lsev ie r.com/ locate /bbab io
Direct electron transfer from graphite and functionalized gold electrodes to T1and T2/T3 copper centers of bilirubin oxidase
Pablo Ramírez a, Nicolas Mano b, Rafael Andreu a, Tautgirdas Ruzgas c, Adam Heller d,Lo Gorton e, Sergey Shleev c,f,e,⁎a Departamento de Química Física, Universidad de Sevilla, 41012 Sevilla, Spainb Centre de Recherche Paul Pascal (CRPP), Université Bordeaux I, UPR 8641, Avenue Albert Schweitzer, 33600 Pessac, Francec Biomedical Laboratory Science, Malmö University, 20506 Malmö, Swedend Department of Chemical Engineering and The Texas Material Institute, The University of Texas, 78712 Austin, USAe Department of Analytical Chemistry, Lund University, 221 00 Lund, Swedenf Laboratory of Chemical Enzymology, Institute of Biochemistry, 119071 Moscow, Russia
Abbreviations: BOD, bilirubin oxidase; NI, native imediate; FR, fully reduced BOD; RF, resting form; DEelectron transfer; IET, intra-molecular electron transfer;midpoint redox potential; ET1, ET2, ET3, and ET2/T3, redox pand the T2/T3 cluster, respectively; ΔEp, peak separationpeaks; k0, standard electron transfer rate constant;constant; Γ, surface concentration; jcat, biocatalytic cubiocatalytic current density; Δjcat(+F−), differences in biocabsence and presence of F−; EDC, N-(3-dimethylaminhydrochloride; NHS, N-hydroxysuccinimide; AMTP, 4mercapto-1-hexanol; MPA, 3-mercaptopropionic acspectrographic graphite electrode; MPA-gold, 3-mergold electrode⁎ Corresponding author. Biomedical Laboratory Scie
Malmö, Sweden.E-mail address: [email protected] (S. Shleev).URL: www.mah.se/shleev (S. Shleev).
0005-2728/$ – see front matter © 2008 Elsevier B.V. Adoi:10.1016/j.bbabio.2008.06.010
a b s t r a c t
a r t i c l e i n f oArticle history:
Direct electron transfer (D Received 8 May 2008Received in revised form 11 June 2008Accepted 17 June 2008Available online 28 June 2008Keywords:Bilirubin oxidaseBioelectrocatalysisO2-electroreductionElectron transfer kinetics
ET) from bare spectrographic graphite (SPGE) or 3-mercaptopropionic acid-modified gold (MPA-gold) electrodes to Trachyderma tsunodae bilirubin oxidase (BOD) was studied underanaerobic and aerobic conditions by cyclic voltammetry and chronoamperometry. On cyclic voltammogramsnonturnover Faradaic signals with midpoint potentials of about 700 mV and 400 mV were clearly observedcorresponding to redox transformations of the T1 site and the T2/T3 cluster of the enzyme, respectively. Theimmobilized BOD was differently oriented on the two electrodes and its catalysis of O2-electroreduction wasalso massively different. On SPGE, where most of the enzyme was oriented with the T1 copper site proximalto the carbon with a quite slow ET process, well-pronounced DET-bioelectroreduction of O2 was observed,starting already at N700 mV vs. NHE. In contrast, on MPA-gold most of the enzyme was oriented with its T2/T3 copper cluster proximal to the metal. Indeed, there was little DET-based catalysis of O2-electroreduction,even though the ET between the MPA-gold and the T2/T3 copper cluster of BOD was similar to that observedfor the T1 site at SPGE. When BOD actively catalyzes the O2-electroreduction, the redox potential of its T1 siteis 690 mV vs. NHE and that of one of its T2/T3 copper centers is 390 mV vs. NHE. The redox potential of theT2/T3 copper cluster of a resting form of BOD is suggested to be about 360 mV vs. NHE. These values,combined with the observed biocatalytic behavior, strongly suggest an uphill intra-molecular electrontransfer from the T1 site to the T2/T3 cluster during the catalytic turnover of the enzyme.
© 2008 Elsevier B.V. All rights reserved.
ntermediate; PI, peroxy inter-T, direct electron transfer; ET,CV, cyclic voltammogram; Em,otentials of the T1, T2, T3 sitesbetween anodic and cathodic
kDET, heterogeneous DET raterrent density; jcat
max, maximumatalytic current densities in theopropyl)-N′-ethylcarbodiimide-aminothiophenol; MHOL, 6-id; DT, 1-decanethiol; SPGE,captopropionic acid-modified
nce, Malmö University, 20506
ll rights reserved.
1. Introduction
Bilirubin oxidase (bilirubin:oxygen oxidoreductase, EC 1.3.3.5) is amulticopper oxidase catalyzing the oxidation of bilirubin to biliverdin,as well as the oxidation of other tetrapyrroles, diphenols and aryldiamines with the concomitant reduction of molecular O2 to H2O [1].Although the crystal structure of BOD is unknown, the structures of itstwo catalytic centers are well understood from biochemical, spectraland kinetic investigations of the wild-type and of mutated forms ofthe enzymes [2–8]. The two centers, one binding and oxidizing theorganic substrate, the other binding and reducing O2, comprise fourcopper ions. In analogy with other blue multicopper oxidases such aslaccase and ascorbate oxidase, the organic substrate binding centercomprises one copper ion and is denoted T1; the O2-binding centercomprises a cluster of three copper centers denoted T2 and T3 [7,9–11]. The T2/T3 copper cluster is one of the few sites encountered innature, which effectively catalyzes the four-electron reduction of O2 toH2O [12,13]. The T1 Cu2+-center accepts electrons not only from the

1365P. Ramírez et al. / Biochimica et Biophysica Acta 1777 (2008) 1364–1369
natural substrates of BOD, but also from artificial electron donors, suchas redox mediators in homogeneous catalysis, or electrodes poised atsufficiently reducing potentials in electrocatalysis. The acceptedelectrons are transferred to the T2/T3 copper cluster, where theyreduce O2 [10]. Interestingly and significantly, when BOD or laccaseare embedded (“wired”) in an electron-conducting redox hydrogel,where electron donors envelope the enzyme such that electrons flowto their T1 centers irrespective of orientation, the copper-enzymes aregreatly superior to platinum as electrocatalysts of the four-electronreduction of O2 to H2O [14]. Here we illuminate some aspects of themechanism of this unique electrocatalytic process.
