
Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy
loss in South Indian Population 1
1.0 Recurrent Pregnancy Loss
Pregnancy is a complex, heterogenous, biological phenomenon in which the embryo
develops into a fetus within the female uterus. The entire process is a vital
immunological paradox, where the semiallogeneic fetus survives by evading maternal
immune recognition and is delivered after the completion of the gestational period.
The duration of gestation is conventionally divided into three trimesters of roughly
three months each. However, due to various etiological factors, the growing embryo
that is unable to survive is expelled from the pregnant mother at different gestational
ages and this is referred to as pregnancy loss or abortion. Pregnancy loss occurs in 10
to 15% of all pregnancies, of which 1-2% are recurrent (1). Recurrent pregnancy loss
(RPL) was initially defined as the loss of three or more clinically recognized
pregnancies spontaneously during early gestation. However, the modern definition
refers to the spontaneous loss of two or more consecutive pregnancies before twenty
weeks of gestation (2). The World Health Organization (WHO) has defined
miscarriage as the loss of a fetus weighing ≤500g, which would normally be at 20-22
complete weeks of gestation (3).
Etiology of recurrent pregnancy loss is among the most studied, yet unresolved issue
in modern gynecology. Among the various proposed etiological factors, abnormal
parental karyotype, antiphospholipid syndrome and uterine anatomic abnormalities
were reported in about 50% of the patients; however, in remaining 50%, the cause is
unknown (4, 5). RPL of unknown etiology provides a fundamental insight into the
processes of embryogenesis and implantation. Epidemiological studies have
suggested that the condition might be multifactorial with a possible genetic
predisposition and environmental factors in its pathogenesis (6, 7). A better
understanding of the role of various gene-gene and gene-environment interactions will
enable identification of high-risk individuals and propose a genetic mechanism to
explain the unknown etiology of RPL. With the completion of human genome project
it is imperative to understand the genetic basis of diseases and to identify the
population and race polymorphism. Since pregnancy is a complex process and in
about 50% of the RPL cases the cause is unidentified, it is essential to explore the
contribution of the genetic variations in RPL.

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy
loss in South Indian Population 2
1.1 Recurrence risk for RPL
About 10–15% of all clinically recognized pregnancies end in a miscarriage globally.
It has been reported that the recurrence risk increases with each successive pregnancy
loss. Risk of pregnancy loss in women with atleast one live born infant without any
prior fetal loss is 12 % for the next pregnancy; whereas the percentage of recurrence
risk for those with at least one, two and three prior fetal loss is 24, 26 and 32%
respectively. However for women with no live born infant and with 2 or more fetal
losses the recurrence risk for the next pregnancy is 40 -45 % (8). The recurrence risk
rates for pregnancy loss are indicated in Fig 1.1
01 2 3 2*
0
5
10
15
20
25
30
35
40
Recurr
ence ris
k (%
)
Recurrence risk in RPL
* - With no live children (Source data – Arjun et al 2000)(8)
Number of fetal Loss
Fig 1.1: Recurrence risk rates for RPL
Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy
loss in South Indian Population 3
1.2 Indian Scenario
A reliable estimate on the rate of pregnancy loss in the Indian context is deficient, as
the registration of marriages, births and deaths are usually incomplete. However an
indirect source that provides information on the incidence of pregnancy loss is the
multicentric study on genetic counseling carried out by the task force of Human
Genetics under the auspices of the Indian Council of Medical Research (ICMR) from
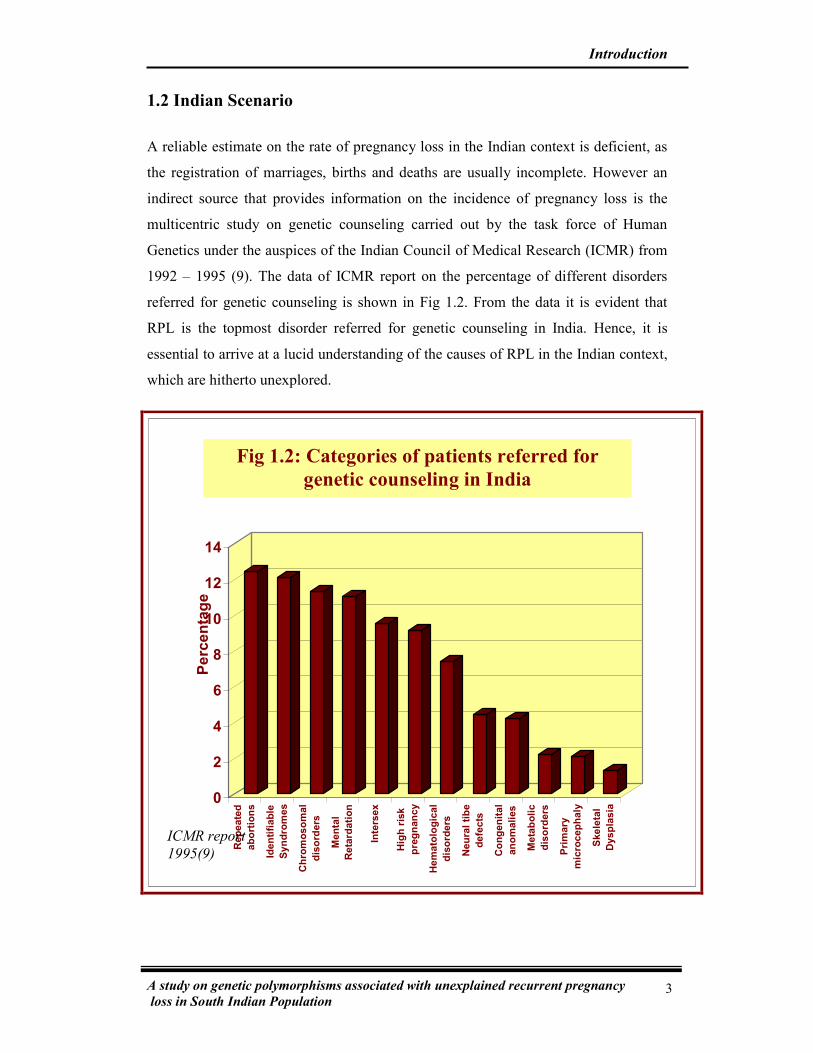
1992 – 1995 (9). The data of ICMR report on the percentage of different disorders
referred for genetic counseling is shown in Fig 1.2. From the data it is evident that
RPL is the topmost disorder referred for genetic counseling in India. Hence, it is
essential to arrive at a lucid understanding of the causes of RPL in the Indian context,
which are hitherto unexplored.
0
2
4
6
8
10
12
14
Repeate
d
abort
ions
Identifiable
Syndro
mes
Chro
mosom
al
dis
ord
ers
Menta
l
Reta
rdation
Inte
rsex
Hig
h r
isk
pre
gnancy
Hem
ato
logic
al
dis
ord
ers
Neura
l tibe
defe
cts
Congenital
anom
alies
Meta
bolic
dis
ord
ers
Pri
mary
mic
rocephaly
Skele
tal
Dyspla
sia
Perc
enta
ge
Fig 1.2: Categories of patients referred for
genetic counseling in India
ICMR report
1995(9)

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy
loss in South Indian Population 4
1.3 Etiological factors for RPL
The established etiological factors that might result in RPL fall into two broad
categories namely embryonal defects like chromosomal abnormality which inhibit the
embryo from implanting or developing and alterations in maternal environment in
which the fetus grows. This includes infection, anatomic, endocrine, genetic and
immunological abnormalities. Fig 1.3 represents in a nutshell these two categories of
etiological factors.
1.3.1 Embryonal defects
Implantation usually occurs after 8-10 days of fertilisation in most healthy
pregnancies. Delay in implantation enhances the proportion of early embryo loss. It
has been shown that chromosomal abnormalities in the conceptus are characteristic of
spontaneous abortions. Chromosomal abnormalities were responsible in about 50% of
first trimester miscarriages, 5% of late pregnancy losses and 0.5% of live births.
Cytogenetically abnormal embryos are usually aneuploid because of sporadic events,
such as meiotic non-disjunction, or polyploid from fertilization abnormalities (11).
Embryonal
defects
Chromosomal
abnormalities
Alterations in
maternal
environment
1) Anatomical 2) Endocrine 3) Infection 4) Genetics 5) Immunology
Recurrent Pregnancy Loss
Fig 1.3: Etiological factors for recurrent pregnancy loss
Meka et al 2006 (10)

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy
loss in South Indian Population 5
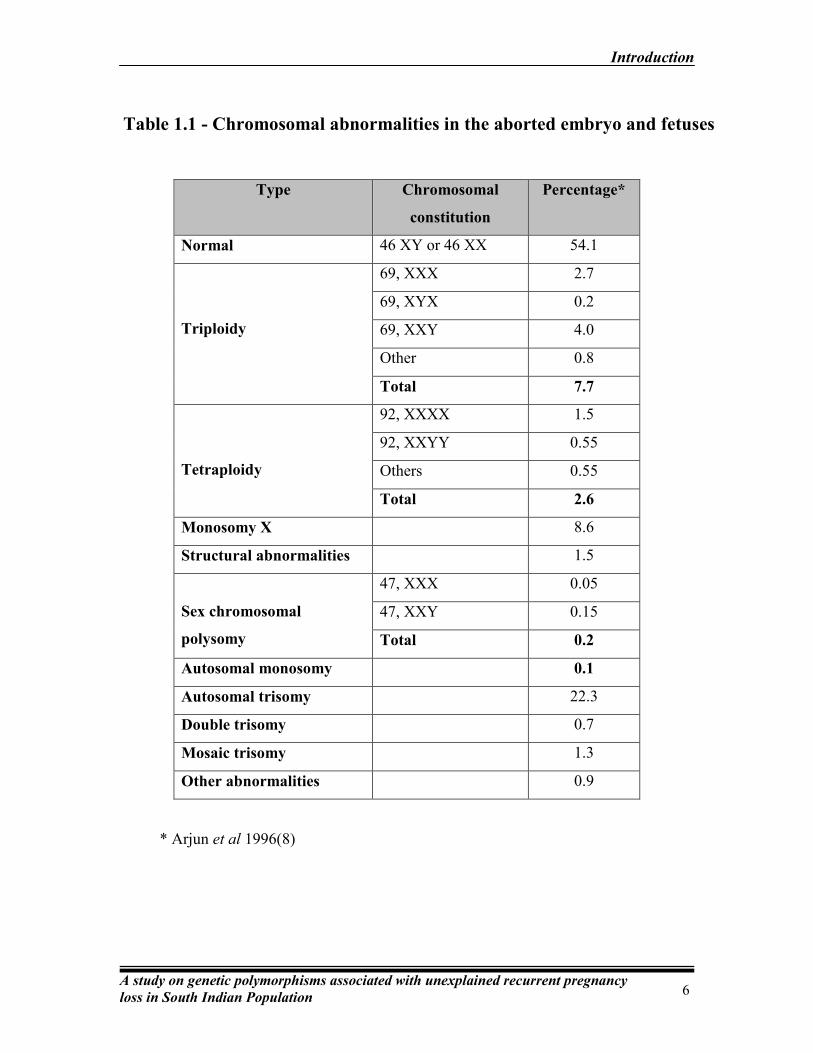
Table 1.1 summarizes the different types of chromosomal abnormalities of the
conceptus associated with recurrent pregnancy loss.
1.3.2 Alterations in the maternal Environment
The maternal environment alterations include anatomic uterine defects, endocrine
anomalies, hormonal imbalances, infection, metabolic dysfunction nutritional
deficiencies, immune disorders and genetic factors.
1.3.2.1 Anatomic uterine defects
Anatomic uterine defects are known to cause obstetric complications including RPL,
preterm labor and delivery. The common anatomic defects associated with RPL are
mullerian duct fusion in the uterus, double uterus, uterine septum, endometrial polyps
and sub-mucous fibroids. Impaired vascularization and fetal growth restriction due to
uterine distortion are common reasons for pregnancy loss. Although congenital
uterine abnormalities have been associated most often with second trimester
pregnancy loss, 15% of women with recurrent early pregnancy loss also exhibit these
abnormalities (12).
1.3.2.2 Hormonal and metabolic disorders
Hormonal imbalances are believed to play a role in at least 10-20% of recurrent
pregnancy losses. Ovulation, implantation and the early stages of pregnancy depend
on an integral maternal endocrine regulatory system. Deficient secretion of
progesterone or a poor endometrial response to progesterone results in Luteal Phase
Deficiency (LPD) which leads to abnormal endometrial development, thus affecting
implantation and thereby enhancing the risk of pregnancy loss. In addition, thyroid-
gland dysfunction, type I diabetes mellitus and poly cystic ovary syndrome have also
been associated with the risk of RPL (13).

0
5
10
15
20
25
30
Percentage
G*01011 G*01012 G*01013 G*01014 G*0103 G*0104 G*0105N
HLA G Alleles
Controls
Cases
Fig 3.5: Distribution of HLA G polymorphism in RPL
and control women
Controls (N = 80) Cases (N =104)
HLA G alleles Number Allele frequency Number Allele frequency
G*01011 22 0.14 22 0.11
G*01012 24 0.15 30 0.14
G*01013 23 0.14 29 0.14
G*01014 16 0.10 17 0.08
G*0103 42 0.26 45 0.22
G*0104 27 0.17 34 0.16
G*0105N 6 0.04 31 0.15
P< 0.001
Table 3.4: Frequency distribution of the various HLA G alleles in
RPL and control women
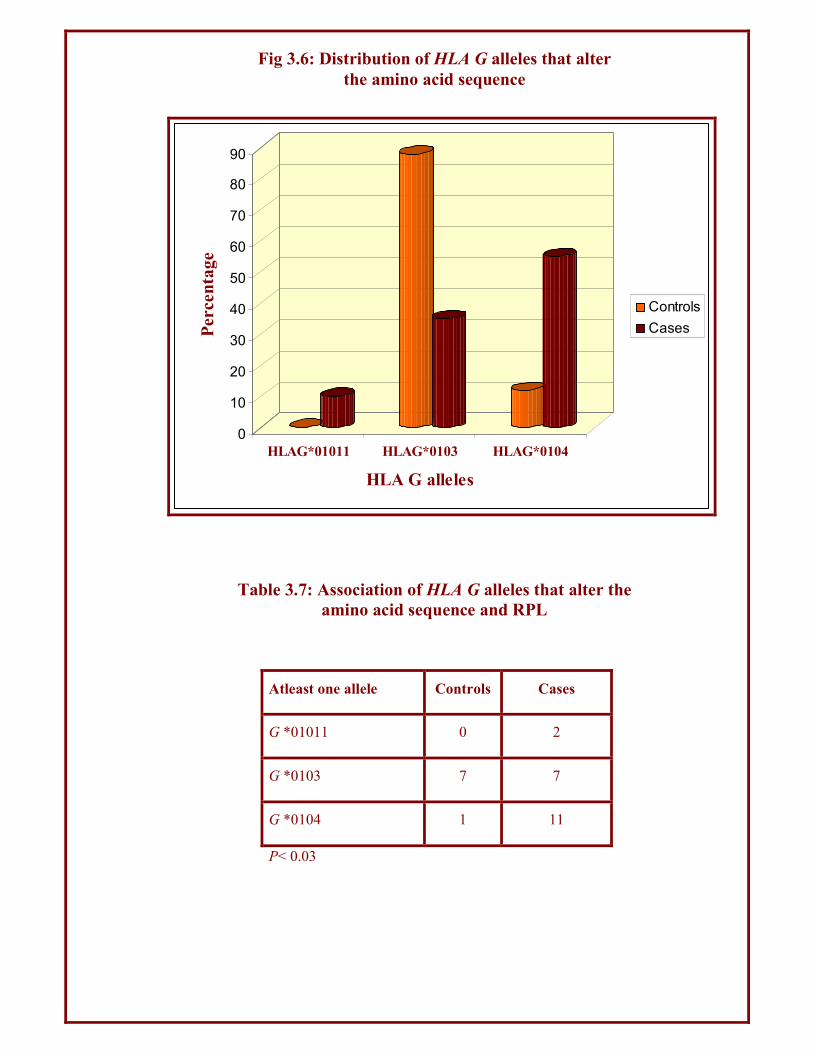
Atleast one allele Controls Cases
G *01011 0 2
G *0103 7 7
G *0104 1 11
P< 0.03
Table 3.7: Association of HLA G alleles that alter the
amino acid sequence and RPL
0
10
20
30
40
50
60
70
80
90
Percentage
HLAG*01011 HLAG*0103 HLAG*0104
HLA G alleles
Controls
Cases
Fig 3.6: Distribution of HLA G alleles that alter
the amino acid sequence

