distributed by: national technical inforhmation service u. s
TRANSCRIPT

AD-753 923
REACTIONS OF AROMATIC NITROCOMPOUNDS.1. PHOTOCHEMISTRY
Oscar Sandus, et al
Picatinny ArsenalDover, New Jersey
December 1972
DISTRIBUTED BY:
National Technical Inforhmation ServiceU. S. DEPARTMENT OF COMMERCE5285 Port Royal Road, Springfield Va. 22151

*I,
' ;" €~OPY NO. •2-
, TECHNICAL REPORT 4385
REACTIONS OF AROMATIC IIITROCOMPOUNDS
"1. PHOTOCHEMISTRY
OSCAR SANDUS
NORMAN SLAGG
DECEMBER 1972
r /
I - APPROVED FOR PUBLIC RELEASE; DISTPIBUTION UNLIMITED.
Rerroduced by ' ",
NATIONAL TECHNICAL ,
INFORMATION SERVICEU S Deporttoen! of Core ,'erce ' '
Springfiefd VA 2 2151
-PICATINNY A NAL.
DOVER. NEW JERSEY
AD

The findings in this report are not, to be construedas an official Department of the Army position:
DISPOSITION
Destory'this report when no longer needed. Do notieturn to the originator.
- I
¾r
:: . , . I- - I I ° I

Pow- --- w-
UNCLASSIFIEDSecurity Classificationa
DOCUMENT CONTROL DATA. -R & D(Soru..lIy classificat ion of title. body of abstract end Indexlng antnotation must be entered when the ovettil report is claeiliedj
1. ORIGINATING ACTIVITY (Colporaft author) 120. REPORT SECURITY CLAtSIFICATION
UnclassifiedPicatinny Arsena2, Dover, New Jersey 2Sb. GROUP
3. REPORT TITLE
REACTIONS OF AROMATIC NITROCONPOUNDS -I. -. otochennistry
4. OESC.AIPTtIVE NOTES (f'o. olrep~ort and InCluele. dotes)
5 AUTHOR(S) (Piret name, smiddle, Initial, feet name)
Oscar Sandus, Norman Slagg
S. REPORT DATE 7a. TOTAL NO. OF PAGES 7b. No. OF REFS
December 1972 46 27S.CONTRACT OR GRANT NO. S.ORIGINATOR'S REPORT NUIIERIS)
AMCMS Cose 501B.11..85661L/ab. PROJECT NO. Technical Report 112
. 1 T)6)1 102A32B01 Sb.e OTHER EPORT NO(S (Any other rninbre Mhat inay be assigned
d.I
10. DISTRIBUTION STATEMENT
Approved for public release; distrilbution unlimited.
II. SUPPLEMEN4TARY NOTES 112. SPONSORING MILITARY ACTIVITY
Picatinny Arsenal, Dover, N.J.13. ABSTRACT
The flash photolysis and continuous photolysis at 2537A (w T r* transitions)of aromatic nitrocompounds in solution were performed.
Flash photolysis revealed intermediates for o-nitrotoluene and its nitorderivatives. The spectrum and mean lifetime of the interxmediate for flash TNT weredetermined., The maximuma absorption of the TNT intermediate occurred in the 470 rimregion and the mean lifetime was 1.5 + 0.3 msec. This intermediate is probably notdirectly related to product formation.
-The continuous phot-olysis experiments reveals that all the aromatic nitro-compounds have disappearance quantum yields of the order 10-3. These low values forthe quantum yields imply that the most important reaction occurring is the deactivationto the ground state of the original molecule. The- presence or absence of oxygen doesnot appear to be of significance.
Product analyses suggest that aromatic nitrosocompounds and nitrophenols areamn products. However, the observed products do not conform in a simple manner
to the constraints of the isosbestic points observed in all photolyses. It appearsthat other products are present, some of which may have low extinction coefficients inthe spectral range studied. The latter may be necessary to acconmmodate the isosbesticpoints.
I RO SS~4It~ OSOLETE POR ARMY USE. UCASFE
-0 laCurity Classification

TINCTA~q9TFTRflSecurity Classification
14 I LINK A LINK 5 LINK CKEY WOROS -,
ROLE WT ROLE WY ROLE WT
Aromatic nitrocompoundsFlash photolysisContinuous photolysisSolution photochemistryDisappearance qua.,•tum yieldsIsosbestic pointsExplosivesIntermediatesPhotochromic (or phototropic) moleculesNitro- and nitrosobenzeneProcedures and apparatusSpectral data
9
UNCLASSITFIEDsecurity Classification
L.

Technical Report 4385
REACTIONS OF AROMATIC NITROCOMPOUNDS
I. PHOTOCHEMISTR)(
by
Oscar SandusNorman Slagg
December 1972
Approved for public release; distributionunlimited.
AMCMS Code 501B. 11. 85661DA Project: 1T06110ZA32B01
Explosives DivisionFeltman Research Laboratory
Picatinny ArsenalDover, New Jersey
- A

ACKNOWLEDGMENT
The authors would like to thank Professor Brian A. Thrush ofCambridge University for several helpful discussions, Mr. RichardJ. Graybusl for purifying the TNT, Dr. Everett E. Gilbert forpreparing the o-nitrosotoluene, and summer students Mr. Alan Mertzand John Lockburner who helped perform some of the experiments.
EV:7

TABLE OF CONTENTS
Page No.
Abstract 1
Introduct •. 2
Photochemical Considerations 2
Review of Past Photochemical Studies 5
Experimental Procedures 9
Materials 9Flash Photolysis Apparatus 10
Continuous Photolysis 12
Results and Discussion 13
Ultraviolet Absorption Spectra of TNT 13and Other Aromatic Nitrocompounds
Spectra and Mean Lifetimes of Intermediates 15Produced by Flash Photolysis
Quantum Yields for the Disappearance of 18Aromatic Nitrocompounds
Pro^duct Formation 20
Conclusion 24
References 27
Distribution List 410

Tables
I ReLationship between mechanism and 4disappearance quantum yield
2 Review of major photochemical studies 6
3 Wavelengths and extinction coefficients 14of the main (7r --> nr*) abscrption bands ofsome aromatic nitrocompounds in theregion 2->220 nm
4 Spectral data for the TNT intermediate 17produced by flash phottolysis
5 Disappearance quantum yields of aromatic 18nitroccmpounds irradiated with 2537A light
6 Isosbestic points and differential spectral 21bands of nitrobenzene, o-nitrotoluene,and TNT
Figures
I Schematic representation of the energy 29levels of an organic molecule containinga nr-electron system
2 Schematic diagram of flash photolysis 30apparatus
3 Spectrum of TNT in cyclohexane
-MOE
4
I I

4 Changes in transmissions of aromatic 32nitrocompounds during continuous
photolysis for one hour at 15-minuteintervals
4a 3.5 x 10- 5 M nitrobenzene in n-heptane 32
4b 5.2 x 10- 5 M o-nitrotolene in n-heptane 33
4c 3. 1 x 10-5 M TNT in cyclohexane 34
5 Differential spectra of photolyzed 35aromatic nitrocompounds. References
are unphotolyzed solutions
5a 3.5 x 10-4 M nitrobenzene in ii-heptane 35continuously photolyzed for 5 hours
5b 2 x 10 4 M o-nitrotoluene in n-heptane 36
photolyzed by flashing 15 times
5c 3 x 10 M TNT in cyclohexane photo- 37lyzed by flashing 10 times
6 Ultraviolet spectrum of 3. 1 x 10"5 M 38o-nitrosotoluene in n-heptane
7 Differential spectrum of a synthetic 39
mixture of 1. 7 x 10" 5 M o-nitro-sotoluene and 1. 7 x 10 4 M o-nitro-toluene in n-heptane. Referencesolution is 2 x 10- 4 M o-nitrotoluenein n-heptane
-~JI7
I - A , i I

ABSTRACT
The flash photolysis and continuous photolysis at 2537A(ir-pr* transitions) of aromatic nitrocompounds in solution wereperformed.
Flash photolysis revea,,., intermediates for o-nitrotoluene andits nitro derivatives. The spectrum and mean lifetime of theintermediate fov flashed TNT were determined. The maximumabsorption of the TNT intermediate occurred in the 470 nm regionand the mean lifetime was 1. 5 ± 0.3 msec. This intermediate ispribably not directly related to product forniation.
The continuous photolysis experiments reveals that all thearomatic nitroccmpounds have disappearance quantum yields ofthe order 10-3 . These low values for the quantum yields implythat the most important reaction occurring is the deactivation tothe ground state of the original molecule. The presence orabsence of oxygen does not appear to be of significance.
Product analyses suggest that aromatic nitrosocompoands and
nitrophenols are main products. However, the observed productsdo not conform in a simple manner to the constraints of theisosbestic points observed in all photolyses. It appears that otherproducts are present, some of which may have low extinctioncoefficients in the spectral range studied. The latter may benecessary to accommodate the isosbestic points.
!I