Electrodes poised at sufficiently reducing potentials transferelectrons directly to fungal BODs from Myrothecium verrucaria andTrachyderma tsunodae [15–17]. Even though M. verrucaria BOD isreadily O2-oxidized when adsorbed on graphite, electron transfer (ET)from graphite to the adsorbed BOD is observed only at 515mV vs. NHEat pH 7.4, about 0.25 V more negative than the reversible potential ofthe O2/H2O redox couple at a low current density [15]. Spectro-electrochemical measurements show that this potential is far from theredox potential of the T1 site (ET1), which is near 670 mV vs. NHE atpH 7.0 [17]. In contrast, M. verrucaria BOD adsorbed on carbonnanotubes is electroreduced already at 685 mV at pH 7.4 [18], but thereduced Cu2+ centers were believed to be those of the T2/T3 cluster,not the T1 site. On gold, very slow DET was observed [17]. Here wereport DET results for T. tsunodae BOD adsorbed on graphite and onmodified gold electrodes. Cyclic voltammograms (CV) exhibit clearlyvisible nonturnover waves with midpoint potentials (Em) of about700 mV and 400 mV, corresponding to redox transformations of theT1 site and the T2/T3 cluster, respectively. This is the first report onwell-pronounced nonturnover signals of electron accepting andoxygen reducing sites of a blue multicopper oxidase coinciding withcorresponding catalytic waves of O2 bioelectroreduction. Importantly,our data strongly suggest an uphill intra-molecular electron transferfrom the T1 site to the T2/T3 cluster during the catalytic turnover ofthe enzyme.
2. Experimental section
2.1. Reagents
Na2HPO4, KH2PO4, KCl, NaCl, H2O2, H2SO4, and NaF were obtainedfrom Merck (Darmstadt, Germany). Citric acid, 2,2′-azinobis-(3-ethylbenzthiazoline-6-sulfonate) (ABTS), and N-(3-dimethylamino-propyl)-N′-ethylcarbodiimide hydrochloride (EDC) were from Sigma(St. Louis, MO, USA).N-hydroxysuccinimide (NHS), 4-aminothiophenol(AMTP), and 6-mercapto-1-hexanol (MHOL) were from Aldrich (St.Louis, MO, USA). 3-Mercaptopropionic acid (MPA) and NaIO4 werepurchased from Janssen Chimica (Geel, Belgium). Absolute ethanol(99.7%) was from Solveco Chemicals AB (Täby, Sweden). 1-Decanethiol(DT) and K3[Fe(CN)6] were from Fluka (Buchs, Switzerland). Allchemicals were of analytical grade. Buffers were prepared with water(18 MΩ) purified with a Milli-Q system (Millipore, Milford, CT, USA).
Anaerobic and aerobic conditions were established using nitrogen(N2) or oxygen (O2) gases from AGAGas AB (Sundbyberg, Sweden) thatwere bubbled through the working solutions.
2.2. Enzymes
BOD from Trachyderma tsunodae was from Amano Enzyme, Inc.(Elgin, IL, USA). Preparations of BOD were stored at −18 °C. Theconcentration of BOD in the stock solution was determined by theestablished method of Ehresmann [19]. The turnover number of BODtowards K3[Fe(CN)6] was determined spectrophotometrically using anUvikon 930 spectrophotometer (Kontron Instruments, Everett, MA,USA) and was found to be 380 s−1, in good agreement with apreviously reported value for a wild-type M. verrucaria BOD [6].
2.3. Electrochemical measurements
Electrochemical measurements were performed using a threeelectrode potentiostat (CV-50W, Bioanalytical Systems, BAS, WestLafayette, IN, USA). The reference electrode was a Hg|Hg2Cl2|KClsatelectrode K401 (SCE, 242 mV vs. NHE) from Radiometer (Copenhagen,Denmark) and the counter electrode was a platinum wire. Thesupporting electrolyte consisted of a 0.1 M phosphate buffer solutionat pH 7.0.
2.3.1. Roughness factor and cleaning of the working electrodesThe gold working electrode, model MF-2014, was purchased from
BAS; its geometrical area was 0.02 cm2. Its microscopic roughnessfactor was calculated from the charge (qreal) associated with the goldoxide reduction process, obtained when running a CV from 0 to1900 mV in 0.5 M H2SO4. The theoretical charge density (σt)associated with the reduction of the gold oxide is 390±10 μC cm−2
[20]. The microscopic area was obtained using the ratio of themeasured charge of gold oxide electroreduction, qreal, and thecalculated theoretical charge for the 0.2 cm2 electrode if it weresmooth, σt (Areal=qreal /σt). The microscopic roughness factor was thusestimated to be 1.4±0.1.