0
5
10
15
20
25
30
35
40
45
50Genotype frequency
I/I I/D D/D
HLAG genotypes
Controls
Cases
Fig 3.7: HLA G I/D polymorphism in RPL and control women
Genotype Genotype frequency OR 95% CI P
Controls
(N=80)
Cases
(N=104)
I/I 27 23 1.0
I/D 28 49 2.34 1.13 - 4.88 <0.05
D/D 25 32 1.65 0.76 to 3.57 NS
NS – Non significant
Table 3.9: Association of HLA G I/D polymorphism and RPL

0
10
20
30
40
50
60
70
80
Percentage
Control Cases
Antipaternal cytotoxic antibodies and RPL
Present
Absent
Fig 3.9 Antipaternal cytotoxic antibodies and RPL
APCA Controls
(N = 30)
Cases
(N = 30) P
Present 22 7
<0.001
Absent 8 23
Table 3.13: Distribution of APCA in RPL
and control women
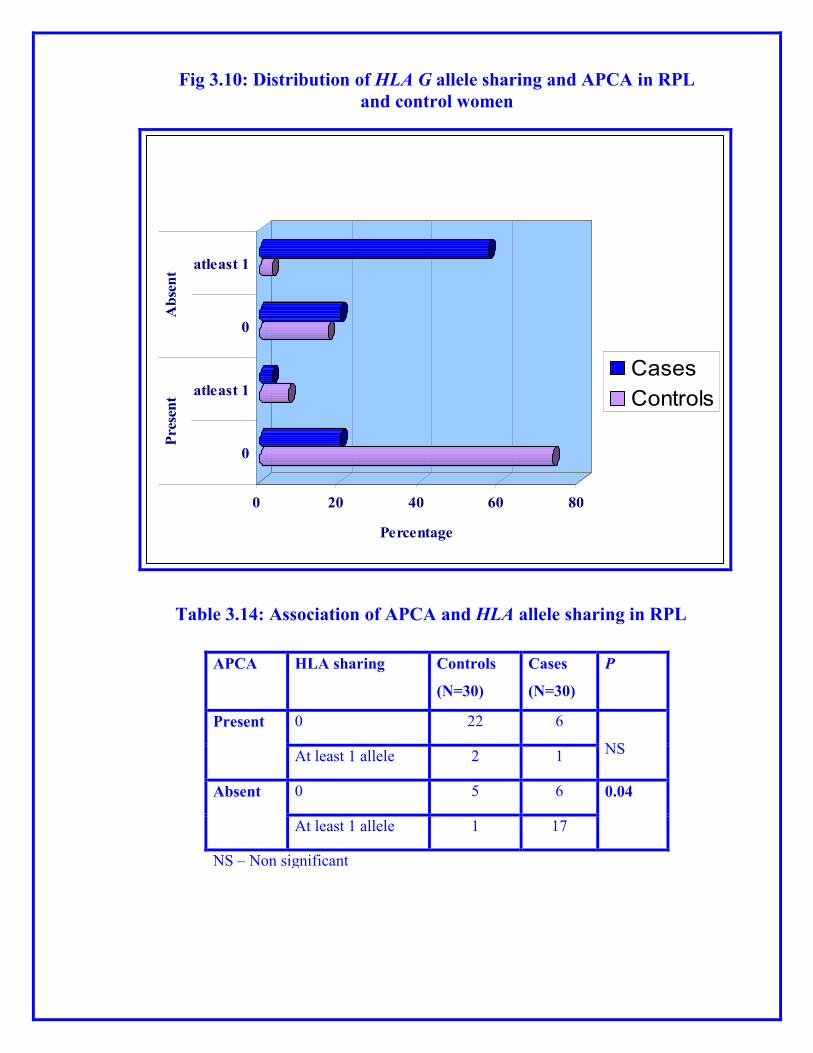
0 20 40 60 80
Percentage
0
atleast 1
0
atleast 1
Present
Absent
Distribution of HLA G allele sharing and APCA in RPL women and
controls
Cases
Controls
Fig 3.10: Distribution of HLA G allele sharing and APCA in RPL
and control women
APCA HLA sharing Controls
(N=30)
Cases
(N=30)
P
Present 0 22 6
NS At least 1 allele 2 1
Absent 0 5 6 0.04
At least 1 allele 1 17
NS – Non significant
Table 3.14: Association of APCA and HLA allele sharing in RPL
Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy
loss in South Indian Population 6
* Arjun et al 1996(8)
Type Chromosomal
constitution
Percentage*
Normal 46 XY or 46 XX 54.1
Triploidy
69, XXX 2.7
69, XYX 0.2
69, XXY 4.0
Other 0.8
Total 7.7
Tetraploidy
92, XXXX 1.5
92, XXYY 0.55
Others 0.55
Total 2.6
Monosomy X 8.6
Structural abnormalities 1.5
Sex chromosomal
polysomy
47, XXX 0.05
47, XXY 0.15
Total 0.2
Autosomal monosomy 0.1
Autosomal trisomy 22.3
Double trisomy 0.7
Mosaic trisomy 1.3
Other abnormalities 0.9
Table 1.1 - Chromosomal abnormalities in the aborted embryo and fetuses

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy
loss in South Indian Population
7
1.3.2.3 Infections
Infected or inflamed uterine lining would be a hostile environment for implantation
and development of the embryo. Microbial infection of the uterus or vagina with
toxoplasma, listeria, cytomegalovirus and parvovirus are known to cause sporadic
pregnancy loss, but no infectious agent has been proven to cause RPL. In addition,
chronic herpes simplex viral infection is a possible cause of RPL. Fetal infection may
lead to either fetal death or malformations incompatible with viability (14).
1.3.2.4 Nutritional factors
The deficiency of vitamin A is a cause of increased infant mortality. Low
concentrations of dietary and circulating folate are associated with increased risks of
preterm delivery, reduced infant birth weight and fetal growth retardation. Folate
deficiency results in elevation of blood homocysteine and increased homocysteine
concentrations have been associated both with spontaneous abortion and pregnancy
complications (15).
1.3.2.5 Immunological causes
The immune factors associated with pregnancy loss are classified as autoimmune and
alloimmune factors. The autoimmune factors include anti-phospholipid and anti-
nuclear antibodies. Phospholipids are fatty molecules on cell membranes that help to
maintain the proper balance between bleeding and clotting. Antiphospholipid
antibodies block the phospholipids from regulating blood flow and clotting, resulting
in the formation of blood clots in the placenta leading to pregnancy loss (16).
Antinuclear antibodies are autoantibodies to the DNA which lead to inflammation in
the placenta. Alloimmune response refers to the maternal immune response to
antigens of placental or fetal tissues. In normal pregnancy, placenta and the growing
embryo are not entirely “self” but rather is a result of both the maternal and paternal
genetic heritages, referred to as a semi-allograft. Though the exact mechanism that
allows the embryo to escape immune rejection is unknown, the human leukocyte
antigen sharing between partners, decrease in natural suppresser cells in the uterus,
serum blocking factors that protect the placenta from paternal antigens and

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy
loss in South Indian Population
8
antileukocytotoxic antibodies against paternal leukocytes have been proposed for
successful pregnancy. Alterations in any of these immune mechanisms could result in
RPL (17).
1.3.2.6 Genetic factors
Genetic factors account for approximately 5% of pregnancy loss (18). Cytogenetic
analyses in blood lymphocytes of couples with RPL in the Indian population have
revealed an overall 4-5% chromosomal abnormality (19, 20). Certain chromosomal
abnormalities seen in couples, which are considered to be a significant cause of RPL
is summarized in Table 1.2.
X chromosome inactivation (XCI), an essential random phenomenon
for dosage compensation in females occurs such that maternally and paternally
derived X chromosomes are inactivated with approximately the same frequency.
Skewed X chromosome inactivation is a condition where there is preferential
inactivation of either the maternal or the paternal derived X chromosome. An
extremely skewed XCI of ≥ 90 % has been reported in females with a history of RPL
(21, 22). Because there are many potential causes of skewed XCI, the reason for an
association between RPL and skewed XCI is not clear and may be heterogeneous. X
linked mutations have been clearly associated with skewed XCI and RPL in some
families (23).
1.3.2.7 Other risk factors
Advanced maternal age is a significant risk factor for pregnancy loss. Fetal loss is
high in women in late thirty years or older, irrespective of reproductive history. The
risk of ectopic pregnancy and stillbirth also increase with increasing maternal age
(24).
Smoking and alcohol are also cited as risk factors for RPL. Organohalide pesticides
and organic solvents have been implicated in RPL. Sporadic miscarriages have also
0
10
20
30
40
50
60
70
80
90
100
H+/W-
(inh)
W+/H-
(inh)
H+/W-
(act)
W+/H-
(act)
Controls
Cases
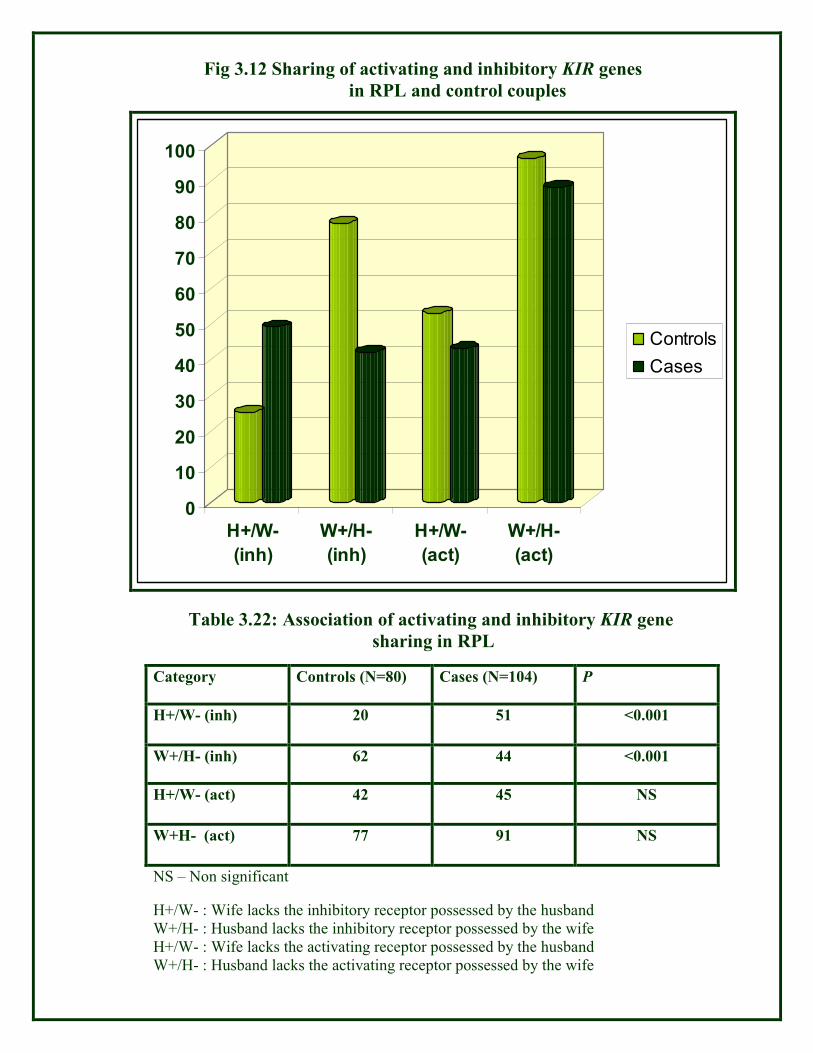
Fig 3.12 Sharing of activating and inhibitory KIR genes
in RPL and control couples
Category Controls (N=80) Cases (N=104) P
H+/W- (inh) 20 51 <0.001
W+/H- (inh) 62 44 <0.001
H+/W- (act) 42 45 NS
W+H- (act) 77 91 NS
NS – Non significant
Table 3.22: Association of activating and inhibitory KIR gene
sharing in RPL
H+/W- : Wife lacks the inhibitory receptor possessed by the husband
W+/H- : Husband lacks the inhibitory receptor possessed by the wife
H+/W- : Wife lacks the activating receptor possessed by the husband
W+/H- : Husband lacks the activating receptor possessed by the wife

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy
loss in South Indian Population 9
* Dubey et al 2005 (20)
Type of abnormality Chromosomal Constitution* Sex
Structural chromosomal abnormalities – (71%)
Reciprocal
Translocation
46,XX t(1;5) (p33:q35) F
46,XX t(2;11) (q21:q24) F
46,XX t(3;11) (q26:11pter) F
46,XX t(3;17) (p22:p13) F
46,XX t(6;X) (q32:q22) F
46,XX t(6;12;13)
(q22:q21:q23:q12)
F
46,XX t(6;18) (q27:q21) F
46,XY t(7;8) (q11:p11) M
46,XY t(7;13) (pter:q13) M
46,XX t(8;15) (q12:p11) F
Robertsonian
translocation
46,XX t(13;14) F
46,XX t(21;22) F
46,XX t(22;22) F
Deletion + marker 46,XYdel(3) (pter → q25) + marker M
46,XYdel(10) (pter → q 22) + marker M
Inversion 46,XY inv (4) (p15:q13) M
Numerical chromosomal abnormalities – 29%
47,XXX / 46,XX F
47,XXY / 46,XY M
47,XXX / 46,XX / 45,XO F
47,XXX / 46,XX F
46,XX / 45,XO F
Other Polymorphic Variants
Polymorphic
chromosomal variants
Yq+, Yq-, Pericentric 1qh+, inv (9), 9qh+, 15p+, 16qh+,
22p+, Fragile sites
Table 1.2 - Chromosomal abnormalities in blood lymphocytes
of couples with RPL

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy
loss in South Indian Population 10
been reported with intrauterine diethylstilbestrol exposure. Though psychological
factors, stress and grief have been implicated in RPL their exact role is unclear. (25)
Though all the above described etiological factors have been proposed to attribute to
the risk of RPL, the unexplained etiology in 50% of the cases stressed the need to
identify the role of additional genetic alterations which might collectively result in
RPL.
1.4 Genetic polymorphisms in RPL
The search for genetic markers of disease susceptibility often utilizes the candidate
gene approach, where a gene is targeted based on the properties and metabolic
pathways of its protein product (26). When these genes are polymorphic and the
variants are distributed differently across populations, interest in them increases since
variation in the DNA sequence could alter protein function and result in variations in
disease risk. Recent studies have revealed an association between RPL and genetic
polymorphisms in metabolic enzymes, cytokines, coagulation factors,
methylenetetrahydrofolate reductase and histocompatibility antigens.
1.4.1 Genetic polymorphisms in Immunomodulatory genes
Human immune system is well known for its ability to discriminate between non-self
antigens like that of infections and solid tissue transplantation over the self antigens.
Over the years it has become evident that highly polymorphic molecules encoded by
genes located on chromosome 6 termed Major Histocompatibility Complex (MHC),
referred to as Human Leukocyte Antigens (HLA) in humans, play a key role in
immune rejection (27, 28). According to the classic transplantation rules, MHC
mismatched transplants are rejected by the recipient, whereas MHC matched
transplants are accepted. A notable, but as yet unresolved exception from the classic
transplantation paradigms is pregnancy, where a semiallogeneic fetus thrives in the
mother’s womb. Though a detailed mechanism behind the maternal–fetal
immunotolerance remains elusive, specific and direct interactions between maternal
and fetal cells is suggested to play an important role (29). Recent evidence strongly

