discussion topic for week 5 : enzyme reactionsserdar/bp/lectures-week5.pdfdiscussion topic for week...
TRANSCRIPT

Discussion topic for week 5 : Enzyme reactions
• The lock in key hypothesis (Emil Fischer) asserts that both the
enzyme and the substrate possess specific complementary
geometric shapes that fit exactly into one another.
What are the problems associated with this hypothesis?

Enzymes and Molecular Machines (Nelson, chap. 10)
Enzymes are biological catalysts that enhance the rate of chem. reactions.
Machines use free energy from an external source (e.g. ATP,
concentration or potential difference) to do useful work. Examples:
• Motors: transduce free energy into linear or rotary motion
– myosin on actin in muscles, kinesin on microtubules in cells.
• Pumps: create concentration differences across membranes
– sodium-potassium pump transports 3 Na+ ions out of the cell and
2 K+ ions into the cell in one cycle.
• Synthases: drive chemical reactions to synthesize biomolecules
– ATP synthase synthesizes the ATP molecules that are used by
most of the molecular machines in the cells.

Enzymes
An extreme example: catalese
Consider the decomposition of hydrogen peroxide: H2O2 H2O + ½ O2
DG0 = -41 kT so the reaction is highly favoured but due to a high
activation barrier it proceeds very slowly:
for 1 M solution the rate is 10-8 M/s (reaction velocity)
Adding 1 mM catalese into the solution increases the rate by 1012 !
10-3 NA catalese molecules perform 104 NA hydrolisis reactions per sec.
So 1 catalese molecule catalyses 107 reactions per sec. (rate: 10-7 s)
H2O2 is produced in cells while eliminating free radicals. Because it is
toxic, its rapid breakdown is important.
More typical rates for enzymes are around 103 s-1

Simple model of enzyme reactions:
Chemical reactions involving biomolecules are extremely complex.
Free energy surface typically involves thousands of coordinates.
Nevertheless a reaction usually proceeds along the path of least
resistance (called reaction coordinate) which allows a simple description.
A simple reaction: H + H2 H2 + H
transition
state

An enzyme facilitates a chemical reaction by binding to the transition
state and thereby reducing the activation energy, DG‡ (but not DG)
Free energy surface along the reaction coordinate
kTEkTEkSkTSTEkTG eeeee D-D--D-D- )(rate
Substrate Enzyme + substrate
DG‡
DG‡
DG DG
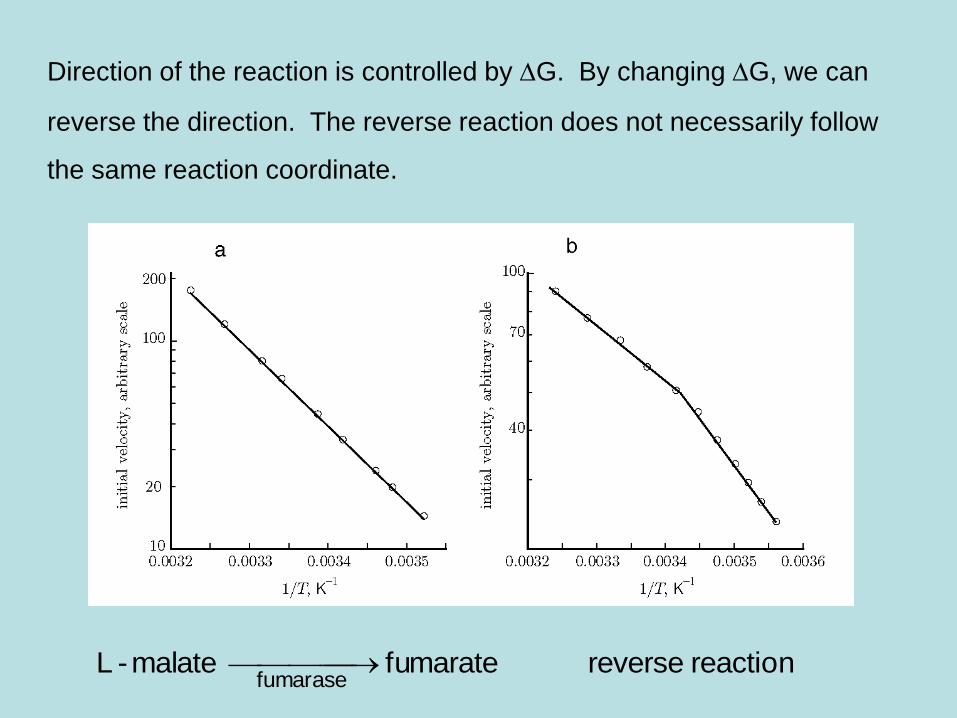
Direction of the reaction is controlled by DG. By changing DG, we can
reverse the direction. The reverse reaction does not necessarily follow
the same reaction coordinate.
reaction reversefumarate malate-Lfumarase

A schematic picture of an enzyme E binding to a substrate S:
E + S ES EP E + P
E+S: The enzyme has a binding site that is a good match for the subst. S
ES: In order to bind, S must deform which stretches a bond to breaking pt.
EP: Thermal fluctuations break the bond producing an EP complex
E+P: The P state is not a good match to the binding site, hence it unbinds,
leaving the enzyme free for binding of the next substrate.