The gold electrode was pre-cleaned by CV scans at a 100 mV/s,between −60 and −1360 mV vs. NHE in 0.5 M NaOH, then polishedwith a DP-suspension (high performance diamond product) and analumina de-agglomerated polishing suspension (1 μm and 0.1 μm,Struers, Copenhagen, Denmark), rinsed with Millipore H2O, andsonicated in after each polishing step for 10 min. The electrode wasthen cleaned by a series of CV scans at a 100 mV/s scan rate between−60 and+1790 mV vs. NHE in 0.5 M H2SO4 and kept in concentrated(96%) H2SO4 till use. Immediately before its use it was rinsedthoroughly with H2O.
The spectrographic graphite electrode (SPGE, Ringsdorff WerkeGmbH, Bonn, Germany, type RW001, 3.05 mm diameter, 13%porosity) was polished with wet fine emery paper (Tufback Durite,P1200), rinsed thoroughly with H2O and allowed to dry. Theadsorptive roughness factor of such an electrode was estimated tobe about 5 [21].
2.3.2. Thiol self-assembled monolayers and BOD deposition on goldThe procedures for physical adsorption of BOD on gold, and for
covalent binding of BOD to AMTP modified gold electrodes werereported earlier [22]. To form DTandMHOL SAMs, the electrodes wererespectively immersed in 1 mM (20:80, v:v, H2O:ethanol) or a 5 mM(absolute ethanol) solution of DT or MHOL, and the monolayers wereallowed to assemble overnight at room temperature. To adsorb BODon bare or modified gold surfaces, the electrode was mounted with itssurface facing up, an aliquot of 5 μl of the BOD solution (10mg/ml) wasplaced on it, and was allowed to react for 3 h, while every 20 min 5 μlof H2O were added to avoid complete drying of the BOD solution.
To form the MPA monolayer, the gold electrode was placed in a5 mM solution (25:75, v:v, H2O:ethanol) of MPA overnight. Toimmobilize on it the BOD the procedure of Rüdiger et al. [23] appliedearlier for the immobilization of Desulfovibrio gigas hydrogenase, wasused. The electrodewas placedwith its surface facing up and 6 μl of ca.25 μM of the BOD solution in 10 mM phosphate buffer pH 7.0 wasadded. Then 4.5 μl of 18 mM NHS and 5.5 μl of 36 mM EDC were alsoadded. After about 90 min the electrode was washed in pH 7.0, 0.1 Mphosphate buffer and was promptly used.
2.3.3. Adsorption of BOD on graphite electrodesA volume of 10 μl of BOD solution (2.5 mg/ml) was placed on the
electrode surface, allowed to adsorb, and after 15 min the SPGE wasrinsed with H2O.
The current densities and enzyme surface concentrations wereestimated using the real area of the electrodes, calculated by

Fig. 1. (A) Cyclic voltammogram of BOD adsorbed on SPGE in the absence of O2. pH 7.0,0.1 M phosphate buffer; 100mV s−1 scan rate; 1000mV starting-scan potential. Curve 1,uncorrected for the background current; Curve 2, circles, background currentsubtracted. The broken line curve represents the calculated theoretical voltammograms.(B) Effect of the scan rate on the background-corrected voltammograms under O2. Curve1, 10 mV s−1; Curve 2, 100 mV s−1.
Fig. 2. Cyclic voltammograms of SPGE covered with a perm selective membrane under1 atm O2. Curve 1, before BOD adsorption; Curve 2, after adsorbing the Trachydermatsunodae BOD. pH 7.0, 0.1 M phosphate buffer; scan rate 10 mV s−1; starting-scanpotential 1000 mV; second scan.
1366 P. Ramírez et al. / Biochimica et Biophysica Acta 1777 (2008) 1364–1369
multiplying the geometrical area by the roughness factor. All reportedpotentials are vs. NHE.
3. Results
3.1. Electrochemistry of BOD adsorbed on graphite
In the absence of O2, two redox processes were observed in the rawCVs of T. tsunodae BOD on SPGE (Fig. 1A); and after subtracting thebackground current (Fig. 1A, curve 2) their voltammetric waves weresymmetrical and well-defined. One wave had an Em of 690 mV and apeak separation (ΔEp) of 155 mV; another had an Em of 390 mV and aΔEp of 73 mV. Calculation of the surface concentration of electroactivespecies (Γ) from the charge associated with the waves and the area ofthe electrodes provided a coverage of 8.3 pmol cm−2 for the first and of5.2 pmol cm−2 for the second, assuming an exchange of one electronper electroactive molecule. The waves fit those expected from theMarcus-DOS theory of ET [24]. The fitting was computed assumingreorganization energy (λ) values within the 0.4–0.8 eV range, asreported for small blue redox proteins containing one T1 site, such asazurin [25] and plastocyanine [26]. The calculated DET standard rateconstant (k0) values were 0.4 s−1 and 1.3 s−1 for the Em=690 mV andEm=390 mV redox processes, respectively. These values are indepen-dent of λ, within the above indicated range, whereas the cathodic rate
constant (kDET) value at 200 mV (the cathodic limit of the usefulelectrocatalytic potential range) strongly depends on λ, and, for theEm=690 mV, it takes values up to 74 s−1 and 437 s−1 for λ of 0.4 and0.8 eV, respectively, i.e. in the order of 100 s−1.