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy
loss in South Indian Population 11
supports active immune tolerance of the fetus during pregnancy (30, 31). The major
genes regulating this immunomodulation include HLA, Natural Killer cell receptors
and cytokines.
1.4.1.1.1 Human Leukocyte Antigen
HLA genes are the main genetic determinants of the repertoire of individual immune
responses. HLA complex plays a critical role throughout pregnancy by influencing
gamete development, embryo cleavage, blastocyst and trophoblast formation,
implantation, fetal development and survival. The HLA complex contains
approximately 4 Mb of DNA on chromosome 6p21 and is composed of three classes
namely class I, II and III (32, 33) (Fig 1.4). The expressed class I genes are
subdivided into class Ia, which includes HLA-A, -B, and -C, and class Ib, which
includes HLA-E, -F, and -G. HLA class II includes the DR, DQ and DP (34, 35).
Human trophoblast cells express one class Ia molecule (HLA-C) and all three class Ib
molecules. Of the HLA class Ib molecules expressed by trophoblast cells, HLA-G
was the first identified antigen of great interest and currently a focus of experimental
evaluation.
A C DR B E G F DQ DP
Class II Class III Class I
Fig 1.4 HLA Complex
(Darkly stained boxes indicate less polymorphic genes)

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy
loss in South Indian Population 12
1.4.1.1.2 Structural features of HLA-G
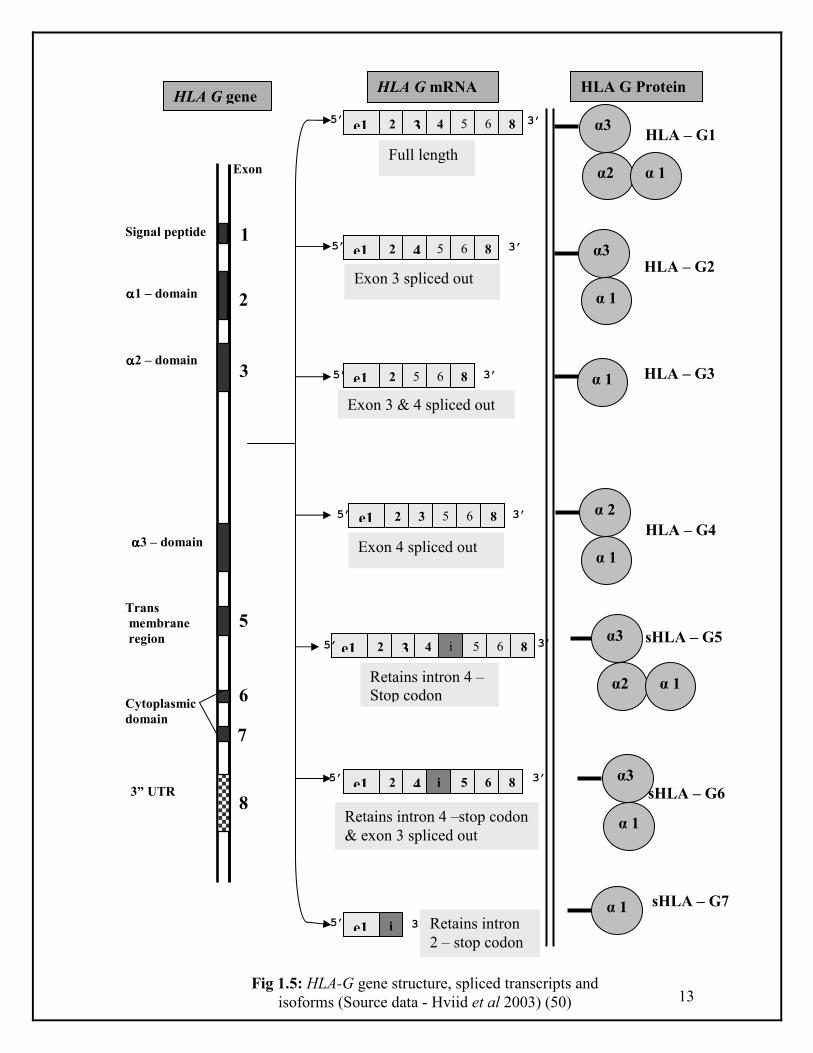
Although the genomic structure of HLA-G is similar to other class I genes, it is unique
in most respects. HLA-G gene consists of eight exons encoding a signal peptide (exon
1), the α1, α2 and α3 domains (exons 2, 3, and 4 respectively), the transmembrane
domain (exon 5) and the intracellular domains (exons 6 and 7) (Fig. 1.5). A premature
stop codon in exon 6 results in a truncated cytoplasmic segment of the HLA-G
protein, which results in the loss of 19 amino acid residues which are highly
conserved in other classical HLA-loci (36). The functional significance of the
shortened cytoplasmic tail of HLA-G is unknown.
Another unique feature of HLA-G is that it encodes multiple isoforms as a result of
alternative splicing. Seven alternatively spliced transcripts have been identified, of
which four are predicted to encode membrane bound proteins (HLA G1, HLA G2,
HLA G3, HLA G4) and three are predicted to encode soluble proteins (HLA G5,
HLA G6, HLA G7). Five of the seven transcripts that result from alternative splicing
are shown in Fig.1.5. The full-length isoform HLA-G1 is structurally similar to the
other class I genes, except for the truncated cytoplasmic tail. The G2 isoform results
from the removal of exon 3 and subsequent homodimerization to form a HLA class II-
like structure (34, 37). These two isoforms with an inclusion of intron 4 sequences in
the mature mRNA with an additional of 21 amino acids are expressed as soluble
secreted proteins known as HLA G5 and G6, respectively (38). HLA G3 results from
the removal of exons 3 and 4. HLA G4 and G7 mRNAs are not abundant in placentas.
Exon 4 (encoding the α 3 domain) is spliced out of the HLA G4 transcript; the HLA
G7 transcript includes exon 2 and part of intron 2 and is predicted to encode a small
soluble isoform. The expression of class Ia antigens is ubiquitous whereas expression
of class Ib antigens may be tissue/ organ-specific and/or conditional (39, 40).
1.4.1.1.3 HLA G Polymorphism
HLA G has relatively little polymorphism in its coding region compared to the
classical class I genes. 13 polymorphisms have been identified in exons 1–4 and one
in the 3’UTR (Fig. 1.6). Polymorphisms at codon 31 in the α 1 domain (Thr3Ser),
13
αααα2 – domain
αααα3 – domain
Cytoplasmic
domain
3” UTR
Exon
1
2
3
5
6
7
8
Signal peptide
αααα1 – domain
Trans
membrane
region
e1 2 3 4 5 6 8
e1 2 4 5 6 8
e1 2 5 6 8
e1 2 3 5 6 8
e1 2 4 4 5 6 i
e1 i
e1 2 3 4 5 6 8 i
5 6 8
α3
α2 α 1
α3
α2 α 1
α3
α 1
α 1
α 2
α 1
α 1
α3
α 1
5’
3’
3’
5’
3’ 5’
Full length
HLA G gene HLA G mRNA HLA G Protein
5’ 3’
5’ 3’
3’ 5’
5’ 3’
Exon 3 spliced out
Exon 3 & 4 spliced out
Exon 4 spliced out
Retains intron 4 –
Stop codon
Retains intron 4 –stop codon
& exon 3 spliced out
Retains intron
2 – stop codon
HLA – G1
HLA – G2
HLA – G3
HLA – G4
sHLA – G5
sHLA – G6
sHLA – G7
Fig 1.5: HLA-G gene structure, spliced transcripts and
isoforms (Source data - Hviid et al 2003) (50)

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy loss in
South Indian Population
14
at codon 110 in the α 2 domain (Leu3Ile), and at codon 258 in the α 3 domain
(Thr3Met) result in an amino acid substitution. Polymorphisms at nucleotide 15 and
36 in exon 1, at codons 35, 57 and 69 in exon 2, at codons 93, 100 and 107 in exon 3,
and at codon 188 in exon 4 do not alter the amino acid sequence of the protein. In
contrast to the class Ia HLA loci, amino acid substitutions at codons 31 and 110 in the
α 1 and α 2 domains, are conservative changes that occur in residues that are not
predicted to interact with bound peptide or T cell receptor (41). The third amino acid
polymorphism at codon 258 is a non-conservative substitution in the α 3 domain that
is highly conserved in the class Ia genes (39). It is located in the pleated sheet
structure of the α 3 domain, where it might affect recognition of CD8 in the HLA G1
and G5 isoforms, or binding to CD4 in the HLA G2 and G6 isoforms (42). A single
base pair (bp) deletion at nucleotide 1597 causes a frameshift at amino acid 130 (43),
resulting in nonfunctional HLA G1 and G5 proteins (41, 44). This null allele has been
associated with increased risk for recurrent pregnancy loss (45, 46) suggesting that
HLA G1 and/or G5 proteins play an important role in the maintenance of pregnancy
and that reduced levels of one or both is a risk factor for RPL.
The polymorphisms that alter the protein sequence define five alleles, called G*0101,
G*0103, G*0104, G*0105N, and G*0106. Silent variation within these allelic classes
defines subtypes, referred to as G*01011, G*01012, etc (Table 1.3). A 14 bp
insertion/deletion polymorphism in the untranslated exon 8 of the HLA G gene was
first described by Harrison and colleagues (47), but has recently been shown to
influence mRNA transcript size and stability. The presence of the 14 bp insertion
allele generates a 92 bp deletion in the 3’UTR of the mRNAs, possibly because it acts
as a cryptic splice site (48). Transcripts with the 92 bp deletion were associated with
more stable mRNA in JEG-3 cells and in an M8 cell line (49). Further, the relative
abundance of the alternatively spliced transcripts may be influenced by
polymorphisms in HLA G (50).

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy loss in South Indian Population
15
** - Polymorphism at codon 188 of Exon 4
Allele Exon 2 Exon 3
Codon 31 Codon 54 Codon 57 Codon 69 Codon 93 Codon 107 Codon 110 Codon 130
G*01011 ACG CAG CCG GCC CAC GGA CTC CTG
G*01012 ACG CAG CCA GCC CAT GGA CTC CTG
G*01013 ACG CAG CCA GCC CAC GGT CTC CTG
G*01014 ACG CAG CCG GCT CAC GGA CTC CTG
G*01015 ACG CAG CCG GCC CAC GGT CTC CTG
G*01016** ACG CAG CCG GCC CAC GGA CTC CTG
G*01017 ACG CAG CCA GCC CAT GGT CTC CTG
G*01018 ACG CAG CCA GCC CAC GGA CTC CTG
G*0102 ACG CGG CCG GCC CAC GGA CTC CTG
G*0103 TCG CAG CCG GCC CAC GGA CTC CTG
G*01041 ACG CAG CCA GCC CAC GGA ATC CTG
G*01043 ACG CAG CCG GCC CAC GGA ATC CTG
G*0105N ACG CAG CCA GCC CAT GGA CTC -TG
Table 1.3: HLA G Polymorphism

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy
loss in South Indian Population 16
1.4.1.2.1 Natural Killer cells
Natural killer (NK) cells represent a subset of lymphocytes found in a variety of
tissues in humans, including deciduas (51). Because of their increased presence and
direct contact with trophoblast, NK cells have been considered to be a crucial cell
population for pregnancy (52). The property of NK cells to recognize and lyse target
cells and discriminate them from normal cells is mediated by the activating and
inhibiting NK receptors (53). The NK cell receptors (NKRs) belong to three main
families: the killer immunoglobulin-like receptors family (KIR) (54, 55), the C-type
lectin family (CD94 ⁄ NKGs) and the immunoglobulin-like transcripts (ILTs or LIRs)
(56, 57). A number of NKRs of each family as well as their ligands have been
identified and it is known that the functions of NK cells are repressed by specific
recognition of HLA class I molecules. Multiple combinations of NKRs are co-
expressed by individual NK cell clones. The NKR repertoire varies among different
individuals and human NK cells employ receptors of all receptor families for the
recognition of different human leukocyte antigen (HLA) class I molecules. Further,
every mature NK cell is predicted to bear at least one type of dominant inhibitory
receptor for a self-MHC class I product, to prevent auto-reactivity against normal
cells (58, 59, 60).
1.4.1.2.2 Killer cell immunoglobulin-like receptors (KIR)
In humans, an important superfamily of NK receptor is KIR (Killer cell Ig-like
receptor). The KIR are members of the immunoglobulin (Ig) super gene family
currently comprised of 15 genes and two pseudogenes encoded within a 100-200 Kb
region of the Leukocyte Receptor Complex (LRC) on human chromosome 19 (Fig
1.6) (61, 62). Based on the number of Ig-domains in the extracellular region, KIR
receptors can be divided into either three Ig domains containing KIR (KIR3D) or two
Ig domains containing KIR (KIR2D) (63, 64). Depending on the cytoplasmic tail
length, they can be further grouped into either long (L) tail KIRs having inhibitory
function, or short (S) tail KIRs with activating function. Long tail KIRs have one or
two immunoreceptor tyrosine-based inhibitory motifs (ITIM) and transducer
inhibitory signals. In contrast, the short tail KIRs have no ITIM motif but possess a
charged residue in the transmembrane region that mediates association with DAP12

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy
loss in South Indian Population 17
(DNAX-activating protein of 12kDa). The DAP12 contains immunoreceptor tyrosine-
based activation motif (ITAM) and transduces activating signals (59).
1.4.1.2.3 KIR Polymorphism
A peculiarity of KIR genetics is their haplotypic polymorphism, i.e. the presence or
absence of some KIR genes on individual chromosomes (57). There is an extensive
polymorphism among KIR haplotypes, which differ not only in nucleotide
sequence but also in gene content. Different haplotypes carry different numbers of
KIR genes, some with few or no activating KIR’s (A haplotypes) and others with
several activating KIR’s (B haplotypes). In addition, each NK cell expresses its
own repertoire of KIR genes.
1.4.1.2.4 KIR Haplotypes
All known KIR haplotypes are flanked at their centromere by KIR3DL3 and at their
telomeric end by KIR3DL2, together with the centric KIR3DP1 and KIR2DL4. These
constitute the framework genes, which limit two regions of variable KIR gene content
where the remaining KIR genes are located (61, 62, 65). All KIR genes are arranged
in a head to tail fashion approximately 2.4 Kb apart from each other (62). Based on
their gene content two kinds of KIR haplotypes, A and B have been described (Fig
1.7). The “A-group haplotypes” are relatively simple, consisting of 7 functional
genes. They include inhibitory KIR genes for all major HLA class I specificities, and
have one or two activating KIRs (KIR2DS2 and/or KIR2DS4). The “B-group
haplotypes” contain 9-12 genes. They are marked by KIR2DL5 gene (66) and most of
the KIR genes in the B-group haplotypes are activating type. B-group haplotypes may
not carry inhibitory KIR genes for some HLA specificities. Both groups of haplotypes
conserve only three ‘framework’ genes (62, 67).

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy
loss in South Indian Population 18
During pregnancy, maternal–fetal interface is characterized by an unusual
combination of HLA and NKR expression on extravillous cytotrophoblasts (EVT) and
the pattern of maternal leukocyte distribution in the decidua. Further, it is reported
that the interaction between the HLA G and the KIR play an important role in the
maintenance of pregnancy.
1.4.1.3 Cytokine gene polymorphisms
1.4.1.3.1. Interleukin 1 (IL – 1 Receptor Antagonist)
Interleukin 1 (IL-1) is a central proinflammatory cytokine, produced by monocytes,
macrophages, and epithelial cells (68). Secretion of IL-1 leads to a proinflammatory
cascade, including TH-1 proliferation, production of tumor necrosis factor (TNF)-α,
interferon (IFN)-γ, IL-2, and IL-12. The level of IL-1 is modulated by an endogenous
factor, IL-1 receptor antagonist (RA). An 86–base pair tandem repeat in intron 2 of
IL-1 RA gene has been associated with increased risk of RPL (69).
3D
L3
3D
P1
2D
L4
3D
L2 2D
S4 3D
L1
2D
L3
2D
P1
2D
L1
Group A
3D
L3
3D
P1
2D
L4
3D
L2 2D
S2
3D
S1 2D
L2
2D
S5
2D
S3
2D
S1
2D
L5
Group B
Fig-1.7: Genomic organization of two groups of KIR haplotypes
Genes conserved in both groups of haplotypes are shown in dark boxes.