Corresponding
free energy
surface

Enzyme Kinetics:
Consider an enzyme reaction with rate constants k1, k2 and k3
Assume:
For a single enzyme, the reaction simplifies to
Let probability of E unoccupied be PE and occupied PES = (1- PE)
The rate of change of PE is
},{,,},,{ 2133322 kkkkkkkccc PES --
ESESE PkkPck
dt
dP211 - -
PEEPESSEk
k
k
k
k
k
--- 3
3
2
2
1
1
PEESSEk
k
ck S
-
2
1
1

Assuming quasi-steady state, the time derivative vanishes, yielding
Rate of production of P per enzyme:
Reaction velocity for a concentration cE of enzymes
1
212max
max
121
122
,k
kkKkcv
cK
cvv
ckkk
ckkcPkcv
ME
SM
S
S
SEESE
-
-
S
SES
ESEsS
ckkk
ckP
PkkPck
121
1
211 0)1(
--
-
-
ESPk2
Michaelis-Menten (MM) rule

Experimental data for pancreatic carboxypeptidase
S
M
SM
S
c
K
vvcK
cvv 1
11
maxmax
vmax=0.085 mM/s
KM=6.4 mM

MM rule displays saturation kinetics, which has very general validity
The key idea is the processing time for S P
At low substrate concentrations, there are more enzymes than S so that
there is no waiting and hence v is proportional to cS
As cS is increased beyond KM, there is competition among S for access
to an enzyme, and they have to queue for processing.
Maximum velocity of the reaction is determined by the number of
enzymes available and the processing rate (the rate limiting step)
Modulation of enzyme activity:
• Regulate the rate of enzyme production
• Competitive inhibition: direct binding of another molecule
• Noncompetitive inhibition: binding of a molecule to a second site
kTGekD-2
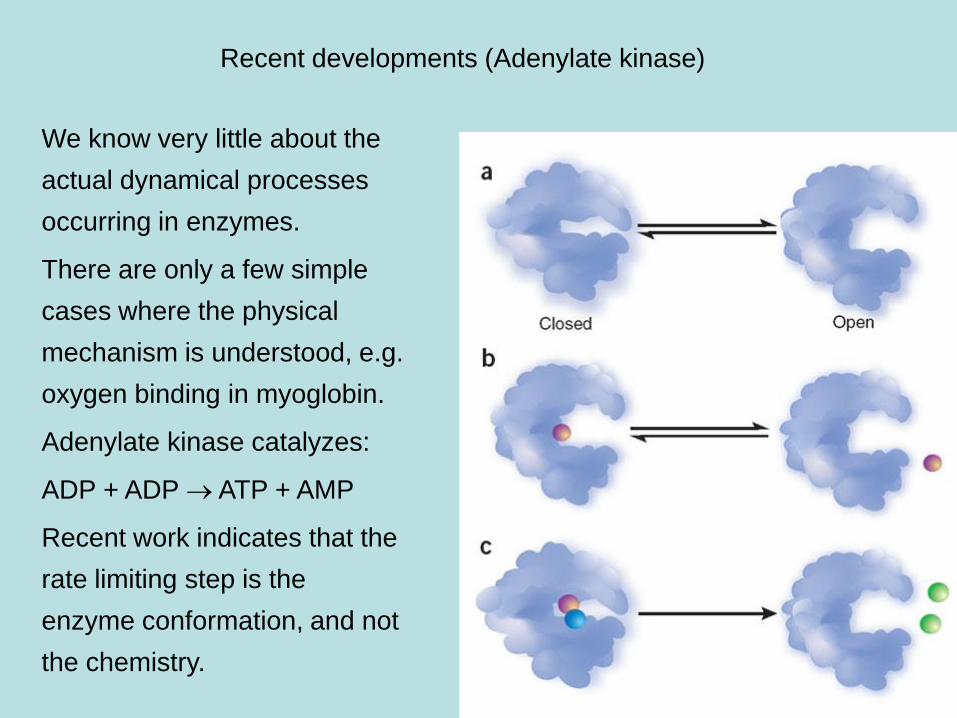
Recent developments (Adenylate kinase)
We know very little about the
actual dynamical processes
occurring in enzymes.
There are only a few simple
cases where the physical
mechanism is understood, e.g.
oxygen binding in myoglobin.
Adenylate kinase catalyzes:
ADP + ADP ATP + AMP
Recent work indicates that the
rate limiting step is the
enzyme conformation, and not
the chemistry.

Molecular motors in muscles: myosin and actin
For structure of the myosin and actin filaments in a myofibril, see
http://distance.stcc.edu/AandP/AP/AP1pages/Units5to9/Unit7/myofibri.htm
Experiment with optical tweezers demonstrates how myosin pulls an actin
filament when 1 mM of ATP is added to the system.
From Finer et al. “Single myosin molecule mechanics” Nature, 1994.