While O2 was not electroreduced on bare SPGE even at a potentialas reducing as 100 mV, it was catalytically reduced on T. tsunodaeBOD-modified SPGE, resembling in this respect M. verrucaria BOD-modified SPGE [15]. The catalytic wave (Fig 1B) started already atN700 mV (Fig. 1B). Moreover, the Em of the first voltammetric waveobserved in the absence of O2 (Fig. 1A) and the mid-wave potential ofthe CV under O2 coincide well and they are close to 690 mV (Fig. 2).The potentials depended quite weakly on the O2 partial pressure, asexpected for a 4-electron reduction process. The maximum bioelec-trocatalytic current density (jmax
cat ) of O2 electroreduction at the BOD-modified SPGEwas ∼40 μA cm−2 (Fig. 1B) based on the true area of theelectrode, i.e. the geometrical area of 0.073 cm2 multiplied by theroughness factor of 5 [15,21]. The engineering current density wastherefore 200 μA cm−2. The BOD-SPGE CVs under O2 were indepen-dent of the scan rate between 10 and 100 mV s−1 in the 730–600 mVpotential range, but did depend on the scan rate at more negativepotential values (Fig. 1B).
The presence of K3[Fe(CN)6] or ABTS did not significantly affect theO2-electroreduction current. When 10 mM F−, a known inhibitor ofBOD [4], was added the true O2-electroreduction current density,observed at 275 mV and 10mV s−1 scan rate, decreased by 17 μA cm−2.
3.2. Electrochemistry of BOD on gold
Attempts to adsorb BOD on bare gold electrode and gold modifiedwith thiol monolayers providing hydrophobic, neutral hydrophilic orcationic-hydrophilic surfaces failed. This was the case when the goldsurface was modified with monolayers of decanethiol; 6-mercapto-1-hexanol or 4-aminothiophenol. Covalent binding of BOD to the anionic-hydrophilic gold surface, by forming amides of chemisorbed 3-mercaptopropionic acid (MPA) and BOD-amines was, however, success-ful. Fig. 3A shows the CVs of anMPA-gold electrode before (curve 1) andafter (curve 2) covalent linking of BOD in the absence of O2. A broadsingle redox process was observed, with an Em of 360 mV, and a 56 mVpeak separation. The area under the anodic peak was somewhat biggerthan the area under its cathodic counterpart, giving an average BODsurface concentration of 10 pmol cm−2, by assuming a one-electrontransfer process. The voltammetric wave was also fitted using theMarcus-DOS theory of ETwith λ values within the 0.4–0.8 eV range. Thedash–dotted voltammogram in Fig. 3A was computed by assuming a
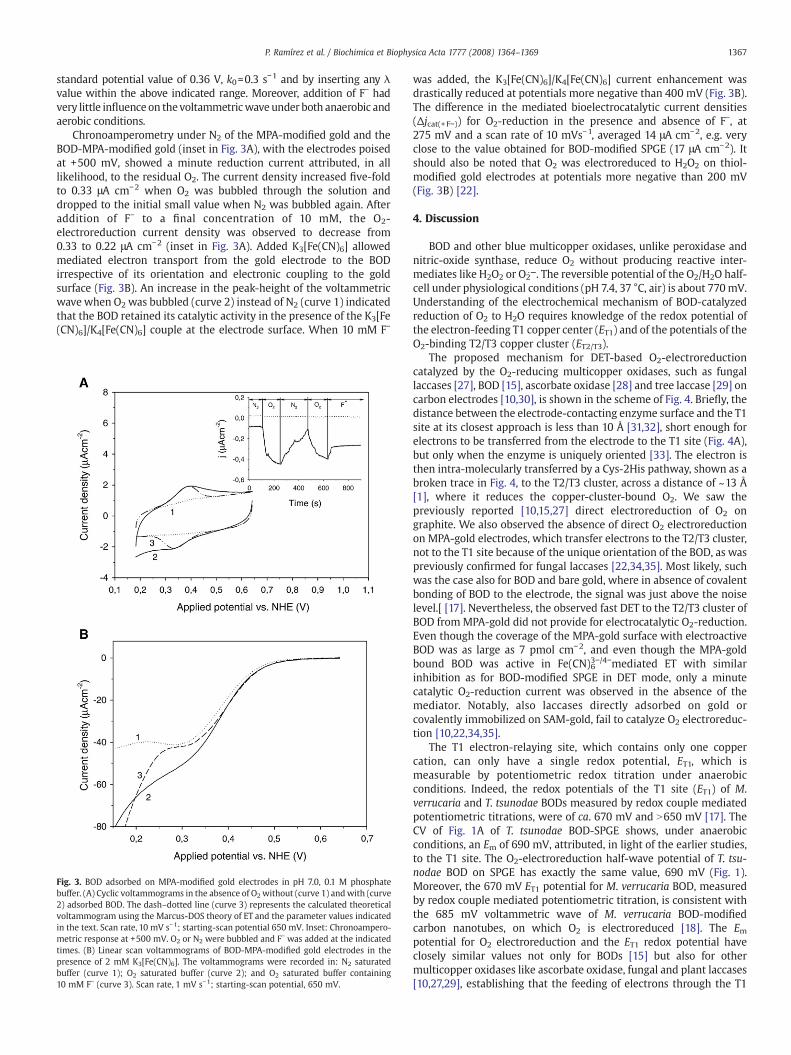
1367P. Ramírez et al. / Biochimica et Biophysica Acta 1777 (2008) 1364–1369
standard potential value of 0.36 V, k0=0.3 s−1 and by inserting any λvalue within the above indicated range. Moreover, addition of F− hadvery little influence on thevoltammetricwave under both anaerobic andaerobic conditions.