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy
loss in South Indian Population 19
1.4.1.3.2. Transforming Growth Factor Beta 1 (TGF Beta 1)
TGF-beta1 is an immunosuppressive cytokine. This cytokine may contribute to
regulation of maternal immune responses directed against the fetal allograft, and
thereby prevent immunological rejection of the fetus. Studies have shown that TGF-
beta1 negatively regulates the primary immune response induced by Th1 but not Th2
memory cells (70, 71). Several polymorphisms have been reported in TGF beta1 and
certain polymorphic alleles are suggested to be associated with an elevated serum
level of TGF beta1 both in vitro and in vivo (72, 73). Polymorphisms at nucleotides
29 and 74 resulting in T to C and G to C substitutions respectively are reported to
affect TGF beta1 secretion and have been implicated in RPL (74)
1.4.1.3.3. Tumor necrosis factor alpha (TNF alpha)
Tumor necrosis factor is a potent cytokine with a wide range of proinflammatory
activities. It is classically produced by monocytes/macrophages although other cell
types, such as T and B cells, also produce significant amounts (75, 76). Circulating
levels of TNF alpha are higher in patients with pregnancy loss compared to those with
a successful pregnancy, suggesting that this cytokine may be an etiologic factor in
RPL (77, 78, 79). Additionally, functional polymorphisms at position -308 and -863
in the promoter region of the human TNF alpha gene have been reported to be
associated with altered TNF alpha activity (80). Though a few studies have reported
these polymorphisms to increase the risk of RPL, others have found no association
(81, 82)
The various candidate genes of the immune system, their function and the effect of
their polymorphisms in RPL have been tabulated (Table 1.4)
1.4.2 Genetic polymorphisms in placental networking and vascular
remodeling
Successful pregnancy requires the development of a complex maternal and fetal
vascular network to support increasing oxygen and metabolic demands of the growing
fetus. There are three stages in human placental vascular development: an initial stage

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy loss in South Indian Population
20
Candidate Genes Gene loci Polymorphism Gene function Effect of Polymorphism
HLA-G
(Aldrich et al 2000) (45)
6p21 HLA G*010103
HLA G*0105N
Immunomodulation at the
feto maternal interface
Low secretor allele
HLA –DR
(Takakuwa et al 2003)(83)
6p21 HLA DRB1*15
HLA DRB1*04
Immune response to the
foreign antigens
Maternal immune response
against the fetus
HLA –B
(Christiansen et al 1997)(84)
6p21 Bw4 Influence both T-cell-
mediated and NK-cell-
mediated rejection
Maternal immune response
against the fetus
NKRs
(Clark et al 2005) (52)
19q13 2DL1, 2DL2, 2DL3 Protect trophoblast from
maternal NK cells attack
and to control trophoblast
migration and placentation.
NK cell mediated cytotoxic
effect on trophobalst cells
TNF – alpha
(Ragupathy et al 2000) (78)
6p21 - 863 C/A
- 308 G/ A
Potent cytokine with a
wide range of
proinflammatory activities.
Associated with altered TNF
promoter activity
Table 1.4: Polymorphisms in immunomodulatory genes reported in RPL

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy loss in South Indian Population
21
TGF Beta –1
(Amani et al 2004)(74)
19q13
-800 G/A
-509 C/T
Modulation of cellular
growth and differentiation,
immunoregulation and
extra cellular matrix
formation
Altered production of TGF
beta
Il-1 RA (Tarlow
et al 1993) (69)
2q14 86bp Tandem
repeat in Intron 2
Competitive antagonist for
IL-1
Altered Function
IL-18
(Dinarello et al 1998)(68)
11q22 -607 (C/A)
-137 (G/C)
Immune regulation at
maternal-fetal interface
Implantation failure in cases
of absence or over activation

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy
loss in South Indian Population 22
of vasculogenesis, subsequent branching and then non branching angiogenesis (85,
86). Vasculogenesis involves the de novo formation of new vessels, whereas
angiogenesis refers to the formation of capillaries from preexisting vessels (87). The
development of a normal functioning placental vascular network requires a
remarkable degree of coordination between different cell-specific growth factors and
signals exchanged between these cells (88). Pregnancy also requires a balance
between coagulation and fibrinolysis in order to avoid excess fibrin accumulation in
placental vessels and intervillous spaces as well as to secure fibrin polymerization and
stabilization of the placental basal plate. Adequate fine-tuning of fibrinolysis is
mandatory to prevent hemorrhage (89, 90). Minimal alterations in the fibrinolysis
cascade leading to either hypo or hyperfibrinolysis are also suspected to interfere with
placentation in early pregnancy (91).
Although numerous factors have been implicated in placental vascular network,
recent observations including gene knockout studies in mice have led to the
identification of the major factors regulating the placental vascular network. It
includes the growth factors like Vascular Endothelial Growth Factor (VEGF), which
helps in the cell proliferation and cell differentiation (92), Angiotensin Converting
Enzyme (ACE), which is involved in the physiological remodeling of spiral arteries
(90) and Methylene Tetrahydrofolate Reductase (MTHFR), the methyl donors for
vascular development (93) (Fig 1.8). The other important genes in this pathway
include Factor V Leiden, PAI1 and prothrombin.
1.4.2.1.1 Vascular endothelial growth factor (VEGF)
VEGF is an angiogenic factor and a prime regulator of endothelial cell proliferation. It
exhibits a crucial role in physiological vasculogenesis and vascular permeability (94).
It belongs to Platelet derived growth factor (PDGF) super family and has more than
10 isoforms namely, VEGF-121, VEGF-165, VEGF-189, VEGF-206, -B, -C, -D, -E,
PIGF-I and PIGF-II, ranging in weight from 35-44 kDa. Each binds to a specific
combination of endothelial cell surface receptor known as VEGFR-1, -2 and -3 (95).
Circulating VEGF binds to VEGFR on endothelial cells, triggering a tyrosine kinase
pathway leading to angiogenesis (96). VEGF also protects endothelial cells from

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy
loss in South Indian Population 23
apoptotic cell death during embryonal development and withdrawal of VEGF by
targeted inactivation of the VEGF gene is found to result in massive endothelial cell
apoptosis, leading to severe hemorrhage (97). Thus VEGF/VEGFR system is one of
the best characterized regulators of angiogenesis (98).
1.4.2.1.2 Polymorphisms in VEGF gene
VEGF gene is located on chromosome 6p21 and is comprised of a 14 kb coding
region with 8 exons and 7 introns (Fig 1.9). Many polymorphisms have been
identified in the VEGF gene and a few of them have been correlated with variation in
VEGF production (99,100).
1.4.2.2.1 Angiotensin converting enzyme (ACE)
ACE is a zinc-metalloproteinase occurring both as a membrane-bound ectoenzyme on
the surface of vascular endothelial and renal epithelial cells as well as a
circulating
enzyme in the plasma (101,102). ACE is a key component of the Renin Angiotensin
system (RAS), which regulates blood pressure, fluid and electrolyte balance (103). In
addition, it plays an important role in activating angiotensin I to angiotensin II.
Evidence from animal models have suggested that angiotensin II stimulates
neovascularization (104,105) by promoting arteriolar smooth muscle cell proliferation
(106). Further, angiotensin II may act as a mitotic factor by inducing or regulating
gene expression in cell cycle progression (107)
Placental and fetal membranes are important sites for the synthesis of the RAS
components. In vivo and in vitro studies suggest the role of RAS in haemostasis
through different mechanisms, including an influence on fibrinolysis, platelet
aggregation and blood clotting activation (108, 109). Studies have reported an
association between the ACE genotypes and increased risk of thrombophilia (110), a
condition predisposing to adverse pregnancy outcomes. Moreover, ACE by
bradykinin degradation reduces nitric oxide levels, thereby contributing to endothelial
dysfunction. In addition to regulating the vasomotor functions, RAS is also involved

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy
loss in South Indian Population 24
in key events of the inflammatory process (111) by increasing vascular permeability
and contributing to the recruitment of inflammatory cells.
1.4.2.2.2 Polymorphisms in ACE gene
ACE gene located on 17q23, spans 21 kb and consists of 26 exons and 25 introns (Fig
1.10). About 47% of interindividual variability of plasma ACE concentration is
determined by the presence [insertion (I)] or absence [deletion (D)] of a 287-bp
Alu-
type sequence in intron 16 of the ACE gene (112). Homozygous I allele displays as
low as half of the ACE level compared with the homozygous
D allele, whereas the I/D
heterozygotes display an intermediate level (107). Though the exact mechanism of
ACE polymorphism on the serum ACE level has not been identified, studies have
suggested that the insertion/deletion may be in linkage disequilibrium with regulatory
elements of the ACE gene, or that the insertion itself might modify the splicing
process of the ACE precursor mRNA by interfering with the lariat formation (113).
1.4.2.3.1 Methylenetetrahydrofolate reductase (MTHFR)
MTHFR catalyzes the reduction of 5, 10-methylenetetrahydrofolate to 5-
methyltetrahydrofolate, the predominant circulatory form of folate and the carbon
donor for remethylation of homocysteine to methionine (114,115). Methionine is
involved in the formation of S-adenosylmethionine, a principal methyl donor in cells
for DNA methylation. S-adenosyl methionine is also an allosteric inhibitor of MTHFR
and an activator of cystathionine ß-synthase. Although the effects of folate
metabolism are still incompletely understood, there is growing evidence that normal
MTHFR activity is essential to maintain the pool of available circulating folate and
methionine as well as prevent accumulation of homocysteine (116).
Hyperhomocysteinemia and reduction in S-adenosylmethionine that might result from
altered MTHFR activity are associated with the impaired DNA methylation and gene
expression leading to defective chorionic villous vascularization.
1.4.2.3.2 Polymorphisms in MTHFR gene
The MTHFR gene is mapped to chromosome 1p36 (Fig 1.11). The cDNA sequence is
2.2 kilobases long and consists of 11 exons. A common functional polymorphism

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy
loss in South Indian Population 25
which alters the MTHFR activity is the C to T substitution in the 677 nucleotide
(117,118). Which results in alanine to valine substitution within the catalytic domain
of MTHFR which results in decreased enzyme activity.
1.4.2.4 Activated protein C and Factor V Leiden
Protein C is a key component in the anticoagulant pathway which when activated
inhibits the actions of coagulation factors V and VIII. Congenital
APC resistance is
almost exclusively due to a single point mutation (G A) at nucleotide position 1691
in the factor V gene, which results in a mutated form of factor V, known as factor V
Leiden (119). Mutated factor V is resistant to inactivation by APC, resulting in
increased thrombin generation and a hypercoagulable state leads to increased
susceptibility for venous thrombosis (120). The prevalence of factor V Leiden and
acquired APC resistance among women with recurrent miscarriage has been variably
reported to be either similar to or increased compared to controls
(121,122).
1.4.2.5 Plasminogen Activator Inhibitor 1 (PAI1)
Plasminogen activator inhibitor-1 is the principal inhibitor of tissue plasminogen
activator (tPA) and urokinase plasminogen activator (uPA), the activators of
plasminogen and hence fibrinolysis. Endothelial PAI-1 expression is modulated by a
4G/5G polymorphism in the PAI-1 promoter, which is 675 bp upstream from the start
site of transcription. Angiotensin II plasma levels also influence PAI-1 expression.
Homozygosity for the 4G allele of the PAI-1 gene increases the risk for pregnancies,
predisposing to prematurity, intrauterine growth retardation, miscarriage and stillbirth
(123).
1.4.2.6 Prothrombin
Prothrombin is the precursor to thrombin in the coagulation cascade. Thrombin is
required to convert fibrinogen into fibrin, which is the primary goal of the coagulation
cascade. G to A transition at nucleotide position 20210 in the 3’untranslated region of
the prothrombin gene has been associated with increased plasma prothrombin
concentrations, which increases risk for venous and arterial thrombosis (124). Though

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy
loss in South Indian Population 26
prothrombin mutation has been identified as a risk factor for RPL in some studies
(125), others have reported lack of association (126).
The various candidate genes of the vascular remodeling, their function and the effect
of their polymorphisms in RPL are summarized in Table 1.5.
1.4.3 Genetic polymorphisms in Xenobiotic metabolism
RPL is believed to be associated with maternal exposure to various xenobiotics like
environmental toxins and teratogens. Heavy metals such as lead and mercury, organic
solvents, alcohol, and ionizing radiation are environmental teratogens, and exposure
to these could contribute to pregnancy loss. Caffeine, cigarette smoking, and
hyperthermia are suspected teratogens, while the teratogenic impact of pesticides
remains unknown (132). Increased caffeine intake and deficient detoxification has
been positively related to an enhanced risk of RPL (133). Further, maternal exposure
to dioxin has also been associated with increase in fetal loss and reduction in birth
weight in experimental animal studies (134). However, there are only a few
epidemiological studies on the relation between the maternal dioxin and pregnancy
outcome in humans (135)
In addition to the lethal effects of the various xenobiotic substances described above,
oxidative stress also results in susceptibility to RPL. The increase in placental blood
circulation towards the end of first trimester results in an enhanced oxidative load,
which facilitates embryonic differentiation and development (136). However, an
excessive oxidative load along with inefficient antioxidant defense generates
abundant free radicals that could induce reproductive toxicity and thus prove lethal to
the embryo, resulting in RPL (137). Hence, the decidual or placental detoxification
system should be efficient and functional so as to protect the conceptus from
increased free radicals as well as xenobiotics.

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy loss in South Indian Population
27
Candidate Genes Gene loci Polymorphism Gene function Effect of
Polymorphism
VEGF
(Papazoglou et al 2005) (127)
6p21 -2578 A/C Growth factor that is active
in angiogenesis and
vasculogenesis
Altered VEGF production
ACE
(Buchholz et al 2003) (123)
17q23 Insertion/ Deletion at
intron 16
Mediates conversion
angiotensin I to angiotensin
II .This enhances
vasoconstrictor activity of
angiotensin
Hypofibrynolysis
MTHFR
(Holmes et al 1999;
Unfried et al 2002) (128,129)
1p36 Codon 119 G to T
transition
Mediates conversion of
homocysteine to
methionine
Accumulation of
homocysteine
Factor V Leiden
(Townson et al 1997) (130) 1q21
G1691A
Produces factor V, which
converts prothrombin to
thrombin.
Resistance to activated
protein C
Table1.5: Polymorphisms in vascular networking and placental remodeling genes reported in RPL

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy loss in South Indian Population
28
PAI1 (Buchholz et al 2003) (123) 7q21 4G/5G in the promoter Regulates fibrynolysis
Hypofibrynolysis
Prothrombin
(Hohlagschwandtner et al 2003)
(131)
11p11 G20210A
Blood clotting protein that is
needed to form fibrin
Increased prothrombin
concentration

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy
loss in South Indian Population 29
1.4.3.1 Phase I metabolism
Phase I metabolism the first enzymatic defense against foreign compounds is
mediated mainly by the CYP450 super gene family of enzymes, such as CYP1A1,
CYP1A2, CYP1B1 etc. In a typical phase I reaction, CYPP450 uses oxygen and
NADH as a cofactor to add a reactive group, such as a hydroxyl radical. As a
consequence of this step reactive molecules that are more toxic than the parent
molecule are produced (138).
1.4.3.2 Phase II metabolism
Phase II metabolism follows phase I activation, in which the reactive intermediates
are detoxified into water-soluble compounds that can be excreted through urine or
bile. Some of the phase II enzymes are acetyltransferases, glutathione S transferases,
uridine 5'-diphosphoglucuronosyl transferases, sulfotransferases, aldoketoreductases,
transaminases and hydrolases (139).
When the body is confronted with a high xenobiotic load, the phase I and/or phase II
enzymes involved in detoxifying this compound are induced, leading to xenobiotic
detoxification. The variations in metabolic activities in each phase or in the
coordination of the two phases affect the clearance of toxic metabolites. Most of the
phase I and phase II enzymes are polymorphic and the variants differ in their ability to
process the xenobiotics. Thus, inherited differences in the effectiveness of activation
and detoxification play a crucial role in RPL Fig 1.12.
1.4.3.3.1 Cytochrome P4501A1 (CYP1A1)
CYP1A1 is a major extra hepatic enzyme involved in the oxidation of the common
environmental toxicants such as polycyclic aromatic hydrocarbons (PAH) like
benzopyrene, polychlorinated biphenyls, etc. (140). Most of these aromatic
hydrocarbons bind to Aryl hydrocarbon (Ah) receptor. Following ligand binding, the
Ah receptor dimerizes with Ah receptor nuclear translocator, and there by acquires the