Translocation of proteins across membrane:
Proteins produced in the cell are exported outside through proteins in the
membrane that form pores. To pass through the pore, the protein has to
unfold. The reverse motion is suppressed because the chemical
asymmetries between inside and outside of the cell leads to a more
stable protein structure outside.
Factors contributing to
asymmetry:
• pH
• ion concentration
• disulfide bonding
• binding of sugars
protein catalyzes translocation

Macroscopic machines are deterministic, there are no random fluctuations
But molecular machines operate in a noisy environment with lots of
random fluctuations.
Consider the ratchets below as possible models for molecular machines.
In G-ratchet the spring retracts during the passage but pops back after
In S-ratchet a latch releases the spring after the passage, which stays up
Can either ratchet pull a load f towards right doing useful work?
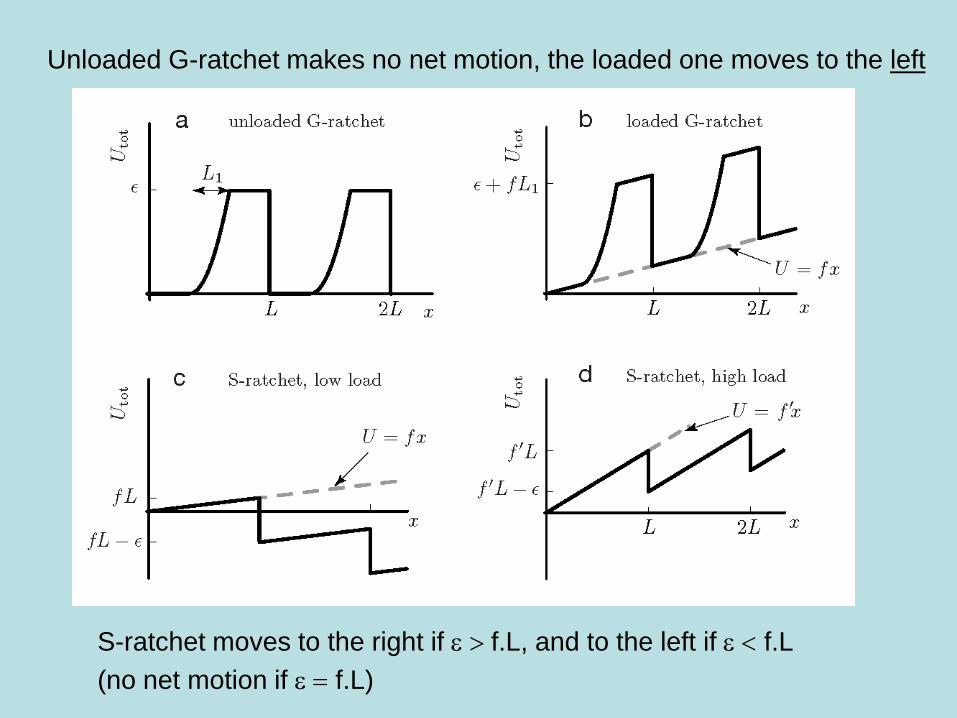
Unloaded G-ratchet makes no net motion, the loaded one moves to the left
S-ratchet moves to the right if e f.L, and to the left if e < f.L
(no net motion if e f.L)

Simple model for a perfect Brownian ratchet: (e kT)
In the absence of any forces, the ratchet diffuses freely until it travels a
distance L. From
Thus the average speed is:
Next we introduce a load f that pulls the ratchet to the left.
The potential energy increases as
in the interval [0, L]
From Boltzmann distribution, the
equilibrium probability will be like
DLtDtx step 22 22
LDtLv step 2
fxU
0)( - kTfxexP

We need an equation to describe the nonequilibrium probability distribution
of the ratchet’s position (cf. Fick’s law and Nerst-Planck Eq.)
| | | | | x Dx L
a-Dx/2 a a+Dx/2 a+Dx
The net flux from a a+Dx depends on (1) the probabilities at those points
and (2) the external forces. If there are N ratchets in our ensemble,
the bins at a and a+Dx have ratchets.
Assuming they move randomly, the net migration from left to right is
xxaNPxaNP DDD )()( and
axax
RL
dx
xdPtND
dx
xdPxN
xxaPaPNN
D-D-
DD-D
)()(
2
1
)()(2
1
2
)1(

Next consider the flux due to an external force,
Drift velocity due to this force:
The number of ratchets moving from left to right:
Adding the two contributions and dividing by Dt, we obtain for the flux
Steady state: flux is constant, and from continuity eq. it is also uniform
)( kTDdx
dU
kT
D
kT
Dffvd -
axdRL
dx
dUt
kT
NDPtvaNPN
D-DD )()2(
dx
dUf -
-
dx
dU
kT
P
dx
dPNDj
00
dx
dU
kT
P
dx
dP
dx
d
dx
dj(Smoluchowski eq.)

Since the potential is periodic, the solutions must be
periodic too.
First consider the equilibrium case:
A possible nonequilibrium solution for the perfect ratchet is
1. vanishes at x = L
2. yields a constant flux
3. hence solves the Smoluchowski eq.
kT
NDCfe
kT
fe
kT
fNDC kTfLxkTfLx
--- ---- 1)()(
1)( )( - -- kTfLxeCxP
)()( xULxU
)( fxU
kTUCexPdx
dU
kT
P
dx
dPj - )(00 (Boltzmann dist.)

