down syndrome (trisomy 21 syndrome) - care of...
TRANSCRIPT

DOWN SYNDROME (TRISOMY 21 SYNDROME)
List cardinal features of down syndrome in newborn period and later
To Construct a health supervision planfor down syndrome in each age group
To identify down syndrome antenatally
To write 4 essential investigation for current management
Trisomy 21 is the most common genetic cause of moderate mental retardation.
The incidence of Down syndrome in live births is approximately 1 in 733; the
incidence at conception is more than twice that rate; the difference is accounted
by early pregnancy losses. In addition to cognitive impairment, Down syndrome is
associated with congenital anomalies and characteristic dysmorphic features).
Although there is variability in the clinical features, the constellation of
phenotypic features is fairly consistent and permits clinical recognition of trisomy
21. Affected individuals are more prone to congenital heart defects (50%) such as
atrioventricular septal defects, ventricular septal defects, isolated secundum atrial
septal defects, patent ductus arteriosus, and tetralogy of Fallot. Congenital and
acquired gastrointestinal anomalies and hypothyroidism are common
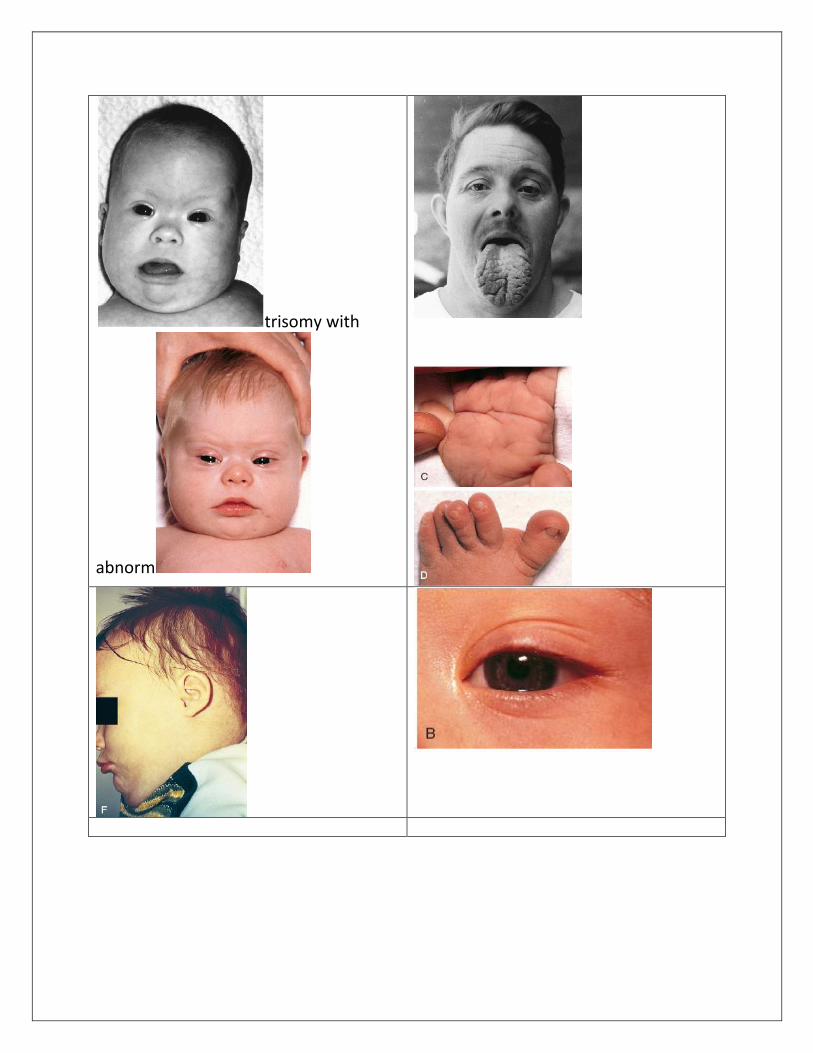
trisomy with
abnorm
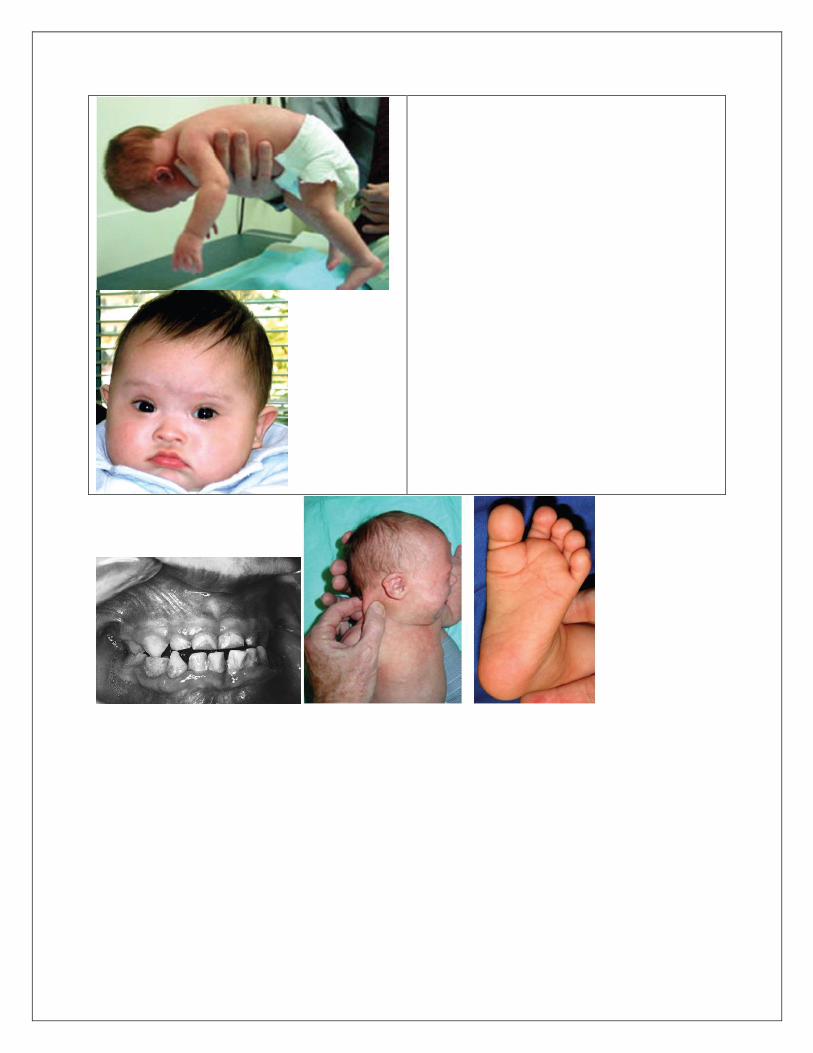

ABNORMALITIES General. Hypotonia with tendency to keep mouth open and protrude the tongue, diastasis recti, hyperflexibility of joints, relatively small stature with awkward gait, increased weight in adolescence. Central Nervous System. Intellectual disability. Craniofacial. Brachycephaly; mild microcephaly with upslanting palpebral fissures; thin cranium with late closure of fontanels; hypoplasia to aplasia of frontal sinuses, short hard palate; small nose with low nasal bridge and tendency to have inner epicanthal folds. Eyes. Speckling of iris (Brushfield spots) with peripheral hypoplasia of iris; fine lens opacities by slit lamp examination (59%); refractive error, mostly myopia (70%); nystagmus (35%); strabismus (45%); blocked tear duct (20%); acquired cataracts in adults (30% to 60%). Ears. Small; overfolding of angulated upper helix; sometimes prominent; small or absent earlobes; hearing loss (66%) of conductive, mixed, or sensorineural type; fluid accumulation in middle ear (60% to 80%). Dentition. Hypoplasia, irregular placement, fewer caries than usual, periodontal disease. Neck. Short with loose folds of skin. Hands. Relatively short metacarpals and phalanges; hypoplasia of midphalanx of fifth finger (60%) with clinodactyly (50%), a single crease (40%), or both; simian

