dna sequencing
TRANSCRIPT
DNA sequencing
The term DNA sequencing refers to methods for determining the order of the nucleotide bases, adenine, guanine, cytosine, and thymine, in a molecule of DNA. The first DNA sequences were obtained by academic researchers, using laborious methods based on 2-dimensional chromatography in the early 1970s. Following the development of dye-based sequencing methods with automated analysis, DNA sequencing has become easier and orders of magnitude faster. Knowledge of DNA sequences of genes and other parts of the genome of organisms has become indispensable for basic research studying biological processes, as well as in applied fields such as diagnostic or forensic research. The advent of DNA sequencing has significantly accelerated biological research and discovery. The rapid speed of sequencing attained with modern DNA sequencing technology has been instrumental in the sequencing of the human genome, in the Human Genome Project. Related projects, often by scientific collaboration across continents, have generated the complete DNA sequences of many animal, plant, and microbial genomes.
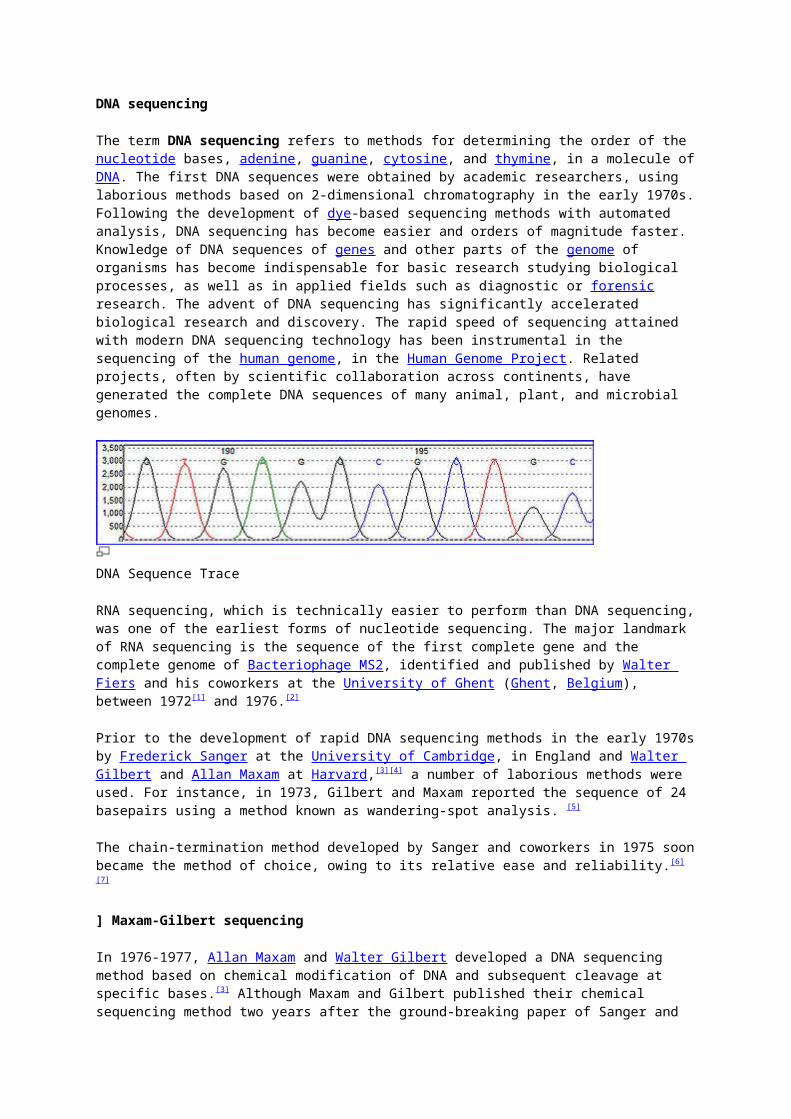
DNA Sequence Trace
RNA sequencing, which is technically easier to perform than DNA sequencing, was one of the earliest forms of nucleotide sequencing. The major landmark of RNA sequencing is the sequence of the first complete gene and the complete genome of Bacteriophage MS2, identified and published by Walter Fiers and his coworkers at the University of Ghent (Ghent, Belgium), between 1972[1] and 1976.[2]
Prior to the development of rapid DNA sequencing methods in the early 1970s by Frederick Sanger at the University of Cambridge, in England and Walter Gilbert and Allan Maxam at Harvard,[3][4] a number of laborious methods were used. For instance, in 1973, Gilbert and Maxam reported the sequence of 24 basepairs using a method known as wandering-spot analysis. [5]
The chain-termination method developed by Sanger and coworkers in 1975 soon became the method of choice, owing to its relative ease and reliability.[6][7]
] Maxam-Gilbert sequencing
In 1976-1977, Allan Maxam and Walter Gilbert developed a DNA sequencing method based on chemical modification of DNA and subsequent cleavage at specific bases.[3] Although Maxam and Gilbert published their chemical sequencing method two years after the ground-breaking paper of Sanger and Coulson on plus-minus sequencing,[6][8] Maxam-Gilbert sequencing rapidly became more popular, since purified DNA could be used directly, while the initial Sanger method required that each read start be cloned for production of single-stranded DNA. However, with the improvement of the chain-termination method (see below), Maxam-Gilbert sequencing has fallen out of favour due to its technical complexity prohibiting its use in standard molecular biology kits, extensive use of hazardous chemicals, and difficulties with scale-up.
The method requires radioactive labelling at one end and purification of the DNA fragment to be sequenced. Chemical treatment generates breaks at a small proportion of one or two of the four nucleotide bases in each of four reactions (G, A+G, C, C+T). Thus a series of labelled fragments is generated, from the radiolabelled end to the first 'cut' site in each molecule. The fragments in the four reactions are arranged side by side in gel electrophoresis for size separation. To visualize the fragments, the gel is exposed to X-ray film for autoradiography, yielding a series of dark bands each corresponding to a radiolabelled DNA fragment, from which the sequence may be inferred.

Also sometimes known as 'chemical sequencing', this method originated in the study of DNA-protein interactions (footprinting), nucleic acid structure and epigenetic modifications to DNA, and within these it still has important applications.
Chain-termination methods
Part of a radioactively labelled sequencing gel
Because the chain-terminator method (or Sanger method after its developer Frederick Sanger) is more efficient and uses fewer toxic chemicals and lower amounts of radioactivity than the method of Maxam and Gilbert, it rapidly became the method of choice. The key principle of the Sanger method was the use of dideoxynucleotide triphosphates (ddNTPs) as DNA chain terminators.
The classical chain-termination method requires a single-stranded DNA template, a DNA primer, a DNA polymerase, radioactively or fluorescently labeled nucleotides, and modified nucleotides that terminate DNA strand elongation. The DNA sample is divided into four separate sequencing reactions, containing all four of the standard deoxynucleotides (dATP, dGTP, dCTP and dTTP) and the DNA polymerase. To each reaction is added only one of the four dideoxynucleotides (ddATP, ddGTP, ddCTP, or ddTTP) which are the chain-terminating nucleotides, lacking a 3'-OH group required for the formation of a phosphodiester bond between two nucleotides, thus terminating DNA strand extension and resulting in various DNA fragments of varying length.
The newly synthesized and labeled DNA fragments are heat denatured, and separated by size (with a resolution of just one nucleotide) by gel electrophoresis on a denaturing polyacrylamide-urea gel with each of the four reactions run in one of four individual lanes (lanes A, T, G, C); the DNA bands are then visualized by autoradiography or UV light, and the DNA sequence can be directly read off the X-ray film or gel image. In the image on the right, X-ray film was exposed to the gel, and the dark bands correspond to DNA fragments of different lengths. A dark band in a lane indicates a DNA fragment that is the result of chain termination after incorporation of a dideoxynucleotide (ddATP, ddGTP, ddCTP, or ddTTP). The relative positions of the different bands among the four lanes are then used to read (from bottom to top) the DNA sequence.

