disorders of the gastrointestinal tract and hepatic disorders · disorders of the gastrointestinal...
TRANSCRIPT

PART 20DISORDERS OF THE GASTROINTESTINAL
TRACT AND HEPATIC DISORDERS
Ch020-F10280.indd 711Ch020-F10280.indd 711 7/27/2007 6:01:27 PM7/27/2007 6:01:27 PM

Ch020-F10280.indd 712Ch020-F10280.indd 712 7/27/2007 6:01:28 PM7/27/2007 6:01:28 PM

713
20.1Abdominal pain and vomiting in children
S. W. Beasley
Acute abdominal pain and vomiting are common symptoms in children and a frequent reason for chil-dren to be taken to the doctor. Their causes are many and diverse; those that require surgery must be dis-tinguished from those with a medical origin. While there is considerable overlap of age in many disor-ders (e.g. gastro-oesophageal refl ux), other condi-tions only occur within a specifi c age range; for example, pyloric stenosis is not seen after the age of 3 months.
Abdominal pain in the fi rst 3 months of lifeAbdominal pain without other symptoms is unusual in early infancy. Severe pain may be accompanied by vomiting, abdominal distension, constipation or other features, in which situation it is more likely to have a surgical cause, e.g. malrotation with volvulus.
Infantile colic is an extremely common condition that usually commences in the fi rst few weeks of life. The cause is poorly understood. The term ‘colic’ is used because of the common assumption that this pattern of behaviour is due to colicky abdominal pain but this explanation is controversial, and there are other hypotheses including an irritable tempera-ment (Ch. 4.1).
The infant:
• has attacks of screaming• draws up the legs• is unable to be comforted.
Vomiting is absent, bowel actions are passed nor-mally and the infant is otherwise thriving well. There is no evidence of a strangulated inguinal hernia. The colic almost invariably disappears by the fourth month of age; until then, treatment is supportive. In some infants, apparent colic may be due to oesopha-gitis from gastro-oesophageal refl ux or to hunger in inadequately breastfed babies. Crying babies may cause stress in the family, which in turn may increase the child’s irritability. In a vulnerable or unstable family situation this may place the infant at risk of abuse.
Abdominal pain later in the fi rst yearThe main surgical cause of abdominal pain between 3 and 12 months of age is intussusception. Vomiting is a frequent accompanying feature, such that, when the colicky abdominal pain is not pronounced, intus-susception must be distinguished from other causes of vomiting in this age group (see below).
Intussusception
In intussusception, the distal ileum (the intussuscep-tum) telescopes into adjoining distal bowel (the intussuscipiens), resulting in intestinal obstruction. It can occur at any age but is most likely in the infant between 3 and 18 months who suddenly develops screaming attacks of pain with vomiting. During each episode of pain the infant becomes pale and may draw up the legs.
The spasms of pain tend to last 2–3 minutes and occur at intervals of about 10–20 minutes, although after a while the pain becomes more persistent. Vom-iting is an early symptom. The passage of a few loose stools early on represents evacuation of the bowel distal to the obstruction. The small volume and limited duration of loose stools in intussusception helps differentiate it from acute gastroenteritis. Con-gestion of the intussusceptum may lead to the passage of bloodstained or ‘redcurrant’ stools. Many infants with intussusception present with little more than pallor, lethargy and vomiting and may have little evidence of abdominal pain. Should these symptoms be ignored, the infant may progress to develop signs of septicaemia or shock.
The infant with intussusception looks pale, lethar-gic, anxious and unwell. A vague mass may be felt in the right or left upper quadrants of the abdomen but, once abdominal distension has developed, the mass becomes obscure and diffi cult to palpate. The apex of the intussusceptum may be palpable on rectal examination in a few, and the examining glove may be bloodstained. A plain X-ray of the abdomen will often be normal but may show an unusual bowel gas distribution or features of bowel obstruction. Ultra-sound examination may be helpful in making the
Ch020-F10280.indd 713Ch020-F10280.indd 713 7/27/2007 6:01:28 PM7/27/2007 6:01:28 PM

714
20.1 DISORDERS OF THE GASTROINTESTINAL TRACT AND HEPATIC DISORDERS
diagnosis. Where intussusception is suspected clini-cally or confi rmed on ultrasonography, a gas or barium enema must be performed unless the child has peritonitis. The enema will demonstrate the posi-tion of the apex of the intussusception.
Treatment
Intussusception can be reduced non-operatively by gas enema or by hydrostatic reduction under ultra-sonographic control; these techniques are successful in 80–90% of patients (Fig. 20.1.1). If gas enema facilities are not available, a barium enema under continuous fl uoroscopic control is a less effective but satisfactory alternative. Peritonitis and septicaemia, which suggest the presence of dead bowel, are the only contraindications to attempted enema reduc-tion. A dehydrated child should have intravenous fl uid resuscitation and be wrapped in warm blankets before commencing an enema reduction. The success of enema reduction is recognized when there is sudden or rapid fl ow of gas or barium into the ileum. If partial reduction is achieved, and the child remains in good clinical condition, a further enema should be attempted after several hours (so called ‘delayed repeat enema’), and in about half of these patients it will be successful. Recurrence of intussusception occurs in about 9% of children after enema reduc-tion, usually within days. Surgery is reserved for:
• those in whom enema reduction has failed• those who have clinical evidence of necrotic
bowel, such as peritonitis and septicaemia• those in whom there is evidence of pathological
lesions at the lead point.
Differential diagnosis
Gastroenteritis is often confused with intussuscep-tion but becomes obvious on clinical grounds by the volume and persistence of the fl uid stools. The plain radiological appearance of the abdomen may be similar in both conditions. Where doubt persists, ultrasonography or a gas or barium enema is indi-cated. Other causes of intestinal obstruction include volvulus secondary to malrotation, a band from a Meckel diverticulum, a duplication cyst or a stran-gulated inguinal hernia. Examination of the groin will detect the irreducible tender lump of a strangu-lated hernia.
Acute abdominal pain in older childrenChildren often present with abdominal pain and in most no specifi c cause is found. Constipation and mesenteric adenitis are probably the most common non-surgical identifi able causes.
Acute appendicitis
Appendicitis may occur at any age, although it is rare under 5 years of age. Early diagnosis is diffi cult in the young child (under 5 years) and in the mentally retarded child; the majority of these children have established peritonitis or an appendix abscess at pre-sentation. Delays in the diagnosis of acute appendi-citis in childhood is related in part to its variable symptomatology. For example, there may be rela-tively little abdominal pain, vomiting may be absent and diarrhoea may be a misleading feature.
Nevertheless, the most important and consistent feature is localized abdominal pain. The pain may be intermittent and colicky initially, or situated in the epigastrium or periumbilical region, but soon shifts to the right iliac fossa. Constant pain that is worse with movement is the result of peritoneal irri-tation (‘peritonism’). Vomiting occurs in the major-ity of children, and some may pass a loose stool. The temperature is usually normal or slightly elevated but occasionally may be in excess of 38°C.
Physical examination of the abdomen should be directed at showing that movement of adjacent peritoneal surfaces exacerbates the pain. The child’s cooperation makes assessment easier, and repeated
Fig. 20.1.1 X-ray demonstration of apex of intussusception during reduction using a gas enema.
Ch020-F10280.indd 714Ch020-F10280.indd 714 7/27/2007 6:01:28 PM7/27/2007 6:01:28 PM

ABDOMINAL PAIN AND VOMITING IN CHILDREN 20.1
715
examination of the abdomen may be required to make the diagnosis. A child with appendicitis usually will exhibit tenderness and guarding localized to the right iliac fossa. Gentle palpation and percus-sion tenderness, performed while observing the child’s face, will provide the most reliable evidence of abdominal tenderness and involuntary guarding. Rebound tenderness is an unreliable sign in children, and attempts to elicit the sign may cause unnecessary pain and destroy the child’s confi dence in the doctor. Rectal examination is required rarely and is primar-ily indicated if a pelvic appendix or pelvic collection is suspected. It should not be performed if examina-tion of the ventral abdominal wall has already enabled a confi dent diagnosis of acute appendicitis to be made. Bowel sounds may be normal or reduced and contribute little to the diagnosis.
Peritonitis should be suspected when the child is acutely ill with abdominal pain and fever and is reluctant to move. On examination, there will be generalized abdominal tenderness and guarding.
Laboratory studies and radiology are rarely helpful in making the diagnosis. However, the urine should be checked routinely.
tis. Pain and tenderness is usually referred to the loin. Urine analysis and radiology will confi rm the diagnosis. In Henoch–Schönlein purpura, the abdominal pain is often severe and colicky, and may be accompanied by vomiting. The characteristic skin lesions over the buttock and legs may be inconspicu-ous or absent when the child is fi rst examined.
In the appropriate ethnic group, sickle cell anaemia is a prominent cause of acute abdominal pain and should be considered in a pale child with splenomegaly.
Children with cystic fi brosis frequently experience episodes of abdominal pain from faecal impaction (called ‘meconium ileus equivalent’), a well known manifestation of this disease. The symptoms resolve following a bowel washout.
It is unusual for constipation in an otherwise normal child to produce suffi cient abdominal pain to suggest a surgical emergency. A plain X-ray of the abdomen will demonstrate the extent of faecal accu-mulation (Fig. 20.1.2). It should be remembered, however, that the diagnosis of constipation is usually made on clinical grounds and that X-ray examina-tion should be reserved for other indications or more complex cases.
Less common causes of abdominal pain include urinary tract infection, haemolytic–uraemic syn-drome and diabetes. Acute hepatitis, cholecystitis and pancreatitis, although all rare in childhood, may
Clinical example
Mark, a 10-year-old boy, had 36 hours of constant lower abdominal pain, which steadily became more severe. He vomited once
initially, and was ‘off his food’. Movement made the pain worse. On examination, he was afebrile but appeared fl ushed. He was tender to gentle palpation in the right iliac fossa and had percussion tenderness in the same region. The urine contained a few white and red cells but no bacteria. No other investigation was performed. At laparoscopy an acutely infl amed appendix was removed.
Differential diagnosis
Mesenteric adenitis is the most diffi cult disorder to distinguish from acute appendicitis. In general, loca-lization of pain and tenderness is variable and less specifi c, and the temperature may be higher. Guard-ing is rarely present in mesenteric lymphadenitis.
Other conditions that may mimic acute appendi-citis are relatively uncommon. Meckel diverticulitis has symptoms identical to those of appendicitis, such that differentiation is possible only at laparoscopy or laparotomy. Pain in the right iliac fossa may repre-sent radiation from torsion of the right testis or a strangulated inguinal hernia, and highlights the importance of examination of the genitalia in all boys with lower abdominal symptoms (Ch. 9.1). Acute abdominal pain may occur with renal colic, pyelonephritis and, at times, acute glomerulonephri-
Fig. 20.1.2 Plain X-ray of the abdomen demonstrating gross faecal overload in a child with severe constipation causing abdominal pain. The child also had soiling of his underwear.
Ch020-F10280.indd 715Ch020-F10280.indd 715 7/27/2007 6:01:28 PM7/27/2007 6:01:28 PM

716
20.1 DISORDERS OF THE GASTROINTESTINAL TRACT AND HEPATIC DISORDERS
also cause abdominal pain. In pancreatitis, vomiting is prominent and epigastric tenderness with guard-ing may be marked. These children often look ill and obtunded. Pancreatitis may follow a blunt injury to the abdomen, e.g. a handlebar injury, and several weeks later may produce a pancreatic pseudocyst. The diagnosis is suggested by estimation of the plasma or urinary amylase or plasma lipase, and is confi rmed with computed tomography (CT) or mag-netic resonance imaging (MRI). The management of acute pancreatitis involves correction of shock, intra-venous fl uid administration, nasogastric suction to keep the stomach empty, and analgesia.
Right lower lobe pneumonia may masquerade as appendicitis. The child is usually febrile, with an increased respiratory rate, and has a cough. Signs of pneumonia may be diffi cult to elicit clinically, so that a chest X-ray will be required.
A general summary of disorders associated with abdominal pain is listed in Table 20.1.1.
Peptic ulceration
The abdominal pain of peptic ulceration is epigastric and usually is unrelated to meals. Nausea and vomit-
ing may occur. Haematemesis and melaena suggest the diagnosis; alternatively it may be made following investigation of iron defi ciency anaemia.
Acute gastritis and acute duodenitis produce abdominal pain with epigastric tenderness. A posi-tive hydrogen breath test is suggestive of Helicobacter pylori infection. Culture of biopsy specimens taken during endoscopic examination of the upper gastro-intestinal tract will confi rm H. pylori (Ch. 20.4). Treatment with ampicillin, metronidazole or tripo-tassium dicitratobismuthate (De-Nol) is usually suc-cessful but relapses are common.
Refl ux oesophagitis
Gastro-oesophageal refl ux is common in infancy but usually resolves with growth. Sometimes it may persist into later childhood, with symptoms of belch-ing, acid eructation and intermittent vomiting. Sub-sternal and epigastric pain (‘heartburn’) suggests refl ux oesophagitis. Oesophageal pH monitoring measures lower oesophageal pH over a period of 24 hours and can establish the relationship of refl ux to symptoms (Ch. 20.4). Oesophagoscopy and biopsy may confi rm oesophagitis. Initial management may involve the administration of H2-receptor antago-nists but, where non-operative measures fail or if an oesophageal stricture is present, surgical correction of the refl ux by laparoscopic fundoplication may be indicated.
Recurrent abdominal pain in childrenRecurrent bouts of abdominal pain is a fairly common paediatric presentation and one that may cause great anxiety to parents. The clinical example is illustrative of this syndrome.
Table 20.1.1 Causes of abdominal pain in childhood
Common• Appendicitis• Mesenteric adenitis• Constipation• Intussusception• Urinary tract infection• Torsion of the testis
Uncommon• Volvulus secondary to malrotation• Meckel diverticulitis• Renal colic• Pyelonephritis• Acute glomerulonephritis• Glandular fever• Drug ingestion, e.g. salicylates, non-steroidal anti-
infl ammatory drugs, corticosteroids, some antibiotics, imipramine, phenytoin, iron preparations
• Peptic ulceration• Refl ux oesophagitis
Rare• Sickle cell anaemia• Henoch–Schönlein purpura• Pancreatitis• Cholecystitis• Acute hepatitis• Diabetes mellitus• Haemolytic–uraemic syndrome• Infl ammatory bowel disease, e.g. Crohn disease
Clinical example
Thomas, aged 7 years, was brought in by his mother, who stated that for the last 4 months he had had severe bouts of abdominal pain.
The attacks occurred at any time, but were more frequent at breakfast time. He was never awakened at night by them. Vomiting was not a feature, and his bowels had been regular. The pain usually was localized to the periumbilical region and usually lasted less than 1 hour. His parents felt that Thomas was pale and had a poor appetite. He was of normal height and weight. Physical examination was unremarkable. The urine was clear. Further questioning elicited the fact that the bouts of abdominal pain had occurred periodically since the age of 3 years.
Ch020-F10280.indd 716Ch020-F10280.indd 716 7/27/2007 6:01:28 PM7/27/2007 6:01:28 PM

ABDOMINAL PAIN AND VOMITING IN CHILDREN 20.1
717
Doctors will be impressed by the concern exhib-ited by parents of these children, who vividly describe the severe pain the child experiences; but there is a disparity between the parents’ description and the physical fi ndings on examination of the abdomen. Investigation almost invariably produces negative results. Enquiry into the personality of the child and into the home situation may reveal that the child is anxious or stressed, but often the pain occurs for no apparent reason. Sometimes the episodes of pain appear to be related to stress within the family. A diagnosis of non-organic recur-rent abdominal pain can be made only after careful appraisal of the child in relation to the environment, and when the physical examination is normal. Most children need no investigations apart from urine culture. Further investigation is required if the abdominal pain is associated with abdominal tenderness or distension, bile stained vomiting, persistent diarrhoea, fever, weight loss or urinary symptoms. This may include full blood examina-tion, erythrocyte sedimentation ratio (ESR), C reactive protein (CRP), radiological and endoscopic studies of the gastrointestinal tract, and specifi c investigations for malabsorption and infl amma-tory bowel disease. The urine should be examined. If the vomitus is bile-stained, malrotation with vol-vulus should be excluded by an urgent barium meal.
The general status of the patient must be assessed. Retardation of height and growth may occur in chronic infl ammatory bowel disease, malabsorption syndromes and tuberculosis. Pallor may be as-sociated with anaemia or conditions such as lead poisoning, sickle cell anaemia and other haemolytic diseases.
Management
Parents will fi nd it helpful to realize that the problem has been taken seriously by the doctor, and the doctor must understand the parents’ perception of the abdominal pain. With this knowledge and the negative physical fi ndings, reassurance can be given more positively. Once parents are convinced that there is no signifi cant organic basis to the recurrent abdominal pain they are usually much relieved. The child should be encouraged in all activities and self-esteem improved. Recurrent pain tends to disappear by the age of 12 years but in females may recur at the time of menarche. However, some children with recurrent pain in childhood present in adult life with symptoms of irritable bowel syndrome. In some, as time goes by the child’s abdominal pain may become associated with and eventually replaced by migraine headaches.
Vomiting in the neonatal periodNeonates frequently vomit small amounts of mucus and blood swallowed during labour. This vomiting usually clears spontaneously within 24 hours. If not, gastric lavage with normal saline will usually relieve it. In the early weeks of life, many normal newborn babies regurgitate after feeds. The cause of this ‘spit-ting up’ or ‘possetting’ is not clear but is presumably related to gastro-oesophageal refl ux.
Systemic infection
Vomiting is one of the many non-specifi c signs of infection in the neonate. Thus, unexplained vomit-ing should be an indication to culture the blood, urine and cerebrospinal fl uid. Urine will usually be obtained by suprapubic aspiration in this age group.
Bowel obstruction
In duodenal obstruction, vomiting appears early and is bile-stained because the site of the obstruction is almost always at the second part of the duodenum, just distal to the ampulla of Vater. In duodenal atresia, there may be other abnormalities such as Down syndrome and imperforate anus. The bile-stained vomiting commences from birth. The diag-nosis is made on plain X-ray of the abdomen (Ch. 11.5). Where there is bowel obstruction beyond the duodenum (e.g. small bowel atresia, Hirschsprung disease and meconium ileus), vomiting commences slightly later and is associated with increasing abdominal distension (Ch. 11.5). A strangulated inguinal hernia may cause a bowel obstruction when a loop of ileum becomes trapped within the hernial sac at the external inguinal ring. The diagnosis becomes evident when a tender irreducible lump is observed in the groin (Ch. 9.1).
Malrotation with volvulus
Volvulus in a neonate or infant with malrotation causes a high bowel obstruction and produces bile-stained vomiting. The volvulus may cut off the blood supply to the midgut and lead to small bowel infarc-tion, septicaemia and death if not treated promptly. Any infant with bile-stained vomiting, otherwise unexplained, should be assumed to have malrotation with volvulus until proven otherwise. A barium meal will confi rm the diagnosis. An urgent laparotomy is required to untwist the bowel (Fig. 20.1.3) and to perform a Ladd procedure to broaden the mesentery of the small bowel: this will prevent subsequent volvulus.
Ch020-F10280.indd 717Ch020-F10280.indd 717 7/27/2007 6:01:29 PM7/27/2007 6:01:29 PM