crease (45%); distal position of palmar axial triradius (84%); ulnar loop dermal ridge pattern on all digits (35%). Feet. Wide gap between first and second toes, plantar crease between first and second toes, open field dermal ridge patterning in hallucal area of sole (50%).
Pelvis. Hypoplasia with outward lateral flare of iliac wings and shallow acetabular angle. Cardiac. Anomaly in approximately 40%; endocardial cushion defect, ventricular septal defect, patent ductus arteriosus, auricular septal defect, and aberrant subclavian artery, in decreasing order of frequency; mitral valve prolapse with or without tricuspid valve prolapse and aortic regurgitation by 20 years of age; risk for regurgitation after 18 years of age. Skin. Cutis marmorata, especially in extremities (43%); dry, hyperkeratotic skin with time (75%); infections in the perigenital area, buttocks, and thighs that begin as follicular pustules in 50% to 60% of adolescents. Hair. Fine, soft, and often sparse; straight pubic hair at adolescence.
Genitalia. Relatively small penis and decreased testicular volume; primary
gonadal deficiency is common and progressive from birth to adolescence and is
definitely present in adults. Although rare, cases of fertility in females have been
reported; no male has reproduced
PRINCIPAL FEATURES IN THE NEONATE A physical examination is the most sensitive test in the first 24 hours of life todiagnose trisomy 21
in an infant. If theclinician feels that enough criteria arepresent on physical examination, then
a blood sample should be sent forchromosome evaluation. The clinicianshould alert the
laboratory and requestrapid results. A study that usesfluorescent in situ hybridization (FISH)
technology should be available within24 to 48 hours to facilitate diagnosisand parent counseling.
A FISH studycan only indicate that an extra copy ofchromosome 21 is present; it cannotdetect
translocations. Therefore, apositive FISH-test result should be confirmed by a complete
chromosome analysis to identify translocations that may have implications for further
reproductive counseling for the parents and possibly other family members.