DNA fragments are labeled with a radioactive or fluorescent tag on the primer (1), in the new DNA strand with a labeled dNTP, or with a labeled ddNTP. (click to expand)
Technical variations of chain-termination sequencing include tagging with nucleotides containing radioactive phosphorus for radiolabelling, or using a primer labeled at the 5’ end with a fluorescent dye. Dye-primer sequencing facilitates reading in an optical system for faster and more economical analysis and automation. The later development by Leroy Hood and coworkers [9][10] of fluorescently labeled ddNTPs and primers set the stage for automated, high-throughput DNA sequencing.
Sequence ladder by radioactive sequencing compared to fluorescent peaks (click to expand)
Chain-termination methods have greatly simplified DNA sequencing. For example, chain-termination-based kits are commercially available that contain the reagents needed for sequencing, pre-aliquoted and ready to use. Limitations include non-specific binding of the primer to the DNA, affecting accurate read-out of the DNA sequence, and DNA secondary structures affecting the fidelity of the sequence.
[edit] Dye-terminator sequencing

Capillary electrophoresis (click to expand)
Dye-terminator sequencing utilizes labelling of the chain terminator ddNTPs, which permits sequencing in a single reaction, rather than four reactions as in the labelled-primer method. In dye-terminator sequencing, each of the four dideoxynucleotide chain terminators is labelled with fluorescent dyes, each of which with different wavelengths of fluorescence and emission. Owing to its greater expediency and speed, dye-terminator sequencing is now the mainstay in automated sequencing. Its limitations include dye effects due to differences in the incorporation of the dye-labelled chain terminators into the DNA fragment, resulting in unequal peak heights and shapes in the electronic DNA sequence trace chromatogram after capillary electrophoresis (see figure to the right). This problem has been addressed with the use of modified DNA polymerase enzyme systems and dyes that minimize incorporation variability, as well as methods for eliminating "dye blobs". The dye-terminator sequencing method, along with automated high-throughput DNA sequence analyzers, is now being used for the vast majority of sequencing projects.
[edit] Challenges
Common challenges of DNA sequencing include poor quality in the first 15-40 bases of the sequence and deteriorating quality of sequencing traces after 700-900 bases. Base calling software typically gives an estimate of quality to aid in quality trimming.
In cases where DNA fragments are cloned before sequencing, the resulting sequence may contain parts of the cloning vector. In contrast, PCR-based cloning and emerging sequencing technologies based on pyrosequencing often avoid using cloning vectors.
[edit] Automation and sample preparation
View of the start of an example dye-terminator read (click to expand)
Automated DNA-sequencing instruments (DNA sequencers) can sequence up to 384 DNA samples in a single batch (run) in up to 24 runs a day. DNA sequencers carry out capillary electrophoresis for size separation, detection and recording of dye fluorescence, and data output as fluorescent peak trace chromatograms. Sequencing reactions by thermocycling, cleanup and re-suspension in a buffer solution before loading onto the sequencer are performed separately. A number of commercial and non-commercial software packages can trim low-quality DNA traces automatically. These programs score the quality of each peak and remove low-quality base peaks (generally located at the ends of the sequence). The accuracy of such algorithms is below visual examination by a human operator, but sufficient for automated processing of large sequence data sets.
[edit] Large-scale sequencing strategies
Current methods can directly sequence only relatively short (300-1000 nucleotides long) DNA fragments in a single reaction.[11] The main obstacle to sequencing DNA fragments above this size limit is insufficient power of separation for resolving large DNA fragments that differ in length by only one nucleotide.

Genomic DNA is fragmented into random pieces and cloned as a bacterial library. DNA from individual bacterial clones is sequenced and the sequence is assembled by using overlapping DNA regions.(click to expand)
Large-scale sequencing aims at sequencing very long DNA pieces, such as whole chromosomes. Common approaches consist of cutting (with restriction enzymes) or shearing (with mechanical forces) large DNA fragments into shorter DNA fragments. The fragmented DNA is cloned into a DNA vector, and amplified in Escherichia coli. Short DNA fragments purified from individual bacterial colonies are individually sequenced and assembled electronically into one long, contiguous sequence. This method does not require any pre-existing information about the sequence of the DNA and is referred to as de novo sequencing. Gaps in the assembled sequence may be filled by primer walking. The different strategies have different tradeoffs in speed and accuracy; shotgun methods are often used for sequencing large genomes, but its assembly is complex and difficult, particularly with sequence repeats often causing gaps in genome assembly.
New sequencing methods
High-throughput sequencing
The high demand for low-cost sequencing has driven the development of high-throughput sequencing technologies that parallelize the sequencing process, producing thousands or millions of sequences at once.[12][13] High-throughput sequencing technologies are intended to lower the cost of DNA sequencing beyond what is possible with standard dye-terminator methods.
In vitro clonal amplification
Molecular detection methods are not sensitive enough for single molecule sequencing, so most approaches use an in vitro cloning step to amplify individual DNA molecules. Emulsion PCR isolates individual DNA molecules along with primer-coated beads in aqueous droplets within an oil phase. Polymerase chain reaction (PCR) then coats each bead with clonal copies of the DNA molecule followed by immobilization for later sequencing. Emulsion PCR is used in the methods by Marguilis et al. (commercialized by 454 Life Sciences), Shendure and Porreca et al. (also known as "polony sequencing") and SOLiD sequencing, (developed by Agencourt, now Applied Biosystems).[14][15][16] Another method for in vitro clonal amplification is bridge PCR, where fragments are amplified upon primers attached to a solid surface. The single-molecule method developed by Stephen Quake's laboratory (later commercialized by Helicos) skips this amplification step, directly fixing DNA molecules to a surface.[17]
Parallelized sequencing
DNA molecules are physically bound to a surface, and sequenced in parallel.Sequencing by synthesis, like dye-termination electrophoretic sequencing, uses a DNA polymerase to determine the base sequence. Reversible terminator methods (used by Illumina and Helicos) use reversible versions of dye-terminators, adding one nucleotide at a time, detect fluorescence at each position in real time, by repeated removal of the blocking group

to allow polymerization of another nucleotide. Pyrosequencing (used by 454) also uses DNA polymerization, adding one nucleotide species at a time and detecting and quantifying the number of nucleotides added to a given location through the light emitted by the release of attached pyrophosphates.[14][18]
Sequencing by ligation
This enzymatic sequencing method uses a DNA ligase to determine the target sequence.[15][16][19] Used in the polony method and in the SOLiD technology, it uses a pool of all possible oligonucleotides of a fixed length, labeled according to the sequenced position. Oligonucleotides are annealed and ligated; the preferential ligation by DNA ligase for matching sequences results in a signal informative of the nucleotide at that position.
Microfluidic Sanger Sequencing
In microfluidic Sanger sequencing the entire thermocycling amplification of DNA fragments as well as their separation by electrophoresis is done on a single chip (approximately 100 cm in diameter) thus reducing the reagent usage as well as cost.[citation needed] In some instances researchers[who?] have shown that they can increase the through-put of conventional sequencing through the use of microchips.[citation needed] Research will still need to be done in order to make this use of technology effective.
Other sequencing technologies
Sequencing by hybridization is a non-enzymatic method that uses a DNA microarray. A single pool of DNA whose sequence is to be determined is fluorescently labeled and hybridized to an array containing known sequences. Strong hybridization signals from a given spot on the array identifies its sequence in the DNA being sequenced.[20] Mass spectrometry may be used to determine mass differences between DNA fragments produced in chain-termination reactions.[21]
DNA sequencing methods currently under development include labeling the DNA polymerase,[22] reading the sequence as a DNA strand transits through nanopores,[23][24] and microscopy-based techniques, such as AFM or electron microscopy that are used to identify the positions of individual nucleotides within long DNA fragments (>5,000 bp) by nucleotide labeling with heavier elements (e.g., halogens) for visual detection and recording.[25]
In October 2006, the X Prize Foundation established an initiative to promote the development of full genome sequencing technologies, called the Archon X Prize, intending to award $10 million to "the first Team that can build a device and use it to sequence 100 human genomes within 10 days or less, with an accuracy of no more than one error in every 100,000 bases sequenced, with sequences accurately covering at least 98% of the genome, and at a recurring cost of no more than $10,000 (US) per genome."[26]
Sanger sequencing