718
20.1 DISORDERS OF THE GASTROINTESTINAL TRACT AND HEPATIC DISORDERS
Cerebral hypoxia
There is often a history of fetal distress during labour and asphyxia at birth requiring resuscitation. Fol-lowing this, some infants remain lethargic and feed poorly, while others may be abnormally wide awake, excessively irritable and cry frequently. The cry may be high-pitched and associated with intermittent twitching and hypertonia; there is head retraction and the thumbs are adducted across the palms with fl exion of the fi ngers. The Moro refl ex may be exag-gerated but in severe cerebral anoxia it may be lost. The fontanelle tension is not increased initially, unless there has been cerebral haemorrhage, but within 24 hours cerebral oedema occurs and causes a rise in the fontanelle tension. In cerebral anoxia the vomiting occurs before or after feeding, and may be forceful.
Treatment
Cerebral haemorrhage is shown on ultrasonography, which may also demonstrate cerebral oedema. Treat-ment is symptomatic: sedation with diazepam 0.1–0.3 mg/kg i.v. or phenobarbital 2 mg/kg i.m. may be required. The pulse, temperature, state of conscious-ness and degree of dehydration should be monitored. Aspiration of vomitus is potentially dangerous. Oral fl uids are given in small volume and are offered fre-quently. An intravenous infusion may be necessary
but fl uid requirements on the fi rst day of life are only 60 ml/kg. This amount increases gradually each day until the end of the fi rst week, when they reach 150 ml/kg. Frequent blood glucose estimations will detect hypoglycaemia early, before it exacerbates the cerebral disturbance and accentuates the vomiting.
Subdural haematoma
With the current high standard of obstetrics, a subdural haematoma is now rare in the neonatal period. In about 50% of infants, vomiting is the only symptom. In others, vomiting is accompanied by developmental delay, convulsions, an expanding head and retinal haemorrhages. The diagnosis is confi rmed on ultrasonography and CT. A subdural haematoma later in childhood must alert the clini-cian to the possibility of child abuse.
Hypoglycaemia
Vomiting may be the only symptom of hypoglycae-mia in the neonatal period. It is more common in ‘small for dates’ babies and in infants of diabetic mothers but may be seen in any stressful situation in the neonatal period, including low birth weight, neo-natal meningitis, septicaemia and severe Rhesus iso-immunization. Symptomatic hypoglycaemia does not usually occur with a blood glucose in excess of 2 mmol/l.
Fig. 20.1.3 Malrotation with volvulus: the small bowel is twisted on its mesentery.
Ch020-F10280.indd 718Ch020-F10280.indd 718 7/27/2007 6:01:29 PM7/27/2007 6:01:29 PM

ABDOMINAL PAIN AND VOMITING IN CHILDREN 20.1
719
Renal disease
In the neonatal period, urinary infection and renal insuffi ciency may present with vomiting and poor weight gain, refl ecting an underlying urinary tract abnormality. Initial urological investigation will include urine culture, renal ultrasonography, mictur-ating cystourethrography and estimation of electro-lytes, urea and creatinine. Renal tubular lesions occasionally present in the neonatal period with vomiting.
Adrenal insuffi ciency
Congenital adrenal hyperplasia, in which there is defi ciency of the enzyme 21-hydroxylase (Ch. 19.3), presents with ambiguous genitalia in the female. If this is not recognized (as in the male), it may lead to unexplained vomiting, dehydration and collapse early in the second week of life. If the adrenal insuf-fi ciency is of the salt-losing type, the diagnosis is further suspected by fi nding low levels of sodium and elevated levels of potassium in the serum, and is confi rmed by appropriate hormonal studies.
Inborn metabolic errors
Although individually rare, there are a number of inborn errors involving, separately, amino acid, car-bohydrate and organic acid metabolism. Most are inherited recessively, and a number can now be treated. Frequently, the presentation is with unex-plained vomiting, lethargy, collapse, seizures and coma (Ch. 10.5).
Vomiting in infancyVomiting is a common non-specifi c symptom in infancy, and disease of almost every system may present with vomiting.
Infection
Vomiting is frequently caused by infections such as tonsillitis, otitis media, pneumonia, meningitis and urinary tract infection. Physical examination will exclude many of these but early signs may be minimal in meningitis and pneumonia, such that a lumbar puncture and chest X-ray will be required if these are suspected. In infants with urinary tract infection, dysuria, frequency of passing urine and loin pain cannot be relied upon for diagnosis, and the urine must always be examined. When infection is con-trolled, the urinary tract should be imaged to exclude underlying structural abnormalities.
Lesions of the gastrointestinal tract
Conditions that produce vomiting in infancy are dif-ferent from those seen in the neonatal period, except for duodenal obstruction from volvulus complicat-ing malrotation, and gastro-oesophageal refl ux. Failure to recognize malrotation with volvulus may result in infarction of the entire midgut (Ch. 11.5). Bowel trapped in a strangulated inguinal hernia in an infant will also produce vomiting. The diagnosis can be made easily if the inguinal orifi ces are exam-ined (Ch. 9.1).
Gastro-oesophageal refl ux
See Chapter 20.4.
Pyloric stenosis
This is one of the most dramatic causes of vomiting in infancy. Typically, the onset is dramatic, commencing between the second and sixth week of life. Males are affected fi ve times more often than females and there is a defi nite familial incidence. Before the onset of vomiting, these infants fed well and were thriving. The vomiting is forceful and rapidly becomes projec-tile. The infant loses weight and becomes dehydrated. Despite vomiting, these infants remain hungry and are keen to feed even immediately after vomiting. The vomitus is not bile-stained but may contain altered blood. The diagnosis is made clinically by feeling the thickened pylorus (‘pyloric tumour’) in the midline in the epigastrium between the rectus abdominis muscles or in the angle between the right rectus and the liver edge. The pyloric tumour is palpable as a hard mobile mass about the size of a small pebble or olive. Peristal-tic waves passing from the left costal margin to the right hypochondrium (‘golf ball waves’) may be visible long after the last feed. Palpation of the tumour is suf-fi cient to establish the diagnosis. Pyloric stenosis can also be shown on ultrasonography (which reveals a thickened pylorus) and barium meal (which shows delayed gastric emptying and a narrow pyloric canal). These infants develop a hypokalaemic, hypochlo-raemic metabolic alkalosis which, together with dehydration, must be corrected before surgery. Pylo-romyotomy is curative (Fig. 20.1.4).
Gastroenteritis
Vomiting in association with fl uid stools is suggestive of gastroenteritis, particularly if the stools contain mucus or blood. However, these features may be seen in a variety of other medical and surgical disorders, which include intussusception and appendicitis. The diagnosis and management of gastroenteritis is dis-cussed in Chapter 20.2.
Ch020-F10280.indd 719Ch020-F10280.indd 719 7/27/2007 6:01:29 PM7/27/2007 6:01:29 PM

720
20.1 DISORDERS OF THE GASTROINTESTINAL TRACT AND HEPATIC DISORDERS
Malabsorption
In the majority of malabsorption syndromes vomit-ing is not a feature. At times, in the more severe cases of coeliac disease (gluten enteropathy, Ch. 20.3) vomiting may be prominent. A gluten-free diet ra -pidly reverses the clinical features of this disorder.
Intussusception
Vomiting commences early in intussusception and is the most consistent symptom. The general features, diagnosis and treatment are discussed in more detail on pages 709–710.
Strangulated inguinal hernia
Strangulation of an inguinal hernia is common in infants and young children. All irreducible inguinal hernias should be assumed to be strangulated. In practice, the vast majority of so-called irreducible hernias can be reduced manually by skilled hands (Ch. 9.1).
Vomiting in older childrenVomiting in older children is usually associated with infection, particularly viral or bacterial infection of the respiratory and gastrointestinal tracts. Neverthe-less, there are some other less frequent but important causes of vomiting.
The possibility of an intracranial neoplasm should always be considered in a child with unexplained vomiting. There may be signs of increased intracra-nial pressure with midline cerebellar tumours, tumours involving the fourth ventricle and tumours involving the pons or medulla. Initially, vomiting tends to occur in the morning before breakfast. There may be remissions for several days but the vomiting invariably returns.
Migraine
In the older child, the association of severe paroxys-mal frontal headache with pallor and vomiting is
Fig. 20.1.4 Thickened pylorus in pyloric stenosis as seen during pyloromyotomy.
Clinical example
Bruce was a healthy 5-month-old baby boy until 24 hours prior to admission, when he commenced vomiting, refused feeds, was
lethargic and had marked pallor. His mother had noted that at times he appeared to be in pain. There was an impression of a slightly tender mass in the right upper quadrant. There was no blood rectally. A provisional diagnosis of intussusception was made, and at gas enema an intussusception in the mid-transverse colon was encountered. This was reduced, with rapid relief of symptoms. Bruce was observed overnight before being discharged the next morning.
Ch020-F10280.indd 720Ch020-F10280.indd 720 7/27/2007 6:01:29 PM7/27/2007 6:01:29 PM

ABDOMINAL PAIN AND VOMITING IN CHILDREN 20.1
721
suggestive of migraine (Ch. 17.5). A positive family history is common. Transient loss of vision, transient hemiparesis, cerebellar ataxia or ophthalmoplegia may be evident. In some children migraine is precipi-tated by minor trauma. In the younger child, attacks of pallor or vomiting may be the only symptom. The diagnosis of migraine is made on clinical history but, where it is diffi cult to exclude an intracranial space-occupying lesion clinically, cerebral CT may be required.
Acute appendicitis and peritonitis
In acute appendicitis in childhood, vomiting is a fre-quent early symptom but is usually preceded by pain. The general features of appendicitis are described on pages 710–712. In the young child (under 5 years), vomiting with or without diarrhoea may be the only obvious symptom. Physical examination in this age group can be diffi cult and unreliable; the child will prefer to lie still, as movement worsens the pain. This pain and the fear of its exacerbation by palpation may make the child appear uncooperative. It is only by repeated examination of the abdomen and an ongoing high index of suspicion that the diagnosis will be made before widespread peritonitis has developed.
Poisoning
Vomiting and respiratory and circulatory collapse in a previously well child should raise the possibility of
poisoning (Ch. 5.3). Non-accidental poisoning is becoming more frequent, and the age incidence of children attempting suicide is decreasing. A history of family discord and emotional problems in the child is not always volunteered.
Psychological causes of vomiting
Psychogenic vomiting may occur in any age group. It can be associated with attempts to force-feed a toddler or a schoolchild, after punishment, and as an attempt to avoid situations perceived as threatening, such as going to preschool or school. Almost any stressful situation may precipitate vomiting in a tense or anxious child. The absence of abnormal physical signs will be a feature.
Cyclical vomiting
Cyclical vomiting is a syndrome of persistent peri-odic vomiting of childhood. The severity varies, but ketosis and metabolic acidosis may develop rapidly. The aetiology is unknown and attacks usually cease spontaneously. Children with cyclical vomiting are often tense and anxious and may develop migraine or psychosomatic disease later in life. Recurring episodes of volvulus from malrota-tion, and metabolic disease, should be excluded before labelling these children as having cyclical vomiting.
Ch020-F10280.indd 721Ch020-F10280.indd 721 7/27/2007 6:01:30 PM7/27/2007 6:01:30 PM

Diarrhoea is defi ned as a measured stool volume greater than 10 ml/kg per day. Both the consistency of the stool (loose or watery) and frequency (usually at least three stools in a 24-hour period) are impor-tant defi ning features of diarrhoea. Acute diarrhoea lasts less than 10 days and has a major impact on both fl uid and electrolyte status, while chronic diar-rhoea suggests that the symptom is present for more than 2–3 weeks and can have a signifi cant effect on the nutritional state of a child. The basic pathologi-cal mechanisms causing diarrhoea include osmotic, secretory and infl ammatory processes (Table 20.2.1). Often more than one mechanism may operate simul-taneously to cause diarrhoea. The commonest cause of acute diarrhoea in children is an enteric infection (acute gastroenteritis).
Acute gastroenteritisAetiology
Rotavirus infection (Fig. 20.2.1) is the most common cause of acute gastroenteritis in children under 5 years of age in developed countries, causing 40–50% of cases where hospital admission is required. It accounts for more severe episodes in infants in devel-oping countries than any other single pathogen; it is more likely to cause dehydration, and is associated with a higher mortality than most other agents. The mucosal damage it causes (Fig. 20.2.2), and hence the need for structural repair, has considerable nutri-tional implications for malnourished children. Asymptomatic reinfection can occur several times and helps maintain immunity.
Enteric adenoviruses (types 40 and 41) cause 5–15% of cases requiring admission to hospital, and several other virus pathogens have been recognized, such as calicivirus, astrovirus and other small viruses, which accounts for a further 15%.
Bacteria cause fewer episodes than viruses in developed countries. Campylobacter jejuni is respon-sible for 5–10% of cases. Salmonella spp., Shigella spp. and various types of Escherichia coli each account for a small percentage. In developing coun-tries, E. coli (enterotoxigenic, enteropathogenic and enteroinvasive) and Shigella spp. are especially
important: E. coli because of the huge number of episodes it causes, and Shigella because it causes prolonged debilitating illness and antibiotic-resistant strains are emerging.
Giardia lamblia rarely causes acute dehydrating diarrhoea but another parasite, Cryptosporidium, is now known to cause 1–4% of cases of acute diar-rhoea in infants admitted to hospital.
Clinical features
Symptoms of acute gastroenteritis include vomit-ing, fever and watery diarrhoea (up to 10–20 stools daily).
Blood, mucus and the passage of small frequent bowel actions accompanied by abdominal pain sug-gests a diagnosis of bacterial gastroenteritis.
Acute gastroenteritis is a diagnosis of exclusion. A few loose stools and vomiting does not necessarily equate with the diagnosis. There are several systemic disorders and surgical emergencies that can mimic infective gastroenteritis (Table 20.2.2).
Management
Once the diagnosis of acute gastroenteritis is made on thorough clinical history and physical examina-tion, the next step is to assess the degree of dehydra-tion and institute an appropriate plan for rehydration. This should be combined with nutritional support that aids the patient during the recovery phase.
Dehydration
This risk is related to the child’s age, with young infants being at greatest risk. This is because infants less than 1 year of age have a high surface area:body volume ratio, resulting in increased insensible fl uid loss. They also have a tendency to more severe vomit-ing and diarrhoea compared with older children and adults.
Fluid loss is usually assessed on the basis of per-centage body weight loss. Physical signs of dehydra-tion are not usually apparent until 4% of body weight is lost.
The signs of dehydration traditionally described are outlined in Table 20.2.3. However, three signs
722
20.2 The child with diarrhoeaG. Alex, M. Oliver
Ch020-F10280.indd 722Ch020-F10280.indd 722 7/27/2007 6:01:30 PM7/27/2007 6:01:30 PM

723
discriminate adequately between dehydration and adequate hydration: deep breathing, decreased skin turgor and poor peripheral perfusion.
Electrolyte loss
This is usually isotonic (water and electrolytes being lost in equal amounts). Hypertonic hypernatraemic dehydration (fl uid loss > electrolyte loss) occurs in 5–10% of cases of acute gastroenteritis, and hypo-tonic hyponatraemic dehydration (electrolyte loss > fl uid loss) can occur if the colon (a major site of sodium reabsorption) is out of circuit, e.g. short gut syndrome.
If corrected too rapidly, hypernatraemic dehydra-tion will result in convulsions due to rapid shifts of water into cells. Hyponatraemic dehydration can also cause signifi cant neurological morbidity and mortality and, in contrast to the hypernatraemic state, requires vigorous replacement of sodium.
Rehydration guidelines
See also Chapter 6.1
No dehydration
• Nutritional intake and fl uids should not be modifi ed but should be offered ad libitum to keep up with ongoing losses
Table 20.2.1 Classifi cation of diarrhoea
Osmotic Secretory Infl ammatory
Clinical features Ceases when enteral Continues when enteral Presence of blood and feeding is ceased feeding is ceased mucus in the faeces
Stool volume <200 ml/day >200 ml/day Variable, usually <200 ml/day
Faecal sodium <60 mosmol/l 90 mosmol/l Variable
Fig. 20.2.1 The rotavirus (electron micrograph).
Table 20.2.2 Differential diagnosis of acute diarrhoea and vomiting in infants and children
Enteric infection• Rotavirus• Other viruses• Bacterial • SaImonella spp. • Shigella spp. • Escherichia coli • Campylobacter jejuni• Protozoa • Cryptosporidium • Giardia lamblia • Entamoeba histolytica• Food poisoning• Staphylococcal toxin
Systemic infection• Urinary tract infection• Pneumonia• Septicaemia
Surgical condition• Appendicitis• Intussusception• Partial bowel obstruction• Hirschsprung disease
Other• Diabetes mellitus• Antibiotic diarrhoea• Haemolytic–uraemic syndrome
THE CHILD WITH DIARRHOEA 20.2
Ch020-F10280.indd 723Ch020-F10280.indd 723 7/27/2007 6:01:30 PM7/27/2007 6:01:30 PM

724
Mild to moderate dehydration
• Oral rehydration solution (ORS) is the cornerstone of successful rehydration and is recommended glob-ally for the management of acute diarrhoea• The success of ORS is based on the basic observa-tion that intestinal sodium transport is enhanced by glucose transport in the small intestine and that this sodium-coupled mechanism for glucose transport remains intact during acute gastroenteritis• To facilitate optimal absorption of sodium, glucose and water, the sodium and glucose must be in the range recommended (Table 20.2.4)
A B
Fig. 20.2.2 Scanning electron microscope appearances of (A) normal and (B) rotavirus-infected calf jejunum. Villi are short, the epithelium is damaged and crypts are deep. (Courtesy of D. G. A. Hall, Institute for Animal Health, Compton, UK. With permission from Walker et al 1991.)
Table 20.2.3 Assessment of dehydration
Mild (£5% body weight loss)• Dry mucous membranes• Decreased peripheral perfusion*• Thirsty, alert, restless
Moderate (6–9% body weight loss)• Exaggeration of the above• Lethargic but irritable• Rapid pulse, normal blood pressure• Sunken eyes, sunken fontanelle• Oliguria is usually obvious• Pinched skin retracts slowly (1–2 s)*
Severe (≥10% body weight loss)• General appearance • Infants: drowsy, limp, cold sweaty, cyanotic limbs,
comatose • Older children: apprehensive, cold, sweaty, cyanotic
limbs• Rapid feeble pulse, low blood pressure• Sunken eyes and fontanelle• Pinched skin retracts slowly (>2 s)*• Deep acidotic breathing*
* These are the only signs proven to discriminate between hydration and dehydration.
Table 20.2.4 Oral rehydration preparations available in Australia
Na K Cl Citrate Glucose
WHO 90 20 80 10 90
Hydralyte 45 20 45 30 90
Gastrolyte 60 20 60 10 90
Repalyte 60 20 60 10 90
Concentration expressed as mmol/l.
20.2 DISORDERS OF THE GASTROINTESTINAL TRACT AND HEPATIC DISORDERS
Ch020-F10280.indd 724Ch020-F10280.indd 724 7/27/2007 6:01:30 PM7/27/2007 6:01:30 PM