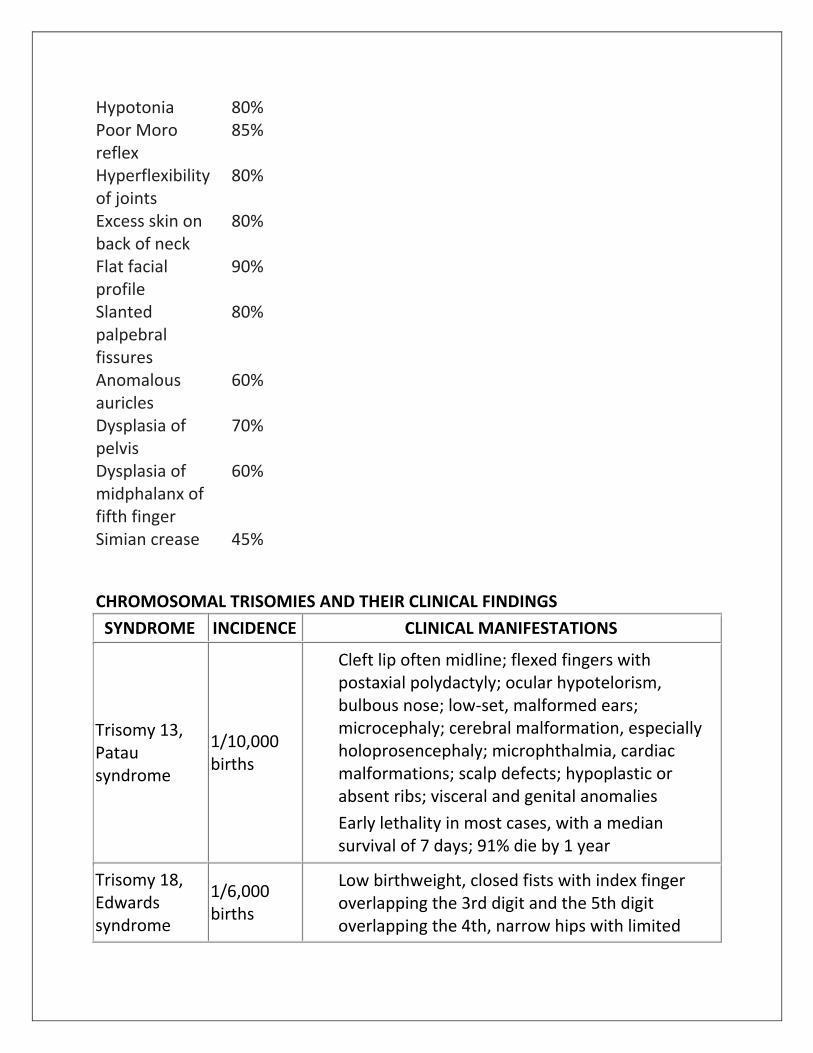
Hypotonia 80% Poor Moro reflex
85%
Hyperflexibility of joints
80%
Excess skin on back of neck
80%
Flat facial profile
90%
Slanted palpebral fissures
80%
Anomalous auricles
60%
Dysplasia of pelvis
70%
Dysplasia of midphalanx of fifth finger
60%
Simian crease 45%
CHROMOSOMAL TRISOMIES AND THEIR CLINICAL FINDINGS
SYNDROME INCIDENCE CLINICAL MANIFESTATIONS
Trisomy 13, Patau syndrome
1/10,000 births
Cleft lip often midline; flexed fingers with postaxial polydactyly; ocular hypotelorism, bulbous nose; low-set, malformed ears; microcephaly; cerebral malformation, especially holoprosencephaly; microphthalmia, cardiac malformations; scalp defects; hypoplastic or absent ribs; visceral and genital anomalies
Early lethality in most cases, with a median
survival of 7 days; 91% die by 1 year
Trisomy 18, Edwards syndrome
1/6,000 births
Low birthweight, closed fists with index finger
overlapping the 3rd digit and the 5th digit overlapping the 4th, narrow hips with limited
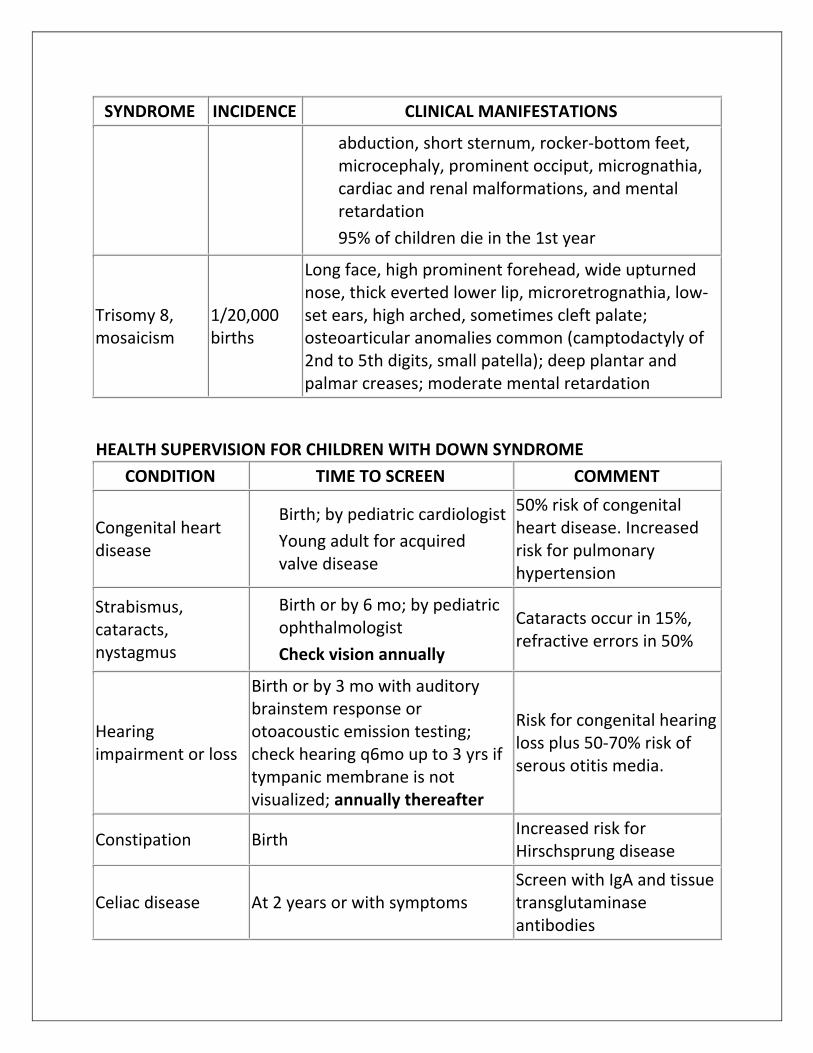
SYNDROME INCIDENCE CLINICAL MANIFESTATIONS
abduction, short sternum, rocker-bottom feet, microcephaly, prominent occiput, micrognathia, cardiac and renal malformations, and mental retardation
95% of children die in the 1st year
Trisomy 8, mosaicism
1/20,000 births
Long face, high prominent forehead, wide upturned nose, thick everted lower lip, microretrognathia, low-set ears, high arched, sometimes cleft palate; osteoarticular anomalies common (camptodactyly of 2nd to 5th digits, small patella); deep plantar and palmar creases; moderate mental retardation
HEALTH SUPERVISION FOR CHILDREN WITH DOWN SYNDROME
CONDITION TIME TO SCREEN COMMENT
Congenital heart disease
Birth; by pediatric cardiologist
Young adult for acquired
valve disease
50% risk of congenital heart disease. Increased risk for pulmonary hypertension
Strabismus, cataracts, nystagmus
Birth or by 6 mo; by pediatric
ophthalmologist
Check vision annually
Cataracts occur in 15%, refractive errors in 50%
Hearing impairment or loss