Average speed of the perfect ratchet:
The average number of ratchets in the interval [0, L]
The time it takes for these ratchets move is
and the speed is
12
1
1
1
-
-
--
--
D
kT
fLe
kT
fL
L
D
kT
fLe
NCkT
f
kT
NCDfL
N
Lj
t
Lv
kTfL
kTfL
jNt D
--
--
-
-
--
kT
fLe
f
kTNCxe
f
kTNC
dxeNCdxxNPN
kTfLL
kTfxL
LkTfLx
L
1
1)(
0
)(
0
)(
0

Too complicated to make sense, so consider the limits:
Plot of the ratchet
speed / (2D/L)
as a function of
z = fL/kT
Activation barrier kicks
in around fL = 5 kT
kTfLekT
fL
L
DvkTfL
L
Dz
zzz
L
Dv
kT
fLz
-
-
--<<
2
122
.2
21
211.1
e
→ activation barrier

Estimate the speed for a typical molecular machine
For small molecules, ions etc.: R 1-3 Å, D 10-9 m2/s
For macromolecules, proteins: R 1-3 nm, D 10-10 m2/s
Typical length scale: L = 1 nm
Average speed: v = 2D/L = 0.2 m/s, (e.g. to move 200 steps takes 1 ms)
The perfect ratchet assumption is that backward rate vanishes
When the forward and backward rates become equal and no
net motion is possible.
In summary:
1. Molecular machines move by random walk over free energy surface
2. Their speed is determined by the activation energy barrier (but not e)
)( fLe
fLe
R
kTkTD
6

Molecular Recognition
Cells contain thousands of different proteins.
Each protein performs a specific task that may require its interaction with
a specific biomolecule, e.g. DNA, another protein or a ligand.
How does a protein distinguish that biomolecule from the thousands of
others that are floating around the cell?
The lock and key hypothesis of Fischer (1894), namely, shape
complementarity of the interacting parts, provided the first clues.
Going beyond the descriptive accounts of protein interactions using
cartoons to a quantitative accounts that can make predictions has only
become possible in the last decade thanks to the advances in
• Structure determination of complexes and single molecule exp’s
• Computer power and simulation methods

Molecular recognition covers a vast area of research
• Enzyme function
• Protein-ligand interactions: binding of a ligand changes the
conformation of a protein enabling its function, e.g. ligand-gated ion
channels, oxygen binding to hemoglobin.
• Protein-protein interactions: e.g. formation of protein complexes
(tertiary structure), signal transduction across membrane, protein transport
and modification
• Protein-DNA (or RNA) interactions: reading and duplication of DNA,
protein manufacturing
• Protein interactions with non-native peptides: e.g. toxins from the
venomous animals (spiders, snakes, scorpions, snails)
• Protein interactions with chemical compounds: e.g. drugs

Experimental methods:
• Structure determination of complexes using x-ray diffraction or NMR
• Measurement of dissociation (or binding) constants.
(mM range: weak binding, μM range: intermediate, nM range: strong)
• High-throughput screening (automated testing of large number of
compounds to discover new drugs)
Theoretical methods:
• Docking methods (popular in “in silico” drug design)
• Monte Carlo methods: search for the free energy minimum using the
Metropolis algorithm
• Brownian dynamics simulations: water is treated as continuum and
protein is rigid, but simulations are fast enough to observe docking
• Molecular dynamics simulations: realistic representation but too slow
to observe docking

Crystal structure of the barnase (blue) - barstar (green) complex
The unbound conformations are superimposed in light blue and orange.

Close up view showing the side chain pairs in the hot spot.
In the complex: barnase (blue) - barstar (green)
Comparison of
the two structures
shows the
importance of side
chain flexibility

Docking methods
There are various docking methods that search for the free energy
minimum of a protein-macromolecule system. The basic ingredients are:
• A phenomenological energy functional. Typically consists of:
electrostatic, Lennard-Jones, hydrogen bond, solvation and entropic
terms. It is parametrized using a training set.
• A search algorithm. Two common methods employed:
1. Random search using the Monte Carlo method
2. Systematic search using a grid over the active site
In the current docking methods, ligand flexibility (mainly torsion angles) is
also taken into account (target protein is still rigid). Here genetic
algorithms provide a very efficient tool (different conformations correspond
to mutations). AutoDock is the most popular method at present.