725
• Rehydration should take place over 4–6 hours and can be given orally or, if either vomiting or fl uid refusal is a problem, a nasogastric tube may be used to achieve a steady infusion of fl uid• Volume required for rehydration = estimated defi cit and maintenance; maintenance for: • 1–3 months of age = 120 ml/kg/24 h • 3–12 months of age = 100 ml/kg/24 h • 12 months onwards = 80 ml/kg/24 h (see Tables 20.2.5 and 20.2.6)
Severe dehydration (10% plus)
• Circulatory insuffi ciency is present and intrave-nous therapy is required. The usual requirement is to fi ll the vascular compartment quickly to restore circulation. This will require rapid rehydration, often using boluses of normal saline by intravenous or intraosseous infusion• Once dehydration is corrected and normal organ perfusion is restored, ORS can be used in conjunc-
tion with intravenous fl uids. The latter is rarely required for longer than 24 hours• Clinical observations must be highlighted: this allows the physician to reassess the patient’s state of hydration and also helps confi rm the diagnosis of acute gastroenteritis• All patients with dehydration require regular checks on pulse, temperature and respiration, and strict fl uid balance charts must be kept. The child should be weighed on admission and, in severe cases, after 6 hours and 24 hours, with an increase in weight being a reliable sign of rehydration. However, in some patients weight may not fall even in the presence of severe dehydration, especially if the child has an ileus, so other signs of dehydration must be sought.
Recommendations on nutritional management
Breastfeeding should continue through rehydration and maintenance phases of treatment, and for mula feeds need to be restarted after rehydration. Use of special formulas or diluted formulas is unjustifi ed.
Pharmacotherapy
• Infants and children with acute gastroenteritis should not be treated with antidiarrhoeal agents• Antibiotic treatment may be indicated in Salmo-nella spp. gastroenteritis in the very young (<3 months), those who are immunocompromised or those who are systemically unwell. It may also be indicated in C. jejuni infection in compromised hosts and Yer-sinia enterocolitica in children with sickle cell disease• Pathogens for which antibacterial therapy is always indicated include Shigella spp. and G. lamblia• Certain types of probiotic have a modest effect in acute infective gastroenteritis; however, their routine use is not recommended until further large-scale trials have been undertaken.
Complications of acute gastroenteritis
Febrile convulsions
These are generally uncommon, but rotavirus infec-tion can cause fevers as high as 39–40°C.
Sugar malabsorption
This is more common in infants less than 6 months of age and recognized by the persistent nature of the diarrhoea when nutrition is reintroduced.
Stools are often watery, frothy and tend to excori-ate the buttocks. If sugar intolerance is suspected, the napkin should be lined with thin plastic material, or a rectal examination should be performed and the
Table 20.2.5 An infant of 10 kg estimated at 8% dehydration has fl uid requirements equal to
Maintenance 100 × 10 kg = 1000 mlDefi cit 8% of 10 kg = 800 mlTotal = 1800 ml
Using oral rehydration the defi cit can be replaced in 6 hours rather than 24 hours, so in the above example the infant would be offered fl uid as follows:
First 6 hoursDefi cit = 800 mlMaintenance 6/24 of 1000 = 250 mlTotal = 1050 ml (175 ml/h)
Next 18 hoursMaintenance 18/24 of 1000 = 750 ml (45 ml/h)
Another simple method which gives about the right answer is to calculate the fl uid defi cit, double it, and give that volume over 6–12 hours.
Table 20.2.6 A 10 kg child with 15% dehydration and shock
Total fl uid defi cit = 15% of 10 kg = 1.5 l = 1500 mlAssume a total of 40 ml/kg (400 ml) normal saline needed
to restore circulationRemaining defi cit = 1500 − 400 = 1100 mlMaintenance fl uid requirement is 100 ml/kg/d = 10 × 100 =
1000 mlFluid in next 24 h = remaining defi cit + maintenance =
1100 + 1000 = 2100 ml = 90 ml/h
Therefore, give 400 ml normal saline quickly, then 90 ml/h of 5% dextrose in N/2 saline with KCl 40 mmol/l for the next 24 h
THE CHILD WITH DIARRHOEA 20.2
Ch020-F10280.indd 725Ch020-F10280.indd 725 7/27/2007 6:01:30 PM7/27/2007 6:01:30 PM

726
fl uid stool collected and tested for reducing sub-stances. It is pointless to test solid stool material.
To test for lactose intolerance, mix 5 drops of liquid stool with 10 drops of water and add a Clinit-est tablet. A positive test of more than 0.5% indicates lactose or glucose malabsorption, but not sucrose, which is not a reducing sugar.
Diarrhoea due to lactose malabsorption resolves rapidly on a lactose-free diet, which should be con-tinued for approximately 4 weeks. A very small pro-portion of infants continue to have diarrhoea despite the exclusion of lactose and sucrose. Under these circumstances, a carbohydrate-free feed is given, with glucose and fructose (different transport mech-anisms across the enterocyte) added to tolerance.
Prevention of acute gastroenteritis
A simple and effective prevention of transmission of the condition is via handwashing when in contact with an index case, especially in the hospital setting.
Prophylactic passive immunization is provided by oral administration of hyperimmune bovine antiro-tavirus colostrums. It reduces nosocomial infection in infants in high-risk settings but its effect stops with cessation of administration. Anti-E.-coli colos-trum tablets are available to help prevent the most common type of traveller’s diarrhoea.
Vaccines against typhoid and cholera infection are available and E. coli vaccines are under development.
Two live attenuated oral rotavirus vaccines are becoming available, one of which is already licensed in several countries. One is a human strain (Rotarix, GSK), which relies on immunity being stimulated across serotypes. Both vaccines appear to be free
Chronic diarrhoeaThis is defi ned as the presence of diarrhoea for more than 2–3 weeks. It can follow a bout of acute gastro-enteritis but usually begins insidiously.
Many causes of chronic diarrhoea are associ-ated with malabsorption of nutrients and are dealt with in detail in Chapter 20.3. Only chronic non-specifi c diarrhoea, postinfective diarrhoea, sucrase–isomaltase defi ciency and infl ammatory bowel disease will be discussed in this chapter.
An approach to diagnosis
Answers to a small number of key questions will usually get very close to a defi nitive diagnosis. Figure 20.2.3 outlines a suggested scheme. Performance of simple stool tests is all that is necessary to guide selection of the appropriate defi nitive tests. Many of the specifi c causes are discussed in Chapter 20.3. Others are dealt with here.
Chronic non-specifi c diarrhoea
• Seen in children between the ages of 12 months and 4 years and current scientifi c evidence suggests that disturbed intestinal motility is pivotal in the patho-genesis of this condition. It is also commonly referred to as toddlers’ diarrhoea• The history is one of frequent, poorly formed and slightly offensive stools. Food material is often rec-ognized in the stool, suggesting rapid gastrointesti-nal transit. The condition often resolves spontaneously at about 3–4 years of age
Practical points
Acute gastroenteritis• Is commonly caused by viruses and is self-limiting• Assess dehydration carefully and correct appropriately,
but reassess constantly• Not all children with diarrhoea have gastroenteritis; be
aware of other conditions that may mimic it• Proper handwashing is the best measure to avoid
transmission
Clinical example
Tim, a previously well 22-month-old infant, suddenly became unwell with onset of vomiting and a temperature of 38.5°C. Within
12 hours he started passing frequent, loose motions. Associated with this he appeared to have bouts of abdominal pain. On the second day the vomiting stopped but the diarrhoea persisted at a rate of more than eight stools per day. On presentation to the emergency department, Tim’s weight was 13.6 kg compared with 14.5 kg 3 weeks previously. He was clinically assessed to be 5–8% dehydrated and was treated with an oral rehydration solution via a nasogastric tube. A provisional diagnosis of acute gastroenteritis was made. His temperature gradually settled over 24 hours days and oral feeds were introduced once rehydration was complete. His stools returned to their normal pattern after 5 days. Rapid stool testing for rotavirus antigen was positive.
20.2 DISORDERS OF THE GASTROINTESTINAL TRACT AND HEPATIC DISORDERS
from the rare serious side effect of intussusception, which led to the withdrawal of an earlier candidate vaccine based on a Rhesus monkey rotavirus strain. Other candidates are under development in develop-ing countries as a strategy to ensure that rotavirus vaccine becomes widely available to children worldwide.
Ch020-F10280.indd 726Ch020-F10280.indd 726 7/27/2007 6:01:30 PM7/27/2007 6:01:30 PM

727
• The child is usually active, with unimpaired growth, appetite is normal and there is a history of increased fl uid intake. Further questioning about diet often reveals a high intake of fruit juices and cordial• The cornerstone of successful treatment includes restriction of fruit juice in the diet, normalizing fl uid intake and (re-)introduction of wholemeal and other dietary fi bres, which add bulk to the stool
• It has also been suggested that many of these chil-dren are on a relatively low fat diet, and normalizing fat content acts to slow proximal gastrointestinal transit and improve symptoms.
Postinfective diarrhoea
• This is defi ned as the persistence of diarrhoea and failure to gain weight for more than 7 days after hospital admission for gastroenteritis
• It is generally due to a sugar intolerance, which can be confi rmed on the basis of stool analysis for reducing sugars and will resolve with elimination of the sugar from the diet
• Other causes include cow’s milk protein hypersensitivity or a persistent gastrointestinal infection.
Sucrase–isomaltase defi ciency
• This is an uncommon inherited disorder (autosomal recessive), with symptoms beginning after sucrose is introduced into the diet
• Symptoms consist of watery diarrhoea and abdominal distension. Growth is usually normal
• Diagnosis is dependent on a positive breath hydrogen test using sucrose as the test sugar. Alternatively, a small bowel biopsy containing very low isomaltase and sucrase levels will establish the diagnosis
• Management is based on dietary restriction of sucrose.
Chronic infl ammatory bowel disease
The incidence of Crohn disease has increased an nually; that of ulcerative colitis has shown an annual fl uctuation without an upward trend. Current opinion regarding the cause of infl ammatory bowel disease (IBD) favours the hypothesis that these two conditions result from an interaction between immu-nological, genetic and environmental factors.
Crohn disease can present in several ways:
• extraintestinal manifestations include growth retardation, anorexia, fatigue, delayed puberty, erythema nodosum, arthritis, clubbing, hepatitis and uveitis
• oropharyngeal involvement include orofacial granulomatosis and recurrent mouth ulcers
• oesophageal, gastric and small bowel Crohn may present as, nausea, vomiting, abdominal pain and diarrhoea
• colonic involvement, presents with passage of blood or mucous per rectum
• perianal involvement include skin tags, fi ssures, fi stulas and abscess.
1. EXCLUDE RETENTION WITH OVERFLOW
2. IS THERE FAILURE TO THRIVE?
3. IS STOOL VOLUME INCREASED?
4. IS THERE FAT MALABSORPTION?
5. IS THERE PROTEIN LOSING ENTEROPATHY?
YES
NO
ALWAYS investigate
Sometimes investigateTime is on your side.
YES NO
WBC, RBC, mucusi.e. Colitis
YES NO
Why morewater?
Digestive orabsorptive?
Fat globules Fatty acid crystals
DIGESTIVE a ABSORPTIVE b
OSMOTIC c
PRIMARY eSECRETAGOGUE d
SECRETION TODDLER
Clinitest, breath,hydrogen stool
electrolytes
Raised alpha -1-AT clearance f
a. Digestive pancreatic insufficiency (confirmed by low trypsin) liver disease obvious)
b. Absorptive mucosal damage: coeliac disease, giardias, proteinhypersensitivity, IBD. lymphatic block: lymphangiectasis.
c. Osmotic disaccharidase deficiency monosaccharide transport:glucose-galatose transport overload: high sugar fluids, fruits, sorbitol
d. Secretagogue bacterial toxin (C. difficile) deconjugated bile salts
e. Primary transport defect (rare) chlorideorrhoea
f. Protein losing enteropathy lymphangiectasia, polyposis
Fig. 20.2.3 Investigation of chronic diarrhoea.
THE CHILD WITH DIARRHOEA 20.2
Ch020-F10280.indd 727Ch020-F10280.indd 727 7/27/2007 6:01:31 PM7/27/2007 6:01:31 PM

728
Children with ulcerative colitis will usually present with lower abdominal pain, urgency, diarrhoea and rectal bleeding; additionally:
• systemic symptoms are less marked• the child can develop arthritis, which usually
correlates with disease activity• pyoderma gangrenosum occurs more commonly
in ulcerative colitis• the child can develop sclerosing cholangitis.
Children may experience the same symptoms, clini-cal presentations, complications and response to treatment as adults with IBD. This chapter will high-light some of the features of IBD that have particular importance in the paediatric patient.
Investigation
Several laboratory tests will support the diagnosis of IBD; however, endoscopy is the gold standard. Gas-troscopy and colonoscopy (with ileoscopy) is essen-tial, taking biopsies at all levels of the gut, whether or not there is macroscopic disease. Biopsies from a normal-appearing stomach or duodenum may contain granulomas, making the diagnosis clear. Pathological alterations above the ileum exclude the diagnosis of ulcerative colitis. A barium meal and follow-through or a labelled white cell scan can be helpful in assessing the area of the gut that is involved in Crohn disease. Capsule endoscopy is a new and evolving technology that is being used to image the small bowel mucosa.
Treatment
Enteral therapy• Provides a similar remission rate to corticosteroids in the treatment of childhood Crohn disease (not ulcerative colitis) and improves growth and infl am-matory markers• Probably exerts its benefi cial effects by alterations in gut fl ora, enterocyte nutrition and modulation of endogenous growth factors• Chronic supplementary enteral therapy reduces relapse rates in Crohn disease but not ulcerative colitis• Often utilizes polymeric and elemental feeds, which are probably equally effective but are not gen-erally palatable and may require nasogastric infu-sion. All other oral intake is ceased during this treatment, which usually lasts 6–8 weeks
Corticosteroids• Used in the dose range of 1–2 mg/kg of
prednisolone for moderate to severe ulcerative
colitis or Crohn disease, usually for a 2–3-month period with a gradual dose reduction
• Can adversely affect growth and have many unpleasant cosmetic and systemic side effects
• Can have a signifi cant effect on bone mineral density and lead to osteoporosis later in life
• Newer corticosteroids, such as budesonide, have fewer side effects and are particularly useful for ileal and right-sided colonic disease
• Steroid enemas are helpful in the management of lower colonic infl ammation in both Crohn disease and ulcerative colitis.
Other pharmacological treatments• Aminosalicylates can be used in both an oral and topical form to manage colitis in both Crohn disease and ulcerative colitis. This is often used in mild ulcerative colitis to achieve remission and in both ulcerative colitis and Crohn colitis as maintenance therapy• Azathioprine is often used in children with both ulcerative colitis and Crohn disease if they are steroid-dependent but will take 12–14 weeks to be clinically effective• Medications such as tacrolimus, ciclosporin and methotrexate may be used in the most severe disease; however, the effectiveness of such agents has only been assessed in open-label studies• Anti-tumour necrosis factor (TNF)α has shown promising results in the management of adults with severe Crohn disease but has been used sparingly in children. Side effects such as severe allergic reactions and the possible development of lymphoproliferative disorders are a major concern• Antibiotics, e.g. metronidazole, are useful in peri-anal and colonic Crohn disease
Surgery• Usually indicated in children with Crohn disease
who have growth failure and have not responded to pharmacological or nutritional therapies. This usually applies to an area of localized disease
• Appropriately timed surgery in children with Crohn disease may accelerate growth and advance puberty
• Colectomy is rarely required in children with ulcerative colitis unless there is uncontrolled bleeding or if toxic megacolon is present
Cancer risk
• Greatest in children with ulcerative colitis, par ticularly those with pancolitis for more than 10 years who also have coexisting sclerosing cholangitis
20.2 DISORDERS OF THE GASTROINTESTINAL TRACT AND HEPATIC DISORDERS
Ch020-F10280.indd 728Ch020-F10280.indd 728 7/27/2007 6:01:31 PM7/27/2007 6:01:31 PM

729
• To avoid colonic cancer, patients with long-stand-ing extensive colitis face either prophylactic colec-tomy or regular surveillance colonoscopy. Neither option is perfect; a more reliable, cheaper process of screening needs to be found• This general approach also applies to children and adults with extensive and long-standing colonic Crohn disease.
Practical points
Infl ammatory bowel disease• Potentially a multisystem chronic relapsing disease• Assessment of distribution of the disease may require
multiple modalities, i.e. endoscopy and biopsy and imaging modalities
• Treatment is individually tailored, based on site and severity of disease
• Be aware of long-term malignant risk, especially in ulcerative colitis, and also side effects of various medication used
Clinical example
Sarah presented at the age of 14 years with a 3-month history of recurrent abdominal pain and diarrhoea. Her stools are described as
watery with variable amounts of blood and mucus. Clinical examination revealed mild pallor and, on abdominal examination, there was tenderness in both the left and the right iliac fossa. Blood test revealed a microcytic anaemia (Hb 90 g/dl and an MCV of 69) and an ESR of 25 mm/h. Stool microscopy showed red blood cells and white blood cells. Repeated stool cultures for viruses, bacteria and also Clostridium diffi cile toxin was negative. At colonoscopy, there was a pancolitis and a normal ileum. A provisional diagnosis of ulcerative colitis was made and this was supported by the histology. Sarah was treated initially with high-dose steroids and later was maintained on sulfasalazine, with good clinical response.
• Observed accumulative incidences range from 5 to 10% after 20 years disease duration and 12–20% after 30 years. This implies that a person with ulcerative colitis has a roughly 12% chance of developing colorectal cancer between 10 and 15 years after the onset of the IBD
THE CHILD WITH DIARRHOEA 20.2
Ch020-F10280.indd 729Ch020-F10280.indd 729 7/27/2007 6:01:31 PM7/27/2007 6:01:31 PM