Birth or by 3 mo with auditory brainstem response or otoacoustic emission testing; check hearing q6mo up to 3 yrs if tympanic membrane is not visualized; annually thereafter
Risk for congenital hearing loss plus 50-70% risk of serous otitis media.
Constipation Birth Increased risk for Hirschsprung disease
Celiac disease At 2 years or with symptoms Screen with IgA and tissue transglutaminase antibodies
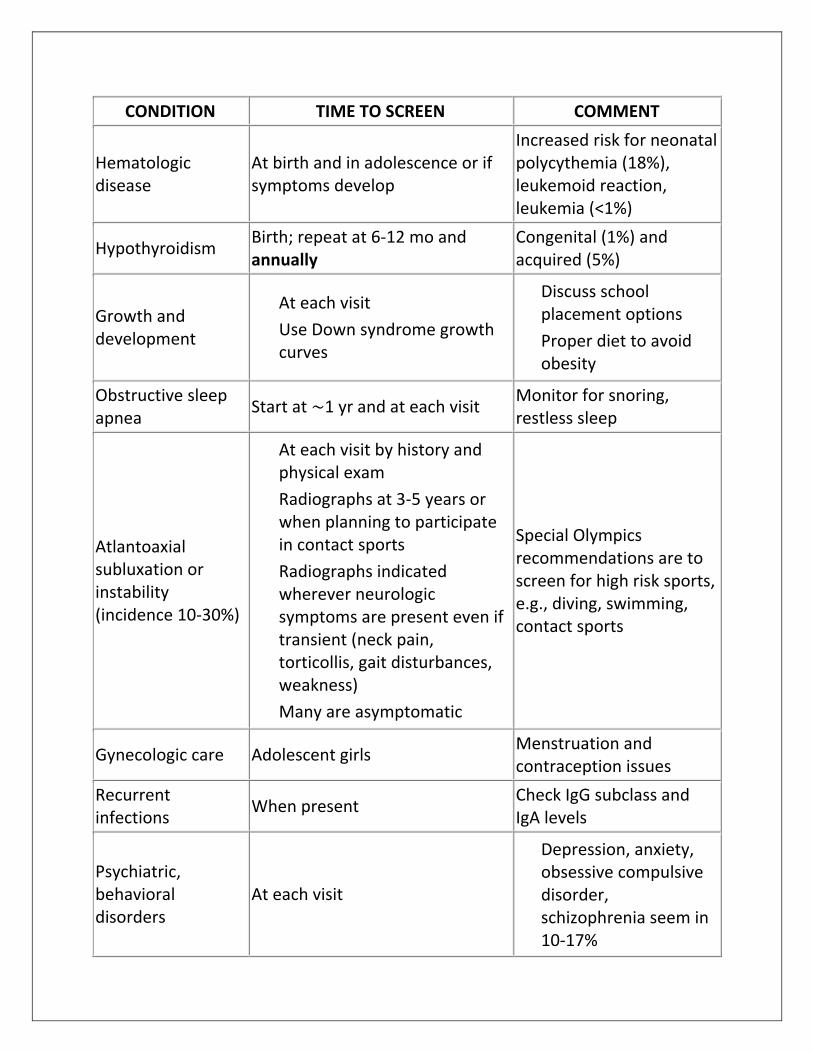
CONDITION TIME TO SCREEN COMMENT
Hematologic disease
At birth and in adolescence or if symptoms develop
Increased risk for neonatal polycythemia (18%), leukemoid reaction, leukemia (<1%)
Hypothyroidism Birth; repeat at 6-12 mo and annually
Congenital (1%) and acquired (5%)
Growth and development
At each visit
Use Down syndrome growth
curves
Discuss school
placement options
Proper diet to avoid
obesity
Obstructive sleep apnea
Start at ∼1 yr and at each visit Monitor for snoring, restless sleep
Atlantoaxial subluxation or instability (incidence 10-30%)
At each visit by history and
physical exam
Radiographs at 3-5 years or
when planning to participate in contact sports
Radiographs indicated wherever neurologic symptoms are present even if transient (neck pain, torticollis, gait disturbances, weakness)
Many are asymptomatic
Special Olympics recommendations are to screen for high risk sports, e.g., diving, swimming, contact sports
Gynecologic care Adolescent girls Menstruation and contraception issues
Recurrent infections
When present Check IgG subclass and IgA levels
Psychiatric, behavioral disorders
At each visit
Depression, anxiety, obsessive compulsive disorder, schizophrenia seem in 10-17%

CONDITION TIME TO SCREEN COMMENT
Autism spectrum
disorder in 5-10%
Early-onset Alzheimer
disease
Extracted from Committee on Genetics: Health supervision for children with Down syndrome, Pediatrics 107:442–449, 2001; and Baum RA, Spader M, Nash PL, et al: Primary care of children and adolescents with Down syndrome: an update, Curr Prob Pediatr Adolesc Health Care 38:235–268, 2008.