Computer simulation of protein interactions
Protein association can be broadly divided into two stages:
1. Diffusional motion until they form an encounter complex
2. Non-diffusional rearrangement process leading to the final bound
complex.
The first stage could take quite a long time (ms), so it is neither possible
nor desirable to use molecular dynamics. Brownian dynamics (BD) is the
natural tool for this stage.
The second stage involves conformational changes in the protein, and
also dehydration and rehydration of water molecules. Thus a microscopic
description that treats all the atoms in the system is necessary at this
stage, which is provided by molecular dynamics (MD).
The focus is, however, on the binding. Can we avoid the BD stage?
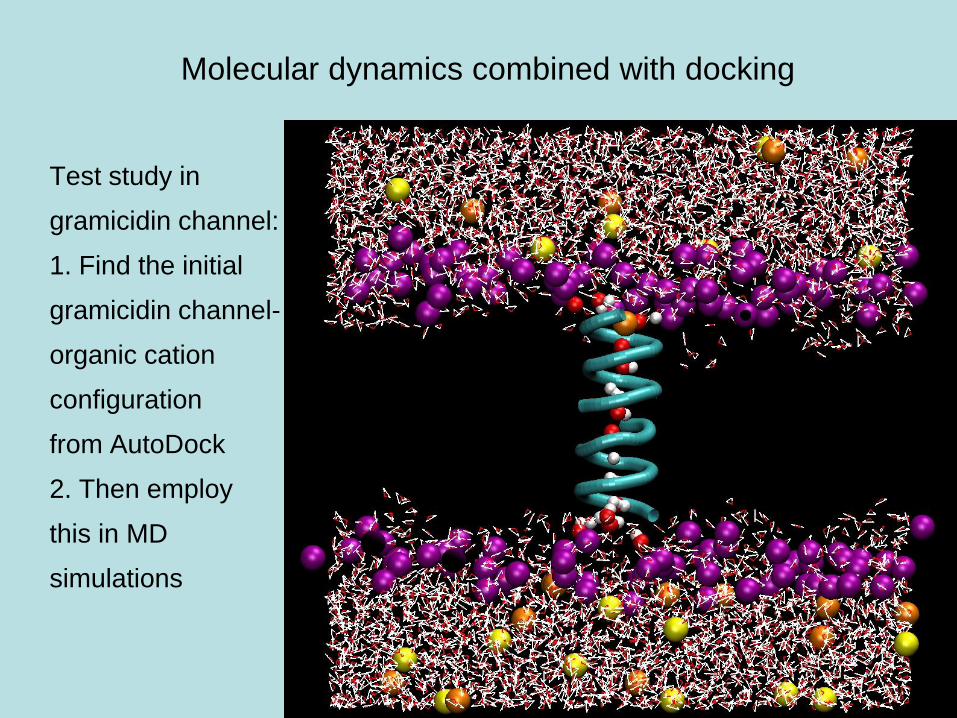
Molecular dynamics combined with docking
Test study in
gramicidin channel:
1. Find the initial
gramicidin channel-
organic cation
configuration
from AutoDock
2. Then employ
this in MD
simulations
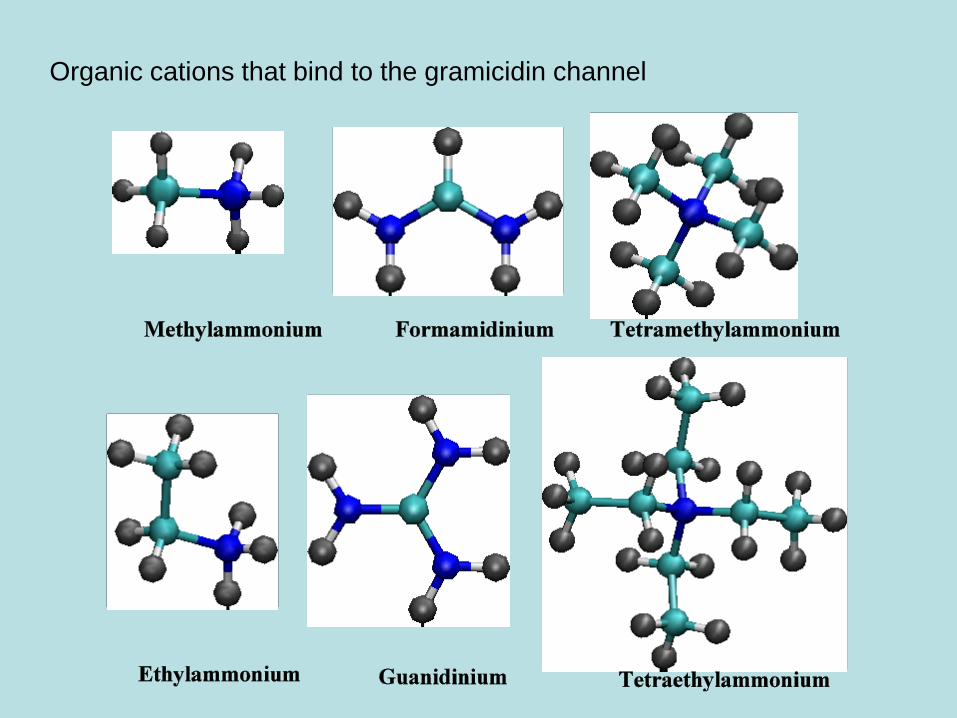
Organic cations that bind to the gramicidin channel
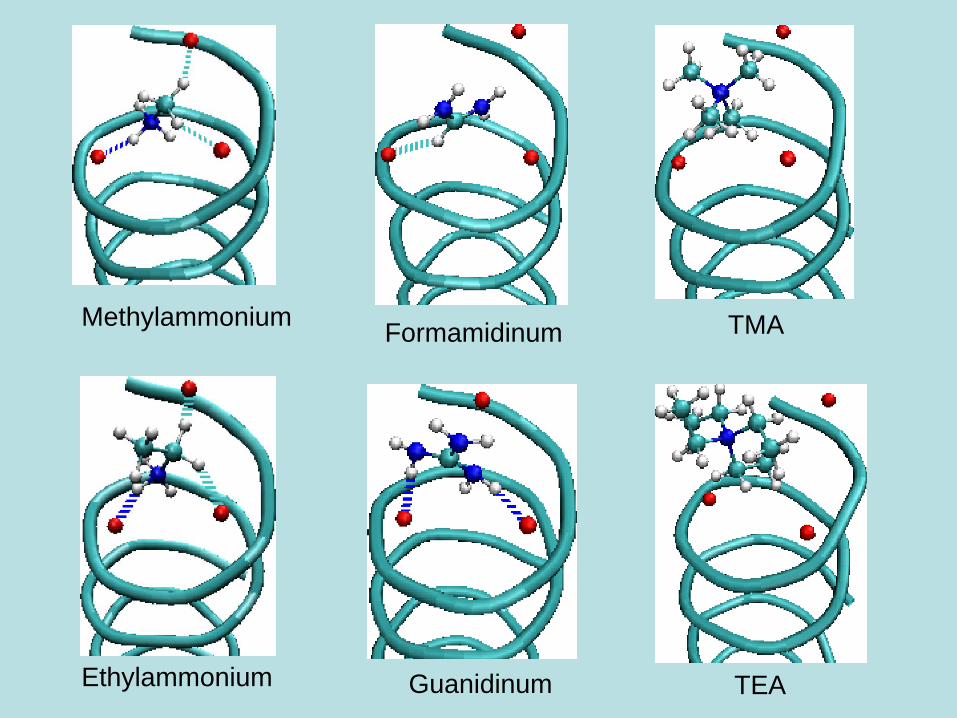
Methylammonium
Ethylammonium
Formamidinum
Guanidinum
TMA
TEA