Malabsorption is not a clinical entity. It can be defi ned as the failure to absorb nutrients. A wide range of intestinal, pancreatic and hepatic disorders can be associated with malabsorption. To under-stand how one approaches the problem of malab-sorption in the clinical setting, an understanding of the normal physiology of nutrient digestion and salt, water and macronutrient and micronutrient absorp-tion is essential. This information is available in general physiology texts.
Diagnostic approachA large number of children have loose stools without having underlying gastrointestinal disease. In young children this is called ‘toddlers’ diarrhoea’. A major clinical challenge is to differentiate well children with loose stools from children who have gastroin-testinal disease. The diagnosis of the majority of children with malabsorption can be established with thorough clinical assessment, stool examination and simple ancillary tests.
Clinical assessment
Initial assessment can reveal whether a child is ill. If so, immediate evaluation will be required. In the well child, a ‘wait and see’ approach may be more reward-ing than immediate investigation.
Malabsorption does not present as malabsorption per se. Rather, individuals with malabsorption can present with a wide array of symptoms and physical signs (Table 20.3.1). Diarrhoea is the most common presentation and may be accompanied by loss of appetite, decreased physical activity, lethargy and growth failure. Children with coeliac disease may have decreased appetite, and are often cranky and irritable. In contrast, children with pancreatic insuf-fi ciency often develop a voracious appetite. In chil-dren with failure to thrive, a detailed dietary history is required. Occasionally, parents manipulate the child’s diet in an attempt to control the diarrhoea, which can lead to signifi cant dietary insuffi ciency with attendant weight loss. Assessment of the age of introduction of various foods into the diet may give insight to the underlying diagnosis. Onset of symp-
toms 3–6 months after the introduction of wheat products suggests the possibility of coeliac disease. Onset shortly after introduction of cow’s milk sug-gests cow’s milk protein intolerance. History of over-seas travel is important, as some unusual infections, such as amoebic dysentery, can cause chronic bloody diarrhoea.
The nature of the loose stool is important to ascer-tain, as it provides important clues to the patho-physiology and thus aetiology. Diarrhoea can be thought of in terms of fatty stools (steatorrhoea), watery diarrhoea (osmotic because of carbohydrate malabsorption or secretory) and bloody diarrhoea. Table 20.3.2 provides a differential diagnosis of chronic diarrhoea and malabsorption categorized by the nature of the stool.
Assessment of general health is important, as many gastrointestinal disorders exhibit extraintesti-nal manifestations. Cystic fi brosis (Ch. 14.6), Shwa-chman syndrome and immunodefi ciency disorders (Ch. 13.2) are associated with infections, particularly sinopulmonary infections. Delayed pubertal devel-opment can accompany many chronic disorders but is particularly prevalent in Crohn disease (Ch. 20.2).
Family history may be of note. Cystic fi brosis, primary disaccharidase defi ciencies and abetalipo-proteinaemia are recessively inherited. Coeliac disease and infl ammatory bowel disease are more frequently observed in fi rst-degree relatives.
Physical examination includes assessment of growth, nutritional status and pubertal development. Plotting percentile charts is mandatory. A child who is growing normally is unlikely to be suffering from serious gastrointestinal disease. Plotting longitudi-nal measurements, if available, is very important as it may give clues to the onset of disease and could indicate the diagnosis. Other physical signs of mal-absorption and specifi c nutritional defi ciencies include: loss of muscle bulk and subcutaneous fat; peripheral oedema (hypoproteinaemia); bruising (vitamin K defi ciency); glossitis and angular stoma-titis (iron defi ciency); fi nger clubbing (cystic fi brosis, Crohn disease, coeliac disease); skin rashes in coeliac disease (dermatitis herpetiformis) and infl ammatory bowel disease (erythema nodosum, pyoderma gan-grenosum); and specifi c skin disorders associated with zinc, vitamin A and essential fatty acid defi ciencies
730
20.3 Chronic diarrhoea and malabsorptionE. O’Loughlin
Ch020-F10280.indd 730Ch020-F10280.indd 730 7/27/2007 6:01:31 PM7/27/2007 6:01:31 PM
731
(Fig. 20.3.1). Rickets (vitamin D defi ciency) is very uncommon in sunny climates, even in condi-tions with severe steatorrhoea. It is important to examine carefully as there are many extraintesti-nal manifestations of gastrointestinal disease and malnutrition.
Stool examination
Stool examination is very simple and provides very important information. The presence of numerous white and red cells indicates colitis. This is usually due to bacterial or parasitic infection, to chronic infl ammatory disorders of the large bowel, or to milk protein intolerance when identifi ed in infants. Leu-kocytes are not increased in the stool of individuals
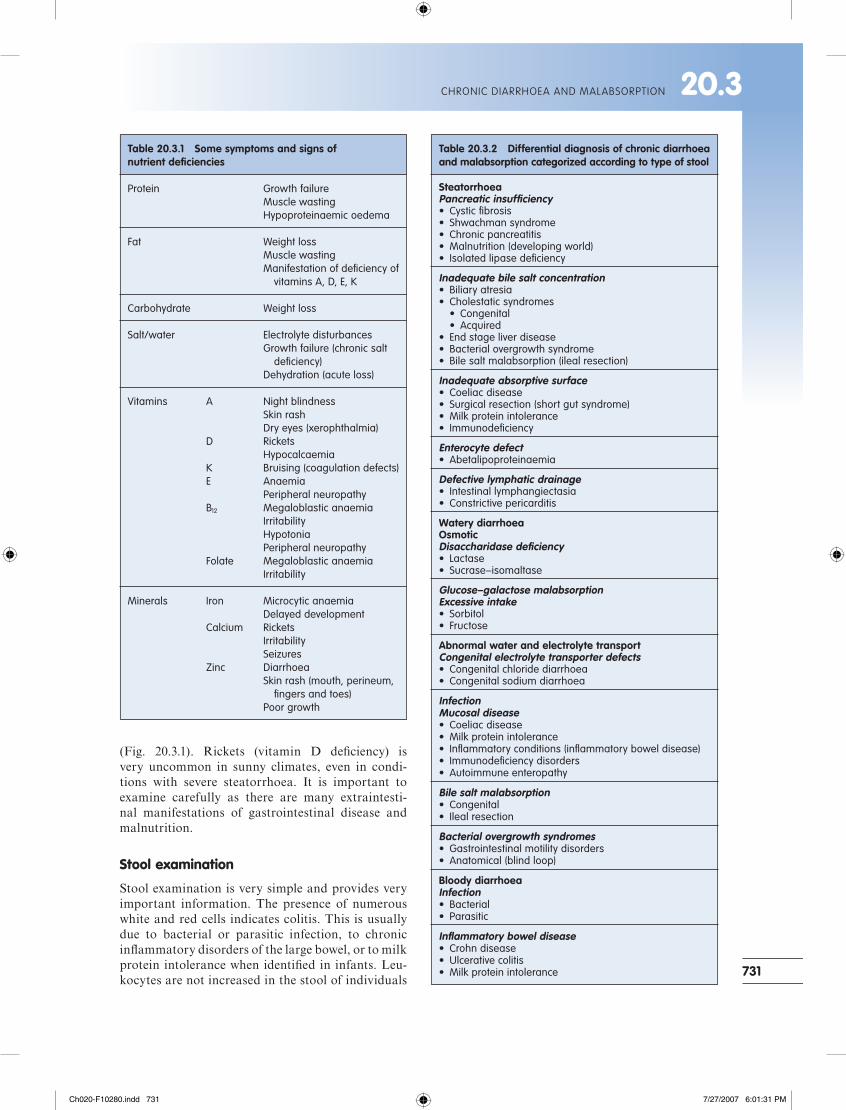
Table 20.3.1 Some symptoms and signs of nutrient defi ciencies
Protein Growth failure Muscle wasting Hypoproteinaemic oedema
Fat Weight loss Muscle wasting Manifestation of defi ciency of vitamins A, D, E, K
Carbohydrate Weight loss
Salt/water Electrolyte disturbances Growth failure (chronic salt defi ciency) Dehydration (acute loss)
Vitamins A Night blindness Skin rash Dry eyes (xerophthalmia) D Rickets Hypocalcaemia K Bruising (coagulation defects) E Anaemia Peripheral neuropathy B12 Megaloblastic anaemia Irritability Hypotonia Peripheral neuropathy Folate Megaloblastic anaemia Irritability
Minerals Iron Microcytic anaemia Delayed development Calcium Rickets Irritability Seizures Zinc Diarrhoea Skin rash (mouth, perineum, fi ngers and toes) Poor growth
Table 20.3.2 Differential diagnosis of chronic diarrhoea and malabsorption categorized according to type of stool
SteatorrhoeaPancreatic insuffi ciency• Cystic fi brosis• Shwachman syndrome• Chronic pancreatitis• Malnutrition (developing world)• Isolated lipase defi ciency
Inadequate bile salt concentration• Biliary atresia• Cholestatic syndromes • Congenital • Acquired• End stage liver disease• Bacterial overgrowth syndrome• Bile salt malabsorption (ileal resection)
Inadequate absorptive surface• Coeliac disease• Surgical resection (short gut syndrome)• Milk protein intolerance• Immunodefi ciency
Enterocyte defect• Abetalipoproteinaemia
Defective lymphatic drainage• Intestinal lymphangiectasia• Constrictive pericarditis
Watery diarrhoeaOsmoticDisaccharidase defi ciency• Lactase• Sucrase–isomaltase
Glucose–galactose malabsorptionExcessive intake• Sorbitol• Fructose
Abnormal water and electrolyte transportCongenital electrolyte transporter defects• Congenital chloride diarrhoea• Congenital sodium diarrhoea
InfectionMucosal disease• Coeliac disease• Milk protein intolerance• Infl ammatory conditions (infl ammatory bowel disease)• Immunodefi ciency disorders• Autoimmune enteropathy
Bile salt malabsorption• Congenital• Ileal resection
Bacterial overgrowth syndromes• Gastrointestinal motility disorders• Anatomical (blind loop)
Bloody diarrhoeaInfection• Bacterial• Parasitic
Infl ammatory bowel disease• Crohn disease• Ulcerative colitis• Milk protein intolerance
CHRONIC DIARRHOEA AND MALABSORPTION 20.3
Ch020-F10280.indd 731Ch020-F10280.indd 731 7/27/2007 6:01:31 PM7/27/2007 6:01:31 PM

732
with small bowel or pancreatic disease. Cysts of parasites such as Giardia lamblia indicate giardiasis.
Oil droplets seen on stool microscopy are always abnormal outside the newborn period and usually indicate fat maldigestion, as occurs with pancreatic insuffi ciency, e.g. in cystic fi brosis. Mucosal disease, such as coeliac disease, in general does not interfere with fat digestion because pancreatic function is usually normal. Mucosal disease interferes with the absorption of triglyceride products. These products are observed as fatty acid crystals on polarizing microscopy.
The presence of carbohydrate in the stool can be detected with Clinitest tablets. This is a commer-cially available bedside test in which the reaction between stool sugars such as lactose causes a colour change when added to the tablets. Greater than 500 mg/dl indicates carbohydrate malabsorption. Measurement of stool electrolytes and osmolality in the stool water is also a very useful test. When the sum of the stool electrolytes, i.e. sodium + potassi-um + chloride + bicarbonate, equals measured osmo-lality, a secretory diarrhoea is present. If the sum of the electrolytes is substantially less than the mea-sured osmolality (>100 mosmol/l), this indicates an osmotic diarrhoea.
Malabsorption with chronic diarrhoeaDiarrhoea is the most common presentation of mal-absorption. Diarrhoea can be defi ned as increased frequency, fl uidity and volume of stool. The follow-ing discussion will provide a systematic approach to
the child with malabsorption and diarrhoea based on the type of stool, i.e.:
• fatty• watery or• bloody.
Some illustrative cases will be provided.
Fatty diarrhoea (steatorrhoea)
The differential diagnosis of fat malabsorption is quite wide ranging (Table 20.3.2); however, if one understands the normal physiology of fat digestion and absorption, the differential diagnosis is much less daunting. Conditions that cause steatorrhoea can also be associated with protein maldigestion
Fig. 20.3.1 Exfoliative rash of zinc defi ciency.
Clinical example
Mary was 9 months old. She presented with poor weight gain, chronic diarrhoea and a history of recurrent respiratory illnesses,
including one admission at age 3 months with ‘bronchiolitis’. Loose stools were found each time her nappy was changed. On occasion mother had noted oil drops in the stool. Despite the poor weight gain, Mary had an excellent appetite and was described as a voracious eater. She consumed a mixed diet, including infant formula, appropriate for age. Cereal was introduced at age 6 months. Mother also commented that she tasted salty when she kissed Mary.
On examination, Mary was found to be a thin wasted girl. Her height was on the 50th percentile and her weight was less than the 3rd percentile. She had mild fi nger clubbing, peripheral oedema, pallor of the tongue and palmar creases but no signs of chronic liver disease. There was no abdominal distension of note, although she had a fi ne scaling rash over her trunk. Respiratory examination was normal. No other abnormal physical signs were present.
Results of investigations included Hb 85 g/l (normal range, 110–140) with a normocytic normochromic fi lm, normal white cell count and differential; albumin 24 g/l (normal range, 34–44) and normal liver function tests. Stool microscopy revealed copious fat droplets. 3-day faecal fat excretion estimation demonstrated an output of 35% of ingested fat (normal <7% of intake).
Mary’s diarrhoea was due to fat malabsorption, as evidenced by her mother’s observation of fat droplets in the stool.
Mary’s sweat test demonstrated a sweat chloride of 80 mmol/l (a result of >60 mmol/l is diagnostic of cystic fi brosis). Genetic testing indicated that she was homozygous ΔF508 (the commonest mutation), consistent with her relatively severe symptoms. Introduction of pancreatic exocrine replacement therapy, a high fat diet and vitamin supplements alleviated her diarrhoea and eventually corrected her failure to thrive, anaemia and skin rash.
20.3 DISORDERS OF THE GASTROINTESTINAL TRACT AND HEPATIC DISORDERS
Ch020-F10280.indd 732Ch020-F10280.indd 732 7/27/2007 6:01:31 PM7/27/2007 6:01:31 PM

733
and/or malabsorption, although symptoms most commonly relate to the malabsorption of fat. The presence of fat in the stool is also more readily observed than protein.
Fat and protein digestion and absorption
Ingested fat in the form of triglycerides, cholesterol and phospholipids is, to a large extent, digested in the lumen of the small intestine and absorbed in the jejunum. This requires bile salts, which form micelles and solubilize the fat; pancreatic enzymes, such as lipase and colipase, which digest the fat; and an intact intestinal mucosa, which is required for absorption of the products of digestion. Following digestion in the micelles, breakdown products diffuse across the enterocyte apical membrane and are reconstituted in the cell into chylomicrons. These are small packets of triglyceride, phospholipid and cholesterol which associate with carrier proteins, such as beta lipopro-tein, essential for cellular traffi cking of the chylomi-crons. After the chylomicrons are reconstituted they exit the mucosa into the lymphatic system and subse-quently pass into the systemic circulation. Some small chain triglycerides can bypass this system and enter the portal venous system directly.
Protein digestion begins in the stomach by the action of pepsin and acid. However, most protein hydrolysis occurs in the lumen of the jejunum by action of pancreatic proteases. These are secreted as inactive precursors. Chymotrypsin is converted to trypsin by the action of the small intestinal enzyme enterokinase. Activated trypsin further activates chymotrypsin and other proteases, such as car-boxypeptidase. The products of protein hydrolysis are amino acids and oligopeptides. The latter are further hydrolysed to mono-, di- and tripeptides by brush border hydrolyases and are absorbed by spe-cifi c membrane transporters. Di- and tripeptides undergo hydrolysis to amino acids in the cytoplasm of the enterocyte. Isolated protein maldigestion/malabsorption is extremely rare. It usually occurs in association with malabsorption of other macronutrients.
Fat malabsorption
Diseases of the pancreas and the small intestine are the usual causes of steatorrhoea in children. Chronic liver disease may cause steatorrhoea but this is in the setting of severe and obvious liver disease (such as the patient who is cirrhotic and jaundiced) and is not usually a diagnostic problem.
Steatorrhoea causes bulky stools and can lead to other nutritional defi cits. Fat is responsible for approximately 40% of caloric intake in the Western
diet. Thus, fat malabsorption can lead to failure to thrive due to an energy-defi cient diet. Some vitamins are fat-soluble and require normal fat digestion for their absorption. These include A, D, E and K. Thus patients with steatorrhoea may also develop signs of fat-soluble vitamin defi ciency, as described above. Essential fatty acids such as arachidonic acid are also malabsorbed in patients with pancreatic malab-sorption. A scaling skin rash is one physical mani-festation of essential fatty acid defi ciency.
Pancreatic and intestinal diseases associated with fat malabsorption can also result in protein and car-bohydrate maldigestion/malabsorption. Thus it is not uncommon to fi nd a mixed picture of malabsorp-tion. Protein maldigestion/malabsorption results in hypoproteinaemia. The main physical manifesta-tions are growth failure, peripheral oedema and ascites.
Pancreatic disease
Cystic fi brosisSee also Chapter 14.6.
Cystic fi brosis:
• is the commonest cause of pancreatic malabsorption in the Caucasian population
• has an incidence in the population of approximately 1 per 2000
• is an inborn error in epithelial chloride secretion (cystic fi brosis transmembrane conductance regulator (CFTR)).
Organs affected include:
• gastrointestinal tract and liver• sinopulmonary tract• pancreas• exocrine portion of the sweat glands• vas deferens• sweat duct (CFTR absorbs rather than secretes
chloride in this organ).
Because of the fl uid and salt transport defects, patients with cystic fi brosis produce more viscous secretions in lung, gut, pancreas and vas deferens, leading to:
• chronic suppurative lung disease• nasal polyps• pancreatic insuffi ciency• intussusception• meconium ileus and distal intestinal obstruction
syndrome• infertility• elevated sweat sodium and chloride, which can
lead to heat prostration in warmer climates.
Chronic liver disease will develop in 10–15% of chil-dren with cystic fi brosis.
CHRONIC DIARRHOEA AND MALABSORPTION 20.3
Ch020-F10280.indd 733Ch020-F10280.indd 733 7/27/2007 6:01:32 PM7/27/2007 6:01:32 PM

734
Malabsorption in cystic fi brosis frequently results in malnutrition and there may be symptoms and signs of specifi c nutrient defi cits such as hypoalbu-minaemic oedema, night blindness due to vitamin A defi ciency or skin rash due to essential fatty acid defi ciency. Median life expectancy is 30 years, with death usually from respiratory failure or haemor-rhage from portal hypertension and oesophageal varices.
Many mutations have been identifi ed in the CFTR. Depending on what part of the channel the mutation affects, the phenotype can vary from mild to severe disease. Individuals with milder mutations have milder lung disease and do not usually have malabsorption, as pancreatic function is normal.
Newborn screening:
• can detect cystic fi brosis in the neonatal period
• involves measurement of immunoreactive trypsinogen and/or CFTR mutations
• is the commonest mode of presentation when it is performed.
In children with the severe phenotype who are missed by screening, or in countries where screening is not performed, presentation is usually in the fi rst year with chronic diarrhoea and failure to thrive, with or without respiratory symptoms. In milder pheno-types, patients may not present until adult life with respiratory disease or infertility.
Diagnostic investigations for cystic fi brosis are:
• elevated sweat sodium and chloride (‘sweat test’) – simplest and cheapest
• CFTR mutation analysis.
Treatment is usually undertaken in a tertiary referral multidisciplinary clinic and involves:
• physiotherapy, inhalation therapy and antibiotics for chest disease
• pancreatic enzyme supplements and nutritional support
• specifi c therapy may be required for the other intestinal/liver complications.
Shwachman syndromeThe features of Shwachman syndrome are:
• agenesis of the pancreatic acinus• short stature• dysplasia of the metaphysis of the long bones• cyclical neutropenia.
There is no specifi c diagnostic test; treatment includes pancreatic exocrine replacement and treatment of infections.
Chronic pancreatitisCauses of chronic pancreatitis include:
• protein energy malnutrition• hereditary pancreatitis (rare)• idiopathic fi brosing pancreatitis (rare).
Small bowel disease
Coeliac disease (gluten enteropathy)Coeliac disease is a disorder characterized by intes-tinal injury induced by the cereal protein gluten. Gluten is a glycoprotein found in wheat, barley and rye and, to a lesser extent, oats. In susceptible individuals, the ingestion of gluten induces a cell-mediated injury of the intestinal mucosa resulting in severe villous atrophy, crypt hyperplasia and infi ltra-tion of the epithelium with lymphocytes (intraepithe-lial lymphocytes). In Western countries, the incidence of coeliac disease in the general population may be as high as 1 in 70, although not all affected individu-als develop the classical manifestations of coeliac disease.
Modes of presentation include:
• ‘Classical’ coeliac disease (Fig. 20.3.2) • between 9 and 18 months of age • anorexia, weight loss, abdominal distension
and wasting
Fig. 20.3.2 Typical physical appearance of a young child with coeliac disease. Note the protuberant abdomen, buttock and shoulder girdle wasting and oedema of the lower limbs. Courtesy of Professor K. Gaskin.
20.3 DISORDERS OF THE GASTROINTESTINAL TRACT AND HEPATIC DISORDERS
Ch020-F10280.indd 734Ch020-F10280.indd 734 7/27/2007 6:01:32 PM7/27/2007 6:01:32 PM

735
• chronic diarrhoea with or without: • iron defi ciency anaemia • hypoproteinaemic oedema • fat-soluble vitamin defi ciency
• The older child with: • growth failure • chronic diarrhoea • iron defi ciency• Positive antibody screening (now the commonest
form of assessment leading to diagnosis).
Examples of antibodies used to screen when there is suspicion of coeliac disease include:
• antigliadin• anti endomysial• antitissue transglutaminase antibodies.
Antiendomysial and antitissue transglutaminase antibodies have sensitivity and specifi city of greater than 95%. However, it is important to note that these are screening tests only.
The following are important points in the approach to the diagnosis of coeliac disease in childhood:
• small bowel biopsy is mandatory for the diagnosis (Fig. 20.3.3)
• small bowel biopsy should be performed while the patient is on an unrestricted diet
• there is no place for an empirical trial of a gluten-free diet
• defi nitive diagnosis is important, as treatment is a lifelong gluten-free diet.
A second biopsy can be undertaken to establish that the intestine has returned to normal on a restricted diet. If there is doubt about the diagnosis, a subse-quent gluten challenge with repeat biopsy can be undertaken.
Enterocyte defectAbetalipoproteinaemia is a recessively inherited defect in chylomicron assembly. Patients develop ste-atorrhoea early in life with:
• fat-soluble vitamin defi ciencies• low serum cholesterol and triglycerides.
Small bowel biopsy reveals fat laden enterocytes.
Impaired lymphatic drainageObstructed lymphatic drainage prevents chylomi-crons from migrating from the gut to the systemic circulation. The main cause is intestinal lymphangi-ectasia. This can lead to:
• fat malabsorption• low serum cholesterol and triglycerides• hypoproteinaemia and lymphopenia (loss of
lymph into gut lumen)• abnormal mucosal biopsy.
Other causes of reduced mucosal surface and reduced contact timeMiscellaneous infl ammatory and surgical conditions can lead to loss of absorptive surface or reduced contact between chyme and the mucosa. Such condi-tions include:
• milk protein intolerance (severe)• infections such as rotavirus infection• severe immunodefi ciency disorders• autoimmune enteropathy• short gut syndrome (surgical removal)• motility disorders causing very rapid intestinal
transit.
Fig. 20.3.3 Micrographs of normal intestine (left) demonstrating normal crypt villus structure, and coeliac disease (right) with marked crypt hyperplasia and villous atrophy.
CHRONIC DIARRHOEA AND MALABSORPTION 20.3
Ch020-F10280.indd 735Ch020-F10280.indd 735 7/27/2007 6:01:32 PM7/27/2007 6:01:32 PM