HEALTH SUPERVISION NEWBORN TO 1 MONTH
● Heart defects (_50% risk). Perform an echocardiogram, to be read by a pediatric cardiologist, regardless ofwhether a fetal echocardiogramwas performed. Refer to a pediatric cardiologist for evaluation any infant whose postnatal echocardiogram results are abnormal. ● Feeding problems. Refer all infantswho have marked hypotonia as wellas infants with slow feeding, choking with feeds, recurrent pneumonia,or other recurrent or persistent respiratory symptoms and unexplained failure to thrive for a radiographic swallowing assessment.14,15 ● Cataracts at birth by looking for ared reflex. Cataracts may progress slowly and, if detected, need prompt evaluation and treatment by an ophthalmologist with experience in managing the child with Down syndrome. ● Congenital hearing loss, with objective testing, such as brainstem auditory evoked response or otoacoustic emission, at birth, according tothe universal newborn hearing screening guidelines. Complete any needed follow-up assessment by 3months.16,17

● Duodenal atresia or anorectal atresia/stenosis by performing a historyand clinical examination. ● Apnea, bradycardia, or oxygen desaturationin a car safety seat forinfants who are at increased risk because they have had cardiac surgeryor are hypotonic. A car safetyseat evaluation should be conductedfor these infants before hospitaldischarge.18 ● Constipation. If constipation is present,evaluate for restricted diet orlimited fluid intake, hypotonia, hypothyroidism,or gastrointestinal tractmalformation, including stenoses or Hirschsprung disease, for whichthere is an increased risk .● Gastroesophageal reflux, which isusually diagnosed and managed clinically. If severe or contributingto cardiorespiratory problems orfailure to thrive, refer for subspecialty intervention. ● Stridor, wheezing, or noisy breathing.If severe or contributing to cardiorespiratory problems or feedingdifficulty, refer to pediatric pulmonologist to assess for airway anomalies. Tracheal anomalies and small tracheal size may also makeintubation more difficult .● Hematologic abnormalities. Obtain a complete blood cell count. Leukemoid reactions, or transient myeloproliferativedisorder (TMD). TMD is found almost exclusively in newborn infantswith Down syndrome and is relatively common in this population (10%).19TMD usually regresses spontaneously within the first 3 months of life, butthere is an increased risk of later onsetof leukemia for these patients (10%–30%).20 Polycythemia is alsocommon in infants with Down syndrome(18%–64%)21 and may require careful management. Infants withTMD and polycythemia should be followed according to subspecialty consultation recommendations.
Parents of infants with TMD should be counseled regarding the risk of leukemia and made aware of the signs, including easy bruising, petechiae, onset oflethargy, or change in feeding patterns.Leukemia is more common inchildren with Down syndrome than inthe general population but still rare
(1%).

● Congenital hypothyroidism (1% risk).Obtain thyroid-stimulating hormone(TSH) concentration if state newbornscreening only measures free thyroxine(T4); congenital hypothyroidism can be missed if only the T4 concentrationis obtained in the newborn screening. Many children with Down syndrome have mildly elevated TSHand normal free T4 levels. Management of children with abnormal thyrotropin or T4 concentrations should be discussed with a pediatric endocrinologist. 1MONTH TO 1 YR
Review the risk of serous otitis media(50%–70%). Review the previous hearingevaluation (brainstem auditoryevoked response [BAER, ABR] or otoacousticemission). If the child passed the screening study, rescreen at 6months of age for confirmation. If theinfant failed to pass screening studies,refer to an otolaryngologist who iscomfortable with examining infants with stenotic external canals to determineif a middle-ear abnormality ispresent.
Tympanometry may be necessaryif the tympanic membrane ispoorly visualized. Middle-ear diseaseshould be treated promptly. Once aclear ear is established, a diagnosticBAER should be performed to accurately establish hearing status. In childrenwith stenotic canals, in whichthe tympanic membranes cannot be seen, refer to an otolaryngologist forexamination under an office microscope.