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy
loss in South Indian Population 30
ability to interact with xenobiotic response elements that enhance transcription of
genes encoding CYP1A1 (141, 142, 143). Oxygenation by P4501A1 serves as an
initial step in the metabolic conversion of the substrates to water-soluble metabolites
for excretion from the body. However, oxygenation of PAHs often generates
electrophilic arene oxide, dioepoxide and other reactive species which damage the
DNA and/or affect protein functions, leading to adverse effects.
1.4.3.3.2 CYP1A1 polymorphism
CYP1A1 gene mapped on 15q22-24 spans 10kb and consists of 7 exons. The CYP1A1
gene is polymorphic and four SNPs have been reported. First and the most important
polymorphism is T 3801 C substitution in the 3’ untranslated region (UTR) referred to
as m1 allele. As this polymorphism creates a restriction site for Msp1 enzyme, it is
also referred to as Msp1 polymorphism. Second, A 2455 G transition in exon 7
referred to as the m2 allele, leads to the substitution of Val for Ile at codon 462 in the
heme-binding region. Both the m1 and m2 allele have been reported to exhibit higher
catalytic activity (144).Third polymorphism is T 3205 C in intron 7 referred to m3
allele and is African-American specific. The fourth one is C 2453 A in exon 7
referred to as m4 allele, which leads to Thr to Asn substitution but its effect on
enzyme activity has not been clarified (145). These SNPs are represented in Fig 1.13.
Fig 1.12 – Xenobiotic metabolism highlighting the genes of interest
AngiogenesisAngiogenesisAngiogenesisAngiogenesis
Free
Radicals
Phase I
Reactive
Intermediates
Phase II
Water
Soluble
Compounds
Excreted
Activation Detoxification
CYP1A1 GSTM1
GSTT1
GSTP1

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy
loss in South Indian Population 31
1.4.3.4 Glutathione S Tranferase (GST) Family Enzymes
The GST family of enzymes are important members of phase II detoxification. They
catalyze the conjugation of a wide range of electrophilic substances to glutathione,
facilitating their elimination from the body. Moreover, GST enzymes also play an
important role in regulation of reduced glutathione levels, and thereby redox reserves
of the individual. Hence any decrease in the GST activity can lead to accumulation of
the products of phase I activity, which can cause severe damage. GSTs are present in
large amounts in many tissues including those of the genital tract, placenta and
deciduas (146, 147). They are expressed very early in the embryonic development and
hence believed to play a crucial role in female reproduction (148).
In humans eight distinct gene families encode GSTs. There are currently five putative
α class genes encoding GSTA1, GSTA2, GSTA3, GSTA4 and GSTw. The GSTP
class contains a single gene encoding the GSTP1 protein, and the θ class consists of
two genes encoding the GSTT1 and GSTT2 proteins and µ class consists of five genes
encoding GSTM1, M2, M3, M4 and M5. Of these GSTM1, GSTT1 and GSTP1
genes are frequently analyzed in disease susceptibility studies (147).
1.4.3.4.1 GSTM1 gene polymorphism
The GSTM1 gene belongs to the GSTµ class gene family, members of which are
located on a 100-kb gene cluster arranged as 5’-GSTM4-GSTM2-GSTM1-GSTM5
GSTM3–3’ at 1p13.3 (149). The GSTM1 gene consists of eight exons and is flanked
by two identical 4.2-kb regions as shown in Fig 1.13. The presence or absence of the
gene constitutes the polymorphism. The GSTM1 deletion caused by homologous
recombination involving the 5’ and 3’ 4.2kb repeats, results in 16kb deletion
containing the GSTM1 gene. The point of deletion has not been localized precisely
because of the high sequence identity repeats (149). Deletion of the GSTM1 gene
frequently affects both the alleles, resulting in the so-called null genotype GSTM-/-.
Distinct ethnic differences have been observed in the distribution of GSTM1 null
genotypes and the GSTM1 deletion frequencies range from 22.4% in South Indians

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy
loss in South Indian Population 32
(150), 28% in Africans (151), 33% in North Indians (152), 46.9% in Italians (153),
48.8% in Caucasians (154), 51% in Chinese (155) and 55.7% in Japanese (156).
1.4.3.4.2 GSTT1 gene polymorphism
GSTT1 gene is 8.1 kb in length and is part of the theta-class GST gene cluster at
22q11.2 (157). GSTT1 and GSTT2 are separated by approximately 50 kb. GSTT2 lies
head-to-head with a gene encoding the D-dopachrome tautomerase (DDCT). The
GSTT1 gene consists of five exons, and is flanked by two 18 kb regions, HA3 and
HA5, which are more than 90% homologous. The GSTT1 null allele (GSTT1-/-)
arises by homologous recombination of the left and right 403bp repeats, which results
in a 54kb deletion of the entire GSTT1 gene (Fig 1.14). The point of deletion has not
been precisely localized because of the sequence identity between the 403bp repeats.
GSTT1 gene is polymorphic in humans and the phenotypic absence of enzyme activity
is due to the deletion of the gene (158). GSTT1 deletion frequently affects both the
alleles and the deletion frequency exhibits ethnic differences. GSTT1 deletion
frequencies vary between 17% and 46% in different population (151, 152, 153, 154,
155, 156).
1.4.3.4.3 GSTP1 gene polymorphism
GSTP1 gene mapped to 11q13 is 2.8 kb long and encodes the GSTP1 protein of 209
amino acids (Fig 1.14). GSTP1 has a polymorphic site in exon 5, where an A to G
transition at nucleotide 313 results in an Ile105Val substitution in the substrate-
binding site of GSTP1 (159, 160, 161). Substitution of the less bulkier and more
hydrophobic valine results in substrate-dependent alterations of GSTP1 activity (160,
162). Although there is an increasing body of evidence suggesting an association
between the Ile105Val polymorphism of GSTP1 and cancer susceptibility, influence
of this polymorphism in RPL remains controversial. C to T transition at nucleotide
341 results in Ala114Val amino acid substitution. However, this polymorphism does
not appear to result in significant alteration in enzymatic function (160).

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy
loss in South Indian Population 33
GSTP1 gene is polymorphic in humans and the frequency of the valine allele exhibits
ethnic differences. The Val allele frequency is 3.1% in South Indians (150), 5.4 in
North Indians (152) and 13.3 in Caucasians (154).
1.4.3.5 Aryl hydrocarbon receptor nuclear translocator gene (ARNT)
polymorphism
ARNT belongs to a family of transcription factors that regulate a wide group of
biological functions including embryonic development and response to low oxygen
tension. ARNT dimerizes with hypoxia inducible factor 1 alpha (HIF1alpha) to
promote angiogenesis and trophoblast invasion in the first trimester of pregnancy. A
to C polymorphism in codon 511 of the ARNT gene, which results in the substitution
of Asn for Asp in the protein, has been implicated in RPL (163).
Allelic variants of GSTs that have impaired detoxification may enhance the rate of
genetic damage and thereby increase the susceptibility to reproductive toxicity, which
could lead to endometriosis, RPL or poor pregnancy outcome (164, 165, 166, 167,
168). Studies on GST gene polymorphisms and RPL have reported contrasting
findings and ethnic differences (166, 169, 170, 171).
The various candidate xenobiotic metabolizing genes, their function and the effect of
their polymorphisms in RPL have been tabulated (Table 1.6)
1.4.4 Genetic polymorphisms in DNA repair genes
In normal pregnancies, the early stages of development take place in a low oxygen
environment. This physiological hypoxia of the early gestational sac protects the
developing
fetus against the deleterious and teratogenic effects of oxygen
free
radicals. Various adaptive changes occur with advancing pregnancy to meet the
increasing demands for proper body functions of the mother to fulfill the requirements
of the fetus. One of the adaptive changes in the respiratory physiology from eight

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy loss in South Indian Population
34
Candidate Genes Gene
loci
Polymorphism Gene function Effect of Polymorphism
CYP1A1
(Suryanarayana et al 2004)(171)
15q24 T to C in 3’ UTR
Oxidation of polycyclic
aromatic hydrocarbons
Increase in enzyme activity
CYP1B1
( Saijo et al 2004)(163)
2p21 432 C/G
Metabolic activation of
benzo[a]pyrene and estrogens 432 G (variant)
enzyme activity
was 3-fold higher than the 432C
CYP2D6
(Suryanarayana et al 2004) (171) 22q13
G1934A
Mixed-function oxidase
system involved in the
metabolism of xenobiotics
Enzyme with poor metabolism
GSTM1, GSTT1
(Zusterzeel et al 2000; (166)
Suryanarayana et al 2004) (171)
1p13, 22q11 Null
Conjugate reduced
glutathione to a number of
elecrophilic substances
including the products of
phase I metabolic
intermediates, thereby
facilitating their elimination
from the body
Loss of enzyme activity
GSTP1
(Zusterzeel et al 2000; (166)
Suryanarayana et al 2004) (171)
11q13 A313G
Ile105Val substitution alters
enzyme activity (101)
Table 1.6: Polymorphisms in xenobiotic metabolism genes reported in RPL

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy loss in South Indian Population
35
Ah receptor
(Saijo et al 2004) (163)
7p15 554C/G
Regulates transcription of the
CYP450 family that
metabolizes many
carcinogens
Functional significance not well
established
NAT2
(Hirvonen et al 1996)(164)
8p22 M1, M2 and M3
M1- C481T and
T342C
M2 -C282T and
G590A
M3- G857A
Responsible for
the
acetylation that determines
whether individuals are slow
or fast acetylators of a number
of drugs and xenobiotics
Altered acetylation activity

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy
loss in South Indian Population 36
week onwards is the increase in ventilation from an initial 36% to a maximum of 50%
or more to meet the increasing demands of oxygen (172). This triggered aerobic
environment should primarily be responsible for raised oxidative stress in pregnancy.
Further, uncontrolled iron supplementation and environmental factors may add to the
oxidative stress. Above all, the feeble antioxidant defense, could lead to an
undesirable level of oxidative stress and oxidative damage of cellular and/or tissue
components. There is mounting evidence that oxidative stress or an imbalance in the
oxidant/antioxidant activity in utero–placental tissues plays a pivotal role in the
development of placental-related diseases (172). In miscarriage, development of the
placento–decidual interface is severely impaired leading to early and widespread
onset
of maternal blood flow and major oxidative degeneration.
This excess oxidative load and the subsequently induced DNA damage have to be
cleared and repaired. Cellular oxidants, such as free radicals and reactive oxygen
species (ROS) are also produced during natural metabolic process. ROS are highly
reactive and potentially damaging to the cells, because they directly affect
macromolecules and organelle functions. Damage to DNA by ROS results in single-
strand and double-strand breaks, apurinic and apyrimidinic sites and adduct
formation. In addition, ROS can catalyze the oxidative modification of proteins,
including enzymes involved in DNA repair (173). If the damage is recognized by the
cell machinery, responses like cell cycle arrest may occur to prevent replication in the
presence of genetic errors (174). Integrity of the so-damaged DNA is typically
restored as a consequence of DNA-repair enzymes, the normal function of which is
important for maintaining genomic integrity (175).
In addition to oxidative stress, transplacental exposure to carcinogenic air pollutants
such as polycyclic aromatic hydrocarbons might cause DNA damage by forming
chemical-DNA adducts. This suggests that DNA repair capacity is essential for the
maintenance of pregnancy, but little is known about the direct effect of DNA repair
capacity on RPL. Complex pathways involving numerous molecules perform DNA
repair. Cells with unrepaired DNA damage either undergo apoptosis or unregulated
growth to malignancy. Integrity of the damaged DNA is typically restored as a
consequence of DNA repair enzymes, the normal function of which is important to
maintain genomic integrity.

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy
loss in South Indian Population 37
1.4.4.1 DNA repair pathways
DNA repair pathways that operate on specific types of DNA damage are: Base
excision repair, Nucleotide excision repair, Mismatch repair, Homologous
recombination and non-homologous end joining.
1.4.4.1.1 Base excision repair
Base excision repair removes simple base modifications, including single-strand
breaks, oxidative DNA damage and non-bulky adducts. The damaged base is removed
by base-specific DNA glycosylases. The abasic site is excised by the endonuclease
action of apurinic/apyrimidinic endonucleases (APEX). DNA synthesis is then
catalyzed by DNA polymerase using the other strand as the template. Following this,
the DNA strands are ligated (176). Molecules involved in the polymerization and
ligation of base excision repair include, polynucleotide kinase, XRCC1 and DNA
ligase (Fig 1.15).
1.4.4.1.2 Nucleotide excision repair
It removes larger lesions, such as pyrimidine dimers, photo-adducts, large chemical
adducts and cross-links which often result from environmental damage, including UV
radiation and external carcinogens. The nucleotide excision repair pathway involves
four steps (Fig 1.16). Damage recognition by a complex of bound proteins including
XPC and XPA; unwinding of the DNA by the TFIIH (transcription Factor II H)
complex that includes XPC, XPA, XPD and XPB; Removal of the damaged single-
stranded fragment (usually about 27–30 bp) by molecules including ERCC1 and XPG
complex; and Polymerization or synthesis of the strand by DNA polymerase and
ligation.
As genetic polymorphisms in DNA-repair enzymes influence DNA adduct levels
(177, 178, 179) DNA repair capacity may be associated with the risk of RPL. The
present study focused on the DNA repair genes, X-ray repair cross complementing
group 1 (XRCC1) and xeroderma pigmentosum group D (XPD). Association of these

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy
loss in South Indian Population 38
polymorphisms in XRCC1 and XPD genes have been shown in various cancers like
lung (180, 181), head and neck (182, 183) and breast (184).
1.4.4.1.3 XRCC1 gene polymorphism
The XRCC1 gene mapped to 19q13.2–13.3, spans 32.25 kb encoding XRCC1 protein
(Fig 1.17). XRCC1 involved in base excision repair, appears to play a scaffolding
role in bringing together a complex of DNA repair proteins, including poly (ADP-
ribose) polymerase (PARP), DNA ligase 3 (LIG3), polynucleotide kinase, AP
endonuclease and DNA polymerase β (185). XRCC1 is believed to form complexes
with DNA polymerase β at its NH2 terminus and with DNA ligase III via the breast
cancer COOH-terminus (BRCT) domain to repair the gaps during base excision
repair.
Two common single nucleotide polymorphisms (SNPs) that have been reported in the
coding region of XRCC1 gene are C to T transition at position 26304 in exon 6 which
leads to an Arg to Trp substitution at codon 194 and G to A transition at position
28152 in exon 10 which leads to an Arg to Gln substitution at codon 399 (Fig 1.17).
These polymorphisms involve an amino acid change at the evolutionarily conserved
regions and could alter the XRCC1 function.
1.4.4.1.4 Xeroderma pigmentosum group D (XPD) gene polymorphism
XPD gene also referred to as ERCC2, mapped to 19q13.3 is comprised of 23 exons,
spans about 54 kbp (Fig 1.17). XPD gene encodes the 86.9 kDa XPD protein which is
a component of the transcription factor TFIIH. XPD possess single strand DNA-
dependent ATPase activity and 5’-3’ helicase activity, thus participating in DNA
unwinding during NER and transcription (186)
Two common SNPs reported in the XPD gene are G to A transition in exon 10, which
results in Asp to Asn substitution at codon 312, and C to A transversion at 35931
nucleotide of exon 23, which results in a Lys to Gln substitution at codon 751.
Mutations in XPD gene can completely prevent DNA unwinding and excision of the

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy
loss in South Indian Population 39
affected strand (187). David Baebes et al 2001 (181) reported that individuals with the
XPD 751 Lys/Lys genotype have a 7-fold increased risk of suboptimal DNA repair.
Qiao et al 2002 (187) observed individuals with 751 Gln/Gln to exhibit sub-optimal
DNA repair to remove UV photoproducts compared to the XPD 751 Lys/Lys
genotypes.
Since the XRCC1 and XPD genes play an important role in DNA repair mechanisms,
the variation in these genes might affect maintenance of pregnancy and thus may be
associated with RPL. Currently, to the best of our knowledge, there are no studies on
DNA repair gene polymorphisms in RPL. The present study is the first of its kind to
analyze the role of polymorphisms in DNA repair genes as a susceptibility marker for
RPL.
1.4.5 Other Genetic polymorphisms in RPL
1.4.5.1 Progesterone Receptor Gene
Progesterone is required for the maintenance of pregnancy and treatment with
progesterone supplementation prevents abortions in some individuals. Three linked
SNPs have been detected in the progesterone receptor gene – exon 1: G 1031 C; Ser
344 Thr, exon 4: G 1978 T; Leu 660 Val, exon 5: C 2310 T; His 770 His (188).
1.4.5.2 Cytochrome P450c17 alpha (CYP 17)
The CYP17 gene encodes the enzyme cytochrome P450c17a, which mediates both
17a-hydroxylase and 17, 20-lyase activity in the steroid biosynthesis pathway. T to C
polymorphism in the 5’ promoter region of CYP17 results in a variant allele (A2).
Women with the CYP17A2 allele have been reported to exhibit increased risk of RPL.
(189).