Part of a radioactively labelled sequencing gel
In chain terminator sequencing (Sanger sequencing), extension is initiated at a specific site on the template DNA by using a short oligonucleotide 'primer' complementary to the template at that region. The oligonucleotide primer is extended using a DNA polymerase, an enzyme that replicates DNA. Included with the primer and DNA polymerase are the four deoxynucleotide bases (DNA building blocks), along with a low concentration of a chain terminating nucleotide (most commonly a di-deoxynucleotide). Limited incorporation of the chain terminating nucleotide by the DNA polymerase results in a series of related DNA fragments that are terminated only at positions where that particular nucleotide is used. The fragments are then size-separated by electrophoresis in a slab polyacrylamide gel, or more commonly now, in a narrow glass tube (capillary) filled with a viscous polymer.
View of the start of an example dye-terminator read (click to expand)
An alternative to the labelling of the primer is to label the terminators instead, commonly called 'dye terminator sequencing'. The major advantage of this approach is the complete sequencing set can be performed in a single reaction, rather than the four needed with the labeled-primer approach. This is accomplished by labelling each of the dideoxynucleotide chain-terminators with a separate fluorescent dye, which fluoresces at a different wavelength. This method is easier and quicker than the dye primer approach, but may produce more uneven data peaks (different heights), due to a template dependent difference in the incorporation of the large dye chain-terminators. This problem has been significantly reduced with the introduction of new enzymes and dyes that minimize incorporation variability.
This method is now used for the vast majority of sequencing reactions as it is both simpler and cheaper. The major reason for this is that the primers do not have to be separately labelled (which can be a significant expense for a single-use custom primer), although this is less of a concern with frequently used 'universal' primers.
Sanger Method for DNA Sequencing
DNA sequencing, first devised in 1975, has become a powerful technique in molecular biology, allowing analysis of genes at the nucleotide level. For this reason, this tool has been applied to many areas of research.

For example, the polymerase chain reaction (PCR), a method which rapidly produces numerous copies of a desired piece of DNA, requires first knowing the flanking sequences of this piece. Another important use of DNA sequencing is identifying restriction sites in plasmids. Knowing these restriction sites is useful in cloning a foreign gene into the plasmid. Before the advent of DNA sequencing, molecular biologists had to sequence proteins directly; now amino acid sequences can be determined more easily by sequencing a piece of cDNA and finding an open reading frame. In eukaryotic gene expression, sequencing has allowed researchers to identify conserved sequence motifs and determine their importance in the promoter region. Furthermore, a molecular biologist can utilize sequencing to identify the site of a point mutation. These are only a few examples illustrating the way in which DNA sequencing has revolutionized molecular biology.
Dideoxynucleotide sequencing represents only one method of sequencing DNA. It is commonly called Sanger sequencing since Sanger devised the method. This technique utilizes 2',3'-dideoxynucleotide triphospates (ddNTPs), molecules that differ from deoxynucleotides by the having a hydrogen atom attached to the 3' carbon rather than an OH group. (Figure 1). These molecules terminate DNA chain elongation because they cannot form a phosphodiester bond with the next deoxynucleotide.
In order to perform the sequencing, one must first convert double stranded DNA into single stranded DNA. This can be done by denaturing the double stranded DNA with NaOH. A Sanger reaction consists of the following: a strand to be sequenced (one of the single strands which was denatured using NaOH), DNA primers (short pieces of DNA that are both complementary to the strand which is to be sequenced and radioactively labelled at the 5' end), a mixture of a particular ddNTP (such as ddATP) with its normal dNTP (dATP in this case), and the other three dNTPs (dCTP, dGTP, and dTTP). The concentration of ddATP should be 1% of the concentration of dATP. The logic behind this ratio is that after DNA polymerase is added, the polymerization will take place and will terminate whenever a ddATP is incorporated into the growing strand. If the ddATP is only 1% of the total concentration of dATP, a whole series of labeled strands will result (Figure 1). Note that the lengths of these strands are dependent on the location of the base relative to the 5' end.
This reaction is performed four times using a different ddNTP for each reaction. When these reactions are completed, a polyacrylamide gel electrophoresis (PAGE) is performed. One reaction is loaded into one lane for a total of four lanes (Figure 2). The gel is transferred to a nitrocellulose filter and autoradiography is performed so that only the bands with the radioactive label on the 5' end will appear. In PAGE, the shortest fragments will migrate the farthest. Therefore, the bottom-most band indicates that its particular dideoxynucleotide was added first to the labeled primer. In Figure 2, for example, the band that migrated the farthest was in the ddATP reaction mixture. Therefore, ddATP must have been added first to the primer, and its complementary base, thymine, must have been the base present on the 3' end of the sequenced strand. One can continue reading in this fashion. Note in Figure 2 that if one reads the bases from the bottom up, one is reading the 5' to 3' sequence of the strand complementary to the sequenced strand. The sequenced strand can be read 5' to 3' by reading top to bottom the bases complementary to the those on the gel.

Figure 1. This figure shows the structure of a dideoxynucleotide (notice the H atom attached to the 3' carbon). Also depicted in this figure are the ingredients for a Sanger reaction. Notice the different lengths of labeled strands produced in this reaction.
Figure 2. This figure is a representation of an acrylamide sequencing gel. Notice that the sequence of the strand of DNA complementary to the sequenced strand is 5' to 3' ACGCCCGAGTAGCCCAGATT while the sequence of the sequenced strand, 5' to 3', is AATCTGGGCTACTCGGGCGT.

Figure 7-29. Sanger (dideoxy) method for sequencing DNA fragments. (a) A single strand of the DNA to be sequenced (blue line) is hybridized to a 5′-end-labeled synthetic deoxyribonucleotide primer. The primer is elongated in four separate reaction mixtures containing the four normal deoxyribonucleoside triphosphates (dNTPs) plus one of the four dideoxyribonucleoside triphosphates (ddNTPs) in a ratio of 100 to 1. A ddNTP molecule can add at the position of the corresponding normal dNTP, but when this occurs, chain elongation stops because the ddNTP lacks a 3′ hydroxyl. In time, each reaction mixture will contain a mixture of prematurely terminated chains ending at every occurrence of the ddNTP (yellow). (b) Three of the labeled chains that would be generated in the presence of ddGTP from the specific DNA sequence shown in blue. (c) An actual autoradiogram of a polyacrylamide gel in which more than 300 bases can be read. Each reaction was carried out in duplicate using Sequenase™, a commercial preparation of the DNA polymerase from bacteriophage T7. [Part (c) courtesy of United States Biochemical Corporation.]
Microfluidic Sanger Sequencing
The completion of the Human Genome Project (HGP) has been a cornerstone in the advancement of biological studies. The outcomes of obtaining a complete reference map (including the sequence) of the human genome have ushered in the post-genome era of studies. Genomics will (if it hasn’t already) revolutionize medicine, forensics, molecular biology, biotechnology, and many other related and even unrelated disciplines in the future.[1][2]
Sequencing of DNA has largely been based on dideoxy chain termination developed by Sanger et al. [3]. However, the ability of the HGP in obtaining the full human genomic sequence meant that modifications were