Interval ear examinationsshould be performed by the otolaryngologist every 3 to 6 months until the tympanic membrane can be visualized by the pediatrician and tympanometrycan be performed reliably.
A behavioral audiogram may be attemptedat 1 year of age, but many children will not be able to complete the study and may need additional testingby BAER.30,31
● At least once during the first 6months of life, discuss with parent ssymptoms of obstructive sleep apnea, including heavy breathing, snoring, uncommon sleep positions, frequent night awakening, daytime sleepiness,apneic pauses, and behavior problemsthat could be associated with poor sleep. Refer to a physicianwith expertise in pediatric sleep disorders for examination and further

evaluation of a possible sleepdisorder if any of the above mentionedsymptoms occur.32,33 ● At each well-child visit, discuss with parents the importance of maintaining the cervical spine in a neutral position during any anesthetic, surgical,or radiographic procedure tominimize the risk of spinal cord injuryand review the signs and symptoms of myelopathy. Perform careful history and physical examination,and pay attention for myelopathicsigns and symptoms. ● Within the first 6 months of life, referto a pediatric ophthalmologistor ophthalmologist with expertiseand experience with infants withdisabilities to evaluate for strabismus,cataracts, and nystagmus.34 Check the infant’s vision at each visit and use developmentally appropriate subjective and objective criteria. If lacrimal duct obstruction is present, refer for evaluation for surgical repair of drainage systemif not resolved by 9 to 12 months ofage.35 ● Verify results of newborn thyroidfunctionscreen if not previouslyperformed. Because of increasedrisk of acquired thyroid disease, repeatmeasurement of TSH at 6 and12 months of age and then annually. ● Monitor infants with cardiac defects,typically ventricular or atrioventricular septal defects thatcause intracardiac left-to-rightshunts, for symptoms and signs ofcongestive heart failure as pulmonaryvascular resistance decreasesand pulmonary blood flow increases. Tachypnea, feeding diffidifficulties,and poor weight gain mayindicate heart failure. Medical management,including nutritional support,may be needed until the infantcan undergo cardiac surgery to repairthe defects. For patients withlarge ventricular septal defects andwithout obstruction to pulmonaryblood flow, repair should be performedbefore 4 months of age tolimit the potential for developmentof pulmonary hypertension and associatedcomplications. Infants and children with Down syndrome arealso at increased risk of pulmonary hypertension even in the absence ofintracardiac structural defects. ● Obtain hemoglobin concentrationbeginning at 1 year of age and annuallythereafter. Children with Down syndrome have been shown to havesignificantly lower dietary intakesof iron than their typically developingpeers.36 Increased erythrocytemean corpuscular volume (MCV)

has been reported in 45% of patients with Down syndrome with andwithout heart disease, and when MCV is decreased, it occurs at approximatelythe same time as anemia.37 Therefore, MCV is not usefulin screening for the diagnoses ofiron deficiency, lead toxicity, or thalassemia in children with Downsyndrome. Serum ferritin concentrationis a sensitive parameter for assessment of iron stores in healthysubjects but is an acute-phase reactantand may be increased in thepresence of chronic inflammationor infection and should be evaluated together with C-reactive protein(CRP) concentration. An elevatedCRP level is an indication that a normal ferritin level may be falsely elevatedand is not a reliable indication of normal iron status. Serum ferritin and CRP or reticulocyte hemoglobin(CHr) concentrations should beobtained at annual visits for patientswho are at increased risk ofiron deficiency on the basis of a historyof decreased iron intake.38–42 ● Monitor for signs of neurologic dysfunctionthat may occur. Childrenwith Down syndrome have an increasedrisk of seizures, includinginfantile spasms (1%–13%)43,44 andother conditions including Moyamoyadisease.45 ● Administer immunizations, includinginfluenza vaccine and othervaccines recommended for all children,unless there are specificcontraindications.46
1 YR TO 5 YRS
● Obtain a history and perform aphysical examination, and give attention to growth and developmental status at every well-child visit.

● Review the risk of hearing loss associated with serous otitis media. Fora child who passed diagnostic hearingtesting, additional screening orbehavioral audiogram and tympanometry should be performed every6 months until normal hearing levelsare established bilaterally by ear-specific testing (usually after 4years of age). Subsequently, behavioralhearing tests should be performedannually. If normal hearingis not established by behavioraltesting, additional screening by otoacoustic emissions or diagnosticBAER should be performed withsedation if necessary. Children who demonstrate a hearing loss should be referred to an otolaryngologist who is comfortable with the examination of children with stenotic ear canals. The risk of serous otitis media between 3 and 5years of age is approximately 50%to 70%. ● Check the child’s vision, and use developmentallyappropriate subjective and objective criteria at eachwell-child visit. Refer the childannually to a pediatric ophthalmologistor ophthalmologist with special expertise and experience withchildren with disabilities. Childrenwith Down syndrome have a 50%risk of refractive errors that leadto amblyopia between 3 and 5years of age. Addressing refractiveerrors and strabismus at anearly age can help prevent amblyopia and encourage normal visualdevelopment.34,49–51 Atlantoaxial Instability Discuss with parents, at least biennially,the importance of cervical spinepositioningprecautions for protection of the cervical spine during any anesthetic,surgical, or radiographic procedure. Perform careful history andphysical examination with attention to myelopathic signs and symptoms at every well-child visit or when symptomspossibly attributable to spinalcord impingement are reported. Parents should also be instructed to contacttheir physician for new onset ofsymptoms of change in gait or use ofarms or hands, change in bowel orbladder function, neck pain, stiff neck, head tilt, torticollis, how the child positionshis or her head, change in general function, or weakness.