INTRODUCT ION
A search of the literature reveals that very little is knownabout the decomposition processes, bond energies, and thermo-dynamic properties of aromatic nitrocompounds. TNT and TNB(trinitrobenzene) are explosives of military interest that fall intothis class of compounds. With the idea of eventually being ableto gain some control of the explosive processes, a study ofaromatic nitrocompounds was initiated. Since a molecule mustdecompose via its ground state or one of its excited states, theinitial phase of the study was concerned with the photochemistryof these compounds using both continuous photolysis at 2537A andflash photolysis. Photochemical studies can reveal the nature ofexcited states, sites of attack, and bond energies. Since thevapor pressure of many of the compounds is low, solution photo-chemistry appeared most suitable for elucidating primary pro-cesses.
Photochemical Considerations
The number of states to which a molecule in the ground statecan go as a result of energy absorption is finite. By usingradiation, the behavior of these upper states can be studied.
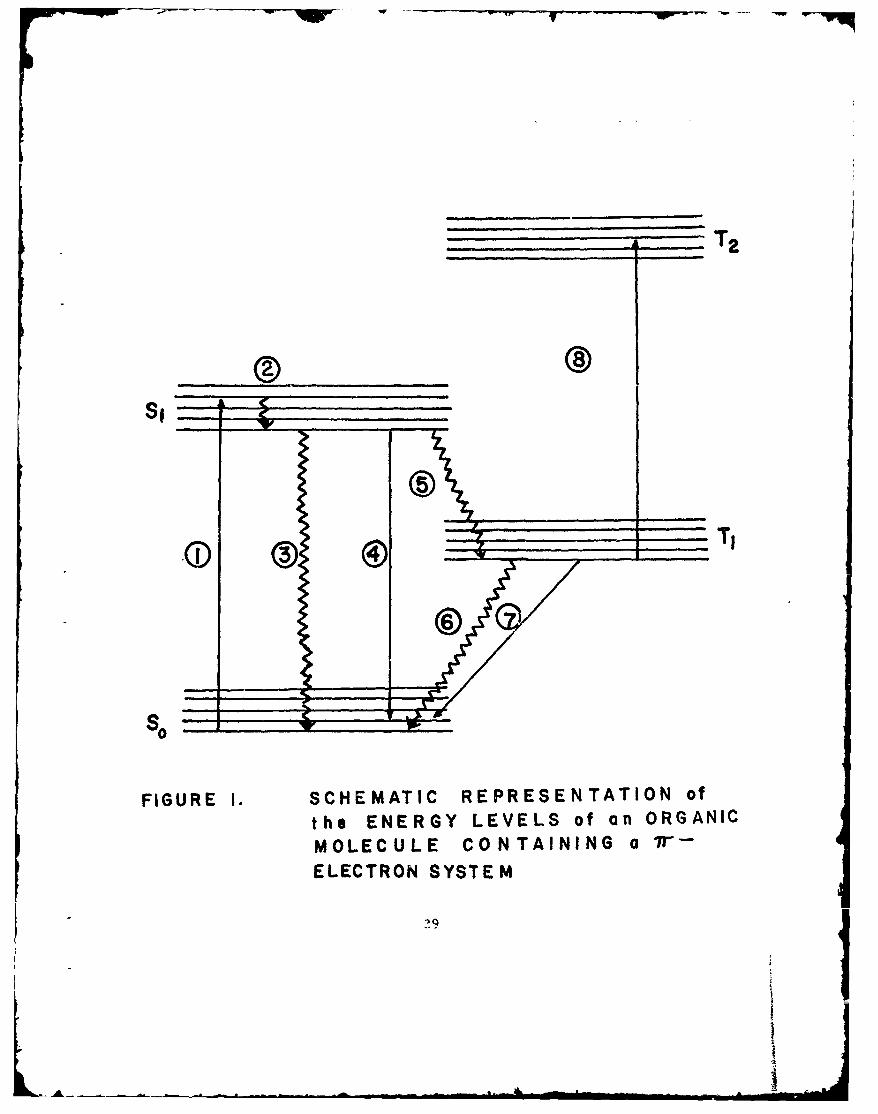
Figure 1 represents the energy levels of a typical organicmolecule containing a ir-electron system. So is the groundelectronic state, which is a singlet; S1 denotes the first excitedsinglet state; and T 1 and T 2 represent the first and secondtriplet states, respectively. Each of the states shows somevibrational energy levels. Some of the processes that occur whenthere is interaction between the molecule and an electromagneticfield, whose energy is in the visible or ultraviolet region of thespectrum, are as follows: In process (1), the molecule absorbs aphoton and is raised to the excited single state (S,). From Sj
the molecule can undergo one of several processes. The mostrapid process, which occurs in solution (-10-12 - 10-13 sec)and is caused by collisions, is a radiationless loss of excessvibrational energy (process (2)). The molecule is now in the lowest
2

-W--- w --FW9
vibrational level of the first excited singlet state whose lifetime is-10-8 seconds. There are three processes by which a moleculecan leave the excited singlet state. Process (3) depicts internalconversion, which is a radiationless deactivation to the groundstate. Process (4) shows a transition to the ground state wherethe molecule emits radiation called fluorescence. Process (5)illustrates intersystem crossing where the molecule crosses fromthe first excited singlet state to the first triplet state. By internalconversion, the molecule reaches the lowest vibrational level to T 1 .The molecule reaches the lowest vibrational level of T1. Themolecule can return to the ground state So by radiationless inter-system crossing (process (6)). Since the selection rules forbiddingradiative transitions between states of different multiplicities arenot strict, the lifetimes are usually-_ 10-3 second or longer,although shorter lifetimes have been observed. When no collisionaldeactivation occurs during this lifetime, deactivation to the groundstate can take place (process (7)) radiatively by phosphorescence,a long-lived luminescence. The advent of flash photolysis hasmade pcssible observation of triplet-triplet absorption due to theusually long lifetime of the triplet state (process (8)).
In addition to the processes described, a molecule may enterinto reactions from either an excited singlet or triplet state. Otherexcited configurations are possible when the molecule consists ofcomplex energy levels.
In the work p.t:,e:nted in this report, two methods were used tostudy the aromatic nitrocompounds. These are: continuous photolysis-..id flash photolysis.
Continuous photolysis involves irradiating a solution with lightof one wavelength for a specific time and determining the numberof molecules reacted and the number cf quanta absorbed. Thequantum yield or quantum efficiency it-r the disappearance of thenitrocompound can be calculated from .as- definition:
No. of molecules chemically reacted
No. of quanta absorbed
3L -- - - - - - -I

1W
The quantum yield is a good indicator as to the possible mechanismsthat are prevalent in a reaction. Suppose molecule A is irradiated andis raised to an excited state, designated A*:
A + hv -), A*
A* can behave in a number of ways to give various quantum yields,as depicted in Table 1.
TABLE I
The relationship between mechanism and disappearance quantum yield
., (Quantum Yield
Mechanism for Disappearance)
(1) A*-)A +hv 0
(2) A* --3B or A* + C --)D + E 1
(3) A* +A--)B 2
(4) "A* -- BIA* + A -vC 1 -2
(5) 'A* -- BA b A + L. or A + -
(6) A*--R +RZS+ A - B + R3? (Propagation)
R3 + A -C + RI) (Repeated) 00.1RI + R3 + (M) -*D + (M) Termination
B,CD, and E represent stable product molecules; RI, R., and R3
represent free radicals, and M represents a third body.
4

" "" ... .
If A* returns to the ground state through fluorescence, asmechanism (1) shows, or by any of the other ways indicatedpreviously, the quantum yield is zero. Now, if A* is rearrangedto a stable molecule or reacts with a molecule other than A andstable products are formed as depicted in mechanism (2), thenthe quantum yield is one. On the other hand, if A* reacts withanother molecule to A to form a stable product or products, thenthe quantum yield equals 2, as mechanism (3) shows. When thereare competing reactions, the quantum yield will not be a wholenumber. In the case of mechanism (4), A* disappears in tworeactions so that the quantum yield is between I and 2 and willdepend upon the relative importance of each of the reactions. Thequantum yield can bc Less than one, if the competing steps indicatedin mechanism (5) occur. In the final case of mechanism (6), A*decomposes into two radicals, R1 and R 2 . R1 reacts with A toproduce a third radical, which in turn reacts with A to produce R 1again. These chain propagation steps are repeated a great manytimes. This chain propagation may be stopped before exhaustionof A by a chain termination step where radicals can be deactivatedby the presence of a third body, if required. It L. i ierally believedthat chain i,'actions are responsible for detonatior. For example,the reaction between H2 and Cl 2 leads to a quantum yield of 10_6107.
Phtochromic (or phototropic) molecules have been studied fora long time. As with other stable molecules, thes m. oleculeq exist instable electronic configurations that give rise to their characteristicabsorption spectra. However, when a photochromic substance isirradiated, usually with ultraviolet light, the molecules absorb theenergy and are excited to an unstable configuration whose absorptionspectrum is in the visible region, so that color is produced. Theoriginal unirradiated substance was either colorless or of a differentcolor; hence the term photochromism. The color persists in asteady state condition as long as the substance is irradiated. However,when the irradiation is removed, the molecules return to theiroriginal stable electronic state. The mechanism for this involvesvery efficient pathways for internal conversion. Charge transfer isanother mechanism that can lead to photochromism, but is mainlyimportant for inorganic substances.
Review of Past Photochemical Studies
Several photochemical studies have been performed. They weremainly concerned with the 3600 A region. The major studies arelisted below.
5