Chronoamperometry under N2 of the MPA-modified gold and theBOD-MPA-modified gold (inset in Fig. 3A), with the electrodes poisedat +500 mV, showed a minute reduction current attributed, in alllikelihood, to the residual O2. The current density increased five-foldto 0.33 μA cm−2 when O2 was bubbled through the solution anddropped to the initial small value when N2 was bubbled again. Afteraddition of F− to a final concentration of 10 mM, the O2-electroreduction current density was observed to decrease from0.33 to 0.22 μA cm−2 (inset in Fig. 3A). Added K3[Fe(CN)6] allowedmediated electron transport from the gold electrode to the BODirrespective of its orientation and electronic coupling to the goldsurface (Fig. 3B). An increase in the peak-height of the voltammetricwave when O2 was bubbled (curve 2) instead of N2 (curve 1) indicatedthat the BOD retained its catalytic activity in the presence of the K3[Fe(CN)6]/K4[Fe(CN)6] couple at the electrode surface. When 10 mM F−
Fig. 3. BOD adsorbed on MPA-modified gold electrodes in pH 7.0, 0.1 M phosphatebuffer. (A) Cyclic voltammograms in the absence of O2without (curve 1) andwith (curve2) adsorbed BOD. The dash–dotted line (curve 3) represents the calculated theoreticalvoltammogram using the Marcus-DOS theory of ET and the parameter values indicatedin the text. Scan rate, 10 mV s−1; starting-scan potential 650 mV. Inset: Chronoampero-metric response at +500 mV. O2 or N2 were bubbled and F− was added at the indicatedtimes. (B) Linear scan voltammograms of BOD-MPA-modified gold electrodes in thepresence of 2 mM K3[Fe(CN)6]. The voltammograms were recorded in: N2 saturatedbuffer (curve 1); O2 saturated buffer (curve 2); and O2 saturated buffer containing10 mM F− (curve 3). Scan rate, 1 mV s−1; starting-scan potential, 650 mV.
was added, the K3[Fe(CN)6]/K4[Fe(CN)6] current enhancement wasdrastically reduced at potentials more negative than 400 mV (Fig. 3B).The difference in the mediated bioelectrocatalytic current densities(Δjcat(+F−)) for O2-reduction in the presence and absence of F−, at275 mV and a scan rate of 10 mVs−1, averaged 14 μA cm−2, e.g. veryclose to the value obtained for BOD-modified SPGE (17 μA cm−2). Itshould also be noted that O2 was electroreduced to H2O2 on thiol-modified gold electrodes at potentials more negative than 200 mV(Fig. 3B) [22].
4. Discussion
BOD and other blue multicopper oxidases, unlike peroxidase andnitric-oxide synthase, reduce O2 without producing reactive inter-mediates like H2O2 or O2
·−. The reversible potential of the O2/H2O half-cell under physiological conditions (pH 7.4, 37 °C, air) is about 770mV.Understanding of the electrochemical mechanism of BOD-catalyzedreduction of O2 to H2O requires knowledge of the redox potential ofthe electron-feeding T1 copper center (ET1) and of the potentials of theO2-binding T2/T3 copper cluster (ET2/T3).
The proposed mechanism for DET-based O2-electroreductioncatalyzed by the O2-reducing multicopper oxidases, such as fungallaccases [27], BOD [15], ascorbate oxidase [28] and tree laccase [29] oncarbon electrodes [10,30], is shown in the scheme of Fig. 4. Briefly, thedistance between the electrode-contacting enzyme surface and the T1site at its closest approach is less than 10 Å [31,32], short enough forelectrons to be transferred from the electrode to the T1 site (Fig. 4A),but only when the enzyme is uniquely oriented [33]. The electron isthen intra-molecularly transferred by a Cys-2His pathway, shown as abroken trace in Fig. 4, to the T2/T3 cluster, across a distance of ~13 Å[1], where it reduces the copper-cluster-bound O2. We saw thepreviously reported [10,15,27] direct electroreduction of O2 ongraphite. We also observed the absence of direct O2 electroreductionon MPA-gold electrodes, which transfer electrons to the T2/T3 cluster,not to the T1 site because of the unique orientation of the BOD, as waspreviously confirmed for fungal laccases [22,34,35]. Most likely, suchwas the case also for BOD and bare gold, where in absence of covalentbonding of BOD to the electrode, the signal was just above the noiselevel.[ [17]. Nevertheless, the observed fast DET to the T2/T3 cluster ofBOD fromMPA-gold did not provide for electrocatalytic O2-reduction.Even though the coverage of the MPA-gold surface with electroactiveBOD was as large as 7 pmol cm−2, and even though the MPA-goldbound BOD was active in Fe(CN)63−/4−mediated ET with similarinhibition as for BOD-modified SPGE in DET mode, only a minutecatalytic O2-reduction current was observed in the absence of themediator. Notably, also laccases directly adsorbed on gold orcovalently immobilized on SAM-gold, fail to catalyze O2 electroreduc-tion [10,22,34,35].