The Asymptomatic Child Children with Down syndrome are atincreased risk of atlantoaxial subluxation.However, the child must be 3 years of age to have adequate vertebra lmineralization and epiphyseal development for accurate radiographic evaluation of the cervical spine.52 Plain radiographs do not predict well which children are at increased risk of developingspine problems, and normal radiographsdo not provide assurance ethat a child will not develop spine problemslater.53,54 For these reasons, routine radiologic evaluation of the cervicalspine in asymptomatic children is not recommended. Current evidencedoes not support performing routinescreening radiographs for assessmentof potential atlantoaxial instability in asymptomatic children.55–64 Parents should be advised that participation in some sports, including contactsports such as football and soccer and gymnastics (usually at olderages),
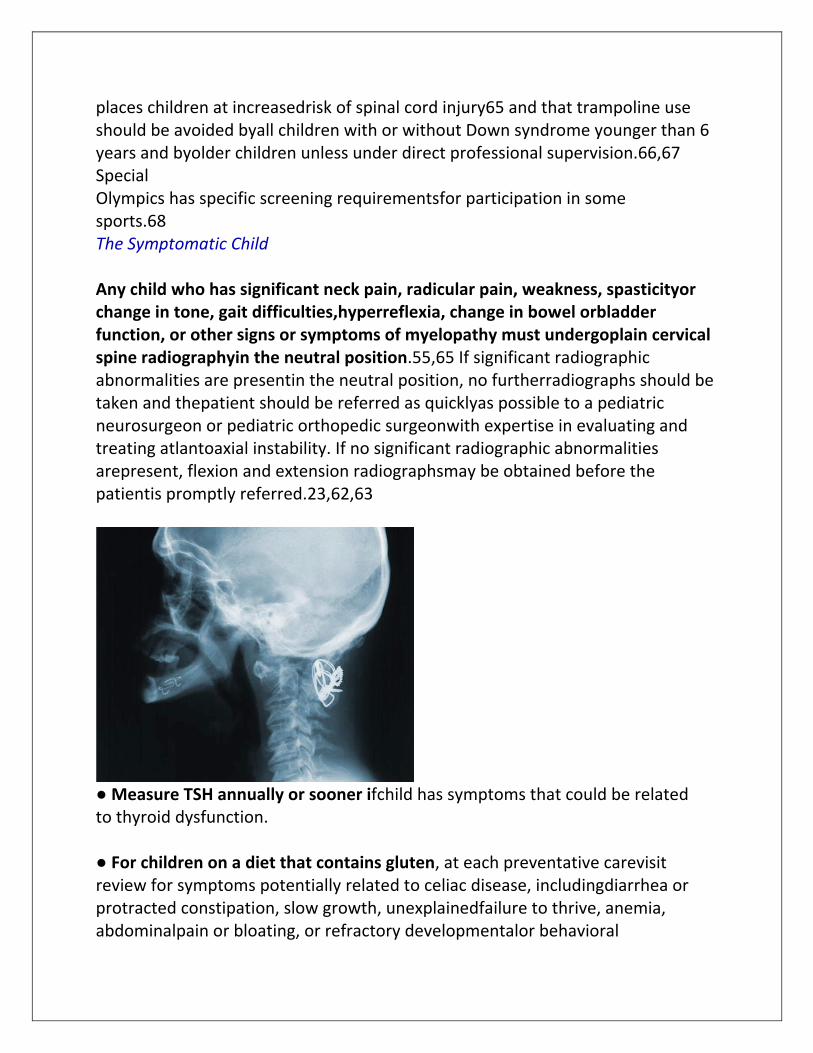
places children at increasedrisk of spinal cord injury65 and that trampoline use should be avoided byall children with or without Down syndrome younger than 6 years and byolder children unless under direct professional supervision.66,67 Special Olympics has specific screening requirementsfor participation in some sports.68 The Symptomatic Child Any child who has significant neck pain, radicular pain, weakness, spasticityor change in tone, gait difficulties,hyperreflexia, change in bowel orbladder function, or other signs or symptoms of myelopathy must undergoplain cervical spine radiographyin the neutral position.55,65 If significant radiographic abnormalities are presentin the neutral position, no furtherradiographs should be taken and thepatient should be referred as quicklyas possible to a pediatric neurosurgeon or pediatric orthopedic surgeonwith expertise in evaluating and treating atlantoaxial instability. If no significant radiographic abnormalities arepresent, flexion and extension radiographsmay be obtained before the patientis promptly referred.23,62,63
● Measure TSH annually or sooner ifchild has symptoms that could be related to thyroid dysfunction. ● For children on a diet that contains gluten, at each preventative carevisit review for symptoms potentially related to celiac disease, includingdiarrhea or protracted constipation, slow growth, unexplainedfailure to thrive, anemia, abdominalpain or bloating, or refractory developmentalor behavioral