0dC 00o. CD.
oD 00-
o ho
C)0N 0).&u 0 0. V
CD -cdI E>. S..
0. 0 0
.21 a, 0 Ld C
Cj 0 t4 14
U~~ 00) 0 ~.~
2O Q%0 0
0 $4CD 4 0 .tN
S4 CD C.CL -4 =
0 -i
CL -t 0 - 0.
~o0 0.CC .0 *0~ 0)0 0 C
0~ *CU00 C d S'0 C a
;.~~ -1q w (D3
0j -cC -0>0 ..' 0 C50Q)' S4.
0.c .0r0 0 g.
F 0 o0 o o q0d '~ 0 0:. . 0k a
0~ ~ 4.N S
Cd 0 r 0 0 0 t 0* C>) Cao
0 .- N
0.-4 -d 0 -
T;C 0OS..a : 1
g~S 0~1 $4 a r: 4 *~-c ~~~ 4 0dr c I a
C,,d.00
N.C CD 00 p.0::" g 0 0 0
z~0 00 d dL0D NC~ >OD .C C>
>f aC.Dwv d l C > '
(Ci CA
- '0
0' N (D*lC
=, 9 0*, .0 oa'0 4
- 0:
> C 002 BP 0 >c.4 0 o 0
Wi (n U2 C. fr
S: - ~ --4.-

0 C0
0
1~. 0 0V ;
- C
Cd 0. m
.0 *...
00. 0Q 0 c0
r_ Q)0 Cs , t:.-0
u S oz
U) 1 ) , 0U
0* - Cd (D $
0) c 0 s.0 0
o0
5) 00r00 o.. 0
03 z~ -0.Q))U
S. . 0 x0D 0 ,0 0 £ 0~' :
c- -- 0
~ .C~ ~ o .00
a ~ 0 .,2 'a .-' m * . U .0 X a)~
0D O-4 4.*a 0
00 or U).C0
00d Z >ý CCC (D (D 44
z0C . ) z:) .- u~ 0 CI) z a)o0
0 C
d CC0 ý4 00. D
0 C) L d .0 r= 0CU o1. 00 B 0 cc .o o N
.0
a) -, c k = C0 J 00 )) S. 0> 0~
00 U 0
0~
0 0 a) C
oco
W , k e 0 U) 41
0'
'0 C; tw 0
U0 -i W0'
>1( -0-Z. s! (6.0 .. b
0~. 0 .-.C.) 0 aE t 0c
S.4 d S. o-0 o
0'00
%7

'So 0 v~2 0
00 > 0
0 0
MD C9n. 4 00r0 >) e.= $4 0-
ct, 02) >=1 0 5 )0
04 0o 00_00 z ) 0) 0 0i
0.2 Q) 0 c
0d 0 0w Y.CU
C)$4c Id
V~~~ 0 s. . t
caN 0 C U 'd 'd 0 o 0
.. :* (L) r 01
M ' 0 4C 0o. C c(; 0-1 0 4OU ~.Cin 0' 0. DC
o S
(71 0 0ncd0 0 '4 54
0 0 0IV. r. - 0
0 0 4CUCD (D~' 0.4 -cad
m- -t 4- . 1 1
- .0 CD .00
a5 a. ao. 0D '0 '0CC 13z W( 000) 0- 0 0 00 c
tCo 00 C: a -0 12
00 t0 ~ 0 o 0D0O~~CD 0 bD 0 $...4
04 r-~ .0 m
0 0 In .0 00o.
CDCUC CU 0 0
5*- 0'Dto0 N oCIS 0* $.0 .0
0 S4 0
-- 4 %4 CIU0m en$4Ln0
0 Cflr-
0.-; u.-, 'S .o
U CU
=8C

The preceding summary illustrates the complexity of thekinetics through the contradictory results obtained by differentworkers. It is seen that Gray, et al, could not repeat the work ofSchultz and Ganguly. Baltrop and Bunce found aniline underconditions similar to those of other workers who did not report it.
One complicating factor in the photolysis of aromatic nitro-compounds in the past was the fact that some products have higherextinction coefficients than the parent molecule. For example, at313 nm the values of e for nitro- and nitrosobenzene are about288 and 2300, respectively. Thus, for this wavelength region, anynitrosobenzene formed will itself be photolyzed. A similar situationexists for nitroso- and nitrotoluene. One may therefore be ablr. topartially explain some of the variations in results of photolyses inthe 3100-3600A/ region. Relative intensities of the wavelengthcomponents of the incident radiation and variations in the extent ofphotolysis of products contribute to the poor reproducibility ofprevious studies. These problems are expected to be less severeat 25371. At 2537A the efor nitro- and nitrosobenzene are 8810 and2600, respectively, and for ortho-nitro- and orthonitrosotoluenethe values are 5790 and 1790, respectively. Thus, products of thistype formed in the photolysis at 2537A are less likely to be furtherphotolyzed and the interpretation of results should be easier.
EXPERIMENTAL PROCEDURES
Materials
The TNT (Eastman Kodak C .)mpany) was purified by the methoddescribed by Gey, Dalbey, and Dolah (Ref 20).
o-Nitrotoluene (Eastman Kodak Company) was fractionallyrecrystallized in a dry-ice bath and vacuum distilled. The middlethird fraction was retained.
The middle third fraction of nitorbenzene (Fisher ScientificCompany) was obtained by vacuum distillation.
9
_
I

o-Nitrosotoluene was prepared according to the method ofLutz and Lytton (Ref 21).
The other nitrocompounds used in the experiments, all ofwhich were obtained from Eastman Kodak Company, were checkedin solution with the Beckman DK-2 spectrophotometer in thevisible and ultraviolet, and some were checked in a gas chromato-graph as well.
The gas chromatograph employed a 9-foot column of 10%DC-25-X-30295 absorbed on 80-90 mesh Anakron ABS support toseparate the nitrocompounds. A flame ionization detector wasused and helium was the carrier gas. The temperature-timeprogram operated between 1000 and 225 0 C. No significantimpurities were evident and the compounds were used as received.
Certified isopropyl alcohol (99 mol %) and spectranalyzed n-heptane and cyclohexane (Fisher Scientific Company) were usedwithout further purification.
Flash Photolysis Apparatus
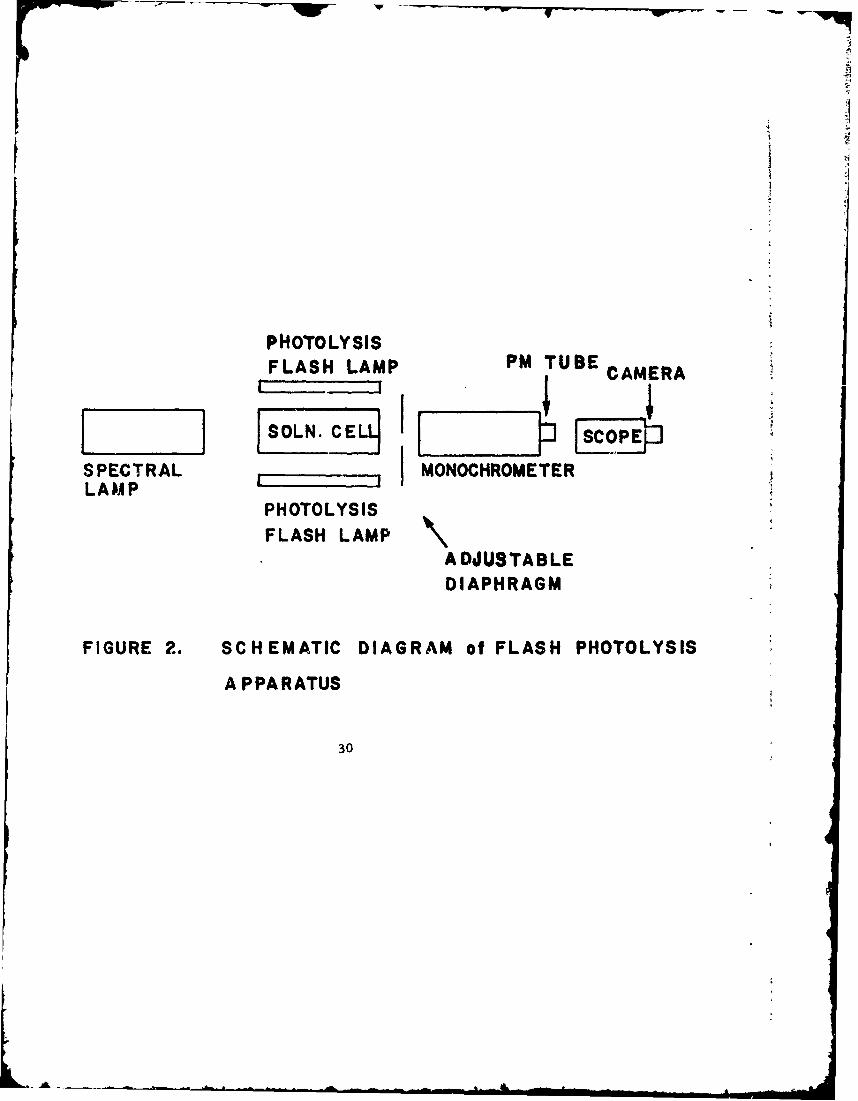
Figure 2 schematically depicts the flash photoly*.'s apparatus.The spectral lamp used initially was a Sylvania R43 ') flash lampthrough which was discharged a 105 pf capacitor charged to 2. 5 KV.The mean lifetime _!of the light pulse was approximately 350 isec.
"Tne photolysis flash lamps were two Xenon Corporation FP-10-IOOAlinear flash lamps connected in series and a 20 lif capacitor chargedto .0 KV was dizcLzu-ged across them. This gave an input energyof 1000 joules and a mean lifetime of about 35 psec for the lightpulse. The delay between flashings of the lamps was obtained withan Electronic Aids time delay generator.
The quartz solution cell between the flash lamps was about11.5 inches long having I-inch-in-diameter optical windows atboth ends. Two tubes were fused to the cell near the ends to allowfor filling. An aluminum reflector partially enclosed the cell.The light passing lengthwise through the cell from the spectral flashlamp was monitored in the visible with a small Bausch and LombHigh Intensity Grating Monochromator with slits adjusted for a
10
LI