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy
loss in South Indian Population 40
1.4.5.3 p53
p53, a tumour suppressor gene encodes a multifunctional transcription factor that is
activated by stress stimuli including DNA damage and hypoxia. It is a well-known
factor regulating apoptosis in a wide variety of cells and also plays a critical role in
angiogenesis (190). In addition, p53 has recently been reported as a potential mediator
of pregnancy by estrogen and progesterone activation (191). A common sequence
polymorphism in p53 is located within the proline-rich domain encoding either
proline or arginine at position 72. The Proline allele has been reported to increase the
risk of RPL (192).
From the literature it is evident that polymorphisms in genes regulating
immunomodulation, angiogenesis, vascular network, xenobiotic detoxification and
DNA repair have an impact on the individual susceptibility to RPL. Further, the
contribution of genetic polymorphisms to the risk of RPL is dependent on the
population studied, as well as environmental and dietary factors that influence the
population. Epidemiological studies have shown that, polymorphisms in the genes
regulating initiation and maintenance of pregnancy attribute to the risk of RPL.
However, these study reports are inconsistent. Moreover, studies on association of
genetic polymorphisms in patients with RPL is very limited in the Indian population,
especially in South Indians. Thus, identification of susceptibility markers is essential
to determine the genetic predisposition to RPL.
For a successful pregnancy outcome, the first step is the modulation of the maternal
immune system to tolerate the semi allogenic fetus. HLA G and KIR alleles have been
proposed to play a major role in this process. The next stage is the establishment of
vascular network and proper angiogenesis between the growing fetus and the uterine
wall of the mother. This process is regulated by the VEGF, MTHFR and ACE gene
products. The excess oxidative stress induced during angiogenesis may prove lethal to
the developing embryo. This surplus oxidative load and the subsequent DNA damage
that could be induced have to be cleared and repaired. This function is mediated by
the enzymes encoded by detoxifying genes such as CYP1A1, GSTM1, GSTT1, GSTP1
and the DNA repair genes like XRCC1 and XPD. Hence the present study adopts a

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy
loss in South Indian Population 41
multigeneic approach and hypothesizes that polymorphisms in genes that alter the
normal function of these pathways might result in an increased susceptibility to
RPL.The study proposition is represented in Fig –1.18.

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy
loss in South Indian Population 42
1.5 Aim and Objectives of the Study
The main aim of the study is to determine the association of the immunological and
genetic factors in the risk of unexplained recurrent pregnancy loss in South Indian
population. In order to investigate the role of immunological factors, gene
polymorphisms were analysed in the spouses. Whereas to assess the other proposed
genetic factors that play a role in embryonal development, only the maternal gene
polymorphisms were examined.
The specific objectives are:
1. To examine association between unexplained recurrent pregnancy loss and
immunomodulatory genes, with specific reference to HLA G and the KIR
(i) To investigate the role of HLA G and KIR polymorphisms in women
with unexplained RPL compared with the control women.
(ii) To determine the role of paternal HLA G and KIR polymorphisms in
RPL.
(iii) To determine the association of sharing of the alleles between the
spouses in the outcome of pregnancy with specific reference to the
HLA G and KIR repertoire.
(iv) To explore the role of anti-paternal cytotoxic antibodies (APCA) in
RPL.
2. To determine the risk of RPL attributed by polymorphism in genes regulating
a. Placental vascular network, with specific reference to
(i) Vascular Endothelial Growth Factor (VEGF) gene
(ii) Angiotensin Converting Enzyme (ACE) gene
(iii) Methylene tetra hydrofolate reductase (MTHFR) gene
b. Xenobiotic metabolism, with specific reference to
(i) Cytochrome P4501A1 (CYP1A1) gene

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy
loss in South Indian Population 43
(ii) Glutathione-S transferase µ (GSTM1) gene
(iii) Glutathione-S transferase pi (GSTP1) gene
(iv) Glutathione-S transferase θ (GSTT1) gene
c. DNA repair pathway, with specific reference to
(i) X-ray repair cross-complementary group 1 (XRCC1) gene
(ii) Xeroderma pigmentosum group D (XPD) gene
3. To correlate the genotypes with age and the number of pregnancy loss at
diagnosis.

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy loss in
South Indian population 44
1.6 Reference
1. Straypedersen B, Straypedersen S. Etiologic factors and subsequent reproductive
performance in 195 couples with a prior history of habitual abortion. Am J Obstet
Gynecol 1984;148:140-146.
2. Definition of Recurrent Pregnancy Loss [online] (October 27th, 2004)
(http://www.rbaonline.com/recurrentloss.asp).
3. World Health Organisation. Recommended definitions, terminology and format for
statistical tables related to perinatal period. Acta Obstet Gynecol Scand
1977;56:247-253.
4. Fenster L, Eskenazi B, Windham GC, Swan SH. Caffeine consumption during
pregnancy and spontaneous abortion. Epidemiology 1991;2:168-174.
5. Stirrat GM. Recurrent abortion. II. Clinical associations, causes, and management.
Lancet 1990;336:728-733.
6. Parazzini F, Bocciolone L, Fedele L, Negri E, La Vecchia C, Acaia B. Risk factors
for spontaneous abortion. Int J Epidemiol 1991;20:157-161.
7. Cramer DW and Wise LA. The epidemiology of recurrent pregnancy loss. Semin
Reprod Med 2000;18:331-339.
8. Arjun G. Recurrent early pregnancy loss, An Introduction to Genetics and Fetal
Medicine 2000. Kamini Rao. 27 - 30 .
9. Genetic counseling and antenatal. Diagnosis of common genetic disorders. Report
of an ICMR task force study, Indian Council of Medical Research, New
Delhi.1996.

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy loss in
South Indian population 45
10. Meka AB and Reddy M. Recurrent spontaneous abortions: An Overview of Genetic
1and Non-Genetic Backgrounds. Int J Hum Genet 2006;6:109-117.
11. Thomas M. Cytogenetic basis of recurrent abortions. Perinatology
1999;1:181 - 187.
12. Srinivas N and Rajangam S. Anatomical Causes of Bad Obstetric History. J Anat
Soc India 2001;50:119-121.
13. Dorman JS, Burke JP, McCarthy BJ, Norris JM, Steenkiste AR, Aarons JH.
Temporal trends in spontaneous abortion associated with Type 1 diabetes. Diabetes
Res Clin Pract 1999;43:41-47.
14. Oumachigui A, Bhatia VN, Nayak PN. Toxoplasma antibodies and fetal wastage.
Indian J Med Res 1980;71:522-525.
15. Scholl TO and Johnson WG. Folic acid: influence on the outcome of pregnancy.
Am J of Clinl Nutr 2000;71:1295-1303.
16. Wilson R, Ling H, MacLean MA, Mooney J, Kinnane D, McKillop JH. Thyroid
antibody titer and avidity in patients with recurrent miscarriage. Fertil Steril
1999;71: 558-561.
17. Choudhury SR and Knapp LA. Human reproductive failure II: Immunogenetic and
interacting factors. Hum Reprod Update 2001;7:135-160.
18. Portnoi MF, Joye N, Van den Akker J, Morlier G, Taillemite JL. Karyotypes of
1142 couples with recurrent abortion. Obstet Gynecol 1988;72:31-34.

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy loss in
South Indian population 46
19. Patel ZM, Sanghavi DA, Adhia RA. Chromosomal rearrangements amongst
couples with recurrent abortions in Indian population. J Clin Genet Tri Res 2000;
5:41-49.
20. Dubey S, Chowdhury MR, Prahlad B, Kumar V, Mathur R, Hamilton S, Kabra M,
Menon PSN, Verma IC. Cytogenetic causes for recurrent spontaneous abortions an
experience of 742 couples (1484 cases) Ind J of Hum Genet 2005; 11: 21-24
21. Sangha KK, Stephenson MD, Brown CJ, Robinson WP. Extremely skewed X-
Chromosome inactivation is increased in women with recurrent spontaneous
abortions. Am J Hum Genet 1999;65:913- 917.
22. Uehara S, Hashiyada M, Sato K, Sato Y, Fujimori K, Okamura K. Preferential X
Chromosome Inactivation in women with idiopathic Recurrent Pregnancy Loss.
Fertil Steril 2001;76:908-914.
23. Pegoraro E, Whitaker J, Rushton PM, Surti U, Lanasa M, Hoffman EP. Familial
Skewed X inactivation: a molecular trait associated with high spontaneous abortion
rate maps on Xq28. Am J Hum Genet 1997;61:160 - 170.
24. Andersen AM, Wohlfahrt J, Christens P, Olsen J, Melbye M. Maternal age and fetal
loss: population based register linkage study. BMJ. 2000; 320:1708-1712.
25. Stirrat GM. Recurrent miscarriage. II. Clinical associations, causes, and
management. Lancet 1990;336:728-733.
26. Ellsworth DL, Manolio TA The emerging importance of genetics in epidemiologic
research. II. Issues in study design and gene mapping. Ann Epidemiol 1999;
9:75-90.
27. Parham, P. Immunology: keeping mother at bay. Curr.Biol 1996;6:638-641.

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy loss in
South Indian population 47
28. Marsh SG, Albert ED, Bodmer WF, Bontrop RE, Dupont B, Erlich HA, Geraghty
DE, Hansen JA, Mach B, Mayr WR. Nomenclature for factors of the HLA system
2002. Tissue Antigens 2002;60:407-464.
29. Hviid TV, Christiansen OB, Johansen JK, Hviid UR, Lundegaard C, Møller C,
Morling N. Characterization of a new HLA-G allele encoding a nonconservative
amino acid substitution in the alpha3 domain (exon 4) and its relevance to certain
complications in pregnancy. Immunogenetics 2001;53:48-53.
30. Hunt JS, Petroff MG, McIntire RH, Ober C. HLA G and immune tolerance in
pregnancy. The FASEB Journal 2005;19:144-149
31. Hviid TV. HLA-G genotype is associated with fetoplacental growth. Hum Immunol
2004;65:586-593.
32. Browning M and Mc Michael A (Eds.). HLA and MHC:Genes, Molecules and
Function. Oxford: Bios Scientific Publishers. 1996
33. Van der Ven K, Fimmers R, Engels G, Van der Ven V, Krebs D. Evidence for
major histocompatibility complex-mediated effects on spermatogenesis in humans.
Hum Reprod 2000;15:189-196.
34. Ishitani A and Geraghty DE. Alternative splicing of HLA-G transcripts yields
proteins with primary structures resembling both class I and class II antigens. Proc
Natl Acad Sci USA 1992;89:3947-3951.
35. Messer G, Zemmour J, Orr HT, Parham P, Weiss EH, Girdlestone J. HLA-J, a
second inactivated class I HLA gene related to HLA-G and HLA-A. Implications
for the evolution of the HLA-A-related genes. J Immunol 1992;148:4043-4053.

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy loss in
South Indian population 48
36. Geraghty DE, Koller BH, Orr HT. A human major histocompatibility complex class
I gene that encodes a protein with shortened cytoplasmic segment. Proc Natl Acad
Sci USA 1987;84:9145-9149.
37. Morales PJ, Pace JL, Platt JS, Phillips TA, Morgan K, Fazleabas AT, Hunt JS.
Placental cell expression of HLA-G2 isoforms is limited to the invasive trophoblast
phenotype. J Immunol 2003;171:6215-6224.
38. Fujii T, Ishitani A, Geraghty DE. A soluble form of the HLA-G antigen is encoded
by a messenger ribonucleic acid containing intron 4. J Immunol 1994;153:5516-
5524.
39. Hviid TV, Christiansen OB, Johansen JK, Hviid UR, Lundegaard C, Moller C
Morling N. Characterization of a new HLA-G allele encoding a nonconservative
amino acid substitution in the alpha3 domain (exon 4) and its relevance to certain
complications in pregnancy. Immunogenetics 2001;53:48-53.
40. Choudhury SR and Knapp LA. Human reproductive failure I: Immunological
factors. Hum Reprod Update 2000;7:113-134.
41. Ober C, Aldrich C, Rosinsky B, Robertson A, Walker MA, Willadsen S, Verp MS,
Geraghty DE, Hunt JS. HLA-G1 protein expression is not essential for fetal
survival. Placenta 1998;19:127-132.
42. Hunt JS, Pace JL, Morales PJ, Ober C. Immunogenicity of the soluble isoforms of
HLA-G. Mol. Hum.Reprod 2003;9:729-735.
43. Suarez MB, Morales P, Fernandez JSC, Varela A, Alvarez M, Laso JM, Villena A.
A new HLA-G allele (HLA-G*0105N) and its distribution in the Spanish
population Immunogenetics 1997; 45:464-465.

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy loss in
South Indian population 49
44. Discorde M, Danff CL, Moreau P, Freiss NR, Carosella ED. HLA-G*0105N null
allele encodes functional HLA-G Isoforms. Biol of Reprod 2005;73:280-288.
45. Aldrich CL, Stephenson MD, Karrison T, Odem RR, Branch DW, Scott JR,
Schreiber JR and Ober C. HLA-G genotypes and pregnancy outcome in couples
with unexplained recurrent miscarriage. Mol Hum Reprod 2001;7:1167-1172.
46. Pfeiffer KA, Fimmers R, Engels G, Van der Ven H, van der Ven K. The HLA-G
genotype is potentially associated with idiopathic recurrent spontaneous abortion.
Mol Hum Reprod 2001;7:373-378.
47. Harrison GA, Humphrey KE, Jakobsen IB, Cooper DW. A 14 bp deletion
polymorphism in the HLA-G gene. Hum Mol Genet 1993;2:2200-2204.
48. Hiby SE, King A, Sharkey A, Loke YW. Molecular studies of trophoblast HLA-G:
polymorphism, isoforms, imprinting and expression in preimplantation embryo.
Tissue Antigens 1999;53:1-13.
49. Rousseau P, Le Discorde M, Mouillot G, Marcou C, Carosella ED, Moreau P. The
14 bp deletion-insertion polymorphism in the 3’ UT region of the HLA-G gene
influences HLA-G mRNA stability. Hum Immunol 2003; 64:1005-1010.
50. Hviid TV, Hylenius S, Rørbye C, Nielsen LG. HLA-G allelic variants are
associated with differences in the HLA-G mRNA isoform profile and HLA-G
mRNA levels. Immunogenetics 2003;55:63-79.
51. King A, Loke YW, Chaouat G. NK cells and reproduction. Immunol Today 1997;
18:64-66.