The T1 electron-relaying site, which contains only one coppercation, can only have a single redox potential, ET1, which ismeasurable by potentiometric redox titration under anaerobicconditions. Indeed, the redox potentials of the T1 site (ET1) of M.verrucaria and T. tsunodae BODs measured by redox couple mediatedpotentiometric titrations, were of ca. 670 mV and N650 mV [17]. TheCV of Fig. 1A of T. tsunodae BOD-SPGE shows, under anaerobicconditions, an Em of 690 mV, attributed, in light of the earlier studies,to the T1 site. The O2-electroreduction half-wave potential of T. tsu-nodae BOD on SPGE has exactly the same value, 690 mV (Fig. 1).Moreover, the 670 mV ET1 potential for M. verrucaria BOD, measuredby redox couple mediated potentiometric titration, is consistent withthe 685 mV voltammetric wave of M. verrucaria BOD-modifiedcarbon nanotubes, on which O2 is electroreduced [18]. The Empotential for O2 electroreduction and the ET1 redox potential haveclosely similar values not only for BODs [15] but also for othermulticopper oxidases like ascorbate oxidase, fungal and plant laccases[10,27,29], establishing that the feeding of electrons through the T1

Fig. 4. Proposed mechanisms of DET from electrodes to BOD connected (A) via the T1 site and (B) via the T2/T3 cluster.
1368 P. Ramírez et al. / Biochimica et Biophysica Acta 1777 (2008) 1364–1369
from electrodes resembles their feeding by dissolved substrates intheir homogeneous catalysis of O2-reduction.
Prior to this work, one of the redox potentials of the BOD T2/T3cluster has been estimated [17], but not actually measured. The earlierattempted measurement of ET2/T3 of BOD by potentiometric titrationcould not provide potentials of the electrochemical states of thecatalytic cycle. Several different redox potentials are expected for thestructurally different T2/T3 cluster intermediates, i.e. its native andperoxy intermediates [1,12], its fully reduced BOD (FR) [17], and itsresting form [36]. For laccase molecules, uniquely oriented for DET onelectrode surfaces, the voltammetrically observed ET2/T3 is about400 mV at pH 6.5 [22,34,35]. In this study, Em values of 390 mV and360 mV are respectively observed for T. tsunodae BOD on SPGE (Fig.1A) and on MPA-gold (Fig. 3A).
One of the possible explanations for the observed efficientmediated, but not DET, electrocatalysis of O2-electroreduction byBOD on MPA-gold, is the formation of a catalytically inactive restingform of the enzyme during DET from gold directly to the T2/T3copper cluster. The redox couple mediated O2-electroreductioncatalyzing enzyme was normally inhibited by F−, just like the BODon graphite that did catalyze the direct electroreduction of O2. Whenthe BOD was oriented for proximity of the T2/T3 cluster to the MPA-gold, which made it non-catalytic in DET mode, the ET rate was asfast as the ET between the T1 copper site proximal to graphite (k0 are0.3 and 0.4 s−1, respectively) in the enzyme catalyzing direct O2-electroreduction (Fig. 1B).
The observed ET2/T3-potential of 360–390 mV values of BOD (Fig.1A) implies uphill IET from the 690 mV (ET1), over an 300 mV barrierbetween the T1 site and the T2/T3 cluster, at least for one intermediateof the enzyme in the catalytic turnover. It is certainly possible for theresting catalytically irrelevant form of BOD [36], which might have avery slow intra-molecular electron transfer rate, in analogy withresting forms of laccases [12,37,38]. It has indeed been suggested forlaccases that the potential of the native intermediate T2/T3 cluster ishigher than that of the resting enzyme; the native intermediate T2/T3cluster slowly decaying to themore stable resting T2/T3 resting cluster[37]. We consider it nevertheless likely that there is an uphill andcatalytically relevant ET step in the electroreduction of O2: as seen inFig. 1B a sigmoidal wave (marked with an arrow) appears in thevoltammogramsmeasured in the O2-saturated solution at 390mV, thepotential of the ET2/T3 cluster of T. tsunodae BOD (Fig. 1). Participation
of the T2/T3 cluster in O2 bioelectroreduction could be detectable onlyunder conditions, where substrate turnover is limited by the rate ofthe supply of electrons and should become effective as the appliedpotential approaches its formal potential, ET2/T3. Indeed, the absenceof flat-topped current-potential CVs, their sensitivity to change in thepotential scan rate, the starting potential of the bioelectrocatalysisvery close to the potential of the T1 site (Figs. 1B and 2), all are verystrong pieces of evidence for DET limiting bioelectrocatalysis.Importantly, because of significant DET limitation, the standardbiocatalytic rate constant of the adsorbed BOD is suggested to bemuch higher than the calculated kDET, e.g. NN74 s−1, taking intoaccount the mechanism of enzyme function during DET-basedbioelectrocatalysis [39,40]. The turnover of BOD towards K4[Fe(CN)6]in homogeneous catalysis was measured to be 380 s−1. Comparison oftwo biocatalytic rate constants obtained in heterogeneous andhomogeneous assays (NN74 s−1 vs. 380 s−1, respectively) allows us toconclude that BOD adsorbed on SPGE is at least quasi-native anddefinitely catalytically active.
Here for the first time we suggest an endergonic tunneling fromthe T1 site to the T2/T3 cluster during the biocatalytic reduction of O2
by blue multicopper oxidases. It is widely held that many biological ETchains contain an uphill ET step and oxidoreductases are built soindividual ET rates do not have to be optimized [41]. For example, atthe 13 Å distance (approximate distance between T1 and T2/T3 in allblue multicopper oxidases [1]) with unremarkable values of otherfactors affecting the ET rate, the tunneling rate can be as high as fewhundreds s−1 for a 0.3 eV endergonic step [41]. Interestingly, this valuecoincides with the maximal reported turnover numbers of BODsdetermined in homogeneous catalysis (ca. 400 s−1). Indeed, furtherstudies of BOD to understand the mechanism of enzyme function arein the scope of our investigations.