nominal bandwidth slightly less than Z nim, an RCA IPZ8 photo-multiplier tube having a 10 K A*) resistor as a load to a Tektronix585A oscilloscope and a Polaroid camera.
An experiment was performed by flashing the spectral flashlamp first and then the photolysis flash lamps 100 psec later, whichis about at the peak intensity of the spectral flash lamp. Bycomparing oscilloscope traces for the spectral flash alone with thoseobtained in the experiments with both spectral and photolytic lampsat various wavelengths, the absorption spectrum and lifetime of anintermediate can be determined.
Although the spectra of the intermediates were determined inthis manner, the lifetime could not be determined since they werelonger than the lifetime of the spectral lamp. Therefore, a con -tinuous light source, Oriel Optics Corporation Z00-watt Xe-HgUniversal Arc Lamp, was used in place of the Sylvania flash lamp.Since this source was much lower in intensity than the photolysisflash lamps, it was necessary to monitor only the strong Hg linesand to interpose two slits between the diaphragm and the mono-chromator in order to increase the ratio of the signal of thespectral lamp to the signal of the scattered light from the photolysisflash lamps reaching the monochromator.
To minimize the effects of heat radiated from the Xe-Hg lampon the solution, a Corning CS 1-58 infrared-absorbing, visible-trans-mitting filter was employed between the lamp and the cell. Theteniipature of the experiments was 25 - 300C.
Some of the solutions were deaerated in the solution cell byflowing nitrogen through them for one hour. A flask containingsome of the pure solvent was put between the cell and the nitrogensupply. Samples removed near the end of the nitrogen flushingindicated that no change in concentration occurs when such samplesare investigated with the spectrophotometer.
The Polaroid records for only the first few flashes were used toobtain the intermediate spectrum and lifetime. This was done becauseproducts having absorption in the same region as the intermediatewere formed, progressively increasing the complications of therecords.
II
I L

For product analysis, the solutions were usually flashed tentimes each in orier to obtain sufficient product concentrations.Differential analyses were performed in the Beckman DK-2 spec -trophotometer using an unflashed solution as reference in mostcases. The concentrations of the solutions were 10- 4M.
Continuous Photolysis
In continuous photolysis, a 1-cm Beckman cell containing 3ml of a 10- 5 M solution of the nitrocompound in n-heptane, isopropylalcohol, or cyclohexane for TNT was placed in front of a lowpressure mercury quartz pencil lamp (Spectronics Corporation).The predominant radiation was 2537A,. A Corning CS9-54 filterwas used to eliminate any 1849K light which was found to photolyzethe solvents. Occasionally, concentrations of 1O- 4 M were usedfor analytical purposes.
After irradiation, normally for 15 minutes, the ,.pectrum ofthe solution was obtained with the Beckman DK-2 spe-trophotometer.The cell was again placed before the Hg lamp, and the process wascontinued for 3 more time intervals. The spectra of the irradiatedsolutions were compared with the spectrum of the unirradiatedsolution to determine what changes had occurred.
The number of quanta absorbed was determined by using apotassium ferrioxalate actinometer according to the method ofHaLchiad and Parker (Ref 22). The number of quanta absorbed bythe solutions per minute was on the order of 1017.
The disappearance quantum yields were calculated from thenumber of molecul.es decomposed per minute and the absorbedintensity.
Experiments were performed both in the presence and in theabsence of oxygen. The latter condition was achieved by employinga Teflon stopper having two holes into which stainless stell tubing0.032 inch in diameter was fitted. One tube was a vent whilenitrogen was bubbled through a small flask containing solvent andthen into the solution using the other tube. The tubes could beclos.ýd-off by slipping on small plastic tubing. It was found that five
12

minutes of rapid N2 - bubbling was sufficient to -emove the dissolvedoxygen, since longer purges gave no difference in results. Thereplacement of the oxygen by nitrogen in the solution caused increasedtransmission at about 250 nm and below, which was taken into accountin the analysis of the results.
RESULTS AND DISCUSSION
Ultraviolet Absorption Spectra of TNT and Other AromaticNitrocom pounds
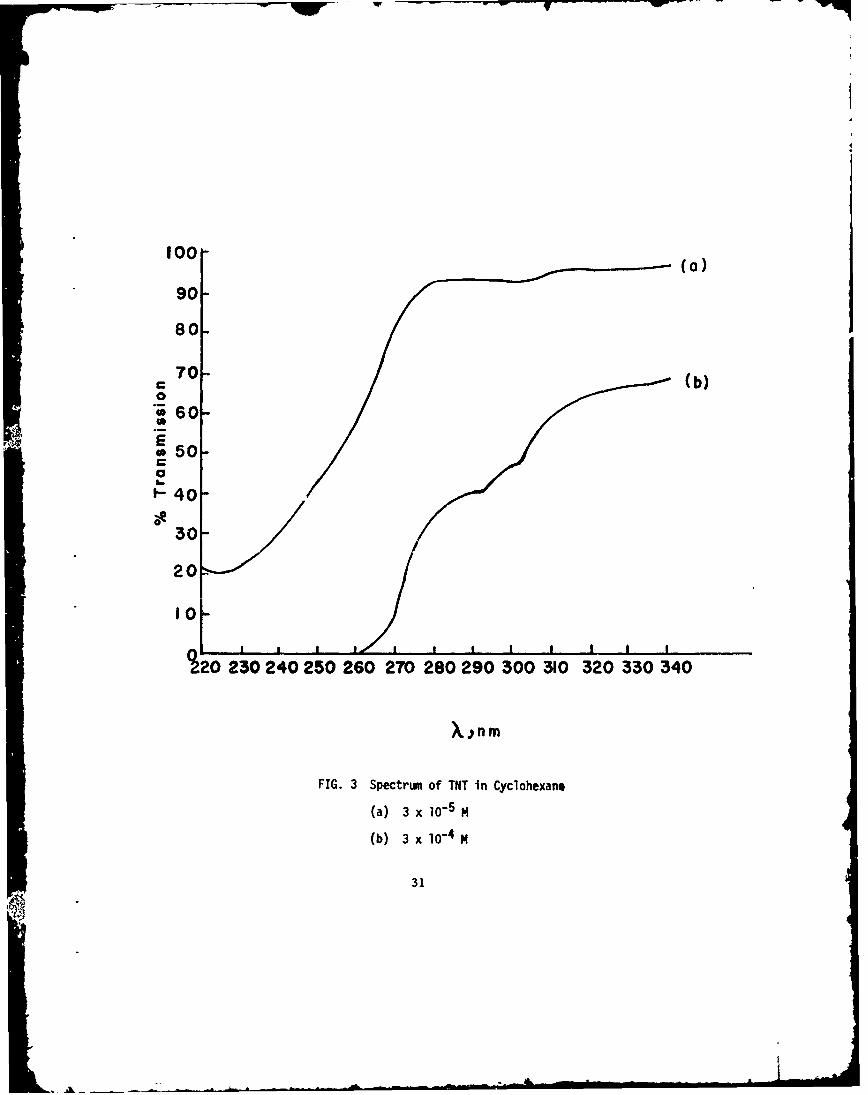
The spectrum of TNT in a nonpolar solvent could not be foundin the literature. Therefore, the ultraviolet 6pectrum in cyclohexanewill be presented and its discussion will also serve as a generalexample for all the aromatic nitrocompounds considered.
Figure 3 depicts the spectrum of TNT in cyclohexane withcyclohexane as the reference.
The shoulder at about 315 nm probably belongs to a class oftransitions in which an electron is promoted from a nonbonding (n)molecular orbital in the ground state to an antibonding ir (W',:)molecular orbital in the excited state. Therefore, this type iscalled an n --> r* transition. This transition occurs at a longerwavelength and has a much lower extinct"--- coefficient than theh-ghly intense Tr --* 7r* transitions in which an electron is promotedfrom a bonding Tr - orbital to an antibonding ir-orbital. For TNT,the n -- irr* band has an extinction coefficient of about 650 litermole- 1 cm- 1 while, for the Tr -9 1T* transition at 224.5 nm,e = 23, 000. The latter is to be compared with the work of Kamlet,Hoffsommer, and Adolph (Ref 23) who obtained e = 19,200 at
'max = 227 nm for TNT in methanol. The bathochromic shijft ingoing from nonpolar to polar solvents is characteristic of 1T --" T$
transitions. As is shown below, this is also true for nitrobenzenein n-heptane and isopropyl alcohol. The shoulders at about 300and 290 nm, with extinction coefficients of about 1000 and 1200respectively, probably also belong to the same or different ,r -, rr*transitions, since the extinction coefficients are somewhat largerthan those expected for n - ir* transitions.
13II
#3
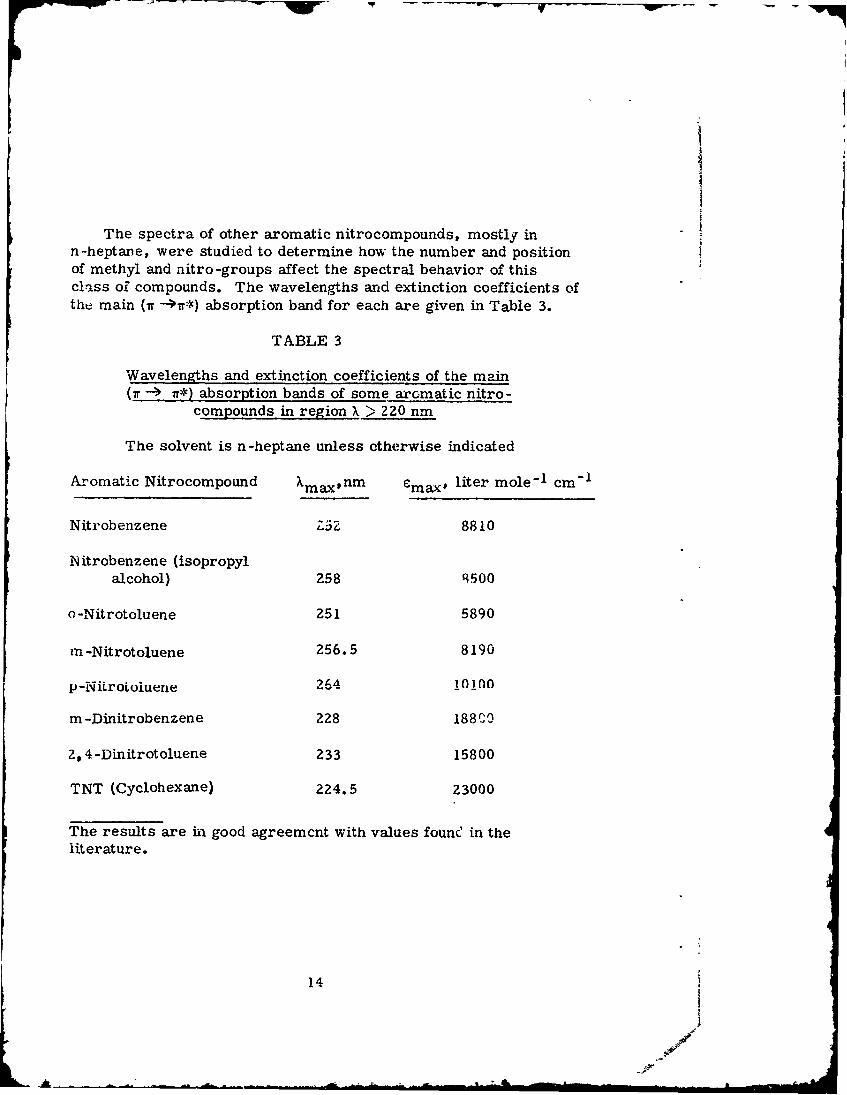
IThe spectra of other aromatic nitrocompounds, mostly in
n-heptane, were studied to determine how the number and positionof methyl and nitro-groups affect the spectral behavior of thisclass oZ compounds. The wavelengths and extinction coefficients ofthe main (1T -i*) absorption band for each are given in Table 3.
TABLE 3
Wavelengths and extinction coefficients of the main(7r -- ) r*) absorption bands of some aromatic nitro-
compounds in region X > 220 nm
The solvent is n-heptane unless otherwise indicated
Aromatic Nitrocompound Xmaxnm emax, liter mole-I cm"1
Nitrobenzene ZSZ 8810
N itrobenzene (isopropyl
alcohol) 258 9500
o-Nitrotoluene 251 5890
m -Nitrotoluene 256.5 8190
p-NiLrotoluene 264 10100
m-Dinitrobenzene 228 188 P0
2,4-Dinitrotoluene 233 15800
TNT (Cyclohexane) 2Z4.5 23000
The results are in good agreemcnt with values founC' in theliterature.
14
i-7