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy loss in
South Indian population 50
52. Clark D, Arck PC, Jalili R, Merali FS, Manuel J, Chaouat G, Underwood J,
Mowbray JF. Psycho-Neuro- Cytokine⁄Endocrine pathways in immunoregulation
during pregnancy. Am J Reprod Immunol 1996; 35:330-337.
53. Yokohama WM: Recognition structures on natural killer cells. Curr Opin Immunol
1993; 5:67-73.
54. Moretta A, Vitale M, Bottino C, Orengo AM, Morelli L, Augugliaro R, Barbarezi
M, Ciccone E, Moretta L. P58 molecules as putative receptors for the major
histocompatibility complex (MHC) class I molecules in human natural killer (NK)
cells. Anti-p58 antibodies reconstitute lysis of the MHC clones displaying different
specificities. J Exp Med 1993;178:597-604.
55. Lanier LL, Phillips JH. Inhibitory MHC class I receptors on NK cells and T cells.
Immunol Today 1996; 17:86-91.
56. Samaridis J, Colonna M. Cloning of novel immunoglobulin superfamily receptors
expressed on human myeloid and lymphoid cells: structural evidence for new
stimulatory and inhibitory pathways. Eur J Immunol 1997; 27:660-665.
57. Hsu KC, Chida S, Geraghty DE, Dupont, B. The killer cell immunoglobulin-like
receptor (KIR) genomic region: gene-order, haplotypes and allelic polymorphism.
Immunol Rev 2002; 190: 40-44.
58. Uhrberg M, Valiante NM, Shum BP, Shilling HG, Lienert-Weidenbach K,
Corliss B, Tyan: Human diversity in killer cell inhibitory receptor genes. Immunity
1997;7:753-763.
59. Lanier LL: NK cell receptors. Annu Rev Immunol 1998;16:359-393

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy loss in
South Indian population 51
60. Lopez-Botet M and Bellon T: Natural killer cell activation and inhibition by
receptors for MHC class I. Curr Opin Immunol 1999; 11:301-307.
61. Suto Y, Ishikawa Y, Kasahara M, Kasai F, Yabe T, Akaza T, Juji T. Gene
arrangement of the killer cell inhibitory receptor family on human chromosome
19q13.4 detected by fiber-FISH. Immunogenetics 1998;48:235-241.
62. Wilson MJ, Torkar M, Haude A, Milne S, Jones T, Sheer D, Beck S, Trowsdale J.
Plasticity in the organization and sequences of human KIR/ILT gene families. Proc
Natl Acad Sci USA 2000;97:4778-4483.
63. Long EO, Colonna M, Lanier LL. Inhibitory MHC class I receptors on NK and T
cells: a standard nomenclature. Immunol Today 1996;17:100-104.
64. Andre P, Biassoni, R, Colonna M, Cosman D, Lanier LL, Long EO, Lopez-Botet
M, Moretta A, Moretta L, Parham P. New nomenclature for MHC receptors. Nat
Immunol 2001;2:661-663.
65. Rajalingam R, Gardiner CM, Canavez F, Vilches C, Parham P. Identification of
seventeen novel KIR variants: fourteen of them from two non-Caucasian donors.
Tissue Antigens 2001;57:22-31.
66. Vilches C, Rajalingam R, Uhrberg M, Gardine, CM, Young NT, Parham P.
KIR2DL5, a novel killer-cell receptor with a D0-D2 configuration of Ig-like
domains. J Immunol 2000;164:5797-5804.
67. Steffens U, Vyas Y, Dupont B, Selvakumar A. Nucleotide and amino acid sequence
alignment for human killer cell inhibitory receptors (KIR). Tissue Antigens
1998;51: 398-413.
68. Dinarello CA. Biology of interleukin-1. FASEB J 1998;2:108 -115.

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy loss in
South Indian population 52
69. Tarlow JK, Blakemore AI, Lennard A, Solari R, Hughes HN, Steinkasserer A.
Polymorphism in human IL-1 receptor antagonist gene intron 2 is caused by
variable numbers of an 86-bp tandem repeat. Hum Genet 1993;91:403- 404.
70. Wegmann TG, Lin H, Guilbert L. Bidirectional cytokine interactions in the
maternal–fetal relationship: is successful pregnancy a Th2 phenomenon? Immunol
Today 1993;14:353-356.
71. Power LL, Popplewell EJ, Holloway JA. Immunoregulatory molecules during
pregnancy and at birth. J Reprod Immunol 2002;56:19-28.
72. Gewaltig J, Mangasser-Stephan K, Gartung C. Association of polymorphisms of
transforming growth factor-beta1 gene with the rate of progression of HCV-induced
liver fibrosis. Clin Chim Acta 2002;316:83-94.
73. Dunning AM, Ellis PD, McBride S. A transforming growth factor-1 signal peptide
variant increases secretion in vitro and is associated with increased incidence of
invasive breast cancer. Cancer Res 2003;63 :2610-2615.
74. Amani D, Zolghadri J, Samsami A. The promoter region (−800, −509)
polymorphisms of transforming growth factor-beta1 (TGFbeta1) gene recurrent
spontaneous abortion. J Reprod Immunol 2004 ;62:159-166.
75. Hill JA, Polgar K, Anderson DJ. T-helper 1-type immunity to trophoblast in women
with recurrent spontaneous abortion. JAMA 1995;273:1933-1936.
76. Hill JA, Choi BC. Maternal immunological aspects of pregnancy success and
failure. J Reprod Fertil 2000;55:91-97.

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy loss in
South Indian population 53
77. Jenkins C, Roberts J, Wilson R, MacLean MA, Shilito J, Walker JJ. Evidence of a
T(H) 1 type response associated with recurrent miscarriage. Fertil Steril
2000;73:1206-1208.
78. Raghupathy R, Makhseed M, Azizieh F, Omu A, Gupta M, Farhat R. Cytokine
production by maternal lymphocytes during normal human pregnancy and in
unexplained recurrent spontaneous abortion. Hum Reprod 2000;15:713-718.
79. Mueller-Eckhardt G, Mallmann P, Neppert J, Lattermann A, Melk A, Heine O,
Pfeiffer R, Zingsem J, Domke N, Mohr-Pennert A. Immunogenetic and serological
investigations in nonpregnant and in pregnant women with a history of recurrent
spontaneous abortions. J.Reprod. Immunol. 1994;27:95-109.
80. Wilson AG, Symons JA, McDowell TL, McDevitt HO, Duff GW. Effects of a
polymorphism in the human tumor necrosis factor alpha promoter on transcriptional
activation. Proc Natl Acad Sci USA 1997;94:3195-3199.
81. Babbage SJ, Arkwright PD, Vince GS, Perrey C, Pravica V, Quenby S, Bates M,
Hutchinson V. Cytokine promoter gene polymorphisms and idiopathic recurrent
pregnancy loss. J Reprod Immunol 2001;51:21-27.
82. Reid JG, Simpson NA,Walker RG, Economidou O, Shillito J, Gooi HC, Duffy
SR,Walker JJ. The carriage of pro-inflammatory cytokine gene polymorphisms in
recurrent pregnancy loss. Am J Reprod Immunol 2001;45:35-40.
83. Takakuwa K, Adachi H, Hataya I, Ishii K, Tamura M, Tanaka K. Molecular genetic
studies of HLA-DRB1 alleles in patients with unexplained recurrent abortion in the
Japanese population. Hum Reprod 2003;18:728-733.

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy loss in
South Indian population 54
84. Christiansen OB , Mohapeloa HP, Steffensen R, Jersild C. HLA-C and -Bw typing
of couples with unexplained recurrent miscarriages. J Reprod Immunol 1997;37:
63-77.
85. Yancopoulos GD, Davis S, Gale NW, Rudge JS, Wiegand SJ, Holash J. Vascular-
specific growth factors and blood vessel formation. Nature 2000;407:242-248.
86. Carmeliet P. Mechanisms of angiogenesis and arteriogenesis. Nat Med 2000;6:389-
395.
87. Ahmed A and Perkins J. Angiogenesis and intrauterine growth restriction.
Baillieres Best Pract Res Clin Obstet Gynaecol. 2000;14:981-998.
88. Ong S, Lash G, Baker PN. Angiogenesis and placental growth in normal and
compromised pregnancies. Baillieres Best Pract Res Clin Obstet Gynaecol
2000;14:969-980.
89. Preston FE, Rosendaal FR, Walker ID. Increased fetal loss in women with heritable
thrombophilia. Lancet 1996;348:913-916.
90. Buchholz T, Lohse P, Rogenhofer N, Kosian E, Pihusch R, Thaler CJ.
Polymorphisms in the ACE and PAI-1 genes are associated with recurrent
spontaneous miscarriages Hum Reprod 2003;18: 2473-2477.
91. Hickey M and Fraser IS. Clinical implications of disturbances of uterine vascular
morphology and function. Baillie Áres Best Pract Res Clin Obstet Gynecol
2000;14: 937-951.
92. Ferrara N. Vascular endothelial growth factor and the regulation of angiogenesis.
Rec Prog Horm Res 2000;55:15-35.

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy loss in
South Indian population 55
93. Lissak A, Sharon A, Fruchter O, Kassel A, Sanderovitz J, Abramovici H.
Polymorphism for mutation of cytosine to thymine at location 677 in the
methylenetetrahydrofolate reductase gene is associated with recurrent early fetal
loss. Am J Obstet Gynecol 1999;181:126-30.
94. Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med
2003;9:669-676.
95. Vuorela P, Carpen O, Tulppala M, Halmesmaki E. VEGF, its receptors and the tie
receptors in recurrent miscarriage. Mol Hum Reprod 2000;6:276-282.
96. Helske S, Vuorela P, Carpen O, Hornig C, Weich H, Halmesmaki E. Expression of
vascular endothelial growth factor receptors 1, 2 and 3 in placentas from normal
and complicated pregnancies. Mol Hum Reprod 2001;7:205-210.
97. Gerber HP, Hillan KJ, Ryan AM, Kowalski J, Keller GA, Rangell L, Wright BD,
Radtke F, Aguet M, Ferrara N. VEGF is required for growth and survival in
neonatal mice. Development 1999;126:1149-1159.
98. Dosiou C and Giudice LC. Natural killer cells in pregnancy and recurrent
pregnancy loss: endocrine and immunologic perspectives. Endocr Rev 2005;26:
44-62.
99. Brogan IJ, Khan N, Isaac K, Hutchinson JA, Pravica V, Hutchinson IV. Novel
polymorphisms in the promoter and 5’UTR regions of the human vascular
endothelial growth factor gene. Hum Immunol 1999; 60:1245-1249.
100. Mohammadi M, Ollier WE, Hutchinson IV. A functional association study of
VEGF gene promoter polymorphisms with VEGF expression by stimulated pbm
cells. Hum Immunol 2003;64:125-128.

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy loss in
South Indian population 56
101. Mattei MG, Hubert C, Alhenc-Gelas F, Roeckel N, Corvol P, Soubrier F.
Angiotensin I converting enzyme gene is on chromosome 17. Cytogenet Cell Genet
1989; 51:1041-1045.
102. Rieder MJ, Taylor SL, Clark AG, Nickerson DA. Sequence variation in the human
angiotensin converting enzyme. Nature Genet 1999; 22:59-62.
103. Malik FS, Lavie CJ, Mehra MR, Milani RV, Re RN. Renin–angiotensin system:
Genes to bedside. Am. Heart J 1997;134:514-527.
104. Otani A, Takagi H, Suzuma K, Yoshihito R, Angiotensin II potentiates vascular
endothelial growth factor-induced angio-genic activity in retinal microcapillary
endothelial cells. Circ Res 1998; 82:619-628.
105. Touyz RM and Berry C. Recent advances in angiotensin II signalling. Braz J Med
Biol Res 2002; 35:1001-1015.
106. Ribichini F. Plasma activity and insertion/deletion poly-morphism of angiotensin I-
converting enzyme. A major risk factor and a marker of risk for coronary
stentrestenosis. Circulation 1998; 97:147-154.
107. Davis GK and Roberts DH. Molecular genetics of the renin–angiotensin system:
Implications for angiotensin II receptor blockage. Pharmacol Ther1997 ;75:43-50.
108. Gayagay G, Yu B, Hambly B, Boston T, Hahn A, Celermajer DS, Trent RJ. Elite
endurance athletes and the ACE I allele–the role of genes in athletic performance.
Hum Genet 1998;103:48-50.
109. Montgomery HE, Marshall R, Hemingway H, Myerson S, Clarkson P, Dollery C,
Hayward M, Holliman DE, Jubb M, World M, Thomas EL, Brynes AE, Saeed N,

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy loss in
South Indian population 57
Barnard M, Bell JD, Prasad K, Rayson M, Talmud PJ, Humphries SE. Human gene
for physical performance. Nature 1998;393:221-222.
110. Girling J and de Swiet M. Inherited thrombophilia and pregnancy. Curr Opin Obstet
Gynecol.1998;10:135-144.
111. Qadar Pasha MA, Khan AP, Kumar R , Grover SK, Ram RB, Norboo T, Srivastava
KK, Selvamurthy W, Brahmachari SK. Angiotensin converting enzyme insertion
allele in relation to high altitude adaptation. Annals Hum Genet 2001;65: 531-536.
112. Tiret L, Rigat B, Visvikis S. Evidence, from combined segregation and linkage
analysis, that a variant of the angiotensin converting enzyme (ACE) gene controls
plasma ACE levels. Am J Hum Genet1992; 51:197-205.
113. Fatini C, Gensini F, Battaglini B. Angiotensin-converting enzyme DD genotype,
angiotensin type 1 receptor CC genotype, and hyperhomocysteinemia increased
first-trimester fetal-loss susceptibility. Blood Coagul Fibrinolysis 2000;11:657-662.
114. Nelen WL, Bulten J, Steegers EA, Blom HJ, Hanselaar AG, Eskes TK. Maternal
homocysteine and chorionic vascularization in recurrent early pregnancy loss. Hum
Reprod 2000;15:954-960.
115. Ray JG, Laskin CA. Folic acid and homocyst(e)ine meta-bolic defects and the risk
of placental abruption, pre-eclampsia and pregnancy loss: A systematic review.
Placenta 1999;20:519-529.
116. Steegers-Theunissen RP, Boers GH, Blom HJ, Trijbels FJ, Eskes TK.
Hyperhomocysteinaemia and recurrent spontaneous abortion or abruptio placentae.
Lancet 1992;339:1122-1123.

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy loss in
South Indian population 58
117. Kang SS, Zhou J, Wong PW, Kowalisyn J, Strokosch G.Intermediate
homocysteinemia: A thermolabile variant of methylenetetrahydrofolate reductase.
Am J Hum Genet 1988;43:414-421.
118. Engbersen AM, Franken DG, Boers GH, Stevens EM, Trijbels FJ, Blom HJ.
Thermolabile 5,10-methylenetetra-hydrofolate reductase as a cause of mild
hyperhomocys-teinemia. Am J Hum Genet 1995;56:142-150.
119. Bertina RM, Koeleman BPC, Koster T, Rosendal FR, Driven RJ, deronde H,
Vandervelden PA, Reitsma PH. Mutation in blood coagulation factor V associated
with resistance to activated protein C. Nature 1994;369:64-67.
120. Da¨hlback B, Carlsson M, Svensson PJ. Familial thrombophilia due to a previously
unrecognized mechanism characterized by poor anticoagulant response to active
protein C: prediction of a cofactor to activated protein C. Proc Natl Acad Sci USA
1993; 90:1004-1008.
121. Brenner B, Vulfsons SL, Lanir N, Nahir M. Coexistence of familial
antiphospholipid syndrome and factor V Leiden-impact on thrombotic diathesis. Br
J Haematol 1996;94:166-169.
122. Souza SS, Ferriani RA, Pontes AG, Zago MA, Franco RF. Factor V Leiden and
factor II G20210A mutations in patients with recurrent abortion. Hum Reprod
1999;14:2448-2450.
123. Buchholz T, Lohse P, Rogenhofer N, Kosian E, Pihusch R, Thaler CJ.
Polymorphisms in the ACE and PAI-1 genes are associated with recurrent
spontaneous miscarriages Hum Reprod 2003;18:2473-2477.