736
Watery diarrhoea facilitated diffusion (non-energy-dependent) by the transporter termed GLUT-5.
Carbohydrate malabsorption
The presence of non-absorbed osmotically active nutrients in the gut lumen results in osmotic retarda-tion of water absorption, leading to watery diar-rhoea. This is referred to as osmotic diarrhoea. Osmotically active compounds are usually low-molecular-weight compounds such as monosaccha-rides and disaccharides. Osmotic diarrhoea is usually due to maldigestion and/or malabsorption of carbo-hydrates but can be caused by the ingestion of laxatives such as sorbitol or MgCl2. Unabsorbed car-bohydrate present in the lumen of the large bowel is fermented to short chain fatty acids such as butyrate. This results in a highly acidic stool, which can cause perianal excoriation. The colon can absorb the anionic forms of these acids in exchange for bicar-bonate, causing a mild hyperchloraemic acidosis.
While stating the obvious, it is important to appre-ciate that one cannot malabsorb a nutrient that has not been ingested. Thus it is useful to obtain a dietary history in patients suspected of osmotic diarrhoea. One needs to ascertain the nature of the carbohy-drates being ingested, and in some instances the age of introduction of the carbohydrate, which can then be compared with the age of onset of symptoms. For example, the onset of osmotic diarrhoea commensu-rate with the introduction of fruit into the diet sug-gests the diagnosis of congenital sucrase–isomaltase defi ciency.
Disaccharidase defi ciencies and monosaccharide malabsorption
CongenitalOntogenic lactase defi ciency:
• occurs in most of the non-Caucasian population of the world
• is dominantly inherited• is physiological (due to the disappearance of
lactase)• presents in late childhood.
Ingesting lactose causes diarrhoea, bloating, exces-sive fl atus and weight loss. Treatment is a low-lactose diet.
Congenital sucrase–isomaltase defi ciency is caused by inactivating mutations in the sucrase–isomaltase gene. These mutations:
• are recessively inherited• lead to similar symptoms as for lactase defi ciency
with the ingestion of sucrose
Clinical example
George was 9 years old. He presented with a 6-month history of intermittent bloating, abdominal pain and diarrhoea up to 6–7
times per day. He had lost 1 kg in weight in the past 2 months. He reported that dairy products such as milk and ice-cream made his symptoms worse. He had no past history of signifi cant illness. George was the oldest son of Greek migrants. His mother reported that she could not drink milk, as it made her feel sick.
On examination he was well looking. His weight was on the 50th percentile and his height was on the 10th percentile. There was no abdominal distension, organomegaly, signs of chronic liver disease or evidence of nutritional defi ciency such as anaemia or peripheral oedema. Examination of his anus did not reveal any evidence of perianal disease.
Investigations included a normal full blood count, differential white cell count and ESR. C reactive protein was less than 1 g/l. Lactose breath hydrogen measurement following oral ingestion of 50 g of lactose increased 100 parts per million above baseline levels within 60 minutes of ingestion of lactose (normal rise 20 parts per million), indicating lactose intolerance.
George was diagnosed as having lactose intolerance. His history suggested ontogenic lactase defi ciency. This was confi rmed by small bowel biopsy, which demonstrated normal morphology, and disaccharidase measurement, which revealed very low lactase activity but normal sucrase and maltase activities. Treatment is a low-lactose diet.
Carbohydrate digestion and absorption
Dietary carbohydrates are primarily starch (poly-saccharides, amylose and amylopectin), disaccha-rides (sucrose, in table sugar; lactose, in milk) and some monosaccharides such as fructose.
Starch polymers are large molecules composed of long chains of glucose. These chains are broken down by the action of salivary and pancreatic amylase, which release a disaccharide (amylose), tri-saccharide (maltotriose) and a series of branched oligosaccharides (alpha limit dextrins). These mole-cules are further digested by the brush border enzymes, sucrase–isomaltase and glucoamylase, to the monosaccharide glucose.
The disaccharides sucrose and lactose are metabo-lized by disaccharidases on the intestinal brush border. Sucrase breaks sucrose down to glucose and fructose and lactase breaks down lactose into glucose and galactose. Glucose and galactose are absorbed by the enterocyte sodium–glucose cotransporter (SGLT), which absorbs the monosaccharides in an energy dependent fashion. Fructose is absorbed by
20.3 DISORDERS OF THE GASTROINTESTINAL TRACT AND HEPATIC DISORDERS
Ch020-F10280.indd 736Ch020-F10280.indd 736 7/27/2007 6:01:32 PM7/27/2007 6:01:32 PM

737
• cause onset of symptoms at the time of weaning when fruit is introduced to the diet.
Treatment is a low-sucrose diet.Congenital monosaccharide malabsorption refers
to defective glucose/galactose malabsorption. Fea-tures are:
• mutations in SGLT1• recessively inherited• present in the neonatal period.
Treatment is substitution of fructose for glucose–galactose.
AcquiredExcept for ontogenic lactase defi ciency, acquired dis-orders are much more common than inherited defi -ciencies. Lactase is more susceptible to injury than sucrase.
Causes of disaccharidase defi ciencies include:
• viral gastroenteritis• coeliac disease• chronic giardiasis• milk protein enteropathy• small bowel bacterial overgrowth syndrome• immunodefi ciency disorders• autoimmune enteropathy.
Monosaccharide transporters are less susceptible to injury because, unlike disaccharidase enzymes, they are deeply embedded in the brush border membrane. However, severe enteropathies can occasionally result in monosaccharide malabsorption. Examples include:
• congential villous atrophy (which presents in newborns)
• severe postinfectious enteritis• milk protein intolerance• autoimmune enteropathy.
Monosaccharide malabsorption is life-threatening and requires a level of care found only in tertiary paediatric centres. The treatment is to remove the offending carbohydrate from the diet and substi-tute an alternative. In acquired disorders, treatment may also be required for the primary mucosal disease.
Disorders of fl uid and electrolyte transport
In the normal child approximately 5 litres (depend-ing on size!) of fl uid and electrolytes enters the upper gastrointestinal tract per day. One litre is ingested and the remaining volume is from normal secretions into the lumen. The majority of this fl uid is absorbed before reaching the colon. Stool weights range from 75 to 150 g per day, of which approximately 75% is
water. Small increases in stool water, as little as 30–40 ml/d are enough to produce diarrhoea.
Water is absorbed by osmosis through paracellu-lar pathways in the mucosa. Electrolytes are absorbed by a variety of active transport or passive transport processes. Anions such as chloride and bicarbonate can be absorbed or actively secreted. This varies according to the region of small or large intestine. Regulation of gastrointestinal fl uid and electrolyte transport is closely integrated by humoral and neural factors involved in fl uid and electrolyte homeostasis. Abnormal fl uid and electrolyte transport can be due to inherited defects in specifi c electrolyte transport-ers, but more commonly it is due to mucosal damage or infl ammation.
CongenitalCongenital sodium diarrhoea and congenital chlo-ride diarrhoea are rare inherited disorders of Na/H exchange and Cl/HCO exchange, respectively. They cause:
• diarrhoea in utero which results in polyhydramnios
• profuse diarrhoea, obvious from birth• systemic electrolyte disturbances.
AcquiredIsolated water and salt malabsorption is very rare in childhood in the developed world. However, defec-tive salt and water transport can contribute to diar-rhoea in:
• disorders which damage or infl ame the mucosa of small or large intestine
• bile salt malabsorption (bile acids irritate the colonic mucosa and act as potent stimulants of secretion).
Excessive salt and water loss in the stool may lead to dehydration and electrolyte disturbances. Treatment may require salt and water replacement in addition to treatment of the underlying disease.
Bloody diarrhoea
Chronic bloody diarrhoea is usually caused by infl ammatory disorders of the colon such as:
• milk colitis in infants• infections such as bacteria or parasites• infl ammatory bowel disease in older children.
The two major forms are: • ulcerative colitis • Crohn disease.
Blood is not always obvious in the stool. How-ever, the presence of leukocytes on stool micro-scopy (Fig. 20.3.4) indicates the presence of colitis.
CHRONIC DIARRHOEA AND MALABSORPTION 20.3
Ch020-F10280.indd 737Ch020-F10280.indd 737 7/27/2007 6:01:32 PM7/27/2007 6:01:32 PM

738
Malabsorption of fl uid and electrolytes by the infl amed colonic mucosa is a major factor contribut-ing to diarrhoea. Malabsorption of nutrients is uncommon in milk colitis and infl ammatory bowel disease. In contrast, excessive blood and protein loss from the infl amed intestinal mucosa can cause iron defi ciency anaemia and hypoproteinaemic oedema. This is called protein-losing enteropathy.
Nutrient malabsorption with little or no diarrhoeaChildren present with symptoms and signs of nutri-ent defi ciency with little or no accompanying diar-rhoea. This is often due to dietary insuffi ciency, e.g. inadequate iron intake, but sometimes it can be due to malabsorption of the specifi c nutrient.
Vitamin B12
Vitamin B12 is ingested in animal protein and is liber-ated by pepsin in the stomach. In the stomach, the free vitamin B12 binds to a binding protein (R protein) which has greater affi nity for the vitamin than intrin-sic factor (carrier protein). Intrinsic factor is pro-duced by epithelial cells in the gastric mucosa. The vitamin B12–R protein complex moves to the duode-num where trypsin cleaves the complex, releasing free vitamin B12, which then binds to intrinsic factor. The intrinsic factor–vitamin B12 complex moves to
Fig. 20.3.4 Microscopic appearance of leukocytes in a stool smear. The large dark structures are polymorphonuclear leukocytes; the small round objects are red blood cells
Clinical example
John was 9 months old. He presented with a 6-week history of poor weight gain, irritability and pallor. His mother was also concerned
about his development. He was able to sit but could not pull himself to standing. His language had not progressed from babbling, which was in stark contrast to his older sibling, who had several single words at this age. John had a poor appetite but no diarrhoea. He was originally breastfed and his mother ingested a normal diet during pregnancy and lactation.
On examination, John was a pale irritable boy. He had moderate abdominal distension but no organomegaly. He could sit up unsupported but was mildly hypotonic and would not weight bear. There were no focal neurological signs.
Investigation results included: Hb of 65 g/l (normal 120–150) with a megaloblastic blood fi lm, serum B12 50 pmol/l (normal 120–600) and red blood cell folate 350 nmol/l (normal 200–1000). A Shilling test revealed urinary excretion of ingested radioactive vitamin B12 (after parenteral administration of a non-radioactive fl ushing dose of 1 mg vitamin B12) of 1% (normal 8%), with no enhancement of urinary excretion with the addition of intrinsic factor.
John’s Shilling test suggested a defect in the ileal vitamin B12 transporter, as the test was abnormal and did not recover with the addition of intrinsic factor. His age of presentation and lack of prior intestinal surgery suggest a congenital defect. His symptoms and megaloblastic anaemia corrected with administration of parenteral vitamin B12.
20.3 DISORDERS OF THE GASTROINTESTINAL TRACT AND HEPATIC DISORDERS
Ch020-F10280.indd 738Ch020-F10280.indd 738 7/27/2007 6:01:32 PM7/27/2007 6:01:32 PM

739
the ileum where it is absorbed into the enterocytes by carrier mediated transport. On entry into the enterocyte, vitamin B12 is separated from intrinsic factor and subsequently exits the enterocyte into the circulation bound to transcobalamin, which carries the vitamin to sites distant from the intestine.
Both congenital and acquired disorders can lead to vitamin B12 malabsorption.
Congenital disorders
Congenital defects in:
• ileal vitamin B12 transporter• intrinsic factor• transcobalamin
can lead to vitamin B12 malabsorption and defi ciency. This usually presents in the second 6 months of life after the vitamin B12 accumulated during intrauter-ine life is exhausted.
Symptoms are due to megaloblastic anaemia and the central nervous system effects of defi ciency. Babies born to vegan mothers (who ingest no animal product and thus can themselves be vitamin-B12-defi cient) and weaned on to a vegan diet can present with a similar picture, although usually in the fi rst 6 months, as they are defi cient from birth. Dietary history is important to differentiate between dietary defi ciency and malabsorption.
Acquired disorders
Acquired disorders that lead to B12 malabsorption are:
• surgical resection of the ileum• atrophic gastritis• gastric surgery• autoimmune pernicious anaemia (blocking
antibodies to intrinsic factor)• pancreatic insuffi ciency (failure to hydrolyse
vitamin B12–R protein)• small bowel bacterial overgrowth (competition
for vitamin B12 by bacteria).
Iron
Iron absorption occurs in the duodenum and proxi-mal jejunum. An apical enterocyte carrier called the divalent metal cation transporter mediates uptake into the enterocyte. Iron is exported to the circula-tion via a basolateral process which has not yet been fully defi ned. In non-breastfed children, only 5–10% of dietary iron is absorbed. The effi ciency of iron absorption is greater in breastfed infants because the iron carrier transferrin is present in breast milk. Iron absorption is fi nely regulated at the level of the
enterocyte so that absorption does not exceed requirements. Excessive iron accumulation can lead to multiple organ damage (haemochromatosis).
Iron defi ciency is the commonest nutritional defi -ciency in humans and is usually due to:
• inadequate dietary intake• excessive gastrointestinal blood loss (bleeding
lesions or infl ammation).
Inherited defects in iron uptake mechanisms leading to iron defi ciency not responsive to oral iron have been described but have not yet been delineated at the molecular or genetic level.
Acquired disordersIron defi ciency anaemia can occasionally be the primary presenting feature of small intestinal disease such as:
• coeliac disease• milk protein intolerance• Crohn disease.
Miscellaneous nutrients
Calcium
Calcium absorption occurs in the duodenum and proximal jejunum and is largely under the regulation of vitamin D:
• hypocalcaemia can be associated with a wide variety of digestive disorders affecting intestinal calcium uptake or the biosynthesis and availability of vitamin D (Ch. 19.5)
• it commonly presents with tetany of the fi ngers and occasionally seizures.
Zinc
Zinc is absorbed by the small intestine. Zinc defi -ciency can be due to:
• low breast milk zinc levels in solely breastfed infants
• an inherited defect in zinc absorption (acrodermatitis enteropathica)
• conditions associated with steatorrhoea• intestinal infl ammatory disorders.
Zinc defi ciency can cause diarrhoea but the most dramatic manifestation is an erythematous scaly rash on the fi nger tips and around the perineum and mouth (Fig. 20.3.1).
Magnesium
Magnesium is absorbed in the proximal small intes-tine. Magnesium malabsorption leading to defi ciency can be:
CHRONIC DIARRHOEA AND MALABSORPTION 20.3
Ch020-F10280.indd 739Ch020-F10280.indd 739 7/27/2007 6:01:33 PM7/27/2007 6:01:33 PM

740
• inherited (primary hypomagnesaemia)• secondary to other conditions leading to
malabsorption.
Hypomagnesaemia causes similar symptoms to calcium defi ciency.
Isolated protein malabsorption
Enterokinase defi ciency:
• is a very rare disorder• presents with diarrhoea, growth failure and
severe hypoproteinaemia.
Amino acids
Defective amino acid absorption due to mutations in amino acid transporters can occur in:
• Hartnup disease• cystinuria• lysinuric protein intolerance.
These defects are rare disorders affecting amino acid transport in gut and kidney and in other organs in the last case. They do not have gastrointestinal symptoms and there are no nutritional consequences because of compensatory absorptive mechanisms for peptide and amino acid absorption.
Summary of the diagnostic approach to suspected malabsorptionInitial clinical assessment and stool examination will suggest the diagnosis in most children. Stool micros-copy and measurement of stool-reducing substances can be performed in the clinician’s offi ce and are readily available ‘bedside’ tests. If the diagnosis is not immediately obvious, the clinician will be in a position to investigate a limited differential list with simple and well directed diagnostic tests.
In patients with steatorrhoea the following will be useful but are not necessarily indicated for each patient:
• full blood count and differential white cell count• serum triglycerides/cholesterol
• sweat test• small bowel biopsy• X-ray of long bones.
In patients with carbohydrate maldigestion/malabsorption the following might be indicated:
• breath hydrogen testing: challenge with the carbohydrate of interest (e.g. lactose)
• small bowel biopsy/mucosal disaccharidase activities
• occasionally with monosaccharide malabsorption • inpatient dietary manipulation with close
observation of stool output.
In patients with bloody diarrhoea (if stool cultures negative for pathogens) consider:
• gastroscopy and colonoscopy• biopsy of small bowel and colon• sometimes radiology looking for infl ammatory
bowel disease in jejunum/ileum.
Sometimes highly specialized investigations will be required to establish the diagnosis of some disorders:
• measurement of micronutrients such as iron, zinc and calcium for suspected defi ciency
• Schilling test is required for the workup of vitamin B12 defi ciency. Abnormally low urinary excretion of the ingested radioactive vitamin B12 indicates vitamin B12 malabsorption
• Schilling test can be used to assess patients with bile salt malabsorption due to ileal resection
• specialized breath tests are used in the workup of bacterial overgrowth syndrome
• immunoglobulins and B- and T-cell subset determination for detection of immunodefi ciency disorders.
Practical points
• Diagnosis is not by exclusion• A thorough history, physical examination and stool
examination will suggest the diagnosis in most disorders
• simple well-directed investigations usually confi rm the clinical diagnosis
• there is no such thing as a ‘malabsorption workup’
20.3 DISORDERS OF THE GASTROINTESTINAL TRACT AND HEPATIC DISORDERS
Ch020-F10280.indd 740Ch020-F10280.indd 740 7/27/2007 6:01:33 PM7/27/2007 6:01:33 PM

This chapter discusses gastro-oesophageal refl ux (GOR) a very common clinical problem in infants and children, and Helicobacter pylori, an infectious agent that colonizes the stomach in more than 50% of the world’s population. H. pylori infection is acquired in early childhood but its disease manifes-tations usually do not occur until adulthood. It is also possible that there may be a relationship between the two, and this will be discussed.
Gastro-oesophageal refl uxGastro-oesophageal refl ux can be defi ned as the spontaneous or involuntary passage of gastric content into the oesophagus. The origin of the gastric content can vary and includes saliva, ingested food and fl uid, gastric secretions and pancreatic or biliary secretions that have fi rst been refl uxed into the stomach (duodenogastric refl ux). The difference between physiological refl ux and gastro-oesophageal refl ux disease (GORD) is often blurred by the anxiety engendered in parents, particularly fi rst-time parents, by symptoms such as vomiting and irritability. Physi-ological refl ux manifested by spilling, regurgitation and occasional vomiting occurs in more than 60% of healthy infants by 4 months of age, resolves in the majority by 12 months and rarely leads to GORD. Conservative management is important, particularly in an otherwise healthy infant, so as not to label the condition as a disease state when in fact it is not.
The symptoms of GORD in children aged 3–18 years ranges from 1.8–22% and are more refractory and associated with complications such as pain, vomiting, haematemesis, oesophagitis, stricture, growth failure, swallowing diffi culties, respiratory symptoms and apnoea.
Pathophysiology (Table 20.4.1)
The main barrier to GOR is the pressure gradient across the lower oesophageal sphincter (LOS) which is formed by the intrinsic LOS (thickened smooth muscle of the lower oesophagus) and the extrinsic
striated muscle of the crural diaphragm. Both com-ponents work together to generate LOS pressure, which can be measured by intraluminal manometry. The current understanding of LOS function suggests that a LOS pressure of 5–10 mmHg above intragas-tric pressure is suffi cient to maintain an antirefl ux barrier. Sphincter incompetence as a pathologi-cal mechanism for GORD is extremely unlikely. Transient lower oesophageal sphincter relaxation (TLOSR) is the major mechanism responsible for GOR in infants, children and adults. A TLOSR is defi ned as an abrupt decrease in LOS pressure unre-lated to swallowing or oesophageal body peristalsis. TLOSRs are signifi cantly longer in duration than swallow-related sphincter relaxation and also have a lower nadir pressure. It is unclear at present whether GORD in children is characterized by either a higher rate of TLOSR or a greater incidence of GOR episodes during TLOSRs. Both have been noted in adults.
Abdominal straining
Abdominal straining, which occurs frequently in infants, probably exacerbates GOR only when there is simultaneous TLOSR, because both LOS tone and the crural diaphragm are inhibited. The neuroregu-lation of TLOSR is controlled via a vagovagal refl ex. The afferent arm of the refl ex is initiated by mecha-noreceptors in the wall of the proximal stomach, and the efferent arm via a brain-stem pattern generator. The presynaptic neurotransmitter is acetylcholine and the postsynaptic neurotransmitter is nitric oxide. Feeding is a potent stimulus for TLOSRs, evidenced by the fact that, in children with GORD, TLOSRs increase from four per hour in the fasting state to eight per hour in the fed state.
Oesophageal body peristalsis
Assessment of oesophageal volume clearance is dif-fi cult because of the lack of defi ned motility criteria. Primary oesophageal body peristalsis following a swallow facilitates clearance. Secondary peristalsis
741
20.4Gastro-oesophageal refl ux and Helicobacter
pylori infectionG. Davidson
Ch020-F10280.indd 741Ch020-F10280.indd 741 7/27/2007 6:01:34 PM7/27/2007 6:01:34 PM