Calculation of free energy profiles for ions
• Potential of Mean Force (PMF) of a molecule is calculated using the
channel axis (z) as the reaction coordinate
• The PMF is obtained from the Boltzmann factor by measuring the z
coordinates of the molecule
• Umbrella sampling
A harmonic potential is used to constrain the molecule at various points
on the channel axis (typical interval, fraction of an Å), and its z
coordinate is sampled during MD simulations
The z distributions are unbiased and combined to obtain the PMF
profile along the z axis.
-
)ρ(z
ρ(z)kTzWzW
00 ln)()(
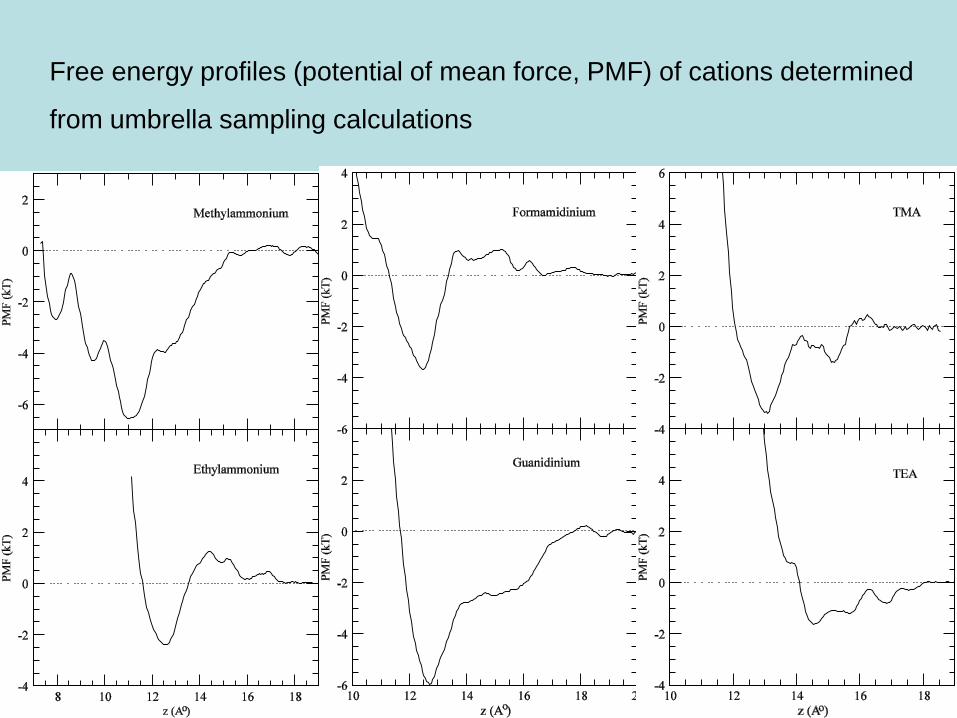
Free energy profiles (potential of mean force, PMF) of cations determined
from umbrella sampling calculations

Binding constants
Binding constant is obtained by integrating the free energy of the ligand
in a volume around the binding site
where we have approximated the volume with a cylinder of radius R.
Using the PMF’s, we can estimate the binding constants:
Methylammonium: K = 4.1 M-1 (exp: 4.4 M-1)
Ethylammonium : K = 0.2 M-1 (exp: ~ 0)
Formamidinium: K = 0.6 M-1 (exp: 23 M-1) (there is a deeper site)
-
--
0
0
0
/)(2
3/])([
zkTzW
V
kTWW
dzeR
rdeK
r
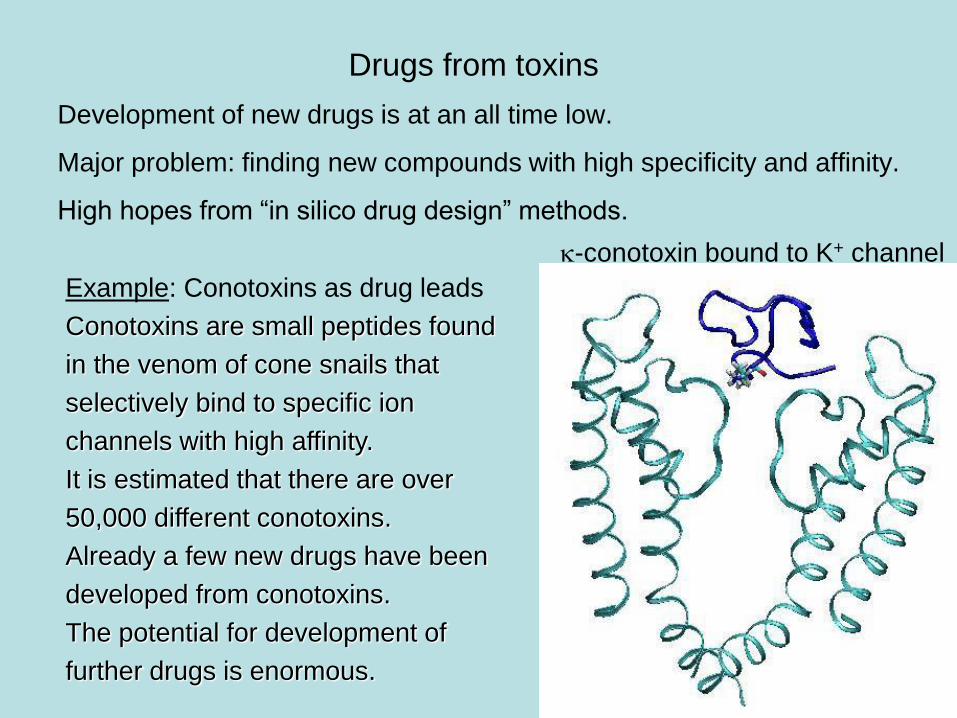
Drugs from toxins
Development of new drugs is at an all time low.
Major problem: finding new compounds with high specificity and affinity.
High hopes from “in silico drug design” methods.
Example: Conotoxins as drug leads
Conotoxins are small peptides found
in the venom of cone snails that
selectively bind to specific ion
channels with high affinity.
It is estimated that there are over
50,000 different conotoxins.
Already a few new drugs have been
developed from conotoxins.
The potential for development of
further drugs is enormous.
k-conotoxin bound to K+ channel