MW lisp,
It can be seen that the introduction of the second nitrogroupcauses Xmax to decrease and emax to increase. The third nitro-
group does not appear to be of great significance in this connection.Not shown here are indications of highly absorbing bands at wave -lengths below 220 nm.
Spectra and Mean Lifetimes of Intermediates Produced by FlashPhotolysis
The results obtained in this work are consistent with theobsecrvations of Margerum et al (Ref 6) in Ehat only those compoundswhich have a nitrogroup ortho to a benzyl hydrogen yield anintermediate which absorbs in the visible region of the spectrum,that is, only for o-nitrotoluene, 2, 4-dinitrotoluene, and TNT wereintermediates observed. It was also found that the spectralcharacteristics and mean lifetimes of the intermediates in nonpolarsolvents were independent of the presence of oxygen, at least foro-nitrotoluene and TNT which were checked (2, 4-dinitrotoluenewas done only in the presence of air). The mean lifetime of aninterme.diate is determined from the oscilloscope trace as follows:
Sinc.e it was found that intermediate decay obeyed first orderkinetics, one can write
C = C e kt (1)
where C is the concentration of the intermediate at time t, CO isthe concentration at zero time and k is the rate constant. Takingcommon logarithins:
log C = log CO - kt (0. 4343) (2)
Now, from Beer's law: I = Io (10) -EC1 (3)
where I is the initial intensity and I is the intensity after transmissionthrough°a path length of I and a solution of concentration C having anextinction coefficient e at a particular wavelength. Taking commonlogarithms of Equation (3) and rearranging the terms gives
I-log (T-)
C = (4)el

Now, taking logarithms of Equation (4),
Ilog C=log C -log -logel (5)
10
Combining Equation (5) with Equation (2) for time t and time zeroleads to
log1[ -log 1 )t" =log[ -log ( 1)o1 -kt(0.4543) (6)10o
In Equation (6), (() and (1 ) are the transmissions at time 1. and
at time zero, respectively. A plot of log [- log (_I)t 1 vs t gives
10a straight line with
k =-slop__ _e (7)
0.4343
so that
- 1 0.4343•m= k =slope
where m is the mean lifetime.
Unfortunately, as is indicated earlier in this report, 10 does
not remain constant, the reason being the formation of a product orproducts that absorb in the same wavelength region. This type ofbehavior has also been observed by Wettermark (Ref 8,9). Therefore,Io was taken as that value for the transmission after the decay of theintermediate. Only the first few flashes were giv a considerationsince the Io decreased rapidly thereafter. The result of this be-havior was a loss of accuracy in determining the mean lifetimes,especially when the intermediate absorption was small. The wave-length of maximum absorption (minimum transmission) for the TNTintermediate appears to be in the 470 nm region. The mean lifetimeis determined to be 1.5 k 0. 3 msec.
16

The experimental transmissions and wavelengtiis observedfor the TNT intermediate are given in Table 4.
TABLE 4
Spectral data for the TNT intermediate produced byflash photolysis*
?'nm % Transmission
400 92.6450 80.8500 81.7550 93.6
*A more complete spectrum for this intermediate, obtainedby C. Capellos and R. Suryanarayanan,will be published soon.
The spectra of the intermediates of o-nitrotoluene (?max -- 370 nm)
and 2,4 dinitrotoluene (Vmax > 350 nm) as well as the lifetime of
the o-nitrotoluene intermediate were consistent with their beingaci-forms of the nitrocompounds; such intermediates were observedin aqueous acid solutions by Wettermark (Ref 8, 9).
0 0
Nitro Form itci FormThe TNT intermediate is also assumed to be a similar aci formby analogy with the above and the long lifetime which is characteristicof isomeric interconversions. Since the concentrations of theintermediates were not known, it was not possible to determinetheir extinction coefficients.
17
AL:• . .. • • • ,. . .