required to be made to this method. In particular, the incorporation of technological innovation, making sequencing automated and high-throughput, made this decade-long worldwide effort successful [4].
Briefly, in its modern inception, high-throughput genome sequencing (also referred to as Whole Genome Shot-gun Sequencing) involves fragmenting the genome into small single-stranded pieces, followed by amplification of the fragments by Polymerase Chain Reaction (PCR). Adopting the Sanger method, each DNA fragment is irreversibly terminated with the incorporation of a fluorescently labeled dideoxy chain-terminating nucleotide, thereby producing a DNA “ladder” of fragments that each differ in length by one base and bear a base-specific fluorescent label at the terminal base. Amplified base ladders are then separated by Capillary Array Electrophoresis (CAE) with automated, in situ “finish-line” detection of the fluorescently labeled ssDNA fragments, which provides an ordered sequence of the fragments. These sequence reads are then computer assembled into overlapping or contiguous sequences (termed "contigs") which resemble the full genomic sequence once fully assembled.[5]
Rapid technological developments have now emerged as a result of the HGP. In particular Massively Parallel Sequencing approaches such as those now in wide commercial use (Illumins/Solexa, Roche/454 Pyrosequencing, and ABI SOLiD) are proving to be attractive tools for sequencing.
Typically MPS methods can only obtain short read lengths (35bp with Illumina platforms to a maximum of 200-300bp by 454 Pyrosequencing). Sanger Methods on the other hand achieve read lengths of approximately 800bp (typically 500-600bp with non-enriched DNA). The longer read lengths in Sanger methods display significant advantages over MPS tools especially in terms of sequencing repetitive regions of the genome. A challenge of short-read sequence data is particularly an issue in sequencing new genomes (de novo) and in sequencing highly rearranged genome segments, typically those seen of cancer genomes or in regions of chromosomes that exhibit structural variation.[6]
Microfluidic Sanger Sequencing
Microfluidic Sanger sequencing is a lab-on-a-chip application for DNA sequencing, in which the Sanger sequencing steps (thermal cycling, sample purification, and capillary electrophoresis) are integrated on a wafer-scale chip using nanoliter-scale sample volumes. This technology generates long and accurate sequence reads, while obviating many of the significant shortcomings of the conventional Sanger method (e.g. high consumption of expensive reagents, reliance on expensive equipment, personnel-intensive manipulations, etc.) by integrating and automating the Sanger sequencing steps.
Applications of Microfluidic Sequencing Technologies
Other useful applications of DNA sequencing include single nucleotide polymorphism (SNP) detection, single-strand conformation polymorphism (SSCP) hetroduplex analysis, and short tandem repeat (STR) analysis. Resolving DNA fragments according to differences in size and/or conformation is the most critical step in studying these features of the genome[5].
Device design
A microfluidic sequencing chip developed by Richard Mathies and colleagues (University of California, Berkeley)[7].
The sequencing chip has a four-layer construction, consisting of three 100-mm-diameter glass wafers (on which device elements are microfabricated) and a polydimethylsiloxane (PDMS) membrane. Reaction chambers and capillary electrophoresis channels are etched between the top two glass wafers, which are thermally bonded. Three-dimensional channel interconnections and microvalves are formed by the PDMS and bottom manifold glass wafer.
The device consists of three functional units, each corresponding to the Sanger sequencing steps. The Thermal Cycling (TC) unit is comprised of a 250-nanoliter reaction chamber with integrated resistive temperature

detector, microvalves, and a surface heater. Movement of reagent between the top all-glass layer and the lower glass-PDMS layer occurs through 500-μm-diameter via-holes. After thermal-cycling, the reaction mixture undergoes purification in the capture/purification chamber, and then is injected into the capillary electrophoresis (CE) chamber. The CE unit consists of a 30-cm capillary which is folded into a compact switchback pattern via 65-μm-wide turns.
Sequencing chemistry
Thermal cycling
In the TC reaction chamber, dye-terminator sequencing reagent, template DNA, and primers are loaded into the TC chamber and thermal-cycled for 35 cycles ( at 95°C for 12 seconds and at 60°C for 55 seconds).
Purification
The charged reaction mixture (containing extension fragments, template DNA, and excess sequencing reagent) is conducted through a capture/purification chamber at 30°C via a 33-Volts/cm electric field applied between capture outlet and inlet ports. The capture gel through which the sample is driven, consists of 40 μM of oligonucleotide (complementary to the primers) covalently bound to a polyacrylamide matrix. Extension fragments are immobilized by the gel matrix, and excess primer, template, free nucleotides, and salts are eluted through the capture waste port. The capture gel is heated to 67-75°C to release extension fragments.
Capillary electrophoresis
Extension fragments are injected into the CE chamber where they are electrophoresed through a 125-167-V/cm field.
Platforms
The Apollo 100 platform (Microchip Biotechnologies Inc., Dublin, CA)[8] integrates the first two Sanger sequencing steps (thermal cycling and purification) in a fully automated system. The manufacturer claims that samples are ready for capillary electrophoresis within three hours of the sample and reagents being loaded into the system. The Apollo 100 platform requires sub-microliter volumes of reagents.
Comparisons to other sequencing techniques
The ultimate goal of high-throughput sequencing is to develop systems that are low-cost, and extremely efficient at obtaining extended (longer) read lengths. Longer read lengths of each single electrophoretic separation, substantially reduces the cost associated with de novo DNA

The Sanger Method
By Sarah Obenrader
_____________________________________________
This web page was produced as an assignment for an undergraduate course at Davidson College.
Background Information
DNA sequencing enables us to perform a thorough analysis of DNA because it provides us with the most basic information of all: the sequence of nucleotides. With this knowledge, for example, we can locate regulatory and gene sequences, make comparisons between homologous genes across species and identify mutations. Scientists recognized that this could potentially be a very powerful tool, and so there was competition to create a method that would sequence DNA. Then in 1974, two methods were independently developed by an American team and an English team to do exactly this. The Americans, lead by Maxam and Gilbert, used a “chemical cleavage protocol”, while the English, lead by Sanger, designed a procedure similar to the natural process of DNA replication. Even though both teams shared the 1980 Nobel Prize, Sanger’s method became the standard because of its practicality (Speed, 1992).
Sanger’s method, which is also referred to as dideoxy sequencing or chain termination, is based on the use of dideoxynucleotides (ddNTP’s) in addition to the normal nucleotides (NTP’s) found in DNA. Dideoxynucleotides are essentially the same as nucleotides except they contain a hydrogen group on the 3’ carbon instead of a hydroxyl group (OH). These modified nucleotides, when integrated into a sequence, prevent the addition of further nucleotides. (Speed, 1992).This occurs because a phosphodiester bond cannot form between the dideoxynucleotide and the next incoming nucleotide, and thus the DNA chain is terminated.
The Method
Before the DNA can be sequenced, it has to be denatured into single strands using heat. Next a primer is annealed to one of the template strands. This primer is specifically constructed so that its 3' end is located next to the DNA sequence of interest. Either this primer or one of the nucleotides should be radioactively or fluorescently labeled so that the final product can be detected on a gel (Russell, 2002). Once the primer is