Acknowledgments
We thank Prof. Ulf Ryde at the Department of TheoreticalChemistry, Lund University (Sweden) for helpful suggestions regard-ing the structures of the intermediates of blue multicopper oxidasesand functioning of the enzymes. T.R., L.G. and S.S. thank the SwedishResearch Council. AH thanks the US Office of Naval Research and theWelch Foundation. N.M. thanks funding from a European YoungInvestigator Award (EURYI) and la Région Aquitaine.

1369P. Ramírez et al. / Biochimica et Biophysica Acta 1777 (2008) 1364–1369
References
[1] E.I. Solomon, U.M. Sundaram, T.E. Machonkin, Multicopper oxidases andoxygenases, Chem. Rev 96 (1996) 2563–2605.
[2] Y. Gotoh, Y. Kondo, H. Kaji, A. Takeda, T. Samejima, Characterization of copperatoms in bilirubin oxidase by spectroscopic analyses, J. Biochem. (Tokyo) 106(1989) 621–626.
[3] K. Hiromi, Y. Yamaguchi, Y. Sugiura, H. Iwamoto, J. Hirose, Bilirubin oxidase fromTrachyderma tsunodae K-2593, a multi-copper enzyme, Biosci. Biotechnol.Biochem 56 (1992) 1349–1350.
[4] J. Hirose, K. Inoue, H. Sakuragi, M. Kikkawa, M. Minakami, T. Morikawa, H.Iwamoto, K. Hiromi, Anions binding to bilirubin oxidase from Trachydermatsunodae K-2593, Inorg. Chim. Acta 273 (1998) 204–212.
[5] K. Kataoka, K. Tanaka, Y. Sakai, T. Sakurai, High-level expression of Myrotheciumverrucaria bilirubin oxidase in Pichia pastoris, and its facile purification andcharacterization, Protein Expr. Purif 41 (2005) 77–83.
[6] A. Shimizu, T. Sasaki, J.H. Kwon, A. Odaka, T. Satoh, N. Sakurai, T. Sakurai, S.Yamaguchi, T. Samejima, Site-directed mutagenesis of a possible type 1 copperligand of bilirubin oxidase; a Met467Gln mutant shows stellacyanin-like proper-ties, J. Biochem. (Tokyo) 125 (1999) 662–668.
[7] A. Shimizu, J.H. Kwon, T. Sasaki, T. Satoh, N. Sakurai, T. Sakurai, S. Yamaguchi, T.Samejima, Myrothecium verrucaria bilirubin oxidase and its mutants for potentialcopper ligands, Biochemistry 38 (1999) 3034–3042.
[8] A. Shimizu, T. Samejima, S. Hirota, S. Yamaguchi, N. Sakurai, T. Sakurai, Type III Cumutants of Myrothecium verrucaria bilirubin oxidase, J. Biochem. (Tokyo) 133(2003) 767–772.
[9] S. Koikeda, K. Ando, H. Kaji, T. Inoue, S. Murao, K. Takeuchi, T. Samejima, Molecularcloning of the gene for bilirubin oxidase from Myrothecium verrucaria and itsexpression in yeast, J. Biol. Chem 268 (1993) 18801–18809.
[10] S. Shleev, J. Tkac, A. Christenson, T. Ruzgas, A.I. Yaropolov, J.W. Whittaker, L.Gorton, Direct electron transfer between copper-containing proteins and electro-des, Biosens. Bioelectron 20 (2005) 2517–2554.
[11] T. Sakurai, K. Kataoka, Structure and function of type 1 copper in multicopperoxidases, Cell. Mol. Life Sci 64 (2007) 2642–2656.
[12] L. Rulisek, E.I. Solomon, U. Ryde, A combined quantum and molecular mechanicalstudy of the O2 reductive cleavage in the catalytic cycle of multicopper oxidases,Inorg. Chem 44 (2005) 5612–5628.
[13] I. Bento, M.A. Carrondo, P.F. Lindley, Reduction of dioxygen by enzymes containingcopper, J. Biol. Inorg. Chem 11 (2006) 539–547.
[14] N. Mano, J.L. Fernandez, Y. Kim, W. Shin, A.J. Bard, A. Heller, Oxygen iselectroreduced to water on a “wired” enzyme electrode at a lesser overpotentialthan on platinum, J. Am. Chem. Soc 125 (2003) 15290–15291.
[15] S. Shleev, A. El Kasmi, T. Ruzgas, L. Gorton, Direct heterogeneous electron transferreactions of bilirubin oxidase at a spectrographic graphite electrode, Electrochem.Commun 6 (2004) 934–939.
[16] S. Tsujimura, T. Nakagawa, K. Kano, T. Ikeda, Kinetic study of direct bioelec-trocatalysis of dioxygen reduction with bilirubin oxidase at carbon electrodes,Electrochemistry (Tokyo) 72 (2004) 437–439.
[17] A. Christenson, S. Shleev, N. Mano, A. Heller, L. Gorton, Redox potentials of theblue copper sites of bilirubin oxidases, Biochim. Biophys. Acta 1757 (2006)1634–1641.
[18] M. Weigel, E. Tritscher, F. Lisdat, Direct electrochemical conversion of bilirubinoxidase at carbon nanotube-modified glassy carbon electrodes, Electrochem.Commun 9 (2007) 689–693.
[19] B. Ehresmann, P. Imbault, J.H. Weil, Spectrophotometric determination of proteinconcentration in cell extracts containing tRNA and rRNA, Anal. Biochem 54 (1973)454–463.
[20] S. Trasatti, O.A. Petrii, Real surface area measurements in electrochemistry, PureAppl. Chem 63 (1991) 711–734.