Exp. structure of the KcsA*- charybdotoxin complex (NMR)
Important pairs:
Y78 (ABCD) – K27
D80 (D) – R34
D64, D80 (C) - R25
D64 (B) - K11
K27 is the pore
inserting lysine –
a common thread in
scorpion and other
toxins.
K11 R34

Developing drugs from ShK toxin for autoimmune diseases
ShK toxin has three
disulfide bonds and
three other bonds:
D5 – K30
K18 – R24
T6 – F27
These bonds confer
ShK toxin an
extraordinary stability
not seen in other toxins
NMR structure of ShK toxin
ShK toxin binds to Kv1.3 channels with picomolar affinity, hence a good
candidate for treatment of autoimmune diseases.

Kv1.3-ShK complex (Docking + MD)
Monomers A and C Monomers B and D
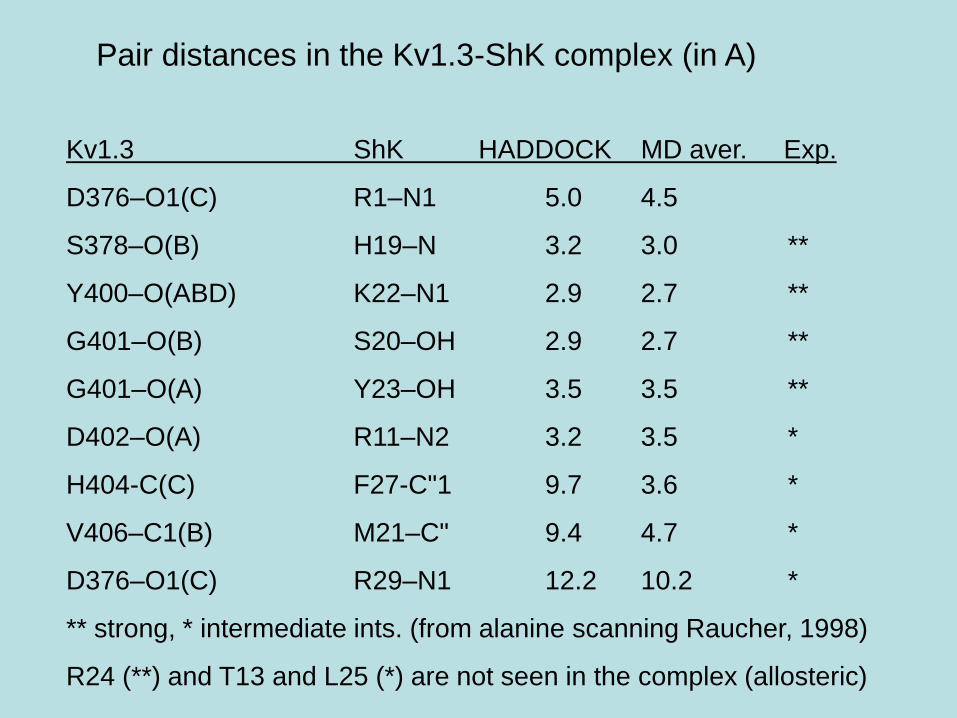
Pair distances in the Kv1.3-ShK complex (in A)
Kv1.3 ShK HADDOCK MD aver. Exp.
D376–O1(C) R1–N1 5.0 4.5
S378–O(B) H19–N 3.2 3.0 **
Y400–O(ABD) K22–N1 2.9 2.7 **
G401–O(B) S20–OH 2.9 2.7 **
G401–O(A) Y23–OH 3.5 3.5 **
D402–O(A) R11–N2 3.2 3.5 *