742
is initiated by an abrupt sustained increase in intra-oesophageal pressure that accompanies a refl ux episode. The frequency of swallowing and type of pressure wave sequence propagated deter-mine the effectiveness of volume clearance. While severe GORD with refl ux oesophagitis is associated with a 30–50% decrease in pressure wave amplitude, this in itself may not impair bolus clearance.
Gastric emptying
The role of gastric emptying in the pathophysiology of GORD is not clear. Delayed gastric emptying could exacerbate GOR by prolonging gastric disten-sion and increasing the frequency of TLOSRs. Studies attempting to correlate a delay in gastric emptying with acid GOR have been inconclusive. While the fi nal answer to this question awaits the development of more sophisticated investigative techniques, there are some children at the severe end of the GORD spectrum in whom delayed gastric emptying may be an issue, especially those with neu-rological or respiratory disease.
Clinical manifestations
There are many causes of regurgitation and vomiting in infants and children, both within the gastrointes-tinal tract and external to it. The more common causes are outlined in Table 20.4.2.
Regurgitation can be defi ned as effortless spilling of gastric content that is usually benign. Vomiting, on the other hand, is a forceful emptying of gastric content that should always be explained. The content of the vomitus is important because of the likely cause, as is the age at onset. Bile staining implies small bowel obstruction and should be examined
immediately. Blood staining implies ulceration or gastritis.
Table 20.4.3 highlights the symptoms suggestive of GORD in infants and children. Symptoms do vary according to age. Infants more frequently regurgi-tate but can also have refl ux-related behaviours, which include apparent discomfort, yawning, stretch-ing, stridor or mouthing. Irritability and crying as the sole manifestation of GOR should be assessed with caution as it has been shown recently that in children with proven GORD there was no associa-tion and no response to proton pump inhibitors. More serious complications include apnoea, acute life-threatening events and recurrent chest disease secondary to aspiration. Chronic cough without associated lung disease is unlikely to be refl ux-related.
Older children, usually over the age of 4 years, can describe common symptoms such as heartburn, chest pain and a sick or sour taste in the mouth, implying refl uxate. Some younger children may com-plain of a hot feeling in the chest, abdomen or throat.
Table 20.4.1 Pathophysiological mechanisms of gastro-oesophageal refl ux in infants, children and adolescents
• Delayed volume clearance • Impaired primary or secondary peristalsis • Reduced pressure wave amplitude• Increased occurrence of GOR: • Transient LOS relaxation • Straining • LOS sphincter hypotonia • LOS pressure drift• Delayed gastric emptying
Modifi ed from Davidson GP, Omari TI 2001 Pathophysiological mechanisms of gastroesophageal refl ux disease in children. Current Gastroenterology Reports 3: 257–262.
Table 20.4.2 Causes of regurgitation and vomiting in infants and children
Gastrointestinal tract• Oesophagus• Achalasia• GOR• Foreign body• Congenital defects• Stomach• Pyloric stenosis• Peptic ulcer disease/gastritis• Duodenum• Malrotation• Duodenal ulcer• Superior mesenteric artery syndrome• Small intestine/colon• Infectious diarrhoea• Intussusception• Soy cow’s milk protein intolerance• Meconium ileus• Infl ammatory bowel disease• Appendicitis• Other organs• Hepatitis• Gallbladder disease• Pancreatitis
Extraintestinal disorders• Generalized sepsis• Rumination• Intoxications• Intracranial lesions, e.g. tumour, hydrocephalus• Adrenal insuffi ciency• Metabolic disorders
20.4 DISORDERS OF THE GASTROINTESTINAL TRACT AND HEPATIC DISORDERS
Ch020-F10280.indd 742Ch020-F10280.indd 742 7/27/2007 6:01:34 PM7/27/2007 6:01:34 PM

743
GORD is a common problem in neurologically impaired children and, while regurgitation is the most likely symptom, problems such as recurrent chest disease, feeding diffi culties and food refusal, anaemia, weight loss and behavioural changes can all be manifestations of GORD.
There are many potential extra-oesophageal man-ifestations of GORD and these are highlighted in Table 20.4.4, and need to be considered as they may be the only presenting symptom or sign. Ear nose and throat manifestations such as otitis media, sinus-itis and dental erosions are now being recognized
Eosinophilic oesophagitis
This condition has only been recognized in the past decade. GORD is distinguished from eosinophilic oesophagitis by the presence of eosinophils in the oesophageal mucosa. Their presence had previously been thought to be due to acid refl ux. The density of eosinophils in the mucosa (<20 per high power fi eld) defi nes the difference between these two conditions.
Dysphagia with solid food, epigastric pain, food impaction, and vomiting. Food allergy is present in more than 60% and at present the only proven effec-tive therapy is strict avoidance of the offending allergen. Topical steroids seem capable of inducing remission if a food is not identifi ed.
Diagnostic tests
Physiological GOR should be diagnosed on clinical grounds and diagnostic tests are not required. There is no single test for the diagnosis of GORD. If there are symptoms or signs of pathological refl ux, such as pain, growth failure or respiratory symptoms, then
Table 20.4.3 Symptoms suggestive of gastro-oesophageal refl ux disease in infants and children
Infants Children
VomitingGastrointestinal Feeding diffi culties Waterbrash Failure to thrive Nausea Malnutrition Dysphagia Cow’s milk protein intoleranceRespiratory Cough, stridor Chronic cough Cyanotic episodes Apnoea Acute life-threatening events
Acid refl uxGastrointestinal Apnoea, cyanotic episodes Heartburn Colic, irritability Oesophageal obstruction Sleep disturbance Dysphagia, odynophagia Flexion patterns after feeds Night waking Hiccoughs Haematemesis Iron defi ciencyRespiratory Apnoea, cyanotic episodes StridorNeurobehavioural Sandifer syndrome Seizure-like events (similar to infantile spasms)
Table 20.4.4 Potential extraintestinal manifestations of gastro-oesophageal refl ux disease
Ear, nose andPulmonary throat Others
Asthma Chronic cough Dental erosions
Chronic bronchitis Laryngitis Non-cardiac chest pain
Bronchiectasis Hoarseness Sleep apnoea
Pulmonary fi brosis Pharyngitis
Pneumonia Sinusitis Vocal cord granuloma Recurrent granuloma
Adapted from Richter JE 2000 Extraesophageal manifestations of gastroesophageal refl ux disease. An overview. American Journal of Gastroenterology 95: 51–53.
GASTRO-OESOPHAGEAL REFLUX AND HELICOBACTER PYLORI INFECTION 20.4
Ch020-F10280.indd 743Ch020-F10280.indd 743 7/27/2007 6:01:34 PM7/27/2007 6:01:34 PM

744
further testing is required (Fig. 20.4.1). The test used will depend on the age of the child, the types of test available and the type and severity of symptoms. The most commonly used tests are outlined in Table 20.4.5.
Barium oesophagram
Most commonly used but least sensitive for the diag-nosis of GOR. Useful for detecting structural abnor-malities such as pyloric stenosis, malrotation and strictures, and may be useful to assess swallowing function or aspiration. Most useful in children with persistent vomiting.
Radionuclide scintigraphy
Radioactive 99Tc–sulphur colloid is added to an age-appropriate liquid meal and can be used as a direct measure of refl ux. It has the benefi t of measuring all refl uxate. It can also be used to evaluate gastric emp-tying and to document aspiration due to refl ux.
Upper gastrointestinal endoscopy and biopsies
Endoscopic examination of the upper gastrointesti-nal tract is indicated in GORD with complications such as chest or epigastric pain, heartburn, hae-matemesis or persistent unexplained iron defi ciency.
Unlike in adult medicine, oesophageal biopsies form an important part of the diagnostic strategy in GORD in children. They can support a refl ux aetiol-ogy and exclude other less common causes of oesoph-agitis, such as infections (cytomegalovirus, herpes simplex virus, candidiasis), Crohn disease or eosino-philic oesophagitis.
24-hour intraoesophageal pH monitoring
This provides an assessment of oesophageal acid exposure. In the majority of infants with GOR, this
Table 20.4.5 Commonly used diagnostic tests for gastro-oesophageal refl ux disease
• Barium oesophagram• Radionuclide scintigraphy (milk scan)• Upper gastrointestinal endoscopy and biopsies• 24-hour intraoesophageal pH monitoring• Oesophageal manometry
History and examination
Physiological reflux (GOR),e.g. regurgitation
Vomiting – irritability
Reassurance,thickened
feeds, posture– antacids
Symptoms resolve
No
response
GORD
Persistent symptoms Atypical symptoms,e.g. chest disease,
asthma, apnoea
e.g. excessive irritabilityabdominal/chest pain
haematemesis
e.g. vomitingFailure to thrive
Diagnostic evaluation
Barium swallow
Abnormal
Treat Appropriatemanagement
Upper gastrointestinalendoscopy
NormalOesophagitis
pH monitoringand/or trial of
therapy
Treat
Normal
Gastrooesophagealscintiscan
pH monitoringchest X-ray
bronchoscopytracheal aspirate
Fig. 20.4.1 Algorithm for diagnostic approach to gastro-oesophageal refl ux disease. (Modifi ed from Youssef NN, Orenstein SR. Clinical Perspectives in Gastroenterology Jan/Feb 2001: 11–17.)
20.4 DISORDERS OF THE GASTROINTESTINAL TRACT AND HEPATIC DISORDERS
Ch020-F10280.indd 744Ch020-F10280.indd 744 7/27/2007 6:01:34 PM7/27/2007 6:01:34 PM

745
test is not required and it should only be carried out if it will alter diagnosis, treatment or outcome. Current indications for its use are outlined in Table 20.4.6.
This is not a simple test and should be carried out only in a specialist centre, as many factors need to be considered, including pretest preparation, inser-tion and positioning of the catheter medication, symptom assessment and analysis of results.
Oesophageal manometry and multichannel intraluminal impedance
Oesophageal manometry is rarely needed clinically in the diagnosis of GORD in children but may be useful prior to fundoplication in children with a sus-pected motility disorder. It also has a place in chil-dren with swallowing diffi culties and in the diagnosis of achalasia.
Multichannel intraluminal impedance measures the fl ow of liquid or gas in the oesophagus and thus has an advantage over pH monitoring, which only recognizes fl uid fl ow with a pH below 4. It is a rela-tively new technique and its role in clinical practice is still being evaluated. In young infants and children where regurgitation is often an issue it may have an important role, particularly in delineating an asso-ciation between refl ux and apnoea.
Diagnostic approach to gastro-oesophageal refl ux disease
The diagnostic approach depends largely on the severity of symptoms and the presence or absence of complications. In the otherwise healthy infant whose main symptoms are vomiting or regurgitation, paren-tal reassurance is all that is required.
If symptoms persist despite simple therapies such as posture and formula thickening, a barium oesoph-agram should be carried out to exclude an anatomi-cal abnormality such as stricture, gastric outlet obstruction or malrotation.
Infants presenting with acid-refl ux-related symp-toms suggestive of oesophagitis require endoscopy
and biopsies. In infants with atypical symptoms, the approach is more diffi cult but initially aspira-tion needs to be considered. Barium oesophagram, chest X-ray and a gastro-oesophageal scintiscan may provide support for this diagnosis. Referral to a respiratory physician may also be indicated for bronchoscopy and computed tomography (CT) scan. Ambulatory 24-hour pH monitoring may detect evi-dence of GOR but this does not prove the associa-tion. Presence of pepsin in a tracheal aspirate taken at bronchoscopy may be a useful adjunct but as yet remains unproven.
Table 20.4.6 Current indications for 24-hour intraoesophageal pH monitoring
• Diagnose occult refl ux in: • Unexplained recurrent pneumonia • Patients with bradycardia, apnoea • Non-gastrointestinal symptoms caused by refl ux,
such as stridor, laryngeal symptoms, atypical chest pain, severe irritability
• Assessment of adequacy of medical therapy in cases of severe intractable GORD
Clinical example
Sophie, the 7-month-old daughter of Greek parents, presented with a history of recurrent haematemesis, with bright and altered blood
noted in regurgitated fl uid and also dark stains on bibs and pillow. She was generally quite happy and thriving, although clinically a little pale. She did not have any evidence of abdominal tenderness.
In view of the recurrent bleeding and the possibility of oesophagitis or gastritis, an upper gastrointestinal endoscopy was carried out and this showed macroscopically ulcerative oesophagitis but normal stomach and duodenum. Sophie was treated with omeprazole 10 mg b.d. and after 4 weeks the bleeding had stopped clinically and the spilling had also decreased. A repeat endoscopy 8 weeks later showed macroscopic healing but still histological evidence of moderate oesophagitis. Sophie remained on omeprazole for a further 4 months but following a trial off therapy her symptoms recurred and she underwent a Nissen fundoplication. When reviewed at the age of 2 years she was well and asymptomatic.
Treatment approach (Table 20.4.7)
The ideal therapy would include the use of a drug that specifi cally reduces the frequency of TLOSRs, but this is currently not available.
Table 20.4.7 Treatment approach to gastro-oesophageal refl ux disease
• General • Reassurance • Positioning • Thickened feeds• Drug therapy • Antacids • Proton pump inhibitors• Continuous nasogastric feeds• Surgery
GASTRO-OESOPHAGEAL REFLUX AND HELICOBACTER PYLORI INFECTION 20.4
Ch020-F10280.indd 745Ch020-F10280.indd 745 7/27/2007 6:01:34 PM7/27/2007 6:01:34 PM

746
General measures
These include reassurance, positioning and thicken-ing feeds. The importance of reassurance in relation to the otherwise healthy infant cannot be overstated, It is important to avoid numerous dietary changes, unnecessary investigations and multiple drug thera-pies, which are often recommended by others or tried by parents.
Previously the only posture proven scientifi cally to be effective was the prone position, but this is no longer recommended because of the increased risk of sudden infant death syndrome (Ch. 3.10). New evidence suggests that laying children on their left side following a meal signifi cantly reduces regurgita-tion and the frequency of TLSORs. Feed thickening has also been shown to reduce symptoms of regurgi-tation and vomiting by reducing the height the refl ux-ate comes up the oesophagus. There are now commercially available infant formulas that contain thickening compounds. The risk is that the attenua-tion of overt symptoms may mask complications of GORD.
Prokinetic drugs
There are no prokinetic agents that have been shown to be benefi cial and thus none can be recommended at present.
Acid suppression
This is effective in reduction of symptoms due to acid irritation of the oesophagus. Acid suppressing agents are:
• antacids. In infants with mild symptoms suggestive of heartburn such as irritability between feeds, a trial of 0.5–1 ml/kg per dose three to six times a day may be worthwhile. Antacids only have a brief dura-tion of action and a response can be noted within several days; if not then do not persist• H2-receptor antagonists. Ranitidine has proved the most effective, in doses often higher than used in adults. It does have potentially serious adverse effects such as fulminant hepatic failure. The dose recom-mended is 3–4 mg/kg per dose three times a day• proton pump inhibitors. These are the most potent acid-suppressing agents and are used if acid-related symptoms fail to respond to other therapies. They are superior to H2-receptor antagonists in effi cacy because of their ability to maintain intragastric pH above 4 for longer periods of time and to inhibit meal-stimulated acid secretion. They are often used as fi rst-line treatment where more complete acid sup-pression is required, e.g. in chronic respiratory disease, neurologically disabled children and repaired
tracheo-oesophageal fi stula. There is also a school of thought that the ‘treat then test’ principle should be used as the medication is so effective and if there is no response then GORD is less likely. Omeprazole has been most extensively studied in adults but there is very little paediatric data. It is used in doses ranging from 0.7–3.5 mg/kg once daily just before the fi rst meal of the day. Twice-daily dosing may be indi-cated in certain situations such as severe oesophagi-tis, peptic stricture, persistent nocturnal refl ux symptoms and extraoesophageal GORD.
Continuous feeding
Children with intractable vomiting and growth failure may respond to continuous nasogastric tube or gastrostomy feeding, with catch-up growth, and surgery may be avoided.
Surgery
The Nissen fundoplication is the most common sur-gical procedure and the indications are shown in Table 20.4.8. It can now be carried out laparoscopi-cally in children. This may work, not by acting as a valve or increasing LOS pressure but by decreasing TLOSRs due to reduction in the fundal surface area. Fundal distension is an important trigger for TLOSRs. There is also evidence that it increases the nadir pressure in the LOS. This option needs very careful consideration in children because of the risk of complications and failure of the effectiveness of surgery.
Summary
It is important to realize that only a small percentage of children with GOR go on to develop GORD. For
Table 20.4.8 Indications for anti-refl ux surgery in children
Absolute• Acute life-threatening event or chronic lung disease due
to aspiration• Severely neurologically impaired children• Continuing severe oesophagitis or oesophageal
ulceration despite adequate therapy• Oesophageal stricture secondary to GOR• Intractable vomiting with growth failure secondary to
GOR
Relative• Persistent symptoms with oesophagitis or growth failure• Severe asthma or respiratory disease unresponsive to
therapy
20.4 DISORDERS OF THE GASTROINTESTINAL TRACT AND HEPATIC DISORDERS
Ch020-F10280.indd 746Ch020-F10280.indd 746 7/27/2007 6:01:34 PM7/27/2007 6:01:34 PM

747
most infants, symptoms resolve completely before 12 months of age. Unfortunately, many of these chil-dren are overdiagnosed and overtreated. It is equally important that those with continuing symptoms are recognized and treated effectively.
lence in breastfed infants. In developed countries, only 10% of all children are infected by the age of 10 years.
The route of transmission is probably similar to other enteric pathogens, being faecal–oral, oral–oral or gastric–oral. H. pylori has been detected in vomitus, saliva, faeces and on children’s dummies, and also in contaminated water and food prepared with contaminated water. The housefl y has also been implicated as a vector. The spread of infection within families is high, most probably from infected mother to child, although there is good evidence of sibling-to-sibling spread, especially in households with high infection rates in all family groups. Risk factors for H. pylori infection in children are shown in Table 20.4.9.
The natural history of H. pylori infection in child-hood remains obscure. A signifi cant fi nding has been spontaneous clearing and reacquisition of gastric infections in preschool children, as spontaneous eradication does not appear to occur in adults.
Helicobacter pylori-associated disease
General
In the past, gastric and duodenal ulcers in children have been described as primary or secondary. Sec-ondary ulcers, which are more common in younger children (10 years) are caused by systemic stresses, such as trauma, burns, septic shock, corticosteroids or non-steroidal anti-infl ammatory drugs. Primary ulcers, which usually occur in older children, give rise to symptoms similar to those in adults, with epigastric nocturnal abdominal pain and vomiting and often a positive family history of peptic ulcer-ation. It is now clear in this latter group that the disease is due to H. pylori infection of gastric mucosa.
All H. pylori strains produce urease, which is thought to be important in the infl ammatory reac-tion in the stomach and also in maintaining the ideal submucous environment for the organism. The urease reaction is also exploited in a number of diag-nostic tests.
Practical points
• GOR in infants is usually a benign, self-limiting condition• Reassurance and minor interventions, e.g. posture, feed
thickening, often suffi ce• GORD always requires further assessment and possibly
investigation• GORD has a number of extra-oesophageal
manifestations that need to be considered• Proton pump inhibitor therapy should be fi rst-line
therapy for treatment of GORD
Helicobacter pylori infection in childrenHelicobacter pylori is the commonest bacterial patho-gen in humans, infecting more than 50% of the world’s population. This infection (initially called Campylobacter pylori), discovered by Warren and Marshall in Perth, Australia in 1982, ranks as one of the most important medical discoveries of the last century and resulted in them being awarded the Nobel Prize for medicine in 2005. They cultured the organism from the gastric antrum of adults with peptic ulcer disease. It meets Koch’s postulates as a human pathogen causing chronic active gastritis. H. pylori as a paediatric infection is usually acquired in the fi rst 2 years of life but the disease consequences rarely arise in childhood. A consensus conference sponsored by the Canadian Helicobacter Study Group met in 2005 to develop evidence-based guide-lines for the approach to H. pylori infection in children.
Epidemiology
Socioeconomic differences are the most important predictor of H. pylori infection prevalence in any population group. In developed countries the preva-lence in children seems to be declining but there is variability in the burden of infection with higher levels in immigrants and indigenous populations. This is particularly the case in aboriginal children, of whom at least 80% are infected. This is similar to developing countries, where up to 80% of children are infected by the age of 2 years, with a lower preva-
Table 20.4.9 Risk factors for Helicobacter pylori infection
• Poor socioeconomic status• Household crowding• Ethnicity• Migration from high prevalence areas• Infected parent, particularly mother• Contaminated water
GASTRO-OESOPHAGEAL REFLUX AND HELICOBACTER PYLORI INFECTION 20.4
Ch020-F10280.indd 747Ch020-F10280.indd 747 7/27/2007 6:01:34 PM7/27/2007 6:01:34 PM