attached to the DNA, the solution is divided into four tubes labeled "G", "A", "T" and "C". Then reagents are added to these samples as follows:
"G" tube: all four dNTP's, ddGTP and DNA polymerase
"A" tube: all four dNTP's, ddATP and DNA polymerase
"T" tube: all four dNTP's, ddTTP and DNA polymerase
"C" tube: all four dNTP's, ddCTP and DNA polymerase
As shown above, all of the tubes contain a different ddNTP present, and each at about one-hundreth the concentration of the the normal precursors (Russell, 2002). As the DNA is synthesized, nucleotides are added on to the growing chain by the DNA polymerase. However, on occasion a dideoxynucleotide is incorporated into the chain in place of a normal nucleotide, which results in a chain-terminating event. For example if we looked at only the "G" tube, we might find a mixture of the following products:
Figure 1: An example of the potential fragments that could be produced in the "G" tube. The fragments are all different lengths due to the random integration of the ddGTP's (Metzenberg).
The key to this method, is that all the reactions start from the same nucleotide and end with a specific base. Thus in a solution where the same chain of DNA is being synthesized over and over again, the new chain will terminate at all positions where the nucleotide has the potential to be added because of the integration of the dideoxynucleotides (Russell, 2002). In this way, bands of all different lengths are produced. Once these reactions are completed, the DNA is once again denatured in preparation for electrophoresis. The contents of each of the four tubes are run in separate lanes on a polyacrylmide gel in order to separate the different sized bands from one another. After the contents have been run across the gel, the gel is then exposed to either UV light or X-Ray, depending on the method used for labeling the DNA.

Figure 2: This is a polyacrylmide gel of the reactions in the "G" tube (the same sequences seen in figure 1). The longer fragments of DNA traveled shorter distances than the smaller fragments because of their heavier molecular weight.The blue section indicates the primer, the black section indicates the newly synthesized strand and the red denotes a ddGTP, which terminated the chain (Metzenberg).
As shown in Figure 2, smaller fragments are produced when the ddNTP is added closer to the primer because the chains are smaller and therefore migrate faster across the gel. If all of the reactions from the four tubes are combined on one gel, the actual DNA sequence in the 5' to 3' direction can be determined by reading the banding pattern from the bottom of the gel up. It is important to remember though that this sequence is complementary to the template strand from the beginning.
Figure 3: This is an autoradiogram of a dideoxy sequencing gel. The letters over the lanes indicate which dideoxy nucleotide was used in the sample being represented by that lane. When you read from the bottom up, you are reading the complementary sequence of the template strand (Metzenberg).
Automated Sequencing
With the many advancements in technology that we have achieved since 1974, it is no surprise that the Sanger method has become outdated. However, the new technology that has emerged to replace this method is based on the same principles of Sanger's method. Automated sequencing has been developed so that more DNA can be sequenced in a shorter period of time. With the automated procedures the reactions are performed in a single tube containing all four ddNTP's, each labeled with a different color dye (Russell, 2002).

Figure 4: In automated sequencing, the oligonucleotide primers can be "end-labeled" with different color dyes, one for each ddNTP. These dyes fluoresce at different wavelengths, which are read via a machine (Metzenberg).
As in Sanger's method, the DNA is separated on a gel, but they are all run on the same lane as opposed to four different ones.
Figure 5: Results of gel electrophoresis for the dye labeled DNA in automated sequencing. The image on the left shows what the gel looks like if the four reactions are run in different lanes, as opposed to the image on the right which shows a gel where all the DNA is run in one lane (Metzenberg).
Since the four dyes fluoresce at different wavelengths, a laser then reads the gel to determine the identity of each band according to the wavelengths at which it fluoresces. The results are then depicted in the form of a chromatogram, which is a diagram of colored peaks that correspond to the nucleotide in that location in the sequence (Russell, 2002).

Figure 6: Results from an automated sequence shown in the form of a chromatogram. The colors represent the four bases: blue is C, green is A, black is G and red is T (Metzenberg).
Sanger Dideoxy Method
This method is based on DNA replication. DNA replication terminated at different sites will produce DNA fragments of variable lengths.
Controlled termination at specific sites can be achieved with the use of 2’,3’-dideoxy analog of the four nucleotides
These analogs lack the 3’- OH group required to form the next phosphodiester bond with the incoming nucleotide.
DNA replication terminates at the site where a dideoxy analog is incorporated.
DNA replication is performed in four separate tubes, each containing:
i. Single stranded DNA to be sequenced

ii. DNA polymerase
iii. Primers
iv. The four dNTPs (dATP, dCTP, dTTP and dGTP)
v. Small amount of one of the four 2’,3’-dideoxy analog (ddATP or ddCTP or ddTTP or ddGTP)
Either the primers or the dNTPs are radiolabeled with 32P .
The amount of dideoxy analog added is small enough (~1% of total dNTP) that termination will occur only occasionally.
The correct nucleotide will be inserted sometimes and the dideoxy analog other times.
In this way, all possible DNA fragments will be produced.
http://www.mcb.mcgill.ca/~hallett/GEP/Lecture15/Image31.gif
The products of all four reactions will be separated by gel electrophoresis in four separate lanes.
Polyacrylamide gels are used to separate fragments containing up to 1000bp.
More porous agarose gels are used to resolve mixtures of larger fragments, up to 20kb.

Smaller DNA fragments runs faster (towards the positive electrode as DNA is negatively charged) and appear at the bottom of the gel.
The base sequence of the new DNA is read from the autoradiogram of the gel in 5’ 3’direction starting from the smallest fragment.
Fluorescent Detection of Oligonucleotides
Fluorescent detection is a highly effective alternative for visualizing DNA.
This method eliminates the use of radioactive reagents and can be readily automated.
Either fluorescent-tagged terminators (dideoxy analogs) or florescent-tagged primers can be used.
When using fluorescent-tagged terminators, each of the four dideoxy nucleotides should carry a tag with a different color.
If fluorescent-tagged primer is the choice, the primers in each of the four separate mixtures should carry tags of different colors.
The DNA mixture will be separated by gel electrophoresis.
Lasers will be used to activate the fluorescent dideoxy analogs or primers and a detector to distinguish the colors.
The last base incorporated into the DNA can be determined from the color of the detected DNA fragment.
Adapted from: http://www.licor.com/bio/Images/IR2Schem.jpg

http://www.dls.ym.edu.tw/ol_biology2/ultranet/FluorDideoxySeq.gif
New Developments
More robust and high-throughput methods are currently being developed to meet the need of whole genome projects, where millions of bases need to be sequenced.
One of the growing techniques developed is pyrosequencing.
Pyrosequencing is based on the detection of released pyrophosphate (PPi) during DNA synthesis.
DNA template can be immobilized on a solid phase and the four nucleotides are added in a stepwise fashione; only one of the four dNTP in the reaction mixture.
If the nucleotide is incorporated, PPi is released.
Subsequent addition of ATP sulfurylase converts PPi to ATP, which provides energy for luciferase to generate light.
The light is easily detected by a photodiode, photomultiplier tube, or a charge-coupled device camera (CCD) camera.
Because the added nucleotide is known, the sequence of nucleotide can be determined.