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy loss in
South Indian population 59
124. Poort SR, Rosendaal FR, Reitsma PH, Bertina RM. A common genetic variation in
the 3’untranslated region of the prothrombin gene is associated with elevated
plasma prothrombin levels and an increase in venous thrombosis. Blood
1996;88:3689-3703.
125. Foka ZJ, Lambropoulos AF, Saravelos H, Karas GB, Karavida A, Agorastos T.
Factor V Leiden and prothrombin G20210A mutations, but not
methylenetetrahydrofolate reductase C677T, are associated with recurrent
miscarriage. Hum Reprod 2000;15:458-462.
126. Kutteh WH, Park VM, Deitcher SR. Hypercoagulable state mutation analysis in
with patients with early first-trimester recurrent pregnancy loss. Fertil Steril
1999;71:1048-1053.
127. Papazoglou D, Galazios G, Koukourakis MI, Panagopoulos I, Kontomanolis EN,
Papatheodorou K, Maltezos E. Vascular endothelial growth factor gene
polymorphisms and preeclampsia. Mol Hum Reprod 2004;10:321-324.
128. Holmes ZR, Regan L, Chilcott I, Cohen H. The C677T MTHFR gene mutation is
not predictive of risk for recurrent fetal loss. Br J Haematol 1999;105:98-101.
129. Unfried G, Griesmacher A, Weismüller W, Nagele F, Huber JC, Tempfer CB. The
C677T Polymorphism of the Methylenetetrahydrofolate Reductase Gene and
Idiopathic Recurrent Miscarriage Obstetrics & Gynecology 2002;99:614-619.
130. Townson DDS, Kinney S, Branch DW, Ward K. The factor V Leiden mutation is
not a common cause of recurrent miscarriage. J Reprod Immunol 1997;34: 217-223.
131. Hohlagschwandtner M, Unfried G, Heinze G, Huber JC, Nagele F, Tempfer C.
Combined thrombophilic polymorphisms in women with idiopathic recurrent
miscarriage. Fertil Steril. 2003;79:1141-1148.

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy loss in
South Indian population 60
132. Gardella JR and Hill JA. Environmental toxins associated with recurrent pregnancy
loss. Semin Reprod Med 2000;18:407-424.
133. Klebanoff MA, Levine RJ, DerSimonian R, Clemens JD, Wilkins DG. Maternal
serum paraxanthine, a caffeine metabolite, and the risk of spontaneous abortion. N
Engl J Med 1999;341:1639-1644.
134. Bjerke D, Sommer R, Moore R, Paterson R. Effects of in utero and lactational
2,3,7,8-tetrachlorodibenzo-p-dioxin exposures on responsiveness of the male rat
reproductive system to testosterone stimulation in adulthood. Toxicol Appl
Pharmacol 1994;127:250-257.
135. Eskenazi B, Gold E, Lasley B, Samuels S, Hammonds S, Wight S. Prospective
monitoring of early fetal loss and clinical spontaneous abortion among female
semiconductor workers. Am J Ind Med 1995;28:833-846.
136. Genbacev O, Zhou Y, John W, Ludlow Y, Fisher SJ. Regulation of human placental
development by oxygen tension. Science 1997;277:1669-1672.
137. Jauniaux E, Watson AL, Hempstock J, Bao YP, Skepper JN, Burton GJ. Onset of
maternal arterial blood flow and placental oxidative stress a possible factor in
human early pregnancy failure. Am J path 2000;157:2111-2122.
138. Nebert DW, McKinnon RA, Puga A. Human drug-metabolizing enzyme
polymorphisms: Effects on risk of toxicity and cancer. DNA Cell Biol 1996; 15:
273-280.

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy loss in
South Indian population 61
139. Hayes JD, Pulford DJ. The glutathione S-transferase supergene family: regulation
of GST and the contribution of the isoenzymes to cancer chemoprotection and drug
resistance. Crit Rev Biochem Mol Biol 1995;30:445-600.
140. Hasler JA, Estabrook R, Murray M. Human cytochromes P450. Mol Aspects Med
1999;20:31-37.
141. Li W, Harper PA, Tang BK and Okey AB. Regulation of cytochrome P450
enzymes by aryl hydrocarbon receptor in human cells: CYP1A2 expression in the
LS180 colon carcinoma cell line after treatment with 2,3,7,8-tetrachlorodibenzo-p-
dioxin or 3-methylcholanthrene. Biochem Pharmacol 1998;56:599-612.
142. Larsen MC, Angus WG, Brake PB, Eltom SE, Sukow KA and Jefcoate CR.
Characterization of CYP1B1 and CYP1A1 expression in human mammary
epithelial cells: role of the aryl hydrocarbon receptor in polycyclic aromatic
hydrocarbon metabolism. Cancer Res 1998;58:2366-2374.
143. Williams JA and Phillips DH. Mammary expression of xenobiotic metabolizing
enzymes and their potential role in breast cancer. Cancer Res 2000;60:4667-4677.
144. Crofts F, Taioli E, Trachman J, Currie D, Toniolo P, Garte SJ. Functional
significance of different human CYP1A1 genotypes. Carcinogenesis 1994;15:
2961-2963.
145. Hoffman EC, Reyes H, Chu FF, Sander F, Conley LH, Brooks BA. Cloning of a
factor required for activity of Ah Dioxin receptor. Science 1991;252:954-958.
146. Knapen MFC, Zusterzeel PL, Peters WH. Glutathione and glutathione S transferase
enzyme in reproduction. A review. Eur J Obstet Gynecol Repro Biol 1999;82:
171-184.

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy loss in
South Indian population 62
147. Zusterzeel PL, Peters WH, de Buryn MAH, Knapen MFCM, Merkus HMWM,
Steegers EA. Glutathione S Transferase isoenzymes in deciduas and placenta in
preeclamptic pregnancies. Obstet Gynecol 1999;94:1033-1038.
148. Van Lieshout EMM, Roelofs HMJ, Dekker S. Polymorphic expression of the
glutathione S-transferase P1 gene and its susceptibity to Barrett's esophagus and
esophageal carcinoma. Cancer Res 1999;59:586-589.
149. Xu SJ, Wang YP, Roe B, Pearson WR. Characterization of the human class
glutathione S-transferase gene cluster and the GSTM1 deletion. J Biol Chem
1998;273: 3517-3527.
150. Vettriselvi V, Vijayalakshmi K, Solomon FD Paul, Venkatachalam P. Genetic
Variation of GSTM1, GSTT1 and GSTP1 Genes in a South Indian Population.
Asian Pacific J Cancer Prev 2006;7:325-328.
151. Millikan R, Pittman G, Tse CK. Glutathione S – transferases M1, T1 and P1 and
breast cancer. Cancer Epidemiol Biomarkers Prev 2000;9:567-573.
152. Mishra DK, Anant Kumar, Srivastava DSL, Mittal RD. Allelic Variation of GSTT1,
GSTM1 and GSTP1 Genes in North Indian Population. Asian Pac J Cancer Prev
2004;5:362-365.
153. Dalo F, Voso MT, Guidi F. Polymorphisms of CYP1A1 and glutathione S-
transferase and susceptibility to adult acute myeloid leukemia. Haematologica
2004;89: 664-670.
154. Gsur A, Haidinger G, Hinteregger S. Polymorphisms of glutathione-S-transferase
genes (GSTP1, GSTM1 and GSTT1) and prostate-cancer risk. Int J Cancer
2001;95:152-155.

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy loss in
South Indian population 63
155. Sctiawan VW, Zhang ZF, Yu GP. GSTT1 and GSTM1 null genotypes and risk of
gastric cancer: A case control study in a Chinese population. Cancer Epidemiol
Biomarkers Prev 2000;9:73-80.
156. Kiyohara C, Yamamura K, Nakanishi Y. Polymorphism in GSTM1, GSTT1 and
GSTP1 and susceptibility to lung cancer in a Japanese population. Asian Pac J
Cancer Prev 2000;1:293-298.
157. Webb G, Vaska V, Coogan M, Board P. Chromosomal localization of the gene for
the human theta class glutathione transferase (GSTT1). Genomics 1996;33:121-
123.
158. Seidegard J, Vorachek WR, Pero RW, Pearson WR. Hereditary differences in the
expression of the human glutathione transferase active on trans-stilbene oxide are
due to a gene deletion. Proc Natl Acad Sci USA 1988;85:7293-7297.
159. Zimniak P, Nanduri B, Pikula S, Bandorowicz-Pikula J, Singhal SS, Srivastava SK.
Naturally occurring human glutathione S-transferase GSTP1–1 isoforms with
isoleucine and valine in position 104 differ in enzymatic properties. Eur J Biochem
1994; 224:893-899.
160. Ali-Osman F, Akande O, Antoun G, Mao Buolamwn JX. Molecular cloning,
characterization, and expression in Escherichia coli of full-length cDNAs of three
human glutathione S-transferase Pi gene variants. Evidence for differential catalytic
activity of the encoded proteins. J Biol Chem 1997;272:10004-10012
161. Henderson CJ, McLaren AW, Moffat GJ, Bacon EJ, Wolf CR. π-class glutathione S-
transferase: regulation and function. Chem Biol Interact 1998;111:69-82.
162. Sundberg K, Johansson AS, Stenberg G, Widersten M, Seidel A, Mannervik B.
Differences in the catalytic efficiencies of allelic variants of glutathione transferase

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy loss in
South Indian population 64
P1-1 towards carcinogenic diol epoxides of polycyclic aromatic hydrocarbons.
Carcinogenesis 1998;19:433-436
163. Saijo Y, Sata F, Yamada H, Suzuki K, Sasaki S, Kondo T. Ah receptor, CYP1A1,
CYP1A2 and CYP1B1 gene polymorphisms are not involved in the risk of
recurrent pregnancy loss. Mol Hum Reprod 2004;10:729-733.
164. Hirvonen A, Taylor JA, Wilcox A, Berkowitz G, Schachter B, Chaparro C, Bell
DA. Xenobiotic metabolism genes and the risk of recurrent spontaneous abortion.
Epidemiology 1996;7:206-208.
165. Baranova H, Canis M, Ivaschenko T, Albuisson E, Bothorishvilli R, Baranov V,
Malet P, Bruhat MA. Possible involvement of arylamine N acetyltransferase 2,
glutathione S-transferases M1 and T1 genes in the development of endometriosis.
Mol. Hum. Reprod 1999;5:636- 641.
166. Zusterzeel PLM, Nelen WLDM, Roelofs HMJ, Peters WHM, Blom HJ, Steegers
EAP. Polymorphisms in biotransformation enzymes and the risk for recurrent early
pregnancy loss Mol Hum Reprod 2000;6:474-478.
167. Hadfield RM, Manek S, Weeks DE. Linkage and association studies of the
relationship between endometriosis and genes encoding the detoxification enzymes
GSTM1, GSTT1 and CYP1A1. Mol Hum Reprod 2001;7:1073-1078.
168. Wang X, Wang M, Niu T, Chen C, Xu X. Microsomal epoxide hydrolase
polymorphism and risk of spontaneous abortion. Epidemiology 1998;9:540-544.
169. Mendola P, Moysich KB, Freudenheim JL, Shields PG, Schisterman EF, Graham S,
Vena JE, Marshall JR, Ambrosone CB. Risk of recurrent spontaneous abortion,
cigarette smoking, and genetic polymorphisms in NAT2 and GSTM1.
Epidemiology 1998;9:666-668.

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy loss in
South Indian population 65
170. Sata F, Yamada H, Suzuki K, Saijo Y, Kato EH, Morikawa M, Minakami H, Kishi
R. Caffeine intake, CYP1A2 polymorphism and the risk of recurrent pregnancy loss
Mol Hum Reprod 2005; 11:357-360.
171. Suryanarayana V, Deenadayal M, Singh L. Association of CYP1A1 gene
polymorphism with recurrent pregnancy loss in the South Indian population. Hum
Reprod 2004;19:2648-2652.
172. Jauniaux E, Watson AL and Burton GJ. Evaluation of respiratory gases and acid-
base gradients in fetal fluids and uteroplacental tissue between 7 and 16 weeks. Am
J Obstet Gynecol 2001;184:998-1003.
173. Stadtman ER. Protein oxidation and aging. Science 1992;257:1220-1224.
174. Vispe S, Cazaux C, Lesca C, Defais M. Overexpression of Rad51 protein stimulates
homologous recombination and increases resistance of mammalian cells to ionizing
radiation. Nucleic Acids Res.1998;26:2859-2864.
175. Sohal RS and Weindruch R. Oxidative stress, caloric restriction, and aging. Science
1996;273:59-63.
176. Lu AL, Li X, Gu Y, Wright PM., Chang DY. Repair of oxidative DNA damage:
mechanisms and functions. Cell Biochem Biophys 2001;35:141-70.
177. Lunn RM, Langlois RG, Hsieh LL, Thompson CL, Bell DA. XRCC1
polymorphisms: effects on aflatoxin B1-DNA adducts and glycophorin A variant
frequency. Cancer Res 1999;59:2557–2561.

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy loss in
South Indian population 66
178. Matullo G, Guarrera S, Carturan S, Peluso M., Malaveille C, Davico L. DNA repair
gene polymorphisms, bulky DNA adducts in white blood cells and bladder cancer
in a case-control study. Int J Cancer 2001;92:562-567.
179. Hu JJ, Smith TR., Miller M., Lohman K, Case LD. Genetic regulation of ionizing
radiation sensitivity and breast cancer risk. Environ Mol Mutagen 2002;39:208-215.
180. Chen S, Tang D, Xue, Xu L, Ma G, Hsu Y. DNA repair gene XRCC1 and XPD
polymorphisms and risk of lung cancer in a Chinese population. Carcinogenesis
2002; 23:1321-1325.
181. David-Beabes GL, Lunn RM, London SJ. No association between the XPD
(Lys751G1n) polymorphism or the XRCC3 (Thr241Met) polymorphism and lung
cancer risk. Cancer Epidemiol Biomark Prev 2001;10:911-912.
182. Sturgis EM, Castillo EJ, Li L, Zheng R, Eicher SA, Clayman GL. Polymorphisms
of DNA repair gene XRCC1 in squamous cell carcinoma of the head and neck.
Carcinogenesis 1999;20:2125-2129.
183. Sturgis EM, Zheng R, Li L, Castillo EJ, Eicher SA, Chen M. XPD/ERCC2
polymorphisms and risk of head and neck cancer: a case-control analysis.
Carcinogenesis 2000;21:2219-2223.
184. Duell EJ, Millikan RC, Pittman GS, Winkel S, Lunn RM., Tse CK. Polymorphisms
in the DNA repair gene XRCC1 and breast cancer. Cancer Epidemiol Biomark Prev
2001;10:217-222.
185. Dianov GL, Sleeth KM, Dianova II, Allinson SL. Repair of abasic sites in DNA.
Mutat Res 2003;531:157-163.

Introduction
A study on genetic polymorphisms associated with unexplained recurrent pregnancy loss in
South Indian population 67
186. Evans E, Moggs JG, Hwang JR, Egly JM, Wood RD. Mechanism of open complex
and dual incision formation by human nucleotide excision repair factors. EMBO J
1997;16:6559-6573.
187. Qiao Y, Spitz MR, Shen H, Guo Z, Shete S, Hedayati M, Grossman L. Modulation
of repair of ultraviolet damage in the host-cell reactivation assay by polymorphic
XPC and XPD/ERCC2 genotypes. Carcinogenesis 2002;23:295-299.
188. Schweikert A, Rau T, Berkholz A. Association of progesterone receptor
polymorphism with recurrent abortions. Eur J Obstet Gynecol Reprod Biol
2004;113:67-72.
189. Sata F, Yamada H, Yamada A, Kato EH, Kataoka S, Saijo Y, Kondo T, Tamaki J,
Minakami H, Kishi R. A polymorphism in the CYP17 gene relates to the risk of
recurrent pregnancy loss. Molecular Human Reproduction 2003;9:725-728.
190. Yuan A, Yu CJ, Luh KT. Aberrant p53 expression correlates with expression of
vascular endothelial growth factor mRNA and interleukin-8 mRNA and
neoangiogenesis in non-small-cell lung cancer. J Clin Oncol 2002;20:900-910.
191. Sivaraman L, Conneely OM, Medina D. p53 is a potential mediator of pregnancy
and hormone-induced resistance to mammary carcinogenesis. Proc Natl Acad Sci
USA 2001;98:12379-12384.
192. Pietrowski D, Bettendorf H, Riener EK, Keck C, Hefler LA, Johannes CH, Tempfer
C. Recurrent pregnancy failure is associated with a polymorphism in the p53
tumour suppressor gene. Hum Reprod 2005;20:848-851.






![Lake District Canine Club [4572] - Dogz Online](https://cdn.vdocument.in/doc/165x107/62d2362b10021d5e6630c60d/lake-district-canine-club-4572-dogz-online.jpg)