Quantum Yields for the Disappearance of Aromatic Nitrocompounds
The disappearance quantum yields determined for the con -
tinuous irradiation with 2537A are given in Table 5.
TABLE 5
Disappearance quantum yields of aromatic nitrocompounds0
irradiated with 2537A light
Aromatic Nitrocompound L x 103
Nitrobenzene 3
Nitrobenzene (isopropyl alcohol) 2
o-Nitrotoluene 4
m -Nitrotoluene 0.8
p -Nitrotoluene z
m -Dinitrobenzene I
2, 4-Dinitrotoluene I
TNT (Cyclohexane) I
It was found that the quantum yields were independent ofoxygen in the solutions, within an experimental error of - 20%.The quantum yields were also independent of the intensity withinthe range investigated: 9 x 1016 to 5 x 1017 quanta/minute.
The formation of products having absorption near the spectralpeaks of the aromatic nitrocompounds, where the quantum yieldswere evaluated, causes the quantum yields to be minimum values.However, the values are not expected to be very far from the truevalues in ,,iew of the small dependence of the quantum yields ontime of ir.'adiation. This indicates that no significant productinterfer,-nce is occurring.
13

For comparison, nitrobenzene in isopropyl alcohol was alsoinvestigated. Anomalous results, apparently due to the photo-decomposition of the products, appeared within the 45-minuteirradiation period. Therefore, 5-minute intervals of irradiationtotaling 20 minutes were used.
As is stated in the Introduction, most of the previous attemptsto study the products obtained from the photolyses of aromaticnitrocompounds were performed with continuous irradiation in the365-366 nm region of the spectrum. Absorption in this regioncorresponds to the n -> Tr* transition, generally but not fully accepted(Ref 24). The lowest triplet state is considered a iT, 7r* state byBaltrop and Bunce (Ref 16), bat Trotter and Testa (Ref 25) giveevidence that this is incorrect and that the state is n, $r* • It hasbeen reported that while most of the molecules apparently returnby internal conversion to the ground state, some of the moleculesundergo intersystem crossing to the triplet state which is thereacting species (Ref 13).
On the other hand, absorption at 2537A. corresponds to the Yr --> I*transition. Since the main absorption bands of the aromatic nitro-compounds are in the 2537K ret;-on or somewhat lower, the essentialeffect of the flash photolysis is the excitation of the same 7r -)- I*transition. Therefore, it should be expected that the primaryprocesses in flash photolysis and in our continuous photolysis shouldbe the same.
When the flashed solutions were held in the dark overnight andtheir differential spectra. observed again, it was found that the over-all transmissions increased somewhat but the shapes of the curveswere essentially the same as before. With the continuous irradiatedsolutions, this generally did mt occur to any significant extent.
A comparison of the quantum yields for the disappearance ofthe aromatic nitrocompounds reveals that the values are smalland essentially riot too different (see Table 5). This indicates thatthe most important reaction (or reactions) occurring is the de-activation to the ground state of the original molecule as exemplifiedby Reaction (5) depicted in Table 1. Since these aromatic nitro-compounds do not usually fluoresce, the deactivation must takeplace by some other means (Ref 13, Z4).
19

Product Formation
As migbt be expected, some product bands were more prominentwith some of the nitrocompounds than with others. Since, in manycases, the reference solutions used in the spectrophotometricdeterminations were the original solutions, the differential curveswere not quite true spectral representations of the products. Thisis due to the fact that the loss of aromatic nitrocompound due tophotolysis is not completely compensated by the reference solution.However, any product or products that are formed having highextinction coefficients shoL ld be easily observed.
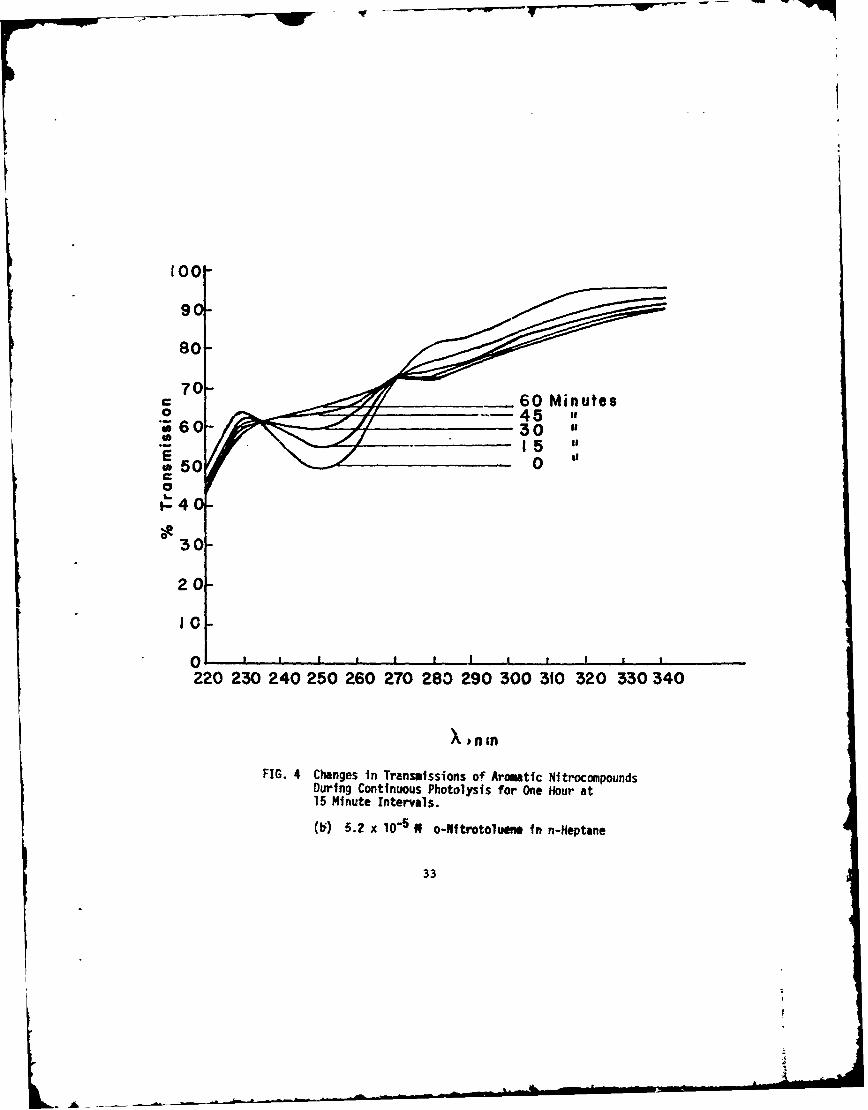
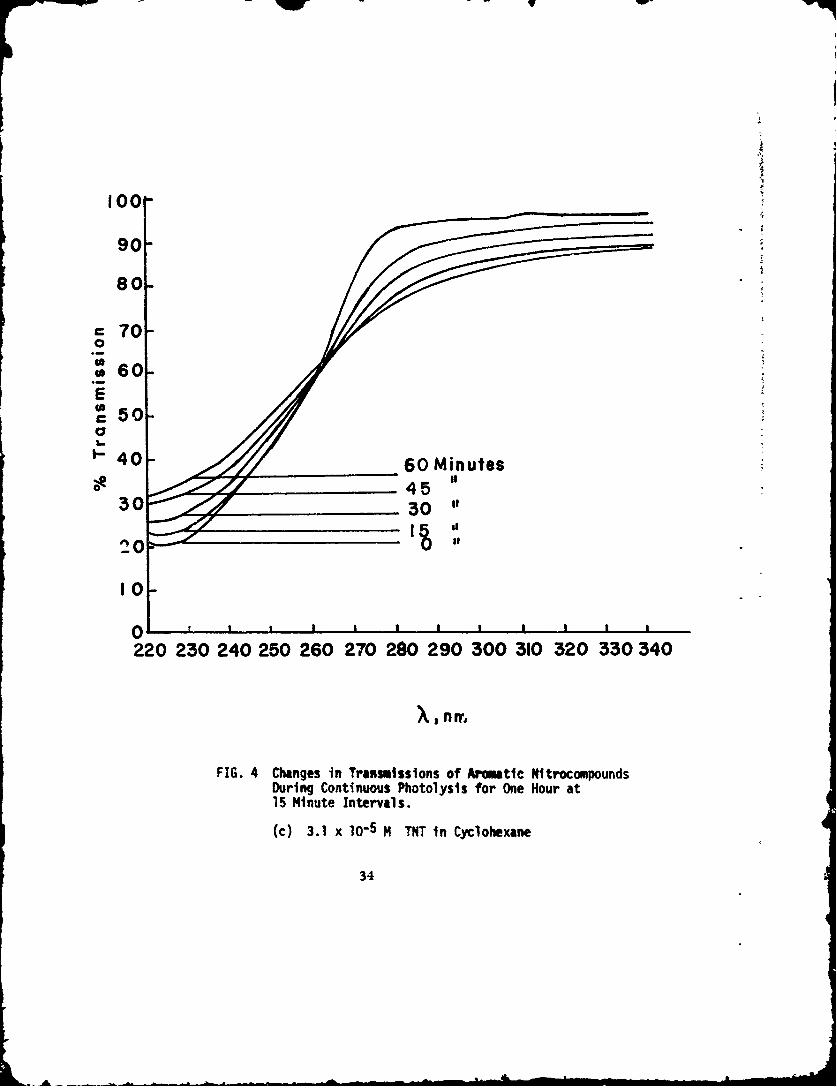
The changes in transmission of photolysis experiments fornitrobenzene in n-heptane, o-nitrotoluene in n-heptane, and TNTin cyclohexane are given in Figure 4. Decreases in the concentrationsof the parent nitrocompounds are seen through increases in trans-mission at their respective absorption bands. Another feature of thesecurves is also quite obvious: there are two points for the nononitro-compounds that do not change with irradiation time. These pointsare called isosbestic points (sometimes this term is applied only toequilibrium conditions and the term iso-absorptive points totime-dependent reactions) (Ref 26). All the photolyses result inisosbestic points and, in the flash photolysis work, each of thefinal flashed solutions crossed the original solution curves at thesesame points. The polynitrocompounds showed only one isosbesticpoint since the main absorption bands were at the extremes of thecurves and were not explored to lower wavelengths. Table 6 comparesthe isosbestic points for nitrobenzene, o-nitrotoluene, and TNT.
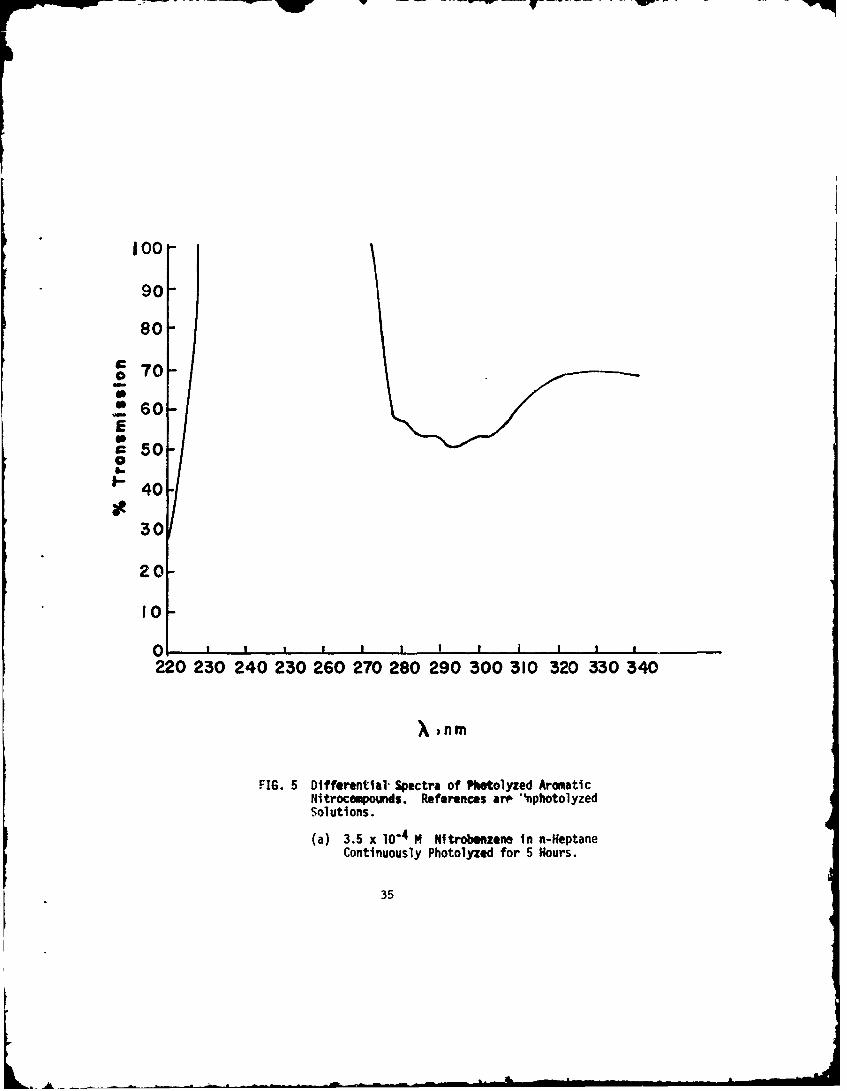
As examples of the differential spectrophotometric curves,those obtained with nitrobenzene in n-heptane, witi. o-nitrotoluenein n-heptane, and with TNT in cyclohexane are given in Figure 5.The differential bands for the three aromatic nitrocompounds arecompared in Table 6. For TNT,the lower wavelengths could notbe investigated because of the overpowering absorption by the TNTitself. Except for different transmission values due to differentamounts of conversion, the curves for the continuously photolyzednitrocompounds were identical to the curves obtained by flashing.
20
ALJ