Genome Research Vol. 11, Issue 1, 3-11, January 2001


The methods described in this chapter provide some useful approaches for DNA sequencing of templates produced by PCR. These procedures have been employed successfully for large-scale DNA sequencing of cosmid fragments subcloned in plasmid or M13 vectors, and for sequence analysis of cDNAs cloned in bacteriophage lambda vectors. In addition, the method describing direct sequencing from PEG-precipitated PCR product has been used successfully for analysis of Caenorhabditis elegans genomic and cDNA sequences. It is important to reiterate that for every combination of amplification primer pair and target DNA, there is an optimal method for PCR amplification; the ability to sequence the products of any PCR experiment directly will also vary. A coupled PCR/DNA sequencing method that works well for one experimental system may work

quite poorly with others. Hence, a few days or hours spent optimizing PCR amplification conditions and selecting the best DNA sequencing method for the target DNA of interest will be time well spent.
Radiolabeled sequencing gel preparation, loading, and electrophoresis (26,29)
To prepare polyacrylamide gels for DNA sequencing, the appropriate amount of urea is dissolved by heating in water and electrophoresis buffer, the respective amount of deionized acrylamide-bisacrylamide solution is added, and ammonium persulfate and TEMED are added to initiate polymerization. Immediately after the addition of the polymerizing agents, the gel solution is poured between two glass plates, taped together and separated by thin spacers corresponding to the desired thickness of the gel, taking care to avoid and eliminate air bubbles. Prior to taping, these glass plates are cleaned with Alconox detergent and hot water, are rinsed with double distilled water, and dried with a Kimwipe. Typically, the notched glass plate is treated with a silanizing reagent and then rinsed with double distilled water. After pouring, the gel immediately is laid horizontally and a well forming comb is inserted into the gel and held in place by metal clamps. The polyacrylamide gels are allowed to polymerize for at least 30 minutes prior to use. After polymerization, the comb and the tape at the bottom of the gel are removed. The vertical electrophoresis apparatus is assembled by clamping the top and bottom buffer wells onto the gel, and adding running buffer to the buffer chambers. The wells are cleaned by circulating buffer into the wells with a syringe and, immediately prior to the loading of each sample, the urea in each well is suctioned out with a mouth pipette.
Each base-specific sequencing reaction terminated with the short termination mix is loaded using a mouth pipette onto a 0.15 mm X 50 cm X 20 cm, denaturing 5% polyacrylamide gel and electrophoresed for 2.25 hours at 22 mA. The reactions terminated with the long termination mix typically are divided in half and loaded onto two 0.15 mm X 70 cm X 20 cm denaturing 4% polyacrylamide gels. One gel is electrophoresed at 15 mA for 8-9 hours and the other is electrophoresed for 20-24 hours at 15 mA. After electrophoresis, the glass plates are separated and the gel is blotted to Whatman paper, covered with plastic wrap, dried by heating on a Hoefer vacuum gel drier, and exposed to X-ray film. Depending on the intensity of the signal and whether the radiolabel is 32-P or 35-S, exposure times varied from 4 hours to several days. After exposure, the films are developed by processing in developer and fixer solutions, rinsed with water, and air dried. The autoradiogram then is placed on a light-box and the sequence is manually read and the data typed into a computer.
C. Taq-polymerase catalyzed cycle sequencing using fluorescent-labeled dye primers (10,26)
Each base-specific fluorescent-labeled cycle sequencing reaction routinely included approximately 100 or 200 ng Biomek isolated single-stranded DNA for A and C or G and T reactions, respectively. Double-stranded cycle sequencing reactions similarly contained approximately 200 or 400 ng of plasmid DNA, isolated using either the standard alkaline lysis or the diatomaceous earth modified alkaline lysis procedures. All reagents except template DNA are added in one pipetting step from a premix of previously aliquotted stock solutions stored at -20degC (see Appendix B). To prepare the reaction premixes, reaction buffer is combined with the base-specific nucleotide mixes. Prior to use, the base-specific reaction premixes are thawed and combined with diluted Taq DNA polymerase and the individual fluorescent end-labeled universal primers (see Appendix C) to yield the final reaction mixes, that are sufficient for 24 template samples.
Once the above mixes are prepared, four aliquots of single or double-stranded DNA are pipetted into the bottom of each 0.2 ml thin-walled reaction tube, corresponding to the A, C, G, and T reactions, and then an aliquot of the respective reaction mixes is added to the side of each tube. These tubes are part of a 96-tube/retainer set tray in a microtiter plate format, which fits into a Perkin Elmer Cetus Cycler 9600. Strip caps are sealed onto the tube/retainer set and the plate is centrifuged briefly. The plate then is placed in the cycler whose heat block had been preheated to 95deg C, and the cycling program immediately started. The cycling protocol consisted of 15-30 cycles of seven-temperatures:
95degC denaturation
55degC annealing
72degC extension
95degC denaturation

72degC extension
95degC denaturation, and
72degC extension, linked to a 4deg C final soak file.
At this stage, the reactions frequently are frozen and stored at -20degC for up to several days. Prior to pooling and precipitation, the plate is centrifuged briefly to reclaim condensation. The primer and base-specific reactions are pooled into ethanol, and the DNA is precipitated and dried. These sequencing reactions could be stored for several days at -20degC.
D. Taq-polymerase catalyzed cycle sequencing using fluorescent-labeled dye terminator reactions
One of the major problems in DNA cycle sequencing is that when fluorescent primers (1) are used the reaction conditions are such that the nested fragment set distribution is highly dependent upon the template concentration in the reaction mix. We have recently observed that the nested fragment set distribution for the DNA cycle sequencing reactions using the fluorescent labelled terminators (8) is much less sensitive to DNA concentration than that obtained with the fluorescent labelled primer reactions as described above. In addition, the fluorescent terminator reactions require only one reaction tube per template while the fluorescent labelled primer reactions require one reaction tube for each of the four terminators. This latter point allows the fluorescent labelled terminator reactions to be pipetted easily in a 96 well format. The protocol used, as described below, is easily interfaced with the 96 well template isolation and 96 well reaction clean-up procedures also described herein. By performing all three of these steps in a 96 well format, the overall procedure is highly reproducable and therefore less error prone.
E. Sequenase[TM] catalyzed sequencing with dye-labeled terminators (29-32)
Single-stranded dye-terminator reactions required approximately 2 ug of phenol extracted M13-based template DNA. The DNA is denatured and the primer annealed by incubating DNA, primer, and buffer at 65degC. After the reaction cooled to room temperature, alpha-thio-deoxynucleotides, fluorescent-labeled dye-terminators, and diluted Sequenase[TM] DNA polymerase are added and the mixture is incubated at 37degC. The reaction is stopped by adding ammonium acetate and ethanol, and the DNA fragments are precipitated and dried. To aid in the removal of unincorporated dye-terminators, the DNA pellet is rinsed twice with ethanol. The dried sequencing reactions could be stored up to several days at -20degC.
Double-stranded dye-terminator reactions required approximately 5 ug of diatomaceous earth modified-alkaline lysis midi-prep purified plasmid DNA. The double-stranded DNA is denatured by incubating the DNA in sodium hydroxide at 65degC, and after incubation, primer is added and the reaction is neutralized by adding an acid-buffer. Reaction buffer, alpha-thio-deoxynucleotides, fluorescent-labeled dye-terminators, and diluted Sequenase[TM] DNA polymerase then are added and the reaction is incubated at 37degC. Ammonium acetate is added to stop the reaction and the DNA fragments similarly are precipitated, rinsed, dried, and stored.
F. Fluorescent-labeled sequencing gel preparation, pre-electrophoresis, sample loading, electrophoresis, data collection, and analysis on the ABI 373A DNA sequencer
Polyacrylamide gels for fluorescent DNA sequencing are prepared as described above except that the gel mix is filtered prior to polymerization. Optically-ground, low fluorescence glass plates are carefully cleaned with hot water, distilled water, and ethanol to remove potential fluorescent contaminants prior to taping. Denaturing 6% polyacrylamide gels are poured into 0.3 mm X 89 cm X 52 cm taped plates and fitted with 36 well forming combs. After polymerization, the tape and the comb are removed from the gel and the outer surfaces of the glass plates are cleaned with hot water, and rinsed with distilled water and ethanol. The gel is assembled into an ABI sequencer, and the checked by laser-scanning. If baseline alterations are observed on the ABI-associated Macintosh computer display, the plates are recleaned. Subsequently, the buffer wells are attached, electrophoresis buffer is added, and the gel is pre-electrophoresed for 10-30 minutes at 30 W.
Prior to sample loading, the pooled and dried reaction products are resuspended in formamide/EDTA loading buffer by vortexing and then heated at 90degC. A sample sheet is created within the ABI data collection software on the Macintosh computer which indicated the number of samples loaded and the fluorescent-labeled