[21] H. Jaegfeldt, T. Kuwana, G. Johansson, Electrochemical stability of catechols with apyrene side chain strongly adsorbed on graphite electrodes for catalytic oxidation
of dihydronicotinamide adenine dinucleotide, J. Am. Chem. Soc 105 (1983)1805–1814.
[22] M. Pita, S. Shleev, T. Ruzgas, V.M. Fernandez, A.I. Yaropolov, L. Gorton, Directheterogeneous electron transfer reactions of fungal laccases at bare and thiol-modified gold electrodes, Electrochem. Commun 8 (2006) 747–753.
[23] O. Rüdiger, J.M. Abad, E.C. Hatchikian, V.M. Fernandez, A.L. De Lacey, Orientedimmobilization of Desulfovibrio gigas hydrogenase onto carbon electrodes bycovalent bonds for nonmediated oxidation of H2, J. Am. Chem. Soc 127 (2005)16008–16009.
[24] K. Weber, S.E. Creager, Voltammetry of redox-active groups irreversibly adsorbedonto electrodes. Treatment using the Marcus relation between rate andoverpotential, Anal. Chem 66 (1994) 3164–3172.
[25] S.J. Corni, The reorganization energy of azurin in bulk solution and in theelectrochemical scanning tunneling microscopy setup, J. Phys. Chem. B 109 (2005)3423–3430.
[26] U. Ryde, M.H.M. Olsson, Structure, strain, and reorganization energy of bluecopper models in the protein, Inter. J. Quant. Chem 81 (2001) 335–347.
[27] S. Shleev, A. Jarosz-Wilkolazka, A. Khalunina, O. Morozova, A. Yaropolov, T. Ruzgas,L. Gorton, Direct electron transfer reactions of laccases from different origins oncarbon electrodes, Bioelectrochemistry 67 (2005) 115–124.
[28] R. Santucci, T. Ferri, L. Morpurgo, I. Savini, L. Avigliano, Unmediated heterogeneouselectron transfer reaction of ascorbate oxidase and laccase at a gold electrode,Biochem. J 332 (1998) 611–615.
[29] A.I. Yaropolov, A.N. Kharybin, J. Emneus, G. Marko-Varga, L. Gorton, Electro-chemical properties of some copper-containing oxidases, Bioelectrochem.Bioenerg 40 (1996) 49–57.
[30] A. Christenson, N. Dimcheva, E. Ferapontova, L. Gorton, T. Ruzgas, L. Stoica, S. Shleev,A. Yaropolov, D. Haltrich, R. Thorneley, S. Aust, Direct electron transfer betweenligninolytic redox enzymes and electrodes, Electroanalysis 16 (2004) 1074–1092.
[31] A. Messerschmidt, R. Ladenstein, R. Huber, M. Bolognesi, L. Avigliano, R.Petruzzelli, A. Rossi, A. Finazzi-Agro, Refined crystal structure of ascorbateoxidase at 1.9 Å resolution, J. Mol. Biol 224 (1992) 179–205.
[32] K. Piontek, M. Antorini, T. Choinowski, Crystal structure of a laccase from thefungus Trametes versicolor at 1.90 Å resolution containing a full complement ofcoppers, J. Biol. Chem 277 (2002) 37663–37669.
[33] H.B. Gray, J.R. Winkler, Electron tunneling through proteins, Quart. Rev. Biophys 36(2003) 341–372.
[34] S. Shleev, A. Christenson, V. Serezhenkov, D. Burbaev, A. Yaropolov, L. Gorton, T.Ruzgas, Electrochemical redox transformations of T1 and T2 copper sites in nativeTrametes hirsuta laccase at gold electrode, Biochem. J 385 (2005) 745–754.
[35] S. Shleev, M. Pita, A.I. Yaropolov, T. Ruzgas, L. Gorton, Direct heterogeneouselectron transfer reactions of Trametes hirsuta laccase at bare and thiol-modifiedgold electrodes, Electroanalysis 18 (2006) 1901–1908.
[36] T. Sakurai, L. Zhan, T. Fujita, K. Kataoka, A. Shimizu, T. Samejima, S. Yamaguchi,Authentic and recombinant bilirubin oxidases are in different resting forms,Biosci. Biotechnol. Biochem 67 (2003) 1157–1159.
[37] S.-K. Lee, S. DeBeer George, W.E. Antholine, B. Hedman, K.O. Hodgson, E.I.Solomon, Nature of the intermediate formed in the reduction of O2 to H2O2 at thetrinuclear copper cluster active site in native laccase, J. Am. Chem. Soc 124 (2002)6180–6193.
[38] J. Yoon, B.D. Liboiron, R. Sarangi, K.O. Hodgson, B. Hedman, E.I. Solomon, The twooxidized forms of the trinuclear Cu cluster in the multicopper oxidases andmechanism for the decay of the native intermediate, Proc. Natl. Acad. Sci. USA 104(2007) 13609–13614.
[39] A. Sucheta, R. Cammack, J. Weiner, F.A. Armstrong, Reversible electrochemistry offumarate reductase immobilized on an electrode surface. Direct voltammetricobservations of redox centers and their participation in rapid catalytic electrontransport, Biochemistry 32 (1993) 5455–5465.
[40] K.A. Vincent, A. Parkin, F.A. Armstrong, Investigating and exploiting theelectrocatalytic properties of hydrogenases, Chem. Rev 107 (2007) 4366–4413.
[41] C.C. Page, C.C. Moser, X. Chen, P.L. Dutton, Natural engineering principles ofelectron tunneling in biological oxidation-reduction, Nature 402 (1999) 47–52.