TABLE 6
Isosbestic points and differential spectral bandsof nitrobenzene, o -nitrotoluene, and TNT
X (nm),Compounds X (nm),isosbestic points Differential Spectral
BandNitrobenzene in
n-heptane 226 219274 286
283292302
o-Nitrotoluene inn-heptane 235 225
269 280285 (Shoulder)316
TNT in cyclohexane 262 274295 (Shoulder)310
The differential uv spectral characteristics of the productsformed in all the photolyses suggest the possibility of aromaticnitrosocompounds. This is particularly true for o-nitrotoluene(and possibly for TNT). Figure 6 is the ultraviolet spectrum ofo-nitrosotoluene in n-heptane. The spectrum indicates shouldersat 310, 280, and 227 nm, as well-defined bands at 287.5 and220 nm (also a further band at -207. 5 nm, which appeared to besomewhat more absorbing than that at 287.5 nm). The extinctioncoefficients were determined to be -9100 at 287.5 nm and - 4310 at310 nm. Also, o-nitrosotoluene has a very weak band at 790 nmwith an observed extinction coefficient of 39.6 due to the nitrosogroup.
21

To check on the identity of the product as o-nitrosotoluene inthe photolysis of o-nitrotoluene, the amount of o-nitrotoluene lostthrough photolysis was calculated and a synthetic solution wasprepared, consisting of the o-nitrotoluene remaining and anequivalent amount of o-nitrosotoluene corresponding to the losto-nitrotoluene. The differential curve is given in Figure 7. Itcan be seen that the overall features are those found in thephotolyzed solution depicted in Figure 5b. The band at 225 nm hasa transmission greater than 100%, indicating at least one otherproduct must be present.
To further substantiate the presence of o-nitro.1otoluene, a 10-4 Mo-nitrotoluene solution was flashed fifteen times and concentratedunder vacuum. A similar solution of nitrobenzene was treated inthe same manner. The concentrated o-nitrotoluer.e solution showeda very weak, but definite band at 790 nm. On the other hand, theconcentrated nitrobenzene solution showed no band il the 700-800 nmregion. Even when flashed 30 times the nitrobenzene solution didnot give any definite indication of a band in this region.
In addition to the ultraviolet region, the visible region between340-900 nm was also observed for the photolyzed aromatic nitro-compounds. For nitrobenzene and p-nitrotoluene, the differentialcurves (90-100% transmission range) indicated for each a weak bandin the 350 nm region. With the other nitrocompounds, this bandwas either absent or indefinite.
The 350 nm band observed in the photolyzed solutions ofnitrobenzene suggests a nitrophenol. This was tested by con-tinuously photolyzing a 10- 4 M nitrobenzene solution for 5 hoursat 2537A (essentially equivalent in percent decomposition to onehour with a 10-5M solution). The solution was extracted withaqueous 0. 01N NaOH, in which the spectra of the nitrophenolshave been studied by Kiss and Howath (Ref 27'. Bands at 405, 282,and 230 nm were observed in the extracted aqueous phase. Thesebands are to be compared with the literature values of 415, 282,and Z25.5 nm for o-nitrophenol.
2 2

In the case of the photolyzed o-nitrotoluene, no such bandswere observed except by extracting the o-nitrotoluene solutionthat was flashed fifteen times and concentrated under vacuum.Here a weak, broad barnd in the 405-410 nm region was obtained!.The lack of spectral data for possible nitrophenols prevents anassignment at this time.
The above results suggest that, in the photolysis of o-nitrotoluene,the main product is o-nitrosotoluene and the secondary product is anitrophenol. In the case of nitrobenzene, the main product appearsto be o-nitrophenol. Thus, an intramolecular process may beoccurring to some extent. If nitrobenzene lost an oxygen atom as aresult of the absorption of a photon, nitrosobenzene would beexpected as a product. For the present conditions, a positiveindication of nitrosobenzene would be absorption at 760 nm. How-ever, the absorption here is weak, and therefore only possible forlarge amounts of nitrosobenzene. Nitrosobenzene has been suggestedas an intermediate product even though it has not been observed(Ref 16). Its failure to appear is attributed to secondary photolysisand chemical reactions. Similarities in the differential spectrasuggest that aromatic nitrosocompounds and nitrophenols are alsoproducts in the photolyses of the other aromatic nitrocompounds.
Any products produced must conform to the constraints of theisosbestic points. The conditions imposed in a photolytic reaction,
aA N bB +cC +dD + ............
where a moles of original material A is photolyzed to products bmoles of B, c moles of C, d moles of D, and so on, is that at eachisosbestic point the relationship among the extinction coefficientsbe as follows:
beB + cCC + deD + ........
a
23
A•m

PPII
Attempts were made to fit solutions containing o-nitrophenoland/or nit~rosobenzene to the isosbestic points observed in thephotolyses of nitrobenzene. These mixtures could not be fitted tothe observed isosbestic points in a simple manner.
The extraction experiments have also indicated other productspresent, some of which may have low extinction coefficients in thespectral range studied. The latter may be necessary to accommodatethe isosbestic points.
With regards to products, the role of oxygen is not clear atthis time. The differential curve of flashed TNT, depicted inFigure 5c was obtained with oxygen present. When oxygen wasremoved, the curve in the region of the 310 nm band had sub -stantially smaller transmission values (greater absorption).Although the curve now had been altered at the higher wavelengths,the general shape was essentially the same and no other bands wereobvious. However, in the case of o-nitrotoluene in n-heptane, onlya slighit change was noted at 340 nm.
CONCLUSIONS
Apparently, the processes are more complex than had beenindicated by a superficial observation of the data. That the photo-chemistry of the aromatic nitrocompounds is quite complex isshown by the review of the literature given in Table 2. Specifically,it is shown by the fact that free radicals have been observed bymeans of electron spin resonance in the photolysis of o-nitrobenzaldehyde,o-nitrocinnamic acid and o-nitrobenzyl alcohol, while negative resultswere obtained with nitrobenzene and o-nitrobenzoic acid (Ref 10). Itwas also observed that the free radicals produced by o-nitroben-zaldehyde wa of such low concentration that it is not expected to be anintermediate in the photochemical rearrangement to a nitroso compound.Free radical intermediates were also observed for o-nitrocumineý,o-nitroethylbenzene and o-nitrotoluene (in the last case, a very smallamount)(Ref 15) by ESR. These radicals were stable for days andcould be produced in the presence or absence of oxygen. Irradiationof the corresponding parasubstituted compounds gave no radicals. Noevidence for the w~esence of radicals has been obtained in the visibleregion.
24
A. -