748
Genetic analysis of H. pylori has demonstrated strains with certain virulence factors, e.g. vacuolat-ing cytotoxin (Vac A), and cytotoxin-associated genes (cagA, cagE). In adult ulcer disease there is a correlation between cagA positivity and peptic ulcer, but this is less clear in children. A study has shown a strong correlation between disease severity and the cagE genotype in children.
Gastrointestinal infection (Table 20.4.10)
GastritisH. pylori colonization of gastric mucosa in children is almost always associated with gastritis, which resolves with eradication of the organism. Endos-copy can be negative and biopsy is essential for diagnosis, although on occasions nodular antral hyperplasia can be seen and is diagnostic of infection.
Duodenal ulcerH. pylori gastritis is found in 90% of children with duodenal ulcers. Ulcers heal faster if anti-H. pylori therapy is given, compared with acid suppression alone. Importantly, ulcers do not recur if the infec-tion is successfully eradicated.
Gastric ulcersH. pylori infection as a cause of gastric ulcers is much less common in children than adults, probably refl ecting the fact that the majority are secondary to systemic causes.
Gastric adenocarcinomaThe epidemiological association between H. pylori infection and gastric cancer has been judged by the World Health Organization to be suffi ciently strong for it to classify H. pylori as the fi rst bacterium to be a human carcinogen. H.-pylori-induced gastric cancer has not been reported in children.
Gastric lymphoma and MALT lymphomaSeroepidemiological studies support an association between long-standing H. pylori infection and lym-phoma and mucosa-associated lymphoid type (MALT) lymphomas. Eradication of H. pylori has resulted in regression of MALT lymphoma in some cases. Both these tumours are rare in children.
Recurrent abdominal painIn adults, a link between non-ulcer dyspepsia (pos-sibly the equivalent of recurrent abdominal pain in childhood) and H. pylori has been suggested by a recent meta-analysis of a large number of controlled studies. A comparable study in children with recur-rent abdominal pain does not support an associa-tion. The major problem with studies in children is the lack of a standardized validated reproducible symptom assessment instrument. It is possible that there is a subset of children in whom H.-pylori-induced gastritis is responsible for recurrent abdomi-nal pain, but more information is required. The current consensus is that recurrent abdominal pain of childhood is not an indication to test for H. pylori infection.
Extragastric disease
Gastro-oesophageal refl uxIt is postulated that certain H. pylori strains cause decreased acid production and atrophic gastritis and that, with eradication of H. pylori, acid rebound occurs, causing GOR disease. This is still a contro-versial area in adults and there is very little support-ing evidence in children.
Iron defi ciency/growth stuntingIron defi ciency has been described in growth-retarded adolescents with H. pylori infection. Eradi-cation of H. pylori infection corrected the defi ciency and led to growth improvement. The current consen-sus is that testing for H. pylori infection should be considered in children with refractory iron defi ciency anaemia where no other cause is found.
A large Australian study has shown a relationship between small-for-gestational-age infants and mater-nal H. pylori infection. Growth delay in height and weight has also been shown in H.-pylori-infected children but this may be biased by socioeconomic status.
Diagnostic tests (Table 20.4.11)
Endoscopy and biopsy is the only method that can provide evidence of disease activity such as gastritis or an ulcer. Urease testing of biopsy material gives indirect identifi cation of infection
Table 20.4.10 Consequences of Helicobacter pylori infection
Gastrointestinal• Gastritis• Duodenal ulcer• Gastric ulcer• Gastric adenocarcinoma• Gastric lymphoma and MALT lymphoma
Extragastric• Gastro-oesophageal refl ux• Iron defi ciency anaemia• Short stature
20.4 DISORDERS OF THE GASTROINTESTINAL TRACT AND HEPATIC DISORDERS
Ch020-F10280.indd 748Ch020-F10280.indd 748 7/27/2007 6:01:34 PM7/27/2007 6:01:34 PM

749
but has only a 50% positive predictive value in children.
The 13C-urea breath test is currently the best non-invasive diagnostic test for H. pylori infection in chil-dren. It has a greater than 95% positive and negative predictive value. The principle of the test is outlined in Figure 20.4.2. Urea can be labelled with either radioactive 14C or stable isotope 13C. In children and women of childbearing age, 13C-urea is recommended.
Serology, while commercially available, is fre-quently unreliable and cannot distinguish between past and present infection. Of the other non-invasive tests under trial the most promising is the use of a monoclonal antibody to detect H. pylori antigen in faeces.
The aim of testing is not to detect the presence of infection but to fi nd the cause of clinical symptoms, and therefore the important question is who should be tested (Table 20.4.12).
Treatment
The discovery of H. pylori as the cause of primary peptic ulceration and gastric ulcers has changed their management, as eradication of the organism cures the disease and prevents recurrence. This has meant that peptic ulcer disease is now a curable con-dition eliminating the need for long-term or destruc-tive surgery.
If endoscopy is indicated to investigate organic disease and H. pylori is found, the child should receive treatment (Table 20.4.13); however, if no ulcer is found the patient/parents should be informed that H. pylori eradication may not relieve the symptoms.
The traditional treatments do not eradicate H. pylori infection and current treatment in children advocates a combined regimen using two anti-biotics and a proton pump inhibitor, based on adult treatment regimens. At present there is only one peer-reviewed controlled study of successful use of the triple therapy treatment regimen in children.
The currently recommended fi rst-line treatment is a combination of a proton pump inhibitor, clarithro-mycin and amoxicillin twice daily for 7 days. Metro-nidazole can be substituted for either amoxicillin or clarithromycin but there is a high resistance to this drug and its use may lead to treatment failure. Failure of eradication leads to the use of second-line options,
*CO2blood
*CO2 breath
*CO2
NH2
NH2
*C=OUrease
inH.pylori
*CO2+NH2
Fig. 20.4.2 The principle of the 13/14C-urea breath test.
Table 20.4.11 Diagnostic tests for Helicobacter pylori
Endoscopic• Biopsy and histology• Rapid urease test• Bacterial culture
Indirect tests• Serum antibody (IgA, IgG)• Stool culture/stool antigen• Urea breath test
Table 20.4.12 Those who should be tested for Helicobacter pylori infection
Yes• Endoscopic/radiologically proven gastric or duodenal
ulcers• Confi rmation of eradication of H. pylori infection
No• Recurrent abdominal pain without ulcer disease• Asymptomatic children• Children in families with history of gastric cancer of
ulcer disease
Table 20.4.13 Who should be treated for Helicobacter pylori infection
• Histologically proven infection with gastrointestinal symptoms
• Duodenal/gastric ulcers• Lymphoma• Atrophic gastritis with intestinal metaplasia
GASTRO-OESOPHAGEAL REFLUX AND HELICOBACTER PYLORI INFECTION 20.4
Ch020-F10280.indd 749Ch020-F10280.indd 749 7/27/2007 6:01:35 PM7/27/2007 6:01:35 PM

750
Clinical example
A 10-year-old Vietnamese child, Than, who had resided in Australia since birth, presented with a long history of recurrent epigastric pain
that woke him from his sleep at night. His appetite was described as poor. There was a family history of peptic ulcer disease. Examination reveals a thin child on the 3rd percentile for weight and 25th centile for height. Epigastric tenderness was present. The signs and symptoms suggested peptic ulcer disease and at endoscopy antral nodular hyperplastic gastritis was noted, as well as an ulcer in the fi rst part of the duodenum. Histology showed evidence of H. pylori infection. Treatment with triple therapy (clarithromycin, amoxicillin and omeprazole) for 1 week led to resolution of symptoms within 4 weeks. 6 weeks after stopping therapy a 13C-urea breath test was carried out and was negative, confi rming eradication. At review 12 months later, Than was well and his height and weight were on the 25th percentile.
which usually include bismuth subsalicylate in a triple or quadruple therapy regimen.
The burden of illness and socioeconomic costs of H.-pylori-related illness is considerable, making the development of a prophylactic vaccine to prevent infection or a therapeutic vaccine to eliminate exist-ing infection desirable, but to date no vaccine is available.
20.4 DISORDERS OF THE GASTROINTESTINAL TRACT AND HEPATIC DISORDERS
Ch020-F10280.indd 750Ch020-F10280.indd 750 7/27/2007 6:01:35 PM7/27/2007 6:01:35 PM

Compared with many other childhood problems, liver disease is infrequent beyond the newborn period; however, it is important because early recog-nition and diagnosis can be critical to the outcome of some disorders and because chronic liver disease carries a high burden of disability for children and their families.
The liver has a central role in:
• intermediary metabolism and homeostasis• synthesis of proteins• bile acid metabolism• bilirubin metabolism (uptake, conjugation and
excretion)• detoxifi cation reactions.
Liver disease disturbs these processes, resulting in one or typically more than one of the following problems:
• abnormalities of liver size and consistency (an enlarged or fi rm liver)
• jaundice• hepatitis (jaundice and/or elevation of liver
transaminases)• metabolic dysfunction (such as hypoglycaemia)• liver failure: • failure of synthetic function (bleeding or
oedema) • failure of detoxifi cation and waste elimination
(encephalopathy)• obstruction to blood fl ow through the portal
system (portal hypertension).
Features that help us understand the nature of liver problems include:
• dark urine (excretion of bilirubin and urobilinogen in urine)
• pale stools (with biliary obstruction or severe impairment of hepatic function)
• tender liver (liver capsule stretched)• hard liver (chronic liver disease with cirrhosis)• splenic enlargement (in portal hypertension)• bruising and bleeding (failure of synthesis of
coagulant proteins)• oedema and ascites (failure of albumin synthesis).
Liver size is proportional to body weight (rather than height) and increases during childhood. The physical
fi ndings of liver examination and the landmarks of the liver change during childhood. Liver size is best determined by percussing the upper border of the liver, usually at the fi fth intercostal space in the mid-clavicular line anteriorly, gently palpating up from the right lower quadrant to determine the liver edge, and then measuring the ‘span’ between the upper and lower borders in the midclavicular line.
Overly fi rm palpation may make it diffi cult to palpate the liver edge. The normal liver span at dif-ferent ages is shown in Table 20.5.1. During infancy the normal liver is usually palpable 2–4 cm below the costal margin. Variants in liver shape that make the liver seem enlarged include a prominent Riedel lobe, felt well below the right costal margin, and a promi-nent left lobe of liver felt in the epigastrium. The normal liver is soft and the surface is smooth and yielding. The consistency of the liver frequently changes when it is abnormal, generally becoming harder. It may also become tender.
The liver enlarges through the accumulation of additional tissue or fl uid because of:
• infl ammation• infi ltration• storage• congestion.
Causes of liver enlargement are shown in Table 20.5.2.
JaundiceJaundice occurs because of failure of the liver to excrete the bilirubin load owing to:
• excess bilirubin load due to haemolysis• defi ciency of conjugating enzymes• obstruction of the biliary tree• secretory defect• damage to liver cells with leakage of bilirubin
into the circulation.
Jaundice in infancy
Jaundice is very common in newborn infants. Up to 50% of Caucasian babies will become jaundiced and
751
20.5Liver diseases in childhood
D. Forbes
Ch020-F10280.indd 751Ch020-F10280.indd 751 7/27/2007 6:01:35 PM7/27/2007 6:01:35 PM

752
even higher proportions of Asian babies may be jaundiced in the fi rst weeks of life.
Physiological jaundice usually develops after the fi rst 24 hours of life and by the fourth day of life and resolves by 10–14 days of age. It is discussed in Chapter 11.2. Jaundice requires further investigation when it:
• develops within the fi rst 24 hours of life• is severe• is associated with fever or other symptoms of
systemic illness• persists beyond the fi rst 2 weeks of life.
The fi rst step is to differentiate between conjugated and unconjugated hyperbilirubinaemia by measur-ing the total bilirubin level and the conjugated frac-tion. Most newborn babies will have unconjugated hyperbilirubinaemia. Non-physiological causes of an elevation of unconjugated bilirubin include:
• breast milk jaundice• haemolysis• duodenal atresia• pyloric stenosis• hypothyroidism• conjugating enzyme defi ciency syndromes.
Breast milk jaundice
Elevation of the unconjugated bilirubin fraction associated with breastfeeding is common, occurring once in every 50–200 breastfed infants. It is a benign disorder recognized by:
• jaundice persisting beyond 2 weeks of life• a thriving healthy baby• no evidence of other disease• resolution of jaundice with temporary
interruption of breastfeeding.
Conjugated hyperbilirubinaemia
Conjugated hyperbilirubinaemia occurs with:
• obstruction to bile fl ow in intrahepatic or extrahepatic ducts, or with a scretory defect
• liver cell damage (hepatitis).
Conjugated hyperbilirubinaemia is considered to be present if more than 20% of the total bilirubin level is conjugated. It may develop soon after birth but may also manifest later in infancy after dietary change exposes a metabolic defect. This is an uncom-mon problem (1 in every 2500 infants) but an impor-tant syndrome to recognize. Every infant with jaundice persisting beyond the fi rst 2 weeks of life should have the conjugated fraction of their bilirubin determined.
Bile duct obstruction and hepatitis are diffi cult to differentiate in this age group because of overlap of the disease processes and clinical features. It is useful, however, to attempt to differentiate these clinical syndromes and identify infants who have treatable disease: obstruction of the large ducts, infections and metabolic diseases. Early recognition and treatment is necessary for satisfactory outcome of these diseases. The clinical approach requires rec-ognition of features suggesting neonatal hepatitis or bile duct obstruction (Table 20.5.3) and the applica-tion of general and then specifi c tests leading to a diagnosis.
Table 20.5.1 Liver span (cm) during childhood
Age Mean Range
Birth 5.5 4–7
1 year 6 5.5–7
2 years 6.5 6–8
3 years 7 6–9
4 years 7.5 6–9
5 years 8 6–9
12 years 9 7–11
Table 20.5.2 Causes of hepatomegaly
Infl ammation• Infectious hepatitis• Autoimmune hepatitis• Drug reactions
Infi ltration• Primary neoplastic liver cancers• Primary non neoplastic liver cancers• Secondary liver cancers: leukaemia, lymphoma,
neuroblastoma
Storage• Glycogen storage disorders• Lipid storage disorders• Steatohepatitis: obesity, steroids, diabetes mellitus,
starvation
Vascular congestion• Congestive heart failure• Pericardial disease• Hepatic vein thrombosis
20.5 DISORDERS OF THE GASTROINTESTINAL TRACT AND HEPATIC DISORDERS
Ch020-F10280.indd 752Ch020-F10280.indd 752 7/27/2007 6:01:35 PM7/27/2007 6:01:35 PM

753
Tests used to distinguish biliary obstruction from neonatal hepatitis include:
• ultrasound examination of the biliary tree to identify dilatation of extrahepatic ducts
• radioisotope (iminodiacetic acid) scanning to demonstrate bile fl ow or obstruction
• liver biopsy to identify characteristic features of hepatitis or cholestasis syndromes
• operative cholangiogram to document the patency of extrahepatic biliary structures.
Most of the identifi able causes of neonatal hepatitis syndrome and cholestatic syndromes are rare (Table 20.5.4). The major clinical issue is differentia-tion of idiopathic neonatal hepatitis from biliary atresia.
The outcome for neonatal hepatitis syndromes varies greatly, depending upon the specifi c cause. Idiopathic neonatal hepatitis and many infections have a good prognosis for spontaneous and complete recovery. Extrahepatic biliary atresia is potentially fatal and is treated surgically. Infants with biliary atresia who are treated within the fi rst 60 days of life have a much greater potential for establishing bile fl ow and restoring liver function than infants who are treated after this time. Late treatment frequently results in biliary cirrhosis and progressive liver failure.
The acutely ill, jaundiced infant should be con-sidered differently from infants with biliary duct obstruction or neonatal hepatitis syndromes, as these babies are likely to have a metabolic defect or an infection (Table 20.5.5). They usually have a con-jugated hyperbilirubinaemia and often manifest all the features of liver failure: jaundice, bleeding, oedema and encephalopathy (drowsiness, irritabil-
ity, deteriorating mental function, convulsions). They require urgent assessment and urgent treat-ment is frequently provided on the basis of a pre-sumed diagnosis, pending confi rmation by specifi c tests.
Table 20.5.3 Clinical features of biliary obstruction and neonatal hepatitis syndromes
Neonatal hepatitis Biliary obstruction
Growth Often impaired Usually normal
Wellbeing Often sickly Usually well
Pale stools Variable Usual
Dysmorphic Common May occurfeatures
Synthetic Often impaired Preserved until latefunction
Hypoglycaemia Common Uncommon
Table 20.5.4 Causes of conjugated hyperbilirubinaemia in infancy
Biliary obstruction syndromes in infancySurgical obstruction of large extrahepatic ductsCholedochal cystExtrahepatic biliary atresiaSpontaneous perforation of the common bile ductPaucity of intrahepatic ducts• Alagille syndrome• Non-syndromic paucity of intrahepatic ducts
Neonatal hepatitis syndromes in infancyInfections• Bacterial • Listeria • Escherichia coli • Syphilis• Protozoan • Toxoplasmosis• Viral • Cytomegalovirus • Rubella • Parvovirus • Herpesvirus • Coxsackie virus • Echovirus family • Hepatitis B virus
Metabolic disordersAlpha–1-Antitrypsin defi ciencyCystic fi brosisCarbohydrate metabolic defects• Galactosaemia• Fructosaemia• Glycogen storage disorder type IVAmino acid metabolic defects• TyrosinaemiaLipid metabolic defects• Cholesterol ester storage disease• Wolman disease• Gaucher disease• Niemann–Pick diseaseDisorders of bile acid metabolism
Endocrine disordersHypopituitarismHypothyroidism
Chromosomal disorders
Toxic disordersParenteral nutrition
Idiopathic neonatal hepatitis
LIVER DISEASES IN CHILDHOOD 20.5
Ch020-F10280.indd 753Ch020-F10280.indd 753 7/27/2007 6:01:35 PM7/27/2007 6:01:35 PM