problems.69–71 For those with symptoms,obtain a tissue transglutaminaseimmunoglobulin A (IgA) level and simultaneousquantitative IgA. Thequantitative IgA is important, becausea low IgA level will result in afalse-negative tissue transglutaminase IgA result. Refer patients withabnormal laboratory values forspecialty assessment. There is noevidence showing routine screeningof asymptomatic individuals as being beneficial. There are neitherdata nor consensus that would indicatewhether patients with persistentsymptoms who had normallaboratory values on initial evaluationshould have further laboratorytests. ● Discuss symptoms of obstructive sleep apnea, including heavybreathing, snoring, restless sleep,uncommon sleep positions, frequentnight awakening, daytimesleepiness, apneic pauses, and behavior problems, that could be associatedwith poor sleep at eachwell-child visit. There is poor correlation between parent report and polysomnogram results.33,72 Therefore,referral to a pediatric sleepl aboratory for a sleep study or polysomnogramfor all children withDown syndrome by 4 years of age isrecommended. Refer to a physician with expertise in pediatric sleep anychild with signs or symptoms of obstructive sleep apnea or abnormal sleep-study results. Discuss obesityas a risk factor for sleep apnea.34 Itis recognized that access to a pediatric sleep laboratory or specialist may be limited for some populationsand geographic areas. ● Maintain follow-up with a pediatric cardiologist for patients with cardiac lesions even after complete repairto monitor for recurrent/residual lesions as well as development of pulmonary hypertension .● Monitor for neurologic dysfunction,including seizures. ● Obtain hemoglobin concentrationannually. Also, obtain serum ferritinand CRP concentrations for anychild at risk of iron deficiency How do you antenatally dx tris0my? Screening options have improved significantly with the introduction of firsttrimester screening, which incorporates maternal age, nuchal translucency ultrasonography, and measurement of maternal serum human chorionic gonadotropin (_-hCG) and pregnancyassociated plasma protein A (PAPP-A).
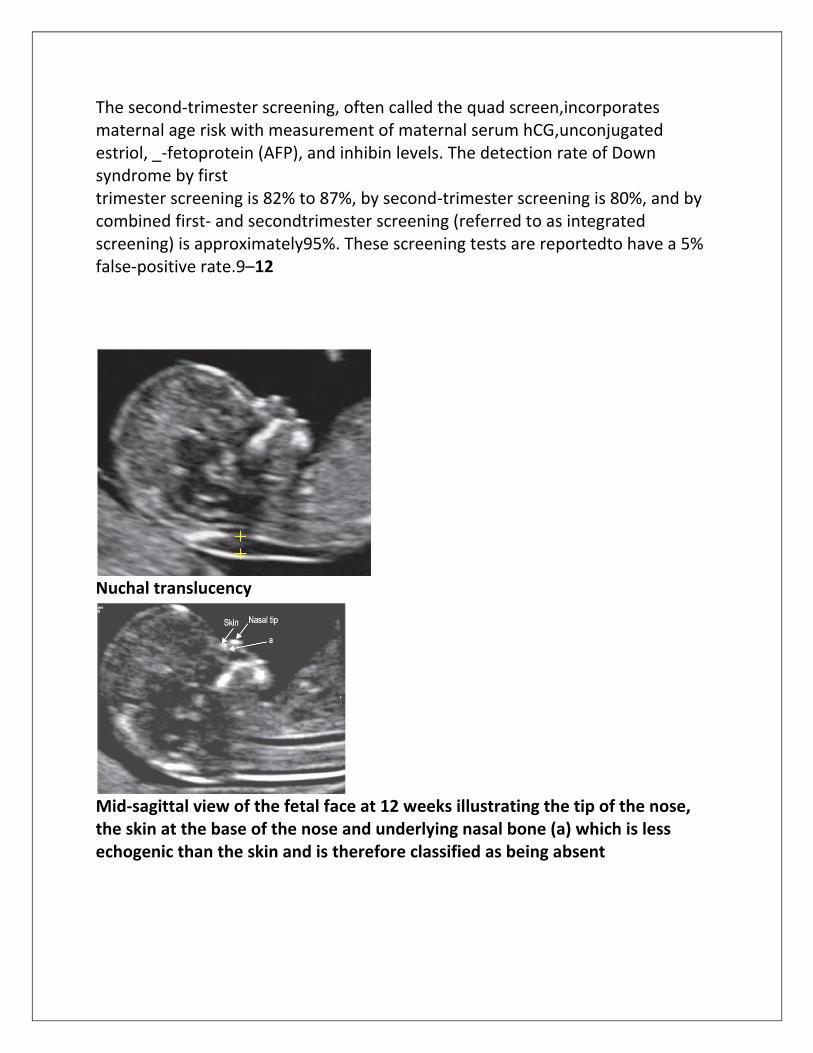
The second-trimester screening, often called the quad screen,incorporates maternal age risk with measurement of maternal serum hCG,unconjugated estriol, _-fetoprotein (AFP), and inhibin levels. The detection rate of Down syndrome by first trimester screening is 82% to 87%, by second-trimester screening is 80%, and by combined first- and secondtrimester screening (referred to as integrated screening) is approximately95%. These screening tests are reportedto have a 5% false-positive rate.9–12
Nuchal translucency
Mid-sagittal view of the fetal face at 12 weeks illustrating the tip of the nose, the skin at the base of the nose and underlying nasal bone (a) which is less echogenic than the skin and is therefore classified as being absent