As is stated in Section B, the intermediates observed by thevisible spectra of the flashed aromatic o-nitrocompounds are con-sistent with the aci form of the compounds. The relativelylong lifetimes of the intermediates show that they are not excitedsinglet states. In addition, it is also unlikely that triplet staesare involved since the same results were obtained in both thepresence and the absence of oxygen, unless the triplet lifetime isless than approximately 20 psec - the experimental limitation. *It is known that oxygen is an excellent quencher of triplet states.
It appear., that the aci and radical intermediates observed aredue to side reactions and are not related directly to the products.(Ref 8 and 10) In addition, the lack of a significant oxygen effecton the quantum yields and products indicates that the intermediatesare not long-lived triplet states. Even at the longer wavelengths,the triplet state is not involved in the photolysis of nitrosobenzene,and in the photolysis of nitrobenzene participation of the triplet statehas not been established unequivocally (Ref 19). Apparently, theintermediates responsible for the products have not been observedas yet.
The results of this study indicate that further progress inelucidating the detailed mechanisms involved in the photolyses ofaromatic nitrocompounds requires the extensive development ofanalytical techniques to determine quantitatively the productsobtained. This study and the previous literature imply that this isnot an easy task.
*Capellos and Suryanarayanan have recently shown, in lowtemperature flash photolysis studies conducted in a PMMA matrix,that the triplet state for trinitrobenzene is not expected to beobserved at room temperature for the time resolution of the flashphotolysis equipment used in this work. The reports listed belowcontains details of this work.
1. C. Capellos "Ruby Laser Nanosecond Flash Photolysis ofa-Nitronaphthalaie Solutions" Picatinny Arsenal Technical Report4341, June 1972.2. C. Capellos and K. Suryanarayanan "Microsecond Flash Photo-lysis of 1, 3, 5-Trinitrobenzene Solutions and Product Formationwith 254 nm Steady State Illumination", Picatinny Arsenal Technical
Report 4342, June 1972.
25
L. II i I :

Further efforts will be concerned with the development ofanalytical techniques, which will include IR and UV spectroscopy,and thin-layer and gas-liquid chromatography. Furthermore,these analytical techniques will be applicable to s1ock decompositionstudies of explosives containing aromatic nitrogroups which are inprogress. Here the attempt is to detect chemical changes as aresult of shockwaves passing through the explosive.
26
JL1

REFERENCES
1. Schultz, G. and Ganguly, K.L., Ber. 58, 7ý)2 (1925)
2a. Leighton, P.A. and Lucy, F.A., J. Chem. Phys. 2756 (1934)
2b. Lucy, F.A. and Leighton, P.A., J. Chem. Phys. 2, 760 (1934)
3. Shelegova, O.M., J. Exptl. Theoret. Pbys. USSR 9, 1527 (1939.)
4. Hastings, S.H. and Matsen, F.A., J. Am. Chem. Soc. 70,3514 (1948)
5. Gray, D.N., Bonomo, F.S., and Denver, R.I., "Study onthe Effect of Temperature Cycling of High Explosives",Report AFSWC TDR 6Z-16. Contract No. AF29(601)2671,March 1962
6. Margerum, J.D., Miller, L.J., Saito, E., Brown, M.S.,Mosher, H..S., and Hardwick, R., J. Phys. Chem. 66,2434 (1962)
7. Wettermark, G., Nature 194, 677 (1962)
8. Wettermark, G., J. Phys. Chem. 66. 2560 (1962)
9. Wettermark, G. and Ricci, R., J. Chem. Phys. 39, 1218(1963)
10. Tench, A.J. and Coppens, P., J. Phys. Chem. 67. 1378(1963)
11. Morrison, H. and Migdalof, B.H., J. Ora. Chem. 30,
3996 (1965)
12a. Ward, R.L., J. Chem. Phys. 38, 2588 (1963)
12b. Brown, J.K. and Williams, W.G., Chemical Communications 495(1966)
13. Hurley, R. and Testa, A.C., J. Am. Chem. Soc. 83, 4330(1966)
27

-wp
7I
II
14. Hurley, R. and Testa, A.C., J. Am. Chem. Soc. 89, 6917(1967)
15. Strom, T., and Weinstein J. J. Org. Chem. 32, 3705(1967)
16. Baltrop, J. A. and Bunce, N.J., J. Chem. Soc. (C) 1467(1968)
17. Hashimoto, S., Sunamoto, J.. Fuyii, H., and Kano, K.,Bull. Chem. Soc. Japan 41, 1249 (1968)
18. Weller, J.W. and Hamilton, G.A., Chemical Communications1390 (1970)
19. Ayscough, P.B., Sealy, R.C., and Woods, D.E., J. Phys.Chem. 75, 3454 (1971)
20. Gey, W.A., Dalbey, E.R., and Van Dolah, J. Am. Chem.Soc. , 78, 1803 (1956)
21. Lutz, R. and Lytton, M.R., Org. Chem. 2, 68 (1937)
22. Hatchard, C.G. and Parker, C.A., Proc. Roy Soc. (London)A235, 518 (1956)
23. Kamlet, M.J., Hoffsommer, J.C. and Adolph, H.G.,J. Am. Chem. Soc. 84. 3925 (1962)
24. McGlynn, S.P., Azumi, T., and Kinoshita, M., MolecularSpectroscopy of the Triplet State, Prentice-Hall, Inc.,Englewood Cliffs, N.J. (1969), p. 251, 252
25. Trotter, W. and Testa, A.C., J. Am. Chem. Soc. 90.7044 (1968)
26. Schlafer, H.L. and Kling. 0., Angew. Chem. 68,. 667 (1956)
27, Kiss, A. 1. and Horvath, G., Acta Chim. Hung. 4, 15 (1964)
28
"li
..... ___, _-_ T 2
- i - T1
S--
0 0
"FIGURE I. SCHEMATIC REPRESENTATION of
the ENERGY LEVELS of an ORGANIC
MOLECULE CONTAINING a ir-
ELECTRON SYSTEM
29
-I ' : •".. . . . . - ' . . .. - "-' • •
PHOTO LYSISFLASH LAMP PM TUBE CAMERA
SPECTRAL ______ MONOCHROMETERLAMAP
PHOTOLYSISFLASH LAMP
ADJUSTABLEDIAPHRAGM
FIGURE 2. SCHEMATIC DIAGRAM of FLASH PHOTOLYSIS
A PPARATUS
30
100- -- (a)
90
80
70- (b)0a "60
* 50
40o
30-
2 0
I0-
120 230 240 250 260 270 280 290 300 310 320 330 340
X,.nm
FIG. 3 Spectrum of TNT in Cyclohexane
(a) 3 x 10-5 M
(b) 3 x 10-4 M
31

1001
90-
80
70 Minutes
#A60 -50 -------
•-4
30
20
I00 - I .I I 80I I .. ,, I I ,,p
220 230 240 250 260 270 280 290 300 310 320 330 340
X*,nm
FIG. 4 Changes in Transmissions of Aromatic NitrocompoundsDuring Continuous Ph.tolysis for Ore Hour at15 Minute Intervals.
(a) 3.5 x 10- 5 H Nitrobenzene in n-IHeptane
32
100
9
80O
70-c 60 Minutes"0 - 45 .
0 0
20-
I0-
220 230 240 250 260 270 283 290 300 310 320 330 340
X ni
FIG. 4 Changes in Transmissions of Aromatic NltrocompoundsDuring Continuous Photolysis for One Hour at15 Minute Intervals.
(b) 5.2 x lO5R 0o-Nitrotoluene in n-Heptane
33
i00
901
c 700
"60
', 50-
•"40- 0Miue
30 30 "
I010
01220 230 240 250 260 270 280 290 300 310 320 330 340
FIG. 4 Changes in Transmissions of Aromatic NitrocompoundsDuring Continuous Photolysis for One Hour at15 Minute Intervals.
(c) 3.1 x 10-5 M TNT in Cyclohexane
34
I00
90
80
70-
60oE
50o
~40-
30
20-
I0-
220 230 240 230 260 270 280 290 300 310 320 330 340
rnm
FIG. 5 Differential Spectra of PItwolyzed AromaticNitrocompounds. References are 'nphotolyzedSolutions.
(a) 3.5 x 10-4 M Nitrobenzene in n-HeptaneContinuously Photolyzed for 5 Hours.
35
K

100
90
80
70
"e60-
E 50-to
S40
S30
20
10
01220 230 240 250 260 270 280 290 300 310 320 330 340
X,nmFIG. 5 DIfferential Spectra of PhotoIyed AromaticNi ocofmPods. efereces are UnPhotolyzed
SOluons.
(b) 2 x 10-4 N o-Nitrotoluene in n-NeptanePhotolyzod by Fleshing IS Times.
36