mobility file to use for sequence data processing. After cleaning the sample wells with a syringe, the odd-numbered sequencing reactions are loaded into the respective wells using a micropipettor equipped with a flat-tipped gel-loading tip. The gel then is electrophoresed for 5 minutes before the wells are cleaned again and the even numbered samples are loaded. The filter wheel used for dye-primers and dye-terminators is specified on the ABI 373A CPU, also where electrophoresis conditions are adjusted. Typically electrophoresis and data collection are for 10 hours at 30W on the ABI 373A that is fitted with a heat-distributing aluminum plate in contact with the outer glass gel plate in the region between the laser stop and the sample loading wells (26).
After data collection, an image file is created by the ABI software which related the fluorescent signal detected to the corresponding scan number. The software then determined the sample lane positions based on the signal intensities. After the lanes are tracked, the cross-section of data for each lane are extracted and processed by baseline subtraction, mobility calculation, spectral deconvolution, and time correction. On the Macintosh computer, the collected data can be viewed in several formats. The overall graphics image of the gel can be displayed to assess the accuracy of lane tracking, and the data from each sample lane can be viewed as either a four-color raw fluorescent signal versus scan number, as a chromatogram of processed sequence data, or as a string of nucleotides. After processing, the sequence data files are transferred to a SPARCstation 2 using NFS Share.
G. Double-stranded sequencing of cDNA clones containing long poly(A) tails using anchored poly(dT) primers
Sequencing double stranded DNA templates has become a common and efficient procedure (10) for rapidly obtaining sequence data while avoiding preparation of single stranded DNA. Double stranded templates of cDNAs containing long poly(A) tracts are difficult to sequence with vector primers which anneal downstream of the poly(A) tail. Sequencing with these primers results in a long poly(T) ladder followed by a sequence which is difficult to read. In an attempt to solve this problem we synthesized three primers which contain (dT)17 and either (dA) or (dC) or (dG) at the 3' end. We reasoned that the presence of these three bases at the 3' end would 'anchor' the primers at the upstream end of the poly(A) tail and allow sequencing of the region immediately upstream of the poly(A) region.
Using this protocol, over 300 bp of readable sequence could be obtained. We have applied this approach to several other poly(A)-containing cDNA clones with similar results. Sequencing of the opposite strand of these cDNAs using insert-specific primers occurred directly upstream of the poly(A) region. The ability to directly obtain sequence immediately upstream from the poly(A) tail of cDNAs should be of particular importance to large scale efforts to generate sequence-tagged sites (STSs) (11) from cDNAs (12,13).
H. cDNA sequencing based on PCR and random shotgun cloning
The following is a rapid and efficient method for sequencing cloned cDNAs based on PCR amplification (14), random shotgun cloning (1,3,15), and automated fluorescent sequencing (16). This method was developed in our laboratory because once the sequence of a genomic DNA containing cosmid is obtained and putative exons are predicted, the corresponding cDNAs should be sequenced in a timely manner. However, the presently implemented directed cDNA sequencing strategies, i.e. primer walking (17) and exonuclease III deletion (18), are both time consuming and labor intensive, while the alternative, i.e. randomly shearing the intact plasmid followed by shotgun sequencing (1,3,15), leads to a significant number of clones containing the original cDNA cloning vector rather than the desired cDNA insert.
This is a PCR-based approach where the "universal" forward and/or reverse priming sites were excluded from the resulting PCR product by choosing a primer pair that lay between the usual "universal" forward and reverse priming sites and the multiple cloning sites of the Stratagene Bluescript vector. These two PCR primers, with the sequence 5'-TCGAGGTCGACGGTATCG-3' for the forward or -16bs primer and 5'-GCCGCTCTAGAACTAG TG-3' for the reverse or +19bs primer, now have been used to amplify sufficient quantities of cDNA inserts in the 1.2 to 3.4 kb size range so that the random shotgun sequencing approach described below could be implemented.
Automated Fluorescent DNA Sequencing - Background
Automated fluorescent DNA sequencing using capillary DNA sequencing machines like the ABI/PRISM 3100 Genetic Analyzers in the CGC is based on the use of a different colored fluorescent dye for each of the four DNA bases. Attaching these dyes to the four dideoxynucleotide terminators used in the standard Sanger chain termination DNA sequencing reaction results in all the fragments ending in one of the four fluorescent dye-labeled terminator corresponding to the dideoxy bases. These fragments are then separated on a liquid denaturing gel pumped into each capillary. The fluorescence is detected as the fragments electrophorese through a transparent section of the capillary which runs in front of a CCD camera, while being excited with laser light. The use of four different dyes allows the sequencing reaction to be performed in a single tube and the resulting fragments to be loaded in a single well, resulting in greater sample capacity per “lane” as compared to what is possible with radioactive labeled fragments. The dyes can also be attached to the primer used to initiate the extension by a DNA polymerase. This method is used less frequently since dye-labeled primers are expensive to manufacture and four separate reactions must be set up for each sequence (they can still be separated in a single lane though). The resulting sequence is, however, generally cleaner than dye-terminator sequencing. The dye-terminator approach can be used with different types of DNA polymerases.
Sequenase (T7) polymerase has been a widely used enzyme for DNA sequencing with radioactive nucleotides because of its high processivity and low error rate. However using this type of enzyme for fluorescent DNA sequencing is not optimal because the amount of fluorescent DNA produced is low and thus a large amount of template is required to produce good fluorescent signals. The enzyme currently in general use for automated fluorescent DNA sequencing is a variant of Taq DNA polymerase. The thermostability of Taq polymerase allows the sequencing reactions to be carried out like PCR reactions, and the reactions are called "cycle sequencing" reactions. Cycle sequencing reactions are analogous to PCRs except only a single primer is used and only single stranded products are generated. The advantage of using the thermostable Taq polymerase over the use of Sequenase is that multiple rounds of sequencing can be performed without the need to add fresh enzyme. This allows the use of much less template DNA, making this the method of choice in the majority of circumstances. Modifications (point mutations affecting amino acid residues in or near the active site) to the Taq DNA polymerase have enabled it to incorporate the fluorescent dye-labeled terminators more evenly and efficiently, resulting in very even peak heights over a sequence read. Attempts have been made to develop polymerases which have some of the advantageous properties of Sequenase but are thermostable like Taq. One of these efforts is described in more detail at http://www.atp.nist.gov/eao/sp950-2/chapt3-2.htm Another important innovation was the introduction of dideoxy terminators that use two fluorescent dye labels to take advantage of fluorescence resonance energy transfer (FRET) (i.e. ABI BigDye). Taking advantage of FRET using the dual-labeled terminators has allowed improvement of signal intensity and spectral separation to the point where very small amounts of templates can be used. Fluorescent sequencing methods are now robust enough that sequencing large templates such as BACS, P1s, etc. that used to be impractical have become reasonably easy, and sequencing plasmids and PCR products is trivial.
A typical processed electropherogram generated using ABI BigDye v3.1 cycle sequencing chemistry and run on a 3100 is shown at right. The G bases are displayed in black, the A's in green, the C's in blue and the T's in red.
The picture below is a screen capture from the data collection software. You can see that the cycle sequencing reaction products from 16 wells of a 96-well plate are being electrophoresed simultaneously. The large pane at the bottom of the screen shows the fluorescent DNA bands which have been separated up to this point in each of