754
Jaundice in older children
Jaundice occurs in children because of:
• hepatitis (liver cell damage)• biliary duct obstruction.
Most older children who develop hepatitis have an infectious illness but drug toxicity, autoimmune and metabolic disorders may also be the cause (Table 20.5.6).
The apparent length of history of jaundice or other symptoms is often not a reliable guide to the duration of liver disease, and children with a short history may
in fact have long-standing liver problems. Because of limited opportunities for the effective treatment of some liver disease it is very important to establish a diagnosis for all children who develop hepatitis.
Chronic liver disease should be suspected in any child who has persistent elevation of liver enzymes 3 months or more after a presumed acute infection. Hepatitis in these circumstances is likely to be asso-ciated with progressive liver damage.
Chronic liver disease is identifi ed by clinical fea-tures in both history and examination:
History
• Recurrent hepatitis• Prolonged jaundice• Lethargy• Anorexia• Bruising• Pruritus• Poor growth.
Examination
• Jaundice• Muscle wasting• Poor growth• Clubbing• Spider naevi• Oedema• Hard liver
Table 20.5.5 The acutely ill jaundiced baby
Infections Metabolic disorders
E. coli bacteraemia Mitochondrial disordersEchovirus GalactosaemiaCoxsackie virus Alpha-1-antitrypsin defi ciencyCytomegalovirus Organic acidaemiasAdenovirus TyrosinaemiaHerpes simplex virus Urea cycle defectsParvovirus Fatty acid oxidation defect Reye syndrome Neonatal iron storage disorder
Table 20.5.6 Hepatitis in older children
Infections• Hepatitis A• Hepatitis B• Hepatitis C• Hepatitis D (Delta agent: coinfection with hepatitis B)• Hepatitis E• Hepatitis G• Infectious mononucleosis/Epstein–Barr virus• Cytomegalovirus• Herpesvirus• Parvovirus
Autoimmune disease• Autoimmune hepatitis• Sclerosing cholangitis
Metabolic disease• Alpha -1-antitrypsin defi ciency• Hereditary fructose intolerance• Tyrosinaemia• Wilson disease• Cystic fi brosis• Reye syndrome
Clinical example
Sarah was a healthy, full-term, breastfed infant who was jaundiced at the third day of life but completely recovered by the eighth
day of life. She gained weight normally. She again became jaundiced at 4 weeks of age. The jaundice increased and she started passing dark urine and had intermittent but increasingly pale stools. When seen at 7 weeks of age she looked healthy but was deeply jaundiced. Her liver was fi rm and had a span of 7 cm. Blood tests revealed that her bilirubin was elevated at 240 μmol/l with a conjugated fraction of 190 μmol/l, her alanine aminotransferase was 210 μmol/l, her gamma-glutamyl transpeptidase was 148 μmol/l and albumin 30 g/l. Ultrasound examination failed to visualize the gallbladder or the common bile duct. A liver biopsy demonstrated bilirubin plugs and bile duct proliferation, together with infl ammation of the hepatic parenchyma. A radioisotope DESIDA scan showed no excretion.
These fi ndings were suggestive of biliary atresia and so an operative cholangiogram was undertaken, with injection of contrast into the small atretic gallbladder found at laparotomy. This showed some dilatation of intrahepatic biliary ducts but no excretion of contrast via the common bile duct. The surgeon therefore proceeded to a portoenterostomy (Kasai procedure). Within a week of surgery Sarah had some pigmented stools, indicating that some biliary fl ow had been established.
20.5 DISORDERS OF THE GASTROINTESTINAL TRACT AND HEPATIC DISORDERS
Ch020-F10280.indd 754Ch020-F10280.indd 754 7/27/2007 6:01:35 PM7/27/2007 6:01:35 PM

755
• Splenomegaly• Ascites.
Evidence of liver fi brosis and cirrhosis may be docu-mented on ultrasound and confi rmed with liver biopsy and histology.
Infectious hepatitis
The clinical features of infectious hepatitis depend upon the age of the child as well as the specifi c infec-tious agent (Table 20.5.6). Younger children may remain asymptomatic despite evidence of signifi cant hepatitis but as children get older they are more likely to have symptoms of nausea, lethargy, fever, vomiting and abdominal pain. A small proportion of children with hepatitis from any cause may develop rapidly progressive, severe hepatitis, known as ful-minant hepatitis, which will result in some deaths.
In Australia and New Zealand most children with acute hepatitis will have an acute viral infection. Typically they are jaundiced, have a tender enlarged liver and variable splenic enlargement. They have elevation of their liver transaminase enzymes (alanine aminotransferase, aspartate aminotransfer-ase) and gamma-glutamyl transpeptidase, and eleva-tion of serum bilirubin (usually). The urine contains bilirubin and urobilinogen. The specifi c viral hepa-titis agents are labeled A–G. Hepatitis A, hepatitis B and hepatitis C are numerically the most important agents.
Hepatitis AThe commonest cause of hepatitis, hepatitis A typically:
• is spread by orofaecal transmission• has an incubation period of around 30 days• causes an acute illness with malaise, nausea,
vomiting and diarrhoea• is associated with examination fi ndings of
jaundice, dark urine and an enlarged, tender liver.
Hepatitis A virus excretion in faeces occurs prior to the onset of jaundice. Complete recovery from infec-tion is usual, although a small proportion of children will develop fulminant hepatitis. Immunity develops following infection, and may be stimulated in unex-posed individuals with hepatitis A vaccine, an inac-tivated virus vaccine. Recent infection can be confi rmed by a rise in antihepatitis A virus IgM antibody, and immunity by the presence of specifi c IgG antibodies.
Hepatitis BInfection with hepatitis B virus (HBV) is a worldwide problem that is more frequent among socially disad-vantaged groups and those Australian and New
Zealand children who come from Pacifi c Island, Asian and African backgrounds. Transmission occurs via body fl uids, and vertical transmission from mother to baby readily occurs. Hepatitis B infection can result in an acute hepatitis, but the majority of acute infections are asymptomatic.
HBV is a DNA-containing hepadnavirus with dis-tinct surface and core proteins, which act as anti-gens. Infection is confi rmed by the presence in serum of these antigens. Antibody to these proteins indi-cates development of immunity. HBV surface antigen (HBsAg) is the fi rst antigen detectable after exposure and persists until recovery occurs. HBV e antigen (HBeAg) also appears in the acute phase of the infec-tion and is indicative of a high viral load and high infectivity (up to 80% of infants of HbeAg-positive mothers will acquire hepatitis B infection). The response of the infected host is initially an anticore antibody (HbcAb), and subsequently HbeAb and HbsAb.
Chronic HBV infection is most likely to occur with perinatal infection. Infection is usually not recog-nized at the time, and may only be identifi ed when the child is found to have elevated liver enzymes at a later date. It may also present as an arteritis, arthri-tis or nephritis. It carries increased risks of cirrhosis and hepatocellular carcinoma later in life.
Passive immunization against hepatitis B using immunoglobulin rich in antihepatitis B antibodies should be initiated at the time of exposure (such as at birth). Active immunization should be undertaken in all high-risk groups. HBV immunization is now part of routine immunization programmes in Australasia (Ch. 3.5).
Treatment of chronic HBV infection is indicated in children with persistent elevation of liver enzymes, carriage of HBV antigens and DNA, and who have biopsy evidence of chronic hepatitis. Treatment is undertaken with alpha-interferon or lamivudine for 4–6 months.
Clinical example
Claire was a 4-year-old girl who had been adopted in Korea in infancy. She was well but during a recent febrile illness she had had
elevation of her transaminases and was subsequently found to be positive for HBsAg and HbeAg. Physical examination was normal. She was not immunized against hepatitis B in the newborn period. Claire was a chronic, asymptomatic carrier of hepatitis B, almost certainly infected in the perinatal period, and was at risk of chronic hepatitis and hepatocellular carcinoma. Hepatitis B can be transmitted to other children and so immunization of all children in her school group was encouraged.
LIVER DISEASES IN CHILDHOOD 20.5
Ch020-F10280.indd 755Ch020-F10280.indd 755 7/27/2007 6:01:35 PM7/27/2007 6:01:35 PM

756
Hepatitis CThe hepatitis C virus (HCV) is an RNA-containing virus that often causes chronic infection. Children acquire HCV infection via blood transfusions, from their mother at or around birth, and from a number of other as yet unknown sources. Children who have received multiple blood product infusions because of thalassaemia, cancer or haemophilia are at increased risk of HCV infection. Shared needles are an impor-tant source of infection in drug-using populations. A high proportion of infected children will develop chronic liver disease. HCV infection should be sus-pected in high-risk individuals who have elevated transaminases and can be confi rmed by detection of HCV antibody or HCV RNA. Children who have progressive liver disease may be treated with alpha-interferon, although the best approach to treatment is still not known.
Infectious hepatitis and day-care and school attendanceChildren with hepatitis A should be excluded from school until asymptomatic, or until at least 1 week after the onset of the jaundice, by which time they have typically stopped excreting the virus. Because of the highly infectious nature of hepatitis and the risks to other children, the school should be informed of the reason for the student’s absence.
Chronic HBV carriage is associated with some risk of spread to other young children in day-care and kindergarten settings. Biting and scratching behav-iour may increase this risk and should be actively discouraged. Despite this it is generally not consid-ered appropriate to exclude children from school. It is important, however, to ensure that all children are immunized against hepatitis B.
Hepatitis C poses similar risks to hepatitis B but to date there is no vaccine to prevent transmission to non-infected individuals. Biting and scratching by young children should be discouraged but exclusion of chronic carriers from day care and school is gener-ally not appropriate.
Drug-induced hepatitis
After infections, drug injury is the commonest cause of hepatitis. The most commonly incriminated agent is paracetamol. Initially the clinical syndrome is often indistinguishable from viral infections, and so drug toxicity should always be considered as a cause of hepatitis. Paracetamol liver toxicity is dose-related. Early recognition, estimation of plasma paracetamol levels to identify those at risk of liver failure and then initiation of treatment with N-acetyl cysteine are important in preventing acute liver failure. Chronic anticonvulsant therapy is often associated with abnormalities in transaminases.
These abnormalities do not carry the same risks as acute drug toxicity.
Steatohepatosis and steatohepatitis
A number of children, particularly adolescents, will be found to have abnormal liver enzymes (elevated alananine aminotransferase and aspartate amino-transferase) and sometimes an enlarged liver, but no evidence of other diseases associated with hepatitis, and with additional testing are found to have increased amounts of fat in their liver cells. Biopsy may show the presence of fat globules in liver cells (steatohepatosis) or the presence of intraheptaocyte fat globules and liver infl ammation (steatohepatitis) This syndrome is known by several names, including non-alcoholic fatty liver disease (NAFLD) and non-alcoholic steatohepatosis (NASH). Children at espe-cial risk of steatohepatosis and steatohepatitis are those who are obese, who have insulin-dependent diabetes and other metabolic diseases, the under-nourished and those on long-term corticosteroid therapy. Steatohepatitis carries risks of progressive liver disease and so needs to be treated seriously. Treament strageies include weight reduction, improved diabetic control and high-dose vitamin E.
Autoimmune hepatitis
This can occur at any age, although it is more likely to occur in the older child and adolescent than in infancy, and can be confi ned to the liver or be part of a systemic autoimmune illness. It may be triggered by viral infections, or drugs, but commonly has no identifi able antecedents. Girls are affected more commonly than boys. The onset is frequently insidi-ous and often comes to light with vague non-specifi c symptoms or with elevated liver enzymes. Different types can be identifi ed, based upon the pattern of auto antibodies:
• type 1: antinuclear antibody and antismooth muscle antibody
• type 2: anti-liver–kidney microsomal antibody• type 3: anti-soluble-liver-antigen antibody.
Treatment involves immunosuppression, usually with steroids and another immunomodulator, such as azathioprine or ciclosporin, often for prolonged periods of time. Liver transplantation is used in chil-dren with progressive, chronic liver disease that results in liver failure.
Alpha-1-antitrypsin defi ciency
This disorder is commonly identifi ed as a cause of neonatal hepatitis but may present at any stage of life
20.5 DISORDERS OF THE GASTROINTESTINAL TRACT AND HEPATIC DISORDERS
Ch020-F10280.indd 756Ch020-F10280.indd 756 7/27/2007 6:01:35 PM7/27/2007 6:01:35 PM

757
with elevated liver enzymes, jaundice or advanced liver disease. It is due to a gene mutation that results in dysfunctional protease inhibitors in the liver and lung, leading to hepatitis and emphysema. Associ-ated hepatitis eventually leads to cirrhosis.
Wilson disease
This disease arises as a result of failure of copper excretion into the bile, secondary to a defect in a transport protein. The disorder leads to accumula-tion of copper in the liver, brain, kidneys and bone. Liver disease typically becomes apparent in late childhood as hepatitis, portal hypertension or liver failure. Patients usually have so called Kayser–Fleischer rings of copper accumulation in the peripheral cornea by the time they manifest liver disease. Diagnosis is established by demonstrating low plasma caeruloplasmin (a copper-containing protein), increased urinary copper excretion and increased liver copper. Although this disorder is uncommon, recognition of Wilson disease is important because it is a treatable cause of chronic liver disease and will often present in childhood. Treatment is with a low-copper diet and long-term penicillamine, which increases the urinary excretion of copper.
Hepatitis, or liver infl ammation, is typically rec-ognized because of the development of jaundice but asymptomatic children may be found to have ele-vated liver enzymes and can have any of the disease processes discussed above.
It is important to remember that jaundice in older children may also be due to obstruction of biliary ducts due to:
• a choledochal cyst• congenital abnormalities of the biliary tree• gallstones• parasites.
These children may have features of hepatitis but may also present with pale stools, dark urine and abdominal pain. They need assessment with liver biochemistry to determine whether they have eleva-tion of alkaline phosphatase and gamma-glutamyl transpeptidase out of proportion to elevation of their transamimases. Imaging with ultrasound, computed tomography (CT) or magnetic resonance imaging (MRI) is necessary to defi ne the anatomy of the biliary ducts.
Liver failureThis is the end result of failure of the metabolic and synthetic functions of the liver. The clinical features of liver failure are relatively common for all causes
Clinical example
Elizabeth was an 8-year-old girl who was seen by her general practitioner because of recurrent hives. She was found to have
elevated immunoglobulin concentrations and then elevation of her transaminases (alanine aminotransferase 320 U/l and aspartate aminotransferase 250 U/l). Screening for alpha-1-antitrypsin defi ciency, Wilson disease, hepatitis B and C was negative, but she had elevated anti-liver–kidney microsomal antibody. A liver biopsy showed evidence of active infl ammation with piecemeal necrosis, and fi brosis. A diagnosis of autoimmune hepatitis was made and Elizabeth was commenced on prednisolone, 2 mg/kg per day, which was tapered to a lower dose over 3 months. There was an initial decrease in the levels of transaminases but these rebounded when the steroid dose was decreased. Azathioprine 1 mg/kg per day was added to her therapy, with subsequent normalization of transaminases. A follow-up liver biopsy showed a marked decrease in the infl ammatory infi ltrate and no progression of fi brosis. Attempts at withdrawing therapy after 2 years resulted in an increase in liver transaminases.
Clinical example
Samuel became jaundiced at about 4 weeks of age. He was otherwise well, was breastfeeding and was gaining weight. His
stools were normally pigmented but he had dark urine. His bilirubin was 180 μmol/1 with a conjugated fraction of 120 μmol/1. His alanine aminotransferase was 260 U/l. Because he had a conjugated hyperbilirubinaemia with evidence of hepatitis, Samuel had serological testing for viral infections (negative), urine testing for non-glucose reducing sugars (negative, making galactosaemia unlikely), a urine microscopy and culture, measurement of serum alpha-1-antitrypsin (very low) and Pi type (ZZ). An ultrasound of his biliary tree showed normal gallbladder and no evidence of duct dilatation. A liver biopsy revealed a giant cell hepatitis with accumulation of bilirubin plugs within bile ducts and accumulation of alpha-1-antitrypsin granules within the liver cells. A diagnosis of neonatal hepatitis due to alpha-1-antitrypsin defi ciency was established. Samuel was commenced on ursodeoxycholic acid to promote bile fl ow, plus the fat-soluble vitamins A, E and K. His jaundice gradually cleared and he grew satisfactorily during early childhood, although his transaminases never returned to normal. In middle childhood he developed easy bruising and prolongation of his prothrombin time, hypoalbuminaemia, muscle wasting and oedema. He received a liver transplant when he was aged 8 years, and remains well, although on long-term immunosuppression.
LIVER DISEASES IN CHILDHOOD 20.5
Ch020-F10280.indd 757Ch020-F10280.indd 757 7/27/2007 6:01:36 PM7/27/2007 6:01:36 PM

758
at different ages. Jaundice may not be seen until late in the course of liver failure. The earliest evidence is usually failing production of the vitamin-K-dependent clotting factors, resulting in prolongation of the prothrombin time and, eventually, easy bruis-ing and bleeding. Oedema due to hypoalbuminaemia is generally a late feature of liver disease. Encepha-lopathy is a late effect of failure of elimination of neurotoxic factors. It may be subtle initially, with drowsiness and then later confusion and tremor.
Portal hypertension
Portal hypertension develops because of increased resistance to blood fl ow through the portal venous system, resulting in distension of the portal vascula-ture and oesophageal, gastric or perianal varices, splenic enlargement, neutropenia and thrombocyto-penia. Portal hypertension occurs with liver disease with cirrhosis but, in up to one-third of cases, with portal vein obstruction in the absence of liver disease. These cases probably arise after neonatal portal venous thrombosis. Portal hypertension should be suspected in children with splenomegaly, especially if associated with thrombocytopenia, and in any child who has a signifi cant haematemesis. Uncom-plicated portal hypertension does not require inter-vention but children should be kept under surveillance and should avoid aspirin or other factors likely to increase the risk of bleeding. Underlying liver disease should be treated in those children who have cirrhosis.
Variceal haemorrhage is a medical emergency treated with resuscitation and then control of haem-orrhage by decreasing portal blood fl ow with vaso-pressin or octreotide or with local compression. Endoscopic injection or banding of varices is fre-quently required to control bleeding and prevent further bleeding. Following variceal bleeding, con-sideration should be given to lowering portal blood pressure with propranolol or by the surgical creation of a ‘shunt’.
Treatment
Treatment of chronic liver disease is aimed at antici-pating and treating the complications of malnutri-tion and defi ciency of energy and fat-soluble vitamins, failure of protein synthesis, portal hypertension and encephalopathy for as long as possible. Treatment involves the following components:
• Nutritional support • increased dietary energy with food and special
supplements • supplementation with vitamins A, D, E and K• Coagulopathy • vitamin K, fresh frozen plasma,
cryoprecipitate, platelets• Fluid balance • avoid excess sodium • diuretics such as spironolactone or furosemide • albumen infusions• Encephalopathy • low-protein diet • lactulose• Portal hypertension • monitoring of white blood cells and platelets • lowering of portal blood pressure with
beta-blockers.
Liver transplantation is life-saving in children with acute or chronic, irreversible end-stage liver failure, evidenced by coagulopathy, hypoalbuminaemia, encephalopathy and variceal haemorrhage. The com-monest problems leading to paediatric liver trans-plantation are biliary atresia, alpha-1-antitrypsin defi ciency and other rarer metabolic disorders, chronic autoimmune hepatitis and fulminant hepa-titis secondary to paracetamol toxicity, or infections. Three-quarters of children undergoing liver trans-plantation will survive at least 4 years, the majority leading healthy lives. They generally need to take immunosuppressive therapy for life.
Practical points
• It is normal to be able to palpate 2–4 cm of liver below the costal margin in infants
• Any newborn with jaundice persisting beyond the fi rst 2 weeks of age must have the conjugated bilirubin fraction measured
• Newborn conjugated hyperbilirubinameia must be assessed as early as possible in life to ensure the best outcome
• Acutely ill infants who are jaundiced are likely to have serious infections or metabolic disorders
• It is important never to assume that abnormal liver biochemistry is due to infection; this may be the only chance for diagnosis of potentially treatable but life-threatening liver disease
• Children with acute hepatitis A should be excluded from school until asymptomatic, while children with chronic carriage of hepatitis B or C rarely need to be excluded
20.5 DISORDERS OF THE GASTROINTESTINAL TRACT AND HEPATIC DISORDERS
Ch020-F10280.indd 758Ch020-F10280.indd 758 7/27/2007 6:01:36 PM7/27/2007 6:01:36 PM