H404-C(C) F27-C"1 9.7 3.6 *
V406–C1(B) M21–C" 9.4 4.7 *
D376–O1(C) R29–N1 12.2 10.2 *
** strong, * intermediate ints. (from alanine scanning Raucher, 1998)
R24 (**) and T13 and L25 (*) are not seen in the complex (allosteric)
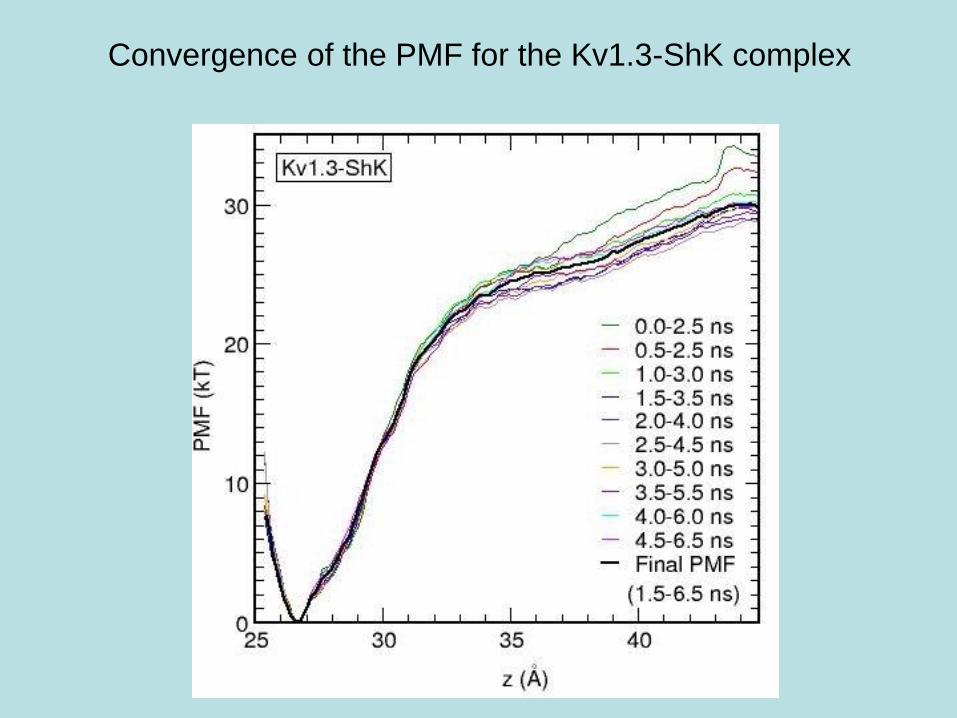
Convergence of the PMF for the Kv1.3-ShK complex
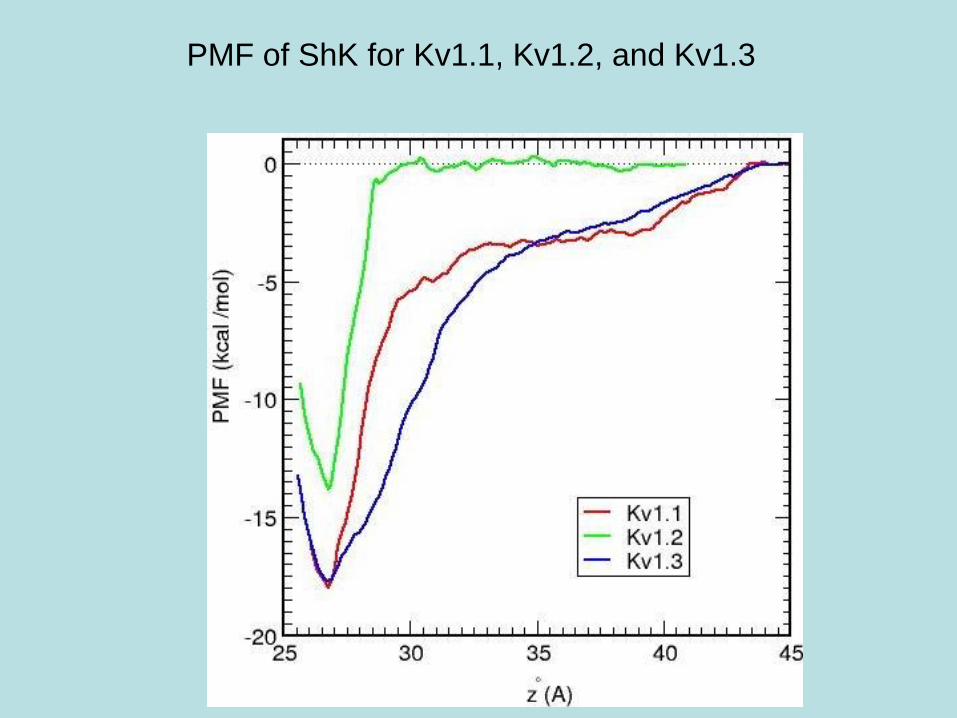
PMF of ShK for Kv1.1, Kv1.2, and Kv1.3

Comparison of binding free energies of ShK to Kv1.x
Binding free energies are obtained from the PMF by integrating it
along the z-axis.
Complex DGwell DGb(PMF) DGb(exp)
Kv1.1–ShK 18.0 14.3 ± 1.1 14.7 ± 0.1
Kv1.2–ShK 13.8 10.1 ± 1.1 11.0 ± 0.1
Kv1.3–ShK 17.8 14.2 ± 1.2 14.9 ± 0.1
Excellent agreement with experiment for all three channels, which
provides an independent test for the accuracy of the complex
models.

Average pair distance as a function of window position
** **
**
**
* * *
** denotes strong coupling and * intermediate coupling