the 16 capillaries lined up next to each other. Since the 3100 is a 16 capillary machine, it takes 6 runs to process a full plate of samples.
Next Section
Abstract
DNA sequencing by synthesis (SBS) on a solid surface during polymerase reaction offers a paradigm to decipher DNA sequences. We report here the construction of such a DNA sequencing system using molecular engineering approaches. In this approach, four nucleotides (A, C, G, T) are modified as reversible terminators by attaching a cleavable fluorophore to the base and capping the 3′-OH group with a small chemically reversible moiety so that they are still recognized by DNA polymerase as substrates. We found that an allyl moiety can be used successfully as a linker to tether a fluorophore to 3′-O-allyl-modified nucleotides, forming chemically cleavable fluorescent nucleotide reversible terminators, 3′-O-allyl-dNTPs-allyl-fluorophore, for application in SBS. The fluorophore and the 3′-O-allyl group on a DNA extension product, which is generated by incorporating 3′-O-allyl-dNTPs-allyl-fluorophore in a polymerase reaction, are removed simultaneously in 30 s by Pd-catalyzed deallylation in aqueous buffer solution. This one-step dual-deallylation reaction thus allows the reinitiation of the polymerase reaction and increases the SBS efficiency. DNA templates consisting of homopolymer regions were accurately sequenced by using this class of fluorescent nucleotide analogues on a DNA chip and a four-color fluorescent scanner.
Shotgun sequencing
In genetics, shotgun sequencing, also known as shotgun cloning, is a method used for sequencing long DNA strands. It is named by analogy with the rapidly-expanding, quasi-random firing pattern of a shotgun.
Since the chain termination method of DNA sequencing can only be used for fairly short strands (100 to 1000 basepairs), longer sequences must be subdivided into smaller fragments, and subsequently re-assembled to give the overall sequence. Two principal methods are used for this: chromosome walking, which progresses through the entire strand, piece by piece, and shotgun sequencing, which is a faster but more complex process, and uses random fragments.
In shotgun sequencing [1] [2], DNA is broken up randomly into numerous small segments, which are sequenced using the chain termination method to obtain reads. Multiple overlapping reads for the target DNA are obtained by performing several rounds of this fragmentation and sequencing. Computer programs then use the overlapping ends of different reads to assemble them into a continuous sequence [1].
Example
For example, consider the following two rounds of shotgun reads:
Strand Sequence
Original AGCATGCTGCAGTCATGCTTAGGCTA
First shotgun sequenceAGCATGCTGCAGTCATGCT--------------------------TAGGCTA
Second shotgun sequence AGCATG--------------------

------CTGCAGTCATGCTTAGGCTA
Reconstruction AGCATGCTGCAGTCATGCTTAGGCTA
In this extremely simplified example, none of the reads cover the full length of the original sequence, however, the four reads can be assembled into the original sequence using the overlap of their ends to align and order them. In reality, this process uses enormous amounts of information that are rife with ambiguities and sequencing errors. Assembly of complex genomes is additionally complicated by the great abundance of repetitive sequence, meaning similar short reads could come from completely different parts of the sequence.
Many overlapping reads for each segment of the original DNA are necessary to overcome these difficulties and accurately assemble the sequence. For example, to complete the Human Genome Project, most of the human genome was sequenced at 12X or greater coverage; that is, each base in the final sequence was present, on average, in 12 reads. Even so, current methods have failed to isolate or assemble reliable sequence for approximately 1% of the (euchromatic) human genome.
Whole genome shotgun sequencing
Whole genome shotgun sequencing for small (4000 to 7000 basepair) genomes was already in use in 1979 [1] broader application benefited from pairwise end sequencing, known colloquially as double-barrel shotgun sequencing. As sequencing projects began to take on longer and more complicated DNAs, multiple groups began to realize that useful information could be obtained by sequencing both ends of a fragment of DNA. Although sequencing both ends of the same fragment and keeping track of the paired data was more cumbersome than sequencing a single end of two distinct fragments, the knowledge that the two sequences were oriented in opposite directions and were about the length of a fragment apart from each other was valuable in reconstructing the sequence of the original target fragment. The first published description of the use of paired ends was in 1990 [3] as part of the sequencing of the human HPRT locus, although the use of paired ends was limited to closing gaps after the application of a traditional shotgun sequencing approach. The first theoretical description of a pure pairwise end sequencing strategy, assuming fragments of constant length, was in 1991[4]. At the time, there was community consensus that the optimal fragment length for pairwise end sequencing would be three times the sequence read length. In 1995 Roach et al.[5] introduced the innovation of using fragments of varying sizes, and demonstrated that a pure pairwise end-sequencing strategy would be possible on large targets. The strategy was subsequently adopted by The Institute for Genomic Research (TIGR) to sequence the genome of the bacterium Haemophilus influenzae in 1995 [6] , and then by Celera Genomics to sequence the drosophila melanogaster (fruit fly) genome in 2000 [7], and subsequently the human genome.
To apply the strategy, high-molecular-weight DNA is sheared into random fragments, size-selected (usually 2, 10, 50, and 150 kb), and cloned into an appropriate vector. The clones are then sequenced from both ends using the chain termination method yielding two short sequences. Each sequence is called an end-read or read and two reads from the same clone are referred to as mate pairs. Since the chain termination method usually can only produce reads between 500 and 1000 bases long, in all but the smallest clones, mate pairs will rarely overlap.
The original sequence is reconstructed from the reads using sequence assembly software. First, overlapping reads are collected into longer composite sequences known as contigs. Contigs can be linked together into scaffolds by following connections between mate pairs. The distance between contigs can be inferred from the mate pair positions if the average fragment length of the library is known and has a narrow window of deviation.
Coverage
Coverage is the average number of reads representing a given nucleotide in the reconstructed sequence. It can be calculated from the length of the original genome (G), the number of reads(N), and the average read length(L) as NL / G. For example, a hypothetical genome with 2,000 base pairs reconstructed from 8 reads with an average length of 500 nucleotides will have 2x redundancy. This parameter also enables one to estimate other quantities, such as the percentage of the genome covered by reads (the coverage). A high coverage in shotgun sequencing is desired because it can overcome errors in base calling and assembly. The subject of DNA sequencing theory addresses the relationships of such quantities.

Proponents of this approach argue that it is possible to sequence the whole genome at once using large arrays of sequencers, which makes the whole process much more efficient than more traditional approaches. Detractors argue that although the technique quickly sequences large regions of DNA, its ability to correctly link these regions is suspect, particularly for genomes with repeating regions. As sequence assembly programs become more sophisticated and computing power becomes cheaper, it may be possible to overcome this limitation[citation
needed].
Next-Generation Sequencing
Although shotgun sequencing was the most advanced technique for sequencing genomes from about 1995-2005, other technologies started surfacing, called next-generation sequencing. These technologies produce shorter reads (anywhere from 25-500bp) but many hundreds of thousands or million reads in a relatively short time (on the order of a day). This results in high coverage, but the assembly process is much more computationally expensive. These technologies are vastly superior to shotgun sequencing due to the high volume of data and the relatively short time it takes to sequence a whole genome. The major disadvantage is that the accuracies are usually lower (although this is compensated by the high coverage).
